

SUMMARY
Gram-negative bacteria release outer membrane vesicles into the external milieu to deliver effector molecules that alter the host and facilitate virulence. Vesicle formation is driven by phospholipid accumulation in the outer membrane and regulated by the phospholipid transporter VacJ/Yrb. We use the facultative human pathogen Vibrio cholerae to show that VacJ/Yrb is silenced early during mammalian infection, which stimulates vesiculation that expedites bacterial surface exchange and adaptation to the host environment. Hypervesiculating strains rapidly alter their bacterial membrane composition and exhibit enhanced intestinal colonization fitness. This adaptation is exemplified by faster accumulation of glycine-modified LPS and depletion of outer membrane porin OmpT, which confer resistance to host-derived antimicrobial peptides and bile, respectively. The competitive advantage of hypervesiculation is lost upon pre-adaptation to bile and antimicrobial peptides, indicating the importance of these adaptive processes. Thus, bacteria use outer membrane vesiculation to exchange cell surface components, thereby increasing survival during mammalian infection.
Keywords: OMV, OmpU, OmpT, porin, lipid A, glycination, Alm pathway, Mla pathway, bile, antimicrobial peptides
Graphical Abstract
eTOC Blurb
Upon infection, Vibrio cholerae must alter its surface profile to evade host defenses and adapt to the gastrointestinal environment. Zingl et al. show that increased release of outer membrane vesicles upon host entry allows the bacteria to rapidly modify its cell surface, thus increasing in vivo adaptation and colonization fitness.
INTRODUCTION
It is widely accepted that all domains of life produce membrane vesicles. Extracellular vesicles released from the outer membrane (OM) of Gram-negative bacteria, generally referred to as outer membrane vesicles (OMVs), belong to the best-studied vesicles (Toyofuku et al., 2019). These spherical, non-living facsimiles of the bacterial donor cell have been mainly characterized as delivery vehicles for effector molecules, such as quorum sensing signals, nucleic acids, degradative enzymes, inflammatory agents or toxins (Jan, 2017; Schwechheimer and Kuehn, 2015). Consequently, OMVs have been attributed a role in intra- and inter-species communication, horizontal gene transfer, nutrient acquisition, immunomodulation, and virulence (Kulp and Kuehn, 2010).
Recently, our group identified a conserved mechanism for OMV release based on phospholipid accumulation in the OM due to inactivation or transcriptional silencing of the VacJ/Yrb ABC transporter, also known as the Mla system [Fig. 1A; (Roier et al., 2016)]. This highly conserved trafficking system of Gram-negative bacteria was originally reported to shuttle phospholipids from the outer to the inner membrane to maintain the lipid asymmetry of the OM (Malinverni and Silhavy, 2009). In contrast to this view, recent data suggests that the VacJ/Yrb transport system can also export phospholipids from the inner membrane (Hughes et al., 2019; Kamischke et al., 2019). While mechanistic details remain to be elucidated, it is evident that inactivation or downregulation of the VacJ/Yrb transporter results in elevated OMV levels (Roier et al., 2016). Importantly, iron depletion in several Gram-negative bacteria (i.e. Escherichia coli, Vibrio cholerae and Haemophilus influenzae) triggers a ferric uptake regulator (Fur)-dependent repression of the VacJ/Yrb transporter, leading to increased OMV release (Roier et al., 2016). Iron limitation is a common stressor for bacterial pathogens during host colonization, therefore, we predicted that transcriptional silencing of the vacJ and yrbF-B genes and the resulting boost in vesiculation should occur upon host entry. In support of this model, H. influenzae was shown to lower expression levels of the VacJ/Yrb transporter during nasopharyngeal colonization (Roier et al., 2016).
Figure 1. Silencing of yrbF-B occurs upon host entry and colonization fitness varies inversely with transporter activity.
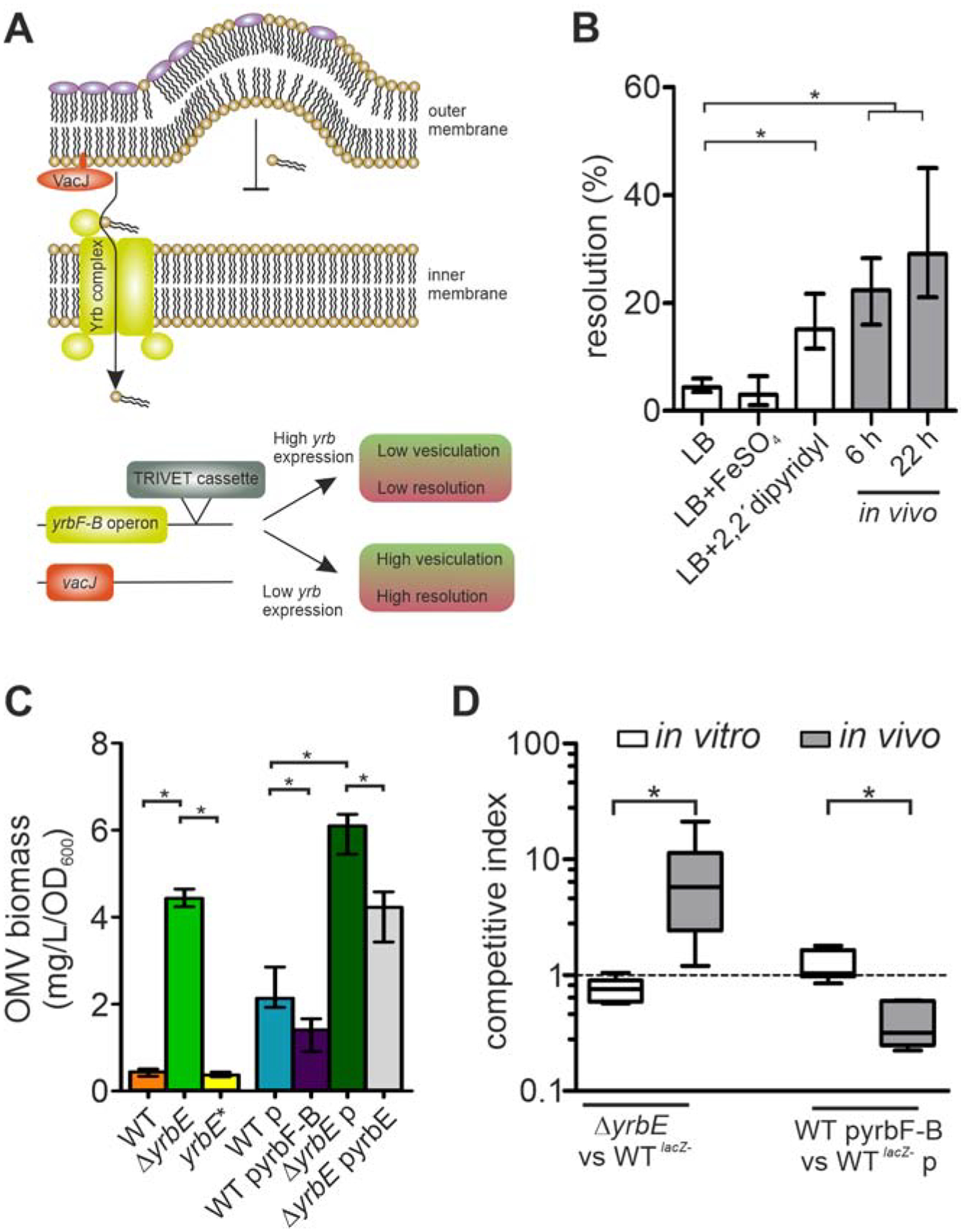
(A) Schematic overview of the phospholipid transporter encoded by the vacJ and yrbFEDCB (yrbF-B operon) genes. The VacJ/Yrb transporter (also known as Mla system) shuttles phospholipids from the outer membrane (OM) back to the inner membrane. High expression is associated with low vesiculation, while low expression or inactivation of the transporter results in phospholipid accumulation in the OM outer leaflet promoting vesiculation. Insertion of the TRIVET reporter cassette downstream of the yrb-locus allows monitoring of its transcriptional activity using excision of an antibiotic resistance cassette (resolution) as a readout.
(B) Transcriptional regulation of the yrbF-B operon was analyzed by the recombination-based in vivo reporter system TRIVET, which results in increased reporter resolution upon transcriptional repression of the yrbF-B operon (Cakar et al., 2018). Shown are resolution frequencies of strain Vc_res1_TRIVET yrbF::tpc grown for 22 h in regular LB, LB with high-(FeSO4) or low-(2,2’-dipyridyl) iron concentrations, as well as in vivo 6 and 22 h post-infection. Shown is median ± interquartile range (IQR) (n = 6; *P < 0.05). See also Figure S1.
(C) OMV quantification (Bradford) for WT, ΔyrbE, yrbE*, WT with empty vector (p), WT with yrbF-B overexpression plasmid (pyrbF-B), ΔyrbE with empty vector (p) and ΔyrbE with expression plasmid (pyrbE) after 8 h cultivation. Shown is median ± IQR (*P < 0.05) with the following number of biological replicates: n = 9 for WT and WT p; n = 6 for ΔyrbE, yrbE*, ΔyrbE p and ΔyrbE pyrbE; n = 8 for WT pyrbF-B.
(D) Competition indices of in vitro (LB) and in vivo (murine model) assays are shown as boxplot with whiskers (*P < 0.05) with the following number of biological replicates: n = 8 for all in vitro competitions, n = 10 for the in vivo competitions of WTlacZ− and ΔyrbE as well as n = 6 for the in vivo competitions of WTlacZ− p and WT pyrbF-B. Strains used for the competition are indicated on the x-axis.
Benefits driving bacteria to alter vesiculation in vivo remain to be elucidated. In the case of H. influenzae, higher OMV levels correlated with increased serum resistance (Roier et al., 2016). Thus, increased vesiculation might facilitate bacterial proliferation in the nasopharynx.
Given that bacteria discharge components of the periplasm and OM into the extracellular milieu via OMVs, increased vesiculation could also represent a previously overlooked adaptation to modulate their cell surface composition. This strategy could be quite valuable as bacteria are frequently challenged to adapt to diverse conditions with different requirements along their lifecycle (Conner et al., 2016; O′Connor and McClean, 2017). The need to change surface components upon host entry, e.g. capsule, flagella, fimbriae, pili, LPS modifications, metabolite uptake systems and porins, is especially apparent for facultative bacterial pathogens (King and Roberts, 2016; Phillips et al., 2019).
Along its lifecycle, the facultative human-pathogen V. cholerae transits between the aquatic reservoir and the human intestinal tract. To assess the impact of vesiculation on surface modulation specifically upon host entry we used V. cholerae as a model organism and focused on the initial stage of infection. Once ingested, V. cholerae modulates its surface profile to adapt to antimicrobial factors present in the gut. For example, the expression of two abundant OM porins OmpU and OmpT are inversely regulated upon host entry. This OmpU/T switch is directly controlled by the virulence regulator ToxR and is decisive for the bacterium to achieve bile resistance in vivo (Provenzano and Klose, 2000). Unlike OmpT, OmpU is an anion-selective porin that restricts the passage of negatively charged compounds (Simonet et al., 2003). Thus, depletion of OmpT in the OM makes V. cholerae less vulnerable to bile salts. Concordantly, upon host entry V. cholerae activates transcription of ompU and represses transcription of ompT to achieve full colonization fitness (Provenzano and Klose, 2000). Another surface modulation of V. cholerae was described for this transitory step, wherein the addition of (di)glycine residues to the lipid A anchor of lipopolysaccharide (LPS) conferred resistance to cationic antimicrobial peptides (Hankins et al., 2012). Importantly, both V. cholerae surface modulations can be reconstituted in vitro by variations in culture conditions. Reconstitution of these processes thus allows temporal monitoring of the surface exchange in strains with differential vesiculation activity.
In this study we used three scenarios - host entry, exposure to host defense mediators [(bile and polymyxin B (PMB)] as well as medium-induced activation of ToxR and/ or LPS modification - to model infection-relevant life cycle transitions for V. cholerae. These models were applied to monitor bacterial cell surface modulation under conditions where transporter activity and therefore vesiculation could be controlled. We show that V. cholerae silences the expression of the VacJ/Yrb transporter inside the host and reduced transporter activity facilitates colonization fitness in the murine infection model. Our findings demonstrate that upregulated vesiculation aids adaptation to both the cationic antimicrobial peptide PMB, via fast accumulation of glycine-modified lipid A, and bile salts, via efficient removal of OmpT. Thus, this study defines a physiological role for OMV production during host colonization where cell surface variation driven by increased OM vesiculation promotes adaptation of a bacterial pathogen upon host entry.
RESULTS
Silencing of VacJ/Yrb ABC transport system in vivo increases colonization fitness of V. cholerae.
The recently established TRIVET (TetR-controlled recombination-based in vivo expression technology) was used to monitor transcriptional silencing of the yrb gene cluster in V. cholerae (Cakar et al., 2018). TRIVET is a highly sensitive, single-cell based reporter technique to detect temporal gene repression by an irreversible switch of an antibiotic resistance phenotype. The system allows detection of gene repression in complex environments with relatively low bacterial load, such as the intestinal tract. In this approach, a promoterless tetR allele is integrated in the V. cholerae chromosome under the sole control of the promoter of the gene of interest. Once produced TetR, in turn, represses TnpR resolvase expression, which excises irreversibly an antibiotic resistance (res)-cassette. In short, silencing of tetR will induce tnpR and cause an irreversible loss of the res-cassette, which can be monitored by a phenotypic change of the antibiotic resistance profile. Hence, resolution frequencies inversely correlate with the expression level of the investigated gene.
Here we integrated the tetR allele in the V. cholerae chromosome downstream of the yrbF-B operon promoter (Fig. 1A). We then examined the resolution frequencies for the corresponding TRIVET strain Vc_res_TRIVET yrb::tpc grown for 22 h in LB supplemented with either FeSO4 or 2,2’-bipyridyl to mimic high or low iron availability, respectively, as well as during colonization of the murine intestinal tract (Fig. 1B). Relatively low-resolution frequencies were detected in regular LB broth, or in LB upon addition of FeSO4, indicating sufficient yrb-promoter activity to silence tnpR. In agreement with previous results (Roier et al., 2016), resolution levels were significantly increased in LB broth supplemented with the iron chelator 2,2’-bipyridyl, reflecting the silencing of the yrb-genes under low iron conditions.
High-resolution frequencies in vivo were also observed during colonization of the mouse intestine 6 and 22 h post-infection (Fig. 1B). In vivo repression of yrbE and vacJ was independently confirmed by quantitative real time PCR (Fig. S1). Overall, the results indicate a significant repression of the yrbF-B operon at early stages of the infection compared to growth in regular LB broth. Based on the current model, inactivation or downregulation of the VacJ/Yrb transporter system results in phospholipid accumulation in the OM promoting OMV release [Fig. 1A; (Roier et al., 2016)]. Concordantly, deletion of yrbE, encoding the permease unit of the VacJ/Yrb transport system, resulted in a hypervesiculating ΔyrbE mutant (Fig. 1C). Expression of yrbE in trans (ΔyrbE pyrbE) reduces the hypervesiculation compared to a ΔyrbE mutant carrying the empty vector (ΔyrbE p) (Fig. 1C). In contrast, reduced vesiculation (hypovesiculation) was achieved upon overexpression of the yrbF-B operon from an IPTG-inducible plasmid in the WT (WT pyrbF-B) compared to WT carrying the empty vector (WT p) (Fig. 1C). Notably, derivatives of this plasmid allow expression even in the absence of IPTG and have been used extensively for V. cholerae studies in vivo (Tamayo et al., 2008; Schild et al., 2007; Butler and Camilli, 2004; Osorio et al., 2004). However, stable plasmid maintenance requires antibiotic selection, which has been reported to elevate OMV levels in bacteria, including V. cholerae [Fig. 1C; (Bauwens et al., 2017; Roier et al., 2016)]. To avoid this effect of antibiotic use, a complementation strain (yrbE*) was constructed by reconstitution of a slightly modified yrbE allele in ΔyrbE on the chromosome (described in “Method details”). The yrbE* complementation strain not only showed significantly reduced vesiculation levels compared to ΔyrbE, but also similar vesiculation levels compared to the WT (Fig. 1C). Thus, yrbE* was chosen for complementation assays throughout this study.
We next investigated the impact of vesiculation on colonization fitness. The ΔyrbE mutant and WT overexpressing the yrbF-B operon (WT pyrbF-B) were challenged, respectively, in a competition assay with a fully virulent LacZ− derivative of the WT (WTlacZ−) or WT carrying the empty vector (WTlacZ− p), in LB broth (in vitro) and in vivo using the infant mouse model. Compared to the in vitro control assay, the hypervesiculating ΔyrbE mutant exhibited a significant advantage over the WT during intestinal colonization (Fig. 1D). In contrast, the hypovesiculating WT pyrbF-B was significantly attenuated in vivo compared to the WTlacZ− p (Fig. 1D). Differential loss of the empty vector (p) and expression vector (pyrbF-B) can be excluded as p and pyrbF-B are maintained at comparable levels during in vitro cultivation as well as during in vivo colonization (Fig. S1B). Thus, increased vesiculation correlates directly with enhanced colonization fitness in the murine model.
Hypervesiculation facilitates exchange of OM structures during transition stages.
A recent report indicated that OMV release allows for the selective loss of certain LPS species from the OM of Salmonella following pH shifts (Bonnington and Kuehn, 2016). Likewise, the observed fitness advantage of hypervesiculating V. cholerae strains might be at least partially explained by alteration of the cell surface via enhanced OMV release upon host entry. In V. cholerae two adaptive surface modulations occurring in the host environment have been reported, (i) removal of the OM porin OmpT to confer bile resistance (Provenzano and Klose, 2000), and (ii) accumulation of (di)glycine-modified lipid A to increase resistance to cationic antimicrobial peptides (Hankins et al., 2012). We aimed to establish a system for inducing each of these surface modifications in vitro to subsequently analyze the impact of vesiculation on the remodeling efficacy. Fortunately, the regulatory pathways and key players for both surface modulations are known and include the virulence gene regulator ToxR, which inversely controls ompU and ompT expression, as well as the Alm-pathway required for (di)glycination of lipid A (Hankins et al., 2012; Provenzano and Klose, 2000).
Previous work indicated that cultivation in minimal media plus the four amino acids asparagine, arginine, glutamate and serine (NRES) significantly increases ToxR levels resulting in activation of ompU and repression of ompT (Mey et al., 2012). To validate differential regulation in our experimental setup, we measured alkaline phosphatase (PhoA) activities in ompU-phoA and ompT-phoA reporter strains harboring a transcriptional fusion of the promoterless phoA to the ompU or ompT promoters (Fig. 2A and B). Upon transition from minimal medium M9 to M9 supplemented with NRES (M9ToxR↑) a significant increase in PhoA activity for ompU-phoA reporter strains was observed, while ompT-phoA reporter strains exhibited a significant decrease (Fig. 2A and B). Thus, the ToxR-dependent OmpU/T switch can also be activated in vitro by shifting V. cholerae from M9 to M9ToxR↑.
Figure 2. Differential expression of almG, ompU, ompT in a transition from minimal media M9 to M9ToxR↑ or M9ToxR↑/Alm↑.
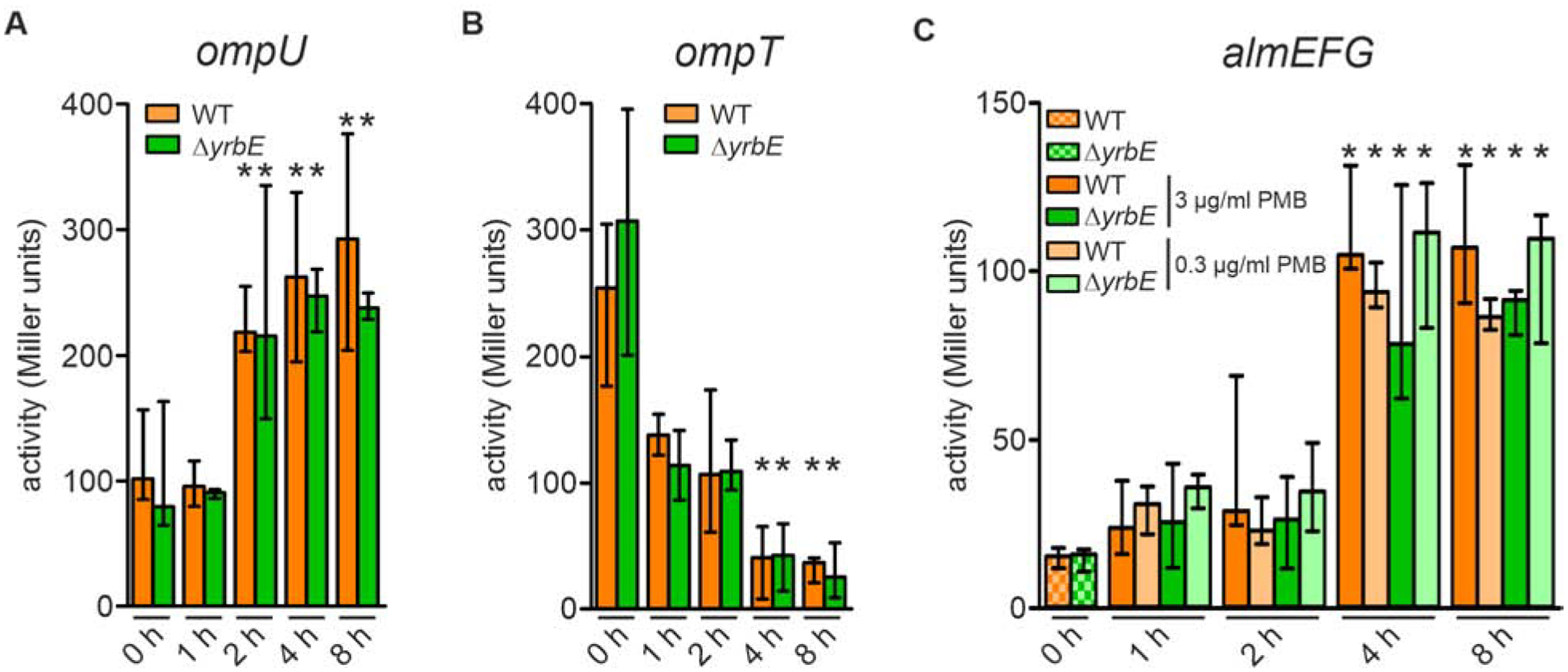
(A and B) Alkaline phosphatase activities of WT and ΔyrbE harboring an ompU-phoA transcriptional fusion (A) and ompT-phoA transcriptional fusion (B) in M9 (0 h) as well as 1, 2, 4 and 8 h after transition to M9ToxR↑. Data presented are median ± IQR with the following number of biological replicates: in case of ompU-phoA (A) n = 17 for WT at 0 h; n = 10 for ΔyrbE at 0 h; n = 4 for WT at 1, 2, 4, and 8 h; n = 6 for ΔyrbE at 1, 2, 4, and 8 h; in case of ompT-phoA (B) n = 9 for WT and ΔyrbE at 0 h; n = 6 for WT and ΔyrbE at 1 h; n = 4 for WT and ΔyrbE at 2, 4, and 8 h.
(C) Alkaline phosphatase activities of WT and ΔyrbE harboring an almG-phoA transcriptional fusion in M9 as well as 1, 2, 4 and 8 h after transition to M9ToxR↑/Alm↑ (including either 3 μg/ml or 0.3 μg/ml final PMB concentrations as indicated). Data presented are median ± IQR with the following number of biological replicates: n = 4 for WT (0 and 2 h with 3 μg/ml PMB) and ΔyrbE (0 h); n = 8 for WT (1 and 4 h with both PMB concentrations, 8 h with 0.3 μg/ml PMB) and ΔyrbE (1 h with both PMB concentrations, 4 and 8 h with 0.3 μg/ml PMB); n = 5 for WT (8 h with 3 μg/ml PMB); n = 6 for WT (2 h with 0.3 μg/ml PMB) and ΔyrbE (2 h with 0.3 μg/ml PMB, 4 and 8 h with 3 μg/ml PMB).
(A to C) Significant differences along the transition (1 – 8 h) to the respective 0 h time points are indicated by an asterisk (*P < 0.05).
The LPS modification system responsible for the addition of (di)glycine residues to lipid A during de novo synthesis at the cytoplasmic side of the inner membrane is encoded by the almEFG operon (Hankins et al., 2012). The Alm-pathway can be activated in the presence of sublethal concentrations of cationic antimicrobial peptides (Herrera et al., 2014). To validate almEFG induction in our setup, we constructed reporter strains harboring a transcriptional fusion of the promoterless phoA to the almEFG promoter. Concordant with previous reports (Herrera et al., 2014), we measured a significant increase in PhoA activity upon growth in M9ToxR↑ with sub-growth inhibitory concentrations of PMB (M9ToxR↑/Alm↑) compared to M9 (Fig. 2C). Notably, comparable temporal induction and maximum levels are observed with 0.3 μg/ml and 3 μg/ml PMB, the two concentrations used throughout this study to activate the Alm-pathway. This suggests that both PMB concentrations have a similar potential to activate the Alm-pathway. We conclude that both surface modulations can be activated in vitro on the WT and ΔyrbE background using minimal media M9 with supplements: (i) The OmpU/T switch is activated via cultivation in M9ToxR↑ allowing bile adaptation and (ii) presence of PMB (M9ToxR↑/Alm↑) additionally activates the Alm-pathway allowing adaptation to cationic antimicrobial peptides.
To investigate whether differential vesiculation affects the dynamics of the surface exchange, regular and hypervesiculating V. cholerae strains were shifted from minimal medium M9 to M9ToxR↑/Alm↑ and samples were collected over time. Analysis of the transcriptional expression pattern of the almEFG operon along the shift experiment showed that induction takes approximately 4 h (Fig. 2C). Therefore, we obtained lipid A extracts of the WT, the hypervesiculating ΔyrbE and the yrbE* complementation strain for liquid chromatography mass spectrometry (LC-MS) analyses at 0, 4, 6 and 8 h after the M9 to M9ToxR↑/Alm↑ shift. The lipid A species were detected as doubly charged ions with m/z 877.7 for the unmodified lipid A (U), 906.2 for mono-glycinated lipid A (M1) and m/z 934.7 for di-glycinated lipid A species (M2), respectively. The peaks of these three species (U, M1, and M2) are shown in the corresponding extracted ion chromatograms (Fig. 3). At 0 h peaks reflecting the unmodified lipid A species were readily detected in all strains, while the (di)glycine-modified lipid A species were below the limit of detection. At 4 h post medium shift peaks reflecting the mono-glycinated lipid A were already prevalent in hypervesiculating ΔyrbE mutant extracts, but only a minor population in the WT or still below limit of detection in the yrbE* complementation strain (Fig. 3). At 6 h post medium shift peaks reflecting the (di)glycine-modified lipid A species dominated in the hypervesiculating ΔyrbE mutant extracts, while still only a minor fraction was modified in WT and yrbE*. However, the extent of surface modulation in the WT still increased at later time points to finally reach a similar abundance of the unmodified and (di)glycine-modified lipid A species at 8 h after the medium shift. In general, the yrbE* complementation strain showed the lowest prevalence of (di)glycine-modified lipid A species with unmodified lipid A species dominating even at the 8 h time point. Extracts from a ΔalmG mutant isolated at the same 8 h time point served as a negative control and lacked peaks reflecting (di)glycine-modified lipid A, as expected (Fig. S2).
Figure 3. Appearance of lipid A modification along the M9 to M9ToxR↑/Alm↑ shift analyzed by mass spectrometry.
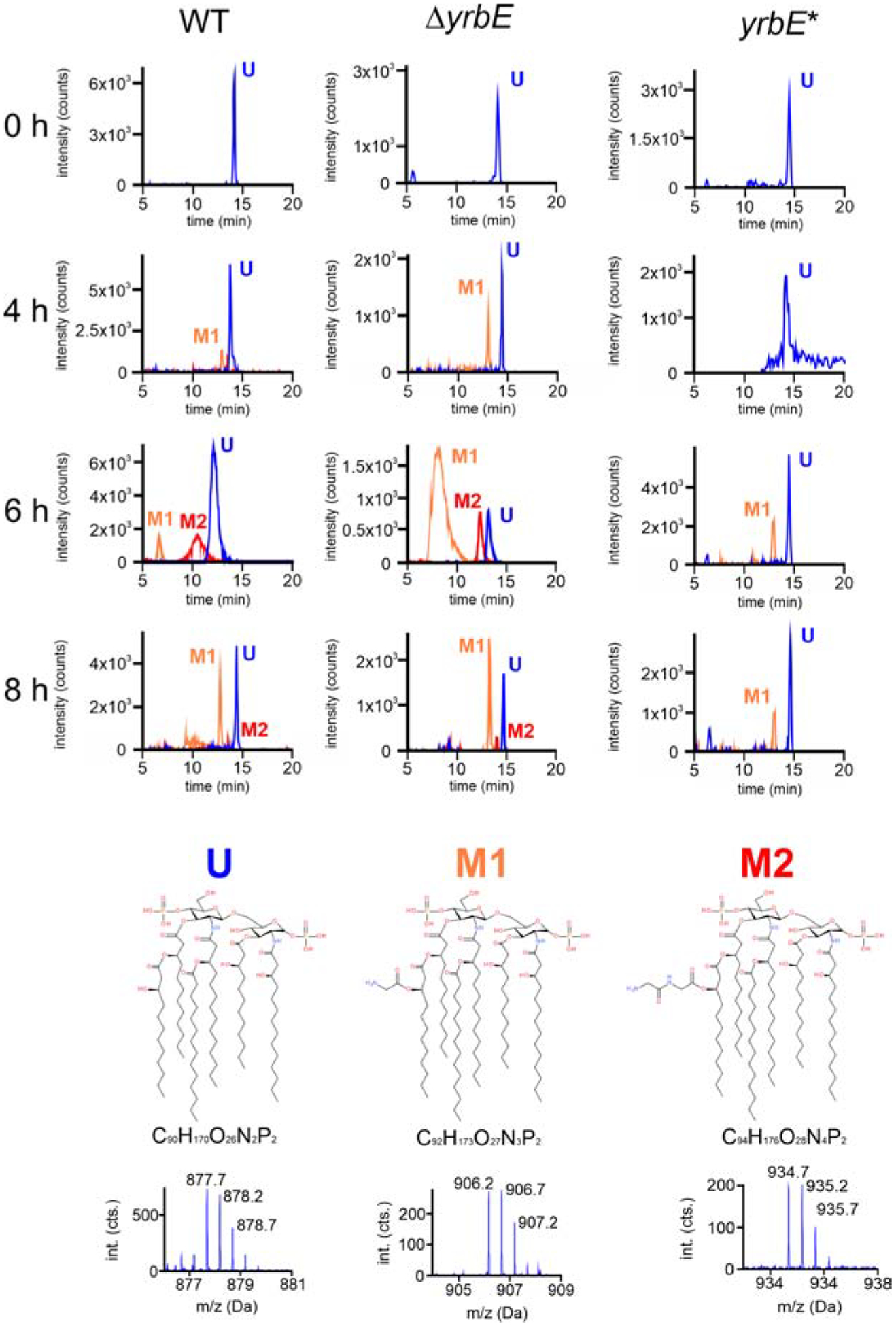
Lipid A extracted from WT, ΔyrbE and yrbE* along the M9 to M9ToxR↑/Alm↑ transition at 0, 4, 6, and 8 h was compared. Extracted ion chromatograms of m/z 877.7 (unmodified lipid A, U) in blue, m/z 1234 906.2 (mono-glycinated lipid A, M1) in orange, and m/z 934.7 (di-glycinated lipid A, M2) in red are shown for indicated strain and time point. At the bottom chemical structures and formulas of the three lipid A species U, M1 and M2 [according to (Hankins et al., 2012)] including the high-resolution mass spectra of the doubly charged lipid A species with their isotope patterns are shown. See also Figure S2.
We conclude that exposure to sub-growth inhibitory concentrations of PMB caused ΔyrbE and WT to accumulate (di)glycine-modified lipid A in the OM in an Alm-dependent manner, but the hypervesiculating ΔyrbE displayed the surface modification more rapidly (Fig. 3). Comparison of the activities obtained using the chromosomal almG-phoA reporter fusions generated in the ΔyrbE and WT backgrounds (Fig. 2C) allowed us to exclude differential expression levels or altered regulation of alm genes during the induced transition as a possible explanation for this accelerated modulation of the hypervesiculating mutant. As expected, PhoA levels in the WT and ΔyrbE background were relatively low in M9, but increased comparably for both strains throughout the shift experiment (Fig. 2C). Thus, the most likely scenario explaining the faster accumulation of modified lipid A in the hypervesiculating ΔyrbE is an enhanced surface turn-over due to increased vesiculation resulting in faster depletion of unmodified lipid A from the OM.
We then monitored OmpT depletion in whole cell lysates or OM preparations for the WT, the hypervesiculating ΔyrbE and the yrbE* complementation strain in response to the M9 to M9ToxR↑ shift as a model for the adaptational response to bile. Immunoblot analyses revealed that OmpU increased slightly over time in whole cell lysates or remained at stable high levels at all time points sampled in OM preparations for all strains (Fig. 4). The increase observed in whole cell lysates follows the activation of ompU expression during the shift experiment (Fig. 2A). Increasing OmpU might not be visible in OM preparations as these samples were normalized to protein equivalents, which can be seen as an adjustment to the most abundant protein, namely OmpU (Chakrabarti et al., 1996). In contrast to OmpU, OmpT was depleted in whole cell lysates and from the OM preparations for all strains during the transition (Fig. 4). We verified that equal amounts of protein were loaded in all lanes by Coomassie-stained SDS gels (Fig. S3) and additionally by immuno-detection of the a-subunit of the RNA Polymerase (RpoA) in whole cell lysates (Fig. 4). Notably, the rate of OmpT depletion differed between strains. In the WT and the yrbE* complementation strain a marked reduction of OmpT signal intensity was observed from 4 h onwards, while OmpT loss was detected after 1 h in the hypervesiculating ΔyrbE strain (Fig. 4). In line with the AlmEFG expression profile, the temporal regulation of ompT and ompU expression during the shift experiment is similar in WT and ΔyrbE (Fig. 2A and B), excluding differential regulation in the two strains.
Figure 4. Vesiculation accelerates depletion of OmpT from the OM.
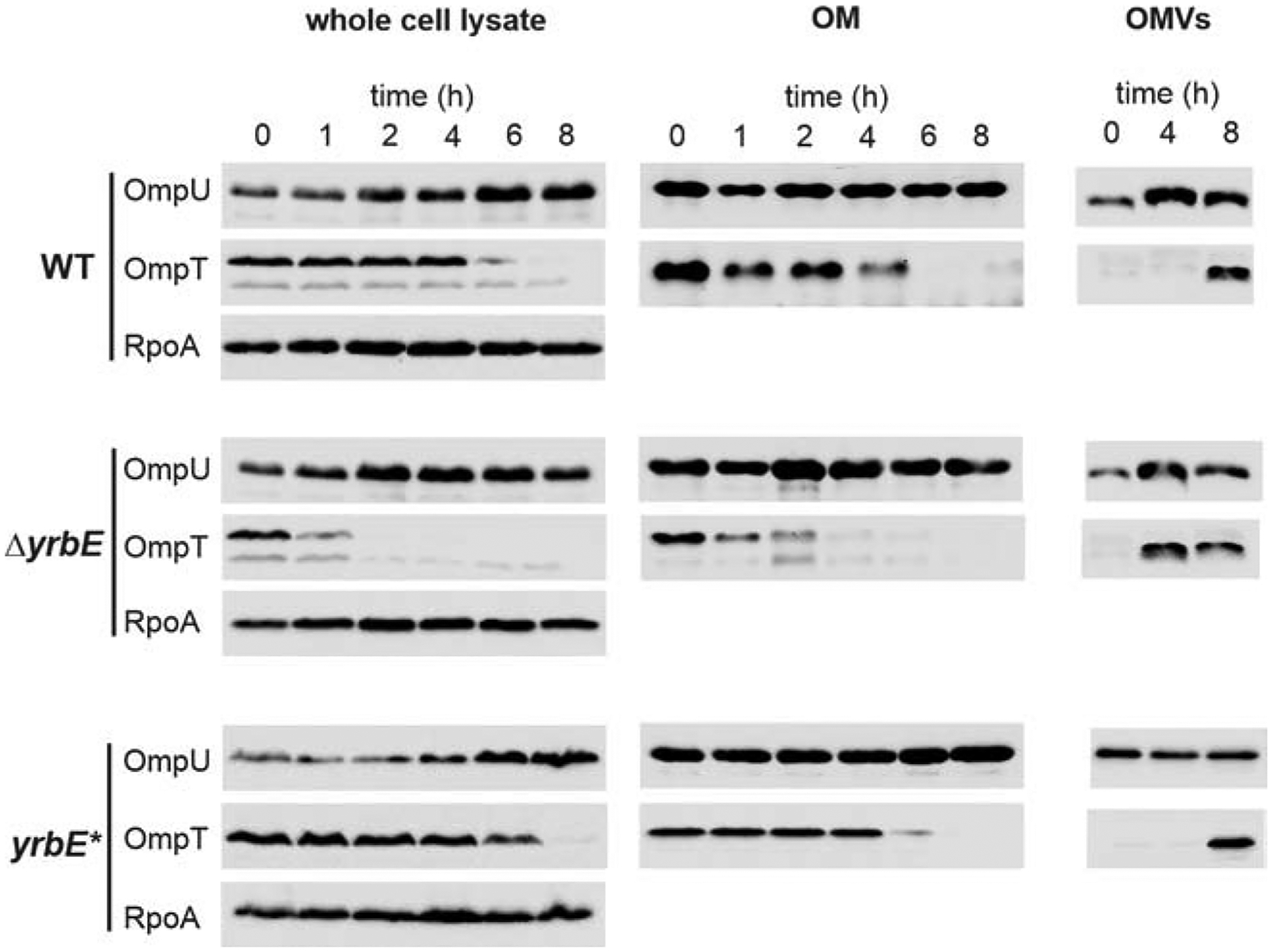
Samples of whole cell lysates, OM preparations and OMVs were isolated from WT, ΔyrbE, and yrbE* at times indicated after transition from minimal medium M9 to M9ToxR↑. Equivalent amounts of total proteins were resolved electrophoretically and representative immunoblots detected OmpU, OmpT and RpoA in case of whole cell lysates (left) or OmpU and OmpT in case of OM preparations (center) and OMVs (right). Coomassie-stained SDS gels executed in parallel additionally controlled loading of equal protein amounts in all lanes. See also Figure S3.
To evaluate a direct role for vesiculation in the OmpU/T switch, OMVs were isolated from culture supernatants obtained at 0, 4 and 8 h of the transition experiment and the relative abundance of these proteins was compared (Fig. 4). OmpT appeared already at 4 h in OMVs released by ΔyrbE, but only after 8 h in the OMVs derived from WT and yrbE*. Hence, the most likely explanation for the OM depletion of OmpT along the transition is a constant removal of OmpT and other components via vesiculation, combined with transcriptional repression of ompT and de novo synthesis. In summary, the adaptive depletion of OmpT from the OM of V. cholerae can be linked to its deposition into OMVs. The hypervesiculating ΔyrbE mutant is thus able to more rapidly discharge OmpT from its OM. In conclusion, both adaptive surface modulations tested here were implemented more rapidly in the hypervesiculating ΔyrbE mutant than in a normally vesiculating WT strain.
Hypervesiculation facilitates faster adaptation to the cationic antimicrobial peptide PMB during transition stages.
As previously reported, accumulation of (di)glycine-modified lipid A facilitates bacterial survival in the presence of cationic antimicrobial peptides, while depletion of OmpT from the OM promotes resistance to bile (Hankins et al., 2012; Provenzano and Klose, 2000). We hypothesized that the faster accumulation of (di)glycine-modified lipid A as well as removal of OmpT observed in a hypervesiculating strain correlates with faster adaptation to these host defenses. To test this premise, we asked whether strains with differential vesiculation exhibit different sensitivity to PMB and bile.
First, the minimal inhibitory concentration (MIC) of PMB affecting growth of the hypervesiculating ΔyrbE mutant and the regular vesiculating WT was assessed for the transition from M9 to M9ToxR↑/Alm↑ as a model for the adaptation phase early in infection. To this end, overnight cultures of the ΔyrbE mutant and WT grown in M9 were allowed to adapt for 2 h in M9ToxR↑/Alm↑ [(di)glycine-modified lipid A activating conditions using sub-MIC PMB (3 μg/ml)] before additional PMB was added to achieve inhibitory concentrations. A 2 h adaptation phase was chosen, as it closely reflects the dynamics of a natural V. cholerae infection, i.e. it reflects the time from oral ingestion to arrival of the bacteria at their primary colonization site in the small intestine (Angelichio et al., 1999). A significantly higher MIC was obtained for the hypervesiculating ΔyrbE mutant compared to WT, implying stronger resistance and better adaptation to PMB (Fig. 5A). A transition of the same M9 overnight cultures to M9 fails to activate the Alm-pathway. As expected under these conditions, ΔyrbE and WT exhibited comparably poor resistance to PMB indicated by equally low MIC values (Fig. 5A). Conversely, overnight cultures grown in M9ToxR↑/Alm↑ shifted to M9ToxR↑/Alm↑ served as fully adapted controls. Accordingly, equally high MIC values were obtained for the ΔyrbE mutant and WT. Thus, the hypervesiculating strains have no advantage in non-adapted or fully adapted stages, but only transiently during the adaptation phase (Fig. 5A). To link the observed advantage to the lipid A modification, we performed the experiment in an ΔalmG background. While deletion of yrbE in an ΔalmG background still resulted in an hypervesiculating phenotype (Fig. S4A), the MICs of PMB were similar for the normally vesiculating ΔalmG and the hypervesiculating ΔalmGΔyrbE following the M9 to M9ToxR↑/Alm↑ transition (Fig. S4B). Notably, a lower adaptive PMB concentration (0.3 μg/ ml instead of 3 μg/ml) had to be used for ΔalmG and ΔalmGΔyrbE compared to the other strains to accommodate the previously reported lower PMB resistance of alm-mutants compared to WT (Hankins et al., 2012). Nonetheless, we observed earlier that induction of the Alm-pathway was similar for 0.3 μg/ml and 3 μg/ml PMB (Fig. 2C) indicating that both PMB concentrations have a comparable potential to activate the Alm-pathway. Thus, the transient advantage of the hypervesiculating strain in the PMB adaptation is AlmG-dependent.
Figure 5. Enhanced vesiculation facilitates adaptation to PMB and bile upon transition from M9 to M9ToxR↑/Alm↑ or M9ToxR↑.
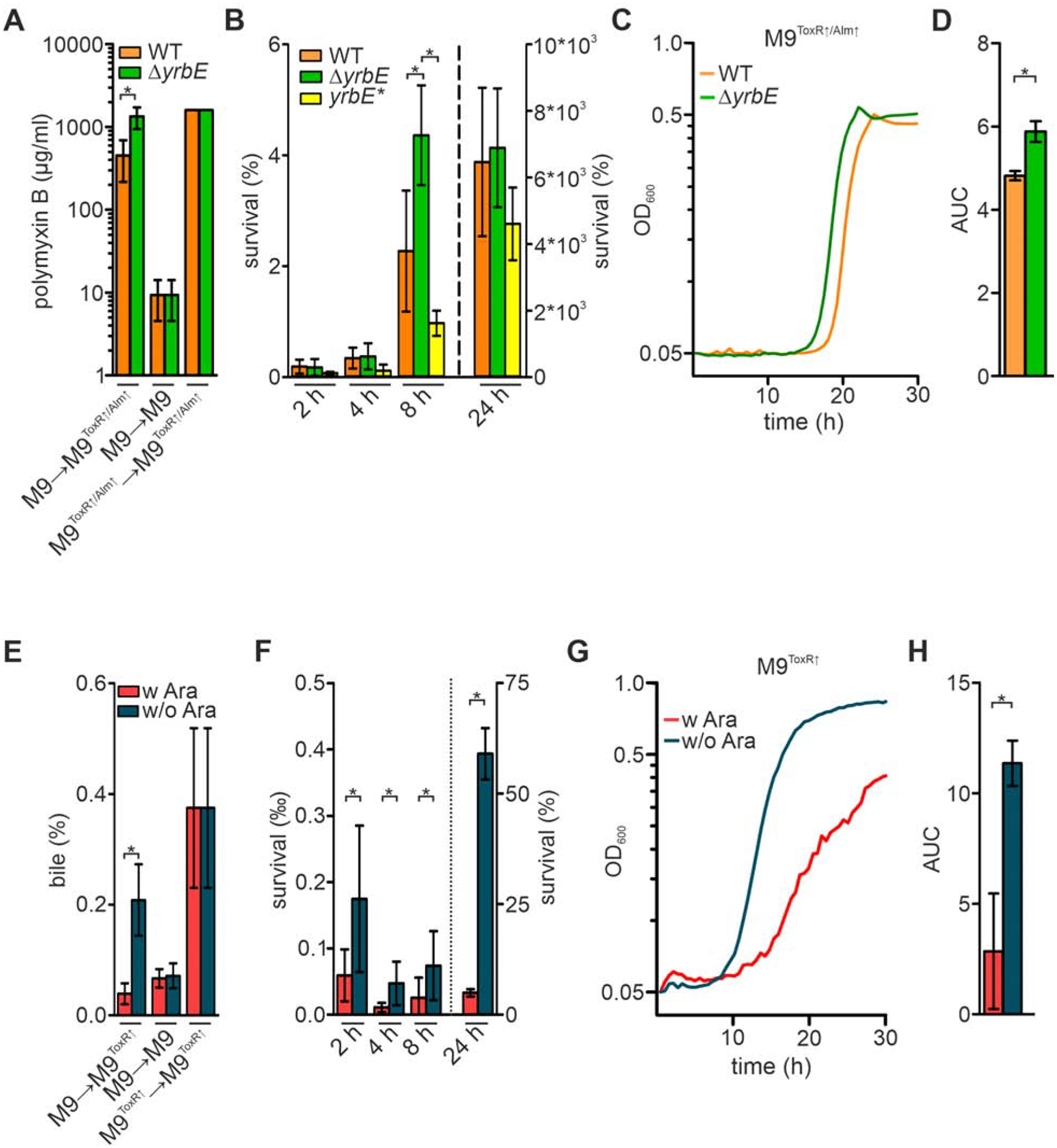
(A) Minimal inhibitory concentrations (MIC) of PMB for WT and ΔyrbE after a M9 to M9ToxR↑/Alm↑, M9 to M9 or M9ToxR↑/Alm↑ to M9ToxR↑/Alm↑ transition. Bacteria were allowed to adapt for 2 h after transition into fresh medium before (additional) PMB was added to achieve diverse concentrations for MIC-determination. Data presented are mean ± standard deviation (SD) with the following number of biological replicates for WT and ΔyrbE: n = 11 for the M9 to M9ToxR↑/Alm↑ transition; n = 6 for the M9 to M9 and the M9ToxR↑/Alm↑ to M9ToxR↑/Alm↑ transition.
(B) Survival of WT, ΔyrbE, and yrbE* was determined by CFU plating along the M9 to M9ToxR↑/Alm↑ [including sub-MIC concentrations of PMB (3 μg/ml)] transition at 2, 4, 8 and 24 h. Data presented are mean ± SD with the following number of biological replicates: n = 18 for WT at 2 and 4 h; n = 10 for ΔyrbE at 2 and 4 h; n = 8 for yrbE* at 2, 4 and 8 h; n = 14 for WT at 8 h; n = 6 for ΔyrbE at 8 and 24 h as well as yrbE* at 24 h; n = 12 for WT at 24 h.
(C) Growth curves reporting mean values (n = 6) of WT and ΔyrbE upon transition from M9 to M9ToxR↑/Alm↑ [including sub-MIC concentrations of PMB (3 μg/ml)].
(D) Mean area under the curve (AUC) values ± SD retrieved from the growth curves presented in panel C (n = 6).
(E) MIC of bile for the arabinose-inducible strain yrbF-BpARA after a M9 to M9ToxR↑, M9 to M9 or M9ToxR↑ to M9ToxR↑ transition with (w) or without (w/o) arabinose (Ara), respectively. Overnight cultures were always grown in presence of arabinose and allowed to adapt for 2 h after transition into the fresh medium (M9 or M9ToxR↑ as indicated). After the 2 h adaptation phase differential amounts of bile were added to determine the MIC. Data presented are mean ± SD with the following number of biological replicates for yrbF-BpARA w and w/o Ara: n = 6 for the M9 to M9ToxR↑ transition, n = 14 for the M9 to M9 transition, n = 4 for the M9ToxR↑ to M9ToxR↑ transition.
(F) Survival of the arabinose-inducible strain yrbF-BpARA was determined by CFU plating along the M9 to M9ToxR↑ transition with (w) or without (w/o) arabinose (Ara) at 2, 4, 8 and 24 h in presence of sub-MIC bile concentrations (0.1%). Data presented are mean ± SD with the following biological replicates for yrbF-BpARA w and w/o Ara: n = 4 for 24 h and n = 8 for all other timepoints.
(G) Growth curves reporting mean values (n = 6) of the arabinose-inducible strain yrbF-BpARA upon transition from M9 to M9ToxR↑ with (w) or without (w/o) arabinose (Ara) in presence of sub-MIC bile concentrations (0.1%).
(H) Mean AUC values ± SD retrieved from the growth curves presented in panel G (n = 6). (A-H) Significant differences are indicated by an asterisk (*P < 0.05). See also Figures S4, S5 and S6.
We next compared the survival fitness of WT, ΔyrbE and yrbE* exposed to a sub-MIC PMB concentration (3 μg/ml) present in M9ToxR↑/Alm↑. Bacterial cultures were grown and shifted using the M9 and M9ToxR↑/Alm↑ model as described above. Upon transition, cell viability dropped massively with equally low survival rates of approximately 1% obtained for all strains at 2 h and 4 h (Fig. 5B). At 8 h average survival rates increased for all strains tested suggesting adaptation to PMB. However, survival rates of ΔyrbE were significantly higher compared to WT and yrbE* highlighting an advantage for the hypervesiculating strain. After 24 h all strains had proliferated to a similar extent, exceeding the starting CFU. Moreover, growth upon transition from M9 to M9ToxR↑/Alm↑ was analyzed to visualize the differential adaptation dynamics. After a relatively long lag phase the hypervesiculating ΔyrbE entered into exponential growth about 2 h earlier than the WT (Fig. 5C). Accordingly, the area under curve (AUC), reflecting the overall biomass produced within the observed time, was significantly higher for the hypervesiculating ΔyrbE compared to WT (Fig. 5D). Similar growth was observed for all strains used in these assays, if bacteria were shifted from M9 to M9ToxR↑ in absence of PMB (Fig. S5A and B). Thus, a general growth advantage of ΔyrbE can be excluded.
Hypervesiculation facilitates faster adaptation to bile salts during transition stages.
Next, we asked whether the faster removal of OmpT from the OM mediated by hypervesiculation promotes a more rapid adaptation to bile. Unexpectedly, the deletion mutant ΔyrbE was three-fold more sensitive to bile compared to WT or yrbE* in M9 (Fig. S6A). All three strains exhibited comparable sensitivity to PMB and SDS treatment, which excludes a general defect in the OM integrity upon hypervesiculation (Fig. 5A and S6B). Characterization of previous loss-of-function mutations of the VacJ/Yrb transporter, however, provide a possible explanation. Complete loss of the retrograde lipid-trafficking results in excessive amounts of phospholipids in the OM (Roier et al., 2016). The normal lipid asymmetry of the OM is lost as a result and large patches of phospholipid bilayers appear (Malinverni and Silhavy, 2009; Roier et al., 2016). Importantly, bile penetrates such phospholipid bilayers more effectively than asymmetric OMs composed of an LPS and phospholipid leaflet (Benz and Bauer, 1988; Hancock, 1984) and may therefore account for increased sensitivity of the ΔyrbE mutant to bile.
To overcome this limitation, a strain allowing finer control of the yrb genes was constructed. The chromosomal promoter of the yrb-operon was replaced by an arabinose-inducible promoter strain and the corresponding regulator araC was inserted into the lacZ–locus generating the arabinose-inducible strain yrbF-BpARA. This allowed a controllable expression of the yrb genes. Basal expression levels in the absence of arabinose and increased production in the presence of arabinose was confirmed by quantitative real time PCR (Fig. S6C). Accordingly, OMV production by a yrbF-BpARA culture was low when grown in M9ToxR↑ with arabinose and significantly higher in M9ToxR↑ lacking arabinose (Fig. S6C). The yrbF-BpARA strain thus allowed us to assess dynamics of vesiculation upon silencing of the yrb-operon. Already 4 h after depletion of arabinose a small, but significant increase in vesiculation could be detected in comparison to a culture grown in presence of arabinose (Fig. S6C). Notably, the highest vesiculation levels achieved for yrbF-BpARA in the absence of arabinose were still two-fold lower compared to the loss-of-function mutant ΔyrbE (Fig. 1C and S6C), indicating a basal yrb operon expression level in yrbF-BpARA even in absence of arabinose. Strain yrbF-BpARA followed the same principles as the regular vesiculating WT and hypervesiculating ΔyrbE mutant for the adaptation to PMB upon shift to M9ToxR↑/Alm↑ (Fig. 5A and S6D). Consistent with the results obtained for WT and ΔyrbE, a higher MIC of PMB was observed for strain yrbF-BpARA under high vesiculation conditions (no arabinose) compared to low vesiculation cultivation (with arabinose). Most importantly, a similar sensitivity to bile was observed for yrbF-BpARA grown in M9 with and without arabinose (Fig. 5E). Thus, the strain yrbF-BpARA is perfectly suited to analyze the impact of differential vesiculation on bile adaptation during transition events.
In general, bile adaptation experiments using the strain yrbF-BpARA in presence or absence of arabinose followed the design described above for the PMB resistance mediated by accumulation of (di)glycine-modified lipid A (Fig. 5). Overnight cultures of the strain yrbF-BpARA were grown in M9 or M9ToxR↑ in the presence of arabinose to promote expression of the yrb genes resulting in moderate vesiculation levels. Subsequently, overnight cultures were shifted from M9 to M9ToxR↑ with or without arabinose (inducing ToxR-dependent ompT repression), from M9 to M9 with or without arabinose (no ompT repression) or M9ToxR↑ to M9ToxR↑ with or without arabinose (ToxR-dependent ompT repression already in the overnight culture). Presence or absence of arabinose triggered moderate or increased vesiculation. Consistent with the PMB transition assays, bacteria were allowed to adapt for 2 h after transition into the fresh media before different bile concentrations were added.
In the M9 to M9ToxR↑ transition experiment, significantly higher MIC values for bile were obtained with the strain grown in absence of arabinose reflecting hypervesiculation than the moderately vesiculating strain grown with arabinose. This finding links increased vesiculation with higher resistance levels along the transition and better adaption to bile (Fig. 5E), in very good agreement with the PMB adaptation data. A M9 to M9 transition prevented the ompT silencing and served as control. Consequently, equally low MIC values were observed in the assays regardless of presence or absence of arabinose (Fig. 5E). Conversely, overnight cultures grown in M9ToxR↑ shifted into M9ToxR↑ with or without arabinose served as controls with full adaptation to bile achieved before the transfer resulting in equally high MIC values (Fig. 5E).
To assess temporal survival in presence of bile the strain yrbF-BpARA was grown overnight in M9 with arabinose and shifted to M9ToxR↑ with or without arabinose for 2 h before a sub-MIC bile concentration (0.1%) was added. Generally, a severe drop in survival rates below 1% could be observed. Survival reached a minimum around 4 h then rose again afterwards (Fig. 5F). Notably, the survival rates of the hypervesiculating strain (no arabinose) were significantly higher compared to the lower vesiculating strain for all time points (Fig. 5F).
Finally, growth curves in presence of a sub-MIC bile concentration (0.1%) were performed to visualize the differential adaptation dynamics (Fig. 5G). Again, the hypervesiculating strain without arabinose entered earlier into exponential phase and showed more robust proliferation compared to the lower vesiculating strain grown with arabinose. This advantage is also indicated by AUC values, which were significantly higher for the strain grown in absence of arabinose than in presence of arabinose (Fig. 5H). Similar growth was observed in M9ToxR↑ with and without arabinose (Fig. S5C and D) excluding an arabinose-dependent growth difference of strain yrbF-BpARA.
Pre-adaptation to bile and PMB negates in vivo advantage of an hypervesiculating strain.
Our data support the model that the faster exchange of cell surface composition observed for hypervesiculating strains is associated with a faster adaptation to antimicrobial compounds. We therefore asked next whether this mechanism underlies the in vivo fitness advantage of hypervesiculating strains. If true, the colonization advantage of a hypervesiculating strain should be diminished if fully adapted strains are used for the in vivo competition experiments. We took advantage of the fact that growth overnight in M9ToxR↑/Alm↑ activates both infection-relevant surface modulations studied thus far. Equally high resistance levels to antibacterial challenge in vitro indicated full adaptation of moderate and hypervesiculating strains (Fig. 5A and E). The in vivo competition assays were then performed using inocula of the hypervesiculating ΔyrbE mutant and the normally vesiculating WT that had been grown in M9 (no adaptation) or M9ToxR↑/Alm↑ (full adaptation). As observed for LB cultures (Fig. 1D), the hypervesiculating strain still outcompeted the WT in vivo when the inoculum was derived from M9 cultures (Fig. 6). In contrast, ΔyrbE and WT competed equally well in the murine model when the inoculum was derived from M9ToxR↑/Alm↑ cultures. Moreover, non-adapted WT (grown in M9) was approximately four-fold attenuated in vivo compared to an adapted WT (grown in M9ToxR↑/Alm↑), while a non-adapted ΔyrbE (grown in M9) was only two-fold attenuated in vivo compared to an adapted ΔyrbE (grown in M9ToxR↑/Alm↑). Thus, non-adapted strains are generally attenuated compared to their adapted counterparts, but hypervesiculation can partially compensate this disadvantage. Hence, we conclude that the in vivo advantage of hypervesiculating strains at early stages of infection is due to accelerated surface modulation, thus promoting adaptation to antimicrobial stressors in the intestine.
Figure 6. Colonization advantage of the hypervesiculating strain is compensated by pre-adaptation to bile and PMB.
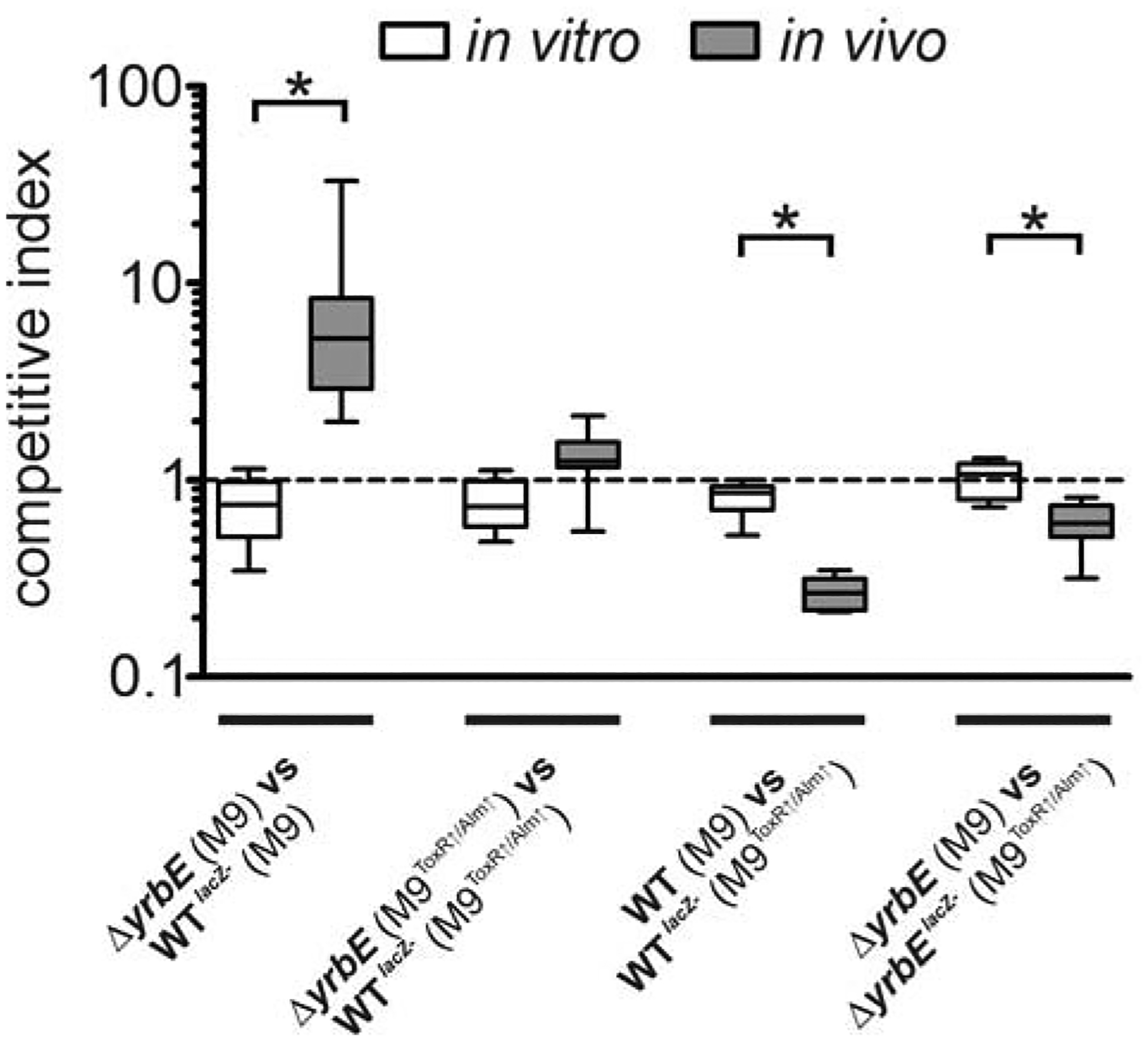
Competition assays were performed in vitro (M9) and in vivo (murine model) between the WT or the hypervesiculating ΔyrbE mutant against their isogenic lacZ− derivatives (WTlacZ− or ΔyrbElacZ−). Strains as well as the cultivation conditions of the inoculum used for the competition are indicated on the x-axis. The data is presented as boxplot with whiskers (*P < 0.05) with the following number of biological replicates: n = 8 for in vitro competitions of WTlacZ− vs ΔyrbE; n = 9 for in vivo competitions of WTlacZ− vs ΔyrbE grown in M9, n = 10 for in vivo competitions of WTlacZ− vs ΔyrbE in M9ToxR↑/Alm↑; n = 12 for in vitro competitions of WT vs WT lacZ− and ΔyrbE vs ΔyrbE lacZ−; n = 7 for in vivo competitions of WT vs WT lacZ− and ΔyrbE vs ΔyrbE lacZ−.
DISCUSSION
This study provides insights into the adaptation strategies of the bacterial pathogen V. cholerae along the environment to host transition. We characterize OMVs as a tool for OM exchange facilitating V. cholerae’s adaptation to antimicrobial intestinal stressors upon host entry.
Upon ingestion by the host, V. cholerae silences the VacJ/Yrb retrograde lipid trafficking system, which results in increased OMV release. In accordance with previous reports and data presented here, iron limitation is a likely signal for the repression of the well conserved vacJ and yrb genes (Roier et al., 2016). Notably, iron limitation - or nutrient limitation in general - could have been a driving evolutionary trigger for OMV release. OMV production generally increases during starvation conditions. Moreover, iron-scavenging proteins have been found in vesicles from several bacterial pathogens, which might imply a role of OMVs in iron acquisition (Gui et al., 2016; Lappann et al., 2013; Lin et al., 2017).
Coming from the aquatic lifestyle V. cholerae needs to quickly change its expression profile in order to adapt to the conditions faced in the intestinal tract of the host. Besides activation of colonization factors and toxins, the adaptation to antimicrobial effectors is crucial for V. cholerae to achieve full virulence. The adaptation processes to cationic antimicrobial peptides and bile require changes in the OM profile, i.e. accumulation of (di)glycine-modified lipid A in LPS molecules and removal of OmpT (Hankins et al., 2011; Provenzano and Klose, 2000). Indeed, regulatory cascades driving both surface modulations are immediately activated upon host entry. Low levels of cationic antimicrobial peptides induce transcription of the AlmEFG system, which catalyzes the (di)glycine-modification along de novo synthesis of LPS (Matson et al., 2017). Moreover, host entry activates ToxR and downregulates ompT transcription (Li et al., 2000; Miller and Mekalanos, 1988). Nonetheless, the time elapsed until such transcriptional changes are manifest in OM alterations can be critical. This is especially true during early stages of the infection when bacterial proliferation is rather low. Besides preventing new OmpT production and activation of (di)glycine-modified LPS synthesis, it is crucial for V. cholerae to deplete disadvantageous structures from the surface. OMVs provide an efficient tool to discharge unfavorable OM compounds, including proteins like OmpT and polysaccharides like unmodified LPS. Increased vesiculation in vivo not only enhances the removal of factors detrimental for colonization, but also creates new space for beneficial compounds. As demonstrated in this study, hypervesiculation of V. cholerae allows an efficient exchange of the surface profile facilitating the adaptation process to cationic antimicrobial peptides and bile along the environment to host transition. Importance of this adaption is highlighted by the observation that hypervesiculation is associated with increased colonization fitness whereas hypovesiculation results in reduced in vivo fitness.
Notably, increased vesiculation only provides a transient advantage during the adaptation process. Once the surface profile change is completed the advantage of hypervesiculation is lost. Previous reports indicate that increased OMV production in E. coli correlates with better bacterial survival in the presence of antimicrobial agents (Kulkarni et al., 2015; Manning and Kuehn, 2011; Urashima et al., 2017). This is likely due to the fact that OMVs can act as a sink for these substances. Notably, these reports rely on OMV amounts present in fully grown cultures. To initiate the transition processes studied here the bacterial cultures are massively diluted. Thus, the initial OMV concentrations in the survival assays of this study are very low (approx. 100-fold below OMV amounts described to be beneficial). This difference might explain why the advantage of the hypervesiculating strain in the present study is only observed during transitions activating the surface exchange and therefore relies on the OM adaptation rather than the OMV amount.
The continuous liberation of compounds of the OM and periplasm via OMVs is highly energy consuming. Thus, it is likely that bacteria have evolved to regulate OMV release in accordance with their needs, e.g. induction of vesiculation upon host entry. Notably, the regulation of vesiculation via transcriptional control of the VacJ/ Yrb lipid trafficking system in response to iron availability seems to be conserved among diverse Gram-negative bacteria (Roier et al., 2016). Moreover, several Gram-negative pathogens modulate the OM composition upon host entry to achieve full colonization fitness. In addition to the surface modifications analyzed in this study, prominent examples include modulation of OM protein abundance involved in serum resistance (e.g. reported for Neisseria gonorrhoeae and Borrelia burgdorferi) or iron uptake (e.g. reported for Klebsiella pneumoniae, Pasteurella haemolytica and H. influenzae), OM-associated proteases of E. coli and Salmonella typhimurium that degrade antimicrobial peptides, LPS modifications such as the lipid A palmitoylation (e.g. reported for S. typhimurium, E. coli and Yersinia enterocolitica), the lipid A aminoarabinose modification (reported for S. typhimurium) as well as the aminoacylation of phospholipids reported for Pseudomonas aeruginosa contributing to antimicrobial peptide resistance (Gunn et al., 1998; Klein et al., 2009; Lin et al., 2002). Additionally, a recent in vitro study suggested that OMV production of S. typhimurium can facilitate infection-relevant LPS remodeling in the OM triggered by pH and magnesium availability (Bonnington and Kuehn, 2016). Based on these observations, we hypothesize that the surface exchange via increased vesiculation upon host entry could be a common principle of bacterial pathogens to quickly adapt to in vivo conditions.
Moreover, OMVs of pathogens have been reported to act as delivery vehicles of bacterial toxins and as a sink for host-derived antimicrobial factors, such as defensins and complement system (Elluri et al., 2014; Kulp and Kuehn, 2010; Roier et al., 2016). Thus, it is becoming evident that enhanced bacterial vesicle production during host colonization can be linked to various beneficial roles and OMVs can increase the colonization fitness of bacterial pathogens in multiple ways.
Notably, hypervesiculation is not only associated with iron availability. The VacJ/Yrb lipid trafficking system might be controlled by additional regulatory elements and alternative OMV biogenesis mechanisms have been reported (Kulp and Kuehn, 2010). Thus, other signals may induce vesiculation, which extend the observations beyond the environment to host transition performed by the facultative human pathogen V. cholerae. Since production of membrane vesicles is observed for Gram-negative and Gram-positive bacteria it is tempting to speculate that varying vesiculation levels could represent a general and efficient adaptation strategy of bacteria during transitions demanding a surface profile modulation.
STAR METHODS
LEAD CONTACT AND MATERIALS AVAILABILITY
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Stefan Schild (stefan.schild@uni-graz.at). All reagents generated in this study are available from the Lead Contact with a completed Materials Transfers Agreement.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Housing conditions for experimental animals
5 to 6 day old CD-1 mice (Crl:CD1, Charles River Laboratories) of both genders were used in all experiments in accordance with the rules of the ethics committee at the University of Graz and the corresponding animal protocol, which has been approved by the Austrian Federal Ministry of Science and Research Ref. II/10b. Mice were housed with food and water ad libitum and monitored under the care of full-time staff.
Competition assay
Competition assays in infant mice (in vivo assay), LB broth or minimal media M9 were performed with WT, deletion mutants, or WT pyrbF-B (lacZ+) competed for ~22 h against isogenic WT, deletion mutants, or WT strain carrying an empty vector (lacZ−) essentially as previously described (Camilli and Mekalanos, 1995; Schild et al., 2007). The individual strains and conditions used for each competition are indicated along the presentation of the respective data set. Briefly, strains were grown on LB plates or in minimal media M9 (M9, M9ToxR↑ or M9ToxR↑/Alm↑) overnight (O/N), diluted to OD600 = 0.002 in a 1:1 ratio, and used to intragastrically inoculate infant mice (5-day old CD-1 mice). Appropriate dilutions of the inoculum were plated on LB-Sm/X-Gal or on LB-Ap/X-Gal plates in case of V. cholerae strains harboring a plasmid to determine the exact input ratio. Approximately 22 h post-infection mice were euthanized, their small bowels were removed and homogenized in 1 ml of LB with 15% glycerol. In vitro competitions in LB or minimal media M9 were performed in parallel by inoculation of 2 ml liquid culture with ~105 CFU from the inoculum and subsequent incubation for ~22 h at 37°C with aeration. CFU were determined by plating appropriate dilutions of the homogenized intestine or culture grown in vitro on LB-Sm/X-Gal or on LB-Ap/X-Gal plates in case of V. cholerae strains harboring a plasmid. Results are given by the competition index (CI), which is the ratio of lacZ+-CFU to lacZ−-CFU normalized for the input ratio. If applicable, plasmid maintenance was determined along the competitions in vivo and in vitro by calculating dividing the respective ApR-CFU (LB-Ap/XGal plates) by the SmR-CFU (LB-Sm/X-Gal).
Resolution assay
Resolution assay was performed as previously described (Cakar et al., 2018). To quantify resolution, strain Vc_res1_TRIVET yrbF::tpc was grown O/N on LB-Sm/Km/Ap plates and adjusted in LB-Sm/Km/Ap to OD600=1, which was used as inoculum. To determine in vitro resolution frequencies, the inoculum was diluted 1:100 in 5 ml of LB-Sm/Ap, LB-Sm/Ap supplemented with 75 μM of 2,2’-bipyridyl or 100 μM of FeSO4 and incubated for 8 h. To determine the in vivo resolution, the inoculum was diluted 1:500 in LB and anesthetized 5-day old CD-1 mice were intragastrically inoculated with 50 l of this dilution (~106 CFU per mouse). Mice were euthanized at 6 or 22 h post-infection time, and bacteria were recovered from intestine. At the given time point, the amount of resolution in vitro and in vivo was determined by plating appropriate dilutions on LB-Sm/Km and LB-Sm/Ap plates. Results were expressed as % resolution, calculated as the SmR/KmS CFU (SmR/ApR CFU minus SmR/KmR CFU) divided by SmR/ApR CFU.
Infection studies to obtain bacterial RNA
For in vivo studies, anesthetized 5-day old CD-1 mice were intragastrically inoculated with 50 L of WT adjusted to OD600 of 0.002 in LB, corresponding to an infection dose of ~106 CFU per mouse. After 22 h, mice were euthanized, and small intestine was removed and mechanically homogenized in 1.5 mL of TRIzol Reagent (ThermoFisher Scientific).
METHOD DETAILS
Bacterial strains and growth conditions
Bacterial strains, plasmids and oligonucleotides used in this study are listed in the “Key resources table”. The clinical isolate V. cholerae O1 El Tor E7946 (Miller et al., 1989) served as wild type (WT) strain in all experiments. Unless stated otherwise, all V. cholerae strains were grown with aeration in Lura Bertani (LB) broth or minimal medium M9 according to standard recipe (Miller, 1972) at 37°C. E. coli strains DH5αλpir and SM10λpir (Miller and Mekalanos, 1988) were used for genetic manipulations and grown with aeration in LB broth at 37°C. Unless stated otherwise, minimal media M9 compositions are abbreviated as follows: M9 for minimal media M9 supplemented with glucose; M9ToxR↑ for minimal media M9 supplemented with glucose, asparagine, arginine, glutamate and serine; M9ToxR↑/Alm↑ for minimal media M9 supplemented with glucose, asparagine, arginine, glutamate, serine and sub-MIC concentrations of polymyxin B (PMB, 3 μg/ml or 0.3 μg/ml for strains with ΔalmG background, respectively). Antibiotics and other supplements were used in the following final concentrations: streptomycin (Sm, 100 μg ml−1), ampicillin (Ap, 50 μg ml−1 in combination with other antibiotics, otherwise 100 μg ml−1), sucrose (10%), glucose (0.2%), arabinose (Ara, 0.2%), asparagine (25 mM) arginine (25 mM), glutamate (25 mM), and serine (25 mM).
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Anti-mouse IgG, HRP-linked Antibody | Jackson ImmunoResearch | Cat#JIM-115-035-003 |
| α-OmpU Mouse serum | (Leitner et al., 2013) | N/A |
| α-OmpT Mouse serum | (Salem et al., 2015) | N/A |
| α-RpoA Mouse serum | BioLegend | Cat#663104; RRID:AB_2687386 |
| Bacterial and Virus Strains | ||
| DH5αλpir | (Kolter et al., 1978) (F− endA1 glnV44 thi-1 recA1 relA1 gyrA96 deoR nupG Φ80dlacZΔM15 Δ(lacZYA-argF) U169 hsdR17(rK− mK+) λpirRK6) | N/A |
| SM10λpir | (Kolter et al., 1978) (thi thr leu tonA lacY supE recA::RPA-2-Te::Mu λpir, KmR) | N/A |
| WT | (Miller et al., 1989) (AC53, wild type V. cholerae strain serogroup, O1; biotype, El Tor; serotype, Ogawa; spontaneous SmR mutant of E7946; clinical isolate from Bahrain 1978; hapR+, SmR) | N/A |
| WTlacZ− | (Tamayo et al., 2008) (insertion of res-cassette (res-neo-sacB-res cassette) in WT, SmR, KmR) | N/A |
| ΔyrbE | (Roier et al., 2016) (Deletion of VC2519 in WT, SmR) | N/A |
| ΔyrbElacZ− | This paper (insertion of res-cassette (res-neo-sacB-res cassette) in ΔyrbE, SmR, KmR) | N/A |
| ΔalmG | This paper (Deletion of VC1577 in WT, SmR) | N/A |
| ΔyrbEΔalmG | This paper (Deletion of VC2519 in ΔalmG, SmR) | N/A |
| lacZ::araC | This paper (Insertion of araC from pBADin lacZ in WT, SmR) | N/A |
| yrbE* | This paper (ΔyrbE with reconstituted yrbE* allele harboring three silent point mutations located around stop codon of yrbE, exchanging AACTGATCA to AATTAAACA SmR) | N/A |
| almG::phoA | This paper (Insertion of pGPphoA in almG of WT, SmR, ApR) | N/A |
| ΔyrbE almG::phoA | This paper (Insertion of pGPphoA in almG of ΔyrbE, SmR, ApR) | N/A |
| ompU::phoA | This paper (Insertion of pGPphoA in ompU of WT, SmR, ApR) | N/A |
| ΔyrbE ompU::phoA | This paper (Insertion of pGPphoA in ompU of ΔyrbE, SmR, ApR) | N/A |
| ompT::phoA | This paper (Insertion of pGPphoA in ompT of WT, SmR, ApR) | N/A |
| ΔyrbE ompT::phoA | This paper (Insertion of pGPphoA in ompT of ΔyrbE, SmR, ApR) | N/A |
| Vc_res1_TRIVET yrbF::tpc | This paper (insertion of PtetA controlled tnpR in Vc_res1 yrbF::tpc, SmR, CmR, KmR) | N/A |
| Vc_res1 yrbF::tpc | This paper (insertion of res-cassette in lacZ in yrbF::tpc, SmR, CmR, KmR) | N/A |
| yrbF::tpc | This paper (insertion of tetRphoAcat downstream of yrbF in WT, SmR, CmR) | N/A |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Polymyxin B sulfate salt | Sigma-Aldrich | Cat#P4932 |
| Bile salts | Sigma-Aldrich | Cat#B8756 |
| Sodium dodecylsulfat (SDS) | Bio-Rad | Cat#161–0302 |
| TRIzol Reagent | ThermoFisher | Cat#15596026 |
| Critical Commercial Assays | ||
| Iscript cDNA synthesis Kit | Biorad | Cat#1708890 |
| RNAse free DNAseI Kit | Promega | Cat#9PIM610 |
| RNA isolation Kit RNeasy | Quiagen | Cat#74106 |
| Monarch Total RNA Miniprep Kit | New England Biolabs | Cat#T2010S |
| PeqGOLD total RNA Kit | VWR | Cat#732–2868 |
| SYBR GreenER™ qPCR SuperMix for ABI PRISM™ Instrument | ThermoFisher | Cat#11760 |
| Experimental Models: Organisms/Strains | ||
| CD-1® IGS Mouse | Charles River | Cat#Crl:CD1(ICR) RRID:IMSR_CRL:022 |
| Oligonucleotides | ||
| For more oligonucleotides, see Table S1 | N/A | N/A |
| qPCR_yrbE_1 GTTCGCCAGTTATACAGCATTG | (Roier et al., 2016) | N/A |
| qPCR_yrbE_2 CTGAGCACCATGCCAATAAAG | (Roier et al., 2016) | N/A |
| qPCR_vacJ_1 AGTTTCGCAAGAGGCCTTATTA | (Roier et al., 2016) | N/A |
| qPCR_vac_2 AGCTTGCCGTTGGAGATAAG | (Roier et al., 2016) | N/A |
| qPCR-VCr001_1 AGGGAGGAAGGTGGTTAAGT | (Fengler et al., 2012) | N/A |
| qPCR-VCr001_2 CGCTACACCTGAAATTCTACCC | (Fengler et al., 2012) | N/A |
| yrbE*_test GCATTGATGTTTGGGAATTAAA | This paper | N/A |
| yrbE*_seq AAACCCGGGCTCCGCCACATCTTCACGTT | This paper | N/A |
| qPCR-VCr001_2 CGCTACACCTGAAATTCTACCC | (Fengler et al., 2012) | N/A |
| Recombinant DNA | ||
| Plasmid pBAD18 | (Guzman et al., 1995) | N/A |
| Plasmid pCVD442 | (Donnenberg and Kaper, 1991) | N/A |
| Plasmid pCVDyrbF::tpc | This paper (pCVD442 with irgA::tetRphoAcat, ApR, CmR) | N/A |
| Plasmid pCVDyrbE* | This paper (pCVD442 with point mutated yrbE fragment, ApR) | N/A |
| Plasmid pCVDΔalmG | This paper (pCVD442 with up- and downstream fragments of almG, ApR) | N/A |
| Plasmid pCVDlacZ::araC | This paper (pCVD442 with araC flanked by up- and downstream fragments of lacZ, ApR) | N/A |
| Plasmid pCVDyrbF-BpARA | This paper (pCVD442 with the arabinose-inducible promoter flanked by up- and downstream fragments of the yrb promoter, ApR) | N/A |
| Plasmid pGPphoA | (Moisi et al., 2009) | N/A |
| Plasmid pGPphoAalmG | This paper (pGPphoA with ‘almG ‘ fragment to generate transcriptional phoA-fusions to the almGEF almEFG promoter, ApR) | N/A |
| Plasmid pGPphoAompU | (Lembke et al., 2018) (pGPphoA with ‘ompU ‘ fragment to generate transcriptional phoA-fusions to the ompU promoter, ApR) | N/A |
| Plasmid pGPphoAompT | (Lembke et al., 2018)(pGPphoA with ‘ompT ‘ fragment to generate transcriptional phoA-fusions to the ompT promoter, ApR) | N/A |
| Plasmid p | (Morales et al., 1991) (pMMB67EH, IncQ broad-host-range low-copy-number cloning vector, IPTG-inducible, Apr) | N/A |
| Plasmid pRes1 | (Osorio et al., 2005) (pSL111 with res1-neo-sacB-res1 cassette, KmR) | N/A |
| Plasmid pyrbF-B | This paper (expression plasmid with yrb operon (VC2516-VC2520) in pMMB67EH, Apr) | N/A |
| Software and Algorithms | ||
| Prism 5.0 | GraphPad | https://www.graphpad.com |
| ImageLab Software | BioRad | https://www.bio-rad.com |
| Corel Draw | Corel Corporation | https://www.coreldraw.com |
Genetic manipulations
The isolation of chromosomal DNA, PCR reactions, the purification of plasmids or PCR products, the construction of suicide and expression plasmids as well as the subsequent generation of deletion mutants were carried out as described previously (Pressler et al., 2016; Seper et al., 2011). Qiagen plasmid kits were used for isolation of plasmid DNA, Qiaquick® Gel extraction and Qiaquick® PCR Purification kits (Qiagen) were used for purifying DNA fragments. PCR reactions for subcloning were carried out using the Q5® High-Fidelity DNA Polymerase (NEB), while Taq DNA Polymerase (NEB) was used for all other PCRs. Constructions of in-frame almG deletion mutants were carried out as described by Donnenberg and Kaper (Donnenberg and Kaper, 1991). Briefly, ~800 bp PCR fragments located up- and downstream of the gene of interest were amplified using the oligonucleotide pairs almG_XbaI_1 and almG _BamHI_2 as well as almG _BamHI_3 and almG _SacI_4. After digestion of the PCR fragments with the appropriate restriction enzyme (NEB) indicated by the name of the oligonucleotide, they were ligated into pCVD442, which was digested with the appropriate restriction enzymes.
Complementation of yrbE was achieved either in trans by using the expression plasmid pyrbE (Roier et al., 2016) or by re-insertion of a slightly modified yrbE* allele (altered nucleotide sequence in the wobble position of some codons, resulting in identical amino acid sequence compared to the WT allele) in the original chromosomal gene locus to avoid the usage of antibiotics for plasmid maintenance. The latter was constructed via PCR fragments amplified using the oligonucleotide pairs VC2519_XbaI_up_fw and comp_yrbE*_1 as well as comp_yrbE*_2 and VC2519_XmaI_down_rv and subsequent splicing by overlap extension (SOE) PCR of the two purified PCR fragments introducing three silent point mutations changing AACTGATCA to AATTAAACA located around stop codon of yrbE. The SOE PCR fragment was digested with XmaI and XbaI (NEB), ligated into a similar digested pCVD442 resulting in pCVDyrbE*.
In case of the suicide vector pCVDlacZ::araC used for araC insertion into the lacZ–locus of V. cholerae ~800 bp PCR fragments flanking lacZ were amplified using the oligonucleotide pairs lacZ_XbaI_1 and lacZ_BamHI_2 as well as lacZ_SphI_3 and lacZ_SacI_4, while araC was amplified from pBAD18 using the oligonucleotide pairs araC_BamHI_1 and araC_EcoRI_2. All fragments were digested using the restriction enzymes, indicated by the name of oligonucleotide To construct the suicide vector pCVDyrbF-BpARA, replacing the natural yrb-promoter with an arabinose-inducible promoter, ~800 bp PCR fragments flanking the yrb-promoter were amplified using the oligonucleotide pairs yrb_SacI_1 and yrb_SphI_2 as well as yrb_EcoRI_3 and yrb_XbaI_4, while the fragment harboring the arabinose-inducible promoter was amplified from pBAD18 using the oligonucleotide pairs pARA_SphI_1 and pARA_EcoRI_2. After digestion with the restriction enzymes, indicated by the name of oligonucleotide, PCR fragments were ligated into pCVD442, restricted with appropriate restriction enzymes (NEB).
The suicide vectors pGPphoA-almG, pGPphoA-ompU and pGPphoA-ompT for chromosomal insertion were constructed to obtain chromosomal transcriptional fusions of promoterless phoA to almG (almG::phoA), ompU (ompU::phoA) and ompT (ompT::phoA), as PhoA acts as a reporter for gene expression in V. cholerae. DNA fragments containing the upstream region of almG, ompU and ompT were amplified by PCR using oligonucleotide pair almG_pGPphoA_XbaI and almG_pGPphoA_KpnI, the PCR product was digested with the restriction enzymes indicated and ligated with similarly digested pGPphoA.
For construction of the expression plasmid pyrbF-B the entire yrb-operon was amplified with oligonucleotides yrb_operon_SacI_1, yrb_operon_XbaI_2, the PCR fragment was digested with the appropriate restriction enzymes and ligated into a similar digested pMMB67EH.
Unless noted otherwise, ligation products were transformed into DH5αλpir and ApR colonies were characterized for the correct constructs by PCR (and restriction analysis).
To obtain insertion and deletion strains generated derivatives of the pGPphoA or pCVD442 were transformed into E. coli Sm10λpir and conjugated into V. cholerae. Exconjugants were purified by SmR/ApR selection. In the case of pCVD442 derivatives sucrose selection was used to obtain ApS colonies and chromosomal deletions/replacements were confirmed by PCR, respectively. To generate the complementation strain yrbE* the pCVDyrbE* was mobilized into yrbE, exconjugants were purified by SmR/ApR selection and finally sucrose selection was used to obtain ApS colonies. Correct chromosomal insertion of the yrbE* allele was confirmed via PCR using a discriminator oligonucleotide pair yrbE*_test and VC2519_XmaI_down_rv as well as via sequencing using yrbE*_ seq.
Resolution assay
Resolution assay was performed as previously described (Cakar et al., 2018). To quantify resolution, strain Vc_res1_TRIVET yrbF::tpc was grown O/N on LB-Sm/Km/Ap plates and adjusted in LB-Sm/Km/Ap to OD600=1, which was used as inoculum. To determine in vitro resolution frequencies, the inoculum was diluted 1:100 in 5 ml of LB-Sm/Ap, LB-Sm/Ap supplemented with 75 μM of 2,2’-bipyridyl or 100 μM of FeSO4 and incubated for 8 h. To determine the in vivo resolution, the inoculum was diluted 1:500 in LB and anesthetized 5-day old CD-1 mice were intragastrically inoculated with 50 l of this dilution (~106 CFU per mouse). Mice were euthanized at 6 or 22 h post-infection time, and bacteria were recovered from intestine. At the given time point, the amount of resolution in vitro and in vivo was determined by plating appropriate dilutions on LB-Sm/Km and LB-Sm/Ap plates. Results were expressed as % resolution, calculated as the SmR/KmS CFU [SmR/ApR CFU minus SmR/KmR CFU] divided by SmR/ApR CFU.
Quantitative real-time PCR
Expression of yrbE and vacJ was determined by quantitative real-time RT-PCR (qRT-PCR). For in vitro studies, WT was grown to an OD600 of 0.5 to 0.8 in LB, while the arabinose-inducible yrbF-B variant was grown to an OD600 of approximately 0.8 in M9ToxR↑ with (w) or without (w/o) arabinose (Ara). Bacterial RNA extraction, DNase digestion, cDNA synthesis, and qRT-PCR were performed as described previously (Lichtenegger et al., 2013). For in vivo studies, anesthetized 5-d-old CD-1 mice were intragastrically inoculated with 50 μL of WT adjusted to OD600 of 0.002 in LB, corresponding to an infection dose of ~106 600 CFU per mouse. After 22 h, mice were euthanized, and small intestine was removed and mechanically homogenized in 1.5 mL of TRIzol Reagent (ThermoFisher Scientific). RNA was isolated using either the Monarch Total RNA Miniprep Kit (New England Biolabs) or the RNA isolation Kit Rneasy (Quiagen) for in vitro samples grown in LB as well as in vivo samples or the PeqGOLD total RNA Kit (Peqlab) for in vitro samples grown in minimal media M9 according to the manufacturer’s protocols, and chromosomal DNA was digested by using RQ1 RNase-Free DNase (Promega). Synthesis of cDNA and qRT-PCR were performed as described previously using the iScript Select cDNA Synthesis Kit (Bio-Rad) and the SYBR GreenER qPCR SuperMix for ABI PRISM [(ThermoFisher Scientific); (Seper et al., 2013)]. Each cDNA sample was tested in triplicate. The sequences of the primers used for qRT-PCR starting with “qPCR_” are listed in the “Key resources table”. Results were analyzed using StepOne Software v2.1, and relative gene expression comparisons were calculated by the mean cycle threshold of samples, which were normalized to the housekeeping gene 16S rRNA (VCr001) and to one randomly selected in vitro reference sample.
Isolation of lipid A
The protocol for large scale lipid A extraction as described by Henderson and coworkers was used (Henderson et al., 2013). Briefly 500 ml of respective culture were harvested, washed with PBS and resuspended in 40 ml PBS. Subsequently 50 ml of chloroform and 100 ml of methanol were added to create a single-phase Bligh-Dyer mixture. Samples were washed with single-phase Bligh-Dyer mixture and resuspended in 54 ml of mild acid hydrolysis buffer. Samples were boiled for 1 h and then converted to a two-phase Bligh-Dyer mixture by adding 60 ml of chloroform and 60 ml of methanol. The lower phase was extracted and a second extraction was performed using the lower phase of a pre-equilibrated two-phase Bligh-Dyer mixture. Both lower phases were pooled and washed with new upper phase of a pre-equilibrated two-phase Bligh-Dyer mixture and dried for subsequent usage.
Analysis of lipid A by LC-MS
Normal phase LC-MS was performed using an Agilent 1200 Quaternary LC system coupled to a high-resolution TripleTOF5600 mass spectrometer (Sciex, Framingham, MA). A Unison UK-Amino column (3 μm, 25 cm × 2 mm) (Imtakt USA, Portland, OR) was used. Mobile phase A consisted of chloroform/methanol/aqueous ammonium hydroxide (800/195/5, v/v/v). Mobile phase B consisted of chloroform/methanol/water/aqueous ammonium hydroxide (600/340/50/5, v/v/v/v). Mobile phase C consisted of chloroform/methanol/water/aqueous ammonium hydroxide (450/450/95/5, v/v/v/v). The elution program consisted of the following: 100% mobile phase A was held isocratically for 2 min and then linearly increased to 100% mobile phase B over 14 min and held at 100% B for 11 min. The LC gradient was then changed to 100% mobile phase C over 3 min and held at 100% C for 3 min, and finally returned to 100% A over 0.5 min and held at 100% A for 5 min. The total LC flow rate was 300 ml/min. The MS settings are as follows: Ion spray voltage (IS) = −4500 V, Curtain gas (CUR) = 20 psi, Ion source gas 1 (GS1) = 20 psi, De-clustering potential (DP) = −55 V, and Focusing Potential (FP) = −150 V. Nitrogen was used as the collision gas for MS/MS experiments. Data acquisition and analysis were performed using Analyst TF1.5 software (Sciex, Framingham, MA). High-resolution mass spectra and isotope patterns allowed a precise identification of the respective lipid A species. It should be noted that preparation of lipid A extracts derived from V. cholerae requires high culture volume and is labor-intensive. Thus, lipid A samples were prepared in batches and subjected to LC-MS analysis within two weeks to minimize artefact formation and sample degradation. Variations in LC retention times for the respective lipid A species can occur between different batches analyzed in different runs due to changes in column and solvent conditions.
Preparation of OMVs
In general, OMV were isolated as described previously with minor adaptations (Schild et al., 2009). O/N cultures of the respective strains were cultivated in M9, diluted 1:100 in M9ToxR↑ or M9ToxR ↑ with arabinose and grown for 4 or 8 h at 37°C and 180 rpm, before the cells were removed from the supernatant by centrifugation (9000 × g, 15 min). The supernatant was filtered through 0.22 μm pore size filters to remove intact cells. OD600 of each culture was determined by photometric measurements using a Beckman Coulter DU730 spectrophotometer for subsequent OMV quantification. To ensure that no bacteria were left in the supernatant, 1 ml of the filtrate was plated on LB-agar plates and incubated at 37°C O/N. The OMVs present in the supernatant were pelleted through subsequent ultracentrifugation (150,000 g, 4°C, 4 h) as previously described (Schild et al., 2009). Protein concentration was determined using Bradford assay (Bio-Rad Laboratories, Protein Assay Dye Reagent) according to the manufacturer’s manual and normalized to the OD600 of the respective culture.
Preparation of outer membrane proteins and whole cell lysates
Proteins of the outer membrane were essentially prepared as previously published (Carlone et al., 1986; Roier et al., 2013; Salem et al., 2015). O/N cultures of the respective strains were cultivated in M9 and diluted 1:100 in M9ToxR↑. At the indicated time point cultures of V. cholerae were harvested by centrifugation (3,200 g, 10 min, 4°C), washed once in HEPES buffer (10 mM, pH 7.4) and resuspended in 0.75 ml HEPES buffer (10 mM, pH 7.4). Then the suspension was transferred in a cryo-tube and cells were disrupted by homogenization with 0.1 mm glass beads in combination with a PowerLyzerTM 24 (MO BIO Laboratories, Inc.), applying three times, 1 min cycles at 3400 rpm with 1 min intervals on ice between each cycle. Unbroken cells were removed by centrifugation (13,000 g, 5 min, 4°C). The supernatant containing the outer membrane material was transferred into a new tube and centrifuged again (16,100 g, 30 min). The membrane pellet was re-suspended in 0.4 ml HEPES buffer (10 mM, pH 7.4). To solubilize the cytoplasmic membrane, 0.4 ml HEPES buffer (10 mM, pH 7.4) with 2% sarcosyl was added and incubated at room temperature and constant shaking for 30 min. After centrifugation (16,100 g, 30 min), the pellet containing proteins of the outer membrane was washed once with 0.5 ml HEPES buffer (10 mM, pH 7.4) and finally re-suspended in 50 μl HEPES buffer (10 mM, pH 7.4). Purified outer membrane preparations were stored at −20°C. The protein concentrations of outer membrane preparations were determined by photometric measurements of the absorbances at 260 nm and 280 nm using a Beckman Coulter DU730 spectrophotometer in combination with a TrayCell (Hellma) and the Warburg-Christian equation given as mg protein/ml = [(1.31 × A280) − (0.57 × A260)] x dilution factor (Warburg and Christian, 1941).
For whole cell lysates, equal amounts of cells (equivalent to 1.3 ml of an OD600=1) were harvested by centrifugation (3,200× g, 10 min, 4°C) from the respective V. cholerae cultures. Cell pellets were directly resuspended in 53 μl SDS-PAGE sample buffer (Laemmli, 1970), boiled for 10 min and subjected to SDS-PAGE.
SDS-PAGE and immunoblot
Proteins were separated by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) using polyacrylamide (15%) gels in combination with the Mini-PROTEAN Tetra cell system (Bio-Rad, Vienna) (Laemmli, 1970). As molecular mass standard Prestained Protein Marker Broad Range (New England Biolab) or PageRulerTM Prestained Protein Ladder 10 to 180 kDa (Thermo Fisher) were used as indicated. Subsequently protein bands were visualized according to Kang et al. (Kang et al., 2002) or further processed for immunoblot analysis as previously described (Schild et al., 2008). As primary antibodies (anti-OmpU, anti-OmpT and anti-RpoA) generated in mice were used [(Leitner et al., 2013; Salem et al., 2015) and BioLegend]. HRP-linked anti-mouse IgG was used as secondary antibody. Chemiluminescence detection was performed by using the Immun-Star™ WesternC™ Kit (Bio-Rad Laboratories) and subsequent exposure in a ChemiDoc XRS system (Bio-Rad Laboratories) in combination with Quantity One software (Bio-Rad Laboratories).
Growth kinetics
Growth kinetics were essentially performed as previously described in transparent 24-well plates (Greiner) with 1 ml culture volume (Gumpenberger et al.; Moisi et al., 2013; Seper et al., 2011). Briefly, the respective strains were grown in a pre-culture for ~16 h in M9 with aeration and shaking at 37°C. For growth assays of the WT, ΔyrbE mutant or yrbE* complementation strain, pre-cultures were adjusted to OD600 = 0.05 in M9 or M9ToxR↑Alm↑ [(di)glycine-modified lipid A activating conditions using sub-MIC PMB concentrations (3 μg/ml)] and allowed to adapt for 2 h before OD600 measurements started. For growth assays of the arabinose-inducible yrbF-B variant, pre-cultures with arabinose were pelleted by centrifugation (6000 × g, 10 min), resuspended in fresh M9 or M9ToxR↑, adjusted to OD600 = 0.05 in either M9 with or without arabinose or M9ToxR↑ with or without arabinose and allowed to adapt for 2 h in the new medium. In case of M9ToxR↑ with or without arabinose, bile was added to final concentration of 0.1% after the 2 h adaptation phase. In all cases, the OD600 was monitored every 30 min in the SPECTROstarNano microplate reader (BMG Labtech) at 37°C with shaking. For presentation of data, mean of at least four independent growth curves were plotted and area under curve (AUC) values were calculated by GraphPad prism version 5.01.
Minimal inhibitory concentration (MIC)-assays
For MIC assays O/N cultures of V. cholerae WT, ΔyrbE mutant or yrbE* complementation strain were either grown in M9 or M9ToxR↑/Alm↑ [(di)glycine-modified lipid A activating conditions using sub-MIC PMB concentrations (3 μg/ml)] as well as ΔalmG or ΔalmGΔyrbE were grown in M9. O/N cultures were shifted into fresh M9 or M9ToxR↑/Alm↑ [(di)glycine-modified lipid A activating conditions using sub-MIC PMB concentrations (3 μg/ml or 0.3 μg/ml for strains with ΔalmG background, respectively)] to a final concentration of 106 CFU/ml. The arabinose-inducible yrbF-B variant was grown O/N in M9 with arabinose or M9ToxR↑ with arabinose. To remove residual arabinose, the O/N cultures were pelleted by centrifugation (6000 × g, 10 min), resuspended in fresh M9 and then diluted into M9 with or without arabinose to a final concentration of 106 CFU/ml. Alternatively, O/N cultures were pelleted by centrifugation (6000 × g, 10 min), resuspended in fresh M9ToxR↑ and diluted into M9ToxR↑ with or without arabinose to a final concentration of 106 CFU/ml.
In all cases, bacteria were allowed to adapt for ~2 h, before 100 μl of the bacterial culture were placed into 96-well plates and 10 μl of appropriate serial dilutions of the respective antimicrobial agent (bile salts, additional PMB, SDS) were added. Two-fold serial dilutions of antimicrobial agents were tested in the following final concentrations: 8% to 0.0078% for bile salts, 3,200 μg/ml to 0.39 μg/ml for PMB and 0.25% to 0.0078% for SDS. After 18 h incubation in a humid chamber at 37°C OD600 was measured using the SpectrostarNANO Microplate Reader (BMG Labtech). The MIC was defined as the lowest antimicrobial agent concentration, which inhibited bacterial growth. Growth was defined by an at least three-fold increase of the OD600 compared to the respective sterile control (M9 with or without arabinose or M9ToxR↑ with or without arabinose).
Survival Assays
V. cholerae WT, yrbE mutant or the complementation strain yrbE* were grown O/N in M9, adjusted to an OD600 = 0.001 in 25 ml M9ToxR↑ before PMB was added to a final concentration of 3 μg/ml. For survival assays in presence of bile, the arabinose-inducible yrbF-B variant was grown O/N in M9 with arabinose, washed once in M9ToxR↑ and then adjusted in 25 ml M9ToxR↑ with or without arabinose to an OD600 = 0.001. After an adaptation for 2 h in M9ToxR↑ (activating OmpU/T switch) bile salts (0.1 %) were added. In general, samples were taken just before addition of PMB or bile salts (0 h), as well as 2, 4, 8 and 24 h after addition of the antimicrobial agent and appropriate dilutions were plated on LB plates. Percent survival for 2, 4, 8 and 24 h was calculated by the determined CFU at the respective time point divided by the determined CFU at 0 h multiplied by 100.
Alkaline phosphatase assays
To determine the enzymatic activities for the transcriptional ompU::phoA, ompT::phoA and almG::phoA fusion, alkaline phosphatase assays were performed as described previously (Manoil, 1991). Briefly, bacterial cultures were shifted from M9 to M9ToxR↑ in case of ompU::phoA and ompT::phoA or shifted from M9 to M9ToxR↑/Alm↑ in case of almG::phoA. Subsequently, bacterial cultures were harvested after 0, 1, 2, 4 and 8 h and subjected to the alkaline phosphatase assays. The activities were expressed in Miller units, given by (A405 × 1,000)/(A600 × ml × min).
QUANTIFICATION AND STATISTICAL ANALYSIS
Data sets are presented as mean with standard deviation (SD) in case of normal distribution or as median with interquartile range (IQR) in case of non-gaussian distribution. Statistical analyses were performed using GraphPad Prism 5.0 as follows: unpaired t-test (single comparison) or one-way Anova followed by Bonferroni’s post test (multiple comparisons) in case of normal distribution (data sets presented as mean with SD) or Mann–Whitney U test (single comparison) or Kruskal–Wallis followed by Dunn’s post test (multiple comparisons) in case of non-gaussian distribution (data sets presented as median with IQR). A P value of less than 0.05 was considered significant and indicated by an asterisk (*P < 0.05). Unless stated otherwise, the number of biological replicates for each data set is given by “n” and is provided in the respective figure legend.
DATA AND CODE AVAILABILITY
This study did not generate/ analyze datasets/ codes.
Supplementary Material
HIGHLIGHTS.
Phospholipid transport of Vibrio cholerae is silenced upon host entry
Reduced transporter activity triggers OMV release and increases colonization fitness
Increased vesiculation accelerates modulation of cell surface composition
Hypervesiculation leads to faster adaptation to host defense mediators
ACKNOWLEDGEMENTS
We are grateful to T. Eichmann (University of Graz) for assistance along the MS data evaluation, L. Schmidberger for assistance with MIC assays and E. L. Zechner for critically reading the manuscript. This work was supported by the BioTechMed-Graz Flagship Project SECRETOME to S.S, the Austrian FWF grants P25691 to S.S., P27654 to S.S., P29404 to J. R. and W901-B12 (DK Molecular Enzymology) to F.G.Z., F.C., and S.S. as well as NIH grants AI-139105 and GM-069338 to Z.G.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
DECLARATION OF INTERESTS
The authors declare no competing interests.
REFERENCES
- Ahmad S, Ahmad M, Khan S, Ahmad F, Nawaz S, and Khan FU (2017). An overview on phase variation, mechanisms and roles in bacterial adaptation. J Pak Med Assoc. 67(2), 285–291. [PubMed] [Google Scholar]
- Angelichio MJ, Spector J, Waldor MK, and Camilli A (1999). Vibrio cholerae intestinal population dynamics in the suckling mouse model of infection. Infect Immun. 67(8), 3733–3739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bauwens A, Kunsmann L, Karch H, Mellmann A, and Bielaszewska M (2017). Antibiotic-Mediated Modulations of Outer Membrane Vesicles in Enterohemorrhagic Escherichia coli O104:H4 and O157:H7. Antimicrob Agents Chemother. 61(9). Published online 2017/06/14 DOI: 10.1128/AAC.00937-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benz R, and Bauer K (1988). Permeation of hydrophilic molecules through the outer membrane of Gram-negative bacteria. review on bacterial porins. Eur. Biol. Chem 176, 1–19. [DOI] [PubMed] [Google Scholar]
- Beveridge TJ (1999). Structures of gram-negative cell walls and their derived membrane vesicles. J Bacteriol. 181(16), 4725–4733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bilecen K, Fong JC, Cheng A, Jones CJ, Zamorano-Sanchez D, and Yildiz FH (2015). Polymyxin B resistance and biofilm formation in Vibrio cholerae are controlled by the response regulator CarR. Infect Immun. 83(3), 1199–1209. Published online 2015/01/15 DOI: 10.1128/IAI.02700-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonnington KE, and Kuehn MJ (2016). Outer Membrane Vesicle Production Facilitates LPS Remodeling and Outer Membrane Maintenance in Salmonella during Environmental Transitions. mBio. 7(5). DOI: 10.1128/mBio.01532-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown L, Wolf JM, Prados-Rosales R, and Casadevall A (2015). Through the wall: extracellular vesicles in Gram-positive bacteria, mycobacteria and fungi. Nat Rev Microbiol. 13(10), 620–630. DOI: 10.1038/nrmicro3480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Butler SM, and Camilli A (2004). Both chemotaxis and net motility greatly influence the infectivity of Vibrio cholerae. Proc Natl Acad Sci USA. 101(14), 5018–5023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cakar F, Zingl FG, Moisi M, Reidl J, and Schild S (2018). In vivo repressed genes of Vibrio cholerae reveal inverse requirements of an H(+)/Cl(−) transporter along the gastrointestinal passage. Proc Natl Acad Sci U S A. 115(10), E2376–E2385. DOI: 10.1073/pnas.1716973115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Camilli A, and Mekalanos JJ (1995). Use of recombinase gene fusions to identify Vibrio cholerae genes induced during infection. Mol Microbiol. 18(4), 671–683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carlone ACY, Myrtle LT, Rumschlag HS, and Sottner FO (1986). Rapid microprocedure for isolating detergent-insoluble outer membrane proteins from Haemophilus-species. J. Clin. Microbiol 24, 330–332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chaban B, Hughes HV, and Beeby M (2015). The flagellum in bacterial pathogens: For motility and a whole lot more. Semin Cell Dev Biol. 46, 91–103. DOI: 10.1016/j.semcdb.2015.10.032. [DOI] [PubMed] [Google Scholar]
- Chakrabarti SR, Chaudhuri K, Sen K, and Das J (1996). Porins of Vibrio cholerae: purification and characterization of OmpU. J Bacteriol. 178(2), 524–530. Published online 1996/01/01 DOI: 10.1128/jb.178.2.524-530.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conner JG, Teschler JK, Jones CJ, and Yildiz FH (2016). Staying Alive: Vibrio cholerae’s Cycle of Environmental Survival, Transmission, and Dissemination. Microbiology spectrum. 4(2). DOI: 10.1128/microbiolspec.VMBF-0015-2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cress BF, Englaender JA, He W, Kasper D, Linhardt RJ, and Koffas MA (2014). Masquerading microbial pathogens: capsular polysaccharides mimic host-tissue molecules. FEMS Microbiol Rev. 38(4), 660–697. DOI: 10.1111/1574-6976.12056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donnenberg MS, and Kaper JB (1991). Construction of an eae deletion mutant of enteropathogenic Escherichia coli by using a positive-selection suicide vector. Infect. Immun 59, 4310–4317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elluri S, Enow C, Vdovikova S, Rompikuntal PK, Dongre M, Carlsson S, Pal A, Uhlin BE, and Wai SN (2014). Outer membrane vesicles mediate transport of biologically active Vibrio cholerae cytolysin (VCC) from V. cholerae strains. PLoS One. 9(9), e106731 DOI: 10.1371/journal.pone.0106731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fengler VH, Boritsch EC, Tutz S, Seper A, Ebner H, Roier S, Schild S, and Reidl J (2012). Disulfide bond formation and ToxR activity in Vibrio cholerae. PLoS One. 7(10), e47756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guerrero-Mandujano A, Hernandez-Cortez C, Ibarra JA, and Castro-Escarpulli G (2017). The outer membrane vesicles: Secretion system type zero. Traffic. 18(7), 425–432. DOI: 10.1111/tra.12488. [DOI] [PubMed] [Google Scholar]
- Gui MJ, Dashper SG, Slakeski N, Chen YY, and Reynolds EC (2016). Spheres of influence: Porphyromonas gingivalis outer membrane vesicles. Molecular oral microbiology. 31(5), 365–378. Published online 2015/10/16 DOI: 10.1111/omi.12134. [DOI] [PubMed] [Google Scholar]
- Gumpenberger T, Vorkapic D, Zingl FG, Pressler K, Lackner S, Seper A, Reidl J, and Schild S (2016). Nucleoside uptake in Vibrio cholerae and its role in the transition fitness from host to environment. Mol Microbiol. 99(3), 470–483. DOI: 10.1111/mmi.13143. [DOI] [PubMed] [Google Scholar]
- Gunn JS, Lim KB, Krueger J, Kim K, Guo L, Hackett M, and Miller SI (1998). PmrA-PmrB-regulated genes necessary for 4-aminoarabinose lipid A modification and polymyxin resistance. Mol. Microbiol 27, 1171–1182. [DOI] [PubMed] [Google Scholar]
- Guzman L-M, Beblin D, Carson MJ, and Beckwith J (1995). Tight regulation, modulation, and high-level expression by vectors containing the arabinose pBAD promotor. J. Bacteriol 177, 4121–4130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hancock RE (1984). Alterations in outer membrane permeability. Annu Rev Microbiol. 38, 237–264. DOI: 10.1146/annurev.mi.38.100184.001321. [DOI] [PubMed] [Google Scholar]
- Hankins JV, Madsen JA, Giles DK, Brodbelt JS, and Trent MS (2012). Amino acid addition to Vibrio cholerae LPS establishes a link between surface remodeling in gram-positive and gram-negative bacteria. Proc Natl Acad Sci USA. 109(22), 8722–8727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hankins JV, Madsen JA, Giles DK, Childers BM, Klose KE, Brodbelt JS, and Trent MS (2011). Elucidation of a novel Vibrio cholerae lipid A secondary hydroxyl-acyltransferase and its role in innate immune recognition. Mol Microbiol. 81(5), 1313–1329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henderson JC, O’Brien JP, Brodbelt JS, and Trent MS (2013). Isolation and chemical characterization of lipid A from gram-negative bacteria. J Vis Exp. (79), e50623 DOI: 10.3791/50623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herrera CM, Crofts AA, Henderson JC, Pingali SC, Davies BW, and Trent MS (2014). The Vibrio cholerae VprA-VprB two-component system controls virulence through endotoxin modification. mBio. 5(6). Published online 2014/12/30 DOI: 10.1128/mBio.02283-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hughes GW, Hall SCL, Laxton CS, Sridhar P, Mahadi AH, Hatton C, Piggot TJ, Wotherspoon PJ, Leney AC, Ward DG, et al. (2019). Evidence for phospholipid export from the bacterial inner membrane by the Mla ABC transport system. Nat Microbiol. 4(10), 1692–1705. Published online 2019/06/27 DOI: 10.1038/s41564-019-0481-y. [DOI] [PubMed] [Google Scholar]
- Jan AT (2017). Outer Membrane Vesicles (OMVs) of Gram-negative Bacteria: A Perspective Update. Frontiers in microbiology. 8, 1053 DOI: 10.3389/fmicb.2017.01053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamischke C, Fan J, Bergeron J, Kulasekara HD, Dalebroux ZD, Burrell A, Kollman JM, and Miller SI (2019). The Acinetobacter baumannii Mla system and glycerophospholipid transport to the outer membrane. eLife. 8 Published online 2019/01/15 DOI: 10.7554/eLife.40171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang D, Gho YS, Suh M, and Kang C (2002). Highly sensitive and fast protein detection with coomassie brilliant blue in sodium dodecyl sulfate-polyacrylamide gel electrophoresis. Bull. Kor. Chem. Soc 23(11), 1511–1512. [Google Scholar]
- Klein S, Lorenzo C, Hoffmann S, Walther JM, Storbeck S, Piekarski T, Tindall BJ, Wray V, Nimtz M, and Moser J (2009). Adaptation of Pseudomonas aeruginosa to various conditions includes tRNA-dependent formation of alanyl-phosphatidylglycerol. Mol Microbiol. 71(3), 551–565. Published online 2008/12/18 DOI: 10.1111/j.1365-2958.2008.06562.x. [DOI] [PubMed] [Google Scholar]
- Kolter R, Inuzuka M, and Helinski DR (1978). Trans-complementation-dependent replication of a low molecular weight origin fragment from plasmid R6K. Cell. 15, 1199–1208. [DOI] [PubMed] [Google Scholar]
- Kulkarni HM, Nagaraj R, and Jagannadham MV (2015). Protective role of E. coli outer membrane vesicles against antibiotics. Microbiol Res. 181, 1–7. Published online 2015/12/08 DOI: 10.1016/j.micres.2015.07.008. [DOI] [PubMed] [Google Scholar]
- Kulp A, and Kuehn MJ (2010). Biological functions and biogenesis of secreted bacterial outer membrane vesicles. Annu Rev Microbiol. 64, 163–184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laemmli UK (1970). Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature. 227, 680–685. [DOI] [PubMed] [Google Scholar]
- Lappann M, Otto A, Becher D, and Vogel U (2013). Comparative proteome analysis of spontaneous outer membrane vesicles and purified outer membranes of Neisseria meningitidis. J Bacteriol. 195(19), 4425–4435. Published online 2013/07/31 DOI: 10.1128/JB.00625-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leitner DR, Feichter S, Schild-Prufert K, Rechberger GN, Reidl J, and Schild S (2013). Lipopolysaccharide modifications of a cholera vaccine candidate based on outer membrane vesicles reduce endotoxicity and reveal the major protective antigen. Infect Immun. 81(7), 2379–2393. Published online 2013/05/01 DOI: 10.1128/IAI.01382-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lembke M, Pennetzdorfer N, Tutz S, Koller M, Vorkapic D, Zhu J, Schild S, and Reidl J (2018). Proteolysis of ToxR is controlled by cysteine-thiol redox state and bile salts in Vibrio cholerae. Mol Microbiol. 110(5), 796–810. Published online 2018/09/16 DOI: 10.1111/mmi.14125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li CC, Crawford JA, DiRita VJ, and Kaper JB (2000). Molecular cloning and transcriptional regulation of ompT, a ToxR-repressed gene in Vibrio cholerae. Mol. Microbiol 35, 189–203. [DOI] [PubMed] [Google Scholar]
- Lichtenegger S, Bina I, Roier S, Bauernfeind S, Keidel K, Schild S, Anthony M, and Reidl J (2013). Characterization of lactate utilization and its implication on the physiology of Haemophilus influenzae. Int J Med Microbiol. 304(3–4), 490–498. Published online 2014/03/29 DOI: 10.1016/j.ijmm.2014.02.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin J, Huang S, and Zhang Q (2002). Outer membrane proteins: key players for bacterial adaptation in host niches. Microbes Infect. 4(3), 325–331. Published online 2002/03/23. [DOI] [PubMed] [Google Scholar]
- Lin J, Zhang W, Cheng J, Yang X, Zhu K, Wang Y, Wei G, Qian PY, Luo ZQ, and Shen X (2017). A Pseudomonas T6SS effector recruits PQS-containing outer membrane vesicles for iron acquisition. Nature communications. 8, 14888 Published online 2017/03/30 DOI: 10.1038/ncomms14888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malinverni JC, and Silhavy TJ (2009). An ABC transport system that maintains lipid asymmetry in the gram-negative outer membrane. Proc Natl Acad Sci U S A. 106(19), 8009–8014. DOI: 10.1073/pnas.0903229106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manning AJ, and Kuehn MJ (2011). Contribution of bacterial outer membrane vesicles to innate bacterial defense. BMC Microbiol. 11, 258 Published online 2011/12/03 DOI: 10.1186/1471-2180-11-258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manoil C (1991). Analysis of membrane protein topology using alkaline phosphatase and beta-galactosidase gene fusions. Methods Cell Biol. 34, 61–75. [DOI] [PubMed] [Google Scholar]
- Matson JS, Livny J, and DiRita VJ (2017). A putative Vibrio cholerae two-component system controls a conserved periplasmic protein in response to the antimicrobial peptide polymyxin B. PLoS One. 12(10), e0186199 Published online 2017/10/12 DOI: 10.1371/journal.pone.0186199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mey AR, Craig SA, and Payne SM (2012). Effects of amino acid supplementation on porin expression and ToxR levels in Vibrio cholerae. Infect Immun. 80(2), 518–528. DOI: 10.1128/IAI.05851-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller JH (1972). Experiments in molecular genetics Cold Spring Harbor Laboratory, Cold Spring Harbor, N. Y. [Google Scholar]
- Miller VL, DiRita VJ, and Mekalanos JJ (1989). Identification of toxS, a regulatory gene whose product enhances toxR-mediated activation of the cholera toxin promoter. J Bacteriol. 171(3), 1288–1293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller VL, and Mekalanos JJ (1988). A novel suicide vector and its use in construction of insertion mutations: osmoregulation of outer membrane proteins and virulence determinants in Vibrio cholerae requires toxR. J. Bacteriol 170(6), 2575–2583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moisi M, Jenul C, Butler SM, New A, Tutz S, Reidl J, Klose KE, Camilli A, and Schild S (2009). A novel regulatory protein involved in motility of Vibrio cholerae. J Bacteriol. 191(22), 7027–7038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moisi M, Lichtenegger S, Tutz S, Seper A, Schild S, and Reidl J (2013). Characterizing the Hexose-6-Phosphate Transport System of Vibrio cholerae, a Utilization System for Carbon and Phosphate Sources. J Bacteriol. 195(8), 1800–1808. Published online 2013/02/19 DOI: 10.1128/JB.01952-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morales VM, Backman A, and Bagdasarian M (1991). A series of wide-host-range low-copy-number vectors that allow direct screening for recombinants. Gene. 97(1), 39–47. [DOI] [PubMed] [Google Scholar]
- O′Connor A, and McClean S (2017). The role of Universal Stress Proteins in Bacterial Infections. Curr Med Chem. November 24;24(36):3970–3979. DOI: 10.2174/0929867324666170124145543. [DOI] [PubMed] [Google Scholar]
- Osorio CG, Crawford JA, Michalski J, Martinez-Wilson H, Kaper JB, and Camilli A (2005). Second-generation recombination-based in vivo expression technology for large-scale screening for Vibrio cholerae genes induced during infection of the mouse small intestine. Infect Immun. 73(2), 972–980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Osorio CG, Martinez-Wilson H, and Camilli A (2004). The ompU Paralogue vca1008 is required for virulence of Vibrio cholerae. J Bacteriol. 186(15), 5167–5171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pressler K, Vorkapic D, Lichtenegger S, Malli G, Barilich BP, Cakar F, Zingl FG, Reidl J, and Schild S (2016). AAA+ proteases and their role in distinct stages along the Vibrio cholerae lifecycle. Int J Med Microbiol. DOI: 10.1016/j.ijmm.2016.05.013. [DOI] [PubMed] [Google Scholar]
- Provenzano D, and Klose KE (2000). Altered expression of the ToxR-regulated porins OmpU and OmpT diminishes Vibrio cholerae bile resistance, virulence factor expression, and intestinal colonization. Proc Natl Acad Sci U S A. 97(18), 10220–10224. DOI: 10.1073/pnas.170219997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roier S, Fenninger JC, Leitner DR, Rechberger GN, Reidl J, and Schild S (2013). Immunogenicity of Pasteurella multocida and Mannheimia haemolytica outer membrane vesicles. Int J Med Microbiol. 303(5), 247–256. Published online 2013/06/05 DOI: 10.1016/j.ijmm.2013.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roier S, Zingl FG, Cakar F, Durakovic S, Kohl P, Eichmann TO, Klug L, Gadermaier B, Weinzerl K, Prassl R, et al. (2016). A novel mechanism for the biogenesis of outer membrane vesicles in Gram-negative bacteria. Nature communications. 7, 10515 DOI: 10.1038/ncomms10515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salem W, Leitner DR, Zingl FG, Schratter G, Prassl R, Goessler W, Reidl J, and Schild S (2015). Antibacterial activity of silver and zinc nanoparticles against Vibrio cholerae and enterotoxic Escherichia coli. Int J Med Microbiol. 305(1), 85–95. DOI: 10.1016/j.ijmm.2014.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schild S, Nelson EJ, Bishop AL, and Camilli A (2009). Characterization of Vibrio cholerae outer membrane vesicles as a candidate vaccine for cholera. Infect Immun. 77(1), 472–484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schild S, Nelson EJ, and Camilli A (2008). Immunization with Vibrio cholerae outer membrane vesicles induces protective immunity in mice. Infect Immun. 76(10), 4554–4563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schild S, Tamayo R, Nelson EJ, Qadri F, Calderwood SB, and Camilli A (2007). Genes induced late in infection increase fitness of Vibrio cholerae after release into the environment. Cell Host Microbe. 2(4), 264–277. DOI: 10.1016/j.chom.2007.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwechheimer C, and Kuehn MJ (2015). Outer-membrane vesicles from Gram-negative bacteria: biogenesis and functions. Nat Rev Microbiol. 13(10), 605–619. DOI: 10.1038/nrmicro3525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seper A, Fengler VH, Roier S, Wolinski H, Kohlwein SD, Bishop AL, Camilli A, Reidl J, and Schild S (2011). Extracellular nucleases and extracellular DNA play important roles in Vibrio cholerae biofilm formation. Mol Microbiol. 82(4), 1015–1037. DOI: 10.1111/j.1365-2958.2011.07867.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seper A, Hosseinzadeh A, Gorkiewicz G, Lichtenegger S, Roier S, Leitner DR, Rohm M, Grutsch A, Reidl J, Urban CF, et al. (2013). Vibrio cholerae Evades Neutrophil Extracellular Traps by the Activity of Two Extracellular Nucleases. PLoS Pathog. 9(9), e1003614 Published online 2013/09/17 DOI: 10.1371/journal.ppat.1003614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simonet VC, Basle A, Klose KE, and Delcour AH (2003). The Vibrio cholerae porins OmpU and OmpT have distinct channel properties. J Biol Chem. 278(19), 17539–17545. [DOI] [PubMed] [Google Scholar]
- Tamayo R, Schild S, Pratt JT, and Camilli A (2008). Role of cyclic Di-GMP during el tor biotype Vibrio cholerae infection: characterization of the in vivo-induced cyclic Di-GMP phosphodiesterase CdpA. Infect Immun. 76(4), 1617–1627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Urashima A, Sanou A, Yen H, and Tobe T (2017). Enterohaemorrhagic Escherichia coli produces outer membrane vesicles as an active defence system against antimicrobial peptide LL-37. Cell Microbiol. 19(11). Published online 2017/06/18 DOI: 10.1111/cmi.12758. [DOI] [PubMed] [Google Scholar]
- van der Woude MW, and Baumler AJ (2004). Phase and antigenic variation in bacteria. Clin Microbiol Rev. 17(3), 581–611, table of contents. DOI: 10.1128/CMR.17.3.581-611.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warburg O, and Christian W (1941). Isolierung und Kristalisation des Gärungsferments Enolase. Biochem. Z 310, 348–421. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
This study did not generate/ analyze datasets/ codes.