

Abstract
Pulsed-field gradient NMR spectroscopy is widely used to measure the translational diffusion and hydrodynamic radius (Rh) of biomolecules in solution. For unfolded proteins, the Rh provides a sensitive reporter on the ensemble-averaged conformation and the extent of polypeptide chain expansion as a function of added denaturant. Hydrostatic pressure is a convenient and reversible alternative to chemical denaturants for the study of protein folding, and enables NMR measurements to be performed on a single sample. While the impact of pressure on the viscosity of water is well known, and our water diffusivity measurements agree closely with theoretical expectations, we find that elevated pressures increase the Rh of dioxane and other small molecules by amounts that correlate with their hydrophobicity, with parallel increases in rotational friction indicated by 13C longitudinal relaxation times. These data point to a tighter coupling with water for hydrophobic surfaces at elevated pressures. Translational diffusion measurement of the unfolded state of a pressure-sensitized ubiquitin mutant (VA2-ubiquitin) as a function of hydrostatic pressure or urea concentration shows that Rh values of both the folded and the unfolded states remain nearly invariant. At ca 23 Å, the Rh of the fully pressure-denatured state is essentially indistinguishable from the urea-denatured state, and close to the value expected for an idealized random coil of 76 residues. The intrinsically disordered protein (IDP) α-synuclein shows slight compaction at pressures above 2 kbar. Diffusion of unfolded ubiquitin and α-synuclein is significantly impacted by sample concentration, indicating that quantitative measurements need to be carried out under dilute conditions.
Keywords: α-synuclein, dioxane, hydration layer, IDP, pressure-induced unfolding, protein compaction, radius of hydration, random coil, translational diffusion, self-diffusion, ubiquitin, unfolded chain, urea
Graphical Abstract
1. Introduction
Pulsed-field gradient (PFG) NMR spectroscopy enables the measurement of translational diffusion of biomolecules in solution [1–4]. Through the Stokes-Einstein equation, the translational diffusion coefficient of a molecule is inversely related to its hydration radius (Rh), which provides insight into size [5, 6], including alterations upon oligomerization [7–12], ligand binding [13], as well as folding and unfolding [14–18]. Translational diffusion coefficients are commonly obtained by recording arrayed 1D or 2D pulsed field gradient (PFG) NMR spectra, from which the signal attenuation caused by diffusion during a fixed delay time is then fit to the Stejskal-Tanner equation [1, 19–23]. PFGs are widely used in modern NMR spectroscopy, and availability of the requisite hardware now enables routine measurement of translational diffusion constants for a wide range of systems, spanning molecules as small as tens of Da [24] to complex protein assemblies [11, 25], amyloid fibrils [26], virus-like particles [27], and phase-separated membrane-less organelles [28, 29]. In addition, pulse sequences have been developed to characterize the diffusion of large proteins [28–31] and those undergoing chemical exchange [32].
PFG translational diffusion measurements also offer insight into structural ensembles populated by denatured, intrinsically disordered, or partially folded proteins [13, 15, 17, 33]. Empirical measurements have established a relationship between the number of amino acids in a fully denatured protein and its expected Rh [33], and comparison to the measured Rh for the protein of interest thus reports on the overall compaction or, in rare cases, expansion of the polypeptide chain. Compaction of the disordered chain can result from the presence of transiently structured elements or weak hydrophobic or electrostatic interactions within the unfolded polypeptide. While studies performed under high denaturant conditions (e.g. 8 M urea or 6 M guanidine hydrochloride) generally yield Rh values that closely follow the empirical relation between chain length and Rh, the magnitude of the change in the structural ensemble populated by unfolded proteins at lower denaturant conditions, approaching the folding threshold, remains controversial: under such conditions, both significantly collapsed and near fully expanded conformations were deduced from Förster resonance energy transfer (FRET) and small-angle X-ray scattering (SAXS) methods [34–37]. Previous solution-state NMR studies demonstrated that Rh of the unfolded state of the N-terminal SH3 domain of drk, which exists in a slow equilibrium with its folded state, is ca 30% larger than its folded state, but ca 10% smaller than the Rh of its fully denatured state in guanidine hydrochloride [17, 38]. Following a recent, renewed debate about discrepancies between the denaturant dependence of the unfolded state measured by SAXS and FRET [39, 40], another group concluded that such differences are consistent with a decoupling of size and shape fluctuations in finite-sized, heteropolymeric disordered systems [41]; i.e., the FRET-derived distance distributions, SAXS-derived radii of gyration, and diffusion-derived Rh contain semi-independent constraints on the ensemble distribution [17].
In comparison to high temperature or addition of denaturing chemicals, elevated hydrostatic pressure represents a relatively benign and often reversible unfolding mechanism [42–45]. Under conditions where there is an appreciably negative volume change between the folded and unfolded states, on the order of ≥ 50–100 mL/mole, a few kilobar of hydrostatic pressure can drive the thermodynamic equilibrium of a folded protein to that of the unfolded state. Hydrophobic cavities (void volume) present in the folded protein but absent in the unfolded state constitute an important component of this volume difference [45, 46], but decreased solvent density at the larger solvent-accessible hydrophobic surface of the unfolded protein may also represent a significant factor [47]. Proteins that unfold at high pressure typically exist in slow exchange with their folded state when studied at intermediate pressures. Under such conditions, PFG NMR can provide simultaneous access to the measurement of translational diffusion coefficients for both the folded and unfolded states that are in equilibrium with one another over a range of pressures. This situation applies for the previously characterized pressure-sensitized double mutant of ubiquitin, V17A/V26A (VA2-ubiquitin [48–50]), used as a model system in the present study.
While high-pressure NMR has been used to study translational diffusion of neat liquids and a handful of small molecules [51–53], relatively little is known about the effect of elevated pressures on the hydrodynamic properties of unfolded proteins. Here, we sought to characterize the impact of increased pressure on the hydrodynamic behavior of three model proteins: the intrinsically disordered protein (IDP) α-synuclein, which remains unfolded at all pressures; the pressure-sensitized VA2-ubiquitin, which can access both folded and unfolded states depending on the pressure; and wild-type (WT) ubiquitin, which remains fully folded at all experimentally accessible pressures. Pressure-induced changes to the viscosity of water become an important factor when performing translational diffusion measurements as a function of pressure. Although the diffusivity of water follows the known pressure dependence of its viscosity (https://webbook.nist.gov/chemistry/fluid/), we find that dioxane and other small hydrophobic molecules show a significant expansion in their Rh values with increasing pressure, and a concomitant decrease in rotational diffusion rate as indicated by 13C T1 times. When using dioxane as an internal reference to derive the Rh of a protein, failure to correct for this effect results in underestimates of the protein Rh. Taking the newly calibrated pressure dependence of the dioxane Rh into account restores accuracy of the internal referencing method. The Rh of α-synuclein and the unfolded chain of VA2-ubiquitin are then found to be nearly independent of pressure up to 2 kbar. At pressures > 2 kbar, a slight compaction of the unfolded chains is observed, whereas WT ubiquitin, which remains fully folded at 3 kbar, shows virtually no change in its Rh at all examined pressures. We compare the high-pressure diffusion data to those collected in the presence of increasing concentrations of urea, and find that the Rh of the unfolded state of VA2-ubiquitin depends minimally on the amount of added urea. Our results suggest that both α-synuclein and the unfolded state of VA2-ubiquitin do not expand significantly between mildly and highly denaturing conditions. However, since NMR diffusion measures an ensemble-averaged property, we cannot rule out a small change in the population of transiently compacted chains as such states would not contribute significantly to the measured signal.
2. Methods
2.1. Protein expression and purification
15N-labeled VA2-ubiquitin and αS were expressed and purified as described previously [54]. αS lacked the N-terminal acetylation present in mammalian cells. For VA2-ubiquitin, a final HPLC purification step was essential to avoid protease contamination, which otherwise presents a problem at high pressures where VA2-ubiquitin but not the protease unfolds, making VA2-ubiquitin an excellent protease substrate. WT ubiquitin was purchased from Sigma-Aldrich (USA). Both VA2- and WT-ubiquitin NMR samples were prepared in 30 mM sodium phosphate at pH 6.4. αS samples were in 30 mM sodium phosphate, 3 mM EDTA, pH 7.
2.2. Rh determination
Determination of Rh from PFG NMR experiments requires accurate knowledge of the solution viscosity and gradient strength in units of G●cm−1. With solvent viscosity being strongly dependent on temperature, and non-linearity of the gradient across the sample making the effective gradient strength sensitive to the sample size and position, in particular when Shigemi tubes are used to restrict the sample volume to the most linear region of the applied gradients, the use of an internal standard is often preferred. In this case, the diffusion of a reference molecule of known Rh is determined alongside that of the protein of interest. For measurements carried out in D2O, diffusion of the residual HDO signal may be used as the internal reference [1], while using a long inter-scan delay to account for the very long T1 of HDO protons. In H2O, radiation damping effects can interfere with measurement of the H2O diffusion rate, making it less suitable as an internal reference. For this reason, a small amount (e.g. 0.1% v/v) of dioxane is commonly used as a reference (Rh = 2.12 Å under ambient conditions) [14]. It conveniently resonates near 3.5 ppm, upfield of most Hα resonances and downfield of most Hβ signals and was found not to interact with folded or unfolded proteins. For all our protein diffusion measurements, samples were doped with 0.1% dioxane (v/v), enabling calculation of the hydration radius for the protein (eq 3), as outlined previously [14]:
| (1) |
| (2) |
| (3) |
where D refers to the diffusion coefficient, kB to Boltzmann’s constant, T to the temperature, η to the solvent viscosity, Ij to the intensity in spectrum j, I0 to the intensity in the reference spectrum, γ to the gyromagnetic ratio, Gj to the gradient strength in spectrum j, δ to the total duration of the encoding gradient pulses, τ to the gradient recovery delay, and Δ is defined as the diffusion time including the total duration of the encoding gradients (δ) plus 2τ for the two short gradient recovery delays, as defined in Fig. 1B of reference [23]. Dreference refers to the diffusion coefficient of the molecule of known Rh, with Dunknown and Rhunknown respectively referring to the diffusion coefficient and hydration radius of the target molecule.
Fig. 1 |.
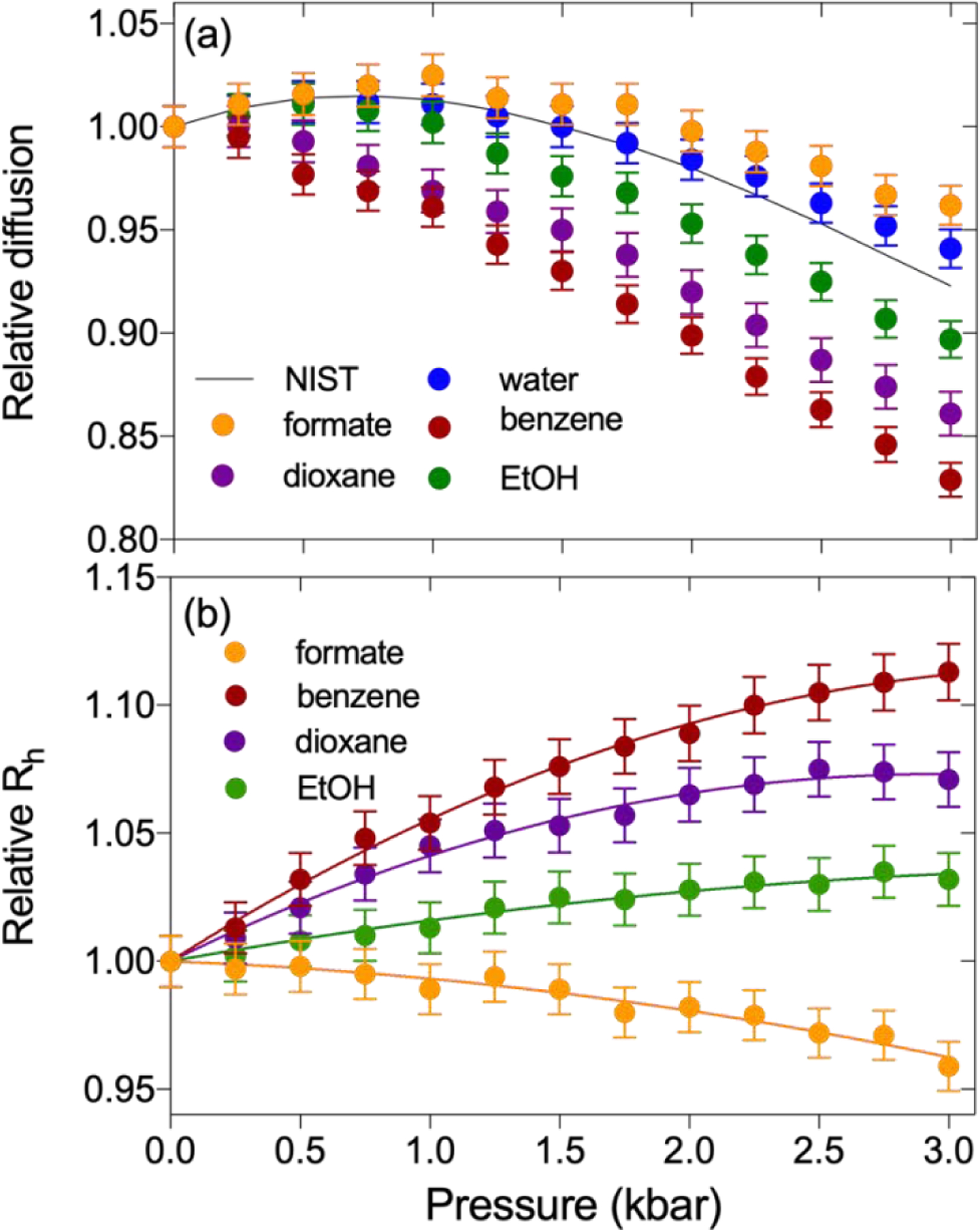
Pressure-dependence of the translational diffusion of tracer molecules. (a) The relative diffusion (Dx-bar / D1-bar) at 293 K, plotted as a function of hydrostatic pressure. Symbols correspond to a sample of 95% H2O, 4% D2O, 0.25% each (v/v) of dioxane, DMSO, methanol and ethanol, plus 150 mM sodium formate, 20 mM benzene, 50 μM DSS, and 50 mM Tris buffer, pH 6.5. The solid black line depicts the inverse relative solvent viscosity (η1-bar / ηx-bar), as obtained from the NIST website for water at 293 K (https://webbook.nist.gov/chemistry/fluid/). (b) Relative Rh values (Rhx-bar / Rh1-bar) of formate, benzene, dioxane, and ethanol as a function of pressure. Rh at elevated pressures were determined from the diffusion rate after correcting for the increased water viscosity, taken from the NIST website. Solid lines represent best-fit second-order polynomials with the coefficients listed in Table 1. Data for Tris, methanol, and DMSO are shown in SI Fig. S3.
2.2. NMR spectroscopy
All 1H-detected NMR spectra were recorded on a 700-MHz Bruker Avance-III spectrometer equipped with a room-temperature, triple-axis gradient TXI probe. 13C relaxation measurements were carried out on a Bruker 600-MHz Avance-II system, with direct 13C detection at 150.9 MHz and using a cryogenic (TCI) probehead. All NMR measurements were carried out using a ceramic NMR tube with a 2.8 mm inner diameter, rated for pressures up to 3000 bar, which was linked to an automatic pump system (Daedalus Innovations, Philadelphia, PA) to regulate the hydrostatic pressure inside the sample cell [55].
To measure self-diffusion coefficients (D), the 1D stimulated echo longitudinal encodedecode pulse sequence with bipolar pulse paired gradients (BPP-LED) [22, 23] was used. BPP gradients [56] strongly reduce eddy current effects and thereby the requisite durations of gradient recovery delays, while also minimizing 1H–1H NOE losses during the diffusion delay [23]. In our BPP-LED experiments, simultaneous x- and y-gradients were employed for encoding and decoding, but the BPP pulse pair was slightly imbalanced, with the first gradient set 12.7% higher than the second, oppositely signed gradient [57]. When both x and y gradients are set simultaneously at their maximum value, this corresponds to a strength of 66.8 G/cm when calibrated based on the measured self-diffusion of dioxane in D2O, using Rh(dioxane) = 2.12 Å, and ηD2O = 1.25 cP at 293 K, 1 bar [58]. Transverse gradients instead of z gradients were employed to minimize convection effects [59]. Encoding and decoding bipolar gradients were applied for 2×1.5 ms, with diffusion delays ranging from 200–400 ms.
For diffusion measurement of the tracer molecules at 293 K, a solution of 95% H2O, 4% D2O, and 0.25% each of dioxane, ethanol, methanol, and DMSO (v/v) was prepared, to which 150 mM sodium formate, 20 mM benzene, 50 μM DSS, and 50 mM tris(hydroxymethyl)aminomethane (Tris), pH 6.5 was added. A second sample contained 96% H2O, 2% D2O, and 0.5% each of dioxane, ethanol, methanol, and DMSO (v/v). Eight 1D spectra with different encoding and decoding gradient strengths were recorded in an interleaved manner, with the diffusion delay set to 200 ms. The encoding/decoding gradients strengths were set to 2.24, 4.34, 5.75, 6.85, 7.78, 8.65, 9.42, and 10.12% of the maximum value of 66.8 G/cm, with the encoding and decoding gradients slightly imbalanced as described above. Strong detuning of the 1H channel of the probe was used to prevent radiation damping effects, thereby removing the need for presaturating or otherwise suppressing the H2O signal. The time domain data were apodized with a 3-Hz exponential line broadening function, zero-filled four-fold, and baseline corrected after Fourier transformation. Data were processed with NMRPipe [50] and analyzed using in-house Python and Nmrglue [60] scripts to automatically extract the attenuation of intensities resulting from translational diffusion (Igrad,/ I0), where I0 refers to the intensity in the spectrum with the weakest encoding/decoding gradients and Igrad to the intensities in the spectra with higher gradient strengths. Errors were obtained from the variance-covariance matrix derived from a fit of ln(Igrad,/I0) versus squared gradient strength.
For 1D PFG NMR diffusion measurements on VA2-ubiquitin, WT ubiquitin, and αS a similar procedure was performed, except for usage of longer diffusion delay, a 7-Hz exponential line broadening function, and data were collected at 288 K. The range of encoding/decoding gradient strengths was adjusted to enable accurate measurement of the diffusion coefficients of both protein and dioxane molecules, and only the data attenuated by less than 10-fold were used for fitting the diffusion decay constants.
2D PFG NMR diffusion data were collected as pseudo-3D spectra, comprising eight interleaved 2D datasets of 40* × 1024* points (where N* indicates N complex data points) with acquisition times of 26 and 113 ms (t1, t2). Depending on the population of the unfolded state, either 8, 16, or 64 scans were acquired per FID, yielding total experimental durations of 2.15 h (8 scans), 4.3 h (16 scans), or 17.2 h (64 scans). The diffusion delay (375 ms), maximum x/y gradient strengths, and gradient durations (2×1.5 ms) were set identically to those in the 1D PFG NMR datasets. To decouple 15N spins during t2 acquisition, WALTZ-16 decoupling with a 1.39 kHz field was employed. Decreasing the decoupling field to 0.69 kHz had no impact on the measured diffusion coefficient, indicating that potential sample heating and convection from radiofrequency irradiation did not affect the results (Supplementary Fig. S1). 2D data were processed with NMRPipe [61] with the time domain of the indirect dimension extended by 50% using the SMILE program [62], followed by apodization with a cosine-squared window. Spectra were visualized with NMRFAM-Sparky [63], and peak shapes were fit with FuDA [64]. Intensities from resonances that arose from the folded or unfolded states were grouped separately and fit to single exponential decays, with errors in the fitted diffusion coefficient estimated from a standard jackknife procedure.
3. Results and Discussion
3.1. Pressure-dependence of translational and rotational diffusion rates of tracer molecules
Prior high-pressure NMR studies have investigated the self-diffusion of neat liquids and a handful of binary mixtures as a function of pressure [51]. Although hydrophobicity was found to impact the pressure dependence of rotational and translational diffusivity, many of these results were obtained in organic solvents. Less is known about the pressure-dependence of the hydrodynamic properties of molecules in aqueous solutions. Here, we evaluate at high precision the rotational and translational diffusion of six, chemically diverse molecules as a function of hydrostatic pressure. Although one might expect the translational diffusivity of small tracer molecules to simply scale with the pressure dependence of the water viscosity, we find this to apply only for highly polar molecules such as sodium formate and tris(hydroxymethyl)aminomethane (Tris). For more hydrophobic solutes, their diffusivity decreases with pressure by amounts that roughly scale with their hydrophobicity (Fig. 1a). Using the pressure dependence of the water viscosity, which is known at very high accuracy https://webbook.nist.gov/chemistry/fluid/, these diffusion coefficients can be converted into apparent radii of hydration relative to those at 1 bar (Fig. 1b). As can be seen, substantial increases in the apparent Stokes radii, far outside the measurement uncertainty, are observed for all hydrophobic solutes. The largest increase is observed for benzene, which increases in Rh by 11.3% at 3 kbar over atmospheric pressure conditions. Importantly, the Rh of dioxane, which often is used as an internal standard also shows a substantial increase of up to 7.5% at 3 kbar.
The diffusivity of water as a function of pressure at a fixed temperature is also readily extracted from the PFG NMR spectra. Note, however, that strong detuning of the 1H coil as well as a weak (ca 1 G/cm) gradient along the z-axis during the diffusion delay were needed to prevent radiation damping. At 293 K, the diffusivity of water as a function of pressure shows a maximum near 800 bar, expected on the basis of the known pressure dependence of its viscosity at this temperature (Fig. 1a). Although the agreement is much closer than seen in early NMR PFG studies of water diffusion [65, 66], a small but systematic difference, approaching 1.5% at 3 kbar is reproducibly seen. This difference relative to the pressure dependence of water viscosity does not arise from the presence of 4% D2O (v/v) and 4% (v/v) tracer molecules (dioxane, methanol, ethanol, and DMSO), as repeating the measurement with 2% D2O (v/v) and 2% tracer molecules (v/v) yielded indistinguishable results (data not shown). Instead, this difference has been attributed to a break-down of the applicability of the Stokes-Einstein equation for describing translational and rotational diffusion of water at high pressures [67, 68].
All tracer molecules also exhibit a slightly non-linear pressure dependence of their 1H chemical shift (Supporting Information Fig. S2; Table S1). However, we note that this apparent pressure dependence is small, with the largest value of −0.03 ppm at 3 kbar observed for methanol, and depends strongly on what is used as the reference value. If chemical shifts are referenced to trimethylsilyl propionate (TSP), for example using the procedure of Vajpai et al. [69], significant non-linearity is observed for the solutes, whereas the water chemical shift to a good approximation increases linearly with pressure. On the other hand, when using the methyl signal of IUPAC-recommended 4,4-dimethyl-4-silapentane-1-sulfonic acid (DSS) as an internal reference, with the exception of water, the non-linearities in 1H chemical shifts are much smaller. To a good approximation, the pressure dependence of both the apparent radii of hydration, as well as the 1H chemical shifts, can be fitted by second order polynomials, with coefficients reported in Table 1.
Table 1.
Fitted parameters obtained for the pressure dependence of 1H chemical shifts and Rh values of tracer molecules, measured in 94% H2O, 293 K, pH 6.5.a
| Molecule | δ0 (ppm) |
B1 (10−5 bar−1) |
B2 (10−9 bar−2) |
Rh,0 (Å) |
C1 (10−4 Å) |
C2 (10−8 Å) |
|---|---|---|---|---|---|---|
| Formate | 8.4402 | −0.584 ± 0.003 | −0.210 ± 0.013 | 1.624 | −0.040 ± 0.017 | −0.284 ± 0.071 |
| Benzene | 7.4298 | 0.035 ± 0.004 | 0.385 ± 0.015 | 2.012 | 0.653 ± 0.019 | −0.937 ± 0.078 |
| Dioxane | 3.7484 | −0.300 ± 0.004 | 0.083 ± 0.017 | 2.120 | 0.497 ± 0.016 | −0.844 ± 0.068 |
| Tris | 3.7206 | −0.613 ± 0.004 | 0.146 ± 0.015 | 3.114 | 0.041 ± 0.019 | −0.447 ± 0.078 |
| MeOH | 3.3481 | −0.898 ± 0.004 | 0.055 ± 0.016 | 1.442 | 0.282 ± 0.017 | −0.462 ± 0.069 |
| DMSO | 2.7167 | 0.901 ± 0.006 | −0.669 ± 0.026 | 1.945 | 0.393 ± 0.013 | −0.631 ± 0.055 |
| EtOH-methyl | 1.1713 | −0.540 ± 0.005 | 0.173 ± 0.019 | 1.883 | 0.166 ± 0.023 | −0.177 ± 0.096 |
| Water | 4.8326 | 0.931 ± 0.009 | 2.049 ± 0.038 | 0.940 | 0.221 ± 0.017 | −1.437 ± 0.071 |
1H chemical shifts (δ) and the effective Rh (Rh,eff) measured at each pressure are fit to second-order polynomials, δ = δ0 + B1(P – P0) + B2(P – P0)2 and . The coefficients B1, B2, C1, and C2 are fitting parameters; P stands for pressure; δ for the measured chemical shift; δ0 for the chemical shift at 1 bar; Rh,eff for the effective Rh; Rh,0 for the Rh at 1 bar; and P0 = 1 bar. Errors represent one standard deviation from the best-fit value.
We find that the change in translational diffusion rate of the hydrophobic solutes with pressure correlates with a decrease in longitudinal 13C relaxation times of these solutes when pressure is increased (Table 2). 13C T1 values are dominated by J (ωC) spectral density and, considering that the rotational diffusion of these small tracer molecules is far into the fast tumbling limit (ωCτr « 1), the decrease in 13C T1 with increasing pressure must result from slower rotational diffusion.
Table 2.
13C longitudinal relaxation times, T1, of various tracer molecules in 96% H2O, 293 K, 14.1 Tesla, at three different pressures.
| Carbon nucleus | T1 (s) | ||
|---|---|---|---|
| 1 bar | 1500 bar | 3000 bar | |
| Formate | 14.8 ± 0.2 | 14.9 ± 0.2 | 14.5 ± 0.2 |
| Benzene | 10.4 ± 0.2 | 9.9 ± 0.2 | 8.8 ± 0.2 |
| Dioxane | 5.91 ± 0.02 | 5.40 ± 0.02 | 4.67 ± 0.02 |
| Tris-methylene | 0.95 ± 0.01 | 0.99 ± 0.01 | 0.96 ± 0.01 |
| Tris-quaternary | 8.80 ± 0.07 | 8.92 ± 0.07 | 8.84 ± 0.05 |
| MeOH | 12.35 ± 0.08 | 13.13 ± 0.08 | 13.13 ± 0.08 |
| DMSO methyl | 5.44 ± 0.04 | 5.15 ± 0.03 | 4.78 ± 0.03 |
| EtOH-methyl | 7.31 ± 0.03 | 7.30 ± 0.03 | 6.91 ± 0.02 |
| EtOH-methylene | 10.17 ± 0.03 | 10.07 ± 0.03 | 9.40 ± 0.03 |
3.2. Rh pressure dependence of a folded and a disordered protein
To assess the impact of pressure on the translational diffusion of proteins, we measured these rates as a function of pressure for two proteins under conditions where pressure does not impact the folding-unfolding equilibrium: WT-ubiquitin and αS. The mid-point for pressure denaturation of WT-ubiquitin is ca 5.4 kbar [70] and this protein therefore remains fully folded under the conditions used in our study. The second protein, αS, is intrinsically disordered and the previously reported pressure dependence of its chemical shifts closely follows that observed for short linear peptides [71], pointing to the absence of any significant secondary structure formation with pressure.
The Rh of αS is rather insensitive to hydrostatic pressure up to ca 2 kbar, but then decreases by ca 3% when the pressure is raised to 3 kbar (Fig. 2a; Supporting Information Table S2). Results also show a small but distinct concentration dependence of the diffusion rate, resulting in an apparent Rh that is ca 1 Å larger at 240 μM than at 120 μM protein concentration. Considering that, at 240 μM αS and using Rh as an approximate radius of the unfolded protein, the apparent volume fraction occupied by αS is only ca 1.7%, the difference in Rh observed at this relatively dilute concentration is larger than expected from obstruction [72]. However, the approximation to consider the protein as an object with a single, constant Rh is clearly an oversimplification. It is likely that obstruction involving extended conformers out of the large ensemble sampled by αS will disproportionately impact the translational diffusion behavior of this IDP. At ca 30 Å, the Stokes radius derived here is smaller than the value originally obtained from gel filtration chromatography (34 Å) [73], and somewhat smaller than the 31.7 Å value derived from NMR diffusion by Baum and co-workers at slightly higher (300 μM) αS concentration [74], while essentially indistinguishable from the value reported more recently by Ruzafa et al. [75], also using NMR diffusion measurements under conditions very similar to ours. We hypothesize that the small, ca 3% decrease in Rh seen at 3 kbar relative to atmospheric conditions may be related to a small shift in the Ramachandran map distribution of random coil residues with pressure, as reflected in the previously noted decrease in 3JHNHα couplings with pressure [71].
Figure 2 |.
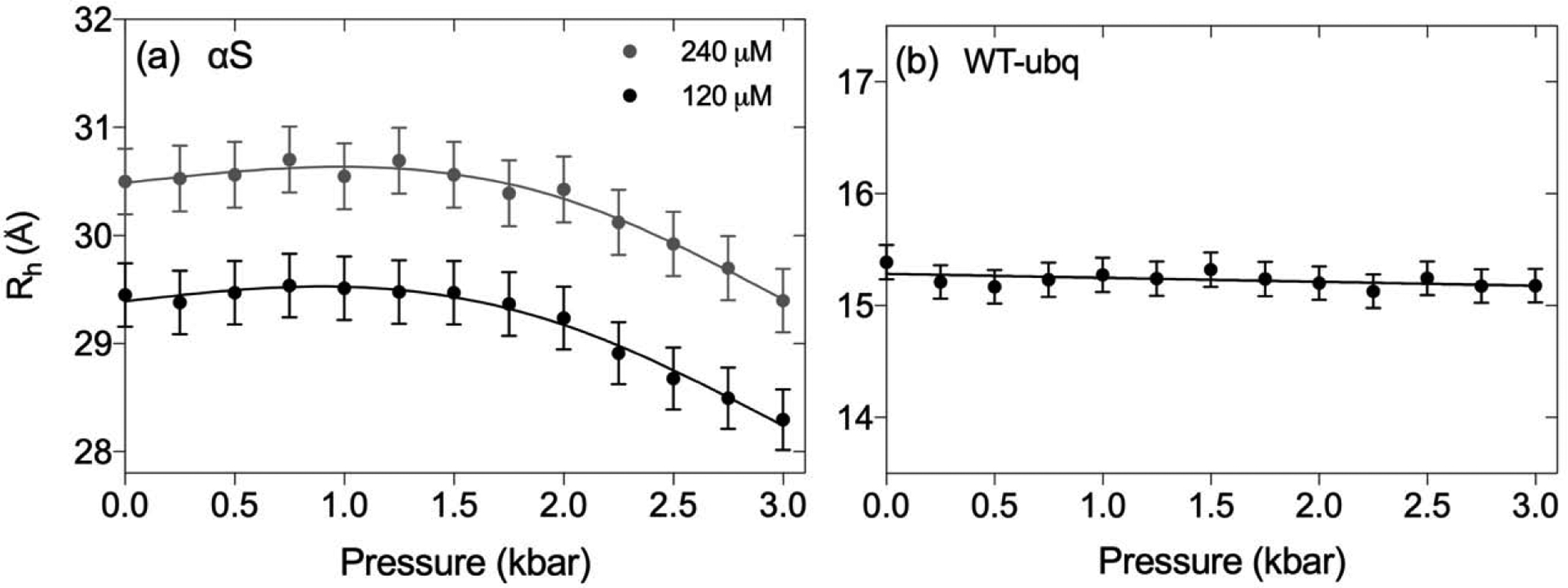
Pressure-dependence of the Rh values of (a) αS and (b) WT-ubiquitin are plotted as a function of hydrostatic pressure. Measurements were carried out at protein concentrations of 0.12 mM (black) and 0.24 mM (grey) αS (pH 7.0), and 0.5 mM WT-ubiquitin (pH 6.5), each in 94% H2O at 288 K. The midpoint of pressure-induced unfolding of WT-ubiquitin is 5.4 kbar [70], and our measurements therefore pertain to the folded state. The Rh measurements are calibrated relative to a value of 2.12 Å for the Rh of internal dioxane at 1 bar and account for the known pressure dependence of the water viscosity.
For WT-ubiquitin, the translational diffusion rates as a function of pressure (Table S2) are found to scale with the inverse of the water viscosity, resulting in a Rh value that is nearly flat (Fig. 2b). A linear fit of the data as a function of pressure yields a very small decrease from 15.39 Å at 1 bar to 15.18 Å at 3 kbar. Using an average compressibility of folded proteins of ca 15 Mbar−1 [76, 77], a volume compression of ca 4.5% is expected at 3 kbar, or a ca 1.5% reduction in radius, in good agreement with our diffusion measurements. The absence of any significant structural rearrangement of ubiquitin at pressures ≤ 3 kbar is consistent with excellent fits of the backbone residual dipolar couplings, measured at these elevated pressures, to the solution NMR structure derived at 1 bar [78], and with minute but detectable changes in the lengths of its backbone H-bonds [79]. Note that both these H-bond and our RDC and diffusion measurements were carried out at near neutral pH. At pH 4.6, hydrostatic pressure was shown to have a substantially larger impact on ubiquitin structure, as judged by changes in backbone amide chemical shifts and measurement of NOEs [80].
3.3. Diffusion rates for mixtures of folded and unfolded protein by 2D PFG NMR
Having validated that accurate translational diffusion coefficients can be obtained at high pressures, we sought to utilize PFG NMR at intermediate pressures to simultaneously study the diffusion of a protein that populates both the folded and unfolded states. For these experiments, we utilized VA2-ubiquitin, which unfolds with a midpoint near 1.6 kbar at 288 K.
In order to sufficiently resolve signals from the folded and unfolded states of VA2-ubiquitin, we used 2D PFG NMR experiments. Exchange between the folded and unfolded states during the long diffusion delay (375 ms) could adversely impact the extracted diffusion rates of each state. However, by evolving the 15N t1 chemical shift evolution right after the gradient encoding, the presence of exchange between the folded and unfolded states, to first order, does not impact the decay of the “diagonal peaks” even if exchange were present [17]. Such exchange would result in ZZ-exchange cross peaks between folded and unfolded species, and the absence of such resonances confirms that the exchange process is slow compared to the 375-ms diffusion delay used in our experiments. Thus, each resonance for folded and unfolded VA2-ubiquitin reports faithfully on the respective state without contamination from exchange. Albeit less sensitive, the 2D dataset has the advantage of yielding a large number of independent intensity measurements, those of the resolved peaks in the 2D NMR spectrum, thereby offering a reliable measure for the statistical uncertainty in the derived diffusion rates.
At 1400 bar, the folded and unfolded species of VA2-ubiquitin are populated at 68% and 32% respectively (Fig. 3a). Diffusion of the unfolded species is considerably slower than for the folded protein, as can be seen from the slower decay of the cross peak of the unfolded G35 resonance compared to A17 of the folded species (Fig. 3b). Well-resolved unfolded 1H–15N correlations were obtained for N=12 amides. To prevent a bias in the intensity measurement at the highest gradient strengths, where the signals become vanishingly weak, it is important to fix the position at which the resonance intensity is measured to that of the most intense spectrum, i.e., that with the weakest gradient [81]. To estimate the statistical uncertainty, resolved resonances then were split into two equal sized groups, and the N/2 resonance intensities for each group within a given spectrum were co-added, thereby decreasing the fractional uncertainty of the sum by √(N/2). Repeating this procedure for a large number of random groupings, the root-mean-square of the pairwise difference between the diffusion rates obtained for such pairs of groups is double the uncertainty of the fit obtained when all intensities are used (SI Fig. S6), thereby yielding an estimate for the uncertainty in the diffusion rate. The decay of such sums for both the folded and unfolded species is well fit by exponential functions, as seen from the linearity of the natural logarithm of these summed intensities plotted against the squared values of the gradient strength (Fig. 3c).
Fig. 3.
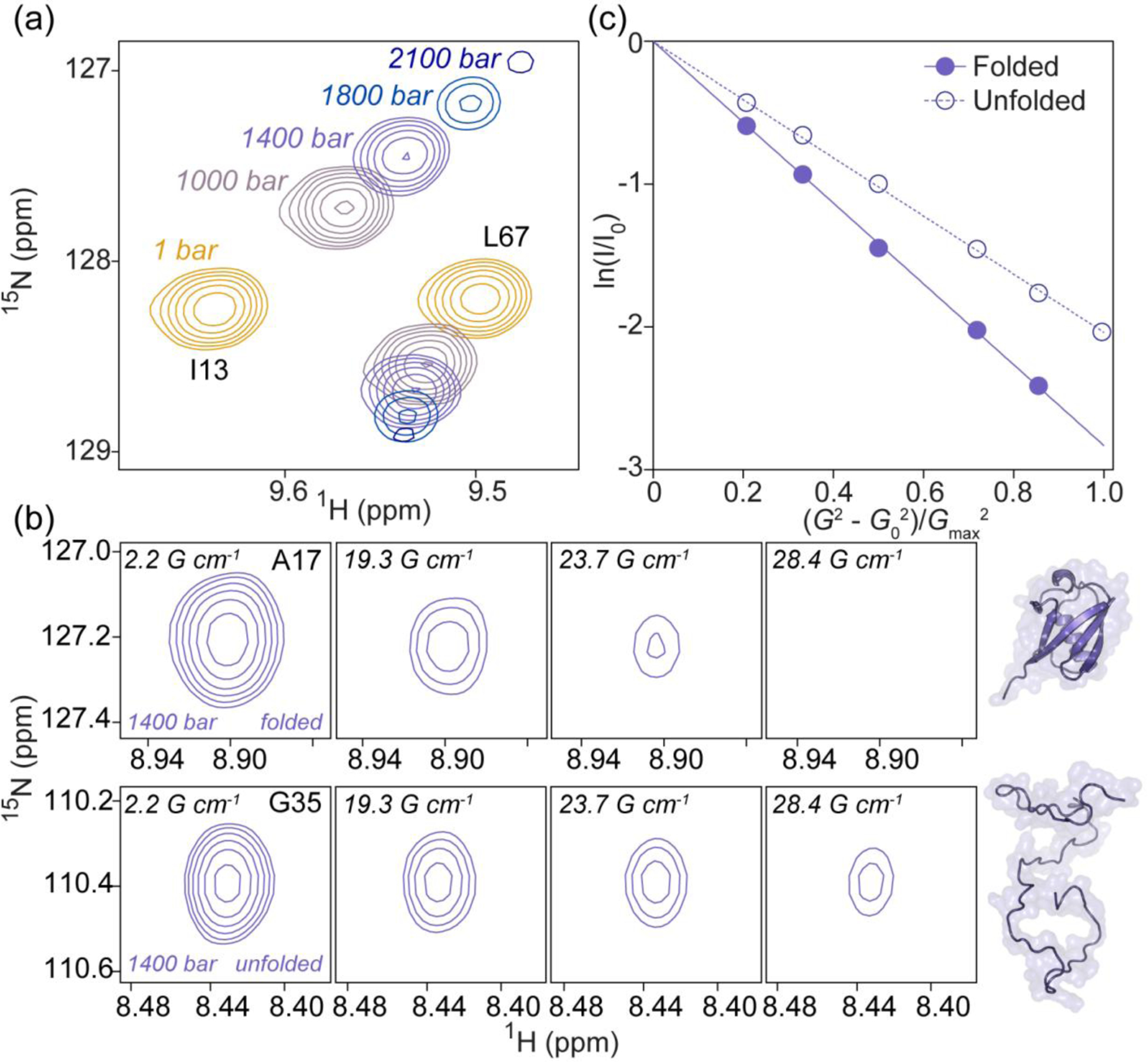
Simultaneous measurement of the diffusion rates of folded and unfolded protein from 2D PFG diffusion experiments. (a) Overlays of a small region of the 2D 1H–15N HSQC spectra of 15N-VA2-ubiquitin recorded at five hydrostatic pressures ranging from 1 bar to 2.1 kbar. Shown are resonances from the folded state (I13, L67) at 1, 1000, 1400, 1800, and 2100 bar. Contours are spaced by factors of 1.4 (b) Example of the decrease in cross peak intensity with increasing gradient strength, G, in 2D PFG NMR spectra of folded (top, A17 resonance) and unfolded (bottom, G35 resonance) VA2-ubiquitin recorded at 1400 bar where spectral intensities of folded and unfolded states are comparable. Corresponding 2D PFG NMR data from the urea-denatured state are shown in Supplementary Figure S6. (c) Intensity decay profiles for the folded (filled) and unfolded (empty circles) states of VA2-ubiquitin at 1400 bar. These are the data from panel B, summed over the N resonances analyzed for each state. The natural logarithm of the intensity decay (I/I0) is plotted vs normalized gradient strength. Gmax = 33.4 G/cm; G0 = 2.1 G/cm.
3.4. Diffusion measurements on equilibria of folded and unfolded protein by 1D PFG NMR
As described above, 2D PFG NMR diffusion experiments are generally required to achieve sufficient resolution to separate signals from the folded and unfold states. However, as a result of relaxation losses during the relatively long diffusion delay, compounded by the loss associated with the stimulated echo which refocuses only half of the initial magnetization, 2D PFG NMR diffusion experiments are intrinsically of rather low sensitivity. Coupled with the requirement to measure the weakest spectrum, i.e., recorded with the strongest gradient, at adequate signal to noise, and reduced sample volumes available in high pressure measurements that require thick wall sample tubes, 2D PFG measurements at elevated pressures are demanding from a sensitivity viewpoint. Moreover, while for small molecules, conventional 1D DOSY measurements are routinely applied to mixtures of molecules that frequently contain resonances free of overlap between molecular species, for a mixture of folded and unfolded protein, the standard 1D method only works well for the folded fraction of the sample. The unfolded protein typically lacks resonances free of overlap with the folded species.
Thus, we explored the feasibility of faster and more sensitive 1D NMR alternatives. The method outlined below requires a 1D reference spectrum, recorded for the folded state (e.g., 1 bar, 0 M urea), henceforth referred to as the reference spectrum. Except for the number of scans, sample concentrations, and pressure or urea concentration, all experimental parameters were kept constant for the measurements on the mixtures of folded and unfolded protein. An in-house Python script that utilizes the software Nmrglue [60] was then used to automatically execute the steps described below.
First, the reference spectrum is shifted to match the position of a unique, folded resonance in the mixed sample (Fig. 4a), e.g. an upfield methyl or downfield amide signal. This is followed by downscaling of the reference spectrum such that the selected resonance (L50-Cδ2H3 for our VA2-ubiquitin sample) in the folded reference spectrum matches the intensity in the mixed sample (Fig. 4b), which then allows generation of a difference spectrum (Fig. 4c) that no longer contains a net integral of folded protein resonance intensity. Of course, it is critical that relative resonance intensities in the 1D reference spectrum are the same as those recorded with the 1D BPP-LED experiment for the folded component of the mixed sample. For this reason, the reference spectrum of the fully folded protein is also recorded with the 1D BPP-LED pulse sequence, using a very weak encoding/decoding gradient combination. Even though resonances of the folded protein may have shifted somewhat relative to the reference spectrum, either due to the effect of pressure or weak interactions of denaturant with the folded protein, thereby generating a difference spectrum that contains negative components (Fig. 4c), the integral of the methyl region is minimally affected by such shifting, provided no resonances move outside of the integrated region. Integration regions were chosen to include most of the methyl signals for the artificially reconstructed unfolded state, typically 0.7 – 0.9 ppm, but results were found rather insensitive to the limits of the chosen region. Using this procedure to generate difference spectra for the unfolded state (Fig. 4d) then permits measuring the integrated unfolded protein intensity over the selected methyl region, at rather high sensitivity. The clean, single exponential decay of the integrated intensity with increased gradient strength (Fig. 4e) implies the method is reliable.
Fig. 4.
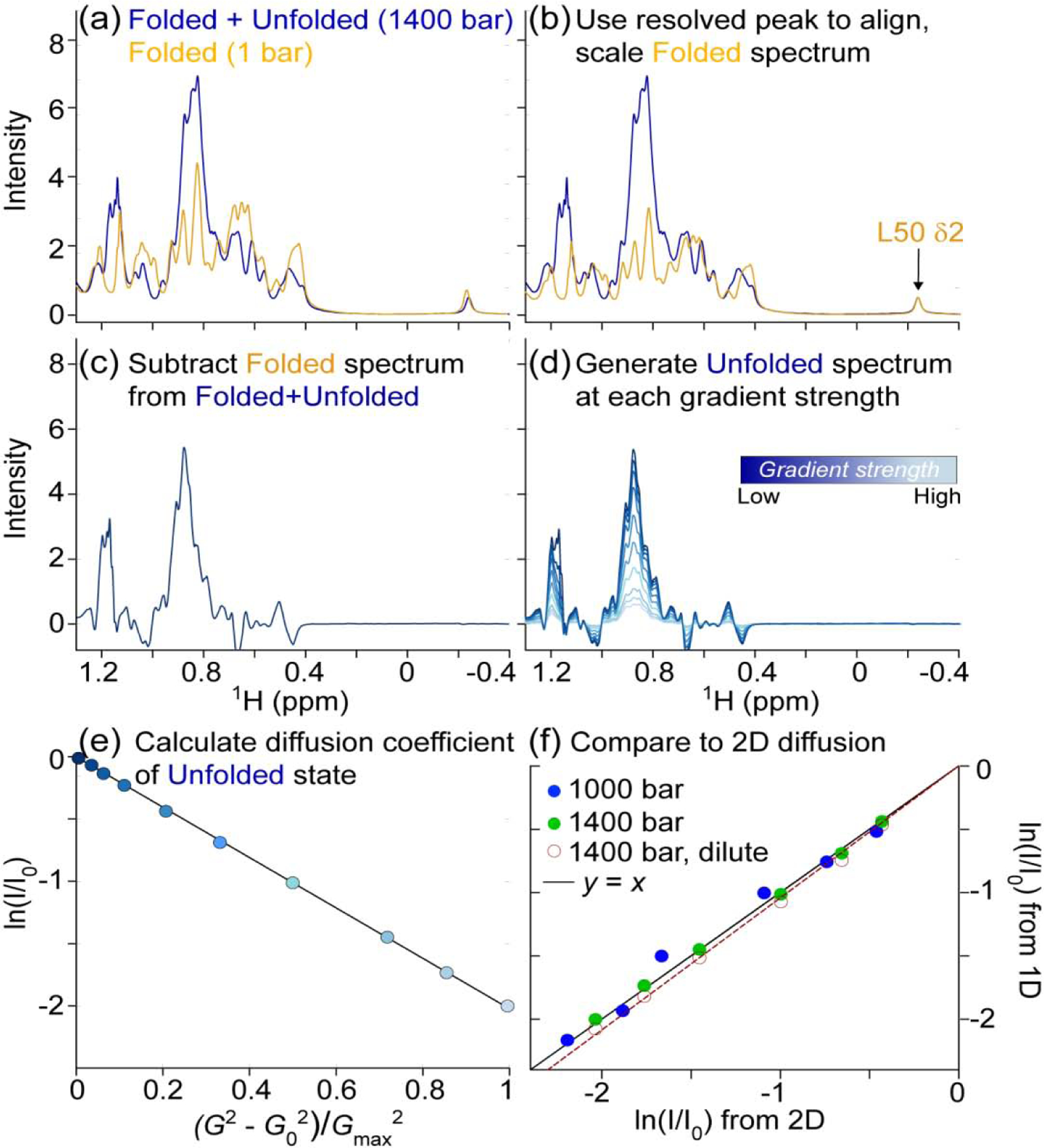
Translational diffusion measurement of unfolded VA2-ubiquitin in 94% H2O. Total protein concentration is 1.3 mM, pH 6.4. (a) The 1D NMR spectrum at 1400 bar (blue) contains signals from both folded and unfolded protein. The L50 Cδ2H3 resonance at −0.25 ppm serves as a reference for the folded protein spectrum (orange). (b) The spectrum of the folded state (1 bar) is aligned and scaled such that L50 Cδ2H3 matches that of the partially unfolded sample. (c) Subtraction of the scaled, folded spectrum then yields an artificial spectrum of the unfolded state. (d) Repeating the procedure for spectra generated at different gradient strengths yields the series of 1D spectra for which integrals over the methyl region are obtained. (e) Plot of the natural logarithm of the integrated methyl resonance intensity, ln(I/I0), at 1400 bar as a function of the squared gradient strength. G0 = 2.2 G/cm; Gmax = 33.4 G/cm. (f) A correlation plot showing the agreement between ln(I/I0) values derived from the unfolded state in 2D PFG NMR diffusion data set (x-axis) and those determined from the 1D method outlined above (y-axis). The I/I0 values from the 1D method were obtained through integration of the 1H chemical shift region between 0.80 and 0.94 ppm after reconstruction of the spectra of the unfolded state. I/I0 values from the 2D diffusion datasets were obtained by summing the intensities of N = 12 unfolded resonances at each gradient strength. Data collected at 1000 bar (blue) and 1400 bar (green) are presented, both at 1.3 mM total protein, and the solid line corresponds to y = x. The empty red circles are from a dilute sample (120 μM total) at 1400 bar and the best-fitted diffusion rate of the 120 μM sample is 5% faster than for the 1.3 mM sample (dashed red line).
The 1D measurement and the relatively short longitudinal relaxation times of protein methyl protons enables extensive signal averaging. As such, we also lowered the total concentration of VA2-ubq to 120 μM and collected 1D diffusion data at 1400 bar where the unfolded protein is present at ca 40 μM (Fig. 4f). Notably, this concentration is lower than that required for typical SAXS measurements. Using the 1D difference method described above, we measured the translational diffusion of both the folded and the unfolded state in the dilute sample. The good linearity of the decay when plotted on a log scale against the square of the gradient strength (Fig. 4e) reflects the robustness of the method. The observation of slightly (ca 5%) faster diffusion at 0.12 vs 1.3 mM VA2-ubiquitin concentrations (Fig. 4f) implies that intermolecular interactions come into play when studying the diffusion behavior of unfolded ubiquitin at elevated concentrations (ca 0.4 mM unfolded protein at 1400 bar).
To validate the accuracy of the 1D PFG based diffusion measurements for mixtures of folded and unfolded protein, these measurements were repeated for a limited number of conditions by the more time-consuming 2D method [17, 23]. The diffusion rates, derived from the 2D PFG diffusion measurements for the unfolded fractions of VA2-ubiquitin at 1000 bar and at 1400 bar are in excellent agreement with those derived from the above described 1D method (Fig. 4f), thereby validating consistency of the independent measurements. The larger scatter in the data collected at 1000 bar, where the fraction of unfolded protein is small (ca 4%), is dominated by the signal-to-noise limited precision of the 2D PFG measurement.
3.5. Minimal compaction of the VA2-ubiquitin unfolded state near its unfolding midpoint
To determine whether the Rh of unfolded VA2-ubiquitin decreases when the conditions are such that the folded and unfolded states are comparable in free energy, we measured the diffusion rates across a range of pressures and urea concentrations where both states are appreciably populated. The diffusion rate of the unfolded state is relatively sensitive to protein concentration, and data of Fig. 4f pointed to a small obstruction effect for the unfolded protein when measured at relatively high concentration of 1.3 mM total protein. As the fraction of unfolded protein changes when increasing pressure or urea concentration, the concomitant change diffusion obstruction could lead to an increase in apparent Rh when the fraction of unfolded protein increases (Fig. 5). To avoid this pitfall, we also collected data at 100 μM total protein concentration, where obstruction effects are negligible. With the denaturation mid-point of VA2-ubiquitin near 3.5 M urea, we measured diffusion data in 250 mM urea increments around this concentration. At 4 M urea, where the protein is ca 68% unfolded, the Rh of the unfolded state (23.1 Å) remains very close to the value measured at 7.6 M urea (23.3 Å) (Fig. 5a). Likewise, at 3 M urea where the unfolded state is populated to ca. 27%, Rh only slightly decreases to 22.9 Å, while the experimental uncertainty in the measurement increases due to the lower concentration of the unfolded species (Fig. 5a). Overall, only a 1.6% decrease in Rh between 5 M and 3 M urea was observed, whereas the population of the unfolded state drops from 93% to ca 27% over this range.
Fig. 5.
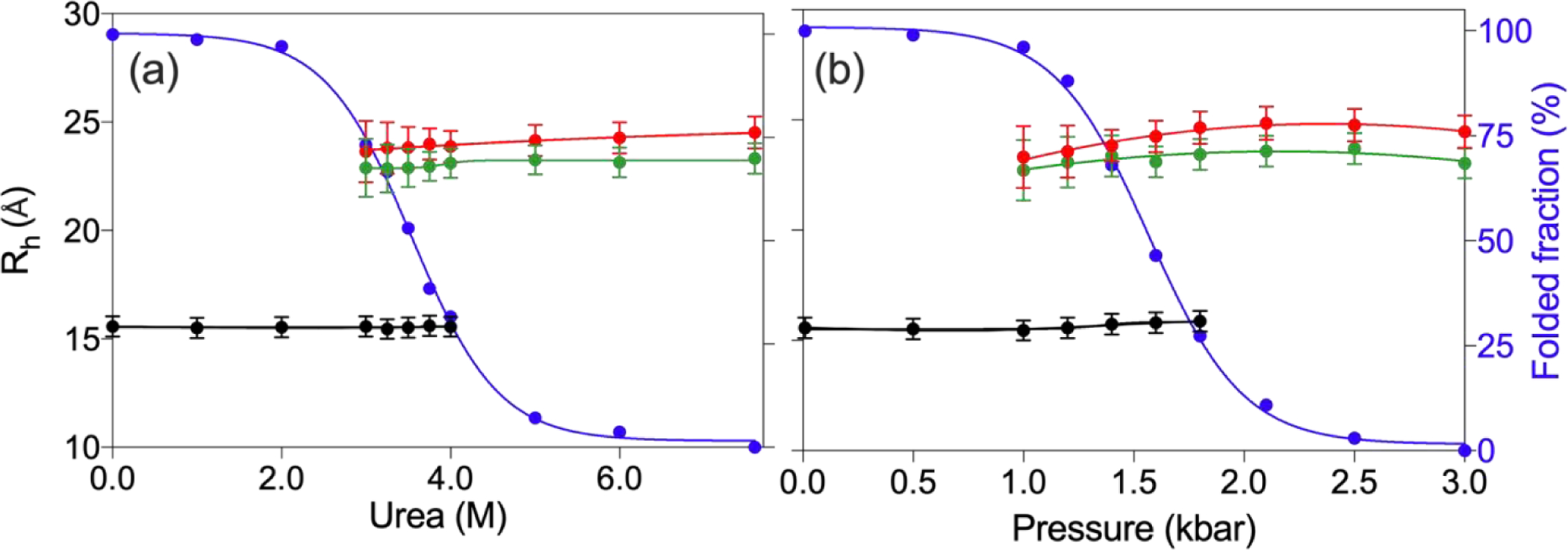
Rh of folded (black) and unfolded (red/green) VA2-ubiquitin under a range of denaturing conditions. (a) Rh values as a function of urea concentration and (b) as a function of hydrostatic pressure. Red and green symbols correspond to high (0.7 mM for urea denaturation; 1.3 mM for pressure unfolding) and dilute (0.1 mM) total protein concentration, respectively. Black symbols correspond to the diffusion of the folded protein at the higher concentration and values are within experimental error from diffusion rates measured at 0.1 mM concentration. Blue symbols correspond to the fraction of folded population. Solvent viscosity as a function of urea concentration for the samples of panel (a), as reflected in the diffusion rate of dioxane, are shown in Fig. S8.
Performing the same experiments on dilute VA2-ubiquitin as a function of pressure yields similar results: comparison of the Rh values measured at 1000 bar (ca. 4% unfolded) and 3000 bar (100% unfolded) shows a ca 1.4% decrease in Rh at the lower pressure, where the folded state is strongly favored (Fig. 5b). Our results therefore indicate that the unfolded state of VA2-ubiquitin shows only a very small degree of collapse when denaturing conditions are adjusted such that both folded and unfolded states are comparably populated. Importantly, our results also show that the pressure-denatured state has a Rh value that is very close to that of the urea-denatured state, confirming that pressure denaturation indeed results in a fully disordered protein with no significant compaction.
At 1 bar, VA2-ubiquitin and the wild-type protein show indistinguishable translational diffusion rates (Supplementary Fig. S5), as expected on the basis of their very similar 3D structures. At 3 kbar, where the folded state of VA2-ubiquitin is no longer appreciably populated, the diffusion-derived Rh of the pressure-denatured chain is 23.0 Å (Fig. 5b). Both the experimentally derived Rh of the folded protein at 1 bar (15.5 Å) and the Rh of the unfolded chain are in good agreement with the empirical equations of Wilkins et al. [14], which predict values of 16.7 and 26.1 Å, respectively for this 76-residue protein. If instead of high pressure, 7.6 M urea is used to denature VA2-ubiquitin, translational diffusion measurements at 1 bar yield Rh = 23.3 Å (Fig. 5a). Using the empirical relation for unfolded chains of Wilkins et al. [14], Rg ≈ 1.06 Rh, between the radius of gyration, Rg, derived from SAXS and the Stokes radius, our value is in good agreement with Rg ≈ 25.2 Å for guanidinium-denatured ubiquitin [82]. However, in contrast to prior SAXS data on urea-unfolded wild-type ubiquitin at pH 2.5 [83], our data show no evidence for the large expansion of the unfolded chain when the urea concentration is raised to 8M. At pH 6.4, we also do not observe the nearly uniform progressive upfield movement of unfolded amide 1H resonances (referenced relative to internal DSS), previously attributed to urea binding [83].
4. Concluding Remarks
Our work confirms that precise measurement of translational diffusion of proteins at elevated hydrostatic pressures is readily feasible, but that care needs to be exercised when following the common procedure of using an internal reference such as dioxane to derive the radius of hydration. Our results for dioxane and a number of other small hydrophobic molecules indicate that their effective Rh values increase substantially with pressure. The increased Rh with pressure is presumed to be caused by changes in their hydration shell. Prior measurements have clearly established the lower density of water molecules at hydrophobic surfaces [47], sometimes discussed as a solvent clathrate shell that can collapse under pressure [84, 85]. Our results suggest that hydrostatic pressure reduces the void space around the hydrophobic solute, thereby impacting the diffusion behavior of the solute. The higher compressibility of water on the surface of solvent-exposed hydrophobic moieties is potentially an important factor in obtaining a more quantitative understanding of the energetics that form the basis of pressure-induced protein unfolding [47]. The tighter coupling between solvent and solute at elevated pressure is also apparent in the 13C T1 values measured for a set of small hydrophobic molecules, which are all in the fast tumbling limit (ωcτc « 1) and decrease significantly with increasing pressure (Table 2). By contrast, the 13C T1 of the polar formate and Tris molecules, as well as their translational diffusion rates, are minimally impacted by pressure.
Probing the translational diffusion of the folded state of VA2-ubiquitin across a range where both folded and unfolded states are significantly populated reveals minimal variation in the Rh value of the folded state, both for the cases where high pressure and urea are used to induce unfolding of the protein. WT ubiquitin, which requires pressures much higher than 3 kbar to unfold, shows an approximate 1.5% reduction in its Rh at 3 kbar, close to the value expected based on the compressibility of folded proteins [76].
More remarkable is the near-absence of compaction of the unfolded VA2-ubiquitin when conditions start favoring the folded state, provided the measurements are made under dilute conditions. A number of single molecule FRET studies provide strong support for compaction when the concentration of denaturant is reduced [86–90], as did two-focus correlation spectroscopy [91]. Such compaction potentially could be attributed to transient formation of secondary structure elements [92]. Our observations, which show an absence of detectable compaction of unfolded VA2-ubiquitin, suggest that transient sampling of such states does not significantly impact the ensemble-averaged hydration radius of the protein. Similar conclusions were reached for several other systems using small angle X-ray scattering (SAXS) measurements [34, 35]. It is important to point out, however, that our study focuses on just a single denaturant- and pressure-unfoldable protein, and our finding that the unfolded chain does not significantly collapse as the ΔΔG between the folded and unfolded states approaches zero may depend strongly on the system studied.
A prior study found no effect of urea on the SAXS-derived Rg of an intrinsically disordered protein [93]. Interestingly, our results show a slight, gradual decrease in Rh of αS at pressures above 2000 bar. The reason for this decrease is not immediately clear but we speculate that it may correlate with a shift in the population of the Ramachandran map of IDPs with pressure, also reflected in a small decrease of 3JHNHα couplings with increasing pressure [71]. The latter suggests a small increase in turn-like conformations, thereby potentially decreasing the persistence length of the unfolded chain, resulting in a decreased Rh. Considering the observed increase in Rh when the αS concentration is increased from 120 to 240 μM, we note that in order to prevent significant inter-particle interference, PFG measurements of Rh for unfolded systems need to be carried out at low concentrations, an effect also noted previously [94].
Our results indicate that the addition of urea or increasing pressures do not cause αS or the unfolded state of VA2-ubiquitin to expand. While αS and VA2-ubiquitin provide two proteins of quite different length and sequence composition, a more systematic analysis of the effect of hydrostatic pressure on the translational diffusion of unfolded proteins is warranted. It appears plausible that the effect of hydrostatic pressure on the unfolded state of a protein will depend on its sequence composition, particularly on its level of enrichment with hydrophobic and aromatic residues.
Supplementary Material
Highlights.
Changes in hydration radii of small molecules with pressure depend on hydrophobicity
Diffusion of folded and unfolded proteins can be deconvolved from overlapping 1D NMR spectra
Pressure- and urea-denatured ubiquitin have very similar hydrodynamic radii
Translational diffusion of unfolded proteins significantly depends on concentration
Hydrodynamic radii of folded and unfolded proteins show little dependence on ΔΔG
ACKNOWLEDGMENT
We thank Y. Shen and J.L. Baber for technical support, and D.A. Torchia and G.M. Clore for useful discussions. This work was supported by the Intramural Research Program of the National Institute of Diabetes and Digestive and Kidney Diseases (grant number DK075141-02).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
References
- 1.Price WS, Pulsed-field gradient nuclear magnetic resonance as a tool for studying translational diffusion: Part II. Experimental aspects. Concepts Magn. Resonance 10 (1998) 197–237. [Google Scholar]
- 2.Price WS, Pulsed-field gradient nuclear magnetic resonance as a tool for studying translational diffusion .1. Basic theory. Concepts Magn. Reson 9 (1997) 299–336. [Google Scholar]
- 3.Johnson CS, Diffusion ordered nuclear magnetic resonance spectroscopy: principles and applications. Prog. Nucl. Magn. Reson. Spectrosc 34 (1999) 203–256. [Google Scholar]
- 4.Price WS, NMR studies of translational motion. (2009) 393. [Google Scholar]
- 5.Kay LE, Torchia DA, Bax A, Backbone dynamics of proteins as studied by 15N inverse detected heteronuclear NMR spectroscopy: application to staphylococcal nuclease. Biochemistry. 28 (1989) 8972–9. [DOI] [PubMed] [Google Scholar]
- 6.Lee D, Hilty C, Wider G, Wuthrich K, Effective rotational correlation times of proteins from NMR relaxation interference. J. Magn. Reson 178 (2006) 72–76. [DOI] [PubMed] [Google Scholar]
- 7.Altieri AS, Hinton DP, Byrd RA, Association of Biomolecular Systems Via PulsedField Gradient Nmr Self-Diffusion Measurements. J. Am. Chem. Soc 117 (1995) 75667567. [Google Scholar]
- 8.Dingley A, Mackay J, Chapman B, Morris M, Kuchel P, Hambly B, King G, Measuring protein self-association using pulsed-field-gradient NMR spectroscopy: Application to myosin light chain 2. J. Biomol. NMR 6 (1995) 321–328. [DOI] [PubMed] [Google Scholar]
- 9.Bernadó P, Åkerud T, de la Torre J. García, Akke M, Pons M, Combined Use of NMR Relaxation Measurements and Hydrodynamic Calculations To Study Protein Association. Evidence for Tetramers of Low Molecular Weight Protein Tyrosine Phosphatase in Solution. J. Am. Chem. Soc 125 (2003) 916–923. [DOI] [PubMed] [Google Scholar]
- 10.Jansma AL, Kirkpatrick JP, Hsu AR, Handel TM, Nietlispach D, NMR analysis of the structure, dynamics, and unique oligomerization properties of the chemokine CCL27. The Journal of biological chemistry. 285 (2010) 14424–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Huang R, Ripstein ZA, Augustyniak R, Lazniewski M, Ginalski K, Kay LE, Rubinstein JL, Unfolding the mechanism of the AAA+ unfoldase VAT by a combined cryoEM, solution NMR study. Proceedings of the National Academy of Sciences. 113 (2016) E4190–E4199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Price WS, Tsuchiya F, Arata Y, Lysozyme aggregation and solution properties studied using PGSE NMR diffusion measurements. J. Am. Chem. Soc 121 (1999) 11503–11512. [Google Scholar]
- 13.Weljie AM, Yamniuk AP, Yoshino H, Izumi Y, Vogel HJ, Protein conformational changes studied by diffusion NMR spectroscopy: application to helix-loop-helix calcium binding proteins. Protein science : a publication of the Protein Society. 12 (2003) 22836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wilkins DK, Grimshaw SB, Receveur V, Dobson CM, Jones JA, Smith LJ, Hydrodynamic radii of native and denatured proteins measured by pulse field gradient NMR techniques. Biochemistry. 38 (1999) 16424–16431. [DOI] [PubMed] [Google Scholar]
- 15.Jones JA, Wilkins DK, Smith LJ, Dobson CM, Characterisation of protein unfolding by NMR diffusion measurements. J. Biomol. NMR 10 (1997) 199–203. [Google Scholar]
- 16.Kamatari YO, Yamada H, Akasaka K, Jones JA, Dobson CM, Smith LJ, Response of native and denatured hen lysozyme to high pressure studied by 15 N/ 1 H NMR spectroscopy. Eur. J. Biochem 268 (2001) 1782–1793. [PubMed] [Google Scholar]
- 17.Choy W-Y, Mulder FAA, Crowhurst KA, Muhandiram DR, Millett IS, Doniach S, Forman-Kay JD, Kay LE, Distribution of molecular size within an unfolded state ensemble using small-angle X-ray scattering and pulse field gradient NMR techniques. J. Mol. Biol 316 (2002) 101–112. [DOI] [PubMed] [Google Scholar]
- 18.Buevich AV,Baum J, Residue-specific real-time NMR diffusion experiments define the association states of proteins during folding. J. Am. Chem. Soc 124 (2002) 7156–7162. [DOI] [PubMed] [Google Scholar]
- 19.Stilbs P, Fourier transform pulsed-gradient spin-echo studies of molecular diffusion. Prog. Nucl. Magn. Reson. Spectrosc 19 (1987) 1–45. [Google Scholar]
- 20.Stejskal EO,Tanner JE, Spin Diffusion Measurements: Spin Echoes in the Presence of a Time-Dependent Field Gradient. The Journal of Chemical Physics. 42 (1965) 288292. [Google Scholar]
- 21.Tanner JE, Use of the Stimulated Echo in NMR Diffusion Studies. The Journal of Chemical Physics. 52 (1970) 2523–2526. [Google Scholar]
- 22.Wu DH, Chen AD, Johnson CS, An improved diffusion-ordered spectroscopy experiment incorporating bipolar-gradient pulses. Journal of Magnetic Resonance Series A. 115 (1995) 260–264. [Google Scholar]
- 23.Chou JJ, Baber JL, Bax A, Characterization of Phospholipid Mixed Micelles by Translational Diffusion. J. Biomol. NMR 29 (2004) 299–308. [DOI] [PubMed] [Google Scholar]
- 24.Nilsson M, Duarte IF, Almeida C, Delgadillo I, Goodfellow BJ, Gil Ana M., Morris GA, High-Resolution NMR and Diffusion-Ordered Spectroscopy of Port Wine. J. Agric. Food Chem 52 (2004) 3736–43. [DOI] [PubMed] [Google Scholar]
- 25.Baldwin AJ, Hilton GR, Lioe H, Bagnéris C, Benesch JLP, Kay LE, Quaternary Dynamics of αB-Crystallin as a Direct Consequence of Localised Tertiary Fluctuations in the C-Terminus. J. Mol. Biol 413 (2011) 310–320. [DOI] [PubMed] [Google Scholar]
- 26.Baldwin AJ, Anthony-Cahill SJ, Knowles TPJ, Lippens G, Christodoulou J, Barker PD, Dobson CM, Measurement of Amyloid Fibril Length Distributions by Inclusion of Rotational Motion in Solution NMR Diffusion Measurements. Angewandte Chemie International Edition. 47 (2008) 3385–3387. [DOI] [PubMed] [Google Scholar]
- 27.Leung RLC, Robinson MDM, Ajabali AAA, Karunanithy G, Lyons B, Raj R, Raoufmoghaddam S, Mohammed S, Claridge TDW, Baldwin AJ, Davis BG, Monitoring the Disassembly of Virus-like Particles by 19 F-NMR. J. Am. Chem. Soc 139 (2017) 5277–5280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Brady JP, Farber PJ, Sekhar A, Lin Y-H, Huang R, Bah A, Nott TJ, Chan HS, Baldwin AJ, Forman-Kay JD, Kay LE, Structural and hydrodynamic properties of an intrinsically disordered region of a germ cell-specific protein on phase separation. Proceedings of the National Academy of Sciences. 114 (2017) E8194–E8203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Huang R, Brady JP, Sekhar A, Yuwen T, Lewis E. Kay, An enhanced sensitivity methyl 1 H triple-quantum pulse scheme for measuring diffusion constants of macromolecules. J. Biomol. NMR 68 (2017) 249–255. [DOI] [PubMed] [Google Scholar]
- 30.Horst R, Horwich AL, Wüthrich K, Translational diffusion of macromolecular assemblies measured using transverse-relaxation-optimized pulsed field gradient NMR. J. Am. Chem. Soc 133 (2011) 16354–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Didenko T, Boelens R, Rudiger SGD, 3D DOSY-TROSY to determine the translational diffusion coefficient of large protein complexes. Protein Engineering Design and Selection. 24 (2011) 99–103. [DOI] [PubMed] [Google Scholar]
- 32.Yuwen T, Sekhar A, Baldwin AJ, Vallurupalli P, Kay LE, Measuring Diffusion Constants of Invisible Protein Conformers by Triple-Quantum H-1 CPMG Relaxation Dispersion. Angewandte Chemie-International Edition. 57 (2018) 16777–16780. [DOI] [PubMed] [Google Scholar]
- 33.Wilkins DK, Grimshaw SB, Receveur V, Dobson CM, Jones Jonathan A., Smith LJ, Hydrodynamic Radii of Native and Denatured Proteins Measured by Pulse Field Gradient NMR Techniques†. Biochemstry. 38 (1999) 16424–16431. [DOI] [PubMed] [Google Scholar]
- 34.Yoo TY, Meisburger SP, Hinshaw J, Pollack L, Haran G, Sosnick TR, Plaxco K, Small-Angle X-ray Scattering and Single-Molecule FRET Spectroscopy Produce Highly Divergent Views of the Low-Denaturant Unfolded State. J. Mol. Biol 418 (2012) 226236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Watkins HM, Simon AJ, Sosnick TR, Lipman EA, Hjelm RP, Plaxco KW, Random coil negative control reproduces the discrepancy between scattering and FRET measurements of denatured protein dimensions. Proc. Natl. Acad. Sci. U. S. A 112 (2015) 6631–6636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Borgia A, Zheng W, Buholzer K, Borgia MB, Schüler A, Hofmann H, Soranno A, Nettels D, Gast K, Grishaev A, Best RB, Schuler B, Consistent View of Polypeptide Chain Expansion in Chemical Denaturants from Multiple Experimental Methods. J. Am. Chem. Soc 138 (2016) 11714–11726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Riback JA, Bowman MA, Zmyslowski AM, Knoverek CR, Jumper JM, Hinshaw JR, Kaye EB, Freed KF, Clark PL, Sosnick TR, Innovative scattering analysis shows that hydrophobic disordered proteins are expanded in water. Science. 358 (2017) 238–241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Mok Y-K, Kay CM, Kay LE, Forman-Kay J, NOE data demonstrating a compact unfolded state for an SH3 domain under non-denaturing conditions. J. Mol. Biol 289 (1999) 619–638. [DOI] [PubMed] [Google Scholar]
- 39.Best RB, Zheng W, Borgia A, Buholzer K, Borgia MB, Hofmann H, Soranno A, Nettels D, Gast K, Grishaev A, Schuler B, Comment on “Innovative scattering analysis shows that hydrophobic disordered proteins are expanded in water”. Science. 361 (2018) eaar7101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Riback JA, Bowman MA, Zmyslowski AM, Knoverek CR, Jumper JM, Kaye EB, Freed KF, Clark PL, Sosnick TR, Response to Comment on “Innovative scattering analysis shows that hydrophobic disordered proteins are expanded in water”. Science. 361 (2018) eaar7949. [DOI] [PubMed] [Google Scholar]
- 41.Fuertes G, Banterle N, Ruff KM, Chowdhury A, Pappu RV, Svergun DI, Lemke EA, Comment on “Innovative scattering analysis shows that hydrophobic disordered proteins are expanded in water”. Science. 361 (2018) eaau8230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Caro JA,Wand AJ, Practical aspects of high-pressure NMR spectroscopy and its applications in protein biophysics and structural biology. Methods. (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Royer CA, Hinck AP, Loh SN, Prehoda KE, Peng XD, Jonas J, Markley JL, Effects of Amino-Acid Substitutions on the Pressure Denaturation of Staphylococcal Nuclease as Monitored by Fluorescence and Nuclear-Magnetic-Resonance Spectroscopy. Biochemistry. 32 (1993) 5222–5232. [DOI] [PubMed] [Google Scholar]
- 44.Nucci NV, Fuglestad B, Athanasoula EA, Wand AJ, Role of cavities and hydration in the pressure unfolding of T4 lysozyme. Proceedings of the National Academy of Sciences. 111 (2014) 13846–13851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Roche J, Royer CA, Roumestand C, Monitoring protein folding through high pressure NMR spectroscopy. Prog. Nucl. Magn. Reson. Spectrosc 102–103 (2017) 15–31. [DOI] [PubMed] [Google Scholar]
- 46.Roche J, Caro JA, Norberto DR, Barthe P, Roumestand C, Schlessman JL, Garcia AE, Garcia-Moreno BE, Royer CA, Cavities determine the pressure unfolding of proteins. Proc. Natl. Acad. Sci. U. S. A 109 (2012) 6945–6950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Chen CR,Makhatadze GI, Molecular determinant of the effects of hydrostatic pressure on protein folding stability. Nature Communications. 8 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Alderson TR, Charlier C, Torchia DA, Anfinrud P, Bax A, Monitoring Hydrogen Exchange During Protein Folding by Fast Pressure Jump NMR Spectroscopy. J. Am. Chem. Soc 139 (2017) 11036–11039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Charlier C, Courtney JM, Alderson TR, Anfinrud P, Bax A, Monitoring 15 N Chemical Shifts During Protein Folding by Pressure-Jump NMR. J. Am. Chem. Soc 140 (2018) 8096–8099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Charlier C, Alderson TR, Courtney JM, Ying J, Anfinrud P, Bax A, Study of protein folding under native conditions by rapidly switching the hydrostatic pressure inside an NMR sample cell. Proc. Natl. Acad. Sci. USA 115 (2018) 201803642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Lang EW,Ludemann HD, Density dependence of rotational and translational molecular dynamics in liquids studied by high-pressure NMR. Progr. NMR Spectrosc 25 (1993) 507–633. [Google Scholar]
- 52.Easteal AJ,Woolf LA, Pressure and temperature dependence of tracer diffusion coefficients of methanol, ethanol, acetonitrile and formamide in water. J. Phys. Chem 89 (1985) 1066–1069. [Google Scholar]
- 53.Lee Y,Jonas J, Pressure and concentration effects on molecular reorientation in waterdioxane mixtures. J. Chem. Phys 59 (1973) 4845–4854. [Google Scholar]
- 54.Alderson TR, Lee JH, Charlier C, Ying J, Bax A, Propensity for cis-Proline Formation in Unfolded Proteins. ChemBioChem. 19 (2018) 37–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Peterson RW,Wand AJ, Self-contained high-pressure cell, apparatus, and procedure for the preparation of encapsulated proteins dissolved in low viscosity fluids for nuclear magnetic resonance spectroscopy. Rev. Sci. Instrum 76 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Wider G, Dotsch V, Wuthrich K, Self-Compensating Pulsed Magnetic-Field Gradients for Short Recovery Times. Journal of Magnetic Resonance Series A. 108 (1994) 255258. [Google Scholar]
- 57.Pelta MD, Morris GA, Stchedroff MJ, Hammond SJ, A one-shot sequence for highresolution diffusion-ordered spectroscopy. Magn. Reson. Chem 40 (2002) S147–S152. [Google Scholar]
- 58.Cho CH, Urquidi J, Singh S, Robinson GW, Thermal offset viscosities of liquid H2O, D2O, and T2O. J. Phys. Chem. B 103 (1999) 1991–1994. [Google Scholar]
- 59.Kiraly P, Swan I, Nilsson M, Morris GA, Improving accuracy in DOSY and diffusion measurements using triaxial field gradients. J. Magn. Reson 270 (2016) 24–30. [DOI] [PubMed] [Google Scholar]
- 60.Helmus JJ,Jaroniec CP, Nmrglue: an open source Python package for the analysis of multidimensional NMR data. J. Biomol. NMR 55 (2013) 355–367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Delaglio F, Grzesiek S, Vuister GW, Zhu G, Pfeifer J, Bax A, NMRpipe - a multidimensional spectral processing system based on Unix pipes. J. Biomol. NMR 6 (1995) 277–293. [DOI] [PubMed] [Google Scholar]
- 62.Ying J, Delaglio F, Torchia DA, Bax A, Sparse multidimensional iterative lineshapeenhanced (SMILE) reconstruction of both non-uniformly sampled and conventional NMR data. J. Biomol. NMR 68 (2017) 101–118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Lee W, Tonelli M, Markley JL, NMRFAM-SPARKY: enhanced software for biomolecular NMR spectroscopy. Bioinformatics (Oxford, England). 31 (2015) 1325–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Vallurupalli P, Hansen DF, Kay LE, Structures of invisible, excited protein states by relaxation dispersion NMR spectroscopy. Proc. Natl. Acad. Sci. U. S. A 105 (2008) 11766–11771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Benedek GB,Purcell EM, NUCLEAR MAGNETIC RESONANCE IN LIQUIDS UNDER HIGH PRESSURE. J. Chem. Phys 22 (1954) 2003–2012. [Google Scholar]
- 66.Defries T,Jonas J, PRESSURE-DEPENDENCE OF NMR PROTON SPIN-LATTICE RELAXATION-TIMES AND SHEAR VISCOSITY IN LIQUID WATER IN TEMPERATURE RANGE-15–10 DEGREESC. J. Chem. Phys 66 (1977) 896–901. [Google Scholar]
- 67.Bove LE, Klotz S, Strassle T, Koza M, Teixeira J, Saitta AM, Translational and Rotational Diffusion in Water in the Gigapascal Range. Phys. Rev. Lett 111 (2013). [DOI] [PubMed] [Google Scholar]
- 68.Dehaoui A, Issenmann B, Caupin F, Viscosity of deeply supercooled water and its coupling to molecular diffusion. Proc. Natl. Acad. Sci. U. S. A 112 (2015) 12020–12025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Vajpai N, Nisius L, Wiktor M, Grzesiek S, High-pressure NMR reveals close similarity between cold and alcohol protein denaturation in ubiquitin. Proc. Natl. Acad. Sci. U. S. A 110 (2013) E368–E376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Herberhold H,Winter R, Temperature- and pressure-induced unfolding and refolding of ubiquitin: A static and kinetic Fourier transform infrared spectroscopy study. Biochemistry. 41 (2002) 2396–2401. [DOI] [PubMed] [Google Scholar]
- 71.Roche J, Ying J, Maltsev AS, Bax A, Impact of Hydrostatic Pressure on an Intrinsically Disordered Protein: A High-Pressure NMR Study of alpha-Synuclein. ChemBioChem. 14 (2013) 1754–1761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Johannesson H,Halle B, Solvent diffusion in ordered macrofluids: A stochastic simulation study of the obstruction effect. J. Chem. Phys 104 (1996) 6807–6817. [Google Scholar]
- 73.Weinreb PH, Zhen WG, Poon AW, Conway KA, Lansbury PT, NACP, a protein implicated in Alzheimer’s disease and learning, is natively unfolded. Biochemistry. 35 (1996) 13709–13715. [DOI] [PubMed] [Google Scholar]
- 74.Wu KP, Weinstock DS, Narayanan C, Levy RM, Baum J, Structural Reorganization of alpha-Synuclein at Low pH Observed by NMR and REMD Simulations. J. Mol. Biol 391 (2009) 784–796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Ruzafa D, Hernandez-Gomez YS, Bisello G, Broersen K, Morel B, Conejero-Lara F, The influence of N-terminal acetylation on micelle-induced conformational changes and aggregation of alpha-Synuclein. PLoS One. 12 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Gavish B, Gratton E, Hardy CJ, ADIABATIC COMPRESSIBILITY OF GLOBULARPROTEINS. Proceedings of the National Academy of Sciences of the United States of America-Biological Sciences. 80 (1983) 750–754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Kharakoz DP, Protein compressibility, dynamics, and pressure. Biophys. J 79 (2000) 511–525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Roche J, Louis JM, Bax A, Best RB, Pressure-induced structural transition of mature HIV-1 protease from a combined NMR/MD simulation approach. Proteins-Structure Function and Bioinformatics. 83 (2015) 2117–2123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Nisius L,Grzesiek S, Key stabilizing elements of protein structure identified through pressure and temperature perturbation of its hydrogen bond network. Nature Chem. 4 (2012) 711–717. [DOI] [PubMed] [Google Scholar]
- 80.Kitahara R, Yokoyama S, Akasaka K, NMR snapshots of a fluctuating protein structure: Ubiquitin at 30 bar-3 kbar. J. Mol. Biol 347 (2005) 277–285. [DOI] [PubMed] [Google Scholar]
- 81.Viles JH, Duggan BM, Zaborowski E, Schwarzinger S, Huntley JJA, Kroon GJA, Dyson HJ, Wright PE, Potential bias in NMR relaxation data introduced by peak intensity analysis and curve fitting methods. J. Biomol. NMR 21 (2001) 1–9. [DOI] [PubMed] [Google Scholar]
- 82.Kohn JE, Millett IS, Jacob J, Zagrovic B, Dillon TM, Cingel N, Dothager RS, Seifert S, Thiyagarajan P, Sosnick TR, Hasan MZ, Pande VS, Ruczinski I, Doniach S, Plaxco KW, Random-coil behavior and the dimensions of chemically unfolded proteins. Proc. Natl. Acad. Sci. U. S. A 101 (2004) 12491–12496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Huang JR, Gabel F, Jensen MR, Grzesiek S, Blackledge M, Sequence-Specific Mapping of the Interaction between Urea and Unfolded Ubiquitin from Ensemble Analysis of NMR and Small Angle Scattering Data. J. Am. Chem. Soc 134 (2012) 44294436. [DOI] [PubMed] [Google Scholar]
- 84.Headgordon T, Is water structure around hydrophobic groups clathrate-like? Proc. Natl. Acad. Sci. U. S. A 92 (1995) 8308–8312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Frank HS,Evans MW, Free volumen and entropy in condensed systems. 3. Entropy in binary liquid mistures - partial molal entropy in dilute solutions - structure and thermodynamics in aqeous electrolytes. J. Chem. Phys 13 (1945) 507–532. [Google Scholar]
- 86.Deniz AA, Laurence TA, Beligere GS, Dahan M, Martin AB, Chemla DS, Dawson PE, Schultz PG, Weiss S, Single-molecule protein folding: Diffusion fluorescence resonance energy transfer studies of the denaturation of chymotrypsin inhibitor 2. Proc. Natl. Acad. Sci. U. S. A 97 (2000) 5179–5184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Schuler B, Lipman EA, Eaton WA, Probing the free-energy surface for protein folding with single-molecule fluorescence spectroscopy. Nature. 419 (2002) 743–747. [DOI] [PubMed] [Google Scholar]
- 88.Michalet X, Weiss S, Jager M, Single-molecule fluorescence studies of protein folding and conformational dynamics. Chem. Rev 106 (2006) 1785–1813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Moglich A, Joder K, Kiefhaber T, End-to-end distance distributions and intrachain diffusion constants in unfolded polypeptide chains indicate intramolecular hydrogen bond formation. Proc. Natl. Acad. Sci. U. S. A 103 (2006) 12394–12399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Aznauryan M, Delgado L, Soranno A, Nettels D, J.-r. Huang, A.M. Labhardt, S. Grzesiek, B. Schuler, Comprehensive structural and dynamical view of an unfolded protein from the combination of single-molecule FRET, NMR, and SAXS. Proc. Natl. Acad. Sci. U. S. A 113 (2016) E5389–E5398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Hofmann H, Soranno A, Borgia A, Gast K, Nettels D, Schuler B, Polymer scaling laws of unfolded and intrinsically disordered proteins quantified with single-molecule spectroscopy. Proc. Natl. Acad. Sci. U. S. A 109 (2012) 16155–16160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Holthauzen LMF, Rosgen J, Bolen DW, Hydrogen Bonding Progressively Strengthens upon Transfer of the Protein Urea-Denatured State to Water and Protecting Osmolytes. Biochemistry. 49 (2010) 1310–1318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Johansen D, Trewhella J, Goldenberg DP, Fractal dimension of an intrinsically disordered protein: Small-angle X-ray scattering and computational study of the bacteriophage lambda N protein. Protein Sci. 20 (2011) 1955–1970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Noppert A, Gast K, MullerFrohne M, Zirwer D, Damaschun G, Reduced-denatured ribonuclease A is not in a compact state. FEBS Lett. 380 (1996) 179–182. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.