

Abstract
Apolipoprotein E (ApoE) is an important modifier of Alzheimer’s disease (AD) pathogenesis, and its abundance has been linked to the clearance of β-amyloid (Aβ) in the brain. The pathways that control the clearance of ApoE in the brain are incompletely understood. We report that Idol, an E3 ubiquitin ligase that targets the low-density lipoprotein receptor (LDLR) for degradation, is a critical determinant of brain ApoE metabolism and Aβ plaque biogenesis. Previous work has shown that Idol contributes minimally to the regulation of hepatic LDLR expression in mice. By contrast, we demonstrate that Idol is a primary physiological regulator of LDLR protein in the brain, controlling the clearance of both ApoE-containing high-density lipoprotein (HDL) particles and Aβ. We studied the consequences of loss of Idol expression in a transgenic mouse model of Aβ amyloidosis. Idol deficiency increased brain LDLR, decreased ApoE, decreased soluble and insoluble Aβ, reduced amyloid plaque burden, and ameliorated neuroinflammation. These findings identify Idol as a gatekeeper of LDLR-dependent ApoE and Aβ clearance in the brain and a potential enzyme target for therapeutic intervention in AD.
INTRODUCTION
Alzheimer’s disease (AD) is a major form of dementia characterized by progressive loss of neurons and neuronal processes and impaired brain function (1, 2). Key pathological features of AD include the accumulation of extracellular amyloid plaques and the formation of intraneuronal neurofibrillary tangles, consisting of β-amyloid (Aβ) peptides and hyperphosphorylated tau proteins, respectively (3). Decades of research suggest that the pathogenesis of AD is linked to interactions between multiple factors, including Aβ, apolipoprotein E (ApoE), tau, and aging (4). However, our understanding is still incomplete, and there are no effective treatments for AD.
ApoE is a major cholesterol carrier in the brain (5). Although several genetic risk factors for AD have been identified, the APOE genotype is the strongest (6). Mounting evidence suggests that ApoE influences AD risk through effects on Aβ metabolism. Genetic deletion of ApoE, anti-ApoE immunotherapy, and increasing ApoE lipidation have all been shown to reduce Aβ aggregation in mouse models (7–10). Thus, expanding our understanding of ApoE and Aβ metabolism and their mechanistic links may identify new opportunities for intervention in AD.
The low-density lipoprotein receptor (LDLR) is a cell surface receptor that facilitates the endocytosis of plasma lipoprotein particles containing ApoB100 or ApoE (11). We identified Idol as a modulator of LDLR function and cholesterol homeostasis (12–15). Idol is an E3 ubiquitin ligase that is transcriptionally regulated by the cholesterol-responsive liver X receptors (LXRs). The Idol-UBE2D complex ubiqui-tinates the LDLR on its cytoplasmic domain, thereby targeting it for lysosomal degradation (16, 17). Previous studies have suggested that ectopic overexpression of LDLR decreases ApoE and inhibits Aβ deposition (18, 19). However, the question of whether increasing endogenous LDLR expression would affect AD pathogenesis remains to be addressed.
Loss of Idol expression in mice increases LDLR in some tissues, such as adipose, but not in others, such as liver (13). Thus, the potential impact of Idol on brain LDLR expression cannot be predicted from previous work. We report here that Idol plays a dominant role in determining LDLR and ApoE protein levels in the brain. Given this regulation, we hypothesized that Idol may affect AD pathogenesis. We assessed the impact of loss of Idol expression on the development of Aβ amyloidosis in the amyloid precursor protein/presenilin-1 (APP/PS1) mouse model of AD. Our results identify the LXR-Idol pathway as an important effector of ApoE and Aβ clearance in the brain and a potential target for therapeutic intervention in AD.
RESULTS
The LXR-Idol pathway regulates LDLR protein expression in the mouse brain
To address the function of the LXR-Idol pathway in the mouse brain, we first characterized Idol expression. Idol mRNA was readily detected in all examined subregions of the mouse central nervous system (CNS) (fig. S1A). Analysis of different cell types present in the CNS revealed abundant Idol mRNA in primary mouse microglia and neurons and lower amounts in astrocytes (Fig. 1A). We targeted the mouse Idol gene with a knockout cassette containing a LacZ reporter (13). Immunohistochemistry confirmed coexpression of β-galactosidase (β-Gal) protein with the microglia marker Iba1 and the neuronal marker NeuN in Idol+/− cells (Fig. 1B).
Fig. 1. The LXR-Idol axis regulates LDLR abundance in the brain.
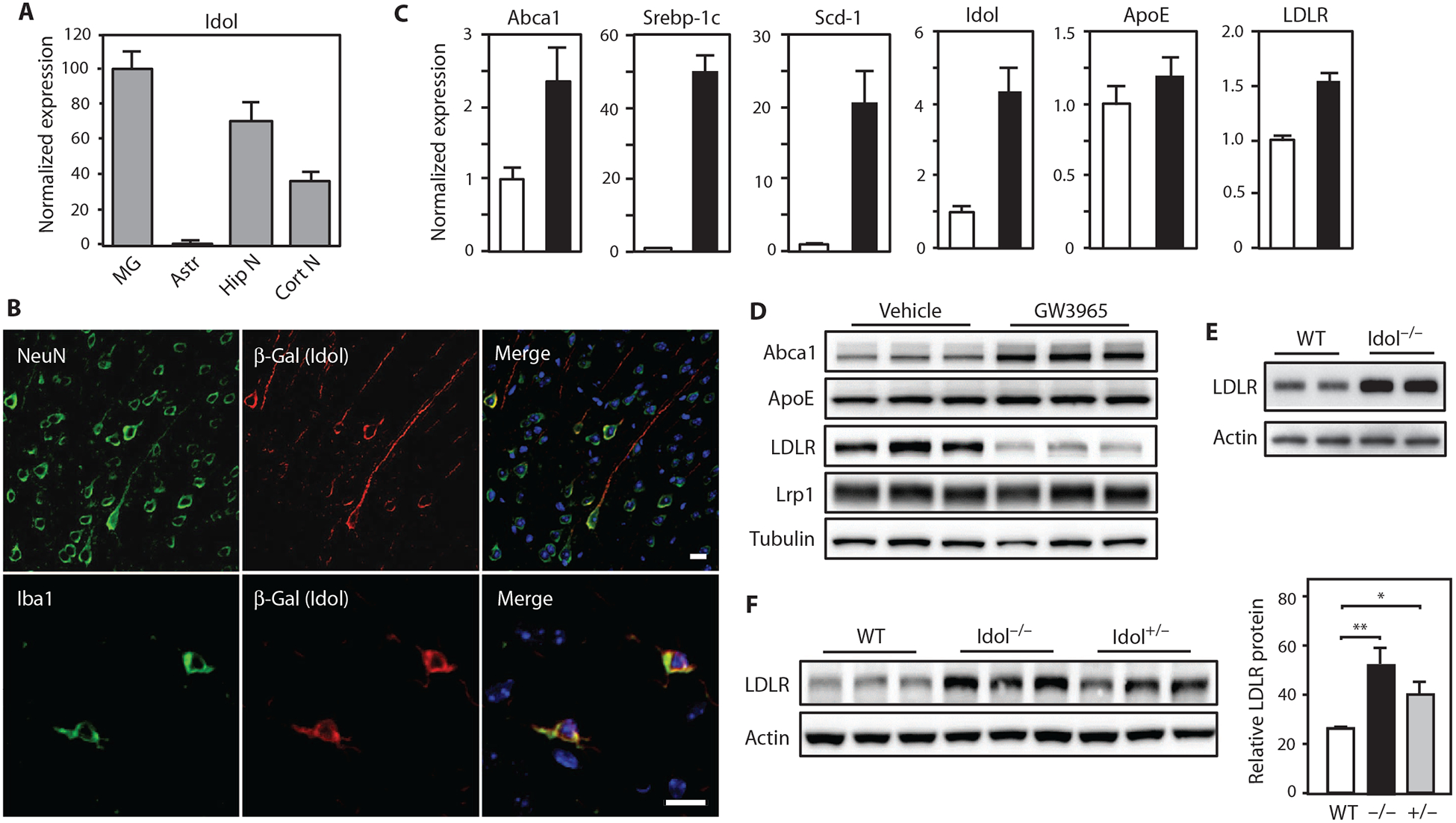
(A) Real-time polymerase chain reaction (PCR) analysis of Idol mRNA expression in different mouse brain cell types, normalized to microglial expression. MG, microglia; Astr, astrocytes; Hip N, hippocampal neurons; Cort N, cortical neurons. Error bars represent SEM. (B) Representative micrographs showing immunofluorescence staining of brain sections from the frontal cortex of Idol+/− mice. Images in top row show Idol expression in neurons. Images in bottom row show Idol expression in microglia. Green, NeuN (neuron) and Iba1 (microglia); red, Idol; blue, 4′,6-diamidino-2-pheny-lindole (DAPI; nucleus). Scale bars, 10 μm. (C) Real-time PCR analysis of LXR target gene expression in primary microglia treated with dimethyl sulfoxide (DMSO) or 1 μM GW3965 for 6 hours, normalized to vehicle (DMSO) control. Error bars represent SEM. (D) Immunoblot analysis of whole-cell lysates from primary microglia treated with DMSO or 1 μM GW3965 for 6 hours. Lanes represent samples from individual cultures. (E) Immunoblot analysis of whole-cell lysates from primary microglia isolated from Idol+/+ or Idol−/− mice. Lanes represent samples from individual animals. WT, wild type. (F) Immunoblot analysis of whole-cell lysates from the frontal cortex of Idol+/+, Idol+/−, or Idol−/− mice. Lanes represent samples from individual animals. Immunoblot signals are quantified on the right. Error bars represent SEM. *P < 0.05, **P < 0.01 by Student’s t test. Each experiment was performed at least twice.
Microglia can exert context-specific effects on AD pathogenesis (20, 21). We tested the ability of Idol to regulate LDLR expression in these cells on the basis of its prominent expression in microglia. Treatment with an LXR agonist robustly increased Idol mRNA in primary mouse microglia (Fig. 1C). Genes encoding the canonical LXR targets Abca1, Srebp-1c, and Scd-1 were up-regulated in parallel, whereas ApoE mRNA expression was unchanged (Fig. 1C). Induction of Idol by an LXR agonist, in turn, resulted in a marked reduction of LDLR protein, consistent with its degradation (Fig. 1D). Abca1 protein was increased by LXR agonist in microglia as expected, but Lrp1 did not change. We also found that overexpression of Idol decreased LDLR and triggered LDLR ubiquitination in Neuro2a mouse neuronal cells (fig. S1, B to D). Conversely, primary microglia from Idol−/− mice exhibited increased LDLR expression compared to wild-type microglia (Fig. 1E).
We next addressed the ability of Idol to regulate LDLR protein in the brain. Immunoblotting revealed striking increases in LDLR in the brains of Idol−/− mice (Fig. 1F). The amount of LDLR protein in Idol+/− mice was intermediate between the amounts of LDLR protein in wild-type and Idol−/− mice, consistent with a gene dosage effect (P < 0.05; Fig. 1F). These observations suggest that Idol plays a dominant role in determining LDLR expression in the brain under physiological conditions.
Loss of Idol decreases Aβ and reduces amyloid plaque burden in a mouse model of Aβ amyloidosis
On the basis of the above findings, we hypothesized that loss of Idol may attenuate the deposition of amyloid plaques. We bred Idol−/− mice with mice carrying APPswe and PSEN1dE9 transgenes (denoted APP/PS1 mice) (22). The extent of Aβ deposition in the brains of the progeny was analyzed at 7 months of age, a time when early amyloidosis is apparent. Because male and female APP/PS1 mice develop amyloid deposits at different rates (19), we analyzed both sexes independently. We first measured Aβ in the cortex by enzyme-linked immunosorbent assay (ELISA). Compared to APP/PS1;Idol+/+ mice, insoluble and soluble Aβ40 and Aβ42 were prominently decreased in the cortex in both male and female APP/PS1;Idol−/− mice (Fig. 2A). The reduction in insoluble and soluble Aβ42 in the cortex of male APP/PS1;Idol+/− mice was also significant versus APP/PS1;Idol+/+ mice (P < 0.01), suggesting that even partial inhibition of Idol activity was sufficient to suppress the accumulation of amyloid plaque.
Fig. 2. Loss of Idol expression inhibits plaque formation in a mouse model of Aβ amyloidosis.
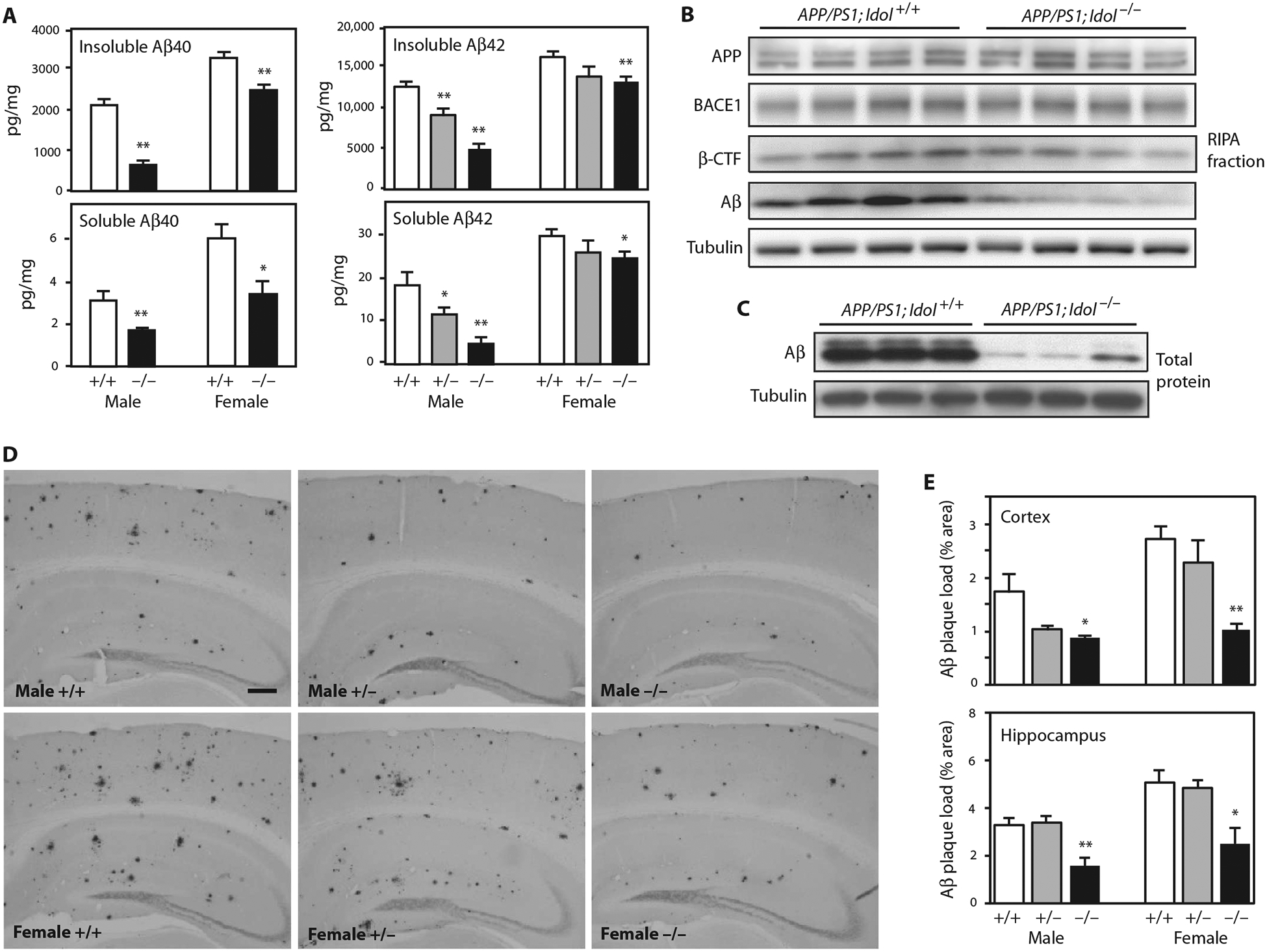
(A) Quantification of Aβ40 and Aβ42 peptides in the insoluble guanidine fraction and the soluble RIPA fraction from the frontal cortex of APP/PS1;Idol+/+, APP/PS1;Idol+/− (for Aβ42), and APP/PS1;Idol−/− mice. Error bars represent SEM. *P < 0.05, **P < 0.01 by Student’s t test. n = 5 to 7. (B) Immunoblot analysis of brain RIPA fractions from APP/PS1; Idol+/+ and APP/PS1;Idol−/− mice. APP was detected with 4G8 antibody. Aβ and β-CTF were detected with 82E1 antibody. (C) Immunoblot analysis of total brain lysates from APP/PS1;Idol+/+ and APP/PS1;Idol−/− mice. (D) Representative micrographs showing immunohistochemical staining of brain sections from APP/PS1;Idol+/+, APP/PS1;Idol+/−, and APP/PS1;Idol−/− mice with Aβ-specific 82E1B antibody. Black stain indicates Aβ plaques. Scale bar, 250 μm. (E) Quantification of Aβ plaque load in the frontal cortex and hippocampus of APP/PS1;Idol+/+, APP/PS1;Idol+/−, and APP/PS1;Idol−/− mice by 82E1B antibody staining for Aβ. Error bars represent SEM. *P < 0.05, **P < 0.01 by Student’s t test. n = 6.
We further analyzed protein expression in radioimmunoprecipitation (RIPA) buffer fractions from brains of APP/PS1;Idol+/+ and APP/PS1;Idol−/− mice by immunoblotting (Fig. 2B). Idol genotype did not affect total APP protein, nor did it affect the expression of the cleavage enzyme BACE1 or its cleavage product β C-terminal fragment (β-CTF). We also assessed Aβ peptide in the RIPA fraction and total brain lysates and found markedly reduced Aβ peptide in APP/PS1;Idol−/− mice (Fig. 2, B and C).
We next characterized amyloid plaque deposition in a second cohort of mice by immunostaining with a biotinylated Aβ-specific monoclonal antibody (82E1B) and quantifying lesion area (Fig. 2, D and E). Consistent with the reduction of Aβ in the setting of Idol deficiency, amyloid plaque burden was reduced in the cortex and hippocampus in both males and females. In this cohort, the cortical plaque burden in male APP/PS1;Idol+/− mice also trended lower compared to male APP/PS1;Idol+/+ mice (P = 0.066).
To further characterize the nature of the deposited plaques, we stained brain sections with thioflavin S, which detects fibrillar amyloid deposits with a β-sheet conformation (Fig. 3). Consistent with the Aβ staining results, male and female APP/PS1 mice lacking Idol also had reduced thioflavin S–positive plaque burden in the cortex and hippocampus. Overall, these results show that Idol expression affects amyloid plaque burden in a gene dosage–dependent manner in a mouse model of AD.
Fig. 3. Reducing Idol expression attenuates the deposition of fibrillar amyloid.

(A) Representative micrographs showing thioflavin S staining of brain sections from APP/PS1;Idol+/+, APP/PS1;Idol+/−, or APP/PS1;Idol−/− mice. White color indicates thioflavin S fluorescence. Scale bar, 250 μm. (B) Quantification of thioflavin S (ThioS)–stained Aβ plaque load in the frontal cortex and hippocampus of male or female APP/PS1;Idol+/+, APP/PS1;Idol+/−, or APP/PS1;Idol−/− mice. Error bars represent SEM. *P < 0.05, **P < 0.01 by Student’s t test. n = 6.
Idol deficiency ameliorates neuroinflammation in a mouse model of Aβ amyloidosis
Activation of microglia is commonly observed in the brains of human AD and mouse models of Aβ amyloidosis (20). Fibrillar amyloid plaque is postulated to trigger detrimental neuroinflammatory responses, and its abundance correlates with the extent of microgliosis (23). To determine whether Idol affected microglia abundance in APP/PS1 mice, we stained brain sections with antibodies against CD45 (marker for activated microglia) and Iba1 (marker for resting and activated microglia) (Fig. 4). Consistent with the decreased Aβ plaque formation, neuroinflammation (as assessed by CD45 staining) was clearly decreased in APP/PS1;Idol−/− compared to APP/PS1;Idol+/+ mice (Fig. 4, A and B; P < 0.01). Furthermore, fewer microglia were present in the brains of APP/PS1;Idol−/− compared to APP/PS1;Idol+/+ mice (Fig. 4, C and D). Microglial abundance in APP/PS1 brains was directly proportional to the Idol gene dosage. Both heterozygous and homozygous knockout mice showed a decrease in the number of microglia in the cortex and hippocampus (Fig. 4, C and D; P < 0.01).
Fig. 4. Reducing Idol expression attenuates microgliosis in a mouse model of Aβ amyloidosis.
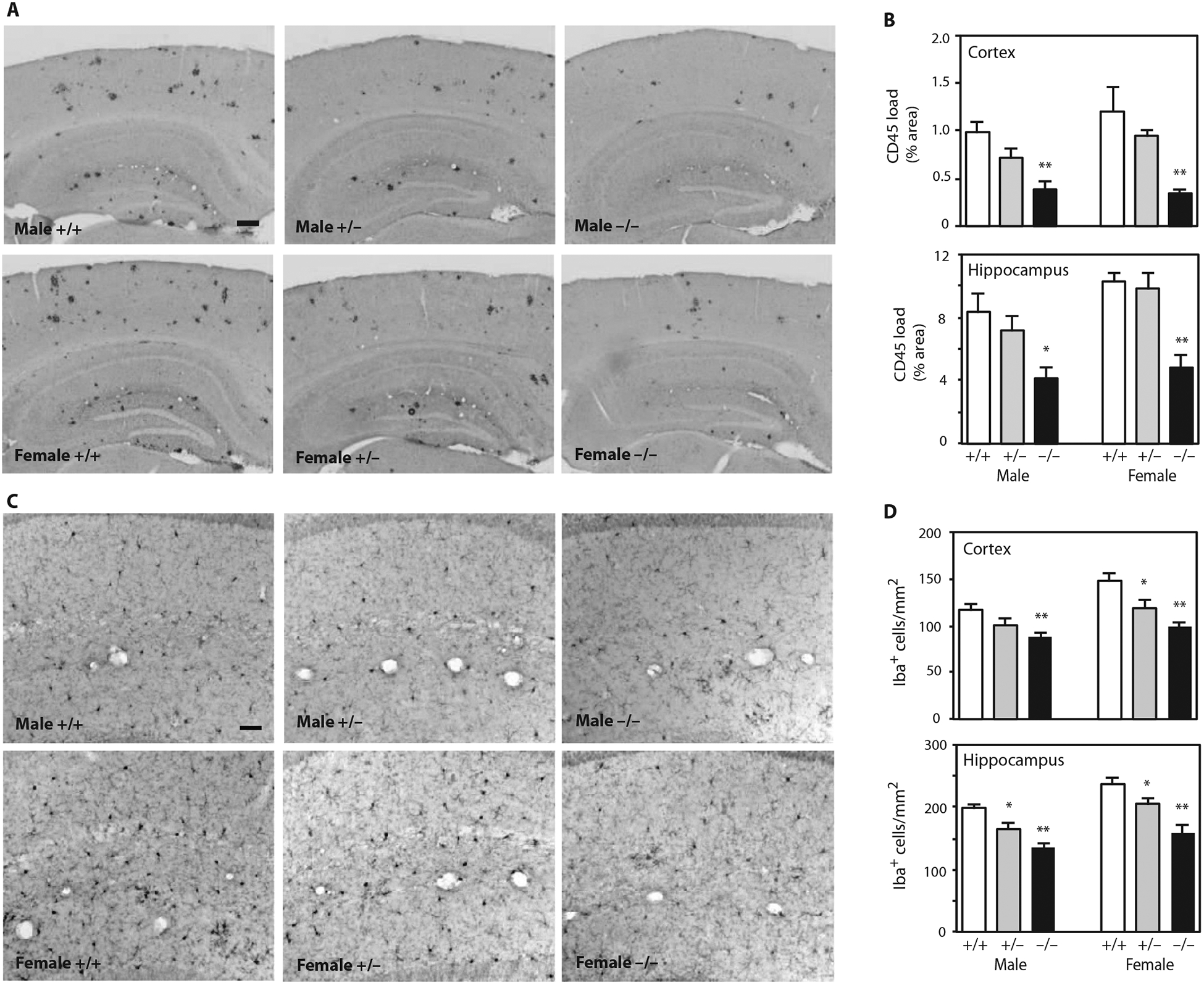
(A) Representative micrographs showing immunohistochemical staining of brain sections from APP/PS1;Idol+/+, APP/PS1;Idol+/−, or APP/PS1;Idol−/− mice with CD45 antibody. Black color indicates CD45-positive lesions. Scale bar, 200 μm. (B) Quantification of CD45 staining load in the frontal cortex or hippocampus of APP/PS1;Idol+/+, APP/PS1;Idol+/−, or APP/PS1;Idol−/− mice. Error bars represent SEM. *P < 0.05, **P < 0.01 by Student’s t test. n = 6. (C) Representative micrographs showing immunohistochemical staining of the hippocampal CA1 regions of APP/PS1;Idol+/+, APP/PS1;Idol+/−, or APP/PS1;Idol−/− mice with Iba1 antibody. Black color indicates Iba1+ cells. Scale bar, 50 μm. (D) Quantification of the number of Iba1+ cells in the frontal cortex or hippocampus of APP/ PS1;Idol+/+, APP/PS1;Idol+/−, or APP/PS1;Idol−/− mice. Error bars represent SEM. *P < 0.05, **P < 0.01 by Student’s t test. n = 6.
To determine whether the differences in microgliosis reflected in-trinsic differences in inflammatory properties between control and Idol-deficient microglia, we examined inflammatory gene expression. Loss of Idol did not change the expression of the signature microglial markers Cd45, Cd68, and Ym1 (fig. S2A). Furthermore, Idol deficiency did not affect the activation of microglia upon lipopolysaccharide (LPS) stimulation, as the induction of genes such as Tnfa, Il1b, Icam1, and Socs3 was not altered (fig. S2B). These data suggest that the difference in microglia abundance in APP/PS1 mice in the presence and absence of Idol is secondary to the difference in Aβ plaque load.
Loss of Idol facilitates ApoE and Aβ uptake and clearance
To investigate a mechanistic link between Idol and AD, we first tested the influence of Idol knockout on brain levels of the LDLR and its ligand ApoE. LDLR protein expression in APP/PS1 mice was higher in the absence of Idol, whereas LRP1 protein was unchanged (Fig. 5A). ApoE protein was correspondingly reduced in APP/PS1;Idol−/− mice compared to APP/PS1;Idol+/+ mice (Fig. 5A). We also observed lower levels of ApoE in phosphate-buffered saline (PBS)–soluble fractions and RIPA fractions of brain lysates from APP/PS1;Idol−/− mice (Fig. 5, B and C). The greater reduction observed in the PBS fraction is consistent with extracellular ApoE being the target of Idol and LDLR. ApoE has been shown to accumulate along with Aβ in amyloid plaques in both AD patients and mouse models of AD (6, 24). In line with our findings of reduced Aβ peptide in the insoluble fraction (Fig. 2A), there was a parallel reduction in ApoE protein in the insoluble fractions from APP/PS1;Idol−/− mouse brains (Fig. 5D; P < 0.05).
Fig. 5. Idol regulates ApoE in the brain.
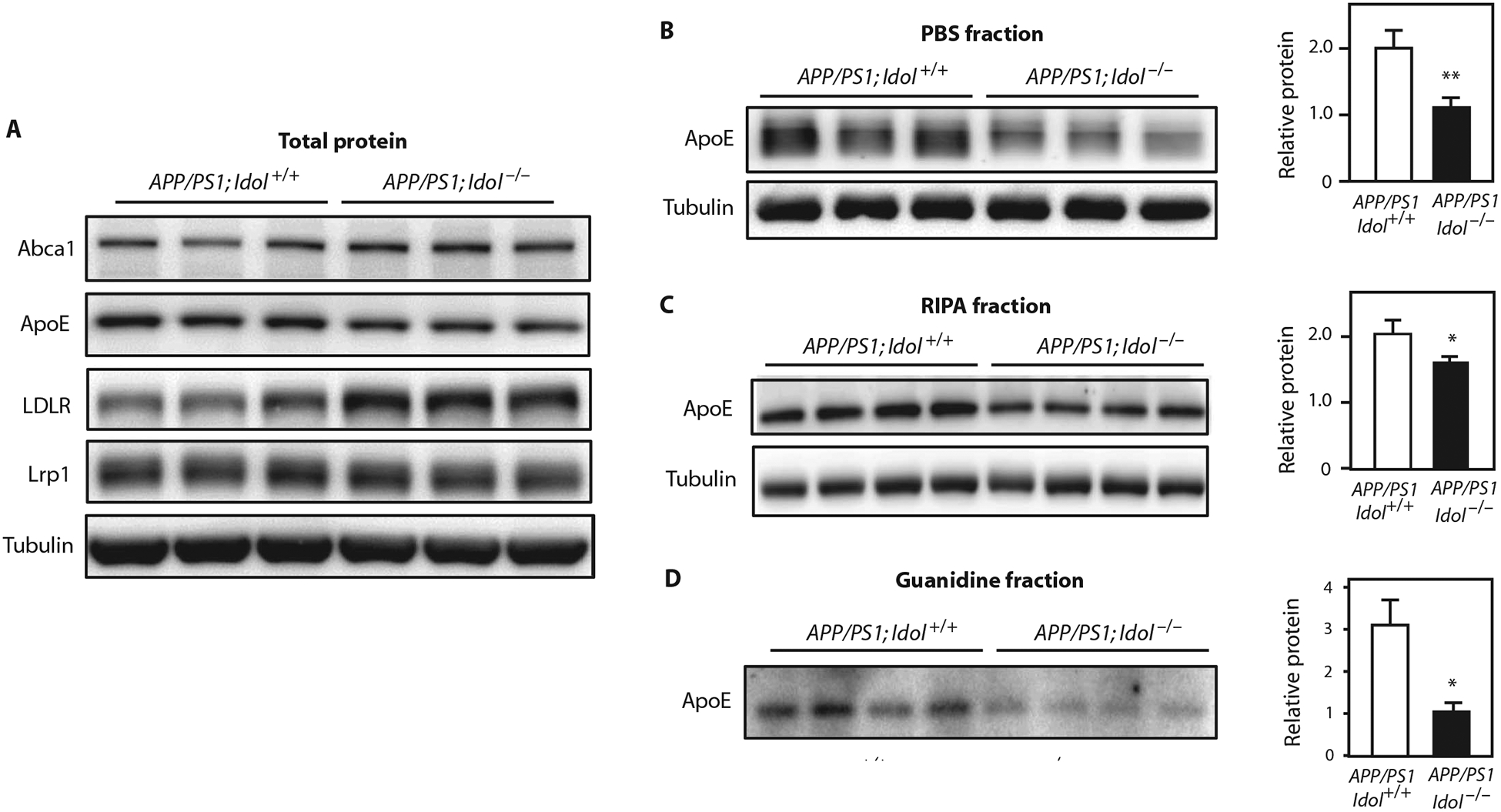
(A) Immunoblot analysis of total brain lysates from the frontal cortex of APP/PS1;Idol+/+ or APP/PS1;Idol−/− mice. Lanes represent samples from individual mice. (B to D) Immunoblot analysis of ApoE and tubulin protein in the (B) PBS-soluble fraction, (C) RIPA fraction, and (D) insoluble (guanidine) fraction from the frontal samples from individual mice. Quantification of the 0.05, **P < 0.01 by Student’s t test. Each experiment was cortex of APP/PS1;Idol+/+ or APP/PS1;Idol−/− mice. Lanes represent signals is presented on the right. Error bars represent SEM. *P < at least twice.
Extracellular Aβ can be cleared by neurons and other cell types in the brain through various receptors on the plasma membrane (25–27). Previous studies have suggested that LDLR and ApoE regulate extracellular Aβ by modulating its clearance by brain cells (28, 29). We therefore examined the effect of Idol on extracellular Aβ clearance. Small interfering RNA (siRNA)–mediated knockdown decreased Idol expression by ~60% and increased LDLR protein in mouse Neuro2a neuronal cells, hApoE3 knock-in astroglial cells, and BV2 microglial cells (Fig. 6A and fig. S3A). To assess the influence of Idol on Aβ uptake and clearance, we inhibited Idol with siRNA and then incubated the cells with synthetic Aβ peptide for 3 hours. We found that Idol knockdown strongly increased Aβ uptake in Neuro2a, hApoE3, and BV2 cells (Fig. 6B). Furthermore, Idol knockdown also decreased the amount of Aβ remaining in the media after 24 hours of incubation (Fig. 6C). We also examined the effects of loss of Idol on uptake of Aβ by primary mouse neurons. In contrast to the results obtained with the immortalized Neuro2a line, there was no difference in the ability of Idolflox/flox control and Cre-infected Idol-deficient neurons to internalize fluorescently labeled aggregated Aβ42 peptide (Fig. 6D).
Fig. 6. Idol modulates cellular Aβ uptake and Aβ clearance in brain-derived cell lines.
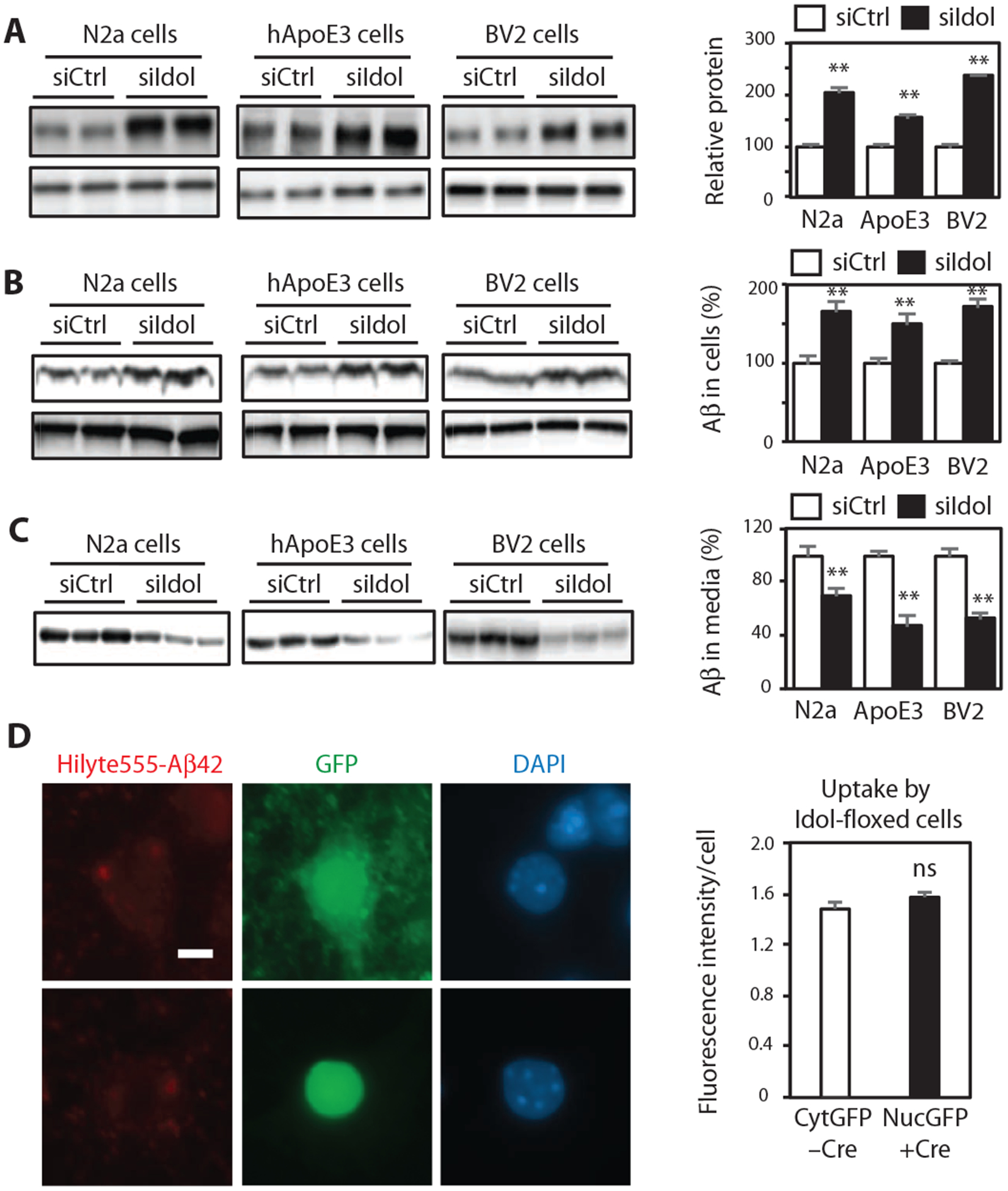
(A) Immunoblot analysis of LDLR protein in Neuro2a (N2a) cells, hApoE3 cells, and BV2 cells transfected with control or Idol-specific siRNA. Quantification of the immunoblot signals is presented on the right. Error bars represent SEM. **P < 0.01 by Student’s t test. (B) Immunoblot analysis of intracellular Aβ protein in Neuro2a cells, hApoE3 cells, and BV2 cells transfected with control or Idol-specific siRNA and then incubated for 3 hours with recombinant Aβ. Quantification of the immunoblot signals is presented on the right. Error bars represent SEM. **P < 0.01 by Student’s t test. (C) Immunoblot analysis of extracellular Aβ protein in the media of cultures of Neuro2a cells, hApoE3 cells, and BV2 cells transfected with control or Idol-specific siRNA and then incubated for 24 hours with recombinant Aβ. Quantification of the immunoblot signals is presented on the right. Error bars represent SEM. **P < 0.01 by Student’s t test. (D) Representative micrographs showing uptake of fluorescently labeled aggregated HiLyte555-Aβ42 after a 4-hour incubation in primary hippocampal neurons from Idolflox/flox mice treated in vitro with control green fluorescent protein (GFP)–expressing or GFP/Cre (fusion protein with nuclear localization signal)–expressing AAV vector. Red, Aβ42; green, GFP; blue, DAPI. Scale bar, 5 μm. GFP or GFP-Cre expression was driven from a neuron-specific calcium/calmodulin-dependent protein kinase IIa (CaMKIIa) promoter. Quantification of the fluorescence intensity is presented on the right. Error bars represent SEM. n ≥ 40. Each experiment was performed at least twice.
We extended these observations by analyzing the clearance capacity of primary microglia. Microglia lacking Idol showed increased ability to internalize fluorescently labeled aggregated Aβ42 peptide (Fig. 7, A and C). Similar results were obtained when Idol was acutely deleted from conditional knockout cells in vitro (fig. S3, B and C). Idolflox/flox primary microglia treated with lentiviral Cre accumulated more Aβ42 peptide upon incubation compared to vector-treated cells. We observed a similar increased uptake activity of Idol-deficient cells toward ApoE-containing lipoproteins. Incubation of primary microglia with fluorescently labeled reconstituted discoidal high-density lipoprotein (HDL) particles containing ApoE revealed accelerated uptake in cells lacking Idol (Fig. 7, B and D). Together, these data suggest that loss of Idol decreases Aβ deposition in the brain by enhancing the clearance of both Aβ- and ApoE-containing lipoproteins, primarily by microglia.
Fig. 7. Idol modulates cellular Aβ and ApoE uptake by microglia.
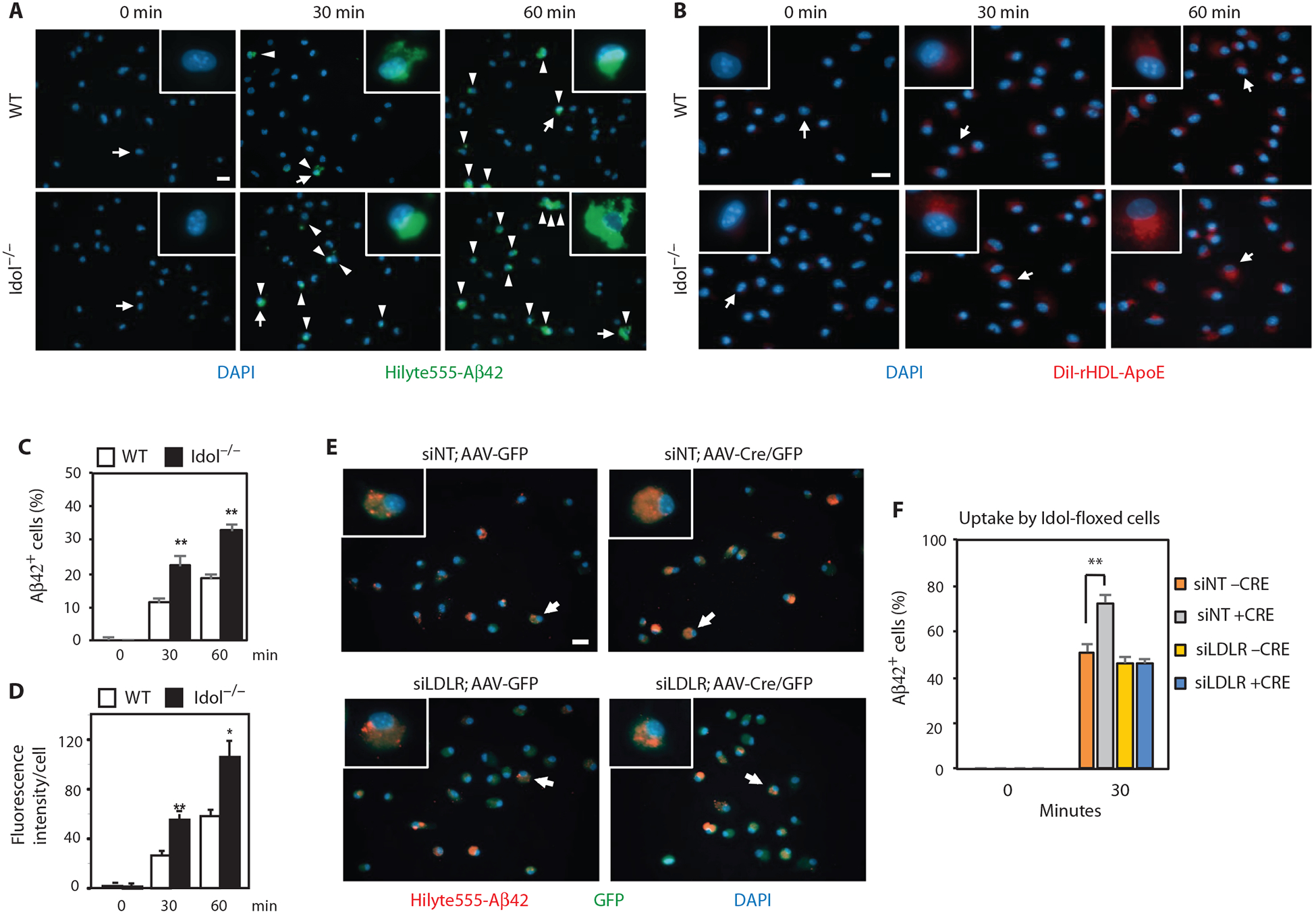
(A) Representative micrographs showing uptake of fluorescently labeled aggregated HiLyte488-Aβ42 in primary microglia from WT and Idol−/− mice. Blue, DAPI; green, Aβ42. Arrowheads denote Aβ42-positive cells. Arrows denote representative cells shown in the magnified field. Scale bar, 20 μm. (B) Representative micrographs showing uptake of 1,1′-dioctadecyl-3,3,3′,3′-tetramethylindocarbocyanine perchlorate (DiI)–labeled reconstituted discoidal HDL particles containing ApoE by primary microglia from WT and Idol−/− mice. Blue, DAPI; red, DiI rHDL-ApoE. Arrows denote representative cells shown in the magnified field. Scale bar, 20 μm. (C) Quantification of the results shown in (A). Error bars represent SEM. **P < 0.01 by Student’s t test. (D) Quantification of the results shown in (B). Error bars represent SEM. *P < 0.05, **P < 0.01 by Student’s t test. (E) Representative micrographs showing uptake of fluorescently labeled aggregated HiLyte555-Aβ42 after a 30-min incubation in primary microglia from Idolflox/flox mice infected with AAV5/CMV-GFP or AAV5/CMV-Cre/GFP and treated with Accell siRNA nontargeting control (siNT) or siRNA against mouse LDLR (siLDLR). Arrows denote representative cells shown in the magnified field. Scale bar, 20 μm. (F) Quantification of the results shown in (E). Error bars represent SEM. **P < 0.01 by Student’s t test. Each experiment was performed at least twice.
To assess directly the contribution of LDLR to increased uptake of Aβ by Idol-deficient microglia, we knocked down LDLR with siRNA in Idolflox/flox microglia treated with adeno-associated viral Cre in vitro and assayed for uptake of Aβ42 peptide (Fig. 7, E and F). LDLR knockdown largely abolished the effect of Idol deficiency on Aβ uptake. These data demonstrate that Idol regulates Aβ uptake in microglia, at least in part, by modulating LDLR levels, and strongly suggest that this mechanism contributes to the effects of Idol on Aβ deposition in APP/PS1 mice.
DISCUSSION
Here, we investigated the impact of Idol, a physiological regulator of LDLR abundance, on the development of Aβ-dependent pathology in mice. Idol is robustly expressed in microglia, and its inactivation increases LDLR protein in the mouse brain. In a transgenic mouse model of amyloidosis, loss of Idol reduced amyloid plaque burden and microglial recruitment in a gene dosage–dependent manner. Mechanistic studies revealed that Idol does not directly modulate neuroinflammatory responses. Rather, Idol regulates the uptake and clearance of ApoE and Aβ by microglia. These results identify the LXR-Idol pathway as a previously unrecognized determinant of brain LDLR and ApoE and the development of AD-like pathology.
ApoE plays a central role in brain lipid homeostasis by mediating the endocytosis of HDL-like lipoprotein particles (30). ApoE is also a crucial factor in the development of AD (5, 6, 31, 32). Reducing ApoE in mouse models of amyloidosis greatly reduces plaque formation (7, 8, 33, 34). ApoE binds to lipoprotein receptors such as LDLR and LRP1. Inactivation of the Ldlr or Lrp1 gene increases, whereas overexpression of LDLR decreases, ApoE protein in the brain (19, 35–37). We have shown here that the LXR-Idol axis targets brain LDLR (but not LRP1) for degradation. Our data suggest that Idol affects amyloid plaque formation by modulating LDLR protein primarily in microglia and, consequently, ApoE protein in the brain.
Our results strengthen and expand previously established links between LDLR and ApoE expression and the amount of Aβ in the brain. However, the relationships between these factors remain controversial and an area of active investigation. For example, the connections between ApoE and Aβ uptake are complex. First, many ApoE receptors, including LDLR and LRP1, have been proposed to be involved in Aβ uptake (31). Second, the mechanisms by which ApoE protein affects Aβ uptake are not fully understood. One model proposes that ApoE binds to insoluble Aβ aggregates (38, 39) and thereby hinders their uptake by brain cells. Other studies suggest that ApoE may compete with soluble Aβ for the same endocytosis pathway (40). Others have suggested that lipidated ApoE facilitates removal of Aβ (29). Another recent study reported that LDLR mediates Aβ uptake through direct binding to Aβ, independent of ApoE (28). Although the exact mechanisms affecting Aβ clearance warrant further investigation, Idol’s ability to regulate LDLR levels and its ability to regulate ApoE clearance would both be predicted to affect Aβ uptake. The combined effect of Idol on both factors may explain its prominent effect on AD-like pathology.
Previous studies of the roles of the LDLR-regulating factors Idol and Pcsk9 were not predictive of the relative importance of these factors in the control of brain LDLR expression. Deletion of Pcsk9 in mice leads to increased LDLR protein in the liver but not in the brain (41, 42). By contrast, loss of Idol expression in mice has little effect on hepatic LDLR (13). Our discovery that Idol plays a major role in determining brain LDLR and ApoE levels under physiological and pathological conditions was thus unexpected.
The LXR signaling pathway also plays an important role in Aβ plaque biogenesis. LXR agonist treatment alleviates AD-like pathology in mouse models of amyloidosis, possibly by inducing Abca1 expression (29, 43–48). Abca1 affects the lipidation status of ApoE in the brain (49), which, in turn, may facilitate the proteolytic degradation of Aβ (29). We previously reported that loss of LXRα or LXRβ increases Aβ plaque burden in the APP/PS1 mouse (50). At first glance, the finding that knockout of the LXR target gene Idol reduces Aβ plaque burden may appear at odds with the consequences of loss of LXR expression. However, it is important to keep in mind that loss of LXRα and LXRβ does not lead to complete loss of Idol expression (15). Furthermore, LXRα and LXRβ control the expression of an array of genes important for cholesterol metabolism, which may exert independent effects on the development of amyloidosis and neuroinflammation (51).
A limitation of the present study is that the relative importance of the three Idol targets for the clearance of ApoE and Aβ in vivo has not yet been tested directly. Although our cellular studies strongly suggest that LDLR is involved, we cannot exclude the possible contribution of the other two Idol targets, ApoER2 (ApoE receptor 2) and VLDLR (very low density lipoprotein receptor), to ApoE and Aβ levels in vivo. Another obvious limitation is that we used a mouse model to study AD-related pathology. We previously reported that the LXR-IDOL-LDLR pathway is highly active in primates (13), but further studies will be needed to translate the current findings to humans.
Previous studies have suggested that Idol inhibition may have beneficial effects on plasma LDL in primates (13). Our present results provide a basis for investigating Idol inhibition as a therapeutic approach for AD. Although targeting E3 ligases with selective small-molecule inhibitors is challenging, there is precedence for this strategy (52). Our studies suggest that pharmacological inhibition of Idol merits further investigation as a means to modulate endogenous ApoE, LDLR, and Aβ in the AD brain. It is possible that modulating physiological LDLR protein expression in microglia by inhibiting Idol-dependent degradation could improve Aβ clearance and possibly delay the onset of AD pathology. The observation that loss of only one copy of the Idol gene affects Aβ-dependent pathology in mice suggests that even partial Idol inhibition could prove beneficial in the setting of AD.
MATERIALS AND METHODS
Study design
This study aimed to uncover the role of Idol in regulating brain LDLR receptor levels and its impact on ApoE clearance and Aβ amyloidosis. To accomplish this aim, Idol was genetically ablated in a mouse model of Aβ amyloidosis as well as in primary microglia, primary hippocampal neurons, and brain-derived cell lines. The effect of Idol deficiency on LDLR, ApoE, and Aβ amyloidosis was assessed by immunohistochemistry, protein/RNA quantitation, and immunocytochemistry. Data were collected from cell culture studies with the investigators aware of the sample identities. Data collection from mouse samples and all the data analysis in both tissue culture and mouse experiments were performed in a blinded fashion.
Animals
APPswe/PSEN1dE9 mice on a C57BL/6 background (APP/PS1 mice) were obtained from The Jackson Laboratory [005864, B6.Cg-Tg(APPswe,PSEN1dE9)85Dbo/J] (22). Idol−/− mice (13) were back-crossed to C57BL/6 mice for eight generations before mating to APP/PS1 mice. For histological analysis, mice were euthanized and brains were collected after transcardial perfusion with 4% paraformaldehyde (PFA). Brains were then postfixed with 4% PFA for 24 hours at 4°C, cryoprotected by 30% sucrose at 4°C, and embedded in optimum cutting temperature compound. Brain specimens for biochemical analysis were directly frozen in liquid nitrogen. All mouse specimens were collected at 7 months of age. All mouse experiments were approved and performed under the guidelines of the Animal Care and Research Advisory Committees at the University of California, Los Angeles (UCLA) and Mayo Clinic.
Primary culture
Primary microglia were isolated from the frontal cortexes of 4- to 5-day-old mouse brains. Two brains per group were collected in ice-cold Hanks’ balanced salt solution, and meninges were removed. Frontal cortexes were dissected out and transferred to Dulbecco’s modified Eagle’s medium (DMEM) containing 10% fetal bovine serum (FBS). Cortexes were then gently triturated with a 1000-μl pipette tip and then centrifuged at 800 rpm for 5 min, and the pellets were resuspended in 30 ml of the above medium to prepare mixed glial cell cultures. Cells were plated in T-225 flasks and incubated at 37°C (5% CO2). After 24 hours, culture medium was changed to DMEM containing 10% FBS and granulocyte-macrophage colony-stimulating factor (25 ng/ml). At 10 and 14 days after the isolation, microglia were detached from mixed glial feeder layers by shaking on an orbital shaker at 220 rpm for 20 min, supernatants were centrifuged at 800 rpm for 3 min, and pellets were resuspended in DMEM containing 10% FBS. Isolated primary microglia were plated in six-well plates at a density of 0.5 million cells per well and incubated at 37°C (5% CO2) until use. Primary neuron and astrocyte cultures were prepared as previously described (53, 54).
Reagents and primary antibodies
GW3965 was synthesized as previously described (55). LPS from Salmonella minnesota R595 (Re) was obtained from Enzo Life Sciences. Thioflavin S was purchased from Sigma (T1892). Aβ(1–42)-HiLyte488 and Aβ (1–42)-HiLyte555 were purchased from AnaSpec. siRNAs for mouse Idol (SasI_Mm02_00343514) and mouse Ldlr (SasI_ Mm01_00084966) were purchased from Sigma (MISSION siRNA). Accell nontargeting siRNA pool and Accell mouse Ldlr (16835) siRNA SMARTpool were purchased from Dharmacon. The antibodies used were ABCA1 (Novus or HJ1, a gift from D. M. Holtzman), Aβ (4G8, Covance or 82E1, IBL International), actin (Abcam), ApoE (K23100R, Meridian Life Science), BACE1 (D10E5, Cell Signaling Technology), β-Gal (Promega), CD45 (MCA1388, Bio-Rad), glyceraldehyde-3-phosphate dehydrogenase (Santa Cruz Bio-technology), Iba1 (019–19741, Wako), LDLR (Cayman Chemical), LRP1 (Abcam), and mouse anti-tubulin (Abcam or EMB Millipore).
RNA analysis
Total RNA was extracted from tissues and cells with TRIzol (Life Technologies). RNAs were reverse-transcribed using the High-Capacity cDNA Reverse Transcription kit (Applied Biosystems). Quantitative PCR was performed with Power SYBR Green PCR Master Mix (Applied Biosystems) or 2× SYBR Green Master Mix (GMO-SG2X-A300, Diagenode). Relative mRNA levels were calculated by the comparative Ct method using GenEx 5.3.2 (MultiD analyses) or QuantStudio 6 Flex (Life Technologies) and normalized to controls as indicated.
Protein analysis
Cultured cells were lysed in RIPA buffer [50 mM tris-HCl (pH 7.4), 150 mM NaCl, 0.25% deoxycholic acid, 1% NP-40, 0.1% SDS, 1 mM EDTA] and protease inhibitor cocktail (Roche). Proteins were sequentially extracted from brain tissues with PBS, RIPA, and 5 M guanidine buffer in the presence of protease inhibitors (19). For Western blots, equal amounts of proteins (10 to 40 μg) were separated on NuPAGE bis-tris gels (Invitrogen) or TGX gels (Bio-Rad), and membranes were probed with primary and secondary antibodies. Signals were visualized by chemiluminescence [ECL Plus (GE Healthcare) or TMA-6 ECL detection kit (Lumigen)]. Blots were quantified by densitometry with ImageJ software [National Institutes of Health (NIH)]. For quantifying Aβ, Aβ40 (KHB3481) and Aβ42 (KHB3441) human ELISA kits from Life Technologies were used.
Histological analysis
Brains were sectioned on a cryostat at 40-μm thickness. For immunofluorescence staining, free-floating sections were blocked with PBS containing 10% normal goat serum (NGS) at room temperature for 30 min, incubated with anti-Iba1 (1:1000) and anti–β-Gal (1:500) in blocking solution at 4°C overnight, and then incubated with Alexa Fluor 488–goat anti-mouse immunoglobulin G (IgG) (for β-Gal; Invitrogen) and Alexa Fluor 565–goat anti-rabbit IgG (for Iba1; Invitrogen) at room temperature for 1 hour. Sections were mounted on slides with ProLong Gold with DAPI (Life Technologies). For immunohistochemistry, free-floating sections were treated with 0.3% H2O2 and 1% SDS (for CD45 only) and blocked with tris-buffered saline containing 3% milk and 0.25% Triton-X (for βb) or 3% NGS (for CD45) or with PBS containing 1.5% NGS and 1% bovine serum albumin (for Iba1). Sections were then incubated with biotinylated 82E1 (1:500) or anti-CD45 (1:500) or anti-Iba1 (1:1000) in blocking solution at 4°C for 24 hours. Sections were incubated with biotinylated anti-rat or anti-rabbit IgG (for CD45 or Iba1, respectively; Vector Laboratories) in blocking solution at room temperature for 1 hour. Antibody binding was detected with Vectastain ABC Elite (Vector Laboratories) and DAB peroxidase (horseradish peroxidase) substrate kits (Fisher Scientific) supplemented with nickel solution. Sections were dehydrated and mounted on slides with Permount (Fisher Scientific). For thioflavin S staining, sections were mounted on Superfrost Plus slides (Fisher Scientific) and dried overnight, incubated in thioflavin S staining solution (50 ml of water + 50 ml of ethanol + 0.025 g of thioflavin S) for 5 min at room temperature, washed twice with 50% ethanol and water, and then mounted with Vectashield (Vector Laboratories). Images were captured on an AxioCam charge-coupled device camera (Zeiss) mounted on an Axioskop2 microscope (Zeiss). Staining was quantified using ImageJ software (19).
Aβ uptake and clearance assay
Neuro2a cells, hApoE3 knock-in astroglial cells, or BV2 cells were cultured at 37°C (5% CO2) in DMEM containing 10% FBS, DMEM/F-12 containing 15% FBS and 1 mM sodium pyruvate, or DMEM containing 2% FBS and 1 mM sodium pyruvate, respectively. Cells were transfected with Idol siRNA or scrambled control using Lipofectamine RNAiMAX (Life Technologies). Forty-eight hours after transfection, cells were washed and cultured in serum-free medium containing 200 nM Aβ40. For Aβ uptake assays, cells were collected 3 hours after medium change, and intracellular Aβ levels were assessed by immunoblotting. For Aβ clearance assays, medium was collected 24 hours after medium change, and Aβ levels in the medium were assessed. All experiments were performed in duplicate or triplicate. Aβ40 monomers were prepared as previously described (56).
Primary microglia Aβ uptake assays were performed as described (38). Aβ(1–42)-HiLyte488 or Aβ(1–42)-HiLyte555 was sequentially resuspended in 10 mM NaOH and in PBS to 1 mg/ml and aggregated at 37°C for 19 hours. For acute Idol knockout in Idolfl/fl microglia, cells were infected with pUltra-hot (Addgene) or pUltra-hot-Cre lentivirus 2 days before the Aβ uptake assay. For LDLR knockdown and acute Idol knockout experiments, primary Idolfl/fl microglia were infected with adeno-associated virus [AAV5/TRUFR–enhanced green fluorescent protein (EGFP) or AAV5/Cre-EGFP] 4 days before and treated with Accell siRNA (nontargeting siRNA pool or mouse Ldlr siRNA SMARTpool, Dharmacon) 3 days before the Aβ uptake assay. Primary microglia plated on PDL-coated coverslips were treated with 0.5 μM aggregated Aβ42-HiLyte488 or Aβ42-HiLyte555 diluted in DMEM. Cells were washed with PBS and warm 0.25% trypsin–EDTA and then fixed with 4% PFA. Coverslips were mounted on slides with ProLong Gold with DAPI. Images were captured and quantified as above.
Primary hippocampal neurons were infected with adeno-associated virus (AAV8/CaMKIIa-EGFP or AAV8/CaMKIIa-GFP/Cre) at 7 days in vitro (DIV), and Aβ uptake was assessed at 14 DIV. Analysis of Aβ uptake was performed as for microglia.
HDL-ApoE uptake assay
Reconstituted discoidal POPC (1-palmitoyl-2-oleoyl-phosphatidylcholine)– ApoE particles were prepared as reported previously (57, 58). The POPC-ApoE particles were stored at 4°C until use. Primary microglia were plated at 5 × 104 cells per well in 24-well plates in complete DMEM. On day 2, the cells were washed once with DMEM without serum, and DMEM containing 10% lipoprotein-deficient serum and 4 mg ApoE/ml POPC-ApoE particles was added to cells. After incubating at 37°C for 30 or 60 min, cells were placed on ice and washed with PBS twice, followed by fixation in 4% PFA for 10 min. Cells were mounted, and images were captured and analyzed as described above.
Statistical analysis
For all analyses in this study, P values were calculated by Student’s t test. Detailed information regarding measurements, sample size, and error bars is presented in Materials and Methods and in figure legends. All data values from in vivo studies are presented in tables S1 to S6.
Supplementary Material
Fig. S1. Idol decreases LDLR protein and promotes LDLR ubiquitination in mouse brain cells.
Fig. S2. Loss of Idol does not directly affect inflammatory responses.
Fig. S3. Idol inhibition promotes the clearance of Ab by microglia.
Table S1. Data for Ab40 shown in Fig. 2A.
Table S2. Data for Ab42 shown in Fig. 2A.
Table S3. Quantification of Ab plaque load in Fig. 2E.
Table S4. Quantification of thioflavin S plaque load in Fig. 3B.
Table S5. Quantification of CD45 load in Fig. 4B.
Table S6. Quantification of Iba+ cells in Fig. 4D.
Acknowledgments:
We thank all members of the Tontonoz and Kim laboratories for helpful advice and reagents.
Funding: Supported by NIH HL066088 (P.T.), BrightFocus Foundation A2012421, Alzheimer’s Association, GHR, and NIH NS069329 (Jungsu K.). P.T. is an Investigator of the Howard Hughes Medical Institute.
Footnotes
Competing interests: UCLA holds a patent related to Idol of which P.T. is a co-inventor.
SUPPLEMENTARY MATERIALS
www.sciencetranslationalmedicine.org/cgi/content/full/7/314/314ra184/DC1
REFERENCES AND NOTES
- 1.Ballard C, Gauthier S, Corbett A, Brayne C, Aarsland D, Jones E, Alzheimer’s disease. Lancet 377, 1019–1031 (2011). [DOI] [PubMed] [Google Scholar]
- 2.Blennow K, de Leon MJ, Zetterberg H, Alzheimer’s disease. Lancet 368, 387–403 (2006). [DOI] [PubMed] [Google Scholar]
- 3.Holtzman DM, Morris JC, Goate AM, Alzheimer’s disease: The challenge of the second century. Sci. Transl. Med 3, 77sr1 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bertram L, Lill CM, Tanzi RE, The genetics of Alzheimer disease: Back to the future. Neuron 68, 270–281 (2010). [DOI] [PubMed] [Google Scholar]
- 5.Mahley RW, Apolipoprotein E: Cholesterol transport protein with expanding role in cell biology. Science 240, 622–630 (1988). [DOI] [PubMed] [Google Scholar]
- 6.Kim J, Basak JM, Holtzman DM, The role of apolipoprotein E in Alzheimer’s disease. Neuron 63, 287–303 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bien-Ly N, Gillespie AK, Walker D, Yoon SY, Huang Y, Reducing human apolipoprotein E levels attenuates age-dependent Aβ accumulation in mutant human amyloid precursor protein transgenic mice. J. Neurosci 32, 4803–4811 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kim J, Jiang H, Park S, Eltorai AEM, Stewart FR, Yoon H, Basak JM, Finn MB, Holtzman DM, Haploinsufficiency of human APOE reduces amyloid deposition in a mouse model of amyloid-b amyloidosis. J. Neurosci 31, 18007–18012 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kim J, Eltorai AEM, Jiang H, Liao F, Verghese PB, Kim J, Stewart FR, Basak JM, Holtzman DM, Anti-apoE immunotherapy inhibits amyloid accumulation in a transgenic mouse model of Aβ amyloidosis. J. Exp. Med 209, 2149–2156 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wahrle SE, Jiang H, Parsadanian M, Kim J, Li A, Knoten A, Jain S, Hirsch-Reinshagen V, Wellington CL, Bales KR, Paul SM, Holtzman DM, Overexpression of ABCA1 reduces amyloid deposition in the PDAPP mouse model of Alzheimer disease. J. Clin. Invest 118, 671–682 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Brown MS, Goldstein JL, A receptor-mediated pathway for cholesterol homeostasis. Science 232, 34–47 (1986). [DOI] [PubMed] [Google Scholar]
- 12.Calkin AC, Lee SD, Kim J, Stijn CMWV, Wu X-H, Lusis AJ, Hong C, Tangirala RI, Tontonoz P, Transgenic expression of dominant-active IDOL in liver causes diet-induced hypercholesterolemia and atherosclerosis in mice. Circ. Res 115, 442–449 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hong C, Marshall SM, McDaniel AL, Graham M, Layne JD, Cai L, Scotti E, Boyadjian R, Kim J, Chamberlain BT, Tangirala RK, Jung ME, Fong L, Lee R, Young SG, Temel RE, Tontonoz P, The LXR–Idol axis differentially regulates plasma LDL levels in primates and mice. Cell Metab. 20, 910–918 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Weissglas-Volkov D, Calkin AC, Tusie-Luna T, Sinsheimer JS, Zelcer N, Riba L, Tino AMV, Ordoñez-Sánchez ML, Cruz-Bautista I, Aguilar-Salinas CA, Tontonoz P, Pajukanta P, The N342S MYLIP polymorphism is associated with high total cholesterol and increased LDL receptor degradation in humans. J. Clin. Invest 121, 3062–3071 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zelcer N, Hong C, Boyadjian R, Tontonoz P, LXR regulates cholesterol uptake through idol-dependent ubiquitination of the LDL receptor. Science 325, 100–104 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Calkin AC, Goult BT, Zhang L, Fairall L, Hong C, Schwabe JWR, Tontonoz P, FERM-dependent E3 ligase recognition is a conserved mechanism for targeted degradation of lipoprotein receptors. Proc. Natl. Acad. Sci. U.S.A 108, 20107–20112 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zhang L, Fairall L, Goult BT, Calkin AC, Hong C, Millard CJ, Tontonoz P, Schwabe JWR, The IDOL–UBE2D complex mediates sterol-dependent degradation of the LDL receptor. Genes Dev. 25, 1262–1274 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Castellano JM, Deane R, Gottesdiener AJ, Verghese PB, Stewart FR, West T, Paoletti AC, Kasper TR, DeMattos RB, Zlokovic BV, Holtzman DM, Low-density lipoprotein receptor overexpression enhances the rate of brain-to-blood Aβ clearance in a mouse model of β-amyloidosis. Proc. Natl. Acad. Sci. U.S.A 109, 15502–15507 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kim J, Castellano JM, Jiang H, Basak JM, Parsadanian M, Pham V, Mason SM, Paul SM, Holtzman DM, Overexpression of low-density lipoprotein receptor in the brain markedly inhibits amyloid deposition and increases extracellular Aβ clearance. Neuron 64, 632–644 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Mosher KI, Wyss-Coray T, Microglial dysfunction in brain aging and Alzheimer’s disease. Biochem. Pharmacol 88, 594–604 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Solito E, Sastre M, Microglia function in Alzheimer’s disease. Front. Pharmacol 3, 14 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Jankowsky JL, Fadale DJ, Anderson J, Xu GM, Gonzales V, Jenkins NA, Copeland NG, Lee MK, Younkin LH, Wagner SL, Younkin SG, Borchelt DR, Mutant presenilins specifically elevate the levels of the 42 residue β-amyloid peptide in vivo: Evidence for augmentation of a 42-specific g secretase. Hum. Mol. Genet 13, 159–170 (2004). [DOI] [PubMed] [Google Scholar]
- 23.Meyer-Luehmann M, Spires-Jones TL, Prada C, Garcia-Alloza M, de Calignon A, Rozkalne A, Koenigsknecht-Talboo J, Holtzman DM, Bacskai BJ, Hyman BT, Rapid appearance and local toxicity of amyloid-β plaques in a mouse model of Alzheimer’s disease. Nature 451, 720–724 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wahrle SE, Jiang H, Parsadanian M, Hartman RE, Bales KR, Paul SM, Holtzman DM, Deletion of Abca1 increases Aβ deposition in the PDAPP transgenic mouse model of Alzheimer disease. J. Biol. Chem 280, 43236–43242 (2005). [DOI] [PubMed] [Google Scholar]
- 25.Lai AY, McLaurin J, Mechanisms of amyloid-b peptide uptake by neurons: The role of lipid rafts and lipid raft-associated proteins. Int. J. Alzheimers Dis 2011, 548380 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Perry VH, Nicoll JAR, Holmes C, Microglia in neurodegenerative disease. Nat. Rev. Neurol 6, 193–201 (2010). [DOI] [PubMed] [Google Scholar]
- 27.Wyss-Coray T, Loike JD, Brionne TC, Lu E, Anankov R, Yan F, Silverstein SC, Husemann J, Adult mouse astrocytes degrade amyloid-β in vitro and in situ. Nat. Med 9, 453–457 (2003). [DOI] [PubMed] [Google Scholar]
- 28.Basak JM, Verghese PB, Yoon H, Kim J, Holtzman DM, Low-density lipoprotein receptor represents an apolipoprotein E-independent pathway of Aβ uptake and degradation by astrocytes. J. Biol. Chem 287, 13959–13971 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Jiang Q, Lee CYD, Mandrekar S, Wilkinson B, Cramer P, Zelcer N, Mann K, Lamb B, Willson TM, Collins JL, Richardson JC, Smith JD, Comery TA, Riddell D, Holtzman DM, Tontonoz P, Landreth GE, ApoE promotes the proteolytic degradation of Aβ. Neuron 58, 681–693 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Pitas RE, Boyles JK, Lee SH, Hui D, Weisgraber KH, Lipoproteins and their receptors in the central nervous system. characterization of the lipoproteins in cerebrospinal fluid and identification of apolipoprotein B,E(LDL) receptors in the brain. J. Biol. Chem 262, 14352–14360 (1987). [PubMed] [Google Scholar]
- 31.Kanekiyo T, Xu H, Bu G, ApoE and Aβ in Alzheimer’s disease: Accidental encounters or partners? Neuron 81, 740–754 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Liu C-C, Kanekiyo T, Xu H, Bu G, Apolipoprotein E and Alzheimer disease: Risk, mechanisms and therapy. Nat. Rev. Neurol 9, 106–118 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bales KR, Verina T, Dodel RC, Du Y, Altstiel L, Bender M, Hyslop P, Johnstone EM, Little SP, Cummins DJ, Piccardo P, Ghetti B, Paul SM, Lack of apolipoprotein E dramatically reduces amyloid β-peptide deposition. Nat. Genet 17, 263–264 (1997). [DOI] [PubMed] [Google Scholar]
- 34.Irizarry MC, Rebeck GW, Cheung B, Bales K, Paul SM, Holzman D, Hyman BT, Modulation of Aβ deposition in APP transgenic mice by an apolipoprotein E null background. Ann. N. Y. Acad. Sci 920, 171–178 (2000). [DOI] [PubMed] [Google Scholar]
- 35.Cao D, Fukuchi K, Wan H, Kim H, Li L, Lack of LDL receptor aggravates learning deficits and amyloid deposits in Alzheimer transgenic mice. Neurobiol. Aging 27, 1632–1643 (2006). [DOI] [PubMed] [Google Scholar]
- 36.Fryer JD, DeMattos RB, McCormick LM, O’Dell MA, Spinner ML, Bales KR, Paul SM, Sullivan PM, Parsadanian M, Bu G, Holtzman DM, The low density lipoprotein receptor regulates the level of central nervous system human and murine Apolipoprotein E but does not modify amyloid plaque pathology in PDAPP mice. J. Biol. Chem 280, 25754–25759 (2005). [DOI] [PubMed] [Google Scholar]
- 37.Liu Q, Zerbinatti CV, Zhang J, Hoe H-S, Wang B, Cole SL, Herz J, Muglia L, Bu G, Amyloid precursor protein regulates brain apolipoprotein E and cholesterol metabolism through lipoprotein receptor LRP1. Neuron 56, 66–78 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Chakrabarty P, Li A, Ceballos-Diaz C, Eddy JA, Funk CC, Moore B, DiNunno N, Rosario AM, Cruz PE, Verbeeck C, Sacino A, Nix S, Janus C, Price ND, Das P, Golde TE, IL-10 Alters immunoproteostasis in APP mice, increasing plaque burden and worsening cognitive behavior, Neuron 85, 519–533 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Namba Y, Tomonaga M, Kawasaki H, Otomo E, Ikeda K, Apolipoprotein E immune-reactivity in cerebral amyloid deposits and neurofibrillary tangles in Alzheimer’s disease and kuru plaque amyloid in Creutzfeldt-Jakob disease. Brain Res 541, 163–166 (1991). [DOI] [PubMed] [Google Scholar]
- 40.Verghese PB, Castellano JM, Garai K, Wang Y, Jiang H, Shah A, Bu G, Frieden C, Holtzman DM, ApoE influences amyloid-β (Aβ) clearance despite minimal apoE/Aβ association in physiological conditions. Proc. Natl. Acad. Sci. U.S.A 110, E1807–E1816 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Liu M, Wu G, Baysarowich J, Kavana M, Addona GH, Bierilo KK, Mudgett JS, Pavlovic G, Sitlani A, Renger JJ, Hubbard BK, Fisher TS, Zerbinatti CV, PCSK9 is not involved in the degradation of LDL receptors and BACE1 in the adult mouse brain. J. Lipid Res 51, 2611–2618 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Rousselet E, Marcinkiewicz J, Kriz J, Zhou A, Hatten ME, Prat A, Seidah NG, PCSK9 reduces the protein levels of the LDL receptor in mouse brain during development and after ischemic stroke. J. Lipid Res 52, 1383–1391 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Burns MP, Vardanian L, Pajoohesh-Ganji A, Wang L, Cooper M, Harris DC, Duff K, Rebeck GW, The effects of ABCA1 on cholesterol efflux and Aβ levels in vitro and in vivo. J. Neurochem 98, 792–800 (2006). [DOI] [PubMed] [Google Scholar]
- 44.Donkin JJ, Stukas S, Hirsch-Reinshagen V, Namjoshi D, Wilkinson A, May S, Chan J, Fan J, Collins J, Wellington CL, ATP-binding cassette transporter A1 mediates the beneficial effects of the liver X receptor agonist GW3965 on object recognition memory and amyloid burden in amyloid precursor protein/presenilin 1 mice. J. Biol. Chem 285, 34144–34154 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Koldamova RP, Lefterov IM, Staufenbiel M, Wolfe D, Huang S, Glorioso JC, Walter M, Roth MG, Lazo JS, The liver X receptor ligand T0901317 decreases amyloid b production in vitro and in a mouse model of Alzheimer’s disease. J. Biol. Chem 280, 4079–4088 (2005). [DOI] [PubMed] [Google Scholar]
- 46.Lefterov I, Bookout A, Wang Z, Staufenbiel M, Mangelsdorf D, Koldamova R, Expression profiling in APP23 mouse brain: Inhibition of Aβ amyloidosis and inflammation in response to LXR agonist treatment. Mol. Neurodegener 2, 20 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Riddell DR, Zhou H, Comery TA, Kouranova E, Lo CF, Warwick HK, Ring RH, Kirksey Y, Aschmies S, Xu J, Kubek K, Hirst WD, Gonzales C, Chen Y, Murphy E, Leonard S, Vasylyev D, Oganesian A, Martone RL, Pangalos MN, Reinhart PH, Jacobsen JS, The LXR agonist TO901317 selectively lowers hippocampal Aβ42 and improves memory in the Tg2576 mouse model of Alzheimer’s disease, Mol. Cell. Neurosci 34, 621–628 (2007). [DOI] [PubMed] [Google Scholar]
- 48.Vanmierlo T, Rutten K, Dederen J, Bloks VW, van LC Vark-van der Zee, F. Kuipers, A. Kiliaan, A. Blokland, E. J. G. Sijbrands, H. Steinbusch, J. Prickaerts, D. Lütjohann, M. Mulder, Liver X receptor activation restores memory in aged AD mice without reducing amyloid. Neurobiol. Aging 32, 1262–1272 (2011). [DOI] [PubMed] [Google Scholar]
- 49.Wahrle SE, Jiang H, Parsadanian M, Legleiter J, Han X, Fryer JD, Kowalewski T, Holtzman DM, ABCA1 is required for normal central nervous system ApoE levels and for lipidation of astrocyte-secreted apoE. J. Biol. Chem 279, 40987–40993 (2004). [DOI] [PubMed] [Google Scholar]
- 50.Zelcer N, Khanlou N, Clare R, Jiang Q, Reed-Geaghan EG, Landreth GE, Vinters HV, Tontonoz P, Attenuation of neuroinflammation and Alzheimer’s disease pathology by liver X receptors. Proc. Natl. Acad. Sci. U.S.A 104, 10601–10606 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Zelcer N, Tontonoz P, Liver X receptors as integrators of metabolic and inflammatory signaling. J. Clin. Invest 116, 607–614 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Skaar JR, Pagan JK, Pagano M, SCF ubiquitin ligase-targeted therapies. Nat. Rev. Drug Discov 13, 889–903 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Beaudoin III GMJ, Lee S-H, Singh D, Yuan Y, Ng Y-G, Reichardt LF, Arikkath J, Culturing pyramidal neurons from the early postnatal mouse hippocampus and cortex. Nat. Protoc 7, 1741–1754 (2012). [DOI] [PubMed] [Google Scholar]
- 54.Viviani B, Preparation and coculture of neurons and glial cells. Curr. Protoc. Cell Biol Chapter 2, Unit 2.7 (2006). [DOI] [PubMed] [Google Scholar]
- 55.Collins JL, Fivush AM, Watson MA, Galardi CM, Lewis MC, Moore LB, Parks DJ, Wilson JG, Tippin TK, Binz JG, Plunket KD, Morgan DG, Beaudet EJ, Whitney KD, Kliewer SA, Willson TM, Identification of a nonsteroidal liver X receptor agonist through parallel array synthesis of tertiary amines. J. Med. Chem 45, 1963–1966 (2002). [DOI] [PubMed] [Google Scholar]
- 56.Kim J, Yoon H, Ramírez CM, Lee S-M, Hoe H-S, Fernández-Hernando C, Kim J, miR-106b impairs cholesterol efflux and increases Aβ levels by repressing ABCA1 expression. Exp. Neurol 235, 476–483 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Li X, Kan H-Y, Lavrentiadou S, Krieger M, Zannis V, Reconstituted discoidal ApoE-phospholipid particles are ligands for the scavenger receptor BI. The amino-terminal 1–165 domain of ApoE suffices for receptor binding. J. Biol. Chem 277, 21149–21157 (2002). [DOI] [PubMed] [Google Scholar]
- 58.Matz CE, Jonas A, Micellar complexes of human apolipoprotein A-I with phosphatidylcholines and cholesterol prepared from cholate-lipid dispersions. J. Biol. Chem 257, 4535–4540 (1982). [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1. Idol decreases LDLR protein and promotes LDLR ubiquitination in mouse brain cells.
Fig. S2. Loss of Idol does not directly affect inflammatory responses.
Fig. S3. Idol inhibition promotes the clearance of Ab by microglia.
Table S1. Data for Ab40 shown in Fig. 2A.
Table S2. Data for Ab42 shown in Fig. 2A.
Table S3. Quantification of Ab plaque load in Fig. 2E.
Table S4. Quantification of thioflavin S plaque load in Fig. 3B.
Table S5. Quantification of CD45 load in Fig. 4B.
Table S6. Quantification of Iba+ cells in Fig. 4D.