

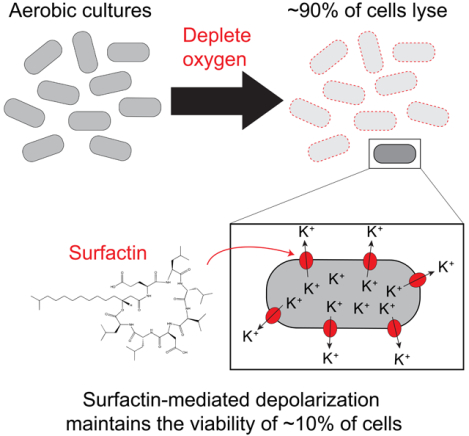
Summary
The presence or absence of oxygen in the environment is a strong effector of cellular metabolism and physiology. Like many eukaryotes and some bacteria, Bacillus subtilis primarily utilizes oxygen during respiration to generate ATP. Despite the importance of oxygen for B. subtilis survival, we know little about how populations adapt to shifts in oxygen availability. Here, we find that when oxygen was depleted from stationary phase B. subtilis cultures, ~90% of cells died while the remaining cells maintained colony-forming ability. We discover that production of the antimicrobial surfactin confers two oxygen-related fitness benefits: it increases aerobic growth yield due to increased oxygen diffusion, and it maintains viability during oxygen depletion by depolarizing the membrane. Strains unable to produce surfactin exhibited a ~50-fold reduction in viability after oxygen depletion. Surfactin treatment of these cells led to membrane depolarization and reduced ATP production. Chemical and genetic perturbations that alter oxygen consumption or redox state support a model in which surfactin-mediated membrane depolarization maintains viability through slower oxygen consumption and/or a shift to a more reduced metabolic profile. These findings highlight the importance of membrane potential in regulating cell physiology and growth, and demonstrate that antimicrobials that depolarize cell membranes can benefit cells when the terminal electron acceptor in respiration is limiting. This foundational knowledge has deep implications for environmental microbiology, clinical anti-bacterial therapy, and industrial biotechnology.
Keywords: surfactin, biosurfactant, oxygen depletion, hypoxia, membrane depolarization, membrane potential, cell lysis, aerobic respiration, oxygen diffusion
Graphical Abstract
eTOC blurb
Cells possess many mechanisms to cope with oxygen deprivation. Arjes et al. show that although oxygen depletion kills the majority of Bacillus subtilis bacteria, a fraction remains viable due to the surfactant surfactin, which mediates survival by depolarizing the membrane. Surfactin also increases oxygen diffusion to promote growth in low oxygen.
Introduction
Many species across all domains of life use oxygen as the terminal electron acceptor during aerobic respiration, which produces ~10-fold more ATP per glucose molecule than glycolysis and fermentation in the absence of respiration. In most human cells, oxygen depletion causes exhaustion of ATP and eventual death, through lysis caused by osmotic stress due to the inability to regulate osmolyte levels [1, 2] or through activation of apoptotic signaling cascades [3]. Thus, maintaining concentration gradients of ions across the membrane is of paramount importance when oxygen is lacking. Like human cells, certain microbes such as the pathogen Mycobacterium tuberculosis and most fungi (with the exception of yeasts) [4] are considered strict aerobes due to their inability to make ATP in the absence of oxygen. After rapidly depleting oxygen, M. tuberculosis cells show a loss in viability [5, 6]. The related soil-dwelling species Mycobacterium smegmatis also loses viability upon oxygen depletion. The remaining viable cells use hydrogen fermentation to maintain the redox balance and activate stress-response genes critical for survival [7, 8]. Despite the clinical importance of M. tuberculosis, few studies have interrogated how strict aerobes respond to oxygen depletion and the mechanisms of survival.
The Gram-positive model bacterium Bacillus subtilis has traditionally been considered a strict aerobe. B. subtilis is naturally found in soil and is used as an additive to prevent infections and promote growth in plants [9, 10]. In soil, B. subtilis undergoes constant shifts in oxygen concentration, as oxygen is present in dry soils but becomes depleted in wet soils following a rain [11]. Early observations of B. subtilis culture lysis upon a shift to anoxic environments have yet to be further characterized [12], and it remains a mystery whether B. subtilis has strategies to cope with oxygen limitation.
B. subtilis has long been domesticated in the lab, leading to a multitude of genetic tools and resources [13–15]. The high level of genetic relatedness between biofilm-forming “wild” strains and derivative non-biofilm-forming laboratory strains has been exploited to identify genetic differences that underlie biofilm community behaviors [16]. The commonly studied laboratory strain 168, which is derived from the biofilm-forming strain 3610, lacks an extrachromosomal plasmid and harbors several point mutations that reduce or abolish social behaviors such as matrix production [17]. Notably, the biofilm strain 3610 produces the small molecule surfactin, a powerful surfactant that has been implicated in permitting cell motility over surfaces [18–23]. Surfactin also has anti-fungal [24] and antibacterial [24–28] properties in vitro. However, fitness benefits of surfactin production in the context of planktonic cultures have yet to be identified.
Here, we characterize the interplay between oxygen availability and surfactin in B. subtilis. We show that oxygen becomes limiting during the transition to stationary phase and that surfactin production improves growth yield in stationary phase by increasing oxygen availability. During a shift to anoxic conditions, we demonstrate that the majority of cells die and lyse due to activity of the LytC autolysin and surfactin. Finally, we discover that surfactin maintains the viability of the remaining cells by causing membrane depolarization, which slows oxygen consumption and allows the cells to transition into a state in which they can survive until oxygen is restored.
Results
Oxygen depletion leads to rapid death and lysis of most B. subtilis cells
When culturing B. subtilis strain 3610 cells in LB, we noticed that when a late-exponential culture was shifted from a shaking incubator to a stationary rack on the bench, the opacity of the tube decreased continuously over a period of 10 hours, suggesting that cells were dying and lysing (Figure 1A). Consistent with lysis, microscopic observation of cultures left on the bench for 10 hours revealed substantial phase-gray cell remnants in addition to phase-dark rod-shaped cells (Figure 1A). We conclude that B. subtilis strain 3610 cultures die and lyse during static incubation after cessation of rapid growth.
Figure 1: B. subtilis strain 3610 lyses due to oxygen depletion.
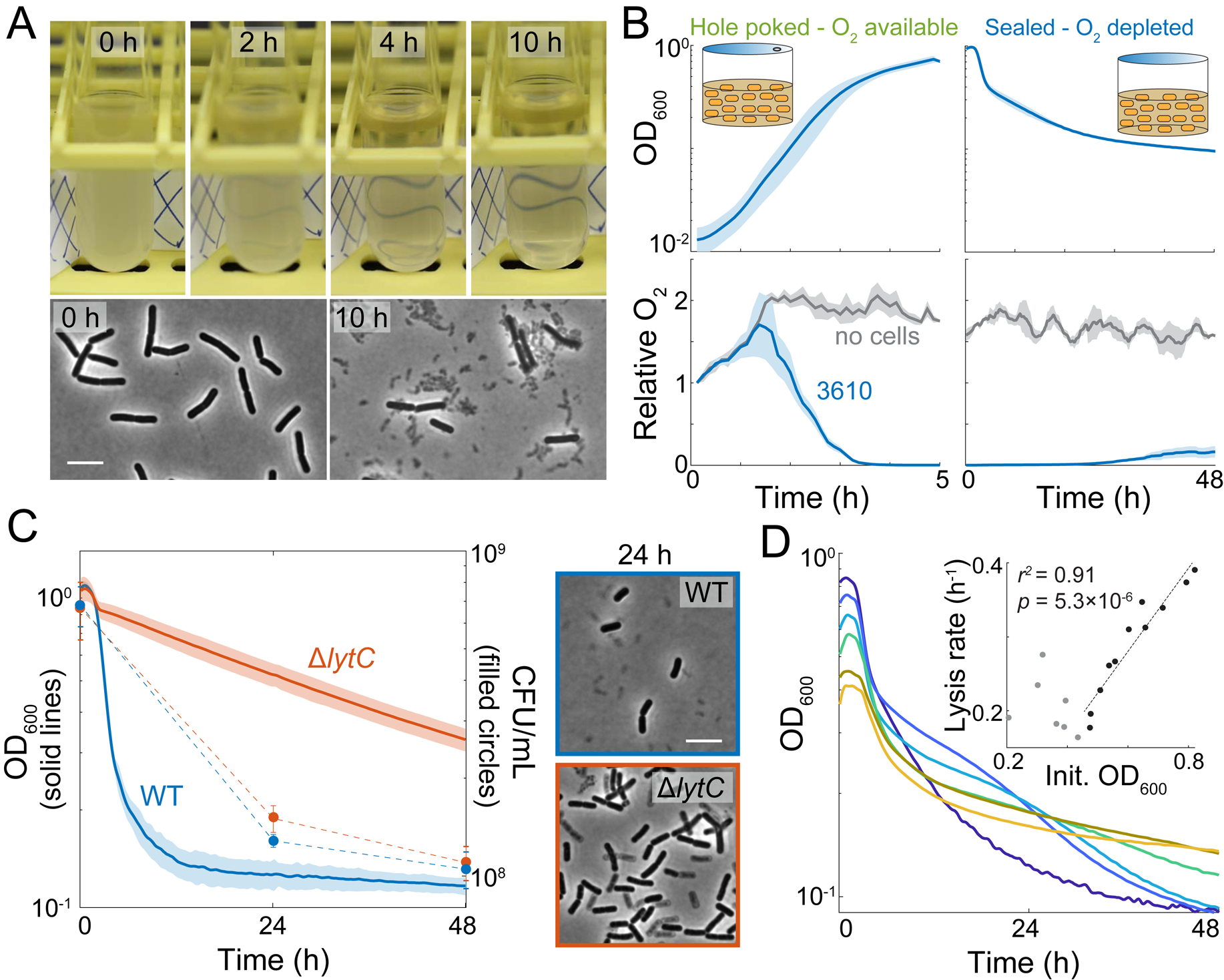
(A) B. subtilis cultures lyse when not shaking. The biofilm-forming wild-type strain 3610 (WT) was grown in a test tube and then incubated at room temperature without shaking for 10 h. Phase-contrast images were acquired at 0 and 10 h. Scale bar: 5 μm; arrowhead points to cell debris.
(B) Oxygen is depleted during exponential growth and remains low throughout subsequent oxygen depletion while cultures are sealed. Cells were cultured with oxygen-sensitive nanoparticles (STAR Methods). OD600 (top) and the relative oxygen levels (bottom, oxygen level at t0growth set to 1) of the cultures were measured. Lines represent the mean and shading represents one standard deviation (SD), n=3.
(C) LytC is necessary for lysis. Left: Despite differences in OD, the ΔlytC 3610 mutant has similar viability to wild type (p=0.07 at 24 h and 0.67 at 48 h, Student’s t-test). Cultures were grown to OD600~1 and then oxygen was depleted at t=0 h and OD600 was monitored. Lines represent the mean and shading represents 1 SD, n=3. Right: ΔlytC cells have phase gray “ghosts” at 24 h post-oxygen depletion. Scale bar: 5 μm; arrowhead points to phase-gray, lysed cell.
(D) Culture lysis is strongly correlated with initial cell density when initial OD600>0.45. Representative lysis curves of 6 cultures that varied in initial OD600 (see Figure S1 for independent replicates). Inset: maximum lysis rate (absolute value of the minimum slope of ln(OD600)) vs. initial OD600. A linear regression analysis was performed on data with initial OD600>0.45.
B. subtilis is considered a strict aerobe that relies on use of oxygen as a terminal electron acceptor during respiration for growth. Despite reports that B. subtilis can grow using nitrate as a terminal electron acceptor [29–31], we were unable to grow either the B. subtilis lab strain 168 or the biofilm-forming strain 3610 in an anaerobic chamber in pre-reduced media even in the presence of nitrate or nitrite (Figure S1). Thus, we infer that previous demonstrations of anaerobic growth on nitrate were likely performed in microaerobic rather than anaerobic conditions [29, 30].
Static cultures would be limited in oxygen diffusion, hence we hypothesized that the death in the culture was due to oxygen limitation. To measure oxygen levels during oxygen growth and depletion, we added phosphorescent oxygen-sensitive nanoparticles (STAR Methods) to cultures in microtiter plates and measured optical density (OD) and light emission over time. To establish a controlled environment, we sealed 96-well microtiter plates and either poked holes above each well to allow oxygen exchange or limited oxygen by leaving wells completely sealed (Figure 1B, top). In wells containing only media, oxygen initially diffused into the medium due to shaking, and then remained at approximately constant levels (Figure 1B, bottom). In cell-containing wells that allow for oxygen exchange, oxygen levels initially increased (due to shaking) but began to decrease when the culture reached OD600~0.05, indicating that cells were consuming the oxygen faster than it dissolved into the medium (Figure 1B). When cultures reached OD600~0.5, oxygen levels became undetectable but the culture continued to grow (Figure 1B), presumably because the cells rapidly consumed any oxygen that dissolved into the medium.
When strain 3610 cultures reached OD600~0.8, we sealed the wells to abolish oxygen exchange in the headspace, and returned the cultures to a shaking environment. As expected, oxygen levels remained low (Figure 1B, lower right). Within 2 hours, OD600 began to decrease, reminiscent of our observations of OD loss in standing test tubes (Figure 1A), and plateaued at ~0.1 after 48 h (Figure 1B, upper right). Upon sealing the wells, cell viability (defined here as the ability of a cell to form a colony on an agar plate) dropped approximately in concert with OD600, decreasing by ~10-fold after 48 h (Figure 1C, S2A). These observations suggest that the loss of OD in standing tubes and in agitated sealed plates was due to cell lysis during oxygen depletion.
To further explore the correlation between lysis and oxygen depletion, we investigated the effect of cell density on hypoxia-induced lysis by growing cultures aerobically to different ODs prior to sealing the wells. Regardless of starting culture density (OD600=0.4–0.8), OD600 either stayed approximately constant or increased slightly for ~1 hour after sealing, and then exhibited large decreases over time (Figure 1D, S2B). The maximum lysis rate increased with increasing starting OD, such that cultures that began at the highest density (OD600~0.8) had a maximum lysis rate of 40% decrease per hour (Figure 1D). Since lysis behavior varied with initial OD, we standardized all further experiments by growing strain 3610 cultures to OD600~0.9–1.1 before cutting off oxygen exchange. Transfer of OD~1 cultures into an anaerobic chamber resulted in an even higher maximum lysis rate of 86% decrease per hour (Figure S2C). From these data, we infer that populations at high density deplete the remaining oxygen more rapidly, and that rapid oxygen depletion more readily triggers cell lysis.
A cell wall autolytic enzyme is necessary for lysis of non-viable cells upon oxygen depletion
The cells that lost viability upon oxygen depletion also lysed and were removed from the population of intact cells, as indicated by the substantial decrease in OD. Given that cellular integrity relies on a robust cell wall, we hypothesized that lysis involves cell-wall autolytic enzymes. Cell-wall growth requires both insertion of new material and cleavage of the existing peptidoglycan by autolysins [32]. In B. subtilis, the major autolysin LytC is under control of the vegetative sigma factor σA and the alternative sigma factor σD [33, 34], preventing unchecked breakdown of the cell wall when insertion is disrupted [35]. We measured the OD of a ΔlytC mutant and found reduced lysis upon oxygen depletion (Figure 1C). As a specificity control, deletion of another σD-dependent autolysin LytD phenocopied wild-type behavior (Figure S2D), suggesting that LytC is the primary autolysin activated in oxygen-depleted conditions. Despite the higher levels of intact biomass in ΔlytC cultures after oxygen depletion, a similar proportion of cells (~10%) retained viability as in wild-type cultures (Figure 1C). Single-cell imaging of oxygen-depleted cultures revealed mixed populations of phase-dark cells and phase-gray “ghosts,” both rod-shaped (Figure 1C). Time-lapse microscopy demonstrated that only a portion of the phase-dark ΔlytC cells were capable of resuming growth (Figure S2E, Video S1, S2), consistent with our plating efficiency data (Figure 1C). These data indicate that lysis and cell viability are genetically separable phenotypes, since many ΔlytC cells remained intact yet still lost viability. Thus, viability loss upon oxygen depletion is independent from cell-wall degradation.
Laboratory-domesticated B. subtilis exhibits more viability loss despite reduced lysis upon oxygen depletion
The B. subtilis lab strain 168, a genetic derivative of the biofilm-forming strain 3610, is commonly used in laboratories as it was selected for dispersed growth in liquid culture and improved genetic tractability [16]. In microtiter plate assays, we found that upon oxygen depletion the OD600 of strain 168 cultures decreased at a lower rate and remained at a higher level throughout 48 hours of monitoring (Figure 2A). Imaging of oxygen-depleted strain 168 cultures revealed the presence of round cells in addition to rod-shaped cells (Figure 2B). Some round cells were intact and did not stain with propidium iodide, while others stained brightly with propidium iodide signifying compromised membranes (Figure 2B). In cell cultures that were first stained with fluorescent D-amino acids (FDAA) to label cell walls, the vast majority of round cells did not retain FDAA staining, indicating they were protoplasts without cell walls (Figure S3A). Indeed, a strain 168 ΔlytC mutant did not form round cells and exhibited less lysis (Figure S3B), indicating that the protoplasts released in strain 168 are more fragile than their walled counterparts. Thus, we conclude that LytC degrades the cell wall in the lab strain 168 during oxygen depletion, releasing membrane-bound protoplasts that account for the higher OD600 in strain 168 cultures compared with the biofilm-forming strain 3610 cultures.
Figure 2: Surfactin production is necessary to maintain viability.
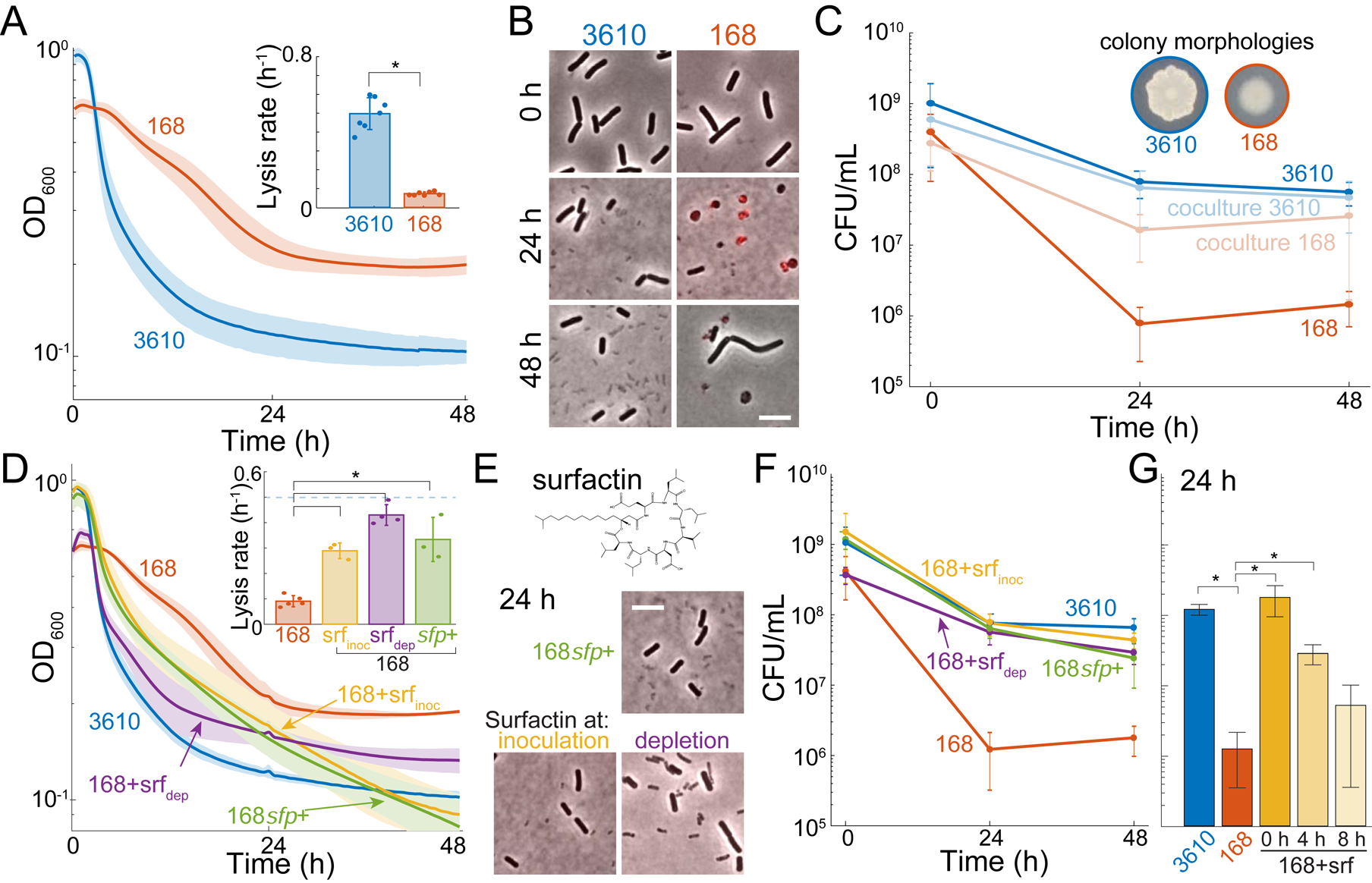
(A) B. subtilis lab strain 168 lyses less upon oxygen depletion than the biofilm-forming strain 3610. B. subtilis strains 3610 and 168 were grown aerobically and then depleted for oxygen at 0 h. Lines represent the mean and shading represents 1 SD, n=7. Inset: maximum lysis rate of strain 3610 was significantly higher than that of strain 168 (*: p=1.7×10−8, Student’s t-test). Circles show individual experiment values, and error bars represent 1 SD.
(B) Many strain 168 cells form cell-wall-less protoplasts upon oxygen depletion. Merge of phase-contrast and fluorescence images of propidium iodide (PI)-stained cells at 0, 24, and 48 h post oxygen depletion. Red indicates membrane-compromised cells.
(C) Co-culturing strain 168 with strain 3610 rescues its viability upon oxygen depletion. 168 viability in monoculture was significantly different than 3610 (*: p<0.005; Student’s t-test). Inset: Strain 3610 and strain 168 have distinct colony morphologies when plated on LB. Error bars represent 1 SD, n=3–5.
(D) Culturing with exogenous surfactin increases lysis of strain 168 cultures upon oxygen depletion. OD curves during oxygen depletion of strains 3610, 168, and 168 genetically rescued for surfactin production (168sfp+) or with 48 μM exogenous surfactin added before growth (srfinoc) or at depletion (srfdep). Lines represent the mean and shading represents 1 SD, n=3–5. Inset: maximum lysis rates (*: p<0.001; Student’s t-test).
(E) Culturing with exogenous surfactin eliminates protoplasts from 168. Top: surfactin molecular structure. Bottom: phase-contrast and PI fluorescence imaging at 24 h post-oxygen depletion of strain 168sfp+ cells or strain 168 cells with 48 μM exogenous surfactin.
(F) Surfactin restores the viability of strain 168 cultures to near strain 3610 levels upon oxygen depletion. Surfactin-treated strain 168 (srfinoc, srfdep, and 168sfp+) cultures were each significantly different from wild-type 168 (p<0.001 at 24 h, p<0.01 at 48 h, Student’s t-test). By contrast, the viability of surfactin-treated 168 cultures was not significantly different from that of strain 3610 at 24 h (p>0.2, Student’s t-test). Error bars represent 1 SD, n=3–5.
(G) Surfactin restores the viability of strain 168 cultures when added at 4 h post depletion but not at 8 h. Surfactin was added anaerobically at 0, 4, and 8 h post-oxygen depletion and viability was assayed at 24 h. *: significant (p<0.01, Student’s t-test). Strain 168 was not significantly different from strain 168 + surfactin at 8 h (p=0.16, Student’s t-test). Error bars represent 1 SD, n=4.
Despite having a higher OD600 than strain 3610 cultures, the colony-forming units (CFU) in strain 168 cultures depleted of oxygen for 24 h decreased ~100-fold relative to strain 3610 (Figure 2C, S2A). Indeed, time-lapse imaging on fresh medium with oxygen showed that despite having intact cell envelopes, only ~1% of rod-shaped strain 168 cells were able to grow and divide, whereas ~95% of strain 3610 cells exhibited growth (Figure S3C, Video S3, S4). We never observed growth or division during 12-hour time-lapse imaging of any protoplasts on fresh LB or filtered spent medium, suggesting that these protoplasts are not viable. We conclude that despite reduced cell lysis, strain 168 cultures experience a much more drastic loss of viability than strain 3610 upon oxygen depletion.
Surfactin increases lysis and restores viability to the domesticated strain 168
Strains 3610 and 168 differ in a number of chromosomal loci, and the lab strain 168 is defective in phenotypes related to secretion of extracellular products that support multicellular behaviors such as biofilm formation and surface motility [17]. To determine whether extracellular products were responsible for oxygen-related phenotypic differences, we subjected a 1:1 volumetric mixture of the strains to oxygen depletion. The viability of strain 168 (which has a distinct colony morphology from strain 3610) increased dramatically in the co-culture (Figure 2C). A candidate compound that could restore viability to strain 168 is surfactin, a strong surfactant that creates K+-permeable pores and solubilizes lipid-bilayer vesicles in vitro [36]. Surfactin is produced by strain 3610 but not strain 168 due to a mutation in sfp, which encodes an enzyme required to activate surfactin biosynthesis [18]. We found that exogenous surfactin addition at either inoculation of the culture or at initiation of oxygen depletion increased the lysis rate of strain 168 and eliminated protoplasts (Figure 2D,E), consistent with surfactin-induced protoplast lysis.
Moreover, exogenous surfactin addition restored the viability of oxygen-depleted strain 168 cultures to similar levels as strain 3610 (Figure 2F). Moreover, a 168 strain with sfp genetically complemented to restore surfactin production phenocopied surfactin-treated strain 168 cultures in terms of lysis, cell morphology, and viability (Figure 2D–F). To determine whether viability loss was reversible, we added surfactin in anaerobic conditions after 4 or 8 hours of oxygen depletion and tested viability dynamics. Adding surfactin at 4 hours post-depletion resulted in significantly higher viability than untreated strain 168 cells at 24 hours post-oxygen depletion (Figure 2G). By contrast, surfactin addition at 8 hours did not significantly improve viability at 24 hours post-oxygen depletion (Figure 2G), indicating that the majority of cells pass a point of no return between 4 and 8 hours after which surfactin can no longer restore viability. Thus, we conclude that surfactin production both promotes protoplast lysis and maintains viability in oxygen-depleted B. subtilis cultures.
Surfactin improves growth yield of strain 168 by increasing oxygen diffusion
One intriguing distinction between the aerobic growth of strains 3610 and 168 was divergence of the growth curves around OD600~0.3; afterward, strain 3610 cultures completed another ~1.5 mass doublings to reach OD600~0.9, while strain 168 cultures underwent 1 mass doubling to reach OD600~0.6 (Figure 3A). We noted that surfactin supplementation increased strain 168 growth yields to levels similar to that of strain 3610 (Figure 3B), despite a slight reduction in maximum growth rate due to surfactin addition (Figure S4). When surfactin was added just prior to the divergence in growth curves, yield was similarly restored (Figure 3B). Thus, we conclude that surfactin production was responsible for the increase in yield of strain 3610 relative to strain 168. The detergent Tween 80 similarly increased yield in strain 168 (Figure 3C), indicating that the detergent properties of surfactin were likely responsible for increasing yield.
Figure 3: Surfactin restores the growth yield of strain 168 due to increased oxygen diffusion.
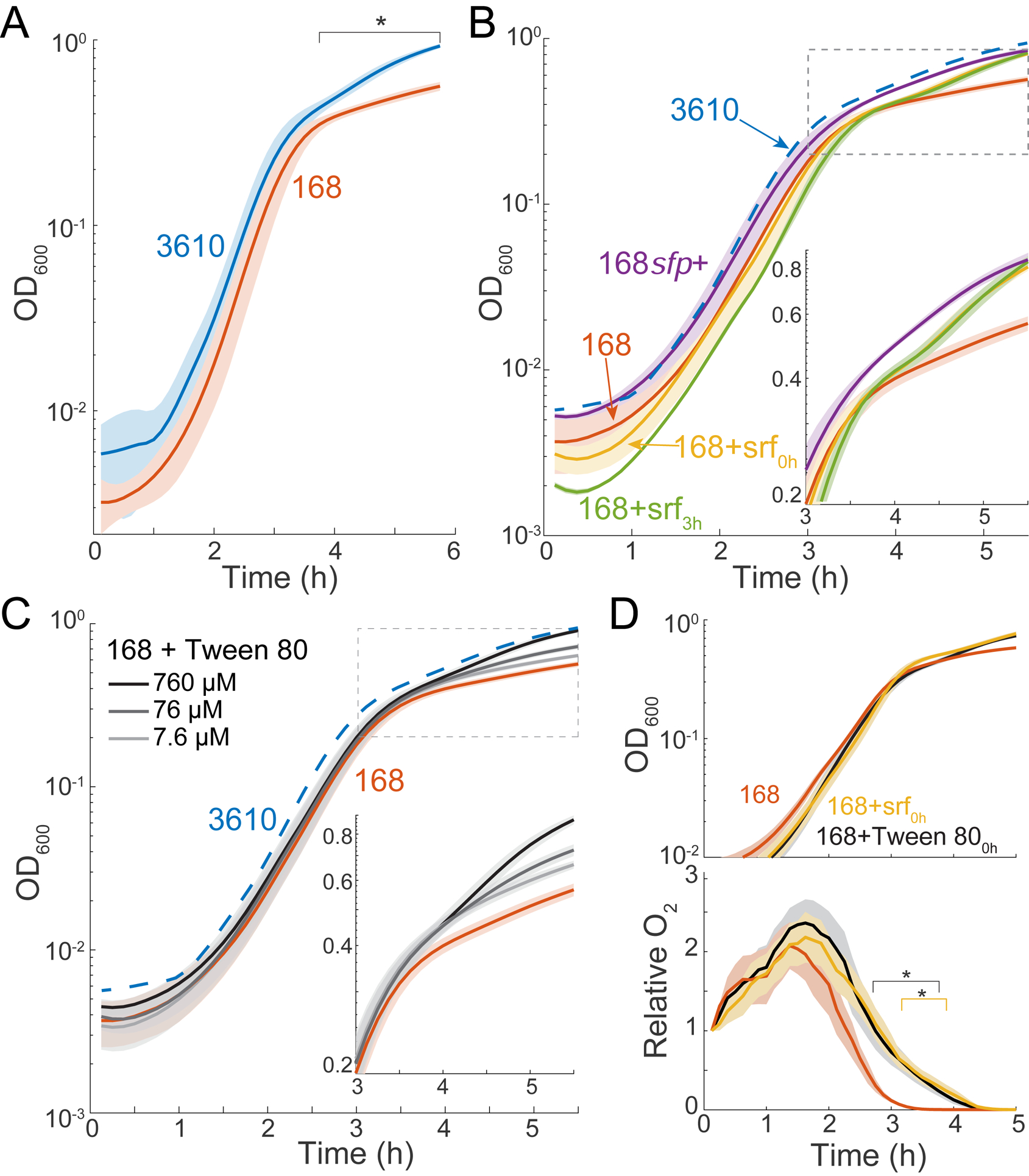
(A) Strain 3610 aerobic cultures achieve a significantly higher growth yield than strain 168. *: time period over which strain 168 growth differed significantly from that of strain 3610 (p<0.05, Student’s t-test). Lines represent the mean and shading represents 1 SD, n=3.
(B) Surfactin addition rescues growth yield. Growth curves of strain 168 with surfactin restored genetically (168sfp+) or 48 μM added exogenously at inoculation (168+srf0h) or at t=3 h (168+srf3h). Lines represent the mean and shading represents 1 SD, n=3. The mean growth curve from strain 3610 (A) is shown as a dotted blue line. Inset: rescaled to highlight growth divergence.
(C) Tween 80 addition rescues growth yield in a concentration-dependent manner. Lines represent the mean and shading represents 1 SD, n=3. The mean growth curve from strain 3610 (A) is shown as a dotted blue line. Inset: rescaled to the period of growth divergence.
(D) Surfactin addition increases oxygen levels during late exponential phase. Relative oxygen levels (bottom) during growth (top) of strain 168 with added surfactin (48 μM) or Tween 80 (0.76 mM). *: time period over which oxygen levels of 168+surfactin (yellow) or 168+Tween 80 (black) were significantly different from untreated 168 cultures (p<0.05, Student’s t-test).
See also Figure S4.
It has been proposed that oxygen diffusivity is rate-limiting for the function of some biological systems [37]. Certain compounds increase oxygen diffusion in liquid [38] or accelerate oxygen diffusion through the air-water interface [39], thus we hypothesized that surfactin enabled higher yield by increasing oxygen diffusivity. To test this hypothesis, we used nanoprobes to compare oxygen levels in strain 168 cultures with and without added surfactin or Tween 80. We found that both detergents increased the oxygen levels in late-exponential phase, when oxygen would normally be depleted lower than our limit of detection (Figure 3D). A biophysical model incorporating diffusion and cellular consumption of oxygen predicted that the observed ~1.2-fold increase in peak oxygen is consistent with a ~1.4-fold increase in diffusivity (Methods). Thus, we infer that surfactin increases growth yield due to enhanced availability of oxygen during the transition to stationary phase when oxygen would otherwise be limiting for growth.
A transposon screen supports surfactin production as the main determinant of survival upon oxygen depletion
Given the large viability decreases after 24 hours of oxygen depletion, we performed a genetic screen to attempt to identify mutations that would increase the survival of strains 168 and 3610 without oxygen. We made 20–30 independent transposon libraries with 5000–10,000 individual transposon mutants per library in each background and subjected these libraries to oxygen depletion, hypothesizing that any mutants with enhanced survival would be enriched (Figure S5A). We could not identify any such mutants in strain 168, suggesting that there is not an easily obtainable loss-of-function mutant in a surfactin-independent pathway that increases viability upon oxygen depletion.
In the 3610 background, we identified several mutants with decreased lysis upon oxygen depletion. In-frame deletions of the genes corresponding to these mutants had lower viability in isolation than the parent after 48 hours of oxygen depletion, consistent with the differences between strains 168 and 3610 (Figure S5C). Many of these mutants were in genes related to surfactin production [18, 40–42] (Figure S3B, Table S1), suggesting that their increased proportion in the pooled library was due to experiencing the benefits of surfactin without the metabolic cost of its production. Upon exogenous surfactin addition, all mutants responded with increased lysis and removal of protoplasts/cell debris (Figure 4B,C, S5D). The facts that we obtained no hits with increased viability in strain 168 and that all hits in strain 3610 respond to surfactin and do not have increased fitness in isolation point to surfactin as the primary determinant of viability in the absence of oxygen.
Figure 4: Transposon mutagenesis identifies genes that impact lysis during oxygen depletion in strain 3610.
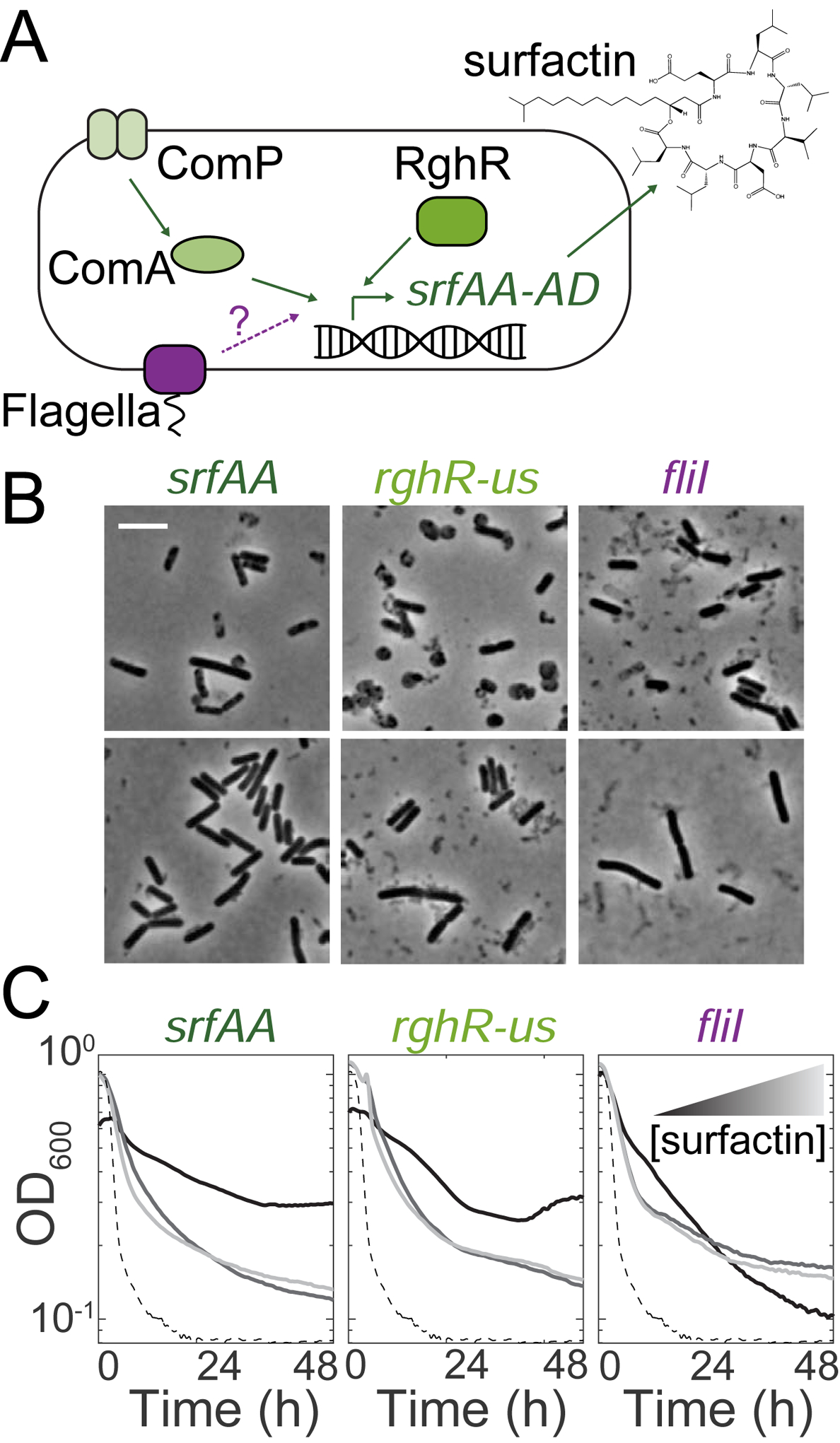
(A) Schematic of regulation of the surfactin synthetase gene operon (srfAA-AD). Known regulators of SrfAA are shown in green. Our data suggests flagellar proteins (purple) might also regulate surfactin.
(B) Surfactin-treated cultures of the transposon-disrupted mutants have fewer protoplasts and cell debris. Phase-contrast images of mutants depleted of oxygen for 24 h with and without 48 μM exogenous surfactin. Top: arrowheads show protoplasts, phase-gray dead cells, and cell debris, all of which were not observed in the surfactin-treated cultures.
(C) Transposon hits exhibit faster lysis when treated with exogenous surfactin. Black curves are without surfactin, medium and light gray curves are with 24 μM and 48 μM surfactin, respectively. The dashed line is the parent (3610).
Surfactin restores viability by depolarizing the membrane
Surfactin creates potassium ion-permeable pores in lipid bilayers in vitro [36], and has been suggested to cause potassium leakage in vivo in B. subtilis [43]. Such pores would reduce the potassium ion gradient across the membrane and alter membrane potential. Thus, we tested cellular energetic state after chemical treatments using the membrane potential-sensitive dyes DiSC3(5) and ThT aerobically in an assay buffer [44, 45]. DiSC3(5) is taken up by cells and fluorescence is quenched. Agents that depolarize cells release DiSC3(5) into the medium, resulting in increased signal [44]. By contrast, the cationic dye ThT enters cells with polarized membranes and fluorescence increases; upon depolarization, ThT exits cells and fluorescence decreases [45]. As expected, we found that treatment of strain 168 cells with valinomycin, a known depolarizing agent that functions as a potassium-specific transporter [46], led to increased DiSC3(5) and decreased ThT signal (Figure 5A, S6A). CCCP, a proton ionophore that dissipates the proton motive force [47], strongly reduced ThT signal and slightly increased DiSC3(5) fluorescence (Figure 5A, S6A). Surfactin treatment led to a large increase in DiSC3(5) fluorescence and a greater decrease in ThT fluorescence than valinomycin (Figure 5A, S6A), demonstrating that surfactin can strongly depolarize B. subtilis cells.
Figure 5: Surfactin maintains viability upon oxygen depletion by depolarizing the membrane.
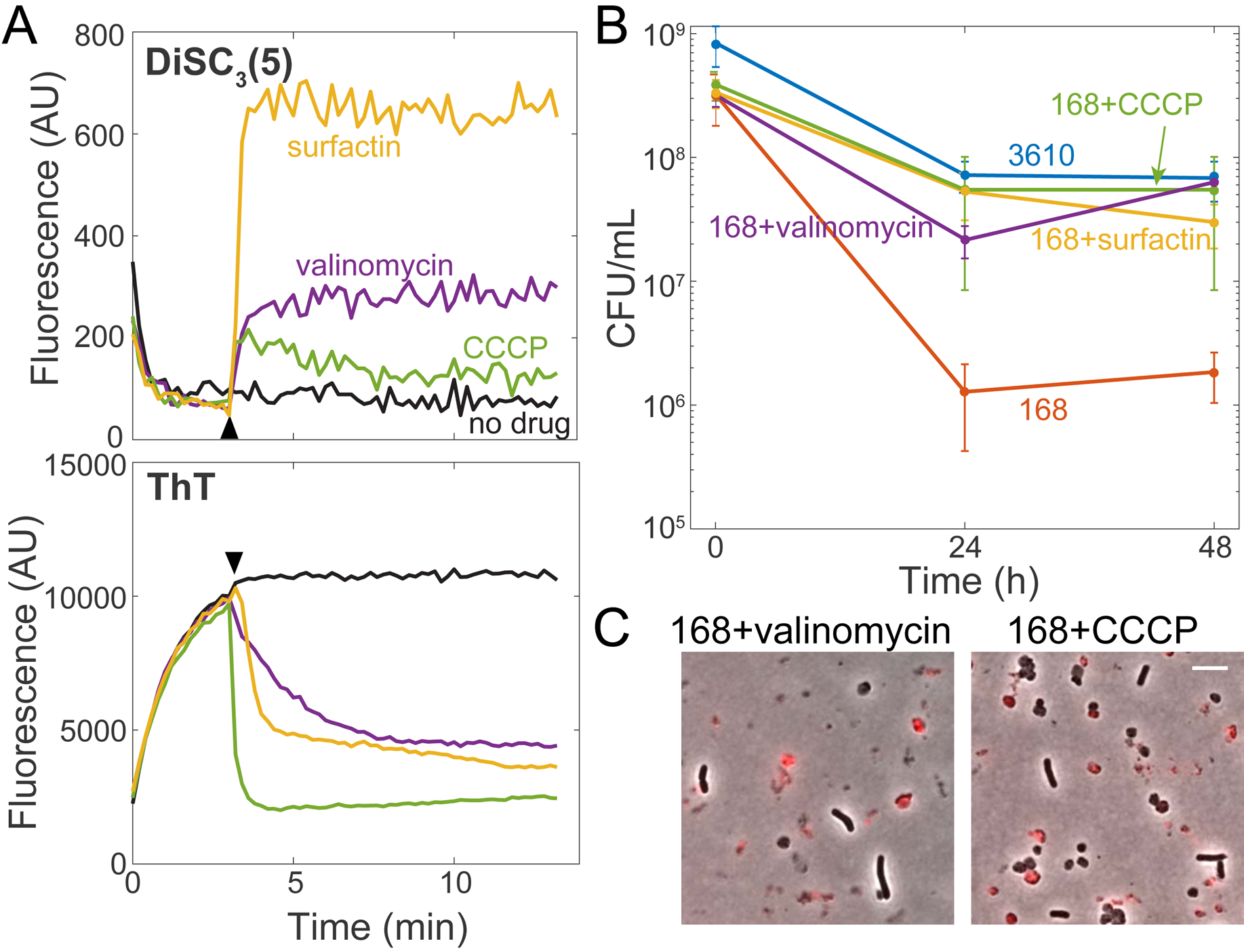
(A) Surfactin depolarizes the membrane in B. subtilis. Membrane potential assays of strain 168 cells using the dyes DiSC3(5) (top) and ThT (bottom). The time of addition of surfactin (48 μM), valinomycin (50 μM), and CCCP (5 μM) is marked by the black arrowhead. One representative experimental replicate is shown (other replicates in Figure S6A).
(B) Treatment with the membrane depolarizing agents valinomycin (5 μM) and CCCP (5 μM) restores plating efficiency of B. subtilis strain 168 after oxygen depletion, similar to surfactin (48 μM). Error bars represent 1 SD, n=3–5. 168 plating efficiency data were significantly different than those of strain 168+surfactin, strain 168+valinomycin, strain 168+CCCP, and strain 3610 (p<0.005, Student’s t-test).
(C) Valinomycin and CCCP-treated strain 168 cultures have protoplasts, demonstrating that protoplast removal is unnecessary for viability enhancement. Overlays of phase-contrast and PI (red) images at 24 h post-oxygen depletion. Scale bar: 5 μm.
See also Figure S6.
Based on these findings, we hypothesized that de-energizing the membrane might rescue the colony-forming ability of strain 168 cultures upon oxygen depletion. Indeed, valinomycin and CCCP both restored viability to a similar extent as surfactin (Figure 5B). Notably, valinomycin- and CCCP-treated cultures had protoplasts after 24 hours of oxygen depletion, consistent with surfactin being necessary for protoplast lysis and indicating that protoplast formation is independent from viability maintenance. By contrast, Tween 80 removed protoplasts but did not significantly restore viability or impact ThT signal (Figure S6B), indicating that viability rescue is due to surfactin-induced membrane depolarization rather than increased oxygen diffusivity. These data indicate that membrane potential dictates the ability of B. subtilis cells to survive oxygen depletion.
Depolarization likely mediates viability rescue through global metabolic changes precipitated by reduced oxygen consumption
To test whether surfactin maintains viability through a transcribed or translated product, we added rifampicin (to block RNA synthesis) or chloramphenicol (to block protein synthesis) to surfactin-treated strain 168 cells. Neither antibiotic reduced viability (Figure S6C). Moreover, we surprisingly found that treatment of strain 168 with either drug in the absence of surfactin significantly restored viability during oxygen depletion (Figure 6A). As neither drug affects membrane potential (Figure S6D), these results suggest that surfactin is mediating a global metabolic change rather than a pathway-specific adaptation that requires transcription or translation.
Figure 6: Surfactin maintains viability upon oxygen depletion by reducing ATP production and oxygen consumption.
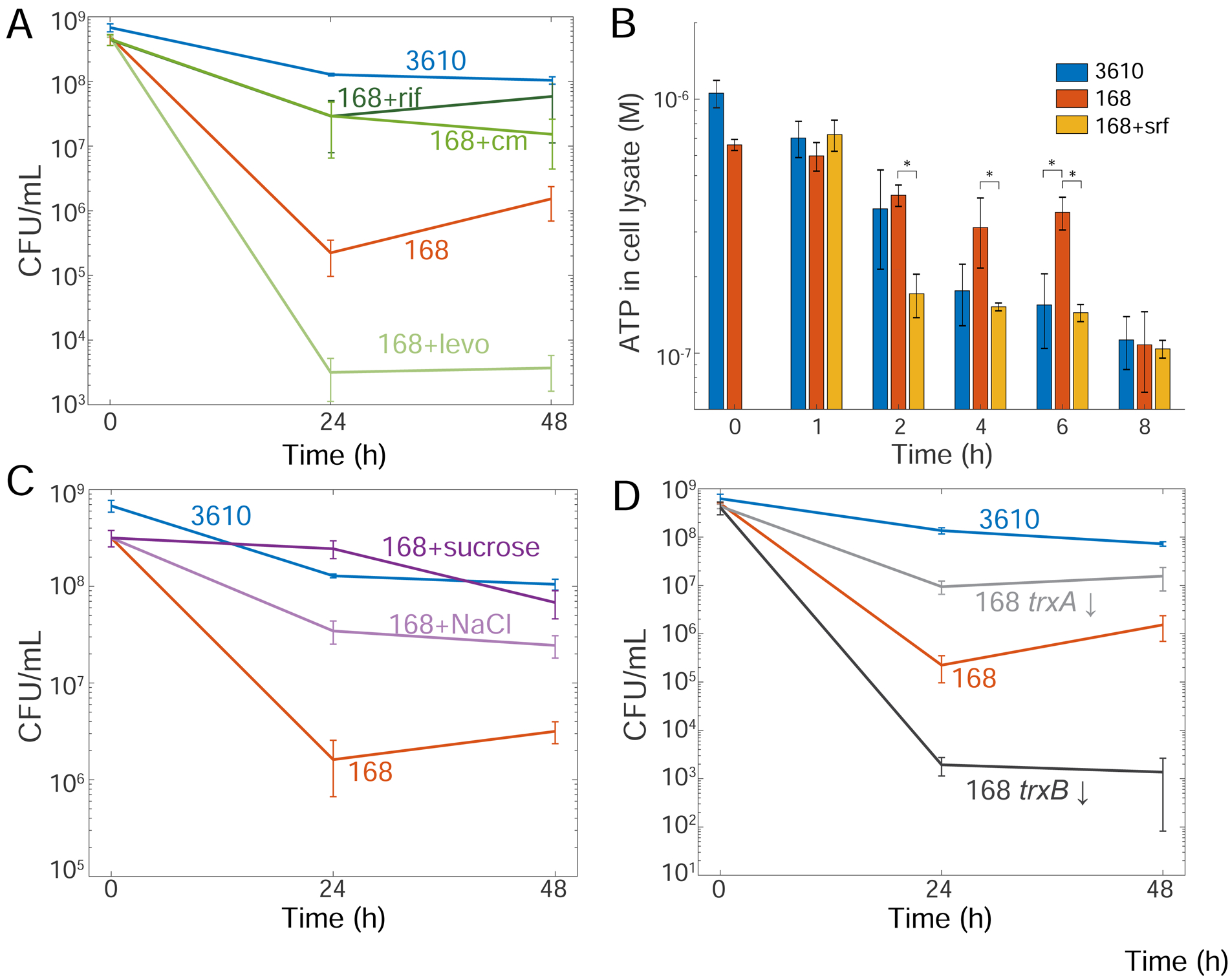
(A) Antibiotics that reduce oxygen consumption (rifampin (rif) and chloramphenicol (cm)) restore the colony-forming ability of strain 168, while an antibiotic that does not reduce oxygen consumption (levofloxacin (levo)) does not. Viability for strain 168+rif and strain 168+cm was significantly higher than untreated strain 168 at 24 h (p<0.05, Student’s t-test). Error bars represent 1 SD, n=3–4.
(B) Surfactin reduces ATP levels in strain 168 cells following oxygen depletion. ATP levels of an equal volume of cell lysate were measured at each time point following oxygen depletion. Error bars represent 1 SD, n=3. *: p< 0.05, Student’s t-test.
(C) Adding 300 mM solutes to reduce oxygen solubility restores viability to strain 168. Viability of strain 168+sucrose and strain 168+NaCl were significantly different from untreated strain 168 at 24 and 48 hours (p < 0.01, Student’s t-test). Error bars represent 1 SD, n=3.
(D) Altering the redox state of the cell by inhibiting thioredoxin (trxA) or thioredoxin reductase (trxB) gene expression using CRISPRi alters the viability of strain 168. 1% xylose was added at the onset of oxygen depletion to block transcription of trxA (trxA ↓) or trxB (trxB ↓). Viability of these mutants was significantly different than wild-type 168 (p< 0.05, Student’s t-test). Error bars represent 1 SD, n=3–4.
See also Figure S6.
Treatment with chloramphenicol and rifampin has been shown to reduce oxygen consumption rates in bacteria [48]. To test whether the viability increase in chloramphenicol- and rifampin-treated strain 168 might be related to oxygen consumption, we treated oxygen-depleted cells with levofloxacin, a fluoroquinolone that does not reduce oxygen consumption rate [48], and found that viability was not restored (Figure 6A). Thus, we hypothesized that surfactin maintains viability during oxygen depletion by reducing oxygen consumption rates.
To determine whether surfactin decreases oxygen consumption during oxygen depletion through a reduction in ATP production, we measured ATP levels after 0, 1, 2, 4, 6, and 8 hours of oxygen depletion in strains 168 and 3610. As a control, we confirmed that treatment with sub-inhibitory levels of CCCP effectively reduced ATP levels at the t=0 time point (Figure S6E). Surfactin addition to strain 168 cultures significantly reduced ATP levels relative to untreated cultures at 2, 4, and 6 hours post-depletion. These levels were similar to those of strain 3610 at 4 and 6 hours post-depletion (Figure 6B). From these results and our observation that surfactin can effectively rescue viability when added at 4 but not 8 hours post-depletion (Figure 2G), we infer that reducing ATP production and the corresponding reduction in oxygen consumption is important for maintaining viability following oxygen depletion.
To further investigate the relationship between viability and oxygen consumption, we reduced the rate cells would encounter oxygen molecules by adding dissolved solutes that limit oxygen solubility [49]. Addition of sucrose or NaCl to reduce oxygen solubility by ~10–20% [49, 50], at the onset of oxygen depletion restored viability to strain 168 independently of surfactin treatment (Figure 6C). Neither solute affected membrane potential when measured by DiSC3(5), and both only slightly affected ThT signal (Figure S6D). Thus, adding solutes that reduce oxygen solubility can restore viability.
To test whether metabolic alterations related to oxygen consumption alter viability upon oxygen depletion, we used CRISPRi [14] to alter redox potential in strain 168 cultures by decreasing expression of the genes for thioredoxin (trxA) or thioredoxin reductase (trxB). Decreased levels of TrxA, which oxidizes NADPH to reduce disulfide bonds [51], should lead to higher levels of NADPH and therefore shift the redox balance to a more reduced state, while decreased levels of TrxB should result in more oxidized TrxA, leading to higher levels of NADP+ and a shift in redox balance to a more oxidized state. Induction of trxA repression at the onset of oxygen depletion increased strain 168’s viability ~80-fold (Figure 6D), while induction of trxB repression reduced strain 168’s viability ~100-fold (Figure 6D). These findings indicate that maintaining a redox balance is critical for survival upon oxygen depletion.
Taken together, our data shows that many interventions (transcription/translation inhibition, solute addition, manipulation of redox balance) restore viability upon oxygen depletion, suggesting that surfactin-mediated membrane depolarization maintains viability through a global metabolic change associated with slower oxygen consumption and/or a shift to a more reduced cytoplasm.
Discussion
While surfactin has been recognized to promote surface motility, sliding motility, and colony expansion in B. subtilis [17, 21, 52], its role in planktonic cultures has remained mysterious. Here, we demonstrate three independent functions of surfactin in planktonic cultures. During aerobic growth, the detergent properties of surfactin increase growth yield by increasing the oxygen available to cells entering stationary phase (Figure 3). During oxygen depletion, surfactin has two independent functions: (1) it acts as a detergent to break down cell membranes and, in tandem with LytC, removes non-viable cells from the culture (Figure 2), and (2) it mediates survival of the viable cell fraction through membrane depolarization (Figure 5).
Our observation that surfactin increases oxygen levels in aerobic cultures through enhancing oxygen diffusivity is the first example of surfactin playing this role. The increase in growth yield due to surfactin suggests that oxygen can be limiting even in aerobic cultures and that increasing oxygen availability increases yield. This finding has industrial implications as B. subtilis is commonly used to produce many enzymes and antibiotics [53], and maintaining oxygen levels is critical to optimize production of the desired compounds [54, 55]. In addition to oxygen, surfactin might also facilitate growth by increasing nutrient diffusion during late exponential phase when nutrients become limiting. As surfactin only impacts growth rate slightly at high concentrations (Figure S4), it is possible that growing B. subtilis cells have mechanisms to protect them from surfactin-induced membrane depolarization and that surfactin is better at depolarizing the membrane upon oxygen limitation.
We also found that surfactin has two independent functions during oxygen starvation that are distinct from its role during aerobic growth. Surfactin causes lysis in cells that experience LytC-mediated cell-wall degradation (Figure S2), presumably through membrane disruption. Moreover, surfactin maintains viability of the remaining intact cells through membrane depolarization. This depolarization reduces ATP levels and oxygen consumption, which might allow these cells to transition to a state with altered redox balance in which they can ride out the stress of oxygen depletion. While it was possible that cell lysis indirectly maintains viability of the remaining cells, our observations that ΔlytC mutants had the same viability as wild-type cultures even though the non-viable cells remain intact (Figure 1C) and that CCCP or valinomycin treatment of 168 cultures rescued viability without removing protoplasts (Figure 5C) demonstrate that surfactin acts directly upon the membrane potential of the surviving cells to maintain viability.
Generally, OD is used as a proxy for cell number, which holds true for our measurements of 3610 cultures undergoing oxygen depletion as the drop in OD reflects cell lysis and quantitatively mirrors the decrease in viability (Figure 1C, S2A). However, our work highlights instances during oxygen depletion when biomass measured by OD is uncoupled from viability: both ΔlytC 3610 cells and wild-type 168 cells remain intact and the cultures have a relatively high OD compared with wild-type 3610, but most cells cannot form colonies (Figure 2C, S1C). This uncoupling between OD and viability has been observed previously in cell-cycle mutants where cells remain intact and metabolically active but cannot divide and form colonies [56], and may be more prevalent than is currently appreciated.
Since membrane depolarization and antibiotic treatment can improve the viability of cells undergoing oxygen depletion, under conditions with limiting terminal electron acceptors certain antibacterial treatments that inhibit growth might actually keep cells viable. This possibility needs to be considered when treating bacterial infections and removing bacteria in low-oxygen clinical and industrial settings, particularly for microbes that respire aerobically. Moreover, it remains generally unclear which antimicrobial treatments disrupt membrane potential or mechanistically how antibiotic treatments generally affect cells in low oxygen environments, motivating further studies of membrane energetics during growth inhibition.
The cell-wall breakdown of B. subtilis cells during oxygen depletion provides further support for a recently discovered regulatory role of membrane potential in cell-wall synthesis [57]. Indeed, LytC is activated upon sodium azide treatment, which deprotonates the cell wall [58, 59], and the activity of peptidoglycan synthesis enzymes in E. coli are regulated by pH [60, 61]. In addition, the recent observation that actively growing and dormant cells in a B. subtilis culture respond oppositely to an electrical pulse and either hyperpolarize or depolarize, respectively [62], suggests that membrane energetics might explain our observations of population heterogeneity in oxygen-depleted 3610 cultures, where ~90% of cells die and ~10% remain viable. Thus, as membrane potential is of utmost importance in metabolism and cell growth, is becoming more appreciated in regulating cell-wall remodeling, and likely feeds back into many additional aspects of cell physiology, bacteria must employ strategies to maintain and/or modulate membrane potential during changing environmental conditions. Moreover, membrane potential could be a key component of other instances of cell-fate heterogeneity, such as the lysis/lysogeny decision during phage infection. The ability of surfactin to both alter membrane energetics to maintain viability during oxygen depletion and to enhance growth in oxygen-limited conditions has likely provided multiple fitness advantages to B. subtilis in spatially structured and complex environments such as native soil communities, and strategies for regulating membrane potential may be an important factor for the survival of other strict aerobes as well.
STAR Methods
Lead Contact and Materials Availability
Further information and requests for reagents may be directed to and will be fulfilled by the Lead Contact, K.C. Huang (kchuang@stanford.edu). All unique/stable reagents generated in this study are available from the Lead Contact without restriction.
Experimental Model and Subject Details
All strains and their genotypes are listed in Table S2. Strains were grown in LB (Lennox broth with 10 g/L tryptone, 5 g/L NaCl, and 5 g/L yeast extract). Antibiotics were used as follows, unless indicated otherwise: kanamycin (5 μg/mL), MLS (a combination of erythromycin at 0.5 μg/mL and lincomycin at 12.5 μg/mL), chloramphenicol (5 μg/mL), rifampin (1 μg/mL), spectinomycin (100 μg/mL), and levofloxacin (2 μg/mL). This concentration of levofloxacin was chosen after testing multiple concentrations (10, 2, 0.4 μg/mL) during oxygen depletion; similar results were obtained with the three concentrations, hence we chose 2 μg/mL to remain consistent with results in [48]. Surfactin was added at a final concentration of 0.05 mg/mL and xylose was added at a final concentration of 1% unless otherwise noted. Strains were cultured either in 5 mL of medium in a test tube on a roller drum or in 200 μL of medium in a 96-well plate in a Biotek Epoch2 spectrophotometer under linear shaking (shaking was set to 567 cycles per minute (cpm), 3-mm magnitude of shaking). For all experiments, the initial inoculum was from a fresh colony struck from a −80 °C freezer stock onto LB 1.5% agar plates and incubated overnight at 37 °C.
Method Details
Strain construction
Strains were constructed using SPP1 phage transduction [63]. The donor strain was grown for >6 h in TY medium (LB supplemented with 0.01 M MgSO4 and 0.1 mM MnSO4 after autoclaving). Ten-fold dilutions of SPP1 phage were added to the culture and 3 mL TY soft (0.5%) agar was mixed with the culture/phage mixture and poured over a TY plate (1.5% agar) overnight. A plate was chosen that exhibited nearly total clearing of cells without a large number of phage-resistant mutants. Five milliliters of TY medium were added to this plate and a 1-mL filter tip was used to scrape up the soft agar. This soft agar/liquid mix was filtered through a 0.4-μm filter. The phage was added to a stationary-phase (grown for 6–10 h) culture in TY medium of the recipient strain (10 μL undiluted phage + 100 μL recipient cells, and optionally 900 μL TY medium) and incubated at 37 °C for 30 min, then plated onto LB + antibiotic and 0.01 M sodium citrate (sodium citrate was omitted for MLS selection). Plates were incubated for 24 h and transductants were struck for single colonies to eliminate the phage.
To generate the ΔlytC in-frame marker-less deletion construct, the region upstream of lytC was PCR-amplified using the primer pair 1427/1428 and digested with SalI and EagI, and the region downstream of lytC was PCR-amplified using the primer pair 1425/1426 and digested with EagI and BamHI. The two fragments were then simultaneously ligated into the SalI and BamHI sites of pMiniMAD, which carries a temperature-sensitive origin of replication and an erythromycin resistance cassette [64], to generate pDP299.
To generate the ΔlytD in-frame marker-less deletion construct, the region upstream of lytD was PCR-amplified using the primer pair 1429/1430 and digested with SalI and EagI, and the region downstream of lytD was PCR-amplified using the primer pair 1431/1432 and digested with EagI and BamHI. The two fragments were then simultaneously ligated into the SalI and BamHI sites of pMiniMAD to generate pDP300.
Deletion plasmids were introduced into strain DK1042 (Table S2) via single cross-over integration by transformation at the restrictive temperature for plasmid replication (37 °C) using MLS resistance as a selection. To evict the plasmid, the strain was incubated in 3 mL LB broth at a permissive temperature for plasmid replication (22 °C) for 14 h, and serially diluted and plated on LB agar at 37 °C. Individual colonies were patched on LB plates and LB plates containing MLS to identify MLS-sensitive colonies that had evicted the plasmid. Chromosomal DNA from colonies that had evicted the plasmid was purified and screened by PCR using primers 1427/1426 or 1430/1431 to determine isolates that retained the ΔlytC or ΔlytD allele, respectively [64, 65].
In-frame deletions of MLS-knockout mutants were constructed as outlined previously [13]. Briefly, the strain of interest was transformed with pDR244 at 30 °C. Several transformants were struck onto LB at 42 °C. Resultant colonies were patched on MLS and spectinomycin plates to confirm that the plasmid and MLS cassette were lost. Colonies were screened for altered oxygen phenotype.
Growth and lysis assays
Strains were struck out for single colonies on the evening prior to the experiment. Colonies were inoculated into fresh LB and grown aerobically at 37 °C, either in 5 mL LB on a roller drum or in 200 μL LB in a Greiner 96-well plate. To deplete oxygen from cultures grown in test tubes, 200 μL were aliquoted into a 96-well plate and the plate was sealed with optical film (Excel Scientific AeraSeal). To grow cultures in the plate reader, 200 μL of stationary-phase inocula were aliquoted into each well of a 96-well plate, with the edge wells containing medium only. Optical film was used to cover the plates, and one hole per well was poked using a 20-gauge needle to allow for air exchange. 168 and 3610 cultures were then grown until the 3610 culture reached an OD600~1.0. To deplete the oxygen, the plate was taken out of the plate reader, the optical seal with holes was removed, and a new seal was placed onto the plate to fully cut off oxygen exchange. We grew and depleted cultures at 37 °C using linear shaking (567 cpm, 3-mm magnitude) and read OD600 every 7.5 min in a Biotek Epoch2 spectrophotometer.
Mariner transposon library construction
The mariner transposon was transduced into the parent strains as described in the “Strain Construction” method’s section. The mariner plasmid has a temperature sensitive origin of replication, transposase, and a transposon that is marked with kanamycin resistance while the backbone plasmid is marked with MLS resistance [66]. The parent strains (HA1235 and HA1414) were struck for single colonies onto MLS plates at 30 °C. One colony per library was grown in 3 mL LB+kanamycin in a roller drum at room temperature overnight. Transposon-insertion libraries were selected by plating 10-fold dilutions of the cultures on LB+kanamycin plates prewarmed to 37 °C and incubating overnight at 37 °C. The dilution that contained very dense individual colonies was used to create the library and the higher dilutions were used to estimate the number of transposon mutants per library.
Screen to enhance for mutants that survive better without oxygen
Each mariner transposon library (~5000–10,000 colonies per library) was inoculated by swabbing a sterile cotton bud over the entire plate and then vigorously shaking the cells into 5 mL LB. The libraries were grown on a roller drum at 37 °C to an OD600~1.0. Libraries were then aliquoted 200 microliters per well in a 96-well microtiter plate. The plates were sealed with optical film to deplete oxygen (Excel Scientific AeraSeal). The sealed cultures were incubated at 37 °C for 2–8 days for strain 168 libraries or at 30 °C for 4–8 days for strain 3610 libraries. Various incubation times were chosen to optimize the chance of recovering hits, as we reasoned that a longer incubation might provide an enhanced selection. At various time points following the start of oxygen depletion, one well of the library was harvested by piercing the well with a sterile scalpel. 10-fold dilutions or dilution streaking were performed to isolate single colonies. One or two colonies per well were tested for either enhanced lysis (strain 3610 background) using the oxygen-depletion methodology described above, or enhanced colony-forming ability (strain 168 background) by growing cultures and depleting oxygen (in 96-well microtiter plates as described above) for 24 h after which 10-fold serial dilutions were plated to identify any colonies with a higher plating efficiency than the parent strain. Any mutants found to have these phenotypes was used as donors in SPP1 phage transduction (described above in Strain Construction) and transduced into the parent strain to ensure that the transposon was the causative agent and that there were no additional mutations contributing to the phenotype.
Mapping the transposon mutations using inverse PCR
To map the mutation, genomic DNA was prepped from each back-crossed mutant using the Promega Wizard® Genomic DNA purification kit. The genomic DNA was digested with Sau3AI (note either Sau3AI or TaqAI can be used to digest) at 37 °C for 90 min and then heat inactivated at 65 °C for 20 min. The reaction mixture was as follows: 15.5 μL milliQH2O, 2 μL NEBuffer1.1, 2 μL digested genomic DNA, 2 μL 10X BSA, 0.5 μL Sau3AI). The digested DNA was then ligated using T4 ligase at room temperature for at least 1 h; the reaction mixture is as follows: 2 μL T4DNA Ligase Buffer, 15.5 μL milliQ H2O, 2 μL digested DNA, 0.5 μL T4 DNA ligase. Inverse PCR was carried out with the ligated DNA using Phusion polymerase with the primers IPCR1 and IPCR2. The reaction mixture is as follows: 33 μL milliQ H2O, 10 μL 5X HF buffer, 2 μL ligated DNA, 2 μL IPRC1, 2 μL IPCR2, 1 μL 10 mM dNTPs, and 0.2 μL Phusion Polymerase. The PCR program is as follows: 98 °C for 30 s, 30 cycles of [98 °C for 10 s, 58 °C for 30 s, 72 °C for 60 s], 72 °C for 10 min, hold at 4 °C. The PCR products were gel-purified and sequenced using the IPCR2 primer. The sequences were mapped onto the B. subtilis genome using BLASTN.
Oxygen nanoprobe measurements
Relative oxygen levels were measured using the oxygen-sensitive nanoprobe (BF2nbm(I)PLA) that emits an oxygen-dependent phosphorescence reading and an oxygen-independent fluorescence reading, which together can be used to calculate the relative oxygen level of the medium [67, 68]. Briefly, the nanoprobe was added at 5% (10 μL into 190 μL) to the inoculum before growth of the cultures. In addition to OD600 readings, fluorescence readings (ex/em: 414/450 nm) and phosphorescence readings (ex/em: 415/560 nm, with a 2-ms delay between excitation and emission) were taken using a Biotek Synergy H1 spectrophotometer. Readings were taken every 7.5 min, with incubation at 37 °C and linear shaking (567 cpm, 3-mm magnitude of shaking).
Propidium iodide staining and phase microscopy
Five hundred nanoliters of cultures were spotted onto LB pads made with 1.5% agar and 10 μM propidium iodide. Once dry, a coverslip was added and cells were imaged on a Nikon Ti-E inverted microscope using a 100X objective (NA: 1.4). Phase and fluorescence (mCherry filter, ex/em: 570/645 nm) images were acquired. Images were processed identically in Adobe Photoshop and merged using FIJI.
Time-lapse microscopy of oxygen-depleted cultures
The bottom of a rectangular Singer PlusPlate culture plate was used to make a large pad [69], in which 35 mL of LB+1.5% agar was pipetted onto the plate ~1 h before imaging so that the agar could solidify completely. Once solid (after ~5–10 min), a second Singer PlusPlate was placed on top of the agar pad to prevent contamination and drying. One microliter of cultures was spotted in the center of the pad and allowed to dry. A large 113 by 77 mm custom-made no. 1.5 glass coverslip (Nexterion) was applied [69]. Imaging was carried out in a heated environmental chamber with a water bubbler and several reservoirs of water to humidify the chamber. Phase images were acquired on a Nikon Ti-E inverted microscope every 5 min using a 40X air objective (NA: 0.95) with 1.5X magnification. Images were compiled into stacks and analyzed using Matlab or FIJI.
Fluorescent D-amino acid staining
HADA [70] was added to cultures at a final concentration of 500 μM during the last mass doubling of growth. To reduce background staining, cultures were diluted 1:10 in MSgg solution (5 mM potassium phosphate buffer + 0.05 M MOPS at pH 7) and then spotted onto a pad of that solution made with 1.5% agar. Cells were imaged using a Nikon Ti-E inverted microscope with a 100X oil objective (NA: 1.4). Phase and fluorescence (DAPI filter, ex/em: 375/460 nm, exposure time 2 s) images were acquired. Phase and fluorescence images were adjusted identically in Adobe Photoshop and merged in FIJI.
Plating efficiency assays
Cultures were harvested by piercing the sealed well with a sterile scalpel. Cultures were diluted 10-fold into LB. For a quantitative plating efficiency assay, 100 microliters of the dilutions were spread onto an LB-agar plate. For a spot plating efficiency assay, 10 microliters of the dilutions were dispensed onto an LB-agar plate and allowed to dry into the plate. These plates were incubated overnight at 37 °C in a single layer (not stacked). CFU/mL values of the original culture were back-calculated from the colony counts of the dilutions that had distinct colonies.
Lysis assay starting at different optical densities
Three colonies were inoculated into a single 10 mL LB culture and mixed well. Six two-fold serial dilutions of the culture were carried out and 5 mL of each dilution were transferred to a test tube and incubated at 37 °C on a roller drum until the undiluted culture reached an OD600~0.8. Two hundred microliters of each culture were then aliquoted into a 96-well plate, the plate was sealed with optical film to eliminate oxygen exchange, and OD600 was monitored in a Biotek Epoch2 spectrophotometer.
Oxygen depletion in an anaerobic chamber
Cultures were grown in aerobic conditions (96-well plate sealed with optical film, with holes poked through the film for each well). Cultures were then transferred directly into a Coy anaerobic chamber where the OD600 was monitored using a Biotek Epoch2 spectrophotometer using the parameters described in the “Growth and Lysis Assays” section above.
Membrane potential measurements
Membrane potential was measured using a protocol modified from [44]. Cells were grown in 5 mL LB in a roller drum to OD600~1. Cells were washed in a buffer containing 10 mM KPO4, 5 mM MgSO4, and 250 mM sucrose (pH 7.0) and then resuspended to a calculated OD600 of 0.085 in that same buffer (pelleting steps were at 5100 rcf for 3 min). Two hundred microliters of this mixture were added to wells in a 96-well plate. 1 μM DiSC3(5) or 10 μM Thioflavin T (ThT) was added to the wells. Fluorescence readings (em/ex: 620/685 for DiSC and 450/482 for ThT) were taken every 12 s with 5 s of linear shaking (567 cpm, 3-mm magnitude) on a Biotek Synergy H1 spectrophotometer. Readings were collected for ~3 min before the drugs were added and then readings were collected for another 10 min.
Surfactin addition after the start of depletion
Cultures were grown aerobically and depleted for oxygen as described in the “Growth and Lysis Assays” section above. At the indicated time points, the plate was removed from the aerobic plate reader and transferred to an anaerobic chamber. In the anaerobic chamber, the seal was removed, surfactin was added, and the plate was resealed. The plate was then brought back to aerobic conditions and incubation was continued in the plate reader.
ATP measurements
ATP levels were measured using an ATP bioluminescence assay kit CLS II (Roche) as described previously [71]. Briefly, 0.2 mL of culture were combined with 0.24 mL of formate solution (0.38 M formic acid, 17 mM EDTA) and rocked in ice for 15 min. Seventy-eight microliters of 2 M KOH were added to neutralize the solution and the culture was mixed well. Cell debris was pelleted by spinning at 21,130 rcf for 1 min. The supernatant was diluted 1:10 in assay buffer (100 mM Tris, pH 7.75, 4 mM EDTA). Fifty microliters of diluted supernatant were mixed with 50 μL of luciferase reagent in a black 96-well plate and luminescence was measured in a Biotek Synergy H1 spectrophotometer with a gain of 135, an integration time of 1 s, and a read height of 1 mm. Values were mapped to an ATP standard curve to calculate the molarity of the cell lysate.
Sucrose and NaCl treatments upon oxygen depletion
Cultures were grown in 96-well microtiter plates as described above. Sterile NaCl and sucrose solutions were added to a separate 96-well microtiter plate to result in a final concentration of 300 mM in 200 μL of culture. This plate was heated at 65 °C until all water evaporated from the solutions, leaving just the solute behind. The plate was then brought back to room temperature. Grown cultures were transferred to the plate with the solutes, and this plate was sealed to deplete oxygen and returned to the plate reader.
Biophysical model linking oxygen diffusivity and concentration in a growing culture
Since the addition of surfactin increased oxygen levels in late-exponential phase of a growing culture (Figure 3D), we sought to understand whether oxygen diffusivity could explain the increase. In a small region of extent Δl at a depth l below the air-liquid interface of the culture, oxygen is depleted at a rate γρ(l)c(l)Δl, where γ is the absorption rate per unit oxygen in close proximity to a cell, ρ(l) is the cell density at depth l, and c(l) is the oxygen concentration at depth l. At steady state, the rate of oxygen uptake in the region of extent Δl must be balanced by the rate of supply via diffusion, that is
| (1) |
where D is the diffusivity of oxygen. The solution to Eq. 1 is , assuming that c0 is the oxygen concentration at the surface, c → 0 as l → ∞, and γρ/D is independent of l. Hence, as D increases, oxygen reaches increased depths as , and the total amount of oxygen in the culture increases as . Thus, a 1.2-fold increase in the total amount of oxygen corresponds to a ~1.4-fold increase in D, assuming that c0 is linked to solubility and γ and ρ remain approximately constant.
Anaerobic culture media and growth conditions
LB-nitrate and LB-nitrite media were made based on the protocol in [29]. Briefly, LB medium (5 g/L NaCl) was supplemented with 20 mM potassium phosphate buffer (stock solution is 0.5 M, 2.72 g K2HPO4 and 1.275 g KH2PO4 in 50 mL) at pH 7.0, 2 mM (NH4)2SO4, 1 mM L-glutamic acid, 1 mM L-tryptophan, 0.8 mM L-phenylalanine, 0.005% ammonium iron (III) citrate, 1 mM glucose, and 10 mM sodium nitrate or sodium nitrite.
Spizizen minimal medium was made based on the protocol in [30], and was supplemented with 1% glycerol and a trace element solution as follows (given as final mass per liter): CaCl2 (5.5 mg), FeCl2•6H2O (13.5 mg), MnCl2•4H2O (1 mg), ZnCl2 (1.7 mg), CuCl2•2H2O (0.43 mg), CoCl2•6H2O (0.6 mg), Na2MoO4•2H2O (0.6 mg) [72]. NaNO3 and glutamate monosodium were added to final concentrations of 0.2%. To allow growth of 168, tryptophan was added at 50 mg/mL.
For anaerobic experiments, media and 96-well plates were pre-reduced in the anaerobic chamber for 48 h. Cells were grown overnight for 10 h in LB at 37 °C and diluted 1:200 into LB, LB-nitrate, LB-nitrite, or Spizizen minimal medium (with and without nitrate). OD600 was measured every 7.5 min in these media in anaerobic and aerobic conditions at 37 °C using the parameters described in the “Growth and Lysis Assays” section above. Holes were poked in the both the anaerobic and aerobic cultures to allow for air exchange. To prevent anaerobic cultures from evaporating, the cultures were completely sealed post-inoculation.
Quantification and Statistical Analysis
Quantification of growing rod-shaped cells
All rod-shaped cells were identified in the first frame of a compiled video of time-lapse microscopy and defined as able to grow if they at least doubled in mass and divided without lysis during the video.
Quantification of oxygen using the oxygen nanoprobe
Fluorescence and phosphorescence measurements were taken as indicated above. Blank fluorescent and phosphorescent values from a well without cells and without a nanoprobe were subtracted from the measurements. The fluorescence data was divided by the phosphorescence data (any timepoints where the phosphorescence data was zero were omitted from the analysis) and the relative oxygen concentrations were calculated by normalizing the first timepoint to 1 in the aerobic culture. Excessively noisy data was smoothed using either the “smoothdata” or “sgolay” functions on matlab and double checked to make sure that the smoothing did not alter the raw data trends.
Calculating doubling time and lysis rates from OD data
OD600 was measured as described above. A blank value from a well with no cells was subtracted from the curves and the OD curve was smoothed using the ‘sgolay’ function on Matlab. For aerobic growth, the maximum growth rate of growth curves was defined as the maximum value of the most positive slope of ln(OD600). For oxygen depletion lysis curves, the maximum lysis rate was defined as the absolute value of the most negative slope of ln(OD600).
Statistical Analysis and software
Matlab was used to analyze and plot all data in this manuscript. Statistical analyses carried out include unpaired Student’s t-test to determine statistical significance between two sets of data (ttest2 function in Matlab) and a linear regression model to determine correlation of the data. The definition of center for the data is typically the mean, and standard deviation or standard error of the mean is shown in figures and indicated in the figure legends. The statistical details including the tests used and the value of n can be found in the figure legends.
Data and Code Availability
This study did not generate any unique datasets or code.
Supplementary Material
Video S1: The majority of oxygen-depleted cells from strain 3610 wild-type cultures grow once oxygen is restored. Related to Figure 1 and S2. Cells from a 24 h oxygen-depleted wild-type 3610 culture were spotted onto an LB pad and imaged every 5 min at 37 °C. This experiment was done in tandem with the 3610 ΔlytC cultures in Video S2.
Video S2: A small minority of oxygen-depleted cells in strain 3610 ΔlytC cultures grow once oxygen is restored. Related to Figure 1 and S2. Cells from a 24 h oxygen-depleted ΔlytC culture were spotted onto an LB pad and imaged every 5 min at 37 °C. This experiment was done in tandem with the 3610 cultures in Video S1.
Video S3: The majority of oxygen-depleted strain 3610 cells grow once oxygen is restored. Related to Figure 2 and S3. This experiment was carried out as in Video S1 and was done in tandem with the 168 cultures in Video S4.
Video S4: A very small subset of oxygen-depleted, rod-shaped strain 168 cells grow once oxygen is restored. Related to Figure 2 and S3. Cells from a 24 h oxygen-depleted culture were spotted onto an LB pad and imaged every 5 min at 37 °C. This experiment was done in tandem with the 3610 cultures in Video S3.
Key Resources Table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Bacterial and Virus Strains | ||
| See Table S2 for all strains used in this study | ||
| Chemicals, Peptides, and Recombinant Proteins | ||
| LB lennox medium | RPI | Cat. #: L24066–1000.0 |
| Kanamycin sulfate | Sigma-Aldrich | SKU: K1377–5G |
| Erythromycin | Sigma-Aldrich | SKU: E5389–5G |
| Lincomycin hydrochloride | Sigma-Aldrich | SKU: L2774–5MU |
| Chloramphenical | Calbiochem | Cat. #: 220551 |
| Rifampin | Sigma-Aldrich | SKU: R3501–1G |
| Spectinomycin dihydrochloride | Sigma-Aldrich | SKU: SB0901–25 |
| Levofloxacin | Sigma-Aldrich | SKU: 28266–1G-F |
| Surfactin | Sigma-Aldrich | SKU: S3523–10MG |
| MnSO4 | Sigma-Aldrich | SKU: 221287–100G |
| MgSO4 | Sigma-Aldrich | SKU: M2643–500G |
| BF2nbm(I)PLA | Cassandra Fraser | N/A |
| Propidium Iodide | Sigma-Aldrich | SKU: P4170–10MG |
| HADA | Erkin Kuru | N/A |
| Sucrose | VWR | Cat. #: BT143635–500G |
| NaCl | Fisher Scientific | Cat. #: BP358–212 |
| Thioflavin T | Sigma-Aldrich | SKU: T3516–5G |
| DiSC3(5) | Sigma-Aldrich | SKU: 43608–100MG |
| Calcium chloride dihydrate | Fisher Scientific | Cat. #: C79–500 |
| Iron (III) chloride hexahydrate | Sigma-Aldrich | SKU: F2877–500G |
| Manganese chloride tetrahydrate | Millipore Sigma | SKU: 21279–100G |
| Zinc chloride | Sigma-Aldrich | SKU: 208086–5G |
| Copper(II) chloride dihydrate | Sigma-Aldrich | SKU: C3279–100G |
| Cobalt (II) chloride hexahydrate | Sigma-Aldrich | SKU: C8661–25 |
| Sodium molybdate dihydrate | Sigma-Aldrich | SKU: 480967–25G |
| Sodium nitrate | Sigma-Aldrich | SKU: S5506–250G |
| Sodium nitrite | Sigma-Aldrich | SKU: 237213–100G |
| L-glutamic acid monosodium salt hydrate | Sigma-Aldrich | SKU: G1626–500G |
| K2HPO4 | Sigma-Aldrich | SKU: P8281–500G |
| KH2PO4 | Fisher Scientific | Cat. #: BP362–500 |
| Ammonium sulfate | Sigma-Aldrich | SKU: A4418–100G |
| L-glutamic acid potassium salt monohydrate | Sigma-Aldrich | SKU: G1501–100G |
| L-phenylalanine | Alfa Aesar | Stock #: A13238 |
| L-tryptophan | Sigma-Aldrich | SKU: T0254–100G |
| Sodium citrate trisodium salt dyhydrate | MP Biomedicals | SKU 0219486890 |
| D-(+)-xylose | Sigma-Aldrich | SKU: X1500–500G |
| Glucose | Fisher Chemical | Cat. #: D16–3 |
| ATP bioluminescence assay kit CLS II | Roche/Sigma Aldrich | SKU: 11699695001 |
| Sau3A1 | NEB | Cat. #: R0169S |
| Phusion | Thermo Scientific | Cat. #: F530S |
| T4 ligase | Thermo Scientific | Cat. #: EL0014 |
| Wizard ® Genomic DNA Purification Kit | Promega | Cat # A1120 |
| Oligonucleotides | ||
| AGGAGGTCGACGAATTAGTCTTGATGGAAAGCAGTAT | This work | 1427 (LytCUF SalI) |
| CTCCTCGGCCGAAGCTGTTGGCACAAAAAGTATGAG | This work | 1428 (lytCUR EagI) |
| AGGAGCGGCCGCATCAGATGCAAGTAAATTGAAGCA | This work | 1425 (lytCDF EagI) |
| CTCCTGGATCCCATGATTTGTTTTGTAATACTTGGCAT | This work | 1426 (lytCDR BamHI) |
| AGGAGCGGCCGAAAAAACTTAGAAAGTTGCAAATAGGCT | This work | 1429 (lytDUR EagI) |
| CTCCTGTCGACTTGATATGAAGAATAGACAGTTGGCA | This work | 1430 (lytDUF SalI) |
| AGGAGGGATCCAGTATTCCTTCAGAATCAACGGGGGAAT | This work | 1431 (lytDDR BamHI) |
| CTCCTCGGCCGTTAGTCTCTTTTTCATTCCTTCTCCTCTT | This work | 1432 (lytDDF EagI) |
| GCTTGTAAATTCTATCATAATTG | This work | 695 (IPRC1) |
| AGGGAATCATTTGAAGGTTGG | This work | 696 (IPCR2) |
| Recombinant DNA | ||
| pDR244 | [13] | N/A |
| pDP299 | This work | N/A |
| pDP300 | This work | N/A |
| pMiniMAD | [63] | N/A |
| Software and Algorithms | ||
| Matlab | MathWorks | R2018a |
| Adobe Photoshop | Adobe, Inc. | CS6 |
| FIJI | ImageJ | 2.0.0-rc-44/1.50e |
Highlights.
The majority of Bacillus subtilis cells die upon oxygen depletion
Surfactin production depolarizes cells to maintain viability upon oxygen depletion
Surfactin promotes growth in early stationary phase by enhancing oxygen diffusion
The autolytic enzyme LytC and surfactin mediate lysis upon oxygen depletion
Acknowledgements
The authors thank John Helmann, Alfred Spormann, Kyler Lugo, and the Huang lab for helpful discussions, Chao Jiang for help with strain construction, and Maya Farha and Eric Brown for advice regarding membrane potential experiments. The authors acknowledge support from the Allen Discovery Center at Stanford on Systems Modeling of Infection (to H.A.A. and K.C.H.), National Institutes of Health (NIH) grants R35 GM131783 (to C.M.D. and D.B.K.) and R01 CA167250 (to C.A.D. and C.L.F.), and the Stanford Summer Research Amgen Scholars Program (to L.V.). K.C.H. is a Chan Zuckerberg Biohub Investigator.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declaration of Interests
The authors declare no competing interests.
References
- 1.Boutilier RG (2001). Mechanisms of cell survival in hypoxia and hypothermia. J Exp Biol 204, 3171–3181. [DOI] [PubMed] [Google Scholar]
- 2.Hochachka PW (1986). Defense strategies against hypoxia and hypothermia. Science 231, 234–241. [DOI] [PubMed] [Google Scholar]
- 3.Sendoel A, and Hengartner MO (2014). Apoptotic cell death under hypoxia. Physiology (Bethesda) 29, 168–176. [DOI] [PubMed] [Google Scholar]
- 4.Prescott LM, Harley JP, and Klein DA (2005). Microbiology, 6th Edition, (Dubuque, IA: McGraw-Hill Higher Education; ). [Google Scholar]
- 5.Wayne LG, and Hayes LG (1996). An in vitro model for sequential study of shiftdown of Mycobacterium tuberculosis through two stages of nonreplicating persistence. Infect Immun 64, 2062–2069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Rao SP, Alonso S, Rand L, Dick T, and Pethe K (2008). The protonmotive force is required for maintaining ATP homeostasis and viability of hypoxic, nonreplicating Mycobacterium tuberculosis. Proc Natl Acad Sci U S A 105, 11945–11950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Berney M, Greening C, Conrad R, Jacobs WR Jr., and Cook GM (2014). An obligately aerobic soil bacterium activates fermentative hydrogen production to survive reductive stress during hypoxia. Proc Natl Acad Sci U S A 111, 11479–11484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.O’Toole R, Smeulders MJ, Blokpoel MC, Kay EJ, Lougheed K, and Williams HD (2003). A two-component regulator of universal stress protein expression and adaptation to oxygen starvation in Mycobacterium smegmatis. J Bacteriol 185, 1543–1554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Pusey PL, and Wilson CL (1984). Postharvest biological control of stone fruit brown rot by Bacillus subtilis. Plant Disease 68, 753–756. [Google Scholar]
- 10.Kumar AS, Lakshmanan V, Caplan JL, Powell D, Czymmek KJ, Levia DF, and Bais HP (2012). Rhizobacteria Bacillus subtilis restricts foliar pathogen entry through stomata. The Plant Journal 72, 694–706. [DOI] [PubMed] [Google Scholar]
- 11.Sierra J, and Renault P (1998). Temporal Pattern of Oxygen Concentration in a Hydromorphic Soil. Soil Science Society of America Journal 62, 1398–1405. [Google Scholar]
- 12.Kaufman W, and Bauer K (1958). Some studies of the mechanism of the“ anaerobic autolysis” of Bacillus subtilis J. Gen. Microbiol 18. [Google Scholar]
- 13.Koo BM, Kritikos G, Farelli JD, Todor H, Tong K, Kimsey H, Wapinski I, Galardini M, Cabal A, Peters JM, et al. (2017). Construction and Analysis of Two Genome-Scale Deletion Libraries for Bacillus subtilis. Cell Syst 4, 291–305 e297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Peters JM, Colavin A, Shi H, Czarny TL, Larson MH, Wong S, Hawkins JS, Lu CHS, Koo BM, Marta E, et al. (2016). A Comprehensive, CRISPR-based Functional Analysis of Essential Genes in Bacteria. Cell 165, 1493–1506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zhu B, and Stulke J (2018). SubtiWiki in 2018: from genes and proteins to functional network annotation of the model organism Bacillus subtilis. Nucleic Acids Res 46, D743–D748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zeigler DR, Pragai Z, Rodriguez S, Chevreux B, Muffler A, Albert T, Bai R, Wyss M, and Perkins JB (2008). The origins of 168, W23, and other Bacillus subtilis legacy strains. J Bacteriol 190, 6983–6995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Julkowska D, Obuchowski M, Holland IB, and Seror SJ (2005). Comparative analysis of the development of swarming communities of Bacillus subtilis 168 and a natural wild type: critical effects of surfactin and the composition of the medium. J Bacteriol 187, 65–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Nakano MM, Corbell N, Besson J, and Zuber P (1992). Isolation and characterization of sfp: a gene that functions in the production of the lipopeptide biosurfactant, surfactin, in Bacillus subtilis. Mol Gen Genet 232, 313–321. [DOI] [PubMed] [Google Scholar]
- 19.Nakano MM, Marahiel MA, and Zuber P (1988). Identification of a genetic locus required for biosynthesis of the lipopeptide antibiotic surfactin in Bacillus subtilis. J Bacteriol 170, 5662–5668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Arima K, Kakinuma A, and Tamura G (1968). Surfactin, a crystalline peptidelipid surfactant produced by Bacillus subtilis: isolation, characterization and its inhibition of fibrin clot formation. Biochem Biophys Res Commun 31, 488–494. [DOI] [PubMed] [Google Scholar]
- 21.Grau RR, de Ona P, Kunert M, Lenini C, Gallegos-Monterrosa R, Mhatre E, Vileta D, Donato V, Holscher T, Boland W, et al. (2015). A Duo of Potassium-Responsive Histidine Kinases Govern the Multicellular Destiny of Bacillus subtilis. MBio 6, e00581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kearns DB, and Losick R (2003). Swarming motility in undomesticated Bacillus subtilis. Mol Microbiol 49, 581–590. [DOI] [PubMed] [Google Scholar]
- 23.Kinsinger RF, Kearns DB, Hale M, and Fall R (2005). Genetic requirements for potassium ion-dependent colony spreading in Bacillus subtilis. J Bacteriol 187, 8462–8469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Płaza GA, Turek A, Król E, and Szczygłowska R (2013). Antifungal and antibacterial properties of surfactin isolated from Bacillus subtilis growing on molasses. African Journal of Microbiology Research 7, 3165–3170. [Google Scholar]
- 25.Korenblum E, de Araujo LV, Guimaraes CR, de Souza LM, Sassaki G, Abreu F, Nitschke M, Lins U, Freire DM, Barreto-Bergter E, et al. (2012). Purification and characterization of a surfactin-like molecule produced by Bacillus sp. H2O-1 and its antagonistic effect against sulfate reducing bacteria. BMC Microbiol 12, 252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Rosenberg G, Steinberg N, Oppenheimer-Shaanan Y, Olender T, Doron S, Ben-Ari J, Sirota-Madi A, Bloom-Ackermann Z, and Kolodkin-Gal I (2016). Not so simple, not so subtle: the interspecies competition between Bacillus simplex and Bacillus subtilis and its impact on the evolution of biofilms. NPJ Biofilms Microbiomes 2, 15027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Snook ME, Mitchell T, Hinton DM, and Bacon CW (2009). Isolation and characterization of leu7-surfactin from the endophytic bacterium Bacillus mojavensis RRC 101, a biocontrol agent for Fusarium verticillioides. J Agric Food Chem 57, 4287–4292. [DOI] [PubMed] [Google Scholar]
- 28.Zhi Y, Wu Q, Du H, and Xu Y (2016). Biocontrol of geosmin-producing Streptomyces spp. by two Bacillus strains from Chinese liquor. Int J Food Microbiol 231, 1–9. [DOI] [PubMed] [Google Scholar]
- 29.Cruz Ramos H, Hoffmann T, Marino M, Nedjari H, Presecan-Siedel E, Dreesen O, Glaser P, and Jahn D (2000). Fermentative metabolism of Bacillus subtilis: physiology and regulation of gene expression. J Bacteriol 182, 3072–3080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Nakano MM, Dailly YP, Zuber P, and Clark DP (1997). Characterization of anaerobic fermentative growth of Bacillus subtilis: identification of fermentation end products and genes required for growth. J Bacteriol 179, 6749–6755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Nakano MM, and Zuber P (1998). Anaerobic growth of a “strict aerobe” (Bacillus subtilis). Annu Rev Microbiol 52, 165–190. [DOI] [PubMed] [Google Scholar]
- 32.Cabeen MT, and Jacobs-Wagner C (2005). Bacterial cell shape. Nat Rev Microbiol 3, 601–610. [DOI] [PubMed] [Google Scholar]
- 33.Lazarevic V, Margot P, Soldo B, and Karamata D (1992). Sequencing and analysis of the Bacillus subtilis lytRABC divergon: a regulatory unit encompassing the structural genes of the N-acetylmuramoyl-L-alanine amidase and its modifier. J Gen Microbiol 138, 1949–1961. [DOI] [PubMed] [Google Scholar]
- 34.Margot P, Mauel C, and Karamata D (1994). The gene of the N-acetylglucosaminidase, a Bacillus subtilis 168 cell wall hydrolase not involved in vegetative cell autolysis. Mol Microbiol 12, 535–545. [DOI] [PubMed] [Google Scholar]
- 35.Koch AL (2001). Autolysis control hypotheses for tolerance to wall antibiotics. Antimicrob Agents Chemother 45, 2671–2675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Sheppard JD, Jumarie C, Cooper DG, and Laprade R (1991). Ionic channels induced by surfactin in planar lipid bilayer membranes. Biochim Biophys Acta 1064, 13–23. [DOI] [PubMed] [Google Scholar]
- 37.Hotez L, Dailey JW, Geelhoed GW, and Gainer JL (1977). The role of oxygen diffusivity in biochemical reactions. Experientia 33, 1424–1425. [DOI] [PubMed] [Google Scholar]
- 38.Laidig KE, Gainer JL, and Daggett V (1998). Altering Diffusivity in Biological Solutions through Modification of Solution Structure and Dynamics. J Am Chem Soc 120, 9394–9395. [Google Scholar]
- 39.Olmeda B, Villen L, Cruz A, Orellana G, and Perez-Gil J (2010). Pulmonary surfactant layers accelerate O(2) diffusion through the air-water interface. Biochim Biophys Acta 1798, 1281–1284. [DOI] [PubMed] [Google Scholar]
- 40.Cosmina P, Rodriguez F, de Ferra F, Grandi G, Perego M, Venema G, and van Sinderen D (1993). Sequence and analysis of the genetic locus responsible for surfactin synthesis in Bacillus subtilis. Mol Microbiol 8, 821–831. [DOI] [PubMed] [Google Scholar]
- 41.Hayashi K, Kensuke T, Kobayashi K, Ogasawara N, and Ogura M (2006). Bacillus subtilis RghR (YvaN) represses rapG and rapH, which encode inhibitors of expression of the srfA operon. Mol Microbiol 59, 1714–1729. [DOI] [PubMed] [Google Scholar]
- 42.Nakano MM, Xia LA, and Zuber P (1991). Transcription initiation region of the srfA operon, which is controlled by the comP-comA signal transduction system in Bacillus subtilis. J Bacteriol 173, 5487–5493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lopez D, Fischbach MA, Chu F, Losick R, and Kolter R (2009). Structurally diverse natural products that cause potassium leakage trigger multicellularity in Bacillus subtilis. Proc Natl Acad Sci U S A 106, 280–285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Farha MA, Verschoor CP, Bowdish D, and Brown ED (2013). Collapsing the proton motive force to identify synergistic combinations against Staphylococcus aureus. Chem Biol 20, 1168–1178. [DOI] [PubMed] [Google Scholar]
- 45.Prindle A, Liu J, Asally M, Ly S, Garcia-Ojalvo J, and Suel GM (2015). Ion channels enable electrical communication in bacterial communities. Nature 527, 59–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Varma S, Sabo D, and Rempe SB (2008). K+/Na+ selectivity in K channels and valinomycin: over-coordination versus cavity-size constraints. J Mol Biol 376, 13–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Strahl H, Burmann F, and Hamoen LW (2014). The actin homologue MreB organizes the bacterial cell membrane. Nat Commun 5, 3442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Lobritz MA, Belenky P, Porter CB, Gutierrez A, Yang JH, Schwarz EG, Dwyer DJ, Khalil AS, and Collins JJ (2015). Antibiotic efficacy is linked to bacterial cellular respiration. Proc Natl Acad Sci U S A 112, 8173–8180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.MacArthur CG (1916). Solubility of Oxygen in Salt Solutions and the Hydrates of These Salts. J. Phys. Chem 20. [Google Scholar]
- 50.Joslyn MA, and Supplee H (1949). Solubility of oxygen in solutions of various sugars. Food Res 14, 209–215. [DOI] [PubMed] [Google Scholar]
- 51.Moller MC, and Hederstedt L (2008). Extracytoplasmic processes impaired by inactivation of trxA (thioredoxin gene) in Bacillus subtilis. J Bacteriol 190, 4660–4665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Thérien M, Kiesewalter HT, Auria E, Charron-Lamoureux V, Wibowo M, Maróti G, Kovács AT, and Beauregard PB (2019). Surfactin Production is not essential for pellicle and root-associated biofilm development of Bacillus subtilis. bioRxiv, 865345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Pham JV, Yilma MA, Feliz A, Majid MT, Maffetone N, Walker JR, Kim E, Cho HJ, Reynolds JM, Song MC, et al. (2019). A Review of the Microbial Production of Bioactive Natural Products and Biologics. Front Microbiol 10, 1404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Hu J, Lei P, Mohsin A, Liu X, Huang M, Li L, Hu J, Hang H, Zhuang Y, and Guo M (2017). Mixomics analysis of Bacillus subtilis: effect of oxygen availability on riboflavin production. Microb Cell Fact 16, 150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Garcia-Ochoa F, and Gomez E (2009). Bioreactor scale-up and oxygen transfer rate in microbial processes: an overview. Biotechnol Adv 27, 153–176. [DOI] [PubMed] [Google Scholar]
- 56.Arjes HA, Kriel A, Sorto NA, Shaw JT, Wang JD, and Levin PA (2014). Failsafe mechanisms couple division and DNA replication in bacteria. Curr Biol 24, 2149–2155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Rojas ER, Huang KC, and Theriot JA (2017). Homeostatic Cell Growth Is Accomplished Mechanically through Membrane Tension Inhibition of Cell-Wall Synthesis. Cell Syst 5, 578–590 e576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Blackman SA, Smith TJ, and Foster SJ (1998). The role of autolysins during vegetative growth of Bacillus subtilis 168. Microbiology 144 (Pt 1), 73–82. [DOI] [PubMed] [Google Scholar]
- 59.Calamita HG, Ehringer WD, Koch AL, and Doyle RJ (2001). Evidence that the cell wall of Bacillus subtilis is protonated during respiration. Proc Natl Acad Sci U S A 98, 15260–15263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Mueller EA, Egan AJ, Breukink E, Vollmer W, and Levin PA (2019). Plasticity of Escherichia coli cell wall metabolism promotes fitness and antibiotic resistance across environmental conditions. Elife 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Peters K, Kannan S, Rao VA, Biboy J, Vollmer D, Erickson SW, Lewis RJ, Young KD, and Vollmer W (2016). The Redundancy of Peptidoglycan Carboxypeptidases Ensures Robust Cell Shape Maintenance in Escherichia coli. MBio 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Stratford JP, Edwards CLA, Ghanshyam MJ, Malyshev D, Delise MA, Hayashi Y, and Asally M (2019). Electrically induced bacterial membrane-potential dynamics correspond to cellular proliferation capacity. Proc Natl Acad Sci U S A 116, 9552–9557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Yasbin RE, and Young FE (1974). Transduction in Bacillus subtilis by bacteriophage SPP1. J Virol 14, 1343–1348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Patrick JE, and Kearns DB (2008). MinJ (YvjD) is a topological determinant of cell division in Bacillus subtilis. Mol Microbiol 70, 1166–1179. [DOI] [PubMed] [Google Scholar]
- 65.Arnaud M, Chastanet A, and Debarbouille M (2004). New vector for efficient allelic replacement in naturally nontransformable, low-GC-content, gram-positive bacteria. Appl Environ Microbiol 70, 6887–6891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Le Breton Y, Mohapatra NP, and Haldenwang WG (2006). In vivo random mutagenesis of Bacillus subtilis by use of TnYLB-1, a mariner-based transposon. Appl Environ Microbiol 72, 327–333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.DeRosa CA, Samonina-Kosicka J, Fan Z, Hendargo HC, Weitzel DH, Palmer GM, and Fraser CL (2015). Oxygen Sensing Difluoroboron Dinaphthoylmethane Polylactide. Macromolecules 48, 2967–2977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.DeRosa CA, Seaman SA, Mathew AS, Gorick CM, Fan Z, Demas JN, Peirce SM, and Fraser CL (2016). Oxygen Sensing Difluoroboron beta-Diketonate Polylactide Materials with Tunable Dynamic Ranges for Wound Imaging. ACS Sens 1, 1366–1373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Shi H, Colavin A, Lee TK, and Huang KC (2017). Strain Library Imaging Protocol: high-throughput, automated single-cell microscopy for large bacterial collections arrayed on multiwell plates. Nature Protocols. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Kuru E, Hughes HV, Brown PJ, Hall E, Tekkam S, Cava F, de Pedro MA, Brun YV, and VanNieuwenhze MS (2012). In Situ probing of newly synthesized peptidoglycan in live bacteria with fluorescent D-amino acids. Angew Chem Int Ed Engl 51, 12519–12523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Zhang S, and Haldenwang WG (2005). Contributions of ATP, GTP, and redox state to nutritional stress activation of the Bacillus subtilis sigmaB transcription factor. J Bacteriol 187, 7554–7560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Spizizen J (1958). Transformation of Biochemically Deficient Strains of Bacillus subtilis by Deoxyribonucleate. Proc Natl Acad Sci U S A 44, 1072–1078. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Video S1: The majority of oxygen-depleted cells from strain 3610 wild-type cultures grow once oxygen is restored. Related to Figure 1 and S2. Cells from a 24 h oxygen-depleted wild-type 3610 culture were spotted onto an LB pad and imaged every 5 min at 37 °C. This experiment was done in tandem with the 3610 ΔlytC cultures in Video S2.
Video S2: A small minority of oxygen-depleted cells in strain 3610 ΔlytC cultures grow once oxygen is restored. Related to Figure 1 and S2. Cells from a 24 h oxygen-depleted ΔlytC culture were spotted onto an LB pad and imaged every 5 min at 37 °C. This experiment was done in tandem with the 3610 cultures in Video S1.
Video S3: The majority of oxygen-depleted strain 3610 cells grow once oxygen is restored. Related to Figure 2 and S3. This experiment was carried out as in Video S1 and was done in tandem with the 168 cultures in Video S4.
Video S4: A very small subset of oxygen-depleted, rod-shaped strain 168 cells grow once oxygen is restored. Related to Figure 2 and S3. Cells from a 24 h oxygen-depleted culture were spotted onto an LB pad and imaged every 5 min at 37 °C. This experiment was done in tandem with the 3610 cultures in Video S3.
Data Availability Statement
This study did not generate any unique datasets or code.