

ABSTRACT
Bispecific antibodies comprise extremely diverse architectures enabling complex modes of action, such as effector cell recruitment or conditional target modulation via dual targeting, not conveyed by monospecific antibodies. In recent years, research on bispecific therapeutics has substantially grown. However, evaluation of binding moiety combinations often leads to undesired prolonged development times. While high throughput screening for small molecules and classical antibodies has evolved into a mature discipline in the pharmaceutical industry, dual-targeting antibody screening methodologies lack the ability to fully evaluate the tremendous number of possible combinations and cover only a limited portion of the combinatorial screening space. Here, we propose a novel combinatorial screening approach for bispecific IgG-like antibodies to extenuate screening limitations in industrial scale, expanding the limiting screening space. Harnessing the ability of a protein trans-splicing reaction by the split intein Npu DnaE, antibody fragments were reconstituted within the hinge region in vitro. This method allows for fully automated, rapid one-pot antibody reconstitution, providing biological activity in several biochemical and functional assays. The technology presented here is suitable for automated functional and combinatorial high throughput screening of bispecific antibodies.
KEYWORDS: High throughput Screening, antibody, bispecific antibody, antibody discovery, split inteins, automation
Introduction
First introduced in the 1990s, high throughput screening (HTS) is now a mature discipline that has brought a veritable boom within the pharmaceutical industry.[1] Screening against biological targets, combined with automation, miniaturization and large-scale data analysis, has become more suitable and cost-efficient.[2] Two approved marketed drugs and 74 leads in clinical development were generated by HTS approaches since 2003.[3-5] Drug discovery starts with HTS since screening against increasing numbers of biological targets to find an optimal lead candidate is necessary.[6] Antibodies are still a rapidly growing field in drug discovery, although in the past their inherent mono-specificity limited applications and their therapeutic potential. Improvements in antibody engineering have paved the way for more complex molecules and modified structures for extended therapeutic applications, focusing on improved target specificity and potency.[6] New Biological Entities (NBEs) with multiple formats like bispecific antibodies (bsAbs) enable extended therapeutic applications due to their ability to bind simultaneously to two epitopes on one molecule, epitopes on two different molecules that are physically interacting, or even on different target cells.[7] Over 100 bsAbs formats are currently described, with more than 85 bsAbs in clinical trials, commonly targeting cancer and redirecting immune cells.[8] Three bsAbs, blinatumomab, catumaxomab and emicizumab are approved by the US Food and Drug Administration.[8,9]
The bispecific format is a combination of two distinct variable regions derived from two different parental monospecific antibodies. The ability to simultaneously bind two different epitopes enables a variety of potential modes of actions, such as an improved cytotoxic potential by bridging cells in-trans, synergistic effects, receptor crosslinking for enhanced inhibition or degradation and higher binding specificity, resulting, for example, in enhanced tumor selectivity.[10-13] Tackling a cancer disease often requires inhibition of more than one signaling pathway. Often, combination therapies or bsAbs can help to overcome the limitations of ordinary monoclonal antibodies (mAbs) restricted to only one epitope.[10,11,14] Although elegant approaches for controlled CH3 heterodimerization like knobs-into-holes, electrostatic steering, DuoMab, strand-exchange engineered domain (SEED), triomab, as well as common LC/HC chain approaches, κλ-bodies or CrossMabs for desired HC/LC pairing have been developed for bsAb production, most of these technologies are not suitable for extensive combinatorial screening campaigns.[6,15,16] BsAb development is usually time- and labor-intensive, involves higher manufacturing costs, and requires safety and efficacy validation of each mAb and in combination.[17]
Approaches used to identify the single binding moieties of an envisioned bsAb tend to focus on reduction of individual clone numbers by filters such as affinity, target specificity, optimally domain or epitope mapping. Most intended modes of action are not conveyed by a single binding moiety, but through the combination to be identified, for example, selective effector cell recruitment, target-specific Fc mediated effector functions or enhanced selectivity binding by avidity or even overall modes of action that are not predictable.[18,19] When attempting to screen a desirable high number of combinations, e.g., 100 times 100 binding moiety combinations, the multiple cloning, expression and purification steps represent a bottleneck for the production, even when multi-parallel small-scale expression methodologies are in place. Therefore, the number of combinations or bsAbs finally generated is usually only a small fraction of the selected repertoire.[20] Moreover, subsequent optimization of each binding moiety, such as affinity maturation, might need to be performed after combining hit candidates and re-engineering into a bispecific format.[21] Finally, most bsAb engineering approaches include comparisons of several formats to further optimize the intended biological mode of action, which also enlarge the screening space or efforts. A broadly applicable method enabling high throughput binding and functional cellular bsAb screenings has to date not been described, but would greatly shorten development times for a wide variety of complex NBEs and enhance the probability of identifying the optimal combination, ultimately leading to the generation of better biotherapeutics.
In this study, we describe a novel screening methodology for bsAbs that bypasses not only chain mispairing issues, but also enables the screening of a large combinatorial space already in the desired format. The platform is based on a combinatorial approach mediated by split inteins. Split inteins are reported to be a powerful tool to avoid LC mispairing during bsAb production.[22,23] Split inteins are auto-processing domains found in nearly every organism.[24] The mechanism mediated by split inteins is called Protein Trans Splicing (PTS), and, since its discovery in 1988, it has been used for biotechnical applications, such as protein ligation,[25] purification or labeling.[26,27] In the case of ligation, a stable peptide bond is formed between two target proteins fused to their flanking exteins. Split inteins are divided into an N-terminal (IntN) and a C-terminal fragment (IntC) and remain inactive when separated from each other. After moving in close proximity, splicing occurs under soft reducing conditions undergoing several steps, as described in detail elsewhere,[28,29] resulting in a reconstituted tag-less (bispecific) antibody product. Biological functionality remains within the mild reducing environment and complex formats can be generated. For example, effector functions can be easily changed or suppressed, by switching the Fc portion. Extensive combinatorial screenings and comparison of several complex formats with comparably low production need is feasible, by applying our intein-based reconstitution approach. Antigen-binding fragment (Fab) binding moieties with different LC can be combined during screening as reconstitution occurs on the protein level, keeping their natural LCs. If needed, specific Fab engineering for retained LC pairing can be conducted subsequently and specifically focused on the identified binder only, resulting in overall faster development times.
In this study, we reconstituted anti-CD40 Fabs with different Fc portions to show reproducibility, isotype switching and potential high throughput amenability in 96- and 384-well microtiter plate, serving as a proof of concept. Anti-CEACAM5 one-armed SEED antibodies and anti-CEACAM6 Fabs fused to their respective intein partners were expressed in mammalian cell culture and reconstituted by PTS for an bsAb approach. The screening potential was confirmed by mixing several c-MET- and epidermal growth factor receptor (EGFR)-targeted bsAbs with different binding affinities. Reconstituted antibodies were compared with classically produced parental antibodies and verified biophysically and biologically in different cellular assays. We were able to show a potential screening approach suitable for HTS, resulting in rapidly reconstituted bsAbs with retained efficacy directly amenable to industry-scale automated cellular functional screenings.
Results
Design, generation and characterization of reconstituted antibodies
In this study, false HC/LC mispairing was avoided by separate expression of a single Fab domain and a one-armed antibody fragment using split inteins for post-translational reconstitution of the single antibody fragments (Figure 1). Split intein parts flanked by their natural exteins were fused to the antibody fragments within the hinge region (Figure 2). The hinge region reveals the most space for PTS, due to the relatively large distance between the Fab and Fc portion.[30,31] Each split intein indicated with IntN and IntC carried a 6xHis-Tag for high-affinity purification. Heterodimeric HC pairing of the bsAb was maintained by using the SEED technology. The sequence divergence of the CH3 portion in the Fc region by combining IgG and IgA species allows correctly assembled HC pairing.[16] Reconstituted antibodies were compared with parental references to estimate biophysical or biochemical differences after antibody reconstitution using split inteins. Fab-IntN fragments and Fc-IntC or oaSEED-IntC portions were produced in Expi293F cells and purified by standard HisTrap or MabSelect affinity chromatography, respectively. Titers were comparable to antibody fragments lacking a split intein part, ranging from 100 mg L−1 to 500 mg L−1 after 5 days post-transfection. The N-terminal part of the split intein consists of 123 aa and is known to decrease solubility based on hydrophobic patches, and it tends to aggregation after protein concentration.[32]
Figure 1.
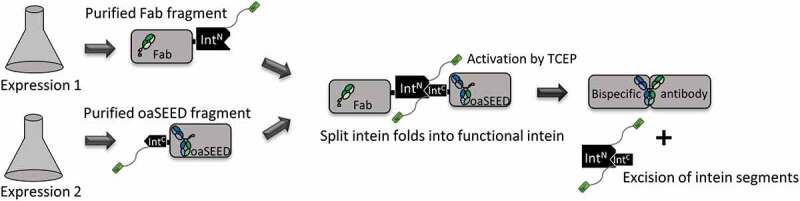
Schematic illustration of the process for generating bsAbs via split intein Npu DnaE. Fab and oaSEED fragments fused to N- or C-terminal split intein were produced separately in mammalian cells and purified. The two fragments were mixed in an equimolar ratio and supplemented with TCEP to activate protein tans splicing (PTS). The bsAb is reconstituted and the mixture was subsequently purified with Ni2+ beads to remove non-reconstituted antibody fragments as well as excised split inteins via hexahistidine tag.
Figure 2.
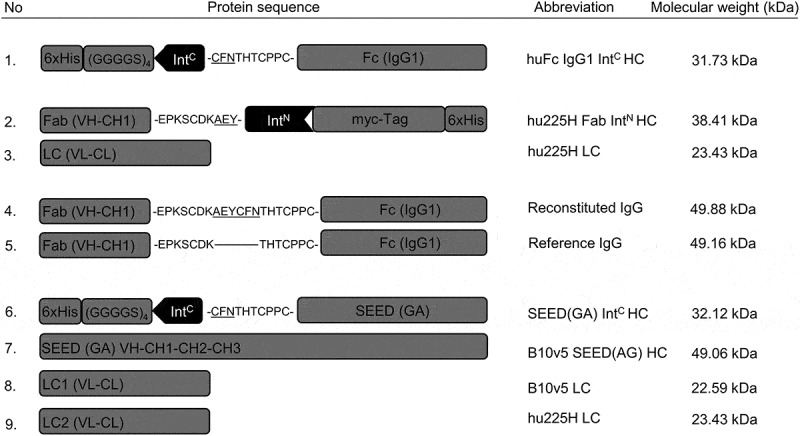
Schematic illustration of protein sequences for heavy and light chains fused to split intein partners IntC and IntN. Protein sequences for heavy and light chains of huFc IgG, Fab and oaSEED fragments with their corresponding molecular weights are exemplarily shown used for antibody reconstitution via split inteins. The partial hinge region is depicted as amino acid sequence fused to ExteinC or ExteinN (underlined) and the corresponding split inteins IntC or IntN. Heavy chains fused to split intein partners were further fused to a hexahistidine tag. Heavy chains fused to IntC were attached to a glycine-serine linker. 1: HC for a huFc IntC fragment. 2 + 3: hu225H Fab IntN HC and corresponding LC. 4 + 5: Reconstituted IgG with modified hinge region aligned to reference IgG. 6–9: B10v5 oaSEED IntC with corresponding HC and LC. Detailed sequence information can be found in the supplements.
Fab-IntN fragments could be produced with high yields and good purity over 95% (Table S1). Aggregation of Fab-IntN was not observed upon protein production using HEK293F (Figure S1). A huFc IntC fragment was reconstituted with two anti-CD40 Fab IntN arms, serving as a proof of concept for antibody reconstitution. The sequences for anti-CD40 Fab IntN fragments and the corresponding LC originated from the reference molecule APX005M for direct head-to-head comparison. Figure 3(a) shows the splicing efficiency over time; a new fusion band of the Fab HC and the Fc HC through PTS (49.8 kDa) was observed based on sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS-PAGE) analysis (Figure 3a). Bands at 14 kDa and 4 kDa occurred after addition of tris(2-carboxyethyl)phosphine (TCEP), indicating spliced IntN and IntC parts.
Figure 3.
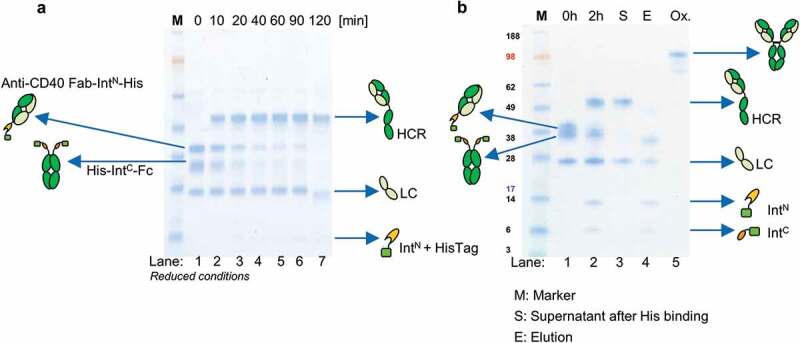
Antibody reconstitution mediated by PTS using split intein Npu DnaE analyzed by SDS-PAGE. (a) Antibody fragments Fab IntN and huFc IntC were mixed in a ratio 2:1 and incubated for 2 h at 37°C in the presence of 0.5 mM reducing agent TCEP. Samples were taken after every 10 min and analyzed by SDS-PAGE (Lane 1–7). Lane 1 shows antibody fragments Fab-IntN (38.3 kDa) and huFc-IntC (31.7 kDa) under reduced conditions at 0 min. The new band at 49.8 kDa in Lanes 2–7 indicates the reconstituted heavy chains (HCR) of Fab and huFc under reduced conditions. Lane 7 shows complete depletion of antibody fragments Fab-IntN and huFc-IntC after 2 h. (b) Lane 1 illustrates antibody reconstitution with a surplus of Fab-IntN over huFc-IntC (3:1 ratio). Non-reconstituted antibody fragments were still present after 2 h (Lane 2). Non-reconstituted antibody fragments were purified by addition of Ni2+ beads after incubation of 1 h at RT, leaving only the reconstituted antibody (Lane 3, S). Non-reconstituted antibodies were captured by Ni2+ beads (Lane 4, E: Elution with 500 mM imidazole). Final antibody reconstitution was achieved after re-oxidation with DHAA for 2 h at 37°C (Lane 5, Ox.).
Combining anti-CD40 Fab IntN and huFc IntC in a molar ratio of 2:1 led to more than 90% PTS efficiency, as monitored by SDS-PAGE analysis (Figure 3a) and by an extrapolated Michaelis Menten kinetic (Figure S2). High performance liquid chromatography (HPLC) measurement supported PTS efficiency analysis by quantifying antibody reconstitution over time, indicating depletion of the antibody fragments and reconstitution to a full-length antibody (Figure S3). Since this reconstitution method is intended for high throughput purposes, additional purification chromatography steps would not be applicable. The purification strategy is depicted in Figure S4a. By design, impurities in the form of non-reconstituted antibody fragments as well as the split intein itself could be depleted by the addition of Ni2+ beads (Figure 4b). The optimal molar ratio for antibody reconstitution was deliberately changed, resulting in an excess of Fab-IntN over IntC, which was not reconstituted at the end of the reaction. After treatment with Ni2+ beads, the residual Fab-IntN as well as the spliced parts IntN and IntC were successfully removed (Figure 3b), showing the proof of principle for the purification strategy, optimal for small-scale antibody reconstitution. No unspecific binding of the final reconstituted antibody to the Ni2+ beads was observed. TCEP was completely oxidized by dehydro-ascorbic acid (DHAA) addition and re-oxidation of the sulfhydryl groups within the antibody to reform cysteine bridges was confirmed by SDS-PAGE (Figure 3b) and HPLC analysis (Figure S3). Antibody reconstitution followed by one-step purification was performed therefore in one reaction well.
Figure 4.
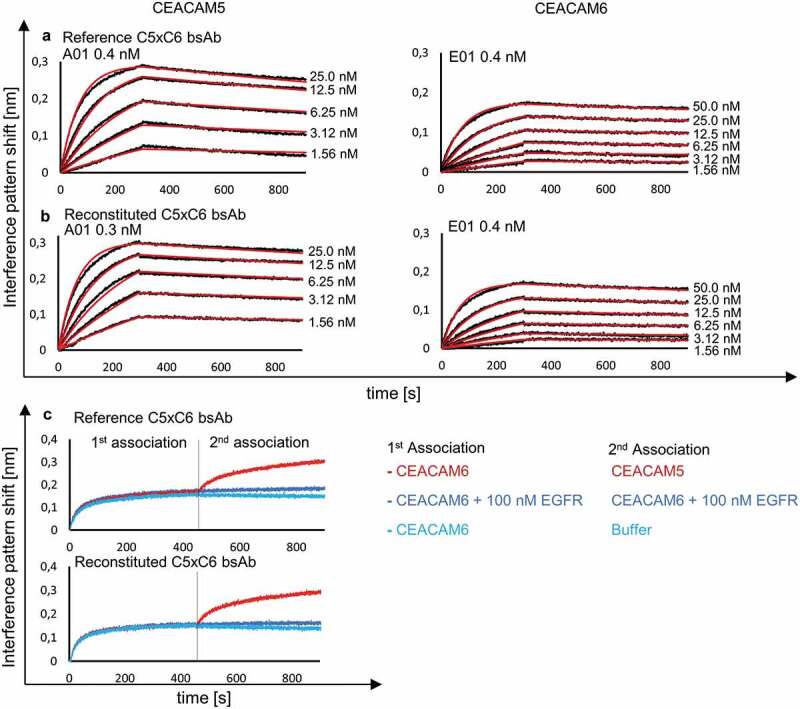
BLI analysis of reconstituted bsAb C5xC6 and comparison to reference C5xC6 binding to soluble CEACAM5 and CEACAM6. KD was determined after association and dissociation of the respective antigens and monitored with varying concentration of analyte over time resulting in an interference pattern shift (nm). (a) Concentration-dependent binding of CEACAM5 and CEACAM6 by reference bsAb version of C5xC6 shows similar kinetic parameters in the nanomolar range compared to reconstituted C5xC6 depicted in (b). (c) Simultaneous binding of soluble recombinant CEACAM5 and CEACAM6 by reconstituted bsAb C5xC6. Both antigens are associated in two steps. EGFR and buffer were used as a negative controls.
Characterization of reconstituted antibodies
Several reconstituted antibodies were characterized by biolayer interferometry (BLI) analysis, differential scanning fluorimetry (DSF) and mass spectrometry (MS). Albeit PTS by split inteins is a well-known mechanism, MS data were used to confirm the correctly assembled antibody after reconstitution and their originating antibody fragments fused to split inteins (Table S2). The intact mass of bsAbs B10v5xhu225L (Figures S5, S6) and B10v5xhu225H (Figures S7, S8) was analyzed by time-of-flight MS and verified correctly reconstituted HCs paired to their corresponding LCs. Reconstituted antibodies were further characterized by BLI analysis and compared against their parental antibodies to demonstrate similar kinetic parameters and simultaneous binding of both antigens. Reconstituted CEACAM5xCEACAM6 (C5xC6) bsAb showed a sub-nanomolar KD value (0.3 nM) for binding soluble recombinant CEACAM5 and 0.4 nM for binding soluble recombinant CEACAM6, which was comparable to the KD values of reference bsAb C5xC6, as previously described (Figure 4a,b).[33] Simultaneous binding of reconstituted C5xC6 to both antigens via stepwise association was retained after PTS compared to reference C5xC6 bsAb (Figure 4c). In congruence, kinetic parameters of several additional reconstituted bsAb were comparable to their genetically fused references, as depicted in Table 1. The PTS reconstituted variants all contain the additional sequence “AEYCFN” in the hinge region. A slightly reduced thermostability for C5xC6 bsAb (Table 1) was observed. A larger data set is required to evaluate whether this is a general feature of these variants.
Table 1.
Kinetic parameters of reconstituted bsAb compared to parental monovalent oaSEEDbodies and bispecific references. Reconstituted bsAb were compared to their parental monovalent antibodies or bispecific references. Antibodies were captured by anti-human Fc biosensors and subjected to respective antigen binding. Melting temperatures were analyzed by thermal shift assays. (ND, Not defined).
| KD |
KD error |
ka |
ka error |
kd |
kd error |
Tm |
||
|---|---|---|---|---|---|---|---|---|
| Antibody | Analyte | [M] | [M] | [M−1 s−1] | [M−1 s−1] | [s−1] | [s−1] | [°C] |
| C5xC6 Reference | Ceacam6 | 4.6E −10 | 4.7E −12 | 2.1E +05 | 1.8E +03 | 9.5E −05 | 4.6E −06 | 68.0 |
| C5xC6 Reconstituted | Ceacam6 | 4.2E −10 | 8.4E −12 | 4.2E +05 | 2.9E +03 | 1.8E −04 | 3.3E −06 | 66.1 |
| C5xC6 Reference | Ceacam5 | 4.3E −10 | 4.5E −12 | 5.9E +05 | 2.8E +03 | 2.6E −05 | 2.4E −06 | 68.0 |
| C5xC6 Reconstituted | Ceacam5 | 2.6E −10 | 4.2E −12 | 5.2E +05 | 2.9E +03 | 1.6E −04 | 2.3E −06 | 66.1 |
| oaC5 | Ceacam5 | 7.3E −10 | 6.3E −12 | 2.7E +05 | 1.2E +03 | 1.9E −04 | 1.5E −06 | ND |
| C6 Fab | Ceacam6 | 1.5E −10 | 1.6E −10 | 3.8E +04 | 6.0E +02 | 3.4E −04 | 2.4E −06 | ND |
| CD40 Reference | CD40 | 5.9E −09 | 5.3E −11 | 4.9E +05 | 4.2E +03 | 2.9E −03 | 8.4E −06 | 69.7 |
| CD40 Reconstituted | CD40 | 6.8E −09 | 4.5E −11 | 5.2E +05 | 3.2E +03 | 3.6E −03 | 7.1E −06 | 69.3 |
| B10v5xhu225L | c-MET | 8.9E −10 | 1.1E −11 | 1.5E +05 | 6.4E +02 | 1.3E −04 | 1.6E −06 | ND |
| B10v5xhu225H | c-MET | 8.6E −10 | 9.6E −12 | 1.8E +05 | 7.8E +02 | 1.5E −04 | 1.6E −06 | ND |
| F06xhu225H | c-MET | 5.4E −08 | 3.4E −11 | 1.5E +06 | 5.4E +04 | 8.3E −02 | 9.9E −04 | ND |
| oaF06 | c-MET | 1.1E −08 | 1.0E −12 | 4.6E +06 | 5.4E +04 | 4.9E −02 | 2.2E −04 | 65.1 |
| oaB10v5 | c-MET | 0.4E −10 | 3.4E −12 | 3.5E +05 | 1.1E +03 | 1.6E −04 | 1.1E −06 | 63.3 |
| B10v5xhu225L | EGFR | 2.2E −07 | 2.9E −10 | 6.1E +05 | 1.1E +04 | 1.2E −01 | 2.8E −03 | ND |
| B10v5xhu225H Trastuzumab Reference Trastuzumab Reconst. |
EGFR Her2 Her2 |
1.9E −10 1.5E −10 2.5E −10 |
3.9E −12 8-5E −12 6.5E −12 |
3.8E +05 8.7E +04 9.3E +04 |
1.9E +03 2.8E +02 2.4E +02 |
7.2E −05 1.3E −05 2.4E −05 |
1.4E −06 7.4E −07 6.1E −07 |
ND ND ND |
Furthermore, potential LC shuffling during antibody reconstitution under mild reducing environment caused by TCEP was debilitated by designing antibody fragments hu225 Fab IntN and Her2 oaSEED IntC with switched LCs, which were not able to bind the respective antigen. We hypothesized that, if LC shuffling occurred during antibody reconstitution, Her2xhu225 should bind either to recombinant Her2 or EGFR or both, according to the shuffling variants depicted in Figure S9. Her2xEGFR did not bind to recombinant Her2 or EGFR after BLI analysis, revealing that no LC shuffling occurred during the reconstitution reaction (Figure S10).
Cellular binding to expressed antigens on the cell surface analyzed by flow cytometry confirmed similar binding levels to CEACAM5 or CEACAM6 by reconstituted and reference bsAb C5xC6 on gastric cancer cell line MKN-45, while no binding was observed to negative cell line HEK293 (Figure 5). Remaining amounts of TCEP and DHAA applied during reconstitution did not toxically affect cells or hamper cellular binding.
Figure 5.
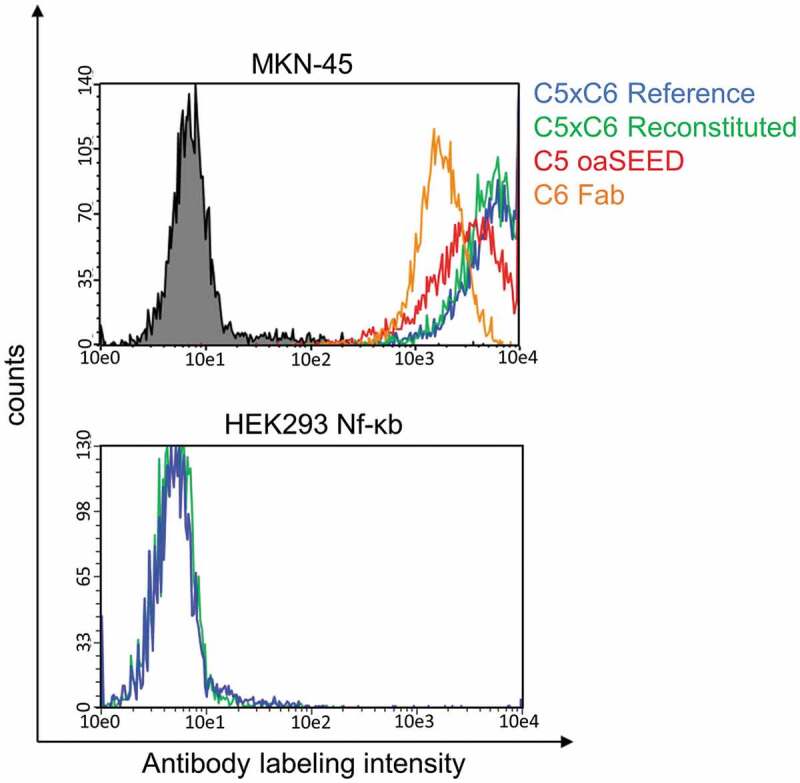
Cellular binding analysis of reconstituted bsAb C5xC6 and corresponding non-reconstituted antibody fragments. Binding to cancer cell line MKN-45 and non-binding to HEK293 cells was analyzed by flow cytometry. MKN-45 cells were incubated with antibodies and detected with Alexa Fluor 488-conjugated anti-human IgG-antibody. Green line: Reconstituted C5xC6; Blue line: C5xC6 Reference; Red line: oaC5-SEED-IntC; orange line: C6-Fab-IntN; Black line: Non-related isotype control (anti-HEL).
Biological functionality of reconstituted antibodies
Biological functionality of the reconstituted antibodies was evaluated in CD40 surface receptor agonism, T-cell activation or c-MET and EGFR receptor phosphorylation cell assays. Reconstituted anti-CD40 mAbs bearing an IgG1 or an IgG2 Fc portion were compared with the published CD40 agonistic antibody APX005M (Apexigen), reproduced as bivalent IgG1. The reconstituted antibodies anti-CD40 IgG1 and anti-CD40 IgG2 showed similar IC50 values, ~3 nM, compared to APX005M, although saturation of dose-response curves was not clearly reached, and antibody concentrations had to be adjusted (Figure 6). Reconstitution of Fabs to different Fc portions offer an easy switch of effector functions by PTS within 1 day. However, the maximum signal absorbance observed for the APX005M reference antibody was higher compared to the reconstituted mAbs. The alteration of the hinge region, especially by including an additional cysteine, opens the possibility for new disulfide bond formations during reduction and re-oxidation needed for antibody reconstitution, and might be the reason for different maximum signal detections.
Figure 6.
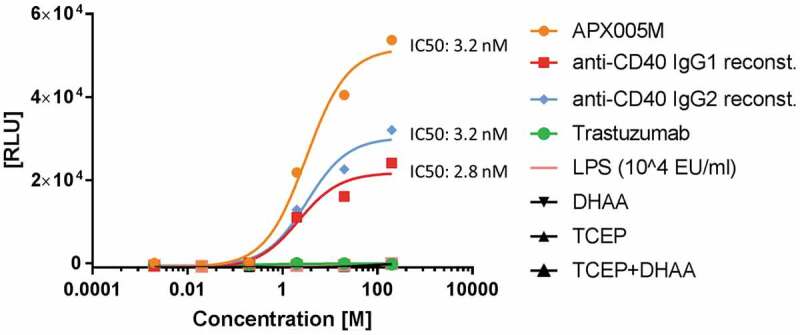
CD40 activation by reconstituted anti-CD40 agonist. CD40-expressing HEK293 cells were treated with different concentrations between 200 nM and 2 pM of reconstituted anti-CD40 IgG1, anti-CD40 IgG2, reference antibody APX005M (used as a positive control) and trastuzumab (used as a negative control) at 37°C, 5% CO2 for 6 h. LPS is known to influence certain cell assays and was used as an assay control up to a concentration of 500 nM. TCEP and DHAA were added in dose concentrations to evaluate false luminescence signal detection. CD40 was activated after treatment with anti-CD40 mAbs and BioGlo Luciferase substrate was added for luminescence readout. Luminescence signal was measured at 0.5 s per well using a sensitivity of 170. The IC50 values were calculated by fitting the dose-response curves using a 4PL with Graphpad Prism software 7.
While lipopolysaccharide (LPS) is known to interfere with cellular assays, LPS was added in different concentrations up to 500 nM to eliminate false-positive signals (Figure 6). Cells were treated with either TCEP or DHAA and in combination to observe possible negative effects in luminescence signaling (Figure 6). None of the agents (LPS, TCEP, DHAA) or mixtures of them negatively influenced the cell assay system.
To assess the ability of reconstituted bsAbs to mediate an often-desired mode of action of effector cell engagement, T cell activation was evaluated by SK-BR-3 cell co-cultivation with reconstituted antibodies and modified Jurkat effector cells bearing a luciferase reporter read out. As a prerequisite, flow cytometric analysis proved specific binding to native antigens on human T-lymphocyte cells expressing CD3 (Jurkat E6.1) and tumor cells (SK-BR-3) expressing Her2 by reconstituted CD3xHer2 bsAb, while no binding was observed for isotype control anti-hen egg lysozyme antibody (Figure S11). Monospecific CD3 displayed binding only to cells expressing CD3 antigen but not to SK-BR-3 cells (Figure S11). Reconstituted CD3xHer2 bispecific T-cell engagers either reconstituted with aHer2 Fab arm or aHer2 VHH binding moiety, showed similar potency of Tcell activation and no bias through TCEP or DHAA with EC50 values of 0.3– 0.5 nM (Figure 7). As expected, T-cell activation was not initiated by treatment with monospecific cetuximab, which lacks an anti-CD3 paratope, but showed aslight luminescence signal detection for monospecific anti-CD3 (Figure 7).
Figure 7.
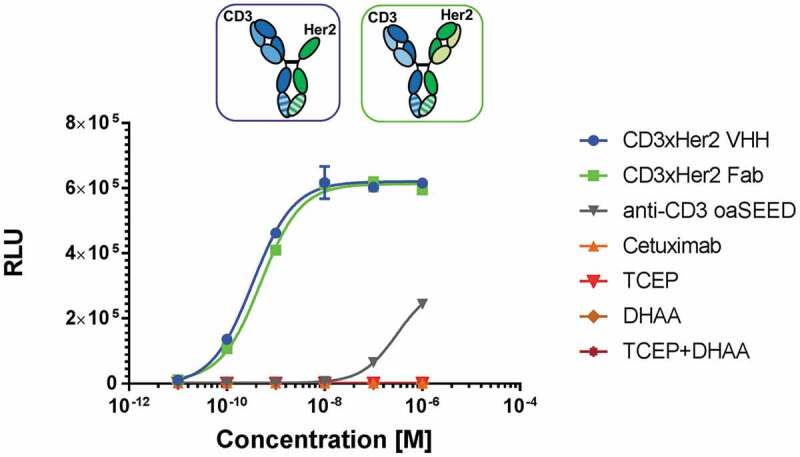
T-cell activation by reconstituted CD3xHer2 bsAbs. SK-BR-3, a Her2-expressing human breast cancer cell line, were used as antigen-presenting cells, and were incubated for 6 h with TCR/CD3 (NFAT) effector cells and reconstituted CD3xHer2 Fab (blue) or CD3xHer2 VHH (green) bsAbs in different concentrations ranging from 0.01 nM to 100 nM. CD3xHer2 binds simultaneously to TCR/CD3 on effector cells and antigen on tumor target cells and stimulates NFAT luciferase activity. BioGlo Luciferase substrate was added for luminescence readout. Luminescence signal was monitored and relative luminescence units (RLU) represent normalized luminescence to untreated cells. Reagents TCEP and DHAA used for bsAb reconstitution were added in different concentration to investigate potential assay interference. Cetuximab (orange): Negative control; monovalent anti-CD3 oaSEED (gray).
Down-scaling automated antibody reconstitution to 96- and 384-well format and HTS suitability
In order to make the split intein antibody reconstitution amenable for HTS, mAb and bsAb reconstitution was performed either in a 96-well plate with a final volume of 200 µL or in a 384-well plate using a final volume of 35 µL. Two anti-CD40 Fab-IntN fragments were fused to an IgG1-Fc-IntC portion and tested for reproducibility in a 96- or 384-well format. Figure 8 depicts the reproducibility of the reconstitution technique as a subset of 24 wells, originating from the whole 96-well plate of n = 80 replicates. The full analysis of all replicates is illustrated in Figure S12. Reconstitution reactions without the addition of Ni2+ and DHAA served as a negative control. All wells containing TCEP for PTS activation showed a fusion band at the expected size of a full-length IgG antibody (145.7 kDa), after treatment with DHAA, as monitored by electronic SDS gel analysis. Impurities in the form of not fully reconstituted Fab-IntN fragments (61.8 kDa) were observed in wells not treated with Ni2+ beads, indicating the importance of the purification step (Figure 8). Slight differences in reconstitution efficiency according to the gel bands after SDS-PAGE analysis could be detected between the wells, containing the full-length reconstituted mAbs (Figure S12). The reconstituted antibody concentrations were normalized, and differences are likely the results of manual pipetting causing volume variations in each well; however, PTS was not influenced by the 96-well format and lower reaction volumes itself. A one-step purification approach was successfully demonstrated by Ni2+ bead addition in a 96-well format.
Figure 8.
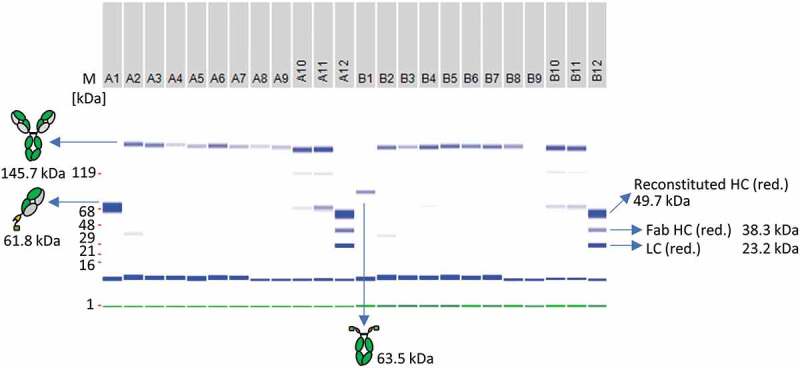
Antibody reconstitution in 96-well format using a surplus of Fab-IntN to demonstrate one-pot purification. A subset of the whole 96-well plate is illustrated. A1: Single anti-CD40 Fab IntN (61.5 kDa). B1: single huFc IntC (63.5 kDa). A2 to A9: Antibody fragments anti-CD40 IntN and huFc IntC were mixed in a ratio 3:1 and PTS was activated by 0.5 mM TCEP. Reconstituted antibodies were separated from non-reconstituted antibodies via Ni2+ beads and further treated with DHAA for reoxidation. Reconstituted anti-CD40 antibodies are indicated at 145.7 kDa under non-reduced conditions. A10-A11: Not treated with Ni2+ beads after reconstitution reaction, resulting in reconstituted anti-CD40 antibody as well as antibody fragments anti-CD40 IntN and huFc IntC. A12 and B12: Antibody fragments anti-CD40 IntN and huFc IntC after TCEP treatment without the addition of Ni2+ beads and DHAA (control). Gel band at 1 kDa (green): Lower Marker (LM). Blue band below 16 kDa: System Peak. The whole gel panel of the 96-well plate analysis is shown in Figure S12.
Binding of reconstituted full-length anti-CD40 antibodies to recombinant human CD40 was confirmed by enzyme-linked immunosorbent assay (ELISA) (Figure S13B). For negative controls and wells without TCEP treatment or lacking the monovalent Fab-IntN or Fc-IntC counterparts, no signals were observed (Figure S13b). To indicate the importance of an automated system, antibody reconstitution generated by the Hamilton MicroLab Starlet liquid handler was compared against a manual approach, producing reconstituted antibodies to avoid errors and variations (Figure S13A, B). Reconstitution reproducibility was further improved up to 15% for semi-automated compared to manually generated reconstituted antibodies in 96-well format by calculating the mean of the overall signal absorbance, estimated after ELISA (Figure S14). Antibody reconstitution was further improved by transferring into a 384-well plate format using a fully automated system. Anti-CD40 Fab-IntN fragments were reconstituted to an IgG1-Fc-IntC portion in each well and tested for reproducibility. Binding of reconstituted full-length anti-CD40 antibodies to recombinant human CD40 was confirmed by ELISA and similar and reproducible detection levels for reconstituted full-length anti-CD40 antibodies were observed compared to the semi-automated approach (Figure 9a). A slight variation pattern in signal detection was observed, which might result from concentration variations in each well upon automatic pipetting. The pipetting for all reconstitution components was row by row starting from E1 to E16 followed by the next row, and might account for these differences (Figure 9a).
Figure 9.
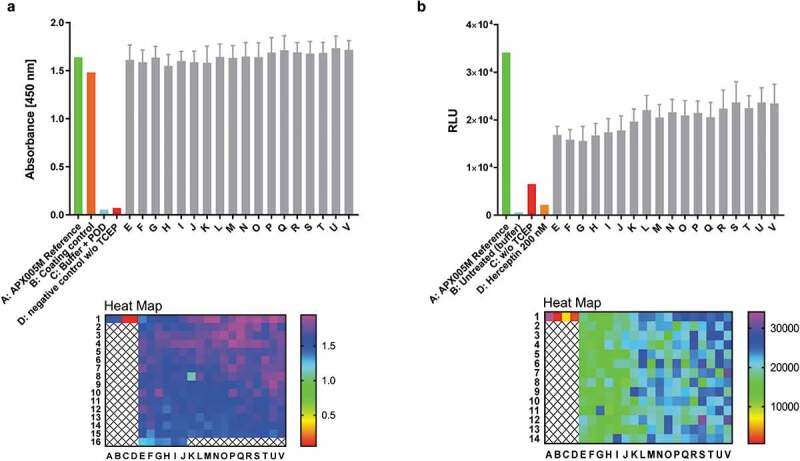
Antibody reconstitution in 384-well format fully automated by BiomekFX HTS platform. Antibody reconstitution was conducted in 386 wells by mixing anti-CD40 Fab IntN with huFc IntC in a 2:1 molar ratio, resulting in a final volume of 40 µL with a total protein amount of 14 µg, followed by addition of 0.5 mM TCEP for PTS activation. Non-reconstituted antibody fragments were removed by magnetic Ni2+ bead addition and reconstituted anti-CD40 antibodies were supplemented with a 10-fold molar excess over TCEP with DHAA for reoxidation. (a) ELISA readout of reconstituted anti-CD40 antibodies (e-v) derived from 384-well plate generated by fully automated BiomekFX HT platform. Each column from E to V represents a cluster of 16 wells including the mean with standard deviation for each cluster. The heat map shows signal detection at 450 nm after ELISA was performed for each well in the 384-well plate. An antibody concentration of 0.1 µg mL−1 was used for ELISA analysis. (b) CD40 activation assay was performed to evaluate cellular functionality of reconstituted anti-CD40 antibodies derived from high throughput reconstitution. Human CD40-expressing HEK293 cells were treated with 4 nM reconstituted antibodies (E-V) and 200 nM APX005M (positive control) for 6 h, activating an engineered NF-κB pathway for luciferase transcription. Luminescence signal was recorded and relative luminescence units (RLU) is representing normalized luminescence to untreated cells. 200 nM trastuzumab was used as a negative control, showing no CD40 activation. The heat map shows CD40 activation for each single well on the plate. Error bar: Mean with standard deviation (SD).
As broad functional screening capacity is highly desired during pharmaceutical antibody development, a feasibility study to perform functional cell assays applying reconstituted antibodies in 384-well plate format without further treatment was conducted by performing a CD40 activation assay (Figure 9b). While parental CD40 agonistic antibody APX005M and trastuzumab as references showed expected activation or no signal, respectively, all reconstituted anti-CD40 antibodies induced stable positive results amenable for high throughput screening purposes (Figure 9b). The process line to generate fully automated high throughput reconstituted antibodies is depicted in Figure S15.
Combinatorial screening to identify possible lead candidates
To evaluate the screening amenability for PTS reconstituted bsAbs, several formats were reconstituted simultaneously in one 96-well microtiter plate (Figure 10a). Reconstituted bsAbs were analyzed for retained binding properties by BLI, flow cytometry or ELISA to confirm reliability of the screening system. As expected, c-METxEGFR bispecifics B10v5xhu225L and B10v5xhu225H showed similar binding kinetics for B10v5 against c-MET (0.8 nM), matching data previously described (S16A, B).[13] Simultaneous binding to c-MET and EGFR was confirmed by stepwise association of both antigens to B10v5xhu225H via BLI analysis and monospecific B10v5 oaSEED was used to investigate unspecific binding to EGFR (Figure S16 C). Cellular binding to expressed antigens c-MET and EGFR on the cell surface of several cancer cell lines was confirmed by flow cytometry (Figure S17). B10v5xhu225L showed no binding to EGFR based on the low affinity (0.2 µM) profile of the hu225L binding moiety after ELISA signal detection (Figure 10a), which was also confirmed by BLI analysis (Figure S16b). B10v5xhu225H showed stronger binding to EGFR after ELISA readout based on its high affinity of 0.18 nM (Figure S16a). A high signal detection was observed after binding c-MET with B10v5 compared to F06 (Figure 10a).
Figure 10.
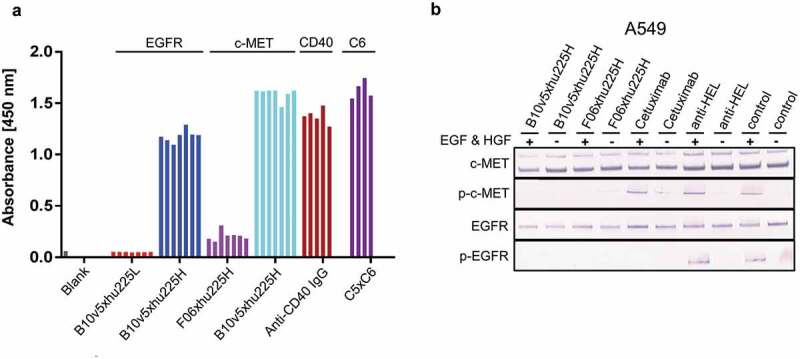
Combinatorial bsAb antibody screening. Several bsAbs B10v5xhu225L, B10v5xhu225H, F06xhu225H, anti-CD40 IgG and C5xC6 have been reconstituted in plate format as described before, to prove suitability as a screening platform based on higher and lower affinity profiles. (a) Reconstituted bsAbs were analyzed by ELISA and detected after binding to corresponding antigens with POD-labeled anti-human Fc secondary antibody. The 4–7 bars in Figure 9a represent replicants for reconstitution reproducibility. (b) Functionality of reconstituted c-METxEGFR bsAbs was demonstrated by inhibition of c-MET and EGFR phosphorylation. A549 cells with high to moderate c-MET and EGFR expression levels were incubated with 300 nM bsAb after starvation and treatment with and without 100 ng mL−1 EGF and HGF. Phosphorylated c-MET and EGFR was detected by specific AP-labeled phospho-c-MET or phospho-EGFR secondary antibodies after cell lysis with RIPA buffer followed by SDS-PAGE and western blot analysis. Cetuximab was used as a positive control for EGFR phosphorylation inhibition, while anti-HEL was used as a negative isotype control.
C-METxEGFR bsAbs F06xhu225H and B10v5xhu225H were additionally applied in functional interrogation of downstream signaling inhibition by incubating bsAbs together with c-MET and EGFR expressing cancer cells A549 after hepatocyte growth factor (HGF) and EGF stimulation, proving retained p-c-MET and p-EGFR inhibition comparable to previously described inhibitory potential (Figure 10b).
Furthermore, C5xC6 bsAbs exhibited strong binding to CEACAM6 and reconstituted anti-CD40 mAbs showed reproducible absorbance signals when compared to the above-mentioned experiments (Figure 10a). Similar ELISA signals of several in parallel reconstituted antibodies in comparison to their respective references, as well as functional read outs such as signal pathway inhibition or T cell engagement, indicated the feasibility for high throughput screening generated bsAbs using PTS.
Discussion
The primary aim of this study was to overcome screening limitations when evaluating a potentially large combinatorial set of biological paratopes as encountered during the pharmaceutical development of bsAbs. Several methods have been developed to screen for bsAb combinations at a high throughput scale. The extensive work of Sampei et al.[34] showed that 200 × 200 mAbs against FIXa and FX resulted in 400 individual cloning steps and 40,000 different bsAb combinations were needed to find the optimal hit candidate.[35] The production of all these different combinations led to a mixture of 10 different species containing false HC/HC or HC/LC pairings, which had to be removed by extensive purification steps. The process of cloning, expression and purification is the major bottleneck in HTS that substantially restricts the screening space. The split intein approach is a promising, powerful technique to overcome those limitations by producing bsAbs via post-translational combinatorial in vitro reconstitution, without the need for a large number of individual expressions and purifications of each combination. Use of the split intein approach would reduce the screening of, for example, 40,000 bsAb combinations to 400. 40,000 combinations could be generated in an automated fashion in individual wells by mix and matching.
The HTS applicability of the novel screening approach has been confirmed by the rapid reconstitution of different bispecific combinations and scales, directly suitable for screening for biological activity without any further treatment. Antibody reconstitution is efficient within 2 h and yields >90% of antibody fragment conversion. As reconstitution efficiency is highly dependent on antibody fragment concentration and the molar ratio of the two monovalent antibody fragments, it has been reported that purification of product-related impurities is very difficult and remains a manufacturing challenge, especially in small-scale screening applications.[14,36] The method presented herein allows for a one-step purification without elution of the desired antibody due to a His-Tag fusion to each antibody fragment as well as the resulting intein side product. Non-reconstituted antibody fragments and processed intein can be extracted via Ni2+ beads in 384-well format. The final antibody product is tag-less, and applicable to functional high throughput screening approaches. However, a non-native sequence addition is required within the hinge region of the antibody fragements for successful reconstitution. Of note, the reducing agent TCEP, unlike dithiothreitol which has also been successfully used for split intein ligation by us and others,[23] is compatible with purification via Ni2+ immobilized metal affinity chromatography.
Bridging target and effector cells is an often applied and thoroughly investigated mode of action for a high percentage of currently developed bsAbs.[37-39] The spatial arrangement of target and effector cell-binding moieties, hence the format with relative paratope distances and their flexibility, are crucial for the intended high potency engagement.[14,40] A screening methodology that allows both high throughput and close in-format functional screening, considering the required extein sequences, would therefore be of great value for rapid and successful development of complex next-generation antibodies such as effector cell engagers. Unlike SpyTag/SpyCatcher technology, which leaves an imprint of 23 amino acids,[41,42] or DuoBodies,[43,44] which are restricted to an IgG format, the split intein approach allows reconstitution of several formats, ranging from bivalent IgGs over heterodimeric Fc bispecifics presented herein to potential fusions of IgGs, Fabs and VHHs in diverse sets of conceivable formats. Han et al.[22] have successfully shown that a reconstituted T-cell engager by split inteins is able to perform as good or even better compared to its reference molecule. They showed cytotoxicity in the presence of both T cells and tumor cells and the dependence of T-cell activation on the presence of tumor cells. These findings support the notion that reconstituted bsAbs are suitable for cellular functional assays and can perform with high potency.
Here, several bsAbs in diverse formats have been characterized compared to their parental antibody references, showing similar KD values, thermostability and similar biological functionality (Table 1). Screening suitability was confirmed by reconstitution of several binder combinations and formats with splicing reproducibly in 96- or 384-well format. Comparable functionality of reconstituted bsAbs was shown for bispecific binding, p-c-MET and p-EGFR inhibition, T-cell engagement and CD40 activation. Over 330 monoclonal anti-CD40 antibodies were reconstituted in a fully automated manner in less than 1 day and showed both reproducible reconstitution yields and CD40 activation in an HTS-compatible functional screening assay. When applied to the full extent of the described industry-scale automation setup, up to 40,000 bsAb reconstituted hit candidates could be functionally screened within 1 week.
The purification method described here is adapted for HTS approaches and might also be amenable for unpurified antibody fragments of unknown concentration present in expression supernatant. Furthermore, the split intein Npu DnaE used in this work is able to undergo PTS directly during expression (data not shown). The expression supernatants of both antibody fragments could be combined prior to purification, generating a full-length bsAb by PTS. Alternatively an already purified oaSEED IntC fragment could be added to the expression supernatant of a Fab IntN and vice versa. Facile purification via Ni2+ beads could be applied as discussed above and safe extensive purification steps.
Different split inteins have been demonstrated to achieve splicing kinetics comparable to Npu DnaE without the need for a reducing environment.[29] Often a pH or a temperature shift is sufficient to initiate the splicing mechanism. These split inteins, like the recently engineered cysteine-less split intein Aes, might be an alternative for generating reconstituted antibodies for sensitive cellular assays not compatible with the reactants TCEP and DHAA as, for instance, applied in effector cell recruitment screenings.[45] TCEP and DHAA do not cause a luminescence signal detection in the CD40 activation assay, but reconstituted antibody variants require the addition of a non-native six amino acid sequence within the hinge for PTS activation, which might alter the geometry and flexibility of the molecule compared to the natural IgG reference molecule. The additional included cysteine might lead to potential new HC – HC or HC – LC disulfide bonds after reduction and reoxidation procedures, as a result of forming a byproduct, and might account for lower maximal extent of CD40 activation compared to the reference antibody. Further investigations and a larger data set are required to evaluate whether this is a general feature of anti-CD40 variants or the reagents used. Functional assays applied in this study were, however, not specifically screened for compatibility but proved applicable for reconstituted antibody screening.
The data described herein represents a technical proof of concept for an industry-scale automated high throughput functional combinatorial antibody screening approach based on split inteins. Diverse exemplary functional screening applications should include the combination of full binder repertoires for optimal bsAb drug discovery for further investigations. Extension of the split intein reconstitution methodology to additional bsAb formats, including VHH-IgG-fusions, could further broaden the application range. Finally, the identification of orthogonal-acting intein variants for reconstitution of three antibody building blocks would enable higher grade combinatorial screening approaches for trispecific antibodies, bispecific Fc effector variants or bispecific antibody-drug conjugates with fixed drug-to-antibody ratios. In summary, the split intein screening approach is a powerful tool for facile combinatorial bsAb generation and broad automated functional screenings. In the future, this approach could support rapid pharmaceutical drug discovery of complex antibodies by increasing the probability for identification of an optimal binder combination.
Materials and methods
Plasmid generation
Subcloning of Npu DnaE genes for secretory expression in mammalia suspension cells: The genes encoding for Npu DnaEN and Npu DnaEC [46] were ordered as gene synthesis from GeneArt (Thermo Fisher Scientific®) as a codon-optimized version for mammalian expression. Genes were modified with a multiple cloning site (mcs) flanking unique restriction sites for antibody fragment subcloning or exchange performed by standard cloning. Enzymes for standard cloning were purchased from New England Biolabs. The Npu DnaEC gene was subcloned into pTT5 vector backbone by using EcoRI and BamHI restriction sites encoding Fc HCs. Npu DnaEC and Npu DnaEN genes were subcloned into pTT5 vector backbone by using BamHI and NotI restriction sites encoding SEED or Fab HCs. Resulting pTT5-NpuDnaEN constructs contained an N-terminal myc-tag followed by a hexahistidine tag downstream of the CH1 domain of Fab HCs. The resulting pTT5-NpuDnaEC construct contained a C-terminal hexahistidine tag followed by a 4x (G4S) linker upstream of the CH2 domain for Fc- or SEED encoding HCs. VH antibody regions encoding anti-CD40, CEACAM5, CEACAM6, c-MET, and EGFR were subcloned into pTT5-NpuDnaEN backbone vector using ApaI and BamHI restriction sites. To avoid homodimer production of SEEDbodies, point mutations were introduced at position 445HY446and 441RF442 to the SEED-GA HC to prevent binding to protein A by the constant region. The mutations allow selective purification of heterodimers, since the SEED-AG HC is known to avoid forming homodimers. All constructs were verified by Sanger sequencing. Plasmids for transient expression were prepared using GenEluteTM HP Plasmid Maxiprep Kit (Sigma-Aldrich) following manufacturer’s instructions.
Expression and purification of bsAb
All antibodies were expressed by transient co-transfection of HCs and LCs for the corresponding antibody or antibody fragment in Expi293FTM cells following the manufacturer’s instructions using the corresponding transfection kit and media from LifeTechnologies. Cells were diluted to 2.9 × 106 vc mL−1 prior to transfection. Cell counting was determined using Vi-CELL XR cell counter (Beckman Coulter). Supernatant was harvested either after 5 days post-transfection, or when cell viability dropped below 80%. Supernatant was filter-sterilized using a 0.22 µm Nalgene Rapid-Flow Bottle Top Filter (Thermo Fisher Scientific) before following purification or analysis. Antibodies were either produced in a small scale of 50 mL or in a midscale of 200 mL. Supernatants with antibodies or antibody fragments containing a His-Tag for purification were dialyzed against phosphate-buffered saline (PBS) pH 7.4 overnight before affinity chromatography (1 or 5 mL HisTrapTM HP, GE Healthcare) using an ÄKTA Pure (GE Healthcare). Supernatants containing antibodies or antibody fragments harboring an Fc portion, were purified by affinity chromatography using HiTrap Mab Select SuRe material (GE Healthcare). A final polishing step of all antibodies or antibody fragments was performed by preparative size exclusion chromatography (HiLoad 16/60 or HiLoad 26/60 Superdex 200 pg, GE Healthcare). Antibodies were concentrated using Amicons (10K MWCO, MilliporeSigma) and protein concentration was determined by UV–VIS spectroscopy at 280 nm (NanoDropTM One, ThermoFisherScientific). Antibody purity was analyzed by analytical size exclusion HPLC using a TSK Super SW3000 column (Tosoh Bioscience) and by SDS gel electrophoresis using 4–12% Bis-Tris Gels (Novex, Invitrogen, LifeTechnologies). Gels were stained with InstantBlueTM Coomassie protein stain (Expedeon) and destained with water.
Antibody reconstitution mediated by split inteins
Fab- and SEED-antibody fragments containing a Npu DnaEN or DnaEC split intein were mixed in an equimolar ratio (1:1) for trans splicing-mediated SEEDbody reconstitution. Fab- and Fc-fragments were mixed in a molar excess ratio of 1:2 in splicing buffer (PBS: 10 mM PO43− 2.7 mM KCl 150 mM NaCl, pH 7.4) for bivalent antibody reconstitution. TCEP was added to a final concentration of 0.5 mM as a splicing catalyst. The mixture was incubated at either 37°C for 2 h or at room temperature (RT) for 4 h. Reconstituted antibodies were supplemented with HisPure Ni-NTA beads (Thermo Fisher Scientific, catalog no. #88,221) and 10 mM imidazole for purification of non-reconstituted antibody fragments. Supernatant was isolated from Ni-NTA beads and reaction was stopped after stated time by adding DHAA in a 20-molar excess (eq.) over TCEP for complete oxidation of reducing agent and reoxidation of the sulfhydryl groups within the antibody to reform cysteine bridges. Samples were taken at specific time intervals and quenched by 4x lithium dodecyl sulfate (LDS) buffer (Invitrogen) supplemented with 0.1% H2O2 for complete oxidation of reducing agent. Samples were boiled at 70°C for 10 min. All reactions were monitored by SDS-PAGE (4–12% Bis-Tris gels from Novex, Invitrogen) followed by Coomassie Instant blue protein staining (Expedeon).
Biolayer interferometry
Kinetic parameters of reconstituted antibodies and antibody references were determined by BLI using the Octet RED96 system (ForteBio, Pall) at 30°C and 1000 rpm agitation speed in a volume of 200 µL in black 96-well microplates. Anti-human IgG Fc capture biosensor tips (AHC, ForteBio, Pall) were equilibrated in Dulbecco´s PBS (Life Technologies) for 60 s before antibody capturing. Antibodies were immobilized for 180 s on AHC biosensor tips using a concentration of 5 µg mL−1 diluted in Dulbecco´s PBS buffer. Biosensors were then transferred into kinetics buffer (PBS, 0.1% Tween-20 and 1% bovine serum albumin (BSA)) and incubated for 45 s followed by an association step to the appropriate target antigen. Antigens were diluted in kinetics buffer in a concentration range varying from 200 nM to 3.13 nM. Association was monitored for 400 s or until the highest concentration showed saturation and followed by a dissociation step in kinetics buffer for 800 s to determine kon and koff values. Antigen was replaced by kinetics buffer, serving as a negative control and reference measurement. A non-binding IgG antibody anti-HEL (made in-house) and an unrelated target protein EGFR (made in-house) were also used as negative controls in each experiment. The buffer reference measurement (control curve) was subtracted from antibody measurements for data fitting and kinetics determined by using ForteBio data analysis software 9.1 using a 1:1 Langmuir binding model after Savitzky-Golay filtering. The standard errors for KD, ka and kd was calculated by using ForteBio data analysis software 9.1.
Simultaneous binding of bispecific SEEDbodies were determined by capturing the antibodies on AHC biosensor tips (ForteBio, Pall) for 180 s followed by a blocking step for 45 s (PBS, 0.1% Tween-20 and 1% BSA). The antigens were added stepwise in two association steps for 400 s using a concentration of 100 nM. Buffer and unrelated target antigens or antibodies were used as a negative control to monitor unspecific binding.
High throughput protein analysis
High throughput protein analysis after split intein-mediated antibody reconstitution was performed by using the LabChipGXII unit (PerkinElmer) and following the manufacturer’s instructions. The chip and reagents used for this assay were equilibrated for 30 min at RT. Antibody samples for labchip analysis were prepared in a 96-well PCR plate (Eppendorf). 5 µL of protein sample was transferred into the 96-well plate for high sensitivity analysis. 35 µL of Milli-Q® water was added to each sample and mixed well by pipetting. Samples and protein express ladder were incubated at 70°C for 10 min for denaturation. High throughput analysis was performed and monitored by using LabChip GX v5.2 software. Sample electropherograms and virtual gel formats were processed and analyzed with LabChip GX Reviewer v5.2.
Enzyme-linked immunosorbent assay
Binding to recombinant target proteins was confirmed by ELISA using Nunc 96-well MaxiSorb flat-bottom plates (Thermo Fisher Scientific). Wells were coated with 100 ng to 1 µg well−1 target protein in 100 µL Dulbecco´s PBS (Life Technologies) for 1 h at RT or at 4°C overnight. The coated plate was washed with 3 × 300 µL PBS (pH 7.4 + 0.05% Tween) using a microplate washer ELx405 (BioTek) and subsequently blocked with 300 µL blocking solution (PBS, pH 7.4 + 2% BSA per well) for 1 h at RT, to saturate unspecific binding sites. The plate was washed with 3 × 300 µL PBS (pH 7.4 + 0.05% Tween) and subsequently treated with 10 to 100 ng mL−1 reconstituted antibodies diluted in blocking buffer using a final volume of 100 µL, for 1 h at RT. Detection antibody Fc-specific goat-anti-human conjugated to peroxidase (Jackson ImmunoResearch, catalog no. #109-035-008) (POD) (g-a-h POD) was added in a 1:5000 dilution after the plate was washed with 3 × 300 µL PBS (pH 7.4 + 0.05% Tween) and incubated for 1 h at RT. The plate was washed with 3 × 300 µL PBS (pH 7.4 + 0.05% Tween). 100 µL of 1step Ultra TMB ELISA solution (Thermo Fisher Scientific) was added to each well and incubated for 1 to max 10 min until solution turned blue. Signal was visualized by adding 100 µL 1 M H2SO4 solution to each well, resulting in a yellow color change. Absorbance was measured at 450 nm using a ParadigmTM plate reader (BeckmanCoulter) and analyzed with the multimode analysis software. Antibody references produced by Merck KGaA, Darmstadt, were used as a reference and positive control. Trastuzumab was used as a negative control and produced in-house. Fc/SEED-IntC and Fab-IntN fragments alone and combined without the addition of TCEP were used as a negative control. Reconstituted antibodies were replaced by PBS buffer to see possible background effects of the g-a-h POD detection antibody. To confirm functional ELISA, target recombinant protein was incubated with anti-penta his (1:1000) and goat-anti-mouse POD (1:5000), serving as an additional positive control, confirming coating of the plate with antigen.
Thermal shift assay
Thermal stability of antibodies was determined by using the Prometheus NT.Plex nano DSF (Nano Temper). Each capillary (Standard, catalog no. #PR-AC002, Nano Temper) was filled with 10 µL of 1 mg mL−1 protein sample to ensure complete filling of the capillaries. Melting curves were monitored starting from 20°C to 95°C with a temperature slope of 1°C min−1 for 1 h and 15 min. Data were analyzed using the PR. ThermControl v2.1 software (Nano Temper).
Cell culture
Human cancer cell lines were obtained from the American Type Culture Collection (A549, MDA-MB-468 and HT1080), and the German Collection of Microorganism and Cell Cultures (MKN45) and maintained according to standard culture conditions (37°C, 5% CO2, 95% humidity). HT1080 human cancer cell line was modified as a full-length human CD40 expression cell line using the PiggyBac system, kindly provided by Prof. H. Wajant, University of Würzburg. The CD40 NF-kB-Luciferase Reporter stable cell line derived from HEK293 cells was obtained from BPS Bioscience (BPS Bioscience, catalog no. #60,626). HT1080 cells were cultured in DMEM high glucose medium supplemented with 10% fetal calf serum (FCS), 2 mM L-glutamine and 1 mM sodium pyruvate (Gibco, Life Technologies). MDA-MBA-468 (human adenocarcinoma) and MKN-45 (human gastric adenocarcinoma) were cultured in Roswell Park Memorial Institute (RPMI) 1640 medium supplemented with 10% FCS, 2 mM L-glutamine and 1 mM sodium pyruvate (Gibco, Life Technologies). CD40 HEK293 stable cell line cells were cultured in MEM medium with earl salts, 10% FCS, 2 mM L-glutamine and 1 mM sodium pyruvate and 1 mM non-essential amino acids (Gibco, Life Technologies). All cell lines were cultured at 37°C in a humidified 5% CO2 atmosphere. Cells were sub-cultured in cell culture adherent T75 flasks (Greiner) at 90% confluence and >90% viability monitored by microscopy (Leitz LABOVERT) and ViCell XR (Beckman Coulter) analysis. To split cells, conditional medium was removed from the flask and cells were washed with pre-warmed PBS. To detach adherent cells from surface, 1 mL of pre-warmed 0.05% trypsin/ethylenediaminetetraacetic acid, was added and incubated for 5 min at 37°C. Trypsinization was stopped by adding fresh culture medium, including FCS. Cell density was determined by ViCell XR (Beckman Coulter) analysis and cells were seeded with an appropriate cell density (1:5 or 1:10 depending on the cell doubling time) to a new T75 flask.
Flow cytometry
Flow cytometric analysis was performed using the Guava easyCyte HT cytometer. Cells were detached at a confluency of 80–90% as described above. Trypsinization was stopped by adding fresh culture medium (Gibco, Invitrogen) including FCS, and cells were counted by Vi-CELL XR (Beckman Coulter). Cellular binding assay was performed with 1 × 105 cells per cavity of a 96-well plate (Greiner). Therefore, cells were washed in fluorescence-activated cell sorting (FACS) binding buffer (PBS pH 7.4, 1% BSA) and distributed to the 96-well cavities in a final volume of 100 µL. Cells were treated with reconstituted antibodies using 10 µg mL−1 and incubated for 1 h on ice. Cells were centrifuged at 250 x g for 10 min at 4°C and washed twice with binding buffer. All washing steps were carried out in a final volume of 200 µL. Cells were treated with secondary fluorescently labeled detection antibody Alexa Fluor® 488-conjugated AffiniPure Goat Anti-Human IgG (Jackson ImmunoResearch, catalog no. #109-547-003) to determine positive or negative cell binding. Detection antibody was incubated for 30 min up to 1 h at 4°C covered from light to prevent fluorescent activation. Cells were washed again in binding buffer twice and resuspended before performing FACS analysis. Software Guava ExpressPro was used to monitor cell binding.
Functional cell assays (CD40 activation)
CD40 activation assays were performed using a CD40 NF-kB Luciferase HEK293 Stable reporter cell line (BPS Bioscience). Cells were detached and seeded with 2.5 × 104 cells per cavity in a 96-well plate suitable for cell culture treatment (Perkin&Elmer). The inner 60 wells were used for CD40 activation, while the outer wells were filled with 75 µL culture medium (Gibco) to avoid edge effects during the assay. Cells were incubated for 1 day overnight at 37°C, 5% CO2 at a humidified atmosphere, allowing the cells to attach. Cells were treated with a concentration between 200 nM and 2 pM antibodies diluted in assay buffer (BPS Bioscience), followed by an incubation step at 37°C, 5% CO2 for 6 h at humidified atmosphere. BioGlo Luciferase substrate was resolved at RT prior to assay and covered from light until usage. After 6 h incubation time, cells were equilibrated to RT for 15 min, followed by the addition of 75 µL per well BioGlo Luciferase substrate. Another incubation step was performed for 5–30 min at RT covered from light. Luminescence signals were measured with the Synergy4 (BioTek) at 0.5 s per well using a sensitivity of 170. Gen5 software was used as an analysis tool. Background luminescence occurring from assay buffer was subtracted from sample wells and the relative luminescence units were plotted against the logarithmic scaled antibody concentration. Dose-response curves were fitted using 4PL model by GraphPad Prism 7 (Graphpad Software, Inc).
T-cell activation bioassay
T-cell activation assays were performed by using a T-Cell Activation Bioassay (NFAT) Kit (Promega) according to the manufacturer’s protocol. Briefly, Her2-expressing SK-BR-3 cells were detached and seeded with 4.0 × 104 cells per cavity in a 96-well plate suitable for cell culture treatment (PerkinElmer). The inner 60 wells were used for T-cell activation, while the outer wells were filled with 75 µL assay buffer (low IgG FBS heat inactivated (Gibco) diluted in RPMI medium) to avoid edge effects during the assay. Cells were incubated for 1 day overnight at 37°C, 5% CO2 at humidified atmosphere, allowing the cells to attach. Medium was removed carefully and replaced by 25 µL assay buffer. 25 µL of diluted antibodies for T-cell activation ranging from 0.01 nM to 100 nM were added to the cells. Jurkat effector cells including a modified nfat-re-luc2 pathway were thawed in water bath at 37°C and diluted with assay buffer according to the manufacturer’s protocol (Promega). 25 µL of effector cells (1 x 105 cells per well cavity) were added to the cells, followed by an incubation step at 37°C, 5% CO2 at humidified atmosphere for 6 h. BioGlo Luciferase substrate was resolved at RT prior to assay and covered from light until usage. After 6 h incubation time, cells were equilibrated to RT for 15 min, followed by the addition of 75 µL per well BioGlo Luciferase substrate. Another incubation step was performed for 5–30 min at RT covered from light. Luminescence signals were measured with the Synergy4 (BioTek) at 0.5 s per well using a sensitivity of 170. The Gen5 software was used as a tool for assay determination. Background luminescence occurring from assay buffer was subtracted from sample wells and the relative luminescence units were plotted against the logarithmic scaled antibody concentration. Dose-response curves were fitted using the 4PL model by GraphPad Prism 7.
Automation
One semi-automated and one fully automated system was set up, to make the antibody reconstitution method accessible for HTS approaches. The Hamilton MicroLab starlet liquid handler (Hamilton) was used for reconstitutions in a 96-well format. Antibody fragments fused to IntC or IntN were mixed in a final volume of 200 µL by the liquid handler. TCEP was added using a final concentration of 0.5 mM. The plate was incubated manually for 2 h at 37°C before addition of Ni2+ magnetic beads (Invitrogen) by the liquid handling system. The plate was incubated at RT for 1 h under shaking conditions at 700 rpm. After incubation, the plate was centrifuged at 4000 x g for 2 min and transferred back to the Hamilton robot, which carried a magnet plate to capture magnetic beads. Two hundred microliters of the reconstitution mixture was transferred into a fresh 96-well plate. DHAA was added in a 10-fold molar excess over TCEP and incubated for another 2 h at 37°C or overnight at 4°C. A fully automated approach was set up for the 384-well format. The protocol was adapted from the Hamilton devices. Briefly, five multidrop dispensers (Thermo Fisher Scientific) were mixing IntN, IntC, TCEP, Ni2+ beads and DHAA at the end of the run. Pipetting was performed by BiomekFX (Beckman Coulter).
A detailed description of the automation in 96- and 384-well plates are described in the supplemental methods section.
Receptor phosphorylation assay
Phosphorylation of c-MET and EGFR was analyzed by western blot with specific detection antibodies against phosphorylated c-MET and EFGR. Target-expressing cancer cells (A549) were seeded in a 24-well tissue plate (Thermo Fisher) overnight. Medium was completely removed and replaced by a serum-starved medium lacking FCS and treated with 300 nM bispecific F06xhu225H and B10v5xhu225H antibodies for 3 h at 37°C, 5% CO2. Cells were stimulated afterward with 100 ng mL−1 HGF and EGFR (R&D Systems) for 10 min at 37°C. Cells without treatment of HGF and EGF served as a negative control. Cells were lysed subsequently with ice-cold RIPA buffer containing protease and phosphatase inhibitors (Calbiochem). Cell lysates were treated with 4x LDS sample buffer and reducing agent (Invitrogen) and boiled for 10 min at 70°C. SDS-PAGE was performed, and gel was blotted on a PVDF membrane for phosphorylation detection using specific anti-phospho-c-MET (Cell Signaling Technologies, catalog no. #3077), and anti-phospho-EGFR antibodies (Abcam, catalog no. #ab32578). Expression levels of c-MET and EGFR were detected by using specific anti-c-MET (Biomol, catalog no. #M3007-13A) and anti-EGFR antibodies (Cell Signaling Technologies, catalog no. #D38B1).
Supplementary Material
Abbreviations
- aa
amino acid
- bsAb
bispecific antibody
- BLI
biolayer interferometry
- c-MET or HGFR
hepatocyte growth factor receptor
- DHAA
Dehydroascorbic acid
- EGFR
EGF receptor
- HTS
High throughput screening
- HGF
hepatocyte growth factor
- HEL
hen egg lysozyme
- IntC
C-terminal part of split intein
- IntN
N-terminal part of split intein
- LPS
Lipopolysaccharide
- MTP
Microtiterplate
- mAb
monoclonal antibody
- Ni
Nickel
- NBE
New Biological Entity
- oa
one- armed
- SEED
strand exchange engineered domain
- POD
Peroxidase
- PTS
Protein trans splicing
- RLU
relative luminescence units
- RT
room temperature
- scFv
single-chain variable fragment
- TCEP
Tris(2-carboxyethyl)phosphine
- VH
variable domain of the heavy chain
- VL
variable domain of the light chain
Acknowledgments
The authors thank Stephan Keller, Alexander Müller, Sigrid Auth and Dirk Müller-Pompalla for advice and laboratory support in mAb purification; Oliver Edler, Marion Sauer, Elvira Meißen for support and advice in mAb expression; Kerstin Hallstein for laboratory instructions and assistance in BLI analysis; Oliver Rammo, Bianca Edelmann, Stefan Zielonka and Friedrich Rippmann for scientific support and construct design; Carolin Sellmann and Simon Krah for sequence and plasmid delivery and scientific input; the manuscript anonymous reviewers for comments and suggestions.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
References
- 1.Wu G, Doberstein SK.. HTS technologies in biopharmaceutical discovery. Drug Discov Today. 2006;11:718–15. doi: 10.1016/j.drudis.2006.06.010. [DOI] [PubMed] [Google Scholar]
- 2.Sundberg SA. High-throughput and ultra-high-throughput screening: solution- and cell-based approaches. Curr Opin Biotechnol. 2000;11:47–53. doi: 10.1016/S0958-1669(99)00051-8. [DOI] [PubMed] [Google Scholar]
- 3.Fox S, Farr-Jones S, Sopchak L, Boggs A, Nicely HW, Khoury R, Biros M. High-throughput screening: update on practices and success. J Biomol Screen. 2006;11:864–69. doi: 10.1177/1087057106292473. [DOI] [PubMed] [Google Scholar]
- 4.Mayr LM, Bojanic D. Novel trends in high-throughput screening. Curr Opin Pharmacol. 2009;9:580–88. doi: 10.1016/j.coph.2009.08.004. [DOI] [PubMed] [Google Scholar]
- 5.Bleicher KH, Böhm HJ, Müller K, Alanine AI. Hit and lead generation: beyond high-throughput screening. Nat Rev Drug Discov. 2003;2:369–78. doi: 10.1038/nrd1086. [DOI] [PubMed] [Google Scholar]
- 6.Sedykh SE, Prinz VV, Buneva VN, Nevinsky GA. Bispecific antibodies: design, therapy, perspectives. Drug Des Devel Ther. 2018;12:195–208. doi: 10.2147/DDDT. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Carter PJ, Lazar GA. Next generation antibody drugs: pursuit of the ‘high-hanging fruit’. Nat Rev Drug Discov. 2018;17:197–223. doi: 10.1038/nrd.2017.227. [DOI] [PubMed] [Google Scholar]
- 8.Labrijn AF, Janmaat ML, Reichert JM, Parren PWHI. Bispecific antibodies: a mechanistic review of the pipeline. Nat Rev Drug Discov. 2019;18:585–608. doi: 10.1038/s41573-019-0028-1. [DOI] [PubMed] [Google Scholar]
- 9.Knight T, Callaghan MU. The role of emicizumab, a bispecific factor IXa- and factor X- directed antibody, for the prevention of bleeding episodes in patients with hemophilia A. Ther Adv Vaccines. 2017;5:39–47. doi: 10.1177/2051013617693753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chames P, van Regenmortel M, Weiss E, Baty D. Therapeutic antibodies: successes, limitations and hopes for the future. Br J Pharmacol. 2009;157:220–33. doi: 10.1111/j.1476-5381.2009.00190.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kontermann RE, Brinkmann U. Bispecific antibodies. Drug Discov Today. 2015;20:838–47. doi: 10.1016/j.drudis.2015.02.008. [DOI] [PubMed] [Google Scholar]
- 12.Mazor Y, Hansen A, Yang C, Chowdhury PS, Wang J, Stephens G, Wu H, Dall´Acqua WF. Insights into the molecular basis of a bispecific antibody’s target selectivity. mAbs. 2015;7:461–69. doi: 10.1080/19420862.2015.1022695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sellmann C, Doerner A, Knuehl C, Rasche N, Sood V, Krah S, Rhiel L, Messemer A, Wesolowski J, Schuette M, et al. Balancing selectivity and efficacy of bispecific epidermal growth factor receptor (EGFR) × c-MET antibodies and antibody-drug conjugates. J Biol Chem. 2016;291:25106–19. doi: 10.1074/jbc.M116.753491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Husain B, Ellerman D. Expanding the boundaries of biotherapeutics with bispecific antibodies. BioDrugs. 2018;32:441–64. doi: 10.1007/s40259-018-0299-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Klein C, Schaefer W, Regula JT, Dumontet C, Brinkmann U, Bacac M, Umaña P. Engineering therapeutic bispecific antibodies using CrossMab technology. Methods. 2019;154:21–31. doi: 10.1016/j.ymeth.2018.11.008. [DOI] [PubMed] [Google Scholar]
- 16.Davis JH, Aperlo C, Li Y, Kurosawa E, Lan Y, Lo KM, Huston JS. SEEDbodies: fusion proteins based on strand-exchange engineered domain (SEED) CH3 heterodimers in an Fc analogue platform for asymmetric binders or immunofusions and bispecific antibodies. Protein Eng Des Sel. 2010;23:195–202. doi: 10.1093/protein/gzp094. [DOI] [PubMed] [Google Scholar]
- 17.Scott MJ, Lee JA, Wake MS, Batt KV, Wattam TA, Hiles ID, Batuwangala TD, Ashman CI, Steward M. ‘In-Format’ screening of a novel bispecific antibody format reveals significant potency improvements relative to unformatted molecules. mAbs. 2017;9:85–93. doi: 10.1080/19420862.2016.1249078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Desjarlais JR, Lazar GA. Modulation of antibody effector function. Exp Cell Res. 2011;317:1278–85. doi: 10.1016/j.yexcr.2011.03.018. [DOI] [PubMed] [Google Scholar]
- 19.Mazor Y, Sachsenmeier KF, Yang C, Hansen A, Filderman J, Mulgrew K, Wu H, Dall´Acqua WF. Enhanced tumor-targeting selectivity by modulating bispecific antibody binding affinity and format valence. Sci Rep. 2017;7:1–11. doi: 10.1038/srep40098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Xiao X, Douthwaite JA, Chen Y, Kemp B, Kidd S, Percival-Alwyn J, Smith A, Goode K, Swerdlow B, Lowe D, et al. A high-throughput platform for population reformatting and mammalian expression of phage display libraries to enable functional screening as full-length IgG. mAbs. 2017;9:996–1006. doi: 10.1080/19420862.2017.1337617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Labrijn AF, Meesters JI, de Goeij BE, van den Bremer ET, Neijssen J, van Kampen MD, Strumane K, Verploegen S, Kundu A, Gramer MJ, et al. Efficient generation of stable bispecific IgG1 by controlled Fab-arm exchange. Proc Natl Acad Sci. 2013;110:5145–50. doi: 10.1073/pnas.1220145110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Han L, Chen J, Ding K, Zong H, Xie Y, Jiang H, Zhang B, Lu H, Yin W, Gilly J, et al. Efficient generation of bispecific IgG antibodies by split intein mediated protein trans-splicing system. Sci Rep. 2017;7:8360. doi: 10.1038/s41598-017-08641-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Han L, Zong H, Zhou Y, Pan Z, Chen J, Ding K, Xie Y, Jiang H, Zhang B, Lu H, et al. Naturally split intein Npu DnaE mediated rapid generation of bispecific IgG antibodies. Methods. 2019;154:32–37. doi: 10.1016/j.ymeth.2018.10.001. [DOI] [PubMed] [Google Scholar]
- 24.Badalà F, Nouri-mahdavi K, Raoof DA. Inteins: nature´s gift to protein chemists. Comput Long Beach Calif. 2008;144:724–32. [Google Scholar]
- 25.Vila-Perelló M, Liu Z, Shah NH, Willis JA, Idoyaga J, Muir TW. Streamlined expressed protein ligation using split inteins. J Am Chem Soc. 2013;135:286–92. doi: 10.1021/ja309126m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wood DW, Camarero JA. Intein applications: from protein purification and labeling to metabolic control methods. J Biol Chem. 2014;289:14512–19. doi: 10.1074/jbc.R114.552653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Guan D, Ramirez M, Chen Z. Split intein mediated ultra-rapid purification of tagless protein (SIRP). Biotechnol Bioeng. 2013;110:2471–81. doi: 10.1002/bit.v110.9. [DOI] [PubMed] [Google Scholar]
- 28.Shah NH, Eryilmaz E, Cowburn D, Muir TW. Extein residues play an intimate role in the rate-limiting step of protein trans -splicing. J Am Chem Soc. 2013;135:5839–47. doi: 10.1021/ja401015p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Carvajal-vallejos P, Pallissé R, Mootz HD, Schmidt SR. Unprecedented rates and efficiencies revealed for new natural split inteins from metagenomic sources. J Biol Chem. 2012;287:28686–96. doi: 10.1074/jbc.M112.372680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Brezski RJ, Jordan RE. Cleavage of IgGs by proteases associated with invasive diseases: an evasion tactic against host immunity? mAbs. 2010;2:212–20. doi: 10.4161/mabs.2.3.11780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Moritz B, Stracke JO. Assessment of disulfide and hinge modifications in monoclonal antibodies. Electrophoresis. 2017;38:769–85. doi: 10.1002/elps.201600425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Zettler J, Schütz V, Mootz HD. The naturally split Npu DnaE intein exhibits an extraordinarily high rate in the protein trans-splicing reaction. FEBS Lett. 2009;583:909–14. doi: 10.1016/j.febslet.2009.02.003. [DOI] [PubMed] [Google Scholar]
- 33.Krah S, Schröter C, Eller C, Riehl L, Rasche N, Beck J, Sellmann C, Günther R, Toleikis L, Hock B, et al. Generation of human bispecific common light chain antibodies by combining animal immunization and yeast display. Protein Eng Des Sel. 2017;30:291–301. doi: 10.1093/protein/gzw077. [DOI] [PubMed] [Google Scholar]
- 34.Sampei Z, Igawa T, Soeda T, Okuyama-Nishida Y, Moriyama C, Wakabayashi T, Tanaka E, Muto A, Kojima T, Kitazawa T, et al. Identification and multidimensional optimization of an asymmetric bispecific IgG antibody mimicking the function of factor VIII cofactor activity. PLoS One. 2013;8:e57479. doi: 10.1371/journal.pone.0057479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kitazawa T, Igawa T, Sampei Z, Muto A, Kojima T, Soeda T, Yoshihashi K, Okuyama-Nishida Y, Saito H, Tsunoda H, et al. A bispecific antibody to factors IXa and X restores factor VIII hemostatic activity in a hemophilia a model. Nat Med. 2012;18:1570–74. doi: 10.1038/nm.2942. [DOI] [PubMed] [Google Scholar]
- 36.Xu Y, Lee J, Tran C, Heibeck TH, Wang WD, Yang J, Stafford RL, Steiner AR, Sato AK, Hallam TJ, et al. Production of bispecific antibodies in ‘knobs-into-holes’ using a cell-free expression system. mAbs. 2015;7:231–42. doi: 10.4161/19420862.2015.989013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Vivier E, Nunès JA, Vély F. Natural killer cell signaling pathways. Science. 2004;306:1517–19. doi: 10.1126/science.1103478. [DOI] [PubMed] [Google Scholar]
- 38.Pahl J, Cerwenka A. Tricking the balance: NK cells in anti-cancer immunity. Immunobiology. 2017;222:11–20. doi: 10.1016/j.imbio.2015.07.012. [DOI] [PubMed] [Google Scholar]
- 39.Baeuerle PA, Reinhardt C. Bispecific T-cell engaging antibodies for cancer therapy. Cancer Res. 2009;69:4941–44. doi: 10.1158/0008-5472.CAN-09-0547. [DOI] [PubMed] [Google Scholar]
- 40.Bluemel C, Hausmann S, Fluhr P, Sriskandarajah M, Stallcup WB, Baeuerle PA, Kufer P. Epitope distance to the target cell membrane and antigen size determine the potency of T cell-mediated lysis by BiTE antibodies specific for a large melanoma surface antigen. Cancer Immunol ImmunoTher. 2010;59:1197–209. doi: 10.1007/s00262-010-0844-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Yumura K, Akiba H, Nagatoishi S, Kusano-Arai O, Iwanari H, Hamakubo T, Tsumoto K. Use of SpyTag/SpyCatcher to construct bispecific antibodies that target two epitopes of a single antigen. J Biochem. 2017;3:203–10. doi: 10.1093/jb/mvx023. [DOI] [PubMed] [Google Scholar]
- 42.Sutherland AR, Alam MK, Geyer CR. Post-translational assembly of protein parts into complex devices by using spytag/spycatcher protein ligase. ChemBioChem. 2019;20:319–28. doi: 10.1002/cbic.v20.3. [DOI] [PubMed] [Google Scholar]
- 43.Brinkmann U, Kontermann RE. The making of bispecific antibodies. mAbs. 2017;9:182–212. doi: 10.1080/19420862.2016.1268307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Garber K. Bispecific antibodies rise again. Nat Rev Drug Discov. 2014;13:799–801. doi: 10.1038/nrd4478. [DOI] [PubMed] [Google Scholar]
- 45.Bhagawati M, Terhorst TME, Füsser F, Hoffmann S, Pasch T. A mesophilic cysteine-less split intein for protein trans - splicing applications under oxidizing conditions. PNAS. 2019;44:22164–72. doi: 10.1073/pnas.1909825116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Stevens AJ, Brown ZZ, Shah NH, Sekar G, Cowburn D, Muir TW. Design of a split intein with exceptional protein splicing activity. J Am Chem Soc. 2016;138:2162–65. doi: 10.1021/jacs.5b13528. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.