

SUMMARY
Unimolecular decomposition pathways are challenging to address in transition-metal catalysis and have previously not been suppressed via incorporation into a solid support. Two robust metal-organic frameworks (IRMOF-10 and bio-MOF-100) are used for the architectural stabilization of a structurally well-defined gold(III) catalyst. The inherent rigidity of these materials is utilized to preclude a unimolecular decomposition pathway – reductive elimination. Through this architectural stabilization strategy, decomposition of the incorporated gold(III) catalyst in the metal-organic frameworks is not observed; in contrast, the homogeneous analogue is prone to decomposition in solution. Stabilization of the catalyst in these metal-organic frameworks precludes leaching and enables recyclability, which is crucial for productive heterogeneous catalysis.
Graphical Abstract
eTOC blurb
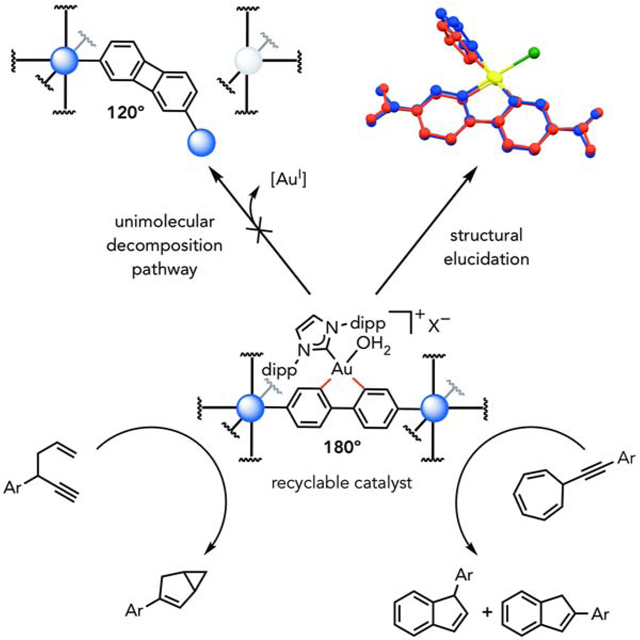
Toste and coworkers present a unique strategy for suppressing a unimolecular decomposition pathway of a transition metal catalyst via architectural stabilization. The structural rigidity of metal-organic frameworks was utilized to constrain the geometry of a gold(III) catalyst to suppress catalyst decomposition by reductive elimination and, therefore, improve catalyst stability. Architectural stabilization is anticipated to serve as a general strategy for the preservation of ligand geometry in otherwise unstable systems.
The Bigger Picture
Catalysis is one of the principles of green chemistry, as catalysts have the potential to promote a chemical reaction without themselves being consumed. However, in many cases catalysts can succumb to undesired processes that limit the number of times they can promote a reaction (turnover number) or their reuse. For example, homogeneous transition metal catalysts can suffer unimolecular or bimolecular decomposition reactions and can be challenging to recycle. This manuscript demonstrates that incorporation of a transition metal catalyst, based on gold(III), into a molecular organic framework MOFs enabled both facile recovery and recyclability, compared to its homogeneous analogue. Moreover, by constraining the geometry of the transition metal catalyst, the architectural rigidity of MOFs suppressed a unimolecular decomposition pathway (reductive elimination). These findings enumerate a strategy for the design of stable and reusable transition metal catalysts that are otherwise prone to unimolecular decomposition pathways.
INTRODUCTION
In mechanochemistry, tensile forces have traditionally been utilized to promote various bond cleavage events,1–3 which can enable productive chemistry through ring opening,4–6 rearrangement,7 and catalyst activation.8,9 More recently, this strategy has been applied towards preserving chemical bonds by suppressing an undesired unimolecular decomposition pathway – a retro-Michael pathway of a maleimide-thiol adduct (Scheme 1A).10 Despite these advances in mechanochemistry, the static force provided by rigid materials has, to the best of our knowledge, previously not been utilized towards the preservation of ligand geometry that are sensitive to bending effects. In cases where reductive elimination is problematic in transition-metal catalysis,11–13 rigidification of ligands could potentially suppress such unimolecular decomposition pathways. Traditionally, solid-state supports have addressed bimolecular decomposition pathways of catalysts;14–16 however, unimolecular decomposition pathways of homogeneous catalysts have previously not been suppressed with solid-state supports. As a model system, we were interested in leveraging architectural stabilization to prevent a unimolecular decomposition pathway of IPrAu(III)(biphenyl)X (where IPr is [1,3-bis(2,6-diisopropylphenyl)imidazole-2-ylidene] and X– is a non-coordinating counteranion), which is known to undergo reductive elimination to yield biphenylene and IPrAu(I)X (Scheme 1B).13 We reasoned that a bifunctionalized IPrAu(III)(biphenyl)X catalyst could be incorporated into a robust porous material to architecturally lock the geometry of the catalyst. Contrary to common solid-state supports, metal-organic frameworks (MOFs) allow for the precise placement of molecules in a well-ordered fashion,17–23 which can constrain the geometry of incorporated guests within the framework. Here, we demonstrate that a unimolecular decomposition pathway of IPrAu(III)(biphenyl)X catalyst is prohibited due to architectural stabilization in MOFs by preserving the geometry of the gold(III) complex such that the linear geometry of the biphenyl ligand is maintained (Scheme 1B). Deviation of its linear geometry is architecturally forbidden because reductive elimination in a rigid MOF would either necessitate the formation of a defect site or result in strain throughout the material. In particular, we demonstrate two strategies for introducing a gold(III) catalyst into MOFs with two distinct biphenyldicarboxylate (BPDC) binding modes, which resulted in no observation of the reductive elimination products in contrast to its homogeneous analogues.
Scheme 1.

Mechanochemical and Architectural Stabilization of Chemical Bonds
RESULTS AND DISCUSSION
In order to probe the architectural stabilization of a gold(III) catalyst in MOFs, we first incorporated a gold(III) precatalyst, IPrAu(III)(BPDC)Cl, into two MOFs through a mixed linker synthesis and a solvent-assisted linker exchange (see Experimental Procedures for details). IPrAu(III)(BPDC)Cl was encompassed into a microporous IRMOF-10 system through a mixed linker synthesis strategy with 5–16% occupancy of gold(III) precatalyst to yield IPrAu(III)Cl-IRMOF-10 (Figure 1A). A partial structure of IPrAu(III)Cl-IRMOF-10 (5% gold occupancy) with primitive cubic (pcu) topology was confirmed via single-crystal X-ray diffraction (SXRD) and powder X-ray diffraction (PXRD). However, the electron density of the gold, chloride, and IPr moieties could not be assigned with the IPrAu(III)Cl-IRMOF-10 SXRD data. The chemical composition of IPrAu(III)Cl-IRMOF-10 was determined by 1H nuclear magnetic resonance (NMR) analysis and inductively coupled plasma-atomic emission spectroscopy (ICP-AES) of the digested samples with an observed IPrAu(III)(BPDC)Cl to BPDC ratio of 16:84 and zinc to gold ratio of 89:11. Mixed linker syntheses with a targeted H2IPrAu(III)(BPDC)Cl/H2BPDC ratio >5:95 in IRMOF-10 yielded crystals that were not suitable for SXRD characterization due to an increase in defects, which affected the overall crystallinity; an increase in defects was attributed to steric clash in the framework between gold(III) complexes bearing bulky IPr ligands. Low gold(III) loading and high symmetry in IRMOF-10 with 5% occupancy of gold(III) complex precluded the possibility of obtaining a full crystal structure of the precatalyst in IPrAu(III)Cl-IRMOF-10 via SXRD to ensure that the catalytic moiety is indeed geometrically constrained between secondary building units (SBUs).
Figure 1. Structures of IPrAu(III)Cl-IRMOF-10 and IPrAu(III)Cl-bio-MOF.

Structure of IPrAu(III)Cl-IRMOF-10 (A) obtained from modeling. Partial structure of IPrAu(III)Cl-bio-MOF-100 (B) identified from single-crystal X-ray diffraction; remainder of structure (dipp groups on IPr) obtained from modeling. Only one gold complex is shown in the structures while the other symmetrically-equivalent positions are omitted for clarity.
In order to mitigate steric clash and increase the occupancy of the gold(III) species in MOFs, bio-MOF-100,24 a mesoporous MOF featuring larger pores (~4.5 nm), was chosen as a support for SXRD characterization of the gold(III) precatalyst and catalyst. Interestingly, bio-MOF-100 features two symmetrically distinct BPDC moieties in its asymmetric unit. This feature offers the opportunity to selectively displace one type of BPDC in the framework and thus make it amenable to low guest loading with high occupancy and as a consequence, enable SXRD characterization (see Supplemental Information). Additionally, bio-MOF-100 possesses a unique binding mode of BPDC to the SBU compared to that of IRMOF-10, which offers an alternative strategy of rigidifying the biphenyl ligand. IPrAu(III)(BPDC)Cl was incorporated into mesoporous bio-MOF-100 through a solvent-assisted linker exchange with up to 40% occupancy of gold(III) precatalyst per substitutable site to yield IPrAu(III)Cl-bio-MOF-100 (Figure 1B), where the occupancy of gold(III) precatalyst was assigned via SXRD. With 40% occupancy of gold(III) precatalyst, gold, chloride, and 7 atoms of the IPr ligand could be assigned in the IPrAu(III)Cl-bio-MOF-100 SXRD crystal structure (Figure S7). The chemical composition of IPrAu(III)Cl-bio-MOF-100 was further confirmed by 1H NMR and ICP-AES analysis of digested samples.
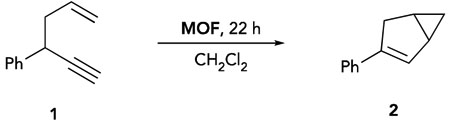
The catalytically active, cationic gold(III) species in the IRMOF-10 system were accessed by treatment of IPrAu(III)Cl-IRMOF-10 with 1 equivalent of AgSbF6 (relative to gold) to access IPrAu(III)SbF6-IRMOF-10, which is analogous to the conditions for activation of homogeneous complex.25 Chloride abstraction was evidenced by the reactivity observed in the IPrAu(III)SbF6-IRMOF-10-catalyzed cycloisomerization reaction of 1,5-enyne substrate 1 to yield the corresponding bicyclohexene product 2 (Table 1, entry 1). In contrast, addition of substrate 1 to IPrAu(III)Cl-IRMOF-10 without AgSbF6 treatment, under otherwise equivalent conditions, resulted in no background reactivity (entry 2). Additionally, subjecting pristine IRMOF, with or without AgSbF6 treatment, to substrate 1 did not yield any product (entries 3 and 4). These observations support the conclusion that the zinc-based SBUs and silver salt are not responsible for the reactivity observed with IPrAu(III)SbF6-IRMOF-10. Additionally, another IPrAu(III)SbF6-IRMOF-10-catalyzed cycloisomerization reaction yielded a product distribution that was consistent with that of the homogeneous gold(III) analogue, which further supports the conclusion that cationic gold(III) species are responsible for the observed reactivity (Table S1).
Table 1.
Control Experiments with IRMOF
 | ||
|---|---|---|
| Entry | MOF | Conversion (%) |
| 1 | IPrAu(III)SbF6-IRMOF-10 (3 mol % Au) | 44 |
| 2 | IPrAu(III)Cl-IRMOF-10 (3 mol % Au) | <1 |
| 3 | IRMOF-9-AgSbF6 | <1 |
| 4 | IRMOF-9 | <1 |
See Experimental Procedures for general reaction conditions.
Efforts towards accessing cationic gold(III) species in bio-MOF-100 through AgSbF6 treatment resulted in a decrease in crystallinity. We posited that this MOF degradation might be attributed to protonation of the BPDC linkers by HSbF6 generated from the hydrolysis of AgSbF6 in the presence of adventitious water. Addition of NaBArF4, a common halide-abstracting agent that is less prone to hydrolysis,26 also yielded poorly crystalline frameworks, presumably due to hard acid – hard base interactions between sodium cations and the carboxylate-based linkers. Thus, TlPF6 was chosen as the halide-abstracting agent, as it is less sensitive towards hydrolysis and features a soft thallium cation. After TlPF6 treatment of IPrAu(III)Cl-bio-MOF-100, the sample retained crystallinity, yielding the desired IPrAu(III)PF6-bio-MOF-100. SXRD measurement revealed preservation of 40% gold(III) occupancy in the framework, which is consistent with that of the precatalyst structure. In the crystal structure of IPrAu(III)PF6-bio-MOF-100, the chloride ligand was no longer observed, which indicates successful halide abstraction from the precatalyst to form the desired cationic catalyst (Figure S8).
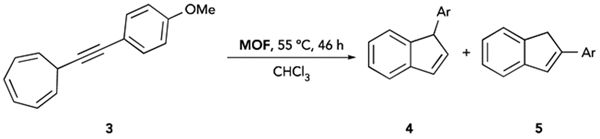
IPrAu(III)PF6-bio-MOF-100 had very low reactivity towards the cycloisomerization reaction of substrate 1 to product 2; this low reactivity was attributed to a potential decrease in rate of diffusion of nonpolar substrates through the intrinsically anionic bio-MOF-100 framework. Raising the temperature to increase the rate of diffusion of 1 was, however, not compatible with this cycloisomerization reaction due to the thermal instability of 1,5-enynes. Thus, alkynyl cylcoheptatriene substrate 3 was chosen as a model substrate, as it has higher thermal stability than 1 and is known to undergo a gold-catalyzed cycloisomerization reaction to yield the corresponding indene products 4 and 5.27 Addition of 3 to IPrAu(III)PF6-bio-MOF-100 resulted in formation of desired products 4 and 5 at elevated temperatures with consistent product selectivity with that of the homogeneous gold(III) analogue (Table 2, entry 1; Table S1). Similar to the IRMOF-based gold(III) reactivity, no product was observed in the corresponding control experiments with bio-MOF-100 (entries 2–4). These data further demonstrate that chloride abstraction from the gold(III) precatalyst was successful by TlPF6 treatment of IPrAu(III)Cl-bio-MOF-100.
Table 2.
Control Experiments with bio-MOF*
 | |||
|---|---|---|---|
| Entry | MOF | Conversion (%) | 4/5 |
| 1 | IPrAu (III)PF6-bio-MOF-100 (7 mol % Au) | 96 | 60:40 |
| 2 | IPrAu(III)Cl-bioMOF-100 (7 mol % Au) | <1 | - |
| 3 | bio-MOF-100-TIPF6 | <1 | - |
| 4 | bio-MOF-100 | <1 | - |
See Experimental Procedures for general reaction conditions.
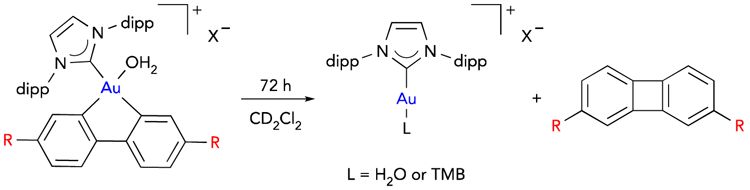
The chemical stability of this cationic gold(III) catalyst in IRMOF-10 and bio-MOF-100 was compared with the stability of their homogeneous counterparts. Reductive elimination of IPrAu(III)(biphenyl)SbF6 is known to be exacerbated in the presence of a trap for cationic gold(I) species, 1,3,5-trimethoxybenzene (TMB), to yield a Au(I)-TMB adduct (Table 3, entry 3).13 In contrast, no 2,7-biphenylene dicarboxylic acid or Au(I)-TMB adduct was observed with the MOF analogues under equivalent conditions in the supernatant or by digestion 1H NMR analysis or ICP-AES (entries 1 and 2). Additionally, we observed 78% decomposition of a homogeneous gold(III) analogue to the corresponding reductive elimination products when heated to 55°C (entry 5). In bio-MOF-100, the gold(III) occupancy remained unperturbed under these conditions and no reductive elimination products were observed in the supernatant by 1H NMR analysis or ICP-AES (entry 4). These results are consistent with the hypothesis that the architectural stabilization afforded by IRMOF-10 and bio-MOF-100 is robust enough to prevent this unimolecular decomposition.
Table 3.
Stability of Homogeneous Gold(III) Complexes vs. MOF Analogues*
 | ||||
|---|---|---|---|---|
| Entry | Catalyst | T (°C) | Additive | Decomposition (%) |
| 1 | IPrAu(III)SbF6-IRMOF-10 | 25 | TMB (10 equiv.) | <5 |
| 2 | IPrAu(III)PF6-bio-MOF-100 | 25 | TMB (10 equiv.) | <5 |
| 3 | IPrAu(III)(biphenyl)SbF6 | 25 | TMB (10 equiv.) | 68 |
| 4 | IPrAu(III)PF6-bio-MOF-100a | 55 | none | <5 |
| 4 | IPrAu(III)(Me2BPDC)SbF6a | 55 | none | 78 |
See Supplemental Information for general reaction conditions.
CDCl3 used instead of CD2Cl2
As a further evaluation that catalysis was occurring in the pores of the framework rather than at the surface or in bulk solution, the impact of substrate size was evaluated in IRMOF-10, as it features smaller pore dimensions than bio-MOF-100 (Figure 2). A Au(III)-IRMOF-10-catalyzed reaction of 1,5-enyne substrate 6, which is slightly larger along one dimension, did not show a substantial decrease in reactivity compared to substrate 1. On the other hand, when the steric bulk was extended along two dimensions with substrate 7, a decrease in reactivity to 2% conversion after 22 h was observed. Further extension of steric bulk along these two dimensions with substrate 8 resulted in no observed product formation by 1H NMR. In contrast, full conversion was observed with substrates 1, 6, 7, and 8 with 4 mol % homogeneous IPrAu(biphenyl)SbF6, after 22 hours (see Supplemental Information). These data are consistent with the hypothesis that the catalysis observed with IPrAu(III)SbF6-IRMOF-10 occurs within the pores, and the leaching of catalytically active species into solution is unlikely. The lack of catalytically active species in solution further highlights the effectiveness of architectural stabilization to prohibit the formation of undesired gold(I) species in bulk solution.
Figure 2. Impact of Substrate Size on Catalysis with IPrAu(III)SbF6-IRMOF-10*.

*See Experimental Procedures for general reaction conditions.
After evaluating the stability of both Au(III)-MOF systems towards reductive elimination, the reuse of both systems was evaluated to further assess the impact of the architectural stability of these frameworks on the catalyst recyclability and longevity. To this end, employing IPrAu(III)SbF6-IRMOF-10 with 3 mol % gold loading as a catalyst, 44% and 46% conversion of enyne 1 to bicyclohexene 2 was observed in cycles 1 and 2, respectively (Figure 3A). Reactivity towards the cycloisomerization of 1 continued to persist in cycles 3–5, albeit at lower conversions. We hypothesized that this decrease in reactivity might be attributed to the slow trapping of Au(III)Cl species in the presence of AgCl within the pores, which is a phenomenon that has previously been observed with solid-supported cationic gold species.28 Indeed, we observed a rebound in reactivity upon treatment of IPrAu(III)SbF6-IRMOF-10 with 1 equivalent of AgSbF6 (relative to gold) in cycle 6 with continued reactivity over the subsequent cycle. Recyclable reactivity over 7 cycles was also observed with IPrAu(III)PF6-bio-MOF-100-catalyzed cycloisomerization reaction of 3 to yield 4 and 5 (Figure 3B), and recyclable reactivity was also observed at shorter reaction durations (Table S3). Additionally, no loss in reactivity of IPrAu(III)PF6-bio-MOF-100 was observed after storing the catalyst for 29 days, demonstrating that catalyst deactivation does not occur even after long-term storage. Recyclability reactivity in both IRMOF-10 and bio-MOF-100 further demonstrates the robustness of these systems engendered by architectural stabilization. This architectural stabilization strategy should prove general to access other immobilized transition-metal catalysts that are otherwise prone to unimolecular decomposition pathways and are consequently unstable or inaccessible in solution.
Figure 3. Recyclability of IPrAu(III)SbF6-IRMOF-10 (A) and Au(III)PF6-bio-MOF-100 (B)*.

*See Experimental Procedures for general reaction conditions.
EXPERIMENTAL PROCEDURES
Synthesis of Au(III)-MOFs for Catalysis
IPrAu(III)Cl-IRMOF-10
To a 2 dram vial was added IPrAu(H2BPDC)Cl (8.6 mg, 0.010 mmol, 0.25 equiv.), H2BPDC (7.3 mg, 0.030 mmol, 0.75 equiv.), Zn(NO3)2•4 H2O (42 mg, 0.16 mmol, 4.0 equiv.), and diethyl formamide (2.14 mL). The reaction mixture was capped, sonicated for 5 minutes, then heated at 90°C in an oven for 24 h. This yielded yellow crystals, which were washed with DMF (6 mL × 5), DCM (6 mL × 15), then MeNO2 (6 mL × 5). Due to a loss of crystallinity of IPrAu(III)Cl-IRMOF-10 in the absence of solvent, the crystals were immersed in solvent prior to AgSbF6 treatment. 16% loading IPrAu(BPDC)Cl vs BPDC was observed by digestion 1H NMR analysis. Zn:Au ratio of 88:12 observed by ICP-AES (expected Zn:Au ratio: 89:11).
IPrAu(III)SbF6-IRMOF-10
To a 2 mL vial was added IPrAu(III)Cl-IRMOF-10 (16% IPrAu(BPDC)Cl loading, 2 mg, 1 equiv.) immersed in MeNO2 (0.1 mL), followed by the addition of a solution of AgSbF6 (7 mM in MeNO2, 0.13 mL, 1 equiv.). After 48 h, the crystals were washed with MeNO2 (2 mL × 3) then DCM (2 mL × 5). Due to a loss of crystallinity of IPrAu(III)SbF6-IRMOF-10 in the absence of solvent, the crystals were left immersed in solvent prior to catalysis.
IPrAu(III)Cl-bio-MOF-100
To a 1 dram vial was added bio-MOF-100 (20 mg), IPrAu(H2BPDC)Cl (20 mg), and DMF (0.9 mL). The reaction mixture was capped, then heated at 50°C in an oven for 96 h. This yielded colorless crystals, which were washed with DMF (3 mL × 7), DCM (3 mL × 15), then MeNO2 (3 mL × 5). Due to a loss of crystallinity of IPrAu(III)Cl-bio-MOF-100 in the absence of solvent, the crystals were immersed in solvent prior to TlPF6 treatment. 15% loading IPrAu(BPDC)Cl vs BPDC was observed by digestion 1H NMR. Zn:Au ratio of 89:11 observed by ICP-AES (expected Zn:Au ratio: 90:10).
IPrAu(III)PF6-bio-MOF-100
To a 2 mL vial was added IPrAu(III)Cl-bio-MOF-100 (15% IPrAu(BPDC)Cl loading, 5 mg, 1 equiv.) immersed in MeNO2 (0.1 mL), followed by the addition of a solution of TlPF6 (15 mM in MeNO2, 0.10 mL, 1 equiv.). After 48 h, the crystals were washed with MeNO2 (2 mL × 3) then CHCl3 (2 mL × 5). Due to a loss of crystallinity of IPrAu(III)PF6-bio-MOF-100 in the absence of solvent, the crystals were left immersed in solvent prior to catalysis.
General Procedures for Catalysis with Au(III)-MOFs
IPrAu(III)SbF6-IRMOF-10-catalyzed 1,5-Enyne Cycloisomerization Reaction
To a 2 mL vial was added IPrAu(III)SbF6-IRMOF-10 (16% IPrAu(BPDC)Cl loading, 0.03 equiv.) immersed in DCM (0.3 mL), followed by the addition of 1,5-enyne (1 equiv.). After 22 h, the organic supernatant was removed with DCM (2 mL × 5). The washed IPrAu(III)SbF6-IRMOF-10 crystals were resubjected to the same conditions for recyclability studies. Conversions were determined by 1H NMR spectroscopy. No additional conversion was detected in the supernatant upon removal of IPrAu(III)SbF6-IRMOF-10. 1H NMR spectra of products 2 and 6 match those previously reported.29 For control experiments, an equivalent amount of IPrAu(III)Cl-IRMOF-10 or IRMOF-9 was used instead of IPrAu(III)SbF6-IRMOF-10. 14% loading IPrAu(BPDC)Cl vs BPDC was observed by digestion 1H NMR analysis for IPrAu(III)SbF6-IRMOF-10 after catalysis. Zn:Au ratio of 88:12 observed for IPrAu(III)SbF6-IRMOF-10 by ICP-AES after catalysis.
IPrAu(III)PF6-bio-MOF-100-catalyzed Alkynyl Cycloheptatriene Cycloisomerization Reaction
To a 2 mL vial was added IPrAu(III)PF6-bio-MOF-100 (15% IPrAu(BPDC)Cl loading, 5 mg, 0.0015 mmol, 0.07 equiv.) immersed in CHCl3 (0.1 mL), followed by the addition of substrate 3 (5 mg, 0.022 mmol, 1 equiv.). After heating the reaction mixture at 55°C for 46 h, the organic supernatant was removed with CHCl3 (2 mL × 5). The washed IPrAu(III)PF6-bio-MOF-100 crystals were resubjected to the same conditions for recyclability studies. Conversions were determined by 1H NMR spectroscopy. 1H NMR spectra of products 4 and 5 match those previously reported.27 For control experiments, an equivalent amount of IPrAu(III)Cl-bio-MOF-100 or bio-MOF-100 was used instead of IPrAu(III)PF6-bio-MOF-100. 14% loading IPrAu(BPDC)Cl vs BPDC was observed by digestion 1H NMR analysis for IPrAu(III)PF6-bio-MOF-100 after catalysis. Zn:Au ratio of 88:12 observed for IPrAu(III)PF6-bio-MOF-100 by ICP-AES after catalysis.
Other experimental details, procedures, and characterization data (Figures S1–S40 and Tables S1–S7) are provided in the Supplemental Information.
Supplementary Material
Highlights.
A structurally well-defined gold(III) precatalyst was introduced into two MOFs
Unimolecular decomposition of a gold(III) catalyst in the MOFs was suppressed
SXRD data revealed that the geometry of the catalyst was architecturally restrained
Incorporated gold(III) catalyst in MOFs demonstrated excellent recyclability
ACKNOWLEDGMENTS
F.D.T. thanks NIHGMS (R35 GM118190), J.S.L. thanks the NSF-GRFP (DGE 1106400 and 1752814), and S.L. thanks predoctoral fellowships from MINECO (BES-2015–072627) for financial support. Financial support for MOF SXRD and PXRD studies in the O.M.Y. laboratory was provided by King Abdulaziz City for Science and Technology as part of a joint KACST-UC Berkeley collaboration (Center of Excellence for Nanomaterials and Clean Energy Applications). We thank Dr. Suhong Kim, Dr. Christian S. Diercks, Banruo Huang, Dr. Cynthia M. Hong, Dr. Patrick T. Bohan, Edward Miller, Danny Q. Thach, and Jeffrey S. Derrick for helpful discussions. O.M.Y. acknowledges the collaboration, input, and support of Prince Turki bin Saud bin Mohammed Al-Saud (President of KACST). This research used resources of the beamline 11.3.1 and 12.2.1 at Advanced Light Source, which is a DOE Office of Science User Facility under contract no. DE-AC02–05-CH11231.
Footnotes
DATA AND SOFTWARE AVAILABILITY
Crystallographic data for the structures reported in this paper are tabulated in the Supplemental Information and have been deposited at the Cambridge Crystallographic Data Center (CCDC) with accession numbers CCDC: 1955738
(IPrAu(III)(H2BPDC)Cl), 1955739 (IPrAu(III)(Me2BPDC)Cl), 1955737
(IPrAu(III)Cl-bio-MOF-100), and 1955736 (IPrAu(III)PF6-bio-MOF-100).
SUPPLEMENTAL INFORMATION
Supplemental Information includes supplemental experimental procedures, 40 figures, 7 tables, and 4 data files.
DECLARATION OF INTERESTS
The authors declare no competing interests.
REFERENCES AND NOTES
- 1.Davis DA, Moore JS, Caruso MM, White SR, Sottos NR, Odom SA, and Shen Q (2009). Mechanically-Induced Chemical Changes in Polymeric Materials. Chem. Rev 109, 5755–5798. [DOI] [PubMed] [Google Scholar]
- 2.May PA and Moore JS (2013). Polymer mechanochemistry: techniques to generate molecular force via elongational flows. Chem. Soc. Rev 42, 7497–7506. [DOI] [PubMed] [Google Scholar]
- 3.Beyer MK, and Clausen-Schaumann H (2005). Mechanochemistry: The Mechanical Activation of Covalent Bonds. Chem. Rev 105, 2921–2948. [DOI] [PubMed] [Google Scholar]
- 4.Hickenboth CR, Moore JS, White SR, Sottos NR, Baudry J, and Wilson SR (2007). Biasing reaction pathways with mechanical force. Nature 446, 423–427. [DOI] [PubMed] [Google Scholar]
- 5.Chen Z, Mercer JAM, Zhu X, Romaniuk JAH, Pfattner R, Cegelski L, Martinez TJ, Burns NZ, and Xia Y (2017). Mechanochemical unzipping of insulating polyladderene to semiconducting polyacetylene. Science 357, 475–479. [DOI] [PubMed] [Google Scholar]
- 6.Kean ZS, Niu Z, Hewage GB, Rheingold AL, and Craig SL (2013). Stress-Responsive Polymers Containing Cyclobutane Core Mechanophores: Reactivity and Mechanistic Insights. J. Am. Chem. Soc 135, 13598–13604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Diesendruck CE, Steinberg BD, Sugai N, Silberstein MN, Sottos NR, White SR, Braun PV, and Moore JS (2012). Proton-Coupled Mechanochemical Transduction: A Mechanogenerated Acid. J. Am. Chem. Soc 134, 12446–12449. [DOI] [PubMed] [Google Scholar]
- 8.Piermattei A, Karthikeyan S, and Sijbesma RP (2009). Activating catalysts with mechanical force. Nat. Chem 1, 133–137. [DOI] [PubMed] [Google Scholar]
- 9.Groote R, Jakobs RTM, and Sijbesma RP (2013). Mechanocatalysis: forcing latent catalysts into action. Polym. Chem 4, 4846–4859. [Google Scholar]
- 10.Huang W, Wu X, Gao X, Yu Y, Lei H, Zhu Z, Shi Y, Chen Y, Qin M, Wang W, and Cao Y (2019). Maleimide–thiol adducts stabilized through stretching. Nat. Chem 11, 310–319. [DOI] [PubMed] [Google Scholar]
- 11.Segelstein BE, Butler TW, and Chenard BL (1995). Equilibration of the Oxidative Addition Product of Tetrakis(triphenylphosphine)palladium and Electron-Rich Aryl Halides Leads to Product Scrambling in the Stille Reaction. J. Org. Chem 60, 12–13. [Google Scholar]
- 12.Kawai H, Wolf WJ, DiPasquale AG, Winston MS, and Toste FD (2016). Phosphonium Formation by Facile Carbon–Phosphorus Reductive Elimination from Gold(III). J. Am. Chem. Soc 138, 587–593. [DOI] [PubMed] [Google Scholar]
- 13.Zhukhovitskiy AV, Kobylianskii IJ, Wu C-Y, and Toste FD (2018). Migratory Insertion of Carbenes into Au(III)–C Bonds. J. Am. Chem. Soc 140, 466–474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Rhers B, Quadrelli EA, Baudouin A, Taoufik M, Copéret C, Lefebvre F, Basset J-M, Fenet B, Sinha A, and Schrock RR (2006). Understanding the reactivity of [W=NAr(CH2tBu)2(=CHtBu)] (Ar=2,6-iPrC6H3) with silica partially dehydroxylated at low temperatures through a combined use of molecular and surface organometallic chemistry. J. Organomet. Chem 691, 5448–5455. [Google Scholar]
- 15.Zhang T, Manna K, and Lin W (2016). Metal–Organic Frameworks Stabilize Solution-Inaccessible Cobalt Catalysts for Highly Efficient Broad-Scope Organic Transformations. J. Am. Chem. Soc 138, 3241–3249. [DOI] [PubMed] [Google Scholar]
- 16.Choi KM, Kim D, Rungtaweevoranit B, Trickett CA, Barmanbek JTD, Alshammari AS, Yang P, and Yaghi OM (2017). Plasmon-Enhanced Photocatalytic CO 2 Conversion within Metal–Organic Frameworks under Visible Light. J. Am. Chem. Soc 139, 356–362. [DOI] [PubMed] [Google Scholar]
- 17.Lee S, Kapustin EA, and Yaghi OM (2016). Coordinative alignment of molecules in chiral metal-organic frameworks. Science 353, 808–811. [DOI] [PubMed] [Google Scholar]
- 18.Kapustin EA, Lee S, Alshammari AS, and Yaghi OM (2017). Molecular Retrofitting Adapts a Metal–Organic Framework to Extreme Pressure. ACS Cent. Sci 3, 662–667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Yuan S, Lu W, Chen Y-P, Zhang Q, Liu T-F, Feng D, Wang X, Qin J, and Zhou H-C (2015). Sequential Linker Installation: Precise Placement of Functional Groups in Multivariate Metal–Organic Frameworks. J. Am. Chem. Soc 137, 3177–3180. [DOI] [PubMed] [Google Scholar]
- 20.Lee J, Farha OK, Roberts J, Scheidt KA, Nguyen ST, and Hupp JT (2009). Metal–organic framework materials as catalysts. Chem. Soc. Rev 38, 1450–1459. [DOI] [PubMed] [Google Scholar]
- 21.Yoon M, Srirambalaji R, and Kim K (2012). Homochiral Metal–Organic Frameworks for Asymmetric Heterogeneous Catalysis. Chem. Rev 112, 1196–1231. [DOI] [PubMed] [Google Scholar]
- 22.Zhang T, and Lin W (2014). Metal–organic frameworks for artificial photosynthesis and photocatalysis. Chem. Soc. Rev 43, 5982–5993. [DOI] [PubMed] [Google Scholar]
- 23.Ma L, Abney C, and Lin W (2009). Enantioselective catalysis with homochiral metal–organic frameworks. Chem. Soc. Rev 38, 1248–1256. [DOI] [PubMed] [Google Scholar]
- 24.An J, Farha OK, Hupp JT, Pohl E, Yeh JI, and Rosi NL (2012). Metal-adeninate vertices for the construction of an exceptionally porous metal-organic framework. Nat. Commun 3, 604. [DOI] [PubMed] [Google Scholar]
- 25.Wu C-Y, Horibe T, Jacobsen CB, and Toste FD (2014). Stable gold(III) catalysts by oxidative addition of a carbon–carbon bond. Nature 517, 449–454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kleinbeck F, and Toste FD (2009). Gold(I)-Catalyzed Enantioselective Ring Expansion of Allenylcyclopropanols. J. Am. Chem. Soc 131, 9178–9179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.McGonigal PR, de León C, Wang Y, Homs A, Solorio-Alvarado CR, and Echavarren AM (2012). Gold for the Generation and Control of Fluxional Barbaralyl Cations. Angew. Chemie Int. Ed 51, 13093–13096. [DOI] [PubMed] [Google Scholar]
- 28.Chen M, Zhang ZM, Yu Z, Qiu H, Ma B, Wu HH, and Zhang J (2015). Polymer-Bound Chiral Gold-Based Complexes as Efficient Heterogeneous Catalysts for Enantioselectivity Tunable Cycloaddition. ACS Catal. 5, 7488–7492. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.