

Abstract
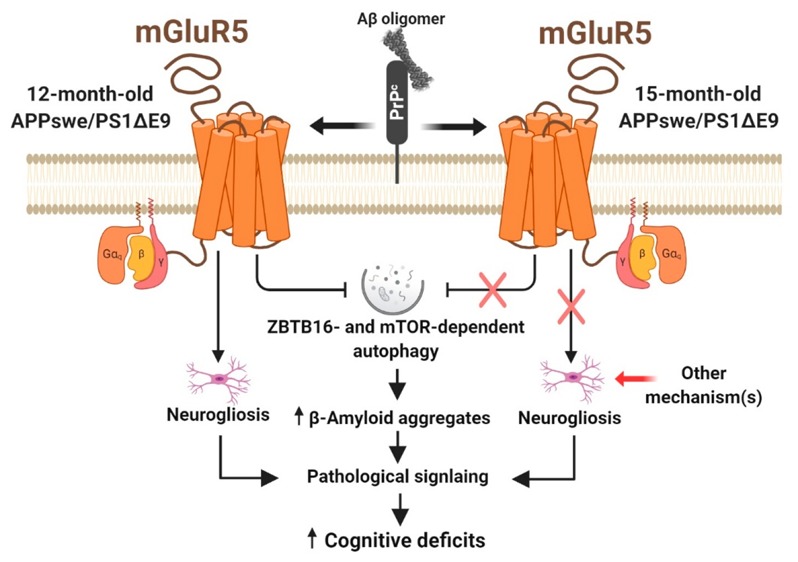
Alzheimer’s disease (AD) is the most prevalent neurodegenerative disease and is characterized by a progressive cognitive decline in affected individuals. Current therapeutic strategies are limited in their efficacy and some have proven to be even less effective at later disease stages or after extended use. We previously demonstrated that chronic inhibition of mGluR5 signaling using the selective negative allosteric modulator (NAM) CTEP in APPswe/PS1ΔE9 mice can rescue cognitive function, activating the ZBTB16-mediated autophagy pathway to reduce Aβ, the principal neurotoxic species in AD brains. Here, we evaluated the efficacy of long-term treatment with CTEP in 6 month old APPswe/PS1ΔE9 mice for either 24 or 36 weeks. CTEP maintained its efficacy in reversing working and spatial memory deficits and mitigating neurogliosis in APPswe/PS1ΔE9 mice when administered for 24 weeks. This was paralleled by a significant reduction in Aβ oligomer and plaque load as a result of autophagy activation via ZBTB16 and mTOR-dependent pathways. However, further extension of CTEP treatment for 36 weeks was found ineffective in reversing memory deficit, neurogliosis, or Aβ-related pathology. We found that this loss in CTEP efficacy in 15 month old APPswe/PS1ΔE9 mice was due to the abolished contribution of ZBTB16 and mTOR-mediated signaling to AD neuropathology at this advanced disease stage. Our findings indicate that the contribution of pathological mGluR5-signaling to AD may shift as the disease progresses. Thus, we provide the first evidence that the underlying pathophysiological mechanism(s) of AD may unfold along the course of the disease and treatment strategies should be modified accordingly to ensure maximal therapeutic outcomes.
Keywords: Alzheimer’s disease, autophagy, mGluR5, neuroglia, age, beta amyloid
Alzheimer’s disease (AD) is a neurodegenerative disorder primarily characterized by progressive memory loss and cognitive decline. It is the most common form of dementia affecting people over 65 years of age1 with more than 40 million people diagnosed with AD worldwide.2 At present, AD has no known cure and the existing treatments only provide symptomatic relief with limited disease-modifying efficacy.3,4 With an aging population the incidence of AD is continuing to rise,1 emphasizing the need for effective, safe, long-term and/or late stage therapeutic strategies for the treatment of AD.
Associated with the neurotoxic effects that characterize AD is the protein amyloid β (Aβ). In its fibrillar plaque form, Aβ forms one of the main hallmarks for AD. However, it is the soluble oligomeric Aβ1–42 that is believed to be the more neurotoxic amyloid species.5,6 AD is known to be associated with a disruption of glutamatergic signaling, and this is believed to be due to the enhanced binding of Aβ oligomers to metabotropic glutamate receptor 5 (mGluR5) in association with cellular prion proteins.7 Specifically, mGluR5 can act as an extracellular scaffold for a Aβ/ cellular prion protein (PrPc) complex that results in impaired lateral diffusion and enhanced clustering of the receptor leading to excessive release of intracellular Ca2+ and neurotoxicity.8−9 We have previously shown that the genetic deletion of mGluR5 in the APPswe/PS1ΔE9 mouse model of AD prevented memory loss and reduced Aβ-related neuropathology in male animals.10 We then showed that the pharmacological inhibition of mGluR5 using a selective negative allosteric modulator (NAM) CTEP (2-chloro-4-[2[2,5-dimethyl-1-[4-(trifluoromethoxy) phenyl] imidazol-4-yl] ethynyl] pyridine) in two male mouse models of AD, APPswe/PS1ΔE9 and 3xTg-AD, rescued deficits in learning and memory and enhanced autophagic clearance of Aβ oligomers and plaques from the brain.11,12 Others have also reported that mGluR5 silent allosteric modulators can improve cognitive impairment but not Aβ deposition in an AD mouse model.13 More so, mGluR5 positive allosteric modulator was proven to reverse Aβ-mediated neurotoxicity but was not successful in reversing cognitive deficits in an AD mouse model.14 Through this work, we and others have demonstrated a contributory role for mGluR5 in AD pathophysiology and highlighted the potential therapeutic applicability of mGluR5 in AD.
More recently, we have shown that mGluR5 does not play a major role in AD pathology in female APPswe/PS1ΔE9, and the use of mGluR5 NAMs does not represent a successful strategy for treating symptomatic female APPswe/PS1ΔE9 mice.15 While these observations clearly indicate that mGluR5 signaling can be regulated in a sex-specific manner, it also points to the fact that mGluR5 contribution to AD neuropathology and therapy may be affected by nonmodifiable risk factors. Given that AD patients are diagnosed at various disease stages and ages, it is imperative that we assess whether age/disease stage, as nonmodifiable risk factors at the time of diagnosis, influence mGluR5-mediated neuropathology and responses to mGluR5 NAM treatment in AD mouse models. In human AD patients, the age of the patient has been found to be a key determinant of the pathogenesis of AD and response to acetylcholine esterase inhibitors, the most prescribed class of medications for symptomatic treatment of AD in patients.3,4 For instance, young symptomatic patients appear to respond better to rivastigmine when compared to donepezil.16 Moreover, head-to-head comparison of donepezil and galantamine showed that although long-term treatment with galantamine is superior to that with donepezil, it offers a temporary initial phase of improvement in memory function followed by a continuation in cognitive decline at later disease stages.17 Taken together, these observations highlight the need to study longitudinal alterations in the pathophysiological mechanisms of AD, as well as the long-term efficacy and tolerability of potential therapies. This is of a particular importance in AD since patients are usually diagnosed at different disease stages and medications are intended to be used for prolonged periods.18
We have previously reported that the mGluR5 NAM, CTEP, improves cognitive deficits and mitigates AD pathology in male APPswe/PS1ΔE9 mice following 12 weeks of treatment at either 6 or 9 months of age.11,12,15 CTEP is an orally bioavailable, blood brain barrier permeable, selective mGluR5 NAM.19 Here, we evaluated the efficacy of CTEP by treating symptomatic 6 month old male APPswe/PS1ΔE9 mice for extended periods (24 and 36 weeks). We show that CTEP retained its efficacy in reversing memory deficits, mitigating neurogliosis, and activating autophagic clearance of Aβ oligomers and plaques in APPswe/PS1ΔE9 mice when administered for 24 weeks. When the treatment was extended to 36 weeks, Aβ oligomers continued to accumulate, but CTEP lost its ability to reverse memory deficits or AD neuropathology. We also found that this loss in CTEP efficacy in 15 month old APPswe/PS1ΔE9 mice was likely due to the abolished contribution of mGluR5 to AD-related neuropathology at this advanced disease stage. This study has critical implications regarding the potential repurposing of mGluR5 NAMs for the long-term treatment of AD. It suggests that age and disease stage should be factored in AD drug trial results and it also highlights the potential usefulness of disease stage-tailored treatment strategies for AD patients.
Results
Chronic Blockade of mGluR5 with CTEP for 24, but not 36, Weeks Ameliorates Cognitive Deficits in APPswe/PS1ΔE9 Mice
We have previously shown that 12 week treatment of either 6 or 9 month old symptomatic male APPswe/PS1ΔE9 with CTEP ameliorated learning deficits in novel object recognition and Morris water maze (MWM) tests.11,15 Here, we tested the efficacy of extended CTEP treatment of 6 month old male APPswe/PS1ΔE9 mice for 24 or 36 weeks in improving working and spatial memory during the novel object recognition and MWM tasks. As previously reported,11,20 vehicle treated 12 and 15 month old APPswe/PS1ΔE9 mice did not discriminate between novel and familiar objects (Figure 1A,B). While treatment of 6 month old APPswe/PS1ΔE9 mice with CTEP for 24 weeks rescued impaired novel object discrimination, the behavioral improvement induced by CTEP treatment was not evident following a 36 week treatment of mice (Figure 1A,B). CTEP did not affect performance of wild-type mice at either time point (Figure 1A,B).
Figure 1.
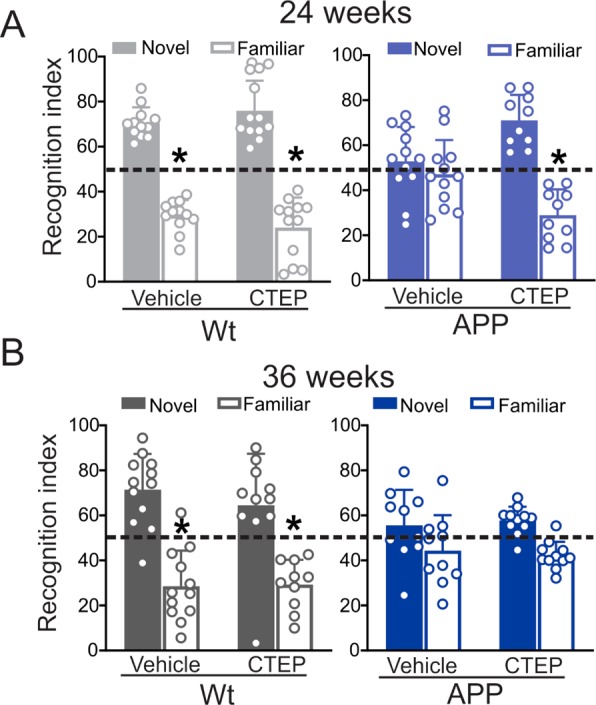
CTEP treatment for 24, but not 36, weeks improved recognition scores of APPswe/PS1ΔE9 mice. Mean ± SD of recognition index, for exploring one novel object versus familiar object in the second day of novel object recognition test following a 24 week (A) or 36 week (B) treatment with either vehicle or CTEP (2 mg/kg) of age-matched wild-type (wt) and APPswe/PS1ΔE9 (APP) mice (n = 10–12). *P < 0.05 versus novel object values. Statistical significance was assessed by two-way ANOVA and Fisher’s LSD comparisons.
When tested in the MWM and Reverse Morris Water (RMWM) tasks, APPswe/PS1ΔE9 mice treated with vehicle for 24 and 36 weeks showed significant impairments in their ability to perform both tasks, with deficits observed in escape latency, path length, and time spent in the target quadrant (Figure 2A–H). Treatment of 6 month old APPswe/PS1ΔE9 mice with CTEP for 24 weeks reversed the behavioral impairment in both the MWM and RMWM, as measured by shorter escape latency, path lengths, and longer time spent in target quadrant (Figure 2A–H). In contrast, APPswe/PS1ΔE9 mice treated with CTEP for 36 weeks showed only modest improvement in the MWM (Figure 2A–D) and were impaired in the RMWM (Figure 2E–H). CTEP treatment of wild-type mice for 24 and 36 weeks had no effect on the performance of these mouse groups in either the MWM or RMWM (Figure 2A–H). The swim speed of the mice during the MWM and RMWM tasks was comparable between all groups indicating that the observed differences are not due to deficits in swimming capabilities of the mice due to aging (Figure 2C,G). Taken together this data shows that while extended mGluR5 antagonism seems to be tolerable in all groups of mice, it only improved working and spatial memory in APPswe/PS1ΔE9 mice up to 12 months of age suggesting that the efficacy of mGluR5 NAM can be limited at an advanced stage of AD.
Figure 2.

CTEP treatment for 24, but not 36, weeks improved performance of APPswe/PS1ΔE9 mice in the Morris Water Maze and Morris Water Maze with reversal. Mean ± SEM of (A) escape latency, (B) path length, (C) swim speed obtained for Morris Water Maze acquisition phase, and (D) mean ± SD of time spent in the target quadrant during the probe trial following a 24 week or 36 week treatment with either vehicle or CTEP (2 mg/kg) of age matched wild-type (wt) and APPswe/PS1ΔE9 (APP) mice. Mean ± SEM of (E) escape latency, (F) path length, and (G) swim speed obtained for Reversal Morris Water Maze acquisition phase, and (H) mean ± SD of time spent in the target quadrant during the probe trial following a 24-week or 36-week treatment with vehicle or CTEP of age matched wt and APP mice (n = 9–11). *P < 0.05 versus age-matched, vehicle-treated wt mice. Statistical significance was assessed by two-way ANOVA and Fisher’s LSD comparisons. Mice were excluded from analysis due to spontaneous death.
Chronic Inhibition of mGluR5 with CTEP for 24, but Not 36, Weeks Reduces Aβ Pathology in APPswe/PS1ΔE9 Mice
To determine whether the differences in cognitive function observed in APPswe/PS1ΔE9 mice treated, beginning at 6 months of age, with CTEP for 24 and 36 weeks were reflected on differences in Aβ pathology, we quantified plaque density in both the cortex and hippocampus of vehicle- and CTEP-treated APPswe/PS1ΔE9 mice. We found the plaque density was significantly lower in both the cortex and hippocampus of APPswe/PS1ΔE9 mice following treatment with CTEP for 24 weeks (Figure 3A). In contrast, CTEP treatment of APPswe/PS1ΔE9 mice for 36 weeks modestly reduced plaque density in the cortex, but did not affect plaque density in the hippocampus (Figure 3B). Moreover, treatment of APPswe/PS1ΔE9 mice with CTEP for 24 weeks significantly reduced soluble Aβ oligomer levels, whereas treatment for 36 weeks did not alter soluble Aβ oligomer levels, and overall soluble Aβ oligomer levels at 36 weeks of treatment were significantly higher than those observed at 24 weeks (Figure 3C). Since the APPswe/PS1ΔE9 mouse model was a mutant amyloid precursor protein (APP) overexpression model,20,21 we tested whether the changes in Aβ burden between the two age groups of APPswe/PS1ΔE9 mice was due to altered APP expression. We found that the expression of APP was not altered by disease progression or CTEP treatment (Figure 3D). Taken together, these observations indicated that extended mGluR5 NAMs could be effective in clearing the Aβ load in APPswe/PS1ΔE9 mice, but this efficacy can be significantly abolished in advanced stages of the disease.
Figure 3.

CTEP treatment for 24, but not 36, weeks reduced Aβ pathology in APPswe/PS1ΔE9 mice. Representative images of Aβ staining and quantification of plaque density in cortical and hippocampal brain slices from age-matched APPswe/PS1ΔE9 (APP) mice following treatment with either vehicle or CTEP (2 mg/kg) for (A) 24 or (B) 36 weeks. Images are representative of four independent experiments (scale bar, 50 μm). Data represent mean ± SD following the quantitation of five different 900 μm2 regions in the cortex and two different 900 μm2 regions in the hippocampus from six brain slices in four independent mice for each group. *P < 0.05 versus the same region of vehicle-treated age-matched APP mice. Statistical significance was assessed by unpaired Student’s t test. (C) Mean ± SD of the whole-brain Aβ oligomer concentrations (pg/mg protein) in age-matched wt and APP mice after either a 24 week or 36 week treatment with either vehicle or CTEP (n = 4–5). The asterisk (∗) denotes significant difference at P < 0.05. (D) Representative Western blots and quantification of folds change in Amyloid precursor protein with the corresponding β-tubulin loading control from age-matched wt and APP mice after either a 24 week or 36 week treatment with either vehicle or CTEP (n = 4). Values represent mean ± SD and were expressed as a fraction of the 24 week, vehicle-treated wt value. *P < 0.05 versus 24 week, vehicle-treated wt mice. Statistical significance for panels C and D was assessed by two-way ANOVA and Fisher’s LSD comparisons.
ZBTB16- and mTOR-Dependent Autophagic Pathways Contribute to Reduced Aβ Pathology in APPswe/PS1ΔE9 Mice Following CTEP Treatment for 24, but Not 36, Weeks
We previously demonstrated that mGluR5 inhibits a GSK3β/Zinc Finger and BTB Domain Containing 16 (ZBTB16)/Autophagy Related 14 (ATG14) autophagic pathway in APPswe/PS1ΔE9 mice.12,22 We found here that GSK3β-pS9 phosphorylation, and both ZBTB16 and the autophagy marker p62 protein expression were increased in brain lysates of 6 month old APPswe/PS1ΔE9 mice treated with vehicle for 24 weeks compared to wild-type mice. CTEP treatment reduced GSK3β-pS9 phosphorylation, ZBTB16, and p62 expression and increased ATG14 expression (Figure 4A–D). In contrast, in 6 month old APPswe/PS1ΔE9 mice treated with either vehicle or CTEP for 36 weeks, we did not observe any changes in either GSK3β-pS9 phosphorylation or protein expression levels of either ZBTB16, ATG14 or p62, when compared to vehicle-treated wild-type mice (Figure 4A-D). However, CTEP treatment induced an increase in ZBTB16 expression in wild-type mice (Figure 4B). The loss of mGluR5-mediated regulation of the GSK3β/ZBTB16/ATG14 autophagic pathway with age was similar to what we previously reported for younger female APPswe/PS1Δ9 mice.15
Figure 4.
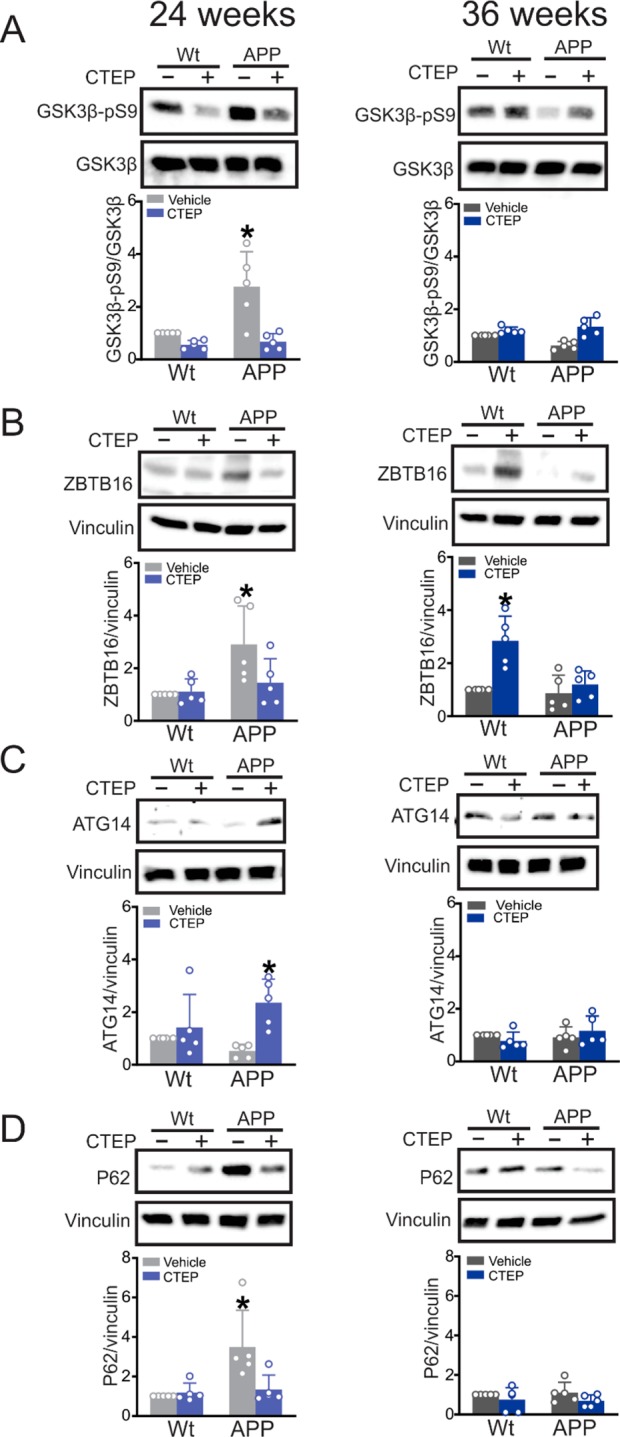
ZBTB16 autophagic pathway was activated in 12 month old APPswe/PS1ΔE9 mice by 24 weeks of CTEP treatment. Representative Western blots and quantification of folds change in (A) GSK3β-pS9, (B) ZBTB16, (C) ATG14, and (D) p62 with the corresponding loading controls in brain lysates from age matched wild-type (wt) and APPswe/PS1ΔE9 (APP) mice after either a 24 week or 36 week treatment with either vehicle or CTEP (2 mg/kg). GSK3β-pS9 was normalized to total GSK3β, ZBTB16, ATG14, and p62 were normalized to vinculin (n = 5 for each group). Values represent mean ± SD and are expressed as a fraction of the age matched, vehicle treated wt value. *P < 0.05 versus age-matched, vehicle-treated wt values. Statistical significance was assessed for GSK3β-pS9, ATG14, and p62 after 24 weeks of treatment with Kruskal–Wallis test and by two-way ANOVA and Fisher’s LSD comparisons for the rest of the panels.
mGluR5 also contributed to the regulation of Akt/mammalian Target of Rapamycin (mTOR) signaling in a zQ175 Huntington’s disease (HD) mouse model.23 mGluR5 can activate phosphoinositide 3-kinase (PI3K) that in turn recruits and activates Akt via direct phosphorylation and results in activation of mTOR downstream signaling to suppress autophagy.23−27 It is worth noting that mTOR phosphorylation at S2448 and the downstream effector p70S6K1 at T389 are both considered hallmarks of mTOR activity.28−31 When tested in AD mice, we found that Akt-pS473, mTOR-pS2448, and p70S6K1-pT389 phosphorylation were increased in APPswe/PS1Δ9 mice treated with vehicle for 24 weeks starting at 6 months of age and that CTEP treatment normalized phosphorylation to wild-type levels (Figure 5A–C). In contrast, no increase in Akt-pS473, mTOR-pS2448, or p70S6K1-pT389 phosphorylation was observed in mice treated with vehicle or CTEP for 36 weeks (Figure 5A–C). Taken together this data indicated that in advanced disease, the contribution of mTOR- and ZBTB16-autophagic pathways to AD-like neuropathology can be diminished, rendering the continued mGluR5 blockade inadequate in reversing Aβ pathology and cognitive deficits.
Figure 5.
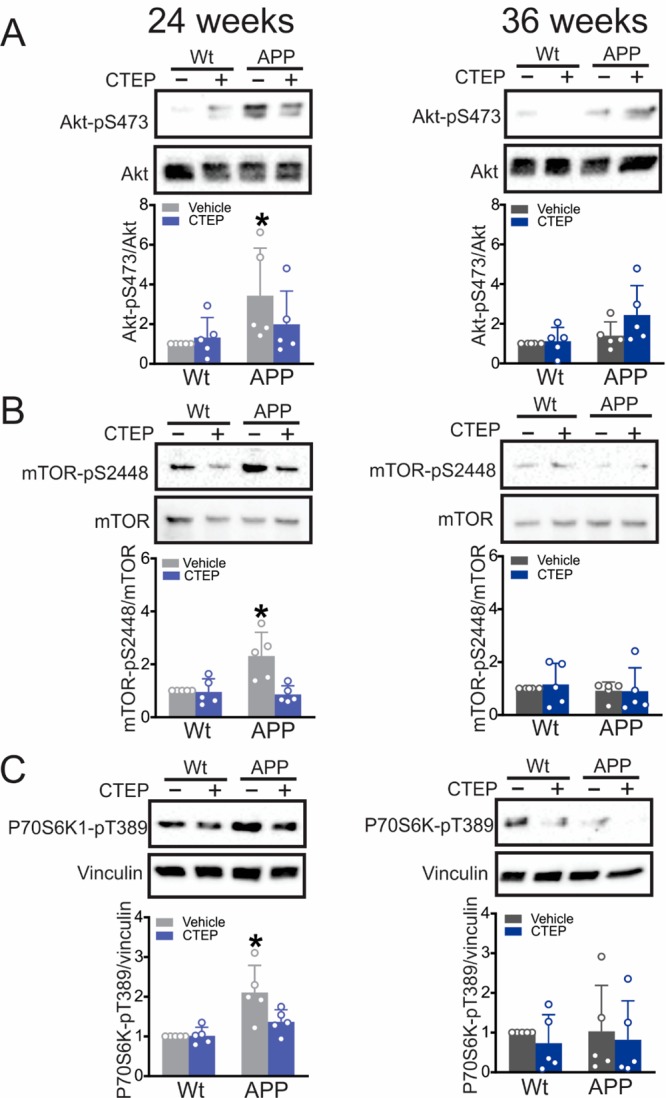
Impaired mTOR signaling in 12 month old APPswe/PS1ΔE9 mice was normalized by 24 weeks of CTEP treatment. Representative Western blots and quantification of folds change in (A) Akt-pS473, (B) mTOR-pS2448, and (C) P70S6K1-pT389 with the corresponding loading controls in brain lysates from age-matched wild-type (wt) and APPswe/PS1ΔE9 (APP) mice after either a 24 week or 36 week treatment with either vehicle or CTEP (2 mg/kg). Akt-pS473 was normalized to total Akt, mTOR-pS2448 was normalized to mTOR, and p70S6K1-pT389 was normalized to vinculin (n = 5 for each group). P70S6K1-pT389 and p62 (Figure 4D) were probed on the same blot. Values represent mean ± SD and expressed as a fraction of the age-matched, vehicle-treated wt value. *P < 0.05 versus age-matched, vehicle-treated wt values. Statistical significance was assessed by two-way ANOVA and Fisher’s LSD comparisons.
mGluR5 Inhibition with CTEP for 24 Weeks, but Not 36, Weeks Reduces Neuroglial Activation in APPswe/PS1ΔE9 Mice
Glial cells, namely astrocytes and microglia, can induce a robust neuroinflammatory response that contributes to synaptic dysfunction and neuronal death in AD.32−34 Specifically, ionized calcium binding adaptor molecule 1 (Iba1)-positive microglia and glial fibrillary acidic protein (GFAP)-positive astrocytes were previously detected around Aβ plaques.32−35 Similarly, we detected a significant increase in the number of Iba1- and GFAP-positives in cortical brain slices derived from 6 month old APPswe/PS1ΔE9 mice treated with vehicle for 24 and 36 weeks (Figure 6A,B). The treatment of 6 month old APPswe/PS1ΔE9 mice with CTEP for 24 weeks reduced the markers of activated microglia and reactive astrocytes (Figure 6A,B). However, continued treatment of the APPswe/PS1ΔE9 mice with CTEP for 36 weeks no longer promoted a reduction in astrogliosis and microgliosis (Figure 6A,B). These findings indicate that neuroglial activation contributes to AD neuropathology in APPswe/PS1ΔE9 mice but, at later disease stages microglial and astrocytic activation is likely meditated by mGluR5-independent mechanisms.
Figure 6.
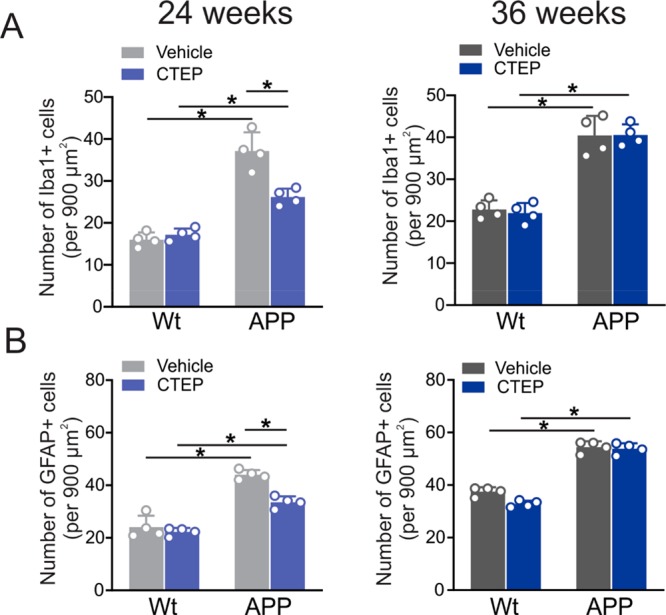
CTEP treatment for 24, but not 36, weeks reduced neuroglial activation in APPswe/PS1ΔE9 mice. Quantification of the number of (A) Iba1 and (B) GFAP positive cells in cortical brain slices from age-matched wild-type (wt) and APPswe/PS1ΔE9 (APP) mice following a 24 week or 36 week treatment with either vehicle or CTEP (2 mg/kg). Data represent the quantification of five different 900 μm2 regions in six cortical slices derived from four independent mouse brains for each group and expressed as the mean ± SD. The asterisk (∗) denotes significant difference at P < 0.05. Statistical significance was assessed by two-way ANOVA and Fisher’s LSD comparisons.
It was important to confirm that the difference in mGluR5 downstream signaling between the two age groups of APPswe/PS1ΔE9 mice was not due to age-related alteration in mGluR5 expression. Thus, we quantified the total expression of mGluR5 in brain lysates of 12 and 15 month old APPswe/PS1ΔE9 mice after extended treatment with either CTEP or vehicle. We detected no significant change in the total expression of mGluR5 between all groups (Figure S-1I) indicating that the shift in AD pathological mechanisms at advanced disease stages is downstream of the receptor.
Discussion
While few targets have been identified as being potential pharmacological targets for AD treatment, it remains less clear which particular target(s) will be successful in maintaining favorable therapeutic outcomes as the disease progresses.3 This is particularly relevant to AD therapy since patients are diagnosed at various disease stages and some of the available drugs have already proved less clinically competent in certain disease stages.16,17,36 This implies a possible shift in the pathophysiological mechanism(s) underlying disease progression that should be studied to ensure that therapeutic goals are met. Moreover, it suggests that we should consider evolving treatment strategies along the course of the disease to maximize therapeutic benefit. Our previous work provides robust evidence for the disease-modifying characteristics of mGluR5 NAMs when tested in preclinical models of AD. In the current study, we challenged mGluR5 NAM for extended periods of treatment and in advanced disease stages to assess its efficacy in APPswe/PS1ΔE9 mice.
We show evidence for a progressive accumulation of Aβ oligomer in brains of APPswe/PS1ΔE9 mice as they age. CTEP treatment of 6 month old APPswe/PS1ΔE9 mice effectively reverses cognitive deficits in the mice following 24 weeks of treatment and this is paralleled by GSK3β/ZBTB16/ATG14- and Akt/mTOR/p70S6K1-mediated activation of autophagy, as well as a reduction in Aβ load and amelioration of neurogliosis. When treatment is extended to 36 weeks, CTEP is no longer effectively mitigating Aβ pathology, neuroinflammation, or cognitive deficits. This loss in the efficacy of CTEP in later disease stages appears to be related to a loss of mGluR5-driven mechanisms of AD neuropathology in APPswe/PS1ΔE9 mice. Thus, it is clear that mGluR5 may not be the best candidate for therapeutic targeting in the late stages of AD and other targets should be considered.
The progression of AD-like neuropathology was evident in our mice as we detected higher levels of Aβ oligomers in vehicle-treated 15 month old compared to 12 month old APPswe/PS1ΔE9 mice. These observations were consistent with previous studies from our group and others reporting that the increase in the extent of Aβ deposition in APPswe/PS1ΔE9 mice was age-dependent and starts as early as 6 months of age and continues up to 17 months of age.11,15,20,21,37 While our study further supported the favorable outcomes of extended mGluR5 inhibition in mitigating AD-like neuropathology and cognitive deficits, it also indicated that the disease progression could also be associated with loss of mGluR5 as a disease modifying target in AD mice.
Aβ is a proteolytic cleavage of APP and is normally found in the brain in a soluble form at low levels. Pathological alterations in the synthesis and/or removal of Aβ increases its level and initiates aggregation that can accelerate neurodegeneration.38,39 In fact, this is the rationale for generating the double transgenic APPswe/PS1ΔE9 mouse line expressing a chimeric mouse/human APP (APP695 swe) and a mutant human presenilin 1 (PS1-ΔE9) resulting in Aβ deposition as early as 6 months of age.21,40 We have previously reported that the ability to induce autophagic clearance of the Aβ burden is a major disease-modifying mechanism that underlies beneficial outcomes of chronic treatment with mGluR5 NAMs in male AD mice.12,15 This is specifically relevant to AD since Aβ is known to induce the clustering and activation of mGluR5, processes that further contribute to neuropathology.8,41 A reduction in autophagy flux rates were also reported in multiple proteinopathies including AD.42−45 Therefore, if mGluR5 NAMs can maintain their ability to activate autophagy and remove Aβ oligomers after an extended treatment regimen, it would represent excellent strategy to slow the process of neurodegeneration and maintain long-term therapeutic efficacy.
We have previously demonstrated that mGluR5 inhibits autophagy via a ZBTB16-Cullin3-Roc1 E3-ubiquitin ligase pathway in male APPswe/PS1ΔE9 and 3xTg-AD mouse models and 12 week treatment with CTEP was able to relieve such inhibition.12,15,46 Similar to 12 week, 24 week treatment of male APPswe/PS1ΔE9 mice starting at 6 months of age with CTEP activated autophagy via the GSK3β/ZBTB16/ATG14 pathway. Interestingly, independent of treatment, we observed no alterations in the markers of the GSK3β/ZBTB16/ATG14 autophagy pathway in 15 month old APPswe/PS1ΔE9 mice. We also detected an upregulation in ZBTB16 in wild-type mice after 36 weeks of treatment that can be explained by either an age-dependent shift in mGluR5-mediated regulation of ZBTB16 expression or a compensatory upregulation in ZBTB16 expression as a consequence of extended mGluR5 inhibition. When these were taken together, we showed that while extended pharmacological inhibition of mGluR5 was capable of activating GSK3β/ZBTB16/ATG14-regulated autophagic pathway to remove Aβ oligomers in APPswe/PS1ΔE9 mice up to 12 months of age, the contribution of this pathway to neuropathology was limited in later disease stages significantly mitigating the therapeutic value of mGluR5 NAMs in AD pathology.
Activation of PI3K/mTOR signaling is another mechanism that allows mGluR5 to regulate autophagy, since mTOR is known to be a master regulator of autophagy.47−49 Canonical mTOR signaling is initiated following mGluR5-dependent activation of PI3K to directly activate Akt via phosphorylation that results in rapid phosphorylation of mTOR at S2448 and subsequently phosphorylation of the mTOR downstream effector, p70S6K1 at T389.25,28 Both mTOR-pS2448 and p70S6K1-pT389 are considered reliable biomarkers of the mTOR complex activity.28−31 We have also shown that in a HD mouse model, mGluR5 inhibition can activate autophagic clearance of mutant huntingtin aggregates by suppressing mTOR signaling.23 Here, we show that CTEP can normalize the mTOR signaling after a 24-week long treatment in APPswe/PS1ΔE9 mice. However, as observed for the ZBTB16 pathway, we did not detect any evidence of mGluR5-mediated mTOR signaling in 15 month old APPswe/PS1ΔE9 mice. These observations indicate that mGluR5-mediated activation of mTOR contributes to the suppression of autophagic clearance of the Aβ load and inhibiting mTOR can be another mechanism by which mGluR5 NAMs initiates autophagy in AD mice. Similar to that of the ZBTB16 pathway, mTOR-regulated autophagy does not seem to contribute to pathology in later stages of AD, hence the inability of mGluR5 NAM to mitigate Aβ pathology in older AD mice.
Microglia and the inflammatory mediators that it releases in response to Aβ oligomers are known to exacerbate neuroinflammation in AD.32,33,50,51 Astrocytes are also activated by Aβ oligomers resulting in glutamate release and inhibition of glutamate uptake from the synaptic cleft, thus contributing to excitotoxity and neuronal apoptosis in AD.52−55 We find that markers of microglia activation and astrogliosis, Iba1 and GFAP, respectively, are elevated in both 12 and 15 month old APPswe/PS1ΔE9 brains. This is consistent with previous findings showing that activated microglia and reactive astrocytes were detected around Aβ plaques as early as 6 months of age in this model.33,51 Treatment with CTEP reduced the number of Iba1- and GFAP-positive cells in APPswe/PS1ΔE9 mice up to 12 months of age, yet was ineffective in 15 month old animals. Thus, it is possible that the autophagic clearance of Aβ oligomers following treatment with CTEP in 12 month old APPswe/PS1ΔE9 mice reduces Aβ oligomer-activated neurogliosis resulting in reduced neuroinflammation and abolishes neurotoxicity caused by glutamate overspill from astrocytes. However, in later disease stages CTEP cannot activate autophagic removal of Aβ oligomers and thus is not capable of mitigating neuroinflammation that can further contribute to therapeutic inefficacy. It is also noteworthy that the effect of CTEP on glial activation in 12 month old APPswe/PS1ΔE9 mice was less robust than its effect on ZBTB16 and mTOR-regulated autophagy. More so, unlike all the autophagy markers that were not altered in 15 month old APPswe/PS1ΔE9 mice, neuroglia was still significantly activated in this age group. This suggests that it is likely that neuroglial activation is less dependent on mGluR5 and there are other pathophysiological mechanism(s) driving neuroglial activation that can not be targeted by mGluR5 NAMs.
One plausible explanation for such difference in mGluR5 NAM efficacy between 12 and 15 month old APPswe/PS1ΔE9 mice could be an age-dependent loss in mGluR5 expression that limits the usefulness of mGluR5 as a therapeutic target; however, we did not detect any significant change in the total expression of mGluR5 with age. These findings suggest that along the disease course there might be abolished contribution of pathological mGluR5 signaling to AD neuropathology in mice and therefore mGluR5 NAM treatment becomes incapable of modifying mGluR5 signaling or disease progression at advanced stages. It is important to acknowledge that this shift in the signaling mechanisms between both 12 and 15 month old mice could be a consequence of the extended treatment paradigm that may upregulate compensatory pathological mechanisms that are mGluR5-independent. It is also noteworthy that we reported a diminished contribution of pathological mGluR5 signaling in young females APPswe/PS1ΔE9 mice that was due to inability of Aβ oligomers to activate mGluR5.15 Thus, it will be imperative to test whether mGluR5 signaling is also regulated in a sex-specific manner in advanced stages of AD, as it will have major implications on drug discovery and interpretation of AD clinical trial results.
In summary, we find that extended mGluR5 antagonism has proven to be well-tolerated and effective in reducing cognitive impairment, Aβ-related pathology and neuroinflammation associated with AD up to a certain disease stage in AD mice. As the disease progresses, mGluR5 contribution to AD pathology is abolished that makes mGluR5 NAMs less therapeutically useful in later stages. Thus, we provide the first evidence that the pathophysiological mechanism(s) underlying AD evolve as the disease progresses and will require further optimization of treatment strategies to ensure maximal therapeutic benefits. Although our study was focused on the age-dependent changes in mGluR5 signaling, we anticipate that other pathophysiological mechanism(s) known to contribute to AD might follow a similar trend that may have major implications on drug discovery and AD clinical trials.
Materials and Methods
Reagents
CTEP was purchased from Axon Medchem (1972). Horseradish peroxidase (HRP)-conjugated antirabbit IgG secondary antibody was from Bio-Rad. HRP-conjugated antimouse secondary and rabbit anti-GSK3β (pS9, 9323), -Akt (pS437, 4060), -mTOR (pS2448, 109268), and -mTOR (2972) and mouse anti-Akt (9272), -GSK3β (9832) antibodies were from Cell Signaling Technology. Rabbit anti- ATG14 (PD026) were from MBL life science. Mouse anti-P62 (56416) and rabbit anti-ZBTB16 (39354), -APP (2072), -GFAP (7260), -vinculin (129002), and -Iba1 (178847) antibodies were from Abcam. Rabbit anti-mGluR5 (AB5675), -β-tubulin (T2200), and mouse anti-P70S6K1 (pT389, 07018I) antibodies were from Sigma-Aldrich. Rabbit anti-β-Amyloid (715800) was from Thermo Scientific. Reagents used for Western blotting were purchased from Bio-Rad, and all other biochemical reagents were from Sigma-Aldrich.
Animals
All animal experimental protocols were approved by the University of Ottawa Institutional Animal Care Committee and were in accordance with the Canadian Council of Animal Care guidelines. STOCK B6C3-Tg (APPswe/PSEN1ΔE9)85Dbo/J mice that carry the human APP with Swedish mutation and the DeltaE9 mutation of the human presenilin 1 gene21 were purchased from Jackson Laboratory (Bar Harbor, ME). Offspring were genotyped using PCR with primers specific for the APP sequence. Animals were bred to establish littermate controlled male wild-type (WT), group-housed in cages of 2–4 animals, received food and water ad libitum, and maintained on a 12 h light/12 h dark cycle at 24 °C. Groups of 48 male wild-type and APPswe/PSEN1ΔE9 mice were aged to 6 months of age, and 12 mice from each group were treated every 48 h with either vehicle (DMSO in chocolate pudding) or CTEP (2 mg/kg in chocolate pudding) for 24 or 36 weeks11,22 based on weekly body weights. The dose was administered in a plastic dish (1.62 cm × 1.62 cm) and the dish was only removed after the experimenter verified that the animal had consumed the full dose. Cognitive and locomotor functions of all animals were assessed prior to and following 24 and 36 weeks of drug treatment. At the end of either 24- or 36-week treatment, mice were sacrificed by exsanguination and brains were collected and randomized for biochemical determinations and immunohistochemical examinations.
Immunoblotting
Freshly dissected brain hemispheres were lysed in ice-cold lysis buffer (50 mM Tris, pH 8.0, 150 mM NaCl, and 1% Triton X-100) containing a protease inhibitors cocktail (100 μM AEBSF, 2 μM leupeptin, 80 nM aprotinin, 5 μM Bestatin, 1.5 μM E-64, and 1 μM pepstatin A) and phosphatase inhibitors (10 mM NaF and 500 μM Na3VO4) and centrifuged twice for 10 min each at 15 000 rpm and 4 °C. Total protein levels were quantified in the supernatant using Bradford Protein Assay (Biorad). Homogenates were diluted in a mix of lysis buffer and β-mercaptoethanol containing 3× loading buffer and boiled for 10 min at 90 °C. Aliquots containing 35 μg total proteins were resolved by electrophoresis on a 7.5% SDS-polyacrylamide gel (SDS-PAGE) and transferred onto nitrocellulose membranes (Bio-Rad). Blots were blocked in Tris-buffered saline, pH 7.6 containing 0.05% of Tween 20 (TBST) and 6% nonfat dry milk for 2 h at room temperature and then incubated overnight at 4 °C with primary antibodies diluted 1:1000 in TBST containing 1% nonfat dry milk. Immunodetection was performed by incubating with the proper secondary antibodies (antirabbit/mouse) diluted 1:5000 in TBST containing 1% of nonfat dry milk for 1 h. Bands were detected and quantified using SuperSignal West Pico PLUS Chemiluminescent Substrate (Thermo Scientific).
Behavioral Analysis
Animals were habituated in the testing room for 30 min prior to testing, and all behavioral testing was blindly performed during the animal’s light cycle.
Novel Object Recognition
Mice were placed in the empty box measuring 45 × 45 × 45 cm3 for 5 min and 5 min later, two identical objects were placed in the box 5 cm from the edges and 5 cm apart. Mice were returned to the box for 5 min, and allowed to explore. Time spent exploring each object was captured using a camera and data were transferred to a computer in a separate room for analysis using Noldus Ethovision 10 software. Mice were considered to be exploring an object if their snout was within 1 cm of the object. The experiment was repeated 1 day after first exposure with one the objects replaced with a novel object. Recognition index was calculated as follows: time spent exploring the familiar object or the novel object over the total time spent exploring both objects multiplied by 100, and was used to measure recognition memory (TA or TB/(TA + TB))×100, where T represents time, A represents the familiar object, and B represents the novel object.
Morris Water Maze (MWM) and Reversal Morris Water Maze (RMWM)
The maze is a white opaque plastic pool (120 cm in diameter), filled with water and maintained at 25 °C. An escape platform (10 cm diameter) was placed 25 cm from the perimeter, hidden one cm beneath the water surface. Visual cues were placed on the walls in the room of the maze as spatial references. Mice were trained for 4 days (four trials per day, 60 s each and 15 min apart) to find the platform at a fixed position from a random start point of the four equally spaced points around the pool. If the mice fail to find the platform within 60 s, they were guided to the platform by the experimenter. Escape latency and swim speed were measured using Ethovision 10 automated video tracking software from Noldus. On day 5, the probe trial (a single trial of 60 s) was performed by removing the platform and allowing the mice to swim freely in the pool and recording the time spent in the target quadrant. RMWM task was initiated 24 h after completion of MWM using the same paradigm as MWM, with 3 days acquisition and probe trial on the fourth day; however, the platform was relocated to a novel position.
Determination of Aβ Oligomer by Sandwich ELISA
ELISA was performed as described previously.11,15 Quantification was performed using a sandwich ELISA kit according to manufacturer’s instructions, KHB3491 for oligomeric Aβ (Thermo Scientific). Briefly, one hemisphere from each mouse was lysed and centrifuged at 4 °C either at 100 000g for 1 h. The supernatant was then diluted with kit buffer 1:10 before performing the ELISA, which was carried out in triplicate and measured as detailed in the manufacturer’s protocol. Protein was quantified using the Bradford protein assay. The final Aβ values were determined after normalization to total protein levels.
β-Amyloid, Iba1, and GFAP Immunohistochemistry
Staining was performed as described previously.11,15 Briefly, brains were coronally sectioned through the cortex and hippocampus and on 40 μm free-floating sections and every sixth section was stained per mouse using a peroxidase-based immunostaining protocol. This yielded approximately eight sections per mouse. Sections were incubated overnight in primary antibody for Aβ (1:200), Iba1 (1:100), or GFAP (1:200) at 4 °C, washed, incubated in biotinylated antibody (biotinylated antirabbit, 1:400, Vector Elite ABC kit mouse) for 90 min at 4 °C, and then incubated in an avidin–biotin enzyme reagent for 90 min at 4 °C (Vector Elite ABC kit mouse, PK-6102, Vector Laboratories). Immunostaining was visualized using a chromogen (Vector SG substrate, Vector Laboratories). Sections were mounted on slides and visualized with a Zeiss AxioObserver epifluorescent microscope with a Zeiss 20× lens, using representative 900 μm2 areas of cortex and hippocampus. Five ROIs were analyzed in the cortex and 2 ROIs in the hippocampus. This number of ROIs prevents the selection of only densely stained regions. Experimenters were blinded to drugging, and analysis and images were analyzed using the cell counter tool in ImageJ (NIH, USA).
Statistical Analysis
Means ± SD or SEM values shown for each of the independent experiments are shown in the various figure legends. GraphPad Prism software was used to analyze data for statistical significance. Data normality tested using the Anderson-Darling test and statistical significance was determined by either a series of 2 (strain) × 2 (drug treatment) ANOVAs followed by Fisher’s LSD comparisons, Student’s t test or Kruskal–Wallis test was applied as appropriate for the significant main effects or interactions.
Acknowledgments
S.S.G.F. is a Tier I Canada Research Chair in Brain and Mind. K.S.A. is a Lecturer in the Department of Pharmacology & Toxicology, Faculty of Pharmacy, Alexandria University, Egypt. Thanks to Shaunessy Hutchinson for breeding the colony and to the Behavior and Physiology core at the University of Ottawa.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsptsci.0c00013.
Uncropped blots corresponding to Figures 3D, 4A–D, and 5A–C, and uncropped blots with quantification of mGluR5 total expression in brain lysates from wildtype and APPswe/PS1ΔE9 mice after either a 24 week or 36 week treatment with vehicle or CTEP (PDF)
Author Contributions
# These authors contributed equally to this work. K.S.A., A.H., and S.S.G.F. were responsible for the conception and design of all experiments. K.S.A., A.H., and A.A. performed the experiments and analyzed the data. K.S.A., A.H., and S.S.G.F. wrote the manuscript. S.S.G.F. supervised the study.
This study was supported by grants from Canadian Institutes for Health Research (CIHR) PJT-148656 and PJT-165967 and Krembil Foundation to S.S.G.F., and Clinician Postdoctoral Fellowship from the Alberta Innovates Health Solutions (AIHS) and CIHR to K.S.A.
The authors declare no competing financial interest.
Supplementary Material
References
- Alzheimer’s Association(2019) 2019 Alzheimer’s disease facts and figures. Alzheimer's Dementia 15, 321–387. 10.1016/j.jalz.2019.01.010 [DOI] [PubMed] [Google Scholar]
- Cahill S. (2020) WHO’s global action plan on the public health response to dementia: some challenges and opportunities. Aging Ment. Health 24, 1–3. 10.1080/13607863.2018.1544213. [DOI] [PubMed] [Google Scholar]
- Casey D. A.; Antimisiaris D.; O’Brien J. (2010) Drugs for Alzheimer’s disease: are they effective?. Pharm. Therap. 35, 208–11. [PMC free article] [PubMed] [Google Scholar]
- Lanctôt K. L.; Rajaram R. D.; Herrmann N. (2009) Therapy for Alzheimer’s Disease: How Effective are Current Treatments?. Ther. Adv. Neurol. Disord. 2, 163–80. 10.1177/1756285609102724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sengupta U.; Nilson A. N.; Kayed R. (2016) The Role of Amyloid-β Oligomers in Toxicity, Propagation, and Immunotherapy. EBioMedicine 6, 42–49. 10.1016/j.ebiom.2016.03.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuperstein I.; Broersen K.; Benilova I.; Rozenski J.; Jonckheere W.; Debulpaep M.; Vandersteen A.; Segers-Nolten I.; Van Der Werf K.; Subramaniam V.; Braeken D.; Callewaert G.; Bartic C.; D’Hooge R.; Martins I. C.; Rousseau F.; Schymkowitz J.; De Strooper B. (2010) Neurotoxicity of Alzheimer’s disease Aβ peptides is induced by small changes in the Aβ42 to Aβ40 ratio. EMBO J. 29, 3408–20. 10.1038/emboj.2010.211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamilton A.; Zamponi G. W.; Ferguson S. S. G. (2015) Glutamate receptors function as scaffolds for the regulation of β-amyloid and cellular prion protein signaling complexes. Mol. Brain 8, 18. 10.1186/s13041-015-0107-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Um J. W.; Kaufman A. C.; Kostylev M.; Heiss J. K.; Stagi M.; Takahashi H.; Kerrisk M. E.; Vortmeyer A.; Wisniewski T.; Koleske A. J.; Gunther E. C.; Nygaard H. B.; Strittmatter S. M. (2013) Metabotropic Glutamate Receptor 5 Is a Coreceptor for Alzheimer Aβ Oligomer Bound to Cellular Prion Protein. Neuron 79, 887–902. 10.1016/j.neuron.2013.06.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Renner M.; Lacor P. N.; Velasco P. T.; Xu J.; Contractor A.; Klein W. L.; Triller A. (2010) Deleterious effects of amyloid beta oligomers acting as an extracellular scaffold for mGluR5. Neuron 66, 739–54. 10.1016/j.neuron.2010.04.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haas L. T.; Salazar S. V.; Kostylev M. A.; Um J. W.; Kaufman A. C.; Strittmatter S. M. (2016) Metabotropic glutamate receptor 5 couples cellular prion protein to intracellular signalling in Alzheimer’s disease. Brain 139, 526–46. 10.1093/brain/awv356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamilton A.; Esseltine J. L.; DeVries R. A.; Cregan S. P.; Ferguson S. S. G. (2014) Metabotropic glutamate receptor 5 knockout reduces cognitive impairment and pathogenesis in a mouse model of Alzheimer’s disease. Mol. Brain 7, 40. 10.1186/1756-6606-7-40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamilton A.; Vasefi M.; Vander Tuin C.; Mcquaid R. J.; Anisman H.; Ferguson S. S. G. (2016) Chronic Pharmacological mGluR5 Inhibition Prevents Cognitive Impairment and Reduces Pathogenesis in an Alzheimer Disease Mouse Model. Cell Rep. 15, 1–7. 10.1016/j.celrep.2016.04.077. [DOI] [PubMed] [Google Scholar]
- Abd-Elrahman K. S.; Hamilton A.; Vasefi M.; Ferguson S. S. G. (2018) Autophagy is increased following either pharmacological or genetic silencing of mGluR5 signaling in Alzheimer’s disease mouse models. Mol. Brain 19. 10.1186/s13041-018-0364-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haas L. T.; Salazar S. V.; Smith L. M.; Zhao H. R.; Cox T. O.; Herber C. S.; Degnan A. P.; Balakrishnan A.; Macor J. E.; Albright C. F.; Strittmatter S. M. (2017) Silent Allosteric Modulation of mGluR5Maintains Glutamate Signaling while Rescuing Alzheimer’s Mouse Phenotypes. Cell Rep. 20, 76–88. 10.1016/j.celrep.2017.06.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bellozi P. M. Q.; Gomes G. F.; da Silva M. C. M.; Lima I. V. de A.; Batista C. R. Á.; Carneiro Junior W. de O.; Dória J. G.; Vieira É. L. M.; Vieira R. P.; de Freitas R. P.; Ferreira C. N.; Candelario-Jalil E.; Wyss-Coray T.; Ribeiro F. M.; de Oliveira A. C. P. (2019) A positive allosteric modulator of mGluR5 promotes neuroprotective effects in mouse models of Alzheimer’s disease. Neuropharmacology 160, 107785. 10.1016/j.neuropharm.2019.10778. [DOI] [PubMed] [Google Scholar]
- Abd-Elrahman K. S., Hamilton A., Souza J. M. de, Albaker A., Ribeiro F. M., and Ferguson S. S. G. (2019) Sex-specific pathophysiological mGluR5-dependent Aβ oligomer signaling in Alzheimer mice. https://www.biorxiv.org/content/10.1101/803262v2.full.
- Bullock R.; Bergman H.; Touchon J.; Gambina G.; He Y.; Nagel J.; Lane R. (2006) Effect of age on response to rivastigmine or donepezil in patients with Alzheimer’s disease. Curr. Med. Res. Opin. 22, 483–494. 10.1185/030079906X89685. [DOI] [PubMed] [Google Scholar]
- Wilcock G.; Howe I.; Coles H.; Lilienfeld S.; Truyen L.; Zhu Y.; Bullock R.; Kershaw P. (2003) GAL-GBR-2 Study Group., A long-term comparison of galantamine and donepezil in the treatment of Alzheimer’s disease. Drugs Aging 20, 777–89. 10.2165/00002512-200320100-00006. [DOI] [PubMed] [Google Scholar]
- Rogers S. L.; Doody R. S.; Pratt R. D.; Ieni J. R. (2000) Long-term efficacy and safety of donepezil in the treatment of Alzheimer’s disease: Final analysis of a US multicentre open-label study. Eur. Neuropsychopharmacol. 10, 195–203. 10.1016/S0924-977X(00)00067-5. [DOI] [PubMed] [Google Scholar]
- Lindemann L.; Jaeschke G.; Michalon A.; Vieira E.; Honer M.; Spooren W.; Porter R.; Hartung T.; Kolczewski S.; Büttelmann B.; Flament C.; Diener C.; Fischer C.; Gatti S.; Prinssen E. P.; Parrott N.; Hoffmann G.; Wettstein J. G. (2011) CTEP: a novel, potent, long-acting, and orally bioavailable metabotropic glutamate receptor 5 inhibitor. J. Pharmacol. Exp. Ther. 339, 474–86. 10.1124/jpet.111.185660. [DOI] [PubMed] [Google Scholar]
- Fu L.; Sun Y.; Guo Y.; Yu B.; Zhang H.; Wu J.; Yu X.; Wu H.; Kong W. (2018) Progressive Spatial Memory Impairment, Brain Amyloid Deposition and Changes in Serum Amyloid Levels as a Function of Age in APPswe/PS1dE9Mice. Curr. Alzheimer Res. 15, 1053–1061. 10.2174/1567205015666180709112327. [DOI] [PubMed] [Google Scholar]
- Jankowsky J. L.; Fadale D. J.; Anderson J.; Xu G. M.; Gonzales V.; Jenkins N. A.; Copeland N. G.; Lee M. K.; Younkin L. H.; Wagner S. L.; Younkin S. G.; Borchelt D. R. (2004) Mutant presenilins specifically elevate the levels of the 42 residue beta-amyloid peptide in vivo: evidence for augmentation of a 42-specific gamma secretase. Hum. Mol. Genet. 13, 159–70. 10.1093/hmg/ddh019. [DOI] [PubMed] [Google Scholar]
- Abd-Elrahman K. S.; Hamilton A.; Hutchinson S. R.; Liu F.; Russell R. C.; Ferguson S. S. G. (2017) mGluR5 antagonism increases autophagy and prevents disease progression in the zQ175 mouse model of Huntington’s disease. Sci. Signaling 10, eaan6387 10.1126/scisignal.aan6387. [DOI] [PubMed] [Google Scholar]
- Abd-Elrahman K. S.; Ferguson S. S. G. (2019) Modulation of mTOR and CREB pathways following mGluR5 blockade contribute to improved Huntington’s pathology in zQ175 mice. Mol. Brain 12, 35. 10.1186/s13041-019-0456-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banerjee S.; Gianino S. M.; Gao F.; Christians U.; Gutmann D. H. (2011) Interpreting Mammalian Target of Rapamycin and Cell Growth Inhibition in a Genetically Engineered Mouse Model of Nf1-Deficient Astrocytes. Mol. Cancer Ther. 10, 279–291. 10.1158/1535-7163.MCT-10-0654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Porta C.; Paglino C.; Mosca A. (2014) Targeting PI3K/Akt/mTOR Signaling in Cancer. Front. Oncol. 4, 64. 10.3389/fonc.2014.00064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hou L.; Klann E. (2004) Activation of the phosphoinositide 3-kinase-Akt-mammalian target of rapamycin signaling pathway is required for metabotropic glutamate receptor-dependent long-term depression. J. Neurosci. 24, 6352–61. 10.1523/JNEUROSCI.0995-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Munson M. J.; Ganley I. G. (2015) MTOR, PIK3C3, and autophagy: Signaling the beginning from the end. Autophagy 11, 2375–2376. 10.1080/15548627.2015.1106668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jung C. H.; Ro S.-H.; Cao J.; Otto N. M.; Kim D.-H. (2010) mTOR regulation of autophagy. FEBS Lett. 584, 1287–95. 10.1016/j.febslet.2010.01.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ikenoue T.; Hong S.; Inoki K. (2009) Chapter 11 Monitoring Mammalian Target of Rapamycin (mTOR) Activity. Methods Enzymol. 452, 165–180. 10.1016/S0076-6879(08)03611-2. [DOI] [PubMed] [Google Scholar]
- Kim J.; Kundu M.; Viollet B.; Guan K.-L. (2011) AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1. Nat. Cell Biol. 13, 132–141. 10.1038/ncb2152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perluigi M.; Di Domenico F.; Butterfield D. A. (2015) mTOR signaling in aging and neurodegeneration: At the crossroad between metabolism dysfunction and impairment of autophagy. Neurobiol. Dis. 84, 39–49. 10.1016/j.nbd.2015.03.014. [DOI] [PubMed] [Google Scholar]
- Chun H.; Marriott I.; Lee C. J.; Cho H. (2018) Elucidating the Interactive Roles of Glia in Alzheimer’s Disease Using Established and Newly Developed Experimental Models. Front. Neurol. 9, 797. 10.3389/fneur.2018.00797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruan L.; Kang Z.; Pei G.; Le Y. (2009) Amyloid deposition and inflammation in APPswe/PS1dE9 mouse model of Alzheimer’s disease. Curr. Alzheimer Res. 6, 531–40. 10.2174/156720509790147070. [DOI] [PubMed] [Google Scholar]
- Kamphuis W.; Orre M.; Kooijman L.; Dahmen M.; Hol E. M. (2012) Differential cell proliferation in the cortex of the appsweps1de9 alzheimer’s disease mouse model. Glia 60, 615–629. 10.1002/glia.22295. [DOI] [PubMed] [Google Scholar]
- Luo R.; Su L.-Y.; Li G.; Yang J.; Liu Q.; Yang L.-X.; Zhang D.-F.; Zhou H.; Xu M.; Fan Y.; Li J.; Yao Y.-G. (2020) Activation of PPARA-mediated autophagy reduces Alzheimer disease-like pathology and cognitive decline in a murine model. Autophagy 16, 1–18. 10.1080/15548627.2019.1596488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rossor M. N.; Iversen L. L.; Reynolds G. P.; Mountjoy C. Q.; Roth M. (1984) Neurochemical characteristics of early and late onset types of Alzheimer’s disease. Br. Med. J. (Clin. Res. Ed). 288, 961–4. 10.1136/bmj.288.6422.961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perez S. E.; Lazarov O.; Koprich J. B.; Chen E. Y.; Rodriguez-Menendez V.; Lipton J. W.; Sisodia S. S.; Mufson E. J. (2005) Nigrostriatal dysfunction in familial Alzheimer’s disease-linked APPswe/PS1ΔE9 transgenic mice. J. Neurosci. 25, 10220–10229. 10.1523/JNEUROSCI.2773-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamenetz F.; Tomita T.; Hsieh H.; Seabrook G.; Borchelt D.; Iwatsubo T.; Sisodia S.; Malinow R. (2003) APP processing and synaptic function. Neuron 37, 925–37. 10.1016/S0896-6273(03)00124-7. [DOI] [PubMed] [Google Scholar]
- Hunter S.; Brayne C. (2018) Understanding the roles of mutations in the amyloid precursor protein in Alzheimer disease. Mol. Psychiatry 23, 81. 10.1038/mp.2017.218. [DOI] [PubMed] [Google Scholar]
- Garcia-Alloza M.; Robbins E. M.; Zhang-Nunes S. X.; Purcell S. M.; Betensky R. A.; Raju S.; Prada C.; Greenberg S. M.; Bacskai B. J.; Frosch M. P. (2006) Characterization of amyloid deposition in the APPswe/PS1dE9 mouse model of Alzheimer disease. Neurobiol. Dis. 24, 516–24. 10.1016/j.nbd.2006.08.017. [DOI] [PubMed] [Google Scholar]
- Sarkar S.; Rubinsztein D. C. (2008) Small molecule enhancers of autophagy for neurodegenerative diseases. Mol. BioSyst. 4, 895–901. 10.1039/b804606a. [DOI] [PubMed] [Google Scholar]
- Rubinsztein D. C.; DiFiglia M.; Heintz N.; Nixon R. A.; Qin Z.-H.; Ravikumar B.; Stefanis L.; Tolkovsky A. (2005) Autophagy and its possible roles in nervous system diseases, damage and repair. Autophagy 1, 11–22. 10.4161/auto.1.1.1513. [DOI] [PubMed] [Google Scholar]
- Nah J.; Yuan J.; Jung Y.-K. (2015) Autophagy in neurodegenerative diseases: from mechanism to therapeutic approach. Mol. Cells 38, 381–9. 10.14348/molcells.2015.0034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nixon R. A. (2013) The role of autophagy in neurodegenerative disease. Nat. Med. 19, 983–997. 10.1038/nm.3232. [DOI] [PubMed] [Google Scholar]
- Zhang T.; Dong K.; Liang W.; Xu D.; Xia H.; Geng J.; Najafov A.; Liu M.; Li Y.; Han X.; Xiao J.; Jin Z.; Peng T.; Gao Y.; Cai Y.; Qi C.; Zhang Q.; Sun A.; Lipinski M.; Zhu H.; Xiong Y.; Pandolfi P. P.; Li H.; Yu Q.; Yuan J. (2015) G-protein-coupled receptors regulate autophagy by ZBTB16-mediated ubiquitination and proteasomal degradation of Atg14L. eLife 4, e06734 10.7554/eLife.06734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim J.; Kundu M.; Viollet B.; Guan K.-L. (2011) AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1. Nat. Cell Biol. 13, 132–141. 10.1038/ncb2152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu Z.; Yang C.; Iyaswamy A.; Krishnamoorthi S.; Sreenivasmurthy S. G.; Liu J.; Wang Z.; Tong B. C.-K.; Song J.; Lu J.; Cheung K.-H.; Li M. (2019) Balancing mTOR Signaling and Autophagy in the Treatment of Parkinson’s Disease. Int. J. Mol. Sci. 20, 728. 10.3390/ijms20030728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Puente C.; Hendrickson R. C.; Jiang X. (2016) Nutrient-regulated Phosphorylation of ATG13 Inhibits Starvation-induced Autophagy. J. Biol. Chem. 291, 6026–35. 10.1074/jbc.M115.689646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGeer P. L.; McGeer E. G. (1995) The inflammatory response system of brain: implications for therapy of Alzheimer and other neurodegenerative diseases. Brain Res. Rev. 21, 195–218. 10.1016/0165-0173(95)00011-9. [DOI] [PubMed] [Google Scholar]
- Kamphuis W.; Mamber C.; Moeton M.; Kooijman L.; Sluijs J. A.; Jansen A. H. P.; Verveer M.; de Groot L. R.; Smith V. D.; Rangarajan S.; Rodríguez J. J.; Orre M.; Hol E. M.; Ikezu T. (2012) GFAP isoforms in adult mouse brain with a focus on neurogenic astrocytes and reactive astrogliosis in mouse models of Alzheimer disease. PLoS One 7, 42823. 10.1371/journal.pone.0042823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abramov A. Y. (2004) -Amyloid Peptides Induce Mitochondrial Dysfunction and Oxidative Stress in Astrocytes and Death of Neurons through Activation of NADPH Oxidase. J. Neurosci. 24, 565–575. 10.1523/JNEUROSCI.4042-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodríguez J. J.; Olabarria M.; Chvatal A.; Verkhratsky A. (2009) Astroglia in dementia and Alzheimer’s disease. Cell Death Differ. 16, 378–85. 10.1038/cdd.2008.172. [DOI] [PubMed] [Google Scholar]
- Matos M.; Augusto E.; Oliveira C. R.; Agostinho P. (2008) Amyloid-beta peptide decreases glutamate uptake in cultured astrocytes: involvement of oxidative stress and mitogen-activated protein kinase cascades. Neuroscience 156, 898–910. 10.1016/j.neuroscience.2008.08.022. [DOI] [PubMed] [Google Scholar]
- Pascual O.; Ben Achour S.; Rostaing P.; Triller A.; Bessis A. (2012) Microglia activation triggers astrocyte-mediated modulation of excitatory neurotransmission. Proc. Natl. Acad. Sci. U. S. A. 109, E197–205. 10.1073/pnas.1111098109. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.