

Abstract
Five new steroid sulfates, sodium 2β,3α-dihydroxy-5α-cholestane 3-sulfate (6), sodium 2β,3α-dihydroxy-5α-cholestane 2-sulfate (7), disodium 2β,3α-dihydroxy-5α-cholestane disulfate (8), sodium 3α-acetoxy-2β-hydroxy-5α-cholestane 2-sulfate (12), and sodium 2β-acetoxy-3α-hydroxy-5α-cholestane 3-sulfate (13), have been synthesized starting from 3β-hydroxy-5α-cholestane (1). The synthetic steroids were completely characterized by one-dimensional and two-dimensional NMR and FABMS spectra. Sulfation was performed using triethylamine–sulfur trioxide complex in dimethylformamide as the sulfating agent. The sulfated steroids were comparatively evaluated for their inhibitory effect on the replication of herpes simplex virus type 2 (HSV-2). Compounds 7 and 8 were the most effective in their inhibitory action against HSV-2. The disulfated steroid 8 also proved to be active against DEN-2 and JV.
Keywords: Sulfated steroids, Synthesis, Antiviral activity
1. Introduction
Sulfated polyhydroxysterols are naturally occurring metabolites in sponges and echinoderms [1], [2]. These compounds have exhibited a broad spectrum of biologic activities, such as anti-HIV effects [3] and inhibition of protein tyrosine kinases [4]. Among echinoderms, ophiuroids are characterized by their content of sulfated steroids with sulfate groups located at C-3 and C-21 and additional hydroxy groups located in rings A and C. Recently, we have demonstrated the antiviral activity of sulfated steroids isolated from the cold water ophiuroids, Ophioplocus januarii [5] and Astrotoma agassizii [6], and synthetic derivatives and analogs against pathogenic viruses of humans [7]. Steroids sulfated at C-21 and C-2 (β) or C-3 (α) with an additional hydroxyl function at C-2 (β) or C-3 (α) were the most effective in their inhibitory action against herpes simplex virus (HSV-2). These results prompted us to synthesize disodium 2β,3α-dihydroxy-5α-cholestane (8) disulfate as well as monosulfated and acetylated derivatives of 2β,3α-dihydroxy-5α-cholestane (6, 7, 12, 13) (Fig. 1 ) in order to evaluate the antiviral activity of these compounds and gain insight into structure–activity correlations.
Fig. 1.
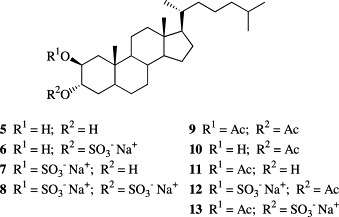
Synthetic intermediates (5, 9–11) and new sulfated steroids (6–8, 12–13).
2. Experimental
Melting points (m.p.) were determined on a Fisher Johns apparatus and are uncorrected. 1H and 13C NMR spectra were recorded on a Bruker AM 500 spectrometer. Chemical shifts (δ) are given in ppm downfield from TMS as the internal standard. Two-dimensional NMR spectra were obtained using standard Bruker software. Mass spectra were collected on a VG-ZAB (HREIMS and FABMS) and on a SHIMADZU QP-5000 (EIMS) mass spectrometer. The FABMS (negative ion mode) were obtained on a glycerol matrix. Reversed-phase chromatography was carried out on octadecyl-functionalized silica gel (Aldrich). Preparative HPLC was carried out on an SP liquid chromatograph equipped with a Spectra Series P100 solvent delivery system, a Rheodyne manual injector and a refractive index detector using a Phenomenex AQUA 5 μ C18 125A column. TLC of sulfated steroids was performed on silica gel F254 (n-BuOH/AcOH/H2O (12:3:5)) and C18 reversed-phase plates (MeOH/H2O (80:20 v/v)) and detected by spraying with sulfuric acid (10% EtOH).
Triethylamine–sulfur trioxide complex was prepared by treatment of triethylamine with chlorosulfonic acid following the procedure of Nair and Bernstein [8]. Commercially available 3β-hydroxy-5α-cholestane (1) was obtained from Aldrich and used as starting material for the synthesis of compounds 5–13. Compound 1: 1H NMR δ (CDCl3): 0.65 (s, 3H, H-18), 0.80 (s, 3H, H-19), 0.87 (d, J=6.9 Hz, 6H, H-26, H-27), 0.90 (d, J=6.9 Hz, 3H, H-21), 3.58 (m, 1H, H-3).
2.1. 3β-Hydroxy-5α-cholestane tosylate (2)
To a solution of 3β-hydroxy-5α-cholestane (1) (2 g, 5.15 mmol) in dry pyridine (6.8 ml) p-toluenesulfonyl chloride (2.22 g, 11.65 mmol) was added. The solution was stirred at room temperature for 24 h and then poured into cold water (20 ml) and extracted with CH2Cl2 (3×15 ml). The combined organic layer was washed successively with 2N HCl solution, water, saturated NaHCO3 solution, and water, and then dried over anhydrous MgSO4 and evaporated in vacuo to give 2.49 g (89%) of 2, m.p. 177–178 °C (acetone–H2O). 1H NMR δ (CDCl3): 0.63 (s, 3H, H-18), 0.77 (s, 3H, H-19), 0.86 (d, J=6.6 Hz, 6H, H-26, H-27), 0.89 (d, J=6 Hz, 3H, H-21), 2.44 (s, 3H, CH 3-Ph), 4.42 (m, 1H, H-3), 7.32 (d, J=8 Hz, 2H, H-3′, H-5′), 7.79 (d, J=8.4 Hz, 2H, H-2′, H-6′). HREIMS m/z=542.3793 [M]+ (C34H54O3S, Δ=2.0 mmu).
2.2. 5α-Cholest-2-ene (3)
Lithium bromide (1.5 g, 17.27 mmol) and lithium carbonate (1.29 g, 17.27 mmol) were added to a solution of 3β-hydroxy-5α-cholestane tosylate (2) (0.93 g, 1.71 mmol) in dry DMF (15 ml), and the mixture was refluxed for 1.5 h. After cooling to room temperature the mixture was slowly poured into 10% HCl solution and extracted with CH2Cl2 (3×15 ml). The combined organic layer was washed successively with water, saturated NaHCO3 solution, and water, and then dried over anhydrous MgSO4 and evaporated to dryness to give 537 mg (85%) of 3 as white solid, m.p. 66 °C (acetone–H2O). 1H NMR δ (CDCl3): 0.67 (s, 3H, H-18), 0.76 (s, 3H, H-19), 0.87 (d, J=6.6 Hz, 6H, H-26, H-27), 0.91 (d, J=8 Hz, 3H, H-21), 5.58 (m, 2H, H-2, H-3). HREIMS m/z=370.3600 [M]+ (C27H46, Δ=3.0 mmu).
2.3. 2,3α-Epoxy-5α-cholestane (4)
To a solution of 5α-cholest-2-ene (3) (481 mg, 1.30 mmol) in CH2Cl2 (33 ml) were added water (22.6 ml) and Na2CO3 (566 mg, 4.77 mmol). The reaction mixture was stirred vigorously and m-chloroperbenzoic acid (310 mg, 1.80 mmol) was added slowly. The mixture was stirred for 4 h at room temperature, and then the aqueous layer was extracted with CH2Cl2 (3×25 ml). The combined CH2Cl2 extracts were washed successively with 5% Na2SO3 solution, saturated NaHCO3 solution, and water, dried over anhydrous MgSO4 and evaporated to dryness to give 497 mg (99%) of 4 as a white powder, m.p. 105–106 °C (acetone). 1H NMR δ (CDCl3): 0.64 (s, 3H, H-18), 0.75 (s, 3H, H-19), 0.86 (d, J=6.6 Hz, 6H, H-26, H-27), 0.90 (d, J=8 Hz, 3H, H-21), 3.13 (m, 2H, H-2, H-3). HREIMS m/z=386.3549 [M]+ (C27H46O, Δ=5.0 mmu).
2.4. 2β,3α-Dihydroxy-5α-cholestane (5)
A solution of epoxide 4 (475 mg, 1.23 mmol) in THF (23 ml) was treated with 1N H2SO4 (1.53 ml, 3.1 mmol) and stirred for 24 h at room temperature. After neutralization with saturated NaHCO3 solution the mixture was evaporated to fifth initial volume, diluted with water (18 ml), and extracted with ethyl acetate (3×15 ml). The combined organic extracts were washed with water, dried over anhydrous MgSO4, filtered and evaporated to dryness. The crude diol 5 (477 mg, 96%) was submitted to vacuum-dry column chromatography on silica gel (32–63 μm), eluted with cyclohexane/acetone (50:50 v/v), evaporated under reduced pressure, and subjected to vacuum-dry column chromatography on silica gel C18 (35–75 μm). Fractions eluted with methanol/water (98:2 v/v) afforded pure 2β,3α-dihydroxy-5α-cholestane (5) (305.1 mg, 62%, m.p. 178–180 °C (acetone–H2O)). 1H NMR δ (CDCl3): 0.66 (s, 3H, H-18), 0.87 (d, J=6.6 Hz, 6H, H-26, H-27), 0.90 (d, J=7.3 Hz, 3H, H-21), 0.99 (s, 3H, H-19), 3.88 (m, 2H, H-2, H-3). EIMS m/z=404 (14%, M+), 386 (2%, M+·H2O), 368 (1%, M+·2H2O).
2.5. Sodium 2β,3α-dihydroxy-5α-cholestane 3-sulfate (6) and sodium 2β,3α-dihydroxy-5α-cholestane 2-sulfate (7)
Triethylamine–sulfur trioxide complex (27 mg, 0.12 mmol) was added to a solution of 2β,3α-dihydroxy-5α-cholestane (5) (24.2 mg, 0.06 mmol) in DMF (0.5 ml), and the mixture was stirred at room temperature for 3 h. Then, the reaction mixture was quenched with water (0.5 ml), evaporated to dryness under reduced pressure, and the residue eluted through Amberlite CG-120 (sodium form) with methanol to afford a mixture of compounds 6 and 7. Preparative HPLC (methanol/water (80:20 v/v)) of the mixture afforded pure 6 (11.84 mg, 39%) and 7 (6.38 mg, 21%) together with the disulfated analog 8 (11.3 mg, 31%). Compound 6: m.p. 124–125 °C. For 1H and 13C NMR see Table 1, Table 2 . FAB− m/z=483 [MNa]−. HRFABMS (negative ion mode) m/z=483.3164 [MNa]− (C27H47O5S, Δ=2.0 mmu). Compound 7: m.p. 151–152 °C. For 1H and 13C NMR see Table 1, Table 2. FAB− m/z=483 [MNa]−. HRFABMS (negative ion mode) m/z=483.3164 [MNa]− (C27H47O5S, Δ=2.0 mmu).
Table 1.
| Proton |
δH multiplet (J) |
||||
| 6 | 7 | 8 | 12 | 13 | |
| H-2 | 4.08 d (2.7) | 4.42 d (2.3) | 4.73 d (2.25) | 4.44 d (2) | 5.14 d (2.5) |
| H-3 | 4.40 d (2.7) | 4.02 d (2.3) | 4.70 d (2.25) | 5.11 d (2) | 4.40 d (2.5) |
| H-18 | 0.68 s | 0.68 s | 0.69 s | 0.69 s | 0.68 s |
| H-19 | 0.99 s | 0.99 s | 0.99 s | 1.01 s | 0.93 s |
| H-21 | 0.93 d (6.6) | 0.93 d (6.6) | 0.93 d (6.6) | 0.93 d (6.6) | 0.93 d (6.6) |
| H-26 | 0.88 d (6.6) | 0.88 d (6.6) | 0.88 d (6.6) | 0.88 d (6.6) | 0.88 d (6.6) |
| H-27 | 0.88 d (6.6) | 0.88 d (6.6) | 0.88 d (6.6) | 0.88 d (6.6) | 0.88 d (6.6) |
| CH3CO | 2.05 s | 2.03 s | |||
Recorded at 500MHz in methanol-d4.
J in Hz.
Table 2.
13C NMR data for compounds 6–8, 12, and 13
| Carbon |
δCa |
||||
| 6 | 7 | 8 | 12 | 13 | |
| 1 | 41.0 t | 38.8 t | 39.2 t | 40.9 t | 38.3 t |
| 2 | 70.0 d | 78.8 d | 76.5 d | 76.0 d | 72.6 d |
| 3 | 78.5 d | 69.0 d | 76.1 d | 72.2 d | 75.5 d |
| 4 | 30.3 t | 32.5 t | 30.5 t | 29.9 t | 30.9 t |
| 5 | 40.6 d | 39.7 d | 40.2 d | 39.6 d | 40.3 d |
| 6 | 29.3 t | 29.3b t | 29.2b t | 29.2b t | 29.4b t |
| 7 | 33.3 t | 33.3 t | 33.2 t | 33.2 t | 33.1 t |
| 8 | 36.3 d | 36.4 d | 36.4b d | 36.4 d | 36.4 d |
| 9 | 56.6 d | 56.7 d | 56.6 d | 56.7 d | 56.4 d |
| 10 | 36.4 s | 36.8 s | 36.4b s | 36.3 s | 36.3 s |
| 11 | 22.0 t | 22.0 t | 22.0 t | 22.0 t | 21.9 t |
| 12 | 41.5 t | 41.5 t | 41.4 t | 41.4 t | 41.4 t |
| 13 | 43.8 s | 43.8 s | 43.8 s | 43.8 s | 43.8 s |
| 14 | 57.9 d | 57.9 d | 57.9 d | 57.9 d | 57.8 d |
| 15 | 25.2 t | 25.2 t | 25.2 t | 25.2 t | 25.2 t |
| 16 | 29.3 t | 29.4b t | 29.3b t | 29.3b t | 29.3b t |
| 17 | 57.7 d | 57.7 d | 57.7 d | 57.7 d | 57.7 d |
| 18 | 12.6 q | 12.6 q | 12.5 q | 12.5 q | 12.6 q |
| 19 | 14.6 q | 14.2 q | 14.3 q | 14.2 q | 14.4 q |
| 20 | 37.1 d | 37.1 d | 37.1 d | 37.1 d | 37.1 d |
| 21 | 19.2 q | 19.2 q | 19.2 q | 19.2 q | 19.2 q |
| 22 | 37.4 t | 37.4 t | 37.4 t | 37.4 t | 37.4 t |
| 23 | 24.9 t | 24.9 t | 24.9 t | 24.9 t | 24.9 t |
| 24 | 40.7 t | 40.7 t | 40.7 t | 40.7 t | 40.7 t |
| 25 | 29.1 d | 29.1 d | 29.1 d | 29.1 d | 29.1 d |
| 27 | 22.9b q | 22.9bq | 22.9b q | 22.9b q | 22.9b q |
| 26 | 23.2b q | 23.2b q | 23.2b q | 23.2b q | 23.2b q |
| CH3 | 21.1 q | 21.3 q | |||
| CO | 171.6 s | 171.6 s | |||
Recorded at 125MHz in methanol-d4; multiplicity by DEPT.
Chemical shifts are interchangeable.
2.6. Disodium 2β,3α-dihydroxy-5α-cholestane disulfate (8)
Triethylamine–sulfur trioxide complex (48 mg, 0.265 mmol) was added to a solution of 2β,3α-dihydroxy-5α-cholestane (5) (17 mg, 0.042 mmol) in DMF (0.5 ml). The reaction mixture was stirred at 95 °C for 13 h and then quenched with water (0.5 ml). After evaporation to dryness the residue was eluted through Amberlite CG-120 (sodium form) with methanol. Further purification by preparative HPLC (methanol/water (80:20 v/v)) afforded pure disodium 2β,3α-dihydroxy-5α-cholestane disulfate (8) (23 mg, 93%), m.p. 147−148 °C. For 1H and 13C NMR see Table 1, Table 2. FAB− m/z=585 [MNa]−. HRFABMS (negative ion mode) m/z=585.2534 [MNa]− (C27H46O8S2Na, Δ=0.2 mmu).
2.7. 2β,3α-Diacetoxy-5α-cholestane (9)
Acetic anhydride (10.5 ml) was added to a solution of 2β,3α-dihydroxy-5α-cholestane (5) (120 mg, 0.297 mmol) in 5 ml of pyridine, and the mixture was stirred at room temperature for 24 h. The mixture was then poured onto ice and 2N HCl solution (20 ml), and extracted with CH2Cl2 (3×30 ml). The combined organic layer was washed successively with water, saturated NaHCO3 solution, and water, dried over anhydrous MgSO4, and evaporated to dryness. The residue was purified by column chromatography on silica gel using cyclohexane/CH2Cl2 (50:50 v/v) as eluent to give 133.1 mg (92%) of 9 as white solid, m.p. 110–111 °C (acetone). 1H NMR δ (CDCl3): 0.64 (s, 3H, H-18), 0.86 (d, J=6.6 Hz, 6H, H-26, H-27), 0.89 (d, J=6.2 Hz, 3H, H-21), 0.90 (s, 3H, H-19), 2.03 (s, 3H, CH 3CO), 2.06 (s, 3H, CH 3CO), 4.88 (m, 2H, H-2, H-3). EIMS m/z=488 (1%, M+), 426 (4%, M+-AcOH-CH2CO).
2.8. 3α-Acetoxy-2β-hydroxy-5α-cholestane (10) and 2β-acetoxy-3α-hydroxy-5α-cholestane (11)
bis(Tributyltin)oxide (BBTO) (0.27 ml, 0.545 mmol) was added to a solution of 2β,3α-diacetoxy-5α-cholestane (9) (133 mg, 0.273 mmol) in toluene (2.7 ml), and the mixture was refluxed for 5 h. The reaction mixture was cooled to room temperature and purified by vacuum-dry column chromatography on silica gel using CH2Cl2/methanol (80:20 v/v) as eluent to give 108.3 mg (89%) of a mixture of monoacetylated compounds 10 and 11. Further purification of this mixture by column chromatography on silica gel using cyclohexane/acetone (90:10 v/v) as eluent afforded pure 10 (29.2 mg, 27%) and 11 (79.1 mg, 73%). Compound 10: m.p. 106–107 °C (acetone). 1H NMR δ (CDCl3): 0.65 (s, 3H, H-18), 0.86 (d, J=6.6 Hz, 6H, H-26, H-27), 0.90 (d, J=7 Hz, 3H, H-21), 0.99 (s, 3H, H-19), 2.06 (s, 3H, CH 3CO), 3.91 (s, 1H, H-2), 4.84 (d, J=2.2 Hz, 1H, H-3). EIMS m/z=446 (1%, M+), 386 (31%, M+-AcOH), 368 (3%, M+-AcOH-H2O). Compound 11: m.p. 85–86 °C (acetone). 1H NMR δ (CDCl3): 0.64 (s, 3H, H-18), 0.86 (d, J=6.8 Hz, 6H, H-26, H-27), 0.89 (d, J=7 Hz, 3H, H-21), 0.90 (s, 3H, H-19), 2.04 (s, 3H, CH 3CO), 3.84 (s, 1H, H-3), 4.87 (d, 1H, J=2.6 Hz, H-2). EIMS m/z=446 (1%, M+), 386 (31%, M+-AcOH), 368 (3%, M+-AcOH-H2O).
2.9. Sodium 3α-acetoxy-2β-hydroxy-5α-cholestane 2-sulfate (12)
Triethylamine–sulfur trioxide complex (8.0 mg, 0.044 mmol) was added to a solution of 3α-acetoxy-2β-hydroxy-5α-cholestane (10) (9.9 mg, 0.022 mmol) in DMF (0.4 ml). The reaction mixture was stirred at room temperature for 13 h, and then quenched with water (0.5 ml). After evaporation to dryness the residue was eluted through Amberlite CG-120 (sodium form) with methanol. Further purification by preparative HPLC (methanol/water (85:15 v/v)) afforded 4.6 mg (38%) of pure 12, m.p. 120–121 °C together with 4 mg of compound 7 (36%). For 1H and 13C NMR see Table 1, Table 2. FAB− m/z=525 [MNa]−. HRFABMS (negative ion mode) m z=525.3253 [MNa]− (C29H49O6S, Δ=0.3 mmu).
2.10. Sodium 2β-acetoxy-3α-hydroxy-5α-cholestane 3-sulfate (13)
By the same procedure used for the preparation of 12, compound 11 was converted into 2β-acetoxy-3α-hydroxy-5α-cholestane 3-sulfate (13) in 75% yield, m.p. 146–148 °C. For 1H and 13C NMR see Table 1, Table 2. FAB− m/z=525 [MNa]−. HRFABMS (negative ion mode) m/z=525.3253 [MNa]− (C29H49O6S, Δ=0.3 mmu).
2.11. Acetylation of compound 7
Acetic anhydride (1 ml) was added to a solution of sodium 2β,3α-dihydroxy-5α-cholestane 2-sulfate (7) (4 mg, 0.0079 mmol) in 1 ml of pyridine, and the mixture was stirred at room temperature for 24 h. The mixture was then poured onto ice and 2N HCl solution (2 ml), and extracted with CH2Cl2 (3×2 ml). The combined organic layer was washed successively with water, saturated NaHCO3 solution, and water, dried over anhydrous MgSO4, and evaporated to dryness. The residue was purified by preparative HPLC to afford 3.85 mg (89%) of pure 12.
2.12. Cells and viruses
Vero (African green monkey kidney) cells were grown in 24-cell culture plates with Eagle’s Minimum Essential Medium (MEM) containing 5% of bovine serum. Maintenance medium (MM), consisted of MEM with 1.5% of bovine serum. HSV-2 strain G was obtained from the American Type Culture Collection (Rockville, USA). Dengue virus type 2 (DEN-2) strain NG was obtained from the Instituto Nacional de Enfermedades Virales Humanas (INEVH), Dr. J. Maiztegui, Pergamino, Argentina. Junin virus (JV) strain IV 4454 was provided by Dr. Marta S. Contigiani from the Instituto de Virología, Córdoba University, Argentina.
2.13. Compounds solutions
Stock solutions of the compounds at a concentration of 10 mg/ml were prepared in DMSO and sterilized by filtration. The solutions were kept at 4 °C until use.
2.14. Cytotoxicity test
Cellular viability was measured with 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT; Sigma-Aldrich) method in Vero cells. Confluent cultures in 96-well plates were exposed to different concentrations of the steroids, with three wells for each concentration, and incubated for 48 h at 37 °C. Then 10 μl of MM containing MTT (5 mg/ml) was added to each well. After 2 h of incubation at 37 °C, the supernatant was removed and 200 μl of ethanol was added to each well to solubilize the formazan crystals. After vigorous shaking, absorbance was measured in a microplate reader at 595 nm. The cytotoxic concentration 50% (CC50) was calculated as the compound concentration required to reduce cell viability by 50%.
2.15. Antiviral assays
Antiviral activity was evaluated by reduction of virus plaque formation. Vero cell monolayers grown in 24-well plates were infected with about 50 plaque forming units (PFU) of virus per well in the absence or presence of various concentrations of the compounds. After 1 h of adsorption, residual inoculum was replaced by MM containing 0.7% methylcellulose and the corresponding dose of each compound. Plaques were counted after 2 days of incubation at 37 °C for HSV-2 and 7 days post-infection for JV and DEN-2. The 50% inhibitory concentration (IC50) was calculated as the compound concentration required to reduce virus plaque by 50%. All determinations were performed twice and each in duplicate.
2.16. Virucidal assay
A virus suspension of HSV-2 containing 4×105 PFU was incubated with an equal volume of MM with or without various concentrations of the compounds for 1 h at 37 °C. The samples were then diluted in cold MM to determine residual infectivity by plaque formation. The sample dilution effectively reduced the drug concentration to be incubated with the cells at least 100-fold to assess that titer reduction was only due to cell-free virion inactivation. The virucidal concentration 50% (VC50), defined as the concentration required to inactivate virions by 50% was then calculated.
3. Results and discussion
Diol 5 was prepared from commercially available 3β-hydroxy-5α-cholestane (1) by the four step synthesis shown in Scheme 1 [9], [10], [11]. Compound 5 was purified by successive vacuum-dry column chromatography on normal and C18 silica gel and used as starting material for the synthesis of compounds 6–13. Triethylamine–sulfur trioxide complex was selected as the sulfating reagent owing to its facile preparation [8] and the stability of the triethylammonium salts of sulfate esters [12].
Scheme 1.
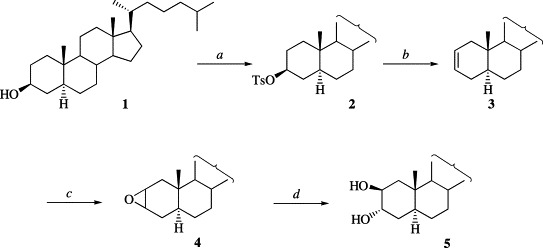
Conditions: (a) p-TsCl, pyridine; (b) LiCO3, LiBr, DMF; (c) m-ClPBA, H2O-CH2Cl2; (d) H2SO4, THF.
Treatment of diol 5 with 6.3 equivalents of triethylamine–sulfur trioxide complex at 95 °C afforded the ammonium sulfate of compound 8, which was transformed via ion exchange into the disodium salt 8 (Scheme 2 ). On the other hand, the reaction of compound 5 with 2 equivalents of the sulfating reagent at room temperature rendered after ion exchange sodium 2β,3α-dihydroxy-5α-cholestane 3-sulfate (6) and sodium 2β,3α-dihydroxy-5α-cholestane 2-sulfate (7) in a 6.5:3.5 ratio, together with minor amounts of the disulfated derivative 8 (Scheme 2). Compounds 6, 7, and 8 were purified by preparative reversed-phase HPLC and characterized by HRFABMS and 1H and 13C NMR spectroscopy (Table 1, Table 2). The assignments of the NMR signals were derived from 1H1H COSY and HETCOR experiments. Disodium 2β,3α-dihydroxy-5α-cholestane disulfate (8) showed two methine signals at δ 4.73 (H-2) and 4.70 (H-3), characteristic of the presence of two sulfate groups at C-2 and C-3 [13]. This is in accordance with the chemical shifts observed for C-2 (76.1) and C-3 (76.1) in the 13C NMR spectrum. The 13C NMR spectra of monosulfated compounds 6 and 7 differed in the chemical shifts of C-1 (δ 41.0 in 6, 38.8 in 7), C-2 (δ 70.0 in 6, 78.8 in 7), C-3 (δ 78.5 in 6, 69.0 in 7), and C-4 (δ 30.3 in 6, 32.5 in 7). The downfield shift of 9.5 ppm for C-3 and the upfield shift of 2.2 ppm for C-4 in compound 6 indicated that the sulfate group was located at C-3. These values confirm the assignment of the position of the sulfate group in ring A for disodium (20R)-2β,3α,21-trihydroxy-5α-cholest-24-ene-3,21-disulfate, isolated previously by us from the antarctic ophiuroid A. agassizii [6]. The presence of the sulfate group at C-2 in 7 was confirmed by the downfield shift of 8.8 ppm for C-2 and the upfield shift of 2.2 ppm for C-1. 1H NMR spectra showed signals at δ 4.08 (H-2) and 4.40 (H-3) for compound 6 and δ 4.42 (H-2) and 4.02 (H-3) for compound 7, confirming the sulfate groups positions in monosulfated derivatives 6 and 7.
Scheme 2.
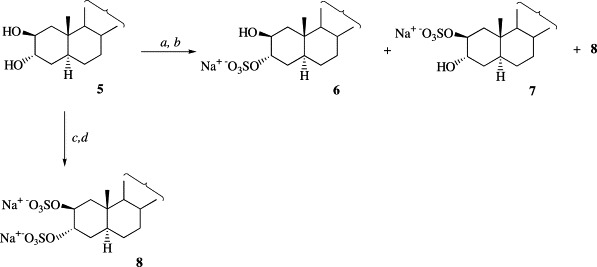
Conditions: (a) 2 equivalents Et3N–SO3, DMF, room temperature; (b) Amberlite CG-120 (MeOH); (c) 6.3 equivalents Et3N–SO3, DMF, 95 °C; (d) Amberlite CG-120 (MeOH).
Looking forward for a major selectivity in the obtention of monosulfated analogs, an alternative synthetic strategy was tried (Scheme 3 ). Diol 5 was treated with acetic anhydride in pyridine rendering the diacetylated compound 9, which was submitted to selective deprotection with BBTO. This reagent has been utilized for the deprotection of a variety of steroid esters in non-hydrolytic conditions, in particular in the selective hydrolysis of the 3β-acetyl group in the presence of the 6α one in 3β,6α-diacetoxy-5α-pregnan-20-one [14]. As shown in Scheme 2, when a solution of diacetate 9 in toluene and BBTO was refluxed for 5 h, a mixture of monoacetylated compounds 10 and 11 was obtained in 89% yield in a 2.7:7.3 ratio. Both monoacetylated derivatives were separated and purified by chromatography and treated separately with triethylamine–sulfur trioxide complex in the same reaction conditions used for the monosulfation of diol 5 to give the sulfated analogs 12 and 13. It is to note that while sulfation of compound 11 rendered only compound 13, compound 10 afforded the expected sulfated analog 12 together with compound 7 derived from the deacetylation of 12. As compounds 7 and 12 were easily separated by HPLC, compound 7 was further transformed into 12 by a simple acetylation reaction.
Scheme 3.
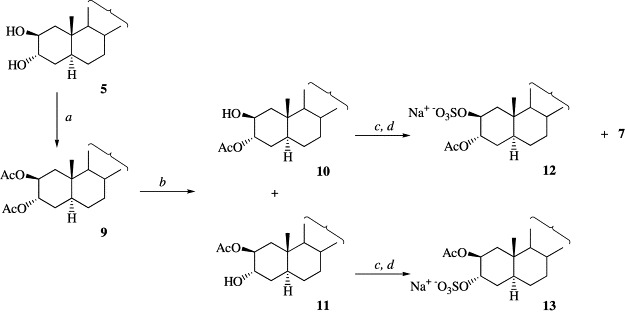
Conditions: (a) Ac2O, pyridine; (b) BBTO, toluene; (c) 2 equivalents Et3N–SO3, DMF, room temperature; (d) Amberlite CG-120 (MeOH).
1H and 13C NMR data of compound 12 (Table 1, Table 2) confirmed that the sulfate group is located at C-2 (δ C 76.0 ppm, δ H 4.44 ppm) and the acetoxy group is attached to C-3 (δ C 72.2 ppm, δ H 5.11 ppm). The spectroscopic data of 13 revealed that the 2β-hydroxy group remained protected (δ C 72.6 ppm, δ H 5.14 ppm) while the sulfation took place at the free 3α-hydroxy group (δ C 75.5 ppm, δ H 4.40 ppm). Again, assignment of proton and carbon resonances was based on the analysis of 1H, 13C, DEPT, 1H1H COSY, and HETCOR NMR experiments.
A great selectivity between positions 2 and 3 was observed for these reactions. Thus, in the monosulfation of diol 5, the analog sulfated at C-3 (6) was obtained as the major product and in the deprotection reaction of the diacetylated compound (9) with BBTO the 2β-acetoxy-3α-hydroxy derivative 13 was the predominant product. These results indicated that, as it was expected, groups attached to C-3 with α configuration were more reactive than those attached to C-2 (β) because of the steric hindrance due to the axial methyl group (C-19) at C-10 (Fig. 2 ).
Fig. 2.
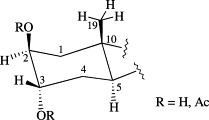
Spatial distribution of A-ring substituents in the synthetic intermediates 5, 9, 10, and 11.
The antiviral activity of the five compounds was tested against HSV-2, the agent responsible for primary and recurrent infections of mucous membranes, genital lesions, neonatal infections, and meningitis [15]. From the data shown in Table 3 , compounds 7 and 8 were the most active against HSV-2 with IC50 values of 19.3 and 23.9 μg/ml, respectively. Monosulfated steroid 6, differing from 7 in the position of the sulfate group as well as acetylated and sulfated compound 13 were not active against HSV-2 at a concentration up to 25 μg/ml. The four steroids also showed virucidal activity against HSV-2 with values of VC50 ranging from 19.4 to 76.0 μg/ml. Due to its low cytotoxic effect on Vero cells, the monosulfated steroid 12 containing an acetyl group at C-2 was assayed for antiviral activity at a higher concentration. Nevertheless, only a weak inhibitory activity against HSV-2 was observed (IC50=90.5 μg/ml). No virucidal effect of this compound was observed up to 100 μg/ml. Of all the synthetic compounds tested, disulfated steroid 8 showed the best selectivity index (SI=2.8). This result enabled us to test the antiviral activity of 8 against two pathogenic viruses in humans: JV and DEN-2. JV causes a severe disease in humans known as Argentine hemorrhagic fever [16] and DEN infection results in acute febrile illness or a more severe manifestation known as DEN hemorrhagic fever-shock syndrome [17]. Derivative 8 was not active against JV at a concentration up to 25 μg/ml, while it elicited a marked DEN-2 inhibitory activity (IC50=16.9±0.2 μg/ml).
Table 3.
Antiviral and virucidal activities of sulfated steroids 6–8 and 12 and 13
| Compound | CC50 (μg/ml)a | IC50 (μg/ml)b | SI (CC50/IC50)c | VC50 (μg/ml)d |
| 6 | 38 | >25 | <1.5 | 29.0 ± 1.9 |
| 7 | 30 | 19.3 ± 2.9 | 1.6 | 20.4 ± 0.1 |
| 8 | 66 | 23.9 ± 1.5 | 2.8 | 19.4 ± 0.2 |
| 12 | >100 | 90.5 ± 13.0 | >1.1 | >100 |
| 13 | 39 | >25 | <1.6 | 76.0 ± 0.9 |
Concentration required to reduce cell viability by 50%.
Concentration that reduced the number of virus plaque by 50%±S.D.
Selectivity index.
Concentration required to inactivate virus by 50%±S.D.
Several sulfated polyhydroxysterols have been recently isolated from sponges [18]. Most of these natural products are characterized by the 2β,3α,6α-tri-O-sulfate functions but several bioactive sterols sulfated at C-2 (β) and C-3 (α) containing additional alkylation in the side chain have been isolated from the sponges Petrosia weinbergi [13], [19] (14–18) and Pachastrella sp. [20] (19) (Fig. 3 ). Weinbesterols A (14) and B (15) and orthoesterol disulfates A (16), B (17), and C (18) exhibited in vitro activity against the feline leukemia virus (FeLV), mouse influenza virus (PR8), and mouse corona virus. Compound 14 was also active in vitro against HIV. Halistanol disulfate B (19) was active in the endothelin-converting enzyme assay while the corresponding diol was totally inactive.
Fig. 3.
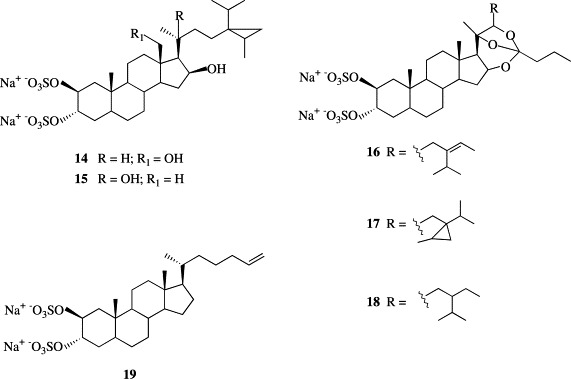
Disulfated steroids isolated from the sponges Petrosia weinbergi and Pachastrella sp.
In order to determine the role of the sulfate groups in the antiviral activity of disulfated steroid 8, we evaluated the in vitro activity of diol 5 against HSV-2. Although compound 5 showed no cytotoxic effect on Vero cells (CC50>200 μg/ml) it was not active against HSV-2 at a concentration up to 50 μg/ml and showed no virucidal effect (VC50>100 μg/ml). These results are indicative of the role of the sulfate groups in the antiviral activity of 8. Due to its antiviral activity, disulfated steroid 8 is a good model compound for further introducing additional functional groups at the steroidal skeleton or its side chain in order to evaluate structure–activity correlations in the inhibitory activity against pathogenic viruses in humans.
Acknowledgements
We thank the International Foundation for Science, ANPCyT and the Universidad de Buenos Aires for financial support of this work. We also wish to thank UMYMFOR (CONICET-FCEN) for spectroscopic analysis. Mass spectrometry was provided by the Washington University Mass Spectrometry Resource, an NIH Research Resource (Grant No. PA1RR00954). M.S.M. and E.B.D. are research members of the National Research Council of Argentina. C.A.P. is a technical member of CONICET.
References
- 1.Kerr R.G., Baker B.J. Marine sterols. Nat. Prod. Rep. 1991;8:456–497. [Google Scholar]
- 2.D’Auria M.V., Minale L., Riccio R. Polyoxygenated steroids of marine origin. Chem. Rev. 1993;93:1839–1895. [Google Scholar]
- 3.McKee T.C., Cardellina J.H., Riccio R., D’Auria M.V., Iorizzi M., Minale L. HIV-inhibitory natural products. 11. Comparative studies of sulfated sterols from marine invertebrates. J. Med. Chem. 1994;37:793–797. doi: 10.1021/jm00032a012. [DOI] [PubMed] [Google Scholar]
- 4.Fu X., Schmitz F.J., Lee R.H., Papkoff J.S., Slate D.L. Inhibition of protein tyrosine kinase pp60v-src: sterol sulfates from the brittle star Ophiarachna incrassata. J. Nat. Prod. 1994;57:1591–1594. doi: 10.1021/np50113a023. [DOI] [PubMed] [Google Scholar]
- 5.Roccatagliata A.J., Maier M.S., Seldes A.M., Pujol C.A., Damonte E.B. Antiviral sulfated steroids from the ophiuroid Ophioplocus januarii. J. Nat. Prod. 1996;59:887–889. doi: 10.1021/np960171a. [DOI] [PubMed] [Google Scholar]
- 6.Roccatagliata A.J., Maier M.S., Seldes A.M. New sulfated polyhydroxysteroids from the antarctic ophiuroid Astrotoma agassizii. J. Nat. Prod. 1998;61:370–374. doi: 10.1021/np970429c. [DOI] [PubMed] [Google Scholar]
- 7.Comin M.J., Maier M.S., Roccatagliata A.J., Pujol C.A., Damonte E.B. Evaluation of the antiviral activity of natural sulfated polyhydroxysteroids and their synthetic derivatives and analogs. Steroids. 1999;64:335–340. doi: 10.1016/s0039-128x(99)00016-1. [DOI] [PubMed] [Google Scholar]
- 8.Nair V., Bernstein S. A convenient procedure for the preparation of triethylamine–sulfur trioxide. Oppi Briefs. 1987;19:466–467. [Google Scholar]
- 9.Robaina Rodríguez C., Coll Manchado F., Pérez Martínez C., Jomarrón Rodiles I. Reacción de Prêvost-Woodward con el (25R)-5α-2-Epirosten-12-ona. Química & Industria. 1995;2:19–22. [Google Scholar]
- 10.Takatsuto S., Ikekawa N. Synthesis of homodolichosterone and related 2-deoxysteroids. Chem. Pharm. Bull. 1987;35:829–832. [Google Scholar]
- 11.Levinson EE, Kuznetsova NA, Podkhalyuzina NY, Traven VF. Synthesis of 2,24-diepicastasterone and its 22S,23S-isomer: novel brassinosteroids with a trans-2,3-diol function. Mendeleev Commun 1994;96–9.
- 12.Dusza J.P., Joseph J.P., Bernstein S. Steroid conjugates. IV. The preparation of steroid sulfate with triethylamine-sulfur trioxide. Steroids. 1968;9:49–61. doi: 10.1016/s0039-128x(68)80079-0. [DOI] [PubMed] [Google Scholar]
- 13.Koehn F.E., Gunasekera M., Cross S.S. New antiviral sterol disulfate ortho esters from the marine sponge Petrosia weinbergi. J. Org. Chem. 1991;56:1322–1325. [Google Scholar]
- 14.Pérez M.G., Maier M.S. Mild deprotection of steroid esters by bis(tributyltin)oxide. Tetrahedron Lett. 1995;36:3311–3314. [Google Scholar]
- 15.Whitley RJ, Gnann JW. The epidemiology and clinical manifestations of herpes simplex virus infections. In: Roizman B, Whitley RJ, López C, editors. The human herpes virus. New York: Raven Press; 1993. p. 69–105 [chapter 3].
- 16.White DO, Fenner FJ. Medical virology. New York: Academic Press; 1994. p. 349, 469.
- 17.Lourdes Muñoz M., Cisneros A., Cruz J., Das P., Tovar R., Ortega A. Putative dengue virus receptors from mosquito cells. FEMS Microbiol. Lett. 1998;168:251–258. doi: 10.1111/j.1574-6968.1998.tb13281.x. [DOI] [PubMed] [Google Scholar]
- 18.Aiello A., Fattorusso E, Menna M., Fattorusso E. Steroids from sponges: recent reports. Steroids. 1999;64:687–714. doi: 10.1016/S0039-128X(99)00032-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sun H.H., Cross S.S., Gunasekera M., Koehn F.E. Weinbersterol disulfates A and B, antiviral steroid sulfates from the sponge Petrosia weinbergi. Tetrahedron. 1991;47:1185–1190. [Google Scholar]
- 20.Patil A.D., Freyer A.J., Breen A., Carte B., Johnson R.K. Halistanol disulfate B, a novel sulfated sterol from the sponge Pachastrella sp.: inhibitor of endothelin converting enzyme. J. Nat. Prod. 1996;59:606–608. doi: 10.1021/np9601770. [DOI] [PubMed] [Google Scholar]