

Abstract
Transglutaminase 2 (TG2) is a ubiquitously expressed enzyme with transamidating activity. We report here that both expression and activity of TG2 are enhanced in mammalian epithelial cells infected with the obligate intracellular bacteria Chlamydia trachomatis. Genetic or pharmacological inhibition of TG2 impairs bacterial development. We show that TG2 increases glucose import by up‐regulating the transcription of the glucose transporter genes GLUT‐1 and GLUT‐3. Furthermore, TG2 activation drives one specific glucose‐dependent pathway in the host, i.e., hexosamine biosynthesis. Mechanistically, we identify the glucosamine:fructose‐6‐phosphate amidotransferase (GFPT) among the substrates of TG2. GFPT modification by TG2 increases its enzymatic activity, resulting in higher levels of UDP‐N‐acetylglucosamine biosynthesis and protein O‐GlcNAcylation. The correlation between TG2 transamidating activity and O‐GlcNAcylation is disrupted in infected cells because host hexosamine biosynthesis is being exploited by the bacteria, in particular to assist their division. In conclusion, our work establishes TG2 as a key player in controlling glucose‐derived metabolic pathways in mammalian cells, themselves hijacked by C. trachomatis to sustain their own metabolic needs.
Keywords: Chlamydia, GFPT, hexosamine biosynthesis, O‐GlcNAcylation, transglutaminase 2
Subject Categories: Metabolism; Microbiology, Virology & Host Pathogen Interaction; Post-translational Modifications, Proteolysis & Proteomics
Chlamydia trachomatis induces transglutaminase 2‐mediated protein transamidation in infected cells to rewire host cell glucose metabolism and sustain bacterial growth.
Introduction
The enzyme transglutaminase 2 (TG2) is an extremely versatile protein exhibiting transamidase, protein disulfide isomerase and guanine and adenine nucleotide binding and hydrolyzing activities (Gundemir et al, 2012). Also designated as “tissue transglutaminase”, it is ubiquitously expressed in the cytoplasm and at the cell surface in association with the extracellular matrix (Eckert et al, 2014). The transamidase activity is the best described activity, and it is regulated by Ca2+ (Folk et al, 1967). It results in the formation of cross‐links between proteins, or of post‐translational modification of a protein substrate through incorporation of a small primary amine, or deamidation of a glutamine into a glutamate. Under steady‐state conditions, TG2 exists in a compact, inactive conformation. Increase in intracellular Ca2+ concentration (upon stress, cell activation, etc) causes a conformational change, and the enzyme becomes catalytically active as a transamidase. Studies of genetically engineered mouse models and/or inherited disorders have implicated TG2 in several pathological conditions (Iismaa et al, 2009). In particular, increased TG2 expression and transamidation activity is a common feature of many inflammatory diseases and events (Eckert et al, 2014). Possibly linked to its increased expression in inflammatory processes, several lines of evidence suggest the involvement of TG2 during cancer development (Huang et al, 2015).
Surprisingly, while the association between TG2 activity and inflammatory situations has been studied in several normal and pathological situations (Iismaa et al, 2009; Di Sabatino et al, 2012; Huang et al, 2015; Ientile et al, 2015; Liu et al, 2017), the implication of this enzyme in a very classical inflammatory process, e.g., the defense response of a tissue to the invasion by a microorganism, has remained very poorly investigated. Chlamydia trachomatis is the most common sexually transmitted bacterial pathogen, and it develops inside a vacuole in a human host cell, typically an epithelial cell of the genital tract (reviewed in ref. AbdelRahman & Belland, 2005). This obligate intracellular bacterium depends on the host to supply several essential metabolites, and in particular glucose (Stephens et al, 1998; Gehre et al, 2016). Epithelial cells respond to the infection with the secretion of proinflammatory cytokines such as interleukin‐6 (IL‐6) and IL‐8 (Rasmussen et al, 1997). The inflammatory response is exacerbated upon reinfection, ultimately leading to tissue damage such as hydrosalpinx and fibrosis (Brunham & Rey‐Ladino, 2005). In this work, we show that TG2 becomes activated during the infection of epithelial cells with C. trachomatis and is required for optimal bacterial growth. The investigation of the consequence of TG2 activation on host metabolism and the identification of targets of TG2 transamidase activity during infection uncovered the control exerted by this enzyme on glucose import and on the hexosamine biosynthesis pathway, two metabolic features that are exploited by C. trachomatis.
Results
TG2 is highly expressed and becomes active during C. trachomatis infection
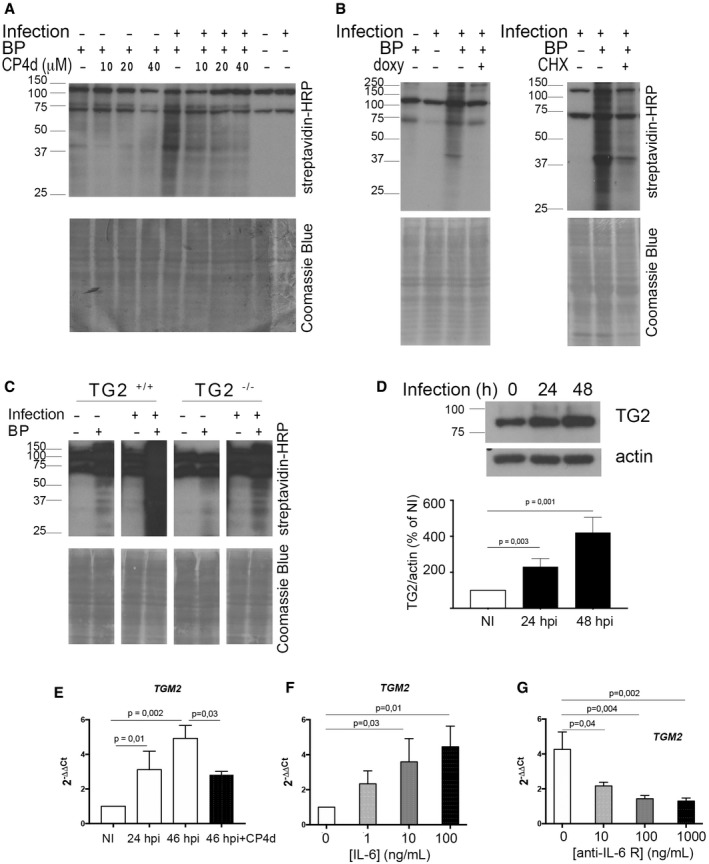
A widely used technique to probe TG2 activation is to measure the incorporation of biotin pentylamine (BP) into proteins. When present in excess, this membrane‐permeable primary amine out‐competes other substrates for the transamidase reaction catalyzed by TG2 and becomes covalently linked to glutamine residues of TG2 substrate proteins. The biotin group is then easily detectable by Western blot using streptavidin coupled to horseradish peroxidase (HRP; Lee et al, 1992). This procedure was applied to HeLa cells infected or not for 48 h with C. trachomatis. In non‐infected samples, BP incorporation was extremely low, as expected since in resting cells low Ca2+ concentration maintains TG2 in an inactive conformation (Gundemir et al, 2012). In contrast, infected cells showed a significant incorporation of BP. CP4d, an inhibitor of TG2 transamidating activity (Caron et al, 2012), abolished BP incorporation in a dose‐dependent manner, indicating that BP incorporation was the result of the transamidase activity of TG2 (Fig 1A). Live proliferating bacteria were needed for TG2 activation since filtered or heat‐inactivated bacteria, or bacteria treated with the antibiotic doxycycline immediately after infection to prevent their proliferation, failed to induce BP incorporation (Fig 1B and Appendix Fig S1). BP incorporation upon infection was also observed in wild‐type mouse embryonic fibroblasts (MEFs) but not in MEFs isolated from TGM2 knocked‐out animals (TG2−/−), further supporting the implication of TG2 in this process (Fig 1C).
Figure 1. TG2 transamidase activity increases during C. trachomatis infection along with its expression.
-
AWhole cell lysates were prepared with HeLa cells infected or not for 48 h with C. trachomatis L2 (multiplicity of infection MOI = 1) in the presence or not of BP. In the indicated samples, CP4d was added 2 h before infection. Cell lysates were run on SDS–PAGE, proteins were transferred to a membrane, and BP incorporation was revealed with HRP‐conjugated streptavidin. BP incorporation is enhanced in infected samples and is inhibited by CP4d. The two main bands present in all samples correspond to naturally biotinylated host proteins (Haneji & Koide, 1989). After blotting, the membrane was stained with Coomassie blue to control for equal loading.
-
BSame as in (A), except that where indicated 250 μM doxycycline (doxy, left) or 7 μM cycloheximide (CHX, right) was added 24 h or 2 hpi, respectively.
-
CWhole cell lysates were prepared with TG2+/+ and TG2−/− MEFs infected or not for 48 h with C. trachomatis L2 in the presence or not of BP, and analyzed as in (A).
-
DWestern blot with anti‐TG2 antibodies on total cell lysates infected or not with C. trachomatis L2 for the indicated time. The histogram displays the mean ± SD of TG2 expression relative to actin from four independent experiments, with the results of the Student's ratio‐paired t‐test. NI: not infected.
-
ECells were infected with C. trachomatis L2 (MOI = 1) for 24 or 46 h. Where indicated, 40 μM CP4d was added 2 hpi. TGM2 transcripts were measured by real‐time RT–qPCR and normalized to actin transcripts following the ΔΔC t method. The data are presented as relative mRNA levels compared to uninfected cells and shown as the mean ± SD. Each experiment was performed in duplicate and repeated at least four times. P‐values of Student's ratio‐paired t‐test < 0.05 are shown.
-
FCells were treated for 18 h with the indicated concentration of human recombinant IL‐6 before measuring TG2 transcription relative to actin like in (E). The data are presented as relative mRNA levels compared to untreated cells and shown as the mean ± SD. Each experiment was performed in duplicate and repeated at least three times. P‐values of Student's ratio‐paired t‐test < 0.05 are shown.
-
GCells were left uninfected or infected with C. trachomatis L2 in the presence of the indicated concentration of anti‐IL‐6 receptor antibodies. Forty‐eight hours later, TG2 transcription relative to actin was measured like in (E). The data are presented as relative mRNA levels compared to uninfected/untreated cells and shown as the mean ± SD. Each experiment was performed in duplicate and repeated at least three times. P‐values of Student's ratio‐paired t‐test < 0.05 are shown.
Source data are available online for this figure.
Probing cellular lysates using anti‐TG2 antibodies showed that activation of TG2 was accompanied with an increased expression of the enzyme (Fig 1D). Consistent with this observation, inhibition of protein synthesis with cycloheximide decreased infection‐induced BP incorporation (Fig 1B). Reverse transcription followed by quantitative PCR (RT–qPCR) measurements revealed a three‐ to fourfold increase in the TGM2 gene transcripts in infected versus non‐infected cells, demonstrating that the increase in TG2 amount during infection is at least partly controlled at the transcriptional level (Fig 1E). Interestingly, a positive feedback loop controls in part TG2 expression since TGM2 transcription was no longer enhanced by infection when cells were treated with CP4d (Fig 1E). TGM2 transcription responds to a number of external stimuli including retinoic acid, hypoxia, and inflammatory cytokines such as IL‐6 (Suto et al, 1993; Eckert et al, 2014). We reasoned that IL‐6 might be implicated in the transcriptional up‐regulation of TG2 in Chlamydia‐infected cells as this cytokine is produced during infection (Rasmussen et al, 1997). We first verified that TGM2 transcription showed a dose‐dependent response to the addition of recombinant IL‐6 in the culture medium (Fig 1F). To test whether IL‐6 contributes to the transcriptional up‐regulation of TGM2 during infection, we next performed the infection in the presence of anti‐IL‐6 receptor antibodies. We observed a reduction of TGM2 transcription in infected cells with increasing concentrations of antibodies in the culture medium (Fig 1G). Altogether, these data indicate that the induction of TGM2 transcription in C. trachomatis infected cells is at least in part a consequence of IL‐6 secretion in response to infection, followed by signaling through the IL‐6 receptor.
In conclusion, C. trachomatis infection increases TG2 levels and activates its transamidase activity.
TG2 activity sustains bacterial growth
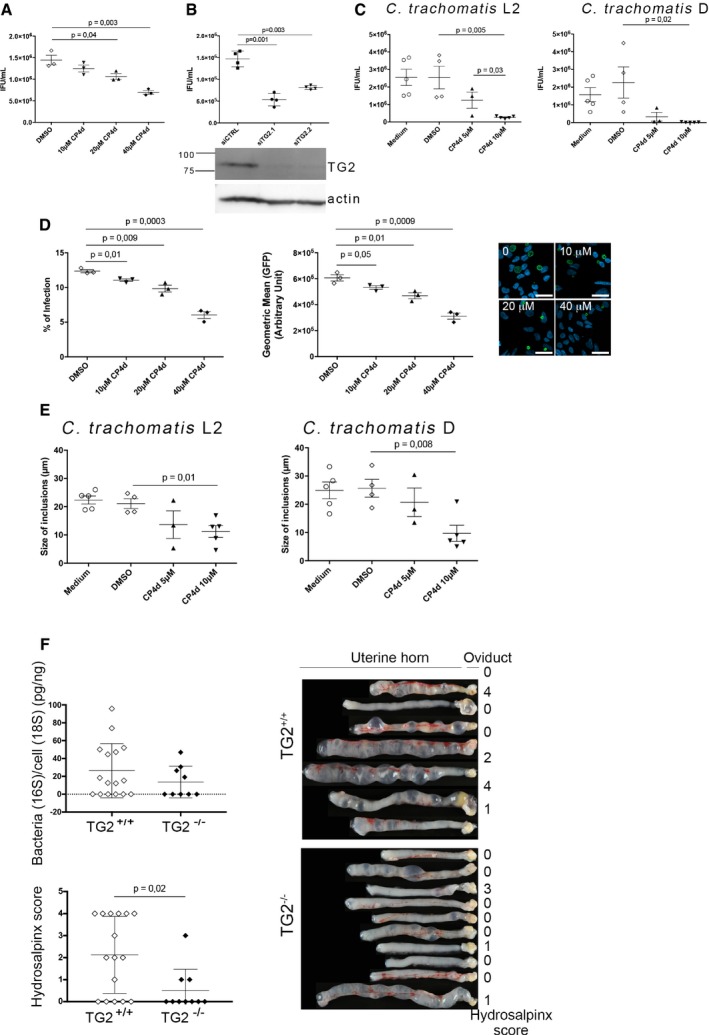
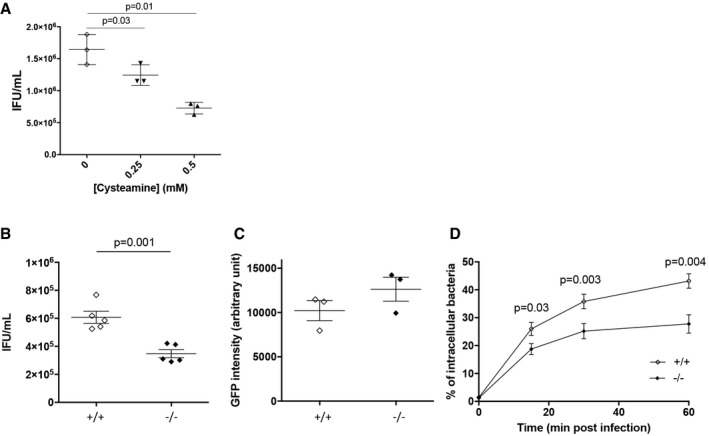
To determine whether TG2 activity affected bacterial development, we infected HeLa cells in the presence or not of the transamidase inhibitor CP4d. Thirty hours later, the progeny was collected and the number of infectious bacteria was determined by infecting fresh cells. Inhibition of TG2 activity with 40 μM CP4d resulted in a twofold decrease in bacterial progeny (Fig 2A). Consistently, a similar reduction in bacterial progeny was observed when TG2 activity was inhibited by cysteamine, another less specific inhibitor of TG2 (Fig EV1A). Progeny was also reduced when the bacteria were grown on TG2−/− MEFs compared to TG2+/+, indicating that the effect we had observed with TG2 inhibitors was not due to a direct toxicity of CP4d or cysteamine on the bacteria (Fig EV1B). Consistent with these observations, silencing TG2 expression with two different siRNA resulted in up to a threefold decrease in progeny (Fig 2B). To confirm these findings in primary cells, we used epithelial cells isolated from fallopian tubes (Roth et al, 2010). CP4d was even more potent at reducing the progeny after one developmental cycle in these cells than in HeLa cells, as a tenfold reduction was observed for 10 μM CP4d (Fig 2C). The negative impact of TG2 inhibition on bacterial development was also observed in primary cells infected with C. trachomatis serovar D, showing that the effect is not restricted to the LGV biovar (Fig 2C).
Figure 2. TG2 activity is needed for optimal C. trachomatis development and enhances hydrosalpinx upon C. muridarum infection in mice.
-
AHeLa cells were pre‐treated with the indicated concentrations of CP4d (or DMSO alone) for 2 h before being infected with L2incDGFP at MOI = 0.15. Thirty hours later, the cells were disrupted and bacterial titers (IFU = inclusion‐forming unit) were determined by re‐infecting fresh HeLa cells as described in the methods. The mean ± SD of three independent experiments is shown. P‐values of Student's paired t‐test are indicated when < 0.05.
-
BHeLa cells were transfected with control siRNA or two siRNAs against TG2. Two days later, the efficiency of the silencing was assessed by Western blot using anti‐TG2 antibodies and anti‐actin antibodies as loading control (bottom). Duplicate wells were infected with L2incDGFP and progeny was analyzed as in (A) (top). The mean ± SD of four independent experiments is shown. P‐values of Student's paired t‐test are indicated when < 0.05.
-
CSame as in (A), except that C. trachomatis serovar L2 (left) or D (right) was grown in primary cells isolated from fallopian tubes. For serovar D, IFUs were determined 48 hpi. The mean ± SD of four to five independent experiments is shown. P‐values of Student's paired t‐test are indicated when < 0.05.
-
DHeLa cells were pre‐treated with the indicated concentrations of CP4d (or DMSO alone) for 2 h before being infected with L2incDGFP at MOI = 0.15. Thirty hours later, the cells were fixed and analyzed by flow cytometry. The percentage of infected cells (left) and the mean fluorescence of the infected population (right) ± SD are shown for three independent experiments. P‐values of Student's paired t‐test are indicated when < 0.05. A representative field for each condition is shown, scale bar = 10 μm.
-
EPrimary epithelial cells isolated from fallopian tubes were pre‐treated with the indicated concentrations of CP4d (or DMSO alone) for 2 h before being infected with C. trachomatis serovar L2 (left) or D (right). Twenty‐four hours later, the cells were fixed, bacteria were stained using FITC‐labeled anti‐Chlamydia‐LPS antibodies, and the mean size of the inclusions was determined manually using ImageJ, on twenty inclusions per experiment. The mean ± SD of three independent experiments is shown. P‐values of Student's paired t‐test are indicated when < 0.05.
-
FMice were infected intravaginally with 105 IFU of C. muridarum. Twenty‐five days later, the mice were sacrificed and the upper genital tract, from the uterine horn to the oviduct, was collected. The right part was used for bacterial burden assessment (top left). The left part was rinsed with PBS and observed with a binocular magnifier (right) to determine the hydrosalpinx score (bottom left). Each dot represents one mouse, and the mean ± SD is shown. P‐values of Mann–Whitney test are indicated when < 0.05.
Figure EV1. TG2 is beneficial for bacterial development (related to Fig 2).
-
AHeLa cells were pre‐treated with the indicated concentrations of cysteamine for 2 h before being infected with L2incDGFP at MOI = 0.15. Thirty hours later, the cells were disrupted and bacterial titers (IFU) were determined by re‐infecting fresh HeLa cells as described in the methods. The mean ± SD of three independent experiments is shown. P‐values of Student's paired t‐test are indicated.
-
BTG2+/+ and TG2−/− MEFs were infected with L2incDGFP at MOI = 0.15. Thirty hours later, the cells were disrupted and bacterial titers were determined by re‐infecting fresh TG2+/+ cells as described in the methods. The mean ± SD of five independent experiments and P‐value from Student's paired t‐test are shown.
-
CTo measure bacterial adhesion, TG2+/+ and TG2−/− MEFs were incubated at 4°C for 4 h with L2incDGFP at MOI = 10 before being washed and fixed as described in the methods. The mean ± SD of three independent experiments is shown.
-
DTG2+/+ and TG2−/− MEFs were infected with L2incDGFP at MOI = 10 and fixed at the indicated time. Extracellular bacteria were differentially labeled as described in the methods. The mean ± SD of three independent experiments, and P‐values from Student's paired t‐test, is shown.
Reduced progeny could result from impairment of one or several of the steps of the chlamydial developmental cycle: adhesion, entry, differentiation into the replicative form, proliferation, and differentiation into the infectious form. We observed that the absence of TG2 in TG2−/− MEFs had no effect on bacterial adhesion (Fig EV1C), but that it decreased the efficiency of bacterial internalization (Fig EV1D). Consistently, 40 μM CP4d decreased the percentage of infected cells by about twofold in HeLa cells (Fig 2D). In addition, inclusions were smaller in cells treated with CP4d, and contained less bacteria, indicating that bacterial growth was slower in the absence of TG2 activity (Fig 2D). The reduction of inclusion size when TG2 was inhibited was also observed in primary cells infected with C. trachomatis L2 or serovar D (Fig 2E).
We next tested the incidence of the absence of TG2 on chlamydial development in a mouse model of infection. Chlamydia muridarum is a mouse‐adapted strain genetically very close to C. trachomatis (Read et al, 2000). Infection of HeLa cells with C. muridarum also activated TG2 (Fig EV2A), and we observed the same effect of silencing TG2 on C. muridarum growth as on C. trachomatis (Fig EV2B). We infected TG2+/+ and TG2−/− mice intravaginally with C. muridarum and 25 days after infection, mice were sacrificed and the genital tract was isolated (Fig 2F). DNA was extracted from the upper genital tract, and bacterial load was determined by qPCR. A slightly higher number of mice retained detectable bacterial DNA in the wild‐type group (10/16, 62%, for the TG2+/+ and 4/9, 44% for the TG2−/− mice). Among the animals in which bacterial DNA was still detected, the trend was for a higher bacterial load in the wild‐type background, but the number of TG2−/− animals we could breed was too low for statistical significance. These data indicate that the absence of TG2 reduces only marginally, if at all, the ability for C. muridarum to establish an infection. It is however possible that in some tissues, the loss of TG2 is compensated by expression of other transglutaminases, limiting the interpretation of these data (Iismaa et al, 2009). One clear difference between the two groups came from anatomical observations: TG2−/− animals showed milder signs of inflammation than their wild‐type littermate, especially when the oviduct hydrosalpinx scores were compared. It thus appears that the presence of TG2 exacerbates the tissue damage in this mouse model of infection, in line with the implication of TG2 in tissue fibrosis (Iismaa et al, 2009; Eckert et al, 2014).
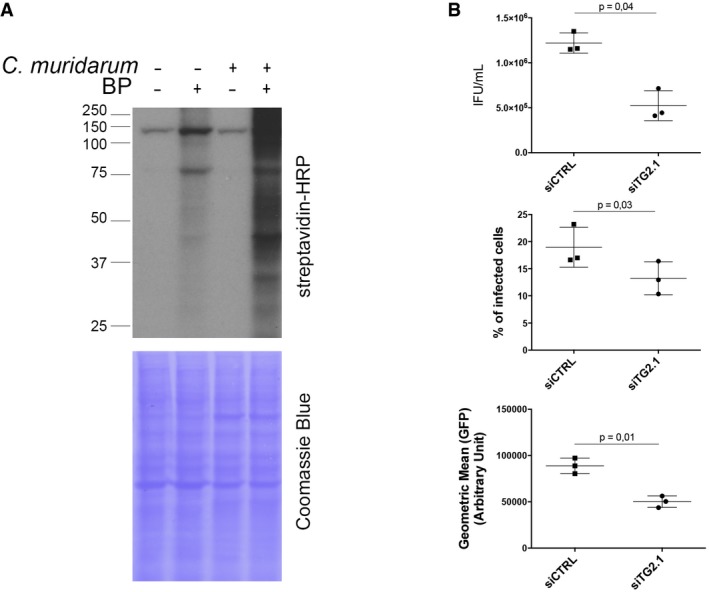
Figure EV2. Infection by C. muridarum activates TG2, which favors bacterial growth (related to Fig 2).
-
AWhole cell lysates were prepared with HeLa cells infected or not for 48 h with C. muridarum (MOI = 1) in the presence or not of BP. Cell lysates were run on SDS–PAGE, proteins were transferred to a membrane, and BP incorporation was revealed with HRP‐conjugated streptavidin.
-
BHeLa cells were transfected with siRNA against TG2 for 48 h before being infected in duplicates with C. muridarum at MOI = 0.15. Thirty hours later, one set of cells were disrupted and bacterial titers (IFU = inclusion‐forming unit) were determined by re‐infecting fresh HeLa cells as described in the methods. The mean ± SD of three independent experiments and P‐values of Student's paired t‐test are shown (top). Duplicate wells were incubated further for a total of 48 h before the cells were fixed, permeabilized with 0.3% Triton X‐100, and stained with rabbit antibodies against C. muridarum GroEL followed with A488‐coupled anti‐rabbit secondary antibodies. Samples were analyzed by flow cytometry, and the percentage of infected cells (middle) and the mean fluorescence of the infected population (bottom) ± SD are shown for three independent experiments; P‐values of Student's paired t‐test are indicated.
TG2 plays a central role in metabolic rewiring
We have recently shown that C. trachomatis acts as a glucose sink (Gehre et al, 2016). The host cell responds to glucose demand by increasing glucose uptake through overexpression of plasma membrane glucose transporters (Ojcius et al, 1998; Wang et al, 2017). Since we observed that TG2 level was increased during C. trachomatis infection, we wondered whether this increase could control the concomitant increase in glucose transporter expression, as it does in mammary epithelial cells (Kumar et al, 2014). If this hypothesis was correct, one prediction that we could make was that low glucose availability in the culture medium should be more detrimental to bacterial growth in TG2−/− MEFs compared to the wild‐type MEFs, as TG2−/− MEFs would be impaired in their ability to adjust their glucose uptake to sustain bacterial growth. To test this hypothesis, we grew MEFs in medium containing decreasing concentrations of glucose and measured the number of infectious bacteria collected 30 hpi. As expected, we observed that decreasing glucose availability resulted in a sharp decrease in bacterial titers, in both cellular backgrounds. However, bacterial titers were more sensitive to glucose deprivation in the TG2−/− MEFs than in the wild‐type cells (Fig 3A). For instance, at 1 mg/ml glucose, the progeny was reduced by 82% in the TG2−/− MEFs, compared to only 35% in the TG2+/+ MEFs.
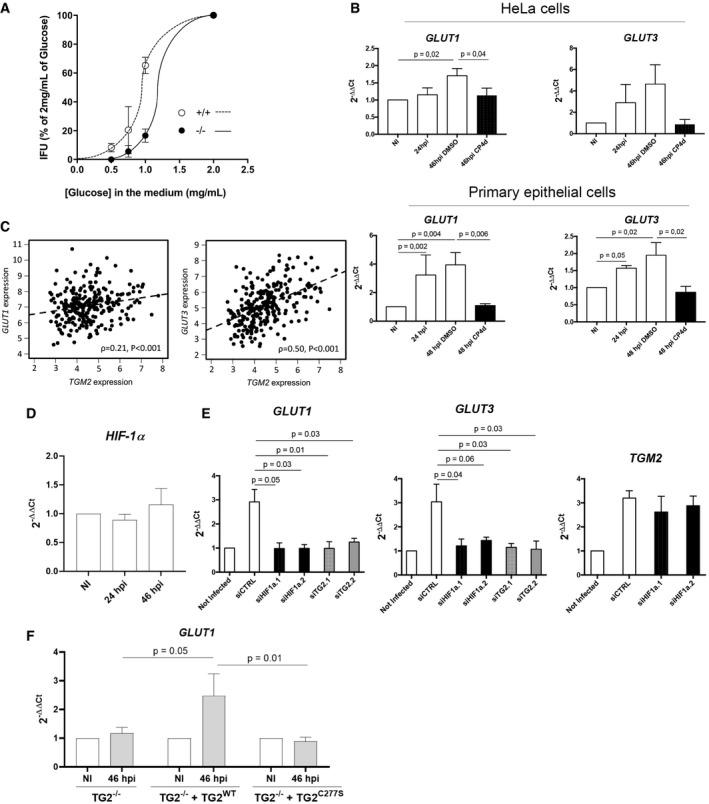
Figure 3. TG2 controls glucose import.
-
AMEFs were grown for 24 h culture medium complemented with the indicated concentration of glucose before being infected with L2 IncDGFP bacteria (MOI = 0.2). Cells were disrupted 30 h later and the bacterial titer determined by re‐infecting fresh wild‐type cells. The mean ± SD of three independent experiments is shown.
-
BCells were infected with C. trachomatis L2 (MOI = 1) for 24 or 48 h. Where indicated, 40 μM CP4d was added 2 hpi. GLUT‐1 and GLUT‐3 transcripts were measured by real‐time RT–qPCR and normalized to actin transcripts following the ΔΔC t method. The data are presented as relative mRNA levels compared to uninfected cells and shown as the mean ± SD. Each experiment was performed in duplicate and repeated four times. P‐values of Student's ratio‐paired t‐test are indicated when < 0.05.
-
CRelationship between TGM2 and GLUT‐1 (top) and GLUT‐3 (bottom) expression across 265 HGSOCs. Expression comparisons were performed using Spearman's rank correlation test.
-
DHeLa cells were infected with C. trachomatis L2 (MOI = 1) for 24 or 46 h. HIF‐1α transcripts were measured by real‐time RT–qPCR and normalized to actin transcripts. The data are presented as relative mRNA levels compared to uninfected cells and shown as the mean ± SD. Each experiment was performed in duplicate and repeated three times. P‐values of Student's ratio‐paired t‐test are > 0.05.
-
EHeLa cells were transfected with control siRNA or two siRNAs against HIF‐1α. Two days later, cells were infected with C. trachomatis L2 (MOI = 1) for 48 h. The indicated transcripts were measured by real‐time RT–qPCR and normalized to actin transcripts. The data are presented as relative mRNA levels compared to uninfected cells and shown as the mean ± SD. Each experiment was performed in duplicate and repeated three times. P‐values of Student's ratio‐paired t‐test are indicated when < 0.05.
-
FTG2−/− MEFs stably transformed or not with the indicated TG2 construct were infected for 2 days with C. trachomatis L2 (MOI = 1). Mouse GLUT‐1 transcripts were measured by real‐time RT–qPCR and normalized to mouse actin transcripts. The data are presented as relative mRNA levels compared to uninfected cells and shown as the mean ± SD. Each experiment was performed in duplicate and repeated three times. P‐values of Student's ratio‐paired t‐test are indicated when < 0.05.
To test the implication of TG2 in the rewiring of host metabolism more directly, we measured the incidence of TG2 inactivation on the cell capacity to uptake glucose. HeLa cells infected with C. trachomatis show increased transcription of GLUT‐1 and GLUT‐3, which allows to increase glucose uptake and meet bacterial needs (Wang et al, 2017). We reproduced this result in HeLa cells, as well as in primary cells isolated from the endocervix (Fig 3B). In contrast, in the presence of the TG2 inhibitor CP4d, the transcription of the glucose transporter genes was no longer induced by infection, indicating that TG2 is necessary for the control of GLUT‐1 and GLUT‐3 transcription. The absence of increase in GLUT‐1 and GLUT‐3 transcripts 48 hpi in the presence of CP4d was not due to the lower bacterial burden because when bacterial proliferation was interrupted 24 hpi by addition of doxycycline, we observed a comparable reduction in bacterial load at 48 hpi as in cells treated with CP4d, but the transcription of the glucose transporter genes remained as high as in non‐treated cells (Appendix Fig S2).
Finally, to explore further the incidence of TG2 expression on that of glucose transporters we examined transcriptional data from a cohort of high‐grade serous ovarian cancer (HGSOC) patients. This population was chosen because clinical and biological data indicate that TG2 overexpression is an adverse prognostic factor in ovarian carcinoma (Hwang et al, 2008; Shao et al, 2009). We observed a significant correlation between expression of TGM2 and GLUT‐3 across the 265 clinical HGSOC specimens (ρ = 0.50, P < 0.001) (Fig 3C). The HGSOC cohort also demonstrated significant correlation between TG2 and GLUT‐1 expression, though the magnitude of correlation was less marked (ρ = 0.21, P < 0.001; Fig 3C). Collectively, these data support the notion that TG2 plays a central role in regulating glucose transporters expression regulation in the context of infection or malignancy, thereby playing a central role in the control of the metabolic balance.
The hypoxia‐inducible factor 1 and the transamidase activity of TG2 are required for the transcriptional up‐regulation of glucose transporters
One major transcriptional regulator of the expression of glucose transporters is the hypoxia‐inducible factor 1 (HIF‐1), which is increased during Chlamydia infection (Sharma et al, 2011). Infection did not result in an increase in HIF‐1α transcripts, indicating that the increase in HIF‐1 α occurs by stabilization of the transcription factor (Fig 3D).To test whether HIF‐1 was implicated in infection‐induced up‐regulation of glucose transport, we silenced HIF‐1α expression before infecting the cells. Under these conditions, we observed a loss of induction of GLUT‐1 and GLUT‐3 transcription in infected cells. In contrast, the increase in TGM2 transcripts upon infection remained, placing HIF‐1α downstream of TGM2 induction (Fig 3E).
In the presence of the TG2 inhibitor CP4d, the transcription of the glucose transporter genes was no longer induced by infection (Fig 3B), indicating that the transamidase activity of TG2 was required. However, this observation could also be accounted for by the inhibition that CP4d exerts on TG2 expression in infection (Fig 1E). To address directly the role of TG2 transamidase activity in the regulation of the expression of glucose transporter genes, we used TG2−/− MEFS in which TG2 wild‐type or mutated for the transamidase activity (C277S mutant) was constitutively expressed (Rossin et al, 2012). We focused on the regulation of GLUT‐1, as we did not observe an increase in GLUT‐3 expression upon infection in this cellular background. Consistent with our previous findings, infection failed to induce an increase in GLUT‐1 transcripts in TG2−/− MEFS (Fig 3F). Constitutive expression of wild‐type TG2, but not of the C277S mutant, restored the induction of GLUT‐1 transcription upon infection. This observation demonstrates that the transamidase activity of TG2 is required for the increase in GLUT‐1 transcription upon infection.
Altogether, these data show that the transcription factor HIF‐1 and TG2 transamidating activity are both required for the up‐regulation of glucose transporters during C. trachomatis infection.
TG2 targets glutamine:fructose‐6‐P amidotransferase and enhances its activity
In addition to its role in the up‐regulation of the transcription of glucose transporter genes, TG2 may confer other benefits to C. trachomatis. To identify TG2 targets in the infectious process, HeLa cells were infected in the presence or absence of BP. Forty‐eight hours later, the cells were lysed and biotinylated proteins were isolated on streptavidin‐coated beads, and identified by mass spectrometry. Sixty‐two proteins were found to be significantly enriched in the infected cell lysates grown in the presence of BP (Appendix Table S1). Fibronectin, galectin 3, RhoA, 40S ribosomal protein SA, immunoglobulin κ chain C region, and hemoglobin beta were already identified as TG2 substrates (Pincus & Waelsch, 1968; Mehul et al, 1995; Orrù et al, 2003; Guilluy et al, 2007; Nelea et al, 2008; Sohn et al, 2010). BAG2 and several other mitochondrial proteins were also enriched in the samples prepared in the presence of BP, in agreement with TG2 being present and active in this compartment (Altuntas et al, 2015).
Among the potential TG2 substrates we identified in C. trachomatis infected cells, the enzyme glutamine:fructose‐6‐P amidotransferase (GFPT) caught our attention because it uses fructose‐6‐P as a substrate, which is derived from glucose‐6‐P. Moreover, both isoforms of the enzyme, GFPT1 (also called GFAT) and GFPT2, had been recovered from the proteomic approach, making it a very strong hit. We first confirmed that GFPT was recovered in the biotinylated fraction of cells infected with C. trachomatis in the presence of BP using anti‐GFPT antibodies. The abundance of GFPT in the biotinylated fraction strongly decreased when infection had been performed in the presence of the TG2 inhibitor CP4d, demonstrating that incorporation of the biotinylated probe in GFPT depended on the activity of TG2 (Fig 4A).
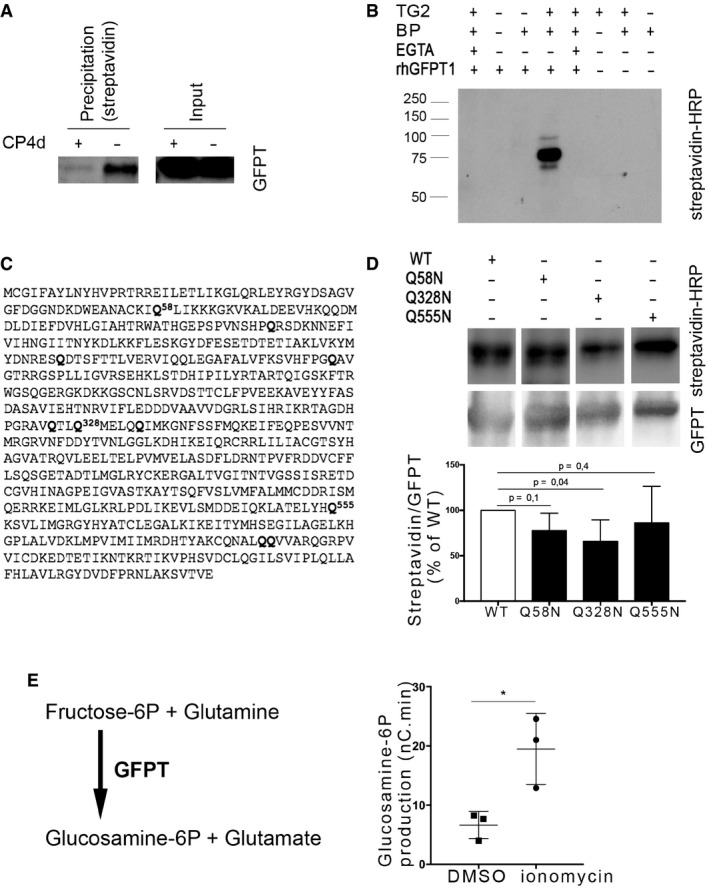
Figure 4. GFPT is a substrate of TG2 transamidase activity.
-
AHeLa cells were infected with C. trachomatis (MOI = 1), and 40 μM CP4d was added or not 2 hpi. After 24 h, 0.5 mM BP was added and cells were lysed at 48 hpi. Lysates were precipitated with streptavidin‐coated beads. After separation with SDS–PAGE, proteins were transferred to a membrane and blotted with anti‐GFPT antibody followed with HRP‐conjugated secondary antibody.
-
BIn vitro assay testing the ability of purified TG2 to cross‐link purified rhGFPT1 with BP. Samples were incubated for 3 h at 37°C before separation by SDS–PAGE. Proteins were transferred to a membrane, and BP was revealed using HRP‐conjugated streptavidin. rhGFPT1 is 77.5 kDa.
-
CGFPT1 sequence: Glutamine residues identified by mass spectrometry as cross‐linked to BP are in bold letter.
-
DIn vitro assay was performed as described in (B) using wild‐type rhGFPT1 (WT), rhGFPT1 Q58N, rhGFPT1 Q328N, or rhGFPT1 Q555N as substrates. The reaction was performed at 37°C for 30 min. After probing with HRP–streptavidin, the membrane was washed and probed with anti‐GFPT antibodies followed with HRP‐conjugated secondary antibodies. The ratio of modified protein (streptavidin signal) to the total GFPT is shown and normalized to its value with WT rhGFPT1. The mean ± SD of five independent experiments is shown, and the P‐value of the Student's ratio‐paired t‐test is indicated.
-
ELysates of cells treated or not for 6 h with ionomycin were incubated at 37°C for 45 min with fructose‐6‐P and glutamine. The production of glucosamine‐6‐P was measured using HPAEC‐PAD. Results of three independent experiments are shown, with mean ± SD, and P‐value of the Student's paired t‐test is indicated (*P < 0.05).
Source data are available online for this figure.
To further validate that GFPT is a novel substrate of TG2, purified TG2 and recombinant human GFPT1 (rhGFPT1) were incubated for 3 h at 37°C in the presence of BP as primary amine donor. The incorporation of the biotinylated probe was analyzed by blotting with HRP‐coupled streptavidin. The biotinylated probe was incorporated into rhGFPT1 in the presence and not in the absence of TG2. Furthermore, chelation of Ca2+ by EGTA inhibited the incorporation of the probe, as expected for a reaction dependent on the transamidase activity of TG2 (Fig 4B). We concluded from these experiments that GFPT is a novel substrate of TG2 that becomes modified by the transamidase activity of the enzyme during C. trachomatis infection.
In order to determine which glutamine residue(s) of GFPT1 was modified by TG2 in vitro, we analyzed the products of the reaction by mass spectrometry. BP incorporation was identified in ten glutamine residues (out of twenty‐eight, Fig 4C), presumably because promiscuous reactions occur in vitro. Among those, two glutamine residues were identified as prone to modification by TG2 using bioinformatics tools designed to score the peptidic environment favorable for TG2 activity, namely Q328 and Q555 (Keresztessy et al, 2006; Sugimura et al, 2006). We thus generated a glutamine to asparagine point mutant for each of these residues to minimize the impact on protein folding. As a control, we also mutated Q58, another candidate target identified by mass spectrometry but not surrounded by a consensus sequence for TG2. Purified recombinant proteins were incubated with TG2 and BP for 30 min at 37°C before stopping the reaction. BP incorporation was significantly reduced only in the rhGFPT1 Q328N, indicating that the Q328 is a prominent glutamine for modification by TG2 (Fig 4D).
The fact that C. trachomatis produce their own GFPT (named GlmS) prevented us from measuring the consequence of TG2 activation on host GFPT activity in infected cells. However, ionomycin is a widely used TG2 activator, as this Ca2+ ionophore increases intracellular Ca2+ concentration, which opens TG2 in its active conformation. We thus measured GFPT activity in lysates of cells treated or not with ionomycin and analyzed the reaction products by high‐performance anion‐exchange chromatography (Appendix Fig S3). We observed a threefold increase in GFPT activity in cells treated with ionomycin, indicating that GFPT modification by TG2 increases the activity of the enzyme (Fig 4E).
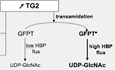
Modification of GFPT by TG2 enhances the hexosamine biosynthesis pathway
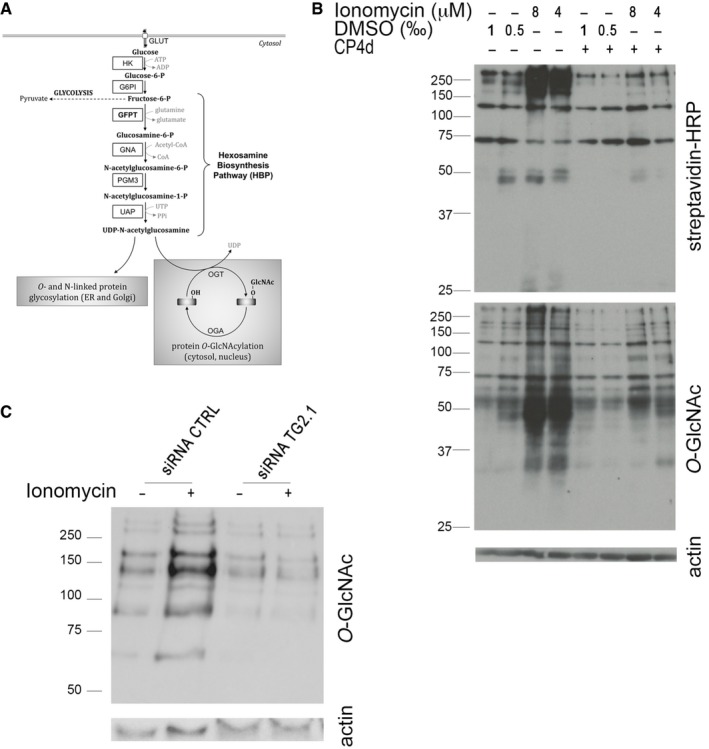
The reaction catalyzed by GFPT is the first and rate‐limiting step of the hexosamine biosynthesis pathway (HBP, Fig 5A). The HBP leads to the formation of uridine 5′‐diphospho‐N‐acetylglucosamine (UDP‐GlcNAc), which is further used for N‐glycosylation, N‐glycan branching, and O‐linked N‐acetylglycosylation (O‐GlcNAcylation) in the ER, Golgi, and nucleus/cytosol, respectively. O‐GlcNAcylation involves the transfer of a single UDP‐GlcNAc moiety to the hydroxyl groups of serine or threonine residues. Two enzymes, O‐GlcNAc transferase (OGT) and O‐GlcNAcase (OGA), catalyze O‐GlcNAc addition and removal, respectively, and the O‐GlcNAc modification level of proteins is directly dependent on the concentration of UDP‐GlcNAc, the donor substrate for OGT (Kreppel & Hart, 1999). To confirm that GFPT modification by TG2 increased its activity and thus hexosamine biosynthesis, we measured the level of O‐GlcNAcylation in primary epithelial cells. We observed an increase in O‐GlcNAcylation in cells treated with ionomycin. This increase was dependent on TG2 activity since it was not observed in the presence of the TG2 inhibitor CP4d (Fig 5B) or in cells in which TGM2 expression had been silenced using siRNA (Fig 5C). Altogether, these experiments show that activation of TG2 transamidase activity enhances the HBP.
Figure 5. TG2 activation results in increased UDP‐GlcNAc production.
-
ASchematic view of the hexosamine biosynthesis pathway. Production of glucosamine‐6‐P by GFPT is the first and rate‐limiting step of the pathway that produces UDP‐GlcNAc. HK: hexokinase; G6PI: glucose‐6‐P isomerase; GFPT: glutamine:fructose‐6‐P amidotransferase; GNA: glucosamine‐6‐P N‐acetyltransferase; PGM3: phosphoglucomutase 3; UAP: UDP‐N‐acetylglucosamine pyrophosphorylase; OGT: O‐GlcNAc transferase OGA: O‐GlcNAcase; GlcNAc: N‐acetylglucosamine.
-
BEndocervical epithelial cells were pre‐treated or not with 40 μM CP4d for 2 h before addition of the indicated concentration of ionomycin (or an equivalent volume of DMSO) and 0.5 μM BP. Six hours later, whole cell lysates were analyzed by Western blot. The membrane was first blotted with HRP‐conjugated streptavidin to detect TG2 activity and then extensively washed and probed with anti‐O‐GlcNAcylation antibody followed with HRP‐conjugated secondary antibodies. Last, the membrane was probed with anti‐actin as a loading control.
-
CThe same experimental procedure as described in (B) was applied to HeLa cells treated for 48 h prior to ionomycin treatment (8 μM) with siRNA control or directed against TG2.
Source data are available online for this figure.
The increase in the hexosamine biosynthetic pathway is hijacked by the bacteria
Surprisingly, we did not observe an increase in O‐GlcNAcylation in cells infected for 48 h by C. trachomatis (Fig 6A). Since O‐GlcNAcylation directly depends on UDP‐GlcNAc concentration, this observation suggests that UDP‐GlcNAc levels in the cytoplasm are not significantly increased in infected cells. We have previously demonstrated that C. trachomatis co‐opts SLC35D2, a host antiporter transporting UDP‐GlcNAc, UDP‐glucose, and GDP‐mannose to import these metabolites into the vacuole in which the bacteria develop (Gehre et al, 2016). We reasoned that UDP‐GlcNAc might not accumulate in the cytoplasm in infected cells because it was relocated to the inclusion lumen. Supporting this hypothesis, we observed that activation of TG2 by ionomycin elicited a lower increase in O‐GlcNAcylation in infected cells compared to non‐infected cells, indicating that less free UDP‐GlcNAc is available for O‐GlcNAcylation in the infected host cytoplasm (Fig 6B). Importantly GFPT expression was stable in all conditions, indicating that the decrease in O‐GlcNAcylation in infected cells is not due to lower GFPT expression (Fig 6A and B).
Figure 6. Optimal bacterial growth requires GFPT and prevents UDP‐GlcNAc accumulation.
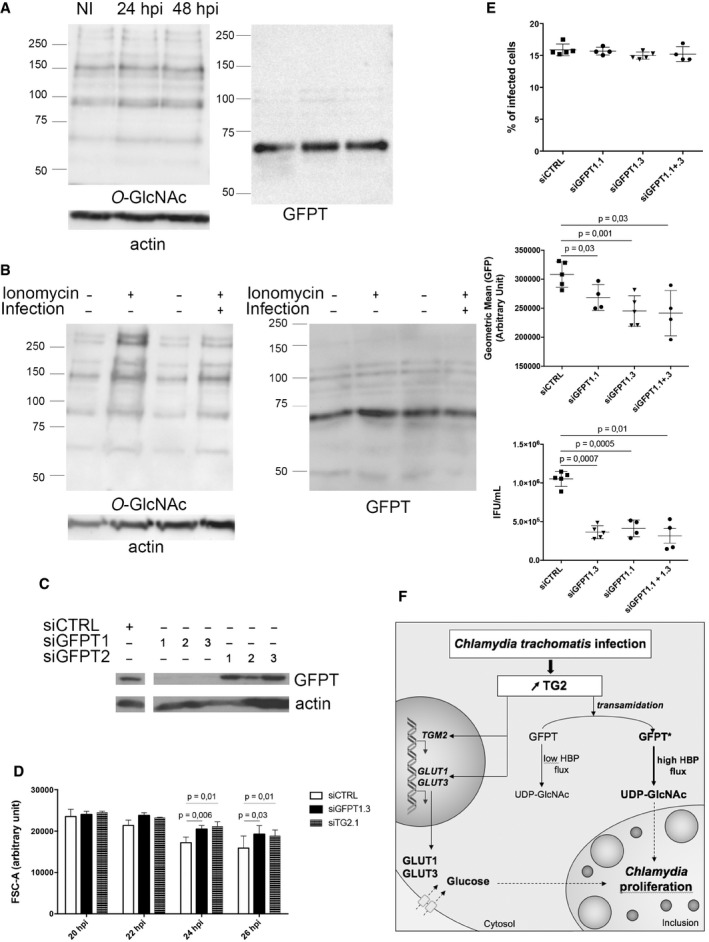
-
AHeLa cells were infected or not (NI) with C. trachomatis (MOI = 1) and then lysed 24 or 48 hpi. After separation with SDS–PAGE, proteins were transferred to a membrane and probed with anti‐O‐GlcNAcylation antibody followed with HRP‐conjugated secondary antibodies. After extensive washes, the membrane was blotted again with anti‐GFPT and anti‐actin antibodies before revelation with HRP‐conjugated secondary antibodies.
-
BHeLa cells were infected or not with C. trachomatis (MOI = 1). Twenty‐four hours later, 8 μM ionomycin (or DMSO alone) and 0.5 mM BP were added. After 6 h of treatment, cells were lysed and proteins revealed as in (A).
-
CHeLa cells treated for 72 h with siRNA targeting GFPT1 or GFPT2 were lysed. After separation with SDS–PAGE, proteins were transferred to a membrane and probed with anti‐GFPT and anti‐actin antibody before revelation with HRP‐conjugated secondary antibodies.
-
DHeLa cells were transfected with control siRNA or siRNAs against GFPT or TG2. Two days later, the cells were infected for the indicated times (MOI = 0.3) before fixation, rupture of the cells, and measurement of bacterial diameter. The mean diameter ± SD and P‐values of Student's paired t‐test on four independent experiments are shown.
-
EHeLa cells treated for 48 h with siRNA targeting GFPT1 or not (siCTRL) were infected with C. trachomatis (MOI = 0.15). Thirty hours later, cells were fixed and analyzed by flow cytometry. The percentage of infected cells (top) and the mean fluorescence of the infected population (middle) ± SD are shown for at least four independent experiments. Duplicate wells were lysed and used to re‐infect fresh HeLa cells to determine the bacterial titer (bottom). P‐values of Student's ratio‐paired t‐test are indicated.
-
FSchematic view of the outcome of TG2 activation in infection. The increase in TG2 expression and activity in cells infected with C. trachomatis results in the up‐regulation of the expression of glucose transporters. Increasing quantities of glucose are thus imported in the host cytoplasm and redirected to the vacuole, where they fuel bacterial growth. Parallel to this transcriptional outcome, the transamidating activity of TG2 targets the host enzyme GFPT, thereby boosting the hexosamine biosynthesis pathway. The bacteria consume the resulting UDP‐GlcNAc, or an intermediate along this pathway, in particular to sustain bacterial division.
Source data are available online for this figure.
In Gram‐negative bacteria, UDP‐GlcNAc supply is mostly used for lipopolysaccharide and peptidoglycan biosynthesis. C. trachomatis do not have a classical cell wall but use peptidoglycan synthesis for bacterial division (Liechti et al, 2016). If UDP‐GlcNAc, or an intermediate along the hexosamine biosynthesis pathway, was consumed at least partly in making bacterial peptidoglycan, lowering hexosamine biosynthesis should delay bacterial division, resulting in larger bacteria being formed. We tested this hypothesis by measuring the consequence of silencing GFPT, the rate‐limiting enzyme in hexosamine biosynthesis, on bacterial size. The mass spectrometry data showed that both isoforms GFPT1 and GFPT2 were expressed in HeLa cells (Appendix Table S1). Comparison of the efficiency of siRNA designed to target specifically one isoform showed that GFPT2 was hardly detectable and targeting GFPT1 was sufficient to strongly decrease GFPT expression (Fig 6C). We thus lowered hexosamine biosynthesis by treating cells with a siRNA against GFPT1 prior to infection, fixed the cells at increasing time of infection, and used flow cytometry to measure bacterial sizes. As expected, the number of replicative bacteria increased with infection time (Appendix Fig S4). Furthermore, it was recently demonstrated that replicative bacteria gradually decrease in size over the course of the developmental cycle (Lee et al, 2017). We indeed observed a decrease in bacterial diameter over a 20–26 hpi time course, thereby validating the use of flow cytometry to measure the size of C. trachomatis (Fig 6D and Appendix Figs S4). In cells treated with a siRNA against GFPT1, the mean bacterial diameter became significantly higher than in control cells 24 hpi (Fig 6D). We confirmed this result by measuring the diameter of bacteria on electron microscopy pictures of cells infected for 30 h (Fig EV3). These kinetics fit well with our observation that TG2 activity increases between 24 and 48 hpi, when bacterial load is high, and access to nutrients might become limiting (Rother et al, 2018). These observations support the hypothesis that a product of the hexosamine biosynthesis pathway is captured by the inclusion to support division. Consistent with a role for GFPT activity in sustaining bacterial growth, we observed a reduction in the number of bacteria per inclusion 24 h post‐infection in the cells treated with siRNA against GFPT1, and the progeny collected was reduced threefold (Fig 6E). Of note, silencing of GFPT1 had no incidence on bacterial entry and the initiation of bacterial development, as the percentage of infected cells was identical to that in control cells (Fig 6E). Altogether, these data strongly support the hypothesis that the increase in hexosamine biosynthesis by the host upon GFPT modification by TG2 is exploited by the bacteria, in particular to assist bacterial division. Interestingly, we observed that silencing TG2 also increased bacterial size (Fig 6D). While silencing TG2 has multiple effects beyond harnessing the HBP, these data are fully consistent with GFPT activation being one of the major outcomes of TG2 up‐regulation during C. trachomatis infection.
Figure EV3. GFPT silencing affects bacterial division (related to Fig 6).
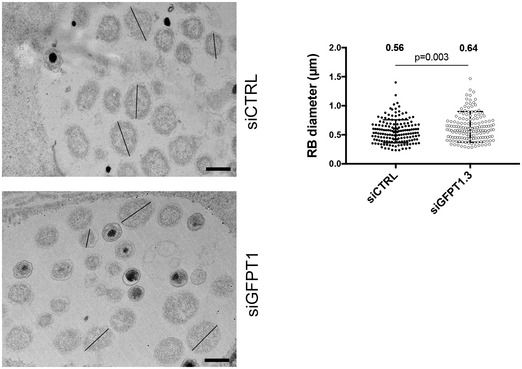
HeLa cells treated for 48 h with siRNA targeting GFPT1 or not (siCTRL) were infected with C. trachomatis (MOI = 1), fixed 30 hpi, and processed for transmission electron microscopy. Lines show example of measured RB diameters. Scale bar = 600 nm. RB diameters were measured using ImageJ on > 300 bacteria in one experiment. Each dot represents one RB; the mean value ± SD and P‐value of Student's paired t‐test are indicated.
Discussion
TG2 transamidase activity is very potent and would be deleterious if not tightly controlled. In basal conditions, it is mostly turned off, and it is thought that under specific stress conditions, the enzyme might be locally turned on and transamidate specific substrates. The infection with C. trachomatis provided a unique physiological situation where the expression and activity of the enzyme were increased over a relatively short period of time in a physiological set‐up. We took advantage of this observation to identify TG2 substrates. Among the 62 candidates identified, we focused on the enzyme GFPT. We showed that GFPT modification by TG2 increased its activity, resulting in higher hexosamine biosynthesis, a process also fueled by the positive control exerted by TG2 on glucose transporters expression. The product of the HBP, UDP‐GlcNAc, is used for post‐translational modification of proteins by O‐GlcNAcylation. Thus, our work uncovered an unsuspected link between TG2 transamidase activity and O‐GlcNAcylation. This link was disrupted in infected cells because the increase in hexosamine biosynthesis in the host was exploited by the bacteria, in particular to assist their division. In conclusion, our work establishes TG2 as a key player in controlling glucose‐derived metabolic pathways in mammalian cells, themselves hijacked by C. trachomatis to sustain their own metabolic needs (Fig 6F).
Several inflammatory conditions are associated with an increase in TG2 expression (Eckert et al, 2014). We confirmed that IL‐6 up‐regulates TGM2 transcription (Suto et al, 1993; Eckert et al, 2014), and we showed that this cytokine is implicated in the increase in TGM2 transcription during C. trachomatis infection (Rasmussen et al, 1997) since anti‐IL‐6 receptor antibodies antagonized the induction of TG2 expression. How the enzyme becomes activated is less clear. In the cell, Ca2+ concentration is high in the endoplasmic reticulum, and this compartment is tightly associated with the inclusion membrane (Derré et al, 2011). The unfolded protein response pathway is activated in infected cells (George et al, 2016), a condition that might be sufficient to activate TG2 (Lee et al, 2014). Indeed accumulation of cytoplasmic Ca2+ around the inclusion has been reported (Majeed et al, 1999), which might be enough to locally activate TG2.
Treatment of cells with a potent inhibitor of TG2, CP4d, reduced progeny tenfold in primary epithelial cells (Fig 2). We have shown that the inhibition of TG2 activity had two distinct effects on C. trachomatis developmental cycle. First, it reduced the ability for the bacteria to enter the cells (Appendix Fig S2). Chlamydia trachomatis use multiple receptors and appear to hijack several entry pathways into epithelial cells (Ford et al, 2018). The positive role played by TG2 on bacterial entry, which could be exerted from its intracellular or extracellular location, remains to be studied in future work. One attractive candidate mechanism is PDGFR signaling, since it is implicated in C. trachomatis entry (Elwell et al, 2008) and it is sensitive to TG2 activity (Nurminskaya et al, 2014). Second, the inclusion developed slower in CP4d‐treated culture, indicating that TG2 is necessary for optimal bacterial growth. We discuss below the two mechanisms we uncovered that account for the link between TG2 activation and bacterial development, and place TG2 as a key regulator of bacterial access to glucose and its derivative UDP‐GlcNAc.
We have shown that TG2 is required for the increase in transcription of GLUT‐1 and GLUT‐3 in infection in HeLa cells and in primary epithelial cells (Fig 2). Glucose is an essential metabolite for C. trachomatis development and preventing the transcription of GLUT‐1 and GLUT‐3 by siRNA led to a twofold decrease in progeny in HeLa cells (Wang et al, 2017). This result shows that glucose import can become limiting for bacterial growth and thus that the control exerted by TG2 on the expression of glucose transporters accounts, at least in part, for the need for this enzyme for optimal bacterial growth. This conclusion is supported by our observation that glucose becomes limiting faster for bacterial progeny in MEFs lacking TG2 than in wild‐type cells. Thus, our study places TG2 as a key regulator for glucose import in infected cells.
Mechanistically, we showed that HIF‐1 was required for Chlamydia‐induced increase in the transcription of glucose transporters. HIF‐1α transcription does not increase during infection. TG2 interacts with HIF‐1β (Filiano et al, 2008), and an increase in TG2 levels upon infection might be sufficient to stabilize the HIF‐1 complex. It is however not sufficient to account for infection‐induced up‐regulation of glucose transporters since we demonstrated that the transamidating activity of TG2 was required in this context. Through a series of elegant experiments, the Johnson Lab showed that the effect of TG2 on transcription was highly cell and context dependent (Gundemir et al, 2013, 2017). TG2 modifies several transcription factors (Gundemir et al, 2012), but none of these known targets were recovered in our proteomic approach for the identification of TG2 targets upon infection. Furthermore, it was shown very recently that TG2 enhanced chromatin binding of the general transcription factor complex TFIID through the serotonylation of histone 3 trimethylated lysine 4 (Farrelly et al, 2019). This, or another transglutaminase‐mediated histone modification, might be implicated in the transcriptional control of glucose transporter genes in the infectious context.
Our proteomic approach identified 62 candidate TG2 substrates in C. trachomatis infection. Six of those were already known substrates of TG2, validating our analysis. We further demonstrated that GFPT was a substrate of TG2 and identified Q328 as a prominent transamidation site. The absence of shift in the migration profile of GFPT suggests that the amine donor is either a small protein, or a small amine, or that deamidation occurs, making identification by mass spectrometry very challenging. Experiments on GFPT immunoprecipitated from ionomycin‐treated samples failed to detect histaminylation or serotonylation. Deamidation was occasionally seen on several glutamine residues, including Q328, raising questions as to the relevance of this observation that will be addressed in future studies.
We showed that GFPT activity was enhanced upon transamidation by TG2. This resulted in an increase in O‐GlcNAcylation, since this post‐translational modification of proteins is directly dependent on the concentration of UDP‐GlcNAc (Kreppel & Hart, 1999). GFPT acts as a tetramer and is negatively regulated by several post‐translational modifications (Chang et al, 2000; Zibrova et al, 2017) and by UDP‐GlcNAc (Assrir et al, 2014). Transamidation on Gln328 could interfere with these down‐regulation mechanisms and thereby unleash the HBP.
Interestingly, the positive correlation between TG2 activation and O‐GlcNAcylation does not hold true in infected cells and our data indicate that this is due to hexosamines being consumed by the infection. Our observation that silencing GFPT expression increases bacterial diameter strongly supports the hypothesis that UDP‐GlcNAc, or an intermediate along the HBP, is hijacked into the inclusion to fuel bacterial division, possibly by feeding peptidoglycan synthesis. Interestingly, a proximity‐based labeling assay recently described an enrichment of TG2 and GFPT1 around the inclusion, suggesting that TG2 activation and GFPT modification might be most efficient in proximity of the bacteria‐containing compartment (Olson et al, 2019). One recent publication showed that UDP‐GlcNAc is also used during infection to post‐translationally modify the intermediate filament vimentin and this also could contribute to significant UDP‐GlcNAc consumption in C. trachomatis infection (Tarbet et al, 2018).
The role played by TG2 in viral or microbial infections is raising increasing interest. Like in the case of C. trachomatis infection, genetic or pharmacological inhibition of TG2 led to a marked reduction in Mycobacterium tuberculosis replicative capacity. However, the mechanism involved might be different, since the data suggest that reduced replication in macrophages lacking TG2 is due to the impairment of autophagy homeostasis (Palucci et al, 2017). Mycobacterium tuberculosis relies largely on lipids and fatty acids as energy source, and glucose availability might not be limiting in this case (Russell et al, 2010). Still, up‐regulation of glucose transporter was also described in a mouse model of M. tuberculosis infection (Shi et al, 2015); thus, the involvement of TG2 in metabolism regulation in this context remains to be investigated.
There are multiple examples of host manipulation by pathogens that shed light on fundamental cellular processes. Here, our work revealed an unsuspected regulation of the HBP by TG2. This discovery has important implications. Like other post‐translational modifications, protein O‐GlcNAcylation dramatically alters the fate and function of target proteins. In particular, transcription factors are modified by O‑GlcNAcylation, which implicates this modification in transcriptional regulation (Jackson & Tjian, 1988). Physiologically, disruption of O‑GlcNAcylation homeostasis has been implicated in the pathogenesis of many human diseases, which include cancer, diabetes, and neurodegeneration (Jóźwiak et al, 2014; Yang & Qian, 2017). The link between TG2 and O‐GlcNAcylation means that TG2 activation is expected to have broad transcriptional consequences. In particular, TG2 is activated in many cancers and future investigation is required to determine the contribution of the TG2/GFPT activation axis to tumorigenesis.
Materials and Methods
Cells and bacteria
HeLa cells (ATCC) and mouse embryonic fibroblasts (MEFs) isolated from KO (TG2−/−) or WT (TG2+/+) C57B6 mice were grown in Dulbecco's modified Eagle's medium with Glutamax (DMEM, Invitrogen), supplemented with 10% (v/v) heat‐inactivated fetal bovine serum (FBS; D'Eletto et al, 2009). TG2−/− cells reconstituted with stable expression of wild‐type or C277S TG2 are described in Rossin et al (2012). Primary cells used for experiments displayed in Fig 2 were isolated from human fallopian tubes and maintained in culture as previously described (Roth et al, 2010). Other primary cells were isolated from endocervix biopsies of female patients and were cultivated in keratinocyte‐SFM (Thermo Fisher Scientific) containing 50 mg/l of bovine pituitary extract (Thermo Fisher Scientific) and 5 μg/l of epidermal growth factor (EGF) human recombinant (Thermo Fisher Scientific; C. Liu, C. Tang, A. Subtil and Y. Wu, in preparation). All cell cultures were maintained at 37°C, in 5% CO2 atmosphere and were routinely tested for mycoplasma using the standard PCR method. C. trachomatis serovar LGV L2 strain 434 and serovar D/UW‐3/CX (ATCC), GFP‐expressing L2 (L2IncDGFP), or C. muridarum MoPn (for in vivo experiments) were propagated on HeLa cells, purified on density gradients as previously described and stored at −80°C (Scidmore, 2005; Vromman et al, 2014).
siRNA treatment
For siRNA experiments, 50,000 cells were plated in a 24‐well plate and immediately mixed with Lipofectamine RNAiMAX (Invitrogen) following the manufacturer's recommendation, using 10 nM of siRNA (Appendix Table S2). For RB size assessment, 300,000 cells were plated in a 6‐well plate. For electron microscopy experiments, 1.5 million cells were plated in a 25‐cm2 flask. For GFPT1 activity assay, 1 million cells were plated in a 10‐cm‐diameter dish. The culture medium was changed the next day, and experiments (infection or treatment with ionomycin) were performed two days post‐treatment with the siRNA.
GFPT1 purification
Recombinant human GFPT1 (rhGFPT1) with an internal 6‐His tag was produced from a plasmid pET28‐rhGFPT1‐6His in Escherichia coli Rosetta (DE3) GlmS::Tc kindly given by Dr. Badet‐Denisot (Centre de Recherche de Gif, France; Li et al, 2007).The mutated form rhGFPT1 Q58N, rhGFPT1 Q328N, and rhGFPT1 Q555N were obtained using QuikChange technology (Agilent) on the plasmid pET28‐rhGFPT1‐6His, with primers listed in Appendix Table S2, following the manufacturer's instructions, and transformed in E. coli Rosetta (DE3) GlmS::Tc.
One liter of culture in 2YT medium supplemented with tetracycline (8 μg/ml, Sigma), kanamycin (50 μg/ml, Sigma), chloramphenicol (15 μg/ml, Sigma), and glucosamine (GlcNH2, 2 mg/ml, Sigma) was incubated with agitation at 37°C until OD600 reached 0.5. Protein expression was induced by addition of 0.5 mM of isopropyl β‐D‐thiogalactopyranoside (Sigma) at 25°C for 24 h before being harvested. The cell pellets were resuspended in lysis buffer (16.27 mM Na2HPO4 and 3.73 mM NaH2PO4 pH 7.5, 200 mM NaCl, 20 mM imidazole, 2 mM Tris(2‐carboxyethyl)phosphine hydrochloride (TCEP), 10% glycerol, 1 mM fructose‐6‐P, Roche EDTA‐free protease inhibitor cocktail) and disrupted by sonication. The recombinant protein was purified by incubation with Qiagen Ni‐NTA agarose beads (Qiagen) for 1 h followed by three washing steps with the lysis buffer before elution with lysis buffer containing increasing concentrations of imidazole: 30, 100, 175 mM, and finally 500 mM. The fractions containing the protein were dialyzed against lysis buffer with 20 mM HEPES replacing the phosphate buffer before storage at −80°C.
TG2 activity assay
In vivo
Cells plated the day before (100,000 cells/well) were infected with C. trachomatis serovar LGV L2 at a MOI of 1, and 0.5 mM biotin pentylamine (BP) (Thermo Fisher Scientific) was added after 24 h. In some experiments, cells were pre‐incubated for 2 h with 40 mM CP4d or DMSO as control before addition of bacteria. CP4d inhibits the transamidase activity of TG2 (Ki = 174 nM) and favors its closed conformation (Caron et al, 2012). For experiments with ionomycin (Sigma), cells pre‐treated with siRNA for 48 h or infected as described above for 24 h were treated with ionomycin and 0.5 mM BP for 6 h.
At the end of the indicated incubation time, cells were lysed using 8 M urea buffer (30 mM Tris, 150 mM NaCl, 8 M urea, 1% SDS, pH = 8.0) and samples subjected to Western blot.
In vitro
1 mU of transglutaminase from guinea pig liver (Sigma) was incubated at 37°C for 15 min or 3 h with 5 μg of rhGFPT1, rhGFPT1 Q58N, or rhGFPT1 Q328N and 1 mM of BP in 20 mM HEPES buffer pH 7.5, 200 mM NaCl, 2 mM TCEP, 10% glycerol, 1 mM fructose‐6‐phosphate, and 10 mM CaCl2. The reaction was stopped by adding ethylene‐bis(oxyethylenenitrilo)tetraacetic acid (EGTA) at a final concentration of 20 mM and boiling the samples 5 min at 95°C.
Streptavidin precipitation of TG2 targets
Twenty million HeLa cells were seeded in a 163‐cm2 flask. One day later, the cells were infected or not with C. trachomatis serovar LGV L2 at a MOI = 1. Two hours post‐infection (hpi), the culture medium was changed and 40 μM CP4d or DMSO was added. BP was added to the culture medium at 0.5 mM 24 h post‐treatment (infection or not), and cells were lysed 46 hpi directly in the well using 8 M urea buffer. DNA was disrupted by sonication, and a dialysis was performed against 2 M urea buffer (Tris 30 mM, NaCl 150 mM, 2 M urea, 1% SDS, pH = 8.0). Samples were incubated overnight at 4°C with streptavidin–agarose beads (Sigma). After three washes with 2 M urea buffer and three washes with phosphate‐buffered saline (PBS), proteins precipitated on the beads were eluted using Laemmli's buffer containing dithiothreitol (Sigma) boiled 5 min at 95°C. Samples were then analyzed by Western blot or by mass spectrometry.
SDS–PAGE and Western blot
Proteins were subjected to sodium dodecyl–sulfate polyacrylamide gel electrophoresis (SDS–PAGE) and transferred to a polyvinylidene difluoride (PVDF) membrane, which was blocked with 1× PBS containing 5% bovine serum albumin (BSA, for biotin revelation only) or milk and 0.01% Tween 20. The membranes were then immunoblotted with primary antibodies diluted in 1× PBS containing 5% milk and 0.01% Tween 20. For analyzing the TG2 activity assay, biotin incorporation was revealed using streptavidin conjugated to HRP (#RPN1231, Sigma). Primary antibodies used in the Western blots were the mouse clone 7D2 against TG2 (#ABIN1109303, Covalab), rabbit anti‐serum against GFPT (kindly given by Dr. C. Weigert, University of Tübingen, Germany), mouse clone RL2 against O‐GlcNAcylation (#MA1‐072 Thermo Fisher Scientific), and mouse clone AC‐74 against β‐actin (#A5441 Sigma). Secondary antibodies were anti‐mouse‐HRP (#NA931, GE Healthcare)‐ or anti‐rabbit‐HRP (#G‐21234, Invitrogen)‐conjugated antibodies. Blots were developed using the Western Lightning Chemiluminescence Reagent (GE Healthcare).
Ovarian cancer cohort and statistical analysis
Expression data for TGM2, GLUT‐1, and GLUT‐3 in 265 high‐grade serous ovarian cancers from Edinburgh were available from previous transcriptomic studies of ovarian cancer (Hollis et al, 2019). Per‐sample expression was calculated as the mean expression of probe‐sets informative for each gene. Expression comparisons were performed using Spearman's rank correlation test. Spearman's rank correlation was chosen over Pearson's correlation following demonstration of non‐normal expression distribution for TG2, GLUT‐1 and GLUT‐3 (Shapiro–Wilk normality test, P < 0.05 for all).
Mass spectrometry
In solution protein digestion
Samples were prepared in triplicate. For streptavidin precipitation of TG2 target samples, tryptic digestion was performed by enhanced filter‐aided sample preparation. All steps were done in‐filter. Briefly, samples were reduced (50 mM TCEP, 30 min at room temperature) and alkylated (50 mM iodoacetamide, 1 h at room temperature in the dark). Then, proteins were incubated overnight at 37°C with 500 ng trypsin (Trypsin Gold Mass Spectrometry Grade, Promega). Peptides were recovered by centrifugation.
After TG2/GFPT1 reactions, in vitro samples were diluted in a large excess of 8 M urea/100 mM Tris–HCl pH 8.5 buffer and then, as previously described, reduced (5 mM TCEP, 30 min at room temperature) and alkylated (10 mM iodoacetamide, 30 min at room temperature in the dark). Proteins were first digested for 5 h at 37°C with 500 ng rLys‐C Mass Spec Grade (Promega, Madison, WI, USA) before being diluted fourfold with 100 mM Tris–HCl pH 8.5 to reach a concentration below 2 M urea. Samples were then incubated overnight at 37°C with 500 ng Sequencing Grade Modified Trypsin (Promega, Madison, WI, USA). To achieve the complete digestion of the peptides, a second incubation with the same amount of trypsin (5 h at 37°C) was performed. Digestion was stopped by adding formic acid to 5% final concentration, and peptides were desalted and concentrated on Sep‐Pak C18SPE cartridge (Waters, Milford, MA, USA) according to the manufacturer's instructions.
Mass spectrometry analysis
Tryptic peptides were analyzed on a Q Exactive Plus instrument (Thermo Fisher Scientific, Bremen) coupled with an EASY nLC 1,000 or 1,200 chromatography system (Thermo Fisher Scientific, Bremen). Sample was loaded on an in‐house packed 50 cm nano‐HPLC column (75 μm inner diameter) with C18 resin (1.9 μm particles, 100 Å pore size, Reprosil‐Pur Basic C18‐HD resin, Dr. Maisch GmbH, Ammerbuch‐Entringen, Germany) and equilibrated in 98% solvent A (H2O, 0.1% FA) and 2% solvent B (ACN, 0.1% FA). 120‐ or 180‐min gradient of solvent B at 250 nL/min flow rates was applied to separated peptides. The instrument method for the Q Exactive Plus was set up in DDA mode (Data Dependent Acquisition). After a survey scan in the Orbitrap (resolution 70,000), the 10 most intense precursor ions were selected for HCD fragmentation with a normalized collision energy set up to 28. Charge state screening was enabled, and precursors with unknown charge state or a charge state of 1 and > 7 were excluded. Dynamic exclusion was enabled for 35 or 45 s, respectively.
Data processing
Data were searched using Andromeda with MaxQuant software 1.4.1.2 or 1.5.3.8 version against, respectively, a Chlamydia trachomatis UniProt reference proteome database concatenated with Homo sapiens UniProt reference proteome database. Data were also searched against usual known mass spectrometry contaminants and reversed sequences of all entries or an E. coli K12 UniProt reference proteome database concatenated with rhGFPT1 and gpTGase proteins (Tyanova et al, 2016). Andromeda searches were performed choosing trypsin as specific enzyme with a maximum number of two missed cleavages. Possible modifications included carbamidomethylation (Cys, fixed), oxidation (Met, variable), N‐ter acetylation (variable), and BP (Gln, variable). The mass tolerance in MS was set to 20 ppm for the first search and then 6 ppm for the main search and 10 ppm for the MS/MS. Maximum peptide charge was set to seven, and five amino acids were required as minimum peptide length. The “match between runs” feature was applied between replicates with a maximal retention time window of 2 or 0.7 min. One unique peptide to the protein group was required for the protein identification. A false discovery rate (FDR) cutoff of 1% was applied at the peptide and protein levels.
Data analysis
To validate the identification of the BP on the glutamine of modified peptides, spectra were manually inspected (or fragment assignments).
For the global quantification, output files from MaxQuant were used for protein quantification. Quantification was performed using the XIC‐based LFQ algorithm with the Fast LFQ mode as described previously (Cox et al, 2014). Unique and razor peptides, included modified peptides, with at least 2 ratio count were accepted for quantification.
For pairwise comparisons, proteins identified in the reverse and contaminant databases and proteins only identified by site were first discarded from the list. Then, proteins exhibiting fewer than 2 LFQ values in at least one condition were discarded from the list to avoid misidentified proteins. After log2 transformation of the leftover proteins, LFQ values were normalized by median centering within conditions (normalizeD function of the R package DAPAR; Wieczorek et al, 2017). Remaining proteins without any LFQ value in one of both conditions have been considered as proteins quantitatively present in a condition and absent in another. They have therefore been set aside and considered as differentially abundant proteins. Next, missing values were imputed using the imp.norm function of the R package norm. Proteins with a fold change under 2 have been considered not significantly differentially abundant. Statistical testing of the remaining proteins (having a fold change over 2) was conducted using a limma t‐test, thanks to the R package limma (Ritchie et al, 2015). An adaptive Benjamini–Hochberg procedure was applied on the resulting p‐values, thanks to the function adjust.p of R package cp4p using the robust method of Pounds and Cheng to estimate the proportion of true null hypotheses among the set of statistical tests (Pounds & Cheng, 2006). The proteins associated with an adjusted P‐value inferior to an FDR level of 1% have been considered as significantly differentially abundant proteins.
Adhesion assay
Adhesion assays were performed as described previously (Vromman et al, 2014). In brief, MEFs cells plated in 24‐well plate the day before (100,000 cells/well) were pre‐cooled 30 min at 4°C and then were incubated for 4 h at 4°C with L2IncDGFP bacteria at a MOI = 10, sonicated prior to infection in order to disrupt bacterial aggregates. Then, cells were washed gently with PBS and detached using 0.5 mM EDTA in PBS. Samples were fixed 30 min in 2% PFA, washed with PBS, and analyzed using flow cytometry.
Bacterial entry assessment
Entry experiments were performed as described previously (Vromman et al, 2014). In brief, MEFs cells plated on coverslips in 24‐well plate the day before (100,000 cells/well) were pre‐cooled 30 min at 4°C and then incubated for 45 min at 4°C with L2IncDGFP bacteria at a MOI = 10, sonicated prior to infection in order to disrupt bacterial aggregates. Then, pre‐warmed medium was added and coverslips were incubated at 37°C before being fixed at different time points in 4% PFA for 20 min. Extracellular bacteria were stained with a mouse anti‐MOMP‐LPS (Argene # 11‐114) antibody followed with Cy5‐conjugated anti‐mouse (#PA45002, Amersham Biosciences) secondary antibody. The dilutions were made in PBS containing 3% of BSA. DNA was stained using 0.5 μg/ml of Hoechst 33342 (Thermo Fisher Scientific) added in the secondary antibody solution. Images were acquired on an Axio observer Z1 microscope equipped with an ApoTome module (Zeiss, Germany) and a 63× Apochromat lens. Images were taken with an ORCAflash4.OLT camera (Hamamatsu, Japan) using the software Zen.
Progeny assay
For glucose privation tests on MEFs cells, 140,000 cells per well were seeded in a glucose‐free DMEM (Invitrogen) supplemented with 10% FBS. The following day, the medium was replaced with glucose‐free DMEM supplemented with 10% FBS and the indicated concentration of glucose (Sigma). The next day, cells were infected with L2IncDGFP bacteria at a MOI = 0.2.
For progeny assays on HeLa cells, primary cells, or MEFs cells, 100,000 cells were seeded in a 24‐well plate. The next day, cells were pre‐treated with CP4d (or DMSO) or cysteamine (Sigma #30078) (or water) for 2 h and infected with L2IncDGFP bacteria at a MOI = 0.15. Cells treated with siRNA for 48 h were directly infected with L2IncDGFP bacteria or C. muridarum at a MOI = 0.2. Thirty hpi, cells were detached and fixed in 2% PFA in PBS prior to flow cytometry analysis in order to evaluate the first round of infection. In duplicate wells, cells were detached and lysed using glass beads and the supernatant was used to infect new untreated cells (or WT cells in the case of MEFs) plated the day before (100,000 cells/well in a 24‐well plate), in serial dilution. The next day, 3 wells per condition with an infection lower than 30% (checked by microscopy) were detached and fixed as described above, before analysis by flow cytometry and determination of the bacterial titer. In the case of C. muridarum infections, bacteria were stained after fixation with a rabbit anti‐CmGroEL antibody followed with Alexa Fluor 488‐conjugated anti‐rabbit secondary antibody (A11034, Invitrogen). Dilutions were made in PBS containing 0.1% of BSA and 0.05% of saponin (Sigma). The anti‐CmGroEL antibody was obtained by AgroBio (La Ferté Saint‐Aubain) by immunizing one New Zealand white rabbit with C. muridarum GroEL kindly prepared by Dr. Lingling Chen (Indiana University) as described in Illingworth et al (2011). Acquisition was performed using a CytoFLEX S (Beckman Coulter), and 50,000 events per sample were acquired and then analyzed using FlowJo (version 10.0.7).
Infection in mice
Female TG2−/− and TG2+/+ KO mice were kindly provided by Dr. C. Papista (INSERM UMR970, Centre de Recherche Cardiovasculaire, Paris) and maintained in the animal facility of the Institut Pasteur, Paris. All animals are treated with 2.5 mg of medroxyprogesterone (Depo‐provera‐SC®, Pfizer) 7 days prior to infection to synchronize the menstrual cycle. Mice were intravaginally inoculated with C. muridarum, 105 IFU per animal. Twenty‐five days after infection, animals were sacrificed and the organs excised including cervix, uterine horn, and oviduct. The bacterial burden in the excised organs, in the right part of the upper genital tract, was measured by qPCR after DNA extraction using the DNeasy Blood and Tissue Kit (Qiagen). The left part of the upper genital tract was excised and rinsed into PBS for the morphological observation. Hydrosalpinx score was determined as described (Peng et al, 2011). Procedures involving mice were previously approved by local Animal Ethics Committees and registered with the French authorities (APAFIS#8635‐2017012314265571).
RT–qPCR and qPCR
One hundred twenty‐five thousand cells in a 24‐well plate were infected or not with C. trachomatis serovar LGV L2 at a MOI = 1. Cells were treated with anti‐IL‐6 receptor antibody (RoActemra®, Roche) CP4d (40 μM) or DMSO 2 hpi, or doxycycline (62.5 ng/ml, Sigma) 24 hpi. Cells pre‐treated with siRNA for 48 h were directly infected with L2IncDGFP bacteria at a MOI = 1. For testing the response of HeLa cells to IL‐6 treatment, 125,000 cells in 24‐well plate were treated with recombinant human IL‐6 (R&D Biosystems) for 18 h.
Total RNAs were isolated 24 or 48 hpi with the RNeasy Mini Kit (Qiagen) with DNase treatment (DNase I, Roche). RNA concentrations were determined with a spectrophotometer NanoDrop (Thermo Fisher Scientific) and normalized to equal contents. Reverse transcription (RT) was performed using the M‐MLV Reverse Transcriptase (Promega) and quantitative PCR (qPCR) undertaken on the complementary DNA (cDNA) with LightCycler 480 system using LightCycler 480 SYBR Green Master I (Roche). For the experiments displayed in Appendix Fig S2, a duplicate well was used to extract genomic DNA (gDNA) of each time point using the DNeasy Blood and Tissue Kit (Qiagen). Data were analyzed using the ΔΔC t method with the actin gene as a control gene (Schmittgen & Livak, 2008). Each RT–qPCR experiment was performed in duplicate and repeated at least three times.
GFPT activity assay
HeLa cells treated with ionomycin (4 μM) or DMSO for 6 h were detached in lysis buffer containing 0.05 M Tris, 0.15 M NaCl, 5% glycerol, 0.5% NP‐40, protease inhibitor cocktail EDTA‐free (Roche), pH 7.5. After lysis at 4°C, NP‐40 concentration was reduced by addition of an excess of reaction buffer (0.05 M Tris, 0.15 M NaCl, 5% glycerol, protease inhibitor cocktail EDTA‐free, pH 7.5) and cell debris was eliminated by centrifugation. Cell lysates were incubated 45 min at 37°C with 0.6 mg/ml fructose‐6‐phosphate (Sigma) and 0.6 mg/ml glutamine (Sigma). The reaction was stopped by incubating the samples for 5 min at 100°C, and precipitates were removed by centrifugation. Analysis of the samples was performed by high‐performance anion‐exchange chromatography (HPAEC, Dionex, model ISC3000) on a CarboPAC‐PA1 column (3.2 × 250 mm, Dionex) using 100 mM NaOH, and 720 mM NaOAc in 100 mM NaOH, as eluent A and B, respectively. The column was pre‐equilibrated for 20 min in 98% A + 2% B. Following sample injection, a gradient run (flow rate 1 ml/min) was performed as follows: 0–2 min, isocratic step (98% A + 2% B), 2–15 min 98% A + 2% B – 80% A + 20% B, 15–20 min 80% A + 20% B – 57% A + 43% B, 20–22 min 57% A + 43% B – 100% B, and 22–25 min 100% B. Samples were detected on a pulsed electrochemical detector.
Bacterial size measurement
HeLa cells in a 6‐well plate, treated with siRNA as described above, were infected every 2 h with L2IncDGFP bacteria at a MOI = 0.3. The next day, all wells were detached and fixed simultaneously in 2% PFA and 2.5% glutaraldehyde (Sigma) in PBS. After 25 min, cells were broken using glass beads, vortexed, and syringed (three times, using 1 ml 26GA × 3/8‐inch syringes). Samples were then analyzed by flow cytometry. An exponential culture of E. coli, purified C. trachomatis elementary bodies (i.e., the infectious non‐replicative form of the bacterium), and non‐infected cells prepared the same way were used to gate successively for particles of equal size or smaller than E. coli (thereby excluding non‐broken cells), larger than elementary bodies (thereby excluding non‐dividing bacteria), and with positive green fluorescence (thereby excluding cell debris). The forward‐scattered light (FSC‐A) was used to compare bacterial diameters. Each data point represents the mean of at least 400 gated events.
Electron microscopy
One million five hundred thousand cells were transfected with siRNA and infected two days later. Cells were fixed 30 hpi with 2.5% glutaraldehyde (v/v) (Electron Microscopy Sciences) in 0.1 M cacodylate buffer pH 7.4, for 1 h at room temperature. After several washes in cacodylate, they were post‐fixed with 1% osmium tetroxide (w/v) in cacodylate for 1 h. After several washes with water, the cells were progressively dehydrated with increasing concentrations of ethanol from 25% to 100%. The cells were then gradually embedded in epoxy resin. After overnight polymerization at 60°C, 50‐ to 70‐nm thin sections were cut in an ultra‐microtome (Ultracut, Leica) and cells were imaged after post‐staining with uranyl acetate and lead citrate in a T12‐FEI transmission EM operated at 120 kV.
Author contributions
AS, BM, ML, and JWK conceived the study and designed the methodology. BM, ML, ST, and SP conducted the experiments and performed data analysis and interpretation. YW conducted the experiments with mice and performed data analysis. MD and MM collected and analyzed the mass spectrometry data. RLH and CG collected and analyzed the HGSOC cohort data. JR collected and analyzed the infection data using cells isolated from the fallopian tube. JWK synthesized TG2 inhibitors. AS and BM wrote the original draft of the manuscript. MM and JWK edited the manuscript. AS and BM revised the manuscript. All authors commented on the manuscript. AS supervised the study and secured funding.
Conflict of interest
The authors declare that they have no conflict of interest.
Supporting information
Appendix
Expanded View Figures PDF
Review Process File
Source Data for Figure 1
Source Data for Figure 4
Source Data for Figure 5
Source Data for Figure 6
Acknowledgements
We thank Anke Hellberg and Béatrice Niragire for technical assistance, Manuela D'Eletto for TG2−/− reconstituted MEFs, Dr Christina Papista for providing mice, Dr Denise Badet‐Denisot for the rhGFPT1 plasmid and for advice, Dr Cora Weigert for anti‐GFPT antibodies, Vishu Aimanianda Bopaiah for help with HPAEC, Dr Lingling Chen for CmGroEL, and Augustin Latourte for RoActemra®. This work was supported by an ERC Starting Grant (NUChLEAR N°282046), the Institut Pasteur (GFP‐LIMNEC METINF), the Centre National de la Recherche Scientifique, and GEFLUC. BM was funded by the Ministère de l'Education Nationale, de la Recherche et de la Technologie, and by Cancéropole Ile‐de‐France.
The EMBO Journal (2020) 39: e102166
Data availability
The mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium via the PRIDE partner repository (http://www.ebi.ac.uk/pride) with the dataset identifier PXD017117.
References
- AbdelRahman YM, Belland RJ (2005) The chlamydial developmental cycle. FEMS Microbiol Rev 29: 949–959 [DOI] [PubMed] [Google Scholar]
- Altuntas S, Rossin F, Marsella C, D'Eletto M, Diaz‐Hidalgo L, Farrace MG, Campanella M, Antonioli M, Fimia GM, Piacentini M (2015) The transglutaminase type 2 and pyruvate kinase isoenzyme M2 interplay in autophagy regulation. Oncotarget 6: 44941–44954 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Assrir N, Richez C, Durand P, Guittet E, Badet B, Lescop E, Badet‐Denisot MA (2014) Mapping the UDP‐N‐acetylglucosamine regulatory site of human glucosamine‐6P synthase by saturation‐transfer difference NMR and site‐directed mutagenesis. Biochimie 97: 39–48 [DOI] [PubMed] [Google Scholar]
- Brunham RC, Rey‐Ladino J (2005) Immunology of Chlamydia infection: implications for a Chlamydia trachomatis vaccine. Nat Rev Immunol 5: 149–161 [DOI] [PubMed] [Google Scholar]
- Caron NS, Munsie LN, Keillor JW, Truant R (2012) Using FLIM‐FRET to measure conformational changes of transglutaminase type 2 in live cells. PLoS One 7: e44159‐7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang Q, Su K, Baker JR, Yang X, Paterson AJ, Kudlow JE (2000) Phosphorylation of human glutamine: fructose‐6‐phosphate amidotransferase by cAMP‐dependent protein kinase at serine 205 blocks the enzyme activity. J Biol Chem 275: 21981–21987 [DOI] [PubMed] [Google Scholar]
- Cox J, Hein MY, Luber CA, Paron I, Nagaraj N, Mann M (2014) Accurate proteome‐wide label‐free quantification by delayed normalization and maximal peptide ratio extraction, termed MaxLFQ. Mol Cell Proteomics 13: 2513–2526 [DOI] [PMC free article] [PubMed] [Google Scholar]
- D'Eletto M, Farrace MG, Falasca L, Reali V, Oliverio S, Melino G, Griffin M, Fimia GM, Piacentini M (2009) Transglutaminase 2 is involved in autophagosome maturation. Autophagy 5: 1145–1154 [DOI] [PubMed] [Google Scholar]
- Derré I, Swiss R, Agaisse H (2011) The lipid transfer protein CERT interacts with the Chlamydia inclusion protein IncD and participates to ER‐Chlamydia inclusion membrane contact sites. PLoS Pathog 7: e1002092 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Sabatino A, Vanoli A, Giuffrida P, Luinetti O, Solcia E, Corazza GR (2012) The function of tissue transglutaminase in celiac disease. Autoimmun Rev 11: 746–753 [DOI] [PubMed] [Google Scholar]
- Eckert RL, Kaartinen MT, Nurminskaya M, Belkin AM, Colak G, Johnson GVW, Mehta K (2014) Transglutaminase regulation of cell function. Physiol Rev 94: 383–417 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elwell CA, Ceesay A, Kim JH, Kalman D, Engel JN (2008) RNA interference screen identifies Abl kinase and PDGFR signaling in Chlamydia trachomatis entry. PLoS Pathog 4: e1000021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farrelly LA, Thompson RE, Zhao S, Lepack AE, Lyu Y, Bhanu NV, Zhang B, Loh Y‐HE, Ramakrishnan A, Vadodaria KC et al (2019) Histone serotonylation is a permissive modification that enhances TFIID binding to H3K4me3. Nature 567: 535–539 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Filiano AJ, Bailey CD, Tucholski J, Gundemir S, Johnson GV (2008) Transglutaminase 2 protects against ischemic insult, interacts with HIF1beta, and attenuates HIF1 signaling. FASEB J 22: 2662–2675 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Folk JE, Mullooly JP, Cole PW (1967) Mechanism of action of guinea pig liver transglutaminase: II. The role of metal in enzyme activation. J Biol Chem 242: 1838–1844 [PubMed] [Google Scholar]
- Ford C, Nans A, Boucrot E, Hayward RD (2018) Chlamydia exploits filopodial capture and a macropinocytosis‐like pathway for host cell entry. PLoS Pathog 14: e1007051 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gehre L, Gorgette O, Perrinet S, Prevost MC, Ducatez M, Giebel AM, Nelson DE, Ball SG, Subtil A (2016) Sequestration of host metabolism by an intracellular pathogen. Elife 5: e12552 [DOI] [PMC free article] [PubMed] [Google Scholar]
- George Z, Omosun Y, Azenabor AA, Partin J, Joseph K, Ellerson D, He Q, Eko F, Bandea C, Svoboda P et al (2016) The roles of unfolded protein response pathways in Chlamydia pathogenesis. J Infect Dis 215: 456–465 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guilluy C, Rolli‐Derkinderen M, Tharaux P‐L, Melino G, Pacaud P, Loirand G (2007) Transglutaminase‐dependent RhoA Activation and Depletion by Serotonin in vascular smooth muscle cells. J Biol Chem 282: 2918–2928 [DOI] [PubMed] [Google Scholar]
- Gundemir S, Colak G, Tucholski J, Johnson GV (2012) Transglutaminase 2: a molecular Swiss army knife. Biochim Biophys Acta 1823: 406–419 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gundemir S, Colak G, Feola J, Blouin R, Johnson GV (2013) Transglutaminase 2 facilitates or ameliorates HIF signaling and ischemic cell death depending on its conformation and localization. Biochim Biophys Acta 1833: 1–10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gundemir S, Monteagudo A, Akbar A, Keillor JW, Johnson GVW (2017) The complex role of transglutaminase 2 in glioblastoma proliferation. Neuro Oncol 19: 208–218 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haneji T, Koide SS (1989) Transblot identification of biotin‐containing proteins in rat liver. Anal Biochem 177: 57–61 [DOI] [PubMed] [Google Scholar]
- Hollis RL, Churchman M, Michie CO, Rye T, Knight L, McCavigan A, Perren T, Williams A, McCluggage WG, Kaplan RS et al (2019) High EMSY expression defines a BRCA‐like subgroup of high grade serous ovarian carcinoma with prolonged survival and hypersensitivity to platinum. Cancer 125: 2772–2781 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang L, Xu A‐M, Liu W (2015) Transglutaminase 2 in cancer. Am J Cancer Res 5: 2756–2776 [PMC free article] [PubMed] [Google Scholar]
- Hwang JY, Mangala LS, Fok JY, Lin YG, Merritt WM, Spannuth WA, Nick AM, Fiterman DJ, Vivas‐Mejia PE, Deavers MT et al (2008) Clinical and biological significance of tissue transglutaminase in ovarian carcinoma. Cancer Res 68: 5849–5858 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ientile R, Curro M, Caccamo D (2015) Transglutaminase 2 and neuroinflammation. Amino Acids 47: 19–26 [DOI] [PubMed] [Google Scholar]
- Iismaa SE, Mearns BM, Lorand L, Graham RM (2009) Transglutaminases and disease: lessons from genetically engineered mouse models and inherited disorders. Physiol Rev 89: 991–1023 [DOI] [PubMed] [Google Scholar]
- Illingworth M, Ramsey A, Zheng Z, Chen L (2011) Stimulating the substrate folding activity of a single ring GroEL variant by modulating the cochaperonin GroES. J Biol Chem 286: 30401–30408 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jackson SP, Tjian R (1988) O‐glycosylation of eukaryotic transcription factors: implications for mechanisms of transcriptional regulation. Cell 55: 125–133 [DOI] [PubMed] [Google Scholar]
- Jóźwiak P, Forma E, Bryś M, Krzeslak A (2014) O‐GlcNAcylation and metabolic reprograming in cancer. Front Endocrinol 5: 145 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keresztessy Z, Csosz E, Harsfalvi J, Csomos K, Gray J, Lightowlers RN, Lakey JH, Balajthy Z, Fesus L (2006) Phage display selection of efficient glutamine‐donor substrate peptides for transglutaminase 2. Protein Sci 15: 2466–2480 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kreppel LK, Hart GW (1999) Regulation of a cytosolic and nuclear O‐GlcNAc transferase: role of the tetratricopeptide repeats. J Biol Chem 274: 32015–32022 [DOI] [PubMed] [Google Scholar]
- Kumar S, Donti TR, Agnihotri N, Mehta K (2014) Transglutaminase 2 reprogramming of glucose metabolism in mammary epithelial cells via activation of inflammatory signaling pathways. Int J Cancer 134: 2798–2807 [DOI] [PubMed] [Google Scholar]
- Lee KN, Maxwell MD, Patterson MK, Birckbichler PJ, Conway E (1992) Identification of transglutaminase substrates in HT29 colon cancer cells: use of 5‐(biotinamido)pentylamine as a transglutaminase‐specific probe. Biochim Biophys Acta 1136: 12–16 [DOI] [PubMed] [Google Scholar]
- Lee JH, Jeong J, Jeong EM, Cho SY, Kang JW, Lim J, Heo J, Kang H, Kim IG, Shin DM (2014) Endoplasmic reticulum stress activates transglutaminase 2 leading to protein aggregation. Int J Mol Med 33: 849–855 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee JK, Enciso GA, Boassa D, Chander CN, Lou TH, Pairawan SS, Guo MC, Wan FYM, Ellisman MH, Sütterlin C et al (2017) Replication‐dependent size reduction precedes differentiation in Chlamydia trachomatis . Nat Commun 9: 45 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y, Roux C, Lazereg S, LeCaer JP, Laprevote O, Badet B, Badet‐Denisot MA (2007) Identification of a novel serine phosphorylation site in human glutamine: fructose‐6‐phosphate amidotransferase isoform 1. Biochemistry 46: 13163–13169 [DOI] [PubMed] [Google Scholar]
- Liechti G, Kuru E, Packiam M, Hsu Y‐P, Tekkam S, Hall E, Rittichier JT, VanNieuwenhze M, Brun YV, Maurelli AT (2016) Pathogenic chlamydia lack a classical sacculus but synthesize a narrow, mid‐cell peptidoglycan ring, regulated by MreB, for cell division. PLoS Pathog 12: e1005590 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu C, Kellems RE, Xia Y (2017) Inflammation, autoimmunity, and hypertension: the essential role of tissue transglutaminase. Am J Hypertens 30: 756–764 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Majeed M, Krause KH, Clark RA, Kihlström E, Stendahl O (1999) Localization of intracellular Ca2+ stores in HeLa cells during infection with Chlamydia trachomatis . J Cell Sci 112: 35–44 [DOI] [PubMed] [Google Scholar]
- Mehul B, Bawumia S, Hughes RC (1995) Cross‐linking of galectin 3, a galactose‐binding protein of mammalian cells, by tissue‐type transglutaminase. FEBS Lett 360: 160–164 [DOI] [PubMed] [Google Scholar]
- Nelea V, Nakano Y, Kaartinen MT (2008) Size distribution and molecular associations of plasma fibronectin and fibronectin crosslinked by transglutaminase 2. Protein J 27: 223–233 [DOI] [PubMed] [Google Scholar]
- Nurminskaya M, Beazley KE, Smith EP, Belkin AM (2014) Transglutaminase 2 promotes PDGF‐mediated activation of PDGFR/Akt1 and beta‐catenin signaling in vascular smooth muscle cells and supports neointima formation. J Vasc Res 51: 418–428 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ojcius D, Degani H, Mispelter J, Dautry‐Varsat A (1998) Enhancement of ATP levels and glucose metabolism during an infection by Chlamydia . J Biol Chem 273: 7052–7058 [DOI] [PubMed] [Google Scholar]
- Olson MG, Widner RE, Jorgenson LM, Lawrence A, Lagundzin D, Woods NT, Ouellette SP, Rucks EA (2019) Proximity labeling to map host‐pathogen interactions at the membrane of a bacterium‐containing vacuole in Chlamydia trachomatis‐infected human cells. Infect Immun 87: e00537‐19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orrù S, Caputo I, D'Amato A, Ruoppolo M, Esposito C (2003) Proteomics identification of Acyl‐acceptor and Acyl‐donor substrates for transglutaminase in a human intestinal epithelial cell line: implications for celiac disease. J Biol Chem 278: 31766–31773 [DOI] [PubMed] [Google Scholar]
- Palucci I, Matic I, Falasca L, Minerva M, Maulucci G, De Spirito M, Petruccioli E, Goletti D, Rossin F, Piacentini M et al (2017) Transglutaminase type 2 plays a key role in the pathogenesis of Mycobacterium tuberculosis infection. J Intern Med 283: 303–313 [DOI] [PubMed] [Google Scholar]
- Peng B, Lu C, Tang L, Yeh IT, He Z, Wu Y, Zhong G (2011) Enhanced upper genital tract pathologies by blocking Tim‐3 and PD‐L1 signaling pathways in mice intravaginally infected with Chlamydia muridarum . BMC Infect Dis 11: 347 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pincus JH, Waelsch H (1968) The specificity of transglutaminase. I. Human hemoglobin as a substrate for the enzyme. Arch Biochem Biophys 126: 34–43 [DOI] [PubMed] [Google Scholar]
- Pounds S, Cheng C (2006) Robust estimation of the false discovery rate. Bioinformatics 22: 1979–1987 [DOI] [PubMed] [Google Scholar]
- Rasmussen SJ, Eckmann L, Quayle AJ, Shen L, Zhang Y‐X, Anderson DJ, Fierer J, Stephens R, Kagnoff M (1997) Secretion of proinflammatory cytokines by epithelial cells in response to Chlamydia infection suggests a central role for epithelial cells in chlamydial pathogenesis. J Clin Invest 99: 77–87 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Read T, Brunham R, Shen C, Gill S, Heidelberg J, White O, Hickey E, Peterson J, Utterback T, Berry K et al (2000) Genome sequences of Chlamydia trachomatis MoPn and Chlamydia pneumoniae AR39. Nucleic Acids Res 28: 1397–1406 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ritchie ME, Phipson B, Wu D, Hu Y, Law CW, Shi W, Smyth GK (2015) limma powers differential expression analyses for RNA‐sequencing and microarray studies. Nucleic Acids Res 43: e47 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rossin F, D'Eletto M, Macdonald D, Farrace MG, Piacentini M (2012) TG2 transamidating activity acts as a reostat controlling the interplay between apoptosis and autophagy. Amino Acids 42: 1793–1802 [DOI] [PubMed] [Google Scholar]
- Roth A, Konig P, van Zandbergen G, Klinger M, Hellwig‐Burgel T, Daubener W, Bohlmann MK, Rupp J (2010) Hypoxia abrogates antichlamydial properties of IFN‐gamma in human fallopian tube cells in vitro and ex vivo . Proc Natl Acad Sci USA 107: 19502–19507 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rother M, Gonzalez E, Teixeira da Costa AR, Wask L, Gravenstein I, Pardo M, Pietzke M, Gurumurthy RK, Angermann J, Laudeley R et al (2018) Combined human genome‐wide RNAi and metabolite analyses identify IMPDH as a host‐directed target against chlamydia infection. Cell Host Microbe 23: 661–671.e8 [DOI] [PubMed] [Google Scholar]
- Russell DG, VanderVen BC, Lee W, Abramovitch RB, Kim M‐j, Homolka S, Niemann S, Rohde KH (2010) Mycobacterium tuberculosis wears what it eats. Cell Host Microbe 8: 68–76 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmittgen TD, Livak KJ (2008) Analyzing real‐time PCR data by the comparative CT method. Nat Protoc 3: 1101–1108 [DOI] [PubMed] [Google Scholar]
- Scidmore MA (2005) Cultivation and laboratory maintenance of Chlamydia trachomatis . Curr Protoc Microbiol Chapter 11: 11A1.1–11A1.25 [DOI] [PubMed] [Google Scholar]
- Shao M, Cao L, Shen C, Satpathy M, Chelladurai B, Bigsby RM, Nakshatri H, Matei D (2009) Epithelial‐to‐mesenchymal transition and ovarian tumor progression induced by tissue transglutaminase. Cancer Res 69: 9192–9201 [DOI] [PubMed] [Google Scholar]
- Sharma M, Machuy N, Bohme L, Karunakaran K, Maurer AP, Meyer TF, Rudel T (2011) HIF‐1alpha is involved in mediating apoptosis resistance to Chlamydia trachomatis‐infected cells. Cell Microbiol 13: 1573–1585 [DOI] [PubMed] [Google Scholar]
- Shi L, Salamon H, Eugenin EA, Pine R, Cooper A, Gennaro ML (2015) Infection with Mycobacterium tuberculosis induces the Warburg effect in mouse lungs. Sci Rep 5: 18176 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sohn J, Chae JB, Lee SY, Kim SY, Kim JG (2010) A novel therapeutic target in inflammatory uveitis: transglutaminase 2 inhibitor. Korean J Ophthalmol 24: 29–34 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stephens RS, Kalman S, Lammel C, Fan J, Marathe R, Aravind L, Mitchell W, Olinger L, Tatusov RL, Zhao Q et al (1998) Genome sequence of an obligate intracellular pathogen of humans: Chlamydia trachomatis . Science 282: 754–755 [DOI] [PubMed] [Google Scholar]
- Sugimura Y, Hosono M, Wada F, Yoshimura T, Maki M, Hitomi K (2006) Screening for the preferred substrate sequence of transglutaminase using a phage‐displayed peptide library: identification of peptide substrates for TGASE 2 and Factor XIIIA. J Biol Chem 281: 17699–17706 [DOI] [PubMed] [Google Scholar]
- Suto N, Ikura K, Sasaki R (1993) Expression induced by interleukin‐6 of tissue‐type transglutaminase in human hepatoblastoma HepG2 cells. J Biol Chem 268: 7469–7473 [PubMed] [Google Scholar]
- Tarbet HJ, Dolat L, Smith TJ, Condon BM, O'Brien ET III, Valdivia RH, Boyce M (2018) Site‐specific glycosylation regulates the form and function of the intermediate filament cytoskeleton. Elife 7: e31807 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tyanova S, Temu T, Cox J (2016) The MaxQuant computational platform for mass spectrometry‐based shotgun proteomics. Nat Protoc 11: 2301–2319 [DOI] [PubMed] [Google Scholar]
- Vromman F, Laverriere M, Perrinet S, Dufour A, Subtil A (2014) Quantitative monitoring of the Chlamydia trachomatis developmental cycle using GFP‐expressing bacteria. Microscopy and flow cytometry. PLoS One 9: e99197 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Hybiske K, Stephens RS (2017) Orchestration of the mammalian host cell glucose transporter proteins‐1 and 3 by Chlamydia contributes to intracellular growth and infectivity. Pathog Dis 75: ftx108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wieczorek S, Combes F, Lazar C, Giai Gianetto Q, Gatto L, Dorffer A, Hesse AM, Coute Y, Ferro M, Bruley C et al (2017) DAPAR & ProStaR: software to perform statistical analyses in quantitative discovery proteomics. Bioinformatics 33: 135–136 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang X, Qian K (2017) Protein O‐GlcNAcylation: emerging mechanisms and functions. Nat Rev Mol Cell Biol 18: 452–465 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zibrova D, Vandermoere F, Göransson O, Peggie M, Mariño KV, Knierim A, Spengler K, Weigert C, Viollet B, Morrice NA et al (2017) GFAT1 phosphorylation by AMPK promotes VEGF‐induced angiogenesis. Biochem J 474: 983–1001 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix
Expanded View Figures PDF
Review Process File
Source Data for Figure 1
Source Data for Figure 4
Source Data for Figure 5
Source Data for Figure 6
Data Availability Statement
The mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium via the PRIDE partner repository (http://www.ebi.ac.uk/pride) with the dataset identifier PXD017117.