

Summary
An outbreak of coronavirus disease 2019 (COVID-19) caused by the 2019 novel coronavirus (SARS-CoV-2) began in the city of Wuhan in China and has widely spread worldwide. Currently, it is vital to explore potential intermediate hosts of SARS-CoV-2 to control COVID-19 spread. Therefore, we reinvestigated published data from pangolin lung samples from which SARS-CoV-like CoVs were detected by Liu et al. [1]. We found genomic and evolutionary evidence of the occurrence of a SARS-CoV-2-like CoV (named Pangolin-CoV) in dead Malayan pangolins. Pangolin-CoV is 91.02% and 90.55% identical to SARS-CoV-2 and BatCoV RaTG13, respectively, at the whole-genome level. Aside from RaTG13, Pangolin-CoV is the most closely related CoV to SARS-CoV-2. The S1 protein of Pangolin-CoV is much more closely related to SARS-CoV-2 than to RaTG13. Five key amino acid residues involved in the interaction with human ACE2 are completely consistent between Pangolin-CoV and SARS-CoV-2, but four amino acid mutations are present in RaTG13. Both Pangolin-CoV and RaTG13 lost the putative furin recognition sequence motif at S1/S2 cleavage site that can be observed in the SARS-CoV-2. Conclusively, this study suggests that pangolin species are a natural reservoir of SARS-CoV-2-like CoVs.
Keywords: pangolin, SARS-CoV-2, COVID-19, origin
Graphical Abstract
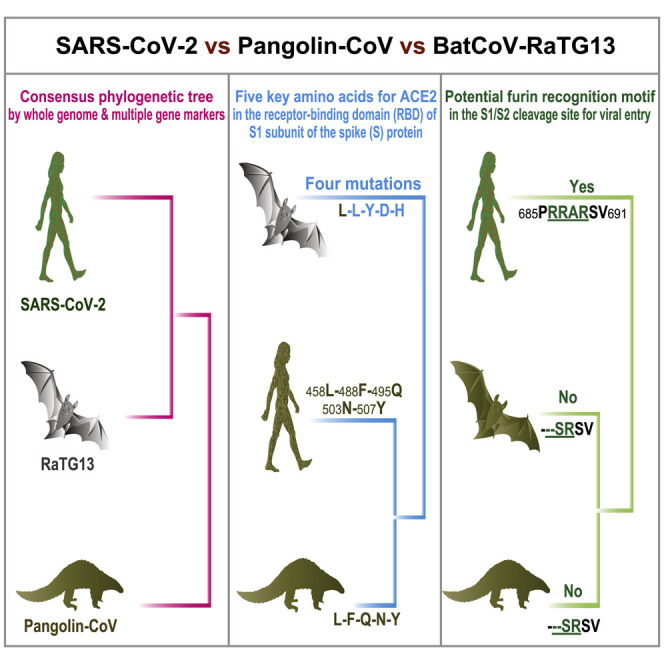
Highlights
-
•
Pangolin-CoV is 91.02% identical to SARS-CoV-2 at the whole-genome level
-
•
Pangolin-CoV is the second closest relative of SARS-CoV-2 behind RaTG13
-
•
Five key amino acids in the RBD are consistent between Pangolin-CoV and SARS-CoV-2
-
•
Only SARS-CoV-2 contains a potential cleavage site for furin proteases
The emerging SARS-coronavirus 2 (SARS-CoV-2) poses tremendous threat to human health. Zhang, Wu et al. show that like bats, pangolin species are a natural reservoir of SARS-CoV-2-like CoVs. This finding might help to find the intermediate host of SARS-CoV-2 for blocking a global coronavirus pandemic.
Results and Discussion
Similar to the case for SARS-CoV and MERS-CoV [2], the bat is still a probable species of origin for 2019 novel coronavirus (SARS-CoV-2) because SARS-CoV-2 shares 96% whole-genome identity with a bat CoV, BatCoV RaTG13, from Rhinolophus affinis from Yunnan Province [3]. However, SARS-CoV and MERS-CoV usually pass into intermediate hosts, such as civets or camels, before leaping to humans [4]. This fact indicates that SARS-CoV-2 was probably transmitted to humans by other animals. Considering that the earliest coronavirus disease 2019 (COVID-19) patient reported no exposure at the seafood market [5], it is vital to find the intermediate SARS-CoV-2 host to block interspecies transmission. On 24 October 2019, Liu and his colleagues from the Guangdong Wildlife Rescue Center of China [1] first detected the existence of a SARS-CoV-like CoV from lung samples of two dead Malayan pangolins with a frothy liquid in their lungs and pulmonary fibrosis, and this fact was discovered close to when the COVID-19 outbreak occurred. Using their published results, we showed that all virus contigs assembled from two lung samples (lung07 and lung08) exhibited low identities, ranging from 80.24% to 88.93%, with known SARSr-CoVs. Hence, we conjectured that the dead Malayan pangolins may carry a new CoV closely related to SARS-CoV-2.
Assessing the Probability of SARS-CoV-2-like CoV Presence in Pangolin Species
To confirm our assumption, we downloaded raw RNA sequencing (RNA-seq) data (SRA: PRJNA573298) for those two lung samples from the SRA and conducted consistent quality control and contaminant removal, as described by Liu’s study [1]. We found 1,882 clean reads from the lung08 sample that mapped to the SARS-CoV-2 reference genome (GenBank: MN908947) [6] and covered 76.02% of the SARS-CoV-2 genome. We performed de novo assembly of those reads and obtained 36 contigs with lengths ranging from 287 bp to 2,187 bp, with a mean length of 700 bp. Via Blast analysis against proteins from 2,845 CoV reference genomes, including RaTG13, SARS-CoV-2s, and other known CoVs, we found that 22 contigs were best matched to SARS-CoV-2s (70.6%–100% amino acid identity; average: 95.41%) and that 12 contigs matched to bat SARS-CoV-like CoV (92.7%–100% amino acid identity; average: 97.48%) (Table S1). These results indicate that the Malayan pangolin might carry a novel CoV (here named Pangolin-CoV) that is similar to SARS-CoV-2.
Draft Genome of Pangolin-CoV and Its Genomic Characteristics
Using a reference-guided scaffolding approach, we created a Pangolin-CoV draft genome (19,587 bp) based on the above 34 contigs. To reduce the effect of raw read errors on scaffolding quality, small fragments that aligned against the reference genome with a length less than 25 bp were manually discarded if they were unable to be covered by any large fragments or reference genome. Remapping 1,882 reads against the draft genome resulted in 99.99% genome coverage (coverage depth range: 1X–47X) (Figure 1 A). The mean coverage depth was 7.71X across the whole genome, which was two times higher than the lowest common 3X read coverage depth for SNP calling based on low-coverage sequencing in the 1000 Genomes Project pilot phase [7]. Similar coverage levels are also sufficient to detect rare or low-abundance microbial species from metagenomic datasets [8], indicating that our assembled Pangolin-CoV draft genome is reliable for further analyses. Based on Simplot analysis [9], Pangolin-CoV showed high overall genome sequence identity to RaTG13 (90.55%) and SARS-CoV-2 (91.02%) throughout the genome (Figure 1B), although there was a higher identity (96.2%) between SARS-CoV-2 and RaTG13 [3]. Other SARS-CoV-like CoVs similar to Pangolin-CoV were bat SARSr-CoV ZXC21 (85.65%) and bat SARSr-CoV ZC45 (85.01%). While this manuscript was under review, two similar preprint studies found that CoVs in pangolins shared 90.3% [10] and 92.4% [11] DNA identity with SARS-CoV-2, approximating the 91.02% identity to SARS-CoV-2 observed here and supporting our findings. Taken together, these results indicate that Pangolin-CoV might be the common origin of SARS-CoV-2 and RaTG13.
Figure 1.

Genome-Related Analysis
(A) Sequence depth of reads remapped to Pangolin-CoV.
(B) Similarity plot based on the full-length genome sequence of Pangolin-CoV. Full-length genome sequences of SARS-CoV-2 (Beta-CoV/Wuhan-Hu-1), BatCoV RaTG13, bat SARSr-CoV 21, bat SARSr-CoV45, bat SARSr-CoV WIV1, and SARS-CoV BJ01 were used as reference sequences.
(C) Comparison of common genome organization similarity among SARS-CoV-2, Pangolin-CoV, and BatCoV RaTG13.
Related to Table S2.
The Pangolin-CoV genome organization was characterized by sequence alignment against SARS-CoV-2 (GenBank: MN908947) and RaTG13. The Pangolin-CoV genome consists of six major open reading frames (ORFs) common to CoVs and four other accessory genes (Figure 1C; Table S2). Further analysis indicated that Pangolin-CoV genes aligned to SARS-CoV-2 genes with coverage ranging from 45.8% to 100% (average coverage 76.9%). Pangolin-CoV genes shared high average nucleotide and amino acid identity with both SARS-CoV-2 (GenBank: MN908947) (93.2% nucleotide/94.1% amino acid identity) and RaTG13 (92.8% nucleotide/93.5% amino acid identity) genes (Figure 1C; Table S2). Surprisingly, some Pangolin-CoV genes showed higher amino acid sequence identity to SARS-CoV-2 genes than to RaTG13 genes, including orf1b (73.4%/72.8%), the spike (S) protein (97.5%/95.4%), orf7a (96.9%/93.6%), and orf10 (97.3%/94.6%). The high S protein amino acid identity implies functional similarity between Pangolin-CoV and SARS-CoV-2.
Phylogenetic Relationships among Pangolin-CoV, RaTG13, and SARS-CoV-2
To determine the evolutionary relationships among Pangolin-CoV, SARS-CoV-2, and previously identified CoVs, we estimated phylogenetic trees based on the nucleotide sequences of the whole-genome sequence, RNA-dependent RNA polymerase gene (RdRp), non-structural protein genes ORF1a and ORF1b, and main structural proteins encoded by the S and M genes. In all phylogenies, Pangolin-CoV, RaTG13, and SARS-CoV-2 were clustered into a well-supported group, here named the “SARS-CoV-2 group” (Figures 2 , S1, and S2). This group represents a novel Betacoronavirus group. Within this group, RaTG13 and SARS-CoV-2 were grouped together, and Pangolin-CoV was their closest common ancestor. However, whether the basal position of the SARS-CoV-2 group is SARSr-CoV ZXC21 and/or SARSr-CoV ZC45 is still under debate. Such debate also occurred in both the Wu et al. [6] and Zhou et al. [3] studies. A possible explanation is a past history of recombination in the Betacoronavirus group [6]. It is noteworthy that the discovered evolutionary relationships of CoVs shown by the whole genome, RdRp gene, and S gene were highly consistent with those exhibited by complete genome information in the Zhou et al. study [3]. This correspondence indicates that our Pangolin-CoV draft genome has enough genomic information to trace the true evolutionary position of Pangolin-CoV in CoVs.
Figure 2.

Phylogenetic Relationship of CoVs Based on the Whole Genome and RdRp Gene Nucleotide Sequences
Red text denotes the Malayan Pangolin-CoV. Pink text denotes SARS-CoV-2. Green text denotes a bat CoV with 96% similarity at the genome level to SARS-CoV-2. Blue text denotes the reference CoVs used in Figure 1B. Detailed information can be found in the STAR Methods. Related to Figures S1–S3.
Dualism of the S Protein of Pangolin-CoV
The CoV S protein consists of two subunits (S1 and S2), mediates infection of receptor-expressing host cells, and is a critical target for antiviral neutralizing antibodies [12]. S1 contains a receptor-binding domain (RBD) that consists of an approximately 193 amino acid fragment, which is responsible for recognizing and binding the cell surface receptor [13, 14]. Zhou et al. experimentally confirmed that SARS-CoV-2 is able to use human, Chinese horseshoe bat, civet, and pig ACE2 proteins as an entry receptor in ACE2-expressing cells [3], suggesting that the RBD of SARS-CoV-2 mediates infection in humans and other animals. To gain sequence-level insight into the pathogenic potential of Pangolin-CoV, we first investigated the amino acid variation pattern of the S1 proteins from Pangolin-CoV, SARS-CoV-2, RaTG13, and other representative SARS/SARSr-CoVs. The amino acid phylogenetic tree showed that the S1 protein of Pangolin-CoV is more closely related to that of 2019-CoV than to that of RaTG13. Within the RBD, we further found that Pangolin-CoV and SARS-CoV-2 were highly conserved, with only one amino acid change (500H/500Q) (Figure 3 ), which is not one of the five key residues involved in the interaction with human ACE2 [3, 14]. These results indicate that Pangolin-CoV could have pathogenic potential similar to that of SARS-CoV-2. In contrast, RaTG13 has changes in 17 amino acid residues, 4 of which are among the key amino acid residues (Figure 3). There are evidences suggesting that the change of 472L (SARS-CoV) to 486F (SARS-CoV-2) (corresponding to the second key amino acid residue change in Figure 3) may make stronger van der Waals contact with M82 (ACE2) [15]. Besides, the major substitution of 404V in the SARS-CoV-RBD with 417K in the SARS-CoV-2-RBD (see 420 alignment position in Figure 3 and without amino acid change between the SARS-CoV-2 and RaTG13) may result in tighter association because of the salt bridge formation between 417K and 30D of ACE2 [15]. Nevertheless, further investigation is still needed about whether those mutations affect the affinity for ACE2. Whether the Pangolin-CoV or RaTG13 are potential infectious agents to humans remains to be determined.
Figure 3.

Amino Acid Sequence Alignment of the S1 Protein and Its Phylogeny
The receptor-binding motif of SARS-CoV and the homologous region of other CoVs are indicated by the gray box. The key amino acid residues involved in the interaction with human ACE2 are marked with the orange box. Bat SARS-CoV-like CoVs had been reported to not use ACE2 and have amino acid deletions at two motifs marked by the yellow box. Detailed information can be found in the STAR Methods.
The S1/S2 cleavage site in the S protein is also an important determinant of the transmissibility and pathogenicity of SARS-CoV/SARS-CoVr viruses [16]. The trimetric S protein is processed at the S1/S2 cleavage site by host cell proteases during infection. Following cleavage, also known as priming, the protein is divided into an N-terminal S1-ectodomain that recognizes a cognate cell surface receptor and a C-terminal S2-membrane anchored protein that drives fusion of the viral envelope with a cellular membrane. We found that the SARS-CoV-2 S protein contains a putative furin recognition motif (PRRARSV) (Figure 4 ) similar to that of MERS-CoV, which has a PRSVRSV motif that is likely cleaved by furin [16, 17] during virus egress. Conversely, the furin sequence motif at the S1/S2 site is missing in the S protein of Pangolin-CoV and all other SARS/SARSr-CoVs. This difference indicates the SARS-CoV-2 might gain a distinct mechanism to promote its entry into host cells [18]. Interestingly, aside from MERS-CoV, similar sequence patterns to the SARS-CoV-2 were also presented in some members of Alphacoronavirus, Betacoronavirus, and Gammcoronavirus [19], raising an interesting question regarding whether this furin sequence motif in SARS-CoV-2 might be derived from those existing S proteins of other coronaviruses or alternatively if the SARS-CoV-2 might be the recombinant of Pangolin-CoV or RaTG13 and other coronaviruses with a similar furin recognition motif in the unknown intermediate host.
Figure 4.

CoV S Protein S1/S2 Cleavage Sites
Four amino acid insertions (SPRRs) unique to SARS-CoV-2 are marked in yellow. Conserved S1/S2 cleavage sites are marked in green.
Amino Acid Variations in the Nucleocapsid (N) Protein for Potential Diagnosis
The N protein is the most abundant protein in CoVs. The N protein is a highly immunogenic phosphoprotein, and it is normally very conserved. The CoV N protein is often used as a marker in diagnostic assays. To gain further insight into the diagnostic potential of Pangolin-CoV, we investigated the amino acid variation pattern of the N proteins from Pangolin-CoV, SARS-CoV-2, RaTG13, and other representative SARS-CoVs. Phylogenetic analysis based on the N protein supported the classification of Pangolin-CoV as a sister taxon of SARS-CoV-2 and RaTG13 (Figure S3). We further found seven amino acid mutations that differentiated our defined “SAR-CoV-2 group” CoVs (12N, 26 G, 27S, 104D, 218A, 335T, 346N, and 350Q) from other known SARS-CoVs (12S, 26D, 27N, 104E, 218T, 335H, 346Q, and 350N). Two amino acid sites (38P and 268Q) are shared by Pangolin-CoV, RaTG13, and SARS-CoVs, which are mutated to 38S and 268A in SARS-CoV-2. Only one amino acid residue shared by Pangolin-CoV and other SARS-CoVs (129E) is consistently different in both SARS-CoV-2 and RaTG13 (129D). The observed amino acid changes in the N protein would be useful for developing antigens with improved sensitivity for SARS-CoV-2 serological detection.
Conclusion
Based on published metagenomic data, this study provides the first report on a potential closely related kin (Pangolin-CoV) of SARS-CoV-2, which was discovered from dead Malayan pangolins after extensive rescue efforts. Aside from RaTG13, the Pangolin-CoV is the CoV most closely related to SARS-CoV-2. Due to unavailability of the original sample, we did not perform further experiments to confirm our findings, including PCR validation, serological detection, or even isolation of the virus particles. Our discovered Pangolin-CoV genome showed 91.02% nucleotide identity with the SARS-CoV-2 genome. However, whether pangolin species are good candidates for SARS-CoV-2 origin is still under debate. Considering the wide spread of SARSr-CoVs in natural reservoirs, such as bats, camels, and pangolins, our findings would be meaningful for finding novel intermediate SARS-CoV-2 hosts to block interspecies transmission.
STAR★Methods
Key Resources Table
Lead Contact and Materials Availability
Requests for further information and data resources should be directed to and will be fulfilled by the Lead Contact, Zhigang Zhang (zhangzhigang@ynu.edu.cn). This study did not generate new unique reagents.
Method Details
Data collection and preprocessing
We downloaded raw data for the lung08 and lung07 samples published in Liu’s study [1] from the NCBI SRA under BioProject SRA: PRJNA573298. Raw reads were first adaptor and quality trimmed using the Trimmomatic program (version 0.39) [20]. To remove host contamination, Bowtie2 (version 2.3.4.3) [21] was used to map clean reads to the host reference genome of Manis javanica (SRA: PRJNA256023). Only unmapped reads were mapped to the SARS-CoV-2 reference genome (GenBank: MN908947) for identifying virus reads.
Genome assembly and gene prediction
Virus-mapped reads were assembled de novo using MEGAHIT (version 1.1.3) [22]. Read remapping to assembled contigs was performed by using Bowtie2 [21]. Mapping coverage and depth were determined using Samtools (version 1.9) [24]. Contigs were taxonomically annotated using BLAST 2.9.0+ [23] against 2845 CoV reference genomes (Table S1). The BatCoV RaTG13 genome was downloaded from the NGDC database (https://bigd.big.ac.cn/) (accession number GWHABKP00000000) [3]. The SARS-CoV-2 reference genome was downloaded from NCBI (accession number MN908947) [6]. Other CoV genomes were downloaded from the ViPR database (https://www.viprbrc.org/brc/home.spg?decorator=corona) on 6 February 2020. We further used a reference-guided strategy to construct a draft genome based on contigs taxonomically annotated to SARS-CoV-2 s, SARS-CoV, and bat SARS-CoV-like CoV. Each contig was aligned against the SARS-CoV-2 reference genome with MUSCLE software (version 3.8.31) [25]. Aligned contigs were merged into consensus scaffolds with BioEdit version 7.2.5 (http://www.mybiosoftware.com/bioedit-7-0-9-biological-sequence-alignment-editor.html) following manual quality checking. Small fragments less than 25 bp in length were discarded if these fragments were not covered by any large fragments. The potential ORFs of the final draft genome obtained were annotated by alignment to the SARS-CoV-2 reference genome (accession number MN908947). SimPlot 3.5.1 [9] was used to analyze whole genome nucleotide identity.
Phylogeny
Sequence alignment was carried out using MUSCLE software [25]. Alignment accuracy was checked manually base by base. Gblocks [26] was used to process the gap in the aligned sequence. Using MegaX (version 10.1.7) [27], we inferred all maximum likelihood (ML) phylogenetic trees.
Quantification and Statistical Analysis
Using MegaX software [27], we constructed all maximum likelihood (ML) phylogenetic trees under the best-fit DNA/amino acid substitution model with 1000 bootstrap replications. Phylogenetic analyses were performed using the nucleotide sequences of various CoV gene datasets: the whole genome, ORF1a, ORF1b, and the membrane (M), S and RdRp genes. The best model of M was GTR+G, and the best for all the others was GTR+G+I. Two additional protein-based trees were constructed under WAG+G (S1 subunit of the S protein) and JTT+G (N protein). Branches with bootstrap values < 70% were hidden in all phylogenetic trees.
Data and Code Availability
The dataset used in this study is provided as supplementary material (Tables S1 and S2). This study did not generate code.
Acknowledgments
This study was supported by the Second Tibetan Plateau Scientific Expedition and Research (STEP) program (no. 2019QZKK0503), the National Key Research and Development Program of China (no. 2018YFC2000500), the Key Research Program of the Chinese Academy of Sciences (no. KFZD-SW-219), and the Chinese National Natural Science Foundation (no. 31970571).
Author Contributions
Z.Z. performed project planning, coordination, execution, and facilitation. T.Z. and Q.W. performed the metagenomic analysis. T.Z. carried out assemblies, gene prediction, and annotation. Q.W. processed data collection and phylogenetic analysis. Z.Z., T.Z., and Q.W. prepared the manuscript.
Declaration of Interests
The authors declare no competing interests.
Published: March 19, 2020; corrected online: March 27, 2020
Footnotes
Supplemental Information can be found online at https://doi.org/10.1016/j.cub.2020.03.022.
Supplemental Information
References
- 1.Liu P., Chen W., Chen J.-P. Viral metagenomics revealed sendai virus and coronavirus infection of Malayan Pangolins (Manis javanica) Viruses. 2019;11:979. doi: 10.3390/v11110979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Li W., Shi Z., Yu M., Ren W., Smith C., Epstein J.H., Wang H., Crameri G., Hu Z., Zhang H. Bats are natural reservoirs of SARS-like coronaviruses. Science. 2005;310:676–679. doi: 10.1126/science.1118391. [DOI] [PubMed] [Google Scholar]
- 3.Zhou P., Yang X.-L., Wang X.-G., Hu B., Zhang L., Zhang W., Si H.-R., Zhu Y., Li B., Huang C.-L. A pneumonia outbreak associated with a new coronavirus of probable bat origin. Nature. 2020 doi: 10.1038/s41586-020-2012-7. Published online February 3, 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Cui J., Li F., Shi Z.-L. Origin and evolution of pathogenic coronaviruses. Nat. Rev. Microbiol. 2019;17:181–192. doi: 10.1038/s41579-018-0118-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Huang C., Wang Y., Li X., Ren L., Zhao J., Hu Y., Zhang L., Fan G., Xu J., Gu X. Clinical features of patients infected with 2019 novel coronavirus in Wuhan, China. Lancet. 2020;395:497–506. doi: 10.1016/S0140-6736(20)30183-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wu F., Zhao S., Yu B., Chen Y.-M., Wang W., Song Z.-G., Hu Y., Tao Z.-W., Tian J.-H., Pei Y.-Y. A new coronavirus associated with human respiratory disease in China. Nature. 2020 doi: 10.1038/s41586-020-2008-3. Published online February 3, 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Abecasis G.R., Altshuler D., Auton A., Brooks L.D., Durbin R.M., Gibbs R.A., Hurles M.E., McVean G.A., 1000 Genomes Project Consortium A map of human genome variation from population-scale sequencing. Nature. 2010;467:1061–1073. doi: 10.1038/nature09534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Albertsen M., Hugenholtz P., Skarshewski A., Nielsen K.L., Tyson G.W., Nielsen P.H. Genome sequences of rare, uncultured bacteria obtained by differential coverage binning of multiple metagenomes. Nat. Biotechnol. 2013;31:533–538. doi: 10.1038/nbt.2579. [DOI] [PubMed] [Google Scholar]
- 9.Lole K.S., Bollinger R.C., Paranjape R.S., Gadkari D., Kulkarni S.S., Novak N.G., Ingersoll R., Sheppard H.W., Ray S.C. Full-length human immunodeficiency virus type 1 genomes from subtype C-infected seroconverters in India, with evidence of intersubtype recombination. J. Virol. 1999;73:152–160. doi: 10.1128/jvi.73.1.152-160.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Xiao K., Zhai J., Feng Y., Zhou N., Zhang X., Zou J.-J., Li N., Guo Y., Li X., Shen X. Isolation and characterization of 2019-nCoV-like coronavirus from Malayan pangolins. bioRxiv. 2020 doi: 10.1101/2020.02.17.951335. [DOI] [Google Scholar]
- 11.Lam T.T.-Y., Shum M.H.-H., Zhu H.-C., Tong Y.-G., Ni X.-B., Liao Y.-S., Wei W., Cheung W.Y.-M., Li W.-J., Li L.-F. Identification of 2019-nCoV related coronaviruses in Malayan pangolins in southern China. bioRxiv. 2020 doi: 10.1101/2020.02.13.945485. [DOI] [Google Scholar]
- 12.Tortorici M.A., Veesler D. Structural insights into coronavirus entry. In: Rey F.A., editor. Advances in Virus Research. Academic Press; 2019. pp. 93–116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ge X.-Y., Li J.-L., Yang X.-L., Chmura A.A., Zhu G., Epstein J.H., Mazet J.K., Hu B., Zhang W., Peng C. Isolation and characterization of a bat SARS-like coronavirus that uses the ACE2 receptor. Nature. 2013;503:535–538. doi: 10.1038/nature12711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wong S.K., Li W., Moore M.J., Choe H., Farzan M. A 193-amino acid fragment of the SARS coronavirus S protein efficiently binds angiotensin-converting enzyme 2. J. Biol. Chem. 2004;279:3197–3201. doi: 10.1074/jbc.C300520200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Yan R., Zhang Y., Li Y., Xia L., Guo Y., Zhou Q. Structural basis for the recognition of the SARS-CoV-2 by full-length human ACE2. Science. 2020 doi: 10.1126/science.abb2762. Published online March 4, 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Millet J.K., Whittaker G.R. Host cell entry of Middle East respiratory syndrome coronavirus after two-step, furin-mediated activation of the spike protein. Proc. Natl. Acad. Sci. USA. 2014;111:15214–15219. doi: 10.1073/pnas.1407087111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Coutard B., Valle C., de Lamballerie X., Canard B., Seidah N.G., Decroly E. The spike glycoprotein of the new coronavirus 2019-nCoV contains a furin-like cleavage site absent in CoV of the same clade. Antiviral Res. 2020;176:104742. doi: 10.1016/j.antiviral.2020.104742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hoffmann M., Kleine-Weber H., Schroeder S., Krüger N., Herrler T., Erichsen S., Schiergens T.S., Herrler G., Wu N.-H., Nitsche A. SARS-CoV-2 cell entry depends on ACE2 and TMPRSS2 and is blocked by a clinically proven protease inhibitor. Cell. 2020 doi: 10.1016/j.cell.2020.02.052. Published online March 4, 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Millet J.K., Whittaker G.R. Host cell proteases: critical determinants of coronavirus tropism and pathogenesis. Virus Res. 2015;202:120–134. doi: 10.1016/j.virusres.2014.11.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bolger A.M., Lohse M., Usadel B. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics. 2014;30:2114–2120. doi: 10.1093/bioinformatics/btu170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Langmead B., Salzberg S.L. Fast gapped-read alignment with Bowtie 2. Nat. Methods. 2012;9:357–359. doi: 10.1038/nmeth.1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Li D., Liu C.-M., Luo R., Sadakane K., Lam T.-W. MEGAHIT: an ultra-fast single-node solution for large and complex metagenomics assembly via succinct de Bruijn graph. Bioinformatics. 2015;31:1674–1676. doi: 10.1093/bioinformatics/btv033. [DOI] [PubMed] [Google Scholar]
- 23.Camacho C., Coulouris G., Avagyan V., Ma N., Papadopoulos J., Bealer K., Madden T.L. BLAST+: architecture and applications. BMC Bioinformatics. 2009;10:421. doi: 10.1186/1471-2105-10-421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Li H., Handsaker B., Wysoker A., Fennell T., Ruan J., Homer N., Marth G., Abecasis G., Durbin R., 1000 Genome Project Data Processing Subgroup The Sequence Alignment/Map format and SAMtools. Bioinformatics. 2009;25:2078–2079. doi: 10.1093/bioinformatics/btp352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Edgar R.C. MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004;32:1792–1797. doi: 10.1093/nar/gkh340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Talavera G., Castresana J. Improvement of phylogenies after removing divergent and ambiguously aligned blocks from protein sequence alignments. Syst. Biol. 2007;56:564–577. doi: 10.1080/10635150701472164. [DOI] [PubMed] [Google Scholar]
- 27.Kumar S., Stecher G., Li M., Knyaz C., Tamura K. MEGA X: molecular evolutionary genetics analysis across computing platforms. Mol. Biol. Evol. 2018;35:1547–1549. doi: 10.1093/molbev/msy096. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The dataset used in this study is provided as supplementary material (Tables S1 and S2). This study did not generate code.