

Physical Examination
Examination of patients with disease of the gastrointestinal tract must include evaluation of the metabolic and cardiovascular status of the patient because acute conditions of the proximal or distal intestinal tract can lead to endotoxemia and sepsis. Examination of the cardiovascular system (heart, peripheral pulse, and mucous membranes), lungs, and abdomen is essential to detect clinical signs of systemic inflammation from endotoxemia, coagulation disorders, dehydration, ileus, shock, and other abnormalities resulting from injury to the small or large intestine. Clinical signs of systemic inflammation from endotoxemia and sepsis are described later in this chapter.
The physical examination of the abdomen should include auscultation, transabdominal ballottement, and transrectal palpation. Abdominal distention often indicates distention of the large intestine; small intestinal distention also can cause visible abdominal distention if a large proportion of the small intestine is involved. Abdominal palpation can be performed in neonatal foals. After several weeks of age the abdominal wall is too rigid to allow effective palpation of intraabdominal structures.
Abdominal auscultation is particularly useful for assessing the motility of the large intestine. Progressive motility of the small intestine, conversely, is difficult to distinguish by auscultation from nonprogressive motility. The distinct character of the borborygmi produced during propulsive contractions of the cecum and ascending colon allows evaluation of the frequency and strength of retropulsion and propulsion. Propulsive contractions of the cecum and ventral colon occur every 3 to 4 minutes and give rise to prolonged rushing sounds heard over long segments of intestine. Retropulsive sounds presumably are similar to propulsive sounds, but they occur less frequently. Distinguishing between propulsion and retropulsion is not important clinically because both types of contractions signify normal motility. Interhaustral and intrahaustral mixing contractions produce nonspecific sounds of fluid and ingesta movement that are difficult to distinguish from other borborygmi, such as small intestinal contractions or spasmodic contractions.1
Auscultation over the right flank and proceeding along the caudal edge of the costal margin toward the xiphoid allows evaluation of the cecal borborygmi. Auscultation over a similar area on the left side allows evaluation of the pelvic flexure and ascending colon. Typical progressive borborygmi heard every 3 to 4 minutes on both sides of the abdomen indicate normal motility of the cecum and ascending colon. Less frequent progressive sounds may indicate a pathologic condition of the large intestine or may result from anorexia, nervousness (sympathetic tone), or pharmacologic inhibition of motility (i.e., α2-adrenergic agonists such as xylazine).2, 3, 4, 5 Absolute absence of any auscultable borborygmi suggests abnormal motility and indicates ileus resulting from a serious pathologic condition but is not specific to any segment of the intestine.3, 6 If borborygmi are audible but progressive sounds are not detectable, determining whether a significant abnormality exists is difficult, and such findings should not be overinterpreted.6 Borborygmi heard more frequently than normal may result from increased motility following feeding; from excessive stimulation from irritation, distention, or inflammation; or after administration of parasympathomimetic drugs such as neostigmine. Large intestinal motility increases in the early stages of intestinal distention regardless of the site.7 Mild inflammation or irritation of the large intestinal mucosa also can stimulate motility.3 Parasympathomimetic drugs stimulate contractions and auscultable borborygmi in the large intestine; an increase in parasympathetic tone may result in segmental contractions, which actually inhibit progressive motility.2
Percussion of the abdomen during auscultation can reveal gas in the large intestine. The characteristic ping produced by simultaneous digital percussion and auscultation over a gas-filled viscus often is associated with abnormal accumulation of gas under pressure. This technique is particularly useful in foals, ponies, and Miniature Horses because of the limitations of rectal palpation.
Transabdominal ballottement can be used to detect large, firm masses or an abnormal volume of peritoneal fluid (PF). The usefulness of this technique is usually limited to animals too small to palpate rectally. Soft tissue masses or fetuses can be detected by bumping the structures with a hand or fist. If excessive PF is present, a fluid wave can be generated by ballottement; however, this technique is not as useful in horses older than 4 weeks because the abdominal wall is rigid.
Transrectal palpation is the most specific physical examination technique for investigation of intestinal disease and is particularly valuable when evaluating obstructive diseases.8 The primary objectives of transrectal palpation are to assess the size, consistency, and position of the segments of the large intestine; to determine the presence of any distention of the small intestine; and to detect intraabdominal masses. Evaluation of the wall thickness and texture and the mesenteric structures (blood and lymphatic vessels and lymph nodes) also may aid in diagnosis of large intestinal disease. The interpretation of transrectal palpation findings in light of clinical signs and laboratory results is an important diagnostic aid for developing appropriate treatment strategies for intestinal diseases manifested by abdominal pain. Enlargement of one or more segments of large intestine detected by transrectal palpation provides evidence of obstruction at or distal to the enlarged segment. By systematically evaluating each segment, the site of obstruction may be determined. Obstruction of the pelvic flexure, for instance, results in enlargement of the pelvic flexure and ventral colon, but the dorsal and descending colons are of normal size. Enlargement of a segment of the large intestine usually is accompanied by abnormal consistency of the contents. It is possible to distinguish among gas, fluid, and ingesta and to detect foreign bodies in palpable segments. Accumulation of gas and fluid suggests complete and acute obstruction, whereas accumulation of ingesta suggests chronic and incomplete obstruction. Accumulation of fluid usually indicates ileus. The practitioner must evaluate the consistency of the contents in light of the size of the segment; ingesta in the ventral colon of a dehydrated patient may be firm, but the size of the ventral colon will be normal. Conversely, if the ingesta is firm because of a distal obstruction, the ventral colon will be enlarged.
Displacement of a segment of the large intestine may create an obstruction detectable by enlargement of the segment and accumulation of gas and fluid, even if the site of obstruction is not palpable. Torsion of the ascending colon at the sternal and diaphragmatic flexures results in acute accumulation of gas and fluid proximal to the torsion, causing distention of the left dorsal and ventral colons. Depending on the degree of torsion, the position of the ventral and dorsal colons may not be significantly abnormal. Displacement of a segment of large intestine often results in incomplete obstruction, and the diagnosis is either confirmed on detection of the displaced segment in an abnormal position or suspected when the segment is not palpable in a normal position. A determination should be made as to whether the segment that appears to be displaced is in a normal position but of smaller than normal size because of a decreased volume of ingesta. The cecum, right dorsal and ventral colons, pelvic flexure, and descending colon are palpable in most horses. The nephrosplenic space should be palpated to detect the presence of intestine, usually pelvic flexure, entrapped within the ligament.
Small intestine is not normally palpable in the horse. Distention indicates ileus with gas or fluid retention, usually following a strangulating or nonstrangulating obstruction. Strangulating obstructions often are accompanied by severe pain, dehydration, PF changes, and a varying degree of gastric fluid accumulation. The small intestine in these cases is turgid and firm on palpation. The mesentery and wall thickness should be assessed in the same manner as for large intestinal disorders. Careful palpation of the inguinal rings in stallions with small intestinal distention is crucial for determining inguinal herniation.
Evaluation of the wall thickness and mesenteric vessels can reveal venous congestion (mural edema and enlarged blood and lymphatic vessels) or inflammation (mural edema with normal vessels). Disruption of arterial blood flow does not cause venous congestion, but the arterial pulse is not detectable. Mesenteric tears may not be palpable, but the entrapped ischemic intestinal segment may be thickened. Enlargement of mesenteric lymph nodes also may be noted. Abnormalities in the wall or vessels should be interpreted in light of the size, consistency, and position of the segment of intestine and the clinical signs. Several conditions involving small intestinal strangulating lesions do not necessarily cause abnormal rectal examination findings until the disease has been present for an extended time. These conditions include diaphragmatic hernias and epiploic foramen entrapments (EFEs). PF analysis can be normal in these cases as well because the fluid is trapped in the thorax or cranial abdomen.
Nonstrangulating causes of small intestinal distention can be divided further into intraluminal and extraluminal obstructions. Ileal impactions are the most common cause of intraluminal obstruction, and on rare occasions the impaction can be palpated in the upper right quadrant, near the ileocecal opening. Intraluminal masses caused by lymphoma, eosinophilic enteritis, foreign bodies, or ascarid impactions often lead to small intestinal distention and are usually indistinguishable from one another on the basis of palpation alone. Small intestine in these cases can be moderately to severely distended, depending on the degree of obstruction. Extraluminal obstructions include abdominal masses and large colon displacement. The rest of the abdomen should be carefully palpated to help rule out these causes. Some cases of small intestinal distention result from physiologic ileus rather than mechanical obstruction. The bowel is usually mildly to moderately distended and almost always is accompanied by significant gastric fluid.
The small colon is easily distinguishable by the presence of normal fecal balls and an antimesenteric band. In horses with impaction of the small colon, a long, hard, tubelike structure is present in the caudal abdomen, and the band is palpable along the length. Fluid stool is often present in the rectum in these horses, as is tenesmus, and the rectal mucosa is often edematous and occasionally roughened. Rectal tears can be detected and evaluated with careful rectal palpation. Also detectable are mural masses in palpable segments of intestine or mesentery; if a mass causes obstruction, then it is possible to detect the result of the obstruction in proximal segments of intestine even if the mass is unreachable. Palpation of the mesenteric vessels may reveal thickening and thrombosis, which can lead to ischemia or infarction.
Visual inspection of the mucosa of the rectum and descending colon can be performed with the aid of a speculum or flexible endoscope. A flexible endoscope is also useful for evaluation of rectal tears or perforations, mural masses, strictures, or mucosal inflammation and obtaining biopsy specimens of the mucosa or masses. The obvious limitations are the amount of fecal material, which can interfere with the examination, and the distance of the lesion of interest from the anus. These techniques offer little advantage over palpation in many cases, unless the patient is too small to palpate.
Examination of the oral cavity in horses with dysphagia or weight loss is an important extension of the physical examination. The horse should be adequately sedated and a full-mouth speculum used to allow palpation and visual examination of all parts of the oral cavity and detection of abnormal dentition, foreign bodies, fractures, abscesses, or mucosal ulceration.
The presence of fluid accumulation in the stomach indicates a functional or mechanical obstruction of gastric outflow. Fluid accumulation in the stomach is assessed by siphoning of the gastric contents with a nasogastric tube and examining the fluid for amount, color, and odor. Normal fluid is green and may contain foamy saliva. The net volume obtained by gastric lavage is usually less than 4 L. Large volumes (≥8–10 L) of foul-smelling fluid may indicate proximal enteritis. Horses with strangulating obstructions or luminal obstructions often accumulate moderate amounts of gastric fluid, but the amount is generally less than in horses with proximal enteritis or postoperative ileus (POI). Distinction between these conditions should not be made based on the volume and character of gastric fluid alone. Hemorrhage in the gastric fluid usually indicates devitalized small intestine, stomach wall, or severe gastric ulceration. Endoscopy or contrast radiography may aid in the diagnosis of gastric outflow obstruction.
Diagnostic Evaluation
Clinical Pathology
Hematologic alterations associated with diseases of the gastrointestinal tract are often nonspecific, reflecting systemic response to inflammation, endotoxemia, or sepsis. Neutrophilic leukocytosis and normochromic, normocytic anemia with or without hyperfibrinogenemia commonly are associated with chronic inflammatory conditions of the intestine. Anemia from chronic blood loss occurs infrequently in adult horses because of the large iron stores and high concentrations of iron in their diet; anemia usually follows chronic inflammation, as do alterations in the leukon and plasma fibrinogen concentrations. Plasma protein concentrations vary depending on gastrointestinal losses of albumin and globulin and elevation of globulin concentration from antigenic stimulation. Protein-losing enteropathy may manifest as a hypoalbuminemia or panhypoproteinemia.
Significant alterations of the hemogram do not accompany acute disease of the intestine unless severe dehydration, endotoxemia, or systemic inflammatory response syndrome (SIRS) is present. During the early stages of SIRS, elevations in circulating concentrations of inflammatory mediators, epinephrine, and cortisol produce characteristic changes in the hemogram. Leukopenia, with neutropenia and a left shift, toxic changes in the neutrophil cytoplasm, and lymphopenia commonly occur early in the disease, but neutrophilic leukocytosis is more common during the later stages of SIRS. Hemoconcentration and hyperfibrinogenemia are also common. Thrombocytopenia and other coagulopathies are also features of SIRS.
Electrolyte imbalances and increased blood lactate are common biochemical abnormalities in horses with acute gastrointestinal disease. Decreased serum calcium concentrations are common and nonspecific.9 Mucosal inflammation can disrupt electrolyte fluxes; diarrhea or gastric reflux greatly exacerbates sodium, potassium, calcium, magnesium, and bicarbonate loss. Large colon ischemia causes increased lactate and potassium concentrations and metabolic acidosis in the colonic vasculature and inflammation in the colonic and systemic vasculature.10 Reduced perfusion of peripheral tissues from hypotensive shock and intestinal ischemia can cause increased blood lactate; intestinal obstruction during ischemia may also result in absorption of lactate from the lumen. Increased blood lactate can result from a variety of causes, including hypovolemia, and blood lactate alone should not be used for diagnostic or prognostic purposes in horses with colic.11 Increases in blood lactate over time are greater in nonsurvivors, relative to survivors, in adult equine emergencies (many of which had gastrointestinal disease).12 Portable lactate analyzers have demonstrated variable intraanalyzer reliability in equine blood so that caution should be exercised in interpretation and comparison of results reported from various searches.13, 14 Metabolic acidosis may accompany lactic acidemia, but an inconsistent association exists between the two, especially when mixed acid-base imbalances are present.15, 16 Increases in hepatic enzymes, specifically γ-glutamyl transferase (GGT), may occur with large colon displacements, duodenal strictures, or proximal enteritis. Increased GTT is more suggestive of right, rather than left, dorsal displacement.17
Relative polycythemia from hemoconcentration or splenic contraction and changes in red blood cell deformability from hypoxia or hypocalcemia may increase blood viscosity. Blood viscosity increases in patients with acute obstructive disease. Hyperviscosity reduces perfusion of capillary beds, exacerbating ischemia and tissue hypoxia.18
Peritoneal Fluid
Abdominocentesis and analysis of PF are performed on many patients with gastrointestinal disease and are especially helpful in differentiating strangulating from nonstrangulating disorders of the small intestine. Important quantifications include white and red blood cell counts and protein, lactate, and glucose concentrations. Cytologic evaluation can reveal cellular abnormalities, especially in horses with intestinal neoplasia. Results of PF analysis may help establish a specific diagnosis and, more important, may reflect inflammatory, vascular, or ischemic injury to the intestine, requiring surgical intervention.
Alteration of PF reflects a sequence of events during acute intestinal vascular injury. The PF protein concentration increases first, followed by increases in red blood cell count and fibrinogen concentration. A transudative process resulting from vascular congestion and increased endothelial permeability allows small macromolecules (albumin) to escape into the PF, followed by larger macromolecules (globulin and fibrinogen), and finally diapedesis of cells (red then white blood cells). Severe ischemic intestinal inflammation or visceral peritonitis result in an exudative process, with large quantities of protein and white blood cells (WBCs), primarily neutrophils, to escape into the PF.19, 20 Eventually, bacteria begin to translocate across the intestinal wall and appear in the PF as the mucosal barrier breaks down. If perforation occurs, bacteria and particles of ingesta appear in the PF, and the neutrophils become degenerate (i.e., pyknotic), with karyorrhexis, karyolysis, and smudge cells.
Increased PF protein concentration is an indicator of early inflammation, whereas increased red blood cell counts in the presence of normal WBC counts suggest vascular damage without significant tissue ischemia.20 Of note, the anticoagulant potassium ethylenediamine tetraacetic acid, but not lithium heparin, can cause an increase in total protein as measured by a refractometer, relative to the value obtained from the same sample without anticoagulant.21 The gross color of the PF can be helpful in detecting injury and necrosis of the intestine. A serosanguineous appearance indicates vascular injury, whereas orange or brown-red indicates necrosis with the release of pigments such as hemosiderin.
Tissue hypoxia and ischemia cause rapid increases in PF lactate dehydrogenase, creatine kinase, and alkaline phosphatase (AP) activity and lactate concentration.22, 23 Phosphate concentration increases when cellular disruption occurs.24 PF enzyme activities, phosphate, and lactate concentration increase faster and higher than serum activities.16, 22, 23, 24 PF pH and glucose concentration tend to decrease during intestinal ischemia but not as dramatically as in septic peritonitis.25 Lactate concentrations in PF are commonly evaluated and are better predictors of strangulating small intestinal obstruction than blood lactate,22 although increases in both lactate (the term by which l-lactate is commonly referred) and d-lactate are likely more accurate for predicting strangulating lesions as opposed to ruling them out.26 Serial sampling of blood and PF lactate may be useful in cases in which clinical and diagnostic findings at initial presentation were not conclusive for either strangulating or nonstrangulating lesions and/or a horse’s clinical condition deteriorates.27
Cytologic examination of PF may reflect chronic inflammatory intestinal conditions or neoplastic diseases.28 Although culturing PF is recommended to distinguish bacterial infections from noninfectious inflammation unless bacteria are visible on cytologic examination, culturing PF is often unrewarding. Decreases in PF glucose concentrations (<30 mg/dL) and pH (<7.3) are early indicators of septic peritonitis. Glucose concentration and pH in PF should approximately equal blood values in normal horses.
Practically, the gross appearance and the total solids of PF in conjunction with a comparison of PF to serum lactate are most useful when distinguishing between strangulating and nonstrangulating small intestinal disorders in a horse presenting for acute colic. Potential risks associated with performing an abdominocentesis should be considered, and this procedure is performed only if results are likely to alter the treatment plan. For example, if other examination findings indicate that an exploratory laparotomy is clearly indicated or not in the case of a horse with acute colic, abdominocentesis is likely not indicated.
Fecal Examination
Gross examination of the feces can provide information about digestion and transit time in the large intestine. Large fiber particles in the feces represent poor mastication or poor digestion in the large intestine. Small, mucus-covered, hard fecal balls indicate prolonged transit through the descending colon, whereas increased fluidity implies decreased transit time. Feces containing sand or gravel are not necessarily abnormal. However, a significant amount of sand implies that large quantities are present in the colon. Alternatively, absence of sand in feces does not confirm an absence of sand in the colon. Frank blood indicates substantial bleeding into the distal colon (right dorsal colon, small colon, or both) resulting from mucosal damage.
Laboratory analysis of the feces can be performed for horses with diarrhea. Fecal cytologic examination and tests for occult blood detect mucosal inflammation, erosion, or ulceration. Increased fecal leukocyte counts have been documented in horses with diarrhea and salmonellosis, but specificity of this test is low.29
Fecal occult blood tests detect blood in the feces, presumably from erosion or ulceration of the mucosa, but do not distinguish the source of the blood. Large volumes of blood (1–2 L) given by nasogastric tube were required to produce a positive test for occult blood in the feces, but the amount of blood originating from the large intestine required to produce a positive test is unknown. Despite initial reports to the contrary,30 there does not appear to be any correlation between endoscopic evidence of glandular or nonglandular gastric ulceration and detection of fecal albumin or hemoglobin.31
Bacteriologic examination of the fecal flora has been used in horses with diarrhea. Quantitation of clostridial species may be beneficial in diagnosing clostridial infection of the large intestine, although tests to detect clostridial toxins in intestinal contents or feces are important for determining whether clostridia cultured from the feces are causing disease. The most common bacterial pathogens isolated from the feces of horses are Salmonella spp. and Clostridium spp. The number of Salmonella organisms isolated from the feces of horses with clinical salmonellosis is usually higher than from horses with asymptomatic infections. However, the volume of feces in many cases of acute diarrhea is high, and the concentration of Salmonella organisms may be lower than would be expected, accounting for many false-negative fecal cultures. The sensitivity of fecal cultures for detecting Salmonella infection may be as low as 20%. Culture of five consecutive daily fecal samples is recommended to increase the sensitivity of the test. Real-time polymerase chain reaction (PCR) assays used on fecal samples can detect DNA from Salmonella spp. and have performed well in recent validation studies; accuracy of point-of-care tests remains elusive.32, 33, 34, 35 Currently, PCR assays can be used for detection of viral (rotavirus and coronavirus), bacterial (Cryptosporidium spp., Salmonella spp., Neorickettsia risticii, Lawsonia intracellularis), or bacterial toxin (Clostridium difficile toxins A and B, Clostridium perfringens enterotoxin A) DNA in feces or blood (N. risticii).
Qualitative fecal examination can detect nematode and cestode ova, protozoan oocysts, parasitic larvae, and protozoan trophozoites. A direct smear of fecal material can rapidly detect parasite larvae and trophozoites and motility of ciliates and parasite larvae. Fecal flotation with zinc sulfate or sucrose solutions is often used to concentrate less dense ova and oocysts. Zinc sulfate produces less distortion of trophozoites and larvae than sucrose solutions. Fecal sedimentation is particularly appropriate for ciliates, Giardia organisms, and trichomonads. Quantitative techniques such as the Cornell–McMaster method allow estimation of the number of eggs per gram of feces and are most appropriate in monitoring parasite control programs.
Radiography
Survey radiography of the normal esophagus is usually unrewarding. It is possible to detect foreign bodies or soft tissue masses and, in cases of esophageal rupture, free air and ingesta in the tissues surrounding the esophagus and pneumomediastinum. Thoracic radiographs may be necessary to detect megaesophagus or cranial mediastinal masses causing extraluminal obstruction. Barium swallows or double-contrast esophagrams may be used after resolution of an esophageal obstruction to determine whether a stricture, diverticulum, or other underlying disorder is present, although endoscopy can provide similar information. Barium sulfate is the usual contrast medium and can be administered orally by way of a dose syringe or nasogastric tube (50–100 mL of a 40% barium sulfate suspension or barium paste). Oral administration is preferred for evaluation of swallowing and lesions in the proximal esophagus. Administration of contrast using a nasogastric tube (preferably cuffed) allows for delivery of larger volumes of barium (up to 500 mL) but should be performed without sedation if possible. Administration of contrast material can be followed with air insufflation to create a double-contrast effect. If rupture of the esophagus is suspected or if the contrast material is likely to be aspirated, barium should be avoided, and iodinated organic compounds in an aqueous solution should be used as contrast material to decrease the potential for adverse effects. When interpreting esophageal radiographs, the veterinarian should take particular care if the horse is sedated. Acepromazine or detomidine administration causes esophageal dilation in normal horses, especially after passage of a nasogastric tube.36
Radiography of the adult equine abdomen is an effective technique in detecting radiodense material in the large intestine, such as enteroliths, sand, and metallic objects.37, 38, 39, 40 One survey demonstrated that radiography has 76.9% sensitivity and 94.4% specificity for diagnosing enterolithiasis.40 Recently, an objective scoring system demonstrated greater efficacy and less interobserver variability than a subjective assessment of radiographic sand accumulation in horses with or without a clinical diagnosis of sand colic.38 The large size and density of the adult abdomen preclude evaluation of soft tissue structures because the detail and contrast of the radiographs are usually poor, and ultrasonography is a much more useful imaging modality in the equine abdomen.
Administration of contrast (barium sulfate 30% at 5 mL/kg) through a nasogastric tube or retrograde (20 mL/kg) through a 24-Fr Foley catheter inserted into the rectum may be helpful for the diagnosis of gastric outflow obstruction or disorders of the rectum, small colon, or transverse colon, respectively, in foals.41, 42, 43, 44
Ultrasonography
Transcutaneous ultrasonographic evaluation of the abdomen is quick and noninvasive and can add valuable information in cases of acute or chronic gastrointestinal disease. Ultrasound has become a virtually indispensable tool for horses with acute or chronic gastrointestinal conditions. This section provides a brief summary. Examination of the adult horse requires a 2.5- to 5.0-MHz transducer, depending on size of the horse; a curvilinear transducer is preferred. A functional examination is often performed with isopropyl alcohol saturation alone, although clipping, with or without coupling gel, can be used to enhance evaluation, especially in large or overweight patients.
A protocol for fast localized abdominal sonography (FLASH) has been described and demonstrates good predictive value of the requirement for surgical intervention in the acute abdomen, even with relatively inexperienced examiners.45 This examination evaluates seven locations (ventral abdomen, gastric window, splenorenal window, left middle third of the abdomen, duodenal window, right middle third of the abdomen, and thoracic window) and can be performed in less than 15 minutes. A complete examination requires a methodical approach to the evaluation of the entire abdomen; details of the examination can be variable based on equipment and experience of the examiner. A complete review of abdominal ultrasound is beyond the scope of this chapter; a thorough, detailed examination is reviewed elsewhere.46
In general terms, a quick examination of the acute abdomen should include estimation of gastric size and assessment of small intestinal diameter, wall thickness, motility, and location. Specific abnormalities, such as ascarid impaction or intussusception, may also be visible. Evaluation of the large colon and cecum should include estimation of mural thickening or increased fluidity of contents, and whether or not the colon obscures viewing of the left kidney in the left paralumbar fossa. It is important to remember that other causes of colonic distention can have the same result; thus, it is always important to combine physical, rectal, and ultrasonographic examination findings for any given case. Sand impactions may appear as hyperechoic bands on the ventral abdominal wall,47 but according to the author and others,46 ultrasound does not allow for consistent diagnosis of sand accumulation. Evaluation of the abdomen always should include assessment of the peritoneal space for any evidence of an increased amount or echogenicity of PF. Ultrasonography also can be useful in determining the ideal location for abdominocentesis.
Nuclear Scintigraphy
Nuclear scintigraphy has several proposed uses for evaluation of the gastrointestinal tract, although most have been replaced with cross-sectional imaging (dental disease48, 49), ultrasonography (right dorsal colitis50), or other modalities (gastric emptying51).
Cross-Sectional Imaging
Computed tomography (CT) and, less commonly, magnetic resonance imaging are extremely useful for evaluating dental disease as well as tumors and masses of the head, larynx, pharynx, and proximal esophagus in adult horses52, 53, 54, 55 and some abdominal disorders in foals.56 Availability, gantry size, and table weight limits provide the biggest limitations for widespread use.
Endoscopy
Endoscopic examination of the gastrointestinal tract begins with evaluation of the pharynx for signs of collapse, dysfunction, or dysphagia. The oral cavity should only be examined endoscopically with the use of heavy sedation or anesthesia and a full-mouth speculum. Indications include examination of the teeth, palate, and tongue for completeness, ulceration, masses, or foreign bodies. The most common gastrointestinal indication is evaluation of the stomach, proximal esophagus, and duodenum, typically with a 3-m flexible endoscope.
The esophagus should be examined aborad to orad because of its collapsible nature. The esophageal mucosa is normally smooth and light pink. Erosion or ulceration can occur secondary to obstruction, reflux esophagitis, or an indwelling nasogastric tube, among other causes. Erosions may be punctate, linear, or circumferential, and their extent (depth, length, etc.) should be evaluated carefully. Distinguishing normal peristaltic contractions from areas of stricture requires observation of the area and its motility over time. Diverticula also may be noted as outpouchings of the mucosa, sometimes associated with a stricture distally. Megaesophagus, although rare, appears as generalized dilation. Reevaluation after resolution of an obstruction is especially important for detecting the presence of complications (ulceration and rupture) or initiating causes (strictures, diverticula, masses).
Gastroscopy is best performed after a minimum 12-hour fast. Complete examination of the stomach, including the antrum and pylorus, and preferably proximal duodenum, is critical to avoid missing lesions in the more aborad regions. The squamous mucosa should resemble the esophageal mucosa. The glandular mucosa should be glistening red and may have a reticulated pattern. The veterinarian should carefully examine it for evidence of ulceration or masses. Transendoscopic biopsy material can be easily obtained from esophageal, pharyngeal, or gastric masses, and because the biopsy size will be small, several samples should be taken for histopathologic examination. A complete description of gastroscopy and evaluation of gastric and gastroduodenal ulceration are available in Gastroduodenal Ulcer Disease.
Tests of Absorption and Digestion
Oral Glucose Tolerance Test
d-Glucose or d-xylose absorption tests are useful in determining malabsorption of carbohydrates from the small intestine in horses. For either test, the horse should be fasted for 14 to 18 hours before testing. Prolonged fasting (≥24 hours) results in a delayed and slightly lower peak plasma glucose concentration.57 A dosage of 1 g/kg of d-glucose is administered as a 20% solution via nasogastric tube. Xylazine sedation does not appear to significantly alter d-xylose results.58 Blood glucose or xylose concentrations are then measured 0, 30, 60, 90, 120, 150, 180, 210, and 240 minutes after administration. Additional samples can be taken up to 6 hours after dosing if the results are questionable. Sodium fluoride and heparin are the preferred anticoagulants for glucose and xylose, respectively.
A normal d-glucose absorption test, also known as an oral glucose tolerance test (OGTT), should peak between 90 and 120 minutes at a glucose concentration >85% above the resting glucose concentration.59 Complete malabsorption is defined as a peak less than 15% above the resting concentration, and partial malabsorption is defined as a peak between 15% and 85% above the resting concentration. Gastric emptying, gastrointestinal transit time, length of fasting, glucose and insulin metabolism, age, diet, and endocrine function can influence glucose absorption curves.59, 60 Higher glucose peaks are recorded from healthy animals eating grass or hay than from those eating concentrates60 and from horses fed a pasture (clover and kikuyu) versus stable (oat hay, complete feed, and alfalfa/oat chaff) diet.61 Results from the OGTT can also be affected by the content of nonstructural carbohydrate and fat in the diet62 and other disorders, such as polysaccharide storage myopathy.63
Oral Xylose Tolerance Test
The xylose absorption test is performed as per the OGTT, except 0.5 g d-xylose per kilogram body weight is administered as a 10% solution via nasogastric tube. The pretest period of fasting and timing of sample collection are identical. The laboratory to which samples will be sent should be contacted before collection to ensure that heparinized plasma is acceptable for their laboratory. Plasma d-xylose should peak between 20 and 25 mg/dL between 60 and 90 minutes following administration, with occasional peaks as late as 120 minutes.64, 65 d-Xylose absorption testing is not confounded by hormonal effects or mucosal metabolism as is glucose, but it is altered by diet, length of fasting (which could also be influenced by recent appetite or degree of cachexia), and age. Horses fed a high-energy (oat chaff, oats, and corn) diet had a lower mean peak plasma d-xylose concentration (14.1 mg/dL versus 24.9 mg/dL) than those consuming a low-energy (alfalfa chaff) diet.61 Healthy mares fasted for up to 96 hours had flatter curves and a slower decrease in plasma xylose than when fasted for 12 to 36 hours.64 Kinetic analysis indicates that prolonged food deprivation does not alter renal or nonrenal excretion of d-xylose; thus, the effect of fasting on the curve is likely related to either intestinal transit, small intestinal absorption, or both.66, 67 Ponies may have lower peak d-xylose concentrations than horses, although the range is wide, and diagnostic discriminatory cutoff points for peak plasma xylose concentrations have not been determined. Foals normally have a higher mean peak xylose concentration at 1 and 2 months of age, but mean peak falls to a level similar to that seen in adults by 3 months of age.68 Gastric emptying rate, intestinal motility, intraluminal bacterial overgrowth, and renal clearance can affect curve shape.69
Lactose Tolerance Testing
Milk intolerance is well documented in children and can result from primary or secondary lactase (neutral β-galactosidase) deficiency. Secondary lactase deficiency can occur when a small intestinal disorder, typically rotavirus, damages the epithelial cells, resulting in decreased brush border disaccharidase activity. Such an association has been reported secondary to clostridial enteritis in the foal.40 In horses, lactase activity peaks at birth and declines slowly, such that activity is approximately 50% at 2 years of age and rapidly decelerates after 3 years of age, and it is barely detectable by 4 years of age.41 The oral lactose tolerance test is performed in foals after a 4-hour fast, with a standard dose of 1 g/kg lactose monohydrate given via nasogastric tube as a 20% solution. Blood sampling is as per the OGTT. Plasma glucose typically peaks 60 minutes following lactose administration, with a range from 30 to 90 minutes, with a mean increase of 77 mg/dL, with a 35-mg/dL increase covering 2 standard deviations.42 Of note, older foals (12 weeks) had a more significant increase in plasma glucose relative to 1-week-old foals.
Evaluation of Gastric Emptying
Assessment of gastric emptying may be useful in cases of gastric and esophageal ulceration or suspected gastric outflow obstruction, although accurate assessment can be challenging.
Contrast radiography can be used in foals. In the normal foal, a significant amount of liquid barium should empty within 2 hours.41 Nuclear scintigraphy can be used for measurement of liquid51 or solid phase emptying70 by use of orally administered 99mTc pentetate (10 mCi) or a 99mTc-labeled pelleted ration, respectively.
Alternatively, absorption testing of glucose or xylose (discussed previously) or acetaminophen can be used as an indirect determination of liquid gastric emptying. Acetaminophen absorption is performed by administering 20 mg/kg of acetaminophen orally, sampling blood, then calculating the time to reach maximum serum concentrations and the absorption constant.71, 72 In humans the proximal small intestine absorbs almost all of the acetaminophen.73 The median time to reach peak plasma levels using acetaminophen absorption in horses is 47.7 minutes.72
The 13C-octane acid breath test offers an easy, noninvasive method of determining gastric emptying of solids.70, 74 This test is performed by feeding a standard 13C-labeled test meal and then collecting breath samples using a modified mask. The breath is then analyzed for the ratio of the novel isotope, 13:CO2, to the normally produced 12:CO2.
Histopathologic Examination
It is often necessary to perform a histopathologic examination of tissues from the intestine to diagnose chronic inflammatory, infiltrative, or neoplastic conditions, and such an examination can be useful in evaluating the extent of injury after obstruction or ischemia. Rectal mucosal biopsies are easy to collect, with few complications. Full-thickness biopsy often provides more thorough analysis and can be directed based on serosal appearance via a flank, ventral midline, or laparoscopic approach. When taking gastric or duodenal biopsies endoscopically for the putative diagnosis of inflammatory bowel disorders, multiple samples are recommended. The likelihood of establishing an accurate diagnosis varies with tissue quality, number of samples, skill of the endoscopist, and submission technique.75
Laparoscopy
Laparoscopic evaluation of the equine abdomen requires specialized equipment and training. The laparoscopic procedure can be done with the horse standing or recumbent. Advantages of this technique over a flank or ventral midline celiotomy include smaller incisions, shorter healing time, and shorter procedure time. Disadvantages include equipment and personnel needs, limited therapeutic potential, and limited visual field, especially relative to midline celiotomy. Potential gastrointestinal applications of abdominal laparoscopy include the correction of rectal tears; percutaneous abscess drainage, assessment of adhesions, displacements, and integrity of the serosa of various bowel segments; biopsy of abdominal masses; and closure of the nephrosplenic space.76, 77, 78, 79, 80
Pathophysiology of Gastrointestinal Inflammation
The inflammatory response of the gastrointestinal tract is a mechanism ultimately aimed at eliminating pathogens, initiating tissue repair, and restoring the gastrointestinal barrier. Blood flow is altered, endothelial permeability increases, cells are rapidly recruited into the tissue, plasma protein cascades are activated, and myriad soluble products are released that coordinate the response, trigger innate and adaptive immunity, and mobilize reparative elements. Although the cellular and vascular response and the secreted mediators of inflammation are important for killing pathogens and limiting invasion of injured tissues by commensal organisms, they can be quite damaging to host cells and proteins if not tightly regulated. If the inciting stimulus is not eliminated quickly, then the inflammatory response itself will cause significant tissue injury. The mechanism regulating inflammation has been the focus of a great deal of research to identify therapeutic targets to modulate the damage to host tissues that occurs in many gastrointestinal diseases. Recent work has provided some of the molecular and cellular details of this complex physiology and has led to novel therapeutic strategies for treating inflammation.
Initiation of the Inflammatory Response
Epithelium
The gastrointestinal epithelium interfaces with a luminal environment that is inhabited by potentially hostile microbial organisms. The epithelium presents a physical barrier to invasion by the flora of the gastrointestinal tract, consisting of the apical cellular membrane, intercellular tight junctions (the permeability of which is highly regulated), and a secreted layer of mucus. When invading pathogens breach the mucosal barrier, potent soluble and neural signals are generated that initiate an inflammatory response.81 The epithelium can be conceptualized as a sensory organ that detects pathogen invasion to trigger an appropriate host defense and reparative response.
Noninfectious mucosal injury or invasion of epithelial cells by pathogenic organisms such as Salmonella activates the synthesis of proinflammatory chemokines (chemoattractants) by epithelial cells that triggers a robust influx of neutrophils into the tissue within hours of the damage.81 Of the chemoattractants produced by epithelium, interleukin-8 (IL-8) has a particularly important role in initiating inflammation by recruiting neutrophils from blood82 and regulating neutrophil migration through tissue matrix adjacent to epithelium.83, 84 Complement fragments such as C5a and bacteria-derived formylated chemotactic peptides also act as potent “end target” chemoattractants that are fully capable of stimulating a robust inflammatory response in the intestine if the epithelial barrier permits invasion of bacteria or the diffusion of bacterial peptides across the mucosa.
Epithelial cells activated during infection produce cytokines such as tumor necrosis factor–α (TNF-α), arachidonic acid metabolites, and other proinflammatory mediators that activate recruited leukocytes.85 Microbial products, particularly lipopolysaccharide (LPS) and other bacterial cell wall components and microbial nucleic acids, are potent activators of leukocytes recruited into the tissue.86 Mast cells are key sentinel leukocytes that sense microbial invasion, releasing TNF-α, which appears to be a critical initiator and regulator of the cellular phase of inflammation.87 Once the inflammatory response has been initiated, TNF-α; IL-1β; and other proinflammatory products of neutrophils, monocytes, mast cells, and epithelial cells amplify the inflammatory response.
The enteric nervous system (ENS) has a key role in sensing and regulating inflammatory responses in the intestine. For example, C. difficile toxin A activates a neural pathway that triggers mast cell degranulation and neutrophil influx into the tissue.88, 89 Blockade of this neural pathway is sufficient to abolish the profound inflammatory response induced by toxin A as well as many of the effects of toxin A on enterocyte secretion. Other pathogens and immune-mediated hypersensitivity reactions similarly stimulate inflammation by mechanisms that involve the ENS. Thus the epithelium interacts in a highly complex manner with the intestinal milieu, the ENS, and inflammatory cells to regulate the tissue response to injury and infection.
Macrophages
Resident macrophages located in the lamina propria, submucosa, and intestinal lymphoid organs are among the first cells beyond the epithelium to respond to infection or injury. Macrophages are activated by microbial products by way of pattern recognition receptors and begin to produce proinflammatory molecules important for recruiting and activating neutrophils and monocytes. Pattern recognition receptors recognize microbial molecules such as LPS, lipoproteins, flagellin, peptidoglycan, and nucleic acids to signal the invasion by pathogens.86 Of the pattern recognition receptors, the LPS receptor complex is perhaps the best defined. LPS activates macrophages by way of the CD14 Toll-like receptor 4 (TLR-4) complex to initiate transcription of the inflammatory cytokines TNF-α and IL-1β, which synergize with LPS to amplify the macrophage response.86 LPS, particularly in concert with inflammatory cytokines, stimulates macrophages to produce copious amounts of nitric oxide, which is both microbicidal and vasoactive.90 Nitric oxide and other nitrogen radicals react with reactive oxygen intermediates (ROIs) generated by the activated oxidase complex to produce some of the most toxic molecules of the host defense system: the peroxynitrites.90 IL-8 is produced as well to recruit neutrophils. As the response progresses, other inflammatory mediators, particularly the arachidonic acid–derived lipids dependent on inflammation-induced cyclooxygenase (COX)-2 and 5-lipoxygenase activity, are produced that have potent vasoactive and proinflammatory effects through the activation of endothelial cells, neutrophils, and platelets.91
Vascular Response During Inflammation
Four important changes occur in the intestinal vasculature during inflammation: (1) alteration of blood flow; (2) increased vascular permeability; (3) increased adhesiveness of endothelial cells, leukocytes, and platelets; and (4) exposure of the basement membrane and activation of the complement, contact, and coagulation cascades.
A wide range of mediators alter blood flow during inflammation in the intestinal tract, ranging from gases such as nitric oxide (a major vasodilator of the intestinal vasculature) to lipids (prostaglandins, leukotrienes, thromboxanes, and platelet-activating factor [PAF]), cytokines, bradykinin, histamine, and others. The major sources for these mediators include activated leukocytes, endothelial cells, epithelial cells, and fibroblasts. The primary determinant of blood flow early in inflammation is vascular caliber, which initially decreases in arterioles but then quickly changes to vasodilation coincident with the opening of new capillary beds, increasing net blood flow. The increase in blood flow is relatively short-lived, because the viscosity of the blood increases from fluid loss and tissue edema resulting from leaky capillaries. Leukocyte margination, platelet adhesion to endothelial cells and exposed matrix, and areas of coagulation protein accumulation further decrease local circulation.
Increased vascular permeability is initially caused by inflammatory mediator actions on the endothelial cells. Histamine, leukotrienes, PAF, prostaglandins, bradykinin, and other mediators stimulate endothelial cell contraction, and interendothelial gaps form.92, 93 This stage of increased vascular permeability is readily reversible. Concurrently, mediators such as the cytokines TNF-α and IL-1β induce a structural reorganization of the interendothelial junctions, resulting in frank discontinuities in the endothelial monolayer.94 Cytokines also stimulate endothelial cells to express adhesion molecules that support adhesion of leukocytes and platelets,95 leading to the next and perhaps most devastating event. Leukocytes (primarily neutrophils) and platelets adhere to exposed basement membranes and activated endothelial cells. Adherent neutrophils and platelets are then exposed to the mediators of inflammation present in the surrounding milieu, which activates the cells to release oxidants and proteases (particularly elastase) that injure the endothelium and have the potential to cause irreparable harm to the microvasculature.96, 97, 98 Marginated neutrophils begin to transmigrate between endothelial cells (as described in later sections), which, if in sufficiently large numbers, disrupts the integrity of the interendothelial junctions, worsening the vascular leakage.97
These stages of enhanced vascular permeability can be conceptualized as a mechanism to allow plasma proteins to enter the tissues and to potentiate the critical influx of leukocytes into tissues. However, if they are not regulated precisely, alterations in both hydrostatic and oncotic forces and irreversible damage to the vascular bed may have devastating consequences. Moreover, inappropriate activation of plasma protein cascades and leukocytes by activated endothelium and exposed matrix proteins can contribute to SIRS (see the later section Gastrointestinal Ileus for more information) characterized by hypotension, generalized vascular leak syndrome, and multiorgan dysfunction, which may be fatal. Phosphodiesterase inhibitors reduce endothelial permeability in ischemia–reperfusion injury and other models of inflammation-induced vascular leakage99, 100 by increasing endothelial tight junction integrity; thus, it may be a viable therapeutic strategy to prevent or reduce the permeability alterations associated with inflammation.
Cellular Effectors of Inflammation
Endothelial Cells
Endothelial cells respond to products of activated epithelial cells and macrophages in the intestinal tissue to recruit cells and humoral mediators of inflammation into the tissue. Activated endothelial cells display a range of molecules critical for neutrophil and platelet adhesion. The role of endothelial cells in mediating neutrophil recruitment is discussed in more detail later in this chapter. Intercellular permeability is increased to expose basement membrane proteins that trigger humoral defense systems (complement, coagulation, and contact system cascades) and to provide access for these macromolecules to the tissue. Endothelial cells are an important source of inflammatory mediators that amplify the response and vasoactive substances (particularly nitric oxide) altering blood flow.
Neutrophils
Recruitment
Infection or injury to the gastrointestinal mucosa causes an influx of leukocytes from the blood that lay the foundation of the inflammatory response. Neutrophils, the first to arrive during inflammation, have a dominant role in the acute response. Within minutes neutrophils are recruited into the tissue, in which they are activated to release products that not only are lethal to pathogens and proinflammatory but also may damage host cells and tissues.101 Not surprisingly, a great deal of attention has been paid to the role of neutrophils in the pathophysiology of many inflammatory conditions.102 Neutrophil depletion is protective in many models of gastrointestinal inflammatory disease. Of interest to clinicians, blockade of neutrophil migration into inflamed tissues prevents many of the pathophysiologic events associated with infectious enteritis, ischemia–reperfusion injury, and other gastrointestinal diseases.98, 103, 104, 105, 106
Neutrophil transendothelial migration is a multistep process that is temporally and spatially regulated and has a degree of cell type specificity (Fig. 12.1 ). The predominant sites of neutrophil transendothelial migration are in the postcapillary venules and, in some tissues, capillaries. Endothelial cells in these vessels respond to cytokines and other soluble signals by expressing molecules that promote neutrophil adhesion and transmigration, including selectins and counterreceptors for integrins. As neutrophils flow through these vessels, they are first tethered to activated endothelium. Tethering is mediated by selectin molecules expressed on neutrophils (L-selectin) and on activated endothelial cells (P- and E-selectins), which bind to PSGL-1, ESL-1, and other mucin counterreceptors.107, 108 The function of tethering is to increase the exposure of the neutrophil to activating chemokines presented on the surface of the endothelial cells.
Fig. 12.1.
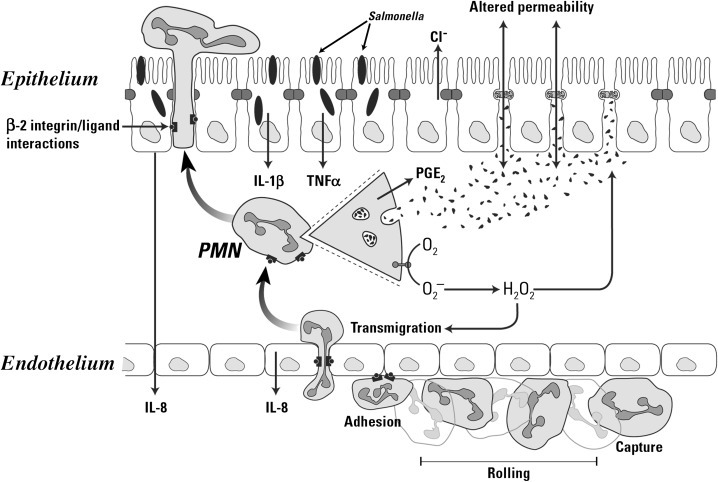
Depiction of neutrophil responses during intestinal inflammation in response to Salmonella infection. Salmonellae infect epithelial cells, stimulating the production of chemokines (interleukin-8 [IL-8]), cytokines (IL-1β and tumor necrosis factor-α [TNF-α]), and other proinflammatory mediators. Endothelial cells stimulated by inflammatory mediators produce chemoattractants (such as IL-8) and display adhesion molecules that promote neutrophil emigration. The three steps of neutrophil (polymorphonuclear [PMN]) emigration capture/rolling (mediated by selectins), adhesion (mediated by β2 integrins), and transendothelial migration (mediated by integrins and platelet/endothelial cellular adhesion molecule) occur on activated endothelium. Chemoattractant molecules such as IL-8 trigger neutrophil emigration. In inflamed tissues cytokines (IL-1β and TNF-α) and a variety of other proinflammatory mediators stimulate the neutrophil oxidase complex to produce reactive oxygen intermediates (ROIs; O2− and H2O2 and their derivatives). Activated neutrophils degranulate to release proteases and other hydrolases, cationic peptides (defensins), myeloperoxidase, and other products into the tissue. Activated neutrophils synthesize a variety of inflammatory mediators, including prostaglandins (PGE2) that modulate the inflammatory response. The products of activated neutrophils (ROIs, proteases, and mediators) stimulate epithelial secretion and alter tight junction permeability, promoting diarrhea. Neutrophils eventually migrate across the infected epithelium by a mechanism that involves integrins, disrupting tight junction integrity and increasing permeability to bacterial products, thus exacerbating the inflammatory response.
Stimulation of neutrophils by IL-8 and other chemokines activate the second step of transendothelial migration. Chemokine binding to their receptors on the neutrophil generates signals that activate the binding of integrin adhesion receptors to their ligands, called intracellular adhesion molecules (ICAMs) or vascular cell adhesion molecules, expressed on endothelial cells in inflamed mucosa. Integrin ligation to ICAMs arrests the tethered neutrophils, resulting in firm adhesion to the endothelium. Of the integrins expressed on neutrophils, the β2-integrins have a particularly important role in transendothelial migration. Calves and people with the disorder leukocyte adhesion deficiency (LAD) illustrate the requirement for β2-integrin–mediated adhesion in neutrophil function. LAD is a result of an autosomal recessive trait resulting in the lack of the β2-integrin expression. The neutrophils from affected individuals cannot migrate into most tissues and do not function normally, resulting in poor tissue healing and profound susceptibility to infection, especially at epithelial barriers.109, 110 Other integrins also have a role in transendothelial migration. β1-Integrins mediate transendothelial migration in some cells and seem to be particularly important for mediating emigration of monocytes into many tissues.111
Following this firm adhesion step, neutrophils migrate through the endothelium along a chemotactic gradient of IL-8 and other chemoattractants, such as C5a and leukotriene B4 (LTB4).82, 97, 112 Neutrophils migrate across the endothelial monolayer at intercellular junctions by way of a mechanism involving a series of integrin-ligand interactions mediated by both β2- and β1-integrins and other adhesion molecules,108 which is generally capable of maintaining the integrity of the endothelial barrier.113 However, a massive flux of neutrophils through the endothelium alters endothelial tight junctions and injures the basement membrane, resulting in increased endothelial permeability to molecules as large as plasma proteins and even endothelial cell detachment from the basement membrane.96, 97 Nonintegrin molecules such as platelet–endothelial cell adhesion molecules (PECAMs) also are involved in transendothelial migration of neutrophils.108 Homotypic binding of PECAMs on adjacent endothelial cells form part of the intercellular junction. Neutrophils express an integrin of the β3-family that can bind PECAMs, and through sequential binding of β3-integrins to PECAMs, the neutrophil can “unzip” the intercellular junction and migrate through, closing it behind itself.
Activation
A key feature of neutrophils and other leukocytes is the requirement for integrin-mediated adhesion to extracellular matrix (ECM) proteins or other cells to achieve an optimal effector phenotype.114 Critical components of the ECM in inflamed tissues include fibronectin, fibrinogen, and vitronectin, deposited in tissues as a result of plasma leakage and by synthesis of new proteins by stromal cells and resident macrophages in response to inflammatory mediator activation. The changing composition of the matrix proteins deposited in tissues during inflammation serves as a clue as to the nature of the tissue environment for recruited inflammatory cells as they become activated. Individual gene expression studies have demonstrated that adhesion to matrix proteins induces the expression of cytokines and chemokines and their receptors, arachidonic acid–derived lipid mediator synthases, metalloproteinases, growth factors, transcription factors, and other genes that influence the differentiation and activation of inflammatory cells.115 ROI production, phagocytosis, degranulation, and other effector functions stimulated by inflammatory mediators and bacterial products are optimal only when neutrophils are adherent to the ECM.114 Adhesion to distinct ECM proteins selectively activates signaling pathways and gene expression of neutrophils, monocytes, and other leukocytes with differing abilities to promote certain functions such that the composition of ECM in many ways controls the development of the ultimate effector phenotype. Thus integrin-mediated adhesion provides a mechanism in which neutrophils and other leukocytes can sense the complex tissue environment and respond appropriately.
Of the activators of neutrophils at sites of inflammation, complement (C3-opsonized particles), cytokines (TNF-α and IL-1β), PAF, immune complexes, and bacterial products are among the most potent stimuli. Other mediators produced during inflammation may modify neutrophil activity, particularly formylated bacterial peptides, chemokines, complement fragments (C5a), LTB4, and prostaglandins. Activated neutrophils are highly phagocytic; produce large amounts of ROI; degranulate to release myeloperoxidase, cationic antimicrobial peptides (defensins), serine proteases (mainly elastase), and metalloproteinases; and secrete inflammatory mediators (TNF-α, IL-1β, prostaglandins, leukotrienes, and others) (see Fig. 12.1).
Mast Cells
Mast cells strategically reside in mucosal tissues, including the submucosa and lamina propria of the gastrointestinal tract, and constitute a crucial first line of defense at epithelial barriers. However, they are also important effector cells of the pathophysiology of inflammatory gastrointestinal diseases.116 Experimental depletion of mast cells, genetic deficiency in the development of mast cells, or pharmacologic stabilization of mast cells to prevent degranulation all have a protective effect in a variety of models of gastrointestinal inflammatory disease, including dextran sulfate sodium–induced or trinitrobenzenesulfonic acid–induced colitis,117, 118 ischemia–reperfusion injury,119, 120 and immediate hypersensitivity responses.121
Mast cells are activated by a wide variety of microbial products and host-derived mediators.122 Among the activators of mast cells, the so-called anaphylatoxins (complement fragments C3a, C5a, and C4a) are extremely potent stimuli causing release of mediators of inflammation. In addition, mast cells are the primary effector cells of IgE-mediated anaphylaxis (type I hypersensitivity reactions) by virtue of their high-affinity receptors for IgE. The cross-linking of a receptor-bound IgE on the mast cell surface by antigens (i.e., food antigens) causes rapid degranulation, which results in the explosive release of granule contents.123 Neural pathways in the intestine also regulate mast cells, which respond to enteric pathogen invasion via neural reflexes that stimulate the release of inflammatory mediators.
Activated mast cells release preformed histamine, 5-hydroxytryptamine (5-HT), proteases, heparin, and cytokines from granules. Activation also stimulates de novo synthesis of a range of inflammatory mediators, including prostaglandins, PAF, and leukotrienes. Transcription of a number of peptide mediators, such as the cytokines TNF-α and IL-1β among many others, also increases stimulation of mast cells. Mast cell products have profound effects on the vasculature, increasing endothelial permeability and causing vasodilation.124 Moreover, mast cell–derived mediators markedly enhance epithelial secretion by a mechanism that involves the activation of neural pathways and direct stimulation of epithelial cells.123 In particular, the mast cell granule protease tryptase operating via the protease-activated receptor-2 is a key regulator of gastrointestinal physiologic responses during inflammation, including epithelial secretion and intercellular junction integrity, motility, and pain responses.125, 126 Mast cell products significantly alter intestinal motility, generally increasing transit and expulsion of intestinal contents. Mast cell–derived leukotrienes and TNF-α also have crucial roles in host defense against bacterial pathogens, acting to recruit and activate neutrophils,127, 128 and are crucial players in the mechanism regulating dendritic cell function and adaptive immune responses.129
Mast cells have a role in host defense and inflammatory responses to bacterial pathogens, partly because of the release of proinflammatory mediators during bacterial infection, which is critical for recruiting and activating other innate host defense cells such as neutrophils.87 Mast cells are also phagocytic, have microbicidal properties, and can act as antigen-presenting cells to the adaptive immune system.87 The role for mast cells in host protective responses appears to be as a sensor of bacterial invasion. Unlike IgE-mediated responses, bacterial products seem to elicit a highly regulated and selective response from mast cells.
Humoral Mediators of Inflammation
Complement
The complement cascade is a fundamental part of the inflammatory response. Activation of the complement cascade, either by immune complexes (classical pathway) or by bacteria or bacterial products, polysaccharides, viruses, fungi, or host cells (alternative pathway), results in the deposition of complement proteins on the activating surface and the release of soluble proteolytic fragments of several complement components.130 In particular, activation of either pathway results in the deposition of various fragments of the complement protein C3, which are potent activators of neutrophils and monocytes.130 Opsonization of particles with C3 fragments constitutes a major mechanism of target recognition and phagocyte activation.131 During the activation of the complement cascade culminating in deposition of C3, soluble fragments of C3 (C3a), C5 (C5a), and C4 (C4a) are liberated. These fragments, termed anaphylatoxins, have potent effects on tissues and cells during inflammation. Perhaps most notably, they are chemotactic for neutrophils (particularly C5a), activate neutrophil and mast cell degranulation, and stimulate reactive oxygen metabolite release from neutrophils.130 The termination of the complement cascade results in the formation of a membrane attack complex in membranes at the site of complement activation. If this occurs on host cells such as endothelium, the cell may be irreversibly injured. Although the primary source of complement is plasma, epithelial cells of the gastrointestinal tract also produce C3, suggesting that local production and activation of the complement cascade during inflammation occurs in intestinal tissues.
It is clear that if the regulatory mechanisms of the complement cascade fail, then the inflammatory response may be inappropriate and tissue injury can occur. The role of complement in gastrointestinal inflammation has been most extensively studied in models of ischemia–reperfusion injury. Activation of the complement cascades has a major role in altered endothelial and epithelial permeability in these models. Several lines of evidence support the importance of complement in intestinal injury. Mice deficient in C3 or C4 are protected against ischemia–reperfusion injury.132 Moreover, administration of monoclonal antibodies against C5 reduced local and remote injury and inflammation during intestinal reperfusion injury in a rat model.133 Administration of a soluble form of complement receptor 1, a regulatory protein that halts the complement cascade by dissociating C3 and C5 on host cell membranes, reduced mucosal permeability, neutrophil influx, and LTB4 production during ischemia–reperfusion injury in rats and mice.132, 134 Although neutrophils and mast cells mediate many of the pathophysiologic effects of the complement cascade, the membrane attack complex may have a primary role in altered vascular permeability during ischemia–reperfusion injury.135
Contact System
The contact system of plasma is initiated by four components: Hageman factor (HF), prekallikrein, factor XI, and high-molecular-weight kininogen. HF is a large plasma glycoprotein that binds avidly to negatively charged surfaces.136 Bacterial cell walls, vascular basement membranes, heparin, glycosaminoglycans, and other negatively charged surfaces in the intestine capture HF and the other three important initiators of the contact system in a large multimolecular complex. Of the surfaces that bind HF, the ECM is an extremely potent activator of the contact system. Once bound, HF is converted to HF-α, which cleaves prekallikrein to kallikrein and factor XI to factor XIa. The ultimate result is further cleavage of HF by kallikrein and triggering of the contact system cascade, activation of intrinsic coagulation by factor XIa, activation of the alternative pathway by HF, and proteolytic cleavage of high-molecular-weight kininogen by kallikrein, releasing biologically active kinins.
The products of the contact system, particularly bradykinin, have several important biologic properties that drive many of the vascular and leukocyte responses during inflammation.136 Bradykinin induces endothelial cell contracture and intracellular tight junction alterations that increase vascular permeability to fluid and macromolecules. Bradykinin also affects vascular smooth muscle contracture, resulting in either vasoconstriction or vasodilation, depending on the location. Bradykinin also increases intestinal motility, enhances chloride secretion by the intestinal mucosa, and intensifies gastrointestinal pain. In neutrophils kinins stimulate the release of many inflammatory mediators, including cytokines, prostaglandins, leukotrienes, and ROIs.137 Kallikrein cleaves C5 to release C5a, a potent chemotactic factor for neutrophils, and thus has a role in recruiting and activating inflammatory leukocytes.
The plasma kallikrein–bradykinin system is activated in a variety of acute and chronic inflammatory diseases of the gastrointestinal tract.138, 139 Blockade of the pathophysiologic effects of bradykinin has clinical applications. Oral or intravenous (IV) administration of the bradykinin receptor antagonist icatibant reduces the clinical signs, onset of diarrhea, and many of the histopathologic changes in experimental models of colitis in mice.140 Inhibition of kallikrein by oral administration of P8720 attenuated the intestinal inflammation, clinical score, and systemic manifestations in a model of chronic granulomatous enterocolitis.138 Thus the contact system is a potential therapeutic target for inflammatory diseases of the intestine.
Tissue Injury During Inflammation
Changes in blood flow to the mucosa and other regions of the intestine that reduce perfusion of the tissues can potentiate the initial damage caused by infection or injury. For example, reperfusion of ischemic tissues is associated with platelet and neutrophil clumping in the small vessels of the mucosa, which can impede blood flow.141 Platelets are activated and adhere to exposed basement membrane and activated endothelial cells and provide a surface for leukocyte adhesion. The accumulation of platelets and leukocytes can significantly reduce vessel diameter and blood flow while potentiating local coagulation and thrombus formation.
Soluble mediators released by activated leukocytes and endothelial cells also affect blood flow. Histamine and the vasoactive lipids derived from arachidonic acid (leukotrienes, prostaglandins, thromboxane, prostacyclin, and PAF) have a prominent role in regulating local perfusion during inflammation and may have systemic effects on blood flow as well. Procoagulant mediators released by inflammatory cells in response to the inflammatory process (i.e., tissue factor produced by macrophages or endothelial cells), exposed basement membrane proteins, and bacterial components can trigger the contact system and the coagulation and complement cascades, the products of which affect blood flow. Nitric oxide, whether produced by endothelial cells or leukocytes (macrophages), is a potent regulator of blood flow and has a significant role in the control of perfusion during inflammation.142 Many of the mediators that affect perfusion also affect endothelial permeability, altering osmotic and hydrostatic balance and tissue edema. In extreme cases local and systemic coagulopathies initiated by vascular injury and absorption of microbial products and inflammatory mediators induce a hypercoagulable state, leading to microthrombus formation, which can reduce blood flow or macrothrombus formation, causing tissue infarction.
The cellular mediators of inflammation have the potential to inflict severe injury to intestinal tissues. Neutrophils have an important role in the pathophysiology of many intestinal diseases, including ischemia–reperfusion injury,98 infectious enterocolitis,105, 143 nonsteroidal antiinflammatory drug–induced mucosal ulceration,106 and others. Depletion of neutrophils, blockade of their emigration into tissues, or inhibition of neutrophil activation reduces the severity of these and other inflammatory diseases.144 Many antiinflammatory therapies are emerging that specifically target neutrophil adhesion, migration, and activation.
Migration of neutrophils through endothelium during emigration into inflamed tissues is remarkable in that the permeability of the endothelial monolayer is preserved under most circumstances. However, there is a limit above which neutrophil migration alters the permeability characteristics of the endothelium. The effect is in part physical in that the mere movement of large numbers of neutrophils through the endothelium is sufficient to mechanically disrupt the tight junctions and in part because of toxic products of neutrophils that damage endothelial cells and basement membranes.141, 145 Serine proteases (particularly elastase) and metalloproteinases released by degranulating neutrophils liquefy tissue matrix proteins and cleave cell-surface proteins that make up endothelial intercellular junctions to ease neutrophil migration to the site of infection.101 These degradative enzymes are particularly damaging to basement membranes and the cellular barriers of the endothelium, thus contributing to vascular permeability (and local tissue edema) and thrombosis. The permeability may be affected to the extent that not only water but also macromolecules (e.g., albumin, matrix proteins, complement) leak into the interstitium. Blockade of neutrophil adhesion to endothelium with anti–β2-integrin antibodies has a sparing effect on the microvasculature in experimental intestinal ischemia–reperfusion injury, reducing the alterations in vascular permeability and histopathologic evidence of microvascular damage.141
Similar to the endothelium of inflamed tissues, massive neutrophil transmigration occurs across the epithelium in response to infection or injury. Neutrophil transepithelial migration increases epithelial permeability by disrupting tight junctions.145 Like the endothelium, neutrophils disrupt the epithelial barrier mechanically as they migrate through (see Fig. 12.1). Proteases, particularly elastase, degrade basement membrane components and tight junction proteins. Protease activated receptor-2 activated by neutrophil granule serine proteases alter epithelial and endothelial tight junction integrity. Proinflammatory products of activated neutrophils (TNF-α and IFN-γ) increase tight junction permeability by direct effects on enterocytes. Prostaglandins released by activated neutrophils stimulate epithelial secretion, thus contributing to diarrhea. Subepithelial accumulation of neutrophils can lead to deadhesion of the epithelial cells from the basement membrane and mild to severe ulceration. The physiologic result of the effects of neutrophils and their products on the epithelial barrier includes protein-losing enteropathy and absorption of bacterial cell wall constituents, which potentiates the local and systemic inflammatory responses.
Neutrophils in inflamed tissues stimulated by potent host-derived activators (such as IL-1β and TNF-α) and bacterial products (LPS) release copious amounts of ROIs (see Fig. 12.1). Although these oxygen and oxyhalide radicals are important for killing pathogens, they are also potentially toxic to epithelial and endothelial cells and matrix proteins. Reactive nitrogen intermediates, produced primarily by macrophages during inflammation, combine with ROIs to form peroxynitrites, which are particularly toxic.90 In addition to injury to mucosal tissues, ROIs also have an as yet ill-defined role in recruiting and activating neutrophils, potentiating the inflammatory response.146 In support of the role of ROIs in inflammatory diseases of the gastrointestinal tract, administration of inhibitors of ROI production or pharmacologic ROI scavengers can be protective in many models of reperfusion injury or enterocolitis. Many therapies are aimed at inhibiting neutrophil activation, and effector functions in tissues have been evaluated for use in intestinal diseases. Phosphodiesterase inhibitors, by causing cyclic adenosine monophosphate (cAMP) accumulation in neutrophils, are antiinflammatory by virtue of their ability to suppress neutrophil activation and ROI production. New phosphodiesterase inhibitors selective for the predominant neutrophil isoform of phosphodiesterase hold promise for use in many inflammatory diseases.
Subepithelial mast cells also have an important role in altering epithelial permeability in inflamed intestine. During the intestinal hypersensitivity response subepithelial mast cell release of mast cell protease tryptase by degranulation increases epithelial permeability via an effect on tight junctions.147, 148 This alteration in tight junction permeability results in enhanced transepithelial flux of macromolecules, including proteins and bacterial products. Cytokines released by mast cells and phagocytes also regulate tight junction permeability. IL-4, a product of mast cells and macrophages, has been demonstrated to increase epithelial permeability.149 Moreover, TNF-α and IFN-γ, products of many inflammatory cells, synergistically increase tight junction permeability.150
Pathophysiology of Diarrhea
Acute equine colitis causes rapid, severe debilitation and, often, death in horses. Diarrhea associated with acute equine colitis occurs sporadically and is characterized by intraluminal sequestration of fluid, moderate to severe colic (abdominal pain), and profuse watery diarrhea with resultant endotoxemia, leukopenia, and hypovolemia.151, 152 Causes of acute colitis and therapeutic options are discussed later in this section.
Although all mechanisms responsible for fluid losses are not known, inflammatory cells likely play an integral role; colitis is characterized by granulocyte infiltration of the large intestinal mucosa.153, 154, 155, 156, 157 Equine cecal and colonic tissues collected during the acute stages of experimentally induced acute equine colitis (Potomac horse fever, lincomycin with and without Clostridium spp. inoculation, nonsteroidal antiinflammatory drug administration) reveal the presence of numerous neutrophils and eosinophils in the lamina propria and submucosa.153, 156, 158, 159 Granulocyte-derived ROIs are crucial to antimicrobial defenses in the gut and stimulate chloride and water secretion by interactions with enterocytes.160, 161 Normal equine intestinal tissue is unique compared with that in most other mammalian species for a preponderance of eosinophils located in the intestinal mucosa and submucosa.162, 163 Production of ROIs by stimulated phagocytic granulocytes following mucosal barrier disruption may be responsible for the massive fluid secretory response that occurs during the early stages of acute equine colitis.
Colitis refers to inflammation and mucosal injury of the colon and cecum (typhlocolitis) that may occur in response to a number of causes.164 The cause of the colonic injury may be well defined such as in naturally occurring infectious or experimentally induced colitis. However, many cases of human and animal diarrhea have a speculative or unknown diagnosis or no diagnosis. Irrespective of the underlying or initiating cause of colonic injury, the colon apparently has a limited repertoire of responses to damage because most forms of colitis demonstrate similarities in histopathologic appearance and clinical presentation. Various degrees of mucosal erosion and ulceration, submucosal/mucosal edema, goblet cell depletion, and the presence of an inflammatory cellular infiltrate within the mucosa and submucosa are common to many types of human and animal colitis.163, 164 Characteristic clinical manifestations include intraluminal fluid sequestration; abdominal discomfort; hypovolemia; and most often profuse, watery diarrhea.
Pathophysiology of Colitis
Large bowel diarrhea results from abnormal fluid and ion transport by cecal and colonic mucosa. Loss of fluid by the large intestine can result from malabsorptive or hypersecretory processes and is often a combination of the two.165 Colonic secretory processes are a function of the crypt epithelium, whereas absorptive processes are limited to surface epithelial cells.166, 167 Under normal baseline conditions, an underlying secretion by crypt epithelium is masked by a greater rate of surface epithelial cell absorption. Abnormal forces influencing the rates of secretion and absorption can result in massive, uncontrolled secretion and malabsorption by large intestinal mucosal epithelial cells, leading to rapid dehydration and death.165, 166, 167
Two intracellular processes control colonic secretion: the cyclic nucleotide (cAMP and cyclic guanosine monophosphate [cGMP]) and the calcium systems.168, 169 Agents may activate adenyl cyclase (vasoactive intestinal peptide, prostaglandin E2 [PGE2]) or guanylyl cyclase (bacterial enterotoxins) and induce increases in cAMP or cGMP, respectively. This reaction causes phosphorylation of specific protein kinases that induce the actual apical and basolateral membrane transport events. Increases in intracellular free calcium may arise from cyclic nucleotide–dependent release of stored calcium within the cell or from increased calcium entry across the cell membrane.165, 166, 167 Calcium may act through calmodulin, which then can activate membrane-phosphorylating protein kinases.
At least four central systems control intestinal secretion: (1) the hormonal system, (2) the ENS, (3) bacterial enterotoxins, and (4) the immune system.169, 170 Hormonal control of colonic electrolyte transport is exerted primarily through the renin–angiotensin–aldosterone axis.171 The ENS controls transport through three separate components: (1) extrinsic nerves of the parasympathetic and sympathetic pathways; (2) intrinsic ganglia and nerves, secreting a variety of neurotransmitters including peptides; and (3) neuroendocrine cells (intraepithelial lymphocytes) that reside in the epithelium and release messengers onto the epithelial cells in a paracrine manner.165, 169, 170, 171 Many bacterial enterotoxins can induce intestinal secretion by cAMP or cGMP signal transduction.172 Bacterial enterotoxins can stimulate release of mediators (such as substance P) from primary afferent neurons, which then affect enteric neurons, often propagating neurogenic inflammation.173
Preformed inflammatory mediators such as histamine, serotonin, or adenosine and newly synthesized mediators such as prostaglandins, leukotrienes, PAF, various cytokines, the inducible form of nitric oxide, and reactive oxygen metabolites can initiate intestinal secretion by directly stimulating the enterocyte and by acting on enteric nerves indirectly to induce neurotransmitter-mediator intestinal secretion.170 Prostaglandins of the E and F series can cause an increase in chloride secretion in intact tissue and isolated colonic cells.174, 175 Leukotrienes, PAF, and a number of cytokines have been shown to have no effect on T84 cell secretion but have a significant effect on electrolyte transport in intact tissue, suggesting that intermediate cell types may be involved in these secretory responses.176, 177, 178
The epithelial cell chloride secretory response occurs via prostaglandin-mediated and adenosine-mediated increases in cellular cAMP, whereas histamine acts by H1 receptor induction of phosphatidylinositol turnover, production of inositol triphosphate, and mobilization of intracellular calcium stores.170 Lipoxygenase products (leukotrienes) are capable of activating a colonic secretory response and do not appear to involve the cyclic nucleotides or calcium ions.176 Phagocyte-derived reactive oxygen mediators (ROMs) can induce colonic electrolyte secretion in vitro, suggesting that oxidants may contribute directly to the diarrhea associated with colitis.179 Reactive oxygen species initiate the secretory response by increasing cellular cAMP or stimulating mesenchymal release of PGE2 or prostacyclin, which in turn stimulates the epithelial cell or enteric neuron, respectively.179, 180, 181, 182 Sodium nitroprusside, an exogenous source of nitric oxide, stimulated an increase in chloride secretion in rat colon that was mediated by COX products and enteric neurons.183 Table 12.1 summarizes inflammatory mediator–induced epithelial cell chloride secretion.
TABLE 12.1.
Inflammatory Mediators That Stimulate Epithelial Cell Chloride Secretion
| Mediator | Action |
|---|---|
| Prostaglandin E2 | Increases Cl secretion |
| Decreases neutral NaCl absorption | |
| Vasoactive intestinal peptide | Increases cAMP-mediated NaCl secretion |
| Activates cholinergic nerves | |
| Endotoxin | Increases Na absorption |
| Increases cell membrane permeability | |
| Serotonin | Increases fluid and electrolyte secretion |
| Interferon-γ | Decreases tight junctions and causes increase in cell membrane permeability |
| Interleukin-1 and interleukin-1β | Increase prostaglandins E2 and F1α and thromboxane B2 |
| Histamine (H1) | Increases Cl secretion via Ca-mediated pathways |
| Bradykinin | Increases Cl secretion through prostaglandin-mediated pathways |
| Reactive oxygen mediators | Increase Cl secretion |
| Thromboxanes | Increase Cl secretion |
| Decrease neutral NaCl absorption | |
| Lipoxygenase products | Increase Cl secretion via prostaglandin-mediated pathways |
| Platelet-activating factor | Increases Isc (Cl secretion) |
| Adenosine | Increases Cl secretion |
cAMP, Cyclic adenosine monophosphate.
Role of Inflammatory Cells
Acute colitis rarely develops by a simple cause or effect phenomenon but is influenced by many extrinsic and intrinsic host and microorganism factors. Inflammatory mediators released from mast cells and monocytic or granulocytic phagocytes cause intestinal chloride and water secretion and inhibit neutral sodium and chloride absorption.169, 170, 184 Inflammatory cells, particularly the phagocytic granulocytes, play an important role in mucosal pathophysiology in cases of colitis.161, 185 Large numbers of these cells are observed on histopathologic examination of tissues from human and animal cases of colitis. Products of cell activation stimulate direct and indirect secretory responses in intestinal cells and tissues.169, 170 Products of phagocyte secretion may amplify the inflammatory signal or have effects on other target cells in intestine such as enterocytes and smooth muscle cells.
Role of Phagocyte-Derived Reactive Oxygen Metabolites
The nicotinamide adenine dinucleotide phosphate (NADPH)-NADPH oxidase system of phagocytes (neutrophils, eosinophils, and monocytes/macrophages) is a potent inducer of superoxide radicals used as a host defense mechanism to kill invading microorganisms.161 During inappropriate stimulation such as inflammation, trauma, or ischemia followed by reperfusion, increased levels of toxic oxygen species are produced, causing damage to host tissues. Engagement of any of several receptor and nonreceptor types including phagocytosis mediators, chemotactic agents, various cytokines, and microbial products can stimulate phagocytes.161 Resident phagocytes or those recruited to colonic mucosa early in the disease process are considered to augment mechanisms causing fluid and electrolyte secretory processes, a so-called amplification process.104, 186
Activation of the respiratory burst results in the production and release of large amounts of superoxide anion (O2 −) and H2O2.187 In addition to these ROMs, activated phagocytes secrete peroxidase enzyme (myeloperoxidase from neutrophils and eosinophil peroxidase from eosinophils) into the extracellular space. The peroxidases catalyze the oxidation of Cl− by H2O2 to yield HOCl, the active ingredient in household bleach products. The peroxidase–H2O2–halide system is the most cytotoxic system of the phagocytes; HOCl is 100 to 1000 times more toxic than O2 − or H2O2. HOCl is a nonspecific oxidizing and chlorinating agent that reacts rapidly with a variety of biologic compounds including DNA, sulfhydryls, nucleotides, amino acids, and other nitrogen-containing compounds. It reacts rapidly with primary amines to produce the cytotoxic N-chloramines. The mechanisms by which these substances damage cells and tissue remain speculative, but possibilities include direct sulfhydryl oxidation, hemoprotein inactivation, protein and amino acid degradation, and inactivation of metabolic cofactors of DNA.188 Luminal perfusion of specific ROMs increased mucosal permeability, and serosal application caused increases in Cl− secretion in vitro.189 Tissue myeloperoxidase activity, an index of tissue granulocyte infiltration, is used experimentally to assess intestinal inflammation.190 Myeloperoxidase activity is elevated in acute flare-ups of human inflammatory bowel disease and various animal models of acute colitis.190, 191, 192 The acute inflammatory response in these conditions is characterized predominantly by neutrophils, although this assay measures total hemoprotein peroxidase, which includes monocyte and eosinophil peroxidase in addition to neutrophils.193 Moreover, levels of peroxidase activity in equine circulating eosinophils are greater than in circulating neutrophils,194 and this may apply to resident tissue eosinophils as well.
Arachidonic acid metabolites are thought to play a role in intestinal inflammation in diarrheal disease.170 Elevated levels of these intermediate metabolites have been demonstrated in natural disease and experimental models of colitis and appear to parallel increases in ROMs in inflamed intestine.195 The addition of H2O2 or HOCl to rat colonic tissue in Ussing chambers induces PGE2 release and active Cl− secretion.182, 196 Prostaglandins can stimulate increases in Cl− secretion in intact intestinal tissue181, 196, 197 and in isolated colonic T84 cells.180, 182 Interactions between ROMs and mesenchymal release of PGE2/PGI2 may be relevant to the mechanisms producing the diarrheic condition. Fibroblasts cocultured or juxtaposed to colonic T84 cells greatly increased the Cl− secretory response to H2O2 in vitro through the release of PGE2.180 In addition, equine colonic mucosa has an increased sensitivity to endogenously released prostaglandin by exhibiting a significant secretory response under in vitro conditions.198
Role of Other Factors
Endotoxin
Endotoxin, the LPS component of the outer cell wall of gram-negative bacteria, is present in large quantities in the large intestine of healthy horses.194, 198 The intact bowel forms an effective barrier to the transport of significant amounts of these highly antigenic toxins, but the diseased gut absorbs these macromolecules in large amounts, causing the subsequent adverse systemic effects that are often life-threatening.199 A complete review of endotoxemia is presented later in this chapter.
Endotoxins trigger mucosal immune cells and subsequent release of inflammatory mediators in cases of colitis. In vitro studies on the effects of endotoxin on intestinal water and electrolyte transport in adult male rats showed a significant decrease in net colonic sodium absorption and increased colonic permeability.200 In horses, endotoxin negatively affects gastrointestinal motility, including in the cecum and right ventral colon, and perfusion.201
Immunodeficiency
The importance of a normal immune system to the defense of the mucosal surface of the gastrointestinal tract is evident in the immunosuppressed state. Primary immunodeficiencies affecting the gastrointestinal tract are well documented. Common agammaglobulinemia is the most frequently reported gastrointestinal immunodeficiency and causes B-cell deficiency–associated giardiasis in other species.202 In horses, severe combined immunodeficiency can result in diarrhea secondary to adenovirus, coronavirus, and/or Cryptosporidium infection.203, 204 Interestingly, selective immunoglobulin A (IgA) deficiency rarely results in intestinal disease because of a speculated increase in mucosal IgM response. However, combined IgA and IgM deficiencies with a higher incidence of intestinal disease occur. A selective deficiency of secretory IgA has been associated with intestinal candidiasis in other species. Certain mucosal pathogens may enhance their pathogenicity by producing IgA proteases.202 Acquired immunodeficiency or immunosuppression in adults can result from infectious diseases (particularly viral), nutritional deficiencies, aging phenomena, and drugs (corticosteroids, azathioprine, and cyclophosphamide). Chronic salmonellosis was documented secondary to adult-onset B-cell deficiency in a Quarter Horse.205
Nutritional Deficiencies
Nutrition is a critical determinant of immunocompetence and risk of illness in many species.206 Impaired systemic and mucosal immunity contributes to an increased frequency and severity of intestinal infections observed in patients with undernourishment. Abnormalities occur in cell-mediated immunity, complement system, phagocytic function, mucosal secretory antibody response, and antibody affinity. Morbidity caused by diarrheal disease is increased, particularly among individuals with stunted growth rate because of malnourishment.207 The interaction between nutritional deficiency and intestinal health in the horse requires further investigation.
Intestinal Microbiota
Recently knowledge regarding the equine intestinal microbiota has expended dramatically. The intestinal microbiome is complex, and additional discussion is provided in Chapter 1 of this text. Briefly, fecal microbiota varies widely between healthy horses and those with colitis,208 and changes can precede episodes of colic in postpartum mares.209 The intestinal microbiota is affected by antimicrobial administration,210 transport, fasting and anesthesia,211 and dietary starch,212 among other factors; many of these have been associated with the development of colitis.
Factors Affecting Motility
Disturbances in motility patterns occur during inflammatory diseases of the colon, but the role of motility alterations in the pathogenesis of diarrhea remains unclear. Invasive bacteria cause characteristic motor patterns in the colon consisting of rapid bursts of motor activity that appear to decrease transit time through the large intestine. The result is reduced clearance of bacteria from the large intestine, which may contribute to the virulence of the organism.213 Absorption of endotoxin and the release of inflammatory mediators such as prostaglandins disrupts the motility patterns of the large intestine, resulting in less coordinated contractions, and may contribute to the alterations in motility seen with invasive bacteria. Although the effect of endotoxin and prostaglandins on transit time is not profound, the disruption of coordinated activity may play a role in causing diarrhea.214 Thorough mixing and prolonged retention time of ingesta are important not only in microbial digestion of nutrients but also in absorption of microbial byproducts and fluid.171 The ingesta is viscous and therefore must be mixed to bring luminal ingesta in contact with the mucosa for absorption. In addition, poor mixing increases the thickness of the unstirred layer, decreasing contact of ingesta with the mucosa and decreasing absorption.171
Progressive motility must be present, however, if a diarrheal state is to occur.171 Ileus may be accompanied by increased fluid in the lumen of the large intestine, but without progressive motility the fluid is not passed. Acute colitis is often associated with a period of ileus characterized by scant stool, which may be the reason for signs of colic in the early stages of colitis. Diarrhea is apparent only when motility returns. Increased progressive motility has been suggested to contribute to diarrhea by decreasing transit time; this is thought to play a role in irritant catharsis and in the mechanism of action of some laxatives.3
Endotoxemia
Endotoxemia is literally defined as the presence of endotoxin in the bloodstream. Most often, however, the term is used to refer to the associated clinical manifestations caused by an excessive and unbalanced inflammatory reaction. Endotoxemia is not a disease in its own right; rather, it is a complication of many septic and nonseptic disease processes affecting horses and other animals. Its diagnosis and management must therefore be discussed in the context of underlying primary pathologic conditions.
In its pathophysiologic consequences the innate immune response to endotoxin (LPS) is similar to the response to other stimuli (e.g., bacterial infection, viral infection, severe trauma). Moreover, the clinical presentation and clinical pathologic abnormalities of patients suffering from severe systemic inflammation are consistent regardless of the etiologic cause, and outcome may be predicted more accurately by the severity of the inflammatory response than the nature of the inciting insult.215 In 1992, therefore, the term systemic inflammatory response syndrome (SIRS) was introduced in a consensus statement of chest physicians and critical care physicians to account for these similarities and provide a case definition for clinical and research purposes. Fairly soon thereafter, however, the 1992 SIRS definition was criticized as being too sensitive while providing little specificity,216 and its usefulness for clinical practice was challenged because it failed to predict outcome and to discriminate patients at a high risk of morbidity and mortality.217 The definition of SIRS may therefore be valuable from a conceptual standpoint, but it has little value in daily clinical practice.
Sepsis was defined as the systemic inflammatory response to infection, and septic shock as “sepsis-induced hypotension, persisting despite adequate fluid resuscitation, along with the presence of hypoperfusion abnormalities or organ dysfunction.”215 According to these definitions the diagnosis of sepsis requires documentation of infection by culture in addition to two or more of the following findings: hypothermia or hyperthermia; tachycardia; tachypnea or hypocapnia; and leukocytosis, leukopenia, or an increased proportion of immature leukocyte forms.
Organ failure is a common sequela of endotoxic or septic shock, and the term multiple organ dysfunction syndrome describes insufficiency of two or more organ systems, as evident by clinical or clinicopathologic changes. In horses the laminae of the feet should be included in the list of organs susceptible to failure.
German scientist Richard Pfeiffer (1858–1945), in working with Vibrio cholerae, first described endotoxin as a toxin “closely attached to, and probably integral of, the bacterial body.”218 He observed this toxin to be distinct from the actively secreted, heat-labile, and proteinaceous bacterial exotoxins. Endotoxin later was found to be a heat-stable LPS structure, and the terms endotoxin and LPS now are often used interchangeably.
LPS is a major structural cell wall component of all gram-negative bacteria, including noninfectious species (Fig. 12.2 ). With 3 to 4 × 106 molecules per cell, LPS makes up about 75% of the outer layer of the outer cell membrane and is a key functional molecule for the bacterial outer membrane, serving as a permeability barrier against external noxious agents. The LPS molecule consists of four domains that are essential for the virulence of gram-negative bacteria.219 Three of the domains (inner core, outer core, and O-specific chain) represent the hydrophilic polysaccharide portion of the molecule, whereas the lipid A portion represents the hydrophobic lipid portion (Fig. 12.3 ). Combined, these domains confer the overall amphiphilic properties of the molecule that lead to the formation of micellar aggregates in aqueous solutions.
Fig. 12.2.
Lipopolysaccharide.
Fig. 12.3.
Three domains of the lipopolysaccharide molecule. FA, fatty acid.
O-specific chains (also called O-antigen polysaccharides or O-chains) are characteristic of any given type of LPS and show enormous structural variability among bacterial serotypes.220 O-chains are synthesized by the addition of preformed oligosaccharide blocks to a growing polymer chain and therefore have a repetitive structure. O-specific chains determine part of the immunospecificity of bacterial cells221 and, on interaction with the host immune system, serve as antigens for the production of species-specific antibodies.222 O-specific chains are further responsible for the smooth appearance of gram-negative bacterial colonies on culture plates,219 and LPS molecules containing an O-chain are termed smooth lipopolysaccharide.
The inner (lipid A-proximal) and outer (O-chain-proximal) core oligosaccharide portion is more conserved among different strains of gram-negative bacteria than the O-specific chain.220 The core of all LPS molecules contains the unusual sugar 3-deoxy-d-manno-oct-2-ulopyranosonic acid (KDO), which links the core region to the lipid A molecule. Synthesis of a minimal core is essential for the survival of bacteria,223 and the smallest naturally occurring LPS structure consists of lipid A and KDO.224 In contrast to the S-form colonies, colonies of gram-negative bacteria with LPS molecules that lack the O-specific chain but contain a core region show a rough appearance on culture plates. Rough LPS molecules are denoted further as Ra, Rb, and so on to indicate the length of the core region. In Re-LPS (also called deep rough LPS), the core region is reduced to a KDO residue. Re-mutants often are used to raise antibodies against the core region in an attempt to provide cross-protection against a variety of bacterial species. The lipid A portion, which serves to anchor the LPS molecule in the bacterial outer membrane, has been identified as the toxic principle of LPS,225 and its structure is highly conserved among gram-negative bacteria. The common structure shared by lipid A molecules is a 1,4′-bisphosphorylated β1,6-linked d-glucosamine disaccharide backbone (lipid A backbone), which is acylated by up to six fatty acids.220 Fig. 12.4 shows the acylation pattern for Escherichia coli LPS. Variation in the lipid A structure between gram-negative bacteria affects the number, length, and position of fatty acids and the backbone structure and the substitution of phosphate by other polar groups.222
Fig. 12.4.
Acylation pattern for Escherichia coli lipopolysaccharide.
Causes of Endotoxemia in Horses
According to its nature as a structural cell wall component, the presence of endotoxin implies the presence of gram-negative bacteria as a source. Depending on the nature of the underlying disease, these bacteria may circulate in the bloodstream in their intact form (i.e., bacteremia), may be confined to a localized infectious process, or may be part of the endogenous bacterial florae colonizing the gastrointestinal tract. In any of these scenarios endotoxin molecules are released as a byproduct of bacterial growth and in large numbers on bacterial cell death.226 Common infectious conditions associated with endotoxemia in horses include neonatal gram-negative sepsis, bacterial pneumonia and pleuropneumonia, endometritis, peritonitis, and infectious colitis with bacteria such as Salmonella spp. that are not part of the normal intestinal florae. In one study, for example, endotoxin was detectable in plasma of 50% of foals evaluated for presumed sepsis.227
The term translocation describes entry of endogenous bacteria and bacterial products from the gastrointestinal tract into tissues and the systemic circulation.228 The natural intestinal flora of horses consists mainly of gram-negative, anaerobic bacteria; thus, large amounts of endotoxin normally exist in the lumen of the equine intestinal tract.229 Even in healthy horse small amounts of endotoxin probably cross the intact mucosal barrier and reach the portal circulation and the liver.230 These molecules are cleared, however, by the mononuclear phagocytic system in the liver and lead only to a localized and restricted activation of the host immune system. For endotoxin translocation to become detrimental, excessive amounts have to cross the intestinal barrier and overwhelm the mononuclear phagocytic system, or the capacity of the liver to detoxify LPS must be compromised. The latter may be a concern in conditions such as hepatitis, cholangiohepatitis, or portosystemic shunting of blood.
Permeability of the intestinal mucosal barrier frequently increases in cases of acute gastrointestinal disease. Colic patients are prime candidates for developing endotoxemia, and plasma endotoxin was detectable in 10% to 40% of colic patients on admission.231, 232 The studies differed in their inclusion criteria, and the higher percentage of horses testing positive for plasma endotoxin was observed when only patients presenting for surgical intervention were evaluated.232 Aside from gastrointestinal rupture, increased permeability to intact bacteria or free endotoxin molecules is thought to be associated most commonly with ischemic insults such as strangulating obstruction and bowel infarction; severe inflammation, as in proximal enteritis and colitis; bacterial overgrowth; and intraluminal acidosis, which occurs with grain overload.233, 234 One study, however, found no difference in plasma endotoxin detection among disease groups, emphasizing the fact that any disease of the abdominal cavity can induce endotoxemia in horses. In the same study endotoxin was approximately three times more likely to be detected in PF as opposed to plasma samples. Similarly, higher cytokine concentrations have been measured in PF than in plasma. The likely explanation for these findings is a local inflammatory response in the peritoneal cavity elicited by translocated bacteria or LPS molecules before their absorption into the systemic circulation.231
Although gastrointestinal disease is likely the most important cause of endotoxin translocation in horses, other conditions may also result in translocation of endotoxin and bacteria. In experimental studies using laboratory animals, entry of gut-associated bacteria into the lymphatic system was demonstrated after hypovolemic shock, burn injuries, trauma, malnutrition, and starvation.235, 236, 237 Furthermore, endotoxin itself caused bacterial translocation into mesenteric lymph nodes after intraperitoneal administration to mice.238 These findings have received much attention in the literature concerning human patients because they serve to explain cases of endotoxic shock in the absence of demonstrable bacterial infection. The veterinarian should keep in mind the possibility of translocation when evaluating cases of presumed SIRS in horses, in which bacterial infection or gastrointestinal disease cannot be demonstrated. Endotoxin translocation also may be associated with strenuous exercise, which results in reduced splanchnic blood flow, hypoxemia, and a higher body temperature. In fit racehorses a significantly increased mean plasma LPS concentration was found after racing, whereas anti-LPS IgG levels were decreased. Fit horses showed significantly higher anti-LPS IgG concentrations at rest than sedentary controls, suggesting leakage of small amounts of endotoxin from the intestinal lumen during training and racing.239 A study in endurance horses demonstrated detectable LPS concentration in approximately 50% of horses, and correlation between plasma endotoxin and lactate concentration suggested that plasma endotoxin concentration may be reflective of the vigor of exercise.240 Surprisingly, the latter study failed to show a correlation between plasma endotoxin concentration and levels of inflammatory mediators such as TNF-α and IL-6,240 such that the clinical significance of endotoxin translocation during exercise requires further investigation. At least experimentally, however, alterations in innate immune function during and after strenuous exercise in horses have been demonstrated.241
Mechanisms of Cellular Activation by Lipopolysaccharide
The initiating event in the pathophysiology of endotoxemia is the activation of LPS-responsive cells by endotoxin, resulting in altered cellular functions and increased expression of inflammatory mediators. Immune cells such as macrophages respond to minute amounts of LPS, which usually allows them to eliminate gram-negative bacteria and free LPS molecules efficiently. An important factor in the exquisite sensitivity to LPS is the presence of LPS-binding protein (LBP).220 LBP is an approximately 60-kDa plasma glycoprotein242 synthesized primarily by hepatocytes243 and belongs to the family of lipid transfer/LBPs. LBP is an acute phase protein, and inflammatory agents and cytokines such as IL-1 increase LBP plasma concentration 10- to 100-fold within 24 to 48 hours of an inflammatory stimulus.244, 245 The main function of LBP is to transfer LPS to endotoxin-responsive cells, which include mononuclear phagocytes, neutrophils, lymphocytes, and endothelial cells. The importance of a highly sensitive response to LPS for protection against gram-negative bacterial infection is demonstrated in experiments using LBP knockout mice—that is, mice that lack the LBP gene and are therefore unable to synthesize LBP. Although these animals are resistant to the effects of purified LPS, they are unable to control infection with viable bacteria and rapidly succumb.246 Despite its crucial importance for an effective host defense, LBP is not essential for LPS-receptor interaction per se, because high concentrations of LPS can activate cells in the absence of LBP.247
Aside from its role as a catalyst of cellular activation by LPS, LBP has opsonizing activity248 and participates in the phagocytosis of LPS by macrophages and neutrophils.249, 250 Although phagocytosis of LPS is receptor dependent, it appears to be uncoupled from intracellular signaling events and occurs in the absence of cell activation.251 LBP further catalyzes transfer of LPS to lipoproteins, such as high-density lipoprotein, which neutralizes LPS activity.252 This detoxifying effect may become important when large amounts of LPS are present. A protective effect of LBP against LPS challenge and infection has been demonstrated in a murine model of septic shock,253 and further studies investigating potential therapeutic uses of LBP are under way. More recently, LBP has also been investigated as a diagnostic and prognostic indicator in human patients, in which it may differentiate between SIRS and sepsis and predict outcome of septic patients.245 One study investigating serum amyloid A (SAA) and LBP in horses with colic254 found no correlation between serum concentration of LBP with outcome, type of disease process, or affected portion of the gastrointestinal tract.
The most important LPS receptors are cluster differentiation antigen 14 (CD14)247 and TLR-4.255 Both are classified as pattern-recognition receptors,256 which means that they recognize LPS as a structural motif common to all gram-negative bacteria. CD14 is a 53-kDa protein that in its membrane-bound form (mCD14) is inserted into the cell membrane via a glycosyl–phosphatidyl–inositol anchor.257 CD14 is expressed primarily on monocytes and tissue macrophages and to a lesser extent on neutrophils.258 It also is found in a soluble form (sCD14)259 that can bind to cell types lacking CD14, such as endothelial cells, and make them LPS responsive. In addition to this proinflammatory effect, high concentrations of sCD14 can sequester and neutralize LPS.260 The amount of circulating sCD14 greatly increases during inflammation, which makes it a useful marker of acute and chronic inflammation.258
Although CD14 is known to be crucial for cellular activation, it cannot transmit signals to the inside of the cell because it lacks a transmembrane domain. The missing link between CD14 and the cytosolic environment is a Toll-like receptor in association with the molecule MD-2.261 The name Toll-like receptor stems from the homology of the mammalian receptor with a receptor type in Drosophila (Toll) that is important for dorsoventral orientation and immune responses in the fly. A number of Toll-like receptors have been identified in mammalian species thus far, but TLR4 appears to be the receptor subtype most important for LPS signaling.255 The importance of CD14 and TLR4 in the cellular response to LPS has been demonstrated in a number of experiments. Mice deficient in CD14 are incapable of mounting a normal inflammatory response to LPS,260 whereas mutation or deletion of the gene encoding for TLR4 causes LPS hyporesponsiveness.262, 263, 264
After binding of LPS to cellular receptors, TLR4 undergoes oligomerization and recruits downstream adaptor proteins to activate intracellular signaling pathways.265 Signaling pathways are characterized by sequential phosphorylation and activation of enzymatic activities and ultimately result in the alterations of cellular metabolism known as cell activation. A typical result of intracellular signaling is the activation of transcription factors—that is, proteins that bind to DNA and promote gene transcription. Translational mechanisms are activated in a similar manner.
TLR4-dependent cell signaling pathways can be differentiated according to the use of adaptor proteins that bind to the intracellular domain of TLR4.265 The myeloid differentiation primary response gene 88 (MyD88)-dependent pathway results in activation of IKK (Iκβ kinase) and mitogen-activated protein kinase (MAPK) pathways and ultimately in the expression of proinflammatory cytokine genes. IKK activates the well-described transcription factor NF-κβ by phosphorylating an inhibitor protein complex (IκB) that sequesters and inactivates NF-κβ in the cytoplasm. On phosphorylation IκB is ubiquitinated and degraded, and NF-κβ is translocated to the nucleus, in which it unfolds its activity.266 The MyD88-independent pathway, on the other hand, is activated by interaction of TLR4 with the adaptor protein Toll-IL-1 receptor domain-containing adaptor inducing interferon-β (TRIF). Although the MyD88-independent pathway also activates MAPK and NF-κβ (in addition to a transcription factor called IRF3), this pathway primarily results in activation of type I interferons, which are important for antiviral and antibacterial responses.265 Despite the characterization of seemingly separate and “ordered” pathways, one should recognize that interaction and synergy between pathways are likely to occur. Similarly, inhibitory pathways are required for regulation of the cell response and can target multiple levels of TLR4 signaling.265 The potential manipulation of signaling pathways for therapeutic use in septic patients is under investigation.
Inflammatory Mediators
Although endotoxin can exert some direct effects, cytokines are a primary mediator of LPS effects. Cytokines are glycoprotein molecules that regulate inflammatory and immune responses by acting as a signal between cells.267 Cytokines of major interest in the pathogenesis of endotoxemia include TNF-α, the interleukins, chemokines, and growth factors such as granulocyte-monocyte colony-stimulating factor. TNF-α is thought of as the most “proximal” cytokine released in response to LPS. Studies corroborate this by showing that administration of recombinant TNF-α mimics the effects of LPS268 and that antibodies directed against TNF-α protect against the lethal effects of endotoxin.269 Increased plasma activity of TNF is associated with increased mortality in equine patients with acute gastrointestinal disease and in septic neonates.231 Despite being a structurally diverse group of proteins, cytokines share several characteristics that allow them to execute their complex functions in the inflammatory response.267 Any individual cytokine generally is produced by several different cell types, can act on different cell types, and has multiple effects on any given cell. Furthermore, cytokine effects are redundant, meaning that different cytokines can share the same effect. In endotoxemia this is particularly true for the effects of IL-1 and TNF-α.270 Many of the biologic activities of cytokines in vivo result from synergistic or antagonistic actions involving two or more cytokines.271 Within itself the cytokine response is highly regulated: cytokines induce or suppress synthesis of other cytokines, including their own (feedback regulation); regulate expression of cytokine receptors; and regulate cytokine activities. Additional regulatory mechanisms include the release of specific cytokine inhibitors such as soluble IL-1 and TNF-α receptors; cytokine receptor antagonists such as the IL-1 receptor antagonist; and antiinflammatory cytokines, including IL-10, IL-4, IL-13, and transforming growth factor-β (TGF-β). Glucocorticoids, which are produced increasingly in response to endotoxin, also inhibit the production of cytokines.272 During a “controlled” inflammatory response, therefore, cytokine secretion is a self-limited event, whereas excessive stimulation of cytokine production can lead to the perpetuation of the inflammatory response even after the initial stimulus has been removed. Aberrations from the controlled, self-regulated inflammatory response have been described as predominantly proinflammatory (SIRS), antiinflammatory or hypoinflammatory (compensatory antiinflammatory response syndrome [CARS]), or combined (mixed antagonist response syndrome) responses.273 That these responses may not represent a continuum in response to infection was demonstrated in a recent study investigating cytokine profiles in an experimental model of sepsis.274 Here, increased plasma concentration of antiinflammatory mediators such as IL-1 receptor antagonist and IL-10 in the early phase of sepsis predicted early mortality almost as accurately as the typical proinflammatory cytokines such as IL-6. Cytokine profiles failed to predict mortality in the later stages of sepsis, thereby suggesting that late outcome is not “preprogrammed” early on in the disease. A progression from proinflammatory (SIRS) to antiinflammatory (CARS), as previously proposed, could not be demonstrated.274 The authors concluded that use of biomarkers may be helpful in determining the inflammatory status of clinical patients suffering from sepsis and that the success of treatments aimed at suppressing or stimulating immune responses may depend on the individual patient’s inflammatory profile.
Interestingly, tolerance to endotoxin develops after repeated exposure to LPS.275 Tolerance can be demonstrated in vitro and in vivo and encompasses decreased production of cytokines and a diminished clinical response.275, 276 Tolerance may be a protective mechanism and was shown to reduce mortality in some experimental models; however, impaired resistance to infectious processes was demonstrated by other investigators.277 Mechanisms that likely are responsible for the development of endotoxin tolerance include receptor downregulation,278 downregulation of intracellular signaling pathways,277, 279 and mediators such as glucocorticoids and IL-10.277 The development of endotoxin tolerance in horses has been reported.280, 281 More recently, the term cellular reprogramming has been introduced to describe altered inflammatory cell functions in septic and SIRS patients. This term better accounts for the observation that cellular LPS-sensing ability appears to be maintained, whereas intracellular signaling and cytokine production are modified toward an antiinflammatory rather than a proinflammatory response.277
Aside from cytokines, a number of other molecules function as inflammatory mediators in the pathogenesis of endotoxemia, the synthesis and release of which are stimulated by endotoxin and by cytokines. These mediators include the arachidonic acid metabolites or prostanoids, PAF, oxygen-derived free radicals, nitric oxide, histamine, kinins, and complement components. Table 12.2 summarizes the origins, targets, and effects of the most important inflammatory mediators involved in the pathogenesis of endotoxemia. Fig. 12.5 shows the pathways of arachidonic acid metabolism by COX and lipoxygenase. COX products are the prostaglandins, prostacyclin (PGI2) and thromboxanes, and the lipoxygenase produces the leukotrienes.
TABLE 12.2.
Important Mediators of the Systemic Inflammatory Response to Endotoxin
| Mediator | Origin | Effects |
|---|---|---|
| TNF | Macrophages | Induces synthesis of TNF, IL-1, IL-6, and GM-CSF |
| Monocytes | Activates neutrophils | |
| Neutrophils | Activates fibrinolysis and coagulation | |
| CD4+ T cells | Activates contact and complement system | |
| Natural killer cells | Induces a catabolic state | |
| Induces insulin resistance | ||
| Is a pyrogen (direct action and via IL-1 induction) | ||
| Induces synthesis of TNF, IL-1, IL-6, PGI2, PAF, and GM-CSF | ||
| IL-1 | Activated macrophages | Activates pyrogen |
| Endothelial cells | Induces malaise | |
| Fibroblasts | Activates neutrophils and chemotaxis | |
| Dendritic cells | Activates fibrinolysis and coagulation | |
| Lymphocytes | Activates contact and complement system | |
| Keratinocytes | Induces acute phase response | |
| Increases activity of lipoprotein lipase | ||
| Mobilizes amino acids | ||
| Induces muscle proteolysis | ||
| IL-6 | Activated macrophages | Induces acute phase response |
| Fibroblasts | Induces stress response | |
| Keratinocytes | Is a weak pyrogen | |
| T lymphocytes | ||
| IL-8 | Macrophages | Activates neutrophils and chemotaxis |
| Endothelial cells | ||
| Thromboxane A2 | Platelets | Induces vasoconstriction |
| Activates platelet aggregation | ||
| PGE2 | Most nucleated cells | Induces vasodilation |
| Activates platelet aggregation | ||
| Induces fever | ||
| PGI2 | Vascular endothelial cells | Induces vasodilation |
| Inhibits platelet aggregation | ||
| PAF | Macrophages | Activates platelet aggregation |
| Platelets | Activates macrophages and neutrophils | |
| Neutrophils | Induces hypotension | |
| Mast cells | Increases vascular permeability | |
| Eosinophils | Aids recruitment of leukocytes | |
| Induces visceral smooth muscle contraction | ||
| Is a negative inotrope and arrhythmogenic | ||
| Induces ileus | ||
| PGF2α | Most nucleated cells | Induces vasoconstriction |
| Activates luteolysis | ||
| Leukotriene B4 | Most nucleated cells | Is a chemoattractant |
| Promotes neutrophil interaction with endothelial cells | ||
| Leukotrienes C4, D4, E4 | Most nucleated cells | Increase vascular permeability |
| Induce bronchoconstriction | ||
| Induce vasoconstriction | ||
| Kinins | Produced from serum precursors | Increase vascular permeability |
| Induce smooth muscle contraction cause pain | ||
| Complement components (C3a, C5a) | Activate neutrophils and chemotaxis | |
| Induce smooth muscle constriction | ||
| Induce mast cell degranulation | ||
| Induce release of histamine and serotonin | ||
| Increase vascular permeability | ||
| Oxygen-derived free radicals | Macrophages | Damage cell membranes |
| Neutrophils | Inactivate enzymes | |
| Damage tissues | ||
| GM-CSF | Induces rebound neutrophilia |
GM-CSF, Granulocyte-monocyte colony-stimulating factor; IL, interleukin; PAF, platelet-activating factor; PG, prostaglandin; TNF, tumor necrosis factor.
Fig. 12.5.
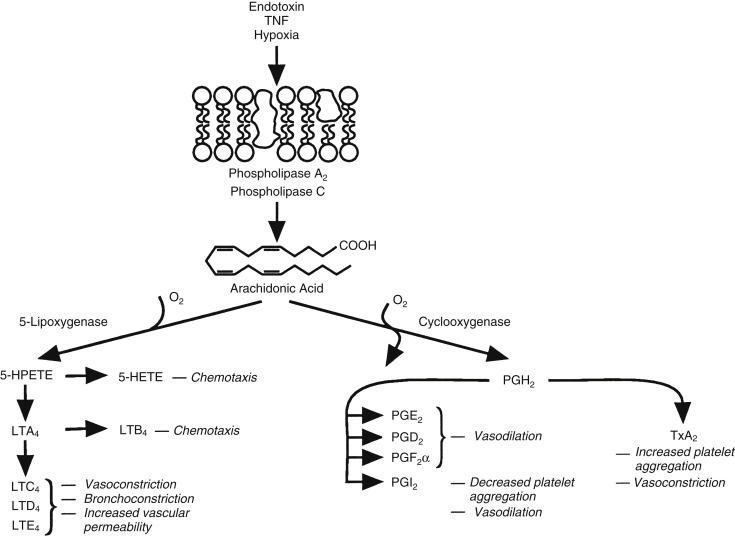
Pathways of arachidonic acid metabolism by cyclooxygenase and lipoxygenase.
Pathogenesis
The innate immune response to LPS is an efficient defense mechanism that provides maintenance of homeostasis and therefore health in the face of an almost continuous exposure to microorganisms and their products.282 Detrimental consequences of this immune response occur only if excessive and uncontrolled mediator output results in endothelial damage, neutrophil-mediated tissue damage, and uncontrolled activation of the coagulation and fibrinolytic cascades and the complement system. Ultimately, the combination of these events culminates in cardiovascular instability, impaired hemostasis, organ failure, shock, and death. The following discussion addresses the various pathophysiologic events in the development of endotoxemia and shock and the role of inflammatory mediators.
Endothelial Dysfunction and Damage
Normal endothelium plays an important role in regulating blood pressure and regional tissue perfusion and provides an anticoagulant surface. Endothelial dysfunction and damage result in a decreased responsiveness to vasoactive agents (vasoplegia), increased vascular permeability, and a tendency for clot formation in the microvasculature. If the basement membrane and underlying matrix are compromised, microvascular hemorrhage can occur. Endothelial cell damage is primarily neutrophil mediated. More specifically, damage is caused by oxygen-derived free radicals, which are produced within endothelial cells through reactions involving neutrophil-derived elastase and hydrogen peroxide molecules, endothelial cell enzymes such as xanthine oxidase, and endothelial cytosolic iron. The hypochloric anion radical (HO˙) is thought to be responsible most directly for endothelial cell cytotoxicity. Nitric oxide, which is produced by constitutively expressed nitric oxide synthase in endothelial cells and scavenges superoxide radicals in a reaction to form peroxynitrite, may afford protection from oxygen radical–induced endothelial cell damage. Variations in the ability to produce nitric oxide may explain why vascular beds in different organs vary in their susceptibility to neutrophil-mediated damage.283 Excessive production of nitric oxide by an inducible form of nitric oxide synthase (iNOS), however, contributes to tissue damage, and increased peroxynitrite concentrations may be responsible in part for PAF-induced increases in vascular permeability.284 In addition to oxygen-derived free radicals, activated neutrophils release matrix metalloproteinases, which contribute to tissue damage.272 Vascular endothelial cells are further susceptible to direct effects of various cytokines, most prominently TNF-α and IL-1. These cytokines are thought to cause damage via the induction of COX activity and production of prostanoids and through generation of free radicals. Endothelial cell damage in response to endotoxin infusion and the association with leukocyte attachment have been demonstrated in horses.285
Neutrophil Activation, Margination, and Transmigration
Neutrophil activation by LPS and cytokines results in stimulation of phagocytosis and the respiratory burst, release of lysosomal enzymes and inflammatory mediators, and expression of adhesion molecules. Perhaps the single most specific clinicopathologic indicator of endotoxemia is pronounced neutropenia,286 which temporally correlates with peak plasma concentrations of TNF.287 Neutropenia is caused primarily by margination of neutrophils in the vasculature, especially of the lungs,288 whereas significant loss through active migration into peripheral tissues likely is limited to the presence of a localized source of infection. In fact, neutrophils exposed to endotoxin or inflammatory mediators exhibit reduced capacity to respond to chemotactic stimuli and extravasate.288 Margination is made possible by adhesion molecules on endothelial cells and leukocytes that interact and allow sticking of leukocytes to the endothelial lining of blood vessels. The details of neutrophil margination and transmigration are reviewed in several excellent texts.289, 290 Recent evidence demonstrates a role for TLR-4 in neutrophil margination in the lung microvasculature during endotoxemia, and suggests that activation and TLR-4 expression of endothelial cells may be more important than that of circulating neutrophils.288 Although neutrophil activation during infections provides the body with an effective defense system against microbial invaders, margination and reduced migratory ability of neutrophils during endotoxemia results in the accumulation of activated cells at the endothelial surface. These cells are then positioned to effect endothelial injury, increase vascular permeability, and in some cases cause parenchymal cell death and organ dysfunction.291 In addition, inhibition of transmigration deprives the body of phagocytic cells at sites of infection, resulting in reduced ability to fight bacterial infection. This latter complication of endotoxemia may have significant clinical impact, because defective neutrophil recruitment has been demonstrated at low endotoxin doses that do not yet result in tissue damage.291, 292 In human patients endotoxemia and impaired neutrophil migration have been associated with infectious complications of surgical procedures.291 Decreased phagocytic function and oxidative burst activity have also been suggested in sick and septic hospitalized foals,293, 294 in which a beneficial effect of plasma transfusion on neutrophil function could be demonstrated. The mechanisms of migration inhibition during endotoxemia are incompletely understood but may include occupation of neutrophil chemotactic receptors by cytokines and complement components, resulting in an inability of activated cells to respond to a chemotactic gradient.291 Rebound neutrophilia, which is observed frequently following episodes of endotoxemia, is caused by neutrophil release from the bone marrow reserve pool and by stimulation of myeloid cell proliferation via granulocyte-macrophage colony-stimulating factor and is mediated primarily by TNF and IL-1.270
Coagulopathy and Disseminated Intravascular Coagulation
In health coagulation and fibrinolysis underlie stringent control mechanisms that allow appropriate clot formation and their resolution. Coagulopathies frequently are observed in horses with colic233, 295, 296 and foals with sepsis227 and are likely attributable to endotoxemia. In human medicine virtually all septic patients are considered to have some degree of coagulopathy, which may range from subclinical abnormalities of the clotting profile to fulminant disseminated intravascular coagulation (DIC).297, 298 DIC results from a widespread activation of the coagulation and fibrinolytic systems and failure of their control mechanisms. Ultimately, this leads to disseminated fibrin deposition in the microvasculature, consumption of platelets and clotting factors, and accumulation of fibrin degradation products (FDPs). Depending on the underlying disease process and the relative impairment of the coagulation and fibrinolytic systems, DIC can manifest as a diffuse thrombotic syndrome leading to ischemic organ failure, a fibrinolytic syndrome with uncontrolled hemorrhage, or a combination of both.299 A procoagulant state characterized by clinicopathologic abnormalities of the clotting profile precedes DIC.
The intrinsic and extrinsic arms of the coagulation cascade are activated in endotoxemia. The intrinsic pathway is initiated by activation of coagulation factor XII (HF), prekallikrein, and high-molecular-weight kininogen, which compose the contact system.300 Although direct activation of coagulation factor XII by endotoxin has been demonstrated,301 the extrinsic pathway likely is more important for the development of coagulopathy in endotoxemia and sepsis.300 Activation of the extrinsic pathway depends on the interaction of coagulation factor VII with tissue factor, which is the only coagulation factor not constitutively present in blood. Tissue factor is present in subendothelial tissues and is exposed on vascular injury but also is expressed on endothelial cells and mononuclear phagocytes in response to LPS.302, 303 Increased expression of monocyte tissue factor (also described as increased procoagulant activity) was significantly associated with coagulopathy and poor prognosis in horses with colic.304 Furthermore, LPS-induced tissue factor expression by equine peritoneal macrophages may be associated with the development of intraabdominal adhesions.280
Regulatory mechanisms of the coagulation cascade include tissue factor pathway inhibitor, antithrombin III (AT III) and the protein C system.300 Protein C acts as an anticoagulant by inactivating clotting factors V and VIII and promotes fibrinolysis by inactivating plasminogen activator inhibitor (PAI).305 Protein C activation by thrombin-thrombomodulin complexes is important for the anticoagulative properties of normal endothelium,300 and downregulation of endothelial thrombomodulin expression by TNF and IL-1 along with decreased expression of AT III and tissue factor pathway inhibitor by damaged endothelial cells contribute to the procoagulant state in endotoxemia and sepsis.306, 307, 308 In addition, activation of vascular endothelial cells leads to a loss of prostacyclin and nitric oxide production and an increased release of thromboxane A2 (TXA2). As a result, platelets are stimulated to aggregate and release TXA2 and PAF, further promoting clot formation.234
The crucial step in the fibrinolytic cascade is the conversion of plasminogen to plasmin, a fibrin-degrading enzyme.300 Tissue-type (tPA) and urokinase-type (uPA) plasminogen activator are the major initiators of fibrinolysis, whereas PAI and α2-antiplasmin are the main regulatory components.309, 310 TNF and IL-1 have been shown to induce the release of uPA and tPA and the synthesis of PAI.300 Activation of fibrinolysis leads to consumption of α2-antiplasmin and accumulation of FDPs, which if present in high concentrations can interfere with platelet aggregation, fibrin polymerization, and thrombin formation and can promote bleeding. Additionally, FDPs mediate an increase in vascular permeability. LPS infusion in rabbits311 and humans312 resulted in an early increase in plasma tPA activity, followed by a later profound rise in PAI activity and fall in tPA activity. Increased plasma PAI concentrations also were found in horses with colic compared with controls.313, 314 Although fibrinolysis may compensate initially for accelerated coagulation, its subsequent inhibition contributes to clot formation.
Cross-activation between the inflammatory and coagulation cascades plays an important role in endotoxemia and sepsis, and the presence of coagulopathies has been associated with an increased risk of organ failure and poor outcome in septic human patients.297, 298 Activated protein C reduces mortality in septic human patients.315 Activated protein C reduces inflammation by inhibiting leukocyte activation and cytokine production and was shown to lower plasma concentration of IL-6.315 Although bleeding complications were increased in treated patients, incidence of severe bleeding was not increased significantly.
Complement Activation
Activation of the complement system in endotoxemia occurs via the alternative pathway through interaction with LPS. Increased concentrations of plasmin and kallikrein (caused by activation of the fibrinolytic and contact system) further promote this pathway by directly activating complement factors C3a and C5a. Aside from being key molecules in the complement cascade, C3a and C5a are anaphylatoxins and cause an increase in vascular permeability via mast cell degranulation. C5a further activates the lipoxygenase pathway in neutrophils and monocytes, acts as a chemotaxin for leukocytes and monocytes, and promotes neutrophil adhesion to endothelial cells.
Acute Phase Response
In response to acute inflammation, synthesis and secretion of a number of proteins called the acute phase proteins increase in hepatocytes, whereas synthesis of albumin decreases. The primary function of this acute phase response may be to suppress and contain inflammatory processes.272 IL-6 and IL-1 are the most important cytokines that induce the acute phase response,316 which typically begins within a few hours of the insult and subsides within 24 to 48 hours,317 unless the initiating cause persists. In horses fibrinogen (the most commonly evaluated acute phase protein), haptoglobin, transferrin, ferritin, ceruloplasmin, coagulation factor VIII:C, SAA, C-reactive protein, α1-acid glycoprotein, and phospholipase A2 are considered part of the acute phase response.318 SAA is a sensitive indicator of experimentally induced inflammation319 and may be helpful in the diagnosis of inflammatory gastrointestinal disease,254 neonatal weakness and diarrhea,320 and various infectious diseases. A commercially available turbidimetric immunoassay for human SAA is reliable for measuring equine SAA.321
The effect of acute inflammation on the serum concentration of several coagulation factors must be considered when evaluating coagulation profiles. Serum fibrinogen concentration is determined primarily by the acute phase response, although fibrinogen is consumed increasingly on activation of the clotting cascade.
Hemodynamic Changes, Development of Shock, and Organ Failure
Shock is characterized by a loss of homeostasis attributable to the breakdown of hemodynamic control mechanisms, decreases in cardiac output and the effective circulating volume, and inadequate perfusion of vital organs. Shock caused by endotoxemia is classified as distributive shock322 and is largely initiated by vascular dysfunction in the periphery. Peripheral vascular beds are of major importance for the regulation of local tissue perfusion and affect systemic blood pressure by regulating total peripheral resistance. Normally, vascular smooth muscle tone is regulated by endothelin-1 (vasoconstriction), nitric oxide, and prostacyclin (vasodilation) released from vascular endothelial cells.323 Detrimental effects of nitric oxide are attributable to overproduction of nitric oxide by iNOS in macrophages and other cell types, rather than endothelial-derived nitric oxide. Peripheral vasomotor effects of endotoxin manifest as vasodilation and vasoplegia and are mediated by PGI2, nitric oxide, and mediators such as bradykinin. Widespread vasodilation leads to vascular blood pooling, decentralization of blood flow, decreased venous return, and in effect decreased effective circulating volume and cardiac output.322 Compensatory responses in the form of an initial hyperdynamic phase include tachycardia, increased cardiac output and central venous pressure, pulmonary hypertension, peripheral vasoconstriction, and increased peripheral vascular resistance.322, 324, 325
The early vasoconstrictive phase corresponds to an increased serum concentration of TXA2,234 but additional vasoconstrictors such as arginine vasopressin, angiotensin II, serotonin, endothelin, and norepinephrine likely are implicated in the pathogenesis of shock and organ failure.272 With progression of disease the animal enters a stage of decompensated shock and progressive systemic hypotension, which corresponds to increased plasma concentrations of prostacyclin, PGE2, and bradykinin.234, 272 Inadequate blood flow and oxygen delivery to tissues caused by hypotension are confounded by direct myocardial suppression via nitric oxide,322 increased vascular permeability,234 intravascular microthrombosis, and impaired tissue oxygen extraction322 and results in progressive metabolic acidosis and inhibition of normal cellular metabolism.
Clinical Signs and Diagnosis
Quantification of endotoxin in plasma samples is possible. The Limulus amebocyte lysate assay is an activity assay based on the endotoxin-sensitive hemolymph coagulation cascade in the horseshoe crab Limulus polyphemus. In Limulus, this reaction is thought to be a defense mechanism against gram-negative infection.326 Although frequently used as a research tool, the assay is not convenient enough to become a routine clinical test. The clinician therefore must appreciate the primary disease processes associated with a high risk of endotoxemia and rely on clinical signs and clinicopathologic data to achieve a diagnosis. In a survey of board-certified internal medicine specialists and surgeons, colitis/enteritis, small intestinal strangulation and obstruction, retained fetal membranes/metritis, grain overload, and pleuropneumonia were among the most frequently cited conditions associated with endotoxemia.327 In some cases, endotoxemia may be the first indication of disease or may be the most overt of otherwise subtle clinical manifestations. With colitis or proximal enteritis, for example, one may detect signs of endotoxemia before the development of colic, diarrhea, or gastric reflux, which more specifically indicate the nature of the primary illness. The presence of neutropenia should always prompt the clinician to investigate causes of endotoxemia.
In vivo LPS challenge experiments in horses clearly show that many of the clinical signs associated with acute gastrointestinal disease and sepsis are attributable to endotoxemia. On administration of sublethal doses of LPS, the clinical response can be divided into the early hyperdynamic and the later hypodynamic or shock phases. Clinical signs during the first phase, which begins within 15 to 45 minutes after LPS administration, include anorexia, yawning, sweating, depression, evidence of abdominal discomfort, muscle fasciculations, and recumbency. Heart and respiratory rates increase, and decreased borborygmi suggest ileus. Hyperemia of the mucous membranes and an accelerated capillary refill time indicate the hyperdynamic state.286 If large amounts of LPS are administered or if exposure is ongoing, depression worsens progressively, anorexia persists, and feces develop a diarrheic character. Signs of colic typically abate after the initial stage. Fever develops as a result of direct action of TNF on the thermoregulatory center and IL-1–induced local production of PGE2 in or near the hypothalamus.328, 329 Because of compromised peripheral perfusion, mucous membrane color changes to brick red or purple, a dark “toxic” line appears, and capillary refill time is prolonged.286 Inadequate peripheral perfusion and compromised organ function finally characterize the hypodynamic shock phase. Body temperature may become subnormal, and the skin, especially on extremities, is cool to the touch. The arterial pulse weakens, and venous fill is decreased. Vascular endothelial damage and increased capillary permeability result in a muddy mucous membrane color and diffuse scleral reddening. Similar changes are evident in horses suffering from endotoxemia associated with natural disease. In the previously mentioned survey,327 tachycardia, fever, abnormal mucous membrane color, and increased capillary refill time were named by most specialists as indicating endotoxemia.
Hemostatic abnormalities can manifest in the form of thrombosis or increased bleeding tendency with mucosal petechiation or ecchymoses and prolonged bleeding from venipuncture sites.299 Bleeding also may occur in the form of spontaneous epistaxis or prolonged hemorrhage after nasogastric intubation.234 Additional clinical signs typically reflect the development of organ failure. Renal failure and laminitis327 appear to be common complications of endotoxemia in horses, and endotoxemia was identified as the only clinical condition significantly associated with the development of acute laminitis in one retrospective case-control study of horses admitted to a referral center.330 Other potential complications include liver failure,299 respiratory failure, colic and ischemia-induced gastrointestinal ulceration,234 cardiac failure, and abortion in pregnant mares.331, 332 Renal failure results from ischemic cortical necrosis and acute tubular necrosis caused by coagulopathy-induced afferent arteriolar obstruction. Clinical signs may include oliguria, anuria, or hematuria caused by renal infarction. Laminitis may lead to lameness, increased digital arterial pulsation, increased warmth of the hoof wall, and sensitivity to hoof tester pressure. The exact nature of the association between endotoxemia and laminitis is not understood, and, interestingly, experimental endotoxin infusion does not reliably induce laminitis. Studies have shown, however, that endotoxin administration decreases digital blood flow and laminar perfusion333 coincident with increased plasma concentrations of 5-HT and thromboxane B2 (TXB2).334 In addition, in vitro vascular reactivity of digital vessels is altered following sublethal endotoxin administration to horses.335 In addition to the response to circulating mediators, LPS exposure also alters the production of vasoactive mediators by digital vascular endothelial cells.336 Alterations in vascular reactivity are, therefore, likely responsible for development of laminitis in endotoxemia; however, other mechanisms may apply depending on the underlying clinical disease.
Clinical Pathologic Testing
Leukopenia caused by neutropenia may be the most specific indicator of acute bacterial sepsis or endotoxemia.286 In prolonged cases an increased proportion of immature neutrophil forms (bands) and toxic changes are observed. Toxic changes resulting from neutrophil activation include vacuolation, cytoplasmic granulation, basophilic cytoplasm, and Döhle bodies. Because neutropenia occurs early in the development of endotoxemia, it also may be a useful parameter for monitoring horses at risk.286 On recovery neutropenia typically is followed by a pronounced rebound neutrophilia. Other alterations in the hemogram and serum biochemical profile mainly reflect the underlying disease process and the occurrence of organ failure.
An elevated hematocrit and total serum protein concentration are frequently interpreted as evidence of dehydration; however, splenic contraction caused by increased sympathetic stimulation, increased production of acute phase proteins, or protein losses also influence these parameters. Hyperproteinemia may be observed as a result of increases in the fibrinogen or globulin concentration, and determination of protein fractions, including protein electrophoresis, is indicated in hyperproteinemic patients. Hypoproteinemia and hypoalbuminemia can occur because of loss via the gastrointestinal or urinary tract or with pleural or peritoneal cavity effusion. Increased vascular permeability and edema formation contribute to hypoproteinemia.
Serum electrolyte abnormalities primarily depend on the nature and duration of underlying disease processes and need to be evaluated individually. In human patients, gram-negative sepsis frequently is associated with hypocalcemia, more specifically a decrease in serum ionized calcium concentration. Endotoxin is thought to be a causative factor, and proposed mechanisms include acquired parathyroid gland insufficiency, dietary vitamin D deficiency, impaired calcium mobilization, and renal 1-hydroxylase insufficiency, leading to decreased 1,25-hydroxylation of vitamin D. Hypocalcemia in septic human patients was associated with hypotension and poor outcome.337 In horses with surgically managed gastrointestinal disease, decreased serum ionized calcium concentration was a common finding and was most severe in patients with strangulating or nonstrangulating infarctions. In some horses, ionized calcium concentration decreased further throughout surgery. Treatment with calcium gluconate resulted in normalization of serum ionized calcium concentrations in all cases.338
Septic neonatal patients are frequently hypoglycemic, which may be attributable to decreased oral intake, generally increased metabolism, glucose use by the infecting bacteria, inhibition of gluconeogenesis by endotoxin, and insulin-like activity produced by macrophages.234 Interestingly, experimental endotoxin administration results in transient hyperglycemia in adult horses,324 whereas profound hypoglycemia occurs in foals.339 Because of the high incidence of coagulopathies in endotoxemic and septic patients, clinicians should consider monitoring coagulation parameters. The most significant changes can be expected with severe inflammatory disease such as colitis,295, 296 devitalized intestine as with strangulating obstruction,296, 340 and with increased duration of disease. In 30 horses with acute gastrointestinal disease, coagulation profiles were considered normal in only two horses.295 Although coagulation times may be shortened during the procoagulant state, commonly observed abnormalities with developing DIC include an increased concentration of FDPs and soluble fibrin monomer, prolonged prothrombin time indicative of factor VII consumption, prolonged activated partial thromboplastin time indicative of factor VIII:C and IX consumption, prolonged thrombin time, decreased AT III activity, thrombocytopenia, and decreased protein C and plasminogen activities. Fibrinogen concentration appears to reflect the acute phase response rather than coagulation abnormalities in horses and is frequently increased.304 Some clinicians make a diagnosis of DIC if three or more coagulation parameters (specifically AT III, FDPs, platelet count, prothrombin time, and activated partial thromboplastin time) are abnormal,340 whereas others require overt clinical signs of hemorrhage and concomitant thrombosis in addition to classic laboratory findings.296 The prognostic value of coagulation parameters has been evaluated.233, 296, 314 Overall, persistence or worsening of abnormalities in the face of treatment appears to be more indicative of poor outcome than alterations in any specific parameter. In one study decreased serum AT III concentration was the parameter most commonly associated with fatal outcome in mature horses with colic.295
Hypoxemia observed in response to endotoxin infusion is thought to be caused by an increase in ventilation-perfusion mismatch rather than pulmonary edema, as occurs in human patients with acute respiratory distress syndrome. Pulmonary edema may occur in patients with associated sepsis or complications such as DIC.341
Management
The ideal treatment for endotoxemia is prevention. Recognition and close monitoring of patients at risk are crucial because doing so allows institution of timely, possibly proactive treatment, which may reverse the effects of endotoxin before the inflammatory response has developed a dynamic of its own. Unfortunately, endotoxemia can develop rapidly, and horses are exquisitely sensitive to the effects of endotoxin; therefore, many equine patients are not presented for evaluation until they have reached more severe stages of endotoxemia or shock. Prognosis and patient outcome then frequently depend on the severity of complications associated with endotoxemia.234
Treatment of endotoxemia involves multiple aspects, and the following strategies have been proposed342:
-
•
Inhibition of endotoxin release into the circulation
-
•
Scavenging of LPS molecules to prevent direct effects and interaction with inflammatory cells
-
•
Inhibition of cellular activation by LPS
-
•
Inhibition of mediator synthesis
-
•
Interference with the effects of inflammatory mediators
-
•
General supportive care
In addition, treatment must also address the primary disease process as well as any complications.
When evaluating reports concerning the efficacy of any one treatment, the clinician should keep in mind differences in underlying disease processes and the complexity of the inflammatory cascade. A “one for all” treatment most likely will not be found; similarly, any one treatment can address only a few pathophysiologic aspects of endotoxemia at most.
Inhibition of Endotoxin Release into the Circulation
Inhibition of endotoxin release requires identification and removal of its source. Identification of responsible microorganisms and their antimicrobial sensitivity spectrum are crucial steps toward effective therapy; however, one should not necessarily delay treatment to obtain culture results. Specimen containers with antimicrobial removal devices may be useful in cases for which initiation of treatment precedes specimen collection. Once a diagnosis is reached, correction of the primary disease process is critical. Examples are removal of devitalized sections of bowel or infected umbilical remnants, drainage of infected pleural or PF, and gastric lavage followed by administration of intestinal adsorbents in cases of grain overload. Di-tri-octahedral smectite (DTO; Bio-Sponge, Platinum Performance Inc., Buellton, CA) was shown to remove endotoxin in an in vitro assay343 and may be useful in preventing endotoxemia of intestinal origin. Septic processes must be addressed with appropriate antimicrobial therapy, and principles of antimicrobial therapy should be followed. Regarding endotoxemia specifically, antimicrobial therapy has been suggested to increase the amount of circulating endotoxin by inducing endotoxin release on cell death of gram-negative bacteria. An in vitro study comparing endotoxin release and inflammatory mediator activity among antimicrobials commonly used to treat E. coli bacteremia in foals evaluated amikacin, ampicillin, amikacin plus ampicillin, ceftiofur, and imipenem. Although these antimicrobials showed no difference in the ability to kill bacteria, amikacin and the amikacin–ampicillin combination resulted in the lowest, and ceftiofur in the greatest, release of endotoxin. Endotoxin release appeared to be dose dependent in that lesser amounts were released at higher antimicrobial concentrations.344 On the basis of these results and clinical experience, combining antimicrobial therapy with endotoxin-binding agents such as polymyxin B may be beneficial, especially when using β-lactam antimicrobials.
Scavenging of Lipopolysaccharide Molecules
Endotoxin typically has a short plasma half-life and is removed rapidly by mononuclear phagocytes or neutralized by binding to serum proteins and lipoproteins. Many conditions responsible for the development of endotoxemia in horses, however, may be associated with an ongoing release of endotoxin. Examples include severe gastrointestinal inflammation as in proximal enteritis or colitis, grain overload, or uncontrolled sepsis. Therapy directed against endotoxin itself may be able to interrupt the continuous activation of the inflammatory cascade in these cases. Further benefits of antiendotoxin treatment may be derived if large amounts of endotoxin have been released before the inciting cause can be addressed.
Immunotherapy
An important consideration regarding the efficacy of immunotherapy is the region of the LPS molecule against which antibodies are raised. The O-chain of LPS acts as a potent antigen222; however, antibodies directed against the O-chain are serotype specific and cannot afford significant cross-protection against heterologous gram-negative bacterial strains. The core and lipid A region, both of which show a much higher degree of homology between LPS derived from different bacterial strains, offer a more promising target for immunotherapy. Active immunization against endotoxins has been reported for horses. Vaccination with a bacterin-toxoid vaccine prepared from rough mutants of Salmonella typhimurium or S. enteritidis protected horses against homologous and heterologous endotoxin challenge345, 346 and carbohydrate overload.346 Despite these encouraging results and the availability of a vaccine for use in horses (Endovac-Equi, Immvac Inc., Columbia, MO), active immunization against endotoxin does not appear to be a common practice. In comparison, passive immunization with anti-LPS antibodies is used widely. Rough bacterial mutants, most commonly J5 of E. coli O111:B4 and S. minnesota Re595, are used to immunize donor horses and subsequently prepare serum or plasma products. Proposed mechanisms of action after binding of the antibodies to LPS include steric blockade of lipid A interaction with cellular receptors and enhanced bacterial clearance by opsonization.347, 348, 349 Studies concerning the efficacy of antibody administration in equine patients vary in their results. Beneficial effects have been described in experimental models of endotoxemia, acute gastrointestinal disease, and neonates with sepsis,346, 350, 351, 352, 353 whereas in other studies antibodies failed to protect foals and horses against endotoxin effects.354, 355, 356 Administration of an S. typhimurium antiserum to foals was associated with an increased respiratory rate and higher serum activities of IL-6 and TNF.354
Various equine serum and plasma products are commercially available. An antiserum raised against the LPS core of S. typhimurium (Endoserum, Immvac Inc., Columbia, MO) is available for administration to endotoxemic horses at a recommended dosage of 1.5 mL/kg body mass. Diluting the serum 10- to 20-fold in crystalloid IV solutions, administering it slowly over 1 to 2 hours, and monitoring the patient for adverse reactions is advisable. Although the product is marketed for use in foals with failure of passive transfer, adverse effects have been reported,354 and one should use caution when administering it to neonates. Plasma from donors inoculated with J5 (E. coli) and S. typhimurium (Re-mutant) is available under a California license (Equiplas J, Plasvacc USA Inc., Templeton, CA). The manufacturer recommends administration of at least 1 to 2 L in cases of endotoxemia. Hyperimmune plasma, which has a guaranteed minimum IgG content but does not contain specific antiendotoxin antibodies (HiGamm Equi, Lake Immunogenics, Inc., Ontario, NY; Equiplas and Equiplas Plus, Plasvacc USA Inc.,), is marketed for treatment of failure of passive transfer, and many clinicians use it to treat endotoxemia and sepsis. In addition to antibodies and protein, plasma contains active constituents such as complement components, fibronectin, clotting factors, and AT III351 and therefore may be particularly useful in patients with endotoxemia-induced coagulopathy. Volumes of 2 to 10 mL/kg body mass of hyperimmune plasma have been recommended for use in endotoxemic patients.272, 357
Polymyxin B
Polymyxin B is a cationic polypeptide antibiotic that binds to the anionic lipid A portion of LPS and neutralizes its endotoxin capacity.358 At dosages required for antimicrobial activity, polymyxin B carries the risk of respiratory paralysis and ototoxic, nephrotoxic, and neurotoxic side effects; however, a much lower dose is required for endotoxin-binding activity. The effects of polymyxin B in horses have been evaluated in different experimental models.354, 358, 359 In an in vivo study in foals, treatment with polymyxin B at a dosage of 6000 U/kg body mass before infusion with S. typhimurium LPS resulted in significantly less severe elevations of body temperature, respiratory rate, and serum activities of TNF and IL-6 compared with untreated controls.354 Similarly, polymyxin B treatment of adult horses given endotoxin ameliorated clinical signs and decreased plasma TNF activity.360 In the latter study benefits of treatment were also evident at lower dosages of polymyxin B (1000 and 5000 U/kg body mass) and administration of polymyxin B 1 hour after the start of endotoxin infusion. Conversely, polymyxin B failed to ameliorate clinical signs of endotoxemia or prevent the development of coagulopathy, acidosis, lameness, and shock in experimental carbohydrate overload.361 Adverse effects suggestive of neurotoxicity appeared after repeated administration of 5 mg/kg body mass (36,000 U/kg) and in a milder form after administration of 2.5 mg/kg body mass (18,000 U/kg). Nephrotoxicity was not observed. Currently, use of polymyxin B in equine patients is recommended at dosages of 1000 to 6000 U/kg body mass every 8 to 12 hours administered as a slow bolus.362, 363 Treatment should be initiated as early in the disease process as possible because the beneficial effects of LPS scavenging may be limited to the first 24 to 48 hours after the onset of endotoxemia, before endotoxin tolerance develops. Adverse effects in the form of neuromuscular blockade and apnea, which necessitate slow infusion of the drug in human patients, have not been observed in horses. If treating horses with hypovolemia, dehydration, or azotemia, the clinician should attempt to improve peripheral tissue perfusion, minimize the polymyxin B dose, and closely monitor patients for nephrotoxicity. Close monitoring is also important if medications such as aminoglycoside antibiotics, which share a similar spectrum of potential side effects, are administered concurrently. Azotemic neonates are more susceptible to the nephrotoxic effects of polymyxin B than adult horses.360
In an attempt to decrease the risk for adverse effects while preserving the LPS-neutralizing ability, a conjugate of polymyxin B with dextran has been developed.364 In conjugated form, polymyxin B is prevented from extravasation into tissues, in which it exerts toxic effects by interaction with cell membranes. In addition, conjugation increases the residence time of polymyxin B in the circulation; therefore, it should prolong the antiendotoxic effect. The polymyxin B–dextran combination was evaluated at a total dose of 5 mg/kg body mass of polymyxin B in 6.6 g/kg body mass dextran, given 15 minutes before administration of endotoxin in horses.365 Treatment blocked the development of tachycardia, tachypnea, fever, and neutropenia completely and prevented increases in serum concentrations of TNF, IL-6, TXB2 (a TXA2 metabolite), and the prostacyclin metabolite 6-keto-PGF1α. Although mild adverse effects in the form of tachypnea, sweating, and increased systolic blood pressure were observed, these were transient and could be prevented by pretreatment with ketoprofen. To the author’s knowledge, the polymyxin B–dextran combination is not commercially available at this time.
Natural Endotoxin-Binding Substances
Natural endotoxin-binding proteins such as LBP, lipoproteins, and sCD14 have been evaluated experimentally. Results of these studies are somewhat contradictory, and detrimental effects occurred in some cases.366 A protein receiving a great deal of attention regarding potential therapeutic efficacy is the bactericidal permeability-increasing (BPI) protein. This protein is structurally similar to LBP but is expressed exclusively in myeloid precursors of polymorphonuclear (PMN) leukocytes.367 BPI is stored in primary granules of mature neutrophils and during inflammation is expressed on their cell membranes and secreted into the extracellular environment.368 BPI has an even higher affinity for LPS than LBP369 and shows antibacterial activity specific for gram-negative bacteria.219 Binding of BPI to the gram-negative bacterial membrane results in growth arrest and is an important factor in the antibacterial activity of intact neutrophils. Furthermore, BPI binding disrupts normal membrane organization and makes bacteria more susceptible to hydrophobic substances, including antimicrobials.370 Experimentally, recombinant BPI protects against the toxic and lethal effects of isolated LPS and intact gram-negative bacteria, and clinical trials in human patients show promising results regarding its therapeutic use.371 The biology and potential use of BPI in horses has not been evaluated.
Phospholipid emulsions have recently been evaluated for treatment of experimentally induced endotoxemia in horses. Phospholipid infusion improved clinical parameters, ameliorated neutropenia, and reduced inflammatory mediator production in response to an endotoxin challenge.372, 373 Because hemolysis was a complication of phospholipid infusion in some horses in these studies, optimization of dose and time of administration will be necessary before evaluating this treatment for potential clinical use.
Inhibition of Cellular Activation by Lipopolysaccharides
Treatments aimed at inhibiting LPS interaction with cells or turning off intracellular signaling pathways are under investigation. Nontoxic LPS or lipid A structures can act as endotoxin antagonists, if they competitively inhibit binding to LBP or cellular receptors or inhibit cellular activation by other mechanisms. Of the potential antagonists that have been evaluated experimentally, LPS and lipid A of the phototrophic bacterium Rhodobacter sphaeroides and the synthetic compounds E5531 and E5564 have been most promising.373, 374, 375, 376, 377, 378, 379 Unfortunately, species differences exist regarding cellular response to these structures, and R. sphaeroides LPS as well as E5531 have been found to have agonist activity in equine cells.380, 381 In cell transfection experiments, TLR-4 was shown to be responsible for this phenotypic variation regarding R. sphaeroides LPS.382 Given these results, any future potential LPS antagonists must be evaluated in equine systems.
Inhibition of Mediator Synthesis
Nonsteroidal Antiinflammatory Drugs
Nonsteroidal antiinflammatory drugs (NSAIDs) are probably the most commonly used drugs to treat endotoxemia in horses. The rationale for their use is inhibition of COX, which inhibits prostanoid production (see Fig. 12.5). Additional beneficial effects may include scavenging of oxygen-derived free radicals and iron chelation; however, side effects may occur at dosages required to achieve these effects.374 Prostanoids have been identified as important mediators in the inflammatory response in a number of studies, and inhibition of their synthesis is associated with beneficial effects. Generally speaking, two COX isoforms are recognized: constitutively expressed COX-1 and inducible COX-2. Upregulation of COX-2 expression results from various proinflammatory stimuli, including LPS, TNF-α, and IL-1.383 Constitutively expressed COX products are likely important for maintenance of homeostasis, whereas increased production of prostanoids by COX-2 is thought to be responsible for detrimental effects during inflammation and shock.
In horses, the most commonly used NSAID to treat endotoxemia is flunixin meglumine. Beneficial effects of flunixin meglumine have been described in experimental models of endotoxemia384, 385, 386 and in clinical cases. In equine colic patients treatment with flunixin meglumine before exploratory surgery resulted in reduced plasma concentrations of TXB2 and PGE2 and had a favorable effect on cardiovascular parameters.387 Flunixin meglumine was shown further to maintain cardiac output and systemic arterial blood pressure, improve blood flow to vital organs, reduce pulmonary endothelial damage, and improve survival on endotoxin challenge.285, 388, 389, 390 Conversely, in vitro studies have suggested that flunixin meglumine impairs recovery of intestinal barrier function in intestinal segments subjected to ischemia–reperfusion injury and may increase mucosal permeability to LPS.391, 392 This effect may be reduced or eliminated by concurrent administration of continuous rate infusion of lidocaine.
NSAID use in horses carries the risk of adverse effects, the most significant of which are development of gastrointestinal ulceration and renal papillary necrosis (renal crest necrosis). Differences may exist among NSAIDs in their propensity to induce adverse effects,393 but all NSAIDs must be used cautiously. Because of the potential concerns for masking of cardiovascular effects of endotoxin in horses with colic, a reduced dose of flunixin meglumine (0.25 mg/kg body mass thrice daily) has been suggested and is used widely in horses.327 At this dosage flunixin meglumine inhibits eicosanoid synthesis efficiently in an in vivo model of endotoxemia.394 Reduction of clinical signs, however, was dose dependent, and lower doses provide minimal, if any, analgesia (see Chapter 2 for further discussion of NSAID therapy in horses).
Some researchers have suggested that ketoprofen offers superior effects because of a proposed dual inhibitory effect on COX and lipoxygenase and may carry a decreased risk of adverse effects compared with flunixin meglumine and phenylbutazone. A comparison of cytokine and eicosanoid production by LPS-stimulated isolated monocytes in vitro, however, showed no significant difference between horses pretreated with flunixin meglumine (1.1 mg/kg body mass) or ketoprofen (2.2 mg/kg body mass), respectively.395
Given 15 minutes before LPS infusion, eltenac at a dosage of 0.5 mg/kg protected against changes in clinical, hemodynamic, and hematologic parameters and blunted the LPS-induced rise in plasma cytokine concentrations compared with controls in an experimental model of endotoxemia.396 Some parameters, however, including heart rate, leukocyte count, lactate concentration, and plasma TNF activity, were not improved. Ibuprofen may have beneficial effects superior to those of the other NSAIDs because it may be possible to achieve tissue concentrations safely that allow iron chelation to occur. According to a study in healthy foals, dosages of ibuprofen up to 25 mg/kg every 8 hours can be given safely for up to 6 days.397 To the author’s knowledge, the COX-2 inhibitor firocoxib has not yet been evaluated critically for treatment of endotoxemia.
Corticosteroids
The use of corticosteroids for antiinflammatory therapy in sepsis and endotoxemia has been controversial in human and equine patients, and beneficial effects superior to the ones achieved by NSAIDs have not been demonstrated consistently. Corticosteroids inhibit the activity of phospholipase A2 and the release of arachidonic acid from cell membrane phospholipids, as well as the production of TNF, IL-1, and IL-6 in response to an LPS stimulus. Experimentally, beneficial effects of dexamethasone in equine endotoxemia have been demonstrated.398, 399 To inhibit TNF production by equine peritoneal macrophages, however, the required concentration of dexamethasone was high and corresponded to an in vivo dosage (approximately 3 mg/kg body mass) greatly exceeding current recommendations.398 Although single doses of corticosteroids are unlikely to carry a disproportionate risk of adverse effects, the clinician should consider the suggested association of laminitis with corticosteroid use in horses. In cases of sepsis immunosuppressive effects could also be detrimental.
In human patients with certain types of septic shock, dysfunction of the hypothalamic–pituitary–adrenal axis has been recognized and successfully treated with hydrocortisone replacement therapy.400 Hypothalamic–pituitary–adrenal axis dysfunction has been suggested to occur in septic foals401; however, the use of low-dose corticosteroids for this indication remains to be investigated in horses.
Pentoxifylline
Pentoxifylline, a methylxanthine derivative and phosphodiesterase inhibitor, has been suggested for use in endotoxemia because of its effects on neutrophil function and its ability to inhibit the production of various cytokines, interferons, and thromboplastin. Decreased production of TNF, IL-6, TXB2, and thromboplastin in response to endotoxin was shown in an equine ex vivo model.402 In horses given endotoxin followed by treatment with pentoxifylline (7.5 mg/kg body mass followed by continuous infusion of 3 mg/kg/h for 3 hours), however, only minimal beneficial effects were observed.403 Treatment significantly improved body temperature, respiratory rate, and whole blood recalcification time, but no effect was observed regarding heart rate, blood pressure, leukocyte count, plasma fibrinogen concentration, and serum cytokine concentrations. The conclusion was that benefits of treatment with pentoxifylline might be restricted to administration of high bolus doses or continuous infusion early in the pathophysiologic process. In an in vivo endotoxemia model in horses, the combination of pentoxifylline (8 mg/kg body mass) and flunixin meglumine (1.1 mg/kg body mass) had greater benefit than each treatment on its own.404 Because of its rheologic properties (i.e., the ability to increase erythrocyte deformability and microvascular blood flow), pentoxifylline has been suggested for use in endotoxemic patients showing evidence of laminitis; however, no effect on blood flow to the hoof was demonstrated after administration to healthy horses.405 An IV preparation of pentoxifylline is not commercially available.
Antioxidants
Dimethyl sulfoxide (DMSO) is used by some clinicians in an attempt to scavenge oxygen-derived radicals. However, DMSO failed to show beneficial effects in an experimental model of intestinal ischemia when administered on reperfusion of the ischemic intestine.406 DMSO at a dosage of 1 g/kg body mass increased mucosal loss after ischemia and reperfusion of the large colon,407 and a reduced dosage of 0.1 g/kg body mass has been proposed for horses with intestinal ischemia. DMSO failed to show significant benefit in an experimental model of endotoxemia in horses, although it ameliorated the effect on fever, and many clinicians do not advocate its use.408 The xanthine oxidase inhibitor allopurinol has been suggested as a treatment to prevent oxygen radical–induced tissue damage. During periods of ischemia tissue xanthine dehydrogenase is converted to xanthine oxidase, which on reperfusion catalyzes the generation of superoxide radicals.409, 410 Evaluation in horses showed beneficial effects of allopurinol 5 mg/kg body mass administered 12 hours before endotoxin challenge.411 In another study mucosal damage attributable to oxygen-derived free radicals was not attenuated by allopurinol in an experimental ischemia–reperfusion model.407
Lidocaine
Lidocaine given IV has been suggested as an antiinflammatory, analgesic, and prokinetic agent. In an experimental endotoxemia model in rabbits, lidocaine inhibited hemodynamic and cytokine responses to endotoxin profoundly if given immediately after LPS infusion.412 Lidocaine further ameliorated the inhibitory effects of flunixin on recovery of mucosal barrier function following ischemic injury in equine small intestine.413 Use of lidocaine therefore may have merit in endotoxemic patients. A common regimen for lidocaine use in horses is the administration of an initial bolus (1.3 mg/kg body mass) followed by continuous infusion at a rate of 0.05 mg/kg/min.
ω-3 Fatty Acids
High concentrations of ω-3 fatty acids can alter the phospholipid composition of cellular membranes toward a decreased ratio of ω-6 to ω-3, affecting membrane functions such as phagocytosis, receptor binding, and activities of membrane-bound enzymes.286 Most important for the treatment of endotoxemia, ω-3 fatty acid incorporation into cell membranes decreases the availability of arachidonic acid (an ω-6 fatty acid) for eicosanoid synthesis414 and provides alternative substrates. Metabolism of ω-3 fatty acids via the COX and lipoxygenase pathway leads to the production of 3-series prostaglandins and 5-series leukotrienes, which have less biologic activity than their 2-series and 4-series counterparts derived from arachidonic acid. Aside from these mechanisms, ω-3 fatty acids prevent LPS-induced upregulation of CD14 in monocytic cells; therefore, they may be able to block transmembrane signaling of LPS.415 Cells from horses given linseed oil (high in ω-3 fatty acids) for 8 weeks before blood collection showed significantly decreased expression of procoagulant activity, TXB2, and TNF in response to LPS stimulation.416, 417 In an in vivo experimental model of endotoxemia in horses, treatment resulted in prolonged activated partial thromboplastin time and whole blood recalcification time, suggesting an anticoagulant effect; however, a significant beneficial effect on clinical response and serum eicosanoid concentrations was not observed.418 Because dietary addition of ω-3 fatty acids requires several weeks of treatment, IV infusion was evaluated and shown to alter the composition of cell membrane phospholipids rapidly.419 Further evaluation of this treatment for use in horses is necessary before specific dosage recommendations can be made.
Interference with the Effects of Specific Inflammatory Mediators
Antibodies Directed Against Tumor Necrosis Factor
Monoclonal and polyclonal antibodies against equine TNF have been evaluated in horses.420, 421, 422Administration of a monoclonal antibody preparation before LPS infusion resulted in significantly reduced plasma TNF activity, improved clinical abnormality scores, lower heart rate, and higher leukocyte count compared with controls.421 Plasma concentrations of lactate and 6-keto-PGF1α were reduced significantly, whereas TXA2 production was not affected.420 In another study422 administration of a rabbit polyclonal antibody against recombinant human TNF did not improve clinical and hematologic parameters when given shortly (15 minutes) after LPS infusion, although inhibition of TNF activity was present in vitro.422, 423 Findings in horses are in agreement with studies in other species and suggest that beneficial effects of TNF inhibition may be limited to administration before LPS exposure. Widespread clinical use therefore is unlikely to become feasible. Clinical trials in septic human patients have not shown significant benefits of TNF antibody treatment.424, 425
Platelet-Activating Factor Receptor Antagonists
The effects of selective PAF receptor antagonists have been evaluated. PAF is implicated in the development of systemic hypotension,426 LPS-induced platelet aggregation,427 ileus,428 and increased vascular permeability429 and may mediate recruitment of leukocytes to inflamed tissues.430, 431 A study in horses using the PAF receptor antagonist SRI 63-441 before LPS infusion showed significant decreases in heart rate and shorter elevation of lactate concentrations in response to the treatment. Although not statistically significant, additional beneficial effects included delayed onset of fever, a shortened period of neutropenia, and reduced maximal platelet aggregation.432
Supportive Care
Fluid Therapy and Cardiovascular Support
Fluid therapy is a mainstay of therapy of most endotoxemic patients suffering from the cardiovascular effects of systemic inflammation. Many endotoxemic equine patients require fluid therapy for treatment of the underlying disease process and correction of dehydration and electrolyte and acid-base abnormalities. Principles of fluid therapy are discussed in Chapter 4 of this text.
Patients with severe hypovolemia and shock present management challenges, especially because increased vascular permeability in endotoxemic patients requires careful consideration of fluid therapy plans. A rapid increase in total body fluid volume may be detrimental in patients with compromised cardiac and peripheral vasomotor function and may increase the severity of vascular pooling in peripheral organs. In these patients hypertonic solutions or colloids may be more appropriate means of stabilization than large volumes of crystalloid solutions. Hypertonic saline solution (7.5% sodium chloride) is the most commonly used hypertonic solution in horses and has beneficial effects in endotoxemic patients.433 A dosage of 4 mL/kg is recommended, which should be given as a bolus infusion over 10 to 15 minutes, followed by administration of an isotonic solution to restore total body fluid volume. The clinician should use hypertonic saline with caution in patients with sodium or chloride derangements and should monitor serum electrolyte concentrations in the case of repeated administration. Failure of urination despite appropriate fluid resuscitation should result in critical evaluation of renal function. In one recent study small-volume resuscitation with hypertonic saline plus hydroxyethyl starch failed to alleviate hemodynamic responses in experimental endotoxin infusion in horses.434
Plasma is an ideal colloid and should be administered to maintain a serum total protein concentration above 4.2 g/dL.357 To raise plasma protein concentration and colloid osmotic pressure significantly, however, horses often require large volumes of plasma (7–10 L or more in a 450-kg horse), and alternative colloids should be considered. High-molecular-weight polymers are thought to provide superior oncotic effects in cases of sepsis and endotoxemia, when vascular permeability is increased. Hetastarch, or hydroxyethyl starch (Hespan), is commercially available as a 6% solution in 0.9% sodium chloride. Hetastarch molecules have a very high molecular weight, and degradation must occur before renal excretion.435 These properties result in a longer plasma half-life and prolonged oncotic effects compared with other colloids; persistence of the oncotic effect for 24 hours was observed in hypoproteinemic horses.436 A dosage of 5 to 15 mL/kg given by slow IV infusion along with an equal or greater volume of crystalloid fluids has been recommended.435, 437 In human patients prolonged activated partial thromboplastin time, decreased factor VIII activity, and decreased serum fibrinogen concentration have been described in association with hetastarch use.438 In the limited number of equine studies, bleeding times were not affected439, 440; however, patients treated with hetastarch should be monitored for coagulopathy.
Metabolic acidosis in endotoxic shock is attributable to lactic acidemia and inadequate tissue perfusion.441 Acid-base balance often improves considerably after fluid resuscitation alone; however, additional sodium bicarbonate may be required in cases in which serum bicarbonate concentration remains below 15 mEq/L.
Foals with sepsis are frequently hypoglycemic, and 5% dextrose solutions are useful as initial resuscitation fluids. The clinician should reduce the glucose concentration of IV solutions according to the blood glucose concentration to avoid prolonged hyperglycemia. Administration of hyperimmune plasma (20–40 mL/kg body mass) is highly recommended in foals with evidence of partial or complete failure of passive transfer.
One should consider positive inotropic and vasomotor agents in patients with persistently inadequate tissue perfusion. Lower dosages of dopamine (0.5–2 μg/kg/min) result in vasodilation of the renal, mesenteric, coronary, and intracerebral vasculature via dopaminergic effects, whereas higher dosages (up to 10 μg/kg/min) also exert stimulation of α1-adrenergic receptors, resulting in increased myocardial contractility and heart rate.442 Dobutamine is a direct α1-adrenergic agonist and does not appear to have significant vasodilator properties. Dosages for dobutamine of 1 to 5 μg/kg/min as a continuous IV infusion have been recommended for use in horses. Norepinephrine was evaluated in hypotensive critically ill foals that were refractory to the effects of dopamine and dobutamine.443 At dosages up to 1.5 μg/kg/min administered concurrently with dobutamine, six of seven foals showed an increase in mean arterial pressure, and all foals had increased urine output. Because of the risk of cardiac side effects, close monitoring of heart rate and rhythm should accompany infusion of inotropes. Indirect blood pressure measurements using a tail cuff may be used to monitor the effects of treatment.
Management of Coagulopathy
More frequently than overt thrombosis or bleeding attributable to DIC, hemostatic abnormalities occur in the form of alterations in the coagulation profile. A procoagulant state with shortened bleeding times or prolonged bleeding times caused by consumption of clotting factors may be evident. One should address abnormalities in the coagulation profile as early as possible but especially if they persist more than 24 hours after initiation of therapy. Because of the complex interactions of coagulation and fibrinolysis during endotoxemia, it might be necessary to combine anticoagulant therapy with the administration of fresh frozen plasma to replace clotting and fibrinolytic factors. Heparin acts as an anticoagulant by activation of AT III and subsequent inhibition of thrombin, release of tissue factor pathway inhibitor from endothelial cells, and inhibition of platelet aggregation.444 Because endogenous AT III levels frequently are decreased in patients with coagulopathy, the addition of heparin to fresh frozen plasma may be the most effective route of administration. An initial dosage of 100 IU/kg body mass followed by 40 to 80 IU/kg body mass thrice daily has been recommended.357 Anemia caused by erythrocyte agglutination occurs in some patients during therapy with unfractionated heparin445, 446 but typically resolves within 96 hours if therapy is discontinued.357 Because of the risk of microthrombosis associated with erythrocyte agglutination, use of low-molecular-weight heparin (50 IU/kg body mass subcutaneously [SC] every 24 hours) has been recommended447 but may be cost prohibitive. Aspirin can be given orally (10–20 mg/kg body mass every 48 hours), which irreversibly inhibits platelet COX activity, to inhibit platelet aggregation and microthrombosis. Platelet hyperaggregability has been implicated in the pathogenesis of carbohydrate-induced laminitis,448 and heparin and aspirin have been recommended to prevent the development of laminitis. In an in vitro study, however, aspirin did not inhibit endotoxin-induced platelet aggregation.449
Other Considerations
Luteolysis caused by increased concentrations of PGF2α may lead to pregnancy loss in endotoxemic mares before day 55 of pregnancy (see Chapter 19).450 Daily administration of altrenogest (Regu-Mate, Hoechst-Roussel Agri-Vet, Somerville, NJ) at a dosage of 44 mg orally consistently prevented fetal loss in mares if administered until day 70 of pregnancy.331 Treatment with flunixin meglumine, by blockade of PGF2α release,332 also may contribute to the maintenance of pregnancy in endotoxemic mares. The pathogenesis of fetal loss and abortion caused by endotoxemia, surgery, or systemic disease later in gestation is not understood completely. Proposed mechanisms include direct effects on the fetus, placental function, or placental progesterone production.451
Decreased nitric oxide production by vascular endothelial cells in response to endotoxin has been suggested as a mechanism for vasoconstriction and decreased blood flow leading to laminitis)452; however, use of nitric oxide donors remains controversial. Maintenance of adequate peripheral perfusion and anticoagulant and antiinflammatory therapy may be helpful in preventing and treating laminitis caused by endotoxemia.
Cryotherapy
Digital cryotherapy is a critical component of supportive care in the equine patient with suspected endotoxemia, including those suffering from many of the inflammatory diseases discussed in this chapter. Cryotherapy has been shown to reduce the severity of laminitis lesions in the oligofructose model,453, 454, 455 including when initiated after the onset of lameness.456 It has also been shown to decrease the incidence of laminitis in horses diagnosed with colitis.457 When initiated, digital cryotherapy should include immersion of the hoof and pastern, at a minimum.458
Inflammatory Diseases of the Small Intestine
Duodentitis-Proximal Jejunitis
Duodenitis-proximal jejunitis (DPJ) is an inflammatory condition affecting the upper small intestine and resulting in distention, abdominal pain, gastric reflux caused by excessive fluid and electrolyte secretion, and increased PF protein concentration without a significant elevated nucleated cell count. Other terms for this condition are anterior enteritis and proximal enteritis. Clinical signs of DPJ mimic those of a small intestinal obstruction. The clinical syndrome of DPJ was well described in the 1980s,19, 459, 460, 461 but the severity of clinical signs, especially duration of disease, is variable. Although not typical, DPJ can occur in conjunction with gastritis, ileitis, typhlitis, and colitis.
Pathophysiology
Typical pathologic findings in horses with DPJ include involvement of the duodenum and usually the proximal jejunum.19 The ileum and large colon usually are grossly normal. Gastric distention is common because of hypersecretion in the proximal small intestine combined with functional ileus. Small intestinal diameter often measures 5 to 7 cm, filled with malodorous, red to brown-red fluid. Duodenal and jejunal serosal surfaces may have varying degrees and distribution of bright red to dark red petechial and ecchymotic hemorrhages and yellow to white streaks. The enteric mucosal surfaces are usually hyperemic with varying degrees of petechiation and ulceration.
Microscopically, the most severe lesions are located in the duodenum and proximal jejunum but may extend proximally to the gastric mucosa and aborally to the large intestinal mucosa and submucosa.19 Microscopic lesions include varying degrees of mucosal and submucosal hyperemia and edema, villous degeneration with necrosis and, more severely, sloughing of villous epithelium. The lamina propria, mucosa, and submucosa may have varying degrees of granulocyte infiltration (predominantly neutrophils), and the muscular layers and serosa may contain small hemorrhages. Proximal small intestinal serosal fibrinopurulent exudate is a common finding in the more severe cases; therefore the term hemorrhagic fibrinonecrotic duodenitis-proximal jejunitis has been suggested as a more descriptive name for this syndrome.461
Horses with DPJ can have hepatic changes including hepatocellular vacuolization, cholestasis, inflammatory infiltrate (either in association with centrilobular necrosis or periportal), and biliary hyperplasia.462 Hepatic disease is thought to result from ascending infection via the common bile duct, local absorption of endotoxin via the portal circulation, systemic consequences of endotoxin absorption, metabolic imbalances, and hypoperfusion or hypovolemia.
In most cases, an underlying etiology cannot be determined. In some cases, Salmonella spp. or Clostridium spp. can be isolated from culture of gastric reflux. Salmonellosis has never been consistently identified in a majority of cases, and many horses with documented infections by these organisms do not develop DPJ. Alternatively, toxigenic strains of C. difficile were isolated from the reflux of five of five horses with DPJ and none of six control horses with other causes of nasogastric reflux.463 To the author’s knowledge, such an association has not been identified in other geographic locations. Another suspected infectious agent is Fusarium spp.464 Alteration of the gastrointestinal microflora caused by recent dietary change has been suggested as a predisposing cause for DPJ; in one report, horses with DPJ were fed significantly more grain and were more likely to have grazed pasture, relative to horses with other forms of colic, but these associations were not deemed strong enough to allow clinical differentiation.465
Two intracellular processes control intestinal secretion: the cyclic nucleotide (cAMP and cGMP) and calcium systems.169 Inflammatory mediators, microorganisms, and toxins can activate adenyl cyclase (vasoactive intestinal peptide and PGE2) or guanylyl cyclase (bacterial enterotoxins) and induce increases in cAMP and cGMP, respectively. This reaction causes phosphorylation of specific protein kinases, which induce the actual mucosal membrane transport events. Increases in intracellular free calcium may arise from cyclic nucleotide–dependent release of stored calcium within the cell or from increased calcium entry across the cell membrane. Calcium may act through calmodulin, which then can activate membrane-phosphorylating protein kinases. The net effect is increased movement of sodium and chloride into the mucosal cell from the interstitium, with secretion of sodium and chloride into the intestinal lumen. Water follows the directional flux of sodium and chloride through highly permeable intercellular spaces. Several bacterial toxins and endogenous mediators can cause active secretion and contribute to a synergistic mucosal secretory response. Passive secretion of protein-rich fluid into the lumen occurs after damage to the mucosal epithelium, capillary endothelium, and submucosal inflammation in the proximal small intestine. The clinically relevant events that result from active and passive fluid secretion are proximal small intestinal distention and nasogastric reflux, dehydration, and circulatory shock.466
The concentration of protein in the PF from horses with DPJ is usually higher than in horses with small intestinal obstruction. A disproportionate increase in total protein concentration relative to nucleated cell count occurs, probably by leakage of blood or plasma into the peritoneal cavity without a significant stimulus for leukocyte chemotaxis. Suggested mechanisms for increased abdominal fluid protein concentration include serositis associated with inflamed intestine and small intestinal distention causing passive congestion and increased capillary hydrostatic pressure of visceral peritoneal vessels.467 Small intestinal ileus is another hallmark sign of DPJ, and the pathophysiology is complicated, involving primary and secondary dysfunction of the central, autonomic, and ENSs and their purported roles in governing intestinal motility. Further detail regarding the effect of inflammation on small intestinal motility is provided in Inflammatory Disease of the Small Intestine later in this chapter.
Clinical and Clinicopathologic Signs
Differentiating horses with DPJ from horses with small intestinal obstructive lesions can be challenging because there is no single distinguishing feature, and all information must be considered collectively. Horses with obstructive lesions of the small intestine usually show consistent signs of abdominal pain until the affected viscus is repaired surgically or ruptures. In contrast, signs of acute abdominal pain typically subside after gastric decompression and volume replacement in horses with DPJ. They are replaced by signs of lethargy and general malaise. On rectal examination the degree of small intestinal distention may be subjectively less with DPJ than with obstructive lesions, particularly following gastric decompression. Although the color and odor of gastric reflux can be similar, horses with DPJ tend to have a larger volume (≥4–20 L with each decompressive effort) of reflux than horses with obstructive lesions. Horses with DPJ often have a mild fever of 38.6°C to 39.1°C (101.5°F–102.5°F), whereas horses with obstructive lesions are typically normothermic or hypothermic.
Abnormalities in the leukogram are more common in horses with DPJ than with an acute obstructive lesion.19, 460 In addition, hyponatremia, hypochloremia, hypokalemia, prerenal azotemia, and elevated hepatic enzymes (GGT, alanine transaminase, and AP) are often evident.462 Hyperlactemia from poor tissue perfusion and hypovolemia is common with both conditions.
PF analysis may be helpful in distinguishing DPJ from an obstructive lesion. The typical findings with DPJ include an increased PF protein concentration (often ≥3.5 g/dL) and a mild to moderate elevation of the peritoneal WBC count, although the count usually is less than 10,000 cells/μL; PF lactate concentration typically mirrors peripheral blood concentrations. The disproportionate increase in PF total protein compared with peritoneal nucleated cell count may be caused by leakage of blood or plasma without a marked leukotactic response.19 The PF is usually yellow and turbid, but in severe cases, diapedesis can occur, resulting in a serosanguineous color. Strangulating lesions typically result in more severe changes in the PF. Serosanguineous PF with increased protein, lactate, and white and red blood cells is typical. Horses with intraluminal obstructions such as ileal impactions typically have grossly normal PF with a mildly elevated protein resulting from intestinal distention.419, 460, 466
Ultrasonographic findings in horses with DPJ typically include gastric and duodenal distention and segments of small intestine containing hypoechoic to anechoic fluid. The wall of the small intestine can be normal or thickened with time. Peristalsis can be decreased, normal, or increased. Differentiation from a small intestinal obstructive lesion remains difficult. More long-standing obstructions typically result in increased wall thickness, sedimentation of ingesta within the small intestinal lumen, and distended small intestine proximal to the lesion with collapsed intestine distally.
In most cases, the diagnosis is suspected on the basis of clinical and clinicopathologic signs and response to therapy; a definitive diagnosis of DPJ requires gross examination of the duodenum and proximal jejunum at surgery or at necropsy
Treatment
Horses with DPJ appear to share a common characteristic clinical presentation. Treatment is supportive and should include volume replacement, analgesic and antiinflammatory therapy, gastric decompression, antiendotoxin therapy, antimicrobial therapy if indicated, nutritional support, and nursing care.
Gastric Decompression
Although the signs of abdominal pain usually resolve after gastric decompression, most horses remain lethargic. Horses with DPJ often require gastric decompression at 2-hour intervals, with 2 to 10 L of fluid recovered each time. Nasogastric tubes left in place for long periods cause varying degrees of pharyngitis (Fig. 12.6 ), laryngitis, and esophagitis, and maintenance of an indwelling nasogastric tube may further delay gastric emptying.466, 468, 469 Thus one should use the smallest, softest nasogastric tube needed for gastric decompression and remove the tube as soon as possible.
Fig. 12.6.
Endoscopic image from a 15-year-old Quarter Horse gelding. The horse was diagnosed with duodenitis-proximal jejunitis (DPJ) and developed mild respiratory stridor and gagging and retching behavior after an indwelling nasogastric tube had been in place for 48 hours to allow for intermittent drainage of gastric reflux. The horse responded well to medical therapy for DPJ and pharyngitis-esophagitis.
Supportive Care
Much of the supportive care for horses with DPJ mirrors that described for other cases of endotoxemia and is discussed in detail in Endotoxemia earlier in this chapter. Specific fluid replacement should take ongoing losses through reflux into consideration. Horses should not receive food or water until they remain comfortable and reflux ceases. Occasional sips of water or rinsing of the mouth may decrease halitosis. The refeeding plan should be conservative, because horses refed quickly may relapse.
Antiinflammatory and Analgesic
NSAIDs are the most frequently used group of drugs for treatment of abdominal pain in horses (flunixin meglumine 0.25–1.1 mg/kg IV every 8–12 hours, with the higher dose given only at the least frequent interval). These agents also have beneficial antiinflammatory and antiendotoxic effects. The clinician must weigh the benefit of these drugs against their negative effects on gastrointestinal mucosa and renal function. Another popular analgesic option is butorphanol, an opioid agonist-antagonist, given at 0.02 to 0.1 mg/kg intramuscularly every 6 to 8 hours or as a constant rate infusion at 13 μg/kg/h.470 This route appears to have minimal effects on gastrointestinal motility.
Antimicrobial
Because Clostridium spp. are suspected as a causative agent of DPJ, penicillin, metronidazole, or both may be administered to affected horses in geographic locations in which said organisms appear prevalent. Broad-spectrum antimicrobial therapy may be considered for horses with DPJ and significant leukopenia.
Nutritional Support
The veterinarian should consider the nutritional needs of horses with DPJ. Most horses have a total body protein loss because of cachexia and a protein-losing enteropathy. Total or partial parenteral nutrition may be indicated in horses that remain anorectic for more than 3 to 4 days. Parenterally administered solutions containing glucose, balanced amino acid solutions, lipid emulsions, balanced electrolyte and trace minerals, and vitamins have been administered to adult horses with small intestinal ileus or enterocolitis. Based on a small number of horses, this therapy has proved promising in terms of minimizing protein losses and decreasing the duration of illness. Postoperative parenteral nutrition ameliorates clinicopathologic evidence of starvation following small intestinal resection and anastomosis in adult horses.471, 472 Providing for part of the nutritional requirements of the horse (8000–12,000 kcal/day) is possible with glucose–amino acid solutions, which are of moderate cost. It is reasonable to suppose that providing nutritional support to an anorectic, severely ill horse will facilitate healing and even shorten the duration of illness. Thus the overall cost of providing parenteral nutritional supplementation to horses with DPJ may be offset by quicker recovery and diminished requirements for other, expensive treatments, but further data are necessary to accurately assess this point.
Prokinetics
Normal (healthy) intestine is necessary for optimal performance of most prokinetic agents in horses. Many motility-modifying agents likely are ineffective in cases of DPJ. Inflamed jejunal tissue has been shown to have downregulation and decreased production of motilin receptors, which may alter the prokinetic response to erythromycin.473 However, some benefit may come of the judicious use of prokinetic agents in inflammatory conditions of the equine intestine, particularly if the agent provides additional effects. Lidocaine has shown particular promise for this use. Although the exact mechanism of action is unknown, the beneficial effects in horses with ileus are likely caused by antiinflammatory effects because a direct prokinetic effect has not been demonstrated.474 In horses with nasogastric reflux attributable to POI or enteritis, a constant rate infusion of lidocaine was shown to reduce the hourly volume of reflux and shorten the time to cessation of reflux compared with saline.475 Prokinetic therapy with erythromycin lactobionate, metoclopramide, or bethanechol can also be considered.476, 477 Motility-modifying agents and the influence of inflammation on their effects are discussed in Gastrointestinal Ileus later in this chapter.
Surgical Therapy
Medical therapy is successful in most cases of DPJ. For horses with DPJ that continue to produce copious enterogastric reflux despite aggressive medical treatment, or when a mechanical obstruction cannot be satisfactorily ruled out, surgery may be considered. Refractory cases have improved with surgical intervention; however, some horses with refractory DPJ have been observed to recover after prolonged (up to 20 days) supportive care and gastric decompression. The latter is not recommended in the author’s experience. For a horse exhibiting abdominal discomfort with small intestinal distention palpable per rectum and more than 2 L of gastric reflux, the veterinarian should recommend referral to an appropriate surgical facility. Typically, the main determinants for surgical intervention are degree and duration of abdominal pain, PF analysis, and results of (often repeated) rectal palpation and ultrasonography.
Short-term mortality rates have been reported to be 37% for horses in which manual evacuation of the small intestine into the cecum was performed surgically, compared with 40% for horses that received medical treatment.478 A 95% recovery rate was reported in horses with DPJ that underwent laparotomy and manual evacuation of the small intestine into the cecum, combined with treatment for C. perfringens consisting of IV metronidazole and intramuscular procaine penicillin.479 Drawbacks of surgery include the expense, risks of anesthesia, prolonged return to training, and risk of incisional complications. However, when an obstruction cannot be ruled out, these concerns are of minor consequence.466
Prognosis
Survival rates for DPJ range between 25%460 and 94%.19, 460 At present the survival of horses with DPJ that undergo surgery is much greater than previously described, and certainly greater than that of horses with small intestinal obstruction that do not have surgery.478, 479 Horses with DPJ that receive appropriate therapy have a reasonably good chance of making a full recovery, and recurrence is rare. Horses that continue to have frequent episodes of voluminous nasogastric reflux and systemic signs of endotoxemia and sepsis have a poorer prognosis for recovery. Frequent complications of DPJ include laminitis, thrombophlebitis, and weight loss. There appears to be geographic variation in the severity and prognosis for DPJ, with horses in the southeastern United States most severely affected.
Equine Coronavirus
Equine coronavirus (ECoV) is classified as a betacoronavirus and is associated with fever, lethargy, anorexia, and enteric disease (colic and/or alteration in fecal consistency) in some horses.480, 481, 482 A definitive association between ECoV and diarrhea in foals was not recognized before the year 2000483, 484 or in adult horses before 2011.480, 481 Diagnosis is confirmed via fecal PCR.480, 482 Mortality is typically low (7%),480, 482 with death caused by suspected482 or confirmed485 hyperammonemia and neurologic disease or severe endotoxemia and sepsis.482, 485 In some outbreaks, the fatality rate has been higher (27%), with fecal viral load higher in nonsurvivors.486 Histologically, ECoV is associated with necrotizing enteritis, particularly in the jejunum and ileum, with hemorrhage in the ventral colon in nonsurviving horses in one report.485
Inflammatory Small Intestinal Diseases Associated with Malabsorption and Maldigestion
Malabsorption and maldigestion are commonly recognized clinical problems in humans and small animals and documented clinical entities in the horse. The term malabsorption implies impairment of digestive and absorptive processes arising from functional or structural disorders of the small intestine and related organs, the pancreas, liver, and biliary tract. The condition can affect absorption of carbohydrates, proteins, fats, vitamins, minerals, and to a lesser extent, water and electrolytes. In horses, the resulting pathophysiologic changes may influence large intestinal function adversely through alterations in the substrate presented for fermentation or through direct infiltration of the large colon.
Differentiation between carbohydrate, protein, or fat malabsorption is not possible in the horse because of the herbivorous diet and the contribution of large intestinal functions. The rarity of pancreatic problems, such as exocrine pancreatic insufficiency, in horses along with their herbivorous diet makes maldigestion less problematic, but maldigestion can contribute to chronic weight loss in horses with severe infiltrative small intestinal disease and exacerbate diarrhea in the suckling foal through reduced intestinal bile salt concentrations from hepatic or ileal dysfunction.
Malabsorption is not synonymous with diarrhea, although diarrhea may be a feature. Adult horses rarely exhibit diarrhea with small intestinal problems unless large intestinal involvement is concomitant. Chronic diarrhea is predominantly a large intestinal disorder that reflects an overload of water and electrolytes and thus may be considered a state of impaired absorption. Primary small intestinal disease is more likely to occur in neonates and young foals. For example, acquired small intestinal brush border lactase deficiency may result in increased lactose fermentation in the large intestine and induction of osmotic diarrhea.487
The clinical signs of chronic wasting and poor body condition, although nonspecific for a diagnosis of malabsorption antemortem, can typically be attributed to proliferative or inflammatory intestinal disorders, often collectively referred to as chronic inflammatory bowel diseases (CIBD).488
Clinical Assessment
The primary clinical sign associated with diseases of malabsorption and maldigestion is weight loss with or without diarrhea. Physical, ultrasonographic, and laboratory assessment is as described for other gastrointestinal diseases and detailed in Diagnostic Evaluation earlier in this chapter.
Rectal biopsy is easy to perform and may provide an indication of cellular infiltration that could be present at more proximal locations, but interpretation is often difficult. In one retrospective study, inflammatory bowel disease was diagnosed from rectal biopsy specimens in approximately 50% of cases.489 In that report, simple proctitis (neutrophils in the crypt or surface epithelium) was associated with inflammatory disorders, whereas only mild scattered neutrophil infiltration was seen in controls. Rectal biopsy aided diagnosis for 3 of 7 horses with lymphocytic-plasmacytic enterocolitis490 and 1 of 2 horses with eosinophilic enterocolitis.491 TABLE 12.3, TABLE 12.4 present the clinicopathologic and pathologic features of the diseases most commonly associated with malabsorption. In the same animal the extent and severity of pathologic changes differ in different regions of the small and large intestines, influencing the severity of clinical signs and abnormalities in tests of intestinal function. Early diagnosis remains a challenge, and even multiple intestinal biopsies taken at exploratory laparotomy may prove unhelpful.
TABLE 12.3.
Predominant Clinical and Clinicopathologic Features of Horses with Proliferative and Infl ammatory Bowel Diseases
| Condition | Breed | Age Range | Clinical Signs | Dermatitis/Coronitis | Hematology | Chemistry | Absorption Tests |
|---|---|---|---|---|---|---|---|
| Alimentary lymphosarcoma | None | 2 yr to aged; Majority ≤4 yr | Weight loss, poor appetite, edema, depression, occadional fever, occasional diarrhea or colic | +/– Scurfy skin | Anemia, neutrophilia; lymphocytosis rare | Decreased albumin; TP normal to increased; increased globulin | Reduced absorption; partial to complete malabsorption |
| Granulomatous enteritis | Standardbred | 1-6 yr; Majority ≤3 yr | Severe wasting, edema, variable appetite, depression, infrequent diarrhea, occasional slight fever | +/– Scurfy skin; severe lesions rare | Anemia; Leukocytes normal to slightly increased or decreased | Decreased albumin; TP normal to decreased; GGT normal, ALP normal to increased | Reduced absorption; partial to complete malabsorption |
| Multisystemic eosinophilic epitheliotropic disease | Standardbred, Thoroughbred | 1 yr to aged; Majority ≤4 yr | Severe wasting, edema, appetite poor to ravenous, slight fever, diarrhea or soft feces common, rare colic, depression, oral ulcers | ++++ Severe skin lesions and ulcerative coronitis prominent | Anemia rare to slight; neutrophilia and eosinophilia rare | Decreased albumin; TP normal to decreased; GGT and ALP normal to increased | Delayed absorption (peak shifted to right); reduced or normal peak concentration |
| Lymphocytic plasmacytic enterocolitis | None | 3 yr to aged | Inappetence, depression, colic, edema | Normal | Decreased albumin and TP; increased fibrinogen | Inadequate absorption | |
| Proliferative enteropathy | None | 3-8 months; sporadic reports older | Depression, colic, diarrhea, edema, appetite often normal, concurrent infection | +/– Scurfy skin | Anemia, leukocytosis | Decreased albumin and TP; increased CK | Often normal |
∗GGT, γ-Glutamyltransferase; ALP, alkaline phosphatase; CK, creatine kinase; none, no predominant breed.
TABLE 12.4.
Pathologic Features of Proliferative and Inflammatory Bowel Diseases of Horses
| Condition | Small Intestine | Large Intestine | Other Organs/Systems | |
|---|---|---|---|---|
| Alimentary lymphosarcoma | G | Constant; extensive thickening, thickened mucosa, fissures, serosal plaques, nodules, congestion | Infrequent; unremarkable to thickened segments | MLNs massively enlarged; occasional enlargement of other LNs |
| H | Villous atrophy (partial to total); crypts disappear with hyperplasia; infiltrate of pleomorphic lymphoid cells, plasma cells; transmural | Nothing evident to diffuse mucosal infiltration | Extensive infiltration of MLNs; moderate in other LNs (liver, spleen, stomach rare) | |
| Granulomatous enteritis | G | Constant; thickened wall and mucosa, fissures, widespread ulceration (tiny ulcers) | Common, generally discrete | MLNs enlarged, edematous; stomach commonly affected (generally discrete); liver/pancreas rare |
| H | Villous atrophy (partial to total), crypt hyperplasia and abscesses, diffuse granulomatous inflammation; mononuclear cells (lymphoid), giant cells, epithelioid foci; lymphangiectasia | Similar infiltrate usually discrete; mucosa, submucosa | Similar infiltrate; stomach discrete; MLNs discrete to florid macrophage infiltration; diffuse cortical hyperplasia | |
| Multisystemic eosinophilic epitheliotropic disease | G | Common; diffusely thickened, especially proximal duodenum and distal ileum; serosal nodules or granularity; ulceration | Constant; severe; segmental or multifocal granuloma; mucosal (predominantly) and transmural thickening; extensive ulcers | MLNs and other LNs enlarged; stomach and esophagus commonly affected; liver/pancreas commonly affected; may be hyperkeratotic Skin: exudative dermatitis, ulcerative coronitis |
| H | Villous atrophy rare; lymphocytic and eosinophilic infiltration most severe in cranial duodenum, ileum, ileocecal junction; infiltrate more widespread than gross lesions | Segmental/multifocal lesions, severe infiltration, reactive fibrosis, tissue eosinophilia, walled-off granulomata, central necrotic core of eosinophilic material | Similar infiltration with fibrosis of MLNs, liver, pancreas Skin: acanthosis, hyperkeratosis, diffuse infiltrate of eosinophils, lymphocytes in dermis; focal eosinophilic accumulations |
|
| Lymphocytic-plasmacytic enterocolitis | G | Constant; mucosal/submucosal edema; prominent folds | Common; edema, congestion, areas of mucosal ulceration | MLNs enlarged |
| H | Villous blunting to atrophy; moderate to severe infiltration of lymphocytes, plasma cells; edema, dilated lymphatics | Similar infiltrate, less remarkable | Minimal evidence | |
| Proliferative enteropathy | G | Constant; significant mucosal thickening, corrugated appearance from proximal jejunum to distal ileum | Uncommon; submucosal edema | MLNs unremarkable |
| H | Villous shortening, severe hyperplasia of crypt epithelium, small curved bacteria in apical cytoplasm, mononuclear infiltrate | No evidence | No evidence |
G, Gross pathologic findings; H, histopathologic findings; LNs, lymph nodes; MLNs, mesenteric lymph nodes.
Biopsies of skin, liver, lymph node, or lung may reveal evidence of multisystemic disease and can be easily obtained in the standing horse, with organ biopsies typically obtained with ultrasonographic guidance. Duodenal mucosal biopsies can be obtained endoscopically. Exploratory laparotomy facilitates rigorous inspection of the gastrointestinal tract and associated organs to obtain multiple biopsies from intestinal sites and lymph nodes. Cost and potential postoperative complications may limit surgical procedures for diagnosis. Laparoscopy may provide an alternative means to facilitate biopsy of certain tissues but is typically more helpful from a diagnostic rather than therapeutic standpoint. From this perspective, surgical exploration should be considered as an option early in the process rather than as a last resort.
Carbohydrate absorption tests, described in Diagnostic Evaluation earlier in this chapter, can provide a practical and inexpensive means to assess the absorptive capability of the small intestine. Pathologic changes in the mucosa and submucosa must be extensive and widely distributed to greatly affect the peak plasma concentration and shape of the curve. In one report of 42 mature horses with chronic weight loss, a normal OGTT (peak glucose concentration at 120 minutes >85% baseline) was not associated with abnormal small intestinal morphology in any of the 5 horses in which it was documented. When peak glucose concentration was between 15% and 85% of baseline at 120 minutes (considered partial malabsorption), approximately 72% had small intestinal infiltrative disease, and when peak concentration at 120 minutes was less than 15% above baseline (total malabsorption), all had severe small intestinal infiltrative disease.59 Other reports have documented horses with flat OGTT curves that subsequently showed more normal OGTT responses and resolved clinical condition.492 Carbohydrate absorption tests probably have poor diagnostic sensitivity to detect small intestinal involvement in horses with chronic diarrhea and predominantly large intestinal problems.493 An abnormal absorption test and weight loss can occur in the horse as a transient event and without significant morphologic changes in the small intestine.
The intestinal sugar active transport system has a low affinity for d-xylose in the equine jejunum in vitro, hence d-xylose absorption likely occurs primarily by convection or diffusion.494 An abnormal d-xylose absorption test likely indicates abnormal mucosal surface area or permeability and has been observed in horses with CIBD, parasitism, and idiopathic villous atrophy.488, 495 Abnormal absorption curves have been detected in the absence of small intestinal histologic changes,496 and interpretation is clouded further by findings from small intestinal resection studies in healthy ponies. One study demonstrated a progressive decline in mean peak xylose concentration after 70% distal small intestinal resection in ponies despite their normal clinical appearance and absence of diarrhea.497 In another study, a similar decline in peak xylose concentration following extensive (≥60%) small intestinal resection was accompanied by weight loss, diarrhea, and ill thrift.498 Peak xylose concentrations were much lower in horses with granulomatous enteritis than those with eosinophilic granulomatosis (EG), whereas in EG the absorption curve shifted to the right, with the peak occurring at 240 minutes.499 This is not surprising given the typical lesion distribution with these disorders. As with the OGTT, results of the xylose absorption test can improve following therapy.500
Alimentary Lymphosarcoma
Alimentary lymphosarcoma of the horse may represent a primary neoplasia of the gut-associated lymphoid tissue with significant cellular infiltration of the small intestine and associated lymph nodes with minimal large intestinal or systemic involvement. Case series and pathology reports indicate that young horses 2 to 4 years of age primarily are affected, although the age range can be broad.501, 502, 503 No breed or sex predilection has been documented, and disease prevalence is unknown. Despite the progressive nature of lymphomata, onset of clinical signs can be rapid, and the animal may become acutely ill. As with all adult cases of CIBD, antemortem diagnosis is by a process of exclusion and usually is confirmed postmortem. Frequent abnormalities include anemia, thrombocytopenia, neutrophilia or neutropenia, and hypoalbuminemia with hyperglobulinemia, resulting in either a normal elevated serum protein. Lymphocytosis is rare. Intraabdominal masses such as enlarged mesenteric lymph nodes may be palpated per rectum. Abdominocentesis and rectal biopsy can provide a diagnosis but are not sensitive indicators of disease. Carbohydrate absorption tests usually reveal partial to total malabsorption indicative of the severely reduced surface area resulting from significant villous atrophy and the extensive mucosal or transmural infiltration. Confirmation of a diagnosis requires exploratory laparotomy to obtain multiple intestinal and lymph node biopsies if rectal biopsy and/or abdominocentesis are normal. Prognosis is poor, especially because most horses are presented in an advanced state of disease. Immunosuppressive drugs or chemotherapy may afford temporary improvement, but long-term outcome is unaffected.
Granulomatous Enteritis
Granulomatous enteritis was first described as a chronic wasting condition in 1974504; 9 of 10 horses were young Standardbreds. Most affected horses are 2 to 3 years of age. Case reports from many countries revealed a predominance of Standardbred over Thoroughbred horses by three to one.499, 505 Some of the Standardbreds were related, implicating a genetic predisposition, but this has not been proven. Prevalence of this disease is low. The condition is sporadic and has an insidious onset, and the course can be protracted. Significant diagnostic features include anemia, slight increases or decreases in WBC counts, hypoalbuminemia, normal serum protein or hypoproteinemia, occasional increases in serum AP activity, normal serum GGT activity, and enlarged mesenteric lymph nodes on rectal palpation. Partial or complete malabsorption is typically documented via carbohydrate absorption testing. One can attribute the low proportion of horses exhibiting diarrhea to the preferential distribution of inflammatory infiltration in the small intestine.506 Rectal biopsy can be a useful aid to diagnosis.489
The cause of granulomatous enteritis is unknown. Several infectious agents have been implicated, including Mycobacterium avium. 507 The condition may represent a granulomatous hypersensitivity reaction. Immunomediated responses to dietary, parasitic, or bacterial antigens may be important initiating factors.488 Six horses purported to have granulomatous enteritis were linked to environmental contamination with aluminum,508 although problems existed regarding the case definition, data, and interpretation.509
Treatment of horses with granulomatous enteritis with a variety of drugs, particularly corticosteroids, has not affected the long-term outcome in the majority of cases.510 Prolonged (5 months) corticosteroid administration produced clinical remission and a favorable athletic outcome in a 6-year-old Standardbred gelding based on improvement in clinical signs and in d-xylose absorption.500 Surgery may be indicated with localized disease. Two young horses underwent resection of the thickened terminal small intestine; one horse died 4 months after surgery, and the other remained clinically normal for at least 10 years.505
Multisystemic Eosinophilic Epitheliotropic Disease
Multisystemic eosinophilic epitheliotropic disease (MEED) encompasses disorders characterized by a predominant eosinophilic infiltrate in the gastrointestinal tract, associated lymph nodes, liver, pancreas, skin, and other structures accompanied by some degree of malabsorption and enteric protein loss. The disorders include chronic eosinophilic gastroenteritis,511 EG,499 chronic eosinophilic dermatitis,512 and probably basophilic enterocolitis.513
Although prevalence is low, MEED appears to be more common than granulomatous enteritis. Most affected horses are 2 to 4 years of age, and Standardbreds and Thoroughbreds are reported to predominate. The condition is sporadic, has an insidious onset, and often a protracted course (duration of 1–10 months). Diarrhea is common. Severe skin lesions with exudative dermatitis and ulcerative coronitis are prominent and frequently are the principal presenting complaint. Despite extensive tissue eosinophilia, systemic eosinophilia is rare, and hematologic values are usually unremarkable. Notable features include hypoalbuminemia and elevations in serum GGT and AP activities. Most reports of carbohydrate absorption test findings indicate a reduced or normal peak concentration delayed for at least 180 minutes. Morphologic changes are less pronounced in the small intestine than in the large intestine,514 and small intestinal lesions predominate segmentally in the proximal duodenum and distal ileum. Significant hyperkeratosis of the fundic region may contribute to gastric muscle contractile disruption. Diarrhea can be a consequence of the severe segmental or multifocal granulomatous lesions in the large intestine with mucosal and transmural thickening and extensive ulceration. Abundant fibrosis is a feature of all affected tissues.
The cause of MEED is unknown, and the disease may represent a chronic ongoing immediate hypersensitivity reaction against undefined antigens ingested or excreted into the lumen from parasitic, bacterial, or dietary sources. Infectious agents have not been identified.511, 512 Eosinophilia is a feature of parasitism in the equine intestinal tract, although nematodes rarely have been identified in any lesions of MEED.511, 515 Failure to detect larval structures in these lesions, however, may be attributable to chronicity of the disease and destruction of the parasites in tissue.505 Biopsies of the rectal mucosa489 or of the skin, liver, intestinal tract, and lymph nodes may assist in diagnosis. Unlike the other conditions associated with malabsorption/maldigestion in horses, MEED has definitive liver and pancreatic involvement; thus, maldigestion may contribute to the wasting disease.
Treatment has been attempted with a variety of drugs, including antibiotics, corticosteroids, and anthelmintics with larvicidal activity. Although some horses can improve briefly, the long-term prognosis is poor.
Eosinophilic Enterocolitis
Idiopathic eosinophilic enterocolitis affects segmental lesions in the small or large intestine, inducing signs of colic, often requiring surgical intervention.505, 516, 517 This problem may not involve evidence of malabsorption and does not have multisystem involvement. Because the problem is often associated with signs of colic and not signs of malabsorption, eosinophilic enterocolitis differs from the other conditions discussed in this section and is often diagnosed at the time of surgery. It carries a much better prognosis than the other inflammatory bowel diseases.
Lymphocytic-Plasmacytic Enterocolitis
The morphologic findings in lymphocytic-plasmacytic enterocolitis reflect the predominant infiltrative cellular elements of this rarely encountered condition. No specific clinical or clinicopathologic features differentiate this condition antemortem from other inflammatory diseases of adult horses. In a retrospective study of 14 horses, carbohydrate absorption was abnormal or delayed in 9 of 12 horses, consistent with the predominance of small intestinal pathologic changes.490 Rectal biopsies were abnormal in 3 of 7 horses, two of which were reported as having lymphocytic-plasmacytic proctitis. Prognosis is typically reported as poor, likely caused by the advanced nature of the condition at the beginning of treatment. In the author’s experience, some horses with small intestinal mural thickening, lymphocytic-plasmacytic infiltrate evident on duodenal biopsy, and signs of abdominal pain and/or weight loss may improve with dietary modification and/or corticosteroid therapy.
Proliferative Enteropathy
Proliferative enteropathy (PE) typically affects weanling foals from 3 to 8 months of age and has been reported in North America, Europe, and Australia, causing disease in individuals or outbreaks of multiple affected animals on the same premise.518, 519, 520, 521, 522 PE is uncommon in yearlings and adult horses.518, 523 The disease affects many other species, namely swine, and is caused by L. intracellularis, an obligate intracellular bacterium found in the cytoplasm of proliferative crypt epithelial cells of the jejunum and ileum.518, 524, 525
Like pigs, horses are affected as weanlings. The incubation period is 2 to 3 weeks in nonequine species and is presumed to be similar in horses. In some epidemiologic investigations, close proximity to swine operations was apparent, but in most instances such an association was not evident.518, 523 Comparisons of epidemiologic findings from the swine disease indicated that overcrowding, feed changes, antibiotic usage, and mixing and transportation were potential risk factors at two of the farms in one study, and recent weaning appears to be a common risk factor.518 The window for exposure appears narrow,526 and multiple cases on a given farm are common. Affected animals shedding the organism in the feces serve as a source of infection for herdmates. It is possible that nonequine species serve as reservoirs contributing to outbreaks on horse farms.
Profound hyperplasia of the mucosa associated with proliferation of crypt epithelium and crypt hyperplasia is induced locally in infected islands of tissue that eventually extend to the entire distal jejunum and ileum. L. intracellularis preferentially infects proliferating cells; thus, it is the tropism for the crypt epithelium. Infected cells proliferate far more rapidly than uninfected cells, suggesting that L. intracellularis directly induces the proliferative response, but the molecular basis for enhanced proliferation is not known. L. intracellularis penetrates epithelial cells in a membrane-bound vesicle but eventually escapes the vacuole and is found free in the cytoplasm, concentrated at the apical pole of the cell. The gross pathologic lesions of equine PE are quite characteristic.518 Lesions may be segmental and are most commonly found in the ileum and terminal jejunum in horses, but the duodenum may also be affected. Severe mucosal hypertrophy is often observed but may wane during the chronic stages of the disease. The mucosa may become corrugated with focal erosions or ulcers. Submucosal edema is often readily identified on cut sections of affected segments. Moderate to severe crypt hyperplasia with atrophy of intestinal villi is a consistent feature. Hyperplastic crypts are branched and may herniate into the submucosa. Necrosis, edema of the submucosal and lamina propria, hemorrhage, mononuclear inflammation, and muscular hypertrophy have been reported in affected intestinal segments but are not consistent. Special stains such as silver stain are required to detect intracellular organisms. The organisms are curved or comma-shaped rods found clustered in the apical cytoplasm of hyperplastic crypt epithelium. The proliferative response of the intestinal mucosa alters absorption of nutrients and fluid secretion by disrupting the architecture of the villi and by altering the maturation of epithelial cells into absorptive cells, accounting for the secretory diarrhea and often severe weight loss. The combined effects of the inflammatory response and malabsorption may account for the clinically observed protein-losing enteropathy.
Clinical signs include depression, rapid and significant weight loss, edema, diarrhea, and colic.518 Poor body condition, a rough hair coat, and potbelly appearance are also reported. Not all clinical signs are present in all cases, and diarrhea has only been observed in approximately half of reported cases. Other problems often were concurrent, including respiratory tract infection, dermatitis, intestinal parasitism, and gastric ulceration. The most significant laboratory finding is profound hypoproteinemia, predominantly characterized by hypoalbuminemia; panhypoproteinemia can also occur.518, 521, 527 Leukocytosis and hyperfibrinogenemia are also common, with occasional alterations in electrolytes (hyponatremia, hypokalemia, and hypochloremia) and elevated serum creatine kinase concentrations. Abdominal ultrasound commonly reveals increased small intestinal mural thickness.521, 527 Although small intestinal mural thickness >3 mm, in conjunction with clinical and clinicopathologic signs, is highly suggestive, PE should not be ruled out in the absence of this finding. Colloid oncotic pressure, if measured, is typically low.527
PE should be considered in a weanling foal with compatible clinical signs and severe hypoalbuminemia with exclusion of common enteric infections. Fecal PCR has very high specificity but variable sensitivity for confirmation of diagnosis.528 Serum immunoperoxidase monolayer assay or indirect enzyme-linked immunosorbent assay (ELISA) are highly specific for exposure; submission of fecal PCR and serologic testing is recommended, although both tests are quite specific, because they can lack sensitivity, especially early in the course of disease (serology) or with prior antimicrobial therapy (fecal PCR).518, 527, 529 Of note, fecal PCR can become negative in affected foals within 4 days of antimicrobial therapy.530 PE is not typically associated with abnormal carbohydrate absorption test results.518, 530 In horses with diarrhea, other infectious causes should be ruled out. A definitive postmortem diagnosis can be confirmed by identifying characteristic mural thickening and intracellular bacteria within the apical cytoplasm of proliferating crypt epithelial cells using silver stains, PCR, and/or immunohistochemical testing.518
Antimicrobial therapy with erythromycin, alone or with rifampin, azithromycin, clarithromycin, oxytetracycline, doxycycline, metronidazole, or chloramphenicol has been reported.518, 519, 527 Macrolides should not be used in adults or older foals because of an increased risk of colitis.529 Recent reports favor the use of IV oxytetracycline, followed by oral doxycycline or minocycline, with apparent success.519, 527 Duration of therapy is typically 2 to 3 weeks. Affected foals often need supportive therapy including crystalloid fluid and electrolyte replacement and, potentially, colloid support. Nonsteroidal antiinflammatory therapy can be used as needed for significant pyrexia. Corticosteroid therapy is not indicated, because inflammation is not a significant pathologic finding. Response to therapy has been good, with reported survival rates between 82% and 93%.518, 519, 527 Rapid improvement in clinical signs, even within 24 hours, precedes the rise in plasma protein concentration.
Foals with PE should be isolated from unaffected animals for at least 1 week after institution of antimicrobial therapy to avoid shedding of organisms into the environment. Intrarectal vaccination with an avirulent live vaccine marketed for use in pigs has demonstrated a protective effect in foals experimentally challenged with L. intracellularis. 531
Miscellaneous Conditions
Abnormal d-xylose absorption has also been noted in association with AA amyloid-associated gastroenteropathy in an 18-year-old Morgan stallion532 and a horse with a gastric mass and secondary small intestinal villous atrophy.533
Management, Therapy, and Outcome
The chronic wasting horse with suspected malabsorption and probable enteric protein loss generally has a guarded to poor prognosis. Diseases with a good prognosis include eosinophilic enteritis and PE. Prognosis may be improved through early and aggressive investigation to achieve a diagnosis. The owner must be cognizant from the start that the outcome may not be altered, even after protracted therapy; only a few case reports of successful responses with long-term follow-up have been documented.
Nutrition
Some level of digestive and absorptive capability remains in the diseased small intestine. Interval feeding of small quantities of easily digestible food may be beneficial. Diet may include feeds with a high fiber, but potentially low bulk. Some commercially available complete pelleted feeds offer high-fiber rations based on beet pulp and soybean hulls. Some affected horses may tolerate increased dietary fat. Consultation with an equine nutritionist may provide additional options for a given horse and, in the author’s experience, can prove beneficial. The objective for an affected horse is to sustain, and preferably increase, dietary intake, value, and efficiency. The owner of an affected horse must be prepared to experiment with feeds slowly and deliberately, must be patient, and must keep records. Exposure to a feed component may contribute to the problem as an allergen eliciting a hypersensitivity reaction. Identifying the potential allergen through immunologic testing or by stepwise removal and outcome assessment over a longer period may be difficult.
Pharmacologic Therapy
Immunosuppressive agents have produced the most promising responses to ameliorate the effects of conditions associated with malabsorption, particularly CIBD. Short-duration, and in some cases more prolonged and sustained, improvements in body condition, weight gain, demeanor, energy, and activity levels have occurred following corticosteroid administration. Treatment should be initiated as early as possible in the course of disease. Initial parenteral (intramuscular or IV) loading doses of dexamethasone (sodium phosphate) should be followed with a series of depot injections, or orally administered prednisolone, on a tapered dose protocol over a period of months. Interval low-dose therapy may be necessary if clinical signs return after treatment ends. The lowest dose necessary to control the clinical signs with alternate-day therapy should be used. Clinical benefits far outweigh concerns over potential adverse effects. Chemotherapeutic agents such as vincristine, cytosine, cyclophosphamide, and hydroxyurea have been tried in a few cases of CIBD or lymphosarcoma with no apparent success, probably related to the advanced stage of the disease when treatment was initiated and the dose selected.
Surgery
Resection of a segment of intestine that is edematous, hemorrhagic, or constricted is an option in localized forms of CIBD,505, 517 particularly if gross changes are not discernible in adjacent or distant parts of the intestinal tract—that is, malabsorption is not a feature. Long-term outcome has been favorable. Removal of a substantial proportion of the diseased small intestine may be indicated in a horse with malabsorption, considering that resection of 70% distal small intestine was performed in healthy animals without inducing adverse effects.497 Because pathologic changes may exist in normal-appearing small or large intestine that is not resected or biopsied, the prognosis remains guarded. Two young horses with granulomatous enteritis had the thickened terminal small intestine resected with positive outcomes; one survived 4 months, the other has a follow-up extending more than 10 years.505
Inflammatory Diseases of the Large Intestine
Acute diarrhea caused by colitis in adult or young horses is a potentially life-threatening disorder with a variety of possible etiologies (Table 12.5 ) characterized by hypersecretion of fluid, motility disturbances, and an impaired mucosal barrier resulting from direct injury or inflammation. Many of the clinical and clinicopathologic features are similar regardless of the underlying cause. Severe dehydration with profound electrolyte abnormalities is common, as is systemic inflammation secondary to absorption of endotoxin or other bacterial products through compromised gastrointestinal mucosa. Severe cases may be complicated by serosal inflammation and mural ischemia and infarction as a direct extension of mucosal inflammation or secondary to coagulopathies. The diagnostic approach for horses with acute diarrhea is aimed at determining the underlying etiology but must be accompanied by clinical and laboratory assessment of hydration, electrolyte and acid-base balance, organ function, and evaluation of the degree of systemic inflammation and the integrity of the intestinal wall. The therapeutic approach for horses with colitis, regardless of cause, consists primarily of controlling local and systemic inflammation, maintaining fluid and electrolyte balance, and promoting mucosal repair. In addition, some horses with acute colitis require specific therapy aimed at the underlying etiology.
TABLE 12.5.
Differentials and Diagnosis of Some Causes of Acute Diarrhea in Adult Horses
| Category | Differentials | Diagnosis |
|---|---|---|
| Infectious | Salmonellosis | Fecal culture (five consecutive) Fecal PCR |
| Clostridium perfringens | Quantitative fecal culture Fecal toxin immunoassay or PCR |
|
| C. difficile | Fecal culture Fecal toxin immunoassay or PCR |
|
| Neorickettsia risticii | Fecal or blood PCR Serology |
|
| Coronavirus | Fecal PCR | |
| Parasitic | Strongylosis | Fecal egg counts Cranial mesenteric artery palpation Serum IgG(T) |
| Cyathostominosis | Fecal egg count Rectal biopsy Cecal or colonic biopsy |
|
| Toxic | NSAID | History and clinical signs Right dorsal colon ultrasonography Laparoscopy or laparotomy |
| Cantharidin | History of exposure Fecal or urine cantharidin concentrations |
|
| Arsenic | History of exposure Fecal, blood, urine, or tissue arsenic concentrations |
|
| Miscellaneous | Carbohydrate overload | History of inappropriate ingestion of carbohydrate Blood lactate concentration |
| Sand enteropathy | Auscultation of ventral colon Fecal sand content Abdominal radiography |
Ig, Immunoglobulin; NSAID, nonsteroidal antiinflammatory drugs; PCR, polymerase chain reaction.
Infectious Diseases
Salmonellosis
Pathogenesis
S. enterica is a species of gram-negative facultatively anaerobic bacteria that is a common gastrointestinal pathogen in horses. Many serovars of S. enterica have been reported to infect horses, but those classified in group B appear to be more commonly associated with disease than those in other groups. Group B includes S. enterica var. Typhimurium and S. enterica var. Agona, two of the species most frequently isolated from horses.534, 535, 536 S. enterica var. Typhimurium is the most pathogenic serotype in horses and is associated with a higher case fatality rate than other serovars of S. enterica. 534 The number of horses that are inapparently infected with and actively shed S. enterica in their feces has been reported to be as high as 10% to 20%, but actual prevalence of S. enterica shedding in the general horse population is likely to be much lower, less than 2%.537 Horses shedding S. enterica are a potential source of infection to susceptible horses,534, 538 as are environmental reservoirs.539, 540, 541 For these reasons salmonellosis is one of the most common nosocomial diseases in horses. Nosocomial salmonellosis significantly affects morbidity and mortality in hospitalized horses.542 The emergence of multidrug resistance in equine S. enterica isolates has been a cause of concern because of the importance of salmonellosis as a nosocomial disease and because a number of serovars of S. enterica are significant zoonotic pathogens.540, 543, 544, 545, 546
The virulence of the bacteria varies tremendously with serotype and even among strains of the same serotype. This is because of the important role of host susceptibility in the pathogenicity of particular organisms. The infective dose is generally on the order of millions of organisms inoculated orally, but various environmental and host factors can reduce the infective dose to a few thousand or even hundreds of organisms.547, 548, 549 Environmental factors or stresses that increase susceptibility to S. enterica infection are not well defined, but it is known that high ambient temperature, for example, can greatly increase the prevalence of salmonellosis in horses.539, 548, 549 Indeed, the peak incidence of salmonellosis in horses occurs in late summer and fall.539, 548, 549 Other environmental and host factors that are associated with salmonellosis or shedding of S. enterica organisms in feces include transportation, antibiotic administration before or during hospitalization, gastrointestinal or abdominal surgery, general anesthesia, preexisting gastrointestinal disease (e.g., colic, diarrhea), the presence of leukopenia or laminitis during hospitalization, prolonged hospital stay, change in diet, and immunosuppression.534, 541, 549, 550, 551 Interestingly, foals with gastrointestinal disease are more likely to shed S. enterica organisms than are adult horses with gastrointestinal disease.548
Host factors that restrict gastrointestinal colonization and invasion by pathogens include gastric pH, commensal gastrointestinal flora, gastrointestinal motility, the mucosal barrier, and mucosal immunity.534, 552 Gastric acidity is an important defense mechanism for preventing live organisms from reaching the intestine.552 Altering the gastric pH, with histamine H2 receptor antagonists, for example, may increase susceptibility to infection. Gastrointestinal flora inhibit the proliferation and colonization of S. enterica by secreting bacteriocins, short-chain fatty acids (SCFAs), and other substances that are toxic to S. enterica. 552 Elements of the normal flora compete for nutrients and space, especially on the mucosa.552 Being predominantly anaerobic, the normal flora maintain a low oxidation-reduction potential in the environment of the large intestine, which inhibits the growth of many bacterial pathogens.553 The importance of normal host gastrointestinal ecology is illustrated by the fact that disturbances of the colonic flora with antibiotics, changes in feed, ileus, or other underlying gastrointestinal disease markedly increases the susceptibility of the host to infection by S. enterica, often resulting in serious disease.
The immune status of the host may be one of the most important factors determining not only the susceptibility to S. enterica infections but also the degree of invasion and subsequent outcome of the infection. Local immunity, such as mucosal antibody secretion and enterocyte-derived cationic peptides, prevents colonization of the mucosa.552, 554, 555 Opsonizing antibodies and activation of the complement cascade are important in fighting systemic invasion by S. enterica by increasing the efficiency of phagocytosis and by direct bactericidal activity. Humoral immunity, however, is often ineffective in preventing disease and dissemination once invasion occurs and S. enterica is established in its intracellular niche. Following invasion, S. enterica is capable of surviving and multiplying within macrophages, rendering the humoral (noncellular) immune systems ineffective.556, 557 Specific cellular immunity may be the most effective defense mechanism in the host arsenal against dissemination and systemic infection by S. enterica. 557, 558 Protective immunity in horses and calves may be induced by oral inoculation with small numbers of virulent organisms, but the duration of the immunity is not known.559, 560 Oral and parenteral vaccines using killed or attenuated organisms and bacterial products have been promising but are effective only against homologous organisms and are usually not cross-protective among different serogroups.559, 560, 561
In adult horses S. enterica primarily infects the cecum and proximal colon, causing enterocolitis, with limited likelihood of dissemination beyond the intestine. In foals, however, salmonellosis is often associated with septicemia. The ability of S. enterica to cause enterocolitis depends on the ability of the bacteria to invade the gastrointestinal mucosa.552, 556 Invasion of the gastrointestinal mucosa occurs preferentially through specialized enterocytes called M cells that overlie intestinal lymphoid tissues such as Peyer’s patches in nonequine species. M cells are exploited by a variety of enteric pathogens during infection of intestinal tissue.562 Invasion of the epithelium occurs by self-induced uptake via the apical membrane of the M cell, often killing the cell in the process.556 S. enterica then invades neighboring cells via the basolateral membrane, eventually spreading the destruction of the epithelium beyond the principal area of attack. Virulent S. enterica have a well-developed invasion mechanism that involves the generation of an apparatus called a type III secretory system that enables virulence gene products to be injected directly into enterocytes.563 Virulence proteins injected by S. enterica into enterocytes engage the cellular machinery and induce the cell to engulf the bacteria by macropinocytosis. S. enterica virulence gene products also induce enterocyte chloride and fluid secretion and upregulate enterocyte transcription of inflammatory cytokines (TNF-α and IL-1β) and chemokines that trigger a mucosal inflammatory response.81, 556, 563
Once S. enterica has invaded the mucosa, organisms are quickly phagocytosed by macrophages and dendritic cells in the lamina propria and in lymphoid tissues. The ability of S. enterica to disseminate systemically and cause enteric fever is associated with the ability to survive and proliferate in macrophages. Indeed, phagocytes have an important role in dissemination to blood, lymph nodes, liver, and spleen.564 The majority of S. enterica in the blood and tissues of animals infected with a strain of S. enterica that is competent to cause enteric fever are within phagocytic cells.564 In adult horses with salmonellosis, dissemination appears to be limited to the intestine and mesenteric lymph nodes, and S. enterica is rarely cultured from blood. However, in foals and in some adults, S. enterica causes an enteric fever–like disease with dissemination to mesenteric lymph nodes, liver, spleen, and blood.
Specific virulence gene clusters called pathogenicity islands encoded on the chromosome or on plasmids confer the main virulence traits of S. enterica: invasion, enteropathogenesis, intracellular survival, and proliferation.556 Some of the genes encoded within these islands or virulence factors are sensors that signal to the bacteria that it has entered an intracellular environment and to turn on other genes required for intracellular survival. Others, such as invasion genes, are transported from the bacteria and injected into macrophage cytosol by a type III secretory system apparatus to prevent phagosome-lysosome fusion and subvert other essential macrophage-killing mechanisms. Virulent S. enterica may also possess multiple genes that enable adhesion to target cells or confer resistance to reactive oxygen and nitrogen metabolites, which is, perhaps, the most lethal antimicrobial mechanisms of macrophages.565
Diarrhea associated with salmonellosis has multiple causes. An S. enterica cytotoxin inhibits protein synthesis in mucosal cells, causing morphologic damage and altered permeability.566 Virulent S. enterica also produce an enterotoxin that is similar to the heat-labile (LT) toxin produced by E. coli. 567, 568 This enterotoxin contributes to, but is not required, in the pathogenesis of diarrhea.569, 570 S. enterica enterotoxin increases secretion of chloride and water by colonic mucosal cells in many species, including horses, by increasing intracellular cAMP concentrations.567, 568, 571
The ability of virulent S. enterica to cause diarrhea appears to be most closely associated with the ability to invade enterocytes and to trigger an inflammatory reaction in the intestinal tissue.103, 556 Gene products injected into enterocyte cytosol by the type III secretory system of invading S. enterica stimulate chloride and fluid secretion.563 S. enterica invasion of enterocytes is also a potent activator of inflammatory chemokine and cytokine production, resulting in the recruitment of leukocytes, particularly neutrophils, and activation of resident macrophages and mast cells. Products of these activated leukocytes, including prostaglandins, leukotrienes, reactive oxygen metabolites, and histamine, are potent stimulators of chloride secretion in the colon of many species.213, 552, 572, 573 The ENS integrates the diverse processes of pathogen recognition, triggering of the inflammatory response, and induction of enterocyte fluid secretion.213
Many of the inflammatory mediators studied stimulate colonic secretion by prostaglandin-dependent mechanisms, resulting in either increased intracellular cAMP or calcium concentrations, or both, in mucosal cells.213 These mediators and the ENS may also stimulate secretion by prostaglandin-independent mechanisms, inhibit sodium and water absorption, cause motility disturbances, and potentiate tissue injury, all of which enhance the pathogenicity and dissemination of S. enterica and contribute to the pathogenesis of diarrhea.213, 573 Neutrophils recruited to the mucosa by signals generated by the infected enterocytes physically contribute to mucosal injury by producing a variety of products that are lethal to pathogens but are also toxic to host cells.84, 574 Neutrophils attracted to infected epithelial cells accumulate beneath the monolayer, lifting it off the basement membrane in sheets. Neutrophils also migrate across the epithelial monolayer in potentially massive numbers—enough to be detectable in feces as a marker of inflammatory diarrhea. Although the transepithelial migration of neutrophils has a benefit, positioning the host defense cell at the apical membrane to ward off attacks by invading bacteria, the mechanical disruption to the epithelial barrier may be significant enough to increase the permeability to macromolecules, bacterial products, and even bacteria.574 Potentially massive losses of electrolytes, water, and protein can occur, depending on bacterial and host factors. Perhaps most devastatingly, mucosal injury and altered permeability allow systemic absorption of bacterial products and dissemination of bacteria, resulting in life-threatening sepsis.
Clinical Signs and Diagnosis
Four clinical syndromes of S. enterica infection have been documented clinically and reproduced experimentally in horses: (1) inapparent infections with latent or active carrier states; (2) depression, fever, anorexia, and neutropenia without diarrhea or colic; (3) fulminant or peracute enterocolitis with diarrhea; and (4) septicemia (enteric fever) with or without diarrhea.575 Inapparent infections can be activated to clinical disease in compromised horses, such as horses with colic or horses being treated with antibiotics, causing mild to severe enterocolitis. Latent infections (nonshedding) can become active infections (shedding) under certain conditions, such as transportation stress and antibiotic treatment. Horses with depression, anorexia, fever, and neutropenia without diarrhea generally have a good prognosis and recover in several days without specific treatment.575 The septicemic form is mostly restricted to neonatal foals and is uncommon in adult horses. The focus of this discussion is acute enterocolitis.
Acute enterocolitis is characterized by severe fibrinonecrotic typhlocolitis, with interstitial edema and variable degrees of intramural vascular thrombosis that may progress to infarction.534 Severe ulceration of the large intestinal mucosa may occur, with serosal ecchymoses and congestion. The earliest signs of enterocolitis are usually fever and anorexia.534, 549 Signs of colic may be seen early in the course of the disease, especially if ileus is present. Clinical signs of endotoxemia are common and range from fever, elevated heart and respiratory rates, poor peripheral perfusion, and ileus to fulminant and rapidly progressive signs of endotoxemic shock. Oral mucous membranes are often pale with perigingival hyperemia (a toxic rim) but may be brick red or cyanotic, with prolonged capillary refill time. Weakness, muscle fasciculations, cold extremities, and other signs suggestive of hypotensive shock; synchronous diaphragmatic flutter; abdominal pain; and marked metabolic and electrolyte abnormalities may be noted in severe cases of enterocolitis. Signs of mild dehydration may be observed before diarrhea is seen. Once diarrhea is evident, dehydration may rapidly become severe. Occasionally, horses die peracutely, without developing diarrhea.
Diarrhea may not occur for several days but usually is evident by 24 to 48 hours after the onset of fever.534, 549 The duration of diarrhea may be days to weeks. The character of the first diarrheal feces is usually watery, with particles of roughage, but may rapidly become fluid without solid material. Finding frank blood and fibrin in the feces is unusual. The volume of feces is often large, with frequent defecation. Straining or signs of colic may be observed when the patient is defecating, and rectal prolapse may occasionally occur. Persistent straining and rectal prolapse may be signs of colonic infarction. Abdominal borborygmi are often absent early in the course of the disease because of ileus but become evident later, usually when diarrhea begins. Fluid and gas sounds are commonly auscultated, but normal progressive motility is less frequently heard. Transrectal palpation may reveal edematous rectal and colonic mucosa and fluid-filled colon and cecum. Gastric reflux may be obtained, especially early in the course, when ileus is evident.
Hematologic abnormalities early in the course of the disease include moderate to severe neutropenia, lymphopenia, and leukopenia; a mild to moderate left shift; and toxic changes in the neutrophils.534, 549 Thrombocytopenia, moderate to severe hemoconcentration, and hyperfibrinogenemia are also common. Neutropenia is an early but nonspecific indicator of salmonellosis, often occurring concurrently with the onset of fever.534 Later in the course of disease, neutrophilic leukocytosis may be seen, indicating recovery. A degenerating left shift, with metamyelocytes and myelocytes seen in the peripheral blood, is a poor prognostic sign.
Serum biochemical analysis may reveal azotemia, increases in serum sorbitol dehydrogenase and γ-glutamine aminotransferase activity, and increased blood lactic acid concentration. Azotemia is often prerenal, but acute hemodynamic renal failure may be seen in severely dehydrated, endotoxemic, or septic patients. Indeed, elevation of creatinine concentration is a poor prognostic indicator in horses with acute colitis.576 Hemodynamic renal disease may be complicated by toxic injury caused by the administration of nephrotoxic drugs. Hyponatremia may also contribute to prerenal azotemia. Elevations in hepatocellular enzymes are usually mild and reflect damage to the hepatocytes from absorbed toxins, such as endotoxin, and from poor perfusion resulting from hypotensive shock, dehydration, or both. Lactic acidemia may be present, reflecting poor tissue perfusion. Plasma protein drops rapidly as protein is lost in the gastrointestinal tract, resulting in moderate to severe hypoalbuminemia and hypoglobulinemia. Peripheral or organ edema (vascular leak syndrome) may occur if hypoproteinemia is severe, coupled with systemic inflammation-induced increases in endothelial permeability.
Hypokalemia, hyponatremia, hypochloremia, and hypocalcemia are common electrolyte abnormalities in patients with enterocolitis. Metabolic acidosis may also be present, and DIC is common. Urinalysis may reveal isosthenuria, proteinuria, hematuria, cylindruria, or glucosuria if hemodynamic or toxic renal injury is present. The number of leukocytes in the feces is usually increased, and occult blood may be detected. PF is usually normal except when severe mural inflammation or colonic infarction occurs.
S. enterica in feces is routinely detected by analyzing five daily cultures of large samples (10–30 g) of feces using enrichment techniques.534, 577, 578 The sensitivity of fecal culture can be as low as 30% to 50%, even if several fecal samples collected daily are cultured.578 Concurrent culture of rectal biopsy specimens and feces increases the sensitivity of culture techniques to 60% to 75%.578 Currently, the PCR test is the most sensitive and rapid way to detect S. enterica in feces. A single PCR test applied early in the course of disease is a more sensitive test for the presence of S. enterica than repeated fecal cultures,579, 580 with as high as 100% sensitivity and 98% specificity for detection of organisms in some reports.581 Although detection of S. enterica organisms in feces does not prove a diagnosis of salmonellosis, the positive predictive value of either a positive PCR or culture result is high in horses with compatible clinical signs. Culture of peripheral blood may allow isolation of the organism if bacteremia or septicemia is present, but blood cultures are not a sensitive test for salmonellosis in adult horses. Foals are more likely than adults to become septicemic, and so blood culture is recommended in all foals with signs of sepsis. Increased numbers of fecal leukocytes suggest an invasive process in the colon but are not specific for salmonellosis.
Early in the course of the disease, dehydration, electrolyte and acid-base imbalances, endotoxemia, and sepsis may be life-threatening. Aggressive treatment during the acute stages to replace fluids lost in the diarrhea and to control sepsis and endotoxemia is often effective in controlling the primary disease. Weight loss and hypoproteinemia are often severe. Possible complications include multiorgan dysfunction, vascular leak syndrome with peripheral and organ edema, laminitis, acute renal failure, venous thrombosis and septic phlebitis, irreversible protein-losing enteropathy or chronic malabsorption, pulmonary aspergillosis, and gastrointestinal infarction. The reader is referred to the section Endotoxemia in this chapter for additional information regarding treatment of horses with severe endotoxemia and SIRS.
In many instances horses recover from acute salmonellosis with aggressive treatment, only to succumb to complications of the disease, which partially explains the high fatality rate of equine salmonellosis compared with that of human salmonellosis. Chronic mild to moderate diarrhea is occasionally seen in horses after a bout of severe salmonellosis, usually with protein-losing enteropathy. If the chronic diarrhea persists beyond 4 to 5 weeks after the onset of signs, the prognosis for recovery is poor.549
Potomac Horse Fever
Pathogenesis
Potomac horse fever is caused by the obligate intracellular rickettsial organism N. risticii (formerly called Ehrlichia risticii).582, 583, 584, 585, 586 The disease is most common from late summer to early fall, with a peak incidence in July and August.583, 584 Potomac horse fever was first described in the northeastern United States but has since been described now in most areas of the continental United States, with a particularly high prevalence in the Northeast and Midwest. The geographic distribution is characterized by a significantly higher percentage of cases found along waterways and rivers.583, 584 The disease occurs sporadically, both temporally and geographically, and can affect any age group of horses. The case fatality rate ranges from 5% to 30%.583
Transmission of N. risticii has been reproduced experimentally by oral, intramuscular, intradermal, SC, and IV routes.583, 587 Attempts to transmit the disease experimentally with ticks (Dermacentor variabilis) or biting flies (Stomoxys calcitrans) were unsuccessful.588, 589 N. risticii infects virgulate cercariae, larval stages of trematodes that use operculate freshwater snails of the family Pleuroceridae (Juga spp. in California and Elimia spp. in Ohio and Pennsylvania), as intermediate hosts in their life cycle.590, 591, 592, 593 Infected virgulate cercariae have been identified in aquatic snails collected in other parts of the world as well.594 Although the trematode species infected with N. risticii remain to be definitively identified, at least two species have been identified as potential vectors,595 and at least two potential definitive hosts were identified when N. risticii DNA was detected in the blood, liver, or spleen of 23 of 53 little and big brown bats harboring gravid trematodes in their intestinal tracts.596
Aquatic snails release large numbers of infected cercariae into water, in which they seek their next intermediate host, which is any of a variety of aquatic insects.593, 597 Successful transmission of N. risticii to horses was accomplished experimentally using trematode stages collected from Juga yrekaensis snails.598 The number of PCR-positive snails in endemic regions corresponds to the seasonal incidence of Potomac horse fever and may be as high as 26%.599 Preliminary studies suggest that N. risticii may in fact be naturally transmitted to horses through the ingestion of caddisflies and mayflies.593, 600
The pathogenesis of N. risticii is not completely understood. The organism infects and survives in monocytes and monocyte-derived leukocytes and can be found in blood monocytes during natural infections, but the sequence of events resulting in enterocolitis remains open to speculation. The organism appears first to infect blood monocytes in experimentally infected horses, which may be the vehicle of organ infection.585, 601 It is unclear whether leukocytes of the monocytic lineage or epithelial cells are infected first in naturally infected horses. The target organ is the gastrointestinal mucosa, with the most severe lesions found in the large intestine.156, 601 Infection of human colonic cells in vitro does not cause major cytopathologic effects for several days.602 Disruption of the microvilli in the region of the plasma membrane in which sodium chloride channels are located has been observed in human colonic cell cultures.602 Infection in horses is associated with variable degrees of morphologic damage.156, 601 Mild morphologic damage and mononuclear cell infiltration of the lamina propria occur early during the infection, but fibrinous, necrotizing typhlocolitis with severe mucosal ulceration and inflammation of the lamina propria may occur later in the disease. Vasculitis and intravascular coagulation are consistent features in the large intestine, with perivascular edema.156 N. risticii can be observed in mucosal cells and macrophages and mast cells of the lamina propria.156, 601 N. risticii can survive and multiply in macrophages by inhibiting the production of ROIs and avoiding lysosomal digestion by blocking phagosome–lysosome fusion.603, 604, 605
Some researchers have suggested that impaired sodium chloride absorption in the colon contributes to diarrhea in infected horses and may be related to destruction of the enterocyte membrane structure in the region of sodium chloride channels.602, 606 Direct injury to the mucosa by N. risticii and colonic inflammation are likely to be prominent features leading to diarrhea, especially later in the disease.156 Loss of fluid, protein, and electrolyte is likely caused by mucosal injury and effects on enterocyte fluid secretion caused by the inflammatory response. Like other inflammatory conditions of the colon, systemic inflammation caused by absorption of bacteria and bacterial products is a potential complication of N. risticii infections if mucosal injury is severe, which contributes to the clinical signs seen during the disease.
Clinical Signs and Diagnosis
N. risticii infection is clinically similar to other forms of enterocolitis and is characterized by anorexia, depression, and fever.156, 583, 607 Experimental infections produce a biphasic fever in which the second febrile phase occurs 6 to 7 days after the first.607, 608 Decreased gastrointestinal motility, manifested as reduced borborygmi, occurs during the early stages, before the onset of diarrhea. Diarrhea is seen in 75% of cases and occurs 2 days after the second fever episode during experimental infections.607, 608 The diarrhea can be moderate to severe and dehydrating. Ileus can develop at any stage of the disease and can cause signs of moderate to severe colic. Systemic signs of endotoxemia, shock, and peripheral edema may occur and are similar to those described for salmonellosis. Experimental and natural infection with N. risticii can cause abortion of infected fetuses in pregnant mares.609, 610 Laminitis is a complication in 20% to 30% of naturally occurring cases and is often severe.584 Other complications include protein-losing enteropathy, thrombosis, and renal failure, as described for salmonellosis.
Hematologic abnormalities reflect endotoxemia, dehydration, and sepsis and are essentially identical to those described for salmonellosis. Neutropenia with a left shift is a consistent feature and occurs concurrently with or soon after the onset of diarrhea.608 Thrombocytopenia is common and often severe.608 Neutrophilic leukocytosis occurs later in the course of the disease. Hyperfibrinogenemia is usually more pronounced than that seen with salmonellosis. Serum electrolyte, acid-base, and biochemical abnormalities are also similar to those described for salmonellosis. Coagulopathies are commonly seen during N. risticii infection and reflect activation of coagulation pathways. DIC is not uncommon and may be the cause of the high frequency of laminitis associated with N. risticii infection.611
Diagnosis of N. risticii infection cannot be based solely on clinical signs because the disease is clinically similar to other forms of enterocolitis. In endemic areas, acute colitis is likely to be caused by N. risticii; thus, the clinical signs of acute inflammatory colitis may in fact have a high predictive value in these areas. Serologic evidence of infection, such as rising antibody titers to N. risticii detected by indirect immunofluorescence (IFA) or ELISA in paired serum samples, may be helpful in establishing a diagnosis.584, 612 Care should be taken when interpreting the IFA serologic test for N. risticii because the test appears to have a high false-positive rate.613 Culture of the organism from blood is possible but difficult and is generally useful only in the research laboratory. Recently developed PCR tests for N. risticii DNA are rapid, highly sensitive (as sensitive as culture), and there are specific tests for N. risticii infection that can be applied to blood or feces.614, 615, 616
Prevention
Prevention of the disease by reducing exposure to the etiologic organism is difficult because the mode of transmission is not known. A killed vaccine has been developed that is relatively effective in preventing clinical illness other than fever in 80% of experimentally challenged horses using the vaccine strain. Field studies, however, suggest the vaccine has limited benefit for preventing natural infection or decreasing its severity.617, 618 Vaccine failures have been attributed to strain differences in antigenicity or to poor antibody responses to the vaccine.617, 618
Equine Intestinal Clostridiosis
Pathogenesis
Clostridiosis is an important cause of acute enterocolitis in foals and adult horses. C. perfringens and C. difficile are most commonly associated with intestinal clostridiosis in horses, but other clostridial species, including C. septicum, C. cadaveris, and C. sordellii have also been isolated from horses with enterocolitis.619, 620, 621, 622, 623, 624 In horses of all ages, clostridial enterocolitis appears to be a common antibiotic-associated and nosocomial cause of enterocolitis.623, 625, 626 Hemorrhagic enterocolitis caused by C. perfringens in neonatal foals is a distinct clinical entity and will be discussed in more detail in Chapter 20. This discussion focuses on adult intestinal clostridiosis.
Clostridium organisms are obligate anaerobic to aerotolerant spore-forming gram-positive rods that are ubiquitous in the environment in the spore form.624 They are elements of the normal flora of horses of all ages and are among the first bacteria acquired after birth. Clostridium organisms inhabiting the gastrointestinal tract are normally found in very low numbers and do not produce enterotoxins. Clostridiosis is associated with an increase in the number of a particular species of Clostridia in the gastrointestinal tract and, perhaps most important, exotoxin production. Although the conditions resulting in exotoxin production are not fully understood, several factors increase clostridial numbers in the gastrointestinal tract. Dietary factors affect the numbers of Clostridium species shed in horse feces.619 Experimental induction of colic increases fecal shedding of Clostridium species in the absence of diarrhea.627 Antibiotics, particularly those administered orally or recycled via the enterohepatic system, increase the recovery of Clostridia colony-forming units (CFUs) in equine feces and clinical clostridiosis.620, 622, 628, 629, 630 Clostridiosis associated with C. difficile is likely to be the most important cause of antibiotic-induced enterocolitis in the horse.
Clostridium perfringens
C. perfringens includes many genetically distinct strains of variable virulence that produce one or more of a large group of exotoxins. The pattern of exotoxin production is used to classify C. perfringens into five types: A, B, C, D, and E. C. perfringens type A is the most common clostridial isolate from healthy and diarrheic horses of all ages. C. perfringens types A, B, C, and D have all been associated with hemorrhagic enteritis in foals younger than 10 days of age, with type C being the most common cause in North America.
The primary toxin produced by C. perfringens type A is α-toxin (phospholipase C), which interferes with glucose uptake and energy production and activates arachidonic acid metabolism and signaling pathways in enterocytes.624 Oral administration of α-toxin does not cause tissue necrosis but causes increased secretion by small intestinal mucosal cells.631, 632 The β-toxin of types B and C is a cytotoxin that causes enterocyte necrosis, ulceration, and ultimately severe intestinal inflammation and hemorrhage.624, 632 A novel toxin designated β2 may also have a role in C. perfringens enterocolitis.633 The biologic activity of the β2-toxin is similar to that of β-toxin, but β2-toxin is not related to β-toxin in its genetic sequence. The β2-toxin was prevalent in two groups of horses with acute enterocolitis but not in healthy horses.634 It is predominantly associated with C. perfringens that would have otherwise been classified as type A but that may in fact represent a previously undescribed type.
Virulent strains of C. perfringens type A and, to a lesser extent, type C may produce enterotoxin. Enterotoxin is a cytotoxin that inserts into cell membranes to form pores, which alter permeability to water and macromolecules and ultimately lead to cellular necrosis.635 Massive desquamation of the intestinal mucosa that is a result of enterotoxin cytotoxicity triggers an inflammatory response, intestinal edema, mural hemorrhage, and systemic inflammation.158 Enterotoxin also alters tight junction integrity, resulting in increased paracellular permeability by a noncytotoxic mechanism.636
Clostridium difficile
C. difficile produces several toxins, only two of which, toxin A and toxin B, have been studied in detail. Toxin B is a potent cytotoxin in vitro, but its role in enterocolitis is less clear than that of toxin A. It does not induce fluid secretion, inflammation, or characteristic alterations in intestinal morphology. C. difficile toxin A is an enterotoxin that induces an inflammatory response with hypersecretory diarrhea.637 Toxin A induces neutrophil influx into intestinal tissue, mast cell degranulation, and secretion of prostaglandins, histamine, cytokines, and 5-HT by these activated leukocytes.142, 637, 638 The products of neutrophils and mast cells have a significant role in the vasodilatory and secretory responses in the intestine during C. difficile infection.
The ENS is central to the induction of intestinal inflammation and mucosal secretion by toxin A. A model for toxin A–induced secretory diarrhea has emerged in which toxin A stimulates substance P–containing afferent sensory nerve fibers, which in turn stimulate mast cell degranulation, recruitment and activation of PMNs, and vasodilation.88, 89, 639 Toxin A–induced stimulation of enterocyte secretion can occur via secretomotor neuronal stimulation by substance P–containing sensory neurons or products of mast cells and PMNs. Mast cell degranulation, PMN influx, and enterocyte secretion are all abolished by neural blockade or depletion of substance P. How toxin A triggers the sensory component of the ENS remains unknown, but it is likely that toxin A–induced necrosis of enterocytes exposes afferent neurons to the noxious milieu of the intestinal contents.
Clinical Signs and Diagnosis
Equine intestinal clostridiosis is clinically similar to other forms of acute enterocolitis in horses.619, 624 Although the clinical course is usually acute, peracute colitis with rapid death may occur. Occasionally, a milder, more prolonged clinical course is seen. Fever, anorexia, and depression may be observed before the onset of gastrointestinal signs, but the absence of prodromal signs is more common. Signs of endotoxemia and shock may accompany acute signs of colic and severe, dehydrating diarrhea. Diarrhea may not be profuse but is usually dark and foul. Like the clinical signs, hematologic and serum biochemical abnormalities are similar to those associated with other forms of enterocolitis and reflect fluid, protein, and electrolyte loss and systemic inflammation resulting from endotoxemia. Neutropenia, leukopenia, and hemoconcentration are common. Hypoproteinemia may be profound. Hyponatremia, hypokalemia, hypochloremia, hypocalcemia, and a mixed prerenal–renal azotemia are often noted, as well as metabolic acidosis and coagulopathies. Serum concentrations of hepatocellular enzymes, such as sorbitol dehydrogenase, may be elevated, and liver function may be reduced.
Preliminary diagnosis of equine intestinal clostridiosis caused by C. perfringens is based on the isolation of greater than 100 CFUs of C. perfringens type A per gram of feces from patients with diarrhea and signs suggestive of toxemia.619, 640 Similar criteria are used to screen human patients for C. perfringens type A infection. Normal horses shed less than 100 CFU/g of feces, and usually horses with intestinal clostridiosis shed greater than 106 CFU/g.619, 640 Identification of high numbers of Clostridium organisms in the feces does not prove infection as the cause of the observed clinical signs. Detection of C. perfringens toxins in feces or intestinal contents in horses with high numbers of fecal CFUs and clinical signs of enterocolitis is more conclusive evidence of an enterotoxigenic infection than that based on culture alone.624 Immunoassays are available to detect C. perfringens enterotoxin,624 but the reliability (specificity) of some immunoassays has come into question. PCR multiplex and gene probe assays are now available for detection of the major lethal toxins in bacterial isolates or fecal samples to determine the pattern of toxin production and are currently the preferred methods of detection.641, 642, 643
Like C. perfringens, diagnosis of C. difficile infection depends on the culture of the organism from feces and identification of toxins in the feces. Bacterial culture of C. difficile may be difficult; therefore, it is an insensitive diagnostic test in horses.644, 645 Enrichment techniques and culture of multiple fecal samples may be required.645, 646 Detection of toxin A or B (or both) in feces by cell cytotoxicity assay or immunoassay is the preferred test for diagnosis of C. difficile infection in humans.624 These tests are more sensitive than bacterial culture for identifying C. difficile infection in adult horses.644, 645 Sensitive PCR methods may also be used to identify genes for toxins A and B in fecal samples from diarrheic horses.624
Strongylosis
Pathogenesis
Strongyle infections in horses are caused by two groups of nematodes: large and small strongyles (see later section Cyathostomiasis). Large strongyles that are pathogenic in horses include Strongylus vulgaris, S. edentatus, and S. equinus. Of these species S. vulgaris is by far the most important cause of disease in the large intestine and is the most pathogenic parasitic infection in horses.647 S. vulgaris infection in horses is manifested as acute or chronic disease.647 The age and resistance of the host, the infective dose, and the size and function of the affected arteries influence the type and degree of disease that occurs. Sudden ingestion of large numbers of infective larvae by a naive host causes acute strongylosis, whereas ingestion of fewer infective larvae over a long period of time by an older, more resistant host causes chronic strongylosis. Acute strongylosis is more likely to cause colic than diarrhea and may be rapidly fatal. Chronic strongylosis tends to cause debilitation and signs of colic but may also cause diarrhea.
Diarrhea associated with acute strongylosis occurs within several days of infection and is likely to be caused by migration of the larvae through the intestinal wall. Fourth-stage larvae migrate through the mucosa and submucosa into the arterioles of the intestine, causing mural edema, hemorrhage, and infiltration of inflammatory cells.647, 648 Increased secretion and decreased absorption of fluid and electrolytes, stimulated by inflammatory mediators such as prostaglandins and histamine, may play a role in the diarrhea induced by S. vulgaris. Interstitial edema and damage to the interstitial matrix and mucosa may occur as a result of inflammation and migration of the parasites, causing increased secretion of fluid and albumin loss. Abnormal gastrointestinal motility may also play a role in the development of diarrhea. Migration of larvae through the intestinal wall early in the course of infection affects myoelectrical activity and motility in the large intestine and may affect retention of ingesta and absorption of fluid.649, 650 The cause of death in acute strongylosis has not been addressed, but it may be related to massive migration through the vasculature, causing thrombosis with ischemia and infarction of the intestine.
Chronic strongylosis causes typical verminous arteritis and is more commonly associated with natural infections in horses than acute strongylosis.647 Lesions of the large intestinal vasculature caused by migration of larvae through the intima are characterized by thrombus formation, narrowing of the arterial lumen, fibrosis, and thickening of the arterial wall.647, 648 Embolization may occur, causing acute segmental infarction of the large intestine, but more commonly, reduced blood flow without embolization causes ischemia and occasionally infarction.648, 651 Postmortem examination of horses with colonic infarction failed to reveal embolization as the cause in the majority of cases.651 Reduced blood flow in the tissues of the intestine usually results from narrowing of the arterial lumen by the thrombus and formation of microthrombi at sites independent of the parasites. Release of vasoconstrictive inflammatory mediators, such as leukotrienes, from platelets, neutrophils, and eosinophils, as well as elaboration of parasitic antigens or toxins, may cause vasoconstriction and ischemia.652 Horses with experimental strongylosis had a 50% reduction of blood flow in the colonic vasculature.653
Clearly, reduced blood flow is an important effect of chronic strongylosis, but the relationship between blood flow and diarrhea is unclear. Disrupted motility resulting from ischemia may lead to diarrhea by reducing the retention of ingesta and absorption of fluid. Acute infarction and mucosal ulceration cause severe, chronic diarrhea in naturally infected horses.654 Release of inflammatory mediators, such as prostaglandins, histamine, and kinins, from inflammatory cells associated with thrombi and inflamed intestine may also affect secretion, absorption, and motility, leading to diarrhea.
Clinical Signs and Diagnosis
The clinical signs of acute strongylosis caused by S. vulgaris infection include depression, moderate to severe colic, and fever.655 Diarrhea is less often a feature of acute strongylosis than colic.647 Most cases of acute strongylosis occur in young naive horses introduced to an infested environment or inoculated experimentally with infective larvae. This form of strongylosis is not often recognized naturally. Chronic strongylosis is most commonly observed as a natural syndrome. Weight loss or poor weight gain; chronic, intermittent colic; fever; poor appetite; and diarrhea are frequently observed.647, 648 Diarrhea may be profuse and watery, or the feces may be soft but of normal volume. Transrectal palpation may reveal thickening and fremitus in the cranial mesenteric artery. Young horses are most commonly affected, but older horses may also be too. Horses with acute infarction or large intestinal ulceration secondary to chronic strongylosis may have signs of severe abdominal pain, sepsis, and endotoxemia, and profuse, watery diarrhea is common.
Hematologic abnormalities associated with strongylosis include neutrophilic leukocytosis and eosinophilia.655, 656, 657 Neutrophilia appears to be an early event during the course of the disease, and eosinophilia tends to appear later.655, 657 Hyperfibrinogenemia may also occur, especially later in the course of the disease. Serum α-globulin and β-globulin and IgG(T) concentrations are characteristically elevated.656, 658 Horses with chronic ulcerative colitis secondary to strongylosis may develop severe hypoalbuminemia.654 PF analysis may reveal an elevated protein concentration and eosinophilia.656, 657 Tentative diagnosis is based on clinical signs, hematologic abnormalities, and PF analysis. Elevated serum α-globulin and β-globulin concentrations and IgG(T) concentration support the diagnosis.658 Fecal analysis may reveal strongyle eggs, but fecal egg counts are often unreliable because nonpatent larvae cause the disease.
Prevention
Appropriate preventive measures are important in controlling this disease, including such management procedures as preventing overcrowding, reducing exposure of susceptible individuals, and instituting proper deworming schedules. Ivermectin is the preferred anthelmintic used to control strongylosis in horses. Monitoring fecal egg counts as a means of evaluating the efficacy of parasite control measures is recommended.
Cyathostomiasis
Pathogenesis
Infection with small strongyles (cyathostomiasis) is well recognized as a cause of diarrhea and large intestinal disease in horses of all ages.659, 660, 661, 662, 663, 664 Clinical disease is caused by intramural larval stages of more than 50 species of small strongyles (cyathostomes). The cyathostome life cycle requires migration by fourth-stage larvae through the mucosa of the large intestine and may include a period of hypobiosis, during which the larvae remain encysted within the mucosal layer of the large intestine.659 After a period of hypobiosis, the larvae emerge in response to a largely unknown stimulus. Most cases occur when larval emergence takes place, classically in the late winter and spring in the northern temperate zones and in the late fall or winter months in the southeastern United States and subtropical regions.659 Sudden emergence of encysted larvae causes mucosal injury, ulceration, and an inflammatory reaction, which are largely responsible for the clinical disease.659, 665 Migration of the larvae as they penetrate the mucosa affects motility patterns and can cause inflammation that may contribute to diarrhea.659 Chronic, eosinophilic, granulomatous colitis and diarrhea with histopathologic evidence of hypobiotic cyathostome larvae in the large intestine have been reported in two horses during a period in which the emergence of larvae would not be expected to occur (early winter).659
Natural emergence of cyathostome larvae causes fibrinous inflammation of the large intestine, focal necrosis, mural hemorrhage, and ulceration of the large intestinal mucosa, which may even result in bleeding into the lumen.648, 665 Mild to moderate eosinophilic and mononuclear inflammation of the lamina propria is seen.648, 665 Moderate to severe interstitial edema is frequently observed.648, 665 Colonic inflammation and interstitial edema may contribute to diarrhea, in conjunction with the loss of the mucosal barrier, by causing increased active and passive secretion of fluid, electrolytes, and protein. Protein loss is often significant, resulting in profound hypoalbuminemia and interstitial edema of skin and other organs. Chronic granulomatous colitis has been reported to occur in response to encysted larvae and may cause diarrhea by increased secretion secondary to granulomatous inflammation or disruption of the interstitium by granulomatous infiltration. Administration of an anthelmintic to horses with a heavy load of encysted larvae may also cause rapid larval death and acute and often severe inflammation similar to natural emergence.
Clinical Signs and Diagnosis
Cyathostomiasis may be the most commonly identified cause of chronic diarrhea in the horse.493, 666, 667 However, an acute syndrome has also been associated with cyathostomiasis.664 Clinical signs of cyathostomiasis are characterized by moderate to severe weight loss or poor weight gain, ill thrift, ventral edema, intermittent fever, and intermittent mild colic.659, 660, 661, 662, 663, 664, 667 Acute onset of diarrhea is typically profuse and progresses to chronic diarrhea that is often mild, the consistency of bovine feces, and may be intermittent.659, 660, 661, 662, 663, 664, 667 Appetite is usually normal, but some affected horses have a ravenous appetite. Transrectal palpation usually does not reveal any abnormalities. Horses of any age may be affected, and clinical signs are more common during periods of emergence of larvae, corresponding to late winter and spring in northern temperate zones. The deworming history may appear to be adequate.
Neutrophilic leukocytosis is typically evident, but the WBC count may be normal.659, 660, 661, 662, 663, 664 Profound hypoalbuminemia is a characteristic feature of cyathostomiasis, manifested clinically by ventral edema. Plasma α-globulin and β-globulin concentrations may be elevated, which can result in a normal total plasma protein concentration in spite of hypoalbuminemia.658, 659, 660 The serum IgG(T) concentration, however, has been reported to be normal, which may help distinguish cyathostomiasis from S. vulgaris infection.659, 661, 662 PF analysis does not usually reveal any abnormalities, in contrast to horses with S. vulgaris infection. Fecal analysis may be unrewarding because the infection is often not patent when clinical signs are apparent. Measurement of plasma fructosamine may provide a measure of protein catabolism or protein loss in the absence of hypoalbuminemia.667, 668 Plasma fructosamine concentrations are significantly lower in horses with experimental cyathostomiasis than in normal controls,667, 668 suggesting that this test may be a useful diagnostic tool. However, the test has not yet been validated in naturally occurring cases, and neither the specificity nor the sensitivity is known. Rectal scrapings or rectal mucosal biopsies may reveal evidence of cyathostome larvae.659, 662 Definitive diagnosis usually requires microscopic examination of biopsy specimens of the cecum and ascending colon, collected by laparotomy. Examination of biopsy specimens collected from the small intestine is recommended to rule out other causes of weight loss and diarrhea. Appropriate diagnostic tests, such as culture of feces for pathogenic bacteria, should be included in the workup to further rule out other causes.
Prevention
Preventive measures are appropriate for other horses on the premises known to have a problem with cyathostomiasis. These include frequent deworming (every 6 weeks) during times of high infectivity (spring and summer in the north and fall, winter, and early spring in the south) to eliminate parasites before they become patent.659 Because of high levels of resistance to benzimidazoles, avermectins (ivermectin or moxidectin) are often the drugs of choice for cyathostome control.669, 670, 671 Resistance to ivermectin has been demonstrated, but the prevalence of ivermectin resistance appears to remain low.669 Although daily pyrantel pamoate administration has also been reported to effectively reduce worm burdens and pasture infectivity in young and mature horses,672 cyathostome resistance has been reported and is a concern for the use of this drug as a routine preventive anthelmintic.670, 673, 674 Because of the rapid emergence of resistant strains to even ivermectin, targeted treatment, based on fecal egg counts and careful monitoring for the development of resistance to any anthelmintics used for cyathostome control, is warranted.674
Toxicologic Diseases
Antibiotic-Associated Diarrhea
Pathogenesis
Antibiotic-associated diarrhea has been reported in many species, including horses.675 Certain antibiotics, such as trimethoprim-sulfonamide combinations, erythromycin, penicillins, tetracyclines, clindamycin, and lincomycin, are associated with naturally occurring and experimental enterocolitis syndromes in horses.620, 675, 676, 677, 678 In some cases, such as those seen with trimethoprim-sulfonamide combinations, the geographic incidence of antibiotic-associated diarrhea appears to differ markedly.
C. perfringens, C. difficile, and serovars of S. enterica are apparently the most common causes of antibiotic-associated diarrhea in horses. Outbreaks of C. difficile have been reported in hospitalized horses being treated with antibiotics.622, 626 In Sweden accidental erythromycin ingestion has been associated with C. difficile enterocolitis in mares in which their foals were being treated for R. equi. 629, 677, 679 Tetracycline administration has been associated with an increase in the numbers of gram-negative enteric bacteria and C. perfringens in the feces of horses as well as reactivation of salmonellosis and prolongation of fecal shedding of serovars of S. enterica. 619, 680
The most common mechanism by which antibiotics cause diarrhea is disruption of the gastrointestinal flora. The normal large intestinal flora, composed of mainly obligate anaerobes and streptococci, protects the host from pathogenic bacteria by colonization resistance.553 Ecologic factors play an important role in colonization resistance. For example, surface bacteria in the large intestine interact with receptors on the mucosal cells, facilitating adherence to the mucosa.545, 681 In doing so, the normal organisms compete more successfully for this important niche. Competition for space and nutrients is an important means of preventing colonization and proliferation of pathogenic bacteria.552, 553, 681 Anaerobic bacteria produce SCFAs and other metabolites that are toxic to facultative anaerobic bacteria, especially in the conditions of the large intestine.552, 553, 681 Organisms of the normal flora produce bacteriocins that inhibit growth of potential pathogens.552
Antibiotics that deplete the population of obligate anaerobes and streptococci efficiently decrease colonization resistance.552 Production of fatty acids is diminished, and competition for space and nutrients is reduced. As a result, gram-negative enteric bacteria, such as S. enterica var. Typhimurium, are able to proliferate. Pathogenic anaerobes normally found in low numbers can also proliferate. Antibiotic-resistant strains of bacteria, especially gram-negative enteric bacteria and possibly clostridia, may be selected by antibiotic administration, allowing proliferation of pathogenic bacteria resistant to many antibiotics.682 Obligate anaerobic commensal organisms, perhaps the most critical group of microbes for maintaining colonization resistance, are usually susceptible to macrolides, tetracyclines, β-lactams, and lincosamides, which may explain the high incidence of diarrhea associated with the administration of these antibiotics.624
In addition to reduction of colonization resistance, depletion of the normal anaerobic microbial population in the intestine decreases carbohydrate fermentation and production of SCFAs, which contributes to the pathogenesis of antibiotic-associated diarrhea by decreasing absorption of sodium and water by the colonic mucosa.171 Ampicillin decreases colonic fermentation of carbohydrates in humans.683 Human patients with antibiotic-associated diarrhea have markedly impaired colonic fermentation and very low production of SCFAs.684 Erythromycin, ampicillin, or metronidazole treatment is associated with decreased production of SCFAs in patients with and without diarrhea.684 Absorption of sodium and water is stimulated by absorption of SCFAs in the equine colon, suggesting that reduction of colonic SCFA content by antibiotic-induced depletion of anaerobic flora has similar effects in horses as in humans.171
Broad-spectrum antibiotics exert a more profound effect on the gastrointestinal flora than narrow-spectrum antibiotics.685 Antibiotics administered orally, especially those that are poorly absorbed, are more likely to cause diarrhea than parenterally administered antibiotics.685 For instance, clindamycin is less likely to cause diarrhea in humans when administered IV than when administered orally.685 Antibiotics with extensive enterohepatic circulation, such as tetracyclines and erythromycin, are excreted in high concentrations in the bile and are more commonly associated with diarrhea than antibiotics that do not undergo enterohepatic circulation.685
Antibiotics may cause diarrhea by other means than by disrupting the normal flora. Direct toxic effects may play a role in producing irritation, increasing secretion, and disrupting motility patterns. Tetracyclines are irritating to the gastrointestinal mucosa and may cause inflammation and increase secretion.685 Erythromycin interacts with smooth muscle cells, stimulating gastrointestinal motility.685, 686 Normal peristalsis plays an important role in suppressing the population size of potentially pathogenic bacteria. Normally, bacteria that are prevented from adhering to the mucosa by colonization resistance are swept aborally by peristalsis and excreted in the feces. Disruption of normal motility patterns may prevent clearance of pathogenic bacteria, contributing to the colonization of mucosal surfaces.
Clinical Signs and Diagnosis
Diarrhea induced by antibiotics usually occurs within 7 days of initiation of antibiotic administration but may occur several days after cessation of antibiotic treatment. The clinical syndrome of antibiotic-associated diarrhea varies from mild diarrhea to fulminant enterocolitis with severe diarrhea. Mild diarrhea is common, especially in foals receiving erythromycin, trimethoprim-sulfa combinations, or rifampin677, 687 and are usually not clinically significant. Acute, severe enterocolitis can occur in horses of all ages receiving antibiotics and can be life-threatening. Clinical signs are identical to those resulting from other causes of acute enterocolitis. Severe, dehydrating diarrhea; endotoxemia; sepsis; and shock may occur. Hemoconcentration, neutropenia, hypoproteinemia, and electrolyte and acid-base imbalances are common. Severe hyponatremia may occur in foals with antibiotic-associated diarrhea, especially if trimethoprim-sulfa and rifampin combinations are the cause.687 More detailed descriptions of the clinical and laboratory findings were given earlier. Diagnosis is presumptive because definitive diagnosis of antibiotic-associated diarrhea is impossible. Fecal culture or PCR testing may reveal S. enterica or Clostridium spp. infection.
Nonsteroidal Antiinflammatory Drug Toxicity
Pathogenesis
Toxicity resulting from NSAID administration has been well documented in several species, including horses, and is discussed in Chapter 2.688, 689, 690, 691, 692, 693, 694 In horses and humans NSAID toxicity is manifested by renal and gastrointestinal disease. Foals are considered to be more susceptible than adult horses to gastrointestinal disease secondary to NSAID administration, and ponies may be more susceptible than horses. NSAID toxicity varies primarily as a result of properties that influence distribution to sensitive tissues and relative selectivity for COX-1 or COX-2. All nonselective NSAIDs are capable of inducing gastrointestinal and renal damage at toxic concentrations. Aspirin has been suggested to be more toxic than other NSAIDs because it irreversibly inactivates COX by acetylation, whereas other NSAIDs reversibly inhibit COX.688 Phenylbutazone is the drug most commonly reported to cause gastrointestinal toxicity in horses, perhaps because of its widespread usage by veterinarians and horse owners or perhaps because of bona fide differences in toxicity in horses compared with other nonselective NSAIDs. Acute phenylbutazone toxicity in horses resulting from overdose is characterized by mucosal ulceration throughout the gastrointestinal tract, oral ulceration, renal papillary necrosis, vasculopathy, thrombosis, and protein-losing enteropathy with hypoalbuminemia.690, 691, 692 COX-2–selective NSAIDs appear to be much less toxic in the equine gastrointestinal tract.392, 695 The focus of this discussion is on the toxic effects of NSAIDs on the large intestine, but this necessarily includes elements of upper gastrointestinal and renal disease.
Horses with large intestinal disease resulting from NSAID toxicity generally are receiving inappropriately large doses or have underlying disorders that predispose the large intestine to the toxic effects of NSAIDs, even at appropriate dosages. The dosage regimen recommended for phenylbutazone (4.4 mg/kg every 12 hours for 1 day, then 2.2 mg/kg every 12 hours) is considered safe. Experimental studies in horses, however, have shown toxicity to occur when amounts exceeding the recommended dosage (6.6 mg/kg/day) are administered for several days.688, 689 Most reported cases of phenylbutazone toxicosis occurred in horses receiving higher than recommended dosages.692, 694, 696 Regardless, administration of phenylbutazone at the recommended dosage has been reported to cause a significant decrease in plasma protein concentration and gastrointestinal disease.691, 697 Moreover, signs of NSAID toxicity have been reported in normovolemic horses treated with appropriate doses of phenylbutazone.697, 698 Dehydration, sepsis, endotoxemia, and other conditions that alter hemodynamic homeostasis exacerbate renal and gastrointestinal toxicity of NSAIDs.688 Underlying inflammation of the intestinal tissues may increase the likelihood of gastrointestinal ulceration resulting from NSAIDs.
Gastrointestinal disease induced by NSAIDs is manifested by mucosal ulceration, inflammation, bleeding, and protein-losing enteropathy.690, 691, 694, 697 In addition to direct effects on the mucosal barrier, NSAID administration causes an acute relapse of preexisting colonic inflammatory disease and worsens colonic inflammation in humans with inflammatory bowel disease.688, 699, 700
It is not clear whether the previously mentioned NSAID effects occur in horses. The mechanism by which NSAIDs induce mucosal damage is probably multifactorial. Direct irritation may play a role in oral and gastric irritation and ulceration; however, parenteral administration of NSAIDs produces oral and gastric ulceration as well. Inhibition of prostaglandin synthesis by inhibition of both COX-1 and COX-2 appears to be the most important mechanism of mucosal injury. Prostaglandins, particularly PGE2 and PGI2, are critical for mucosal health and repair after injury.701, 702 PGE2 increases mucosal blood flow; increases secretion of mucus, water, and bicarbonate; increases mucosal cell turnover rate and migration; stimulates adenyl cyclase activity; and exerts other protective effects in the gastric mucosa of several species.688, 701, 702 Perhaps most important, PGE2 and PGI2 have roles in maintaining epithelial tight junction integrity, which is indispensable for mucosal barrier function and repair after mucosal injury.701
In spite of the overwhelming amount of information about the role of prostaglandins in maintaining the mucosal barrier in other species and clear clinical and experimental evidence that NSAIDs injure the equine colonic mucosa, the role of prostaglandins in mucosal protection in the equine colon is not yet well defined. Inhibition of COX-1 and COX-2 in equine colonic mucosa with flunixin meglumine results in reduced electrical resistance of the mucosa and increased permeability to macromolecules in vitro,703 suggesting that flunixin treatment disrupts the epithelial tight junctions in the equine colon. This was correlated with a profound inhibition of PGE2 and PGI2 concentrations in the treated tissues. Administration of a PGE2 analog prevents the gastrointestinal manifestations of phenylbutazone toxicosis in ponies.691
The recent development of NSAIDs that specifically inhibit COX-2 has markedly reduced the frequency and severity of gastrointestinal side effects in humans taking NSAIDs for chronic musculoskeletal conditions.704 COX-2–specific NSAIDs such as firocoxib hold promise for use in horses to treat arthritis705 and other conditions, with reduced incidence of toxicity. For example, the relatively COX-2–specific inhibitors meloxicam and firocoxib are less harmful to equine intestinal mucosa than flunixin meglumine in vitro.392, 695 Moreover, COX-2–selective inhibitors are significantly more permissive than flunixin for recovery of the mucosa in equine ischemic-injured intestinal tissues; recovery is no different from that for control tissues.392, 695
NSAID-induced mucosal injury is associated with a marked inflammatory response to microbial products exposed to the lamina propria.706 This inflammation exacerbates mucosal dysfunction and injury associated with NSAID toxicity. For example, depletion of neutrophils or blockade of neutrophil influx into gastrointestinal tissues or inhibition of neutrophil activation and release of toxic products prevents many of the pathophysiologic effects of NSAID toxicity in the gastrointestinal tract.106, 707, 708, 709 The inflammatory response alone may result in moderate to severe gastrointestinal ulceration, mural vascular thrombosis and edema, fluid secretion, protein-losing enteropathy, and mucosal hemorrhage.
Clinical Signs and Diagnosis
NSAID colitis manifests as two clinical syndromes: right dorsal ulcerative colitis (RDUC) and generalized NSAID toxicity. As its name implies, RDUC is a disorder isolated to the right dorsal segment of the large intestine.693, 694, 698 The most prominent clinical signs of RDUC are anorexia, lethargy, and colic. Anorexia, depression, diarrhea, fever, and signs of endotoxemia may also be features. If RDUC is chronic, weight loss, intermittent colic, lethargy, anorexia, and ventral edema are common clinical signs with soft and unformed feces. Ulceration of the right dorsal colonic mucosa results in protein-losing enteropathy and significant hypoproteinemia attributable mainly to hypoalbuminemia. Hypoproteinemia may be one of the earliest clinical manifestations of RDUC and can be sufficiently severe to cause peripheral (usually ventral) edema. In some horses, dehydration, electrolyte abnormalities, neutropenia or anemia, azotemia, and biochemical abnormalities may be noted if the ulceration and diarrhea are severe or if systemic inflammation is present.
Clinical signs of generalized NSAID toxicity vary from mild diarrhea with no systemic signs to severe dehydrating diarrhea with anorexia, fever, depression, peripheral edema, oral ulceration, and colic.691, 692, 696
Clinical signs of systemic inflammation caused by endotoxemia may occur, manifested as poor peripheral perfusion, tachycardia and tachypnea, weakness, trembling, and cyanotic or hyperemic oral mucous membranes. Hematuria or oliguria may be present with renal involvement. Complications associated with other forms of severe enterocolitis, such as laminitis, thrombophlebitis, and severe weight loss, may occur.
Although phenylbutazone has been associated specifically with bone marrow depression resulting in abnormalities in one or more blood cell lines,710 hematologic abnormalities of generalized NSAID toxicity are usually nonspecific and include neutropenia with a left shift or leukocytosis and hemoconcentration. Serum biochemical analysis is characterized by profound hypoproteinemia, hyponatremia, and metabolic acidosis.696, 697 Hypocalcemia, hypokalemia, hypochloremia, and elevated hepatocellular enzyme activities may also be seen. Hypoproteinemia may occur without signs of diarrhea. Azotemia may be prerenal as a result of dehydration, but it is frequently caused by renal failure that results from a combination of hemodynamic effects of NSAIDs and direct toxic renal injury. Urinalysis frequently reveals hematuria, proteinuria, cylindruria, and isosthenuria. Fecal occult blood is frequently detected.
Diagnosis of either form of NSAID colitis is often presumptive, with a history of overdose of NSAIDs being strong evidence of NSAID toxicity. As discussed earlier, toxicity may occur with dosage regimens that are not considered inappropriate, particularly if the horse experiences a concurrent period of dehydration. Ultrasonographic examination of the right dorsal colon can be used to confirm a diagnosis of RDUC, but the sensitivity of this method appears to be low.711 Ultrasonography (3.5- to 5-MHz transducer at the right twelfth through fifteenth intercostal spaces below the margin of the lung axial to the liver) may reveal a thickened right dorsal colon (>0.5 cm) and evidence of colonic edema in horses with RDUC.711, 712 Nuclear scintigraphy of horses after infusion with technetium-99–labeled WBCs can be used to document inflammation of the right dorsal colon.50 Laparotomy or laparoscopic examination of the right dorsal colon may be required for definitive diagnosis of RDUC. Other causes of enterocolitis, such as salmonellosis, Potomac horse fever, clostridiosis, and antibiotic-associated diarrhea, must be ruled out.
Cantharidin Toxicity
Pathogenesis
Cantharidin is the toxic substance found in beetles of the genus Epicauta, commonly known as blister beetles. 713, 714, 715 Ingestion of the beetles causes release of the toxin and absorption through the gastrointestinal tract. Transcutaneous absorption may occur but appears to be rare in horses. Blister beetles feed on the flowers of alfalfa and may be incorporated into processed alfalfa hay if the hay is cut and processed simultaneously, as by crimping.713, 714, 715 The beetles often swarm, and large numbers of beetles may be found in relatively small portions of hay. The lethal dose of cantharidin is less than 1 mg/kg, but the concentration of cantharidin varies among species of blister beetles and between sexes.713, 714 As many as 100 to as few as 6 beetles may be lethal. Usually, only one or a few horses fed contaminated hay will ingest beetles because they are concentrated in a small portion of the hay. However, outbreaks involving many horses on a farm have occurred. Most cases have occurred in Texas and Oklahoma, but horses in other states may be affected as well, especially if hay is imported from states in which blister beetles are common. Peak incidence is in late summer and fall.716 The fatality rate may be 50% or greater,713, 717 but if the patient survives several days, recovery is probable.
Cantharidin is absorbed from the gastrointestinal tract and excreted by the kidneys. It is a potent irritant, causing acantholysis and vesicle formation when applied topically.713, 715, 717 The chemical is thought to disrupt the oxidative metabolism in the mitochondria, causing mitochondrial swelling, plasma membrane damage, and changes in membrane permeability.713 The mucosa of the gastrointestinal tract is most commonly affected in horses because they ingest the toxin. Cell swelling and necrosis occur, resulting in mucosal ulceration. Oral, esophageal, gastric, and small and large intestinal ulceration have been observed in natural and experimental canthariasis.713, 715, 717 Severe fibrinous to pseudomembranous inflammation and submucosal edema of the intestine have also been reported. Diarrhea probably results from the severe ulceration and inflammation of the large intestine, causing increased secretion of water, electrolytes, and protein and decreased absorption of fluid. Large volumes of fluid and protein are lost in the gastrointestinal tract, causing hemoconcentration and profound hypoalbuminemia in some affected horses.713, 715, 717
Cystitis and myocarditis occur in natural and experimentally induced cases of cantharidin toxicity.713, 715, 717 The toxin is excreted by the kidneys, and high concentrations of cantharidin in the urine induce cystitis. Occasionally, hemorrhagic cystitis may occur, resulting in hematuria or frank hemorrhage into the bladder.713 The cause of myocarditis and myocardial necrosis is unknown but may be a direct effect of toxin on the myocardium. Increased plasma creatine kinase activity is often observed and has been postulated to arise from the damaged myocardium.713, 714 Affected horses have a characteristically stiff gait, but histopathologic evidence of skeletal muscle injury that explains the elevated plasma creatine kinase activity has not been observed.714 The kidneys are often pale, swollen, and moist, with occasional infarcts.715
Hypocalcemia and hypomagnesemia are biochemical features of cantharidin toxicity in horses that have not been explained.713, 714, 717 Hypocalcemia may result from hypoalbuminemia, but the ionized calcium concentration is often decreased, indicating that hypoalbuminemia is not responsible for the hypocalcemia.714
Clinical Signs and Diagnosis
Cantharidin toxicity can cause a range of clinical signs, from mild depression and abdominal discomfort to fulminant signs of toxemia and rapid death, depending on the ingested dose of toxin.713, 714, 717 Most commonly, clinical signs include depression, sweating, irritability, abdominal pain, elevated heart and respiratory rates, fever, polyuria, polydipsia, and profuse diarrhea.713, 714, 717 Blood is rarely seen in the feces. Stranguria and pollakiuria are common.713 Signs of hypocalcemia include synchronous diaphragmatic flutter and tremors. A stiff and stilted gait may be evident. Neurologic signs such as head pressing, swaying, and disorientation may be noted.717 Signs of systemic inflammation resulting from endotoxemia may be seen in severe cases. Some horses develop severe depression and toxemia and may die within hours of ingesting cantharidin without developing diarrhea.713, 717
Hematologic abnormalities include hemoconcentration and neutrophilic leukocytosis.713, 714 Occasionally, neutropenia and leukopenia may accompany endotoxemia. Serum biochemical analysis usually reveals increased creatine kinase activity, hypocalcemia, and hypoalbuminemia.713, 714 Biochemical abnormalities include hypocalcemia (both ionized and total calcium concentrations), hypomagnesemia, and azotemia.713, 714, 717 Urine specific gravity is characteristically in the hyposthenuric range.713, 714 Microscopic hematuria and mild proteinuria may be evident. Fecal occult blood is often present, but hematochezia is unusual.
A tentative diagnosis can be made on the basis of clinical signs and identification of blister beetles in the hay. Determining the species of the insects may be necessary to estimate the amount of cantharidin ingested. All species of Epicauta contain cantharidin, but some have small amounts. Definitive diagnosis requires the measurement of the cantharidin concentration in gastric or intestinal contents and urine.713, 716
Arsenic Toxicosis
Pathogenesis
Arsenic toxicosis is an unusual cause of diarrhea in horses, resulting from ingestion of arsenic-containing herbicides, insecticides, and other pest-control products contaminating water or roughage used as a food source.718 The toxicity of arsenic depends on the valence of the element.718, 719 Arsenate may be reduced to arsenite in mammalian systems.719 It is thought to be more toxic than arsenate and less rapidly excreted in urine.719 Arsenate and arsenite uncouple oxidative phosphorylation, leading to the breakdown of energy metabolism in the cells of many tissues.719 Widespread cellular injury and death occur rapidly during acute arsenic toxicosis. Multiorgan failure is usually the result. Cardiomyopathy and pulmonary disease are common causes of death in humans.720 Damage to the large intestine is probably caused in part by direct cellular toxicity and corrosion by the compound. However, vasculitis is a hallmark of the disease in humans and horses and is thought to be the most important mechanism of large intestinal disease in humans.718, 721 Acute hemorrhagic colitis is a feature of arsenic toxicosis, with severe mural edema and mucosal ulceration.718 Profuse hemorrhagic diarrhea and abdominal pain result. Chronic arsenic toxicity can occur but appears to be rare in horses.
Clinical Signs and Diagnosis
Depression, weakness, abdominal pain, hemorrhagic diarrhea, and shock are characteristic of acute arsenic toxicosis in horses.718 Death may occur before diarrhea is evident. Initial clinical signs may be difficult to distinguish from other peracute forms of colitis and are related to endotoxic shock, metabolic disturbances, and dehydration. Later, cardiac arrhythmias, pulmonary edema, acute renal failure, and neurologic deficits (ataxia and stupor) may develop.718 Anuria or polyuria may be observed. Hemolytic anemia caused by preferential binding of arsenic compounds to red blood cells is a feature of arsenic poisoning in humans.720 Hematologic abnormalities resulting from injury to bone marrow cells and ongoing hemolysis may be seen after the peracute stage. Leukopenia and thrombocytopenia have been described in human patients.720 Serum biochemical analysis may reveal azotemia, hepatocellular enzyme activities higher than generally attributed to endotoxemia, and increased creatine kinase activity.718 Urine specific gravity may be in the isosthenuric range, with hematuria, cylindruria, and proteinuria evident by urinalysis.
Diagnosis may be possible by measuring blood and urine arsenic concentration, but these tests may not be diagnostic. Postmortem diagnosis is confirmed by measuring arsenic concentrations in liver and kidney samples.718 History of exposure and clinical signs remain the primary means of diagnosis.
Miscellaneous Inflammatory Disorders of the Large Intestine
Intestinal Anaphylaxis
Pathogenesis
Severe intestinal anaphylaxis is a syndrome in horses characterized by peracute, rapidly fatal colitis.722 The severe syndrome is clinically and pathologically similar to other known causes of peracute colitis. Some cases are less severe and manifest as mild to moderate diarrhea or colic (or both). The syndrome of intestinal anaphylaxis can be produced by either an IgE-mediated type I hypersensitivity or an IgE-independent anaphylactoid reaction.723, 724 Intestinal anaphylaxis is usually induced by local gastrointestinal exposure to a food, environmental, drug, or other allergen723, 725 but may also occur with systemic exposure to an allergen.726, 727, 728 Massive mast cell degranulation, secretion of inflammatory mediators, and activation of enteric neural reflexes in the intestine cause profound alterations in blood flow, increased vascular permeability and interstitial edema, recruitment of neutrophils, altered motility, mucosal injury, absorption of microbial products, and mucosal hypersecretion.729, 730, 731, 732, 733 Systemic signs may be caused by the anaphylactic reaction or may be associated with SIRS.
The peracute form is characterized by severe intramural edema and hemorrhagic inflammation of the large intestine, often producing submucosal thickening on the order of many centimeters.162, 722 Vascular thrombosis may be widespread, with mucosal and serosal petechiae and ecchymoses. Less severe forms of intestinal anaphylaxis may manifest as patchy areas of intestinal edema and congestion.726 Diarrhea results from intestinal inflammation initiated by the type I hypersensitivity response. Many of the mediators of type I hypersensitivity, such as histamine and 5-HT, have well-documented stimulatory effects on mucosal secretory activity, vascular and epithelial permeability, and motility729, 730, 731 in the intestine. Systemic inflammation resulting from endotoxemia may be overwhelming once the mucosal barrier breaks down. Infarction of intestinal segments and other organs may result from intravascular coagulation. Ileus, abdominal distention, and moderate to severe abdominal pain may result from motility disturbances and infarction of the large intestine.
Clinical Signs and Diagnosis
The clinical signs are similar to those described for other forms of peracute colitis. Severe diarrhea is possible, but death may occur before diarrhea is evident. Multiorgan failure resulting from DIC is not unusual. The rapid onset of weakness, staggering, and trembling commonly precedes death. The syndrome may cause death in 4 to 24 hours.
Diagnosis is based on clinical signs, postmortem findings, and exclusion of other causes. Most diagnostic tests are inconclusive. If an antigen is suspected to be the trigger of the anaphylaxis, a Prausnitz-Kustner passive cutaneous anaphylaxis sensitization test can confirm the presence of antigen-specific IgE in the patient’s serum.726
Carbohydrate Overload
Pathogenesis
Overeating of soluble carbohydrates, especially so-called hot grains such as corn, overwhelms the digestive capability of the small intestine, resulting in a high percentage of soluble carbohydrates entering the large intestine. The amount of soluble carbohydrates that will produce diarrhea varies according to the previous dietary history of the individual. Horses fed diets higher in soluble carbohydrates are more resistant to the deleterious effects of carbohydrate overload. Gradual accommodation to a diet high in carbohydrates can be accomplished over several weeks. Horses fed an unusually large amount of grains or other form of soluble carbohydrates often develop diarrhea and may, depending on the amount ingested, develop severe colitis, systemic inflammation resulting from endotoxemia, metabolic acidosis, and laminitis.229, 734, 735, 736
The pathogenesis of colitis from carbohydrate overload is primarily caused by the toxic effects on the microbial flora in the large intestine.735 A sudden delivery of soluble carbohydrates to the large intestine causes rapid fermentation by gram-positive lactic acid–producing bacteria and a sudden increase in organic acid production.229 The cecal pH level rapidly decreases, and the lactic acid concentration rapidly increases.229 Rapid organic acid production overwhelms the buffering capacity of the large intestine, not only by directly depleting the buffers found in the contents but also by reducing the efficiency of buffer secretion. Bicarbonate secretion is linked to absorption of volatile fatty acids, which are produced in low amounts by fermentation of soluble carbohydrates. The contents of the large intestine become profoundly acidic, resulting in unfavorable conditions for the microbial flora. Lactic acid–producing bacteria flourish, whereas the gram-negative bacteria, especially the Enterobacteriaceae, are killed in large numbers by the acids. Large quantities of endotoxin are released from the dying bacteria.229
The osmotic load from lactic acid produced in the large intestine is an important factor in the development of diarrhea because organic acids such as lactic acid are poorly absorbed. Mild cases of carbohydrate overload may result purely from osmotic diarrhea. In more severe cases the acidic contents of the large intestine are toxic to the mucosa, causing necrosis of the mucosal tissues similar to that seen in ruminal acidosis. Mucosal ulceration allows absorption of large quantities of endotoxin and lactic acid produced by the massive die-off of acid-intolerant microbes and fermentation of soluble carbohydrates, normally poorly absorbed by intact mucosa.736 Systemic inflammation resulting from endotoxemia may be overwhelming; clinical and laboratory findings are consistent with that described for endotoxemia. Laminitis is a frequent complication; carbohydrate overload is used to induce laminitis as an experimental model because of the consistency of laminitis produced.229, 735, 736
Clinical Signs and Diagnosis
Clinical signs of colitis from carbohydrate overload can vary according to the amount of carbohydrate ingested and accommodation of the flora to a high-carbohydrate diet. Mild cases may result in a transient osmotic diarrhea, with no systemic effects. Severe cases are characterized by signs similar to those described for other forms of colitis, including abdominal pain, moderate to severe diarrhea, and dehydration. Signs of endotoxemia and sepsis are frequently present in severe cases. Nasogastric intubation may yield significant acidic gastric reflux. Particles of grain may be noted in the gastric reflux and the feces if grain overload is the source of the carbohydrate overload. Laminitis is a common complication.
Sand Enteropathy
Sand enteropathy is often associated with acute intestinal obstruction due to abnormally large amounts of sand in the large intestine.737 However, chronic sand-induced diarrhea is a distinct syndrome that can occur at any age as a result of the abnormal accumulation of sand in the large intestine.738, 739 Chronic diarrhea and signs of colic may be seen without obstruction. Diagnosis is usually based on the presence of abnormal amounts of sand in the feces, although some horses with sand enteropathy may not be passing sand in the feces at any given moment. Occasionally, radiography may be required to detect sand in the colon.738
Principles of Therapy for Acute Diarrhea
The principles of therapy for acute diarrhea resulting from colitis are similar regardless of the cause and include replacement of fluid and electrolyte losses, control of colonic inflammation, reduction of fluid secretion, promotion of mucosal repair, control of endotoxemia and sepsis, and reestablishment of normal flora. The reader is referred elsewhere in this chapter for a discussion of general treatment of endotoxemia, for which many of the principles of therapy overlap.
Fluid Replacement and Circulatory Support
Replacement of fluid and electrolyte losses is of primary concern in treating horses with salmonellosis. Depending on the severity of the disease, fluid losses may be minimal or massive. Fluid and electrolytes can be administered orally or IV. Some horses with mild to moderate diarrhea may maintain hydration and electrolyte balance by consuming water and electrolytes voluntarily. Fresh water and water containing electrolytes should be available in all cases. In many instances, periodic nasogastric intubation and administration of water and electrolytes through the tube may be sufficient to maintain hydration.740 IV administration of fluids is preferred in cases requiring significant quantities of fluid to replace and maintain hydration and electrolyte balance.357 It is not unusual for patients with severe diarrhea to require large volumes (50–100 L/day) of IV fluids to maintain hydration. Monitoring of packed cell volume, serum electrolyte concentration, venous blood gases, blood urea nitrogen and creatinine, urine protein and cytology, and body weight is important to evaluate hydration, electrolyte and acid-base balance, and renal function.
Isotonic sodium chloride or lactated Ringer’s solution is frequently used to restore and maintain fluid and electrolyte balance. Potassium chloride can be added to the fluids and administered at a rate up to 0.5 to 1.0 mEq/kg/h. Generally, a rate of less than 0.5 mEq/kg/h is used. Hypertonic NaCl solutions (1–2 L of 5% or 7.5% NaCl) have been used in horses with acute, severe hyponatremia (<120 mEq/dL) and those with hemodynamic shock resulting from sepsis. The beneficial effects of hypertonic NaCl are short-lived (30–60 minutes). Isotonic solutions should be administered concurrently or immediately after administration of hypertonic NaCl solutions. Isotonic (1.3%) or hypertonic (5.0%) sodium bicarbonate solutions are used to correct metabolic acidosis. Prolonged administration of sodium-containing fluids may promote diuresis and renal water loss or accumulation of peripheral edema and should be used conservatively when a relative free water loss is noted. Administration of isotonic dextrose (5%) or 2.5% dextrose/0.45% NaCl solutions may be beneficial when free water loss (relative sodium excess) is evident.
Many horses with acute colitis are concurrently hypoproteinemic because of gastrointestinal losses and are absorbing bacterial products that induce a systemic inflammatory response. Plasma oncotic pressures are abnormally low in the face of increased vascular permeability. Interstitial edema formation is a clinical problem in these patients, which contributes to organ dysfunction. Crystalloid fluids, although critical for replacing water and electrolyte losses resulting from diarrhea, may actually contribute to a decline in plasma oncotic pressure as a result of hemodilution.436, 439 Administration of colloid solutions is helpful for volume expansion and maintenance of plasma oncotic pressures. Discussion of colloid replacement is included in the section Endotoxemia.
Antiinflammatory Therapy
Control of colonic inflammation and secretion is a difficult and poorly studied aspect of equine acute colitis. The role of inflammation and mediators such as prostaglandins as a cause of fluid loss is well known during S. enterica and clostridial infection.103, 142, 198, 552, 571, 637, 638, 741 COX inhibitors (NSAIDs) have antisecretory effects on the inflamed intestinal tract,91, 169 including in the equine colon.742, 743 NSAIDs are commonly administered to horses with colitis to reduce inflammation-associated fluid secretion. However, prostaglandins such as PGE2 and PGI2 also have cytoprotective effects on gastrointestinal mucosa and are critical for mucosal repair.91, 701 NSAIDs used pharmacologically to inhibit colonic inflammation and secretion may be detrimental to mucosal integrity and healing if not used judiciously. NSAIDs exacerbate colonic inflammation in humans with inflammatory colitis, impede mucosal healing in several models of mucosal injury, and have well-documented detrimental effects on colonic mucosa in horses.91, 690, 698, 701 In addition to toxicity to the colonic mucosa, gastric ulceration is not unusual in horses with enterocolitis and may be related to treatment with NSAIDs. Although the use of COX-2–selective NSAIDs should, in theory, be less likely to harm or impede repair of the colonic mucosal in patients with colonic inflammation causing diarrhea, the safety and efficacy of these medications has not been evaluated in this application. The prothrombotic adverse effects of COX-2–selective drugs744 suggest that very cautious use of highly COX-2–selective medications is warranted in patients with systemic inflammation at risk for thrombosis (e.g., endotoxemia and sepsis).
In addition to NSAIDs, other drugs are occasionally used as antiinflammatory and antisecretory therapy. Bismuth subsalicylate is commonly used in adults and foals with diarrhea. The volume required for any effect in adults with colitis is quite high (1–4 L by nasogastric tube every 4–8 hours), which often precludes its use. Metronidazole has beneficial effects in experimental models of gastrointestinal inflammation, including NSAID toxicity,706 and may be useful for treating horses with colitis; however, evidence supporting its use is lacking.
Mucosal Repair and Protection
Sucralfate (20 mg/kg by mouth every 6 hours) has been advocated to aid in healing of the colonic mucosa in patients with NSAID toxicity. There is evidence in experimental phenylbutazone toxicosis in foals suggesting that sucralfate administration lessens ulceration and other histopathologic lesions throughout the alimentary tract and lessens protein loss.745 There is no evidence supporting the use of sucralfate to treat colonic ulceration in adult horses.
Misoprostol (5 μg/kg by mouth every 12 hours or 2 μg/kg by mouth every 6–8 hours) and other synthetic PGE analogs enhance mucosal healing in the intestine and promote recovery in experimental models of colitis.746 Misoprostol may be particularly useful for treating NSAID toxicity, either the generalized form or RDUC. However, the efficacy of misoprostol in hastening mucosal healing is clinically unproven in equine colitis. The primary drawbacks of prostaglandin analogs such as misoprostol are the adverse effects of the drug, including abdominal cramping, diarrhea, sweating, and abortion in pregnant mares.
Psyllium mucilloid can be added to the diet (5 tablespoons every 12–24 hours) to increase the production of SCFAs in the colon. Amylase-resistant fermentable fiber such as psyllium is hydrolyzed by colonic bacteria to SCFAs such as butyrate, which represent a major energy source for colonocytes. Butyrate and other SCFAs hasten epithelial maturation and stimulate salt (and thus fluid) absorption in the colon, improve the clinical course of ulcerative colitis, and hasten colon healing.747 Psyllium is itself a source of butyrate in the colon and also promotes the movement of amylase-sensitive carbohydrates into the distal colon, which are then fermented to SCFAs. Psyllium is thought to be clinically useful for promoting mucosal healing in colitis.
Pain Control
Many horses with colitis have mild to severe signs of abdominal pain. Analgesia can be accomplished with NSAIDs such as flunixin, but the potential for worsening mucosal injury or nephrotoxicity may prevent the use of analgesic doses, especially in horses with suspected NSAID toxicity. Relative COX-2–selective inhibitors may spare the gastrointestinal mucosa. For example, meloxicam and firocoxib have analgesic properties in horses in experimental models of ischemic colic and spare the intestinal mucosa from the detrimental effects associated with nonselective COX inhibitor treatment.695, 748
Xylazine, detomidine, or butorphanol (bolus dosing) may provide temporary analgesia. Constant rate infusions of butorphanol, lidocaine, and/or ketamine (alone or in combination) may provide more profound analgesia and can be useful if one is certain that an obstructive or infarctive process is not present. Pain management is discussed in Chapter 3 of this text.
Antimicrobial Therapy
Broad-spectrum antibiotic treatment is often recommended in neutropenic horses or horses with signs of sepsis. Broad-spectrum antibiotics lessen septic complications in human patients. Evidence supporting this principle in horses with colitis is lacking.
Treatment with antibiotics is not thought to alter the course of the enterocolitis, but it may lessen dissemination and severity of disease. Treatment with antibiotics directly targeted at S. enterica is often reserved for patients with sepsis. Lipid-soluble antibiotics are ideally suited for S. enterica infections because the bacteria persist intracellularly. Enrofloxacin is often preferred, but antimicrobial sensitivity of the isolate should dictate directed therapy.
In patients in which the likelihood of N. risticii infection is high, treatment with directed antimicrobial therapy is often indicated before a definitive diagnosis. Lipid-soluble drugs are preferred because the organism can live within cells. Oxytetracycline (6.6 mg/kg/day IV), often followed by doxycycline (10 mg/kg PO every 12 hours), appear to be the most effective antibiotics for treatment of Potomac horse fever and are considered the treatment of choice. Treatment is most successful if initiated before the onset of diarrhea.584, 749
If antibiotics are being administered at the onset of enterocolitis, they should be discontinued if possible. Specific treatment with metronidazole is effective for treating clostridiosis in humans and appears to be effective in horses.624, 750 Metronidazole resistance in clinical isolates of C. difficile has been reported in one outbreak but appears to be rare in most human and equine cases.751 Metronidazole-resistant isolates were sensitive to vancomycin, which may be effective for treating clinical cases if metronidazole resistance is suspected. Vancomycin use is not recommended in horses because of concerns for antimicrobial stewardship. C. perfringens type C antitoxin has been recommended for treatment of neonatal clostridiosis, but there is no evidence of its efficacy.752 Antitoxin preparations are not advocated for use in adult horses with clostridiosis.
Anticoagulation
Hypercoagulability is a common complication of enterocolitis, associated with systemic inflammation resulting from endotoxemia. Administration of heparin (20–80 IU/kg SC or IV every 6–12 hours) may prevent thrombosis in these patients, provided antithrombin III concentrations are adequate in the plasma. Concentrated sources of antithrombin III are not available for use in horses, but whole plasma may provide an important source. Treatment with heparin is thought to decrease thrombosis, especially of the jugular vein, which is a serious complication of salmonellosis. Low-dose aspirin treatment (10 mg/kg PO every 24–48 hours) in conjunction with heparin treatment may provide added benefit by irreversibly inhibiting platelet function.753 Heparin and aspirin may have protective effects on the digital lamina.753, 754 Heparin may enhance the phagocytic activity of the reticuloendothelial system by enhancing the efficiency of opsonins, such as fibronectin and immunoglobulin, stimulating phagocytosis of products of coagulation and possibly other particles, including bacteria.755, 756
Probiotics
Maintenance of the bacterial flora in the gastrointestinal tract is an important defense mechanisms preventing colonization by pathogenic bacteria. Little work has been done to investigate the efficacy of these products in horses. Preparations of Saccharomyces boulardii were shown to decrease the duration and severity of disease in horses with colitis.757 In foals, probiotic use was associated with increased risk of diarrhea and, especially, diarrhea requiring clinical intervention. In humans, fecal transplantation (known to most veterinarians as transfaunation) has been proven effective for intestinal clostridiosis. As the intestinal microbiota are extremely diverse,756 restoration of the overall flora is more likely to be effective than restoration of single bacterial species.
Absorbent Powders and Mineral Oil
Absorbent powders are often used to reduce the bioactivity and absorption of bacterial toxins produced by bacterial or toxic metabolites produced by bacteria and other microorganisms (e.g., lactic acid in grain overload). In a rat model of cantharidin toxicosis, mineral oil, which is often used in equine cantharidin toxicosis, increased cantharidin absorption and also increased morbidity and mortality.758 In that study, rats treated with charcoal or DTO smectite had improved survival. DTO smectite powder (Bio-Sponge, Platinum Performance, Buellton, California) binds C. difficile and C. perfringens exotoxins in vitro343, 759 and may be useful for treating intestinal clostridiosis, or unspecified colitis, in horses. DTO smectite is available as a powder or paste and should be administered according to the manufacturer’s instructions for 3 to 5 days.
Nutrition
Good nursing care and adequate nutrition are vital to the treatment of horses with colitis. Normal intake of roughage to provide energy may be inadequate; however, feeding of grains should be avoided to prevent delivery of highly fermentable carbohydrate to the colon. Dietary management usually consists of restricting or eliminating long-stem roughage (hay) from the diet and feeding exclusively a complete pelleted diet (at least 30% dietary fiber). The rationale behind this recommendation is to reduce the mechanical and physiologic load on the colon. Frequent meals (4–6 times a day) are recommended. Corn oil (1 cup every 12–24 hours) can be added to the pellets to increase the caloric intake without adding roughage or grain. It is important to note that if a horse with colitis refuses to eat pelleted feed, then high-quality grass hay should be fed. In anorectic or severely catabolic patients, enteral and parenteral nutrition (total and partial) has been used successfully to provide calories and nutritional support.
Specific Therapies
Strongylosis
Treatment of S. vulgaris infection requires treatment of the migrating parasite larvae and the lesions produced by the parasite. Fenbendazole (10 mg/kg PO every 24 hours for 3 days or 10 mg/kg PO every 24 hours for 5 days) and ivermectin (200 mg/kg, PO) are effective in killing fourth-stage larvae.647 Other anthelmintics may also be effective when given at higher doses than those required to kill adult worms. The efficacy of these anthelmintics against larvae within thrombi is not known.
Thrombolytic and antithrombotic therapy has been advocated in horses with suspected strongylosis.647, 654 Heparin (20–80 IU IV or SC every 6–12 hours) may be administered as an anticoagulant. High-molecular-weight heparin causes anemia by inducing aggregation of red blood cells, which is an undesirable effect in sepsis. Low-molecular-weight heparins appear to be less likely to have this effect in horses. Aspirin (10–30 mg/kg PO every 12–48 hours) is usually combined with heparin to inhibit platelet adhesion. Aspirin may also inhibit release of platelet products, such as thromboxane, which affect the motility of the large intestine. Low-molecular-weight dextrans have been advocated as antithrombotics that act by inhibiting platelet function and coagulation.654 The clinical efficacy of dextran administration appears to be good, but no controlled studies have been performed.
Cyathostomiasis
Anthelmintic administration is usually the only treatment necessary for mild to moderate cases of cyathostomiasis treated early in the course of the disease (within 1–3 weeks of onset). Fenbendazole is effective against many larval stages, but resistance is high in some populations. Although the reported efficacy of ivermectin is variable against certain stages,760 one study reported an overall efficacy of 75%.761 Currently, fenbendazole (7.5–10 mg/kg PO every 24 hours for 5 days) followed on day 6 by ivermectin (200 mg/kg PO) is the most commonly advocated treatment regimen.659, 762 Moxidectin (400 μg/kg PO every 24 hours) may also be effective against adult organisms and L3 and L4 larval stages763 and may be useful for treating cyathostomiasis. Antiinflammatory therapy may be beneficial, especially in severe or refractory cases or before treatment with larvicidal medications. Pretreatment with dexamethasone or prednisolone is indicated before anthelmintic administration if heavy larval loads are suspected to prevent an acute exacerbation of the disease by rapid death of encysted larvae. Larvicidal treatment with moxidectin appears to be less likely than fenbendazole to result in tissue inflammation resulting from larval death.764 NSAID administration may have limited value, but dexamethasone appears to be efficacious in refractory cases when used in conjunction with larvicidal anthelmintics.659, 662 Bismuth subsalicylate is often administered orally as an antisecretory agent in young animals. Supportive care may be necessary in severe cases, particularly if hypoproteinemia is severe. Administration of IV crystalloid fluids and plasma or other colloids is occasionally required. Proper nutritional support is also important.
Arsenic Toxicity
Reduction of arsenic absorption by administration of cathartics such as activated charcoal should be initiated immediately.718 Chelation therapy with sodium thiosulfate (20–30 g in 300 mL of water, administered orally) and dimercaprol (British anti-Lewisite; 3 mg/kg PO every 4 hours) is indicated.718 Dimercaprol is a specific antidote for trivalent arsenicals, but its efficacy in horses is questionable.
Intestinal Anaphylaxis
Treatment of intestinal anaphylaxis is in principle similar to treatment of other forms of colitis, but it is often unsuccessful because of the rapidly progressive nature of the syndrome.162, 722, 727 Early treatment with prednisolone sodium succinate (10–20 mg/kg IV) or dexamethasone (0.1–0.2 mg/kg IV) may be essential for successful treatment.722
Peritonitis
Structure and Function
The peritoneum consists of a single layer of mesothelial cells. The mesothelial lining of the diaphragm, abdominal walls, and pelvic cavity is termed the parietal peritoneum. The visceral peritoneum forms the serosal surfaces of the intraabdominal organs. Caudally, the peritoneum reflects over the surfaces of the pelvic organs (portions of the urogenital tract and rectum), excluding them from the peritoneal space, and defining the retroperitoneal space. The peritoneal space communicates with the lumen of the uterus (and thus the external environment) via the fallopian tubes in females. In males the peritoneum forms a true blind sac. The vascular supply and nervous innervation of the visceral peritoneum are supplied by the splanchnic vessels and visceral autonomic nerves, respectively. Branches of the intercostal, lumbar, and iliac arteries supply the parietal peritoneum, and the phrenic and intercostal nerves provide nervous innervation. Inflammation of the parietal peritoneum is perceived as somatic pain, resulting in a splinted abdominal wall, pain on external palpation, and reluctance to move.
The peritoneal lining functions as a semipermeable barrier to the diffusion of water and low-molecular-weight solutes between the blood and the abdominal cavity.765 The peritoneum secretes a serous fluid that lubricates the abdominal cavity, inhibits adhesion formation, and has minor antibacterial properties.765, 766 Macrophages, mast cells, mesothelial cells, and lymphocytes provide immune function within the peritoneum.767 The peritoneal surface maintains a high level of fibrinolytic activity through the production of plasminogen activators by mesothelial cells. This function, together with the lubricant properties of the PF, helps to maintain gliding surfaces within the peritoneum and prevent adhesion formation. PF produced by the mesothelium tends to move ventrally and cranially, aided largely by diaphragmatic movement. PF, waste products, and foreign material exit the peritoneal cavity to enter the lymphatic system through diffusely distributed subendothelial pores or via the large diaphragmatic stomata, depending on particle size. Large molecules and particles greater than approximately 40,000 MW (such as bacteria) exit through the diaphragmatic stomata and ultimately enter the thoracic duct. The term peritonitis refers to inflammation of the mesothelial lining of the peritoneal cavity.
Etiopathogenesis
Peritonitis can occur in association with any insult—mechanical, chemical, or infectious—that results in disruption or irritation of the peritoneal lining, inflammation or infection of abdominal organs, or compromise of the intestinal wall.765 Common mechanical injuries include blunt or perforating trauma to the abdominal wall, breeding and foaling accidents, and abdominal surgery. A variety of iatrogenic insults can cause peritonitis, such as abdominocentesis, enterocentesis, splenic puncture, bowel trocarization, liver biopsy, uterine biopsy, castration, and rectal tear. Chemical insults of endogenous origin include blood, urine, pancreatic enzymes, bile, gastric juice, chyme, and chyle. Talc, contrast agents, antibiotics, and lavage solutions are additional examples of chemical insults. Traumatic events often involve bacterial contamination at the time of injury, and mechanical and chemical injuries can become infected secondarily.
The most common manifestation of peritonitis is acute, diffuse, septic peritonitis following inflammation, vascular insult, perforation, or surgical manipulation (enterotomy, resection, and anastomosis) of the gastrointestinal tract. The septic process in such cases involves mixed bacteria of gastrointestinal origin. Penetrating abdominal wounds also result in mixed infections. Less commonly, singular bacterial forms gain access to the peritoneum through hematogenous spread, extension from a contiguous organ, or through the female genital tract. Primary monomicrobial infections have been reported involving Streptococcus equi subsp. equi, 768 S. equi subsp. zooepidemicus,769 Rhodococcus equi, 770 and Corynebacterium pseudotuberculosis. 771 Several case series involving peritonitis associated with Actinobacillus equuli have been reported.772, 773, 774, 775 Sepsis, septic omphalitis, ascending urinary tract infections, and uterine infections are additional causes of monomicrobial infection.
Most cases of trauma or intestinal perforation result in contamination of the peritoneum with large numbers of many types of bacteria. The intestinal tract contains a mixed population of bacteria, and the quantity of bacteria and prevalence of anaerobic species increase in the distal segments. Not surprisingly, mortality associated with contamination from the lower bowel is high. Hirsch and Jang776 reported isolation of an infective agent from exudative equine PF in approximately 25% of attempts. Obligate anaerobic bacteria were cultured most frequently, followed by members of the Enterobacteriaceae family (predominantly E. coli). Penicillin-resistant Bacteroides fragilis was isolated from 10% to 20% of cases. In another study in which bacteria were identified in equine abdominal fluid by cytologic examination or culture, E. coli was the organism most commonly isolated.777 In human beings and laboratory animals, despite the variety of organisms initially introduced by polymicrobial contamination, established infections are characterized by only a few types of bacteria, which are often gram-negative aerobes and anaerobic bacteria. This selectivity occurs through the processes of selective reduction of bacterial populations and bacterial synergism. An example of synergism in human beings and laboratory animals is peritonitis involving E. coli and B. fragilis. The presence of each organism is beneficial to the survival of the other, and each is important in the overall pathogenesis of the disease. E. coli is associated with septicemia and early mortality, whereas B. fragilis infection tends to result in chronic abscessation with delayed morbidity and mortality.766
Other causes of equine peritonitis include parasites, viral disorders (influenza, equine viral arteritis, equine infectious anemia, African horse sickness), and neoplasia. Verminous arteritis caused by strongylosis can lead to vascular damage (thromboembolism and infarction) to the intestine. The activities of strongyles, ascarids, and tapeworms can result in perforation of the bowel and damage to other abdominal organs. There has been one report of septic peritonitis caused by colonic perforation associated with aberrant migration of Gasterophilus intestinalis larva.778
Biologic events resulting from contamination of the abdomen or injury to the mesothelial cells include release of catecholamines, histamine, and serotonin from peritoneal mast cells; vasodilation and hyperemia; an increase in peritoneal vascular permeability; secretion of protein-rich fluid into the peritoneum; transformation of mesothelial cells into macrophages; and influx of PMN cells, humoral opsonins, natural antibodies, and serum complement into the peritoneal cavity.765 Other possible events include depression of the peritoneal fibrinolytic activity, fibrin deposits on the peritoneal surface, and inflammatory-mediated and sympathetic-mediated ileus. These processes serve to confine contamination and infection and, with clean, minimally invasive procedures such as enterocentesis or trocarization, are effective. However, with greater severity of peritoneal contamination or irritation, these processes are magnified and become deleterious. Consequences include hypovolemia, hypoproteinemia, gastrointestinal ileus, ischemia of the bowel wall with subsequent absorption of bacteria and toxins, and ultimately adhesion and abscess formation. Equine peritoneal macrophages release a multitude of inflammatory mediators when exposed to bacterial LPS, and endotoxemia contributes the clinical picture. A complete review of endotoxemia is presented in Endotoxemia.
A complete pathologic description of peritonitis includes origin (primary or secondary), onset (peracute, acute, and chronic), distribution (localized versus diffuse), and presence of bacteria (septic versus nonseptic). Clinically, viewing the pathogenesis of peritonitis as a series of stages is useful. The contamination stage, lasting 3 to 6 hours, involves introduction of bacteria into the peritoneum and initiation of the acute inflammatory response previously described. If the organisms are not eliminated, the process evolves to the stage of acute diffuse peritonitis as, regardless of the location of the initial contamination, bacteria spread throughout the peritoneum within several hours. The stage of acute diffuse peritonitis lasts up to 5 days. The inflammatory response persists and escalates with continued exudation of proteinaceous fluid and influx of inflammatory cells. Offending organisms are delivered to the lymphatic system and may be eliminated by the immune system. Alternatively, organisms can gain access to the systemic circulation in sufficient numbers to result in clinically relevant bacteremia. This stage of the disease process has the highest mortality because of the effects of severe peritoneal inflammation, endotoxemia, and sepsis. If the animal survives this stage but fails to eliminate infection from the peritoneal cavity, the disease enters a transitional phase referred to as the acute adhesive (or localizing) stage, typically occurring 4 to 10 days after the initial insult. Neutrophils are still active, macrophages are increasing in numbers, and fibrin aggregates are being organized or lysed. If infection persists beyond this point, organization of fibrin proceeds and organisms become isolated from host defenses. At this point, the disease process enters the stage of chronic abscessation. This stage can begin as early as 8 days after inoculation and persist indefinitely.
Clinical Signs
Clinical signs of peritonitis depend on the primary disease process, the duration of the problem, and the extent of peritoneal inflammation. Localized peritonitis may have few or no systemic manifestations, whereas severe localized or generalized peritonitis often is accompanied by severe toxemia, sepsis, or both. Septic peritonitis usually causes more severe clinical signs because of the systemic inflammatory response and endotoxemia. Most clinical signs are nonspecific and include fever, lethargy, inappetence, decreased borborygmi, and dehydration. Additional signs include colic, ileus, weight loss, and diarrhea.768
Horses with peracute peritonitis, as occurs with rupture of the bowel or rectal tear, have clinical signs associated with severe endotoxemia, weakness, lethargy, colic, and/or circulatory failure. Fever may not be present depending on the degree of shock. Parietal pain, characterized by reluctance to move, splinting of the abdominal wall, and sensitivity to external abdominal pressure occur in some acute cases. With extensive abdominal fecal contamination, rectal examination may reveal a gritty feeling of the serosal and parietal surface of the peritoneum because of fibrin deposition.
In horses with more chronic peritonitis, clinical signs include intermittent colic, lethargy, anorexia, weight loss, intermittent fever, ventral edema, exercise intolerance, decreased or absent intestinal sounds, and mild dehydration. Heart and respiratory rates may be normal. Fecal output may be normal; however, horses with chronic diarrhea and weight loss have been reported. Rectal examination findings can include pain on palpation of fibrinous or fibrous adhesions, intestinal distention, an abdominal mass, or an impression of bowel floating in fluid. In many cases, rectal examination does not reveal significant abnormalities.768
In cases of A. equuli peritonitis, clinical signs in most horses included lethargy, inappetence, and mild to moderate abdominal pain if acute or weight loss if chronic.773, 774 Postpartum mares with peritonitis secondary to a uterine perforation typically present with fever and depression, with or without abdominal pain. Septic tenosynovitis of the tarsal sheath secondary to bacterial peritonitis from gastrointestinal perforation has been reported.779 Foals with peritonitis usually exhibit signs of colic (acute or chronic) and are febrile, depressed, and inappetent. In young foals, peritonitis can cause rapid metabolic deterioration, and determination and correction of the primary problem require immediate attention. In older foals, peritonitis may occur insidiously in association with S. equi subsp. equi or R. equi infections.
Clinicopathologic Findings
Clinicopathologic abnormalities vary depending on the time of onset and severity. In the acute stage, leukopenia, hemoconcentration, metabolic acidosis, and azotemia predominate. After several days, leukocytosis and hyperfibrinogenemia are more typical. In chronic peritonitis, hyperproteinemia with hyperglobulinemia may be present. SAA is also increased.254 Neonates with uroperitoneum tend to develop azotemia, hyponatremia, hypochloremia, hyperkalemia, and acidemia.
PF analysis is principal to the diagnosis of peritonitis. Detailed description of PF analysis is provided in the Examination section of this chapter. PF WBC counts in acute peritonitis are reportedly higher than those in chronic peritonitis,768 but this is not always the case, and the WBC count does not always correlate with disease severity or prognosis. The PF WBC count can be greater than 100,000/μL following enterocentesis, with no clinical signs or problems.780 Conversely, peritoneal WBC counts of fewer than 100,000/μL may be found in foals or horses with intraabdominal abscesses.781 The peritoneal WBC count can increase to greater than 150,000/μL following celiotomy782 and can be higher if an enterotomy is done. Postoperatively, the WBC count normally continues to decline and returns to near normal by 5 to 7 days. Failure of the WBC count to decrease suggests peritonitis resulting from a postoperative complication. Finally, PF WBC counts greater than 500,000/μL indicate severe focal or generalized peritoneal sepsis. With acute peritonitis, PMN cells typically increase to a greater degree than mononuclear cells, but this depends on the cause. In horses with gastrointestinal disease and endotoxemia, the number of peritoneal mononuclear cells increases, as does transformation of mesothelial cells to macrophages. In chronic cases, one easily may mistake transforming mesothelial cells for neoplastic cells, which can make diagnosis difficult.
The presence of free and phagocytosed bacteria in PF indicates generalized suppuration, abscessation, or compromised bowel. If one observes numerous microorganisms of mixed types free in the PF, especially in conjunction with plant material, bowel rupture likely has occurred. The presence of toxic or degenerate neutrophils and bacteria within PMN cells helps to distinguish PF from intestinal contents in such horses. Fluid obtained from enterocentesis is largely devoid of WBCs but is discolored and contains mixed microorganisms and plant material. Bacterial contamination of a sample can occur during collection of the sample, and iatrogenic contamination of a sample can result in free and intracellular bacteria, particularly if processing is delayed. In such cases the bacterial numbers are few and the neutrophils appear healthy. In some cases of gastrointestinal perforation the luminal material, inflammatory cells, and protein may be sequestered by the omentum and further contained by fibrinous adhesions. Abdominal fluid obtained via standard ventral paracentesis may have low cellularity and protein content but large numbers of mixed bacteria, indicating bowel rupture.768 Examples include gastric rupture along the greater curvature of the stomach between the omental layers (omental bursa) and perforated gastric or duodenal ulcers in foals. Correlating all cytologic findings with clinical and clinicopathologic findings is important for interpreting the results of PF cytologic examination.
Biochemical analysis of PF may be useful in detecting sepsis when cytologic examination and culture are negative or otherwise unavailable. PF pH and glucose concentrations from horses with septic peritonitis were significantly lower than in horses with nonseptic peritonitis and healthy horses.783 PF pH less than 7.3, glucose less than 30 mg/dL, and fibrinogen concentration greater than 200 mg/dL were considered highly predictive of septic peritonitis. Serum to peritoneal glucose concentration differences of greater than 50 mg/dL were considered the most diagnostically useful test for septic peritonitis in the study. PF lactate concentration is also increased because of septic peritonitis, and a blood-to-peritoneal lactate concentration difference may have diagnostic significance. In a small study in dogs, a blood-to-fluid lactate difference of ≤2.0 mmol/L was shown to be 100% sensitive and specific for a diagnosis of septic peritonitis.784
PF samples should be submitted for aerobic and anaerobic cultures in appropriate media (i.e., BBL Port-A-Cul tubes, Becton, Dickinson & Co., Franklin Lakes, NJ, in an attempt to identify the pathogenic organism(s). To enhance recovery of bacteria, PF may be inoculated into blood culture medium (i.e., Septi-Chek Columbia, Hoffmann-LaRoche Inc., Nutley, NJ). If the horse has received prior antimicrobial treatment, the fluid sample should be passed through an antimicrobial removal device before culture (i.e., A.R.D., Becton Dickinson & Co., Franklin Lakes, NJ).
Treatment
Early and aggressive therapy is important for a successful outcome. Treatment goals are to resolve the primary problem, minimize inflammation, and prevent long-term complications. In the acute phase, analgesia and therapy as described for endotoxemia are important. Flunixin meglumine is advocated for its local and systemic antiinflammatory effects and may be effective in retarding adhesion formation.785
Initial antimicrobial selection should be broad spectrum based on the presumption that mixed infection is present. IV administration is preferred over oral or intramuscular routes in acute, diffuse, septic peritonitis because more reliable concentrations of drugs are achieved in the tissues and PF.786 The combination of a β-lactam antibiotic with an aminoglycoside, such as potassium penicillin (22,000–44,000 IU/kg IV every 6 hours) combined with gentamicin (6.6 mg/kg every 24 hours), is appropriate in most circumstances. Metronidazole (25 mg/kg orally every 12 hours) can be added to the regimen given the strong possibility of infection involving penicillin-resistant B. fragilis. This regimen can be modified when culture and antimicrobial sensitivity results become available. Peritonitis caused by A. equuli generally responds well to therapy with penicillin either alone or in combination with gentamicin. Aminoglycosides and NSAIDs have the potential to induce acute renal tubular damage, particularly with concurrent dehydration and decreased renal perfusion. Therefore adequate restoration and maintenance of hydration and monitoring of renal function are important. Monitoring of the WBC count, plasma fibrinogen, SAA, and abdominal fluid analysis often guide duration of therapy. Horses with abdominal abscessation resulting from monomicrobial infections typically require weeks to months of therapy, whereas polymicrobial infection may require many months of antibiotic treatment. Further details regarding principles of antimicrobial therapy are provided elsewhere in this text (Chapter 2).
Abdominal drainage and lavage can help remove excess fluid, foreign materials, fibrin, and bacterial products from horses with peritonitis. Postoperative lavage decreases the incidence of experimentally induced abdominal adhesions.787 Open surgical exploration provides the most effective and thorough examination of all peritoneal surfaces and is recommended if gastrointestinal perforation or ischemia is suspected or in any other case in which correction of a primary lesion is indicated. A ventral abdominal drain can either be placed at the time of surgery or in the standing horse with sedation and local anesthesia. Techniques have been described in detail elsewhere.788, 789
Peritoneal lavage is typically performed with 10 to 20 L of a balanced isotonic electrolyte solution twice a day for 3 to 5 days, until the lavage solution becomes clear or until the catheter becomes clogged with fibrin or omentum. Hypertonic solutions should be avoided because they can result in fluid shifts into the peritoneum. The addition of povidone iodine to a balanced solution should be avoided because concentrations as low as 3% can induce peritoneal inflammation.790 Other agents such as antibiotics or heparin have also been suggested as components of the lavage solution, but data demonstrating their benefit are not currently available. Active (or closed suction) abdominal drains have also been advocated, with similar benefits and potential complications to other methods.789 Lavage with a plain isotonic solution did not alter the pharmacokinetics of gentamicin administered systemically.791 Thus alteration of antimicrobial dosing does not appear necessary if lavage with plain solutions is part of the therapeutic regimen. Complications associated with the use of abdominal drains or repeated peritoneal drainage include retrograde infection, local irritation, pneumoperitoneum, and SC seepage around the drain and resultant cellulitis. If the patient is hypovolemic or hypoproteinemic, volume replacement and administration of plasma or synthetic colloids should be considered before removing large quantities of fluid from the abdomen.
In horses with suspected parasitic involvement, one should give larvicidal doses of an anthelmintic once the condition of the horse is stabilized. Ivermectin, fenbendazole, and thiabendazole have been recommended as larvicidal therapies.
The decision to perform surgical versus medical treatment is controversial in horses with peritonitis and needs to be made on a case-by-case basis. Although surgical exploration may allow diagnosis and resolution of the inciting cause and more thorough lavage of the abdomen, the risks of anesthesia, possible additional cost of surgery, and potential for a prolonged return to performance may be significant. In a recent study of horses and foals with peritonitis, survival to discharge without surgery was associated with lack of signs of abdominal pain, normal or improved rectal temperature, normal or improved borborygmi, normal fecal production, no abnormal findings on palpation per rectum, no nasogastric reflux, and yellow/orange PF.792 A retrospective comparison of medical and surgical treatment of postpartum mares with peritonitis secondary to a uterine tear found no significant difference between admission variables, survival rate, hospital bill, duration of hospital stay, and likelihood to foal following discharge.793 A limitation of this study is the ability to definitively diagnose a uterine tear in those mares treated medically; if a tear was not palpable, the diagnosis was based on the exclusion of other causes.
Prognosis
The prognosis is grave for peritonitis associated with gastrointestinal rupture. Reported survival rates for horses with peritonitis vary but can be as high as 78%.792, 794 In one study with a 78% overall survival rate, 68% of horses treated medically survived792; in another, 93% of those treated medically survived.795 Some of the variability in reported survival percentages can be related to inclusion criteria, mainly whether or not horses with gastrointestinal rupture were included. Septic peritonitis following abdominal surgery was associated with high mortality (56%) in some reports,777 although no difference in short-term survival was seen in another study.796 Peritonitis associated with A. equuli carries a very favorable prognosis, and all horses in these reports responded to medical therapy if attempted.773, 774
Pathophysiology of Mucosal Injury and Repair
Mucosal Barrier Function
To gain an appreciation of the mechanisms in which the mucosa is injured and subsequently repaired, it is important to understand how the integrity of the mucosa is physiologically regulated. Regulation of mucosal integrity is referred to as mucosal barrier function, which is vital because it prevents bacteria and associated toxins from gaining access to subepithelial tissues and the circulation. However, the mucosa has two conflicting functions: it must serve as a protective barrier while continuing to absorb solutes necessary to maintain the well-being of the host. This conflict is most notable at the intercellular (paracellular) space, which allows passage of select solutes and water797, 798, 799, 800 but does not admit large molecules, including bacterial toxins.801 The paracellular space is almost exclusively regulated by the tight junction,574 which is the interepithelial junction at the apical-most aspect of the paracellular space. Although these tight junctions were originally viewed as inert cellular adhesion sites, it has become clear in recent years that tight junction permeability is dependent on tissue-specific molecular structure and regulated by a complex array of intracellular proteins and the cytoskeleton. Tight junctions consist of a group of transmembrane proteins that interdigitate from adjacent cells. Although occludin was originally thought to be the predominant tight junction transmembrane protein, a group of proteins termed claudins appear to fine-tune the function of the tight junction. For example, select claudins are responsible for the relative porosity of the barrier to select electrolytes based on their charge within the paracellular space.802 These transmembrane proteins interact with the cytoskeleton via a series of intracellular proteins, including zonula occludens (ZO)-1, ZO-2, ZO-3, cingulin, and others.803 In addition, local regulatory proteins such as the small GTPase Rho are critical to tight junction function. Generally, the relative contractile state of the actin cytoskeleton determines the degree to which tight junctions are open or closed, but the complexities of regulation of this process are poorly understood.804, 805
The most sensitive measure of mucosal barrier function is transepithelial electrical resistance, which is measured by mounting mucosa in an in vitro system called an Ussing chamber, because this measurement is largely a reflection of the permeability of mucosa to ions.806, 807 There are two routes ions it may follow when traversing the epithelium: transcellular and paracellular.801 Because cell membranes have a resistance to the passive flow of ions 1.5 to 3 log units greater than that of the epithelium as a whole, measurements of transepithelial resistance largely reflect the resistance of the paracellular space, particularly the tight junctions that regulate the paracellular flow of ions.807 Because tight junctions differ in structure from different portions of the mucosa,808 measurements of transepithelial resistance reflect the net resistance of the epithelium of variable permeability within a given tissue. For example, tight junctions in the intestinal glandular structures called crypts are leakier than those in the surface epithelium because of fewer and less organized tight junction strands.806, 809 Conversely, surface epithelium has a greater number of well-organized tight junction strands that result in epithelium with a relatively high resistance.806 This correlates well with the absorptive function of epithelium located on the mucosal surface and the secretory function of crypt epithelium. The structure of tight junctions also varies with the segment of intestine. For example, tight junctions have more strands in the ileum than the jejunum, which is reflected by a higher transepithelial resistance in the ileum.810 In addition, cells are more closely apposed at the level of the tight junction within the colon. This is in keeping with the largely absorptive role of the colon and is advantageous given the hostile microbial environment of the colon.
Gastric Mucosal Barrier Function
There are four regions of the stomach based on the type of mucosal lining (in an orad to aborad order): nonglandular stratified squamous epithelium, cardiac epithelium, proper gastric mucosa, and pyloric mucosa.811 Stratified squamous epithelium has distinct differences in terms of barrier function compared with the remainder of the gastrointestinal tract. This epithelium has baseline transepithelial resistance measurements of approximately 2 to 3000 Ω∙cm2, which is an order of magnitude higher than the adjacent cardiac mucosa.812, 813 Thus the stratified squamous mucosa is exceptionally impermeable. This is the only mechanism this mucosa has to defend itself against injury. The stratified squamous epithelium consists of four layers: the outer stratum corneum, stratum transitionale, stratum spinosum, and the basal stratum germinativum. However, not all layers contribute equally to barrier function, which is largely composed of interepithelial tight junctions in the stratum corneum and mucosubstances secreted by the stratum spinosum. 812, 814 The relative impermeability of stratified squamous mucosa can be demonstrated by the effects of HCl on this type of epithelium in vitro, which has very little effect until it reaches a pH of 2.5 or below.813 Although the majority of the literature on equine ulceration pertains to the effects of HCl and inhibitors of HCl secretion,815, 816, 817, 818 other factors may be critical to the development of gastric ulcer disease.
The site of HCl secretion (proper gastric mucosa) is protected from so-called back diffusion of H+ ions by a relatively high transepithelial electrical resistance (compared with cardiac mucosa), but there are also a number of other critical mechanisms to prevent acid injury. The gastric mucosa secretes both mucus and bicarbonate, which together form an HCO3 − containing gel that titrates acid before it reaches the lumen.819, 820 The mucous layer is principally formed by glycoproteins (mucins) secreted by goblet cells but also includes other gastric secretions and sloughed epithelial cells. Mucins consist of core peptides with a series of densely packed O-linked polysaccharide side chains that, once secreted, become hydrated and form a viscoelastic gel. The mucous layer, however, does not form an absolute barrier to back diffusion of acid. For acid that does back diffuse into the gastric mucosa, epithelial Na+/H+ exchangers are capable of expelling H+ once the cell reaches a critical pH.820
Recent studies have renewed interest in the protective mechanisms of mucus because of the discovery of a group of compounds secreted by goblet cells called the trefoil peptides. The name of these peptides is derived from a highly conserved cloverleaf structural motif, which confers substantial resistance to degradation by proteases including pepsin. There are three known members of this group, pS2, SP, and intestinal trefoil factor (ITF), the latter of which is solely secreted by goblet cells in the small and large intestine. Both pS2 and SP are secreted by goblet cells within the stomach and are believed to intercalate with mucous glycoproteins, possibly contributing to the barrier properties of mucus.821 These peptides also play a critical role in repair of injured mucosa.
An additional mucosal function that serves to reduce the level of injury is adaptive cytoprotection, in which application of topical irritants to gastric mucosa results in subsequent protection of mucosa in response to repeated exposure to damaging agents. For example, pretreatment with 10% ethanol protected against mucosal damage in response to subsequent application of absolute ethanol, and this effect was abolished by treatment with the COX inhibitor indomethacin.822 The cytoprotective effects of prostaglandins have been demonstrated directly in studies in which preadministration of prostaglandins protected gastric mucosa from damage by agents such as concentrated hydrochloric acid and hypertonic saline.823 Prostaglandins appear to be cytoprotective in the stomach at doses less than those used to inhibit gastric acid secretion, ruling out a simple antacid mechanism.824 Although not fully characterized, cytoprotection has been attributed in part to prostaglandin-stimulated mucous production.825 An associated beneficial effect of prostaglandins is the increased production of bicarbonate, which is trapped within mucus on the surface of the mucosa.826, 827 Interestingly, PGE2 appears to lose its cytoprotective activity in the presence of the mucolytic agent N-acetylcysteine. Attention has also been directed at enhanced mucosal blood flow as a potential mechanism for prostaglandin-mediated cytoprotection—for example, pretreatment with PGI2 protected against ethanol-induced mucosal damage as a result of increased mucosal blood flow.828 In addition PGE2, which is also cytoprotective despite the fact that it does not increase blood flow,829 prevents vascular stasis associated with irritant-induced vascular damage by inhibiting neutrophil adherence to damaged endothelium.830
Sensory nerves distributed throughout gastrointestinal mucosa have also been implicated in cytoprotective mechanisms. As an example of their importance in mucosal cytoprotection, pretreatment of newborn rats with capsaicin (which dose dependently destroys sensory nerves) renders the mature rats more susceptible to gastric injury.831 Alternatively, the use of a low dose of capsaicin, which stimulates rather than destroys sensory nerves, protects gastric mucosa against injurious agents.832, 833 Sensory nerves contain neuropeptides such as calcitonin-gene–related peptide (CGRP) and substance P, which may play a protective role via vascular mechanisms. For instance, CGRP stimulates increased gastric blood flow, which is theorized to reduce injury similarly to prostaglandins. Recent studies suggest that the roles of prostaglandins and CGRP in gastric cytoprotection are intimately intertwined. In particular, PGI2 is believed to sensitize sensory nerves following treatment with a mild irritant, with resultant increases in CGRP release and mucosal flow. Similar studies have shown that antagonists of CGRP inhibit the cytoprotective action of PGE2.834 Another neural mediator, nitric oxide, has also been implicated in adaptive cytoprotection. Interestingly, nitric oxide has a number of actions that are similar to those of prostaglandins, including maintenance of mucosal blood flow.835
Intestinal Barrier Function
Regulation of barrier function in the intestine is not as well characterized as that of the stomach, although mechanisms of barrier function, including secretion of mucus and regulation of mucosal blood flow, are presumed to be similar. The proximal duodenum also has to protect itself from acid damage as it receives gastric contents, and this involves secretion of mucus and bicarbonate similarly to the stomach. One other mechanism that helps both the stomach and the intestine to maintain mucosal barrier function is the speed with which the mucosa repairs. For a defect to develop in the mucosal barrier, injurious factors have to outpace mucosal recovery. Such recovery initially involves epithelial migration across denuded regions of basement membrane (restitution).821 This process is so rapid that epithelial defects may be resurfaced within minutes. For example, in bile salt–injured equine colon, denuded surface mucosa was completely covered by restituting epithelium within 180 minutes.207 In the small intestine, intestinal villi greatly amplify the surface area of the mucosal luminal surface, which in turn takes far longer to resurface with restituting epithelium once it has become denuded.836 Intestinal villi, however, are able to dramatically reduce the denuded surface area by contracting.837
Mechanisms of Gastric Injury
Although the stratified squamous epithelium is relatively impermeable to HCl, there are a number of factors that can dramatically enhance the damaging effects of HCl in this epithelium. In particular, bile salts and SCFAs are capable of breaking down the squamous epithelial barrier at an acid pH, exposing deep layers to HCl, with subsequent development of ulceration.813, 838 Relatively high concentrations of SCFA normally exist within the equine stomach as a result of microbial fermentation.812 These weak acids penetrate squamous mucosa and appear to damage Na+ transport activity principally located in the stratum germinativum. Bile salts may also be present in the proximal stomach as a result of reflux from the duodenum. Although such reflux has a relatively high pH, it appears that bile salts adhere to stratified squamous epithelium, becoming lipid soluble and triggering damage once the pH falls below 4.839 Diet and management (e.g., periods of fasting) also play crucial roles in the development of conditions conducive to gastric ulceration. Typically, there is a pH gradation in horses from proximal to distal compartments of the stomach, with the lowest pH values in the distal stomach.840 During periods of fasting, this stratification is disrupted such that low pH values may be recorded in the proximal stomach.841 Fasting conditions also increase the concentration of duodenal contents within the proximal stomach, particularly bile.839
Proper gastric mucosa is exposed to injurious agents, including pepsin, bile, and acid. The latter is constantly secreted by parietal cells in the horse as an adaptation to near-continuous intake of roughage,811 but it is tightly regulated by enterochromaffin (ECL)-like cells within the proper gastric mucosa and G and D cells, which are present within the pyloric mucosa. Acid secretion is amplified by ECL-released histamine, which interacts with H2 receptors on parietal cells, and G cells, which release the prosecretory hormone gastrin. A combination of histamine and gastrin can have a synergistic effect on parietal cell gastric secretion, because these mediators have distinct receptors and second messengers. On the other hand, D cells are sensitive to an acidic environment and release somatostatin, which inhibits acid secretion.842 Nonetheless, gastric mucosa may be exposed to acid for prolonged periods of time, particularly in horses that are extensively meal fed and do not have the benefit of roughage, which tends to buffer stomach contents.839, 842
Aside from peptic ulceration, induced by combinations of acid and pepsin, research in the human field has revealed the tremendous importance of Helicobacter pylori in inducing ulceration. Infection with this organism has the effect of raising gastric pH because of disruption of gastric glands but also induces an inflammatory reaction that causes damage.843 However, there is very little evidence that this organism is involved in gastric ulcers in horses. In the absence of a known role for infectious agents in gastric ulceration in animals, ulceration likely develops from injurious factors similar to those found in the proximal stomach, including gastric acid and bile. Some factors that are important to the induction of squamous epithelial ulceration may not be important in the development of proper gastric mucosal ulceration. For example, feed deprivation and intensive training reproducibly induce squamous epithelial ulceration in horses but have little effect on proper gastric mucosa in horses.844 Gastric acid likely plays a key role, whereas other factors such as NSAIDs serve to reduce gastric defense mechanisms. In particular, inhibition of prostaglandin production would reduce mucous and bicarbonate secretion while reducing gastric mucosal blood flow.845 Some of the NSAIDs also have a topical irritant effect, although this appears to be of minor significance, because the route of administration (oral or parenteral) seems to have little influence on the development of ulceration.846
The source of prostaglandins responsible for gastric protection was originally assumed to be COX-1, because this COX isoform is constitutively expressed in gastric mucosa, whereas COX-2 is not expressed in the stomach unless it is induced by inflammatory mediators. However, mice in which the COX-1 gene has been knocked out fail to develop spontaneous gastric lesions,847 possibly because of compensatory increases in prostaglandin production by COX-2.848 This concept agrees with recent data indicating that inhibition of both COX isoforms is required to induce gastric ulceration.849 From a clinical perspective, these data indicate that drugs selective for either COX-1 or COX-2 may be less ulcerogenic in the horse because they allow the uninhibited COX isoforms to continue to produce protective prostanoids. Because COX-2 elaborates prostaglandins induced by inflammatory stimuli, preferential or selective inhibitors of COX-2 may be particularly useful because of their ability to serve as antiinflammatory agents that are less ulcerogenic.392
Intestinal Ischemia/Reperfusion Injury
The most notable cause of intestinal mucosal injury in horses, particularly those suffering from colic, is ischemia. Initially, it seems intuitive that by reducing gastrointestinal blood supply, the mucosa becomes injured. The anatomy of the gastrointestinal tract, and the differing structure of the intestinal mucosa at various anatomic locations, has a significant influence on the extent of mucosal injury. Ischemic injury may be induced by several different mechanisms, including occlusion of arterial supply by a thrombus, strangulation of intestinal vasculature, and generalized reduction in blood flow associated with various shock states. A number of seemingly distinct mechanisms of intestinal injury, such as intestinal distention, also trigger mucosal injury via an ischemic mechanism. Reperfusion injury may also influence the extent of mucosal injury following an ischemic episode and has been proposed as a potential site of therapeutic intervention.850, 851 It is critical that the mechanisms of ischemia/reperfusion injury be understood to develop an understanding of the severity of various clinical conditions, and begin to formulate a therapeutic approach to diseases characterized by this devastating form of injury.
Regulation of Intestinal Blood Flow
The intestinal circulation is capable of closely regulating blood flow during periods of low systemic perfusion pressure.852, 853 In particular, local regulation of resistance vessels within the microvasculature is particularly prominent; metabolic end products of ATP result in continued dilatation of resistance vessels despite reductions in systemic arterial pressure. This results in continued perfusion of gastrointestinal tissues during the early stages of shock, while other organs such as skeletal muscle undergo massive shunting of blood as a result of marked increases in the resistance of resistance vessels. The reasons for these differences in regulation are not entirely clear but may relate to the relatively high level of energy required to fuel the intestinal mucosa and the serious systemic effects of breaches in the mucosal barrier. When blood flow falls below a critical level, regulatory systems are no longer effective and oxygen uptake by the gastrointestinal tissue decreases, culminating in tissue damage.852
The villous tip is the most susceptible region affected by hypoxia in the equine small intestine, largely because of the countercurrent exchange mechanism of blood flow in the small intestinal villus.852 This countercurrent exchange mechanism is attributable to the vascular architecture, which consists of a central arteriole that courses up the core villus, arborizes at the tip, and is drained by venules coursing down the periphery of the villus.854 As oxygenated blood flows into the central arteriole, oxygen tends to diffuse across to the adjacent venules, which are flowing in the opposite direction. This series of events takes place along the length of the villus, resulting in a villous tip that is relatively hypoxic even under normal conditions. Furthermore, when blood flow is reduced, as occurs in hypovolemic or septic shock, the countercurrent exchange of oxygen is enhanced, and the tip becomes absolutely hypoxic.852 This mechanism might explain why the small intestinal mucosa is more susceptible to ischemic injury, compared with the colon, which has no villi. The duration of ischemia required to produce severe morphologic damage to the equine colon is approximately 25% longer than the small intestine.855
Ischemic Epithelial Injury
Intestinal mucosal epithelium is very susceptible to hypoxia because of the relatively high level of energy required to fuel the Na+/K+-ATPase that directly or indirectly regulates ion and nutrient flux. The first biochemical event to occur during hypoxia is a loss of oxidative phosphorylation. The resulting diminished ATP concentration causes failure of the energy-dependent Na+/K+-ATPase and the accumulation of sodium and subsequently intracellular water. The pH of the cytosol drops as lactic acid and inorganic phosphates accumulate from anaerobic glycolysis. The falling pH damages cell membranes, including lysosomal membranes, resulting in the release and activation of lysosomal enzymes into the cytosol, further damaging cellular membranes. Damage to the cell membrane allows the accumulation of high concentrations of calcium in the cytosol, which activates calcium-dependent degradative enzymes.856 These events result in cytoplasmic blebbing of the basal membrane, with subsequent detachment of cells from the underlying basement membrane.
Recent studies on epithelial injury during ischemia suggest that the majority of epithelial cells undergo programmed cell death (apoptosis) during ischemia and reperfusion rather than necrosis, allowing retention of reusable components of irreversibly injured cells.857 In one study, 80% of detached epithelium during small intestinal ischemia/reperfusion underwent apoptosis.858 Although the most obvious result of apoptosis is loss of surface epithelium, a number of cells on the lower portion of the villus (in the small intestine) and cells within the crypts may also undergo apoptosis that only becomes evident up to 24 hours following reperfusion of ischemic tissue.859
Morphologic changes observed in ischemic-injured small intestinal mucosa follow a similar sequence regardless of whether injury is induced by ischemia alone or ischemia/reperfusion (Table 12.6 ).860 Initially, epithelium separates from the underlying basement membrane, forming a fluid-filled space termed Grüenhagen’s space (Fig. 12.7 ). The mechanism of fluid accumulation in this space is not entirely understood but may result from continued epithelial absorption of NaCl and water before it has fully detached from neighboring epithelial cells. This fluid accumulation likely exacerbates epithelial separation from the basement membrane. Subsequently, epithelium progressively sloughs from the tip of the villus toward the crypts, which are the last components of the intestinal mucosa to become injured.163, 406, 861 This likely relates to the vascular architecture, because crypts receive a blood supply that is separate from the vasculature involved in the villous countercurrent exchange mechanism. The early morphologic changes observed in the equine large colon during ischemia are somewhat different from those described in the equine small intestine because of the lack of intestinal villi. As might be expected, the more superficially located surface cells are sloughed before those in crypts.855, 862 The orderly progression of tissue injury has been used by one group of investigators to accurately predict survival in horses with large colon volvulus. Biopsies were taken from the pelvic flexure, which has been previously shown to accurately reflect mucosal changes along the length of the colon,863 and histologically examined for the width of the crypts and intercrypt interstitial space. The latter measurements were expressed as an interstitium: crypt width (I:C) ratio. Nonviable colon was defined as that which had >60% loss of crypt and an I:C ratio >3. Using this methodology, survival was correctly predicted in 94% of horses.864
TABLE 12.6.
Grading System for Ischemia/Reperfusion Injury in Small Intestinal Mucosa
| Grade | Description |
|---|---|
| 1 | Separation of epithelium at the tip of the villus, creating a small space between the epithelium and basement membrane called Grüenhagen’s space |
| 2 | Loss of epithelium from the tip of the villus |
| 3 | Loss of epithelium from the upper third of the villus |
| 4 | Complete loss of villous epithelium |
| 5 | Injury or loss of epithelium within the crypt in addition to complete loss of villous epithelium |
Adapted from Chiu CJ, McArdle AH, Brown R, et al. Intestinal mucosal lesion in low-flow states. I. A morphological, hemodynamic, and metabolic reappraisal. Arch Surg. 1970; 101:478-483.
Fig. 12.7.
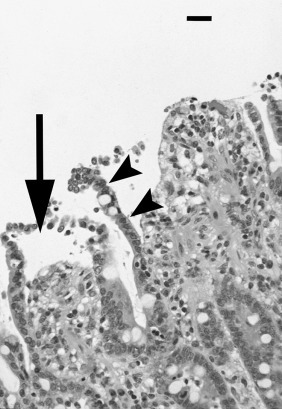
Histologic appearance of Grüenhagen’s space in ischemic injured ileal mucosa. Note separation of epithelium at the tip of the villus from its basement membrane, creating a space (arrows). Epithelium subsequently sloughs into the lumen (arrowheads). 1-cm bar = 100 μm.
Strangulating Obstruction
Since the dramatic decline in S. vulgaris–induced colic, which was frequently associated with infarction of intestinal arterial blood supply,865 the vast majority of ischemic lesions are associated with strangulating obstruction. Therefore it is important to consider mechanisms of ischemic injury in horses with naturally occurring strangulating lesions. The majority of experimental work has either assessed complete ischemia (complete occlusion of the arterial blood supply)855 or low-flow ischemia (reduction of arterial blood flow).866, 867 During intestinal strangulation, however, a disparity between the degree of occlusion of the veins and arteries occurs in which veins are occluded before arteries because of differences in the compliance of vascular walls. Strangulating lesions are typically hemorrhagic (hemorrhagic strangulating obstruction), because the arteries continue to supply blood to tissues that have little or no venous drainage. This results in ischemic injury, as previously outlined, but also in tremendous congestion of the tissues. Such hemorrhagic congestion has two opposing effects: it disrupts tissue architecture, including the mucosa and its epithelium, but it continues to provide oxygenated blood to the tissues during much of the ischemic episode. In contrast, when strangulation results in sudden cessation of arterial blood flow (ischemic strangulating obstruction), tissues appear pale, and the mucosa rapidly degenerates because of a complete lack of oxygenated blood.163 From a clinical standpoint, this makes it difficult to assess the degree of mucosal injury in horses with strangulating injuries, because intestine that may look nonviable (dark red) may in fact have less mucosal injury than that of ischemic strangulated intestine.868
An additional consideration in clinical strangulating obstruction is the degree of ischemia that may be induced by intestinal distention. For example, experimental distention (18 cm of H2O for 2 hours) and decompression (2 hours) of the jejunum resulted in a significant increase in microvascular permeability and a significant decrease in tissue oxygenation similar to what would be expected with low-flow ischemia.869, 870 In particular, microscopic evaluation of vasculature revealed capillary endothelial cell damage and local edema formation.871 These data suggest that distended intestine proximal to an obstruction may undergo mucosal injury despite its relatively normal appearance. Indeed, in one study, intraluminal pressures greater than 15 cm H2O in naturally occurring cases of colic correlated with a poor prognosis for survival.872
Reperfusion Injury
Although it has recently been taken for granted that reperfusion of ischemic tissues results in exacerbation of mucosal injury, it should be remembered that mechanisms underlying intestinal reperfusion injury have been largely defined in laboratory animals under specific conditions.104, 410, 873, 874, 875 On the other hand, studies on reperfusion injury in horses have had some conflicting results.861, 867, 876 This may be attributed to the way in which the studies have been performed. In particular, the type of ischemia used in most laboratory animal studies has been “low-flow ischemia” (in which the blood flow is typically reduced to 20% of baseline flow), whereas studies in horses have used a number of different ischemic models, including various types of strangulating obstruction. Although strangulating obstruction is of great clinical relevance, this type of ischemic insult is less likely to develop reperfusion injury.861, 877, 878 Conversely, low-flow ischemia appears to prime tissues for subsequent injury once the tissue is reperfused, and there is considerable evidence to support the presence of reperfusion injury in horses following low-flow ischemia.866, 867, 871, 879 Nonetheless, low-flow ischemia may not be a common clinical entity.
In addition to the type of ischemia, there are other factors involved in priming tissues for reperfusion injury, including species and anatomic-specific variation in oxidant enzyme and neutrophil levels. For example, the foal appears to have very low levels of small intestinal xanthine oxidase, an enzyme that plays a critical role in triggering reperfusion injury in laboratory animals,410, 875, 880 whereas adult levels are much greater, particularly in the proximal small intestine.881 In addition, horses appear to have low numbers of resident neutrophils in the intestinal mucosa,882 and it is this population of neutrophils (rather than those recruited from the circulation) that appear to be most critical for induction of reperfusion injury.104 Studies demonstrating reperfusion injury in the equine colon following low-flow ischemia have shown significant accumulation of neutrophils within the mucosa.866 A complete understanding of the mechanisms of neutrophilic infiltration and the mechanisms by which they damage tissue will require further study.
Reperfusion injury is initiated during ischemia when the enzyme xanthine dehydrogenase is converted to xanthine oxidase, and its substrate, hypoxanthine, accumulates simultaneously because of ATP utilization (Fig. 12.8 ).850, 883 There is little xanthine oxidase activity during ischemia, because oxygen is required as an electron acceptor. During reperfusion, xanthine oxidase rapidly degrades hypoxanthine in the presence of oxygen, producing the superoxide radical as a byproduct.850 The superoxide radical contributes to oxidative tissue damage and, most importantly, activates neutrophil chemoattractants.410, 875 Inhibition of xanthine oxidase in feline studies of intestinal ischemia/reperfusion injury prevents infiltration of neutrophils and subsequent mucosal injury.410, 874 Inhibition of xanthine oxidase has had no effect on ischemia/reperfusion injury in equine small intestine876 and colon,407 suggesting that either reperfusion injury is simply a continuation of injury initiated during ischemia, as suggested in some equine studies,856 or that the classic reperfusion injury pathway is activated by alternate sources of ROMs. The latter has been suggested by studies in feline models of ischemia/reperfusion injury, in which the source of a significant proportion of ROMs is unknown, and independent of xanthine oxidase and neutrophils.874
Fig. 12.8.
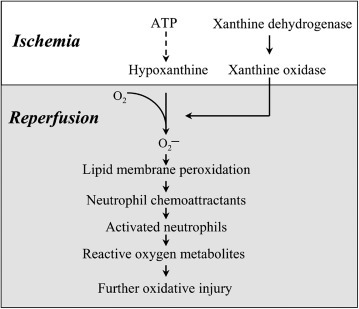
Intestinal reperfusion injury cascade. Reperfusion injury is initiated by elaboration of superoxide by metabolism of hypoxanthine by xanthine oxidase and subsequent infiltration of neutrophils.
In a veterinary review of the pathogenesis of intestinal reperfusion injury in the horse, the concept of a therapeutic window in which treatment of reperfusion injury would be beneficial was suggested.850 The basis of this concept is that there are certain conditions under which ischemic injury is minimal and that tissues are severely damaged during reperfusion.877 Thus under conditions of low-flow ischemia, very little injury is demonstrated during 3 hours of ischemia, but remarkable injury occurs during 1 hour of reperfusion.410, 874, 875 A therapeutic window may not exist under conditions of strangulating obstruction in which severe injury occurs during ischemia and minimal injury occurs during reperfusion.884 This in turn greatly reduces clinicians’ abilities to ameliorate ischemia/reperfusion injury with treatments such as antioxidants at the time of reperfusion.
Mechanisms of Gastrointestinal Mucosal Repair
Gastric Reparative Mechanisms
Mechanisms of gastric repair are highly dependent on the extent of injury. For instance, superficial erosions can be rapidly covered by migration of epithelium adjacent to the wound, a process termed epithelial restitution. Ulceration (full-thickness disruption of mucosa and penetration of the muscularis mucosa), however, requires repair of submucosal vasculature and ECM. This is initiated by the formation of granulation tissue, which supplies connective tissue elements and microvasculature necessary for mucosal reconstruction. Connective tissue elements include proliferating fibroblasts that accompany newly produced capillaries that form from proliferating endothelium. Recent studies indicate that nitric oxide is critical to both of these processes,835, 885 which likely explains the reparative properties of it in the stomach.886
Once an adequate granulation bed has been formed, newly proliferated epithelium at the edge of the wound begins to migrate across the wound. Gastric glands at the base of the ulcer begin to bud and migrate across the granulation bed in a tubular fashion.887 Epidermal growth factor (EGF) is expressed by repairing epithelium and appears to facilitate these processes.888 These events are facilitated by a mucoid cap, which retains reparative factors and serum adjacent to the wound bed.845 Once the ulcer crater has been filled with granulation tissue, and the wound has been reepithelialized, the subepithelial tissue remodels by altering the type and amount of collagen. Despite the remodeling process, ulcers tend to recur at sites of previous ulceration, and there is concern that this remodeling can result in excessive deposition of collagen and fibrosis.821
Intestinal Reparative Mechanisms
Reparative mechanisms are similar in the intestine, except that in the small intestine, mucosal villi contribute to mucosal repair. Once intestinal epithelium is disrupted, there are two events that occur almost immediately to reduce the size of the denuded portion of the villus: contraction of the villus and epithelial restitution (Fig. 12.9 ). For example, in porcine ileum subjected to 2 hours of ischemia, villi were 60% of their former height, and 50% of the denuded villous surface area was covered in flattened epithelium within 6 hours.836 Villous contraction appears to be regulated by enteric nerves, because inhibition of enteric nerve conduction prevents villous shortening following injury. The contractile component of the villus is a network of myofibroblasts distributed throughout the lamina propria of the villus and along the central lacteal. Inhibition of villous contraction results in retarded epithelial repair because of the larger denuded surface that remains to be covered by migrating epithelium compared with similarly injured villi that have contracted.837 PGE2 has also been implicated in regulating villous contraction, because application of PGE2 resulted in villous contraction when perfused through normal rat ileum.889 As villi contract, assuming there is an intact basement membrane, epithelium from the margins of the wound migrates in a centripetal direction to resurface toward the tip of the villus.837 The process of restitution is similar in denuded colonic mucosa, except that it may proceed more rapidly because of the lack of villi.207 Epithelial restitution is solely a migratory event that does not depend on provision of new enterocytes by proliferation. Cellular migration is initiated by extension of cellular lamellipodia that receive signals from the basement membrane via integrins. Intracellular signaling converges on the actin cytoskeleton, which is responsible for movement of lamellipodia. Specific components of the basement membrane appear to be critical to the migratory process. For example, application of antibodies to collagen types III and IV, which are important components of intestinal mucosal basement membrane, impeded epithelial restitution.890, 891 Other elements of the basement membrane, including proteoglycans, hyaluronic acid, and noncollagenous proteins, such as fibronectin and laminin, may also provide important signals.892 The subepithelial matrix components that facilitate restitution may form the basis for clinical treatments designed to speed up the repair process, analogous to the administration of matrix components to horses with articular cartilage damage.
Fig. 12.9.
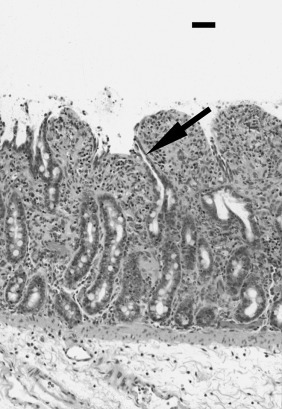
Histologic appearance of repairing intestinal mucosa 6 hours after a 2-hour ischemic episode. Note blunting of the villus, attributable to villous contraction, and evidence of epithelial restitution (arrows). 1-cm bar = 100 μm.
Although epithelial restitution results in gross closure of previously denuded regions of gastrointestinal mucosa, the closure of interepithelial spaces is ultimately required to restore normal epithelial barrier resistance.893 Because the tight junction is principally responsible for regulating the permeability of the interepithelial space, it is likely that repair and closure of this structure is critical to restore intestinal barrier function. Recent research indicates that prostaglandins play a vital role in the recovery of tight junction resistance,893 indicating that the administration of nonselective COX inhibitors to horses with colic, particularly those recovering from strangulating obstruction, may be deleterious. Judicious use of NSAIDs is appropriate until more selective drugs that allow continued production of reparative prostaglandins are available for use in horses. Recent studies have shown that NSAIDs preferential for COX-2 allow for optimal repair of injured intestine compared with traditional nonselective NSAIDs.392
Once the epithelial barrier has been restored, normal mucosal architecture must be reestablished to allow normal gut absorptive and digestive function. In porcine ileum subjected to 2 hours of ischemia, the epithelial barrier was restored within 18 hours, but villi were contracted and covered in epithelium with a squamous appearance. Restoration of normal villous architecture required a further 4 days.836 The flattened villous epithelium that characterizes restitution is replaced by newly proliferated crypt epithelium. Under normal circumstances, new enterocytes are formed by the division of stem cells, of which there are approximately four at the base of each mucosal crypt. Newly divided enterocytes migrate from the crypt onto the villus.894 During migration, enterocytes differentiate and acquire specific absorptive and digestive functions. Fully differentiated enterocytes reside on the upper third of the villus for 2 to 3 days and are then sloughed into the intestinal lumen.895 This process is accelerated during mucosal repair, which requires increased proliferative rates. Increased proliferation may be stimulated within 12 to 18 hours by a variety of locally available gut-derived factors, including luminal nutrients, polyamines, and growth factors.836 The return of the normal leaf-like shape of the villus occurs subsequent to the appearance of normal columnar epithelium.
Mediators of Repair
Prostaglandins
Although prostaglandins have been strongly implicated in mucosal cytoprotective function, relatively few studies have assessed their importance in mucosal repair. One study implicated prostaglandins in growth factor–stimulated restitution,896 but a more prominent role of prostaglandins in mucosal repair is their ability to close interepithelial tight junctions.893, 897, 898 For instance, ischemic-injured small intestine rapidly recovered barrier function (as measured in vitro as transepithelial resistance) in the presence of prostaglandins I2 and E2, despite the fact that these prostanoids had relatively little effect on villous contraction and epithelial restitution. However, electron microscopic examination of tissues reveals dilation of tight junctions in tissues treated with NSAIDs,898 whereas those additionally treated with prostaglandins have closely apposed tight junctions (Fig. 12.10 ). Prostaglandins stimulate closure of tight junctions via the second messengers cAMP and Ca2+,893 which interestingly were among the first mediators found to modulate tight junction permeability.899, 900 Such tight junction closure is of considerable importance to patients with intestinal injury that are treated with nonsteroidal antiinflammatory agents, because reduced prostaglandin levels may result in increased intestinal permeability. For example, in a study on ischemic-injured porcine ileum, treatment with the NSAID indomethacin resulted in a significant increase in intestinal permeability to inulin and lipopolysaccharide compared with tissues that were additionally treated with PGI2 and PGE2.893
Fig. 12.10.
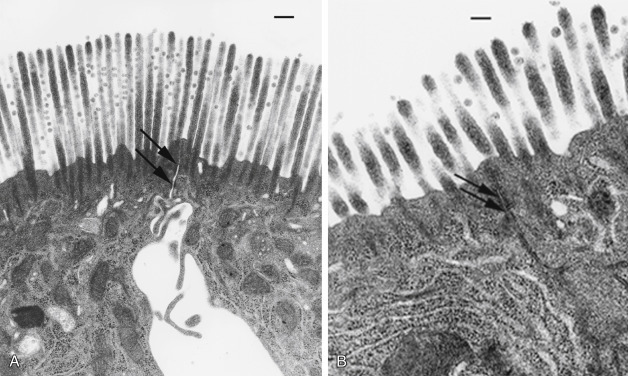
Ultrastructural appearance of repairing ischemic-injured mucosa. (A) Restituting epithelium 2 hours after a 1-hour ischemic episode in the presence of the nonselective cyclooxygenase inhibitor indomethacin. Note dilation of the interepithelial space and the apical tight junction (arrows), which correlates with a “leaky” intestinal barrier. (B) Similar restituting epithelium that has been additionally treated with PGE2 and PGI2. Note the close apposition of the tight junction (arrows) and the interepithelial space correlated with normalization of intestinal barrier function. 1-cm bar = 6 μm.
Polyamines
The process of restitution is absolutely dependent on a group of compounds called polyamines.901, 902 The rate-limiting enzyme in the formation of the polyamines spermine, spermidine, and putrescine is ornithine decarboxylase (ODC). In rats with stress-induced duodenal ulcers, systemic administration of the ODC inhibitor dl-alpha-difluoromethyl ornithine significantly reduced polyamine levels and markedly reduced epithelial restitution. Intragastric treatment of these same rats with putrescine, spermidine, and spermine prevented the delayed mucosal repair induced by dl-alpha-difluoromethylornithine.901 Interestingly, gastric tissue levels of ODC were increased in rats with stress-induced gastric ulcers, suggesting that polyamine production is enhanced during tissue injury and may contribute to the normal rapid rate of epithelial restitution.903
The mechanisms by which polyamines stimulate epithelial restitution are not clear. McCormack et al. hypothesized that polyamines increased transglutaminase activity, an enzyme that catalyzes the cross-linking of cytoskeletal and basement membrane proteins.904 Further investigation of the role of polyamines in IEC-6 cell migration showed that depletion of polyamines resulted in disruption of the cytoskeleton and reduced the physical extension of lamellipodia.905 More recent studies have clarified this pathway. Polyamines regulate cytoskeletal cellular migration via activation of the small GTPase Rho-A by elevating intracellular Ca2+ levels. These elevations in Ca2+ result from polyamine regulation of expression of voltage-gated K+ channels and altered membrane electrical potential.906
Polyamines also play a role in the normal physiologic regulation of crypt cell proliferation and differentiation.907, 908 They are produced by fully differentiated enterocytes at the villous tip and may reach the crypt either within sloughed luminal epithelium or via local villous circulation.909 Following intestinal injury, polyamines appear to stimulate enhanced proliferation by increasing the expression of protooncogenes, which control the cell cycle.910 The mechanism by which polyamines influence gene expression likely relates to the cationic nature of these compounds, which may influence the tertiary structure of negatively charged DNA and RNA.901
Growth Factors
Locally produced growth factors, including EGF, TGF-α, TGF-β, and hepatocyte growth factor, have the ability to modulate mucosal recovery. The most important of these growth factors in early mucosal repair events is TGF-β, which is a potent stimulus of epithelial restitution and modulator of the ECM.821 Neutralization of TGF-β retards epithelial migration in vitro, and it appears that TGF-β may serve as a point of convergence for mediators of restitution, because neutralizing TGF-β also inhibits the effects of other peptides. However, TGF-β paradoxically inhibits epithelial proliferation, reducing the supply of new enterocytes for mucosal repair. Conversely, EGF, produced by the salivary glands and duodenal Brunner’s glands, and the related TGF-α, produced by small intestinal enterocytes, are potent stimulants of enterocyte proliferation. These growth factors share approximately 30% of their amino acid structure, bind to the same receptor on the basolateral surface of enterocytes, and are not related to TGF-β.911 The physiologic role of EGF is somewhat difficult to discern because it is present in the intestinal lumen, with no apparent access to its basally located receptor.912 It has been proposed that EGF acts as a “surveillance agent” that gains access to its receptor during epithelial injury (when the EGF receptor would likely be exposed) to stimulate proliferation.912 TGF-α presumably has a similar role, but it is present in greater concentrations in the small intestine because it is produced by differentiated villous enterocytes. The mature peptide is cleaved from the extracellular component of the transmembrane TGF-α precursor and released into the lumen.911
Trefoil Peptides
Another group of proreparative peptides that are locally produced within the gastrointestinal tract are the trefoil peptides. Under physiologic conditions, trefoil peptides are secreted by mucus-producing cells at distinct anatomic sites. For example, the trefoil peptide pS2 is produced by gastric epithelium, whereas ITF is produced by small and large intestinal mucosa.701 However, any of the trefoil peptides may be upregulated within repairing epithelium regardless of the anatomic site.821, 913 In addition, trefoil peptides have the ability to induce their own expression, amplifying the level of these reparative factors at sites of mucosal repair.914 Trefoil peptides are the most potent stimulants of epithelial migration in vitro, and their effects are independent of growth factors, including TGF-β.915 Recent evidence suggests that EGF receptor activation is required for the induction of pS2 and another of the trefoil peptides called spasmolytic peptide in gastric epithelium in vitro. The importance of trefoil peptides to the mucosal repair response in vivo is illustrated by gene knockout studies, in which mice deficient in ITF have a dramatically reduced ability to repair intestinal injury.916 Detergent-induced mucosal injury was lethal because of a lack of restitution compared with wild-type mice that fully recovered from similar mucosal injury. The fact that restitution was restored by administration of ITF has important therapeutic implications. The mechanism by which trefoil peptides stimulate epithelial migration has yet to be fully characterized, but it appears to involve translocation of the adherens junction protein E-cadherin, allowing cells to become untethered from neighboring cells.821
Intestinal Nutrients
The principal metabolic fuel of enterocytes is glutamine, and for colonocytes it is butyrate. However, recent studies suggest that glutamine and butyrate have more specific proliferative actions aside from their role as nutrients. For example, in the piglet IPEC-J2 enterocyte cell line, glutamine enhanced gene transcription by increasing MAPK activity.917, 918 Similarly, butyrate stimulated mucosal growth following colonic infusion in the rat.919 Because of such growth-promoting actions, glutamine was shown to prevent the intestinal mucosal atrophy and dysfunction that accompany starvation920, 921 and long-term total parenteral nutrition.922, 923 Glutamine improves the function of transplanted small intestine924, 925 and protects intestinal mucosa from injury if administered before chemotherapy926 and radiation.927, 928 Intestinal nutrients may also synergize with other proliferative agents. For example, administration of glutamine and TGF-α to porcine ileum that had been subjected to 2 hours of ischemia resulted in a synergistic increase in MAPK activity, enterocyte proliferation, and villous surface area.836 Although there has been a concern that such early return to normal surface area may result in dysfunctional mucosal digestive and absorptive function because of resurfacing denuded mucosa with immature epithelium, nutrients and growth factors also appear to promote early differentiation. In the case of glutamine and TGF-α restoration of postischemic small intestine, rapid recovery of digestive enzymes was also documented.929
Gastrointestinal Ileus
Effective gastrointestinal motility involves a complex interaction between the ENS, muscular wall, and luminal contents. Additional factors that influence the net transit of digesta include gravity, the volume and viscosity of the contents, and pressure gradients created by simultaneous contraction and relaxation of adjacent segments of bowel. Casual use of the term intestinal motility in veterinary medicine often underestimates the complexity of the processes involved in the transit of intestinal contents. This is particularly true when the term is used to describe the frequency and or intensity of intestinal sounds, or borborygmi. The existence of borborygmi does not always equate with progressive movement of intestinal contents.
Disruption to normal motility occurs commonly in horses for a variety of reasons. Examples of diseases in which altered motility may be present include grass sickness, gastroduodenal ulceration, intraluminal obstruction or impaction, excessive wall distention, strangulating obstruction, peritonitis, and inflammatory disorders such as duodenitis proximal jejunitis or colitis. Ineffective intestinal motility is also a feature of several neonatal diseases, including prematurity, systemic sepsis, and perinatal asphyxia. Certain parasitic infections, electrolyte derangements, and endotoxemia can modify digesta transit in horses of all ages. General anesthesia and specific sedatives, such as xylazine, romifidine, or detomidine, also disturb motility.
Manifestations of Ileus
The inhibition of propulsive bowel activity usually is referred to as ileus. Ileus is ascribed most frequently to the condition that occurs after laparotomy and is termed simple or uncomplicated POI. The term complicated or paralytic ileus describes intestinal motility disturbed for longer periods after surgery. POI in horses is associated most commonly with surgery of the small intestine, particularly after resection and anastomosis,930, 931 and can have a negative effect on short-term postoperative survival.932, 933, 934, 935 Motility dysfunction likely is present in all horses after laparotomy, but many are affected subclinically and require minimal or no specific intervention. In symptomatic animals, clinical signs are apparent shortly after recovery and include colic, tachycardia, dehydration, decreased borborygmi and fecal output, and sequestration of fluid within the stomach. Rectal examination and ultrasound reveal small intestinal distention with rare or absent wall movement. The severity and duration of intestinal stasis varies, lasting from minutes to days.
A specific motility disorder involving the cecum or ileocecocolic region occurs sporadically in horses.936, 937, 938 The condition most commonly occurs after general anesthesia and extraabdominal surgery, particularly orthopedic and upper airway procedures, and often is categorized as a form of POI. Other cases occur spontaneously, often in animals with painful primary conditions such as uveitis or septic tenosynovitis. In a study of 114 horses diagnosed with cecal impaction, 12 were hospitalized for a condition other than colic at the time of diagnosis, and 9 others were being treated with phenylbutazone, most for a musculoskeletal injury.939 Eight of the 114 horses had undergone general anesthesia in the 8 days preceding diagnosis. The syndrome is frustrating in that clinical signs are often subtle unless cecal perforation has occurred. In horses with a cecal emptying defect after anesthesia, overt signs are usually apparent 3 to 5 days after the procedure. The earliest detectable signs include depression and a reduction in feed intake and fecal output. Ineffective emptying results in overfilling of the cecum with moist contents, which is manifested by signs of mild to moderate colic. If the condition is recognized late or untreated, the cecum may rupture and result in fatal peritonitis.
Physiology
Current understanding of motility throughout the equine gastrointestinal tract is remarkably limited, and much of our presumptive knowledge comes from work in other species. The ENS is involved in all aspects of motility, either directly via neurotransmitters or indirectly via interstitial cells of Cajal (ICC) or immune or endocrine regulation. The inherent rhythmicity of electric activity in the intestine is controlled by the ICC, which is specialized cells that are electrically coupled to myocytes via gap junctions.940 These cells are responsible for generating and propagating slow-wave activity; hence, they are deemed the pacemaker cells of the intestine. A decrease in ICC density has been observed in horses with obstructive disorders of the large intestine941 and in the ileum and pelvic flexure of horses diagnosed with equine grass sickness (dysautonomia),942 although such a decrease was not evident in a horse with dysautonomia that recovered.943 This alteration in ICC infrastructure appears to result in reduced slow-wave activity in vitro.944
The ENS primarily controls and coordinates intestinal contraction. A combination of central and autonomic innervation influences events, but contraction does not require external neural input. The parasympathetic supply to the gastrointestinal tract is via the vagus and pelvic nerves, and the sympathetic supply is through postganglionic fibers of the cranial and caudal mesenteric plexuses. A complex network of interneurons within each plexus integrates and amplifies neural input; the intensity and frequency of resultant smooth muscle contractions are proportional to the amount of sympathetic and parasympathetic input. Additional binding sites for a number of other endogenous chemicals, including dopamine, motilin, and serotonin, exist within the ENS and on smooth muscle cells.945 Acetylcholine is the dominant excitatory neurotransmitter in the gastrointestinal tract and exerts its action through muscarinic type 2 receptors on smooth muscle cells. Sympathetic fibers innervating the gastrointestinal tract are adrenergic, postganglionic fibers with cell bodies located in the prevertebral ganglia. Activation of α2-adrenergic receptors on cholinergic neurons within enteric ganglia inhibits the release of acetylcholine, reducing intestinal contraction. β1-, β2-, and β-atypical receptors are directly inhibitory to the intestinal smooth muscle.946 Inhibitory nonadrenergic, noncholinergic neurotransmitters include adenosine triphosphate, vasoactive intestinal peptide, and nitric oxide.947, 948 These neurotransmitters are critical for mediating descending inhibition during peristalsis and receptive relaxation. Substance P is a nonadrenergic, noncholinergic neurotransmitter that may be involved in contraction of the large colon.949, 950
The rate and force of intestinal contractions along the small intestine and large colon of the horse are important determinants of intestinal motility; of even greater importance to the net propulsion of digesta are the cyclical patterns of contractile activity. These patterns are known as the small intestinal and colonic migrating motility (or myoelectric) complexes (MMCs).951, 952 The colonic complex usually originates in the right ventral colon and variably traverses the ascending and descending colons. Many of these complexes are related temporally to a specialized motility event of the ileum, the migrating action potential complex.953
Pathophysiology
Inflammation
Local inflammation within the intestinal muscularis and inhibitory neural events are important initiators of intestinal ileus.954, 955 Intestinal inflammation not only is important in primary intestinal diseases in horses, such as DPJ and colitis, but also is induced after simple intestinal handling during laparotomy. In rodents, simple intestinal manipulation causes a cascade of inflammation within the muscularis, resulting in leukocyte infiltration and subsequent suppression of muscle contractility. Similar inflammatory effects were evident with mechanical manipulation of the equine jejunum.956 The inflammatory response to bowel manipulation is not limited to the affected tissue, but it can also result in global inflammation and ileus throughout the gastrointestinal tract.957
The associated inflammatory events are extremely complex, involving a milieu of proinflammatory cytokines, prostaglandins, and leukocytes. Depletion or inactivation of muscularis macrophage function can prevent inflammation associated with intestinal manipulation and associated decreased contractility.958 Mast cell activation is involved in intestinal manipulation-associated POI in humans.959 Inflammation associated with colonic manipulation may involve gut-derived bacterial products.960 Another factor in the development of intestinal stasis after inflammation is the local overproduction of nitric oxide caused by the upregulation of iNOS by resident macrophages.961 The iNOS upregulation was important for initiation of the inflammatory response and inhibition of motility. Nitric oxide is a key inhibitory neurotransmitter of the nonadrenergic, noncholinergic system.948
In the horse, significant neutrophilic inflammation is apparent in the jejunum from clinical cases necessitating resection and following a period of recovery in the jejunum subjected to 1 or 2 hours of ischemia.962 The ischemic tissue also had evidence of leukocyte activation, as demonstrated by calprotectin-positive cells in associated tissue histologically.
Pharmacologic Alteration
The inhibitory effects of α2-adrenergic agonists such as xylazine and detomidine on duodenal, cecal, and large colon motility are well described, because these drugs activate presynaptic receptors within the ENS.4, 963, 964, 965, 966, 967, 968 IV-administered xylazine inhibits cecal and large colon motility for 20 to 30 minutes without seriously disrupting small intestinal myoelectric activity, and detomidine can reduce large intestinal myoelectric activity for up to 3 hours. Detomidine decreases duodenal motility in a dose-dependent fashion.969 The α2-antagonist yohimbine has a weak but positive effect on cecal emptying in normal ponies, suggesting that normal motility is under constant α2-adrenergic tone.4
Several opioid agonists also have documented inhibitory effects on equine gastrointestinal motility at both a central and peripheral level. Morphine administration decreased frequency of defecation and fecal moisture content while increasing gastrointestinal transit time in normal horses at a dose of 0.5 mg/kg twice daily for 6 days.970 Single doses of fentanyl or morphine decrease jejunal and colonic MMC activity in ponies, whereas the antagonist naloxone elicited increased propulsive activity in the colon.971 Fentanyl administered as an IV constant rate infusion did not have an apparent deleterious effect on duodenal motility.972 Butorphanol, an opioid agonist-antagonist, decreases myoelectrical activity in the jejunum but not pelvic flexure.973 In another series of experiments, butorphanol alone did not decrease gastric or duodenal motility,974 but administration in combination with xylazine resulted in a synergistic inhibitory effect, which was more pronounced than that obtained by administration of xylazine alone.968 Administration of butorphanol as a constant rate infusion appears to have a minimal to no effect on global gastrointestinal470, 975 or duodenal motility.976 When lidocaine, ketamine, and butorphanol were administered alone or in combination as constant rate infusions to healthy horses, combinations containing butorphanol (butorphanol/lidocaine, or a combination of all three drugs) delayed total gastrointestinal transit time.977
N-Butylscopolammonium bromide has a profound but very short-lived negative effect on duodenal motility, but this effect was not significant between groups.976 Atropine is a postganglionic blocking agent that binds to muscarinic receptors. When administered at 0.04 mg/kg, atropine inhibits individual small intestinal, cecal, and colonic contractions for about 120 minutes but suppresses small intestinal and colonic migrating complexes for up to 8 hours.978
Neural Reflexes
Neural reflexes may mediate inhibition of motility associated with peritoneal inflammation.979, 980 The afferent segment is composed partly of capsaicin-sensitive visceral afferent C fibers that terminate in the dorsal horn of the spinal cord, in which they can activate inhibitory sympathetic fibers or synapse directly on the sympathetic ganglia. Consequently, the efferent limb of the reflex expresses increased sympathetic outflow, primarily mediated through stimulation of α2-adrenoreceptors, and inhibition of acetylcholine release, which provides the rationale for α2-blockade in treating ileus. Intraluminal infusion of capsaicin before abdominal surgery ameliorated the severity of POI in experimental rats. This finding highlights the importance of visceral afferent fibers in the development of POI.981
Distention
Ileus also can occur in association with intestinal obstruction or displacement. Mild to moderate distention of the bowel, such as that occurring in the early stages of an intraluminal obstruction, evokes an increase in local contractile activity.982, 983 Excessive distention results in the inhibition of motility within the distended segment of bowel. Intestinal stasis is not always detrimental and under certain conditions may be protective. Repeated distention for determination of nociceptive threshold results in an overall decrease in duodenal motility over time, irrespective of other interventions.972, 976
Endotoxin
Endotoxemia is a clinical feature of many diseases of the equine gastrointestinal tract, and endotoxins independently can exert a negative effect on intestinal motility and transit.214 A variety of mediators likely are involved, but the activation of α2-adrenoreceptors and production of prostanoids appear to be important, because pretreatment with yohimbine or NSAIDs (phenylbutazone or flunixin), respectively, ameliorates the inhibitory effects of experimental endotoxin infusion.201, 984, 985, 986 Pretreatment with metoclopramide or cisapride had a similar effect.71, 987 Endotoxin infusion induced an inflammatory response in the intestine of rats that mimicked the response induced by handling during laparotomy.988 The similarity of the responses was highlighted in a recent study that demonstrated that prior exposure of the muscularis to endotoxin protected the intestine from the effects of manipulation.989 In rats, colonic manipulation alone causes transference of intraluminal LPS to the muscularis, which likely contributes to the global gastrointestinal inflammatory response and decrease in contractility associated with tissue manipulation.960 In response to endotoxin alone, the inflammatory response within the jejunal muscularis is predominantly monocytic, whereas the response to polymicrobial sepsis is predominantly neutrophilic.990
Other Effects
Temperature also appears to affect in vitro slow-wave activity in the horse. In a recent study, the slow-wave frequency was approximately linearly related to temperature in the range studied (27–41°C) and was highly temperature sensitive in isolated equine ileal segments.991
The pathophysiology of cecal emptying defect is not known. This syndrome may best mimic POI in human beings, which is generally considered a large intestinal disorder. An important difference in horses is that laparotomy is a rare predisposing factor, and most cases occur in horses undergoing routine extraabdominal surgical procedures. The disorder, therefore, is probably not appropriately considered as a form of POI in horses. General anesthesia itself is a potent inhibitor of gastrointestinal motility in horses, but these effects are short-lived and reversible within hours of anesthetic withdrawal.952 The return of normal motility in horses after experimental ileus was most delayed in the cecum, suggesting that this may be a common site of ileus in horses.992 A link between routine postoperative medications, such as phenylbutazone and aminoglycoside antibiotics, has been suspected but not established. An inhibitory effect of NSAIDs on large colon contractility has been demonstrated using in vitro techniques.993 Primary sympathetic overstimulation could be involved, because many of the affected animals are young, male horses or animals with painful diseases.
The duration of surgery influences the development of small intestinal POI but not cecal emptying dysfunction.938, 994 Technique may have a weak influence on small intestinal POI after jejunojejunostomy. The duration of intestinal ileus was shorter in animals that received a side-to-side stapled anastomosis than those that had a hand-sewn end-to-end procedure.932 The duration of ileus after stapled end-to-end anastomosis was not different from that after either procedure. Jejunocecostomy more commonly results in POI than other types of small intestinal resection and anastomosis, whether related to diseases necessitating this procedure or the procedure itself.995
Other reported risk factors for the development of POI include age (>10 years), small intestinal resection and anastomosis, breed (Arabians had a greater risk than other breeds), and duration of surgery.994 A prospective study found small intestinal lesion, high packed cell volume, and duration of anesthesia to increase the risk of POI, whereas performance of a pelvic flexure enterotomy and intraoperative administration of lidocaine may have a modest protective effect against POI.996
Diagnosis
The diagnosis of ileus is based on history and physical examination findings. Case inclusion criteria for clinical studies of POI have varied.994, 995, 996, 997 Recent surveys of the Diplomates of the European Colleges of Equine Internal Medicine and Veterinary Surgery and the American Colleges of Veterinary Surgery, Veterinary Internal Medicine, and Veterinary Emergency and Critical Care revealed that the presence of reflux on passage of a nasogastric tube, evidence of multiple fluid-distended loops of small intestine on ultrasonographic examination, and the presence of multiple fluid-distended loops of small intestine on rectal palpation were the features most commonly identified as “extremely important” for a diagnosis of POI.998, 999 One can palpate cecal distention with digesta in horses with advanced cecal dysfunction.
Distinguishing functional ileus from mechanical obstruction is important and can be difficult, but horses with mechanical obstruction typically have sustained high volumes of gastric reflux that vary little over time and abdominal pain, which is typically not relieved by gastric decompression. Abdominal ultrasound in horses with ileus typically reveals mild to moderately fluid-filled hypomotile to immotile small intestine, without alteration in the amount or character of PF or small intestinal wall thickness. Horses with reflux from other causes (peritonitis and mechanical obstruction) will have changes reflective of their disease process. This differentiation is important for appropriate case management, because horses with a mechanical obstruction often need either primary or repeat laparotomy, which should not be delayed.
Treatment
The management of intestinal ileus depends on the segment of gastrointestinal tract involved. Therapy for ileus of the proximal gastrointestinal tract involves a combination of gastric decompression, fluid and electrolyte therapy, and antiinflammatory therapy. Electrolyte therapy is critical, particularly for maintaining adequate extracellular concentrations of potassium, calcium, and magnesium. Calculation of the volume of fluid to be administered should include maintenance requirements plus an estimate of losses, especially those lost through gastric decompression. Parenteral provision of calories should be considered when feed has been withheld for more than 96 hours, particularly after surgery. Hand walking also may provide some benefit to these animals but is not likely to have a direct effect on intestinal motility.
Drugs that can have an inhibitory effect on motility should be avoided or used sparingly. Horses with primary cecal impaction or impaction caused by an emptying defect may require surgery to prevent fatal rupture. The surgical management of these cases is controversial and may include typhlotomy alone, typhlotomy with a bypass procedure such as ileocolic or jejunocolic anastomosis, or a bypass without typhlotomy.1000 Most horses that undergo simple typhlotomy have an uneventful recovery.1001 In a large retrospective study, 44/54 horses treated medically survived to discharge, whereas 37 of 49 horses treated surgically were allowed to recover, 35 of which survived to discharge.939 Ileocolostomy was only performed in two of the 37 horses treated surgically (one of which survived to discharge), with the remainder receiving typhlotomy without bypass. Survival to 1 year was not statistically different between horses treated medically (18/19) or surgically (25/28), although 6 horses had a recurrence of cecal impaction.939
Experimental and anecdotal evidence provides a strong rationale for using antiinflammatory drugs to prevent and treat gastrointestinal ileus, particularly in animals that may have endotoxemia.1002 Flunixin meglumine is used widely in equine practice as an analgesic and antiinflammatory agent, and it ameliorates many of the adverse systemic effects of endotoxin, particularly those on the cardiovascular system. A potential negative effect of NSAIDs on large intestinal contractility has been suggested. A differential effect on contractility between selective and nonselective COX inhibitors is currently unknown. Broad-spectrum antimicrobials are indicated when one suspects sepsis or in cases of profound neutropenia. High concentrations of aminoglycoside antimicrobials inhibited intestinal contractions in exposed sections of intestine in vitro, but this inhibitory effect is unlikely to occur at clinically relevant doses.1003
Motility-enhancing drugs have been advocated to treat gastrointestinal ileus. Unfortunately, information directly pertinent to horses is limited and must be extrapolated cautiously from that of other species because of the differences in intestinal anatomy and physiology. Prokinetic drugs potentially can shorten the length of hospitalization, reducing the cost of treatment and the number of potential complications such as weight loss, thrombophlebitis, and laminitis. Experimental evidence indicates that prokinetic drugs can minimize the development of postoperative abdominal adhesions.1004 Most prokinetic drugs require a healthy gut wall to enhance intestinal contraction, and downregulation of motilin receptors has been demonstrated in the inflamed equine jejunum.473 Therefore one should not assume that many of these drugs would be effective in the presence of an inflammatory injury such as that which can occur after intestinal manipulation at surgery or that associated with DPJ.
Cholinomimetics
Bethanechol is a parasympathomimetic agent that acts at the level of the myenteric plexus and directly on intestinal smooth cells through muscarinic receptors. In the horse, this effect is mediated predominantly by the M3 receptor, but the M2 receptor may also play a role.1005 Bethanechol is a synthetic ester of acetylcholine and is not degraded by anticholinesterase. Bethanechol has cholinergic side effects, including abdominal discomfort, sweating, and salivation, although these are minimal when the drug is administered at 0.025 mg/kg body mass SC or orally. Bethanechol has efficacy in diseases that involve abnormal gastric emptying and delayed small intestinal transit and increases gastric contractility and hastens the emptying of liquid and solid phase markers from the stomach of normal horses.1006 Bethanechol also increases the strength and duration of wall contractions in the cecum and right ventral colon, consequently speeding up cecal emptying.4
Neostigmine increases receptor concentration of acetylcholine by inhibiting cholinesterase. The drug (0.022–0.025 mg/kg IV) promotes cecal and colonic contractile activity and hastens the emptying of radiolabeled markers from the cecum.4 Neostigmine has been used to manage small intestinal ileus, but it significantly delayed the emptying of 6-mm beads from the stomach of normal adult horses.1007
Benzamides and Dopamine Antagonists
Metoclopramide acts principally as a 5-hydroxytryptamine 4-receptor (5HT-4) agonist and 5HT-3–receptor antagonist. In contrast to newer generation benzamides, metoclopramide is also an antagonist at dopamine 1 (DA1) and dopamine 2 (DA2) receptors. Antagonism of prejunctional DA2 receptors facilitates acetylcholine release and smooth muscle contraction. Metoclopramide crosses the blood-brain barrier, where its antagonist properties on central DA2 receptors can result in extrapyramidal signs, including seizure. Metoclopramide increased contractility of muscle strips in vitro in the pyloric antrum, proximal duodenum, and midjejunum.1008 These in vitro data support previous work in which metoclopramide administration restored gastroduodenal coordination of motility in a model of POI.1009 In another study, metoclopramide had no effect on jejunal or pelvic flexure myoelectrical activity.973 Constant IV infusion (0.04 mg/kg/h) of metoclopramide was well tolerated in a population of postoperative horses and significantly decreased the volume and duration of gastric reflux over control and intermittent drug infusion groups.1010
Cisapride is a second-generation benzamide that acts as a 5HT-4 agonist and 5HT-3–receptor antagonist but is without antidopaminergic action. Stimulation of 5HT-4 receptors within the ENS enhances release of acetylcholine from the myenteric plexus. Several reports suggest the efficacy of cisapride in managing intestinal disease in horses, including the resolution of persistent large colon impaction, treatment of equine grass sickness, and as a preventative for POI in horses after small intestinal surgery (0.1 mg/kg body mass intramuscularly during the postoperative period).1011, 1012, 1013, 1014 The horse erratically absorbs tablets administered rectally, but a method for preparing a parenteral form of the drug from tablets has been described.1015 Cisapride has the potential to cause adverse cardiac side effects mediated through blockage of the rapid component of the delayed rectifier potassium current that include lengthening of the QT interval and development of torsades de pointes, a potentially fatal arrhythmia.1016 These adverse effects have resulted in withdrawal of the drug in the United States but have not been reported in the horse.
Tegaserod, a 5HT-4 agonist, increases pelvic flexure smooth muscle contractility (0.27 mg/kg PO)1017 and hastens gastrointestinal transit time (0.02 mg/kg IV) in healthy horses.1018 It has not, to the author’s knowledge, been objectively evaluated in abnormal horses, but it may prove useful. In humans, this drug was marketed for women with constipation-predominant or mixed symptom irritable bowel syndrome and demonstrated clear benefits in quality of life and gastrointestinal symptoms but is currently only available in a restricted fashion because of an association with ischemic colitis and cardiovascular disease.1019
Domperidone acts as a competitive antagonist at peripheral DA2 receptors. The drug is a therapeutic agent (1.1 mg/kg/day) for mares grazing endophyte-infected tall fescue, principally because of drug-enhanced prolactin release. Modest efficacy of domperidone (0.2 mg/kg IV) has been demonstrated in experimental ileus in ponies.1011 In another study, a much higher oral dosage (5 mg/kg) was required to increase gastric emptying; 1.1 mg/kg orally had limited effects on the gastrointestinal tract in healthy horses.1020
Antimicrobials
Erythromycin, a macrolide antibiotic, is a direct motilin receptor agonist on smooth muscle cells and may act within the ENS to facilitate the release of acetylcholine and motilin. Erythromycin displaces motilin from its receptor in the equine duodenum, jejunum, cecum, and pelvic flexure.476 Erythromycin enhances gastric emptying in normal horses but has a more pronounced effect on the hindgut.1006, 1021 Erythromycin lactobionate (1.0 mg/kg IV) hastens cecal emptying in normal animals and induces colonic MMC-like activity across the colon. Administration often is associated with defecation and abdominal discomfort. The prokinetic effect of erythromycin apparent in the ileum, cecum, and pelvic flexure documented in normal horses was reduced in the immediate postoperative period.686 Luminal distention and decompression resulted in inflammation and a decreased response to erythromycin.1022 A decrease in motilin receptors in response to luminal distention has been documented in the equine jejunum,473 and this may explain the difference in response between normal and clinically affected horses. Repeated dosing can cause downregulation of motilin receptors in other species.1023 Erythromycin can induce diarrhea in adults; therefore, one should avoid dosing over many days.
Potassium penicillin (20 million IU IV to adult horses) can stimulate defecation and increase myoelectrical activity in the cecum and pelvic flexure, and these effects are not produced by an equimolar amount of potassium ion given IV as potassium chloride.1024
Opioid and α2-Adrenoreceptor Antagonists
Naloxone (0.05 mg/kg IV) induces contractile activity in the cecum and left colon.971 Defecation commonly follows administration of naloxone within 15 to 20 minutes. N-methylnaltrexone increases jejunal and pelvic flexure contractility in vitro1025 and prevents the negative effects of morphine on fecal output and intestinal transit time when administered concurrently.1026
α2-Adrenoreceptor antagonists such as yohimbine or tolazoline counteract increased sympathetic outflow in response to nociceptive stimulation. Yohimbine infusion (75 μg/kg) also may attenuate the negative effects of endotoxin on motility.201, 985
Local Anesthetics
The use of IV lidocaine as a prokinetic has gained tremendous popularity and was reported to be the agent most commonly used by equine surgeons for treatment of POI.1027 In more recent surveys, flunixin meglumine and lidocaine were used most frequently for the treatment of POI by specialists in Europe and North America.998, 999 Lidocaine may exert prokinetic effects by suppressing primary afferent neurons, limiting reflex efferent inhibition of motility.1028 Other proposed mechanisms of action include antiinflammatory properties, potentially through NF-κB signaling1029 or improving mucosal repair.413 IV lidocaine also has analgesic effects, although it was shown to alter somatic but not visceral antinociception in clinically normal horses in one study.1030 Lidocaine increased contractile activity in isolated strips of proximal duodenum in vitro.1008 The most commonly cited dosage is a 1.3-mg/kg bolus, typically over 15 minutes, followed by a 0.05-mg/kg/min constant rate infusion. This dosage did not alter MMC duration or spiking activity, reset the MMC in the jejunum in clinically normal horses1031 or significantly alter a variety of indicators of POI after colic surgery.1032 Significantly more horses with POI stopped refluxing within 30 hours following the institution of lidocaine infusion, relative to saline infusion.475 Lidocaine administration did not affect the prevalence of POI, duration or volume of reflux, or survival in horses requiring surgery for treatment of small intestinal colic.1033 Lidocaine infusion can be associated with reversible side effects that include muscle fasciculations, ataxia, and seizure. Consequently, the rate of infusion requires close monitoring. Prolonged infusion of lidocaine in the horse appears safe, although accumulation of the GX metabolite has been documented.1034
Ischemic Disorders of the Intestinal Tractg
Pathophysiology Of Strangulating Obstruction
Strangulating obstruction of the intestine is characterized by simultaneous occlusion of the intestinal lumen and its blood supply. Although strangulation of the intestinal lumen results in clinical signs similar to those of simple obstruction, occlusion of the blood supply results in a more rapid deterioration of the intestinal mucosa and subsequent onset of sepsis. Although there has been a great deal of interest in the relevance and treatment of intestinal reperfusion injury,851, 861, 882 the lesion that develops during strangulation is often severe, leaving little viable bowel for further injury during reperfusion.882 Although extensive lengths of strangulated small intestine may be resected, strangulation of the large colon presents a much greater treatment dilemma because strangulated intestine usually extends beyond the limits of surgical resection.1035 Therefore horses with large intestinal strangulation are often recovered with extensive intestinal injury left in place. As a result, subtle degrees of reperfusion injury may be very important in horses with large colon disease, warranting further work in this area in an attempt to reduce mortality.851
Strangulating obstruction may be divided into hemorrhagic and ischemic forms.163 In hemorrhagic strangulating obstruction, which is most common, the veins become occluded before the arteries because of the greater stiffness of arterial walls. This lesion is noted by a darkened appearance in affected bowel and increased thickness as blood is pumped into the lesion. Ischemic strangulating obstruction occurs if the intestine is twisted tightly enough to simultaneously occlude both arteries and veins. In the case of the colon, some researchers suggest that this may be determined by how much ingesta is in the colon, because intestinal contents may prevent the intestine from twisting tightly.1036 Tissue involved in ischemic strangulating obstruction appears pale and of normal or reduced thickness because of a complete lack of blood flow (Fig. 12.11 ). Bowel peripheral to strangulating lesions may also become injured as a result of distention, which reduces mural blood flow once it reaches critical levels. As this intestine is decompressed, it may also undergo reperfusion injury.879, 1037, 1038
Fig. 12.11.
Ischemic strangulating obstruction of the small colon by a mesenteric lipoma. (A) Note the lipoma (arrow), which has tightly encircled a segment of small colon. (B) After resection of the lipoma, a pale area of strangulated small colon is clearly demarcated (arrows), the appearance of which is consistent with ischemic strangulating obstruction.
Small Intestinal Strangulation
Clinical Signs
Horses with small intestinal strangulating obstruction typically have moderate to severe signs of abdominal pain that is only intermittently responsive to analgesic medications. During the latter stages of the disease process, horses may not experience much pain but rather become profoundly depressed as affected intestine undergoes necrosis. Affected horses have progressive signs of sepsis, including congested mucous membranes, delayed capillary refill time, and an elevated heart rate (>60 beats/min in most cases). Reflux is typically obtained after passage of a stomach tube, and loops of distended small intestine are usually detected on rectal palpation of the abdomen. These latter findings are variable, depending on the duration and location of the obstruction. For example, horses with ileal obstructions tend to reflux later in the course of the disease process than horses with jejunal obstructions. A horse that has an entrapment of small intestine in the epiploic foramen or a rent in the proximal small intestinal mesentery may not have palpable loops of small intestine because of the cranial location of these structures.1039 Abdominocentesis can provide critical information on the integrity of the intestine and is indicated in horses with suspected strangulation of the small intestine. Affected horses typically have serosanguineous abdominal fluid with an elevated protein concentration (>2.5 mg/dL) and an increase in PF:plasma lactate, although these cases must be differentiated from proximal enteritis.11, 22, 783 When an initial sample is inconclusive, repeated measurement of PF lactate may provide additional support for a diagnosis of a strangulating lesion.27 Generally, horses with small intestinal strangulation show continued signs of abdominal pain, whereas horses with proximal enteritis (discussed in Duodenitis-Proximal Jejunitis previously in this chapter) tend to be depressed after initial episodes of mild abdominal pain.1040 In addition, horses with small intestinal strangulation continue to deteriorate clinically despite appropriate medical therapy and begin to show elevated WBC counts (>10,000 cells/μL) in the abdominal fluid as the duration of strangulation increases. There are horses in which small intestinal strangulation and proximal enteritis cannot be not readily distinguished, at which point surgery may be elected rather than prolonging the decision to perform abdominal exploration on a horse with a potential strangulating lesion.1040
Prognosis
The prognosis for survival in horses with small intestinal strangulating lesions is generally lower than for most forms of colic,995 but some reports indicate that more than 80% of horses with small intestinal strangulating lesions will survive to discharge from the hospital.1041 Owners should be warned that long-term survival rates are less than 70%,1042 in part because of long-term complications such as adhesions.1043, 1044 The prognosis is particularly poor for some forms of strangulation, including entrapment of small intestine within a mesenteric rent.1045 Some horses with small intestinal strangulation can be managed surgically without resection, with favorable short- and long-term prognosis.1046 Geriatric horses have a higher likelihood of small intestinal strangulation, relative to mature counterparts undergoing colic surgery, but carry a similar prognosis.1047 In one hospital, horses with gastrosplenic ligament entrapment (GLE) had higher short-term survival (72.7%), relative to horses with other small intestinal strangulating lesions (50%),1048 whereas short-term survival was similar between horses with GLE (88%) and EFE (85%) at another hospital.1049 Short-term survival in horses with EFE was higher (95%) than for other causes of small intestinal strangulation in another report.1050
Epiploic Foramen Entrapment
The epiploic foramen is a potential opening (because the walls of the foramen are usually in contact) to the omental bursa located within the right cranial quadrant of the abdomen. It is bounded dorsally by the caudate process of the liver and caudal vena cava and ventrally by the pancreas, the hepatoduodenal ligament, and the portal vein.1039 Intestine may enter the foramen from the visceral surface of the liver toward the right body wall or the opposite direction. Studies differ as to which is the most common form. In the case of entrapments that enter the foramen in a left-to-right direction, the omental bursa is ruptured as the intestine migrates through the epiploic foramen, which may contribute to the intraabdominal hemorrhage often seen with this condition. Clinical signs include acute onset of severe colic with examination findings compatible with small intestinal obstruction. The stereotypic behavior of crib biting is a significant risk factor for EFE,1051 possibly because of changes in abdominal pressure as the horse prepares the esophagus to ingest air. Other risk factors include increased height of the horse and previous colic surgery.1051 The condition was once believed to be more prevalent in older horses,1039 but this has been refuted.1051 The disorder has also been recognized in foals as young as 4 months of age.1052 The diagnosis is definitively made at surgery, although ultrasonographic findings of distended loops of edematous small intestine adjacent to the right middle body wall are suggestive of EFE.1039 Generally, thickened, immotile intestine on ultrasonographic examination is highly predictive for small intestinal strangulating obstruction.1053, 1054 Small intestine entrapped in the epiploic foramen may be limited to a portion of the intestinal wall (parietal hernia),1055 and the large colon may become entrapped within the epiploic foramen.1051 In treating EFE, the epiploic foramen must not be enlarged either by blunt force or with a sharp instrument, because rupture of the vena cava or portal vein and fatal hemorrhage may occur. Prognosis has substantially improved over the past decade, with current short-term survival rates (discharge from the hospital) ranging from 74% to 95%.1039, 1049, 1050, 1051, 1056 Survival to 1 year (50.6%) and 2 years (34.3%) postoperatively declines substantially, with a median survival time of 397 days in one report.1051 Preoperative abdominocentesis,1039, 1056 packed cell volume, length of small intestine resected, and POI1051 have been associated with postoperative survival.
Strangulation by Pedunculated Mesenteric Lipoma
As horses age, lipomas form between the leaves of the mesentery and develop mesenteric stalks as the weight of the lipoma tugs on the mesentery. The stalk of the lipoma may subsequently wrap around a loop of small intestine or small colon, causing strangulation. Strangulating lipomas should be suspected in aged (>15 years) geldings with acute colic referable to the small intestinal tract.1057, 1058, 1059 Ponies also appear to be at risk for developing disease,1059 suggesting alterations in fat metabolism may predispose certain horses to the development of mesenteric lipomas. The diagnosis is usually made at surgery, although on rare occasions a lipoma can be palpated rectally. Treatment involves surgical resection of the lipoma and strangulated bowel, although strangulated intestine is not always nonviable.1059 Studies indicate that approximately 50%1058 to 78%1059 of horses are discharged from the hospital after surgical treatment.
Small Intestinal Volvulus
A volvulus is a twist along the axis of the mesentery, whereas torsion is a twist along the longitudinal axis of the intestine. Small intestinal volvulus is theoretically initiated by a change in local peristalsis or the occurrence of a lesion around which the intestine and its mesentery may twist (e.g., an ascarid impaction).1060 It is reportedly one of the most commonly diagnosed causes of small intestinal obstruction in foals.1061, 1062 It has been theorized that young foals may be at risk for small intestinal volvulus because of changing feed habits and adaptation to a bulkier adult diet. Onset of acute, severe colic; a distended abdomen; and radiographic evidence of multiple loops of distended small intestine in a young foal would be suggestive of small intestine volvulus. However, it is not possible to differentiate volvulus from other causes of small intestinal obstruction preoperatively. In adult horses volvulus frequently occurs in association with another disease process, during which small intestinal obstruction results in distention and subsequent rotation of the small intestinal around the root of the mesentery. Although any segment of the small intestine may be involved, the distal jejunum and ileum are most frequently affected because of their relatively longer mesenteries.1060 The diagnosis is made at surgery by palpating a twist at the origin of the cranial mesenteric artery. Treatment includes resection of devitalized bowel, which may not be an option because of the extent of small intestinal involvement (similar to large colon volvulus). Prognosis is based on the extent of small intestine involved and its appearance after surgical correction of the lesion. Generally, horses in which more than 50% of the small intestine is devitalized are considered to have a grave prognosis.1063
Strangulation by Way of Mesenteric or Ligamentous Rents
There are a number of structures that, when torn, may incarcerate a segment of intestine (typically the small intestine), including intestinal mesentery,1045 the gastrosplenic ligament,1048, 1049, 1064 the broad ligament,1065 and the cecocolic ligament.1066 Horses with such incarcerations present signs typical of a horse with strangulating small intestine. The prognosis for many of these cases appears to be worse than for horses with other types of small intestinal strangulations. Of horses with small intestine entrapped in a mesenteric rent, only 7 of 15 horses were discharged from the hospital, and only 2 of 5 horses for which follow-up information was available survived long term (>5 months).1045 Poor outcome may result from the difficulty in releasing the incarcerated intestine, the degree of hemorrhage, and the length of intestine affected.
Inguinal Hernia
Inguinal hernias are more common in Standardbred and Tennessee Walking Horses that tend to have congenitally large inguinal canals.1060 Inguinal hernias may also occur in neonatal foals but differ from hernias in mature horses in that they are typically nonstrangulating. The nature of the hernia (direct versus indirect) is determined on the basis of the integrity of the parietal vaginal tunic. In horses in which the bowel remains within the parietal vaginal tunic, the hernia is referred to as indirect because, strictly speaking, the bowel remains within the peritoneal cavity. Direct hernias are those in which strangulated bowel ruptures through the parietal vaginal tunic and occupies an SC location. These direct hernias most commonly occur in foals and should be suspected when a congenital inguinal hernia is associated with colic, swelling that extends from the inguinal region of the prepuce, and intestine that may be palpated SC.1067, 1068 Although most congenital indirect inguinal hernias resolve with repeated manual reduction or application of a diaper, surgical intervention is recommended for congenital direct hernias.1068
Historical findings in horses with strangulating inguinal hernias include acute onset of colic in a stallion that had recently been used for breeding. A cardinal sign of inguinal herniation is a cool, enlarged testicle on one side of the scrotum.1069, 1070, 1071 However, inguinal hernias, including of the large colon, have also been reported in geldings.1072, 1073 Inguinal hernias can be detected on rectal palpation, and manipulation of herniated bowel per rectum has been used to reduce a hernia, but this procedure is not recommended because of the risk of rectal tears. In many horses the short segment of herniated intestine will markedly improve in appearance once it has been reduced and does not always have to be resected.1074 The affected testicle will be congested because of vascular compromise within the spermatic cord, and although it may remain viable, it is generally recommended that it be resected.1074 The prognosis in adult horses is good, with up to 75% of horses surviving to 6 months of age.1069, 1070, 1071 Horses that have been treated for inguinal hernias may be used for breeding. In these horses the remaining testicle will have increased sperm production, although an increased number of sperm abnormalities will be noticed after surgery because of edema and increased temperature of the scrotum.
Strangulating Umbilical Hernias
Although umbilical hernias are common in foals, strangulation of herniated bowel is rare. In one study, 6 of 147 (4%) horses with umbilical hernias had incarcerated intestine.1075 Clinical signs include a warm, swollen, firm, and painful hernia sac associated with signs of colic. The affected segment of bowel is usually small intestine, but herniation of cecum or large colon has also been reported.1076 In rare cases a hernia that involves only part of the intestinal wall may be found; this is termed a Richter’s hernia. In foals that have a Richter’s hernia, an enterocutaneous fistula may develop.1076 In one study, 13 of 13 foals with strangulating umbilical hernias survived to discharge, although at least 3 were lost to long-term complications.1076
Intussusceptions
An intussusception involves a segment of bowel (intussusceptum) that invaginates into an adjacent aboral segment of bowel (intussuscipiens). The reason for such invagination is not always clear, but it may involve a lesion at the leading edge of the intussusception, including small masses, foreign bodies, or parasites. In particular, tapeworms (Anoplocephala perfoliata) have been implicated.1077 Ileocecal intussusceptions are the most common intestinal intussusceptions in the horse and typically affect young animals. In one study evaluating 26 cases of ileocecal intussusception, the median age of affected horses was 1 year.1078 Acute ileocecal intussusceptions are those in which the horses have a duration of colic of less than 24 hours and involve variable lengths of intestine, which ranged in one study from 6 cm to 457 cm in length. In acute cases the involved segment of ileum typically has a compromised blood supply. Chronic ileocecal intussusceptions typically involve short segments of ileum (up to 10 cm in length), and the ileal blood supply is frequently intact.1078 Abdominocentesis results are variable because strangulated bowel is contained within the adjacent bowel. There is often evidence of obstruction of the small intestine, including nasogastric reflux and multiple distended loops of small intestine on rectal palpation. Horses with chronic ileocecal intussusceptions have mild, intermittent colic, often without evidence of small intestinal obstruction. In one study a mass was palpated in the region of the cecal base in approximately 50% of affected horses.1078 Transabdominal ultrasound may be helpful in discerning the nature of the mass. The intussusception has a characteristic target appearance on cross section1079, 1089, 1081 and has been reported as a frequent incidental finding in Standardbred foals.1082 Other segments of the small intestine may also be intussuscepted, including the jejunum (Fig. 12.12 ). In one study of 11 jejunojejunal intussusceptions, the length of bowel involved ranged between 0.4 and 9.1 m.1083 Attempts at reducing intussusceptions at surgery are usually futile because of intramural swelling of affected bowel. Jejunojejunal intussusceptions should be resected.
Fig. 12.12.
Jejunojejunal intussusception in a horse with colic. Note the intussusceptum, which has become ischemic as a result of invagination of intestine and its mesenteric blood supply into the intussuscipiens.
For acute ileocecal intussusceptions the small intestine should be transected as far distally as possible, and a jejunocecal anastomosis should be performed. In cases with particularly long intussusceptions (up to 10 m has been reported), an intracecal resection may be attempted.1084 For horses with chronic ileocecal intussusceptions, a jejunocecal bypass without small intestinal transection should be performed. The prognosis is good for horses with chronic ileocecal intussusceptions and guarded to poor for horses with acute ileocecal intussusceptions, depending on the length of bowel involved.1078
Diaphragmatic Hernias
Herniation of intestine through a rent in the diaphragm is rare in the horse. Any segment of bowel may be involved, although small intestine is most frequently herniated.1085 Diaphragmatic rents may be congenital or acquired, but acquired hernias are more common.1085 Congenital rents may result from incomplete fusion of any of the four embryonic components of the diaphragm: pleuroperitoneal membranes, transverse septum, body wall, and esophageal mesentery.1085 Abdominal compression of the foal at parturition may result in a congenital hernia.1085 Acquired hernias are presumed to result from trauma to the chest or a sudden increase in intraabdominal pressure, such as might occur during parturition, distention of the abdomen, a sudden fall, and strenuous exercise.1086 Hernias have been described in a number of different locations; large congenital hernias are typically present at the ventral-most aspect of the diaphragm, and most acquired hernias are located at the junction of the muscular and tendinous portions of the diaphragm.1085 A peritoneopericardial hernia has been documented in at least one horse.1087
The clinical signs are usually associated with intestinal obstruction rather than respiratory distress.1086 Careful auscultation may reveal an area of decreased lung sounds associated with obstructed intestine and increased fluid within the chest cavity.1088 Such signs may prompt thoracic radiography or ultrasound, both of which can be used to make a diagnosis. Auscultation may also reveal thoracic intestinal sounds, but it is typically not possible to differentiate these from sounds referred from the abdomen. In one report, two of three horses diagnosed with small intestinal strangulation by diaphragmatic hernia had respiratory acidemia, attributable to decreased ventilation.1089 Treatment of horses with diaphragmatic hernia is fraught with complications because of the need to reduce and resect strangulated bowel and the need to repair the defect in the diaphragm; prognosis has not appeared to improve over time.1089, 1090, 1091, 1092 Because dorsal defects in the diaphragm are among the most common forms of diaphragmatic defect, it may not be possible to close the diaphragmatic hernia by way of the approach used for abdominal exploratory. Because herniation is likely to recur, it is appropriate to schedule a second surgery using an appropriate approach to resolve the diaphragmatic defect.
Large Colon Volvulus
Clinical Signs
Horses with large colon volvulus have rapid onset of severe, unrelenting abdominal pain. Postpartum broodmares appear to be at risk for this form of colic.1035 Once the large colon is strangulated (>270-degree volvulus), gas distention is marked, leading to gross distention of the abdomen, compromised respiration as the distended bowel presses up against the diaphragm, and visceral pooling of blood as the caudal vena cava is compressed. Horses with this condition are frequently refractory to even the most potent of analgesics. These horses may prefer to lie in dorsal recumbency, presumably to take weight off the strangulated colon. An abbreviated physical examination is warranted in these cases because the time from the onset of strangulation to surgical correction is critical. Under experimental conditions the colon is irreversibly damaged within 3 to 4 hours of a 360-degree volvulus of the entire colon.855 Despite severe pain and hypovolemia, horses may have a paradoxically low heart rate, possibly related to increased vagal tone. Results of abdominocentesis often do not indicate the degree of colonic compromise,1035, 1093 and in many cases it is not worth attempting to obtain abdominal fluid because of extreme colonic distention.1093 Rectal palpation reveals severe gas distention of the large colon, frequently associated with colonic bands traversing the abdomen. Severe colonic distention may restrict access to the abdomen beyond the pelvic brim. One study has shown that plasma lactate levels below 6.0 mmol/L had a sensitivity of 84% and a specificity of 83% in predicting survival in horses with large colon volvulus.1094
Surgical Findings
At surgery the volvulus is typically located at the mesenteric attachment of the colon to the dorsal body wall, and the most common direction of the twist is dorsomedial when the right ventral colon is used as a reference point.1035 However, the colon may twist in the opposite direction, twist greater than 360 degrees (up to 720 degrees has been reported), or twist at the level of the diaphragmatic and sternal flexures.1035 In all cases the colon should be decompressed as much as possible, and in many cases a colonic evacuation by way of a pelvic flexure enterotomy will greatly aid correction of the volvulus. After correction of the volvulus, a determination must be made as to whether the colon has been irreversibly injured. This should be based on mucosal color and bleeding (if an enterotomy has been performed), palpation of a pulse in the colonic arteries, serosal color, and appearance of colonic motility.1036 If the colon is judged to be irreversibly damaged, the feasibility of a large colon resection can be considered. Although 95% of the colon can be resected (that part of the colon distal to the level of the cecocolic fold), damage from the volvulus may exceed that which can be resected. In these cases surgeons may elect to resect as much damaged bowel as possible or advise euthanasia.1036
Prognosis
Although early reports of short-term survival were low (35%),1093 survival can vary with degree of volvulus. Short-term survival was reported as 36% for horses with 360-degree volvulus compared with 71% for horses with 270-degree volvulus.1035 More recent studies report higher short-term survival (88%), with duration of disease as a major factor associated with survival.1095 Impression of improved prognosis was echoed in a recent survey of the American Colleges of Veterinary Surgery Diplomates, who also note time to surgery as the most important factor associated with survival.1096 Another study reports much higher short-term survival (74%) with large colon resection, along with positive survival rates at 1 year (67.8%), 2 years (66%), and 3 years (63.5%) postoperatively.1097 Postoperative complications include hypovolemic and endotoxemic shock, extensive loss of circulating protein, DIC, diarrhea, and laminitis. Large colon volvulus may have a propensity to recur. Although one study documented a recurrence rate of less than 5%,1093 some authors believe recurrence may be as high as 50%.1036 Methods to prevent recurrence may be considered in patients at risk for recurrence, particularly broodmares that tend to suffer from the disease recurrently during the foaling season.1098
Intussusceptions
The most common intussusceptions of the large intestine are cecocecal and cecocolic.1099 Both are likely attributable to the same disease process, with variable inversion of the cecum. These conditions tend to occur in young horses (63% were younger than 3 years old in one study) and may be associated with intestinal tapeworms.1099 Clinical signs are variable, including acute, severe colic, intermittent pain over a number of days, and chronic weight loss.1099 These variable presentations likely relate to the degree to which the cecum has intussuscepted. Initially, the cecal tip inverts, creating a cecocecal intussusception, which does not obstruct the flow of ingesta. As the intussusception progresses, the cecum inverts into the right ventral colon (cecocolic intussusception), which obstructs the flow of ingesta and often causes severe colic (Fig. 12.13 ). The cause of abdominal pain is often difficult to differentiate in these cases, although it is sometimes possible to detect a mass on the right side of the abdomen by either rectal palpation or ultrasound examination.1099 Treatment involves manual surgical reduction by retracting the intussusceptum directly or by way of an enterotomy in the right ventral colon.1099, 1100 Sometimes the cecum cannot be readily reduced because of severe thickening, and in other cases surgical procedures result in fatal contamination. In one report 8 of 11 horses were euthanized in the perioperative period because of complications,1101 and in another report 12 of 30 horses were euthanized either before or during surgery. The latter included all of the horses with chronic disease because of irreversible changes to the cecum.1099 One report on cecocolic intussusceptions indicated that 7 of 8 horses that underwent right ventral colon enterotomy and cecal resection survived long term,1100 suggesting that continued improvements in surgical techniques may improve the prognosis.
Fig. 12.13.
Cecocolic intussusception in a horse with colic. An enterotomy has been made in the right ventral colon (short arrows) to reveal an intussuscepted cecum (arrows). Although this picture was taken at necropsy, an enterotomy such as the one shown in this figure can be used to exteriorize and resect the majority of the compromised cecum. Note the ileum adjacent to the colon (double arrow).
Colocolic intussusceptions are exceptionally rare but have reportedly affected the pelvic flexure and the left colon.1102, 1103, 1104 Although the condition is reportedly more common in young horses,1102, 1104 older horses may be affected.1103 Clinical findings may include a palpable mass on the left side of the abdomen.1102 Ultrasonography may also be useful. Treatment requires manual reduction of the intussusception at surgery or resection of affected bowel. Because the left colons can be extensively exteriorized and manipulated at surgery, the prognosis is fair.1102, 1103, 1104
Rectal Prolapse
Rectal prolapse may occur secondary to any disease that causes tenesmus, including diarrhea, rectal neoplasia, and parasitism,1105 or it can occur secondary to elevations in intraabdominal pressure during parturition or episodes of coughing.1106 Rectal prolapse is classified into four categories (Table 12.7 ) depending on the extent of prolapsed tissue and the level of severity.1107 Type I rectal prolapse is most common and is characterized by a doughnut-shaped prolapse of rectal mucosa and submucosa (Fig. 12.14 ). Type II prolapse involves full-thickness rectal tissue, and type III prolapse additionally has invagination of small colon into the rectum. Type IV prolapse involves intussusception of proximal rectum or small colon through the anus in the absence of prolapse of tissue at the mucocutaneous junction at the anus.1107 These can be differentiated from other forms of prolapse by their appearance and a palpable trench between prolapsed tissue and the anus.
TABLE 12.7.
Classification of Rectal Prolapse
| Grade | Description | Prognosis |
|---|---|---|
| I | Prolapse of rectal mucosa | Good |
| II | Prolapse of full-thickness rectum | Fair |
| III | Grade 2 prolapse with additional protrusion of small colon | Guarded |
| IV | Intussusception of rectum and small colon through the anus | Poor |
Fig. 12.14.
Type I rectal prolapse in a horse. Note circumferential protrusion of partial-thickness rectal tissue (arrows) that is becoming congested as a result of pressure from the surrounding anus.
Type I prolapses are most frequently seen in horses with diarrhea, and the rectal mucosa becomes irritated and protrudes intermittently during episodes of tenesmus. If tenesmus persists, rectal mucosa can remain prolapsed. Rectal mucosa rapidly becomes congested and edematous under these conditions and should be treated with osmotic agents such as glycerin or magnesium sulfate and by massaging and reducing the prolapse.1108 A purse-string suture may be necessary to keep the mucosa inside the rectum. Topical application of lidocaine solution or jelly, epidural anesthesia, and sedation may help reduce tenesmus that incites and exacerbates rectal prolapse. Similar treatments can be applied with type II rectal prolapses. However, these more severe prolapses may not be reducible without surgical resection of mucosa and submucosa from the prolapsed bowel.1105, 1106
Types III and IV rectal prolapses are more serious injuries because the small colon is involved.1109 In horses with type III prolapse, an abdominocentesis should be performed to determine whether the injury to the small colon has resulted in peritonitis. The small colon component should be reduced manually if possible, whereas prolapsed rectal tissue typically requires mucosal or submucosal resection. Surgical exploration of the abdomen should be performed to determine the status of the small colon, although serial abdominocenteses can be used in lieu of surgery to detect progressive necrosis of the bowel. Type IV prolapses are seen most commonly in horses with dystocia.1106 This type of prolapse is almost always fatal because of stretching and tearing of mesenteric vasculature, with subsequent infarction of affected bowel. Euthanasia is often warranted on the basis of physical examination findings. Confirmation of severe small colon injury requires abdominal exploration using either a midline approach or laparoscopy.1110 It is conceivable that a horse with a compromised small colon could undergo a colostomy of the proximal small colon, but the compromised small colon will typically necrose beyond that which can be resected using a midline abdominal approach.1108
Nonstrangulating Infarction
Nonstrangulating infarction occurs secondary to cranial mesenteric arteritis caused by migration of S. vulgaris 1111 and has become a relatively rare disorder since the advent of broad-spectrum anthelmintics. Although thromboemboli have been implicated in the pathogenesis of this disease, careful dissection of naturally occurring lesions has not revealed the presence of thrombi at the site of intestinal infarctions in most cases.1111 These findings suggest that vasospasm plays an important role in this disease.1060 Clinical signs are highly variable, depending on the extent to which arterial flow is reduced and the segment of intestine affected. Any segment of intestine supplied by the cranial mesenteric artery or one of its major branches may be affected, but the distal small intestine and large colon are more commonly involved.1111 There are no clinical variables that can be used to reliably differentiate this disease from strangulating obstruction.1099 In some cases massive infarction results in acute, severe colic.1111 Occasionally, an abnormal mass and fremitus may be detected on rectal palpation of the root of the cranial mesenteric artery. This disease should be considered a differential diagnosis in horses with a history of inadequate anthelmintic treatment and the presence of intermittent colic that is difficult to localize. Although fecal parasite egg counts should be performed, they are not indicative of the degree of parasitic infestation.
In addition to routine treatment of colic, dehydration, and endotoxemia, medical treatment may include aspirin (20 mg/kg/day) to decrease thrombosis.11 Definitive diagnosis requires surgical exploration. Affected horses can be difficult to treat because of the patchy distribution of the lesions and the possibility of lesions extending beyond the limits of surgical resection. Further infarction may occur after surgery. The prognosis is fair for horses with intermittent mild episodes of colic that may be amenable to medical therapy but poor in horses that require surgical intervention.1111
Obstructive Disorders of the Gastrointestinal Tract
Examination and approach to the horse with colic are discussed in Chapter 7; pain management is discussed in Chapter 3. These are critical factors in the diagnosis and treatment of a horse with obstructive disease.
Small Intestinal Simple Obstruction
Simple obstruction involves intestinal obstruction of the lumen without obstruction of vascular flow. Because there is a tremendous volume of fluid that enters the small intestinal lumen on a daily basis, the obstructed intestine tends to become distended, which in turn may cause reduced mural blood flow.879 Ultimately, such distention can result in necrosis of tissues, particularly in the immediate vicinity of the obstruction.1112 There are relatively few causes of simple obstruction in the small intestine, and the incidence of these types of obstructions is low (approximately 3% of all referred horses in one large hospital-based study).1041 In some geographic regions this type of obstruction has a higher prevalence. For example, in the southeastern United States, ileal impactions are relatively common.1113, 1114, 1115
Ascarid Impaction
Impactions caused by Parascaris equorum typically occur in foals younger than 6 months of age that have been on a poor deworming program and have a heavy parasite burden. Products that cause sudden ascarid death, including organophosphates, ivermectin, and pyrantel pamoate, have been incriminated in triggering acute intestinal obstruction by dead parasites.1116 This is a particular problem with ascarids because of the relatively large size of the adult parasite. Clinical signs include acute onset of colic after administration of an anthelmintic and signs compatible with small intestinal obstruction, including nasogastric reflux. Occasionally, dead parasites are present in the reflux. The onset of the disease varies according to the degree of obstruction.1116 A tentative diagnosis may be made on the basis of the history and signs referable to small intestinal obstruction. Abdominal ultrasound may indicate the presence of multiple loops of distended small intestine and can be used to estimate ascarid burden within the small intestine.1117 Initial medical treatment should include pain management and supportive care. Surgical treatment may involve an enterotomy removal of ascarids, although manual reduction without enterotomy was associated with improved survival in one report.1118 The prognosis is fair in cases that are rapidly addressed but poor in foals with evidence of hypovolemia and septic shock. In one study, long-term survival of 25 affected horses was 33%.1116 Another reported 80% short-term survival and 60% survival to 1 year.1118
Ileal Impaction
Ileal impactions occur most commonly in adult horses in the southeastern United States. Although feeding of coastal Bermuda hay has been implicated in this regional distribution,1115 it has been difficult to separate geographic location from regional hay sources as risk factors.1119 Nonetheless, it is likely that feeding suboptimal quality coastal Bermuda hay puts horses at risk for ileal impaction, possibly because this type of hay may have a high fiber content and thin strands that can lead to premature swallowing. The relationship between fiber content and eating patterns is theoretical and remains to be proved. Sudden changes in feed from an alternative type of hay to coastal Bermuda hay likely put a horse at risk for ileal impaction.1119 Studies in the United Kingdom have revealed tapeworm infection as an important risk factor for ileal impaction. Based on risk analysis, the data suggested that more than 80% of the ileal impaction cases studied were associated with serologic or fecal evidence of tapeworm infection.1120 Because of the poor sensitivity of fecal analysis for tapeworms, a serologic test (ELISA) has been developed by Proudman et al. with a sensitivity of approximately 70% and a specificity of 95%.1121, 1122
Clinical signs of horses with ileal impaction are typical for a horse with small intestinal obstruction, including onset of moderate to severe colic and rectally palpable loops of distended small intestine as the condition progresses. Because the ileum is the distal-most aspect of the small intestinal tract, nasogastric reflux may take a considerable time to develop and is found in only approximately 50% to 60% of horses requiring surgical correction of impacted ileum1113, 1123; reflux is more likely in horses with ileal impaction taken to surgery.1113 A definitive diagnosis is usually made at surgery, although an impacted ileum may on occasion be palpated rectally.1114 Multiple loops of distended small intestine make the impaction difficult to palpate. More than 50% of ileal impactions may resolve with medical treatment.1113 Most reports indicate a good to excellent prognosis for short-term survival113, 114, 115, 1123 with 1-year survival of 91% to 92% in horses treated surgically or medically.1113
Ileal Hypertrophy
Ileal hypertrophy is a disorder in which the muscular layers (both circular and longitudinal) of the ileum thicken for unknown reasons (idiopathic) or secondary to an incomplete or functional obstruction. A proposed mechanism for idiopathic ileal hypertrophy is parasympathetic neural dysfunction resulting in chronically increased muscle tone and subsequent hypertrophy of the muscular layers of the ileal wall. Such neural dysfunction possibly results from parasite migration.1124 Alternative hypotheses include chronic increases in the muscular tone of the ileocecal valve, leading to muscular hypertrophy of the ileum as it contracts against a partially occluded ileocecal valve. The jejunum may also be hypertrophied, either alone or in combination with the ileum.1124 Clinical signs include chronic intermittent colic as the ileum hypertrophies and gradually occludes the lumen. Partial anorexia and chronic weight loss (1–6 months) were documented in 45% of affected horses, most likely because of intermittent colic and reduced appetite.1124 Because the ileal mucosa is not affected by this condition, there is no reason to believe that these horses experience malabsorption of nutrients. The diagnosis is usually made at surgery, although the hypertrophied ileum may be palpated rectally in some cases.1124 An ileocecal or jejunocecal anastomosis to bypass the hypertrophied ileum is usually performed in affected horses. Without surgical bypass intermittent colic persists, and the thickened ileum may ultimately rupture.1124 The prognosis is fair with surgical treatment.1125
Secondary ileal hypertrophy is most commonly noted in horses that have previously had colic surgery and that may have a partial or functional obstruction at an anastomotic site. For example, in one case report a horse developed ileal hypertrophy after surgical correction of an ileocecal intussusception.1126 Ileal hypertrophy was also observed in a horse in which an ileocolic anastomosis was incorrectly oriented during surgical treatment of a cecal impaction.1000 Horses are typically reexamined for recurrence of colic in these cases. Surgical therapy is directed at addressing the cause of small intestinal obstruction and resecting hypertrophied intestine.
Meckel’s Diverticulum
Meckel’s diverticulum is an embryonic remnant of the vitelloumbilical duct, which fails to completely atrophy and becomes a blind pouch projecting from the antimesenteric border of the ileum.1127, 1128 Similar diverticula have also been noted in the jejunum.1129 These diverticula may become impacted, resulting in partial luminal obstruction, or may wrap around an adjacent segment of intestine, causing strangulation.1128 Occasionally, an associated mesodiverticular band may course from the diverticulum to the umbilical remnant and serve as a point around which small intestine may become strangulated. Mesodiverticular bands may also originate from the embryonic ventral mesentery and attach to the antimesenteric surface of the bowel, forming a potential space within which intestine may become entrapped.1038 Clinical signs range from chronic colic, for an impacted Meckel’s diverticulum, to acute, severe colic if a mesodiverticular band strangulates intestine. The diagnosis is made at surgery, and treatment requires resection of the diverticulum and any associated bands.1038 The prognosis is good for horses with simple impaction of a Meckel’s diverticulum and guarded for horses with an associated small intestinal strangulation.1038
Adhesions
Adhesions of one segment of bowel to another or of a segment of intestine to other organs and the body wall typically occur after abdominal surgery and may be clinically silent, cause chronic colic attributable to partial obstruction, or result in acute obstruction. These differing clinical syndromes are attributable to the type of adhesions that develop. For example, a fibrous adhesion that does not by itself obstruct the intestinal lumen might serve as the pivot point for a volvulus, whereas an adhesion between adjacent segments of the intestinal tract may create a hairpin turn that causes chronic partial obstruction.1130 The number of adhesions that develop may also vary dramatically from horse to horse. Some horses may develop a single adhesion adjacent to an anastomotic site or a discrete segment of injured intestine, whereas other horses may develop diffuse adhesions involving multiple segments of intestine, likely because of widespread inflammatory disease at the time of the original surgery.
The mechanisms by which adhesions develop are complex but likely involve injury to the serosa initiated by intestinal ischemia, reperfusion injury, and luminal distention.1037 Such injury involves infiltration of neutrophils into the serosa, accompanied by loss of mesothelial cells. In one study assessing the margins of resected small intestine, extensive neutrophil infiltration was documented in the serosa, particularly in the proximal resection margin that had been distended before correction of a variety of strangulating lesions.868 Regions of serosal injury and inflammation subsequently undergo reparative events similar to those of any wound, including local production of fibrin, de novo synthesis of collagen by infiltrating fibroblasts, and ultimately maturation and remodeling of fibrous tissue. Unfortunately, during this process fibrin may result in injured intestinal surfaces adhering to adjacent injured bowel or an adjacent organ. Once a fibrinous adhesion has developed, new collagen synthesis may result in a permanent fibrous adhesion. Alternatively, fibrinous exudate may be lysed by proteases released by local phagocytes, reversing the adhesive process. Formation of adhesions may be viewed as an imbalance of fibrin deposition and fibrinolysis.1131
Prevention of adhesions relies on inhibition of the mechanisms involved in adhesion formation, including reduction of serosal injury with early intervention and good surgical technique, reduction of inflammation by administration of antiinflammatory medications, physical separation of inflamed serosal surfaces (e.g., carboxymethylcellulose, hyaluronan),1132, 1133, 1134 and pharmacologic modulation of fibrinous adhesion formation (e.g., heparin1135). Early return of motility in the small intestine after surgery may reduce contact time between inflamed surfaces of intestine, reducing the chances of adhesion formation.1131
Horses at greatest risk of developing adhesions after colic surgery appear to be those that have small intestinal disease.1130 In one study of horses undergoing surgical correction of small intestinal obstruction, 22% developed a surgical lesion associated with adhesions. Foals appear to have an increased incidence of adhesions compared with mature horses, regardless of the nature of the abdominal surgery.1130 One study indicated that 17% of foals developed lesions attributable to adhesions regardless of the type of the initial surgery.1136 Studies conflict as to whether the degree of surgical intervention influences adhesion formation,1130 but horses that require enterotomy or resection and anastomosis were at greatest risk of developing adhesions in one study.1137 Adhesions are among the most important reasons for repeat laparotomy in postoperative colic patients.1137, 1138
Clinical signs in horses with adhesions are highly variable, depending on whether the adhesion is causing partial obstruction, complete luminal obstruction, or involvement of intestinal vasculature. Adhesions are an important differential for intermittent colic in the postoperative period, particularly if such colic was not relieved by nasogastric decompression of the stomach. Continued intermittent colic should prompt abdominocentesis to determine whether there is evidence of septic peritonitis, which may contribute to adhesion formation. If postoperative colic persists, repeat laparotomy or laparoscopy may be elected. In one study on adhesions, 70% of repeat laparotomies were performed within 60 days, suggesting that surgical colic attributable to adhesions typically occurs within 2 months of an initial surgical procedure. Unfortunately, the prognosis for horses with colic attributable to adhesions is poor, with only 16% of horses surviving adhesion-induced colic in one study.1130
Large Intestinal Simple Obstruction
Simple obstructions of the large intestine, such as impaction, tend to have a more gradual onset than those of the small intestine, although horses may experience acute and severe pain with some forms of colon displacement. In fact, some of these cases mimic and may progress toward large colon volvulus. Medical therapy is frequently successful in correcting large colon impactions. However, cecal impactions present much more of a dilemma because of the greater propensity of this organ to rupture, the relative difficulty of surgically manipulating the cecum, and the onset of cecal dysfunction that may prevent the cecum from emptying after surgical resolution of impaction.
Cecal Impaction
Cecal impaction may be divided into two syndromes: primary cecal impactions that result from excessive accumulation of ingesta in the cecum and secondary cecal impactions that develop while a horse is being treated for a separate problem.936, 1139 Although primary impactions typically consist of impacted, relatively dry fecal material and secondary cecal impactions tend to have very fluid contents, there is considerable overlap between the two syndromes. In horses with primary cecal impactions, there is a gradual onset of abdominal pain over a number of days reminiscent of the development of a large colon impaction. Cecal impactions should be differentiated from large colon impactions on the basis of rectal palpation findings. These impactions have a propensity to rupture before the development of severe abdominal pain or systemic deterioration and therefore must be closely monitored.1139 Secondary cecal impactions typically develop after unrelated surgical procedures that result in postoperative pain (particularly orthopedic surgeries).1140 Secondary cecal impactions may be even more difficult to detect because postoperative depression and decreased fecal output may be attributed to the operative procedure rather than colic. By the time horses with secondary cecal impactions show noticeable signs of colic, the cecum may be close to rupture. In many cases there will be no signs of impending rupture.1140 Therefore the feed intake and manure production of all horses that undergo surgery in which considerable postoperative pain may develop should be closely monitored. A recent study indicated that horses producing less than three piles of manure daily in the postoperative period are at risk of developing a large intestinal impaction. Horses that underwent prolonged (>1 hour) orthopedic surgery that received inadequate treatment with phenylbutazone were at considerable risk of reduced postoperative fecal output.1141
A diagnosis of cecal impaction is based on rectal palpation of a firm, impacted or fluid-filled cecum. In some cases, cecal impactions may be difficult to differentiate from large colon impactions. Careful palpation, however, will reveal the inability to move the hand completely dorsal to the impacted viscus because of the cecum’s attachment to the dorsal body wall.
Treatment may include initial medical therapy with IV or oral fluids and analgesics, or surgical intervention with typhlotomy alone or jejunocecostomy. In two recent larger scale retrospective studies, the prognosis appeared to vary significantly by geographic region.1139, 939 In one, a very favorable short-term outcome was achieved with either medical (81%) or surgical (95%) management.939 In the majority of surgical cases, typhlotomy alone was performed. In another, short-term outcome was less favorable with either medical (61%) or surgical (82%) management.1139 Many (68%) cases in the latter report appeared to have secondary cecal impactions as they had recent illness or surgery unrelated to gastrointestinal disease. This report underscores the importance of prompt diagnosis and supports the benefit of early surgical intervention.
Large Colon Impaction
Ingesta impactions of the large colon occur at sites of anatomic reductions in luminal diameter, particularly the pelvic flexure and the right dorsal colon.1142 Although there are a number of reported risk factors, most have not been proved. A sudden restriction in exercise associated with musculoskeletal injury appears to be frequently associated with onset of impaction.1143 Twice-daily feeding of concentrate results in large fluxes of fluid into and out of the colon, associated with readily fermentable carbohydrate in the colon and subsequent increases in serum aldosterone, respectively.1144 These fluid fluxes, which may cause dehydration of ingesta during aldosterone-stimulated net fluid flux out of the colon, may be prevented with frequent small feedings.
Impaction of the ascending colon can be induced by the drug amitraz, an acaricide associated with clinical cases of colon impaction,1145, 1146 providing some clues as to the pathogenesis of large colon impaction. Amitraz appears to alter pelvic flexure pacemaker activity, resulting in uncoordinated motility patterns between the left ventral and left dorsal colon and excessive retention of ingesta. Absorption of water from the ingesta increases with retention time, dehydrating the contents of the colon and resulting in impaction. It is conceivable that parasite migration in the region of a pacemaker may have a similar action.653 Other factors implicated in large colon impaction include limited exercise, poor dentition, coarse roughage, and dehydration.
Clinical signs of large colon impaction include slow onset of mild colic. Fecal production is reduced, and the feces are often hard, dry, and covered with mucus because of delayed transit time. The heart rate may be mildly elevated during episodes of pain but is often normal. Signs of abdominal pain are typically well controlled with administration of analgesics but become increasingly more severe and refractory if the impaction does not resolve. The diagnosis is based on rectal palpation of a firm mass in the large colon. The extent of the impaction may be underestimated by rectal palpation alone because much of the colon remains out of reach.1142 Adjacent colon may be distended if the impaction has resulted in complete obstruction. Initial medical treatment should include pain management and enteral fluid therapy if reflux is not obtained on nasogastric intubation. Hydration of colonic contents is superior with enteral fluid therapy compared with IV fluid therapy.1147, 1148 Saline cathartics such as magnesium sulfate (0.1 mg/kg in 2–4 L by nasogastric tube) may also be useful. Access to feed should not be permitted. If the impaction remains unresolved, the horse’s pain becomes uncontrollable, or extensive gas distention of the colon occurs, surgery is indicated. At surgery the contents of the colon are evacuated by way of a pelvic flexure enterotomy. Long-term survival is good with medical management (95%) and fair (58%) in horses that require surgical intervention.1143
Enteroliths
Enteroliths are mineralized masses typically composed of ammonium magnesium phosphate (struvite).37 However, magnesium vivianite has also been identified in enteroliths, along with variable quantities of Na, S, K, and Ca. The formation of Mg-based minerals is puzzling because of the relative abundance of Ca in colonic fluids, which would favor the formation of Ca phosphates (apatite) rather than struvite. Elevated dietary intake of magnesium and protein may play a role. Feeding of alfalfa hay and decreased dietary proportions of grass hay and pasture grass have been consistently identified as risk factors in horses with enterolithiasis.1149, 1150, 1151 Alfalfa hay has a concentration of magnesium approximately six times the daily requirements of the horse.1152 The high protein concentration in alfalfa hay may contribute to calculus formation by increasing the ammonia nitrogen load in the large intestine. Enteroliths most commonly form around a nucleus of silicon dioxide (a flintlike stone), but nidi have included nails, rope, and hair that have been ingested.1153 Enteroliths are usually found in the right dorsal and transverse colons.1152 Although enterolithiasis has a wide geographic distribution, horses in California have the highest incidence. In one California study, horses with enterolithiasis represented 28% of the surgical colic population, and Arabians, Morgans, American Saddlebreds, and donkeys were at greatest risk of this disease.37 In a study of enterolithiasis in Texas, risk factors also included feeding of alfalfa hay and the Arabian breed. In that study, Miniature Horses were also found to be at risk.1149 Horses with enteroliths are rarely younger than 4 years old, with a median age of 11 in one report37; an enterolith has been reported in an 11-month-old Miniature Horse.1154
The most common reported clinical signs are episodic mild to moderate abdominal pain.37, 1152 Enteroliths may be diagnosed by abdominal radiography or at surgery.40, 1155 On rare occasions an enterolith may be palpated rectally, particularly if it is present in the distal small colon.
Generally, surgery is required, although there are reports of enteroliths being retrieved rectally. In one study 14% of horses that required treatment of enterolithiasis had a history of passing an enterolith in the feces.37 Enteroliths are typically located in the right dorsal colon, transverse colon, or small colon. After removal of an enterolith, further exploration must be conducted to determine whether other enteroliths are present. Solitary enteroliths are usually round, whereas multiple enteroliths have flat sides. The prognosis is good (92% 1-year survival in 900 horses), unless the colon is ruptured during removal of an enterolith, which was reported in 15% of cases.37
Sand Impaction of the Large Colon
Sand impactions are common in horses with access to sandy soils, particularly horses whose feed is placed on the ground. Some horses, especially foals, deliberately eat sand. Fine sand tends to accumulate in the ventral colon, whereas coarse sand may accumulate in the dorsal colon.737, 1156 Individual differences in colonic function may contribute to accumulation of sand because some horses can clear consumed sand, whereas others cannot. Distention resulting from the impaction itself, or gas proximal to the impaction, causes abdominal pain. Sand may also trigger diarrhea, presumably as a result of irritation of the colonic mucosa.738 In horses with sand impactions, clinical signs are similar to those of horses with large colon impactions. Sand may be found in the feces, and auscultation of the ventral abdomen may reveal sounds of sand moving within the large colon.1157 The diagnosis is made via radiography or surgery but may be tentatively based on clinical signs compatible with a large colon impaction and evidence of sand in the feces. Sand sedimentation is performed by mixing feces with water in a rectal palpation sleeve or other container. Abdominal radiographs can be used to detect mineral opacity within the ventral colon.38, 1158 Ultrasonography can also be used to support the diagnosis but is not as accurate as radiography.1159 Abdominal paracentesis is not recommended in suspected cases, because large quantities of sand in the ventral colon make inadvertent perforation of the colon more likely.737
Medical management typically offers a good prognosis.1160 Administration of psyllium hydrophilic mucilloid (0.25–0.5 kg/500 kg in 2 L mineral oil by stomach tube) may facilitate passage of sand. If mixed with water, it should be administered rapidly because of formation of a viscous gel. The psyllium leaves the oil phase and mixes with the water, forming a gel within the stomach. Psyllium is thought to act by stimulating motility or agglutinating the sand. However, one experimental study failed to show a benefit of this treatment.1161 If a severe impaction is present, then psyllium should not be given until the impaction is softened by administering IV or oral fluids and other laxatives. Perforation is a potential complication in horses with sand impactions because the sand stretches and irritates the intestinal wall and causes inflammation. If colic becomes intractable, surgical evacuation of the large colon should be performed. The prognosis is generally regarded as good.737, 1156
Large Colon Displacement
Displacement of the ascending colon is a common cause of large intestinal obstruction. The ascending colon is freely movable except for the right dorsal and ventral colons. Contact with adjacent viscera and the abdominal wall tends to inhibit movement of the ascending colon from a normal position; accumulation of gas and fluid or ingesta, however, may cause the colon to migrate.1162 Feeding behavior, including feeding of large concentrate meals, likely plays a role in initiating displacement of the large colon. Large concentrate meals increase the rate of passage of ingesta, allowing a greater percentage of soluble carbohydrates to reach the large intestine.1163 This in turn increases the rate of fermentation and the amount of gas and volatile fatty acids that are produced. The production of large amounts of volatile fatty acids stimulates the secretion of large volumes of fluid into the colon.2 The association between feeding concentrate and development of displacements of the large colon is illustrated by studies indicating that ascending colon displacement is more prevalent in horses fed a high-concentrate, low-roughage diet.1164 Abnormal motility patterns of the ascending colon may also contribute to the development of colonic displacement. Feeding stimulates colonic motility by way of the gastrocolic reflex, but large meals may alter normal motility patterns and concurrently allow rapid accumulation of gas and fluid resulting from fermentation.1163, 1165 Migration of parasite larvae (strongyles) through the intestinal wall also alters colonic motility patterns.650 S. vulgaris infection results in reduced blood flow to segments of the large intestine without necessarily causing infarction. Electrical activity of the colon and cecocolic junction increases after infection with S. vulgaris and cyathostome larvae, probably reflecting a direct effect of migration through the intestine and an early response to reduced blood flow.650
Displacements of the ascending colon are generally divided into three types: left dorsal displacement, right dorsal displacement, and retroflexion.1162, 1166 Left dorsal displacement is characterized by entrapment of the ascending colon in the renosplenic space. The colon is often twisted 180 degrees, such that the left ventral colon is situated in a dorsal position relative to the left dorsal colon. The entrapped portion may be only the pelvic flexure or may involve a large portion of the ascending colon, with the pelvic flexure situated near the diaphragm. The colon may become entrapped by migrating dorsally between the left abdominal wall and the spleen or may migrate in a caudodorsal direction over the nephrosplenic ligament.1166 Occasionally, the ascending colon can be palpated between the spleen and abdominal wall, lending support to the first mechanism of displacement. Gastric distention is thought to predispose horses to left dorsal displacement of the ascending colon by displacing the spleen medially, allowing the colon room to migrate along the abdominal wall.1166 Right dorsal displacement begins by movement of the colon cranially, either medial (medial flexion) or lateral (lateral flexion) to the cecum. According to one author, the proportion of right dorsal displacements with medial versus lateral flexion is approximately 1:15.1166 In either case the pelvic flexure ends up adjacent to the diaphragm. Retroflexion of the ascending colon occurs by movement of the pelvic flexure cranially without movement of the sternal or diaphragmatic flexures.
Displacement of the ascending colon partially obstructs the lumen, resulting in accumulation of gas or ingesta and causing distention. The distention may be exacerbated by the secretion of fluid in response to the distention. Tension and stretch of the visceral wall are important sources of the pain associated with colonic displacement. Tension on mesenteric attachments and the root of the mesentery by the enlarged colon may also cause pain.1162 Ischemia is rarely associated with nonstrangulating displacement of the colon. Congestion and edema, however, are often seen in the displaced segments of colon because of increased hydrostatic pressure from reduced venous outflow. Morphologic damage to tissues is usually minor.
Clinically, displacement of the ascending colon is often characterized by intermittent signs of mild to moderate abdominal pain of acute onset, but an insidious onset may also be noted.1166 Dehydration may occur if the duration of the displacement is prolonged. The heart rate may be increased in conjunction with abdominal pain but is often normal. Abdominal distention may be present, and fecal production is reduced. Left dorsal displacements are often diagnosed by rectal palpation, because the ascending colon can be traced to the nephrosplenic space, and the spleen may be displaced medially. Alternatively, a tentative diagnosis can be reached using abdominal ultrasonography.1167 The spleen can be imaged on the left side of the abdomen, but the left kidney will be obscured by gas-distended bowel. Evaluation of this technique indicates that there are very few instances of false-positive results, although false-negative results may occasionally occur.1167 A definitive diagnosis may require surgery. Right dorsal displacements are characterized by the presence of the distended ventral colon running across the pelvic inlet and may be felt between the cecum and the body wall if a lateral flexion is present. The pelvic flexure is usually not palpable. Retroflexion of the ascending colon may produce a palpable kink in the colon. If the displaced colons are not distended by gas in the instance of right dorsal displacement and retroflexion, the ascending colon may not be palpable and is conspicuous by its absence from a normal position. Horses with right dorsal displacement often have an increased GGT, relative to horses with left dorsal displacement, which is presumptively caused by compression of the bile duct and temporary extrahepatic obstruction.17
For nephrosplenic entrapment of the large colon, medical management is often effective.1167, 1168, 1169 Medical therapy with phenylephrine and rolling under anesthesia (84%) was significantly more effective than phenylephrine (3–6 μg/kg/min over 15 minutes) and exercise (63.2%) in a recent report.1169 There are reports of fatal internal hemorrhage caused by rupture of large blood vessels after treatment of older horses with phenylephrine, and the drug should probably be used with caution in horses older than 15 years.1170
Medical therapy has also been reported effective (64%) in horses with right dorsal displacement,1171 although definitive diagnosis of that condition is challenging without surgical confirmation. If medical management is not successful, horses should undergo surgery promptly. Surgical prognosis for horses with large colon displacement is good. A number of horses will suffer recurrence of nephrosplenic entrapment of the colon. Currently, the least invasive method of preventing this complication is laparoscopic closure of the nephrosplenic space.78, 1172, 1173
Foreign Body and Fecalith Obstruction
Foreign material such as bedding, rope, plastic, fence material, and feedbags can cause obstruction and may be ingested, particularly by young horses. These foreign bodies may result in impaction with ingesta and distention of the intestine, typically in the transverse or descending colon. Young horses are usually affected. In one study the obstructing mass could be rectally palpated in three of six horses.1174 Fecaliths are common in ponies, miniature horses, and foals.1175 Older horses with poor dentition may also be predisposed to fecaliths because of the inability to fully masticate fibrous feed material. Fecaliths commonly cause obstruction in the descending colon and may cause tenesmus.1174 Other clinical signs are similar to those of enterolithiasis. Abdominal radiography may be useful in smaller patients to identify the obstruction, especially if gas distention around the foreign body or fecalith provides contrast. Surgical treatment is usually required.
Mural Masses and Strictures
Mural masses such as abscesses, tumors (adenocarcinoma and lymphosarcoma), granulomas, and hematomas can cause luminal obstruction and impaction, typically in older horses. Impaction may result from obstruction of the lumen or impaired motility in the segment of intestine with the mass. Abscesses may originate from the lumen of the intestine or may extend from the mesentery or mesenteric lymph nodes. Intramural hematomas form most commonly in the descending colon and cause acute abdominal pain.1176 Once the acute pain caused by the hematoma subsides, impaction proximal to the hematoma develops as a result of impaired motility through the affected portion of the colon. Trauma, ulceration of the mucosa, and parasitic damage are speculated causes of intramural hematomas.1176, 1177 Stricture of the large intestine occurs when fibrous tissue forms in a circular pattern around or within the intestine, reducing the luminal diameter and the ability of the wall to stretch. Strictures may be congenital or secondary to peritonitis, previous abdominal surgery, or inflammatory bowel disease. In a report of 11 horses with inflammatory bowel disease, 6 horses had strictures, 4 of which were in the small intestine and 2 of which were in the large colon.517
Clinical signs vary according to the degree of luminal obstruction. Partial obstruction and impaction tend to produce mild to moderate abdominal pain of insidious onset. Mural hematomas tend to produce signs of acute abdominal pain.1176, 1177 Rectal palpation of the abdomen may reveal the presence of a mass or simply the impacted segment, without the mass itself being felt. Fever, weight loss, and anorexia may be noted if an abscess or tumor is the cause. An elevated WBC count; hyperfibrinogenemia; hyperglobulinemia; or normocytic, normochromic anemia may be seen with abscesses or tumors. PF may reflect the cause of the mass. Tumor cells may infrequently be seen. Evidence of inflammation with bacteria may be noted if the cause of colic is an abscess or granuloma, in which case the fluid should be cultured. Hematomas may cause hemorrhage into the PF. Treatment usually requires surgical resection of the mass. Abscesses may be treated with appropriate antibiotics if the impaction can be resolved medically with oral or IV analgesics and laxatives. Streptococcus spp., Actinomyces pyogenes, C. pseudotuberculosis, R. equi, anaerobic bacteria, and gram-negative enteric organisms are commonly involved in abscesses.
Small Colon Impaction
Small colon impaction is distinct from other forms of impaction in its predispositions and clinical appearance. In one study the key risk factor for impaction of this segment of the intestine was diarrhea.1178 This paradoxic finding may be explained by edema of the colonic mucosa associated with proinflammatory causes of diarrheal disease that is usually noted in the ascending colon but may extend into the transverse and small colons. Once diarrheal disease is initiated, large volumes of ingesta are rapidly expelled from the ascending colon into the small colon, which has a far smaller diameter, especially if it is edematous. This may result in the initial appearance of diarrhea, followed by intermittent episodes of colic that may be explained by impaction. Diagnosis is by rectal examination, during which the rectum typically feels edematous and gritty. The most important point to remember is that horses should be closely assessed for impaction even if diarrheal disease is present. Other parameters that are typically helpful for assessing the severity of colic, such as heart rate, are not predictive of obstruction in horses with small colon impaction.1178 Horses may be treated medically during the early stages with fluids, laxatives, and analgesics. The key clinical sign that indicates the need for surgery appears to be abdominal distention, associated with distention of the large colon. Other clinical signs, such as elevations in heart rate and refractory colic, are less pronounced in this disease. Postdiarrheal disease is not the only form of small colon impaction. These impactions can be formed as simple collections of ingesta or in response to luminal narrowing.
Atresia Coli
Atresia of a segment of the colon is a rare congenital abnormality in horses.1179 The heritability and causes of the condition are unknown. One potential mechanism for development of the lesion is intestinal ischemia during fetal life, which secondarily results in necrosis of a segment of intestine.1179 Clinical signs include a failure to pass meconium and colic within the first 12 to 24 hours of life. Secondary abdominal distention results from complete intestinal obstruction, and abdominal radiographs may reveal a gas-distended colon. The diagnosis is made at surgery. Any portion of the colon may be absent, but the distal segment of the large colon or the proximal small colon (or both) is usually most severely affected. If sufficient tissue is present, anastomosis to the proximal blind end of the colon may be attempted.1179 The prognosis depends on the segment of the colon that is absent but is usually poor because of an absence of distal colon.
Ileocolonic Aganglionosis
Ileocolonic aganglionosis, commonly known as lethal white foal syndrome (LWFS), occurs in white foals with overo-spotted parents. Affected foals are either completely white or have very little pigmented hair around the muzzle, base of the tail, or hooves. They are homozygous for an abnormal endothelin receptor B (EDNRB) gene that results in altered neural crest cell migration or survival, which affects progenitor cells for melanocytes and intestinal ganglia.1180, 1181 The EDNRB genotype is highly correlated with white patterning; frame overo, highly white calico overo, and frame blend overo have the highest incidence of heterozygotes.1182 Rarely, an LWFS-affected foal may be born to a solid-colored mare. Homozygous foals have aganglionosis of the submucosal and myenteric ganglia of the distal portion of the ileum and large intestine, and extrinsic innervation of the ileum and pelvic flexure has been identified.1183, 1184 This results in a foal that appears normal at birth but develops signs of intestinal ileus and colic within 12 to 24 hours. The eyes are blue, and the skin is pink. A genetic test is available to identify horses that are heterozygous for the defective EDNRB gene.
Diseases of the Oral Cavity, Esophagus, and Stomach
Oral Cavity
The mouth is bounded laterally by the cheeks, dorsally by the palate, and ventrally by the body of the mandible and by the mylohyoideus muscles. The caudal margin is the soft palate. The mouth of the horse is long and cylindric, and when the lips are closed, the contained structures almost fill the cavity. A small space remains between the root of the tongue and the epiglottis and is termed the oropharynx. The cavity of the mouth is subdivided into sections by the teeth. The space external to the teeth and enclosed by the lips is termed the vesicle of the mouth, and in the resting state the lateral margins of the vesicle, that is, the buccal mucosa, are in close contact with the cheek teeth. Caudally, the external space communicates with the pharynx through the aditus pharyngis. The mucous membrane of the mouth is continuous at the margin of the lips with the skin and during life is chiefly pink but can be more or less pigmented, depending on the skin color and the breed type.
Morphology and Function
The lips are two muscular membranous folds that unite at angles close to the first cheek teeth. Each lip presents an outer and an inner surface. The upper lip has a shallow median furrow (philtrum); the lower lip has a rounded prominence or chin (mentum). The internal surface is covered with a thick mucous membrane that contains small, pitted surfaces that are the openings of the ducts of the labial glands. Small folds of the mucous membrane called the frenulum labii pass from the lips to the gum.
The free border of the lip is dense and bears short, stiff hairs. The arteries of the mouth are derived from the maxillary, mandibular, labial, and sphenopalatine arteries of the major palatine artery. The veins drain chiefly to the lingual facial vein. Sensory nerves originate from the trigeminal nerve (cranial nerve V) and the motor nerves from the facial nerve (cranial nerve VII). The cheeks spread back from the lips and form both sides of the mouth and are attached to the alveolar borders of the bones of the jaws. The cheeks are composed of skin and muscular and glandular layers and then the internal mucous membrane. The skin is thin and pliable. In contrast, the oral mucous membrane is dense and in many areas of the oral cavity is attached firmly to the periosteum so that construction of oral mucosal flaps can be achieved only by horizontal division of the periosteal attachment. Such a feature is important in reconstructive techniques applied to the oral cavity. The blood supply to the cheeks comes from the facial and buccal arteries and the sensory nerves from the trigeminal and motor nerves from the facial nerve.
The hard palate (palatum durum) is bounded rostrally and laterally by the alveolar arches and is continuous with the soft palate caudally. The hard palate has a central raphe that divides the surface into two equal portions. From the line of the rostral cheek tooth, the hard palate is concave to the line of the caudal cheek tooth. Paired transverse ridges (about 18) traverse the concavity and have their free edges directed caudally. The incisive duct is a small tube of mucous membrane that extends obliquely through the palatine fissure. The dorsal component communicates by a slitlike opening in the rostral portion of the ventral nasal meatus, and its palatine end is blind and lies in the submucosa of the palate. When stallions display their flehmen response, watery secretions enter the nose from the glands of the vomeronasal duct. To what extent these secretions aid in pheromone reception is not known.1185
That portion of the palatine mucosa immediately behind the incisor teeth frequently is swollen (lampas) during eruption of the permanent teeth. This swelling is physiologic, not pathologic.
The tongue is situated on the floor of the mouth between the bodies of the mandible and is supported by the sling formed by the mylohyoideus muscles. The root of the tongue is attached to the hyoid bone, soft palate, and pharynx. The upper surface and the rostral portion of the tongue are free; the body of the tongue has three surfaces. The apex of the tongue is spatulate and has a rounded border. The mucous membrane adheres intimately to the adjacent structure and on the dorsum is dense and thick. From the lower surface of the free part of the tongue, a fold of mucous membrane passes to the floor of the mouth, forming the lingual frenulum. Caudally, a fold passes on each side of the dorsum to join the soft palate, forming the palatoglossal arch. Dorsally from the soft palate the palatopharyngeal arch attaches and circumvents the aditus laryngis and attaches to the roof of the nasopharynx. The mucous membrane of the tongue presents four kinds of papillae:
-
1.
Filiform papillae are fine threadlike projections across the dorsum of the tongue. They are absent on the root of the tongue and are small on the rostral portion of the tongue.
-
2.
The fungiform papillae are larger and easily seen at the rounded free end. They occur principally on the lateral portion of the tongue.
-
3.
Vallate papillae are usually two or three in number and are found on the caudal portion of the dorsum of the tongue. The free surface bears numerous small, round secondary papillae.
-
4.
Foliate papillae are situated rostral to the palatoglossal arches of the soft palate on which they form a rounded eminence about 2 or 3 cm in length marked by transverse fissures.
Foliate, vallate, and fungiform papillae are covered with taste buds and secondary papillae. The lingual and sublingual arteries supply the tongue from the linguofacial trunk and matching veins. The linguofacial trunk drains into the linguofacial vein. The lingual muscles are innervated by the hypoglossal nerve (cranial nerve XII), and the sensory supply is from the lingual and glossopharyngeal (cranial nerve IX) nerves.
Teeth
The formula for the deciduous teeth of the horse is 2 times I3-3 C0-0 P3-3 for a total of 24. The permanent dental formula is 2 times I3-3 C1-1 P3-3 or P4-3 M3-3 for a total of 40 or 42. In the mare the canine teeth are usually small or do not erupt, reducing the number to 36 or 38. The first premolar tooth (wolf tooth) is often absent and has been reported as occurring in only 20% of the upper dentition of Thoroughbred horses.1186 The teeth of the horse are complex in shape and are compounded of different materials (dentin, cementum, and enamel). They function as grinding blades to masticate and macerate cellulose food in the important first stage of the digestive process. The cheek teeth in the horse are a well-documented feature of the evolution of Equus caballus.
The first deciduous incisor is present at birth or the first week of life. The second incisor erupts at 4 to 6 weeks of age; the third incisor, at 6 to 9 months of age; the first and second premolars, at birth to 2 weeks of age; and the third premolar, at 3 months of age.
The eruption times for the permanent teeth are as follows: first incisor, 2½ years of age; second incisor, 3½ years of age; third incisor, 4½ years of age; the canine tooth, 4 to 5 years of age; the first premolar (wolf tooth), 5 to 6 months of age; the second premolar, 2½ years of age; the third premolar, 3 years of age; the fourth premolar, 4 years of age; the first molar, 10 to 12 months of age; the second molar, 2 years of age; and the third molar, 3½ to 4 years of age. This eruption sequence clearly indicates that the eruption of the second and third permanent premolar teeth have the potential for dental impaction.
The modern horse has six incisor teeth in each jaw that are placed close together so that the labile edges form a semicircle. The occlusal surface has a deep enamel invagination (infundibulum) filled only partially with cementum. As the incisor teeth wear, a characteristic pattern forms in which the infundibulum is surrounded by rings of enamel, dentin, enamel, and crown cementum in a concentric pattern. Each incisor tooth tapers from a broad crown to a narrow root so as the midportion of the incisor is exposed to wear, the cross-sectional diameters are about equal; that is, at 14 years of age, the central incisor tooth of the horse has an occlusal surface that is an equilateral triangle. Observations on the state of eruption, the angles of incidence of the incisor teeth, and the pattern of the occlusal surfaces are used as guides for the aging of horses. The canine teeth are simple teeth without complex crowns and are curved. The crown is compressed and is smooth on its labial aspect but carries two ridges on its lingual aspect. No occlusal contact occurs between the upper and lower canine teeth.
When erupted, the six cheek teeth of the horse function as a single unit in the mastication of food. Each arcade consists of three premolar and three molar teeth. The maxillary arcade is slightly curved, and the teeth have a square occlusal surface. The occlusal surfaces of the mandibular teeth are more oblong, and each arcade is straighter. The horse is anisognathic—that is, the distance between the mandibular teeth is narrower (one-third) than the distance between the upper cheek teeth. This anatomic arrangement affects the inclination of the dental arcade as the jaws slide across each other in the food preparation process. The unworn upper cheek tooth presents a surface with two undulating and narrow ridges, one of which is lateral and the other medial. On the rostral and lingual side of the medial style is an extra hillock. The central portion of these surfaces is indented by two depressions that are comparable with, but much deeper than, the infundibula of the incisor teeth. When the teeth have been subjected to wear, the enamel that closed the ridges is worn through and the underlying dentin appears on the surface. After a time the chewing surface displays a complicated pattern that may be likened to the outline of an ornate letter B, and the upright stroke of the B is on the lingual aspect. Dentin supports the enamel internally, cementum supports the enamel lakes, and the peripheral cementum fills in the spaces between the teeth so that all six teeth may function as a single unit—that is, the dental arcade. Transverse ridges cross each tooth so that the entire maxillary arcade consists of a serrated edge. The serrations are formed so that a valley is present at the area of contact with adjacent teeth. These serrations match fitting serrations on the mandibular arcade. One should note that the mediolateral mandibular motion while chewing pellets does not provide full occlusal contact as it does when chewing hay.1187
The true roots of the cheek teeth are short compared with the total length of the tooth. Cheek teeth have three roots: two small lateral roots and one large medial root. That portion of the crown embedded within the dental alveolus is referred to as the reserve crown, and the term root is confined to that area of the tooth that is comparatively short and enamel free. Wear on the tooth gradually exposes the reserve crown, and the roots lengthen. In an adult 1000-lb horse the maxillary cheek teeth are between 8.0 and 8.5 cm in length. Dental wear accounts for erosion and loss of tooth substance at a rate of 2 mm/year. The pulp chambers of the teeth are also complex. The incisors and canines have a single pulp chamber. The mandibular cheek teeth have two roots and two separate pulp chambers. The maxillary cheek teeth, although they have three roots, have five pulp chambers.
As occlusal wear proceeds, deposition of secondary dentin within the pulp chambers protects the chambers (e.g., the dental star, medial to the infundibulum on the incisor teeth). In the mandibular cheek teeth the transverse folding of the enamel anlage (during morphogenesis of the tooth) does not take place, and the occlusal surface is a simple surface of central dentin surrounded by enamel. Each tooth then is conformed to a single arcade by the presence of peripheral crown cementum.
Examination of the Oral Cavity
The oral cavity and oropharynx are subject to a variety of diseases, many of which produce the same clinical signs, regardless of their cause. The classic signs of dental disease in the horse include difficulty and slowness in feeding, together with a progressive unthriftiness and loss of body condition. In some instances, the horse may quid (drop food while chewing), and halitosis may be obvious. Additional problems reported by owners include bitting and riding problems and headshaking or head shyness. Facial or mandibular swelling, nasal discharge (dental disease associated with maxillary sinus), and mandibular fistulae (lower cheek tooth apical infections) are also possible. Some correlation exists between the age of the animal and clinical signs.
One can examine a considerable portion of the mouth and teeth from the outside by palpation of the structures through the folds of the cheek. Most horses allow a cursory oral examination without sedation or the use of an oral speculum. In many horses, however, a detailed oral examination is best achieved after sedation and with the use of an oral speculum and a light source. The mouth should be irrigated to remove retained food material so as to be able to inspect and palpate the lips, cheeks, teeth, and gums.
Ancillary aids for a complete examination of the oral cavity of the horse may include radiology, endoscopic examination, fluoroscopy, biopsy, and culture. Oral endoscopy should be performed with either sedation and an oral speculum or under general anesthesia to prevent inadvertent mastication of the endoscope. CT allows detailed imaging of the oral cavity and sinuses.
Dysphagia
The lips of the horse are mobile and prehensile. Consequently, loss of motor function (e.g., facial palsy) affects the efficiency of the prehensile system. The lips grasp food in grazing or browsing, and the incisor teeth section the food. With mastication and lubrication with saliva, the bolus of food forms and is manipulated from side to side across the mouth, assisted by the tight cheeks of the horse and the palatine ridges. Swallowing begins as the food bolus contacts the base of the tongue and the pharyngeal walls. During swallowing, the soft palate elevates to close the nasopharynx, the base of the tongue elevates, and the hyoid bone and the larynx move rostrally following contraction of the hyoid muscles. During this process, the rima glottidis closes and the epiglottis tilts dorsally and caudally to protect the airway so that food is swept through lateral food channels around the sides of the larynx into the laryngoesophagus. Fluoroscopic studies in nursing foals in the dorsoventral view showed that contact occurs between the lateral food channels in the midline so that in outline the food bolus achieves a bow-tie shape.1188
Dysphagia is defined as a difficulty or inability to swallow. Anatomic classifications for dysphagia include prepharyngeal, pharyngeal, and esophageal (postpharyngeal) dysphagias. The site of the cause for dysphagia influences the clinical signs. Prepharyngeal dysphagia is characterized by dropping food (quidding) or water from the mouth, reluctance to chew, hypersalivation, or abnormalities in prehension. Pharyngeal and esophageal dysphagias are characterized by coughing; nasal discharge containing saliva, water, or food material; gagging; anxiousness; and neck extension during attempts to swallow. The following section describes esophageal dysphagia in more detail. Causes of dysphagia can be divided into four types: painful, muscular, neurologic, or obstructive (Table 12.8 ). Pain and obstruction cause dysphagia by interfering with the mechanics of prehension, bolus formation and transfer to the pharynx, and deglutition. Muscular and neurologic causes of dysphagia impede prehension and swallowing by affecting the motor function of the lingual or buccal musculature, muscles of mastication (temporal and masseters), and pharyngeal and cranial esophageal muscles. Sensory loss to the lips, buccal mucous membranes, pharynx, or tongue also may cause dysphagia. Neurologic causes of dysphagia may affect the forebrain, brainstem, or peripheral nerves that control prehension (cranial nerves Vm, Vs, VII, and XII), transfer of the food bolus to the pharynx (cranial nerves Vs and XII), and swallowing (cranial nerves IX and X). The latter point was classical thinking, but recent evidence suggests that, while stimulation of cranial nerve IX stimulates swallowing, bilateral blockade of that nerve does not prevent normal swallowing of either liquid or solid material.1189
TABLE 12.8.
Differential Diagnoses for Dysphagia
| Class of Dysphagia | Differential Diagnoses |
|---|---|
| Painful | Tooth root abscess or periodontal disease Broken teeth Abnormal dentition or wear Stomatitis, glossitis, or pharyngitis Nonsteroidal antiinflammatory drug toxicity Chemical irritation Thrush (candidiasis) Influenza Streptococcus equi subsp. equi Vesicular stomatitis virus Actinobacillus lignieresii Buccal, gingival, or glossal trauma (bits or chains) Foreign bodies Retropharyngeal lymphadenopathy or abscess Mandibular trauma Temporohyoid osteoarthropathy Temporomandibular osteopathy |
| Muscular | Hyperkalemic periodic paralysis Nutritional myopathy (white muscle disease) Polysaccharide storage disease Glycogen branching enzyme deficiency Masseter myositis Hypocalcemia tetany or eclampsia Myotonia Rectus capitis ventralis rupture White snakeroot toxicity Megaesophagus |
| Obstructive | Retropharyngeal abscess and lymphadenopathy Oral, pharyngeal, retropharyngeal, laryngeal, or esophageal malformations, injury, edema, or neoplasia Pharyngeal or epiglottic cysts Pharyngeal abscess or foreign body Dorsal displacement of the soft palate Cleft palate |
| Neurologic forebrain disease; generalized neuropathy; disorders of cranial nerves V, VII, IX, X, or XII | Guttural pouch tympany or empyema Follicular pharyngitis Esophageal obstruction Pharyngeal cicatrix Retropharyngeal abscess or neoplasia Guttural pouch empyema, mycosis, or neoplasia Stylohyoid osteopathy Lead poisoning Petrous temporal bone osteomyelitis or fracture Retropharyngeal abscess Botulism Yellow star thistle toxicity Viral encephalitis Cerebral edema Cerebral or brainstem hemorrhage Intracranial masses (hematoma, neoplasia, and abscess) Meningitis Verminous encephalitis Equine protozoal myeloencephalitis Equine herpesvirus 1 Equine dysautonomia Hepatoencephalopathy Tetanus Polyneuritis equi |
Diagnosis of the cause of dysphagia is based on physical examination including a careful oral examination; neurologic examination; clinical signs; and endoscopy of the pharynx, esophagus, and guttural pouches. Radiology may be useful to assess the bony structures of the head and throat. Ultrasonography is valuable for examining the retropharyngeal space and esophagus to detect and evaluate masses. Pharyngeal or esophageal causes of dysphagia may be detected with routine endoscopic examination or with contrast radiography. Endoscopy may also be used to assess deglutition, but this function may be adversely affected by sedation of the horse. Deglutition may also be assessed using fluoroscopy1190 or manometry,1191 but these techniques require specialized equipment. Specific diagnostic procedures for nonalimentary causes of dysphagia are covered elsewhere in this text (see Chapter 7).
Specific treatments aimed at resolving the underlying disorder causing dysphagia are discussed in detail elsewhere. Most horses with dysphagia should not be fed roughage with long fiber length (hay or grass). Dietary modifications that promote swallowing, such as feeding slurries made from complete pelleted feeds, may be sufficient to manage some cases of partial dysphagia. Aspiration pneumonia is a potential complication in horses with pharyngeal or esophageal dysphagia. Foals may be managed by feeding mare’s milk or a suitable substitute via nasogastric tube. Pellet slurries or formulated liquid diets may be fed via nasogastric tube to older horses. Prolonged nutritional management of dysphagic horses may require extraoral feeding using a tube placed through an esophagostomy.1192
Formulated pelleted diets are often easy to administer through a tube as slurry and are balanced to meet the nutritional requirements for healthy horses. Sufficient quantities must be fed to deliver adequate calories (16–17 Mcal/day for a 500-kg horse). Adjustments may be necessary for horses that are cachectic or have extra metabolic demand (such as pregnancy). Adding corn oil to the ration (1 cup every 12 or 24 hours) is a common method of increasing fed calories. Equine-specific enteral formulations are also available (Well-Gel, Land O’Lakes Purina Feed LLC, Arden Hills, MN). Regardless of the method of nutritional management, salivary losses of electrolytes should be monitored and electrolyte replacement therapy provided as needed. Saliva contains high concentrations of Na, K, and Cl. A group of ponies with experimental esophagostomies1193 and a horse with esophageal squamous cell carcinoma1194 were fed a complete pelleted diet through esophagostomy tubes but developed metabolic acidosis, hyponatremia, and hypochloremia apparently because of salivary losses. Surprisingly, salivary losses of potassium did not result in hypokalemia in these cases, presumably because of replacement in the diet. Electrolyte replacement may be accomplished by adding NaCl and KCl to the diet. Horses can be maintained for months with frequent feedings through an esophagostomy tube.1194 Parenteral nutrition (total or partial) may be useful in the short term but is not often feasible for long-term management.
Dental Diseases
Eruption Disorders
Tooth eruption is a complex phenomenon involving the interplay of dental morphogenesis and those vascular forces responsible for creating the eruption pathway. These changes are responsible for osteitis and bone remodeling within the maxilla and mandible. Young horses frequently show symmetric bony swelling resulting from these eruption cysts. In some cases, additional clinical signs of nasal obstruction with respiratory stridor or nasal discharges may be apparent.
Pathologic problems associated with maleruption include a variety of dental diseases.1195 Oral trauma can displace or damage erupting teeth or the permanent tooth buds. As a result, teeth may be displaced and erupt in abnormal positions or may have abnormal shapes. Supernumerary teeth, incisors and molars, can develop, as well as palatal displacement of impacted teeth (maxillary P3-3, or third cheek tooth). In almost all of these conditions some form of surgical treatment is necessary, but depending on the number and location, conservative therapy can be successful.1196
Dental impaction is a major cause of dental disease in the horse. In a series of 142 extracted teeth, 63 were P3-3 or P4-4 (cheek tooth 2 or 3, respectively).1197 Early observations had indicated that the first molar (M1, or cheek tooth 4) was the most commonly diseased tooth, and an “open infundibulum” in this tooth has been suggested as the cause.1198 In a later study, the mandibular cheek teeth 2 and 3 were the most commonly affected, whereas cheek teeth 2 and 4 were most commonly affected in the maxillary arcade, which was the more commonly affected arcade.1199 Studies on cementogenesis of the maxillary cheek teeth have shown, however, that most maxillary cheek teeth have a greater or lesser degree of hypoplasia of cementum within the enamel lakes and that this “lesion” rarely expands into the pulp. The central infundibular hole is the site of its vascular supply to the unerupted cement lake. On those occasions in which caries of cementum occurs—that is, secondary inflammatory disease and acid necrosis of the cementum—apical osteitis may develop.
Dental Decay
Pulpitis is key to the pathogenesis of dental decay in the horse. The initiation of inflammatory pulp changes may be a sequela to dental impaction or dental caries or may result from fracture of a tooth. If the onset of the inflammatory process is slow, then formation of secondary dentin within the pulp chambers may protect the pulp and the tooth. Secondary dentin formation occurs from stimulation of odontoblasts within the pulp chamber. Such changes are the normal process of protection during dental wear and attrition as crown substances wear away and the reserve crown comes into wear. In acute disease, however, this defense mechanism is ineffective, and the changes that occur and that are sequelae to pulpitis reflect the location of each affected tooth. For example, pulpitis and apical osteitis of the third mandibular cheek tooth most commonly results in the development of a mandibular dental fistula. Pulpitis of the third maxillary cheek tooth, however, results in an inflammatory disease within the rostral maxillary sinus and in development of chronic maxillary sinus empyema.
Oblique radiographs greatly assist the diagnosis of dental decay by demonstrating sinus tract formation, sequestration of bone, mandibular osteitis, hyperplasia of cementum, and new bone formation (so-called alveolar periostitis).1200 Nuclear scintigraphy and CT can aid in an accurate diagosis.49, 53 The management of dental decay in the horse usually involves surgical extraction of the diseased tooth. In some horses, apicoectomy and retrograde endodontic techniques may be used to save the diseased tooth. Care must be taken, however, in selection of patients. In most cases of apical osteitis in the horse that result from dental impaction, immature root structures make achieving an apical seal of the exposed pulp difficult.
Periodontal Disease
Gingival hyperemia and inflammation occur during the eruption of the permanent teeth and are common causes of a sore mouth in young horses (particularly 3-year-olds as the first dental caps loosen). Such periodontal changes usually resolve as the permanent dental arcade is established. During normal mastication, the shearing forces generated by the occlusal contact of the cheek teeth essentially clean the teeth of plaque and effectively inhibit deposition of dental calculus. Wherever occlusal contact is ineffective, periodontal changes and calculus buildup occur; for example, the deposition of calculus on the canine teeth of mature geldings and stallions is common. Routine dental prophylaxis forms an important component of maintaining normal occlusal contact, and for this reason arcade irregularities that result in enamel point formation on the buccal edges of the maxillary cheek teeth and the lingual edges of the mandibular cheek teeth should be corrected. These edges may be smoothed annually in horses that are at grass and twice yearly in young horses, aged horses, and stabled horses. Horses at grass have a greater range of occlusal contact and therefore better periodontal hygiene than stabled horses. In stabled horses the range of occlusal contact is narrower, and the formation of enamel points occurs more frequently with subsequent buccal ulceration and the initiation of a cycle of altered occlusal contact and, hence, irregular arcade formation. This process leads to severe forms of periodontal disease and wave mouth formation.
Periodontal disease occurs with abnormal occlusal contact and initiation of the cycle of irregular wear and abnormal contact. Such changes progress to loss of alveolar bone, gross periodontal sepsis, and loss of tooth support. In this sense periodontal disease truly is the scourge of the equine mouth and results in tooth loss.1201
Congenital and Developmental Abnormalities
Cleft Palate
Palatine clefts may result from an inherited defect and are caused by failure of the transverse palatal folds to fuse in the oral cavity. Harelip accompanies few palatine clefts in the horse. The degree of palatine clefting depends on the stage at which interruption in the fusion of the palatopalatal folds occurs. Toxic or teratogenic effects are documented in other species, but few data are available in the horse.
Treatment for repair of uncomplicated palatine defects has been recommended, but prognosis is generally poor because of the considerable nursing care required and the high incidence of surgical failures. Early surgery and the use of mandibular symphysiotomy in affording surgical exposure should be emphasized. The combination of mandibular symphysiotomy and trans-hyoid pharyngotomy to approach the caudal margins of the soft palate affords surgical access, and mucosal flaps can be constructed to repair the defects. The incidence of breakdown in the surgical repair is high, and healing by first intention is the exception rather than the rule. A surgical report documented the successful closure of a median cleft of the lower lip and mandible in a donkey.1202
Campylorhinus Lateralis
Foals born with a severely deviated premaxilla and palate have a wry nose. One can achieve a good functional and cosmetic outcome with surgical correction.1203 Circumstantial evidence indicates that such a defect has a genetic cause, and the defect occurs most frequently in the Arabian breed.
Cysts
Subepiglottic cysts are congenital abnormalities resulting from cystic distortion of remnants of the thyroglossal duct, which may cause dyspnea and choking in foals. Surgical removal of these cysts results in normal function.
Brachygnathism
The most significant developmental defect of dental origin is a maxilla that is longer than the mandible (parrot mouth). An overbite of 2 cm in the incisor arcade may be present in a horse with a mismatch of less than 1 cm between the first upper and lower cheek teeth. Parrot mouth and monkey or sow mouth are thought to be inherited conditions. Some correction of minor incisor malocclusion occurs up to 5 years of age. Recognition and detection of parrot mouth are important in the examination of potential breeding stock. Surgical attempts to inhibit overgrowth of the premaxilla by wiring or by the application of dental bite plate procedures have been documented.1204
Oral Wounds
Wounds of the lips, incisive bone, and the mandibular incisor area occur commonly in the horse and usually result from the horse getting the lips, jaw, or teeth caught in feeding buckets, in fence posts, or in halters or having a segment of tongue encircled with hair in tail chewing. As the horse panics and pulls away from its oral entrapment, considerable trauma can occur to the lips, teeth, and gums. Most wounds repair satisfactorily, provided they are identified early after the injury and basic principles of wound hygiene, excision of necrotic tissue, and wound closure are observed. Oral mucosal defects should be closed and effective oral seals made before external wounds are closed. In some horses, offering specially constructed diets or feeding the horse by nasogastric tube or esophagostomy during the healing processes may be necessary.
Stomatitis and Glossitis
Foreign body penetration of the tongue, cheek, or palate has been reported in grazing and browsing horses and in particular in horses that have certain hay sources that contain desiccated barley awns or yellow bristle grass.1205 Other plant material and grass awns occasionally may penetrate the tongue, gingiva, or cheek, causing inflammation or abscesses. Metallic foreign bodies have been reported in the tongue, and a history of feeding hay or the use of cable-framed tractor tires was often reported as part of the history.1206 Ulcerative stomatitis may occur as the result of phenylbutazone toxicity.1207 Vesicular stomatitis is a highly contagious viral blistering disease of horses and other animals. Treatment of glossitis and stomatitis primarily aims at removing the inciting cause. A. lignieresii, the causative agent of actinobacillosis, has been isolated and identified from ulcers on the free border of the soft palate and oral and laryngeal granulomata. The bacterium also was reported in a sublingual caruncle in a horse with a greatly swollen tongue.1208 Therapy with 150 mL of 20% sodium iodide and 5 g of ampicillin every 8 to 12 hours effected a clinical cure.
Salivary Glands
Saliva is important for lubricating and softening food material. The horse has paired parotid, mandibular, and polystomatic sublingual salivary glands. The parotid gland is the largest of the salivary glands in the horse and is situated in the space between the ramus of the mandible and the wing of the atlas. The parotid duct is formed at the ventral part of the gland near the facial crest by the union of three or four smaller ducts. The duct leaves the gland above the linguofacial vein, crosses the tendon of the sternocephalicus muscle, and enters the mouth obliquely in the cheek opposite the third upper cheek tooth. The parotic duct orifice is small, but some dilation of the duct and a circular mucous fold (the parotid papillae) exist at this point. The mandibular gland is smaller than the parotid gland and extends from the atlantal fossa to the basihyoid bone. For the most part, the mandibular gland is covered by the parotid gland and by the lower jaw. The mandibular duct is formed by the union of a number of small duct radicles that emerge along the concave edge of the gland and run rostral to the border of the mouth opposite the canine tooth.
The orifice is at the end of a sublingual caruncle. The mandibular gland possesses serous, mucous, and mixed alveolar glandular components. The parotid gland is a compound alveolar serous gland. The parotid salivary gland can secrete saliva to yield rates of 50 mL/min, and a total daily parotid secretion can be as much as 12 L in a 500-kg horse. Parotid secretion only occurs during mastication, and administration of atropine or anesthesia of the oral mucosa can block secretion. Parotid saliva is hypotonic compared with plasma, but at high rates of flow, concentrations of sodium, chloride, and bicarbonate ions increase.
Parotid saliva of the horse has a high concentration of calcium, and occasionally calculi (sialoliths) form within the duct radicles of the parotid salivary gland.1209 Congenital parotid duct atresia, acquired stricture from trauma to the duct, or obstruction by plant material (sticks or foxtails and other seeds) also may occur. The clinical signs of sialolithiasis or other forms of ductule obstruction include a fluid swelling in the form of a mucocele proximal to the stone and occasionally inflammation of the parotid gland. Ultrasonography is useful to diagnose salivary mucoceles and to detect foreign bodies or sialoliths. Measurement of electrolyte concentrations in aspirates from suspected mucoceles might be helpful to distinguish them from hematomas. Salivary potassium and calcium concentrations are higher than plasma. Treatment may require surgical removal of the stone or plant material in the case of sialolithiasis or foreign body obstructions. Other causes of obstruction may require resection of the affected portion of the duct or chemical ablation of the gland.1210
Primary sialadenitis is unusual but can occur in one or both glands. The condition is painful and may be associated with a fever and anorexia. Secondary sialadenitis is more common and usually is associated with trauma. Infectious sialadenitis from C. pseudotuberculosis 1211 or other bacterial pathogens also may occur. Diagnosis is by physical examination and by finding an enlarged edematous parotid gland tissue on ultrasonographic examination. Culture and cytologic examination of aspirates may be useful for diagnostic purposes. Treatment in usually palliative, consisting of NSAIDs. Appropriate antibiotic therapy is indicated as directed by culture and sensitivity results.
Chemical irritation, glossitis, stomatitis, or other causes of prepharyngeal dysphagia cause ptyalism or excessive salivation in horses. Specific therapy for the ptyalism usually is not required as long as salivary losses are not excessive, resulting in dehydration and electrolyte imbalances. Ingestion of the fungal toxin slaframine also causes hypersalivation in horses.1212 The fungus Rhizoctonia leguminicola, which produces slaframine, causes black patch disease in red clover. Slaframine is a parasympathomimetic compound that stimulates exocrine secretion in the parotid gland. Slaframine toxicosis most commonly occurs in the spring or early summer and rarely requires treatment other than removal from the pasture. Mowing removes the source in most cases because regrowth in pastures often has less fungal contamination.1213
Esophagus
The esophagus is a musculomembranous tube that originates from the pharynx dorsal to the larynx and terminates at the cardia of the stomach.1214 In adult Thoroughbred horses the esophagus is approximately 120 cm long. The cervical portion is approximately 70 cm long; the thoracic portion, approximately 50 cm long; and the short abdominal portion, only approximately 2 cm long. The cervical esophagus generally lies dorsal and to the left of the trachea in the cervical region. In the thorax the esophagus courses through the mediastinum lying dorsal to the trachea and crosses to the right of the aortic arch dorsal to the heart base.
The esophagus has no digestive or absorptive functions and serves as a conduit to the stomach for food, water, and salivary secretions. The esophageal mucosa is a keratinized stratified squamous epithelium.1214 The submucosa contains elastic fibers that contribute to the longitudinal folds of the esophagus and confer elasticity to the esophageal wall. A transition occurs in the muscle type composing the tunica muscularis from striated skeletal muscle in the proximal two thirds of the esophagus to smooth muscle in the distal third. In the proximal esophagus the skeletal muscle layers spiral across one another at angles. Within the smooth muscle layers of the distal esophagus the outer layer becomes more longitudinal, whereas the inner layer thickens and becomes circular. The wall of the terminal esophagus can be 1 to 2 cm thick. Deep cervical fascia, pleura, and peritoneum contribute to the thin fibrous tunica adventitia of the esophagus. Motor innervation to the striated skeletal muscle of the esophagus includes the pharyngeal and esophageal branches of the vagus nerve, which originate in the nucleus ambiguus of the medulla oblongata. Parasympathetic fibers of the vagus nerve supply the smooth muscle of the distal esophagus. Sympathetic innervation of the esophagus is minimal.
Passage of ingesta through the esophagus can be considered part of the swallowing process, which consists of oral, pharyngeal, and esophageal stages. The oral stage is voluntary and involves transport of the food bolus from the mouth into the oropharynx. During the involuntary pharyngeal stage the food bolus is forced through the momentarily relaxed upper esophageal sphincter by simultaneous contractions of the pharyngeal muscles. In the esophageal phase of swallowing the upper esophageal sphincter closes immediately, the lower esophageal sphincter opens, and esophageal peristalsis propels the bolus into the stomach.1215 Unlike a food bolus, liquids do not require peristalsis to reach the lower esophageal sphincter and may precede the food bolus during swallowing.
The upper esophageal sphincter prevents esophagopharyngeal reflux during swallowing and air distention of the esophagus during inspiration. Upper esophageal pressure increases in response to pressure from a food bolus and to increased intraluminal acidity, as would occur with gastroesophageal reflux. The lower esophageal sphincter is a smooth muscle located at the gastroesophageal junction that is morphologically ill defined but forms an effective functional barrier.1215 Normally the lower esophageal sphincter is closed in response to gastric distention to restrict gastroesophageal reflux. Relaxation of the lower esophageal sphincter permits passage of ingested material from the esophagus to the stomach. Distention of the stomach with ingesta mechanically constricts the lower esophageal sphincter. Gastric distention also triggers a vagal reflex that increases lower esophageal sphincter tone, which is a safety mechanism against gastroesophageal reflux. The mechanical and vagal mechanisms that promote lower esophageal sphincter tone prevent spontaneous decompression of the stomach, which along with a lack of a vomiting reflex in the horse, increases the risk of gastric rupture during episodes of severe distention.
A wide variety of congenital and acquired disorders of the esophagus have been described in horses. These are summarized in Table 12.9 and discussed in detail in the following section.
TABLE 12.9.
Esophageal Disorders of Horses
| Disorder | Presenting Complaints | Diagnosis | Treatment | Selected References |
|---|---|---|---|---|
| Acquired Disorders | ||||
| Choke | Nasal discharge of saliva and food; retching; excessive salivation; cough; sweating; extension of head and neck | Passage of a nasogastric tube; endoscopy | Medical and surgical treatment options as described in text | 1187, 1188, 1189 |
| Foreign bodies | Acute or recurrent choke | Endoscopy; radiography | Manual retrieval or removal; endoscopic removal; surgery | 1190, 1191, 1192, 1193, 1194 |
| External compression | Acute or recurrent choke | Endoscopy; radiography; ultrasound | Removal of the obstructive mass | 1195 |
| Muscular hypertrophy | No clinical signs observed in most affected horses; may predispose to esophageal diverticula | Incidental finding at necropsy | None | 1196 |
| Gastroesophageal reflux disease | Inappetence; bruxism; ptyalism; colic; gastric reflux; weight loss; exercise intolerance | Esophageal and gastric endoscopy | Correct primary problem; decrease gastric acidity; gastric protectants; surgery | 1197, 1198, 1199 |
| Stricture | Recurrent choke; weight loss | Endoscopy; contrast radiography | Bougienage; surgery | 49, 53, 1200, 1201, 1202, 1203, 1204, 1205, 1206, 1207 |
| Diverticulaa | Recurrent choke; weight loss | Endoscopy; contrast radiography | Surgery | 1206, 1208, 1209, 1210, 1211 |
| Perforation, trauma | Salivation; bruxism; cough; nasal discharge; sepsis | Endoscopy | Enteral feeding; supportive care | 1212, 1213 |
| Megaesophagusa | Recurrent choke; intermittent food and saliva from nares; pneumonia; weight loss; colic | Endoscopy; contrast radiography | Nutritional modification as described in text | 1214, 1215 |
| Neoplasia | Recurrent choke; weight loss | Endoscopy; biopsy | Surgical resection | 1216, 1217 |
| Granulation tissue | Recurrent choke; swelling in the region of the cervical esophagus | Endoscopy; biopsy | Laser surgical resection | 1218 |
| Congenital Disorders | ||||
| Tubular duplication of the esophagus | Young horse; mass caudal to mandible; dyspnea; dysphagia; nasal regurgitation of food and saliva | Radiography and ultrasonography of mass; endoscopy; contrast radiography | Surgical excision | 1219, 1220 |
| Cystic duplication of the esophagus | Young horse; mass in cervical or throatlatch area; recurrent choke; bruxism; excessive salivation; nasal regurgitation of saliva and feed; weight loss | Endoscopy; ultrasonography; aspiration of cyst; contrast radiography | Surgical excision; marsupialization of cyst | 1224, 1225, 1226, 1227, 1228, 1229, 1230, 1231 |
| Vascular ring anomaly | Cervical swelling after introduction to solid feed; chronic respiratory disease | Endoscopy; contrast radiography; computed tomography; magnetic resonance imaging | Surgical correction | 1224, 1225, 1226, 1227, 1228, 1229, 1230, 1231 |
| Congenital stenosis | Nasal regurgitation of milk; cough | Endoscopy; contrast radiography | Dietary management as described in text | 36 |
| Ectasia | Nasal regurgitation of milk | Histologic evaluation | None described | 1232, 1233 |
May occur as congenital or acquired lesions.
Esophageal Obstruction
Esophageal obstruction has many causes (Table 12.10 ) and most often is manifested clinically by impaction of food material and resulting esophageal dysphagia. Esophageal obstruction may be caused by primary impactions (simple choke) of roughage, particularly leafy alfalfa hay; coarse grass hay; bedding; and even grass.1216 Prior esophageal trauma or poor mastication caused by dental abnormalities may predispose horses to primary esophageal impaction.1217 “Wolfing” or gulping food may precipitate primary impactions, particularly if the horse is exhausted or mildly dehydrated after a long ride or is weakened from chronic debilitation. Impactions also may result from disorders that physically impede the passage of food material and fluid by narrowing the luminal diameter, reduce the compliance of the esophageal wall, or alter the conformation of the esophageal wall such that food material accumulates in a pocket or diverticulum. Foreign bodies, intramural or extramural masses, or acquired or congenital anomalies cause these so-called secondary impactions. Intramural causes of esophageal obstruction include tumors (squamous cell carcinoma [SCC]), strictures, diverticula, and cysts.1218, 1219, 1220, 1221, 1222, 1223, 1224, 1225, 1226 Mediastinal or cervical masses (tumors or abscesses) may cause extramural obstructions. Congenital anomalies are covered in detail later.
TABLE 12.10.
Causes of Complete or Partial Esophageal Obstruction in the Horse
| Category | Differential | Examples |
|---|---|---|
| Intraluminal | Foreign body | Apples, potatoes |
| Feed material | ||
| Extramural | Neoplasia | Squamous cell carcinoma, lymphoma |
| Vascular ring anomaly | Persistent right aortic arch | |
| Granuloma | ||
| Intramural | Esophageal abscess | |
| Granuloma | ||
| Neoplasia | Squamous cell carcinoma, leiomyosarcoma | |
| Cysts | Intramural cysts, duplication cysts | |
| Diverticula | ||
| Stenosis | ||
| Functional disorders | Dehydration | |
| Exhaustion | ||
| Pharmacologic | Acepromazine, detomidine | |
| Primary megaesophagus | Congenital ectasia | |
| Esophagitis | ||
| Autonomic dysautonomia | ||
| Vagal neuropathies |
Clinical Signs and Diagnosis
A thorough physical examination, including complete oral and neurologic examination, can help rule out causes of dysphagia and nasal discharge other than esophageal obstruction. Clinical signs associated with esophageal obstructions are related primarily to regurgitation of food, water, and saliva caused by esophageal (postpharyngeal) dysphagia.1227 Horses with esophageal obstruction are often anxious and stand with their neck extended. Gagging or retching may be observed, particularly with acute proximal obstructions. Bilateral frothy nasal discharge containing saliva, water, and food material; coughing; odynophagia; and ptyalism are characteristic clinical signs, the severity of which varies with the degree and location of the obstruction. Distention in the jugular furrow may be evident at the site of obstruction. Other clinical signs related to regurgitation of saliva, water, and food material, such as dehydration, electrolyte, or acid-base imbalances; weight loss; and aspiration pneumonia may be observed. In extreme cases, pressure necrosis from the impaction or trauma to the esophagus may cause esophageal rupture. If the rupture is in the cervical esophagus, crepitus or cellulitis may be evident along with signs of systemic inflammation. Thoracic auscultation is important to determine whether aspiration pneumonia is present. Intrathoracic esophageal rupture may result in pleuritis and its associated clinical signs.
Passage of a nasogastric tube is an effective way to detect and localize an obstruction but provides little information about the nature of the obstruction or the condition of the esophagus. The most direct method for diagnosis of esophageal obstructions is endoscopic examination. Esophageal obstruction occurs most commonly at sites of natural narrowing of the esophageal lumen, such as the cervical esophagus, the thoracic inlet, base of the heart, or the terminal esophagus; an endoscope greater than 1 m in length is necessary for complete evaluation. Endoscopic evaluation is useful before relief of an impaction to localize the obstruction and to investigate the nature of the impaction if one suspects a foreign body. Foreign bodies may be retrievable via transendoscopic tethering.1228 Critical diagnostic and prognostic information is also obtained via endoscopy after resolution of the impaction. Assessing the affected esophagus for mucosal ulceration, rupture, masses, strictures, diverticula, and signs of functional abnormalities is important (Fig. 12.15, Fig. 12.16 ).
Fig. 12.15.
Endoscopic view of the cervical esophagus in an adult horse 6 months after an episode of choke that caused circumferential ulceration of the esophageal mucosa. The area of luminal narrowing (stricture) is at the upper right of the image, and the proximal dilation forms an outpouching of the esophageal wall. A contrast esophagram revealed that the outpouching was a pulsion diverticulum.
Fig. 12.16.
Endoscopic view of the proximal esophagus in a yearling filly with recurrent esophageal obstruction. Circumferential mucosal ulceration is evident proximal to incomplete stricture formation.
Ultrasonography of the cervical region is useful not only to confirm a cervical esophageal impaction but also to provide critical information about the location and extent of the impaction and esophageal wall thickness and integrity. Ultrasonography may provide information about the cause.1194 Radiographic assessment of the esophagus can confirm the presence of esophageal obstruction in cases in which one cannot view the affected area adequately using endoscopy. Impacted food material in the esophagus may be visualized as a typical granular pattern with gas accumulation proximal to the obstruction. Air or barium contrast radiographic studies are most useful for evaluating the esophagus following relief of the impaction if one suspects a stricture. Esophageal dilation, diverticula, rupture, functional disorder (megaesophagus), or luminal narrowing caused by extraluminal compression are detected more easily using contrast radiographic studies than endoscopy (Fig. 12.17 ).1229, 1230, 1231 Radiographic studies of the esophagus should be interpreted cautiously in sedated horses, particularly after passage of a nasogastric tube or other esophageal manipulations that may contribute to esophageal dilation.36
Fig. 12.17.
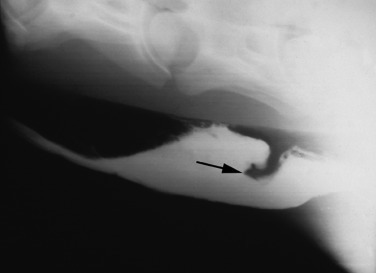
Contrast esophagram in a horse with circumferential esophageal stricture (arrow) and a pulsion diverticulum proximal to the stricture.
Treatment
The primary goal of treatment for esophageal impaction is to relieve the obstruction. A variety of approaches have been described, ranging from minimal conservative therapy to aggressive intervention to relieving the obstruction. Parenteral administration of acepromazine (0.05 mg/kg IV), xylazine (0.25–0.5 mg/kg IV) or detomidine (0.01–0.02 mg/kg IV), oxytocin (0.11–0.22 IU/kg intramuscularly), and/or esophageal instillation of lidocaine (30–60 mL of 1% lidocaine) may reduce esophageal spasms caused by pain or may decrease esophageal tone.36, 1232, 1233 N-Butylscopolammonium bromide may reduce smooth muscle tone and has been used to help resolve some obstructions.
In many horses with esophageal obstruction secondary to a feed impaction, the problem may be resolved with conservative management. To facilitate examination, relieve anxiety, and relax the esophagus, one should begin by sedating the horse. A nasogastric tube is then passed to confirm the diagnosis. When an obstruction is encountered, gentle pressure is applied in an attempt to dislodge and move distally the offending feed material. If the choke is not easily dislodged with gentle pressure, the tube is removed. Appropriate sedative, antiinflammatory/analgesic, and smooth muscle–relaxing drugs are administered. The horse is moved to an unbedded stall with absolutely no food or water within reach and muzzled if necessary. The horse is left alone in the stall for several hours. If there is evidence of dehydration, appropriate IV fluid therapy is provided. If there is evidence of aspiration pneumonia, appropriate IV antimicrobial therapy is provided. On reexamination, a stomach tube is passed and gentle pressure is again applied to the area of obstruction. In many horses, the impaction will have softened and can be easily dislodged, if it has not resolved already, with minimal pressure from the stomach tube.
Some clinicians prefer a more interventional approach to the resolution of an impaction, and some impactions are severe enough to require physical dispersal of the material.1232 A nasogastric tube can be used to displace the impacted material along with external massage if the obstruction is in the cervical region. Careful lavage of the esophagus with water via an uncuffed or a cuffed nasogastric tube while the head is lowered may be a useful aid in breaking up the impaction. Some clinicians advocate a dual tube method in which a tube is placed through each nasal passage into the esophagus for ingress and egress of the lavage fluid. Because of the risk of aspiration of water and food material, esophageal lavage sometimes is done under general anesthesia with a cuffed nasotracheal tube. In refractory cases, IV administration of polyionic fluids provides hydration and alleviates any electrolyte or acid-base imbalances resulting from salivary losses of chloride, sodium, and potassium.1193 Oxytocin may or may not provide a direct effect for the resolution of an esophageal obstruction. Oxytocin reduced the area under the curve for esophageal smooth muscle strip contractions but had no effect on skeletal muscle strips in vitro.1234 In one in vivo report, oxytocin administration resulted in decreased esophageal tone in the proximal esophagus (aborad to the larynx and thoracic inlet).1233 In a separate study, oxytocin administration did not affect esophageal manometric recordings.1235 Rarely, esophageal obstruction ultimately may require esophagotomy to relieve the impaction. Strict restriction of food and water, including access to bedding material, must be enforced until the obstruction is resolved and the esophagus has regained function. Surgical removal of esophageal foreign bodies can be considered if the size and/or orientation of the object is such that transendoscopic retrieval is considered unlikely to be successful.1226, 1236 In one report, an intraluminal mass comprised of exuberant granulation tissue was removed via serial transendoscopic Nd:YAG laser ablation.1237
Systemic effects of dysphagia associated with esophageal impaction include dehydration, hyponatremia, hypochloremia, and metabolic alkalosis from prolonged loss of salivary free water and electrolytes.1193
Esophageal endoscopy should be performed after relief of the impaction to determine whether any complications of the impaction have developed or if a primary cause of the obstruction is present. Endoscopic examination is critical to determine the postobstruction treatment plan and for follow-up evaluation of esophageal healing. The horse should be reevaluated every 2 to 4 weeks following resolution of the impaction if esophageal dilation or mucosal injury is observed. In one report, male sex, age >15 years, and need for general anesthesia to relieve the obstruction were associated with an increased risk of complications.1238
Dilation proximal to the site of obstruction, mucosal injury from trauma, stricture formation, formation of a diverticulum, megaesophagus, and esophagitis are sequelae to esophageal obstruction that predispose patients to reobstruction. Underlying functional or morphologic abnormalities were much more likely in cases of recurrent, relative to first time, obstruction in a retrospective study.1216
The rate of reobstruction may be as high as 37%. Depending on the duration of the obstruction and the degree of trauma or dilation, the risk of reobstruction is high for 24 to 48 hours or longer. Food should be withheld for at least 24 to 48 hours after resolution of the obstruction. Sucralfate (20 mg/kg orally every 6 hours) may hasten healing if esophageal ulceration is evident, but the efficacy of sucralfate for this purpose is not established. Some clinicians suggest that administration of an NSAID may reduce the development of strictures, although judicious use of NSAIDs is recommended to prevent worsening of esophageal mucosal injury. Orally administered NSAIDs should be avoided if esophagitis is present. After 48 to 72 hours or when the esophageal mucosa has recovered as assessed by endoscopy, the horse may be fed soft food (moistened pellets and bran mashes) then transitioned gradually to a high-quality roughage diet over 7 to 21 days, depending on the degree of esophageal damage induced by the impaction and the nature of any underlying disease. The prognosis for survival is good (78–88%), but some horses may require permanent dietary modification if persistent chronic obstruction is a problem.1216, 1220, 1238
Aspiration pneumonia is a potential complication in every case of esophageal obstruction, and perforation is possible with severe or prolonged obstruction. In one report, duration of obstruction before presentation was a good predictor of pneumonia, whereas endoscopic evidence of tracheal food contamination was not.1216 Administration of broad-spectrum antibiotics that are effective against gram-positive and gram-negative organisms, including metronidazole for anaerobes, is highly recommended if the duration of obstruction is either unknown or prolonged or if aspiration is suspected.
Esophagitis
Esophagitis refers to a clinical syndrome of esophageal inflammation that may or may not be ulcerative. The major protective mechanisms of the esophageal mucosa include salivary and food material buffers, normal peristaltic motility, and the barrier formed by the gastroesophageal sphincter. Reflux esophagitis is caused by repeated episodes of gastric fluid regurgitation into the distal esophagus and subsequent chemical injury to the mucosa.1239 Esophageal mucosal ulceration also can occur if the clearance of gastric fluid from the esophagus is delayed, such as in functional disorders of the esophagus. Like ulceration of the squamous portion of the stomach in horses, gastric acid and bile salt chemical injury is a major mechanism of esophageal squamous epithelial ulceration.838, 1239 Reflux esophagitis may occur along with gastric ulcer disease, motility disorders, increased gastric volume from gastric outflow obstructions, gastric paresis, intestinal ileus, or impaired lower esophageal sphincter function.1222, 1239 Other causes of esophagitis in horses include trauma (foreign bodies, food impactions, and nasogastric tubes), infection (mural abscesses), or chemical injury (pharmaceuticals and cantharidin; Fig. 12.18 ).1236, 1240, 1241
Fig. 12.18.
Endoscopic view of the cervical esophagus of a horse that had repeated passages of a stiff nasogastric tube. The deep, linear ulceration of the esophageal mucosa is notable.
Clinical Signs and Diagnosis
The clinical signs of esophagitis are nonspecific and similar to esophageal obstruction and gastric ulceration. Gagging or discomfort when swallowing may be evident, along with hypersalivation and bruxism. Esophageal (postpharyngeal) dysphagia may be evident. Partial or complete anorexia with resultant weight loss may be observed in affected horses. Esophageal hypomotility dysfunction caused by the inflammatory process may result in esophageal impaction. Clinical signs of underlying diseases that predispose to esophagitis may predominate or mask the signs of esophagitis. Horses with gastrointestinal motility disorders such as proximal enteritis or gastric outflow obstruction are at a high risk of developing reflux esophagitis because of the presence of gastric acid and bile salts in the fluid reflux. Foals with obstructive gastroduodenal ulcer disease (GDUD) commonly have reflux esophagitis.
Diagnosis requires endoscopic examination, with diffuse, patchy, linear, or coalescing erosion or ulcerations or significant edema or hyperemia. Determining whether an underlying disease, such as infection, neoplasia, esophageal strictures, or diverticula, is present is important. Evaluation of the stomach is important to rule out gastric outflow obstruction. Contrast radiography may be helpful to assess esophageal motility and transit time.1230
Treatment
The principles of therapy for reflux esophagitis include control of gastric acidity, mucosal protection, and correction of any underlying disorder contributing to gastroesophageal reflux. Reduction of gastric acid production with proton pump antagonists or H2 receptor antagonists is critical for resolution. Some clinicians advocate using sucralfate to promote healing of ulcerated esophageal mucosa. The ability of sucralfate to bind ulcerated esophageal mucosa or hasten ulcer healing, however, is not proven in the horse.
If a primary gastric outflow obstruction is present, surgical intervention is warranted. Horses with delayed gastric outflow without obstruction may benefit from prokinetics such as metoclopramide (0.02–0.1 mg/kg SC every 4–12 hours) or bethanechol (0.025–0.035 mg/kg SC every 4–24 hours). For esophagitis from trauma or pressure injury after esophageal impaction, judicious use of NSAIDs may be warranted to reduce inflammation and pain.
Dietary modification may be necessary for patients with esophagitis, depending on the degree of ulceration or if motility is impaired. Horses with mild esophagitis should be fed frequent small meals of moistened pellets and fresh grass. Severe esophagitis may necessitate withholding food and complete esophageal rest for several days. Although the prognosis for esophagitis is good in the absence of underlying disease, the risk of stricture formation is high if severe circumferential or coalescing ulcerations are present. Esophagitis from severe trauma or infection may be prone to stricture formation.
Motility Disorders
Motility dysfunction of the equine esophagus is most often manifest as hypomotility resulting in esophageal dilation (ectasia) or megaesophagus. Although megaesophagus in horses most commonly is acquired, reports indicate idiopathic megaesophagus in young horses may be congenital.1191, 1242, 1243, 1244, 1245 Acquired megaesophagus in horses may be a consequence of chronic or recurrent esophageal obstruction.1220, 1222 Esophageal impactions of a short duration cause a proximal dilation of the esophagus that is generally reversible.1230 If the duration of the obstruction is long enough, the motility of the esophagus proximal to the site of obstruction may be impaired permanently. Other causes of acquired megaesophagus include extraesophageal obstruction by tumors or abscesses, pleuropneumonia, and vascular ring anomalies.1220, 1223 A retrospective report of horses with megaesophagus reveals an overrepresentation of Friesian horses (14 of 18 cases) suggesting the possibility of a predisposition for esophageal disorders in this breed.1214 The authors state that preliminary pedigree analyses suggest the possibility of a recessive pattern of inheritance.1214
Megaesophagus also may result from neurologic, neuromuscular, and muscular disorders. Neurologic diseases that cause vagal neuropathy, such as equine protozoal myeloencephalitis, equine herpesvirus myeloencephalitis, and idiopathic vagal neuropathy, have been associated with megaesophagus in horses. Pleuropneumonia may be associated with a vagal neuropathy resulting in megaesophagus. Megaesophagus is an early sign of equine dysautonomia1246 and may be observable in patients with botulism. Myasthenia gravis is a well-known cause of megaesophagus in nonequine species but has not been reported in horses. Also in other species, electrolyte disorders, cachexia, primary myopathies, myositis, and Addison’s disease may affect esophageal motility but have not been associated with megaesophagus in horses. Iatrogenic megaesophagus can be induced by the α2-adrenergic agonist detomidine but is transient and reversible.36, 1247 Acepromazine, detomidine, and a combination of xylazine and butorphanol can alter proximal esophageal motility by disrupting coordinated peristalsis and decreasing spontaneous swallowing, but only acepromazine affected the manometric profile of the distal esophagus.1235 None of these drugs altered contractility of isolated esophageal smooth or striated muscle strips in vitro.1234 Nonetheless, the use of these drugs may complicate clinical evaluation of esophageal motility. Esophageal disorders including megaesophagus, esophageal diverticulum, and esophageal rupture appear to be more common in young Friesian horses, suggesting the possibility of a genetic predisposition in this breed.
Esophageal inflammation, particularly reflux esophagitis, may affect motility and cause megaesophagus. Because esophageal hypomotility affects the tone and function of the lower esophageal sphincter, reflux esophagitis also may be a complication of a primary functional disorder. Assessing esophageal motility in horses with esophagitis that is not responding appropriately to treatment is important.
Clinical Signs and Diagnosis
Thorough neurologic examination is important to help rule out primary neurologic causes of megaesophagus. Because esophageal hypomotility is a functional obstruction, the clinical signs are similar to esophageal obstruction, although the onset is typically insidious. The clinical signs include those associated with esophageal dysphagia.1191, 1222, 1223, 1243, 1244, 1245 The cervical esophagus may be dilated enough to be evident externally, and weight loss is common.
Diagnosis of esophageal hypomotility requires evaluation of transit via fluoroscopy or contrast radiography.1230, 1246 Endoscopy may reveal a dilated esophagus and an absence of peristaltic waves1191, 1222 or evidence of underlying disease causing obstruction or esophageal dilation.1220, 1222 Esophageal manometry may be useful to document abnormal postdeglutition contraction pressures, contraction time, and propagation times but is not often available for routine clinical application.1248 If neurologic or neuromuscular disease is suspected, cerebrospinal fluid analysis or electromyography may be indicated.
Treatment
Treatment of an underlying cause should be initiated, if applicable. Dietary modification should include a soft/slurry diet and feeding from an elevated position to promote transit by gravity flow. Metoclopramide or bethanechol may benefit patients with reflux esophagitis associated with megaesophagus. The prognosis depends on the underlying cause and the degree of dilation. Although many cases of megaesophagus associated with reflux esophagitis respond well to treatment, many other forms of megaesophagus including congenital megaesophagus have a poor prognosis.
Esophageal Stricture
Strictures most commonly are caused by pressure necrosis from esophageal impactions that induce circumferential erosion or ulceration of the esophageal mucosa, although esophageal injury caused by oral administration of corrosive medicinal agents and trauma to the neck may also result in stricture formation.1249 Congenital strictures also have been reported.1250 Strictures caused by mucosal and submucosal trauma are termed esophageal webs or rings. Strictures may also originate in the muscular layers and adventitia of the esophagus (mural strictures) or in all of the layers of the esophagus (annular stenosis).1217, 1250 Horses with these lesions have a presentation similar to those with simple obstructions, because strictures result in partial obstruction and impaction of food material in the lumen. Esophageal webs or rings may be detected with endoscopy (see Fig. 12.15, Fig. 12.16), whereas identification of mural strictures or annular stenosis may require a double-contrast esophagram (see Fig. 12.17). In a retrospective study of horses with esophageal stricture following simple obstruction, maximal reduction in esophageal lumen diameter occurred within 30 days of the esophageal obstruction. Although surgery has been used to relieve such strictures, initial medical management is warranted because strictures may resolve with conservative therapy, and the esophagus continues to remodel for up to 60 days following ulceration. In one report, seven horses with esophageal obstruction–induced stricture were treated conservatively by feeding a slurry diet and administering antiinflammatory and antimicrobial medications, and five of seven were clinically normal within 60 days.1249 One of the five successfully treated horses had a 10-cm area of circumferential ulceration, suggesting that the potential exists for extensive mucosal injury to resolve without permanent stricture formation.
If resolution of strictures within 60 days is insufficient, other methods to increase esophageal diameter should be investigated. Balloon dilation has been used successfully in several reports.1251, 1252, 1253, 1254, 1255, 1256 Commercial balloon dilators have been adapted for use in the horse, although it is important to note that repeated dilation is necessary.1251, 1252, 1257 Corticosteroid injection was used successfully in conjunction with this procedure in foals.1251 Alternatively, a number of surgical techniques have been used to resolve strictures, including resection and anastomosis,1258, 1259 temporary esophagostomy with fenestration of the stricture,1217 esophagomyotomy for strictures of the muscularis and adventitia,1260, 1261 or patch grafting with local musculature.1262 Such surgeries, however, are fraught with complications, largely because of the propensity of the traumatized esophagus to restricture.1220, 1249 The esophagus lacks a serosal layer and does not rapidly form a fibrin seal as does the remainder of the intestinal tract, so anastomoses tend to leak.1259 In addition, tension on the esophagus during swallowing and movement of the neck impairs healing of anastomoses.1217, 1258 In spite of these difficulties, the long-term prognosis for horses with chronic esophageal strictures treated surgically is better than for those treated nonsurgically.1220 Two recent reviews describe surgical approaches to the esophagus in detail.1263, 1264
Esophageal Diverticula
Esophageal diverticula may be classified as traction (true) diverticula and pulsion (false) diverticula. Traction diverticula result from wounding and subsequent contraction of periesophageal tissues, with resultant tenting of the wall of the esophagus. Pulsion diverticula arise from protrusion of esophageal mucosa through defects in the muscular wall of the esophagus and usually result from trauma or acute changes in intraluminal pressure.1250 Traction diverticula appear as a dilation with a broad neck on contrast esophagography, whereas pulsion diverticula typically have a flask shape with a small neck on an esophagram (see Fig. 12.17).1225, 1265 Although traction diverticula are usually asymptomatic and of little clinical significance, pulsion diverticula may fill with feed material, ultimately leading to esophageal obstruction and rupture.1265, 1266, 1267
A movable mass in the midcervical region may be noticeable before onset of complete obstruction.1250 Pulsion diverticula may be corrected surgically by inverting or resecting prolapsed mucosa and closing the defect in the wall of the esophagus.1225, 1265, 1266 Inversion of excessive mucosa may reduce the diameter of the esophageal lumen and predispose horses to esophageal obstruction; therefore, it should be reserved for small diverticula.1225
Congenital Disorders
Congenital disorders of the esophagus are rare. Reported congenital abnormalities include congenital stenosis,1268 persistent right aortic arch,1223, 1269, 1270, 1271, 1272, 1273 other vascular anomalies,1274 esophageal duplication cysts,1275, 1276, 1277 intramural inclusion cysts,1224, 1278 and idiopathic megaesophagus.1243, 1244, 1245 In the one report of congenital stenosis, double-contrast radiography revealed concentric narrowing of the thoracic esophagus in the absence of any vascular abnormalities at the base of the heart. Successful treatment included having the foal stand with the forelimbs elevated off the ground following each feeding.1268
Persistent right aortic arch is a congenital anomaly in which the right fourth aortic arch becomes the definitive aorta instead of the left aortic arch, which results in constriction of the esophagus by the ligamentum arteriosum as it extends between the anomalous right aorta and the left pulmonary artery.1279 Clinical signs may include those associated with esophageal (postpharyngeal) dysphagia, drooling, and distention of the cervical esophagus resulting from partial obstruction of the thoracic esophagus.1223, 1269 Endoscopic examination typically reveals dilation of the esophagus cranial to the obstruction with evidence of diffuse esophagitis. Surgical treatment of persistent right aortic arch has been reported in foals, with varying degrees of success.1269, 1271, 1273 In one report, preoperative CT was used to identify the exact anatomic location of the offending lesion and guide the surgical approach.1273
Esophageal duplication cysts and intramural inclusion cysts cause typical signs of esophageal obstruction, including salivation, esophageal dysphagia, and swelling of the cervical esophagus as the cysts enlarge.1275, 1277, 1278 Such signs can make them difficult to differentiate from other forms of esophageal obstruction. Endoscopic examination may reveal compression of the esophageal lumen and communication with the esophageal lumen if it exists.
Ultrasonographic examination may be the most useful method of antemortem diagnosis if the cyst is in the cervical esophagus. Examination of an aspirate of the mass may aid in the diagnosis by revealing the presence of keratinized squamous cells.1275, 1278 Surgical treatments have included complete surgical resection and surgical marsupialization.1275, 1277, 1278 The latter appears to be more successful and results in fewer complications.1277, 1280 Possible complications of surgical resection include laryngeal hemiplegia following surgical trauma to the recurrent laryngeal nerve in the region of the esophagus and esophageal fistula formation.1278
Esophageal Perforation
Esophageal perforation typically occurs in the cervical region in response to external trauma, necrosis of the esophageal wall caused by a food impaction, or rupture of an esophageal lesion such as an impacted diverticulum. The esophagus is particularly vulnerable to external trauma in the distal third of the neck because only a thin layer of muscle covers it at this point.1281 Iatrogenic perforation may occur in response to excessive force with a stomach tube against an obstruction or a compromised region of the esophagus.1240 Esophageal perforations may be open or closed and tend to cause extensive cellulitis and necrosis of tissues surrounding the wound because of drainage of saliva and feed material within fascial planes. Systemic inflammation from septic cellulitis may occur. Closed perforations of the esophagus are particularly troublesome because food material, water, saliva, and air may migrate to the mediastinum and pleural space via fascial planes.1240, 1281 Because of the leakage of air into the tissues surrounding the rupture, extensive SC and fascial emphysema frequently develops and is usually evident clinically and on cervical radiographs. Pneumomediastinum and pneumothorax are potentially fatal complications of esophageal ruptures. If the esophagus ruptures into the mediastinum, horses most commonly present with signs of acute SIRS that may be mistaken for colic.
Treatment should include converting closed perforations to open perforations if possible,1282 extensive debridement and lavage of affected tissues, broad-spectrum antibiotics, tetanus prophylaxis, and esophageal rest. The clinician may achieve the latter by placing a feeding tube into the esophagus via the wound. Alternatively, a small nasogastric tube (12-Fr diameter) may be placed.1240 For open perforations, once the wound has granulated and contracted to a small size, oral feeding may be attempted.1281 Extensive loss of saliva via esophageal wounds may lead to hyponatremia and hypochloremia. Transient metabolic acidosis may occur because of salivary bicarbonate loss, followed by progressive metabolic alkalosis.1193 Although reports of esophageal wounds healing well by second intention exist, healing takes a prolonged time.1283 In addition, some perforations never completely heal and form permanent esophagocutaneous fistulae that may require surgical correction. The development of esophageal strictures is not common because wounds are usually linear and not circumferential. However, traction diverticula may develop. Other potential complications of esophageal wounds include Horner’s syndrome and left laryngeal hemiplegia.1281
In a retrospective study on esophageal disorders, only 2 of 11 horses with esophageal perforations survived long term,1220 and in a report of esophageal trauma following nasogastric intubation, 4 of 5 horses were euthanized.1240 The prognosis is poor in horses with esophageal perforations, largely because of the extent of cellulitis, tissue necrosis, shock, and local wound complications.
Diseases of the Stomach
Gastroduodenal Ulceration
Pathophysiology of mucosal injury and repair is detailed previously in this chapter in Mucosal Injury and Repair. Of importance, ulceration can occur in either the stratified squamous or glandular epithelium, although different clinical syndromes and pathophysiologic mechanisms apply. As a result, the broad term “equine gastric ulcer syndrome” (EGUS) has been used to encompass the wide array of associated clinical syndromes,1284 although separation into equine squamous gastric disease (ESGD) and equine glandular gastric disease (EGGD) has recently been proposed.1285 GDUD primarily affects weanling/suckling foals and is typically considered a separate entity.1286 Regardless of terminology, EGUS in its various forms is arguably the most clinically and economically important clinical entity of the equine stomach.
Prevalence
The prevalence of gastric ulceration has been reported for a variety of breeds and usages. The prevalence of squamous ulceration in horses in race training varies from 70% to 95%1287, 1288, 1289, 1290, 1291, 1292, 1293, 1294, 1295 and can be as high as 100% when limited to animals actively racing.1291 Horses performing in other disciplines have also been evaluated, including active show horses (58% prevalence),1296 endurance horses (67% overall1297; 48% interseason and 93% during competition season1298), Western performance horse (40%),1299 Thoroughbred broodmares (67% pregnant; 77% nonpregnant),1300 and nonracing performance horses (17% precompetition; 56% postcompetition).1301 In one large retrospective study (3715 adult horses from the years 1924–1996) evaluating incidences of gastric ulceration identified at necropsy, an overall prevalence of 10.3% was found, with the highest prevalence in Thoroughbreds (including Arabians) and Standardbred trotters (19%).1302 Horses in a university riding program demonstrated a low squamous ulceration prevalence (11%).1303 Approximately 49% of horses presented to a referral hospital for colic had evidence of gastric ulceration.1304 The reported prevalence of gastric ulceration in foals varies from 25% to 57%.1305, 1306, 1307
Many earlier studies investigating prevalence of gastric ulceration do not differentiate between nonglandular and glandular lesions, and many evaluate only the nonglandular region of the stomach, but this trend is changing. In 1 of 162 horses in a hospital setting, 58% had antral or pyloric erosions or ulcerations, 58% had squamous mucosal lesions, and 8% had lesions involving the glandular body.1308 In other studies, 56% of Thoroughbreds had EGGD,1293 and 47% of racehorses (Thoroughbred and Standardbred) had EGGD.1295 In the former,1293 all horses with glandular lesions also had squamous disease, whereas such an association was not seen in the latter study.1295 In one report of endurance horses with a 67% overall lesion prevalence, 27% had EGGD.1297 In another, prevalence varied by season, with 16% EGGD prevalence out of competition and 33% during competition season.1298 In Danish horses, the prevalence of EGGD was 57% in two separate reports.1309, 1310 In a postmortem evaluation, lesions were most commonly located in the squamous mucosa along the margo plicatus, followed by the glandular body, proximal squamous mucosa, and antrum.1302 Overall, glandular lesions tend to occur near the pylorus.
Pathophysiology
Because the pathophysiology of mucosal injury and repair was documented earlier, it will not be reviewed here. Acid secretion clearly plays a role in squamous mucosal ulceration, and key components of acid regulation will be summarized. Horses secrete acid continuously, and measured pH of equine gastric contents is variable from less than 2 to greater than 6 depending on the horse’s dietary state (fed/fasted).818, 1311 A protocol of repeated 24-hour periods of fasting and feeding has been shown to induce squamous erosion and ulceration.841 Because this protocol results in periods of prolonged gastric acidity (pH <2.0) and concurrent administration of the H2 receptor antagonist ranitidine reduces lesion severity, it supports the role of acid exposure in the pathogenesis of squamous ulcer disease. This effect was also noted as pH of the proximal, but not ventral, stomach decreased in early morning hours, corresponding to periods of decreased hay intake.1312
Several peptides can stimulate or inhibit the secretion of acid by the parietal cell. The predominant stimuli to hydrochloric acid secretion are gastrin, histamine, and acetylcholine via the vagus nerve.1313 Gastrin is released by G cells within the antral mucosa, whereas histamine is released by mast cells and ECL-like cells in the gastric gland. Histamine binds to type 2 receptors on the parietal cell membrane, causing an increase in adenosine monophosphate (cAMP), resulting in phosphorylation of enzymes that activate the proton pump. Gastrin and acetylcholine can act via calcium-mediated intracellular pathways and also stimulate histamine release directly.1314 Isolated equine parietal cells respond maximally to histamine stimulation and only minimally to carbachol and pentagastrin.1315 In vivo, histamine or pentagastrin infusion can stimulate similar maximal acid output.1316 Interestingly, pentagastrin stimulation also induces a marked duodenal secretion of a sodium chloride–rich fluid, which can reflux back into the stomach under fasting conditions.1316 Gastrin release is primarily controlled by gastrin-releasing peptide, which is stimulated by gastric distention and increased luminal pH, but the interaction between gastrin and histamine has not been fully elucidated in the horse.
Gastric acid secretion by parietal cells is primarily inhibited by somatostatin, which is released by fundic and antral D cells. The inhibitory effect of somatostatin is primarily paracrine, but plasma levels of somatostatin negatively correlate with gastric luminal acidity.1317 Gastric acid secretion also is inhibited by EGF, a peptide produced in saliva.1318
Foals can produce significant amounts of gastric acid by the second day of life, with consistent periods of acidity (pH <2.0) in clinically normal animals.1319, 1320 In one study, foals tended to have a high gastric pH at day 1 of age,1319 but in a study of critically ill foals, some foals demonstrated periods of gastric acidity on the first day of life.1321 Suckling was associated with an immediate rise in gastric pH, whereas periods of rest in which foals did not suck for more than 20 minutes were associated with prolonged periods of acidity.1320 Premature human infants are capable of gastric acid production at 28 weeks of gestation.1322 Only 1 of 7 premature foals demonstrated an acidic pH recording in a study of gastric pH profiles in critically ill foals.1321 Although multiple factors were likely involved in those foals, the true ontogeny of gastric acid production in foals is currently unknown.
Equine squamous mucosa is very thin at birth but becomes hyperplastic and parakeratotic within days (Fig. 12.19 ).814 The parallel between decreasing pH and proliferation of squamous epithelium correlates with that seen in other species.1323 The combination of a relatively thin gastric epithelium with a high acid output may leave neonatal foals susceptible to ulcer formation at a very young age. The difference in the normal appearance of the squamous mucosa should be considered when interpreting gastric endoscopy in a neonatal population.
Fig. 12.19.
Endoscopic image of the greater curvature of the stomach in a 3-day-old Thoroughbred colt.
Overall, acid remains the major contributing factor to nonglandular mucosal damage, although other factors such as pepsin and bile salts may play an important role as well, either in the initiation or perpetuation of disease.
Risk Factors
Many aspects of diet and management are associated with the development of nonglandular ulceration in adult horses, although some data are conflicting. Risk factors associated with ESGD are clearer than are those associated with EGGD.
Horses in race training have a high incidence of ESGD and are frequently fed high-concentrate, low-roughage diets. A diet high in concentrate and either high or low in forage, in conjunction with stall confinement but no exercise, induce ESGD within 2 weeks.1324 In one study, higher volatile fatty acid concentrations, higher gastric juice pH, and lower number and severity of ESGD were documented after feeding an alfalfa hay–grain diet compared with a bromegrass hay diet.1325 Many factors differed between the diets, however, such as digestible energy, bulk, crude protein, and mineral content (especially calcium). These findings were supported by a study in which an alfalfa hay/pelleted concentrate diet significantly reduced ESGD severity scores and/or prevented ulcer development relative to a coastal hay/concentrate diet in horses managed with dry lot housing and regular exercise.1326 Intermittent feeding has clearly been shown to induce ESGD and is a consistent model of ulcer induction.841, 1311, 1327 Similarly, quality of forage (straw versus other types) and increased time between forage meals was associated with higher incidence of ESGD in one report.1328
The pathophysiologic correlation between exercise and ESGD has not yet been defined despite the high prevalence of disease in performance horses. In one large epidemiologic study, there was a significant association between ESGD and individual trainer, urban versus rural area, lack of direct contact with other horses, solid barriers, and talk versus music radio.1329 ESGD can develop within 8 days of exercise varying from light halter to active race training,1330 and time in work was found to be a risk factor for ESGD in racing Thoroughbreds.1331 Elevations in postfeeding serum gastrin concentration have been demonstrated after treadmill exercise.1332 During treadmill exercise at gaits faster than a walk, proximal gastric size decreases in conjunction with an increase in intraabdominal pressure, resulting in a simultaneous decrease in proximal gastric pH.1333 Both proximal gastric size and pH return to baseline levels as soon as the horse returned to a walk, and the resultant theory was that gastric contracture could result in increased acid exposure to the squamous mucosa by raising the level of liquid gastric contents.
Stall confinement1327 and transport1334 are associated with ESGD, but a distinct mechanism for these associations has not been definitively determined. A recent study did not detect a difference in pH in the proximal or ventral stomach in response to three different environmental situations (stall confinement alone in a barn, stall confinement with a horse in an adjacent stall, and paddock turnout with a companion horse), each for 24 hours.1312 This work suggests that increased acid exposure to the proximal stomach alone is not causative.
Several studies have failed to document a correlation between NSAID administration and naturally occurring ulcer disease.1287, 1288, 1290, 1291, 1302 NSAID administration, however, is a well-known cause of gastric ulceration under experimental conditions.393, 691, 692, 1335, 1336 NSAID-related ulceration is typically described as predominantly glandular in nature, although nonglandular ulceration can also occur by a mechanism that has not yet been fully characterized. NSAIDs cause a decrease in PGE2 synthesis caused by inhibition of the COX pathway. A resultant decrease in glandular mucosal protection, most notably via decreased mucosal blood flow and mucous production, is the most likely mechanism of action. In one study, however, phenylbutazone administration resulted in ulceration of the glandular mucosa at the pyloric antrum but did not significantly alter mucosal PGE2 concentration.1335
Other risk factors associated with gastric ulceration include gender and age, and the reported prevalence of gastric ulcers has increased over time. In one study, the frequency of gastric ulceration increased from <6% before 1945 to approximately 18% after 1975.1302 The association between sex or age and ulceration has not been consistent among studies.1291, 1293, 1302, 1337 Crib biting has also been discussed as a risk factor for ESGD.1331, 1338 In foals, risk factors are less clearly defined and will be discussed along with the clinical syndrome in each age group. Limited access to water has been associated with ESGD.1328
Clinical Syndrome: Neonatal Foals
Clinical signs typically associated with gastric ulceration in foals include poor appetite, diarrhea, and colic. Many foals probably never exhibit clinical signs, and some do not exhibit clinical signs until ulceration is severe or fatal perforation has occurred. Glandular ulceration is typically considered the most clinically significant type of disease in this population.
The physiologic stress of a concurrent illness has been associated with gastric ulceration in foals. Retrospectively, 14 (23%) of 61 foals up to 85 days of age with a clinical disorder had lesions in the gastric glandular mucosa,1307 and prospectively 8 (40%) of 20 foals up to 30 days of age with a clinical disorder had glandular ulceration.1339 In contrast, only 4% to 9% of clinically normal foals examined in endoscopic surveys had lesions in the gastric glandular mucosa.1306, 1340
Critically ill neonatal foals can have a markedly different pH profile compared with that seen in clinically normal foals, potentially caused by alterations in gastric motility and acid secretion.1321 Gastric ulceration was not identified in any animals at necropsy in that study; however, ulceration has been documented in a similar population.1305 Thus factors other than acid exposure, most notably mucosal perfusion, may play an important role in the “stress”-related ulceration seen in neonates. Gastric ulceration and rupture in the hospitalized neonatal population appears to occur less commonly now than in previous reports, despite a decline in the use of ulcer prophylaxis in one hospital.1341 Advances in overall neonatal care, especially supportive care, have likely contributed to this decline.
Gastroduodenal Ulcer Disease
GDUD occurs almost exclusively in suckling and early weanling foals. Clinical signs of duodenal ulceration are similar to those described for gastric ulceration (bruxism, colic, ptyalism, and diarrhea), but the consequences are often more severe. Lesions occur primarily in the proximal duodenum, ranging from diffuse inflammation to severe ulceration, but affected foals typically have severe squamous and/or glandular ulceration as well (Fig. 12.20 ). Foals with duodenal ulceration often have delayed gastric emptying and may have gastroesophageal reflux. Complications can include gastric or duodenal rupture, pyloric or duodenal stricture (Fig. 12.21 ), and ascending cholangitis. Severe squamous and esophageal ulceration and aspiration pneumonia can occur secondary to gastroesophageal reflux.1222, 1342, 1343, 1344, 1345
Fig. 12.20.

Endoscopic image of the lesser curvature in a 3-month-old foal diagnosed with gastroduodenal ulcer disease and a gastric outflow obstruction.
Fig. 12.21.
Endoscopic image of the pylorus in the foal depicted in Fig. 2, 2 months after surgery. Note the extremely small pyloric opening, which had formed a mechanical obstruction to gastric outflow.
GDUD syndrome can occur in outbreaks and is most commonly identified in intensive breeding operations. The cause of duodenal lesions in foals is not known. One theory is that the problem begins with diffuse duodenal inflammation that can coalesce down to a focal area of ulceration (G.D. Lester and A.M. Merritt, personal communication). A temporal relationship between GDUD and rotaviral diarrhea has been suggested, but an infectious etiology remains unproven. Although lesion location and severity associated with rotaviral infection varies among species, duodenal ulceration has not been reported.1346
Clinical Signs: Adults
Reported clinical signs attributable to EGUS in adult horses are variable and classically include anorexia and intermittent or recurrent postprandial colic of varying severity.1347 Overall, clinical signs are nonspecific and poorly associated with the presence of EGUS.1285 Many horses with endoscopic evidence of disease may appear to be clinically normal. Vague signs including decreased consumption of concentrates, postprandial episodes of colic, poor performance or failure to train up to expectations, poor quality hair coat, and diminished condition or failure to thrive have been reported. Diarrhea is not typically associated with gastric ulceration in adult horses, although ulceration can occur concurrently with other causes of diarrhea. Bleeding from ulcers in the gastric squamous mucosa is typically not associated with anemia or hypoproteinemia.
Diagnosis
Gastroscopy is the only reliable method for diagnosis of EGUS in the live animal.1285 Evaluation of the entire stomach and, preferably, proximal duodenum is critical to a diagnosis, because there is no relationship between the presence of squamous and glandular ulceration.1295, 1308, 1309 Urine1348 and blood1349 sucrose absorption testing or fecal albumin or hemoglobin31 is not reliable. Various ulcer scoring systems have been promoted, but the system proposed by the Equine Gastric Ulcer Council1284 (Table 12.11 ) has been recommended because of its repeatability and ease of use.1285, 1350
TABLE 12.11.
Equine Gastric Ulcer Syndrome Lesion Scoring System1284
| Lesion Grade | Description |
|---|---|
| 0 | Intact epithelium with no appearance of hyperemia or hyperkeratosis |
| 1 | Intact mucosa with areas of reddening or hyperkeratosis (squamous) |
| 2 | Small single or multifocal lesions |
| 3 | Large single or multifocal lesions or extensive superficial lesions |
| 4 | Extensive lesions with areas of deep ulceration |
Adapted from Chiu CJ, McArdle AH, Brown R, et al. Intestinal mucosal lesion in low-flow states. I. A morphological, hemodynamic, and metabolic reappraisal. Arch Surg. 1970; 101:478-483; Baker GJ. Diseases of the teeth. In: Colohan PT, Mayhew IG, Merritt AM, et al., eds. Equine medicine and surgery, 4th ed., Vol. 1, Goleta, CA: American Veterinary Publications; 1991.
Duodenal ulceration can be difficult to confirm, and duodenoscopy is the most specific means of diagnosis, although the procedure is more difficult than gastroscopy. Diffuse reddening or inflammation may be the only recognizable lesion in foals with early duodenal disease. In older foals with GDUD, detection of gastric outflow obstruction is critical to the therapeutic plan and appropriate prognosis. A distended stomach is typically evident with ultrasound. Abdominal radiography without contrast in foals with outflow obstruction typically reveals a distinctly enlarged, gas-filled stomach. Liquid barium contrast will either have markedly delayed (with incomplete obstruction) or no (complete obstruction) outflow. Clinically, foals with outflow obstruction will develop reflux after suckling, or marked reflux even with limited to no suckling if the duodenal obstruction is distal to the common bile duct.
Treatment
Multiple pharmacologic treatments have been suggested for the treatment of EGUS, and these can vary for ESGD, EGGD, and GDUD. Because acid has been implicated as the most important pathophysiologic component of ESGD, acid suppression is critical. The principal therapeutic options for ulcer treatment include H2 antagonists (cimetidine, ranitidine, famotidine, and nizatidine), proton pump inhibitors (PPIs; omeprazole, pantoprazole, rabeprazole, and esomeprazole), the mucosal adherent sucralfate, and antacids.
The H2 antagonists suppress hydrochloric acid secretion through competitive inhibition of the parietal cell histamine receptor that can be partially overcome with exogenous pentagastrin.1351 Use of H2 antagonists has been successful in raising gastric pH and resolving gastric lesions in both foals and adult horses.1320, 1344, 1352 Clinical and experimental evidence has demonstrated greater individual variability with lower dosages of H2 antagonists.1339 Thus dosage recommendations are based on levels necessary to increase gastric pH and promote ulcer healing in a majority of horses. Commonly recommended ranitidine dosages are 6.6 mg/kg orally every 8 hours or 1.5 to 2 mg/kg IV every 6 hours. Although clinically normal foals respond predictably to ranitidine,1320 sick neonates have shown variability in pH response to IV ranitidine, with a much shorter duration of action and, in some cases, no noticeable response.1321
PPIs block secretion of H+ at the parietal cell membrane by irreversibly binding to the H+, K+-ATPase proton pump. The powder form of omeprazole is rapidly degraded in an acidic environment; an enteric-coated capsule or a specially formulated paste must be used to allow delivery of the active drug to the small intestine for absorption. An increase in gastric pH and a decrease in acid output are evident 5 to 8 hours after omeprazole paste administration.1353 Omeprazole (GastroGard, Merial, Ltd., Duluth, GA) is the only currently FDA-approved agent for the treatment of EGUS in the United States; other preparations, including enteric-coated granules and buffered formulations, are available elsewhere.1354, 1355, 1356 In the United States, compounded preparations have shown limited to no efficacy in pharmacodynamic and clinical trials,1357, 1358 and GastroGard was the only product (including buffers, H2-receptor antagonists, sucralfate, and compounded preparations) that decreased the odds of gastric ulceration in a population of racehorses.1359 Several studies have documented the safety of oral omeprazole in foals and adult horses.1360, 1361 Omeprazole appears more effective for ulcer healing than either ranitidine or cimetidine.1362, 1363 Omeprazole (4 mg/kg PO q 24 h) has consistently demonstrated efficacy of (70%–80% healing) for ESGD, including Thoroughbreds maintained in race training.816, 815, 1362, 1364, 1365, 1366
Treatment with 1 mg/kg, 2 mg/kg, or 4 mg/kg orally every 24 hours decreases or prevents disease or the recurrence of disease in horses maintained in training.815, 1330, 1367, 1368 Other preparations have shown efficacy for ESGD healing at lower dosages (1 mg/kg or 2 mg/kg PO q 24 h) in clinical trials,1354, 1369, 1370 although a lower dosage is not yet recommended for treatment. Preexercise versus postexercise administration did not have a significant effect on healing of ESGD or EGGD in a recent trial.1371 Duration of therapy for ESGD has been recommended for 28 days, although most healing likely occurs within 21 days.1364 In summary, the primary current recommendation for treatment of ESGD is omeprazole, 4 mg/kg PO q 24 h for 21 to 28 days. In locations in which the enteric-coated granule formation is available, 1 mg/kg may be substituted. Alternate recommendations include ranitidine or lower dosages of omeprazole.1285
Lower healing rates (25%) have recently been reported following omeprazole treatment for EGGD.1354, 1370, 1371
Omeprazole (4 mg/kg) also has demonstrated efficacy for raising intragastric pH in clinically normal1372 or critically ill1373 foals and for ulcer healing in foals.1366 The definitive reason for this is unknown but may be related to timing or duration of acid suppression. A bacterial role in EGGD has not been established, despite solid efforts, and the addition of trimethoprim-sulfa did not have apparent benefit in ulcer healing.1374 Because these factors and basic principles of responsible antimicrobial use, antimicrobial therapy is not recommended for treatment of EGUS in general or EGGD specifically.1285
Sucralfate is effective in the treatment of peptic ulcers and prevention of stress-induced ulcers in humans, likely via adherence to ulcerated mucosa, stimulation of mucous secretion, enhanced PGE synthesis, and concentration of growth factor at the site of ulceration.1375 These are all factors relevant to glandular mucosa; thus, its use seems reasonable for treatment of EGGD. In one study, sucralfate did not promote subclinical ulcer healing in foals, relative to corn syrup.1376 In a recent study, sucralfate (12 mg/kg PO q 12 h) combined with omeprazole (4 mg/kg PO q 24 h) resulted in a 67.5% EGGD healing rate.1377 Current recommendations for EGGD include omeprazole plus sucralfate at the previously mentioned dosages for a minimum of 4 weeks, followed by repeat gastroscopy.1285 Therapy for at least 8 weeks should be pursued before the addition of adjunctive therapy.
The use of synthetic PGE1 analogs, such as misoprostol, has been effective in the treatment of gastric and duodenal ulcers in humans, and the proposed mechanism of action involves both inhibition of gastric acid secretion and mucosal cytoprotection.1378 In horses, misoprostol (5 μg/kg) increases gastric pH1379 and ameliorates deleterious effects of flunixin on mucosal recovery after ischemic injury in vitro,391 but data supporting its use in clinical trials are currently lacking. Misoprostol is contraindicated in pregnant mares.
Prokinetic drugs should be considered in foals with duodenal disease, gastroesophageal reflux, and when delayed gastric emptying without a physical obstruction is suspected. Bethanechol and erythromycin have been shown to increase the rate of gastric emptying in horses.1006 In foals with acute gastric atony, bethanechol 0.025 to 0.030 mg/kg SC every 3 to 4 hours has been effective in promoting gastric motility and emptying, followed by oral maintenance dosages of 0.35 to 0.45 mg/kg three to four times daily. Adverse effects can include diarrhea, inappetence, salivation, and colic, but at the dosages stated, adverse effects have been infrequent and mild.
For foals with severe GDUD that have developed duodenal stricture, surgical therapy is necessary.1343, 1380 These animals require a serious financial commitment because intensive perioperative medical therapy is critical for a successful outcome. Prognosis in two recent abstracts has improved over that previously reported. In one, 98% of surgically treated foals survived to discharge from the hospital, with 68% survival 8 months following discharge from the hospital.1381 In that study, of the surviving Thoroughbreds of racing age, 71% had started a race. In another, short-term survival was reported as 80% for those foals treated surgically and 50% for those treated medically.976
A compounded IV preparation of omeprazole (0.5 mg/kg) increases gastric juice pH and decrease the number of nonglandular lesions in horses.1382 From a regulatory standpoint, omeprazole (4 mg/kg PO q 24 h) does not appear to affect quantitative measurements of performance in Standardbreds.1383
Prophylaxsis
Because gastric perforation caused by glandular ulcer disease has been reported in hospitalized neonates, many clinicians routinely use prophylactic antiulcer therapy in this population. Because some critically ill foals have a predominantly alkaline gastric pH profile, and because gastric acidity may be protective against bacterial translocation in neonates, the need for prophylactic ulcer therapy is controversial. In critically ill human neonates, although IV ranitidine therapy increases gastric pH and gastric bacterial colonization, it does not increase the risk of sepsis.1384 In a retrospective study of 85 hospitalized foals <30 days of age, no difference in the frequency of gastric ulceration at necropsy was found between those foals that received prophylactic treatment for gastric ulcers and those that did not; none died as a result of gastric ulcer disease.1341 In a multicenter retrospective study, acid prophylaxis was associated with an increased incidence of diarrhea but not with alteration in clinical outcome.1385 Many clinicians no longer recommend routine ulcer prophylaxis in all ill neonates. Exceptions could potentially include those foals requiring significant doses of NSAIDs for painful orthopedic disorders.
In adults, dietary and environmental management can help prevent gastric ulceration. Pasture turnout and continuous access to good quality forage, especially alfalfa, are currently recommended. For horses at high risk, the best proven pharmacologic approach to prevention involves administration of omeprazole 1 to 2 mg/kg orally every 24 hours.815, 1363, 1367, 1368 Further work on these recommendations will likely be forthcoming.
Neutraceuticals and Antacids
Although appealing, most agents have not yet been shown to promote healing of EGUS. Some products—including those containing sea buckthorn berries1386; organic acids and B vitamins1387; and a combination of yeast, magnesium hydroxide, and Apolectol1388—have shown promise for prevention of EGUS. Various other products, including a pectin-lecithin complex, have not demonstrated efficacy despite initial promise.1389
The use of antacids in the treatment of gastric ulcers has not been critically examined in the horse. Research in horses has shown that 30 g aluminum hydroxide/15 g magnesium hydroxide will result in an increase in gastric pH above 4 for approximately 2 hours.1390 Although antacids may be useful for the treatment of ulcers in horses, a dose of approximately 180 to 200 mL every 2 to 4 hours is necessary for a standard adult horse, and the use of antacids is not justified or recommended.
Other Disorders of the Stomach
Pyloric Obstruction and Delayed Gastric Emptying
Pyloric stenosis is a structural resistance to gastric outflow. Congenital pyloric stenosis has been reported in foals and one yearling and results from hypertrophy of the pyloric musculature.1061, 1391, 1392 Acquired pyloric stenosis can result from neoplasia or duodenal ulceration.1393, 1394, 1395, 1396 Clinical signs are dependent on the degree of obstruction but include abdominal pain, salivation, and teeth grinding. Complete or near complete obstruction can result in gastric reflux and reflux esophagitis. In foals with congenital pyloric hypertrophy, clinical signs may begin with the consumption of solid feed. In foals, a presumptive diagnosis can be made via gastric endoscopy and radiography (plain and contrast studies). Depending on the cause and severity of disease, gastric endoscopy may provide a presumptive diagnosis in the adult horse. Measurement of gastric emptying can aid the diagnosis. Several methods of measurement are currently available, including nuclear scintigraphy, acetaminophen absorption, and postconsumption [13C]octanoic acid blood or breath testing.986, 1006, 1397 During an exploratory laparotomy, a distended stomach and thickened pylorus are accompanied by a relatively empty intestinal tract.
If complete obstruction is not present, medical therapy with a prokinetic such as bethanechol can increase the rate of gastric emptying.1006 Phenylbutazone and cisapride have also been shown to attenuate the delay in gastric emptying caused by endotoxin administration.986, 987 Surgical repair is necessary for definitive treatment of complete or near-complete obstruction and consists of either gastroenterostomy or pyloroplasty.1075, 1380 Pyloric-duodenal intussusception has been reported in an adult horse presenting for colic.1398
Gastric Dilatation and Rupture
Gastric dilatation can be classified as primary, secondary, or idiopathic. Causes of primary gastric dilatation include gastric impaction, grain engorgement, excessive water intake after exercise, aerophagia, and parasitism.1396, 1399 Secondary gastric dilatation occurs more commonly and can result from primary intestinal ileus or small or large intestinal obstruction. Time to development of gastric reflux is proportional to the distance to the intestinal segment involved, with duodenal obstruction resulting in reflux within 4 hours.1400 Clinical signs of gastric dilatation include those associated with acute colic and, in severe cases, ingesta appearing at the nares. Associated laboratory abnormalities include hemoconcentration, hypokalemia, and hypochloremia.1396
The most common cause of gastric rupture in horses varies among reports. In a retrospective study of 54 horses, gastric rupture occurred most commonly as a secondary phenomenon (65%), usually caused by small intestinal obstruction, with primary gastric dilatation and idiopathic rupture occurring almost equally (15% and 17%, respectively).1399 In another retrospective study of 50 horses in combination with a search of the Veterinary Medical Database (VMDB), 60% of the gastric rupture cases were classified as idiopathic.1401 Risk factors for gastric rupture include feeding grass hay, not feeding grain, gelding, and a nonautomatic water source.1399, 1401 Nasogastric intubation does not preclude the possibility of gastric rupture, and the amount of reflux obtained before rupture is highly variable.1399 Because of the retrospective nature of these reports, one cannot rule out confounding factors with certainty.
Regardless of the initiating cause, gastric rupture usually occurs along the greater curvature. In horses with rupture caused by gastric dilatation, tears in the seromuscular layer are frequently larger than the corresponding tears in the mucosal layer, indicating that the seromuscularis likely weakens and tears before the mucosa.1399, 1401 In contrast, horses with gastric rupture secondary to gastric ulceration usually demonstrate full-thickness tears of equal size in all layers. Gastric rupture is usually fatal because of widespread contamination of the peritoneal cavity, septic peritonitis, and septic shock. Initial clinical signs vary with the primary disease; however, when rupture occurs, an animal previously in pain can exhibit signs of relief. Subsequent signs are consistent with peritonitis and shock, including tachypnea, tachycardia, sweating, and muscle fasciculations. Surgical repair is difficult but has been reported for partial-thickness tears,1402 and, in one case of a combined tear of the mucosa and muscularis with only a focal serosal tear, a full-thickness repair was performed with a favorable outcome.1403
Gastric Impaction
Gastric impaction can result in either acute or chronic signs of colic in the horse. Although a specific cause is not always evident, ingestion of coarse roughage (straw bedding and poor-quality forage), foreign objects (rubber fencing material), and feed that may swell after ingestion or improper mastication (persimmon seeds, mesquite beans, wheat, barley, and sugar beet pulp) has been implicated.1404, 1405, 1406, 1407 Possible predisposing factors include poor dentition, poor mastication and rapid consumption of feedstuffs, and inadequate water consumption. Clinical signs can vary from anorexia and weight loss to those consistent with severe abdominal pain. In severe cases, spontaneous reflux may occur, with gastric contents visible at the nares. In cases presenting with acute severe abdominal pain, a diagnosis is often made during exploratory celiotomy. In animals not exhibiting signs of colic warranting surgical intervention, an endoscopic finding of a full stomach after a normally adequate fast (18–24 hours) can often confirm the diagnosis. Abdominal radiographs are reserved for smaller horses and ponies. In addition to pain management, specific treatment consists of gastric lavage via nasogastric intubation or massage and injection of fluid to soften the impaction during laparotomy.1404, 1405, 1406
Miscellaneous Causes of Gastritis
Nonulcerative gastritis rarely appears to be a clinical problem in the horse, but it has been reported at necropsy in a large retrospective study.1302 Emphysematous gastritis caused by C. perfringens 1408 and C. septicum 1409 has been reported.
Neoplasia of the Alimentary Tract
Neoplasia in the alimentary tract of the horse is uncommon.1410 Primary and metastatic neoplasia can affect multiple locations within the oral cavity and gastrointestinal tract, and occurrence is not isolated to geriatric horses. The alimentary form of lymphoma occurs commonly in horses less than 5 years of age.1411 In one study, Arabian horses were 4.5 times more likely to have intestinal neoplasia relative to other breeds.1412 Identification of tumor type is important for formulating a prognosis and therapeutic plan.
Clinical Signs
The clinical signs associated with alimentary neoplasia are related to the tumor location. For example, oral tumors can cause cutaneous enlargement; tumors of the tongue can cause quidding, dysphagia, and halitosis1413; tumors of the esophagus cause dysphagia, ptyalism, intermittent colic, weight loss, and halitosis1219, 1414, 1415; and gastric neoplasia is associated with anorexia, weight loss, abdominal distention, and intermittent fever.1416 Abdominal neoplasia has been implicated in 4% of horses with intermittent or chronic colic.1417, 1418 Weight loss is the most common clinical sign of horses with intestinal neoplasia.1412 Acute signs of colic can occur with intestinal obstructions from malignant or benign tumors. Paraneoplastic syndromes can occur in the horse. These include weight loss despite adequate caloric intake (cancer cachexia), ectopic hormone production, anemia, leukocytosis, thrombocytopenia, hypergammaglobulinemia, fever, and neurologic abnormalities.1419
Diagnostic Evaluation
Diagnosis of alimentary neoplasia can be challenging. Data collected from a complete blood count, biochemistry panel and urinalysis are rarely confirmatory. Normocytic normochromic anemia of chronic disease is the most common red cell abnormality associated with abdominal neoplasia; blood loss and hemolytic anemia occur less frequently.1420 Peripheral eosinophilia and MEED have been reported with lymphoma.1421 Leukocytosis and hyperfibrinogenemia are common. The most common biochemical abnormalities include hypoalbuminemia, hyperglobulinemia, and hypercalcemia.1422, 1423 Hypoglycemia can occur with pancreatic or hepatic neoplasia.1423
Rectal examination may detect an abdominal mass, thickening of the intestinal wall, or lymph node enlargement. Rectal biopsy can detect diffuse lymphoma in some cases.489 Some tumors can exfoliate into the peritoneal cavity, allowing diagnosis via abdominocentesis.1414, 1424, 1425, 1426 Collecting a large volume of fluid followed by Cytospin and subsequent cytologic evaluation may improve the diagnostic potential; characterization as an inflammatory exudate or modified transudate without any neoplastic cells present is common. Neoplasia was the apparent cause of hemoperitoneum in 13% of cases in one report of 54 cases.1427 Abdominocentesis accurately predicted neoplasia in 11 of 25 cases in another study.769
Specific immunoglobulin testing for IgM deficiency may aid in the diagnosis of lymphoma.1428 DNA cell cycle analysis of suspect neoplastic cells has been used to detect lymphoma in equine patients confirmed with the disease. This method of evaluating fluid or tissue aspirates may increase the accuracy for diagnosing neoplasia in the future.1429
Previously mentioned diagnostics, such as endoscopy, contrast radiography, and cross-sectional imaging may be helpful depending on tumor location. For abdominal neoplasia, transcutaneous ultrasonography is often most useful. An experienced sonographer and thorough examination are likely to improve diagnostic yield.46 Laparoscopy or exploratory laparotomy may be required to obtain a final diagnosis.
Specific Tumors
Lymphoma is the most common neoplasia in the horse and has been divided into four categories1412; only the intestinal/alimentary form will be discussed. The term lymphoma is preferred over lymphosarcoma because there is no benign form of this disease.1423 Lymphoma originates from lymphoid tissue and predominantly affects intestinal lymph nodes. Chronic weight loss, intermittent colic, and fever are the most common clinical findings502, 1430; chronic diarrhea,1431 pruritus, and alopecia1432 have been reported. Peripheral lymphadenopathy is not generally noted, but enlargement of mesenteric lymphadenopathy may be palpable rectally. Therapeutic large colon resection has been reported in two horses with lymphoma.1433
SCC is the second most common neoplasia in the horse and is the most common tumor of the proximal gastrointestinal tract.1434 In the oral cavity SCC may affect the lips, tongue, hard palate, pharynx, and oral mucosa.1435, 1436 Metastasis beyond the regional lymph nodes is rare for oral, esophageal, and SCC, although it is possible. Abnormal masses were palpable rectally in 4 of 5 cases of gastric SCC.1416 Most discussions of SCC treatment about the head are based on dermal, ocular, and adnexal SCC, which are more common. Available treatment includes surgical resection, iridium-192 brachytherapy, 5-fluorouracil, and intralesional cisplatin or carboplatin.1437, 1438, 1439, 1440, 1441 The prognosis for survival is good if complete resection is possible, although it typically is not for gastric SCC.1423 Local recurrence is possible. One report of recurring SCC on the lip with metastasis to the lymph nodes was successfully treated with piroxicam over 58 months.1442 Hypertrophic osteopathy secondary to gastric SCC has been documented in one horse.1443
Adenocarcinoma has been reported in the small intestine, cecum, and large colon.1412, 1434, 1444 The tumor arises from the glandular crypts of the gastrointestinal tract and has been reported in middle-aged and older horses. Metastasis to the lymph nodes, liver, and lungs is possible; osseous metaplasia has also been reported.1445 In a retrospective study of intestinal neoplasia, adenocarcinoma represented 32% of cases, 82% of which involved the small intestine.1412 The short-term prognosis for resectable adenocarcinoma is fair; long-term prognosis remains poor.1412, 1446
Leiomyosarcoma and leiomyoma are malignant and benign, respectively, tumors of the smooth muscle lining the gastrointestinal tract. Both have been reported in the stomach, small intestine, and small colon/rectum; leiomyoma has also been reported in the omentum.1434, 1447, 1448, 1449, 1450, 1451, 1452, 1453 Clinical signs are consistent with intestinal obstruction, and prognosis is favorable with resection of the affected intestinal segment.
Lipoma is a benign tumor that occurs in older horses arising from mesenteric adipocytes. Clinical signs do not typically occur unless the tumor stalk results in intestinal obstruction.
Oral cavity neoplasia may involve the dental tissue (odontogenic tumors), bone (osteogenic tumors), or soft tissues. Ameloblastoma occurs in horses >10 years old, and it mainly affects the mandible. Ameloblastic odontoma affects younger horses and usually involves the maxilla. Both are benign but locally invasive. Radiographs may distinguish between an ameloblastoma (radiolucent lesion) and ameloblastic odontoma (radiolucent lesion with partially mineralized density). The best treatment option is surgical resection and/or radiation therapy regardless of the type.1454
Juvenile mandibular ossifying fibroma occurs in the rostral mandible of young horses between the age of 2 months and 2 years. It may cause significant distortion of the bone. Surgical excision of the mass has a good prognosis if diagnosed early.1455
Melanomas, sarcoids, SCC, and papilloma can occur on the mouth and lips. Melanomas are commonly found in the commissure of the lips and parotid salivary glands and can metastasize to regional lymph nodes. Sarcoids can cause local ulcerations of the buccal mucosa, which are difficult to treat. Intralesional cisplatin, cryosurgery, radiation, and laser excision have been tried with limited success. Equine papilloma virus is responsible for the common skin wart that is found on the lips and muzzle of young horses. These lesions are typically self-limiting, but cryosurgery or excision can be used for removal, if desired.
Footnotes
Previous version by Anthony T. Blikslager
References
- 1.Sellers A., Lowe J. Visualization of auscultation sounds of the large intestine. Proc Am Assoc Equine Pract. 1983;29:359–364. [Google Scholar]
- 2.Argenzio R.A. Functions of the equine large intestine and their interrelationship in disease. Cornell Vet. 1975;65:303–330. [PubMed] [Google Scholar]
- 3.Adams S.B. Equine intestinal motility: an overview of normal activity, changes in disease, and effects of drug administration. Proc Am Assoc Equine Pract. 1987;33:539–553. [Google Scholar]
- 4.Lester G.D., Merritt A.M., Neuwirth L. Effect of alpha 2-adrenergic, cholinergic, and nonsteroidal anti-inflammatory drugs on myoelectric activity of ileum, cecum, and right ventral colon and on cecal emptying of radiolabeled markers in clinically normal ponies. Am J Vet Res. 1998;59:320–327. [PubMed] [Google Scholar]
- 5.Naylor J.M., Poirier K.L., Hamilton D.L. The effects of feeding and fasting on gastrointestinal sounds in adult horses. J Vet Intern Med. 2006;20:1408–1413. doi: 10.1892/0891-6640(2006)20[1408:teofaf]2.0.co;2. [DOI] [PubMed] [Google Scholar]
- 6.Parry B.W., Anderson G.A., Gay C.C. Prognosis in Equine Colic—A Study of Individual Variables Used in Case Assessment. Equine Vet J. 1983;15:337–344. doi: 10.1111/j.2042-3306.1983.tb01818.x. [DOI] [PubMed] [Google Scholar]
- 7.King J.N., Gerring E.L. Observations on the colic motor complex in a pony with a small intestinal obstruction. Equine Vet J Suppl. 1989:43–45. doi: 10.1111/j.2042-3306.1989.tb05654.x. [DOI] [PubMed] [Google Scholar]
- 8.Blikslager A.T., Roberts M.C. Accuracy of clinicians in predicting site and type of lesion as well as outcome in horses with colic. J Am Vet Med Assoc. 1995;207:1444–1447. [PubMed] [Google Scholar]
- 9.Navarro M., Monreal L., Segura D. A comparison of traditional and quantitative analysis of acid-base and electrolyte imbalances in horses with gastrointestinal disorders. J Vet Intern Med. 2005;19:871–877. doi: 10.1892/0891-6640(2005)19[871:acotaq]2.0.co;2. [DOI] [PubMed] [Google Scholar]
- 10.Grosche A., Morton A.J., Graham A.S. Effect of large colon ischemia and reperfusion on concentrations of calprotectin and other clinicopathologic variables in jugular and colonic venous blood in horses. Am J Vet Res. 2013;74:1281–1290. doi: 10.2460/ajvr.74.10.1281. [DOI] [PubMed] [Google Scholar]
- 11.Delesalle C., Dewulf J., Lefebvre R.A. Determination of lactate concentrations in blood plasma and peritoneal fluid in horses with colic by an Accusport analyzer. J Vet Intern Med. 2007;21:293–301. doi: 10.1892/0891-6640(2007)21[293:dolcib]2.0.co;2. [DOI] [PubMed] [Google Scholar]
- 12.Tennent-Brown B.S., Wilkins P.A., Lindborg S. Sequential plasma lactate concentrations as prognostic indicators in adult equine emergencies. J Vet Intern Med. 2010;24:198–205. doi: 10.1111/j.1939-1676.2009.0419.x. [DOI] [PubMed] [Google Scholar]
- 13.Nieto J.E., Dechant J.E., le Jeune S.S. Evaluation of 3 handheld portable analyzers for measurement of L-lactate concentrations in blood and peritoneal fluid of horses with colic. Vet Surg. 2015;44:366–372. doi: 10.1111/j.1532-950X.2014.12231.x. [DOI] [PubMed] [Google Scholar]
- 14.Saulez M.N., Cebra C.K., Dailey M. Comparative biochemical analyses of venous blood and peritoneal fluid from horses with colic using a portable analyser and an in-house analyser. Vet Rec. 2005;157:217–223. doi: 10.1136/vr.157.8.217. [DOI] [PubMed] [Google Scholar]
- 15.Gossett K.A., Cleghorn B., Adams R. Contribution of whole blood L-lactate, pyruvate, D-lactate, acetoacetate, and 3-hydroxybutyrate concentrations to the plasma anion gap in horses with intestinal disorders. Am J Vet Res. 1987;48:72–75. [PubMed] [Google Scholar]
- 16.Gossett K.A., Cleghorn B., Martin G.S. Correlation between anion gap, blood L lactate concentration and survival in horses. Equine Vet J. 1987;19:29–30. doi: 10.1111/j.2042-3306.1987.tb02573.x. [DOI] [PubMed] [Google Scholar]
- 17.Gardner R.B., Nydam D.V., Mohammed H.O. Serum gamma glutamyl transferase activity in horses with right or left dorsal displacements of the large colon. J Vet Intern Med. 2005;19:761–764. doi: 10.1892/0891-6640(2005)19[761:sggtai]2.0.co;2. [DOI] [PubMed] [Google Scholar]
- 18.Andrews F.M., Hamlin R.L., Stalnaker P.S. Blood viscosity in horses with colic. J Vet Intern Med. 1990;4:183–186. doi: 10.1111/j.1939-1676.1990.tb00895.x. [DOI] [PubMed] [Google Scholar]
- 19.Johnston J.K., Morris D.D. Comparison of Duodenitis Proximal Jejunitis and Small Intestinal-Obstruction in Horses—68 Cases (1977-1985) J Am Vet Med Assoc. 1987;191:849–854. [PubMed] [Google Scholar]
- 20.Hunt E., Tennant B., Whitlock R.H. Interpretation of peritoneal fluid erythrocyte counts in horses with abdominal disease. Proc Equine Colic Res Symp. 1986;2:168–174. [Google Scholar]
- 21.Estepa J.C., Lopez I., Mayer-Valor R. The influence of anticoagulants on the measurement of total protein concentration in equine peritoneal fluid. Res Vet Sci. 2006;80:5–10. doi: 10.1016/j.rvsc.2005.03.007. [DOI] [PubMed] [Google Scholar]
- 22.Latson K.M., Nieto J.E., Beldomenico P.M. Evaluation of peritoneal fluid lactate as a marker of intestinal ischaemia in equine colic. Equine Vet J. 2005;37:342–346. doi: 10.2746/0425164054529319. [DOI] [PubMed] [Google Scholar]
- 23.Turner A., McIlwraith C., Trotter G. Biochemical analysis of serum and peritoneal fluid in experimental colonic infarction in horses. Proc Equine Colic Res Symp. 1984:79–87. [Google Scholar]
- 24.Arden W.A., Stick J.A. Serum and peritoneal fluid phosphate concentrations as predictors of major intestinal injury associated with equine colic. J Am Vet Med Assoc. 1988;193:927–931. [PubMed] [Google Scholar]
- 25.Parry B.W. Use of clinical pathology in evaluation of horses with colic. Vet Clin North Am Equine Pract. 1987;3:529–542. doi: 10.1016/s0749-0739(17)30663-6. [DOI] [PubMed] [Google Scholar]
- 26.Yamout S.Z., Nieto J.E., Beldomenico P.M. Peritoneal and Plasma D-lactate Concentrations in Horses with Colic. Vet Surg. 2011;40:817–824. doi: 10.1111/j.1532-950X.2011.00859.x. [DOI] [PubMed] [Google Scholar]
- 27.Peloso J.G., Cohen N.D. Use of serial measurements of peritoneal fluid lactate concentration to identify strangulating intestinal lesions in referred horses with signs of colic. J Am Vet Med Assoc. 2012;240:1208–1217. doi: 10.2460/javma.240.10.1208. [DOI] [PubMed] [Google Scholar]
- 28.Bach L.G., Ricketts S.W. Paracentesis as an aid to the diagnosis of abdominal disease in the horse. Equine Vet J. 1974;6:116–121. doi: 10.1111/j.2042-3306.1974.tb03943.x. [DOI] [PubMed] [Google Scholar]
- 29.Morris D.D., Whitlock R.H., Palmer J.E. Fecal Leukocytes and Epithelial—Cells in Horses with Diarrhea. Cornell Vet. 1983;73:265–274. [PubMed] [Google Scholar]
- 30.Pellegrini F.L. Results of a large-scale necroscopic study of equine colonic ulcers. J Equine Vet Sci. 2005;25:113–117. [Google Scholar]
- 31.Sykes B.J.J., Hallowell G.D. Evaluation of a commercial faecal blood test for the diagnosis of gastric ulceration in Thoroughbred racehorses: a preliminary report. Proc Equine Colic Res Symp. 2014;11:4. [Google Scholar]
- 32.Burgess B.A., Weller C.B., Pabilonia K.L. Detection of different serotypes of Salmonella enterica in experimentally inoculated equine fecal samples by commercially available rapid tests. J Vet Intern Med. 2014;28:1853–1859. doi: 10.1111/jvim.12440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ekiri A.B., Long M.T., Hernandez J.A. Diagnostic performance and application of a real-time PCR assay for the detection of Salmonella in fecal samples collected from hospitalized horses with or without signs of gastrointestinal tract disease. Vet J. 2016;208:28–32. doi: 10.1016/j.tvjl.2015.11.011. [DOI] [PubMed] [Google Scholar]
- 34.Pusterla N., Byrne B.A., Hodzic E. Use of quantitative real-time PCR for the detection of Salmonella spp in fecal samples from horses at a veterinary teaching hospital. Vet J. 2010;186:252–255. doi: 10.1016/j.tvjl.2009.08.022. [DOI] [PubMed] [Google Scholar]
- 35.Ward M.P., Alinovi C.A., Couetil L.L. Evaluation of a PCR to detect Salmonella in fecal samples of horses admitted to a veterinary teaching hospital. J Vet Diagn Invest. 2005;17:118–123. doi: 10.1177/104063870501700204. [DOI] [PubMed] [Google Scholar]
- 36.King J.N., Davies J.V., Gerring E.L. Contrast radiography of the equine oesophagus: effect of spasmolytic agents and passage of a nasogastric tube. Equine Vet J. 1990;22:133–135. doi: 10.1111/j.2042-3306.1990.tb04225.x. [DOI] [PubMed] [Google Scholar]
- 37.Hassel D.M., Langer D.L., Snyder J.R. Evaluation of enterolithiasis in equids: 900 cases (1973-1996) J Am Vet Med Assoc. 1999;214:233–237. [PubMed] [Google Scholar]
- 38.Keppie N.J., Rosenstein D.S., Holcombe S.J. Objective radiographic assessment of abdominal sand accumulation in horses. Vet Radiol Ultrasound. 2008;49:122–128. doi: 10.1111/j.1740-8261.2008.00337.x. [DOI] [PubMed] [Google Scholar]
- 39.Ruohoniemi M., Kaikkonen R., Raekallio M. Abdominal radiography in monitoring the resolution of sand accumulations from the large colon of horses treated medically. Equine Vet J. 2001;33:59–64. doi: 10.2746/042516401776767403. [DOI] [PubMed] [Google Scholar]
- 40.Yarbrough T.B., Langer D.L., Snyder J.R. Abdominal radiography for diagnosis of enterolithiasis in horses: 141 cases (1990-1992) J Am Vet Med Assoc. 1994;205:592–595. [PubMed] [Google Scholar]
- 41.Campbell M.L., Ackerman N., Peyton L.C. Radiographic Gastrointestinal Anatomy of the Foal. Veterinary Radiol. 1984;25:194–204. [Google Scholar]
- 42.Fischer A.T., Kerr L.Y., Obrien T.R. Radiographic Diagnosis of Gastrointestinal Disorders in the Foal. Veterinary Radiol. 1987;28:42–48. [Google Scholar]
- 43.Fischer A.T., Yarbrough T.Y. Retrograde contrast radiography of the distal portions of the intestinal tract in foals. J Am Vet Med Assoc. 1995;207:734–737. [PubMed] [Google Scholar]
- 44.Lester G.D., Lester N.V. Abdominal and thoracic radiography in the neonate. Vet Clin North Am Equine Pract. 2001;17:19–46. doi: 10.1016/s0749-0739(17)30073-1. v. [DOI] [PubMed] [Google Scholar]
- 45.Busoni V., De Busscher V., Lopez D. Evaluation of a protocol for fast localised abdominal sonography of horses (FLASH) admitted for colic. Vet J. 2011;188:77–82. doi: 10.1016/j.tvjl.2010.02.017. [DOI] [PubMed] [Google Scholar]
- 46.le Jeune S., Whitcomb M.B. Ultrasound of the Equine Acute Abdomen. Vet Clin North Am Equine Pract. 2014;30:353–381. doi: 10.1016/j.cveq.2014.04.011. [DOI] [PubMed] [Google Scholar]
- 47.Korolainen R., Kaikkonen R., Ruohoniemi M. Ultrasonography in monitoring the resolution of intestinal sand accumulations in the horse. Equine Vet Educ. 2003;15:331–336. [Google Scholar]
- 48.Weller R., Cauvin E.R., Bowen I.M. Comparison of radiography, scintigraphy and ultrasonography in the diagnosis of a case of temporomandibular joint arthropathy in a horse. Vet Rec. 1999;144:377–379. doi: 10.1136/vr.144.14.377. [DOI] [PubMed] [Google Scholar]
- 49.Weller R., Livesey L., Maierl J. Comparison of radiography and scintigraphy in the diagnosis of dental disorders in the horse. Equine Vet J. 2001;33:49–58. doi: 10.2746/042516401776767458. [DOI] [PubMed] [Google Scholar]
- 50.East L.M., Trumble T.N., Steyn P.F. The application of technetium-99m hexamethylpropyleneamine oxime (99mTc- HMPAO) labeled white blood cells for the diagnosis of right dorsal ulcerative colitis in two horses. Vet Radiol Ultrasound. 2000;41:360–364. doi: 10.1111/j.1740-8261.2000.tb02088.x. [DOI] [PubMed] [Google Scholar]
- 51.Lohmann K.L., Roussel A.J., Cohen N.D. Comparison of nuclear scintigraphy and acetaminophen absorption as a means of studying gastric emptying in horses. Am J Vet Res. 2000;61:310–315. doi: 10.2460/ajvr.2000.61.310. [DOI] [PubMed] [Google Scholar]
- 52.Morrow K.L., Park R.D., Spurgeon T.L. Computed tomographic imaging of the equine head. Vet Radiol Ultrasound. 2000;41:491–497. doi: 10.1111/j.1740-8261.2000.tb01876.x. [DOI] [PubMed] [Google Scholar]
- 53.Tietje S., Becker M., Bockenhoff G. Computed tomographic evaluation of head diseases in the horse: 15 cases. Equine Vet J. 1996;28:98–105. doi: 10.1111/j.2042-3306.1996.tb01599.x. [DOI] [PubMed] [Google Scholar]
- 54.Carmalt J.L., Kneissl S., Rawlinson J.E. Computed tomographic appearance of the temporomandibular joint in 1018 Asymptomatic Horses: A Multi-Institution Study. Vet Radiol Ultrasound. 2016;57:237–245. doi: 10.1111/vru.12334. [DOI] [PubMed] [Google Scholar]
- 55.Manso-Diaz G., Dyson S.J., Dennis R. Magnetic resonance imaging characteristics of equine head disorders: 84 cases (2000-2013) Vet Radiol Ultrasound. 2015;56:176–187. doi: 10.1111/vru.12210. [DOI] [PubMed] [Google Scholar]
- 56.Beccati F., Cercone M., Angeli G. Imaging diagnosis—use of multiphase computed tomographic urography in the diagnosis of ureteral tear in a 6-day-old foal. Vet Radiol Ultrasound. 2016;57:E10–E15. doi: 10.1111/vru.12287. [DOI] [PubMed] [Google Scholar]
- 57.Breukink H.J. Oral mono- and disaccharide tolerance tests in ponies. Am J Vet Res. 1974;35:1523–1527. [PubMed] [Google Scholar]
- 58.Fintl C., Ihler C.F. The effect of sedation on D(+)-xylose absorption tests in 6 normal horses. Equine Vet J Suppl. 2011:149–152. doi: 10.1111/j.2042-3306.2011.00374.x. [DOI] [PubMed] [Google Scholar]
- 59.Mair T.S., Hillyer M.H., Taylor F.G. Small intestinal malabsorption in the horse: an assessment of the specificity of the oral glucose tolerance test. Equine Vet J. 1991;23:344–346. doi: 10.1111/j.2042-3306.1991.tb03735.x. [DOI] [PubMed] [Google Scholar]
- 60.Murphy D., Reid S.W., Love S. The effect of age and diet on the oral glucose tolerance test in ponies. Equine Vet J. 1997;29:467–470. doi: 10.1111/j.2042-3306.1997.tb03160.x. [DOI] [PubMed] [Google Scholar]
- 61.Jacobs K.A., Norman P., Hodgson D.R.G. Effect of Diet on the Oral D-Xylose Absorption Test in the Horse. Am J Vet Res. 1982;43:1856–1858. [PubMed] [Google Scholar]
- 62.Pratt S.E., Geor R.J., McCutcheon L.J. Effects of dietary energy source and physical conditioning on insulin sensitivity and glucose tolerance in Standardbred horses. Equine Vet J Suppl. 2006:579–584. doi: 10.1111/j.2042-3306.2006.tb05608.x. [DOI] [PubMed] [Google Scholar]
- 63.De La Corte F.D., Valberg S.J., MacLeay J.M. Glucose uptake in horses with polysaccharide storage myopathy. Am J Vet Res. 1999;60:458–462. [PubMed] [Google Scholar]
- 64.Freeman D.E., Ferrante P.L., Kronfeld D.S. Effect of food deprivation on d-xylose absorption test results in mares. Am J Vet Res. 1989;50:1609–1612. [PubMed] [Google Scholar]
- 65.Bolton J.R., Merritt A.M., Cimprich R.E. Normal and abnormal xylose absorption in the horse. Cornell Vet. 1976;66:183–197. [PubMed] [Google Scholar]
- 66.Ferrante P.L., Freeman D.E., Ramberg C.F. Kinetic analysis of d-xylose absorption after its intragastric administration to mares deprived of food. Am J Vet Res. 1993;54:2110–2114. [PubMed] [Google Scholar]
- 67.Ferrante P.L., Freeman D.E., Ramberg C.F. Kinetic-Analysis of d-Xylose Distribution After Intravenous Administration to Mares. Am J Vet Res. 1993;54:147–151. [PubMed] [Google Scholar]
- 68.Merritt T., Mallonee P.G., Merritt A.M. d-xylose absorption in the growing foal. Equine Vet J. 1986;18:298–300. doi: 10.1111/j.2042-3306.1986.tb03634.x. [DOI] [PubMed] [Google Scholar]
- 69.Roberts M.C. Carbohydrate digestion and absorption studies in the horse. Res Vet Sci. 1975;18:64–69. [PubMed] [Google Scholar]
- 70.Sutton D.G., Bahr A., Preston T. Validation of the 13C-octanoic acid breath test for measurement of equine gastric emptying rate of solids using radioscintigraphy. Equine Vet J. 2003;35:27–33. doi: 10.2746/042516403775467423. [DOI] [PubMed] [Google Scholar]
- 71.Doherty T.J., Andrews F.M., Abraha T.W. Metoclopramide ameliorates the effects of endotoxin on gastric emptying of acetaminophen in horses. Can J Vet Res. 1999;63:37–40. [PMC free article] [PubMed] [Google Scholar]
- 72.Lohmann K.L., Bahr A., Cohen N.D. Evaluation of acetaminophen absorption in horses with experimentally induced delayed gastric emptying. Am J Vet Res. 2002;63:170–174. doi: 10.2460/ajvr.2002.63.170. [DOI] [PubMed] [Google Scholar]
- 73.Clements J.A., Heading R.C., Nimmo W.S. Kinetics of acetaminophen absorption and gastric emptying in man. Clin Pharmacol Ther. 1978;24:420–431. doi: 10.1002/cpt1978244420. [DOI] [PubMed] [Google Scholar]
- 74.Wyse C.A., Murphy D.M., Preston T. The(13)C-octanoic acid breath test for detection of effects of meal composition on the rate of solid-phase gastric emptying in ponies. Res Vet Sci. 2001;71:81–83. doi: 10.1053/rvsc.2001.0488. [DOI] [PubMed] [Google Scholar]
- 75.Day M.J., Bilzer T., Mansell J. Histopathological standards for the diagnosis of gastrointestinal inflammation in endoscopic biopsy samples from the dog and cat: a report from the World Small Animal Veterinary Association Gastrointestinal Standardization Group. J Comp Pathol. 2008;138(suppl 1):S1–S43. doi: 10.1016/j.jcpa.2008.01.001. [DOI] [PubMed] [Google Scholar]
- 76.Hendrickson D.A., Wilson D.G. Instrumentation and techniques for laparoscopic and thoracoscopic surgery in the horse. Vet Clin North Am Equine Pract. 1996;12:235–259. doi: 10.1016/s0749-0739(17)30281-x. [DOI] [PubMed] [Google Scholar]
- 77.Trostle S. Gastrointestinal endoscopic surgery. Vet Clin North Am Equine Pract. 2000;16:329–341. doi: 10.1016/s0749-0739(17)30108-6. [DOI] [PubMed] [Google Scholar]
- 78.Albanese V., Hanson R.R., McMaster M.A. Use of a Barbed Knotless Suture for Laparoscopic Ablation of the Nephrosplenic Space in 8 Horses. Vet Surg. 2016;45:824–830. doi: 10.1111/vsu.12520. [DOI] [PubMed] [Google Scholar]
- 79.Bracamonte J.L., Duke-Novakovski T. A pilot study evaluating laparoscopic closure of the nephrosplenic space using an endoscopic suturing device in standing horses. Can Vet J. 2016;57:651–654. [PMC free article] [PubMed] [Google Scholar]
- 80.van Bergen T., Wiemer P., Schauvliege S. Laparoscopic Evaluation of the Epiploic Foramen after Celiotomy for Epiploic Foramen Entrapment in the Horse. Vet Surg. 2016;45:596–601. doi: 10.1111/vsu.12493. [DOI] [PubMed] [Google Scholar]
- 81.Kagnoff M.F., Eckmann L. Epithelial cells as sensors for microbial infection. J Clin Invest. 1997;100:6–10. doi: 10.1172/JCI119522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Huber A.R., Kunkel S.L., Todd R.F., 3rd Regulation of transendothelial neutrophil migration by endogenous interleukin-8. Science. 1991;254:99–102. doi: 10.1126/science.1718038. [DOI] [PubMed] [Google Scholar]
- 83.McCormick B.A., Colgan S.P., Delp-Archer C. Salmonella typhimurium attachment to human intestinal epithelial monolayers: transcellular signalling to subepithelial neutrophils. J Cell Biol. 1993;123:895–907. doi: 10.1083/jcb.123.4.895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.McCormick B.A., Miller S.I., Carnes D. Transepithelial signaling to neutrophils by salmonellae: a novel virulence mechanism for gastroenteritis. Infect Immun. 1995;63:2302–2309. doi: 10.1128/iai.63.6.2302-2309.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Jung H.C., Eckmann L., Yang S.K. A distinct array of proinflammatory cytokines is expressed in human colon epithelial cells in response to bacterial invasion. J Clin Invest. 1995;95:55–65. doi: 10.1172/JCI117676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Akira S. Toll-like receptors and innate immunity. Adv Immunol. 2001;78:1–56. doi: 10.1016/s0065-2776(01)78001-7. [DOI] [PubMed] [Google Scholar]
- 87.Malaviya R., Abraham S.N. Mast cell modulation of immune responses to bacteria. Immunol Rev. 2001;179:16–24. doi: 10.1034/j.1600-065x.2001.790102.x. [DOI] [PubMed] [Google Scholar]
- 88.Pothoulakis C., Castagliuolo I., LaMont J.T. CP-96,345, a substance P antagonist, inhibits rat intestinal responses to Clostridium difficile toxin A but not cholera toxin. Proc Natl Acad Sci U S A. 1994;91:947–951. doi: 10.1073/pnas.91.3.947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Castagliuolo I., LaMont J.T., Letourneau R. Neuronal involvement in the intestinal effects of Clostridium difficile toxin A and Vibrio cholerae enterotoxin in rat ileum. Gastroenterology. 1994;107:657–665. doi: 10.1016/0016-5085(94)90112-0. [DOI] [PubMed] [Google Scholar]
- 90.Bogdan C., Rollinghoff M., Diefenbach A. Reactive oxygen and reactive nitrogen intermediates in innate and specific immunity. Curr Opin Immunol. 2000;12:64–76. doi: 10.1016/s0952-7915(99)00052-7. [DOI] [PubMed] [Google Scholar]
- 91.Blikslager A.T., Moeser A.J., Gookin J.L. Restoration of barrier function in injured intestinal mucosa. Physiol Rev. 2007;87:545–564. doi: 10.1152/physrev.00012.2006. [DOI] [PubMed] [Google Scholar]
- 92.Joris I., Cuenoud H.F., Doern G.V. Capillary leakage in inflammation. A study by vascular labeling. Am J Pathol. 1990;137:1353–1363. [PMC free article] [PubMed] [Google Scholar]
- 93.Joris I., Majno G., Corey E.J. The mechanism of vascular leakage induced by leukotriene E4. Endothelial contraction. Am J Pathol. 1987;126:19–24. [PMC free article] [PubMed] [Google Scholar]
- 94.Brett J., Gerlach H., Nawroth P. Tumor necrosis factor/cachectin increases permeability of endothelial cell monolayers by a mechanism involving regulatory G proteins. J Exp Med. 1989;169:1977–1991. doi: 10.1084/jem.169.6.1977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Springer T.A. Traffic signals for lymphocyte recirculation and leukocyte emigration: the multistep paradigm. Cell. 1994;76:301–314. doi: 10.1016/0092-8674(94)90337-9. [DOI] [PubMed] [Google Scholar]
- 96.Harlan J.M., Killen P.D., Harker L.A. Neutrophil-mediated endothelial injury in vitro mechanisms of cell detachment. J Clin Invest. 1981;68:1394–1403. doi: 10.1172/JCI110390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Rosengren S., Olofsson A.M., von Andrian U.H. Leukotriene B4-induced neutrophil-mediated endothelial leakage in vitro and in vivo. J Appl Physiol. 1985;1991(71):1322–1330. doi: 10.1152/jappl.1991.71.4.1322. [DOI] [PubMed] [Google Scholar]
- 98.Hernandez L.A., Grisham M.B., Twohig B. Role of neutrophils in ischemia-reperfusion-induced microvascular injury. Am J Physiol. 1987;253:H699–H703. doi: 10.1152/ajpheart.1987.253.3.H699. [DOI] [PubMed] [Google Scholar]
- 99.Coe D.A., Freischlag J.A., Johnson D. Pentoxifylline prevents endothelial damage due to ischemia and reperfusion injury. J Surg Res. 1997;67:21–25. doi: 10.1006/jsre.1996.4940. [DOI] [PubMed] [Google Scholar]
- 100.Nakagawa K., Miller F.N., Knott A.W. Pentoxifylline inhibits FMLP-induced macromolecular leakage. Am J Physiol. 1995;269:H239–H245. doi: 10.1152/ajpheart.1995.269.1.H239. [DOI] [PubMed] [Google Scholar]
- 101.Nathan C. Neutrophils and immunity: challenges and opportunities. Nat Rev Immunol. 2006;6:173–182. doi: 10.1038/nri1785. [DOI] [PubMed] [Google Scholar]
- 102.Dallegri F., Ottonello L. Tissue injury in neutrophilic inflammation. Inflamm Res. 1997;46:382–391. doi: 10.1007/s000110050208. [DOI] [PubMed] [Google Scholar]
- 103.Giannella R.A. Importance of the intestinal inflammatory reaction in salmonella-mediated intestinal secretion. Infect Immun. 1979;23:140–145. doi: 10.1128/iai.23.1.140-145.1979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Kubes P., Hunter J., Granger D.N. Ischemia/reperfusion-induced feline intestinal dysfunction: importance of granulocyte recruitment. Gastroenterology. 1992;103:807–812. doi: 10.1016/0016-5085(92)90010-v. [DOI] [PubMed] [Google Scholar]
- 105.Elliott E., Li Z., Bell C. Modulation of host response to Escherichia coli o157: H7 infection by anti-CD18 antibody in rabbits. Gastroenterology. 1994;106:1554–1561. doi: 10.1016/0016-5085(94)90410-3. [DOI] [PubMed] [Google Scholar]
- 106.Wallace J.L., Keenan C.M., Granger D.N. Gastric ulceration induced by nonsteroidal anti-inflammatory drugs is a neutrophil-dependent process. Am J Physiol. 1990;259:G462–G467. doi: 10.1152/ajpgi.1990.259.3.G462. [DOI] [PubMed] [Google Scholar]
- 107.Ley K., Tedder T.F. Leukocyte interactions with vascular endothelium. New insights into selectin-mediated attachment and rolling. J Immunol. 1995;155:525–528. [PubMed] [Google Scholar]
- 108.Brown E.J., Lindberg F.P. Leucocyte adhesion molecules in host defence against infection. Ann Med. 1996;28:201–208. doi: 10.3109/07853899609033121. [DOI] [PubMed] [Google Scholar]
- 109.Nagahata H., Kehrli M.E., Jr., Murata H. Neutrophil function and pathologic findings in Holstein calves with leukocyte adhesion deficiency. Am J Vet Res. 1994;55:40–48. [PubMed] [Google Scholar]
- 110.Anderson D.C., Springer T.A. Leukocyte adhesion deficiency: an inherited defect in the Mac-1, LFA-1, and p150,95 glycoproteins. Annu Rev Med. 1987;38:175–194. doi: 10.1146/annurev.me.38.020187.001135. [DOI] [PubMed] [Google Scholar]
- 111.Issekutz A.C., Issekutz T.B. Monocyte migration to arthritis in the rat utilizes both CD11/CD18 and very late activation antigen 4 integrin mechanisms. J Exp Med. 1995;181:1197–1203. doi: 10.1084/jem.181.3.1197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Shuster D.E., Kehrli M.E., Jr., Ackermann M.R. Neutrophilia in mice that lack the murine IL-8 receptor homolog. Science. 1995;269:1590–1591. doi: 10.1126/science.7667641. [DOI] [PubMed] [Google Scholar]
- 113.Huang A.J., Furie M.B., Nicholson S.C. Effects of human neutrophil chemotaxis across human endothelial cell monolayers on the permeability of these monolayers to ions and macromolecules. J Cell Physiol. 1988;135:355–366. doi: 10.1002/jcp.1041350302. [DOI] [PubMed] [Google Scholar]
- 114.Berton G., Yan S.R., Fumagalli L. Neutrophil activation by adhesion: mechanisms and pathophysiological implications. Int J Clin Lab Res. 1996;26:160–177. doi: 10.1007/BF02592978. [DOI] [PubMed] [Google Scholar]
- 115.Rosales C., O’Brien V., Kornberg L. Signal transduction by cell adhesion receptors. Biochim Biophys Acta. 1995;1242:77–98. doi: 10.1016/0304-419x(95)00005-z. [DOI] [PubMed] [Google Scholar]
- 116.Wershil B.K. Lessons from genetically engineered animal models—IX. Mast cell-deficient mice and intestinal biology. Am J Phys Gastrointest Liver Physiol. 2000;278:G343–G348. doi: 10.1152/ajpgi.2000.278.3.G343. [DOI] [PubMed] [Google Scholar]
- 117.Araki Y., Andoh A., Fujiyama Y. Development of dextran sulphate sodium-induced experimental colitis is suppressed in genetically mast cell-deficient Ws/Ws rats. Clin Exp Immunol. 2000;119:264–269. doi: 10.1046/j.1365-2249.2000.01094.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Stein J., Ries J., Barrett K.E. Disruption of intestinal barrier function associated with experimental colitis: possible role of mast cells. Am J Physiol. 1998;274:G203–G209. doi: 10.1152/ajpgi.1998.274.1.G203. [DOI] [PubMed] [Google Scholar]
- 119.Andoh A., Kimura T., Fukuda M. Rapid intestinal ischaemia-reperfusion injury is suppressed in genetically mast cell-deficient Ws/Ws rats. Clin Exp Immunol. 1999;116:90–93. doi: 10.1046/j.1365-2249.1999.00851.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Kimura T., Fujiyama Y., Sasaki M. The role of mucosal mast cell degranulation and free-radical generation in intestinal ischaemia-reperfusion injury in rats. Eur J Gastroenterol Hepatol. 1998;10:659–666. [PubMed] [Google Scholar]
- 121.Yang P.C., Berin M.C., Yu L. Mucosal pathophysiology and inflammatory changes in the late phase of the intestinal allergic reaction in the rat. Am J Pathol. 2001;158:681–690. doi: 10.1016/S0002-9440(10)64010-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Galli S.J., Maurer M., Lantz C.S. Mast cells as sentinels of innate immunity. Curr Opin Immunol. 1999;11:53–59. doi: 10.1016/s0952-7915(99)80010-7. [DOI] [PubMed] [Google Scholar]
- 123.Castro G.A., Harari Y., Russell D. Mediators of anaphylaxis-induced ion transport changes in small intestine. Am J Physiol. 1987;253:G540–G548. doi: 10.1152/ajpgi.1987.253.4.G540. [DOI] [PubMed] [Google Scholar]
- 124.Metcalfe D.D., Baram D., Mekori Y.A. Mast cells. Physiol Rev. 1997;77:1033–1079. doi: 10.1152/physrev.1997.77.4.1033. [DOI] [PubMed] [Google Scholar]
- 125.Kawabata A., Matsunami M., Sekiguchi F. Gastrointestinal roles for proteinase-activated receptors in health and disease. Br J Pharmacol. 2008;153(suppl 1):S230–S240. doi: 10.1038/sj.bjp.0707491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Bueno L., Fioramonti J. Protease-activated receptor 2 and gut permeability: a review. Neurogastroenterol Motil. 2008;20:580–587. doi: 10.1111/j.1365-2982.2008.01139.x. [DOI] [PubMed] [Google Scholar]
- 127.Malaviya R., Abraham S.N. Role of mast cell leukotrienes in neutrophil recruitment and bacterial clearance in infectious peritonitis. J Leukoc Biol. 2000;67:841–846. doi: 10.1002/jlb.67.6.841. [DOI] [PubMed] [Google Scholar]
- 128.Malaviya R., Ikeda T., Ross E. Mast cell modulation of neutrophil influx and bacterial clearance at sites of infection through TNF-alpha. Nature. 1996;381:77–80. doi: 10.1038/381077a0. [DOI] [PubMed] [Google Scholar]
- 129.McLachlan J.B., Shelburne C.P., Hart J.P. Mast cell activators: a new class of highly effective vaccine adjuvants. Nat Med. 2008;14:536–541. doi: 10.1038/nm1757. [DOI] [PubMed] [Google Scholar]
- 130.Goldstein I.M. Complement: biologically active products. In: Gallin J.I., Goldstein I.M., Snyderman R., editors. Inflammation: Basic Principles and Clinical Correlates. Raven Press; New York: 2008. p. 63. [Google Scholar]
- 131.Brown E.J. Complement receptors and phagocytosis. Curr Opin Immunol. 1991;3:76–82. doi: 10.1016/0952-7915(91)90081-b. [DOI] [PubMed] [Google Scholar]
- 132.Williams J.P., Pechet T.T., Weiser M.R. Intestinal reperfusion injury is mediated by IgM and complement. J Appl Physiol (1985) 1999;86:938–942. doi: 10.1152/jappl.1999.86.3.938. [DOI] [PubMed] [Google Scholar]
- 133.Wada K., Montalto M.C., Stahl G.L. Inhibition of complement C5 reduces local and remote organ injury after intestinal ischemia/reperfusion in the rat. Gastroenterology. 2001;120:126–133. doi: 10.1053/gast.2001.20873. [DOI] [PubMed] [Google Scholar]
- 134.Eror A.T., Stojadinovic A., Starnes B.W. Antiinflammatory effects of soluble complement receptor type 1 promote rapid recovery of ischemia/reperfusion injury in rat small intestine. Clin Immunol. 1999;90:266–275. doi: 10.1006/clim.1998.4635. [DOI] [PubMed] [Google Scholar]
- 135.Austen W.G., Jr., Kyriakides C., Favuzza J. Intestinal ischemia-reperfusion injury is mediated by the membrane attack complex. Surgery. 1999;126:343–348. [PubMed] [Google Scholar]
- 136.Cochrane C.G., Griffin J.H. The biochemistry and pathophysiology of the contact system of plasma. Adv Immunol. 1982;33:241–306. doi: 10.1016/s0065-2776(08)60837-8. [DOI] [PubMed] [Google Scholar]
- 137.Bockmann S., Paegelow I. Kinins and kinin receptors: importance for the activation of leukocytes. J Leukoc Biol. 2000;68:587–592. [PubMed] [Google Scholar]
- 138.Stadnicki A., Sartor R.B., Janardham R. Specific inhibition of plasma kallikrein modulates chronic granulomatous intestinal and systemic inflammation in genetically susceptible rats. FASEB J. 1998;12:325–333. doi: 10.1096/fasebj.12.3.325. [DOI] [PubMed] [Google Scholar]
- 139.Stadnicki A., Sartor R.B., Janardham R. Kallikrein-kininogen system activation and bradykinin (B2) receptors in indomethacin induced enterocolitis in genetically susceptible Lewis rats. Gut. 1998;43:365–374. doi: 10.1136/gut.43.3.365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Arai Y., Takanashi H., Kitagawa H. Effect of icatibant, a bradykinin B2 receptor antagonist, on the development of experimental ulcerative colitis in mice. Dig Dis Sci. 1999;44:845–851. doi: 10.1023/a:1026694732602. [DOI] [PubMed] [Google Scholar]
- 141.Thiagarajan R.R., Winn R.K., Harlan J.M. The role of leukocyte and endothelial adhesion molecules in ischemia-reperfusion injury. Thromb Haemost. 1997;78:310–314. [PubMed] [Google Scholar]
- 142.Mashimo H., Goyal R.K. Lessons from genetically engineered animal models. IV. Nitric oxide synthase gene knockout mice. Am J Physiol. 1999;277:G745–750. doi: 10.1152/ajpgi.1999.277.4.G745. [DOI] [PubMed] [Google Scholar]
- 143.Kelly C.P., Becker S., Linevsky J.K. Neutrophil recruitment in Clostridium difficile toxin A enteritis in the rabbit. J Clin Invest. 1994;93:1257–1265. doi: 10.1172/JCI117080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Brown E. Neutrophil adhesion and the therapy of inflammation. Semin Hematol. 1997;34:319–326. [PubMed] [Google Scholar]
- 145.Edens H.A., Parkos C.A. Modulation of epithelial and endothelial paracellular permeability by leukocytes. Adv Drug Deliv Rev. 2000;41:315–328. doi: 10.1016/s0169-409x(00)00049-1. [DOI] [PubMed] [Google Scholar]
- 146.Suzuki M., Asako H., Kubes P. Neutrophil-derived oxidants promote leukocyte adherence in postcapillary venules. Microvasc Res. 1991;42:125–138. doi: 10.1016/0026-2862(91)90081-l. [DOI] [PubMed] [Google Scholar]
- 147.Scudamore C.L., Jepson M.A., Hirst B.H. The rat mucosal mast cell chymase, RMCP-II, alters epithelial cell monolayer permeability in association with altered distribution of the tight junction proteins ZO-1 and occludin. Eur J Cell Biol. 1998;75:321–330. doi: 10.1016/s0171-9335(98)80065-4. [DOI] [PubMed] [Google Scholar]
- 148.Scudamore C.L., Thornton E.M., McMillan L. Release of the mucosal mast cell granule chymase, rat mast cell protease-II, during anaphylaxis is associated with the rapid development of paracellular permeability to macromolecules in rat jejunum. J Exp Med. 1995;182:1871–1881. doi: 10.1084/jem.182.6.1871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Colgan S.P., Resnick M.B., Parkos C.A. IL-4 directly modulates function of a model human intestinal epithelium. J Immunol. 1994;153:2122–2129. [PubMed] [Google Scholar]
- 150.Mullin J.M., Snock K.V. Effect of tumor necrosis factor on epithelial tight junctions and transepithelial permeability. Cancer Res. 1990;50:2172–2176. [PubMed] [Google Scholar]
- 151.Whitlock R.H. Colitis: differential diagnosis and treatment. Equine Vet J. 1986;18:278–283. doi: 10.1111/j.2042-3306.1986.tb03627.x. [DOI] [PubMed] [Google Scholar]
- 152.Merritt A.M., Bolton J.R., Cimprich R. Differential diagnosis of diarrhoea in horses over six months of age. J S Afr Vet Assoc. 1975;46:73–76. [PubMed] [Google Scholar]
- 153.Johnson C.M., Cullen J.M., Roberts M.C. Morphologic Characterization of Castor Oil-Induced Colitis in Ponies. Vet Pathol. 1993;30:248–255. doi: 10.1177/030098589303000305. [DOI] [PubMed] [Google Scholar]
- 154.Roberts M.C., Clarke L.L., Johnson C.M. Castor-oil induced diarrhoea in ponies: a model for acute colitis. Equine Vet J Suppl. 1989:60–67. doi: 10.1111/j.2042-3306.1989.tb05658.x. [DOI] [PubMed] [Google Scholar]
- 155.Rikihisa Y., Johnson G.C., Wang Y.Z. Loss of absorptive-capacity for sodium and chloride in the colon causes diarrhea in potomac horse fever. Res Vet Sci. 1992;52:353–362. doi: 10.1016/0034-5288(92)90037-3. [DOI] [PubMed] [Google Scholar]
- 156.Cordes D.O., Perry B.D., Rikihisa Y. Enterocolitis Caused by Ehrlichia Sp in the Horse (Potomac Horse Fever) Vet Pathol. 1986;23:471–477. doi: 10.1177/030098588602300418. [DOI] [PubMed] [Google Scholar]
- 157.Umemura T., Ohishi H., Ikemoto Y. Histopathology of colitis X in the horse. Nippon Juigaku Zasshi. 1982;44:717–724. doi: 10.1292/jvms1939.44.717. [DOI] [PubMed] [Google Scholar]
- 158.Ochoa R., Kern S.R. The effects of Clostridium perfringens type A enterotoxin in Shetland ponies—clinical, morphologic and clinicopathologic changes. Vet Pathol. 1980;17:738–747. doi: 10.1177/030098588001700609. [DOI] [PubMed] [Google Scholar]
- 159.Lees P., Higgins A.J. Effects of a phenylbutazone paste in ponies: model of acute nonimmune inflammation. Am J Vet Res. 1986;47:2359–2363. [PubMed] [Google Scholar]
- 160.Keshavarzian A., Morgan G., Sedghi S. Role of reactive oxygen metabolites in experimental colitis. Gut. 1990;31:786–790. doi: 10.1136/gut.31.7.786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Weiss S.J. Tissue destruction by neutrophils. N Engl J Med. 1989;320:365–376. doi: 10.1056/NEJM198902093200606. [DOI] [PubMed] [Google Scholar]
- 162.Rooney J.R., Bryans J.T., Prickett M.E. Exhaustion shock in the horse. Cornell Vet. 1966;56:220–235. [PubMed] [Google Scholar]
- 163.Meschter C.L., Tyler D.E., White N.A. Histologic findings in the gastrointestinal tract of horses with colic. Am J Vet Res. 1986;47:598–606. [PubMed] [Google Scholar]
- 164.Guerrant R.L., Bobak D.A. Bacterial and protozoal gastroenteritis. N Engl J Med. 1991;325:327–340. doi: 10.1056/NEJM199108013250506. [DOI] [PubMed] [Google Scholar]
- 165.Argenzio R.A. Pathophysiology of diarrhea. In: Anderson N.V., editor. Veterinary Gastroenterology. Lea & Febiger; Philadelphia, PA: 1992. [Google Scholar]
- 166.Field M., Rao M.C., Chang E.B. Intestinal electrolyte transport and diarrheal disease (1) N Engl J Med. 1989;321:800–806. doi: 10.1056/NEJM198909213211206. [DOI] [PubMed] [Google Scholar]
- 167.Field M., Rao M.C., Chang E.B. Intestinal electrolyte transport and diarrheal disease (2) N Engl J Med. 1989;321:879–883. doi: 10.1056/NEJM198909283211307. [DOI] [PubMed] [Google Scholar]
- 168.Musch M.W., Kachur J.F., Miller R.J. Bradykinin-stimulated electrolyte secretion in rabbit and guinea pig intestine. Involvement of arachidonic acid metabolites. J Clin Invest. 1983;71:1073–1083. doi: 10.1172/JCI110857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Perdue M.H., McKay D.M. Integrative immunophysiology in the intestinal mucosa. Am J Physiol. 1994;267:G151–G165. doi: 10.1152/ajpgi.1994.267.2.G151. [DOI] [PubMed] [Google Scholar]
- 170.Powell D.W. Immunophysiology of intestinal electrolyte transport. In: Field M., Frizzell R.A., editors. Handbook of Physiology: the Gastrointestinal System. American Physiology Society; Rockville, MD: 1991. [Google Scholar]
- 171.Argenzio R.A. Physiology of diarrhea—large intestine. J Am Vet Med Assoc. 1978;173:667–672. [PubMed] [Google Scholar]
- 172.Field M., Graf L.H., Jr., Laird W.J. Heat-stable enterotoxin of Escherichia coli: in vitro effects on guanylate cyclase activity, cyclic GMP concentration, and ion transport in small intestine. Proc Natl Acad Sci USA. 1978;75:2800–2804. doi: 10.1073/pnas.75.6.2800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Mantyh C.R., Pappas T.N., Lapp J.A. Substance P activation of enteric neurons in response to intraluminal Clostridium difficile toxin A in the rat ileum. Gastroenterology. 1996;111:1272–1280. doi: 10.1053/gast.1996.v111.pm8898641. [DOI] [PubMed] [Google Scholar]
- 174.Racusen L.C., Binder H.J. Effect of prostaglandin on ion transport across isolated colonic mucosa. Dig Dis Sci. 1980;25:900–904. doi: 10.1007/BF01308038. [DOI] [PubMed] [Google Scholar]
- 175.Weymer A., Huott P., Liu W. Chloride secretory mechanism induced by prostaglandin E1 in a colonic epithelial cell line. J Clin Invest. 1985;76:1828–1836. doi: 10.1172/JCI112175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Jett M.F., Marshall P., Fondacaro J.D. Action of peptidoleukotrienes on ion transport in rabbit distal colon in vitro. J Pharmacol Exp Ther. 1991;257:698–705. [PubMed] [Google Scholar]
- 177.Hanglow A.C., Bienenstock J., Perdue M.H. Effects of platelet-activating factor on ion transport in isolated rat jejunum. Am J Physiol. 1989;257:G845–G850. doi: 10.1152/ajpgi.1989.257.5.G845. [DOI] [PubMed] [Google Scholar]
- 178.Chang E.B., Musch M.W., Mayer L. Interleukins 1 and 3 stimulate anion secretion in chicken intestine. Gastroenterology. 1990;98:1518–1524. doi: 10.1016/0016-5085(90)91084-j. [DOI] [PubMed] [Google Scholar]
- 179.Grisham M.B., Gaginella T.S., von Ritter C. Effects of neutrophil-derived oxidants on intestinal permeability, electrolyte transport, and epithelial cell viability. Inflammation. 1990;14:531–542. doi: 10.1007/BF00914274. [DOI] [PubMed] [Google Scholar]
- 180.Berschneider H.M., Powell D.W. Fibroblasts modulate intestinal secretory responses to inflammatory mediators. J Clin Invest. 1992;89:484–489. doi: 10.1172/JCI115610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Tamai H., Kachur J.F., Baron D.A. Monochloramine, a neutrophil-derived oxidant, stimulates rat colonic secretion. J Pharmacol Exp Ther. 1991;257:887–894. [PubMed] [Google Scholar]
- 182.Tamai H., Gaginella T.S., Kachur J.F. Ca-mediated stimulation of Cl secretion by reactive oxygen metabolites in human colonic T84 cells. J Clin Invest. 1992;89:301–307. doi: 10.1172/JCI115576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Wilson K.T., Xie Y., Musch M.W. Sodium nitroprusside stimulates anion secretion and inhibits sodium chloride absorption in rat colon. J Pharmacol Exp Ther. 1993;266:224–230. [PubMed] [Google Scholar]
- 184.Perdue M.H., Masson S., Wershil B.K. Role of mast cells in ion transport abnormalities associated with intestinal anaphylaxis. Correction of the diminished secretory response in genetically mast cell-deficient W/Wv mice by bone marrow transplantation. J Clin Invest. 1991;87:687–693. doi: 10.1172/JCI115047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Verspaget H.W., Mulder T.P., van dSV Reactive oxygen metabolites and colitis: a disturbed balance between damage and protection. A selective review. Scand J Gastroenterol Suppl. 1991;188:44–51. doi: 10.3109/00365529109111229. [DOI] [PubMed] [Google Scholar]
- 186.Buell M.G., Berin M.C. Neutrophil-independence of the initiation of colonic injury. Comparison of results from three models of experimental colitis in the rat. Dig Dis Sci. 1994;39:2575–2588. doi: 10.1007/BF02087693. [DOI] [PubMed] [Google Scholar]
- 187.Petrone W.F., English D.K., Wong K. Free radicals and inflammation: superoxide-dependent activation of a neutrophil chemotactic factor in plasma. Proc Natl Acad Sci USA. 1980;77:1159–1163. doi: 10.1073/pnas.77.2.1159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Granger R.N., Rutili G. Neutrophil-mediated mucosal injury: role of reactive oxygen metabolites. Dig Dis Sci. 1988;33:S6–S15. doi: 10.1007/BF01538126. [DOI] [PubMed] [Google Scholar]
- 189.Miller M.J., Zhang X.J., Barkemeyer B. Rabbit gut permeability in response to histamine chloramines and chemotactic peptide. Gastroenterology. 1992;103:1537–1546. doi: 10.1016/0016-5085(92)91175-4. [DOI] [PubMed] [Google Scholar]
- 190.Krawisz J.E., Sharon P., Stenson W.F. Quantitative assay for acute intestinal inflammation based on myeloperoxidase activity. Assessment of inflammation in rat and hamster models. Gastroenterology. 1984;87:1344–1350. [PubMed] [Google Scholar]
- 191.Wardle T.D., Hall L., Turnberg L.A. Inter-relationships between inflammatory mediators released from colonic mucosa in ulcerative colitis and their effects on colonic secretion. Gut. 1993;34:503–508. doi: 10.1136/gut.34.4.503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.McConnico R.S., Roberts M.C., Poston M.B. The interrelationship between arachidonic acid and reactive oxygen metabolites with neutrophilic infiltration in the large intestine in a pony model of acute colitis. Proc ACVIM Forum. 1994;12:A1016. [Google Scholar]
- 193.Grisham M.B., Benoit J.N., Granger D.N. Assessment of leukocyte involvement during ischemia and reperfusion of intestine. Methods Enzymol. 1990;186:729–742. doi: 10.1016/0076-6879(90)86172-r. [DOI] [PubMed] [Google Scholar]
- 194.Moore J.N., Morris D.D. Endotoxemia and septicemia in horses: experimental and clinical correlates. J Am Vet Med Assoc. 1992;200:1903–1914. [PubMed] [Google Scholar]
- 195.Hinterleitner T.A., Powell D.W. Immune system control of intestinal ion transport. Proc Soc Exp Biol Med. 1991;197:249–260. doi: 10.3181/00379727-197-43252. [DOI] [PubMed] [Google Scholar]
- 196.Karayalcin S.S., Sturbaum C.W., Wachsman J.T. Hydrogen peroxide stimulates rat colonic prostaglandin production and alters electrolyte transport. J Clin Invest. 1990;86:60–68. doi: 10.1172/JCI114715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197.Bern M.J., Sturbaum C.W., Karayalcin S.S. Immune system control of rat and rabbit colonic electrolyte transport. Role of prostaglandins and enteric nervous system. J Clin Invest. 1989;83:1810–1820. doi: 10.1172/JCI114086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198.Clarke L.L., Argenzio R.A. NaCl transport across equine proximal colon and the effect of endogenous prostanoids. Am J Physiol. 1990;259:G62–G69. doi: 10.1152/ajpgi.1990.259.1.G62. [DOI] [PubMed] [Google Scholar]
- 199.van Deventer S.J., ten Cate J.W., Tytgat G.N. Intestinal endotoxemia. Clinical significance. Gastroenterology. 1988;94:825–831. doi: 10.1016/0016-5085(88)90261-2. [DOI] [PubMed] [Google Scholar]
- 200.Ciancio M.J., Vitiritti L., Dhar A. Endotoxin-induced alterations in rat colonic water and electrolyte transport. Gastroenterology. 1992;103:1437–1443. doi: 10.1016/0016-5085(92)91162-w. [DOI] [PubMed] [Google Scholar]
- 201.Eades S.C., Moore J.N. Blockade of endotoxin-induced cecal hypoperfusion and ileus with an alpha 2 antagonist in horses. Am J Vet Res. 1993;54:586–590. [PubMed] [Google Scholar]
- 202.Katz A.J., Rosen F.S. Gastro Intestinal Complications of Immuno Deficiency Syndromes. Ciba Found Symp. 1977:243–261. doi: 10.1002/9780470720288.ch12. [DOI] [PubMed] [Google Scholar]
- 203.Mair T.S., Taylor F.G., Harbour D.A. Concurrent cryptosporidium and coronavirus infections in an Arabian foal with combined immunodeficiency syndrome. Vet Rec. 1990;126:127–130. [PubMed] [Google Scholar]
- 204.Bjorneby J.M., Leach D.R., Perryman L.E. Persistent cryptosporidiosis in horses with severe combined immunodeficiency. Infect Immun. 1991;59:3823–3826. doi: 10.1128/iai.59.10.3823-3826.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205.MacLeay J.M., Ames T.R., Hayden D.W. Acquired B lymphocyte deficiency and chronic enterocolitis in a 3-year-old quarter horse. Vet Immunol Immunopathol. 1997;57:49–57. doi: 10.1016/s0165-2427(96)05778-9. [DOI] [PubMed] [Google Scholar]
- 206.Chandra R.K., Kumari S. Nutrition and Immunity—An Overview. J Nutr. 1994;124:S1433–S1435. doi: 10.1093/jn/124.suppl_8.1433S. [DOI] [PubMed] [Google Scholar]
- 207.Chandra R.K. Nutrition and the immune system from birth to old age. Eur J Clin Nutr. 2002;56:S73–S76. doi: 10.1038/sj.ejcn.1601492. [DOI] [PubMed] [Google Scholar]
- 208.Costa M.C., Arroyo L.G., Allen-Vercoe E. Comparison of the fecal microbiota of healthy horses and horses with colitis by high throughput sequencing of the V3-V5 region of the 16S rRNA gene. PLoS One. 2012;7:e41484. doi: 10.1371/journal.pone.0041484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209.Weese J.S., Holcombe S.J., Embertson R.M. Changes in the faecal microbiota of mares precede the development of post partum colic. Equine Vet J. 2015;47:641–649. doi: 10.1111/evj.12361. [DOI] [PubMed] [Google Scholar]
- 210.Costa M.C., Stampfli H.R., Arroyo L.G. Changes in the equine fecal microbiota associated with the use of systemic antimicrobial drugs. BMC Vet Res. 2015;11:19. doi: 10.1186/s12917-015-0335-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 211.Schoster A., Mosing M., Jalali M. Effects of transport, fasting and anaesthesia on the faecal microbiota of healthy adult horses. Equine Vet J. 2016;48:595–602. doi: 10.1111/evj.12479. [DOI] [PubMed] [Google Scholar]
- 212.Harlow B.E., Lawrence L.M., Hayes S.H. Effect of Dietary Starch Source and Concentration on Equine Fecal Microbiota. PLoS One. 2016;11:e0154037. doi: 10.1371/journal.pone.0154037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 213.O’Loughlin E.V., Scott R.B., Gall D.G. Pathophysiology of infectious diarrhea: changes in intestinal structure and function. J Pediatr Gastroenterol Nutr. 1991;12:5–20. doi: 10.1097/00005176-199101000-00004. [DOI] [PubMed] [Google Scholar]
- 214.King J.N., Gerring E.L. The action of low dose endotoxin on equine bowel motility. Equine Vet J. 1991;23:11–17. doi: 10.1111/j.2042-3306.1991.tb02705.x. [DOI] [PubMed] [Google Scholar]
- 215.American College of Chest Physicians/Society of Critical Care Medicine Consensus Conference: definitions for sepsis and organ failure and guidelines for the use of innovative therapies in sepsis. Crit Care Med. 1992;20(6):864–874. [PubMed] [Google Scholar]
- 216.Vincent J.-L. Dear SIRS, I’m sorry to say that I don’t like you. Crit Care Med. 1997;25:372–374. doi: 10.1097/00003246-199702000-00029. [DOI] [PubMed] [Google Scholar]
- 217.Opal S.M. The uncertain value of the definition for SIRS. Systemic inflammatory response syndrome. Chest. 1998;113:1442–1443. doi: 10.1378/chest.113.6.1442. [DOI] [PubMed] [Google Scholar]
- 218.Pfeiffer R. Untersuchungen ueber das Choleragift. Z Hyg. 1892;11:393–412. [Google Scholar]
- 219.Vaara M. Lipopolysaccharide and the permeability of the bacterial outer membrane. In: Brade H., Opal S.M., Vogel S.N., editors. Endotoxin in Health and Disease. Marcel Dekker; New York: 1999. [Google Scholar]
- 220.Rietschel E.T., Brade H., Holst O. Bacterial endotoxin: chemical constitution, biological recognition, host response, and immunological detoxification. In: Rietschel E.T., Wagner H., editors. Pathology of Septic Shock. Springer; Berlin: 1996. [DOI] [PubMed] [Google Scholar]
- 221.Jansson P.-E. The chemistry of O-polysaccharide chains in bacterial lipopolysaccharides. In: Brade H., Opal S.M., Vogel S.N., editors. Endotoxin in Health and Disease. Marcel Dekker; New York: 1999. [Google Scholar]
- 222.Zahringer U., Lindner B., Rietschel E.T. Molecular structure of lipid A, the endotoxic center of bacterial lipopolysaccharides. Adv Carbohydr Chem Biochem. 1994;50:211–276. [PubMed] [Google Scholar]
- 223.Holst O. Chemical structure of the core region of lipopolysaccharides. In: Brade H., Opal S.M., Vogel S.N., editors. Endotoxin in Health and Disease. Marcel Dekker; New York: 1999. [Google Scholar]
- 224.Poxton I.R. Antibodies to lipopolysaccharide. J Immunol Methods. 1995;186(1):1–15. doi: 10.1016/0022-1759(95)00123-r. [DOI] [PubMed] [Google Scholar]
- 225.Galanos C., Luderitz O., Rietschel E.T. Synthetic and natural Escherichia coli free lipid A express identical endotoxic activities. Eur J Biochem. 1985;148(1):1–5. doi: 10.1111/j.1432-1033.1985.tb08798.x. [DOI] [PubMed] [Google Scholar]
- 226.Bradley S.G. Cellular and molecular mechanisms of action of bacterial endotoxins. Annu Rev Microbiol. 1979;33:67–94. doi: 10.1146/annurev.mi.33.100179.000435. [DOI] [PubMed] [Google Scholar]
- 227.Barton M.H., Morris D.D., Norton N. Hemostatic and fibrinolytic indices in neonatal foals with presumed septicemia. J Vet Intern Med. 1998;12(1):26–35. doi: 10.1111/j.1939-1676.1998.tb00493.x. [DOI] [PubMed] [Google Scholar]
- 228.Alexander J.W., Boyce S.T., Babcock G.F. The process of microbial translocation. Ann Surg. 1990;212(4):496–510. doi: 10.1097/00000658-199010000-00012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 229.Moore J.N., Garner H.E., Berg J.N. Intracecal endotoxin and lactate during the onset of equine laminitis: a preliminary report. Am J Vet Res. 1979;40(5):722–723. [PubMed] [Google Scholar]
- 230.Triger D.R., Boyer T.D., Levin J. Portal and systemic bacteraemia and endotoxaemia in liver disease. Gut. 1978;19:935–939. doi: 10.1136/gut.19.10.935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 231.Barton M.H., Collatos C. Tumor necrosis factor and interleukin-6 activity and endotoxin concentration in peritoneal fluid and blood of horses with acute abdominal disease. J Vet Intern Med. 1999;13(5):457–464. doi: 10.1892/0891-6640(1999)013<0457:tnfaia>2.3.co;2. [DOI] [PubMed] [Google Scholar]
- 232.Steverink P.J.G.M., Sturk A., Rutten V.P.M.G. Endotoxin, interleukin-6 and tumor necrosis factor concentrations in equine acute abdominal disease: relation to clinical outcome. J Endotoxin Res. 1995;2:289–299. [Google Scholar]
- 233.Henry M.M., Moore J.N. Whole blood re-calcification time in equine colic. Equine Vet J. 1991;23(4):303–308. doi: 10.1111/j.2042-3306.1991.tb03723.x. [DOI] [PubMed] [Google Scholar]
- 234.Morris D.D. Endotoxemia in horses: a review of cellular and humoral mediators involved in its pathogenesis. J Vet Intern Med. 1991;5(3):167–181. doi: 10.1111/j.1939-1676.1991.tb00944.x. [DOI] [PubMed] [Google Scholar]
- 235.Schlag G., Redl H., Dinges H.P. Bacterial translocation in a baboon model of hypovolemic-traumatic shock. In: Schlag G., Redl H., Siegel J.H., editors. Shock, Sepsis and Organ Failure. Springer; Berlin: 1991. [Google Scholar]
- 236.Deitch E.A., Maejima K., Berg R. Effect of oral antibiotics and bacterial overgrowth on the translocation of the GI tract microflora in burned rats. J Trauma. 1985;25(5):385–392. doi: 10.1097/00005373-198505000-00002. [DOI] [PubMed] [Google Scholar]
- 237.Deitch E.A., Winterton J., Berg R. Effect of starvation, malnutrition, and trauma on the gastrointestinal tract flora and bacterial translocation. Arch Surg. 1987;122(9):1019–1024. doi: 10.1001/archsurg.1987.01400210057008. [DOI] [PubMed] [Google Scholar]
- 238.Deitch E.A., Berg R., Specian R. Endotoxin promotes the translocation of bacteria from the gut. Arch Surg. 1987;122(2):185–190. doi: 10.1001/archsurg.1987.01400140067008. [DOI] [PubMed] [Google Scholar]
- 239.Baker B., Gaffin S.L., Wells M. Endotoxaemia in racehorses following exertion. J S Afr Vet Assoc. 1988;59(2):63–66. [PubMed] [Google Scholar]
- 240.Barton M.H., Williamson L., Jacks S., Norton N. Effects on plasma endotoxin and eicosanoid concentrations and serum cytokine activities in horses competing in a 48-, 83-, or 159-km endurance ride under similar terrain and weather conditions. Am J Vet Res. 2003;64(6):754–761. doi: 10.2460/ajvr.2003.64.754. [DOI] [PubMed] [Google Scholar]
- 241.Donovan D.C., Jackson C.A., Colahan P.T. Assessment of exercise-induced alterations in neutrophil function in horses. Am J Vet Res. 2007;68(11):1198–1204. doi: 10.2460/ajvr.68.11.1198. [DOI] [PubMed] [Google Scholar]
- 242.Tobias P.S., Soldau K., Ulevitch R.J. Isolation of a lipopolysaccharide-binding acute phase reactant from rabbit serum. J Exp Med. 1986;164(3):777–793. doi: 10.1084/jem.164.3.777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 243.Ramadori G., Meyer zum Buschenfelde K.H., Tobias P.S. Biosynthesis of lipopolysaccharide-binding protein in rabbit hepatocytes. Pathobiology. 1990;58(2):89–94. doi: 10.1159/000163569. [DOI] [PubMed] [Google Scholar]
- 244.Schumann R.R., Leong S.R., Flaggs G.W. Structure and function of lipopolysaccharide binding protein. Science. 1990;249(4975):1429–1431. doi: 10.1126/science.2402637. [DOI] [PubMed] [Google Scholar]
- 245.Zweigner J., Schumann R.R., Weber J.R. The role of lipopolysaccharide-binding protein in modulating the innate immune response. Microbes Infect. 2006;8:946–952. doi: 10.1016/j.micinf.2005.10.006. [DOI] [PubMed] [Google Scholar]
- 246.Jack R.S., Fan X., Bernheiden M. Lipopolysaccharide-binding protein is required to combat a murine gram-negative bacterial infection. Nature. 1997;389(6652):742–745. doi: 10.1038/39622. [DOI] [PubMed] [Google Scholar]
- 247.Wright S.D., Ramos R.A., Tobias P.S. CD14, a receptor for complexes of lipopolysaccharide (LPS) and LPS binding protein. Science. 1990;249(4975):1431–1433. doi: 10.1126/science.1698311. [DOI] [PubMed] [Google Scholar]
- 248.Wright S.D., Tobias P.S., Ulevitch R.J. Lipopolysaccharide (LPS) binding protein opsonizes LPS-bearing particles for recognition by a novel receptor on macrophages. J Exp Med. 1989;170(4):1231–1241. doi: 10.1084/jem.170.4.1231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 249.Schiff D.E., Kline L., Soldau K. Phagocytosis of gram-negative bacteria by a unique CD14-dependent mechanism. J Leukoc Biol. 1997;62(6):786–794. doi: 10.1002/jlb.62.6.786. [DOI] [PubMed] [Google Scholar]
- 250.Grunwald U., Fan X., Jack R.S. Monocytes can phagocytose gram-negative bacteria by a CD14-dependent mechanism. J Immunol. 1996;157(9):4119–4125. [PubMed] [Google Scholar]
- 251.Tobias P.S. Lipopolysaccharide-binding protein. In: Brade H., Opal S.M., Vogel S.N., editors. Endotoxin in Health and Disease. Marcel Dekker; New York: 1999. [Google Scholar]
- 252.Wurfel M.M., Hailman E., Wright S.D. Soluble CD14 acts as a shuttle in the neutralization of lipopolysaccharide (LPS) by LPS-binding protein and reconstituted high density lipoprotein. J Exp Med. 1995;181(5):1743–1754. doi: 10.1084/jem.181.5.1743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 253.Lamping N., Dettmer R., Schroder N.W. LPS-binding protein protects mice from septic shock caused by LPS or gram-negative bacteria. J Clin Invest. 1998;101(10):2065–2071. doi: 10.1172/JCI2338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 254.Vanderplas M.L., Moore J.N., Barton M.H. Concentrations of serum amyloid A and lipopolysaccharide-binding protein in horses with colic. Am J Vet Res. 2005;66(9):1509–1516. doi: 10.2460/ajvr.2005.66.1509. [DOI] [PubMed] [Google Scholar]
- 255.Chow J.C., Young D.W., Golenbock D.T. Toll-like receptor-4 mediates lipopolysaccharide-induced signal transduction. J Biol Chem. 1999;274(16):10689–10692. doi: 10.1074/jbc.274.16.10689. [DOI] [PubMed] [Google Scholar]
- 256.Janeway C.A., Jr. The immune system evolved to discriminate infectious nonself from noninfectious self. Immunol Today. 1992;13(1):11–16. doi: 10.1016/0167-5699(92)90198-G. [DOI] [PubMed] [Google Scholar]
- 257.Haziot A., Chen S., Ferrero E. The monocyte differentiation antigen, CD14, is anchored to the cell membrane by a phosphatidylinositol linkage. J Immunol. 1988;141(2):547–552. [PubMed] [Google Scholar]
- 258.Stelter F. Structure/function relationships of CD14. Chem Immunol. 2000;74:25–41. [PubMed] [Google Scholar]
- 259.Durieux J.J., Vita N., Popescu O. The two soluble forms of the lipopolysaccharide receptor, CD14: characterization and release by normal human monocytes. Eur J Immunol. 1994;24(9):2006–2012. doi: 10.1002/eji.1830240911. [DOI] [PubMed] [Google Scholar]
- 260.Jack R.S. Introduction: hunting devils. Chem Immunol. 2000;74:1–4. [PubMed] [Google Scholar]
- 261.Shimazu R., Akashi S., Ogata H. MD-2, a molecule that confers lipopolysaccharide responsiveness on Toll-like receptor 4. J Exp Med. 1999;189(11):1777–1782. doi: 10.1084/jem.189.11.1777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 262.Poltorak A., He X., Smirnova I. Defective LPS signaling in C3H/HeJ and C57BL/10ScCr mice: mutations in Tlr4 gene. Science. 1998;282(5396):2085–2088. doi: 10.1126/science.282.5396.2085. [DOI] [PubMed] [Google Scholar]
- 263.Qureshi S.T., Lariviere L., Leveque G. Endotoxin-tolerant mice have mutations in Toll-like receptor 4 (Tlr4) J Exp Med. 1999;189(4):615–625. doi: 10.1084/jem.189.4.615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 264.Arbour N.C., Lorenz E., Schutte B.C. TLR4 mutations are associated with endotoxin hyporesponsiveness in humans. Nat Genet. 2000;25(2):187–191. doi: 10.1038/76048. [DOI] [PubMed] [Google Scholar]
- 265.Lu Y.-C., Yeh W.-C., Ohashi P.S. LPS/TLR4 signal transduction pathway. Cytokine. 2008;42:145–151. doi: 10.1016/j.cyto.2008.01.006. [DOI] [PubMed] [Google Scholar]
- 266.Maniatis T. Catalysis by a multiprotein IkappaB kinase complex. Science. 1997;278(5339):818–819. doi: 10.1126/science.278.5339.818. [DOI] [PubMed] [Google Scholar]
- 267.Tizard I.R. Veterinary Immunology: An Introduction. WB Saunders; Philadelphia: 1996. Cytokines and the immune system. [Google Scholar]
- 268.Beutler B., Cerami A. Cachectin and tumour necrosis factor as two sides of the same biological coin. Nature. 1986;320(6063):584–588. doi: 10.1038/320584a0. [DOI] [PubMed] [Google Scholar]
- 269.Beutler B., Milsark I.W., Cerami A.C. Passive immunization against cachectin/tumor necrosis factor protects mice from lethal effect of endotoxin. Science. 1985;229(4716):869–871. doi: 10.1126/science.3895437. [DOI] [PubMed] [Google Scholar]
- 270.Le J., Vilcek J. Biology of disease; tumor necrosis factor and interleukin 1: cytokines with multiple overlapping biological activities. Lab Invest. 1987;56:234–248. [PubMed] [Google Scholar]
- 271.Le J.M., Vilcek J. Interleukin 6: a multifunctional cytokine regulating immune reactions and the acute phase protein response. Lab Invest. 1989;61(6):588–602. [PubMed] [Google Scholar]
- 272.MacKay R.J. Treatment of endotoxemia and SIRS. Proc Am Coll Vet Int Med. 2001;11:283–285. [Google Scholar]
- 273.Bone R.C. Sir Isaac Newton, sepsis, SIRS, and CARS. Crit Care Med. 1996;24(7):1125–1128. doi: 10.1097/00003246-199607000-00010. [DOI] [PubMed] [Google Scholar]
- 274.Osuchowski M.F., Welch K., Siddiqui J., Remick D.G. Circulating cytokine/inhibitor profiles reshape the understanding of the SIRS/CARS continnuum in sepsis and predict mortality. J Immunol. 2006;177:1967–1974. doi: 10.4049/jimmunol.177.3.1967. [DOI] [PubMed] [Google Scholar]
- 275.Schade U., Flach R., Hirsch T. Endotoxin as an inducer of cytokines. In: Redl H., Schlag G., editors. Cytokines in Severe Sepsis and Septic Shock. Birkhauser Verlag; Basel: 1999. [Google Scholar]
- 276.Mengozzi M., Ghezzi P. Cytokine down-regulation in endotoxin tolerance. Eur Cytokine Netw. 1993;4(2):89–98. [PubMed] [Google Scholar]
- 277.Cavaillon J-M, Adib-Conquy M: Bench-to-bedside review: endotoxin tolerance as a model of leukocyte reprogramming in sepsis. Crit Care 10(5):233 [DOI] [PMC free article] [PubMed]
- 278.Nomura F., Akashi S., Sakao Y. Cutting edge: endotoxin tolerance in mouse peritoneal macrophages correlates with down-regulation of surface Toll-like receptor 4 expression. J Immunol. 2000;164(7):3476–3479. doi: 10.4049/jimmunol.164.7.3476. [DOI] [PubMed] [Google Scholar]
- 279.Heagy W., Hansen C., Nieman K. Impaired mitogen-activated protein kinase activation and altered cytokine secretion in endotoxin-tolerant human monocytes. J Trauma. 2000;49(5):806–814. doi: 10.1097/00005373-200011000-00003. [DOI] [PubMed] [Google Scholar]
- 280.Barton M.H., Collatos C., Moore J.N. Endotoxin induced expression of tumour necrosis factor, tissue factor and plasminogen activator inhibitor activity by peritoneal macrophages. Equine Vet J. 1996;28(5):382–389. doi: 10.1111/j.2042-3306.1996.tb03109.x. [DOI] [PubMed] [Google Scholar]
- 281.Allen G.K., Campbell-Beggs C., Robinson J.A. Induction of early-phase endotoxin tolerance in horses. Equine Vet J. 1996;28(4):269–274. doi: 10.1111/j.2042-3306.1996.tb03090.x. [DOI] [PubMed] [Google Scholar]
- 282.Elsbach P. Bactericidal/permeability-increasing protein, p15s and phospholipases A2, endogenous antibiotics in host defense against bacterial infections. In: Brade H., Opal S.M., Vogel S.N., editors. Endotoxin in Health and Disease. Marcel Dekker; New York: 1999. [Google Scholar]
- 283.Lentsch A.B., Ward P.A. Regulation of inflammatory vascular damage. J Pathol. 2000;190(3):343–348. doi: 10.1002/(SICI)1096-9896(200002)190:3<343::AID-PATH522>3.0.CO;2-M. [DOI] [PubMed] [Google Scholar]
- 284.Klabunde R.E., Anderson D.E. Role of NO and ROS in platelet activating factor-induced microvascular leakage. FASEB J. 2001;15:A47. doi: 10.1016/s0014-2999(00)00632-4. [DOI] [PubMed] [Google Scholar]
- 285.Turek J.J., Templeton C.B., Bottoms G.D. Flunixin meglumine attenuation of endotoxin-induced damage to the cardiopulmonary vascular endothelium of the pony. Am J Vet Res. 1985;46(3):591–596. [PubMed] [Google Scholar]
- 286.Barton M.H. Endotoxemia. In: White N.A., Moore J.N., editors. Current Techniques in Equine Surgery and Lameness. 2nd ed. WB Saunders; Philadelphia: 1998. [Google Scholar]
- 287.Morris D.D., Crowe N., Moore J.N. Correlation of clinical and laboratory data with serum tumor necrosis factor activity in horses with experimentally induced endotoxemia. Am J Vet Res. 1990;51(12):1935–1940. [PubMed] [Google Scholar]
- 288.Andonegui G., Goyert S.M., Kubes P. Lipopolysaccharide-induced leukocyte-endothelial cell interactions: a role for CD14 versus toll-like receptor 4 within microvessels. J Immunol. 2002;169:2111–2119. doi: 10.4049/jimmunol.169.4.2111. [DOI] [PubMed] [Google Scholar]
- 289.Tizard I.R. Veterinary Immunology: An Introduction. WB Saunders; Philadelphia: 2000. Innate immunity: inflammation. [Google Scholar]
- 290.Meager A. Cytokine regulation of cellular adhesion molecule expression in inflammation. Cytokine Growth Factor Rev. 1999;10(1):27–39. doi: 10.1016/s1359-6101(98)00024-0. [DOI] [PubMed] [Google Scholar]
- 291.Wagner J.G., Roth R.A. Neutrophil migration during endotoxemia. J Leukocyte Biol. 1999;66:10–24. doi: 10.1002/jlb.66.1.10. [DOI] [PubMed] [Google Scholar]
- 292.Wagner J.G., Harkema J.R., Roth R.A. Temporal relationship between intravenous and intratracheal administrations of lipopolysaccharide (LPS) on pulmonary neutrophil (PMN) recruitment. Am J Respir Crit Care Med. 1996;153:A837. (abstract) [Google Scholar]
- 293.Gardner A.B., Nydam D.V., Luna J.A. Serum opsonization capacity, phagocytosis, and oxidative burst activity in neonatal foals in the intensive care unit. J Vet Intern Med. 2007;21(4):797–805. doi: 10.1892/0891-6640(2007)21[797:socpao]2.0.co;2. [DOI] [PubMed] [Google Scholar]
- 294.McTaggart C., Penhale J., Raidala S.L. Effect of plasma transfusion on neutrophil function in healthy and septic foals. Aust Vet J. 2005;83(8):499–505. doi: 10.1111/j.1751-0813.2005.tb13304.x. [DOI] [PubMed] [Google Scholar]
- 295.Johnstone I.B., Crane S. Hemostatic abnormalities in equine colic. Am J Vet Res. 1986;47(2):356–358. [PubMed] [Google Scholar]
- 296.Prasse K.W., Topper M.J., Moore J.N. Analysis of hemostasis in horses with colic. J Am Vet Med Assoc. 1993;203(5):685–693. [PubMed] [Google Scholar]
- 297.Levi M., de Jonge E., van der Poll T. Sepsis and disseminated intravascular coagulation. J Throm Thrombolysis. 2003;16:43–47. doi: 10.1023/B:THRO.0000014592.27892.11. [DOI] [PubMed] [Google Scholar]
- 298.Amaral A., Opal S.M., Vincent J.L. Coagulation in sepsis. Intensive Care Med. 2004;30:1032–1040. doi: 10.1007/s00134-004-2291-8. [DOI] [PubMed] [Google Scholar]
- 299.Morris D.D. Recognition and management of disseminated intravascular coagulation in horses. Vet Clin North Am Equine Pract. 1988;4(1):115–143. doi: 10.1016/s0749-0739(17)30654-5. [DOI] [PubMed] [Google Scholar]
- 300.Hack C.E. Cytokines, coagulation and fibrinolysis. In: Redl H., Schlag G., editors. Cytokines in Severe Sepsis and Septic Shock. Birkhauser Verlag; Basel: 1999. [Google Scholar]
- 301.Kalter E.S., Daha M.R., ten Cate J.W. Activation and inhibition of Hageman factor-dependent pathways and the complement system in uncomplicated bacteremia or bacterial shock. J Infect Dis. 1985;151(6):1019–1027. doi: 10.1093/infdis/151.6.1019. [DOI] [PubMed] [Google Scholar]
- 302.Lyberg T. Clinical significance of increased thromboplastin activity on the monocyte surface: a brief review. Haemostasis. 1984;14(5):430–439. doi: 10.1159/000215101. [DOI] [PubMed] [Google Scholar]
- 303.Drake T.A., Cheng J., Chang A. Expression of tissue factor, thrombomodulin, and E-selectin in baboons with lethal Escherichia coli sepsis. Am J Pathol. 1993;142(5):1458–1470. [PMC free article] [PubMed] [Google Scholar]
- 304.Henry M.M., Moore J.N. Clinical relevance of monocyte procoagulant activity in horses with colic. J Am Vet Med Assoc. 1991;198(5):843–848. [PubMed] [Google Scholar]
- 305.Welles E.G., Prasse K.W., Moore J.N. Use of newly developed assays for protein C and plasminogen in horses with signs of colic. Am J Vet Res. 1991;52(2):345–351. [PubMed] [Google Scholar]
- 306.Nawroth P.P., Stern D.M. Modulation of endothelial cell hemostatic properties by tumor necrosis factor. J Exp Med. 1986;163(3):740–745. doi: 10.1084/jem.163.3.740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 307.Pober J.S., Gimbrone M.A., Jr., Lapierre L.A. Overlapping patterns of activation of human endothelial cells by interleukin 1, tumor necrosis factor, and immune interferon. J Immunol. 1986;137(6):1893–1896. [PubMed] [Google Scholar]
- 308.Hack C.E., Zeerleder S. The endothelium in sepsis: source of and a target for inflammation. Crit Care Med. 2001;29(suppl 7):S21–S27. doi: 10.1097/00003246-200107001-00011. [DOI] [PubMed] [Google Scholar]
- 309.Kruithof E.K. Plasminogen activator inhibitors: a review. Enzyme. 1988;40(2-3):113–121. doi: 10.1159/000469153. [DOI] [PubMed] [Google Scholar]
- 310.Travis J., Salvesen G.S. Human plasma proteinase inhibitors. Annu Rev Biochem. 1983;52:655–709. doi: 10.1146/annurev.bi.52.070183.003255. [DOI] [PubMed] [Google Scholar]
- 311.Krishnamurti C., Barr C.F., Hassett M.A. Plasminogen activator inhibitor: a regulator of ancrod-induced fibrin deposition in rabbits. Blood. 1987;69(3):798–803. [PubMed] [Google Scholar]
- 312.Suffredini A.F., Harpel P.C., Parrillo J.E. Promotion and subsequent inhibition of plasminogen activation after administration of intravenous endotoxin to normal subjects. N Engl J Med. 1989;320(18):1165–1172. doi: 10.1056/NEJM198905043201802. [DOI] [PubMed] [Google Scholar]
- 313.Collatos C., Barton M.H., Schleef R. Regulation of equine fibrinolysis in blood and peritoneal fluid based on a study of colic cases and induced endotoxaemia. Equine Vet J. 1994;26(6):474–481. doi: 10.1111/j.2042-3306.1994.tb04053.x. [DOI] [PubMed] [Google Scholar]
- 314.Collatos C., Barton M.H., Prasse K.W. Intravascular and peritoneal coagulation and fibrinolysis in horses with acute gastrointestinal tract diseases. J Am Vet Med Assoc. 1995;207(4):465–470. [PubMed] [Google Scholar]
- 315.Bernard G.R., Vincent J.L., Laterre P.F. Efficacy and safety of recombinant human activated protein C for severe sepsis. N Engl J Med. 2001;344:699–709. doi: 10.1056/NEJM200103083441001. [DOI] [PubMed] [Google Scholar]
- 316.Ramadori G., Christ B. Cytokines and the hepatic acute-phase response. Semin Liver Dis. 1999;19(2):141–155. doi: 10.1055/s-2007-1007106. [DOI] [PubMed] [Google Scholar]
- 317.Tizard I.R. Veterinary Immunology: An Introduction. WB Saunders; Philadelphia: 1996. Inflammation. [Google Scholar]
- 318.Topper M.J., Prasse K.W. Analysis of coagulation proteins as acute-phase reactants in horses with colic. Am J Vet Res. 1998;59(5):542–545. [PubMed] [Google Scholar]
- 319.Nunokawa Y., Fujunaga T., Taira T. Evaluation of serum amyloid A protein as an acute-phase reactive protein in horses. J Vet Med Sci. 1993;55(6) doi: 10.1292/jvms.55.1011. 1011–6. [DOI] [PubMed] [Google Scholar]
- 320.Hultén C., Demmers S. Serum amyloid A (SAA) as an aid in the management of infectious disease in the foal: comparison with total leucocyte count, neutrophil count and fibrinogen. Equine Vet J. 2002;34(7) doi: 10.2746/042516402776250360. 693–8. [DOI] [PubMed] [Google Scholar]
- 321.Jacobsen S., Kjelgaard-Hansen M., Hagbard Petersen H., Jensen A.L. Evaluation of a commercially available human serum amyloid A (SAA) turbidometric immunoassay for determination of equine SAA concentrations. Vet J. 2006;172(2):315–319. doi: 10.1016/j.tvjl.2005.04.021. [DOI] [PubMed] [Google Scholar]
- 322.Muir W.W. Shock. Compend Cont Educ Pract Vet. 1998;20(5):549–566. [Google Scholar]
- 323.Thiemermann C.T.D.W. Nitric oxide and endothelin-1 in circulatory shock involving cytokines. In: Redl H., Schlag G., editors. Cytokines in Severe Sepsis and Septic Shock. Birkhauser Verlag; Basel: 1999. [Google Scholar]
- 324.Burrows G.E. Escherichia coli endotoxemia in the conscious pony. Am J Vet Res. 1971;32(2):243–248. [PubMed] [Google Scholar]
- 325.Clark E.S., Collatos C. Hypoperfusion of the small intestine during slow infusion of a low dosage of endotoxin in anesthetized horses. Cornell Vet. 1990;80(2):163–172. [PubMed] [Google Scholar]
- 326.Armstrong G.P. Cellular and humoral immunity in the horseshoe crab. In: Gupta A.P., editor. Limulus polyphemus: Immunology of Insects and Other Arthropods. CRC Press; Boca-Raton: 1991. [Google Scholar]
- 327.Shuster R., Traub-Dargatz J., Baxter G. Survey of diplomates of the American College of Veterinary Internal Medicine and the American College of Veterinary Surgeons regarding clinical aspects and treatment of endotoxemia in horses. J Am Vet Med Assoc. 1997;210(1):87–92. [PubMed] [Google Scholar]
- 328.Dinarello C.A., Cannon J.G., Wolff S.M. Tumor necrosis factor (cachectin) is an endogenous pyrogen and induces production of interleukin 1. J Exp Med. 1986;163(6):1433–1450. doi: 10.1084/jem.163.6.1433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 329.Coceani F., Bishai I., Dinarello C.A. Prostaglandin E2 and thromboxane B2 in cerebrospinal fluid of afebrile and febrile cat. Am J Physiol. 1983;244(6):R785–R793. doi: 10.1152/ajpregu.1983.244.6.R785. [DOI] [PubMed] [Google Scholar]
- 330.Parsons C.S., Orsini J.A., Krafty R. Risk factors for development of acute laminitis in horses during hospitalization: 73 cases (1997-2004) J Am Vet Med Assoc. 2007;230(6):885–889. doi: 10.2460/javma.230.6.885. [DOI] [PubMed] [Google Scholar]
- 331.Daels P.F., Stabenfeldt G.H., Hughes J.P. Evaluation of progesterone deficiency as a cause of fetal death in mares with experimentally induced endotoxemia. Am J Vet Res. 1991;52(2):282–288. [PubMed] [Google Scholar]
- 332.Daels P.F., Stabenfeldt G.H., Hughes J.P. Effects of flunixin meglumine on endotoxin-induced prostaglandin F2 alpha secretion during early pregnancy in mares. Am J Vet Res. 1991;52(2):276–281. [PubMed] [Google Scholar]
- 333.Ingle-Fehr J.E., Baxter G.M. Evaluation of digital and laminar blood flow in horses given a low dose of endotoxin. Am J Vet Res. 1998;59(2):192–196. [PubMed] [Google Scholar]
- 334.Menzies-Gow N.J., Bailey S.R., Katz L.M. Endotoxin-induced digital vasoconstriction in horses: associated changes in plasma concentrations of vasoconstrictor mediators. Equine Vet J. 2004;36(3):273–278. doi: 10.2746/0425164044877260. [DOI] [PubMed] [Google Scholar]
- 335.Zerpa H., Verga F.M., Vasquez J. Effect of sublethal endotoxemia on in vitro digital vascular reactivity in horses. J Vet Med A. 2005;52:67–73. doi: 10.1111/j.1439-0442.2004.00684.x. [DOI] [PubMed] [Google Scholar]
- 336.Menzies-Gow N.J., Bailey S.R., Berhane Y. Evaluation of the induction of vasoactive mediators from equine digital vein endothelial cells by endotoxin. Am J Vet Res. 2008;69(3):349–355. doi: 10.2460/ajvr.69.3.349. [DOI] [PubMed] [Google Scholar]
- 337.Zaloga G.P., Chernow B. The multifactorial basis for hypocalcemia during sepsis: studies of the parathyroid hormone-vitamin D axis. Ann Intern Med. 1987;107(1):36–41. doi: 10.7326/0003-4819-107-1-36. [DOI] [PubMed] [Google Scholar]
- 338.Dart A.J., Snyder J.R., Spier S.J. Ionized calcium concentration in horses with surgically managed gastrointestinal disease: 147 cases (1988-1990) J Am Vet Med Assoc. 1992;201(8):1244–1248. [PubMed] [Google Scholar]
- 339.Lavoie J.P., Madigan J.E., Cullor J.S. Haemodynamic, pathological, haematological and behavioural changes during endotoxin infusion in equine neonates. Equine Vet J. 1990;22(1):23–29. doi: 10.1111/j.2042-3306.1990.tb04198.x. [DOI] [PubMed] [Google Scholar]
- 340.Welch R.D., Watkins J.P., Taylor T.S. Disseminated intravascular coagulation associated with colic in 23 horses (1984-1989) J Vet Intern Med. 1992;6(1):29–35. doi: 10.1111/j.1939-1676.1992.tb00982.x. [DOI] [PubMed] [Google Scholar]
- 341.Olson N.C. Effects of endotoxin on lung water, hemodynamics, and gas exchange in anesthetized ponies. Am J Vet Res. 1985;46(11):2288–2293. [PubMed] [Google Scholar]
- 342.Moore J.N., Barton M.H. An update on endotoxemia part 2: treatment and the way ahead. Equine Vet Educ. 1999;11(1):30–34. [Google Scholar]
- 343.Weese J.S., Cote N.M., DeGannes R.V.G. Evaluation of in vitro properties of di-tri-octahedral smectite on clostridial toxins and growth. Equine Vet J. 2003;35(7):638–641. doi: 10.2746/042516403775696384. [DOI] [PubMed] [Google Scholar]
- 344.Bentley A.P., Barton M.H., Norton N. Antimicrobial-induced endotoxin and cytokine activity in an in vitro model of septicemia in foals. Am J Vet Res. 2002;63(5):660–668. doi: 10.2460/ajvr.2002.63.660. [DOI] [PubMed] [Google Scholar]
- 345.Sprouse R.F., Garner H.E., Lager K. Protection of ponies from heterologous and homologous endotoxin challenges via Salmonella typhimurium bacterin-toxoid. Equine Pract. 1989;11(2):34–40. [Google Scholar]
- 346.Garner H.E., Sprouse R.F., Green E.M. Active and passive immunization for blockade of endotoxemia. Proc Amer Assoc Equine Pract. 1985;31:525–532. [Google Scholar]
- 347.Ziegler E.J., Fisher C.J., Jr., Sprung C.L. Treatment of gram-negative bacteremia and septic shock with HA-1A human monoclonal antibody against endotoxin: a randomized, doubleblind, placebo-controlled trial, The HA-1A Sepsis Study Group. N Engl J Med. 1991;324(7):429–436. doi: 10.1056/NEJM199102143240701. [DOI] [PubMed] [Google Scholar]
- 348.Ziegler E.J., McCutchan J.A., Fierer J. Treatment of gram-negative bacteremia and shock with human antiserum to a mutant Escherichia coli. N Engl J Med. 1982;307(20):1225–1230. doi: 10.1056/NEJM198211113072001. [DOI] [PubMed] [Google Scholar]
- 349.Sakulramrung R., Domingue G.J. Cross-reactive immunoprotective antibodies to Escherichia coli O111 rough mutant J5. J Infect Dis. 1985;151(6):995–1004. doi: 10.1093/infdis/151.6.995. [DOI] [PubMed] [Google Scholar]
- 350.Garner H.E., Sprouse R.F., Lager K. Cross-protection of ponies from sublethal Escherichia coli endotoxemia by Salmonella typhimurium antiserum. Equine Pract. 1988;10(4):10–17. [Google Scholar]
- 351.Spier S.J., Lavoie J.P., Cullor J.S. Protection against clinical endotoxemia in horses by using plasma containing antibody to an Rc mutant E. coli (J5) Circ Shock. 1989;28(3):235–248. [PubMed] [Google Scholar]
- 352.Gaffin S.L., Baker B., DuPreez J. Prophylaxis and therapy with anti-endotoxin hyperimmune serum against gastroenteritis and endotoxemia in horses. Proc Amer Assoc Equine Pract. 1982;28:335–340. [Google Scholar]
- 353.Peek S.F., Semrad S., McGuirk S.M. Prognostic value of clinicopathologic variables obtained at admission and effect of antiendotoxin plasma on survival in septic and critically ill foals. J Vet Int Med. 2006;20:569–574. doi: 10.1892/0891-6640(2006)20[569:pvocvo]2.0.co;2. [DOI] [PubMed] [Google Scholar]
- 354.Durando M.M., MacKay R.J., Linda S. Effects of polymyxin B and Salmonella typhimurium antiserum on horses given endotoxin intravenously. Am J Vet Res. 1994;55(7):921–927. [PubMed] [Google Scholar]
- 355.Morris D.D., Whitlock R.H., Corbeil L.B. Endotoxemia in horses: protection provided by antiserum to core lipopolysaccharide. Am J Vet Res. 1986;47(3):544–550. [PubMed] [Google Scholar]
- 356.Morris D.D., Whitlock R.H. Therapy of suspected septicemia in neonatal foals using plasma-containing antibodies to core lipopolysaccharide (LPS) J Vet Intern Med. 1987;1(4):175–182. doi: 10.1111/j.1939-1676.1987.tb02012.x. [DOI] [PubMed] [Google Scholar]
- 357.Cohen N.D., Divers T. Acute colitis in horses. 2. Initial management. Compend Cont Educ Pract Vet. 1998;20:228–234. [Google Scholar]
- 358.Coyne C.P., Fenwick B.W. Inhibition of lipopolysaccharide-induced macrophage tumor necrosis factor-alpha synthesis by polymyxin B sulfate. Am J Vet Res. 1993;54(2):305–314. [PubMed] [Google Scholar]
- 359.Parviainen A.K., Barton M.H., Norton N.N. Evaluation of polymyxin B in an ex vivo model of endotoxemia in horses. Am J Vet Res. 2001;62(1):72–76. doi: 10.2460/ajvr.2001.62.72. [DOI] [PubMed] [Google Scholar]
- 360.Barton M.H. Use of polymyxin B for treatment of endotoxemia in horses. Compend Cont Educ Pract Vet. 2000;11:1056–1059. [Google Scholar]
- 361.Raisbeck M.F., Garner H.E., Osweiler G.D. Effects of polymyxin B on selected features of equine carbohydrate overload. Vet Hum Toxicol. 1989;31(5):422–426. [PubMed] [Google Scholar]
- 362.Barton M.H., Parviainen A.K. Proceedings of the American College of Veterinary Internal Medicine Forum. Wash; Seattle: May 25-28, 2000. Use of polymyxin B for equine endotoxemia. [Google Scholar]
- 363.Morresey P.R., MacKay R.J. Endotoxin-neutralizing activity of polymyxin B in blood after IV administration in horses. Am J Vet Res. 2006;67:642–647. doi: 10.2460/ajvr.67.4.642. [DOI] [PubMed] [Google Scholar]
- 364.Coyne C.P., Moritz J.T., Fenwick B.W. Inhibition of lipopolysaccharide-induced TNF-alpha production by semisynthetic polymyxin-B conjugated dextran. Biotechnol Ther. 1994;5(3-4):137–162. [PubMed] [Google Scholar]
- 365.MacKay R.J., Clark C.K., Logdberg L. Effect of a conjugate of polymyxin B-dextran 70 in horses with experimentally induced endotoxemia. Am J Vet Res. 1999;60(1):68–75. [PubMed] [Google Scholar]
- 366.Hellman J., Warren H.S. Antiendotoxin strategies. Infect Dis Clin North Am. 1999;13(2):371–386. doi: 10.1016/s0891-5520(05)70080-5. [DOI] [PubMed] [Google Scholar]
- 367.Weiss J., Olsson I. Cellular and subcellular localization of the bactericidal/permeability-increasing protein of neutrophils. Blood. 1987;69(2):652–659. [PubMed] [Google Scholar]
- 368.Weersink A.J., van Kessel K.P., van den Tol M.E. Human granulocytes express a 55-kDa lipopolysaccharide-binding protein on the cell surface that is identical to the bactericidal/permeability-increasing protein. J Immunol. 1993;150(1):253–263. [PubMed] [Google Scholar]
- 369.Abrahamson S.L., Wu H.M., Williams R.E. Biochemical characterization of recombinant fusions of lipopolysaccharide binding protein and bactericidal/permeability-increasing protein: implications in biological activity. J Biol Chem. 1997;272(4):2149–2155. doi: 10.1074/jbc.272.4.2149. [DOI] [PubMed] [Google Scholar]
- 370.Vaara M. Lipid A: target for antibacterial drugs. Science. 1996;274(5289):939–940. doi: 10.1126/science.274.5289.939. [DOI] [PubMed] [Google Scholar]
- 371.Levin M., Quint P.A., Goldstein B. Recombinant bactericidal/permeability-increasing protein (rBPI21) as adjunctive treatment for children with severe meningococcal sepsis: a randomised trial, rBPI21 Meningococcal Sepsis Study Group. Lancet. 2000;356(9234):961–967. doi: 10.1016/s0140-6736(00)02712-4. [DOI] [PubMed] [Google Scholar]
- 372.Winchell W.W., Hardy J., Levine D.M. Effect of administration of a phospholipid emulsion on the initial response of horses administered endotoxin. Am J Vet Res. 2002;63(10):1370–1378. doi: 10.2460/ajvr.2002.63.1370. [DOI] [PubMed] [Google Scholar]
- 373.Moore J.N., Norton N., Barton M.H. Rapid infusion of a phospholipid emulsion attenuates the effects of endotoxaemia in horses. Equine Vet J. 2007;39(3):243–248. doi: 10.2746/042516407x173343. [DOI] [PubMed] [Google Scholar]
- 374.Lei M.G., Qureshi N., Morrison D.C. Lipopolysaccharide (LPS) binding to 73-kDa and 38-kDa surface proteins on lymphoreticular cells: preferential inhibition of LPS binding to the former by Rhodopseudomonas sphaeroides lipid A. Immunol Lett. 1993;36(3):245–250. doi: 10.1016/0165-2478(93)90096-k. [DOI] [PubMed] [Google Scholar]
- 375.Golenbock D.T., Hampton R.Y., Qureshi N. Lipid A-like molecules that antagonize the effects of endotoxins on human monocytes. J Biol Chem. 1991;266(29):19490–19498. [PubMed] [Google Scholar]
- 376.Zuckerman S.H., Qureshi N. In vivo inhibition of lipopolysaccharide-induced lethality and tumor necrosis factor synthesis by Rhodobacter sphaeroides diphosphoryl lipid A is dependent on corticosterone induction. Infect Immun. 1992;60(7):2581–2587. doi: 10.1128/iai.60.7.2581-2587.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 377.Christ W.J., Asano O., Robidoux A.L. E5531, a pure endotoxin antagonist of high potency. Science. 1995;268(5207):80–83. doi: 10.1126/science.7701344. [DOI] [PubMed] [Google Scholar]
- 378.Bunnell E., Lynn M., Habet K. A lipid A analog, E5531, blocks the endotoxin response in human volunteers with experimental endotoxemia. Crit Care Med. 2000;28(8):2713–2720. doi: 10.1097/00003246-200008000-00005. [DOI] [PubMed] [Google Scholar]
- 379.Rossignol D.P., Lynn M. Antagonism of in vivo and ex vivo response to endotoxin by E5564, a synthetic lipid A analogue. J Endotoxin Res. 2002;8(6):483–488. doi: 10.1179/096805102125001127. [DOI] [PubMed] [Google Scholar]
- 380.Lohmann K.L., Vandenplas M., Barton M.H., Moore J.N. Lipopolysaccharide from Rhodobacter sphaeroides is an agonist in equine cells. J Endotoxin Res. 2003;9(1) doi: 10.1179/096805103125001315. 33–7. [DOI] [PubMed] [Google Scholar]
- 381.Bryant C.E., Oullette A., Lohmann K. The cellular Toll-like receptor 4 antagonist E5531 can act as an agonist in horse whole blood. Vet Immunol Immunopathol. 2007;116(3-4) doi: 10.1016/j.vetimm.2007.01.013. 182–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 382.Lohmann K.L., Vandenplas M.L., Barton M.H. The equine TLR4/MD-2 complex mediates recognition of lipopolysaccharide from Rhodobacter sphaeroides as an agonist. J Endotoxin Res. 2007;13(4) doi: 10.1177/0968051907083193. 235–42. [DOI] [PubMed] [Google Scholar]
- 383.Fink M.P. Eicosanoids and platelet activating factor in the pathogenesis of sepsis and organ dysfunction. In: Williams J.G., editor. Multiple Organ Dysfunction Syndrome: Examining the Role of Eicosanoids and Procoagulants. RG Landes; Austin, TX: 1996. [Google Scholar]
- 384.Ewert K.M., Fessler J.F., Templeton C.B. Endotoxin-induced hematologic and blood chemical changes in ponies: effects of flunixin meglumine, dexamethasone, and prednisolone. Am J Vet Res. 1985;46(1):24–30. [PubMed] [Google Scholar]
- 385.Moore J.N., Garner H.E., Shapland J.E. Prevention of endotoxin-induced arterial hypoxaemia and lactic acidosis with flunixin meglumine in the conscious pony. Equine Vet J. 1981;13(2):95–98. doi: 10.1111/j.2042-3306.1981.tb04122.x. [DOI] [PubMed] [Google Scholar]
- 386.Moore J.N., Hardee M.M., Hardee G.E. Modulation of arachidonic acid metabolism in endotoxic horses: comparison of flunixin meglumine, phenylbutazone, and a selective thromboxane synthetase inhibitor. Am J Vet Res. 1986;47(1):110–113. [PubMed] [Google Scholar]
- 387.Gerdemann R., Deegen E., Kietzmann M. Effect of flunixin meglumine on plasma prostanoid concentrations in horses with colic in the perioperative period. Dtsch Tierarztl Wochenschr. 1997;104(9):365–368. [PubMed] [Google Scholar]
- 388.Templeton C.B., Bottoms G.D., Fessler J.F. Effects of repeated endotoxin injections on prostanoids, hemodynamics, endothelial cells, and survival in ponies. Circ Shock. 1985;16(3):253–264. [PubMed] [Google Scholar]
- 389.Bottoms G.D., Fessler J.F., Roesel O.F. Endotoxin-induced hemodynamic changes in ponies: effects of flunixin meglumine. Am J Vet Res. 1981;42(9):1514–1518. [PubMed] [Google Scholar]
- 390.Fessler J.F., Bottoms G.D., Roesel O.F. Endotoxin-induced change in hemograms, plasma enzymes, and blood chemical values in anesthetized ponies: effects of flunixin meglumine. Am J Vet Res. 1982;43(1):140–144. [PubMed] [Google Scholar]
- 391.Tomlinson J.E., Blikslager A.T. Effects of cyclooxygenase inhibitors flunixin and deracoxib on permeability of ischaemic-injured equine jejunum. Equine Vet J. 2005;37(1):75–80. doi: 10.2746/0425164054406865. [DOI] [PubMed] [Google Scholar]
- 392.Little D., Brown S.A., Campbell N.B. Effects of the cyclooxygenase inhibitor meloxicam on recovery of ischemia-injured equine jejunum. Am J Vet Res. 2007;68(6):614–624. doi: 10.2460/ajvr.68.6.614. [DOI] [PubMed] [Google Scholar]
- 393.MacAllister C.G., Morgan S.J., Borne A.T. Comparison of adverse effects of phenylbutazone, flunixin meglumine, and ketoprofen in horses. J Am Vet Med Assoc. 1993;202(1):71–77. [PubMed] [Google Scholar]
- 394.Semrad S.D., Hardee G.E., Hardee M.M. Low dose flunixin meglumine: effects on eicosanoid production and clinical signs induced by experimental endotoxaemia in horses. Equine Vet J. 1987;19(3):201–206. doi: 10.1111/j.2042-3306.1987.tb01380.x. [DOI] [PubMed] [Google Scholar]
- 395.Jackman B.R., Moore J.N., Barton M.H. Comparison of the effects of ketoprofen and flunixin meglumine on the in vitro response of equine peripheral blood monocytes to bacterial endotoxin. Can J Vet Res. 1994;58(2):138–143. [PMC free article] [PubMed] [Google Scholar]
- 396.MacKay R.J., Daniels C.A., Bleyaert H.F. Effect of eltenac in horses with induced endotoxaemia. Equine Vet J Suppl. 2000;(32):26–31. doi: 10.1111/j.2042-3306.2000.tb05330.x. [DOI] [PubMed] [Google Scholar]
- 397.Breuhaus B.A., DeGraves F.J., Honore E.K. Pharmacokinetics of ibuprofen after intravenous and oral administration and assessment of safety of administration to healthy foals. Am J Vet Res. 1999;60(9):1066–1073. [PubMed] [Google Scholar]
- 398.Morris D.D., Moore J.N., Crowe N. Dexamethasone reduces endotoxin-induced tumor necrosis factor activity production in vitro by equine peritoneal macrophages. Cornell Vet. 1991;81(3):267–276. [PubMed] [Google Scholar]
- 399.Frauenfelder H.C., Fessler J.F., Moore A.B. Effects of dexamethasone on endotoxin shock in the anesthetized pony: hematologic, blood gas, and coagulation changes. Am J Vet Res. 1982;43(3):405–411. [PubMed] [Google Scholar]
- 400.Annane D. Corticosteroids for septic shock. Crit Care Med. 2001;29(suppl 7):S117–S120. doi: 10.1097/00003246-200107001-00036. [DOI] [PubMed] [Google Scholar]
- 401.Gold J.R., Divers T.J., Barton M.H. Plasma adrenocorticotropin, cortisol, and adrenocorticotropin/cortisol ratios in septic and normal-term foals. J Vet Intern Med. 2007;21(4) doi: 10.1892/0891-6640(2007)21[791:pacacr]2.0.co;2. 791–6. [DOI] [PubMed] [Google Scholar]
- 402.Barton M.H., Moore J.N. Pentoxifylline inhibits mediator synthesis in an equine in vitro whole blood model of endotoxemia. Circ Shock. 1994;44(4):216–220. [PubMed] [Google Scholar]
- 403.Barton M.H., Moore J.N., Norton N. Effects of pentoxifylline infusion on response of horses to in vivo challenge exposure with endotoxin. Am J Vet Res. 1997;58(11):1300–1307. [PubMed] [Google Scholar]
- 404.Baskett A., Barton M.H., Norton N. Effect of pentoxifylline, flunixin meglumine, and their combination on a model of endotoxemia in horses. Am J Vet Res. 1997;58(11):1291–1299. [PubMed] [Google Scholar]
- 405.Ingle-Fehr J.E., Baxter G.M. The effect of oral isoxsuprine and pentoxifylline on digital and laminar blood flow in healthy horses. Vet Surg. 1999;28(3) doi: 10.1053/jvet.1999.0154. 154–60. [DOI] [PubMed] [Google Scholar]
- 406.Arden W.A., Slocombe R.F., Stick J.A. Morphologic and ultrastructural evaluation of effect of ischemia and dimethyl sulfoxide on equine jejunum. Am J Vet Res. 1990;51(11):1784–1791. [PubMed] [Google Scholar]
- 407.Moore R.M., Muir W.W., Bertone A.L. Effects of dimethyl sulfoxide, allopurinol, 21-aminosteroid U-74389G, and manganese chloride on low-flow ischemia and reperfusion of the large colon in horses. Am J Vet Res. 1995;56(5):671–687. [PubMed] [Google Scholar]
- 408.Kelmer G., Doherty T.J., Elliott S. Evaluation of dimethyl sulphoxide effects on initial response to endotoxin in the horse. Equine Vet J. 2008;40(4):358–363. doi: 10.2746/042516408X293501. [DOI] [PubMed] [Google Scholar]
- 409.Weisiger R.A. Oxygen radicals and ischemic tissue injury. Gastroenterology. 1986;90(2):494–496. doi: 10.1016/0016-5085(86)90955-8. [DOI] [PubMed] [Google Scholar]
- 410.Grisham M.B., Hernandez L.A., Granger D.N. Xanthine oxidase and neutrophil infiltration in intestinal ischemia. Am J Physiol. 1986;251(4 Pt 1):G567–G574. doi: 10.1152/ajpgi.1986.251.4.G567. [DOI] [PubMed] [Google Scholar]
- 411.Lochner F., Sangiah S., Burrows G. Effects of allopurinol in experimental endotoxin shock in horses. Res Vet Sci. 1989;47(2):178–184. [PubMed] [Google Scholar]
- 412.Taniguchi T., Shibata K., Yamamoto K. Effects of lidocaine administration on hemodynamics and cytokine responses to endotoxemia in rabbits. Crit Care Med. 2000;28(3):755–759. doi: 10.1097/00003246-200003000-00025. [DOI] [PubMed] [Google Scholar]
- 413.Cook V.L., Jones Shults J., McDowell M. Attenuation of ischaemic injury in the equine jejunum by administration of systemic lidocaine. Equine Vet J. 2008;40(4):353–357. doi: 10.2746/042516408X293574. [DOI] [PubMed] [Google Scholar]
- 414.Carrick J.B., McCann M.E. Proceedings of the American College of Veterinary Internal Medicine Forum. Lake Buena Vista; FL: 1997. The effect of short-term administration of omega 3 fatty acids on endotoxemia. [Google Scholar]
- 415.Chu A.J., Walton M.A., Prasad J.K. Blockade by polyunsaturated n-3 fatty acids of endotoxin-induced monocytic tissue factor activation is mediated by the depressed receptor expression in THP-1 cells. J Surg Res. 1999;87(2):217–224. doi: 10.1006/jsre.1999.5762. [DOI] [PubMed] [Google Scholar]
- 416.Morris D.D., Henry M.M., Moore J.N. Effect of dietary alpha-linolenic acid on endotoxin-induced production of tumor necrosis factor by peritoneal macrophages in horses. Am J Vet Res. 1991;52(4):528–532. [PubMed] [Google Scholar]
- 417.Henry M.M., Moore J.N., Feldman E.B. Effect of dietary alpha-linolenic acid on equine monocyte procoagulant activity and eicosanoid synthesis. Circ Shock. 1990;32(3):173–188. [PubMed] [Google Scholar]
- 418.Henry M.M., Moore J.N., Fischer J.K. Influence of an omega-3 fatty acid-enriched ration on in vivo responses of horses to endotoxin. Am J Vet Res. 1991;52(4):523–527. [PubMed] [Google Scholar]
- 419.McCann M.E., Moore J.N., Carrick J.B. Effect of intravenous infusion of omega-3 and omega-6 lipid emulsions on equine monocyte fatty acid composition and inflammatory mediator production in vitro. Shock. 2000;14(2):222–228. doi: 10.1097/00024382-200014020-00024. [DOI] [PubMed] [Google Scholar]
- 420.Cargile J.L., MacKay R.J., Dankert J.R. Effects of tumor necrosis factor blockade on interleukin 6, lactate, thromboxane, and prostacyclin responses in miniature horses given endotoxin. Am J Vet Res. 1995;56(11):1445–1450. [PubMed] [Google Scholar]
- 421.Cargile J.L., MacKay R.J., Dankert J.R. Effect of treatment with a monoclonal antibody against equine tumor necrosis factor (TNF) on clinical, hematologic, and circulating TNF responses of miniature horses given endotoxin. Am J Vet Res. 1995;56(11):1451–1459. [PubMed] [Google Scholar]
- 422.Barton M.H., Bruce E.H., Moore J.N. Effect of tumor necrosis factor antibody given to horses during early experimentally induced endotoxemia. Am J Vet Res. 1998;59(6):792–797. [PubMed] [Google Scholar]
- 423.MacKay R.J., Socher S.H. Anti-equine tumor necrosis factor (TNF) activity of antisera raised against human TNF-alpha and peptide segments of human TNF-alpha. Am J Vet Res. 1992;53(6):921–924. [PubMed] [Google Scholar]
- 424.Abraham E., Wunderink R., Silverman H. Efficacy and safety of monoclonal antibody to human tumor necrosis factor alpha in patients with sepsis syndrome: a randomized, controlled, double-blind, multicenter clinical trial, TNF-alpha MAb Sepsis Study Group. J Am Med Assoc. 1995;273(12):934–941. [PubMed] [Google Scholar]
- 425.Fisher C.J., Jr., Opal S.M., Dhainaut J.F. Influence of an anti-tumor necrosis factor monoclonal antibody on cytokine levels in patients with sepsis, The CB0006 Sepsis Syndrome Study Group. Crit Care Med. 1993;21(3):318–327. doi: 10.1097/00003246-199303000-00006. [DOI] [PubMed] [Google Scholar]
- 426.Wilson D.V., Eberhart S.W., Robinson N.E. Cardiovascular responses to exogenous platelet-activating factor (PAF) in anesthetized ponies, and the effects of a PAF antagonist, WEB 2086. Am J Vet Res. 1993;54(2):274–279. [PubMed] [Google Scholar]
- 427.Jarvis G.E., Evans R.J. Platelet-activating factor and not thromboxane A2 is an important mediator of endotoxin-induced platelet aggregation in equine heparinised whole blood in vitro. Blood Coagul Fibrinolysis. 1996;7(2):194–198. doi: 10.1097/00001721-199603000-00021. [DOI] [PubMed] [Google Scholar]
- 428.King J.N., Gerring E.L. Antagonism of endotoxin-induced disruption of equine gastrointestinal motility with the platelet-activating factor antagonist WEB 2086. J Vet Pharmacol Ther. 1990;13(4):333–339. doi: 10.1111/j.1365-2885.1990.tb00786.x. [DOI] [PubMed] [Google Scholar]
- 429.Mills P.C., Ng J.C., Seawright A.A. Kinetics, dose response, tachyphylaxis and cross-tachyphylaxis of vascular leakage induced by endotoxin, zymosan-activated plasma and platelet-activating factor in the horse. J Vet Pharmacol Ther. 1995;18(3):204–209. doi: 10.1111/j.1365-2885.1995.tb00579.x. [DOI] [PubMed] [Google Scholar]
- 430.Dawson J., Lees P., Sedgwick A.D. Platelet activating factor as a mediator of equine cell locomotion. Vet Res Commun. 1988;12(2-3):101–107. doi: 10.1007/BF00362788. [DOI] [PubMed] [Google Scholar]
- 431.Foster A.P., Lees P., Cunningham F.M. Platelet activating factor is a mediator of equine neutrophil and eosinophil migration in vitro. Res Vet Sci. 1992;53(2):223–229. doi: 10.1016/0034-5288(92)90114-h. [DOI] [PubMed] [Google Scholar]
- 432.Carrick J.B., Morris D.D., Moore J.N. Administration of a receptor antagonist for platelet-activating factor during equine endotoxaemia. Equine Vet J. 1993;25(2):152–157. doi: 10.1111/j.2042-3306.1993.tb02927.x. [DOI] [PubMed] [Google Scholar]
- 433.Bertone J.J., Gossett K.A., Shoemaker K.E. Effect of hypertonic vs isotonic saline solution on responses to sublethal Escherichia coli endotoxemia in horses. Am J Vet Res. 1990;51(7):999–1007. [PubMed] [Google Scholar]
- 434.Pantaleon L.G., Furr M.O., McKenzie H.C., 2nd, Donaldson L. Cardiovascular and pulmonary effects of hetastarch plus hypertonic saline solutions during experimental endotoxemia in anesthetized horses. J Vet Int Med. 2006;20(6):1422–1428. doi: 10.1892/0891-6640(2006)20[1422:capeoh]2.0.co;2. [DOI] [PubMed] [Google Scholar]
- 435.McFarlane D. Hetastarch: a synthetic colloid with potential in equine patients. Compend Cont Educ Pract Vet. 1999;21(9):867–877. [Google Scholar]
- 436.Jones P.A., Bain F.T., Byars T.D. Effect of hydroxyethyl starch infusion on colloid oncotic pressure in hypoproteinemic horses. J Am Vet Med Assoc. 2001;218(7):1130–1135. doi: 10.2460/javma.2001.218.1130. [DOI] [PubMed] [Google Scholar]
- 437.Cohen N.D., Divers T. Proceedings of the Fifteenth American College of Veterinary Internal Medicine Forum. Lake Buena Vista; Fla: 1997. Equine colitis. [Google Scholar]
- 438.Turkan H., Ural A., Beyan C. Effects of hydroxyethyl starch on blood coagulation profile. Eur J Anaesthesiol. 1999;16(3):156–159. doi: 10.1046/j.1365-2346.1999.00407.x. [DOI] [PubMed] [Google Scholar]
- 439.Jones P.A., Tomasic M., Gentry P.A. Oncotic, hemodilutional, and hemostatic effects of isotonic saline and hydroxyethyl starch solutions in clinically normal ponies. Am J Vet Res. 1997;58(5):541–548. [PubMed] [Google Scholar]
- 440.Meister D., Hermann M., Mathis G.A. Kinetics of hydroxyethyl starch in horses. Schweiz Arch Tierheilkd. 1992;134(7):329–339. [PubMed] [Google Scholar]
- 441.Moore J.N., Garner H.E., Shapland J.E. Lactic acidosis and arterial hypoxemia during sublethal endotoxemia in conscious ponies. Am J Vet Res. 1980;41(10):1696–1698. [PubMed] [Google Scholar]
- 442.Hosgood G. Pharmacologic features and physiologic effects of dopamine. J Am Vet Med Assoc. 1990;197(9):1209–1211. [PubMed] [Google Scholar]
- 443.Corley K.T., McKenzie H.C., Amoroso L.M. Initial experience with norepinephrine infusion in hypotensive critically ill foals. J Vet Emerg Crit Care. 2000;10(4):267–276. [Google Scholar]
- 444.Moore B.R., Hinchcliff K.W. Heparin: a review of its pharmacology and therapeutic use in horses. J Vet Intern Med. 1994;8(1):26–35. doi: 10.1111/j.1939-1676.1994.tb03192.x. [DOI] [PubMed] [Google Scholar]
- 445.Mahaffey E.A., Moore J.N. Erythrocyte agglutination associated with heparin treatment in three horses. J Am Vet Med Assoc. 1986;189(11):1478–1480. [PubMed] [Google Scholar]
- 446.Moore J.N., Mahaffey E.A., Zboran M. Heparin-induced agglutination of erythrocytes in horses. Am J Vet Res. 1987;48(1):68–71. [PubMed] [Google Scholar]
- 447.Monreal L., Villatoro A.J., Monreal M. Comparison of the effects of low-molecular-weight and unfractioned heparin in horses. Am J Vet Res. 1995;56(10):1281–1285. [PubMed] [Google Scholar]
- 448.Weiss D.J., Evanson O.A., McClenahan D. Evaluation of platelet activation and platelet-neutrophil aggregates in ponies with alimentary laminitis. Am J Vet Res. 1997;58(12):1376–1380. [PubMed] [Google Scholar]
- 449.Jarvis G.E., Evans R.J. Endotoxin-induced platelet aggregation in heparinised equine whole blood in vitro. Res Vet Sci. 1994;57(3):317–324. doi: 10.1016/0034-5288(94)90124-4. [DOI] [PubMed] [Google Scholar]
- 450.Daels P.F., Starr M., Kindahl H. Effect of Salmonella typhimurium endotoxin on PGF-2 alpha release and fetal death in the mare. J Reprod Fertil Suppl. 1987;35:485–492. [PubMed] [Google Scholar]
- 451.Immegart H.M. Abnormalities of pregnancy. In: Youngquist R.S., editor. Current Therapy in Large Animal Theriogenology. WB Saunders; Philadelphia: 1997. [Google Scholar]
- 452.Baxter G.M. Alterations of endothelium-dependent digital vascular responses in horses given low-dose endotoxin. Vet Surg. 1995;24(2):87–96. doi: 10.1111/j.1532-950x.1995.tb01301.x. [DOI] [PubMed] [Google Scholar]
- 453.Pollitt C.C., van Eps A.W. Prolonged, continuous distal limb cryotherapy in the horse. Equine Vet J. 2004;36:216–220. doi: 10.2746/0425164044877152. [DOI] [PubMed] [Google Scholar]
- 454.van Eps A.W. Progress towards effective prevention and therapy for laminitis. Equine Vet J. 2012;44:746–748. doi: 10.1111/j.2042-3306.2012.00667.x. [DOI] [PubMed] [Google Scholar]
- 455.Van Eps A.W., Pollitt C.C. Equine laminitis model: cryotherapy reduces the severity of lesions evaluated seven days after induction with oligofructose. Equine Vet J. 2009;41:741–746. doi: 10.2746/042516409x434116. [DOI] [PubMed] [Google Scholar]
- 456.van Eps A.W., Pollitt C.C., Underwood C. Continuous digital hypothermia initiated after the onset of lameness prevents lamellar failure in the oligofructose laminitis model. Equine Vet J. 2014;46:625–630. doi: 10.1111/evj.12180. [DOI] [PubMed] [Google Scholar]
- 457.Kullmann A., Holcombe S.J., Hurcombe S.D. Prophylactic digital cryotherapy is associated with decreased incidence of laminitis in horses diagnosed with colitis. Equine Vet J. 2014;46:554–559. doi: 10.1111/evj.12156. [DOI] [PubMed] [Google Scholar]
- 458.van Eps A.W., Orsini J.A. A comparison of seven methods for continuous therapeutic cooling of the equine digit. Equine Vet J. 2016;48:120–124. doi: 10.1111/evj.12384. [DOI] [PubMed] [Google Scholar]
- 459.Blackwell R.B., White N.A. Duodenitis-proximal jejunitis in the horse. Proc Equine Colic Res Symp. 1982;1:106. [Google Scholar]
- 460.Seahorn T.L., Cornick J.L., Cohen N.D. Prognostic indicators for horses with duodenitis-proximal jejunitis—75 Horses (1985–1989) J Vet Intern Med. 1992;6:307–311. doi: 10.1111/j.1939-1676.1992.tb00360.x. [DOI] [PubMed] [Google Scholar]
- 461.White N.A., Tyler D.E., Blackwell R.B. Hemorrhagic fibrinonecrotic duodenitis-proximal jejunitis in horses: 20 cases (1977-1984) J Am Vet Med Assoc. 1987;190:311–315. [PubMed] [Google Scholar]
- 462.Davis J.L., Blikslager A.T., Catto K. A retrospective analysis of hepatic injury in horses with proximal enteritis (1984–2002) J Vet Intern Med. 2003;17:896–901. doi: 10.1111/j.1939-1676.2003.tb02530.x. [DOI] [PubMed] [Google Scholar]
- 463.Arroyo L.G., Staempfli H., Rousseau J.D. Culture evaluation of Clostridium spp. in the nasogastric reflux of horses with duodenitis proximal jejunitis. Proc Equine Colic Res Symp. 2005;8:51–52. [Google Scholar]
- 464.Schumacher J., Mullen J., Shelby R. An investigation of the role of Fusarium moniliforme in duodenitis/proximal jejunitis of horses. Vet Hum Toxicol. 1995;37:39–45. [PubMed] [Google Scholar]
- 465.Cohen N.D., Toby E., Roussel A.J. Are feeding practices associated with duodenitis-proximal jejunitis? Equine Vet J. 2006;38:526–531. doi: 10.2746/042516406x155975. [DOI] [PubMed] [Google Scholar]
- 466.Freeman D.E. Duodenitis-proximal jejunitis. Equine Vet Educ. 2000;12:322–332. [Google Scholar]
- 467.Morris D.D., Johnston J.K. Peritoneal fluid constituents in horses with colic due to small intestinal disease. Proc Equine Colic Res Symp. 1986;2 [Google Scholar]
- 468.Desrochers A.M. Abdominal ultrasonography of normal and colicky adult horses. Proc AAEP Focus on Colic. 2005:20–26. [Google Scholar]
- 469.Lammers T.W., Roussel A.J., Boothe D.M. Effect of an indwelling nasogastric tube on gastric emptying rates of liquids in horses. Am J Vet Res. 2005;66:642–645. doi: 10.2460/ajvr.2005.66.642. [DOI] [PubMed] [Google Scholar]
- 470.Sellon D.C., Roberts M.C., Blikslager A.T. Effects of continuous rate intravenous infusion of butorphanol on physiologic and outcome variables in horses after celiotomy. J Vet Intern Med. 2004;18:555–563. doi: 10.1892/0891-6640(2004)18<555:eocrii>2.0.co;2. [DOI] [PubMed] [Google Scholar]
- 471.Durham A.E., Phillips T.J., Walmsley J.P. Nutritional and clinicopathological effects of post operative parenteral nutrition following small intestinal resection and anastomosis in the mature horse. Equine Vet J. 2004;36:390–396. doi: 10.2746/0425164044868369. [DOI] [PubMed] [Google Scholar]
- 472.Durham A.E., Phillips T.J., Walmsley J.P. Study of the clinical effects of postoperative parenteral nutrition in 15 horses. Vet Rec. 2003;153:493–498. doi: 10.1136/vr.153.16.493. [DOI] [PubMed] [Google Scholar]
- 473.Koenig J.B., Sawhney S., Cote N. Effect of intraluminal distension or ischemic strangulation obstruction of the equine jejunum on jejunal motilin receptors and binding of erythromycin lactobionate. Am J Vet Res. 2006;67:815–820. doi: 10.2460/ajvr.67.5.815. [DOI] [PubMed] [Google Scholar]
- 474.Cook V.L., Blikslager A.T. Use of systemically administered lidocaine in horses with gastrointestinal tract disease. J Am Vet Med Assoc. 2008;232:1144–1148. doi: 10.2460/javma.232.8.1144. [DOI] [PubMed] [Google Scholar]
- 475.Malone E., Ensink J., Turner T. Intravenous continuous infusion of lidocaine for treatment of equine ileus. Vet Surg. 2006;35:60–66. doi: 10.1111/j.1532-950X.2005.00113.x. [DOI] [PubMed] [Google Scholar]
- 476.Koenig J.B., Cote N., LaMarre J. Binding of radiolabeled porcine motilin and erythromycin lactobionate to smooth muscle membranes in various segments of the equine gastrointestinal tract. Am J Vet Res. 2002;63:1545–1550. doi: 10.2460/ajvr.2002.63.1545. [DOI] [PubMed] [Google Scholar]
- 477.Cohen N.D., Faber N.A., Brumbaugh G.W. Use of bethanechol and metoclopramide in horses with duodenitis proximal jejunitis: 13 cases (1987–1993) J Equine Vet Sci. 1995;15:492–494. [Google Scholar]
- 478.Leeth B., Robertson J.T. A retrospective comparison of surgical to medical management of proximal enteritis in the horse. Proc Am Assoc Equine Pract. 1989;34:69–79. [Google Scholar]
- 479.Edwards G.B. Duodenitis-proximal jejunitis (anterior enteritis) as a surgical problem. Equine Vet Educ. 2000;12:318–321. [Google Scholar]
- 480.Oue Y., Ishihara R., Edamatsu H. Isolation of an equine coronavirus from adult horses with pyrogenic and enteric disease and its antigenic and genomic characterization in comparison with the NC99 strain. Vet Microbiol. 2011;150:41–48. doi: 10.1016/j.vetmic.2011.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 481.Oue Y., Morita Y., Kondo T. Epidemic of equine coronavirus at Obihiro Racecourse, Hokkaido, Japan in 2012. J Vet Med Sci. 2013;75:1261–1265. doi: 10.1292/jvms.13-0056. [DOI] [PubMed] [Google Scholar]
- 482.Pusterla N., Mapes S., Wademan C. Emerging outbreaks associated with equine coronavirus in adult horses. Vet Microbiol. 2013;162:228–231. doi: 10.1016/j.vetmic.2012.10.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 483.Davis E., Rush B.R., Cox J. Neonatal enterocolitis associated with coronavirus infection in a foal: a case report. J Vet Diagn Invest. 2000;12:153–156. doi: 10.1177/104063870001200210. [DOI] [PubMed] [Google Scholar]
- 484.Guy J.S., Breslin J.J., Breuhaus B. Characterization of a coronavirus isolated from a diarrheic foal. J Clin Microbiol. 2000;38:4523–4526. doi: 10.1128/jcm.38.12.4523-4526.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 485.Giannitti F., Diab S., Mete A. Necrotizing enteritis and hyperammonemic encephalopathy associated with equine coronavirus infection in equids. Vet Pathol. 2015;52:1148–1156. doi: 10.1177/0300985814568683. [DOI] [PubMed] [Google Scholar]
- 486.Fielding C.L., Higgins J.K., Higgins J.C. Disease associated with equine coronavirus infection and high case fatality rate. J Vet Intern Med. 2015;29:307–310. doi: 10.1111/jvim.12480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 487.Roberts M.C., Kidder D.E., Hill F.W. Small intestinal beta-galactosidase activity in the horse. Gut. 1973;14:535–540. doi: 10.1136/gut.14.7.535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 488.Roberts M.C. Malabsorption-syndromes in the horse. Comp Contin Educ Pract Vet. 1985;7:S637–S646. [Google Scholar]
- 489.Lindberg R., Nygren A., Persson S.G. Rectal biopsy diagnosis in horses with clinical signs of intestinal disorders: a retrospective study of 116 cases. Equine Vet J. 1996;28:275–284. doi: 10.1111/j.2042-3306.1996.tb03091.x. [DOI] [PubMed] [Google Scholar]
- 490.Kemper D.L., Perkins G.A., Schumacher J. Equine lymphocytic-plasmacytic enterocolitis: a retrospective study of 14 cases. Equine Vet J Suppl. 2000:108–112. doi: 10.1111/j.2042-3306.2000.tb05346.x. [DOI] [PubMed] [Google Scholar]
- 491.Gibson K.T., Alders R.G. Eosinophilic enterocolitis and dermatitis in two horses. Equine Vet J. 1987;19:247–252. doi: 10.1111/j.2042-3306.1987.tb01397.x. [DOI] [PubMed] [Google Scholar]
- 492.Church S., Middleton D.J. Transient glucose malabsorption in two horses—fact or artefact? Aus Vet J. 1997;75:716–718. doi: 10.1111/j.1751-0813.1997.tb12251.x. [DOI] [PubMed] [Google Scholar]
- 493.Love S., Mair T.S., Hillyer M.H. Chronic diarrhea in adult horses—a review of 51 referred cases. Vet Rec. 1992;130:217–219. doi: 10.1136/vr.130.11.217. [DOI] [PubMed] [Google Scholar]
- 494.Freeman D.E. In vitro concentrative accumulation of D-xylose by jejunum from horses and rabbits. Am J Vet Res. 1993;54:965–969. [PubMed] [Google Scholar]
- 495.Brown C.M. The diagnostic value of the d-xylose absorption test in horses with unexplained chronic weight loss. Brit Vet Jl. 1992;148:41–44. doi: 10.1016/0007-1935(92)90065-9. [DOI] [PubMed] [Google Scholar]
- 496.Roberts M.C. Small intestinal malabsorption in horses. Equine Vet Educ. 2000;12:214–219. [Google Scholar]
- 497.Haven M., Roberts M., Argenzio R. Intestinal adaptation following 70% small bowel resection in the horse. Pferdeheilkunde. 1992:86–87. [Google Scholar]
- 498.Tate L.P., Jr., Ralston S.L., Koch C.M. Effects of extensive resection of the small intestine in the pony. Am J Vet Res. 1983;44:1187–1191. [PubMed] [Google Scholar]
- 499.Lindberg R., Persson S.G., Jones B. Clinical and pathophysiological features of granulomatous enteritis and eosinophilic granulomatosis in the horse. Zentralbl Veterinarmed A. 1985;32:526–539. doi: 10.1111/j.1439-0442.1985.tb01973.x. [DOI] [PubMed] [Google Scholar]
- 500.Duryea J.H., Ainsworth D.M., Mauldin E.A. Clinical remission of granulomatous enteritis in a Standardbred gelding following long-term dexamethasone administration. Equine Vet J. 1997;29:164–167. doi: 10.1111/j.2042-3306.1997.tb01662.x. [DOI] [PubMed] [Google Scholar]
- 501.Roberts M.C., Pinsent P.J. Malabsorption in the horse associated with alimentary lymphosarcoma. Equine Vet J. 1975;7:166–172. doi: 10.1111/j.2042-3306.1975.tb03259.x. [DOI] [PubMed] [Google Scholar]
- 502.van den Hoven R., Franken P. Clinical aspects of lymphosarcoma in the horse: a clinical report of 16 cases. Equine Vet J. 1983;15:49–53. doi: 10.1111/j.2042-3306.1983.tb01702.x. [DOI] [PubMed] [Google Scholar]
- 503.Platt H. Alimentary lymphomas in the horse. J Comp Pathol. 1987;97:1–10. doi: 10.1016/0021-9975(87)90121-6. [DOI] [PubMed] [Google Scholar]
- 504.Cimprich R.E. Equine granulomatous enteritis. Vet Pathol. 1974;11:535–547. doi: 10.1177/030098587401100608. [DOI] [PubMed] [Google Scholar]
- 505.Schumacher J., Edwards J.F., Cohen N.D. Chronic idiopathic inflammatory bowel diseases of the horse. J Vet Intern Med. 2000;14:258–265. doi: 10.1892/0891-6640(2000)014<0258:ciibdo>2.3.co;2. [DOI] [PubMed] [Google Scholar]
- 506.Lindberg R. Pathology of equine granulomatous enteritis. J Comp Pathol. 1984;94:233–247. doi: 10.1016/0021-9975(84)90043-4. [DOI] [PubMed] [Google Scholar]
- 507.Merritt A.M., Cimprich R.E., Beech J. Granulomatous enteritis in nine horses. J Am Vet Med Assoc. 1976;169:603–609. [PubMed] [Google Scholar]
- 508.Fogarty U., Perl D., Good P. A cluster of equine granulomatous enteritis cases: the link with aluminium. Vet Hum Toxicol. 1998;40:297–305. [PubMed] [Google Scholar]
- 509.Collery P., McElroy M., Sammin D. Equine granulomatous enteritis linked with aluminum? Vet Hum Toxicol. 1999;41:49–50. [PubMed] [Google Scholar]
- 510.Meuten D.J., Butler D.G., Thomson G.W. Chronic enteritis associated with malabsorption and protein-losing enteropathy in the horse. J Am Vet Med Assoc. 1978;172:326–333. [PubMed] [Google Scholar]
- 511.Pass D.A., Bolton J.R. Chronic eosinophilic gastroenteritis in the horse. Vet Pathol. 1982;19:486–496. doi: 10.1177/030098588201900504. [DOI] [PubMed] [Google Scholar]
- 512.Nimmo Wilkie J.S., Yager J.A., Nation P.N. Chronic eosinophilic dermatitis: a manifestation of a multisystemic, eosinophilic, epitheliotropic disease in five horses. Vet Pathol. 1985;22:297–305. doi: 10.1177/030098588502200401. [DOI] [PubMed] [Google Scholar]
- 513.Pass D.A., Bolton J.R., Mills J.N. Basophilic enterocolitis in a horse. Vet Pathol. 1984;21:362–364. doi: 10.1177/030098588402100318. [DOI] [PubMed] [Google Scholar]
- 514.Lindberg R., Karlsson L. Topography and enterocyte morphology of the small bowel mucosal surface in equine granulomatous enteritis. J Comp Pathol. 1985;95:65–78. doi: 10.1016/0021-9975(85)90078-7. [DOI] [PubMed] [Google Scholar]
- 515.Platt H. Chronic inflammatory and lymphoproliferative lesions of the equine small intestine. J Comp Pathol. 1986;96:671–684. doi: 10.1016/0021-9975(86)90063-0. [DOI] [PubMed] [Google Scholar]
- 516.Edwards G.B., Kelly D.F., Proudman C.J. Segmental eosinophilic colitis: a review of 22 cases. Equine Vet J Suppl. 2000:86–93. doi: 10.1111/j.2042-3306.2000.tb05341.x. [DOI] [PubMed] [Google Scholar]
- 517.Scott E.A., Heidel J.R., Snyder S.P. Inflammatory bowel disease in horses: 11 cases (1988–1998) J Am Vet Med Assoc. 1999;214:1527–1530. [PubMed] [Google Scholar]
- 518.Lavoie J.P., Drolet R., Parsons D. Equine proliferative enteropathy: a cause of weight loss, colic, diarrhoea and hypoproteinaemia in foals on three breeding farms in Canada. Equine Vet J. 2000;32:418–425. doi: 10.2746/042516400777591110. [DOI] [PubMed] [Google Scholar]
- 519.Frazer M. Lawsonia intracellularis infection in horses: 2005-2007. J Vet Intern Med. 2008;22:1243–1248. doi: 10.1111/j.1939-1676.2008.0160.x. [DOI] [PubMed] [Google Scholar]
- 520.McClintock S.A., Collins A.M. Lawsonia intracellularis proliferative enteropathy in a weanling foal in Australia. Aust Vet J. 2004;82:750–752. doi: 10.1111/j.1751-0813.2004.tb13236.x. [DOI] [PubMed] [Google Scholar]
- 521.McGurrin M.K., Vengust M., Arroyo L.G. An outbreak of Lawsonia intracellularis infection in a standardbred herd in Ontario. Can Vet J. 2007;48:927–930. [PMC free article] [PubMed] [Google Scholar]
- 522.Pusterla N., Gebhart C. Lawsonia intracellularis infection and proliferative enteropathy in foals. Vet Microbiol. 2013 doi: 10.1016/j.vetmic.2013.06.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 523.Wilson J.H., Gebhart C.J. Lawsonia proliferative enteropathy in foals: clinical features and piglet parallels. Proc AAEP Focus on the First Year of Life. 2008 [Google Scholar]
- 524.Williams N.M., Harrison L.R., Gebhart C.J. Proliferative enteropathy in a foal caused by Lawsonia intracellularis-like bacterium. J Vet Diagn Invest. 1996;8:254–256. doi: 10.1177/104063879600800220. [DOI] [PubMed] [Google Scholar]
- 525.Cooper D.M., Swanson D.L., Gebhart C.J. Diagnosis of proliferative enteritis in frozen and formalin-fixed, paraffin-embedded tissues from a hamster, horse, deer and ostrich using a Lawsonia intracellularis-specific multiplex PCR assay. Vet Microbiol. 1997;54:47–62. doi: 10.1016/s0378-1135(96)01264-3. [DOI] [PubMed] [Google Scholar]
- 526.Page A.E., Stills H.F., Jr., Horohov D.W. The effect of passively acquired antibodies on Lawsonia intracellularis infection and immunity in the horse. Equine Vet J. 2015;47:655–661. doi: 10.1111/evj.12335. [DOI] [PubMed] [Google Scholar]
- 527.Sampieri F., Hinchcliff K.W., Toribio R.E. Tetracycline therapy of Lawsonia intracellularis enteropathy in foals. Equine Vet J. 2006;38:89–92. doi: 10.2746/042516406775374270. [DOI] [PubMed] [Google Scholar]
- 528.Jacobson M., Aspan A., Königsson M.H. Routine diagnostics of Lawsonia intracellularis performed by PCR, serological and post mortem examination, with special emphasis on sample preparation methods for PCR. Vet Microbiol. 2004;102:189–201. doi: 10.1016/j.vetmic.2004.04.014. [DOI] [PubMed] [Google Scholar]
- 529.Page A.E., Slovis N.M., Horohov D.W. Lawsonia intracellularis and equine proliferative enteropathy. Vet Clin North Am Equine Pract. 2014;30:641–658. doi: 10.1016/j.cveq.2014.08.001. [DOI] [PubMed] [Google Scholar]
- 530.Dauvillier J., Picandet V., Harel J. Diagnostic and epidemiological features of Lawsonia intracellularis enteropathy in 2 foals. Can Vet J. 2006;47:689–691. [PMC free article] [PubMed] [Google Scholar]
- 531.Pusterla N., Vannucci F.A., Mapes S.M. Efficacy of an avirulent live vaccine against Lawsonia intracellularis in the prevention of proliferative enteropathy in experimentally infected weanling foals. Am J Vet Res. 2012;73:741–746. doi: 10.2460/ajvr.73.5.741. [DOI] [PubMed] [Google Scholar]
- 532.Hayden D.W., Johnson K.H., Wolf C.B. AA amyloid-associated gastroenteropathy in a horse. J Comp Pathol. 1988;98:195–204. doi: 10.1016/0021-9975(88)90018-7. [DOI] [PubMed] [Google Scholar]
- 533.MacKay R.J., Iverson W.O., Merritt A.M. Exuberant granulation tissue in the stomach of a horse. Equine Vet J. 1981;13:119–122. doi: 10.1111/j.2042-3306.1981.tb04135.x. [DOI] [PubMed] [Google Scholar]
- 534.Smith B.P. Salmonella infection in horses. Comp Cont Ed Pract Vet. 1981;3:S4–S17. [Google Scholar]
- 535.Smith B.P., Reina-Guerra M., Hardy A.J. Prevalence and epizootiology of equine salmonellosis. J Am Vet Med Assoc. 1978;172:353. [PubMed] [Google Scholar]
- 536.Donahue J.M. Emergence of antibiotic-resistant Salmonella agona in horses in Kentucky. J Am Vet Med Assoc. 1986;188:592. [PubMed] [Google Scholar]
- 537.Traub-Dargatz J.L., Garber L.P., Fedorka-Cray P.J. Fecal shedding of Salmonella spp by horses in the United States during 1998 and 1999 and detection of Salmonella spp in grain and concentrate sources on equine operations. J Am Vet Med Assoc. 2000;217:226. doi: 10.2460/javma.2000.217.226. [DOI] [PubMed] [Google Scholar]
- 538.Traub-Dargatz J.L., Salman M.D., Jones R.L. Epidemiologic study of salmonellae shedding in the feces of horses and potential risk factors for development of the infection in hospitalized horses. J Am Vet Med Assoc. 1990;196:1617. [PubMed] [Google Scholar]
- 539.House J.K., Mainar-Jaime R.C., Smith B.P. Risk factors for nosocomial Salmonella infection among hospitalized horses. J Am Vet Med Assoc. 1999;214:1511. [PubMed] [Google Scholar]
- 540.Schott H.C., Ewart S.L., Walker R.D., R.M An outbreak of salmonellosis among horses at a veterinary teaching hospital. J Am Vet Med Assoc. 2001;218:1152. doi: 10.2460/javma.2001.218.1152. [DOI] [PubMed] [Google Scholar]
- 541.Tillotson K., Savage C.J., Salman M.D. Outbreak of Salmonella infantis infection in a large animal veterinary teaching hospital. J Am Vet Med Assoc. 1997;211:1554. [PubMed] [Google Scholar]
- 542.Mainar-Jaime R.C., House J.K., Smith B.P. Influence of fecal shedding of Salmonella organisms on mortality in hospitalized horses. J Am Vet Med Assoc. 1998;213:1162. [PubMed] [Google Scholar]
- 543.Hartmann F.A., Callan R.J., McGuirk S.M. Control of an outbreak of salmonellosis caused by drug-resistant Salmonella anatum in horses at a veterinary hospital and measures to prevent future infections. J Am Vet Med Assoc. 1996;209:629. [PubMed] [Google Scholar]
- 544.Bucknell D.G., Gasser R.B., Irving A., Whithear K. Antimicrobial resistance in Salmonella and Escherichia coli isolated from horses. Aust Vet J. 1997;75:355. doi: 10.1111/j.1751-0813.1997.tb15713.x. [DOI] [PubMed] [Google Scholar]
- 545.Hartmann F.A., West S.E. Utilization of both phenotypic and molecular analyses to investigate an outbreak of multidrug-resistant in horses. Can J Vet Res. 1997;61:173. [PMC free article] [PubMed] [Google Scholar]
- 546.Dargatz D.A., Traub-Dargatz J.L. Multidrug-resistant Salmonella and nosocomial infections. Vet Clin North Am Equine Pract. 2004;20:587. doi: 10.1016/j.cveq.2004.07.008. [DOI] [PubMed] [Google Scholar]
- 547.Smith B.P. Understanding the role of endotoxins in gram-negative sepsis. Vet Med. 1986;12:1148. [Google Scholar]
- 548.Carter J.D., Hird D.W., Farver T.B., Hjerpe C.A. Salmonellosis in hospitalized horses: seasonality and case fatality rates. J Am Vet Med Assoc. 1986;188:163. [PubMed] [Google Scholar]
- 549.Morse E.V., Duncan M.A., Page E.A., Fessler J.F. Salmonellosis in Equidae: a study of 23 cases. Cornell Vet. 1976;66:198. [PubMed] [Google Scholar]
- 550.Ernst N.S., Hernandez J.A., MacKay R.J. Risk factors associated with fecal Salmonella shedding among hospitalized horses with signs of gastrointestinal tract disease. J Am Vet Med Assoc. 2004;225:275. doi: 10.2460/javma.2004.225.275. [DOI] [PubMed] [Google Scholar]
- 551.Kim L.M., Morley P.S., Traub-Dargatz J.L., Salman M.D., Gentry-Weeks C. Factors associated with Salmonella shedding among equine colic patients at a veterinary teaching hospital. J Am Vet Med Assoc. 2001;218:740. doi: 10.2460/javma.2001.218.740. [DOI] [PubMed] [Google Scholar]
- 552.Giannella R.A. Pathogenesis of acute bacterial diarrheal disorders. Annu Rev Med. 1981;32:341. doi: 10.1146/annurev.me.32.020181.002013. [DOI] [PubMed] [Google Scholar]
- 553.Hirsh D.C. The alimentary canal as a microbial habitat. In: Biberstein E.L., Zee Y.C., editors. Review of Veterinary Microbiology. Blackwell Scientific; Boston: 1990. p. 93. [Google Scholar]
- 554.Selsted M.E., Miller S.I., Henschen A.H., Ouellette A.J. Enteric defensins: antibiotic peptide components of intestinal host defense. J Cell Biol. 1992;118:929. doi: 10.1083/jcb.118.4.929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 555.Brandtzaeg P., Baekkevold E.S., Farstad I.N. Regional specialization in the mucosal immune system: what happens in the microcompartments? Immunol Today. 1999;20:141. doi: 10.1016/s0167-5699(98)01413-3. [DOI] [PubMed] [Google Scholar]
- 556.Ohl M.E., Miller S.I. Salmonella: a model for bacterial pathogenesis. Annu Rev Med. 2001;52:259. doi: 10.1146/annurev.med.52.1.259. [DOI] [PubMed] [Google Scholar]
- 557.Clarke R.C., Gyles C.L. Salmonella. In: Gyles C.L., Thoen C.O., editors. Pathogenesis of Bacterial Infections in Animals. Iowa State University Press; Ames, IA: 2001. p. 95. [Google Scholar]
- 558.Hirsh D.C. Salmonella. In: Biberstein E.L., Zee Y.C., editors. Review of veterinary microbiology. Blackwell Scientific; Boston: 2001. p. 110. [Google Scholar]
- 559.Smith B.P., Hardy A.J., Reina-Guerra M. A preliminary evaluation of some preparations of Salmonella typhimurium vaccines in horses. In: Moore J.N., White N.A., Becht J.L., editors. Proceedings of the first Equine Colic Symposium. Veterinary Learning Systems; Lawrenceville, NJ: 1982. p. 211. [Google Scholar]
- 560.Smith B.P., Reina-Guerra M., Hoiseth S.K. Aromatic-dependent Salmonella typhimurium as modified live vaccines for calves. Am J Vet Res. 1984;45:59. [PubMed] [Google Scholar]
- 561.Sheoran A.S., Timoney J.F., Tinge S.A., Sundaram P., Curtiss R. Intranasal immunogenicity of a Delta cya Delta crp-pabA mutant of Salmonella enterica serotype Typhimurium for the horse. Vaccine. 2001;19:3787. doi: 10.1016/s0264-410x(01)00091-3. [DOI] [PubMed] [Google Scholar]
- 562.Sansonetti P.J., Phalipon A. M cells as ports of entry for enteroinvasive pathogens: mechanisms of interaction, consequences for the disease process. Semin Immunol. 1999;11:193. doi: 10.1006/smim.1999.0175. [DOI] [PubMed] [Google Scholar]
- 563.Galan J.E., Collmer A. Type III secretion machines: bacterial devices for protein delivery into host cells. Science. 1999;284:1322. doi: 10.1126/science.284.5418.1322. [DOI] [PubMed] [Google Scholar]
- 564.Vazquez-Torres A., Jones-Carson J., Baumler A.J. Extraintestinal dissemination of Salmonella by CD18-expressing phagocytes. Nature. 1999;401:804. doi: 10.1038/44593. [DOI] [PubMed] [Google Scholar]
- 565.Vazquez-Torres A., Fang F.C. Oxygen-dependent anti-Salmonella activity of macrophages. Trends Microbiol. 2001;9:29. doi: 10.1016/s0966-842x(00)01897-7. [DOI] [PubMed] [Google Scholar]
- 566.Koo F.C., Peterson J.W., Houston C.W., Molina N.C. Pathogenesis of experimental salmonellosis: inhibition of protein synthesis by cytotoxin. Infect Immun. 1984;43:93. doi: 10.1128/iai.43.1.93-100.1984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 567.Giannella R.A., Gots R.E., Charney A.N., Greenough W.B., Formal S.B. Pathogenesis of Salmonella-mediated intestinal fluid secretion. Activation of adenylate cyclase and inhibition by indomethacin. Gastroenterology. 1975;69:1238. [PubMed] [Google Scholar]
- 568.Peterson J.W., Molina N.C., Houston C.W., Fader R.C. Elevated cAMP in intestinal epithelial cells during experimental cholera and salmonellosis. Toxicon. 1983;21:761. doi: 10.1016/0041-0101(83)90065-x. [DOI] [PubMed] [Google Scholar]
- 569.Chopra A.K., Huang J.H., Xu X. Role of Salmonella enterotoxin in overall virulence of the organism. Microb Pathog. 1999;27:155. doi: 10.1006/mpat.1999.0294. [DOI] [PubMed] [Google Scholar]
- 570.Watson P.R., Galyov E.E., Paulin S.M. Mutation of invH, but not stn, reduces Salmonella-induced enteritis in cattle. Infect Immun. 1432;66 doi: 10.1128/iai.66.4.1432-1438.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 571.Murray M.J. Enterotoxin activity of a Salmonella typhimurium of equine origin in vivo in rabbits and the effect of Salmonella culture lysates and cholera toxin on equine colonic mucosa in vitro. Am J Vet Res. 1986;47:769. [PubMed] [Google Scholar]
- 572.Murray M.J. Digestive physiology of the large intestine in adult horses. Part II: pathophysiology of colitis. Comp Cont Ed Pract Vet. 1988;10:1309. [Google Scholar]
- 573.Powell D.W. Neuroimmunophysiology of the gastrointestinal mucosa: implications for inflammatory diseases. Trans Am Clin Climatol Assoc. 1994;106:124. [PMC free article] [PubMed] [Google Scholar]
- 574.Madara J.L. Review article: pathobiology of neutrophil interactions with intestinal epithelia. Aliment Pharmacol Ther. 2000;11(suppl 3):57. doi: 10.1111/j.1365-2036.1997.tb00809.x. [DOI] [PubMed] [Google Scholar]
- 575.Smith B.P., Reina-Guerra M., Hardy A.J., Habasha F. Equine salmonellosis: experimental production of four syndromes. Am J Vet Res. 1979;40:1072. [PubMed] [Google Scholar]
- 576.Cohen N.D., Woods A.M. Characteristics and risk factors for failure to survive of horses with acute diarrhea: 122 cases (1990-1996) J Am Vet Med Assoc. 1999;214:382. [PubMed] [Google Scholar]
- 577.van Duijkeren E., Flemming C., van Oldruitenborgh-Oosterbaan M.S., H Diagnosing salmonellosis in horses. Culturing of multiple versus single faecal samples. Vet Q. 1995;17:63. doi: 10.1080/01652176.1995.9694534. [DOI] [PubMed] [Google Scholar]
- 578.Palmer J.E., Whitlock R.H., Benson C.E. Comparison of rectal mucosal cultures and fecal cultures in detecting Salmonella infection in horses and cattle. Am J Vet Res. 1985;46:697. [PubMed] [Google Scholar]
- 579.Cohen N.D., Martin L.J., Simpson R.B. Comparison of polymerase chain reaction and microbiological culture for detection of salmonellae in equine feces and environmental samples. Am J Vet Res. 1996;57:780. [PubMed] [Google Scholar]
- 580.Amavisit P., Browning G.F., Lightfoot D. Rapid PCR detection of Salmonella in horse faecal samples. Vet Microbiol. 2001;79:63. doi: 10.1016/s0378-1135(00)00340-0. [DOI] [PubMed] [Google Scholar]
- 581.Kurowski P.B., Traub-Dargatz J.L., Morley P.S., Gentry-Weeks C.R. Detection of Salmonella spp in fecal specimens by use of real-time polymerase chain reaction assay. Am J Vet Res. 2002;63:1265. doi: 10.2460/ajvr.2002.63.1265. [DOI] [PubMed] [Google Scholar]
- 582.Dumler J.S., Barbet A.F., Bekker C.P. Reorganization of genera in the families Rickettsiaceae and Anaplasmataceae in the order Rickettsiales: unification of some species of Ehrlichia with Anaplasma, Cowdria with Ehrlichia and Ehrlichia with Neorickettsia, descriptions of six new species combinations and designation of Ehrlichia equi and ‘HGE agent’ as subjective synonyms of Ehrlichia phagocytophila. Int J Syst Evol Microbiol. 2001;51:2145. doi: 10.1099/00207713-51-6-2145. [DOI] [PubMed] [Google Scholar]
- 583.Palmer J.E. Potomac horse fever. Vet Clin North Am Equine Pract. 1993;9:399. doi: 10.1016/s0749-0739(17)30406-6. [DOI] [PubMed] [Google Scholar]
- 584.Mulville P. Equine monocytic ehrlichiosis (Potomac horse fever): a review. Equine Vet J. 1991;23:400. doi: 10.1111/j.2042-3306.1991.tb03749.x. [DOI] [PubMed] [Google Scholar]
- 585.Dutta S.K., Myrup A.C., Rice R.M. Experimental reproduction of Potomac horse fever in horses with a newly isolated Ehrlichia organism. J Clin Microbiol. 1985;22:265. doi: 10.1128/jcm.22.2.265-269.1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 586.Rikihisa Y., Perry B.D. Causative ehrlichial organisms in Potomac horse fever. Infect Immun. 1985;49:513. doi: 10.1128/iai.49.3.513-517.1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 587.Madigan J.E., Pusterla N. Ehrlichial diseases. Vet Clin North Am Equine Pract. 2000;16:487. doi: 10.1016/s0749-0739(17)30091-3. ix. [DOI] [PubMed] [Google Scholar]
- 588.Levine J.F., Levy M.G., Nicholson W.L., Gager R.B. Attempted Ehrlichia risticii transmission with Dermacentor variabilis (Acari: Ixodidae) J Med Entomol. 1990;27:931. doi: 10.1093/jmedent/27.5.931. [DOI] [PubMed] [Google Scholar]
- 589.Burg J.G., Roberts A.W., Williams N.M. Attempted transmission of Ehrlichia risticii (Rickettsiaceae) with Stomoxys calcitrans (Diptera: Muscidae) J Med Entomol. 1990;27:874. doi: 10.1093/jmedent/27.5.874. [DOI] [PubMed] [Google Scholar]
- 590.Reubel G.H., Barlough J.E., Madigan J.E. Production and characterization of Ehrlichia risticii, the agent of Potomac horse fever, from snails (Pleuroceridae: Juga spp.) in aquarium culture and genetic comparison to equine strains. J Clin Microbiol. 1998;36:1501. doi: 10.1128/jcm.36.6.1501-1511.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 591.Barlough J.E., Reubel G.H., Madigan J.E. Detection of Ehrlichia risticii, the agent of Potomac horse fever, in freshwater stream snails (Pleuroceridae: Juga spp.) from northern California. Appl Environ Microbiol. 1998;64:2888. doi: 10.1128/aem.64.8.2888-2893.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 592.Kanter M., Mott J., Ohashi N. Analysis of 16S rRNA and 51-kilodalton antigen gene and transmission in mice of Ehrlichia risticii in virgulate trematodes from Elimia livescens snails in Ohio. J Clin Microbiol. 2000;38:3349. doi: 10.1128/jcm.38.9.3349-3358.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 593.Mott J., Muramatsu Y., Seaton E. Molecular analysis of Neorickettsia risticii in adult aquatic insects in Pennsylvania, in horses infected by ingestion of insects, and isolated in cell culture. J Clin Microbiol. 2002;40:690. doi: 10.1128/JCM.40.2.690-693.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 594.Park B.K., Kim M.J., Kim E.H. Identification of trematode cercariae carrying Neorickettsia risticii in freshwater stream snails. Ann N Y Acad Sci. 2003;990:239–247. doi: 10.1111/j.1749-6632.2003.tb07371.x. [DOI] [PubMed] [Google Scholar]
- 595.Pusterla N., Johnson E.M., Chae J.S., Madigan J.E. Digenetic trematodes, Acanthatrium sp. and Lecithodendrium sp., as vectors of Neorickettsia risticii, the agent of Potomac horse fever. J Helminthol. 2003;77:335. doi: 10.1079/joh2003181. [DOI] [PubMed] [Google Scholar]
- 596.Gibson K.E., Rikihisa Y., Zhang C., Martin C. Neorickettsia risticii is vertically transmitted in the trematode Acanthatrium oregonense and horizontally transmitted to bats. Environ Microbiol. 2005;7:203. doi: 10.1111/j.1462-2920.2004.00683.x. [DOI] [PubMed] [Google Scholar]
- 597.Chae J.S., Pusterla N., Johnson E. Infection of aquatic insects with trematode metacercariae carrying Ehrlichia risticii, the cause of Potomac horse fever. J Med Entomol. 2000;37:619. doi: 10.1603/0022-2585-37.4.619. [DOI] [PubMed] [Google Scholar]
- 598.Pusterla N., Madigan J.E., Chae J.S. Helminthic transmission and isolation of Ehrlichia risticii, the causative agent of Potomac horse fever, by using trematode stages from freshwater stream snails. J Clin Microbiol. 2000;38:1293. doi: 10.1128/jcm.38.3.1293-1297.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 599.Pusterla N., Johnson E., Chae J. Infection rate of Ehrlichia risticii, the agent of Potomac horse fever, in freshwater stream snails (Juga yrekaensis) from northern California. Vet Parasitol. 2000;92:151. doi: 10.1016/s0304-4017(00)00309-5. [DOI] [PubMed] [Google Scholar]
- 600.Madigan J.E., Pusterla N., Johnson E. Transmission of Ehrlichia risticii, the agent of Potomac horse fever, using naturally infected aquatic insects and helminth vectors: preliminary report. Equine Vet J. 2000;32:275. doi: 10.2746/042516400777032219. [DOI] [PubMed] [Google Scholar]
- 601.Rikihisa Y., Perry B.D., Cordes D.O. Ultrastructural study of ehrlichial organisms in the large colons of ponies infected with Potomac horse fever. Infect Immun. 1985;49:505. doi: 10.1128/iai.49.3.505-512.1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 602.Rikihisa Y. Growth of Ehrlichia risticii in human colonic epithelial cells. Ann N Y Acad Sci. 1990;590:104. doi: 10.1111/j.1749-6632.1990.tb42212.x. [DOI] [PubMed] [Google Scholar]
- 603.Williams N.M., Cross R.J., Timoney P.J. Respiratory burst activity associated with phagocytosis of Ehrlichia risticii by mouse peritoneal macrophages. Res Vet Sci. 1994;57:194. doi: 10.1016/0034-5288(94)90057-4. [DOI] [PubMed] [Google Scholar]
- 604.Williams N.M., Timoney P.J. In vitro killing of Ehrlichia risticii by activated and immune mouse peritoneal macrophages. Infect Immun. 1993;61:861. doi: 10.1128/iai.61.3.861-867.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 605.Wells M.Y., Rikihisa Y. Lack of lysosomal fusion with phagosomes containing Ehrlichia risticii in P388D1 cells: abrogation of inhibition with oxytetracycline. Infect Immun. 1988;56:3209. doi: 10.1128/iai.56.12.3209-3215.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 606.Rikihisa Y., Johnson G.C., Cooke H.J. Pathophysiological changes in the large colon of horses infected with Ehrlichia risticii. In: Moore J.N., White S., Morris D.D., editors. Proceedings of the third equine colic symposium. Veterinary Learning Systems; Lawrenceville NJ: 1988. p. 93. [Google Scholar]
- 607.Dutta S.K., Penney B.E., Myrup A.C. Disease features in horses with induced equine monocytic ehrlichiosis (Potomac horse fever) Am J Vet Res. 1988;49:1747. [PubMed] [Google Scholar]
- 608.Ziemer E.L., Whitlock R.H., Palmer J.E., Spencer P.A. Clinical and hematologic variables in ponies with experimentally induced equine ehrlichial colitis (Potomac horse fever) Am J Vet Res. 1987;48:63. [PubMed] [Google Scholar]
- 609.Long M.T., Goetz T.E., Kakoma I. Evaluation of fetal infection and abortion in pregnant ponies experimentally infected with Ehrlichia risticii. Am J Vet Res. 1995;56:1307. [PubMed] [Google Scholar]
- 610.Long M.T., Goetz T.E., Whiteley H.E. Identification of Ehrlichia risticii as the causative agent of two equine abortions following natural maternal infection. J Vet Diagn Invest. 1995;7:201. doi: 10.1177/104063879500700206. [DOI] [PubMed] [Google Scholar]
- 611.Morris D.D., Messick J., Whitlock R.H. Effect of equine ehrlichial colitis on the hemostatic system in ponies. Am J Vet Res. 1988;49:1030. [PubMed] [Google Scholar]
- 612.Dutta S.K., Rice R.M., Hughes T.D. Detection of serum antibodies against Ehrlichia risticii in Potomac horse fever by enzyme-linked immunosorbent assay. Vet Immunol Immunopathol. 1987;14:85. doi: 10.1016/0165-2427(87)90077-8. [DOI] [PubMed] [Google Scholar]
- 613.Madigan J.E., Rikihisa Y., Palmer J.E. Evidence for a high rate of false-positive results with the indirect fluorescent antibody test for Ehrlichia risticii antibody in horses. J Am Vet Med Assoc. 1995;207:1448. [PubMed] [Google Scholar]
- 614.Pusterla N., Leutenegger C.M., Sigrist B. Detection and quantitation of Ehrlichia risticii genomic DNA in infected horses and snails by real-time PCR. Vet Parasitol. 2000;90:129. doi: 10.1016/s0304-4017(00)00227-2. [DOI] [PubMed] [Google Scholar]
- 615.Mott J., Rikihisa Y., Zhang Y. Comparison of PCR and culture to the indirect fluorescent-antibody test for diagnosis of Potomac horse fever. J Clin Microbiol. 1997;35:2215. doi: 10.1128/jcm.35.9.2215-2219.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 616.Biswas B., Mukherjee D., Mattingly-Napier B.L., Dutta S.K. Diagnostic application of polymerase chain reaction for detection of Ehrlichia risticii in equine monocytic ehrlichiosis (Potomac horse fever) J Clin Microbiol. 1991;29:2228. doi: 10.1128/jcm.29.10.2228-2233.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 617.Atwill E.R., Mohammed H.O. Evaluation of vaccination of horses as a strategy to control equine monocytic ehrlichiosis. J Am Vet Med Assoc. 1996;208:1290. [PubMed] [Google Scholar]
- 618.Dutta S.K., Vemulapalli R., Biswas B. Association of deficiency in antibody response to vaccine and heterogeneity of Ehrlichia risticii strains with Potomac horse fever vaccine failure in horses. J Clin Microbiol. 1998;36:506. doi: 10.1128/jcm.36.2.506-512.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 619.Wierup M. Equine intestinal clostridiosis. An acute disease in horses associated with high intestinal counts of Clostridium perfringens type A. Acta Vet Scand suppl. 1977;1 [PubMed] [Google Scholar]
- 620.Prescott J.F., Staempfli H.R., Barker I.K. A method for reproducing fatal idiopathic colitis (colitis X) in ponies and isolation of a clostridium as a possible agent. Equine Vet J. 1988;20:417. doi: 10.1111/j.2042-3306.1988.tb01563.x. [DOI] [PubMed] [Google Scholar]
- 621.Jones R.L., Adney W.S., Alexander A.F. Hemorrhagic necrotizing enterocolitis associated with Clostridium difficile infection in four foals. J Am Vet Med Assoc. 1988;193:76. [PubMed] [Google Scholar]
- 622.Madewell B.R., Tang Y.J., Jang S. Apparent outbreaks of Clostridium difficile-associated diarrhea in horses in a veterinary medical teaching hospital. J Vet Diagn Invest. 1995;7:343. doi: 10.1177/104063879500700308. [DOI] [PubMed] [Google Scholar]
- 623.Weese J.S., Staempfli H.R., Prescott J.F. A prospective study of the roles of clostridium difficile and enterotoxigenic Clostridium perfringens in equine diarrhoea. Equine Vet J. 2001;33:403. doi: 10.2746/042516401776249534. [DOI] [PubMed] [Google Scholar]
- 624.Jones R.L. Clostridial enterocolitis. Vet Clin North Am Equine Pract. 2000;16:471. doi: 10.1016/s0749-0739(17)30090-1. [DOI] [PubMed] [Google Scholar]
- 625.Donaldson M.T., Palmer J.E. Prevalence of Clostridium perfringens enterotoxin and Clostridium difficile toxin A in feces of horses with diarrhea and colic. J Am Vet Med Assoc. 1999;215:358. [PubMed] [Google Scholar]
- 626.Baverud V., Gustaffsson A., Franklin A. Clostridium difficile associated with acute colitis in mature horses treated with antibiotics. Equine vet j. 1997;29:279. doi: 10.1111/j.2042-3306.1997.tb03124.x. v. [DOI] [PubMed] [Google Scholar]
- 627.Linerode P.A., Goode R.L. The effect of colic on the microbial activity of the equine intestine. Proc Am Assoc Eq Pract. 1970;16:219. [Google Scholar]
- 628.White G., Prior S.D. Comparative effects of oral administration of trimethoprim/sulphadiazine or oxytetracycline on the faecal flora of horses. Vet Rec. 1982;111:316. doi: 10.1136/vr.111.14.316. [DOI] [PubMed] [Google Scholar]
- 629.Baverud V., Franklin A., Gunnarsson A. Clostridium difficile associated with acute colitis in mares when their foals are treated with erythromycin and rifampicin for Rhodococcus equi pneumonia. Equine Vet J. 1998;30:482. doi: 10.1111/j.2042-3306.1998.tb04523.x. [DOI] [PubMed] [Google Scholar]
- 630.Staempfli H.R., Prescott J.F., Brash M.L. Lincomycin-induced severe colitis in ponies: association with Clostridium cadaveris. Can J Vet Res. 1992;56:168. [PMC free article] [PubMed] [Google Scholar]
- 631.Samuel S.C., Hancock P., Leigh D.A. An investigation into Clostridium perfringens enterotoxin-associated diarrhoea. J Hosp Infect. 1991;18:219. doi: 10.1016/0195-6701(91)90146-y. [DOI] [PubMed] [Google Scholar]
- 632.Niilo L. Enterotoxigenic Clostridium perfringens. In: Gyles C.L., Thoen C.O., editors. Pathogenesis of bacterial infections in animals. Iowa State University Press; Ames, IA: 1986. p. 321. [Google Scholar]
- 633.Gibert M., Jolivet-Reynaud C., Popoff M.R., Jolivet-Renaud C. Beta2 toxin, a novel toxin produced by Clostridium perfringens. Gene. 1997;203:65. doi: 10.1016/s0378-1119(97)00493-9. [DOI] [PubMed] [Google Scholar]
- 634.Herholz C., Miserez R., Nicolet J. Prevalence of beta2-toxigenic Clostridium perfringens in horses with intestinal disorders. J Clin Microbiol. 1999;37:358. doi: 10.1128/jcm.37.2.358-361.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 635.McClane B.A. An overview of Clostridium perfringens enterotoxin. Toxicon. 1996;34:1335. doi: 10.1016/s0041-0101(96)00101-8. [DOI] [PubMed] [Google Scholar]
- 636.Sarker M.R., Singh U., McClane B.A. An update on Clostridium perfringens enterotoxin. J Nat Toxins. 2000;9:251. [PubMed] [Google Scholar]
- 637.Kelly C.P., LaMont J.T. Clostridium difficile infection. Annual Review of Medicine. 1998;49:375. doi: 10.1146/annurev.med.49.1.375. [DOI] [PubMed] [Google Scholar]
- 638.Wershil B.K., Castagliuolo I., Pothoulakis C. Direct evidence of mast cell involvement in Clostridium difficile toxin A-induced enteritis in mice. Gastroenterology. 1998;114:956. doi: 10.1016/s0016-5085(98)70315-4. [DOI] [PubMed] [Google Scholar]
- 639.Castagliuolo I., Keates A.C., Qiu B. Vol. 94. 1997. Increased substance P responses in dorsal root ganglia and intestinal macrophages during Clostridium difficile toxin A enteritis in rats; p. 4788. (Proceedings of the National Academy of Sciences of the United States of America). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 640.Wierup M., DiPietro J.A. Bacteriologic examination of equine fecal flora as a diagnostic tool for equine intestinal clostridiosis. Am J Vet Res. 1981;42:2167. [PubMed] [Google Scholar]
- 641.Daube G., Simon P., Limbourg B. Hybridization of 2,659 Clostridium perfringens isolates with gene probes for seven toxins (alpha, beta, epsilon, iota, theta, mu, and enterotoxin) and for sialidase. Am J Vet Res. 1996;57:496. [PubMed] [Google Scholar]
- 642.Netherwood T., Wood J.L., Mumford J.A., Chanter N. Molecular analysis of the virulence determinants of Clostridium perfringens associated with foal diarrhoea. Vet J. 1998;155:289. doi: 10.1016/s1090-0233(05)80025-5. [DOI] [PubMed] [Google Scholar]
- 643.Meer R.R., Songer J.G. Multiplex polymerase chain reaction assay for genotyping Clostridium perfringens. Am J Vet Res. 1997;58:702. [PubMed] [Google Scholar]
- 644.Weese J.S., Staempfli H.R., Prescott J.F. Survival of Clostridium difficile and its toxins in equine feces: implications for diagnostic test selection and interpretation. J Vet Diagn Invest. 2000;12:332. doi: 10.1177/104063870001200406. [DOI] [PubMed] [Google Scholar]
- 645.Jones R.L. Diagnostic procedures for isolation and characterization of Clostridium difficile associated with enterocolitis in foals. J Vet Diagn Invest. 1989;1:84. doi: 10.1177/104063878900100125. [DOI] [PubMed] [Google Scholar]
- 646.Marler L.M., Siders J.A., Wolters L.C. Comparison of five cultural procedures for isolation of Clostridium difficile from stools. J Clin Microbiol. 1992;30:514. doi: 10.1128/jcm.30.2.514-516.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 647.Drudge J.H. Clinical aspects of Strongylus vulgaris infection in the horse. Emphasis on diagnosis, chemotherapy, and prophylaxis. Vet Clin North Am Large Anim Pract. 1979;1:251. doi: 10.1016/s0196-9846(17)30183-0. [DOI] [PubMed] [Google Scholar]
- 648.Owen J., Slocombe D. Pathogenesis of helminths in equines. Vet Parasitol. 1985;18:139. doi: 10.1016/0304-4017(85)90063-9. [DOI] [PubMed] [Google Scholar]
- 649.Bueno L., Ruckebusch Y., Dorchies P. Disturbances of digestive motility in horses associated with strongyle infection. Vet Parasitol. 1979;5:253. [Google Scholar]
- 650.Lester G.D., Bolton J.R., Cambridge H., Thurgate S. The effect of Strongylus vulgaris larvae on equine intestinal myoelectrical activity. Equine Vet J Suppl. 1989:8–13. doi: 10.1111/j.2042-3306.1989.tb05646.x. [DOI] [PubMed] [Google Scholar]
- 651.White N.A. Intestinal infarction associated with mesenteric vascular thrombotic disease in the horse. J Am Vet Med Assoc. 1981;178:259. [PubMed] [Google Scholar]
- 652.Becht J.L. The role of parasites in colic. Proc Am Assoc Eq Pract. 1987;33:301. [Google Scholar]
- 653.Sellers A.F., Lowe J.E., Drost C.J. Retropulsion-propulsion in equine large colon. Am J Vet Res. 1982;43:390. [PubMed] [Google Scholar]
- 654.Greatorex J.C. Diarrhoea in horses associated with ulceration of the colon and caecum resulting from S vulgaris larval migration. Vet Rec. 1975;97:221. doi: 10.1136/vr.97.12.221. [DOI] [PubMed] [Google Scholar]
- 655.Patton S., Drudge J.H. Clinical response of pony foals experimentally infected with Strongylus vulgaris. Am J Vet Res. 1977;38:2059. [PubMed] [Google Scholar]
- 656.Amborski G.F., Bello T.R., Torbert B.J. Host response to experimentally induced infections of Strongylus vulgaris in parasite-free and naturally infected ponies. Am J Vet Res. 1974;35:1181. [PubMed] [Google Scholar]
- 657.Klei T.R., Torbert B.J., Ochoa R., Bello T.R. Morphologic and clinicopathologic changes following Strongylus vulgaris infections of immune and nonimmune ponies. Am J Vet Res. 1982;43:1300. [PubMed] [Google Scholar]
- 658.Patton S., Mock R.E., Drudge J.H., Morgan D. Increase of immunoglobulin T concentration in ponies as a response to experimental infection with the nematode Strongylus vulgaris. Am J Vet Res. 1978;39:19. [PubMed] [Google Scholar]
- 659.Lyons E.T., Drudge J.H., Tolliver S.C. Larval cyathostomiasis. Vet Clin North Am Equine Pract. 2000;16:501. doi: 10.1016/s0749-0739(17)30092-5. [DOI] [PubMed] [Google Scholar]
- 660.Chiejina S.N., Mason J.A. Immature stages of Trichonema spp as a cause of diarrhoea in adult horses in spring. Vet Rec. 1977;100:360. doi: 10.1136/vr.100.17.360. [DOI] [PubMed] [Google Scholar]
- 661.Giles C.J., Urquhart K.A., Longstaffe J.A. Larval cyathostomiasis (immature trichonema-induced enteropathy): a report of 15 clinical cases. Equine Vet J. 1985;17:196. doi: 10.1111/j.2042-3306.1985.tb02469.x. [DOI] [PubMed] [Google Scholar]
- 662.Church S., Kelly D.F., Obwolo M.J. Diagnosis and successful treatment of diarrhoea in horses caused by immature small strongyles apparently insusceptible to anthelmintics. Equine Vet J. 1986;18:401. doi: 10.1111/j.2042-3306.1986.tb03666.x. [DOI] [PubMed] [Google Scholar]
- 663.Mair T.S. Recurrent diarrhoea in aged ponies associated with larval cyathostomiasis. Equine Vet J. 1993;25:161. doi: 10.1111/j.2042-3306.1993.tb02929.x. [DOI] [PubMed] [Google Scholar]
- 664.Mair T.S. Outbreak of larval cyathostomiasis among a group of yearling and two-year-old horses. Vet Rec. 1994;135:598. [PubMed] [Google Scholar]
- 665.Love S., Murphy D., Mellor D. Pathogenicity of cyathostome infection. Vet Parasitol. 1999;85:113. doi: 10.1016/s0304-4017(99)00092-8. [DOI] [PubMed] [Google Scholar]
- 666.Mair T.S., de Westerlaken L.V., Cripps P.J., Love S. Diarrhoea in adult horses: a survey of clinical cases and an assessment of some prognostic indices. Vet Rec. 1990;126:479. [PubMed] [Google Scholar]
- 667.Murphy D., Love S. The pathogenic effects of experimental cyathostome infections in ponies. Vet Parasitol. 1997;70:99. doi: 10.1016/s0304-4017(96)01153-3. [DOI] [PubMed] [Google Scholar]
- 668.Murphy D., Reid S.W., Graham P.A., Love S. Fructosamine measurement in ponies: validation and response following experimental cyathostome infection. Res Vet Sci. 1997;63:113. doi: 10.1016/s0034-5288(97)90002-3. [DOI] [PubMed] [Google Scholar]
- 669.Klei T.R., Rehbein S., Visser M. Re-evaluation of ivermectin efficacy against equine gastrointestinal parasites. Vet Parasitol. 2001;98:315. doi: 10.1016/s0304-4017(01)00436-8. [DOI] [PubMed] [Google Scholar]
- 670.Tarigo-Martinie J.L., Wyatt A.R., Kaplan R.M. Prevalence and clinical implications of anthelmintic resistance in cyathostomes of horses. J Am Vet Med Assoc. 2001;218:1957. doi: 10.2460/javma.2001.218.1957. [DOI] [PubMed] [Google Scholar]
- 671.Jacobs D.E., Hutchinson M.J., Parker L., Gibbons L.M. Equine cyathostome infection: suppression of faecal egg output with moxidectin. Vet Rec. 1995;137:545. doi: 10.1136/vr.137.21.545. [DOI] [PubMed] [Google Scholar]
- 672.Monahan C.M., Chapman M.R., Taylor H.W. Experimental cyathostome challenge of ponies maintained with or without benefit of daily pyrantel tartrate feed additive: comparison of parasite burdens, immunity and colonic pathology. Vet Parasitol. 1998;74:229. doi: 10.1016/s0304-4017(97)00095-2. [DOI] [PubMed] [Google Scholar]
- 673.Chapman M.R., French D.D., Monahan C.M., Klei T.R. Identification and characterization of a pyrantel pamoate resistant cyathostome population. Vet Parasitol. 1996;66:205. doi: 10.1016/s0304-4017(96)01014-x. [DOI] [PubMed] [Google Scholar]
- 674.Little D., Flowers J.R., Hammerberg B.H., Gardner S.Y. Management of drug-resistant cyathostominosis on a breeding farm in central North Carolina. Equine Vet J. 2003;35:246. doi: 10.2746/042516403776148264. [DOI] [PubMed] [Google Scholar]
- 675.Andersson G., Ekman L., Mansson I. Lethal complications following administration of oxytetracycline in the horse. Nord Vet Med. 1971;23:2. [PubMed] [Google Scholar]
- 676.Raisbeck M.F., Holt G.R., Osweiler G.D. Lincomycin-associated colitis in horses. J Am Vet Med Assoc. 1981;179:362. [PubMed] [Google Scholar]
- 677.Stratton-Phelps M., Wilson W.D., Gardner I.A. Risk of adverse effects in pneumonic foals treated with erythromycin versus other antibiotics: 143 cases (1986–1996) J Am Vet Med Assoc. 2000;217:68. doi: 10.2460/javma.2000.217.68. [DOI] [PubMed] [Google Scholar]
- 678.Wilson D.A., MacFadden K.E., Green E.M. Case control and historical cohort study of diarrhea associated with administration of trimethoprim-potentiated sulphonamides to horses and ponies. J Vet Intern Med. 1996;10:258. doi: 10.1111/j.1939-1676.1996.tb02059.x. [DOI] [PubMed] [Google Scholar]
- 679.Gustafsson A., Baverud V., Gunnarsson A. The association of erythromycin ethylsuccinate with acute colitis in horses in Sweden. Equine Vet J. 1997;29:314–318. doi: 10.1111/j.2042-3306.1997.tb03129.x. [DOI] [PubMed] [Google Scholar]
- 680.Owen R.A., Fullerton J., Barnum D.A. Effects of transportation, surgery, and antibiotic therapy in ponies infected with Salmonella. Am J Vet Res. 1983;44:46. [PubMed] [Google Scholar]
- 681.Borriello S.P. The influence of the normal flora on Clostridium difficile colonisation of the gut. Ann Med. 1990;22:61. doi: 10.3109/07853899009147244. [DOI] [PubMed] [Google Scholar]
- 682.Owen R., Fullerton J.N., Tizard I.R. Studies on experimental enteric salmonellosis in ponies. Can J Comp Med. 1979;43:247. [PMC free article] [PubMed] [Google Scholar]
- 683.Rao S.S., Edwards C.A., Austen C.J. Impaired colonic fermentation of carbohydrate after ampicillin. Gastroenterology. 1988;94:928. doi: 10.1016/0016-5085(88)90549-5. [DOI] [PubMed] [Google Scholar]
- 684.Clausen M.R., Bonnen H., Tvede M., Mortensen P.B. Colonic fermentation to short-chain fatty acids is decreased in antibiotic-associated diarrhea. Gastroenterology. 1991;101:1497. doi: 10.1016/0016-5085(91)90384-w. [DOI] [PubMed] [Google Scholar]
- 685.Grossman R.F. The relationship of absorption characteristics and gastrointestinal side effects of oral antimicrobial agents. Clin Ther. 1991;13:189. [PubMed] [Google Scholar]
- 686.Roussel A.J., Hooper R.N., Cohen N.D. Prokinetic effects of erythromycin on the ileum, cecum, and pelvic flexure of horses during the postoperative period. Am J Vet Res. 2000;61:420. doi: 10.2460/ajvr.2000.61.420. [DOI] [PubMed] [Google Scholar]
- 687.Lakritz J., Madigan J., Carlson G.P. Hypovolemic hyponatremia and signs of neurologic disease associated with diarrhea in a foal. J Am Vet Med Assoc. 1992;200:1114. [PubMed] [Google Scholar]
- 688.Kore A.M. Toxicology of nonsteroidal antiinflammatory drugs. Vet Clin North Am Small Anim Pract. 1990;20:419. doi: 10.1016/s0195-5616(90)50036-4. [DOI] [PubMed] [Google Scholar]
- 689.Gibson G.R., Whitacre E.B., Ricotti C.A. Colitis induced by nonsteroidal anti-inflammatory drugs. Report of four cases and review of the literature. Arch Intern Med. 1992;152:625. [PubMed] [Google Scholar]
- 690.Meschter C.L., Gilbert M., Krook L. The effects of phenylbutazone on the intestinal mucosa of the horse: a morphological, ultrastructural and biochemical study. Equine Vet J. 1990;22:255. doi: 10.1111/j.2042-3306.1990.tb04264.x. [DOI] [PubMed] [Google Scholar]
- 691.Collins L.G., Tyler D.E. Experimentally induced phenylbutazone toxicosis in ponies: description of the syndrome and its prevention with synthetic prostaglandin E2. Am J Vet Res. 1985;46:1605. [PubMed] [Google Scholar]
- 692.Collins L.G., Tyler D.E. Phenylbutazone toxicosis in the horse: a clinical study. J Am Vet Med Assoc. 1984;184:699. [PubMed] [Google Scholar]
- 693.Karcher L.F., Dill S.G., Anderson W.I., King J.M. Right dorsal colitis. J Vet Intern Med. 1990;4:247. doi: 10.1111/j.1939-1676.1990.tb03117.x. [DOI] [PubMed] [Google Scholar]
- 694.Hough M.E., Steel C.M., Bolton J.R., Yovich J.V. Ulceration and stricture of the right dorsal colon after phenylbutazone administration in four horses. Aust Vet J. 1999;77:785. doi: 10.1111/j.1751-0813.1999.tb12945.x. [DOI] [PubMed] [Google Scholar]
- 695.Cook, V. L., C. T. Meyers, N. B. Campbell, and A. T. Blikslager. 2008. Effect of firocoxib or flunixin meglumine on recovery of ischemic-injured equine jejunum. 70:992–1000. [DOI] [PubMed]
- 696.Murray M.J. Phenylbutazone toxicity in a horse. Comp Cont Ed Pract Vet. 1985;7:S389–S394. [Google Scholar]
- 697.Lees P., Creed R.F., Gerring E.E. Biochemical and haematological effects of phenylbutazone in horses. Equine Vet J. 1983;15:158. doi: 10.1111/j.2042-3306.1983.tb01745.x. [DOI] [PubMed] [Google Scholar]
- 698.Cohen N.D., Carter G.K., Mealey R.H., Taylor T.S. Medical management of right dorsal colitis in 5 horses: a retrospective study (1987–1993) J Vet Intern Med. 1995;9:272. doi: 10.1111/j.1939-1676.1995.tb01079.x. [DOI] [PubMed] [Google Scholar]
- 699.Kaufmann H.J., Taubin H.L. Nonsteroidal anti-inflammatory drugs activate quiescent inflammatory bowel disease. Ann Intern Med. 1987;107:513. doi: 10.7326/0003-4819-107-4-513. [DOI] [PubMed] [Google Scholar]
- 700.Hovde O., Farup P.G. NSAID-induced irreversible exacerbation of ulcerative colitis. J Clin Gastroenterol. 1992;15:160. [PubMed] [Google Scholar]
- 701.Blikslager A.T., Roberts M.C. Mechanisms of intestinal mucosal repair. J Am Vet Med Assoc. 1998;211:1437. [PubMed] [Google Scholar]
- 702.Semble E.L., Wu W.C. Prostaglandins in the gut and their relationship to non-steroidal anti-inflammatory drugs. Baillieres Clin Rheumatol. 1989;3:247. doi: 10.1016/s0950-3579(89)80020-2. [DOI] [PubMed] [Google Scholar]
- 703.Campbell N.B., Jones S.L., Blikslager A.T. The effects of cyclo-oxygenase inhibitors on bile-injured and normal equine colon. Equine Vet J. 2002;34:493. doi: 10.2746/042516402776117737. [DOI] [PubMed] [Google Scholar]
- 704.Jones S.L., Blikslager A.T. The future of antiinflammatory therapy. Vet Clin North Am Equine Pract. 2001;17:245. doi: 10.1016/s0749-0739(17)30060-3. [DOI] [PubMed] [Google Scholar]
- 705.Doucet M.Y., Bertone A.L., Hendrickson D. Comparison of efficacy and safety of paste formulations of firocoxib and phenylbutazone in horses with naturally occurring osteoarthritis. J Am Vet Med Assoc. 2008;232:91. doi: 10.2460/javma.232.1.91. [DOI] [PubMed] [Google Scholar]
- 706.Yamada T., Deitch E., Specian R.D. Mechanisms of acute and chronic intestinal inflammation induced by indomethacin. Inflammation. 1993;17:641. doi: 10.1007/BF00920471. [DOI] [PubMed] [Google Scholar]
- 707.Beck P.L., Xavier R., Lu N. Mechanisms of NSAID-induced gastrointestinal injury defined using mutant mice. Gastroenterology. 2000;119:699. doi: 10.1053/gast.2000.16497. [DOI] [PubMed] [Google Scholar]
- 708.Wallace J.L., Granger D.N. Pathogenesis of NSAID gastropathy: are neutrophils the culprits? Trends Pharm Sci. 1992;13:129. doi: 10.1016/0165-6147(92)90046-9. [DOI] [PubMed] [Google Scholar]
- 709.Morise Z., Komatsu S., Fuseler J.W. ICAM-1 and P-selectin expression in a model of NSAID-induced gastropathy. Am J Physiol Cell Physiol. 1998;274:G246–G252. doi: 10.1152/ajpgi.1998.274.2.G246. [DOI] [PubMed] [Google Scholar]
- 710.Brune K., Beck W.S. Towards safer nonsteroidal anti-inflammatory drugs. Agents Actions Suppl. 1991;32:13–25. doi: 10.1007/978-3-0348-7405-2_1. [DOI] [PubMed] [Google Scholar]
- 711.Davis J.L., Gardner S.Y., Jones S.L., Schwabenton B.A., Papich M.G. Pharmacokinetics of azithromycin in foals after I.V. and oral dose and disposition into phagocytes. J Vet Pharmacol Ther. 2002;25:99. doi: 10.1046/j.1365-2885.2002.00387.x. [DOI] [PubMed] [Google Scholar]
- 712.Cohen N.D., Mealey R.H., Chaffin M.K., Carter G.K. The recognition and medical management of right dorsal colitis in horses. Vet Med. 1995;9:687. doi: 10.1111/j.1939-1676.1995.tb01079.x. [DOI] [PubMed] [Google Scholar]
- 713.Schmitz D.G. Cantharidin toxicosis in horses. J Vet Intern Med. 1989;3:208. doi: 10.1111/j.1939-1676.1989.tb00859.x. [DOI] [PubMed] [Google Scholar]
- 714.Shawley R.V., Rolf L.L.J. Experimental cantharidiasis in the horse. Am J Vet Res. 1984;45:2261. [PubMed] [Google Scholar]
- 715.Schoeb T.R., Panciera R.J. Pathology of blister beetle (Epicauta) poisoning in horses. Vet Pathol. 1979;16:18. doi: 10.1177/030098587901600102. [DOI] [PubMed] [Google Scholar]
- 716.Ray A.C., Kyle A.L., Murphy M.J., Reagor J.C. Etiologic agents, incidence, and improved diagnostic methods of cantharidin toxicosis in horses. Am J Vet Res. 1989;50:187. [PubMed] [Google Scholar]
- 717.Helman R.G., Edwards W.C. Clinical features of blister beetle poisoning in equids: 70 cases (1983-1996) J Am Vet Med Assoc. 1997;211:1018. [PubMed] [Google Scholar]
- 718.Osweiler G.D., Carron J.L., Buck W.B. Kendal Hunt; Dubuque, IA: 1985. Clinical and diagnostic veterinary toxicology; p. 253. [Google Scholar]
- 719.Tamaki S., Frankenberger W.T.J. Environmental biochemistry of arsenic. Rev Environ Contam Toxicol. 1992;124:79. doi: 10.1007/978-1-4612-2864-6_4. [DOI] [PubMed] [Google Scholar]
- 720.Louria D.B. Trace metal poisoning. In: Wyndgaarden J.B., Smith L.H., editors. Cecil Textbook of Medicine. WB Saunders; Philadelphia: 1988. p. 2385. [Google Scholar]
- 721.Mack R.B. Gee, honey, why does the iced tea have a garlic taste? Arsenic intoxication. N C Med J. 1983;44:753. [PubMed] [Google Scholar]
- 722.Olson N.E. Acute diarrheal disease in the horse. J Am Vet Med Assoc. 1966;148:418. [PubMed] [Google Scholar]
- 723.Wershil B.K., Walker W.A. The mucosal barrier, IgE-mediated gastrointestinal events, and eosinophilic gastroenteritis. Gastroenterol. Clin North Am. 1992;21:387. [PubMed] [Google Scholar]
- 724.Strobel S. IgE-mediated (and food-induced) intestinal disease. Clin Exp Allergy. 1995;25(suppl 1):3. doi: 10.1111/j.1365-2222.1995.tb01123.x. [DOI] [PubMed] [Google Scholar]
- 725.Ohtsuka Y., Naito K., Yamashiro Y. Induction of anaphylaxis in mouse intestine by orally administered antigen and its prevention with soluble high affinity receptor for IgE. Pediatr Res. 1999;45:300. doi: 10.1203/00006450-199903000-00002. [DOI] [PubMed] [Google Scholar]
- 726.Zimmel D.N., Blikslager A.T., Jones S.L. Vaccine-associated anaphylactic-like reaction in a horse. Comp Cont Ed Pract Vet. 2000;1:81. [Google Scholar]
- 727.Mansmann R.A. Equine anaphylaxis. J Am Vet Med Assoc. 1972;161:438. [PubMed] [Google Scholar]
- 728.McGavin M.D., Gronwall R.R., Mia A.S. Pathologic changes in experimental equine anaphylaxis. J Am Vet Med Assoc. 1972;160:1632. [PubMed] [Google Scholar]
- 729.Stenton G.R., Vliagoftis H., Befus A.D. Role of intestinal mast cells in modulating gastrointestinal pathophysiology. Ann Allergy Asthma Immunol. 1998;81:1. doi: 10.1016/S1081-1206(10)63105-5. [DOI] [PubMed] [Google Scholar]
- 730.Mourad F.H., O’Donnell L.J., Ogutu E. Role of 5-hydroxytryptamine in intestinal water and electrolyte movement during gut anaphylaxis. Gut. 1995;36:553. doi: 10.1136/gut.36.4.553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 731.Catto-Smith A.G., Patrick M.K., Hardin J.A., Gall D.G. Intestinal anaphylaxis in the rat: mediators responsible for the ion transport abnormalities. Agents Actions. 1989;28:185. doi: 10.1007/BF01967399. [DOI] [PubMed] [Google Scholar]
- 732.Scott R.B., Diamant S.C., Gall D.G. Motility effects of intestinal anaphylaxis in the rat. Am J Physiol. 1988;255:G505–G511. doi: 10.1152/ajpgi.1988.255.4.G505. [DOI] [PubMed] [Google Scholar]
- 733.Baron D.A., Baird A.W., Cuthbert A.W., Margolius H.S. Intestinal anaphylaxis: rapid changes in mucosal ion transport and morphology. Am J Physiol. 1988;254:G307–G314. doi: 10.1152/ajpgi.1988.254.3.G307. [DOI] [PubMed] [Google Scholar]
- 734.Garner H.E., Hutcheson D.P., Coffman J.R. Lactic acidosis: a factor associated with equine laminitis. J Anim Sci. 1977;45:1037. doi: 10.2527/jas1977.4551037x. [DOI] [PubMed] [Google Scholar]
- 735.Garner H.E., Moore J.N., Johnson J.H. Changes in the caecal flora associated with the onset of laminitis. Equine Vet J. 1978;10:249. doi: 10.1111/j.2042-3306.1978.tb02273.x. [DOI] [PubMed] [Google Scholar]
- 736.Sprouse R.F., Garner H.E., Green E.M. Plasma endotoxin levels in horses subjected to carbohydrate induced laminitis. Equine Vet J. 1987;19:25. doi: 10.1111/j.2042-3306.1987.tb02571.x. [DOI] [PubMed] [Google Scholar]
- 737.Ragle C.A., Meagher D.M., Lacroix C.A., Honnas C.M. Surgical treatment of sand colic. Results in 40 horses. Vet Surg. 1989;18:48. doi: 10.1111/j.1532-950x.1989.tb01042.x. [DOI] [PubMed] [Google Scholar]
- 738.Bertone J.J., Traub-Dargatz J.L., Wrigley R.W. Diarrhea associated with sand in the gastrointestinal tract of horses. J Am Vet Med Assoc. 1988;193:1409. [PubMed] [Google Scholar]
- 739.Ramey D.W., Reinertson E.L. Sand-induced diarrhea in a foal. J Am Vet Med Assoc. 1984;185:537. [PubMed] [Google Scholar]
- 740.McGuinness S.G., Mansmann R.A., Breuhaus B.A. Nasogastric electrolyte replacement in horses. Comp Cont Ed Pract Vet. 1996;18:942. [Google Scholar]
- 741.Duebbert I.E., Peterson J.W. Enterotoxin-induced fluid accumulation during experimental salmonellosis and cholera: involvement of prostaglandin synthesis by intestinal cells. Toxicon. 1985;23:157. doi: 10.1016/0041-0101(85)90118-7. [DOI] [PubMed] [Google Scholar]
- 742.Freeman D.E., Inoue O.J., Eurell T.E. Effects of flunixin meglumine on short circuit current in equine colonic mucosa in vitro. Am J Vet Res. 1997;58:915. [PubMed] [Google Scholar]
- 743.Richter R.A., Freeman D.E., Wallig M. In vitro anion transport alterations and apoptosis induced by phenylbutazone in the right dorsal colon of ponies. Am J Vet Res. 2002;63:934. doi: 10.2460/ajvr.2002.63.934. [DOI] [PubMed] [Google Scholar]
- 744.Joshi G.P., Gertler R., Fricker R. Cardiovascular thromboembolic adverse effects associated with cyclooxygenase-2 selective inhibitors and nonselective antiinflammatory drugs. Anesth Analg. 2007;105:1793. doi: 10.1213/01.ane.0000286229.05723.50. [DOI] [PubMed] [Google Scholar]
- 745.Geor R.J., Petrie L., Papich M.G., Rousseaux C. The protective effects of sucralfate and ranitidine in foals experimentally intoxicated with phenylbutazone. Can J Vet Res. 1989;53:231. [PMC free article] [PubMed] [Google Scholar]
- 746.Fedorak R.N., Empey L.R., MacArthur C., Jewell L.D. Misoprostol provides a colonic mucosal protective effect during acetic acid-induced colitis in rats. Gastroenterology. 1990;98:615. doi: 10.1016/0016-5085(90)90280-e. [DOI] [PubMed] [Google Scholar]
- 747.Wachtershauser A., Stein J. Rationale for the luminal provision of butyrate in intestinal diseases. Eur J Nutr. 2000;39:164. doi: 10.1007/s003940070020. [DOI] [PubMed] [Google Scholar]
- 748.Rhoads J.M., Chen W., Gookin J. Arginine stimulates intestinal cell migration through a focal adhesion kinase dependent mechanism. Gut. 2004;53:514. doi: 10.1136/gut.2003.027540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 749.Palmer J.E., Benson C.E., Whitlock R.H. Effect of treatment with oxytetracycline during the acute stages of experimentally induced equine ehrlichial colitis in ponies. Am J Vet Res. 1992;53:2300. [PubMed] [Google Scholar]
- 750.McGorum B.C., Dixon P.M., Smith D.G. Use of metronidazole in equine acute idiopathic toxaemic colitis. Vet Rec. 1998;142:635. doi: 10.1136/vr.142.23.635. [DOI] [PubMed] [Google Scholar]
- 751.Jang S.S., Hansen L.M., Breher J.E. Antimicrobial susceptibilities of equine isolates of Clostridium difficile and molecular characterization of metronidazole-resistant strains. Clin Infect Dis. 1997;25(suppl 2):S266–S267. doi: 10.1086/516235. [DOI] [PubMed] [Google Scholar]
- 752.MacKay R.J. Equine neonatal clostridiosis: treatment and prevention. Comp Cont Ed Pract Vet. 2001;23:280. [Google Scholar]
- 753.Cambridge H., Lees P., Hooke R.E., Russell C.S. Antithrombotic actions of aspirin in the horse. Equine Vet J. 1991;23:123. doi: 10.1111/j.2042-3306.1991.tb02736.x. [DOI] [PubMed] [Google Scholar]
- 754.Belknap J.K., Moore J.N. Evaluation of heparin for prophylaxis of equine laminitis: 71 cases (1980–1986) J Am Vet Med Assoc. 1989;195:505. [PubMed] [Google Scholar]
- 755.Fuller R. Probiotics in man and animals. J Appl Bacteriol. 1989;66:365. [PubMed] [Google Scholar]
- 756.Schoster A., Staempfli H.R., Abrahams M. Effect of a probiotic on prevention of diarrhea and Clostridium difficile and Clostridium perfringens shedding in foals. J Vet Intern Med. 2015;29:925–931. doi: 10.1111/jvim.12584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 757.Desrochers A.M., Dolente B.A., Roy M.F. Efficacy of Saccharomyces boulardii for treatment of horses with acute enterocolitis. J Am Vet Med Assoc. 2005;227:954. doi: 10.2460/javma.2005.227.954. [DOI] [PubMed] [Google Scholar]
- 758.Qualls H.J., Holbrook T.C., Gilliam L.L. Evaluation of efficacy of mineral oil, charcoal, and smectite in a rat model of equine cantharidin toxicosis. J Vet Intern Med. 2013;27:1179–1184. doi: 10.1111/jvim.12164. [DOI] [PubMed] [Google Scholar]
- 759.Lawler J.B., Hassel D.M., Magnuson R.J. Adsorptive effects of di-tri-octahedral smectite on Clostridium perfringens alpha, beta, and beta-2 exotoxins and equine colostral antibodies. Am J Vet Res. 2008;69:233. doi: 10.2460/ajvr.69.2.233. [DOI] [PubMed] [Google Scholar]
- 760.Xiao L., Herd R.P., Majewski G.A. Comparative efficacy of moxidectin and ivermectin against hypobiotic and encysted cyathostomes and other equine parasites. Vet Parasitol. 1994;53:83. doi: 10.1016/0304-4017(94)90020-5. [DOI] [PubMed] [Google Scholar]
- 761.Love S., Duncan J.L., Parry J.M., Grimshaw W.T. Efficacy of oral ivermectin paste against mucosal stages of cyathostomes. Vet Rec. 1995;136:18. doi: 10.1136/vr.136.1.18. [DOI] [PubMed] [Google Scholar]
- 762.Duncan J.L., Bairden K., Abbott E.M. Elimination of mucosal cyathostome larvae by five daily treatments with fenbendazole. Vet Rec. 1998;142:268. doi: 10.1136/vr.142.11.268. [DOI] [PubMed] [Google Scholar]
- 763.Hutchens D.E., Paul A.J. Moxidectin: spectrum of activity and uses in an equine anthelmintic program. Comp Cont Ed Pract Vet. 2000;22:373. [Google Scholar]
- 764.Steinbach T., Bauer C., Sasse H. Small strongyle infection: consequences of larvicidal treatment of horses with fenbendazole and moxidectin. Vet Parasitol. 2006;139:115. doi: 10.1016/j.vetpar.2006.03.028. [DOI] [PubMed] [Google Scholar]
- 765.Peritonitis Hosgood G. 1. A Review of the Pathophysiology and Diagnosis. Aust Vet Pract. 1986;16:184–190. [Google Scholar]
- 766.Trent A.M. The peritoneum and peritoneal cavity. In: Kobluk C.N., Ames T.R., Geor R.J., editors. The horse: diseases and clinical management. WB Saunders; Philadelphia: 1995. pp. 373–404. [Google Scholar]
- 767.Dabareiner R.M. Peritonitis in horses. In: Smith B.P., editor. Large Animal Internal Medicine. Mosby; St. Louis, MO: 1996. pp. 742–749. [Google Scholar]
- 768.Dyson S. Review of 30 cases of peritonitis in the horse. Equine Vet J. 1983;15:25–30. doi: 10.1111/j.2042-3306.1983.tb01693.x. [DOI] [PubMed] [Google Scholar]
- 769.Zicker S.C., Wilson W.D., Medearis I. Differentiation between intra-abdominal neoplasms and abscesses in horses, using clinical and laboratory data: 40 cases (1973–1988) J Am Vet Med Assoc. 1990;196:1130–1134. [PubMed] [Google Scholar]
- 770.Reuss S.M., Chaffin M.K., Cohen N.D. Extrapulmonary disorders associated with Rhodococcus equi infection in foals: 150 cases (1987–2007) J Am Vet Med Assoc. 2009;235:855–863. doi: 10.2460/javma.235.7.855. [DOI] [PubMed] [Google Scholar]
- 771.Pratt S.M., Spier S.J., Carroll S.P. Evaluation of clinical characteristics, diagnostic test results, and outcome in horses with internal infection caused by Corynebacterium pseudotuberculosis: 30 cases (1995–2003) J Am Vet Med Assoc. 2005;227:441–448. doi: 10.2460/javma.2005.227.441. [DOI] [PubMed] [Google Scholar]
- 772.Gay C.C., Lording P.M. Peritonitis in horses associated with Actinobacillus equuli. Aust Vet J. 1980;56:296–300. doi: 10.1111/j.1751-0813.1980.tb05727.x. [DOI] [PubMed] [Google Scholar]
- 773.Golland L.C., Hodgson D.R., Hodgson J.L. Peritonitis associated with Actinobacillus equuli in horses: 15 cases (1982–1992) J Am Vet Med Assoc. 1994;205:340–343. [PubMed] [Google Scholar]
- 774.Matthews S., Dart A.J., Dowling B.A. Peritonitis associated with Actinobacillus equuli in horses: 51 cases. Aust Vet J. 2001;79:536–539. doi: 10.1111/j.1751-0813.2001.tb10741.x. [DOI] [PubMed] [Google Scholar]
- 775.Watts A., Johnson A., Felippe M. Recurrent Actinobacillus peritonitis in an otherwise healthy Thoroughbred horse. Aust Vet J. 2011;89:143–146. doi: 10.1111/j.1751-0813.2011.00693.x. [DOI] [PubMed] [Google Scholar]
- 776.Hirsh D.C., Jang S.S. Antimicrobic susceptibility of bacterial pathogens from horses. Vet Clin North Am Equine Pract. 1987;3:181–190. doi: 10.1016/s0749-0739(17)30697-1. [DOI] [PubMed] [Google Scholar]
- 777.Hawkins J.F., Bowman K.F., Roberts M.C. Peritonitis in horses: 67 cases (1985–1990) J Am Vet Med Assoc. 1993;203:284–288. [PubMed] [Google Scholar]
- 778.Lapointe J.M., Celeste C., Villeneuve A. Septic peritonitis due to colonic perforation associated with aberrant migration of a Gasterophilus intestinalis larva in a horse. Vet Pathol. 2003;40:338–339. doi: 10.1354/vp.40-3-338. [DOI] [PubMed] [Google Scholar]
- 779.Archer D.C., Clegg P.D., Edwards G.B. Septic tenosynovitis of the tarsal sheath of an Arab gelding and suspected sepsis of the lateral digital flexor tendon subsequent to bacterial peritonitis. Vet Rec. 2004;155:485–489. doi: 10.1136/vr.155.16.485. [DOI] [PubMed] [Google Scholar]
- 780.Schumacher J., Spano J.S., Moll H.D. Effects of enterocentesis on peritoneal fluid constituents in the horse. J Am Vet Med Assoc. 1985;186:1301–1303. [PubMed] [Google Scholar]
- 781.Rumbaugh G.E., Smith B.P., Carlson G.P. Internal abdominal abscesses in the horse: a study of 25 cases. J Am Vet Med Assoc. 1978;172:304–309. [PubMed] [Google Scholar]
- 782.Blackford J.T., Schneiter H.L., Van Steenehouse J.L. Equine peritoneal fluid analysis following celiotomy. Proc Equine Colic Res Symp. 1986;3:130. [Google Scholar]
- 783.Van Hoogmoed L., Rodger L.D., Spier S.J. Evaluation of peritoneal fluid pH, glucose concentration, and lactate dehydrogenase activity for detection of septic peritonitis in horses. J Am Vet Med Assoc. 1999;214:1032–1036. [PubMed] [Google Scholar]
- 784.Bonczynski J.J., Ludwig L.L., Barton L.J. Comparison of peritoneal fluid and peripheral blood pH, bicarbonate, glucose, and lactate concentration as a diagnostic tool for septic peritonitis in dogs and cats. Vet Surg. 2003;32:161–166. doi: 10.1053/jvet.2003.50005. [DOI] [PubMed] [Google Scholar]
- 785.Sullins K.E., White N.A., Lundin C.S. Prevention of ischaemia-induced small intestinal adhesions in foals. Equine Vet J. 2004;36:370–375. doi: 10.2746/0425164044868431. [DOI] [PubMed] [Google Scholar]
- 786.Kunesh J.P. Therapeutic strategies involving antimicrobial treatment of large animals with peritonitis. J Am Vet Med Assoc. 1984;185:1222–1225. [PubMed] [Google Scholar]
- 787.Hague B.A., Honnas C.M., Berridge B.R. Evaluation of postoperative peritoneal lavage in standing horses for prevention of experimentally induced abdominal adhesions. Vet Surg. 1998;27:122–126. doi: 10.1111/j.1532-950x.1998.tb00107.x. [DOI] [PubMed] [Google Scholar]
- 788.Davis J.L. Treatment of peritonitis. Vet Clin North Am Equine Pract. 2003;19:765–778. doi: 10.1016/j.cveq.2003.08.007. [DOI] [PubMed] [Google Scholar]
- 789.Nieto J.E., Snyder J.R., Vatistas N.J. Use of an active intra-abdominal drain in 67 horses. Vet Surg. 2003;32:1–7. doi: 10.1053/jvet.2003.50013. [DOI] [PubMed] [Google Scholar]
- 790.Schneider R.K., Meyer D.J., Embertson R.M. Response of pony peritoneum to four peritoneal lavage solutions. Am J Vet Res. 1988;49:889–894. [PubMed] [Google Scholar]
- 791.Easter J.L., Hague B.A., Brumbaugh G.W. Effects of postoperative peritoneal lavage on pharmacokinetics of gentamicin in horses after celiotomy. Am J Vet Res. 1997;58:1166–1170. [PubMed] [Google Scholar]
- 792.Southwood L.L., Russell G. The use of clinical findings in the identification of equine peritonitis cases that respond favorably to medical therapy. J Vet Emerg Crit Care. 2007;17:382–390. [Google Scholar]
- 793.Javsicas L.H., Giguere S., Freeman D.E. Comparison of Surgical and Medical Treatment of 49 Postpartum Mares with Presumptive or Confirmed Uterine Tears. Veterinary Surgery. 2010;39:254–260. doi: 10.1111/j.1532-950X.2010.00645.x. [DOI] [PubMed] [Google Scholar]
- 794.Nogradi N., Slovis N.M., Gebhart C.J. Evaluation of the field efficacy of an avirulent live Lawsonia intracellularis vaccine in foals. Vet J. 2012;192:511–513. doi: 10.1016/j.tvjl.2011.05.026. [DOI] [PubMed] [Google Scholar]
- 795.Nogradi N., Toth B., Macgillivray K.C. Peritonitis in horses: 55 cases (2004–2007) Acta Vet Hung. 2011;59:181–193. doi: 10.1556/AVet.2011.011. [DOI] [PubMed] [Google Scholar]
- 796.Mair T.S., Smith L.J. Survival and complication rates in 300 horses undergoing surgical treatment of colic. Part 4: early (acute) relaparotomy. Equine Vet J. 2005;37:315–318. doi: 10.2746/0425164054529454. [DOI] [PubMed] [Google Scholar]
- 797.Pappenheimer J.R. Paracellular intestinal absorption of glucose, creatinine, and mannitol in normal animals: relation to body size. Am J Physiol. 1990;259:G290–G299. doi: 10.1152/ajpgi.1990.259.2.G290. [DOI] [PubMed] [Google Scholar]
- 798.Pappenheimer J.R. Physiological regulation of epithelial junctions in intestinal epithelia. Acta Physiol Scand Suppl. 1988;571:43–51. [PubMed] [Google Scholar]
- 799.Pappenheimer J.R. Physiological regulation of transepithelial impedance in the intestinal mucosa of rats and hamsters. J Membr Biol. 1987;100:137–148. doi: 10.1007/BF02209146. [DOI] [PubMed] [Google Scholar]
- 800.Pappenheimer J.R., Reiss K.Z. Contribution of solvent drag through intercellular junctions to absorption of nutrients by the small intestine of the rat. J Membr Biol. 1987;100:123–136. doi: 10.1007/BF02209145. [DOI] [PubMed] [Google Scholar]
- 801.Madara J.L. Warner-Lambert/Parke-Davis Award lecture. Pathobiology of the intestinal epithelial barrier. Am J Pathol. 1990;137:1273–1281. [PMC free article] [PubMed] [Google Scholar]
- 802.Van Itallie C.M., Anderson J.M. Claudins and epithelial paracellular transport. Annu Rev Physiol. 2006;68 doi: 10.1146/annurev.physiol.68.040104.131404. 403–29. [DOI] [PubMed] [Google Scholar]
- 803.Itoh M., Furuse M., Morita K., Kubota K., Saitou M., Tsukita S. Direct binding of three tight junction-associated MAGUKs, ZO-1, ZO-2, and ZO-3, with the COOH termini of claudins. J Cell Biol. 1999;147:1351–1363. doi: 10.1083/jcb.147.6.1351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 804.Karczewski J., Groot J. Molecular physiology and pathophysiology of tight junctions III. Tight junction regulation by intracellular messengers: differences in response within and between epithelia. Am J Physiol Gastrointest Liver Physiol. 2000;279:G660–G665. doi: 10.1152/ajpgi.2000.279.4.G660. [DOI] [PubMed] [Google Scholar]
- 805.Mitic L.L., Van Itallie C.M., Anderson J.M. Molecular physiology and pathophysiology of tight junctions I. Tight junction structure and function: lessons from mutant animals and proteins. Am J Physiol Gastrointest Liver Physiol. 2000;279:G250–G254. doi: 10.1152/ajpgi.2000.279.2.G250. [DOI] [PubMed] [Google Scholar]
- 806.Madara J.L., Trier J.S. The functional morphology of the mucosa of the small intestine. In: Johnson L.R., editor. Physiology of the Gastrointestinal Tract. Raven Press; New York: 1994. pp. 1577–1622. [Google Scholar]
- 807.Madara J.L. Loosening tight junctions. Lessons from the intestine. J Clin Invest. 1989;83:1089–1094. doi: 10.1172/JCI113987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 808.Madara J.L., Marcial M.A. Structural correlates of intestinal tight-junction permeability. Kroc Found Ser. 1984;17:77–100. [PubMed] [Google Scholar]
- 809.Tice L.W., Carter R.L., Cahill M.B. Changes in tight junctions of rat intestinal crypt cells associated with changes in their mitotic activity. Tissue Cell. 1979;11:293–316. doi: 10.1016/0040-8166(79)90043-0. [DOI] [PubMed] [Google Scholar]
- 810.Marcial M.A., Carlson S.L., Madara J.L. Partitioning of paracellular conductance along the ileal crypt-villus axis: a hypothesis based on structural analysis with detailed consideration of tight junction structure-function relationships. J Membr Biol. 1984;80:59–70. doi: 10.1007/BF01868690. [DOI] [PubMed] [Google Scholar]
- 811.Stevens C.E., Hume I.D. Cambridge University Press; New York: 1995. Comparative Physiology of the Vertebrate Digestive System. [Google Scholar]
- 812.Argenzio R.A. Comparative pathophysiology of nonglandular ulcer disease: a review of experimental studies. Equine Vet J Suppl. 1999;29:19–23. doi: 10.1111/j.2042-3306.1999.tb05163.x. [DOI] [PubMed] [Google Scholar]
- 813.Argenzio R.A. Mechanisms of acid injury in porcine gastroesophageal mucosa. Am J Vet Res. 1996;57:564–573. [PubMed] [Google Scholar]
- 814.Murray M.J., Mahaffey E.A. Age-related characteristics of gastric squamous epithelial mucosa in foals. Equine Vet J. 1993;25:514–517. doi: 10.1111/j.2042-3306.1993.tb03003.x. [DOI] [PubMed] [Google Scholar]
- 815.Andrews F.M., Sifferman R.L., Bernard W. Efficacy of omeprazole paste in the treatment and prevention of gastric ulcers in horses. Equine Vet J Suppl. 1999;29:81–86. doi: 10.1111/j.2042-3306.1999.tb05176.x. [DOI] [PubMed] [Google Scholar]
- 816.Vatistas N.J., Snyder J.R., Nieto J. Acceptability of a paste formulation and efficacy of high dose omeprazole in healing gastric ulcers in horses maintained in race training. Equine Vet J Suppl. 1999;29:71–76. doi: 10.1111/j.2042-3306.1999.tb05174.x. [DOI] [PubMed] [Google Scholar]
- 817.Murray M.J. Suppression of gastric acidity in horses. J Am Vet Med Assoc. 1997;211:37–40. [PubMed] [Google Scholar]
- 818.Campbell-Thompson M.L., Merritt A.M. Basal and pentagastrin-stimulated gastric secretion in young horses. Am J Physiol. 1990;259:R1259–R1266. doi: 10.1152/ajpregu.1990.259.6.R1259. [DOI] [PubMed] [Google Scholar]
- 819.Schreiber S., Nguyen T.H., Stuben M., Scheid P. Demonstration of a pH gradient in the gastric gland of the acid-secreting guinea pig mucosa. Am J Physiol Gastrointest Liver Physiol. 2000;279:G597–G604. doi: 10.1152/ajpgi.2000.279.3.G597. [DOI] [PubMed] [Google Scholar]
- 820.Flemstrom G. Gastric and duodenal mucosal secretion of bicarbonate. In: Johnson L.R., editor. Physiology of the Gastrointestinal Tract. Raven Press; New York: 1994. pp. 1285–1309. [Google Scholar]
- 821.Podolsky D.K. Mucosal immunity and inflammation. V. Innate mechanisms of mucosal defense and repair: the best offense is a good defense. Am J Physiol. 1999;277:G495–G499. doi: 10.1152/ajpgi.1999.277.3.G495. [DOI] [PubMed] [Google Scholar]
- 822.Robert A., Nezamis J.E., Lancaster C. Mild irritants prevent gastric necrosis through “adaptive cytoprotection” mediated by prostaglandins. Am J Physiol. 1983;245:G113–G121. doi: 10.1152/ajpgi.1983.245.1.G113. [DOI] [PubMed] [Google Scholar]
- 823.Robert A. Cytoprotection by prostaglandins in rats. Prevention of gastric necrosis produced by alcohol, HCl, NaOH, hypertonic NaCl, and thermal injury. Gastroenterology. 1979;77:433–443. [PubMed] [Google Scholar]
- 824.Robert A. Prostaglandins: effects on the gastrointestinal tract. Clin Physiol Biochem. 1984;2:61–69. [PubMed] [Google Scholar]
- 825.Ruppin H., Person B., Robert A., Domschke W. Gastric cytoprotection in man by prostaglandin E2. Scand J Gastroenterol. 1981;16:647–652. doi: 10.3109/00365528109182025. [DOI] [PubMed] [Google Scholar]
- 826.Mutoh H., Ota S., Hiraishi H. Adaptive cytoprotection in cultured rat gastric mucus-producing cells. Role of mucus and prostaglandin synthesis. Dig Dis Sci. 1995;40:872–878. doi: 10.1007/BF02064994. [DOI] [PubMed] [Google Scholar]
- 827.Wallace J.L. Increased resistance of the rat gastric mucosa to hemorrhagic damage after exposure to an irritant. Role of the “mucoid cap” and prostaglandin synthesis. Gastroenterology. 1988;94:22–32. doi: 10.1016/0016-5085(88)90605-1. [DOI] [PubMed] [Google Scholar]
- 828.Konturek S.J., Robert A. Cytoprotection of canine gastric mucosa by prostacyclin: possible mediation by increased mucosal blood flow. Digestion. 1982;25:155–163. doi: 10.1159/000198824. [DOI] [PubMed] [Google Scholar]
- 829.Leung F.W., Robert A., Guth P.H. Gastric mucosal blood flow in rats after administration of 16,16-dimethyl prostaglandin E2 at a cytoprotective dose. Gastroenterology. 1985;88:1948–1953. doi: 10.1016/0016-5085(85)90024-1. [DOI] [PubMed] [Google Scholar]
- 830.Asako H., Kubes P., Wallace J. Modulation of leukocyte adhesion in rat mesenteric venules by aspirin and salicylate. Gastroenterology. 1992;103:146–152. doi: 10.1016/0016-5085(92)91107-f. [DOI] [PubMed] [Google Scholar]
- 831.Holzer P., Sametz W. Gastric mucosal protection against ulcerogenic factors in the rat mediated by capsaicin-sensitive afferent neurons. Gastroenterology. 1986;91:975–981. doi: 10.1016/0016-5085(86)90702-x. [DOI] [PubMed] [Google Scholar]
- 832.Holzer P., Pabst M.A., Lippe I.T. Afferent nerve-mediated protection against deep mucosal damage in the rat stomach. Gastroenterology. 1990;98:838–848. doi: 10.1016/0016-5085(90)90005-l. [DOI] [PubMed] [Google Scholar]
- 833.Holzer P., Pabst M.A., Lippe I.T. Intragastric capsaicin protects against aspirin-induced lesion formation and bleeding in the rat gastric mucosa. Gastroenterology. 1989;96:1425–1433. doi: 10.1016/0016-5085(89)90508-8. [DOI] [PubMed] [Google Scholar]
- 834.Merchant N.B., Dempsey D.T., Grabowski M.W. Capsaicin-induced gastric mucosal hyperemia and protection: the role of calcitonin gene-related peptide. Surgery. 1994;116:419–425. [PubMed] [Google Scholar]
- 835.Wallace J.L., Miller M.J. Nitric oxide in mucosal defense: a little goes a long way. Gastroenterology. 2000;119:512–520. doi: 10.1053/gast.2000.9304. [DOI] [PubMed] [Google Scholar]
- 836.Blikslager A.T., Rhoads J.M., Bristol D.G. Glutamine and transforming growth factor-alpha stimulate extracellular regulated kinases and enhance recovery of villous surface area in porcine ischemic-injured intestine. Surgery. 1999;125:186–194. [PubMed] [Google Scholar]
- 837.Moore R., Carlson S., Madara J.L. Villus contraction aids repair of intestinal epithelium after injury. Am J Physiol. 1989;257:G274–G283. doi: 10.1152/ajpgi.1989.257.2.G274. [DOI] [PubMed] [Google Scholar]
- 838.Lang J., Blikslager A., Regina D. Synergistic effect of hydrochloric acid and bile acids on the pars esophageal mucosa of the porcine stomach. Am J Vet Res. 1998;59:1170–1176. [PubMed] [Google Scholar]
- 839.Berschneider H.M., Blikslager A.T., Roberts M.C. Role of duodenal reflux in nonglandular gastric ulcer disease of the mature horse. Equine Vet J Suppl. 1999;29:24–29. doi: 10.1111/j.2042-3306.1999.tb05164.x. [DOI] [PubMed] [Google Scholar]
- 840.Baker S.J., Gerring E.L. Technique for prolonged, minimally invasive monitoring of intragastric pH in ponies. Am J Vet Res. 1993;54:1725–1734. [PubMed] [Google Scholar]
- 841.Murray M.J. Equine model of inducing ulceration in alimentary squamous epithelial mucosa. Dig Dis Sci. 1994;39:2530–2535. doi: 10.1007/BF02087686. [DOI] [PubMed] [Google Scholar]
- 842.Merritt A.M. Normal equine gastroduodenal secretion and motility. Equine Vet J Suppl. 1999;29:7–13. doi: 10.1111/j.2042-3306.1999.tb05161.x. [DOI] [PubMed] [Google Scholar]
- 843.Peek RMJ I.V. Helicobacter pylori strain-specific activation of signal transduction cascades related to gastric inflammation. Am J Physiol Gastrointest Liver Physiol. 2001;280:G525–G530. doi: 10.1152/ajpgi.2001.280.4.G525. [DOI] [PubMed] [Google Scholar]
- 844.Murray M.J. Pathophysiology of peptic disorders in foals and horses: a review. Equine Vet J Suppl. 1999;29:14–18. doi: 10.1111/j.2042-3306.1999.tb05162.x. [DOI] [PubMed] [Google Scholar]
- 845.Wallace J.L. Nonsteroidal anti-inflammatory drugs and gastroenteropathy: the second hundred years. Gastroenterology. 1997;112:1000–1016. doi: 10.1053/gast.1997.v112.pm9041264. [DOI] [PubMed] [Google Scholar]
- 846.Henry D., Dobson A., Turner C. Variability in the risk of major gastrointestinal complications from nonaspirin nonsteroidal anti-inflammatory drugs. Gastroenterology. 1993;105:1078–1088. doi: 10.1016/0016-5085(93)90952-9. [DOI] [PubMed] [Google Scholar]
- 847.Langenbach R., Morham S.G., Tiano H.F. Prostaglandin synthase 1 gene disruption in mice reduces arachidonic acid-induced inflammation and indomethacin-induced gastric ulceration. Cell. 1995;83:483–492. doi: 10.1016/0092-8674(95)90126-4. [DOI] [PubMed] [Google Scholar]
- 848.Smith W.L., Langenbach R. Why there are two cyclooxygenase isozymes. J Clin Invest. 2001;107:1491–1495. doi: 10.1172/JCI13271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 849.Wallace J.L., McKnight W., Reuter B.K., Vergnolle N. NSAID-Induced gastric damage in rats: requirement for inhibition of both cyclooxygenase 1 and 2. Gastroenterology. 2000;119:706–714. doi: 10.1053/gast.2000.16510. [DOI] [PubMed] [Google Scholar]
- 850.Moore R.M., Muir W.W., Granger D.N. Mechanisms of gastrointestinal ischemia-reperfusion injury and potential therapeutic interventions: a review and its implications in the horse. J Vet Intern Med. 1995;9:115–132. doi: 10.1111/j.1939-1676.1995.tb03285.x. [DOI] [PubMed] [Google Scholar]
- 851.Moore R.M. Clinical relevance of intestinal reperfusion injury in horses. J Am Vet Med Assoc. 1997;211:1362–1366. [PubMed] [Google Scholar]
- 852.Shepherd A.P., Granger D.N. Metabolic regulation of intestinal circulation. In: Shepherd A.P., Granger D.N., editors. Physiology of Intestinal Circulation. Raven Press; New York: 2001. [Google Scholar]
- 853.Bulkley G.B., Kvietys P.R., Parks D.A. Relationship of blood flow and oxygen consumption to ischemic injury in the canine small intestine. Gastroenterology. 1985;89:852–857. doi: 10.1016/0016-5085(85)90583-9. [DOI] [PubMed] [Google Scholar]
- 854.Dart A.J., Snyder J.R., Julian D., Hinds D.M. Microvascular circulation of the small intestine in horses. Am J Vet Res. 1992;53:995–1000. [PubMed] [Google Scholar]
- 855.Snyder J.R., Olander H.J., Pascoe J.R. Morphologic alterations observed during experimental ischemia of the equine large colon. Am J Vet Res. 1988;49:801–809. [PubMed] [Google Scholar]
- 856.McAnulty J.F., Stone W.C., Darien B.J. The effects of ischemia and reperfusion on mucosal respiratory function, adenosine triphosphate, electrolyte, and water content in the ascending colon of ponies. Vet Surg. 1997;26:172–181. doi: 10.1111/j.1532-950x.1997.tb01481.x. [DOI] [PubMed] [Google Scholar]
- 857.Noda T., Iwakiri R., Fujimoto K. Programmed cell death induced by ischemia-reperfusion in rat intestinal mucosa. Am J Physiol. 1998;274:G270–G276. doi: 10.1152/ajpgi.1998.274.2.G270. [DOI] [PubMed] [Google Scholar]
- 858.Ikeda H., Suzuki Y., Suzuki M. Apoptosis is a major mode of cell death caused by ischaemia and ischaemia/reperfusion injury to the rat intestinal epithelium. Gut. 1998;42:530–537. doi: 10.1136/gut.42.4.530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 859.Coopersmith C.M., O’Donnell D., Gordon J.I. Bcl-2 inhibits ischemia-reperfusion-induced apoptosis in the intestinal epithelium of transgenic mice. Am J Physiol. 1999;276:G677–G686. doi: 10.1152/ajpgi.1999.276.3.G677. [DOI] [PubMed] [Google Scholar]
- 860.Chiu C.J., McArdle A.H., Brown R. Intestinal mucosal lesion in low-flow states. I. A morphological, hemodynamic, and metabolic reappraisal. Arch Surg. 1970;101:478–483. doi: 10.1001/archsurg.1970.01340280030009. [DOI] [PubMed] [Google Scholar]
- 861.Laws E.G., Freeman D.E. Significance of reperfusion injury after venous strangulation obstruction of equine jejunum. J Invest Surg. 1995;8:263–270. doi: 10.3109/08941939509031600. [DOI] [PubMed] [Google Scholar]
- 862.Meschter C.L., Craig D., Hackett R. Histopathological and ultrastructural changes in simulated large colonic torsion and reperfusion in ponies. Equine Vet J. 1991;23:426–433. doi: 10.1111/j.2042-3306.1991.tb03755.x. [DOI] [PubMed] [Google Scholar]
- 863.van Hoogmoed L., Snyder J.R., Pascoe J.R., Olander H.J. Evaluation of uniformity of morphological injury of the large colon following severe colonic torsion. Equine Vet J Suppl. 2000;32:98–100. doi: 10.1111/j.2042-3306.2000.tb05343.x. [DOI] [PubMed] [Google Scholar]
- 864.van Hoogmoed L., Snyder J.R., Pascoe J.R., Olander H. Use of pelvic flexure biopsies to predict survival after large colon torsion in horses. Vet Surg. 2000;29:572–577. doi: 10.1053/jvet.2000.17836. [DOI] [PubMed] [Google Scholar]
- 865.White N.A., Moore J.N., Douglas M. SEM study of Strongylus vulgaris larva-induced arteritis in the pony. Equine Vet J. 1983;15:349–353. doi: 10.1111/j.2042-3306.1983.tb01822.x. [DOI] [PubMed] [Google Scholar]
- 866.Moore R.M., Bertone A.L., Bailey M.Q. Neutrophil accumulation in the large colon of horses during low-flow ischemia and reperfusion. Am J Vet Res. 1994;55:1454–1463. [PubMed] [Google Scholar]
- 867.Moore R.M., Bertone A.L., Muir W.W. Histopathologic evidence of reperfusion injury in the large colon of horses after low-flow ischemia. Am J Vet Res. 1994;55:1434–1443. [PubMed] [Google Scholar]
- 868.Gerard M.P., Blikslager A.T., Roberts M.C. The characteristics of intestinal injury peripheral to strangulating obstruction lesions in the equine small intestine. Equine Vet J. 1999;31:331–335. doi: 10.1111/j.2042-3306.1999.tb03826.x. [DOI] [PubMed] [Google Scholar]
- 869.Dabareiner R.M., White N.A., Donaldson L.L. Effects of intraluminal distention and decompression on microvascular permeability and hemodynamics of the equine jejunum. Am J Vet Res. 2001;62:225–236. doi: 10.2460/ajvr.2001.62.225. [DOI] [PubMed] [Google Scholar]
- 870.Dabareiner R.M., Sullins K.E., Snyder J.R. Evaluation of the microcirculation of the equine small intestine after intraluminal distention and subsequent decompression. Am J Vet Res. 1993;54:1673–1682. [PubMed] [Google Scholar]
- 871.Dabareiner R.M., Snyder J.R., White N.A. Microvascular permeability and endothelial cell morphology associated with low-flow ischemia/reperfusion injury in the equine jejunum. Am J Vet Res. 1995;56:639–648. [PubMed] [Google Scholar]
- 872.Allen D.J., White N.A., Tyler D.E. Factors for prognostic use in equine obstructive small intestinal disease. J Am Vet Med Assoc. 1986;189:777–780. [PubMed] [Google Scholar]
- 873.Schoenberg M.H., Poch B., Younes M. Involvement of neutrophils in postischaemic damage to the small intestine. Gut. 1991;32:905–912. doi: 10.1136/gut.32.8.905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 874.Nilsson U.A., Schoenberg M.H., Aneman A. Free radicals and pathogenesis during ischemia and reperfusion of the cat small intestine. Gastroenterology. 1994;106:629–636. doi: 10.1016/0016-5085(94)90695-5. [DOI] [PubMed] [Google Scholar]
- 875.Granger D.N. Role of xanthine oxidase and granulocytes in ischemia-reperfusion injury. Am J Physiol. 1988;255:H1269–H1275. doi: 10.1152/ajpheart.1988.255.6.H1269. [DOI] [PubMed] [Google Scholar]
- 876.Horne M.M., Pascoe P.J., Ducharme N.G. Attempts to modify reperfusion injury of equine jejunal mucosa using dimethylsulfoxide, allopurinol, and intraluminal oxygen. Vet Surg. 1994;23:241–249. doi: 10.1111/j.1532-950x.1994.tb00478.x. [DOI] [PubMed] [Google Scholar]
- 877.Park P.O., Haglund U., Bulkley G.B., Falt K. The sequence of development of intestinal tissue injury after strangulation ischemia and reperfusion. Surgery. 1990;107:574–580. [PubMed] [Google Scholar]
- 878.Haglund U. Gut ischaemia. Gut. 1994;35:S73–S76. doi: 10.1136/gut.35.1_suppl.s73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 879.Dabareiner R.M., Snyder J.R., Sullins K.E., White N.A., Gardner I.A. Evaluation of the microcirculation of the equine jejunum and ascending colon after ischemia and reperfusion. Am J Vet Res. 1993;54:1683–1692. [PubMed] [Google Scholar]
- 880.Grisham M.B., Granger D.N. Neutrophil-mediated mucosal injury. Role of reactive oxygen metabolites. Dig Dis Sci. 1988;33:6S–15S. doi: 10.1007/BF01538126. [DOI] [PubMed] [Google Scholar]
- 881.Prichard M., Ducharme N.G., Wilkins P.A. Xanthine oxidase formation during experimental ischemia of the equine small intestine. Can J Vet Res. 1991;55:310–314. [PMC free article] [PubMed] [Google Scholar]
- 882.Blikslager A.T., Roberts M.C., Gerard M.P., Argenzio R.A. How important is intestinal reperfusion injury in horses? J Am Vet Med Assoc. 1997;211:1387–1389. [PubMed] [Google Scholar]
- 883.Parks D.A., Williams T.K., Beckman J.S. Conversion of xanthine dehydrogenase to oxidase in ischemic rat intestine: a reevaluation. Am J Physiol. 1988;254:G768–G774. doi: 10.1152/ajpgi.1988.254.5.G768. [DOI] [PubMed] [Google Scholar]
- 884.Blikslager A.T., Roberts M.C., Rhoads J.M., Argenzio R.A. Is reperfusion injury an important cause of mucosal damage after porcine intestinal ischemia? Surgery. 1997;121:526–534. doi: 10.1016/s0039-6060(97)90107-0. [DOI] [PubMed] [Google Scholar]
- 885.Schaffer M.R., Efron P.A., Thornton F.J. Nitric oxide, an autocrine regulator of wound fibroblast synthetic function. J Immunol. 1997;158:2375–2381. [PubMed] [Google Scholar]
- 886.Konturek S.J., Brzozowski T., Majka J. Inhibition of nitric oxide synthase delays healing of chronic gastric ulcers. Eur J Pharmacol. 1993;239:215–217. doi: 10.1016/0014-2999(93)90997-v. [DOI] [PubMed] [Google Scholar]
- 887.Tarnawski A., Tanoue K., Santos A.M., Sarfeh I.J. Cellular and molecular mechanisms of gastric ulcer healing. Is the quality of mucosal scar affected by treatment? Scand J Gastroenterol Suppl. 1995;210:9–14. doi: 10.3109/00365529509090261. [DOI] [PubMed] [Google Scholar]
- 888.Tarnawski A., Stachura J., Durbin T. Increased expression of epidermal growth factor receptor during gastric ulcer healing in rats. Gastroenterology. 1992;102:695–698. doi: 10.1016/0016-5085(92)90123-g. [DOI] [PubMed] [Google Scholar]
- 889.Erickson R.A. 16,16-Dimethyl prostaglandin E2 induces villus contraction in rats without affecting intestinal restitution. Gastroenterology. 1990;99:708–716. doi: 10.1016/0016-5085(90)90959-5. [DOI] [PubMed] [Google Scholar]
- 890.Moore R., Madara J.L., MacLeod R.J. Enterocytes adhere preferentially to collagen IV in a differentially regulated divalent cation-dependent manner. Am J Physiol. 1994;266:G1099–G1107. doi: 10.1152/ajpgi.1994.266.6.G1099. [DOI] [PubMed] [Google Scholar]
- 891.Moore R., Madri J., Carlson S., Madara J.L. Collagens facilitate epithelial migration in restitution of native guinea pig intestinal epithelium. Gastroenterology. 1992;102:119–130. doi: 10.1016/0016-5085(92)91791-2. [DOI] [PubMed] [Google Scholar]
- 892.McCormack S.A., Viar M.J., Johnson L.R. Migration of IEC-6 cells: a model for mucosal healing. Am J Physiol. 1992;263:G426–G435. doi: 10.1152/ajpgi.1992.263.3.G426. [DOI] [PubMed] [Google Scholar]
- 893.Blikslager A.T., Roberts M.C., Rhoads J.M., Argenzio R.A. Prostaglandins I2 and E2 have a synergistic role in rescuing epithelial barrier function in porcine ileum. J Clin Invest. 1997;100:1928–1933. doi: 10.1172/JCI119723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 894.Bjerknes M., Cheng H. Clonal analysis of mouse intestinal epithelial progenitors. Gastroenterology. 1999;116:7–14. doi: 10.1016/s0016-5085(99)70222-2. [DOI] [PubMed] [Google Scholar]
- 895.Jankowski J.A., Goodlad R.A., Wright N.A. Maintenance of normal intestinal mucosa: function, structure, and adaptation. Gut. 1994;35:S1–S4. doi: 10.1136/gut.35.1_suppl.s1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 896.Zushi S. Role of prostaglandins in intestinal epithelial restitution stimulated by growth factors. Am J Physiol. 1996;270:G757–G762. doi: 10.1152/ajpgi.1996.270.5.G757. [DOI] [PubMed] [Google Scholar]
- 897.Blikslager A.T., Roberts M.C., Young K.M. Genistein augments prostaglandin-induced recovery of barrier function in ischemia-injured porcine ileum. Am J Physiol Gastrointest Liver Physiol. 2000;278:G207–G216. doi: 10.1152/ajpgi.2000.278.2.G207. [DOI] [PubMed] [Google Scholar]
- 898.Moeser A.J., Haskell M.M., Shifflett D.E. ClC-2 chloride secretion mediates prostaglandin-induced recovery of barrier function in ischemia-injured porcine ileum. Gastroenterology. 2004;127:802–815. doi: 10.1053/j.gastro.2004.06.004. [DOI] [PubMed] [Google Scholar]
- 899.Duffey M.E., Hainau B., Ho S., Bentzel C.J. Regulation of epithelial tight junction permeability by cyclic AMP. Nature. 1981;294:451–453. doi: 10.1038/294451a0. [DOI] [PubMed] [Google Scholar]
- 900.Palant C.E., Duffey M.E., Mookerjee B.K. Ca2+ regulation of tight-junction permeability and structure in Necturus gallbladder. Am J Physiol. 1983;245:C203–C212. doi: 10.1152/ajpcell.1983.245.3.C203. [DOI] [PubMed] [Google Scholar]
- 901.Wang J.Y., Johnson L.R. Luminal polyamines substitute for tissue polyamines in duodenal mucosal repair after stress in rats. Gastroenterology. 1992;102:1109–1117. [PubMed] [Google Scholar]
- 902.Wang J.Y., Johnson L.R. Polyamines and ornithine decarboxylase during repair of duodenal mucosa after stress in rats. Gastroenterology. 1991;100:333–343. doi: 10.1016/0016-5085(91)90200-5. [DOI] [PubMed] [Google Scholar]
- 903.Wang J.Y., Johnson L.R. Role of ornithine decarboxylase in repair of gastric mucosal stress ulcers. Am J Physiol. 1990;258:G78–G85. doi: 10.1152/ajpgi.1990.258.1.G78. [DOI] [PubMed] [Google Scholar]
- 904.McCormack S.A., Wang J.Y., Viar M.J. Polyamines influence transglutaminase activity and cell migration in two cell lines. Am J Physiol. 1994;267:C706–C714. doi: 10.1152/ajpcell.1994.267.3.C706. [DOI] [PubMed] [Google Scholar]
- 905.McCormack S.A., Wang J.Y., Johnson L.R. Polyamine deficiency causes reorganization of F-actin and tropomyosin in IEC-6 cells. Am J Physiol. 1994;267:C715–C722. doi: 10.1152/ajpcell.1994.267.3.C715. [DOI] [PubMed] [Google Scholar]
- 906.Rao J.N., Li L., Golovina V.A. Ca2+-RhoA signaling pathway required for polyamine-dependent intestinal epithelial cell migration. Am J Physiol Cell Physiol. 2001;280:C993–1007. doi: 10.1152/ajpcell.2001.280.4.C993. [DOI] [PubMed] [Google Scholar]
- 907.Caco-2 cells by different mechanisms. Am J Physiol Gastrointest Liver Physiol. 2001;281:G37–G43. doi: 10.1152/ajpgi.2001.281.1.G37. [DOI] [PubMed] [Google Scholar]
- 908.Ray R.M., Zimmerman B.J., McCormack S.A. Polyamine depletion arrests cell cycle and induces inhibitors p21(Waf1/Cip1), p27(Kip1), and p53 in IEC-6 cells. Am J Physiol. 1999;276:C684–C691. doi: 10.1152/ajpcell.1999.276.3.C684. [DOI] [PubMed] [Google Scholar]
- 909.Johnson L.R., Tseng C.C., Wang P. Mucosal ornithine decarboxylase in the small intestine: localization and stimulation. Am J Physiol. 1989;256:G624–G630. doi: 10.1152/ajpgi.1989.256.3.G624. [DOI] [PubMed] [Google Scholar]
- 910.Wang J.Y., Johnson L.R. Expression of protooncogenes c-fos and c-myc in healing of gastric mucosal stress ulcers. Am J Physiol. 1994;266:G878–G886. doi: 10.1152/ajpgi.1994.266.5.G878. [DOI] [PubMed] [Google Scholar]
- 911.Barnard J.A., Beauchamp R.D., Russell W.E. Epidermal growth factor-related peptides and their relevance to gastrointestinal pathophysiology. Gastroenterology. 1995;108:564–580. doi: 10.1016/0016-5085(95)90087-x. [DOI] [PubMed] [Google Scholar]
- 912.Playford R.J., Wright N.A. Why is epidermal growth factor present in the gut lumen? Gut. 1996;38:303–305. doi: 10.1136/gut.38.3.303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 913.Khulusi S., Hanby A.M., Marrero J.M. Expression of trefoil peptides pS2 and human spasmolytic polypeptide in gastric metaplasia at the margin of duodenal ulcers. Gut. 1995;37:205–209. doi: 10.1136/gut.37.2.205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 914.Taupin D., Wu D.C., Jeon W.K. The trefoil gene family are coordinately expressed immediate-early genes: EGF receptor- and MAP kinase-dependent interregulation. J Clin Invest. 1999;103:R31–R38. doi: 10.1172/JCI3304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 915.Goke M., Zuk A., Podolsky D.K. Regulation and function of extracellular matrix intestinal epithelial restitution in vitro. Am J Physiol. 1996;271:G729–G740. doi: 10.1152/ajpgi.1996.271.5.G729. [DOI] [PubMed] [Google Scholar]
- 916.Mashimo H., Wu D.C., Podolsky D.K., Fishman M.C. Impaired defense of intestinal mucosa in mice lacking intestinal trefoil factor. Science. 1996;274:262–265. doi: 10.1126/science.274.5285.262. [DOI] [PubMed] [Google Scholar]
- 917.Rhoads J.M., Argenzio R.A., Chen W. Glutamine metabolism stimulates intestinal cell MAPKs by a cAMP-inhibitable, Raf-independent mechanism. Gastroenterology. 2000;118:90–100. doi: 10.1016/s0016-5085(00)70417-3. [DOI] [PubMed] [Google Scholar]
- 918.Rhoads J.M., Argenzio R.A., Chen W. l-glutamine stimulates intestinal cell proliferation and activates mitogen-activated protein kinases. Am J Physiol. 1997;272:G943–G953. doi: 10.1152/ajpgi.1997.272.5.G943. [DOI] [PubMed] [Google Scholar]
- 919.Kripke S.A., Fox A.D., Berman J.M. Stimulation of intestinal mucosal growth with intracolonic infusion of short-chain fatty acids. J Parenter Enteral Nutr. 1989;13:109–116. doi: 10.1177/0148607189013002109. [DOI] [PubMed] [Google Scholar]
- 920.Inoue Y., Grant J.P., Snyder P.J. Effect of glutamine-supplemented total parenteral nutrition on recovery of the small intestine after starvation atrophy. J Parenter Enteral Nutr. 1993;17:165–170. doi: 10.1177/0148607193017002165. [DOI] [PubMed] [Google Scholar]
- 921.Souba W.W., Herskowitz K., Salloum R.M. Gut glutamine metabolism. J Parenter Enteral Nutr. 1990;14:45S–50S. doi: 10.1177/014860719001400403. [DOI] [PubMed] [Google Scholar]
- 922.Platell C., McCauley R., McCulloch R., Hall J. The influence of parenteral glutamine and branched-chain amino acids on total parenteral nutrition-induced atrophy of the gut. J Parenter Enteral Nutr. 1993;17:348–354. doi: 10.1177/0148607193017004348. [DOI] [PubMed] [Google Scholar]
- 923.Tremel H., Kienle B., Weilemann L.S. Glutamine dipeptide-supplemented parenteral nutrition maintains intestinal function in the critically ill. Gastroenterology. 1994;107:1595–1601. doi: 10.1016/0016-5085(94)90797-8. [DOI] [PubMed] [Google Scholar]
- 924.Frankel W.L., Zhang W., Afonso J. Glutamine enhancement of structure and function in transplanted small intestine in the rat. J Parenter Enteral Nutr. 1993;17:47–55. doi: 10.1177/014860719301700147. [DOI] [PubMed] [Google Scholar]
- 925.Zhang W., Frankel W.L., Singh A. Improvement of structure and function in orthotopic small bowel transplantation in the rat by glutamine. Transplantation. 1993;56:512–517. doi: 10.1097/00007890-199309000-00005. [DOI] [PubMed] [Google Scholar]
- 926.Fox A.D., Kripke S.A., De Paula J. Effect of a glutamine-supplemented enteral diet on methotrexate-induced enterocolitis. J Parenter Enteral Nutr. 1988;12:325–331. doi: 10.1177/0148607188012004325. [DOI] [PubMed] [Google Scholar]
- 927.Klimberg V.S., Salloum R.M., Kasper M. Oral glutamine accelerates healing of the small intestine and improves outcome after whole abdominal radiation. Arch Surg. 1990;125:1040–1045. doi: 10.1001/archsurg.1990.01410200104017. [DOI] [PubMed] [Google Scholar]
- 928.Klimberg V.S., Souba W.W., Dolson D.J. Prophylactic glutamine protects the intestinal mucosa from radiation injury. Cancer. 1990;66:62–68. doi: 10.1002/1097-0142(19900701)66:1<62::aid-cncr2820660113>3.0.co;2-e. [DOI] [PubMed] [Google Scholar]
- 929.Ahdieh N., Blikslager A.T., Bhat B.G. l-glutamine and transforming growth factor-alpha enhance recovery of monoacylglycerol acyltransferase and diacylglycerol acyltransferase activity in porcine postischemic ileum. Pediatr Res. 1998;43:227–233. doi: 10.1203/00006450-199802000-00012. [DOI] [PubMed] [Google Scholar]
- 930.Adams S.B. Recognition and management of ileus. Vet Clin North Am Equine Pract. 1988;4:91–104. doi: 10.1016/s0749-0739(17)30652-1. [DOI] [PubMed] [Google Scholar]
- 931.Becht J.L., Richardson D.W. Ileus in the horse: clinical significance and management. Proc Am Assoc Equine Pract. 1981;27:291–297. [Google Scholar]
- 932.Semevolos S.A., Ducharme N.G., Hackett R.P. Clinical assessment and outcome of three techniques for jejunal resection and anastomosis in horses: 59 cases (1989–2000) J Am Vet Med Assoc. 2002;220:215–218. doi: 10.2460/javma.2002.220.215. [DOI] [PubMed] [Google Scholar]
- 933.van den Boom R., van der Velden M.A. Short-and long-term evaluation of surgical treatment of strangulating obstructions of the small intestine in horses: a review of 224 cases. Vet Q. 2001;23:109–115. doi: 10.1080/01652176.2001.9695095. [DOI] [PubMed] [Google Scholar]
- 934.Morton A.J., Blikslager A.T. Surgical and postoperative factors influencing short-term survival of horses following small intestinal resection: 92 cases (1994–2001) Equine Vet J. 2002;34:450–454. doi: 10.2746/042516402776117700. [DOI] [PubMed] [Google Scholar]
- 935.Mair T.S., Smith L.J. Survival and complication rates in 300 horses undergoing surgical treatment of colic. Part 2: short-term complications. Equine Vet J. 2005;37:303–309. doi: 10.2746/0425164054529364. [DOI] [PubMed] [Google Scholar]
- 936.Campbell M.L., Colahan P.C., Brown M.P. Cecal impaction in the horse. J Am Vet Med Assoc. 1984;184:950–952. [PubMed] [Google Scholar]
- 937.Ross M.W., Martin B.B., Donawick W.J. Cecal perforation in the horse. J Am Vet Med Assoc. 1985;187:249–253. [PubMed] [Google Scholar]
- 938.Hilbert B.J., Little C.B., Bolton J.R. Caecal overload and rupture in the horse. Aust Vet J. 1987;64:85–86. doi: 10.1111/j.1751-0813.1987.tb09624.x. [DOI] [PubMed] [Google Scholar]
- 939.Plummer A.E., Rakestraw P.C., Hardy J. Outcome of medical and surgical treatment of cecal impaction in horses: 114 cases (1994–2004) J Am Vet Med Assoc. 2007;231:1378–1385. doi: 10.2460/javma.231.9.1378. [DOI] [PubMed] [Google Scholar]
- 940.Horowitz B., Ward S.M., Sanders K.M. Cellular and molecular basis for electrical rhythmicity in gastrointestinal muscles. Annu Rev Physiol. 1999;61:19–43. doi: 10.1146/annurev.physiol.61.1.19. [DOI] [PubMed] [Google Scholar]
- 941.Fintl C., Hudson N.P., Mayhew I.G. Interstitial cells of Cajal (ICC) in equine colic: an immunohistochemical study of horses with obstructive disorders of the small and large intestines. Equine Vet J. 2004;36:474–479. doi: 10.2746/0425164044877314. [DOI] [PubMed] [Google Scholar]
- 942.Hudson N., Mayhew I., Pearson G. A reduction in interstitial cells of Cajal in horses with equine dysautonomia (grass sickness) Auton Neurosci. 2001;92:37–44. doi: 10.1016/S1566-0702(01)00316-2. [DOI] [PubMed] [Google Scholar]
- 943.Milne E.M., Fintl C., Hudson N.P. Observations on the interstitial cells of Cajal and neurons in a recovered case of equine dysautonomia (grass sickness) J Comp Pathol. 2005;133:33–40. doi: 10.1016/j.jcpa.2005.01.004. [DOI] [PubMed] [Google Scholar]
- 944.Hudson N., Mayhew I., Pearson G. Presence of in vitro electrical activity in the ileum of horses with enteric nervous system pathology: equine dysautonomia (grass sickness) Auton Neurosci. 2002;99:119–126. doi: 10.1016/s1566-0702(02)00065-6. [DOI] [PubMed] [Google Scholar]
- 945.Bertaccini G., Coruzzi G. Receptors in the gastrointestinal tract. Pharmacol Res Commun. 1987;19:87–118. doi: 10.1016/0031-6989(87)90001-4. [DOI] [PubMed] [Google Scholar]
- 946.Re G., Belloli C., Badino P. Identification of beta-adrenergic receptor subtypes mediating relaxation in isolated equine ileum. Am J Vet Res. 1997;58:621–625. [PubMed] [Google Scholar]
- 947.Malone E.D., Kannan M.S., Brown D.R. Adrenergic, cholinergic, and nonadrenergic-noncholinergic intrinsic innervation of the jejunum in horses. Am J Vet Res. 1999;60:898–904. [PubMed] [Google Scholar]
- 948.Rakestraw P.C., Snyder J.R., Woliner M.J. Involvement of nitric oxide in inhibitory neuromuscular transmission in equine jejunum. Am J Vet Res. 1996;57:1206–1213. [PubMed] [Google Scholar]
- 949.Sellers A.F., Lowe J.E., Cummings J.F. Trials of serotonin, substance P and alpha 2-adrenergic receptor effects on the equine large colon. Cornell Vet. 1985;75:319–323. [PubMed] [Google Scholar]
- 950.Sonea I.M., Wilson D.V., Bowker R.M. Tachykinin receptors in the equine pelvic flexure. Equine Vet J. 1997;29:306–312. doi: 10.1111/j.2042-3306.1997.tb03128.x. [DOI] [PubMed] [Google Scholar]
- 951.Merritt A.M., Panzer R.B., Lester G.D. Equine pelvic flexure myoelectric activity during fed and fasted states. Am J Physiol. 1995;269:G262–G268. doi: 10.1152/ajpgi.1995.269.2.G262. [DOI] [PubMed] [Google Scholar]
- 952.Lester G.D., Bolton J.R., Cullen L.K. Effects of general anesthesia on myoelectric activity of the intestine in horses. Am J Vet Res. 1992;53:1553–1557. [PubMed] [Google Scholar]
- 953.Ross M.W., Cullen K.K., Rutkowski J.A. Myoelectric activity of the ileum, cecum, and right ventral colon in ponies during interdigestive, nonfeeding, and digestive periods. Am J Vet Res. 1990;51:561–566. [PubMed] [Google Scholar]
- 954.Kalff J.C., Carlos T.M., Schraut W.H. Surgically induced leukocytic infiltrates within the rat intestinal muscularis mediate postoperative ileus. Gastroenterology. 1999;117:378–387. doi: 10.1053/gast.1999.0029900378. [DOI] [PubMed] [Google Scholar]
- 955.Turler A., Moore B.A., Pezzone M.A. Colonic postoperative inflammatory ileus in the rat. Ann Surg. 2002;236:56–66. doi: 10.1097/00000658-200207000-00010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 956.Hopster-Iversen C.C., Hopster K., Staszyk C. Effects of experimental mechanical manipulations on local inflammation in the jejunum of horses. Am J Vet Res. 2014;75:385–391. doi: 10.2460/ajvr.75.4.385. [DOI] [PubMed] [Google Scholar]
- 957.Schwarz N.T., Kalff J.C., Turler A. Selective jejunal manipulation causes postoperative pan-enteric inflammation and dysmotility. Gastroenterology. 2004;126:159–169. doi: 10.1053/j.gastro.2003.10.060. [DOI] [PubMed] [Google Scholar]
- 958.Wehner S., Behrendt F.F., Lyutenski B.N. Inhibition of macrophage function prevents intestinal inflammation and postoperative ileus in rodents. Gut. 2007;56:176–185. doi: 10.1136/gut.2005.089615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 959.The F.O., Bennink R.J., Ankum W.M. Intestinal handling-induced mast cell activation and inflammation in human postoperative ileus. Gut. 2008;57:33–40. doi: 10.1136/gut.2007.120238. [DOI] [PubMed] [Google Scholar]
- 960.Turler A., Schnurr C., Nakao A. Endogenous endotoxin participates in causing a panenteric inflammatory ileus after colonic surgery. Ann Surg. 2007;245:734–744. doi: 10.1097/01.sla.0000255595.98041.6b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 961.Kalff J.C., Schraut W.H., Billiar T.R. Role of inducible nitric oxide synthase in postoperative intestinal smooth muscle dysfunction in rodents. Gastroenterology. 2000;118:316–327. doi: 10.1016/s0016-5085(00)70214-9. [DOI] [PubMed] [Google Scholar]
- 962.Little D., Tomlinson J.E., Blikslager A.T. Post operative neutrophilic inflammation in equine small intestine after manipulation and ischaemia. Equine Vet J. 2005;37:329–335. doi: 10.2746/0425164054529472. [DOI] [PubMed] [Google Scholar]
- 963.Roger T., Ruckebusch Y. Colonic alpha 2-adrenoceptor-mediated responses in the pony. J Vet Pharmacol Ther. 1987;10:310–318. doi: 10.1111/j.1365-2885.1987.tb00107.x. [DOI] [PubMed] [Google Scholar]
- 964.Merritt A.M., Campbell-Thompson M.L., Lowrey S. Effect of xylazine treatment on equine proximal gastrointestinal tract myoelectrical activity. Am J Vet Res. 1989;50:945–949. [PubMed] [Google Scholar]
- 965.Clark E.S., Thompson S.A., Becht J.L. Effects of xylazine on cecal mechanical activity and cecal blood flow in healthy horses. Am J Vet Res. 1988;49:720–723. [PubMed] [Google Scholar]
- 966.Adams S.B., Lamar C.H., Masty J. Motility of the distal portion of the jejunum and pelvic flexure in ponies: effects of six drugs. Am J Vet Res. 1984;45:795–799. [PubMed] [Google Scholar]
- 967.Rutkowski J.A., Ross M.W., Cullen K. Effects of xylazine and/or butorphanol or neostigmine on myoelectric activity of the cecum and right ventral colon in female ponies. Am J Vet Res. 1989;50:1096–1101. [PubMed] [Google Scholar]
- 968.Merritt A.M., Burrow J.A., Hartless C.S. Effect of xylazine, detomidine, and a combination of xylazine and butorphanol on equine duodenal motility. Am J Vet Res. 1998;59:619–623. [PubMed] [Google Scholar]
- 969.Elfenbein J.R., Sanchez L.C., Robertson S.A. Effect of detomidine on visceral and somatic nociception and duodenal motility in conscious adult horses. Vet Anaesth Analg. 2009;36:162–172. doi: 10.1111/j.1467-2995.2008.00441.x. [DOI] [PubMed] [Google Scholar]
- 970.Boscan P., Van Hoogmoed L.M., Farver T.B. Evaluation of the effects of the opioid agonist morphine on gastrointestinal tract function in horses. Am J Vet Res. 2006;67:992–997. doi: 10.2460/ajvr.67.6.992. [DOI] [PubMed] [Google Scholar]
- 971.Roger T., Bardon T., Ruckebusch Y. Colonic motor responses in the pony: relevance of colonic stimulation by opiate antagonists. Am J Vet Res. 1985;46:31–35. [PubMed] [Google Scholar]
- 972.Sanchez L.C., Robertson S.A., Maxwell L.K. Effect of fentanyl on visceral and somatic nociception in conscious horses. J Vet Intern Med. 2007;21:1067–1075. doi: 10.1892/0891-6640(2007)21[1067:eofova]2.0.co;2. [DOI] [PubMed] [Google Scholar]
- 973.Sojka J.E., Adams S.B., Lamar C.H. Effect of butorphanol, pentazocine, meperidine, or metoclopramide on intestinal motility in female ponies. Am J Vet Res. 1988;49:527–529. [PubMed] [Google Scholar]
- 974.Merritt A.M., Campbell-Thompson M.L., Lowrey S. Effect of butorphanol on equine antroduodenal motility. Equine Vet J Suppl. 1989:21–23. doi: 10.1111/j.2042-3306.1989.tb05649.x. [DOI] [PubMed] [Google Scholar]
- 975.Sellon D.C., Monroe V.L., Roberts M.C. Pharmacokinetics and adverse effects of butorphanol administered by single intravenous injection or continuous intravenous infusion in horses. Am J Vet Res. 2001;62:183–189. doi: 10.2460/ajvr.2001.62.183. [DOI] [PubMed] [Google Scholar]
- 976.Sanchez L.C., Elfenbein J.R., Robertson S.A. Effect of acepromazine, butorphanol, or N-butylscopolammonium bromide on visceral and somatic nociception and duodenal motility in conscious horses. Am J Vet Res. 2008;69:579–585. doi: 10.2460/ajvr.69.5.579. [DOI] [PubMed] [Google Scholar]
- 977.Elfenbein J.R., Robertson S.A., MacKay R.J. Systemic and anti-nociceptive effects of prolonged lidocaine, ketamine, and butorphanol infusions alone and in combination in healthy horses. BMC Vet Res. 2014;10(suppl 1):S6. doi: 10.1186/1746-6148-10-S1-S6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 978.Lester G.D. Murdoch University; Perth, Australia: 1990. The development and application of a computer system for the recording and analysis of intestinal myoelectrical activity in the horse. [Google Scholar]
- 979.Sjoqvist A., Hallerback B., Glise H. Reflex adrenergic inhibition of colonic motility in anesthetized rat caused by nociceptive stimuli of peritoneum. An alpha 2-adrenoceptor-mediated response. Dig Dis Sci. 1985;30:749–754. doi: 10.1007/BF01320489. [DOI] [PubMed] [Google Scholar]
- 980.Pairet M., Ruckebusch Y. On the relevance of non-steroidal anti-inflammatory drugs in the prevention of paralytic ileus in rodents. J Pharm Pharmacol. 1989;41:757–761. doi: 10.1111/j.2042-7158.1989.tb06360.x. [DOI] [PubMed] [Google Scholar]
- 981.Zittel T.T., Meile T., Huge A. Preoperative intraluminal application of capsaicin increases postoperative gastric and colonic motility in rats. J Gastrointest Surg. 2001;5:503–513. doi: 10.1016/s1091-255x(01)80088-3. [DOI] [PubMed] [Google Scholar]
- 982.Lowe J.E., Sellers A.F., Brondum J. Equine pelvic flexure impaction. A model used to evaluate motor events and compare drug response. Cornell Vet. 1980;70:401–412. [PubMed] [Google Scholar]
- 983.MacHarg M.A., Adams S.B., Lamar C.H. Electromyographic, myomechanical, and intraluminal pressure changes associated with acute extraluminal obstruction of the jejunum in conscious ponies. Am J Vet Res. 1986;47:7–11. [PubMed] [Google Scholar]
- 984.King J.N., Gerring E.L. Antagonism of endotoxin-induced disruption of equine bowel motility by flunixin and phenylbutazone. Equine Vet J Suppl. 1989:38–42. doi: 10.1111/j.2042-3306.1989.tb05653.x. [DOI] [PubMed] [Google Scholar]
- 985.Meisler S.D., Doherty T.J., Andrews F.M. Yohimbine ameliorates the effects of endotoxin on gastric emptying of the liquid marker acetaminophen in horses. Can J Vet Res. 2000;64:208–211. [PMC free article] [PubMed] [Google Scholar]
- 986.Valk N., Doherty T.J., Blackford J.T. Phenylbutazone prevents the endotoxin-induced delay in gastric emptying in horses. Can J Vet Res. 1998;62:214–217. [PMC free article] [PubMed] [Google Scholar]
- 987.Valk N., Doherty T.J., Blackford J.T. Effect of cisapride on gastric emptying in horses following endotoxin treatment. Equine Vet J. 1998;30:344–348. doi: 10.1111/j.2042-3306.1998.tb04108.x. [DOI] [PubMed] [Google Scholar]
- 988.Eskandari M.K., Kalff J.C., Billiar T.R. Lipopolysaccharide activates the muscularis macrophage network and suppresses circular smooth muscle activity. Am J Physiol. 1997;273:G727–G734. doi: 10.1152/ajpgi.1997.273.3.G727. [DOI] [PubMed] [Google Scholar]
- 989.Schwarz N.T., Engel B., Eskandari M.K. Lipopolysaccharide preconditioning and cross-tolerance: the induction of protective mechanisms for rat intestinal ileus. Gastroenterology. 2002;123:586–598. doi: 10.1053/gast.2002.34777. [DOI] [PubMed] [Google Scholar]
- 990.Overhaus M., Togel S., Pezzone M.A. Mechanisms of polymicrobial sepsis-induced ileus. Am J Physiol Gastrointest Liver Physiol. 2004;287:G685–G694. doi: 10.1152/ajpgi.00359.2003. [DOI] [PubMed] [Google Scholar]
- 991.Fintl C., Hudson N.P., Handel I. The effect of temperature changes on in vitro slow wave activity in the equine ileum. Equine Vet J. 2016;48:218–223. doi: 10.1111/evj.12401. [DOI] [PubMed] [Google Scholar]
- 992.Hooper R.N., Roussel A.J., Cohen N.D. Erythromycin stimulates myoelectric activity in the ileum and pelvic flexure of horses in the post-operative period. Proc Equine Colic Res Symp. 1998;6:42. [Google Scholar]
- 993.Van Hoogmoed L., Rakestraw P.C., Snyder J.R. In vitro effects of nonsteroidal anti-inflammatory agents and prostaglandins I2, E2, and F2alpha on contractility of taenia of the large colon of horses. Am J Vet Res. 1999;60:1004–1009. [PubMed] [Google Scholar]
- 994.Roussel A.J., Jr., Cohen N.D., Hooper R.N. Risk factors associated with development of postoperative ileus in horses. J Am Vet Med Assoc. 2001;219:72–78. doi: 10.2460/javma.2001.219.72. [DOI] [PubMed] [Google Scholar]
- 995.Freeman D.E., Hammock P., Baker G.J. Short- and long-term survival and prevalence of postoperative ileus after small intestinal surgery in the horse. Equine Vet J Suppl. 2000:42–51. doi: 10.1111/j.2042-3306.2000.tb05333.x. [DOI] [PubMed] [Google Scholar]
- 996.Cohen N.D., Lester G.D., Sanchez L.C. Evaluation of risk factors associated with development of postoperative ileus in horses. J Am Vet Med Assoc. 2004;225:1070–1078. doi: 10.2460/javma.2004.225.1070. [DOI] [PubMed] [Google Scholar]
- 997.Blikslager A.T., Bowman K.F., Levine J.F. Evaluation of factors associated with postoperative ileus in horses: 31 cases (1990–1992) J Am Vet Med Assoc. 1994;205:1748–1752. [PubMed] [Google Scholar]
- 998.Lefebvre D., Hudson N.P., Elce Y.A. Clinical features and management of equine post operative ileus (POI): Survey of Diplomates of the American Colleges of Veterinary Internal Medicine (ACVIM), Veterinary Surgeons (ACVS) and Veterinary Emergency and Critical Care (ACVECC) Equine Vet J. 2016;48:714–719. doi: 10.1111/evj.12520. [DOI] [PubMed] [Google Scholar]
- 999.Lefebvre D., Pirie R.S., Handel I.G. Clinical features and management of equine post operative ileus: survey of diplomates of the European Colleges of Equine Internal Medicine (ECEIM) and Veterinary Surgeons (ECVS) Equine Vet J. 2016;48:182–187. doi: 10.1111/evj.12355. [DOI] [PubMed] [Google Scholar]
- 1000.Gerard M.P., Bowman K.F., Blikslager A.T. Jejunocolostomy or ileocolostomy for treatment of cecal impaction in horses: nine cases (1985–1995) J Am Vet Med Assoc. 1996;209:1287–1290. [PubMed] [Google Scholar]
- 1001.Roberts C.T., Slone D.E. Caecal impactions managed surgically by typhlotomy in 10 cases (1988-1998) Equine Vet J Suppl. 2000:74–76. doi: 10.1111/j.2042-3306.2000.tb05338.x. [DOI] [PubMed] [Google Scholar]
- 1002.Collins S.M. The immunomodulation of enteric neuromuscular function: implications for motility and inflammatory disorders. Gastroenterology. 1996;111:1683–1699. doi: 10.1016/s0016-5085(96)70034-3. [DOI] [PubMed] [Google Scholar]
- 1003.Paradelis A.G. Inhibition of the pendular movements of the intestine by aminoglycoside antibiotics. Methods Find Exp Clin Pharmacol. 1981;3:173–177. [PubMed] [Google Scholar]
- 1004.Sparnon A.L., Spitz L. Pharmacological manipulation of postoperative intestinal adhesions. Aust N Z J Surg. 1989;59:725–729. doi: 10.1111/j.1445-2197.1989.tb01665.x. [DOI] [PubMed] [Google Scholar]
- 1005.Marti M., Mevissen M., Althaus H. In vitro effects of bethanechol on equine gastrointestinal contractility and functional characterization of involved muscarinic receptor subtypes. J Vet Pharmacol Ther. 2005;28:565–574. doi: 10.1111/j.1365-2885.2005.00693.x. [DOI] [PubMed] [Google Scholar]
- 1006.Ringger N.C., Lester G.D., Neuwirth L. Effect of bethanechol or erythromycin on gastric emptying in horses. Am J Vet Res. 1996;57:1771–1775. [PubMed] [Google Scholar]
- 1007.Adams S.B., MacHarg M.A. Neostigmine methylsulfate delays gastric emptying of particulate markers in horses. Am J Vet Res. 1985;46:2498–2499. [PubMed] [Google Scholar]
- 1008.Nieto J.E., Rakestraw P.C., Snyder J.R. In vitro effects of erythromycin, lidocaine, and metoclopramide on smooth muscle from the pyloric antrum, proximal portion of the duodenum, and middle portion of the jejunum of horses. Am J Vet Res. 2000;61:413–419. doi: 10.2460/ajvr.2000.61.413. [DOI] [PubMed] [Google Scholar]
- 1009.Gerring E.E., Hunt J.M. Pathophysiology of equine postoperative ileus: effect of adrenergic blockade, parasympathetic stimulation and metoclopramide in an experimental model. Equine Vet J. 1986;18:249–255. doi: 10.1111/j.2042-3306.1986.tb03618.x. [DOI] [PubMed] [Google Scholar]
- 1010.Dart A.J., Peauroi J.R., Hodgson D.R. Efficacy of metoclopramide for treatment of ileus in horses following small intestinal surgery: 70 cases (1989–1992) Aust Vet J. 1996;74:280–284. doi: 10.1111/j.1751-0813.1996.tb13775.x. [DOI] [PubMed] [Google Scholar]
- 1011.Gerring E.L., King J.N. Cisapride in the prophylaxis of equine post operative ileus. Equine Vet J Suppl. 1989:52–55. doi: 10.1111/j.2042-3306.1989.tb05656.x. [DOI] [PubMed] [Google Scholar]
- 1012.Milne E.M., Doxey D.L., Woodman M.P. An evaluation of the use of cisapride in horses with chronic grass sickness (equine dysautonomia) Br Vet J. 1996;152:537–549. doi: 10.1016/s0007-1935(96)80006-6. [DOI] [PubMed] [Google Scholar]
- 1013.Steinebach M.A., Cole D. Use of cisapride in the resolution of pelvic flexure impaction in a horse. Can Vet J. 1995;36:624–625. [PMC free article] [PubMed] [Google Scholar]
- 1014.Valden M.A., Klein W.R. The effects of cisapride on the restoration of gut motility after surgery of the small intestine in horses: a clinical trial. Vet Q. 1993;15:175–179. doi: 10.1080/01652176.1993.9694400. [DOI] [PubMed] [Google Scholar]
- 1015.Cable C.S., Ball M.A., Schwark W.S. Preparation of a parenteral formulation of cisapride from propulsid tablets and pharmacokinetic analysis after its intravenous administration. J Equine Vet Sci. 1999;18:616–621. [Google Scholar]
- 1016.Cubeddu L.X. QT prolongation and fatal arrhythmias: a review of clinical implications and effects of drugs. Am J Ther. 2003;10:452–457. doi: 10.1097/00045391-200311000-00013. [DOI] [PubMed] [Google Scholar]
- 1017.Delco M.L., Nieto J.E., Craigmill A.L. Pharmacokinetics and in vitro effects of tegaserod, a serotonin 5-hydroxytryptamine 4 (5-HT4) receptor agonist with prokinetic activity in horses. Vet Ther. 2007;8:77–87. [PubMed] [Google Scholar]
- 1018.Lippold B.S., Hildebrand J., Straub R. Tegaserod (HTF 919) stimulates gut motility in normal horses. Equine Vet J. 2004;36:622–627. doi: 10.2746/0425164044864543. [DOI] [PubMed] [Google Scholar]
- 1019.Hammerle C.W., Surawicz C.M. Updates on treatment of irritable bowel syndrome. World J Gastroenterol. 2008;14:2639–2649. doi: 10.3748/wjg.14.2639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1020.Nieto J.E., Maher O., Stanley S.D. In vivo and in vitro evaluation of the effects of domperidone on the gastrointestinal tract of healthy horses. Am J Vet Res. 2013;74:1103–1110. doi: 10.2460/ajvr.74.8.1103. [DOI] [PubMed] [Google Scholar]
- 1021.Lester G.D., Merritt A.M., Neuwirth L. Effect of erythromycin lactobionate on myoelectric activity of ileum, cecum, and right ventral colon, and cecal emptying of radiolabeled markers in clinically normal ponies. Am J Vet Res. 1998;59:328–334. [PubMed] [Google Scholar]
- 1022.Nieto J.E., Van Hoogmoed L.M., Spier S.J. Use of an extracorporeal circuit to evaluate effects of intraluminal distention and decompression on the equine jejunum. Am J Vet Res. 2002;63:267–275. doi: 10.2460/ajvr.2002.63.267. [DOI] [PubMed] [Google Scholar]
- 1023.Bologna S.D., Hasler W.L., Owyang C. Down-regulation of motilin receptors on rabbit colon myocytes by chronic oral erythromycin. J Pharmacol Exp Ther. 1993;266:852–856. [PubMed] [Google Scholar]
- 1024.Roussel A.J., Hooper R.N., Cohen N.D. Evaluation of the effects of penicillin G potassium and potassium chloride on the motility of the large intestine in horses. Am J Vet Res. 2003;64:1360–1363. doi: 10.2460/ajvr.2003.64.1360. [DOI] [PubMed] [Google Scholar]
- 1025.Van Hoogmoed L.M., Boscan P.L. In vitro evaluation of the effect of the opioid antagonist N-methylnaltrexone on motility of the equine jejunum and pelvic flexure. Equine Vet J. 2005;37:325–328. doi: 10.2746/0425164054529346. [DOI] [PubMed] [Google Scholar]
- 1026.Boscan P., Van Hoogmoed L.M., Pypendop B.H. Pharmacokinetics of the opioid antagonist N-methylnaltrexone and evaluation of its effects on gastrointestinal tract function in horses treated or not treated with morphine. Am J Vet Res. 2006;67:998–1004. doi: 10.2460/ajvr.67.6.998. [DOI] [PubMed] [Google Scholar]
- 1027.Van Hoogmoed L.M., Nieto J.E., Snyder J.R. Survey of prokinetic use in horses with gastrointestinal injury. Vet Surg. 2004;33:279–285. doi: 10.1111/j.1532-950X.2004.04041.x. [DOI] [PubMed] [Google Scholar]
- 1028.Rimback G., Cassuto J., Tollesson P.O. Treatment of postoperative paralytic ileus by intravenous lidocaine infusion. Anesthesia Analgesia. 1990;70:414–419. [PubMed] [Google Scholar]
- 1029.Lahat A., Horin S.B., Lang A. Lidocaine down-regulates nuclear factor-kappaB signalling and inhibits cytokine production and T cell proliferation. Clin Exp Immunol. 2008;152:320–327. doi: 10.1111/j.1365-2249.2008.03636.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1030.Robertson S.A., Sanchez L.C., Merritt A.M. Effect of systemic lidocaine on visceral and somatic nociception in conscious horses. Equine Vet J. 2005;37:122–127. doi: 10.2746/0425164054223723. [DOI] [PubMed] [Google Scholar]
- 1031.Milligan M., Beard W., Kukanich B. The effect of lidocaine on postoperative jejunal motility in normal horses. Vet Surg. 2007;36:214–220. doi: 10.1111/j.1532-950X.2007.00255.x. [DOI] [PubMed] [Google Scholar]
- 1032.Brianceau P., Chevalier H., Karas A. Intravenous lidocaine and small-intestinal size, abdominal fluid, and outcome after colic surgery in horses. J Vet Intern Med. 2002;16:736–741. doi: 10.1892/0891-6640(2002)016<0736:ilassa>2.3.co;2. [DOI] [PubMed] [Google Scholar]
- 1033.Salem S.E., Proudman C.J., Archer D.C. Has intravenous lidocaine improved the outcome in horses following surgical management of small intestinal lesions in a UK hospital population? BMC Vet Res. 2016;12:157. doi: 10.1186/s12917-016-0784-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1034.Dickey E.J., McKenzie H.C., 3rd, Brown J.A. Serum concentrations of lidocaine and its metabolites after prolonged infusion in healthy horses. Equine Vet J. 2008 doi: 10.2746/042516408X284664. [DOI] [PubMed] [Google Scholar]
- 1035.Snyder J.R., Pascoe J.R., Olander H.J. Strangulating volvulus of the ascending colon in horses. J Am Vet Med Assoc. 1989;195:757–764. [PubMed] [Google Scholar]
- 1036.Hughes F.E., Slone D.E., Jr. Large colon resection. Vet Clin North Am Equine Pract. 1997;13:341–350. doi: 10.1016/s0749-0739(17)30243-2. [DOI] [PubMed] [Google Scholar]
- 1037.Lundin C., Sullins K.E., White N.A. Induction of peritoneal adhesions with small intestinal ischaemia and distention in the foal. Equine Vet J. 1989;21:451–458. doi: 10.1111/j.2042-3306.1989.tb02195.x. [DOI] [PubMed] [Google Scholar]
- 1038.Freeman D.E., Koch D.B., Boles C.L. Mesodiverticular bands as a cause of small intestinal strangulation and volvulus in the horse. J Am Vet Med Assoc. 1979;175:1089–1094. [PubMed] [Google Scholar]
- 1039.Vachon A.M., Fischer A.T. Small intestinal herniation through the epiploic foramen: 53 cases (1987–1993) Equine Vet J. 1995;27:373–380. doi: 10.1111/j.2042-3306.1995.tb04073.x. [DOI] [PubMed] [Google Scholar]
- 1040.White N.A., Lessard P. Determining the diagnosis and prognosis of the acute abdomen. In: White N.A., editor. The Equine Acute Abdomen. Lea & Febiger; Philadelphia, PA: 1990. pp. 101–152. [Google Scholar]
- 1041.White N.A., Lessard P. Risk factors and clinical signs associated with cases of equine colic. Proc Am Assoc Equine Pract. 1986:637–644. [Google Scholar]
- 1042.Mair T.S., Smith L.J. Survival and complication rates in 300 horses undergoing surgical treatment of colic. Part 1: short-term survival following a single laparotomy. Equine Vet J. 2005;37:296–302. doi: 10.2746/0425164054529409. [DOI] [PubMed] [Google Scholar]
- 1043.MacDonald M.H., Pascoe J.R., Stover S.M. Survival after small intestine resection and anastomosis in horses. Vet Surg. 1989;18:415–423. doi: 10.1111/j.1532-950x.1990.tb01116.x. [DOI] [PubMed] [Google Scholar]
- 1044.Proudman C.J., Edwards G.B., Barnes J. Factors affecting long-term survival of horses recovering from surgery of the small intestine. Equine Vet J. 2005;37:360–365. doi: 10.2746/0425164054529481. [DOI] [PubMed] [Google Scholar]
- 1045.Gayle J.M., Blikslager A.T., Bowman K.F. Mesenteric rents as a source of small intestinal strangulation in horses: 15 cases (1990–1997) J Am Vet Med Assoc. 2000;216:1446–1449. doi: 10.2460/javma.2000.216.1446. [DOI] [PubMed] [Google Scholar]
- 1046.Freeman D.E., Schaeffer D.J., Cleary O.B. Long-term survival in horses with strangulating obstruction of the small intestine managed without resection. Equine Vet J. 2014;46:711–717. doi: 10.1111/evj.12216. [DOI] [PubMed] [Google Scholar]
- 1047.Gazzerro D.M., Southwood L.L., Lindborg S. Short-term complications after colic surgery in geriatric versus mature non-geriatric horses. Vet Surg. 2015;44:256–264. doi: 10.1111/j.1532-950X.2014.12281.x. [DOI] [PubMed] [Google Scholar]
- 1048.Bergren A.L., Credille B.C., Epstein K.L. Retrospective Comparison of Gastrosplenic Entrapment of the Small Intestine to Other Strangulating Small Intestinal Lesions in Adult Horses. Vet Surg. 2015;44:535–539. doi: 10.1111/j.1532-950X.2014.12235.x. [DOI] [PubMed] [Google Scholar]
- 1049.Kilcoyne I., Dechant J.E., Nieto J.E. Comparison of clinical findings and short-term survival between horses with intestinal entrapment in the gastrosplenic ligament and horses with intestinal entrapment in the epiploic foramen. J Am Vet Med Assoc. 2016;249:660–667. doi: 10.2460/javma.249.6.660. [DOI] [PubMed] [Google Scholar]
- 1050.Freeman D.E., Schaeffer D.J. Short-term survival after surgery for epiploic foramen entrapment compared with other strangulating diseases of the small intestine in horses. Equine Vet J. 2005;37:292–295. doi: 10.2746/0425164054529436. [DOI] [PubMed] [Google Scholar]
- 1051.Archer D.C., Pinchbeck G.L., Proudman C.J. Factors associated with survival of epiploic foramen entrapment colic: a multicentre, international study. Equine Vet J Suppl. 2011;39:56–62. doi: 10.1111/j.2042-3306.2011.00409.x. [DOI] [PubMed] [Google Scholar]
- 1052.Freeman D.E., Schaeffer D.J. Age distributions of horses with strangulation of the small intestine by a lipoma or in the epiploic foramen: 46 cases (1994–2000) J Am Vet Med Assoc. 2001;219:87–89. doi: 10.2460/javma.2001.219.87. [DOI] [PubMed] [Google Scholar]
- 1053.Beccati F., Pepe M., Gialletti R. Is there a statistical correlation between ultrasonographic findings and definitive diagnosis in horses with acute abdominal pain? Equine Vet J Suppl. 2011:98–105. doi: 10.1111/j.2042-3306.2011.00428.x. [DOI] [PubMed] [Google Scholar]
- 1054.Klohnen A., Vachon A.M., Fischer A.T. Use of diagnostic ultrasonography in horses with signs of acute abdominal pain. J Am Vet Med Assoc. 1996;209:1597–1601. [PubMed] [Google Scholar]
- 1055.Hammock P.D., Freeman D.E., Magid J.H. Parietal hernia of the small intestine into the epiploic foramen of a horse. J Am Vet Med Assoc. 1999;214:1354–1356. 1334-1355. [PubMed] [Google Scholar]
- 1056.Engelbert T.A., Tate L.P., Jr., Bowman K.F. Incarceration of the small intestine in the epiploic foramen. Report of 19 cases (1983-1992) Vet Surg. 1993;22:57–61. doi: 10.1111/j.1532-950x.1993.tb00370.x. [DOI] [PubMed] [Google Scholar]
- 1057.Garcia-Seco E., Wilson D.A., Kramer J. Prevalence and risk factors associated with outcome of surgical removal of pedunculated lipomas in horses: 102 cases (1987–2002) J Am Vet Med Assoc. 2005;226:1529–1537. doi: 10.2460/javma.2005.226.1529. [DOI] [PubMed] [Google Scholar]
- 1058.Edwards G.B., Proudman C.J. An analysis of 75 cases of intestinal obstruction caused by pedunculated lipomas. Equine Vet J. 1994;26:18–21. doi: 10.1111/j.2042-3306.1994.tb04324.x. [DOI] [PubMed] [Google Scholar]
- 1059.Blikslager A.T., Bowman K.F., Haven M.L. Pedunculated lipomas as a cause of intestinal obstruction in horses: 17 cases (1983–1990) J Am Vet Med Assoc. 1992;201:1249–1252. [PubMed] [Google Scholar]
- 1060.Robertson J.T. Diseases of the small intestine. In: White N.A., editor. The Equine Acute Abdomen. Lea & Febiger; Philadelphia, PA: 1990. pp. 347–368. [Google Scholar]
- 1061.Crowhurst R.C., Simpson D.J., McEnery R.J. Intestinal surgery in the foal. J S Afr Vet Assoc. 1975;46:59–67. [PubMed] [Google Scholar]
- 1062.Orsini J.A. Abdominal surgery in foals. Vet Clin North Am Equine Pract. 1997;13:393–413. doi: 10.1016/s0749-0739(17)30247-x. [DOI] [PubMed] [Google Scholar]
- 1063.Tate L.P., Jr., Ralston S.L., Koch C.M. Effects of extensive resection of the small intestine in the pony. Am J Vet Res. 1983;44:1187–1191. [PubMed] [Google Scholar]
- 1064.Jenei T.M., Garcia-Lopez J.M., Provost P.J. Surgical management of small intestinal incarceration through the gastrosplenic ligament: 14 cases (1994–2006) J Am Vet Med Assoc. 2007;231:1221–1224. doi: 10.2460/javma.231.8.1221. [DOI] [PubMed] [Google Scholar]
- 1065.Becht J.L., McIlwraith C.W. Jejunal displacement through the mesometrium in a pregnant mare. J Am Vet Med Assoc. 1980;177:436. [PubMed] [Google Scholar]
- 1066.Gayle J.M., Macharg M.A., Smallwood J.E. Strangulating obstruction caused by intestinal herniation through the proximal aspect of the cecocolic fold in 9 horses. Vet Surg. 2001;30:40–43. doi: 10.1053/jvet.2001.20342. [DOI] [PubMed] [Google Scholar]
- 1067.Spurlock G.H., Robertson J.T. Congenital inguinal hernias associated with a rent in the common vaginal tunic in five foals. J Am Vet Med Assoc. 1988;193:1087–1088. [PubMed] [Google Scholar]
- 1068.van der Velden M.A. Ruptured inguinal hernia in new-born colt foals: a review of 14 cases. Equine Vet J. 1988;20:178–181. doi: 10.1111/j.2042-3306.1988.tb01492.x. [DOI] [PubMed] [Google Scholar]
- 1069.Schneider R.K., Milne D.W., Kohn C.W. Acquired inguinal hernia in the horse: a review of 27 cases. J Am Vet Med Assoc. 1982;180:317–320. [PubMed] [Google Scholar]
- 1070.van der Velden M.A. Surgical treatment of acquired inguinal hernia in the horse: a review of 51 cases. Equine Vet J. 1988;20:173–177. doi: 10.1111/j.2042-3306.1988.tb01491.x. [DOI] [PubMed] [Google Scholar]
- 1071.Weaver A.D. Acquired incarcerated inguinal hernia: a review of 13 horses. Can Vet J. 1987;28:195–199. [PMC free article] [PubMed] [Google Scholar]
- 1072.Ivens P.A., Piercy R.J., Eliashar E. Inguinal herniation of the large colon in a cob gelding four weeks after castration. Vet Rec. 2009;165:380–381. doi: 10.1136/vr.165.13.380. [DOI] [PubMed] [Google Scholar]
- 1073.van der Velden M.A., Stolk P.W. Different types of inguinal herniation in two stallions and a gelding. Vet Q. 1990;12:46–50. doi: 10.1080/01652176.1990.9694241. [DOI] [PubMed] [Google Scholar]
- 1074.Freeman D.E. Surgery of the small intestine. Vet Clin North Am Equine Pract. 1997;13:261–301. doi: 10.1016/s0749-0739(17)30240-7. [DOI] [PubMed] [Google Scholar]
- 1075.Freeman D.E., Orsini J.A., Harrison I.W. Complications of umbilical hernias in horses: 13 cases (1972–1986) J Am Vet Med Assoc. 1988;192:804–807. [PubMed] [Google Scholar]
- 1076.Markel M.D., Pascoe J.R., Sams A.E. Strangulated umbilical hernias in horses: 13 cases (1974–1985) J Am Vet Med Assoc. 1987;190:692–694. [PubMed] [Google Scholar]
- 1077.Edwards G.B. Surgical management of intussusception in the horse. Equine Vet J. 1986;18:313–321. doi: 10.1111/j.2042-3306.1986.tb03640.x. [DOI] [PubMed] [Google Scholar]
- 1078.Ford T.S., Freeman D.E., Ross M.W. Ileocecal intussusception in horses: 26 cases (1981–1988) J Am Vet Med Assoc. 1990;196:121–126. [PubMed] [Google Scholar]
- 1079.Bernard W.V., Reef V.B., Reimer J.M. Ultrasonographic diagnosis of small-intestinal intussusception in three foals. J Am Vet Med Assoc. 1989;194:395–397. [PubMed] [Google Scholar]
- 1089.Edens L.M., White N.A., Dabareiner R.M. Transrectal ultrasonographic diagnosis of an ileocaecal intussusception in a horse. Equine Vet J. 1996;28:81–83. doi: 10.1111/j.2042-3306.1996.tb01594.x. [DOI] [PubMed] [Google Scholar]
- 1081.Fontaine-Rodgerson G., Rodgerson D.H. Diagnosis of small intestinal intussusception by transabdominal ultrasonography in 2 adult horses. Can Vet J. 2001;42:378–380. [PMC free article] [PubMed] [Google Scholar]
- 1082.Abraham M., Reef V.B., Sweeney R.W. Gastrointestinal ultrasonography of normal Standardbred neonates and frequency of asymptomatic intussusceptions. J Vet Intern Med. 2014;28:1580–1586. doi: 10.1111/jvim.12413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1083.Gift L.J., Gaughan E.M., DeBowes R.M. Jejunal intussusception in adult horses: 11 cases (1981–1991) J Am Vet Med Assoc. 1993;202:110–112. [PubMed] [Google Scholar]
- 1084.Beard W.L., Byrne B.A., Henninger R.W. Ileocecal intussusception corrected by resection within the cecum in two horses. J Am Vet Med Assoc. 1992;200:1978–1980. [PubMed] [Google Scholar]
- 1085.Bristol D.G. Diaphragmatic hernias in horses and cattle. Comp Contin Ed Pract Vet. 1986;8:S407–S411. [Google Scholar]
- 1086.Wimberly H.C., Andrews E.J., Haschek W.M. Diaphragmatic hernias in the horse: a review of the literature and an analysis of six additional cases. J Am Vet Med Assoc. 1977;170:1404–1407. [PubMed] [Google Scholar]
- 1087.Orsini J.A., Koch C., Stewart B. Peritoneopericardial hernia in a horse. J Am Vet Med Assoc. 1981;179:907–910. [PubMed] [Google Scholar]
- 1088.Everett K.A., Chaffin M.K., Brinsko S.P. Diaphragmatic herniation as a cause of lethargy and exercise intolerance in a mare. Cornell Vet. 1992;82:217–223. [PubMed] [Google Scholar]
- 1089.Santschi E.M., Juzwiak J.S., Moll H.D. Diaphragmatic hernia repair in three young horses. Vet Surg. 1997;26:242–245. doi: 10.1111/j.1532-950x.1997.tb01492.x. [DOI] [PubMed] [Google Scholar]
- 1090.Dabareiner R.M., White N.A. Surgical repair of a diaphragmatic hernia in a racehorse. J Am Vet Med Assoc. 1999;214:1517–1518. 1496. [PubMed] [Google Scholar]
- 1091.Hart S.K., Brown J.A. Diaphragmatic hernia in horses: 44 cases (1986–2006) J Vet Emerg Crit Care (San Antonio) 2009;19:357–362. doi: 10.1111/j.1476-4431.2009.00439.x. [DOI] [PubMed] [Google Scholar]
- 1092.Romero A.E., Rodgerson D.H. Diaphragmatic herniation in the horse: 31 cases from 2001–2006. Can Vet J. 2010;51:1247–1250. [PMC free article] [PubMed] [Google Scholar]
- 1093.Harrison I.W. Equine large intestinal volvulus. A review of 124 cases. Vet Surg. 1988;17:77–81. doi: 10.1111/j.1532-950x.1988.tb00281.x. [DOI] [PubMed] [Google Scholar]
- 1094.Johnston K., Holcombe S.J., Hauptman J.G. Plasma lactate as a predictor of colonic viability and survival after 360 degrees volvulus of the ascending colon in horses. Vet Surg. 2007;36:563–567. doi: 10.1111/j.1532-950X.2007.00305.x. [DOI] [PubMed] [Google Scholar]
- 1095.Hackett E.S., Embertson R.M., Hopper S.A. Duration of disease influences survival to discharge of Thoroughbred mares with surgically treated large colon volvulus. Equine Vet J. 2015;47:650–654. doi: 10.1111/evj.12358. [DOI] [PubMed] [Google Scholar]
- 1096.Fiege J.K., Hackett E.S., Rao S. Current treatment of ascending colon volvulus in horses: a survey of ACVS Diplomates. Vet Surg. 2015;44:398–401. doi: 10.1111/j.1532-950X.2014.12195.x. [DOI] [PubMed] [Google Scholar]
- 1097.Ellis C.M., Lynch T.M., Slone D.E. Survival and complications after large colon resection and end-to-end anastomosis for strangulating large colon volvulus in seventy-three horses. Vet Surg. 2008;37:786–790. doi: 10.1111/j.1532-950X.2008.00449.x. [DOI] [PubMed] [Google Scholar]
- 1098.Hance S.R., Embertson R.M. Colopexy in broodmares: 44 cases (1986-1990) J Am Vet Med Assoc. 1992;201:782–787. [PubMed] [Google Scholar]
- 1099.Martin B.B., Jr., Freeman D.E., Ross M.W. Cecocolic and cecocecal intussusception in horses: 30 cases (1976–1996) J Am Vet Med Assoc. 1999;214:80–84. [PubMed] [Google Scholar]
- 1100.Hubert J.D., Hardy J., Holcombe S.J. Cecal amputation within the right ventral colon for surgical treatment of nonreducible cecocolic intussusception in 8 horses. Vet Surg. 2000;29:317–325. doi: 10.1053/jvet.2000.5598. [DOI] [PubMed] [Google Scholar]
- 1101.Gaughan E.M., Hackett R.P. Cecocolic intussusception in horses: 11 cases (1979–1989) J Am Vet Med Assoc. 1990;197:1373–1375. [PubMed] [Google Scholar]
- 1102.Dyson S., Orsini J. Intussusception of the large colon in a horse. J Am Vet Med Assoc. 1983;182:720. [PubMed] [Google Scholar]
- 1103.Robertson J.T., Tate L.P., Jr. Resection of intussuscepted large colon in a horse. J Am Vet Med Assoc. 1982;181:927–928. [PubMed] [Google Scholar]
- 1104.Wilson D.G., Wilson W.D., Reinertson E.L. Intussusception of the left dorsal colon in a horse. J Am Vet Med Assoc. 1983;183:464–465. [PubMed] [Google Scholar]
- 1105.Turner T.A., Fessler J.F. Rectal prolapse in the horse. J Am Vet Med Assoc. 1980;177:1028–1032. [PubMed] [Google Scholar]
- 1106.Blythman W.G. Rectal prolapse in a foaling mare. Vet Rec. 1988;122:471–472. doi: 10.1136/vr.122.19.471. [DOI] [PubMed] [Google Scholar]
- 1107.Rick M.C. Management of rectal injuries. Vet Clin North Am Equine Pract. 1989;5:407–428. doi: 10.1016/s0749-0739(17)30597-7. [DOI] [PubMed] [Google Scholar]
- 1108.Freeman D.E. Rectum and anus. In: Auer J.A., Stick J.A., editors. Equine Surgery. WB Saunders; Philadelphia, PA: 2005. pp. 286–293. [Google Scholar]
- 1109.Jacobs K.A., Barber S.M., Leach D.H. Disruption of the blood supply to the small colon following rectal prolapse and small colon intussusception in a mare. Can Vet J. 1982;23:132–134. [PMC free article] [PubMed] [Google Scholar]
- 1110.Ragle C.A., Southwood L.L., Galuppo L.D. Laparoscopic diagnosis of ischemic necrosis of the descending colon after rectal prolapse and rupture of the mesocolon in two postpartum mares. J Am Vet Med Assoc. 1997;210:1646–1648. [PubMed] [Google Scholar]
- 1111.White N.A., 2nd Intestinal infarction associated with mesenteric vascular thrombotic disease in the horse. J Am Vet Med Assoc. 1981;178:259–262. [PubMed] [Google Scholar]
- 1112.Allen D., Jr., White N.A., 2nd, Tyler D.E. Morphologic effects of experimental distention of equine small intestine. Vet Surg. 1988;17:10–14. doi: 10.1111/j.1532-950x.1988.tb00269.x. [DOI] [PubMed] [Google Scholar]
- 1113.Fleming K., Mueller P.O. Ileal impaction in 245 horses: 1995-2007. Can Vet J. 2011;52:759–763. [PMC free article] [PubMed] [Google Scholar]
- 1114.Hanson R.R., Schumacher J., Humburg J. Medical treatment of horses with ileal impactions: 10 cases (1990–1994) J Am Vet Med Assoc. 1996;208:898–900. [PubMed] [Google Scholar]
- 1115.Little D., Blikslager A.T. Factors associated with development of ileal impaction in horses with surgical colic: 78 cases (1986–2000) Equine Vet J. 2002;34:464–468. doi: 10.2746/042516402776117773. [DOI] [PubMed] [Google Scholar]
- 1116.Cribb N.C., Cote N.M., Boure L.P. Acute small intestinal obstruction associated with Parascaris equorum infection in young horses: 25 cases (1985–2004) N Z Vet J. 2006;54:338–343. doi: 10.1080/00480169.2006.36721. [DOI] [PubMed] [Google Scholar]
- 1117.Nielsen M.K., Donoghue E.M., Stephens M.L. An ultrasonographic scoring method for transabdominal monitoring of ascarid burdens in foals. Equine Vet J. 2016;48:380–386. doi: 10.1111/evj.12478. [DOI] [PubMed] [Google Scholar]
- 1118.Tatz A.J., Segev G., Steinman A. Surgical treatment for acute small intestinal obstruction caused by Parascaris equorum infection in 15 horses (2002–2011) Equine Vet J Suppl. 2012:111–114. doi: 10.1111/j.2042-3306.2012.00607.x. [DOI] [PubMed] [Google Scholar]
- 1119.Parks A.H., Allen D. The purported role of coastal Bermuda hay in the etiology of ileal impactions: results of a questionnaire (abstract) Equine Colic Res Symp. 1988;37 [Google Scholar]
- 1120.Proudman C.J., French N.P., Trees A.J. Tapeworm infection is a significant risk factor for spasmodic colic and ileal impaction colic in the horse. Equine Vet J. 1998;30:194–199. doi: 10.1111/j.2042-3306.1998.tb04487.x. [DOI] [PubMed] [Google Scholar]
- 1121.Proudman C.J., Trees A.J. Correlation of antigen specific IgG and IgG(T) responses with Anoplocephala perfoliata infection intensity in the horse. Parasite Immunol. 1996;18:499–506. doi: 10.1046/j.1365-3024.1996.d01-18.x. [DOI] [PubMed] [Google Scholar]
- 1122.Proudman C.J., Trees A.J. Use of excretory/secretory antigens for the serodiagnosis of Anoplocephala perfoliata cestodosis. Vet Parasitol. 1996;61:239–247. doi: 10.1016/0304-4017(95)00837-3. [DOI] [PubMed] [Google Scholar]
- 1123.Hanson R.R., Wright J.C., Schumacher J. Surgical reduction of ileal impactions in the horse: 28 cases. Vet Surg. 1998;27:555–560. doi: 10.1111/j.1532-950x.1998.tb00531.x. [DOI] [PubMed] [Google Scholar]
- 1124.Chaffin M.K., Fuenteabla I.C., Schumacher J. Idiopathic muscular hypertrophy of the equine small intestine: 11 cases (1980–1991) Equine Vet J. 1992;24:372–378. doi: 10.1111/j.2042-3306.1992.tb02858.x. [DOI] [PubMed] [Google Scholar]
- 1125.Edwards G.B. Obstruction of the ileum in the horse: a report of 27 clinical cases. Equine Vet J. 1981;13:158–166. doi: 10.1111/j.2042-3306.1981.tb03474.x. [DOI] [PubMed] [Google Scholar]
- 1126.Mair T.S., Lucke V.M. Ileal muscular hypertrophy and rupture in a pony three years after surgery for ileocaecal intussusception. Vet Rec. 2000;146:472–473. doi: 10.1136/vr.146.16.472. [DOI] [PubMed] [Google Scholar]
- 1127.Grant B.D., Tennant B. Volvulus associated with Meckel’s diverticulum in the horse. J Am Vet Med Assoc. 1973;162:550–551. [PubMed] [Google Scholar]
- 1128.Hooper R.N. Small intestinal strangulation caused by Meckel’s diverticulum in a horse. J Am Vet Med Assoc. 1989;194:943–944. [PubMed] [Google Scholar]
- 1129.Yovich J.V., Horney F.D. Congenital jejunal diverticulum in a foal. J Am Vet Med Assoc. 1983;183:1092. [PubMed] [Google Scholar]
- 1130.Baxter G.M., Broome T.E., Moore J.N. Abdominal adhesions after small intestinal surgery in the horse. Vet Surg. 1989;18:409–414. doi: 10.1111/j.1532-950x.1990.tb01115.x. [DOI] [PubMed] [Google Scholar]
- 1131.Southwood L.L., Baxter G.M. Current concepts in management of abdominal adhesions. Vet Clin North Am Equine Pract. 1997;13:415–435. doi: 10.1016/s0749-0739(17)30248-1. [DOI] [PubMed] [Google Scholar]
- 1132.Hay W.P., Mueller P.O., Harmon B. One percent sodium carboxymethylcellulose prevents experimentally induced abdominal adhesions in horses. Vet Surg. 2001;30:223–227. doi: 10.1053/jvet.2001.17849. [DOI] [PubMed] [Google Scholar]
- 1133.Mueller P.O., Harmon B.G., Hay W.P. Effect of carboxymethylcellulose and a hyaluronate-carboxymethylcellulose membrane on healing of intestinal anastomoses in horses. Am J Vet Res. 2000;61:369–374. doi: 10.2460/ajvr.2000.61.369. [DOI] [PubMed] [Google Scholar]
- 1134.Mueller P.O., Hunt R.J., Allen D. Intraperitoneal use of sodium carboxymethylcellulose in horses undergoing exploratory celiotomy. Vet Surg. 1995;24:112–117. doi: 10.1111/j.1532-950x.1995.tb01304.x. [DOI] [PubMed] [Google Scholar]
- 1135.Parker J.E., Fubini S.L., Car B.D. Prevention of intraabdominal adhesions in ponies by low-dose heparin therapy. Vet Surg. 1987;16:459–462. doi: 10.1111/j.1532-950x.1987.tb00988.x. [DOI] [PubMed] [Google Scholar]
- 1136.Vatistas N.J., Snyder J.R., Wilson W.D. Surgical treatment for colic in the foal (67 cases): 1980–1992. Equine Vet J. 1996;28:139–145. doi: 10.1111/j.2042-3306.1996.tb01606.x. [DOI] [PubMed] [Google Scholar]
- 1137.Phillips T.J., Walmsley J.P. Retrospective analysis of the results of 151 exploratory laparotomies in horses with gastrointestinal disease. Equine Vet J. 1993;25:427–431. doi: 10.1111/j.2042-3306.1993.tb02985.x. [DOI] [PubMed] [Google Scholar]
- 1138.Parker J.E., Fubini S.L., Todhunter R.J. Retrospective evaluation of repeat celiotomy in 53 horses with acute gastrointestinal disease. Vet Surg. 1989;18:424–431. doi: 10.1111/j.1532-950x.1990.tb01118.x. [DOI] [PubMed] [Google Scholar]
- 1139.Aitken M.R., Southwood L.L., Ross B.M. Outcome of Surgical and Medical Management of Cecal Impaction in 150 Horses (1991–2011) Vet Surg. 2015;44:540–546. doi: 10.1111/j.1532-950X.2014.12286.x. [DOI] [PubMed] [Google Scholar]
- 1140.Dart A.J., Dowling B.A., Hodgson D.R. Caecal disease. Equine Vet Educ. 1999;1:21–28. [Google Scholar]
- 1141.Little D., Redding W.R., Blikslager A.T. Risk factors for reduced postoperative fecal output in horses: 37 cases (1997–1998) J Am Vet Med Assoc. 2001;218:414–420. doi: 10.2460/javma.2001.218.414. [DOI] [PubMed] [Google Scholar]
- 1142.White N.A., 2nd, Dabareiner R.M. Treatment of impaction colics. Vet Clin North Am Equine Pract. 1997;13:243–259. doi: 10.1016/s0749-0739(17)30239-0. [DOI] [PubMed] [Google Scholar]
- 1143.Dabareiner R.M., White N.A. Large colon impaction in horses: 147 cases (1985–1991) J Am Vet Med Assoc. 1995;206:679–685. [PubMed] [Google Scholar]
- 1144.Clarke L.L., Argenzio R.A., Roberts M.C. Effect of meal feeding on plasma volume and urinary electrolyte clearance in ponies. Am J Vet Res. 1990;51:571–576. [PubMed] [Google Scholar]
- 1145.Roberts M.C., Argenzio A. Effects of amitraz, several opiate derivatives and anticholinergic agents on intestinal transit in ponies. Equine Vet J. 1986;18:256–260. doi: 10.1111/j.2042-3306.1986.tb03620.x. [DOI] [PubMed] [Google Scholar]
- 1146.Roberts M.C., Seawright A.A. Amitraz induced large intestinal impaction in the horse. Aust Vet J. 1979;55:553–554. doi: 10.1111/j.1751-0813.1979.tb07043.x. [DOI] [PubMed] [Google Scholar]
- 1147.Lopes M.A., Walker B.L., White N.A., 2nd Treatments to promote colonic hydration: enteral fluid therapy versus intravenous fluid therapy and magnesium sulphate. Equine Vet J. 2002;34:505–509. doi: 10.2746/042516402776117782. [DOI] [PubMed] [Google Scholar]
- 1148.Lopes M.A., White N.A., 2nd, Donaldson L. Effects of enteral and intravenous fluid therapy, magnesium sulfate, and sodium sulfate on colonic contents and feces in horses. Am J Vet Res. 2004;65:695–704. doi: 10.2460/ajvr.2004.65.695. [DOI] [PubMed] [Google Scholar]
- 1149.Cohen N.D., Vontur C.A., Rakestraw P.C. Risk factors for enterolithiasis among horses in Texas. J Am Vet Med Assoc. 2000;216:1787–1794. doi: 10.2460/javma.2000.216.1787. [DOI] [PubMed] [Google Scholar]
- 1150.Hassel D.M., Aldridge B.M., Drake C.M. Evaluation of dietary and management risk factors for enterolithiasis among horses in California. Res Vet Sci. 2008;85:476–480. doi: 10.1016/j.rvsc.2008.03.001. [DOI] [PubMed] [Google Scholar]
- 1151.Hassel D.M., Rakestraw P.C., Gardner I.A. Dietary risk factors and colonic pH and mineral concentrations in horses with enterolithiasis. J Vet Intern Med. 2004;18:346–349. doi: 10.1892/0891-6640(2004)18<346:drfacp>2.0.co;2. [DOI] [PubMed] [Google Scholar]
- 1152.Lloyd K., Hintz H.F., Wheat J.D. Enteroliths in horses. Cornell Vet. 1987;77:172–186. [PubMed] [Google Scholar]
- 1153.Blue M.G., Wittkopp R.W. Clinical and structural features of equine enteroliths. J Am Vet Med Assoc. 1981;179:79–82. [PubMed] [Google Scholar]
- 1154.Peloso J.G., Coatney R.W., Caron J.P. Obstructive enterolith in an 11-month-old miniature horse. J Am Vet Med Assoc. 1992;201:1745–1746. [PubMed] [Google Scholar]
- 1155.Maher O., Puchalski S.M., Drake C. Abdominal computed radiography for the diagnosis of enterolithiasis in horses: 142 cases (2003–2007) J Am Vet Med Assoc. 2011;239:1483–1485. doi: 10.2460/javma.239.11.1483. [DOI] [PubMed] [Google Scholar]
- 1156.Specht T.E., Colahan P.T. Surgical treatment of sand colic in equids: 48 cases (1978–1985) J Am Vet Med Assoc. 1988;193:1560–1564. [PubMed] [Google Scholar]
- 1157.Ragle C.A., Meagher D.M., Schrader J.L. Abdominal auscultation in the detection of experimentally induced gastrointestinal sand accumulation. J Vet Intern Med. 1989;3:12–14. doi: 10.1111/j.1939-1676.1989.tb00322.x. [DOI] [PubMed] [Google Scholar]
- 1158.Kendall A., Ley C., Egenvall A. Radiographic parameters for diagnosing sand colic in horses. Acta Vet Scand. 2008;50:17. doi: 10.1186/1751-0147-50-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1159.Korolainen R., Ruohoniemi M. Reliability of ultrasonography compared to radiography in revealing intestinal sand accumulations in horses. Equine Vet J. 2002;34:499–504. doi: 10.2746/042516402776117764. [DOI] [PubMed] [Google Scholar]
- 1160.Hart K.A., Linnenkohl W., Mayer J.R. Medical management of sand enteropathy in 62 horses. Equine Vet J. 2013;45:465–469. doi: 10.1111/evj.12014. [DOI] [PubMed] [Google Scholar]
- 1161.Hammock P.D., Freeman D.E., Baker G.J. Failure of psyllium mucilloid to hasten evaluation of sand from the equine large intestine. Vet Surg. 1998;27:547–554. doi: 10.1111/j.1532-950x.1998.tb00530.x. [DOI] [PubMed] [Google Scholar]
- 1162.Hackett R.P. Nonstrangulated colonic displacement in horses. J Am Vet Med Assoc. 1983;182:235–240. [PubMed] [Google Scholar]
- 1163.Clarke L.L., Roberts M.C., Argenzio R.A. Feeding and digestive problems in horses. Physiologic responses to a concentrated meal. Vet Clin North Am Equine Pract. 1990;6:433–450. doi: 10.1016/s0749-0739(17)30550-3. [DOI] [PubMed] [Google Scholar]
- 1164.Morris D.D., Moore J.N., Ward S. Comparison of age, sex, breed, history and management in 229 horses with colic. Equine Vet J Suppl. 1989:129–132. doi: 10.1111/j.2042-3306.1989.tb05672.x. [DOI] [PubMed] [Google Scholar]
- 1165.Ruckebusch Y. Motor functions of the intestine. Adv Vet Sci Comp Med. 1981;25:345–369. [PubMed] [Google Scholar]
- 1166.Huskamp B. Displacement of the large colon. In: Robinson N.E., editor. Current Therapy in Equine Medicine. W.B. Saunders; Philadelphia, PA: 1987. pp. 60–65. [Google Scholar]
- 1167.Santschi E.M., Slone D.E., Jr., Frank W.M. Use of ultrasound in horses for diagnosis of left dorsal displacement of the large colon and monitoring its nonsurgical correction. Vet Surg. 1993;22:281–284. doi: 10.1111/j.1532-950x.1993.tb00398.x. [DOI] [PubMed] [Google Scholar]
- 1168.Baker W.T., Frederick J., Giguere S. Reevaluation of the effect of phenylephrine on resolution of nephrosplenic entrapment by the rolling procedure in 87 horses. Vet Surg. 2011;40:825–829. doi: 10.1111/j.1532-950X.2011.00879.x. [DOI] [PubMed] [Google Scholar]
- 1169.Fultz L.E., Peloso J.G., Giguere S. Comparison of phenylephrine administration and exercise versus phenylephrine administration and a rolling procedure for the correction of nephrosplenic entrapment of the large colon in horses: 88 cases (2004–2010) J Am Vet Med Assoc. 2013;242:1146–1151. doi: 10.2460/javma.242.8.1146. [DOI] [PubMed] [Google Scholar]
- 1170.Frederick J., Giguere S., Butterworth K. Severe phenylephrine-associated hemorrhage in five aged horses. J Am Vet Med Assoc. 2010;237:830–834. doi: 10.2460/javma.237.7.830. [DOI] [PubMed] [Google Scholar]
- 1171.McGovern K.F., Bladon B.M., Fraser B.S. Attempted medical management of suspected ascending colon displacement in horses. Vet Surg. 2012;41:399–403. doi: 10.1111/j.1532-950X.2011.00915.x. [DOI] [PubMed] [Google Scholar]
- 1172.Rocken M., Schubert C., Mosel G. Indications, surgical technique, and long-term experience with laparoscopic closure of the nephrosplenic space in standing horses. Vet Surg. 2005;34:637–641. doi: 10.1111/j.1532-950X.2005.00098.x. [DOI] [PubMed] [Google Scholar]
- 1173.Zekas L.J., Ramirez S., Brown M.P. Ablation of the nephrosplenic space for treatment of recurring left dorsal displacement of the large colon in a racehorse. J Am Vet Med Assoc. 1999;214:1361–1363. 1335. [PubMed] [Google Scholar]
- 1174.Gay C.C., Speirs V.C., Christie B.A. Foreign body obstruction of the small colon in six horses. Equine Vet J. 1979;11:60–63. doi: 10.1111/j.2042-3306.1979.tb01302.x. [DOI] [PubMed] [Google Scholar]
- 1175.McClure J.T., Kobluk C., Voller K. Fecalith impaction in four miniature foals. J Am Vet Med Assoc. 1992;200:205–207. [PubMed] [Google Scholar]
- 1176.Speirs V.C., van Veenendaal J.C., Christie B.A. Obstruction of the small colon by intramural haematoma in three horses. Aust Vet J. 1981;57:88–90. doi: 10.1111/j.1751-0813.1981.tb00454.x. [DOI] [PubMed] [Google Scholar]
- 1177.Pearson H., Waterman A.E. Submucosal haematoma as a cause of obstruction of the small colon in the horse: a review of four cases. Equine Vet J. 1986;18:340–341. doi: 10.1111/j.2042-3306.1986.tb03647.x. [DOI] [PubMed] [Google Scholar]
- 1178.Frederico L.M., Jones S.L., Blikslager A.T. Predisposing factors for small colon impaction in horses and outcome of medical and surgical treatment: 44 cases (1999–2004) J Am Vet Med Assoc. 2006;229:1612–1616. doi: 10.2460/javma.229.10.1612. [DOI] [PubMed] [Google Scholar]
- 1179.Benamou A.E., Blikslager A.T., Sellon D.C. Intestinal atresia in foals. Compend Contin Educ Pract Vet. 1995;17:1510. [Google Scholar]
- 1180.McCabe L., Griffin L.D., Kinzer A. Overo lethal white foal syndrome: equine model of aganglionic megacolon (Hirschsprung disease) Am J Med Genet. 1990;36:336–340. doi: 10.1002/ajmg.1320360319. [DOI] [PubMed] [Google Scholar]
- 1181.Metallinos D.L., Bowling A.T., Rine J. A missense mutation in the endothelin-B receptor gene is associated with Lethal White Foal Syndrome: an equine version of Hirschsprung disease. Mamm Genome. 1998;9:426–431. doi: 10.1007/s003359900790. [DOI] [PubMed] [Google Scholar]
- 1182.Santschi E.M., Vrotsos P.D., Purdy A.K. Incidence of the endothelin receptor B mutation that causes lethal white foal syndrome in white-patterned horses. Am J Vet Res. 2001;62:97–103. doi: 10.2460/ajvr.2001.62.97. [DOI] [PubMed] [Google Scholar]
- 1183.Santschi E.M., Purdy A.K., Valberg S.J. Endothelin receptor B polymorphism associated with lethal white foal syndrome in horses. Mamm Genome. 1998;9:306–309. doi: 10.1007/s003359900754. [DOI] [PubMed] [Google Scholar]
- 1184.Giancola F., Gentilini F., Romagnoli N. Extrinsic innervation of ileum and pelvic flexure of foals with ileocolonic aganglionosis. Cell Tissue Res. 2016;366:13–22. doi: 10.1007/s00441-016-2422-x. [DOI] [PubMed] [Google Scholar]
- 1185.Lindsay F.E., Burton F.L. Observational study of “urine testing” in the horse and donkey stallion. Equine Vet J. 1983;15:330–336. doi: 10.1111/j.2042-3306.1983.tb01816.x. [DOI] [PubMed] [Google Scholar]
- 1186.Baker G.J. Oral examination and diagnosis: management of oral disease. In: Harvey C.E., editor. Veterinary dentistry. WB Saunders; Philadelphia, PA: 1985. [Google Scholar]
- 1187.Bonin S.J., Clayton H.M., Lanovaz J.L. Comparison of mandibular motion in horses chewing hay and pellets. Equine Vet J. 2007;39:258–262. doi: 10.2746/042516407x157792. [DOI] [PubMed] [Google Scholar]
- 1188.Baker G.J. Fluroscopic investigations of swallowing in the horse. Vet Radiol. 1982;23:84–88. [Google Scholar]
- 1189.Klebe E.A., Holcombe S.J., Rosenstein D. The effect of bilateral glossopharyngeal nerve anaesthesia on swallowing in horses. Equine Vet J. 2005;37:65–69. doi: 10.2746/0425164054406900. [DOI] [PubMed] [Google Scholar]
- 1190.Heffron C.J., Baker G.J., Lee R. Fluoroscopic Investigation of Pharyngeal Function in the Horse. Equine Veterinary Journal. 1979;11:148–152. doi: 10.1111/j.2042-3306.1979.tb01327.x. [DOI] [PubMed] [Google Scholar]
- 1191.Clark E.S., Morris D.D., Whitlock R.H. Esophageal dysfunction in a weanling thoroughbred. Cornell Vet. 1987;77:151–160. [PubMed] [Google Scholar]
- 1192.Freeman D.E., Naylor J.M. Cervical esophagostomy to permit extraoral feeding of the horse. J Am Vet Med Assoc. 1978;172:314–320. [PubMed] [Google Scholar]
- 1193.Stick J.A., Robinson N.E., Krehbiel J.D. Acid-base and electrolyte alterations associated with salivary loss in the pony. Am J Vet Res. 1981;42:733–737. [PubMed] [Google Scholar]
- 1194.Jones S.L., Zimmel D., Tate L.P. Case presentation—Dysphagia caused by squamous cell carcinoma in two horses. Compend Contin Educ Pract Vet. 2001;23:1020–1024. [Google Scholar]
- 1195.Dixon P.M., Tremaine W.H., Pickles K. Equine dental disease part 2: a long-term study of 400 cases: disorders of development and eruption and variations in position of the cheek teeth. Equine Vet J. 1999;31:519–528. doi: 10.1111/j.2042-3306.1999.tb03862.x. [DOI] [PubMed] [Google Scholar]
- 1196.Quinn G.C., Tremaine W.H., Lane J.G. Supernumerary cheek teeth (n = 24): clinical features, diagnosis, treatment and outcome in 15 horses. Equine Vet J. 2005;37:505–509. doi: 10.2746/042516405775314808. [DOI] [PubMed] [Google Scholar]
- 1197.Baker G.J. Diseases of the teeth. In: Colahan P.C., Mayhew I.G., Merrit A.M., editors. Equine medicine and surgery. American Veterinary Publications; Goleta, CA: 1991. [Google Scholar]
- 1198.Hormeyr C.B. Comparative dental pathology (with particular reference to caries and paradental disease in the horse and the dog) J S Afr Vet Med Assoc. 1960;29:471–475. [Google Scholar]
- 1199.Dixon P.M., Tremaine W.H., Pickles K. Equine dental disease part 4: a long-term study of 400 cases: apical infections of cheek teeth. Equine Vet J. 2000;32:182–194. doi: 10.2746/042516400776563581. [DOI] [PubMed] [Google Scholar]
- 1200.Baker G.J. Some aspects of equine dental radiology. Equine Vet J. 1971;3:46–51. doi: 10.1111/j.2042-3306.1971.tb04439.x. [DOI] [PubMed] [Google Scholar]
- 1201.Baker G.J. Some aspects of equine dental decay. Equine Vet J. 1974;6:127–130. doi: 10.1111/j.2042-3306.1974.tb03945.x. [DOI] [PubMed] [Google Scholar]
- 1202.Farmand M., Stohler T. The median cleft of the lower lip and mandible and its surgical correction in a donkey. Equine Vet J. 1990;22:298–301. doi: 10.1111/j.2042-3306.1990.tb04274.x. [DOI] [PubMed] [Google Scholar]
- 1203.Schumacher J., Brink P., Easley J. Surgical correction of wry nose in four horses. Vet Surg. 2008;37:142–148. doi: 10.1111/j.1532-950X.2007.00362.x. [DOI] [PubMed] [Google Scholar]
- 1204.Gift L.J., DeBowes R.M., Clem M.F. Brachygnathia in horses: 20 cases (1979–1989) J Am Vet Med Assoc. 1992;200:715–719. [PubMed] [Google Scholar]
- 1205.Bankowski R.A., Wichmann R.W., Stuart E.E. Stomatitis of cattle and horses due to yellow bristle grass (Setaria lutescens) J Am Vet Med Assoc. 1956;129:149–152. [PubMed] [Google Scholar]
- 1206.Pusterla N., Latson K.M., Wilson W.D. Metallic foreign bodies in the tongues of 16 horses. Vet Rec. 2006;159:485–488. doi: 10.1136/vr.159.15.485. [DOI] [PubMed] [Google Scholar]
- 1207.Snow D.H., Bogan J.A., Douglas T.A. Phenylbutazone toxicity in ponies. Vet Rec. 1979;105:26–30. doi: 10.1136/vr.105.2.26. [DOI] [PubMed] [Google Scholar]
- 1208.Baum K.H., Shin S.J., Rebhun W.C. Isolation of Actinobacillus-Lignieresii from Enlarged Tongue of A Horse. J Am Vet Med Assoc. 1984;185:792–793. [PubMed] [Google Scholar]
- 1209.Freestone J.F., Seahorn T.L. Miscellaneous conditions of the equine head. Vet ClinNorth Am Equine Pract. 1993;9:235–242. doi: 10.1016/s0749-0739(17)30426-1. [DOI] [PubMed] [Google Scholar]
- 1210.Schmotzer W.B., Hultgren B.D., Huber M.J. Chemical involution of the equine parotid salivary gland. Vet Surg. 1991;20:128–132. doi: 10.1111/j.1532-950x.1991.tb00320.x. [DOI] [PubMed] [Google Scholar]
- 1211.Aleman M., Spier S.J., Wilson W.D. Corynebacterium pseudotuberculosis infection in horses: 538 cases (1982–1993) J Am Vet Med Assoc. 1996;209:804–809. [PubMed] [Google Scholar]
- 1212.Sockett D.C., Baker J.C., Stowe C.M. Slaframine (Rhizoctonia leguminicola) intoxication in horses. J Am Vet Med Assoc. 1982;181:606. [PubMed] [Google Scholar]
- 1213.Plumlee K.H., Galey F.D. Neurotoxic mycotoxins: a review of fungal toxins that cause neurological disease in large animals. J Vet InternMed. 1994;8:49–54. doi: 10.1111/j.1939-1676.1994.tb03195.x. [DOI] [PubMed] [Google Scholar]
- 1214.Sisson S. Equine digestive system. In: Getty R., editor. Sisson and Grossman’s the anatomy of domestic animals. WB Saunders; Philadelphia, PA: 1975. [Google Scholar]
- 1215.Green E.M., MacFadden K.E. Esophageal disorders of the horse. In: Smith B.P., editor. Large animal internal medicine. Mosby; St. Louis, MO: 1996. pp. 698–709. [Google Scholar]
- 1216.Feige K., Schwarzwald C., Furst A. Esophageal obstruction in horses: a retrospective study of 34 cases. Can Vet J. 2000;41:207–210. [PMC free article] [PubMed] [Google Scholar]
- 1217.Craig D., Todhunter R. Surgical repair of an esophageal stricture in a horse. Vet Surg. 1987;16:251–254. doi: 10.1111/j.1532-950x.1987.tb00948.x. [DOI] [PubMed] [Google Scholar]
- 1218.Roberts M.C., Kelly W.R. Squamous cell carcinoma of the lower cervical oesophagus in a pony. Equine Vet J. 1979;11:199–201. doi: 10.1111/j.2042-3306.1979.tb01343.x. [DOI] [PubMed] [Google Scholar]
- 1219.Moore J.N., Kintner L.D. Recurrent esophageal obstruction due to squamous cell carcinoma in a horse. Cornell Vet. 1976;66:590–597. [PubMed] [Google Scholar]
- 1220.Craig D.R., Shivy D.R., Pankowski R.L. Esophageal disorders in 61 horses. Results of nonsurgical and surgical management. Vet Surg. 1989;18:432–438. doi: 10.1111/j.1532-950x.1990.tb01120.x. [DOI] [PubMed] [Google Scholar]
- 1221.Green S., Green E.M., Aronson E. Squamous-Cell Carcinoma—An Unusual Cause of Choke in A Horse. Modern Veterinary Practice. 1986;67:870–875. [Google Scholar]
- 1222.Murray M.J., Ball M.M., Parker G.A. Megaesophagus and aspiration pneumonia secondary to gastric ulceration in a foal. J Am Vet Med Assoc. 1988;192:381–383. [PubMed] [Google Scholar]
- 1223.Butt T.D., MacDonald D.G., Crawford W.H. Persistent right aortic arch in a yearling horse. Can Vet J. 1998;39:714–715. [PMC free article] [PubMed] [Google Scholar]
- 1224.Scott E.A., Snoy P., Prasse K.W. Intramural esophageal cyst in a horse. J Am Vet Med Assoc. 1977;171:652–654. [PubMed] [Google Scholar]
- 1225.Hackett R.P., Dyer R.M., Hoffer R.E. Surgical correction of esophageal diverticulum in a horse. J Am Vet Med Assoc. 1978;173:998–1000. [PubMed] [Google Scholar]
- 1226.Jansson N. Foreign body obstruction of the esophagus in a foal. Comp Contin Educ Vet Equine. 2006;1:147–150. [Google Scholar]
- 1227.MacKay R.J. On the true definition of dysphagia. Compend Contin Educ Pract Vet. 2001;23:1024–1028. [Google Scholar]
- 1228.Traver D.S., Egger E., Moore J.N. Retrieval of an esophageal foreign body in a horse. Vet Med Small Anim Clin. 1978;73:783–785. [PubMed] [Google Scholar]
- 1229.Alexander J.E. Radiologic findings in equine choke. J Am Vet Med Assoc. 1967;151:47–53. [PubMed] [Google Scholar]
- 1230.Greet T.R. Observations on the potential role of oesophageal radiography in the horse. Equine Vet J. 1982;14:73–79. doi: 10.1111/j.2042-3306.1982.tb02341.x. [DOI] [PubMed] [Google Scholar]
- 1231.Quick C.B., Rendano V.T. Equine radiology: the esophagus. Mod Vet Pract. 1978;59:625–631. [PubMed] [Google Scholar]
- 1232.Hillyer M. Management of oesophageal obstruction (‘choke’) in horses. In Practice. 1995;17:450–457. [Google Scholar]
- 1233.Meyer G.A., Rashmir-Raven A., Helms R.J. The effect of oxytocin on contractility of the equine oesophagus: a potential treatment for oesophageal obstruction. Equine Vet J. 2000;32:151–155. doi: 10.2746/042516400777591660. [DOI] [PubMed] [Google Scholar]
- 1234.Wooldridge A.A., Eades S.C., Hosgood G.L. In vitro effects of oxytocin, acepromazine, detomidine, xylazine, butorphanol, terbutaline, isoproterenol, and dantrolene on smooth and skeletal muscles of the equine esophagus. Am J Vet Res. 2002;63:1732–1737. doi: 10.2460/ajvr.2002.63.1732. [DOI] [PubMed] [Google Scholar]
- 1235.Wooldridge A.A., Eades S.C., Hosgood G.L. Effects of treatment with oxytocin, xylazine butorphanol, guaifenesin, acepromazine, and detomidine on esophageal manometric pressure in conscious horses. Am J Vet Res. 2002;63:1738–1744. doi: 10.2460/ajvr.2002.63.1738. [DOI] [PubMed] [Google Scholar]
- 1236.Meagher D.M., Spier S. Foreign-Body Obstruction in the Cervical Esophagus of the Horse—A Case-Report. J Equine V Sci. 1989;9:137–140. [Google Scholar]
- 1237.Erkert R.S., MacAllister C.G., Higbee R. Use of a neodymium:yttrium-aluminum-garnet laser to remove exuberant granulation tissue from the esophagus of a horse. J Am Vet Med Assoc. 2002;221:403–407. doi: 10.2460/javma.2002.221.403. 368. [DOI] [PubMed] [Google Scholar]
- 1238.Chiavaccini L., Hassel D.M. Clinical features and prognostic variables in 109 horses with esophageal obstruction (1992-2009) J Vet Intern Med. 2010;24:1147–1152. doi: 10.1111/j.1939-1676.2010.0573.x. [DOI] [PubMed] [Google Scholar]
- 1239.Crawford J.M. The gastrointestinal tract: esophagus. In: Contran R.S., Kumar V., Robbins S.L., editors. Pathologic basis of disease. WB Saunders; Philadelphia, PA: 1994. [Google Scholar]
- 1240.Hardy J., Stewart R.H., Beard W.L. Complications of nasogastric intubation in horses: nine cases (1987–1989) J Am Vet Med Assoc. 1992;201:483–486. [PubMed] [Google Scholar]
- 1241.Appt S.A., Moll H.D., Scarratt W.K. Esophageal foreign body obstruction in a Mustang. Equine Practice. 1996;18:8–11. [Google Scholar]
- 1242.Broekman L.E., Kuiper D. Megaesophagus in the horse. A short review of the literature and 18 own cases. Vet Q. 2002;24:199–202. doi: 10.1080/01652176.2002.9695136. [DOI] [PubMed] [Google Scholar]
- 1243.Bowman K.F., Vaughan J.T., Quick C.B. Megaesophagus in a colt. J Am Vet Med Assoc. 1978;172:334–337. [PubMed] [Google Scholar]
- 1244.Barber S.M., McLaughlin B.G., Fretz P.B. Esophageal Ectasia in A Quarterhorse Colt. Can Vet J Rev Vet Canadienne. 1983;24:46–49. [PMC free article] [PubMed] [Google Scholar]
- 1245.Rohrbach B.W. Congenital esophageal ectasia in a thoroughbred foal. J Am Vet Med Assoc. 1980;177:65–67. [PubMed] [Google Scholar]
- 1246.Greet T.R., Whitwell K.E. Barium swallow as an aid to the diagnosis of grass sickness. Equine Vet J. 1986;18:294–297. doi: 10.1111/j.2042-3306.1986.tb03633.x. [DOI] [PubMed] [Google Scholar]
- 1247.Watson T.D.G., Sullivan M. Effects of Detomidine on Equine Esophageal Function As Studied by Contrast Radiography. Veterinary Record. 1991;129:67–69. doi: 10.1136/vr.129.4.67. [DOI] [PubMed] [Google Scholar]
- 1248.Clark E.S., Morris D.D., Whitlock R.H. Esophageal manometry in horses, cows, and sheep during deglutition. Am J Vet Res. 1987;48:547–551. [PubMed] [Google Scholar]
- 1249.Todhunter R.J., Stick J.A., Trotter G.W. Medical management of esophageal stricture in seven horses. J Am Vet Med Assoc. 1984;185:784–787. [PubMed] [Google Scholar]
- 1250.Fubini S.L., Starrack G.S., Freeman D.E. Esophagus. In: Auer J.A., Stick J.A., editors. Equine surgery. WB Saunders; Philadelphia: 1999. [Google Scholar]
- 1251.Chidlow H.B., Robbins E.G., Slovis N.M. Balloon dilation to treat oesophageal strictures in five foals. Equine Vet Educ. 2015 [Google Scholar]
- 1252.Prutton J.S., Marks S.L., Aleman M. Endoscopic Balloon Dilation of Esophageal Strictures in 9 Horses. J Vet Intern Med. 2015;29:1105–1111. doi: 10.1111/jvim.13572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1253.Reichelt U., Hamann J., Lischer C. Balloon dilation of oesophageal strictures in two horses. Equine Vet Educ. 2012;24:379–384. [Google Scholar]
- 1254.Tillotson K., Traub-Dargatz J.L., Twedt D. Balloon dilation of an oesophageal stricture in a one-month-old Appaloosa colt. Equine Vet Educ. 2003;15:67–71. [Google Scholar]
- 1255.Borchers-Collyer P., Wong D., Sponseller B. Esophageal strictures. Compendium Equine. 2007;2:144–156. [Google Scholar]
- 1256.Knottenbelt D.C., Harrison L.J., Peacock P.J. Conservative treatment of oesophageal stricture in five foals. Vet Rec. 1992;131:27–30. doi: 10.1136/vr.131.2.27. [DOI] [PubMed] [Google Scholar]
- 1257.Hawkins J.F. Balloon dilation of oesophageal strictures. Equine Vet Educ. 2012;24:385–386. [Google Scholar]
- 1258.Suann C.J. Oesophageal resection and anastomosis as a treatment for oesophageal stricture in the horse. Equine Vet J. 1982;14:163–164. doi: 10.1111/j.2042-3306.1982.tb02377.x. [DOI] [PubMed] [Google Scholar]
- 1259.Gideon L. Esophageal anastomosis in two foals. J Am Vet Med Assoc. 1984;184:1146–1148. [PubMed] [Google Scholar]
- 1260.Nixon A.J., Aanes W.A., Nelson A.W. Esophagomyotomy for relief of an intrathoracic esophageal stricture in a horse. J Am Vet Med Assoc. 1983;183:794–796. [PubMed] [Google Scholar]
- 1261.Wagner P.C., Rantanen N.W. Myotomy As A Treatment for Esophageal Stricture in A Horse. Equine Practice. 1980;2:40–45. [Google Scholar]
- 1262.Hoffer R.E., Barber S.M., Kallfelz F.A. Esophageal patch grafting as a treatment for esophageal stricture in a horse. J Am Vet Med Assoc. 1977;171:350–354. [PubMed] [Google Scholar]
- 1263.Freeman D.E. Surgery for obstruction of the equine oesophagus and trachea. Equine Vet Educ. 2005;17:135–141. [Google Scholar]
- 1264.Waguespack R.W., Bolt D.M., Hubert J.D. Esophageal strictures and diverticula. Compendium Equine. 2007;2:194–206. [Google Scholar]
- 1265.Ford T.S., Schumacher J., Chaffin M.K. Surgical repair of an intrathoracic esophageal pulsion diverticulum in a horse. Vet Surg. 1991;20:316–319. doi: 10.1111/j.1532-950x.1991.tb01274.x. [DOI] [PubMed] [Google Scholar]
- 1266.Frauenfelder H.C., Adams S.B. Esophageal diverticulectomy in a horse. J Am Vet Med Assoc. 1982;180:771–772. [PubMed] [Google Scholar]
- 1267.MacDonald M.H., Richardson D.W., Morse C.C. Esophageal phytobezoar in a horse. J Am Vet Med Assoc. 1987;191:1455–1456. [PubMed] [Google Scholar]
- 1268.Clabough D.L., Roberts M.C., Robertson I. Probable congenital esophageal stenosis in a thoroughbred foal. J Am Vet MedAssoc. 1991;199:483–485. [PubMed] [Google Scholar]
- 1269.Mackey V.S., Large S.M., Breznock E.M. Surgical-Correction of A Persistent Right Aortic-Arch in A Foal. Vet Surg. 1986;15:325–328. [Google Scholar]
- 1270.Bartels J.E., Vaughan J.T. Persistent right aortic arch in the horse. J Am Vet Med Assoc. 1969;154:406–409. [PubMed] [Google Scholar]
- 1271.Petrick S.W., Roos C.J., van Niekerk J. Persistent right aortic arch in a horse. J S Afr Vet Assoc. 1978;49:355–358. [PubMed] [Google Scholar]
- 1272.van der Linde-Sipman J.S., Goedegebuure S.A., Kroneman J. Persistent right aortic arch associated with a persistent left ductus arteriosus and an interventricular septal defect in a horse. Tijdschr Diergeneeskd. 1979;104(suppl 4):189–194. [PubMed] [Google Scholar]
- 1273.Bauer S., Livesey M.A., Bjorling D.E. Computed tomography assisted surgical correction of persistent right aortic arch in a neonatal foal. Equine VetEduc. 2006;8:40–46. [Google Scholar]
- 1274.Smith T.R. Unusual vascular ring anomaly in a foal. CanVet J. 2004;45:1016–1018. [PMC free article] [PubMed] [Google Scholar]
- 1275.Orsini J.A., Sepesy L., Donawick W.J. Esophageal duplication cyst as a cause of choke in the horse. J Am Vet Med Assoc. 1988;193:474–476. [PubMed] [Google Scholar]
- 1276.Peek S.F., De Lahunta A., Hackett R.P. Combined oesophageal and tracheal duplication cyst in an Arabian filly. Equine Vet J. 1995;27:475–478. doi: 10.1111/j.2042-3306.1995.tb04430.x. [DOI] [PubMed] [Google Scholar]
- 1277.Gaughan E.M., Gift L.J., Frank R.K. Tubular duplication of the cervical portion of the esophagus in a foal. J Am Vet Med Assoc. 1992;201:748–750. [PubMed] [Google Scholar]
- 1278.Sams A.E., Weldon A.D., Rakestraw P. Surgical treatment of intramural esophageal inclusion cysts in three horses. Vet Surg. 1993;22:135–139. doi: 10.1111/j.1532-950x.1993.tb01687.x. [DOI] [PubMed] [Google Scholar]
- 1279.Grevemeyer B., Kainer R.A., Morgan J.P. Persistent right aortic arch in foals. Compendium Equine. 2008;3:95–102. [Google Scholar]
- 1280.Wilcke J.R., Crisman M.V., Sams R.A. Pharmacokinetics of phenylbutazone in neonatal foals. AmJVetRes. 1993;54:2064–2067. [PubMed] [Google Scholar]
- 1281.Freeman D.E. Wounds of the esophagus and trachea. Vet Clin North Am Equine Pract. 1989;5:683–693. doi: 10.1016/s0749-0739(17)30582-5. [DOI] [PubMed] [Google Scholar]
- 1282.Digby N.J., Burguez P.N. Traumatic oesophageal rupture in the horse. Equine Vet J. 1982;14:169–170. doi: 10.1111/j.2042-3306.1982.tb02380.x. [DOI] [PubMed] [Google Scholar]
- 1283.Lunn D.P., Peel J.E. Successful treatment of traumatic oesophageal rupture with severe cellulitis in a mare. Vet Rec. 1985;116:544–545. doi: 10.1136/vr.116.20.544. [DOI] [PubMed] [Google Scholar]
- 1284.Andrews F.M., Bernard W.V., Byars T.D. Recommendations for the diagnosis and treatment of equine gastric ulcer syndrome (EGUS) Equine Vet Educ. 1999;1:122–134. [Google Scholar]
- 1285.Sykes B.W., Hewetson M., Hepburn R.J. European College of Equine Internal Medicine Consensus Statement—Equine Gastric Ulcer Syndrome in Adult Horses. J Vet Intern Med. 2015;29:1288–1299. doi: 10.1111/jvim.13578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1286.Murray M.J. Gastroduodenal ulceration in foals. Equine Vet Educ. 1999;1:43–52. [Google Scholar]
- 1287.Hammond C.J., Mason D.K., Watkins K.L. Gastric ulceration in mature thoroughbred horses. Equine Vet J. 1986;18:284–287. doi: 10.1111/j.2042-3306.1986.tb03629.x. [DOI] [PubMed] [Google Scholar]
- 1288.Vatistas N.J., Snyder J.R., Carlson G. Epidemiological study of gastric ulceration in the thoroughbred racehorse: 202 horses 1992–1993. Proc Am Assoc Equine Pract. 1994;40:125–126. [Google Scholar]
- 1289.Vatistas N.J., Snyder J.R., Carlson G. Cross-sectional study of gastric ulcers of the squamous mucosa in thoroughbred racehorses. Equine Vet J Suppl. 1999:34–39. doi: 10.1111/j.2042-3306.1999.tb05166.x. [DOI] [PubMed] [Google Scholar]
- 1290.Murray M.J., Grodinsky C., Anderson C.W. Gastric ulcers in horses: a comparison of endoscopic findings in horses with and without clinical signs. Equine Vet J Suppl. 1989:68–72. doi: 10.1111/j.2042-3306.1989.tb05659.x. [DOI] [PubMed] [Google Scholar]
- 1291.Murray M.J., Schusser G.F., Pipers F.S. Factors associated with gastric lesions in thoroughbred racehorses. Equine Vet J. 1996;28:368–374. doi: 10.1111/j.2042-3306.1996.tb03107.x. [DOI] [PubMed] [Google Scholar]
- 1292.Orsini J.A., Pipers F.S. Endoscopic evaluation of the relationship between training, racing, and gastric ulcers. Vet Surg. 1997;26:424. [Google Scholar]
- 1293.Bell R.J.W., Kingston J.K., Mogg T.D. The prevalence of gastric ulceration in racehorses in New Zealand. N Z Vet J. 2007;55:13–18. doi: 10.1080/00480169.2007.36729. [DOI] [PubMed] [Google Scholar]
- 1294.Jonsson H., Egenvall A. Prevalence of gastric ulceration in Swedish Standardbreds in race training. Equine Vet J. 2006;38:209–213. doi: 10.2746/042516406776866390. [DOI] [PubMed] [Google Scholar]
- 1295.Begg L.M., O’Sullivan C.B. The prevalence and distribution of gastric ulceration in 345 racehorses. Aust Vet J. 2003;81:199–201. doi: 10.1111/j.1751-0813.2003.tb11469.x. [DOI] [PubMed] [Google Scholar]
- 1296.McClure S.R., Glickman L.T., Glickman N.W. Prevalence of gastric ulcers in show horses. J Am Vet Med Assoc. 1999;215:1130–1133. [PubMed] [Google Scholar]
- 1297.Nieto J.E., Snyder J.R., Beldomenico P. Prevalence of gastric ulcers in endurance horses—a preliminary report. Vet J. 2004;167:33–37. doi: 10.1016/j.tvjl.2003.09.005. [DOI] [PubMed] [Google Scholar]
- 1298.Tamzali Y., Marguet C., Priymenko N. Prevalence of gastric ulcer syndrome in high-level endurance horses. Equine Vet J. 2011;43:141–144. doi: 10.1111/j.2042-3306.2010.00129.x. [DOI] [PubMed] [Google Scholar]
- 1299.Bertone J. Prevalence of gastric ulcers in elite, heavy use western performance horses. J Vet Intern Med. 2000;14:366. [Google Scholar]
- 1300.Jeune S., Nieto J., Dechant J. Prevalence of gastric ulcers in Thoroughbred broodmares in pasture. Vet J. 2009;181:251–255. doi: 10.1016/j.tvjl.2008.03.020. [DOI] [PubMed] [Google Scholar]
- 1301.Hartmann A.M., Frankeny R.L. A preliminary investigation into the association between competition and gastric ulcer formation in non-racing performance horses. J Equine Vet Sci. 2003;23:560–561. [Google Scholar]
- 1302.Sandin A., Skidell J., Haggstrom J. Postmortem findings of gastric ulcers in Swedish horses older than age one year: a retrospective study of 3715 horses (1924–1996) Equine Vet J. 2000;32:36–42. doi: 10.2746/042516400777612044. [DOI] [PubMed] [Google Scholar]
- 1303.Chameroy K.A., Nadeau J.A., Bushmich S.L. Prevalence of non-glandular gastric ulcers in horses involved in a university riding program. J Equine Vet Sci. 2006;26:207–211. [Google Scholar]
- 1304.Dukti S.A., Perkins S., Murphy J. Prevalence of gastric squamous ulceration in horses with abdominal pain. Equine Vet J. 2006;38:347–349. doi: 10.2746/042516406777749164. [DOI] [PubMed] [Google Scholar]
- 1305.Wilson J.H. Gastric and duodenal ulcers in foals: a retrospective study. Proc 2nd Symp Equine Colic Res. 1985:126–128. [Google Scholar]
- 1306.Murray M.J., Grodinsky C., Cowles R.R. Endoscopic evaluation of changes in gastric lesions of Thoroughbred foals. J Am Vet Med Assoc. 1990;196:1623–1627. [PubMed] [Google Scholar]
- 1307.Murray M.J. Endoscopic appearance of gastric lesions in foals: 94 cases (1987–1988) J Am Vet Med Assoc. 1989;195:1135–1141. [PubMed] [Google Scholar]
- 1308.Murray M.J., Nout Y.S., Ward D.L. Endoscopic findings of the gastric antrum and pylorus in horses: 162 cases (1996–2000) J Vet Intern Med. 2001;15:401–406. [PubMed] [Google Scholar]
- 1309.Luthersson N., Nielsen K.H., Harris P. The prevalence and anatomical distribution of equine gastric ulceration syndrome (EGUS) in 201 horses in Denmark. Equine Vet J. 2009;41:619–624. doi: 10.2746/042516409x441910. [DOI] [PubMed] [Google Scholar]
- 1310.Husted L., Jensen T.K., Olsen S.N. Examination of equine glandular stomach lesions for bacteria, including Helicobacter spp by fluorescence in situ hybridisation. BMC Microbiol. 2010;10:84. doi: 10.1186/1471-2180-10-84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1311.Murray M.J., Schusser G.F. Measurement of 24-h gastric pH using an indwelling pH electrode in horses unfed, fed and treated with ranitidine. Equine Vet J. 1993;25:417–421. doi: 10.1111/j.2042-3306.1993.tb02983.x. [DOI] [PubMed] [Google Scholar]
- 1312.Husted L., Sanchez L.C., Olsen S.N. Effect of paddock vs. stall housing on 24 hour gastric pH within the proximal and ventral equine stomach. Equine Vet J. 2008 doi: 10.2746/042516408X284673. [DOI] [PubMed] [Google Scholar]
- 1313.Mertz H.R., Walsh J.H. Peptic ulcer pathophysiology. Med Clin North Am. 1991;75:799–814. doi: 10.1016/s0025-7125(16)30412-6. [DOI] [PubMed] [Google Scholar]
- 1314.Wolfe M.M., Soll A.H. The physiology of gastric acid secretion. N Engl J Med. 1988;319:1707–1715. doi: 10.1056/NEJM198812293192605. [DOI] [PubMed] [Google Scholar]
- 1315.Campbell-Thompson M. Secretagogue-induced [14C]aminopyrine uptake in isolated equine parietal cells. Am J Ve tRes. 1994;55:132–137. [PubMed] [Google Scholar]
- 1316.Kitchen D.L., Burrow J.A., Heartless C.S. Effect of pyloric blockade and infusion of histamine or pentagastrin on gastric secretion in horses. Am J Vet Res. 2000;61:1133–1139. doi: 10.2460/ajvr.2000.61.1133. [DOI] [PubMed] [Google Scholar]
- 1317.Schubert M.L., Edwards N.F., Makhlouf G.M. Regulation of gastric somatostatin secretion in the mouse by luminal acidity: a local feedback mechanism. Gastroenterology. 1988;94:317–322. doi: 10.1016/0016-5085(88)90418-0. [DOI] [PubMed] [Google Scholar]
- 1318.Lewis J.J., Goldenring J.R., Asher V.A. Effects of epidermal growth factor on signal transduction in rabbit parietal cells. Am J Physiol. 1990;258:G476–G483. doi: 10.1152/ajpgi.1990.258.3.G476. [DOI] [PubMed] [Google Scholar]
- 1319.Baker S.J., Gerring E.L. Gastric pH monitoring in healthy, suckling pony foals. Am J Vet Res. 1993;54:959–964. [PubMed] [Google Scholar]
- 1320.Sanchez L.C., Lester G.D., Merritt A.M. Effect of ranitidine on intragastric pH in clinically normal neonatal foals. J Am Vet Med Assoc. 1998;212:1407–1412. [PubMed] [Google Scholar]
- 1321.Sanchez L.C., Lester G.D., Merritt A.M. Intragastric pH in critically ill neonatal foals and the effect of ranitidine. J Am Vet Med Assoc. 2001;218:907–911. doi: 10.2460/javma.2001.218.907. [DOI] [PubMed] [Google Scholar]
- 1322.Kuusela A.L. Long-term gastric pH monitoring for determining optimal dose of ranitidine for critically ill preterm and term neonates. Arch Dis Child Fetal Neonatal Ed. 1998;78:F151–F153. doi: 10.1136/fn.78.2.f151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1323.De Backer A., Haentjens P., Willems G. Hydrochloric acid. A trigger of cell proliferation in the esophagus of dogs. Dig Dis Sci. 1985;30:884–890. doi: 10.1007/BF01309520. [DOI] [PubMed] [Google Scholar]
- 1324.McGowan C.M., McGowan T.W., Andrews F.M. Induction and recovery of dietary induced gastric ulcers in horses. J Vet Intern Med. 2007;21 603–603. [Google Scholar]
- 1325.Nadeau J.A., Andrews F.M., Mathew A.G. Evaluation of diet as a cause of gastric ulcers in horses. Am J Vet Res. 2000;61:784–790. doi: 10.2460/ajvr.2000.61.784. [DOI] [PubMed] [Google Scholar]
- 1326.Lybbert T., Gibbs P., Cohen N. Feeding alfalfa hay to exercising horses reduces the severity of gastric squamous mucosal ulceration. Proc Am Assoc Equine Pract. 2007;53:525–526. [Google Scholar]
- 1327.Murray M.J., Eichorn E.S. Effects of intermittent feed deprivation, intermittent feed deprivation with ranitidine administration, and stall confinement with ad libitum access to hay on gastric ulceration in horses. Am J Vet Res. 1996;57:1599–1603. [PubMed] [Google Scholar]
- 1328.Luthersson N., Nielsen K.H., Harris P. Risk factors associated with equine gastric ulceration syndrome (EGUS) in 201 horses in Denmark. Equine Vet J. 2009;41:625–630. doi: 10.2746/042516409x441929. [DOI] [PubMed] [Google Scholar]
- 1329.Lester G.D., Robinson I., Secombe C. Canberra: Australian Government: Rural industries Research and Development Corporation. 2008. Risk factors for gastric ulceration in Thoroughbred racehorses; pp. 1–42. [Google Scholar]
- 1330.White G., McClure S.R., Sifferman R. Effects of short-term light to heavy exercise on gastric ulcer development in horses and efficacy of omeprazole paste in preventing gastric ulceration. J Am Vet Med Assoc. 2007;230:1680–1682. doi: 10.2460/javma.230.11.1680. [DOI] [PubMed] [Google Scholar]
- 1331.Lester G.D., Robertson I.D., Secombe C. Risk factors for gastric ulceration in thoroughbred racehorses. Proc Am Assoc Equine Pract. 2007;53:529. [Google Scholar]
- 1332.Furr M., Taylor L., Kronfeld D. The effects of exercise training on serum gastrin responses in the horse. Cornell Vet. 1994;84:41–45. [PubMed] [Google Scholar]
- 1333.Lorenzo-Figueras M., Merritt A.M. Effects of exercise on gastric volume and pH in the proximal portion of the stomach of horses. Am J Vet Res. 2002;63:1481–1487. doi: 10.2460/ajvr.2002.63.1481. [DOI] [PubMed] [Google Scholar]
- 1334.McClure S.R., Carithers D.S., Gross S.J. Gastric ulcer development in horses in a simulated show or training environment. J Am Vet Med Assoc. 2005;227:775–777. doi: 10.2460/javma.2005.227.775. [DOI] [PubMed] [Google Scholar]
- 1335.Meschter C.L., Gilbert M., Krook L. The effects of phenylbutazone on the morphology and prostaglandin concentrations of the pyloric mucosa of the equine stomach. Vet Pathol. 1990;27:244–253. doi: 10.1177/030098589002700405. [DOI] [PubMed] [Google Scholar]
- 1336.MacKay R.J., French T.W., Nguyen H.T. Effects of large doses of phenylbutazone administration to horses. Am J Vet Res. 1983;44:774–780. [PubMed] [Google Scholar]
- 1337.Roy M.A., Vrins A., Beauchamp G. Prevalence of ulcers of the squamous gastric mucosa in Standardbred horses. J Vet Intern Med. 2005;19:744–750. doi: 10.1892/0891-6640(2005)19[744:pouots]2.0.co;2. [DOI] [PubMed] [Google Scholar]
- 1338.Nicol C.J., Davidson H.P.D., Harris P.A. Study of crib-biting and gastric inflammation and ulceration in young horses. Vet Rec. 2002;151:658–662. doi: 10.1136/vr.151.22.658. [DOI] [PubMed] [Google Scholar]
- 1339.Furr M.O., Murray M.J., Ferguson D.C. The effects of stress on gastric ulceration, T3, T4, reverse T3 and cortisol in neonatal foals. Equine Vet J. 1992;24:37–40. doi: 10.1111/j.2042-3306.1992.tb02776.x. [DOI] [PubMed] [Google Scholar]
- 1340.Murray M.J., Murray C.M., Sweeney H.J. Prevalence of gastric lesions in foals without signs of gastric disease: an endoscopic survey. Equine Vet J. 1990;22:6–8. doi: 10.1111/j.2042-3306.1990.tb04193.x. [DOI] [PubMed] [Google Scholar]
- 1341.Barr B.S., Wilkins P.A., Del Piero F. Is prophylaxis for gastric ulcers necessary in critically ill equine neonates?: a retrospective study of necropsy cases 1989–1999. JVetInternMed. 2000;14:328. [Google Scholar]
- 1342.Nappert G., Vrins A., Larybyere M. Gastroduodenal ulceration in foals. Comp Contin Ed. 1989;11:338–345. [Google Scholar]
- 1343.Orsini J.A., Donawick W.J. Surgical treatment of gastroduodenal obstructions in foals. Vet Surg. 1986;15:205–213. [Google Scholar]
- 1344.Becht J.L., Byars T.D. Gastroduodenal ulceration in foals. Equine Vet J. 1986;18:307–312. doi: 10.1111/j.2042-3306.1986.tb03638.x. [DOI] [PubMed] [Google Scholar]
- 1345.Campbell-Thompson M.L., Merritt A.M. Gastroduodenal ulceration in foals. Proc Am Assoc Equine Pract. 1987;33:29–40. [Google Scholar]
- 1346.Morris A.P., Estes M.K. Microbes and microbial toxins: paradigms for microbial-mucosal interactions. VIII. Pathological consequences of rotavirus infection and its enterotoxin. Am J Physiol Gastrointest Liver Physiol. 2001;281:G303–G310. doi: 10.1152/ajpgi.2001.281.2.G303. [DOI] [PubMed] [Google Scholar]
- 1347.Murray M.J. Gastric ulceration in horses: 91 cases (1987–1990) J Am Vet Med Assoc. 1992;201:117–120. [PubMed] [Google Scholar]
- 1348.O’Conner M.S., Steiner J.M., Roussel A.J. Evaluation of urine sucrose concentration for detection of gastric ulcers in horses. AmJ Vet Res. 2004;65:31–39. doi: 10.2460/ajvr.2004.65.31. [DOI] [PubMed] [Google Scholar]
- 1349.Hewetson M., Cohen N.D., Love S. Sucrose concentration in blood: a new method for assessment of gastric permeability in horses with gastric ulceration. J Vet Intern Med. 2006;20:388–394. doi: 10.1892/0891-6640(2006)20[388:sciban]2.0.co;2. [DOI] [PubMed] [Google Scholar]
- 1350.Bell R.J.W., Kingston J.K., Mogg T.D. A comparison of two scoring systems for endoscopic grading of gastric ulceration in horses. N Z Vet J. 2007;55:19–22. doi: 10.1080/00480169.2007.36730. [DOI] [PubMed] [Google Scholar]
- 1351.Campbell-Thompson M.L., Merritt A.M. Effect of ranitidine on gastric acid secretion in young male horses. Am J Vet Res. 1987;48:1511–1515. [PubMed] [Google Scholar]
- 1352.Furr M.O., Murray M.J. Treatment of gastric ulcers in horses with histamine type 2 receptor antagonists. Equine Vet J Suppl. 1989:77–79. doi: 10.1111/j.2042-3306.1989.tb05661.x. [DOI] [PubMed] [Google Scholar]
- 1353.Daurio C.P., Holste J.E., Andrews F.M. Effect of omeprazole paste on gastric acid secretion in horses. Equine Vet J Suppl. 1999:59–62. doi: 10.1111/j.2042-3306.1999.tb05171.x. [DOI] [PubMed] [Google Scholar]
- 1354.Sykes B.W., Sykes K.M., Hallowell G.D. A comparison of three doses of omeprazole in the treatment of equine gastric ulcer syndrome: a blinded, randomised, dose-response clinical trial. Equine Vet J. 2015;47:285–290. doi: 10.1111/evj.12287. [DOI] [PubMed] [Google Scholar]
- 1355.Sykes B.W., Underwood C., Greer R. Pharmacokinetics and bioequivalence testing of five commercial formulations of omeprazole in the horse. J Vet Pharmacol Ther. 2016;39:78–83. doi: 10.1111/jvp.12240. [DOI] [PubMed] [Google Scholar]
- 1356.Sykes B.W., Underwood C., McGowan C.M. The effect of feeding on the pharmacokinetic variables of two commercially available formulations of omeprazole. J Vet Pharmacol Ther. 2015;38:500–503. doi: 10.1111/jvp.12210. [DOI] [PubMed] [Google Scholar]
- 1357.Merritt A.M., Sanchez L.C., Burrow J.A. Effect of GastroGard and three compounded oral omeprazole preparations on 24 h intragastric pH in gastrically cannulated mature horses. Equine Vet J. 2003;35:691–695. doi: 10.2746/042516403775696339. [DOI] [PubMed] [Google Scholar]
- 1358.Nieto J.E., Spier S., Pipers F.S. Comparison of paste and suspension formulations of omeprazole in the healing of gastric ulcers in racehorses in active training. J Am Vet Med Assoc. 2002;221:1139–1143. doi: 10.2460/javma.2002.221.1139. [DOI] [PubMed] [Google Scholar]
- 1359.Orsini J.A., Haddock M., Stine L. Odds of moderate or severe gastric ulceration in racehorses receiving antiulcer medications. J Am Vet Med Assoc. 2003;223:336–339. doi: 10.2460/javma.2003.223.336. [DOI] [PubMed] [Google Scholar]
- 1360.Plue R.E., Wall H.G., Daurio C. Safety of omeprazole paste in foals and mature horses. Equine Vet J Suppl. 1999:63–66. doi: 10.1111/j.2042-3306.1999.tb05172.x. [DOI] [PubMed] [Google Scholar]
- 1361.Murray M.J., Eichorn E.S., Holste J.E. Safety, acceptability and endoscopic findings in foals and yearling horses treated with a paste formulation of omeprazole for twenty-eight days. Equine Vet J Suppl. 1999:67–70. doi: 10.1111/j.2042-3306.1999.tb05173.x. [DOI] [PubMed] [Google Scholar]
- 1362.Lester G.D., Smith R.L., Robertson I.D. Effects of treatment with omeprazole or ranitidine on gastric squamous ulceration in racing Thoroughbreds. J Am Vet Med Assoc. 2005;227:1636–1639. doi: 10.2460/javma.2005.227.1636. [DOI] [PubMed] [Google Scholar]
- 1363.Nieto J.E., Spier S.J., Van Hoogmoed L. Comparison of omeprazole and cimetidine in healing of gastric ulcers and prevention of recurrence in horses. Equine Vet Educ. 2001;13:260–264. [Google Scholar]
- 1364.Murray M.J., Haven M.L., Eichorn E.S. Effects of omeprazole on healing of naturally-occurring gastric ulcers in thoroughbred racehorses. Equine Vet J. 1997;29:425–429. doi: 10.1111/j.2042-3306.1997.tb03153.x. [DOI] [PubMed] [Google Scholar]
- 1365.Doucet M.Y., Vrins A.A., Dionne R. Efficacy of a paste formulation of omeprazole for the treatment of naturally occurring gastric ulcers in training standardbred racehorses in Canada. Can Vet J. 2003;44:581–585. [PMC free article] [PubMed] [Google Scholar]
- 1366.MacAllister C.G., Sifferman R.L., McClure S.R. Effects of omeprazole paste on healing of spontaneous gastric ulcers in horses and foals: a field trial. Equine Vet J Suppl. 1999:77–80. doi: 10.1111/j.2042-3306.1999.tb05175.x. [DOI] [PubMed] [Google Scholar]
- 1367.McClure S.R., White G.W., Sifferman R.L. Efficacy of omeprazole paste for prevention of recurrence of gastric ulcers in horses in race training. J Am Vet Med Assoc. 2005;226:1685–1688. doi: 10.2460/javma.2005.226.1685. [DOI] [PubMed] [Google Scholar]
- 1368.McClure S.R., White G.W., Sifferman R.L. Efficacy of omeprazole paste for prevention of gastric ulcers in horses in race training. J Am Vet Med Assoc. 2005;226:1681–1684. doi: 10.2460/javma.2005.226.1681. [DOI] [PubMed] [Google Scholar]
- 1369.Sykes B.W., Sykes K., Hallowell G.D. Comparison of the effect of two doses of omeprazole on the squamous gastric mucosa in thoroughbred racehorses. Vet Rec. 2014;175:249. doi: 10.1136/vr.102622. [DOI] [PubMed] [Google Scholar]
- 1370.Sykes B.W., Sykes K.M., Hallowell G.D. A comparison of two doses of omeprazole in the treatment of equine gastric ulcer syndrome: a blinded, randomised, clinical trial. Equine Vet J. 2014;46:416–421. doi: 10.1111/evj.12191. [DOI] [PubMed] [Google Scholar]
- 1371.Sykes B.W., Sykes K.M., Hallowell G.D. A comparison between pre- and post exercise administration of omeprazole in the treatment of equine gastric ulcer syndrome: a blinded, randomised, clinical trial. Equine Veterinary Journal. 2014;46:422–426. doi: 10.1111/evj.12191. [DOI] [PubMed] [Google Scholar]
- 1372.Sanchez L.C., Murray M.J., Merritt A.M. Effect of omeprazole paste on intragastric pH in clinically normal neonatal foals. Am J Vet Res. 2004;65:1039–1041. doi: 10.2460/ajvr.2004.65.1039. [DOI] [PubMed] [Google Scholar]
- 1373.Javsicas L.H., Sanchez L.C. The effect of omeprazole paste on intragastric pH in clinically ill neonatal foals. Equine Vet J. 2008;40:41–44. doi: 10.2746/042516407X235803. [DOI] [PubMed] [Google Scholar]
- 1374.Sykes B.W., Sykes K.M., Hallowell G.D. Administration of trimethoprim-sulphadimidine does not improve healing of glandular gastric ulceration in horses receiving omeprazole: a randomised, blinded, clinical study. BMC Veterinary Research. 2014;10 doi: 10.1186/s12917-014-0180-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1375.Ogihara Y., Okabe S. Effect and mechanism of sucralfate on healing of acetic acid-induced gastric ulcers in rats. J Physiol Pharmacol. 1993;44:109–118. [PubMed] [Google Scholar]
- 1376.Borne A.T., MacAllister C.G. Effect of sucralfate on healing of subclinical gastric ulcers in foals. J Am Vet Med Assoc. 1993;202:1465–1468. [PubMed] [Google Scholar]
- 1377.Hepburn R.J., Proudman C.J. Treatment of ulceration of the gastric glandular mucosa: retrospective evaluation of omeprazole and sucralfate combination therapy in 204 sport and leisure horses [abstract] International Equine Colic Research Symposium. 2014:108. [Google Scholar]
- 1378.Leandro G., Pilotto A., Franceschi M. Prevention of acute NSAID-related gastroduodenal damage: a meta- analysis of controlled clinical trials. Dig Dis Sci. 2001;46:1924–1936. doi: 10.1023/a:1010687115298. [DOI] [PubMed] [Google Scholar]
- 1379.Sangiah S., MacAllister C.C., Amouzadeh H.R. Effects of misoprostol and omeprazole on basal gastric pH and free acid content in horses. Res Vet Sci. 1989;47:350–354. [PubMed] [Google Scholar]
- 1380.Campbell-Thompson M.L., Brown M.P., Slone D.E. Gastroenterostomy for treatment of gastroduodenal ulcer disease in 14 foals. J Am Vet Med Assoc. 1986;188:840–844. [PubMed] [Google Scholar]
- 1381.Zedler S.T., Embertson R.M., Bernard W.V. Surgical treatment of gastric outflow obstruction in 40 foals. Vet Surg. 2009;38:623–630. doi: 10.1111/j.1532-950X.2009.00539.x. [DOI] [PubMed] [Google Scholar]
- 1382.Andrews F.M., Frank N., Sommardahl C.S. Effects of intravenously administrated omeprazole on gastric juice pH and gastric ulcer scores in adult horses. J Vet Intern Med. 2006;20:1202–1206. doi: 10.1892/0891-6640(2006)20[1202:eoiaoo]2.0.co;2. [DOI] [PubMed] [Google Scholar]
- 1383.McKeever J.M., McKeever K.H., Albeirci J.M. Effect of omeprazole on markers of performance in gastric ulcer-free standardbred horses. Equine Vet J Suppl. 2006:668–671. doi: 10.1111/j.2042-3306.2006.tb05624.x. [DOI] [PubMed] [Google Scholar]
- 1384.Cothran D.S., Borowitz S.M., Sutphen J.L. Alteration of normal gastric flora in neonates receiving ranitidine. JPerinatol. 1997;17:383–388. [PubMed] [Google Scholar]
- 1385.Furr M., Cohen N.D., Axon J.E. Treatment with histamine-type 2 receptor antagonists and omeprazole increase the risk of diarrhoea in neonatal foals treated in intensive care units. Equine Vet J. 2012;44:80–86. doi: 10.1111/j.2042-3306.2011.00499.x. [DOI] [PubMed] [Google Scholar]
- 1386.Huff N.K., Auer A.D., Garza F., Jr. Effect of sea buckthorn berries and pulp in a liquid emulsion on gastric ulcer scores and gastric juice pH in horses. J Vet Intern Med. 2012;26:1186–1191. doi: 10.1111/j.1939-1676.2012.00975.x. [DOI] [PubMed] [Google Scholar]
- 1387.Hellings I.R., Larsen S. ImproWin® in the treatment of gastric ulceration of the squamous mucosa in trotting racehorses. Acta Vet Scand. 2014;56:13. doi: 10.1186/1751-0147-56-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1388.Sykes B.W., Sykes K.M., Hallowell G.D. Efficacy of a Combination of Apolectol, Live Yeast (Saccharomyces cerevisiae [CNCM I-1077]), and Magnesium Hydroxide in the Management of Equine Gastric Ulcer Syndrome in Thoroughbred Racehorses: A Blinded, Randomized, Placebo-Controlled Clinical Trial. J Equine Vet Sci. 2014;34:1274–1278. [Google Scholar]
- 1389.Venner M., Lauffs S., Deegen E. Treatment of gastric lesions in horses with pectin-lecithin complex. Equine Vet J Suppl. 1999:91–96. doi: 10.1111/j.2042-3306.1999.tb05178.x. [DOI] [PubMed] [Google Scholar]
- 1390.Clark C.K., Merritt A.M., Burrow J.A. Effect of aluminum hydroxide/magnesium hydroxide antacid and bismuth subsalicylate on gastric pH in horses. J Am Vet Med Assoc. 1996;208:1687–1691. [PubMed] [Google Scholar]
- 1391.Munroe G.A. Pyloric stenosis in a yearling with an incidental finding of Capillaria hepatica in the liver. Equine Vet J. 1984;16:221–222. doi: 10.1111/j.2042-3306.1984.tb01913.x. [DOI] [PubMed] [Google Scholar]
- 1392.Barth A.D., Barber S.M., McKenzie N.T. Pyloric stenosis in a foal. CanVet J. 1980;21:234–236. [PMC free article] [PubMed] [Google Scholar]
- 1393.Church S., Baker J.R., May S.A. Gastric retention associated with acquired pyloric stenosis in a gelding. Equine Vet J. 1986;18:332–334. doi: 10.1111/j.2042-3306.1986.tb03644.x. [DOI] [PubMed] [Google Scholar]
- 1394.McGill C.A., Bolton J.R. Gastric retention associated with a pyloric mass in two horses. Aust Vet J. 1984;61:190–191. doi: 10.1111/j.1751-0813.1984.tb07239.x. [DOI] [PubMed] [Google Scholar]
- 1395.Laing J.A., Hutchins D.R. Acquired pyloric stenosis and gastric retention in a mare. Aust Vet J. 1992;69:68–69. doi: 10.1111/j.1751-0813.1992.tb07455.x. [DOI] [PubMed] [Google Scholar]
- 1396.Campbell-Thompson M.L., Merritt A.M. Alimentary System: diseases of the stomach. In: Colahan P.T., Mayhew I.G., Merritt A.M., editors. Equine Medicine and Surgery. Mosby; St. Louis, MO: 1999. pp. 699–715. [Google Scholar]
- 1397.Wyse C., Murphey D., Preston T. Use of the [13 C]octanoic acid breath test for assessment of gastric emptying in ponies: a preliminary study. Proc 6th Equine Colic Res Symp. 1998;40 [Google Scholar]
- 1398.Buchanan B.R., Sommardahl C.S., Moore R.R. What is your diagnosis? Pyloric-duodenal intussusception. J Am Vet Med Assoc. 2006;228:1339–1340. doi: 10.2460/javma.228.9.1339. [DOI] [PubMed] [Google Scholar]
- 1399.Todhunter R.J., Erb H.N., Roth L. Gastric rupture in horses: a review of 54 cases. Equine Vet J. 1986;18:288–293. doi: 10.1111/j.2042-3306.1986.tb03631.x. [DOI] [PubMed] [Google Scholar]
- 1400.Puotunen-Reinert A., Huskamp B. Experimantal duodenal obstruction in the horse. Vet Surg. 1986;15:420–428. [Google Scholar]
- 1401.Kiper M.L., Traub-Dargatz J., Curtis C.R. Gastric rupture in horses: 50 cases (1979–1987) JAm VetMed Assoc. 1990;196:333–336. [PubMed] [Google Scholar]
- 1402.Steenhaut M., Vlaminck K., Gasthuys F. Surgical repair of a partial gastric rupture in a horse. Equine Vet J. 1986;18:331–332. doi: 10.1111/j.2042-3306.1986.tb03643.x. [DOI] [PubMed] [Google Scholar]
- 1403.Hogan P.M., Bramlage L.R., Pierce S.W. Repair of a full-thickness gastric rupture in a horse. J Am Vet Med Assoc. 1995;207:338–340. [PubMed] [Google Scholar]
- 1404.Owen R.A., Jagger D.W., Jagger F. Two cases of equine primary gastric impaction. Vet Rec. 1987;121:102–105. doi: 10.1136/vr.121.5.102. [DOI] [PubMed] [Google Scholar]
- 1405.Barclay W.P., Foerner J.J., Phillips T.N. Primary gastric impaction in the horse. J Am Vet Med Assoc. 1982;181:682–683. [PubMed] [Google Scholar]
- 1406.Honnas C.M., Schumacher J. Primary gastric impaction in a pony. J Am Vet Med Assoc. 1985;187:501–502. [PubMed] [Google Scholar]
- 1407.Kellam L.L., Johnson P.J., Kramer J. Gastric impaction and obstruction of the small intestine associated with persimmon phytobezoar in a horse. J Am Vet Med Assoc. 2000;216:1279–1281. doi: 10.2460/javma.2000.216.1279. [DOI] [PubMed] [Google Scholar]
- 1408.Weldon A.D., Rowland P.H., Rebhun W.C. Emphysematous gastritis in a horse. Cornell Vet. 1991;81:51–58. [PubMed] [Google Scholar]
- 1409.Delesalle C., Deprez P., Vanbrantegem L. Emphysematous gastritis associated with Clostridium septicum in a horse. J Vet Intern Med. 2003;17:115–118. doi: 10.1892/0891-6640(2003)017<0115:egawcs>2.3.co;2. [DOI] [PubMed] [Google Scholar]
- 1410.Pascoe R.R., Summers P.M. Clinical survey of tumours and tumour-like lesions in horses in south east Queensland. Equine Vet J. 1981;13:235–239. doi: 10.1111/j.2042-3306.1981.tb03504.x. [DOI] [PubMed] [Google Scholar]
- 1411.Carlson G.P. Lymphosarcoma in horses. Leukemia. 1995;9(suppl 1):S101. [PubMed] [Google Scholar]
- 1412.Taylor S.D., Pusterla N., Vaughan B. Intestinal neoplasia in horses. J Vet Intern Med. 2006;20:1429–1436. doi: 10.1892/0891-6640(2006)20[1429:inih]2.0.co;2. [DOI] [PubMed] [Google Scholar]
- 1413.Rhind S.M., Hawe C., Dixon P.M. Oral metastasis of renal cell carcinoma in a horse. J Comp Pathol. 1999;120:97–103. doi: 10.1053/jcpa.1998.0255. [DOI] [PubMed] [Google Scholar]
- 1414.McKenzie E.C., Mills J.N., Bolton J.R. Gastric squamous cell carcinoma in three horses. Aust Vet J. 1997;75:480–483. doi: 10.1111/j.1751-0813.1997.tb14376.x. [DOI] [PubMed] [Google Scholar]
- 1415.Ford T.S., Vaala W.E., Sweeney C.R. Pleuroscopic diagnosis of gastroesophageal squamous cell carcinoma in a horse. J Am Vet Med Assoc. 1987;190:1556–1558. [PubMed] [Google Scholar]
- 1416.Olsen S.N. Squamous cell carcinoma of the equine stomach: a report of five cases. Vet Rec. 1992;131:170–173. doi: 10.1136/vr.131.8.170. [DOI] [PubMed] [Google Scholar]
- 1417.Mair T.S., Hillyer M.H. Chronic colic in the mature horse: a retrospective review of 106 cases. Equine Vet J. 1997;29:415–420. doi: 10.1111/j.2042-3306.1997.tb03151.x. [DOI] [PubMed] [Google Scholar]
- 1418.Hillyer M.H., Mair T.S. Recurrent colic in the mature horse: a retrospective review of 58 cases. Equine Vet J. 1997;29:421–424. doi: 10.1111/j.2042-3306.1997.tb03152.x. [DOI] [PubMed] [Google Scholar]
- 1419.Ogilvie G.K. Paraneoplastic syndromes. Vet Clin North Am Equine Pract. 1998;14:439–449. doi: 10.1016/s0749-0739(17)30179-7. v. [DOI] [PubMed] [Google Scholar]
- 1420.Reef V.B., Dyson S.S., Beech J. Lymphosarcoma and associated immune-mediated hemolytic anemia and thrombocytopenia in horses. J Am Vet Med Assoc. 1984;184:313–317. [PubMed] [Google Scholar]
- 1421.La Perle K.M., Piercy R.J., Long J.F. Multisystemic, eosinophilic, epitheliotropic disease with intestinal lymphosarcoma in a horse. Vet Pathol. 1998;35:144–146. doi: 10.1177/030098589803500209. [DOI] [PubMed] [Google Scholar]
- 1422.McCoy D.J., Beasley R. Hypercalcemia associated with malignancy in a horse. J Am Vet Med Assoc. 1986;189:87–89. [PubMed] [Google Scholar]
- 1423.East L.M., Savage C.J. Abdominal neoplasia (excluding urogenital tract) Vet Clin North Am Equine Pract. 1998;14:475–493. doi: 10.1016/s0749-0739(17)30181-5. v-vi. [DOI] [PubMed] [Google Scholar]
- 1424.Harps O., Brumhard J., Bartmann C.P. Ascites as a result of peritoneal mesotheliomas in a horse. Tierarztl Prax. 1996;24:270–274. [PubMed] [Google Scholar]
- 1425.Ricketts S.W., Peace C.K. A case of peritoneal mesothelioma in a thoroughbred mare. Equine Vet J. 1976;8:78–80. doi: 10.1111/j.2042-3306.1976.tb03298.x. [DOI] [PubMed] [Google Scholar]
- 1426.Fulton I.C., Brown C.M., Yamini B. Adenocarcinoma of intestinal origin in a horse: diagnosis by abdominocentesis and laparoscopy. Equine Vet J. 1990;22:447–448. doi: 10.1111/j.2042-3306.1990.tb04314.x. [DOI] [PubMed] [Google Scholar]
- 1427.Conwell R.C., Hillyer M.H., Mair T.S. Haemoperitoneum in horses: a retrospective review of 54 cases. Vet Rec. 2010;167:514–518. doi: 10.1136/vr.c4569. [DOI] [PubMed] [Google Scholar]
- 1428.Perkins G.A., Nydam D.V., Flaminio M.J. Serum IgM concentrations in normal, fit horses and horses with lymphoma or other medical conditions. J Vet Intern Med. 2003;17:337–342. doi: 10.1111/j.1939-1676.2003.tb02458.x. [DOI] [PubMed] [Google Scholar]
- 1429.Davis E.G., Wilkerson M.J., Rush B.R. Flow cytometry: clinical applications in equine medicine. J Vet Intern Med. 2002;16:404–410. doi: 10.1892/0891-6640(2002)016<0404:fccaie>2.3.co;2. [DOI] [PubMed] [Google Scholar]
- 1430.Rebhun W.C., Bertone A. Equine lymphosarcoma. J Am Vet Med Assoc. 1984;184:720–721. [PubMed] [Google Scholar]
- 1431.Wiseman A., Petrie L., Murray M. Diarrhoea in the horse as a result of alimentary lymphosarcoma. Vet Rec. 1974;95:454–457. doi: 10.1136/vr.95.20.454. [DOI] [PubMed] [Google Scholar]
- 1432.Finley M.R., Rebhun W.C., Dee A. Paraneoplastic pruritus and alopecia in a horse with diffuse lymphoma. J Am Vet Med Assoc. 1998;213:102–104. [PubMed] [Google Scholar]
- 1433.Dabareiner R.M., Sullins K.E., Goodrich L.R. Large colon resection for treatment of lymphosarcoma in two horses. J Am Vet Med Assoc. 1996;208:895–897. [PubMed] [Google Scholar]
- 1434.Taylor S.D., Haldorson G.J., Vaughan B. Gastric neoplasia in horses. J Vet Intern Med. 2009;23:1097–1102. doi: 10.1111/j.1939-1676.2009.0356.x. [DOI] [PubMed] [Google Scholar]
- 1435.Schuh J.C. Squamous cell carcinoma of the oral, pharyngeal and nasal mucosa in the horse. Vet Pathol. 1986;23:205–207. doi: 10.1177/030098588602300217. [DOI] [PubMed] [Google Scholar]
- 1436.Tuckey J.C., Hilbert B.J., Beetson S. Squamous cell carcinoma of the pharyngeal wall in a horse. Aust Vet J. 1995;72:227. doi: 10.1111/j.1751-0813.1995.tb03527.x. [DOI] [PubMed] [Google Scholar]
- 1437.Hewes C.A., Sullins K.E. Use of cisplatin-containing biodegradable beads for treatment of cutaneous neoplasia in equidae: 59 cases (2000–2004) J Am Vet Med Assoc. 2006;229:1617–1622. doi: 10.2460/javma.229.10.1617. [DOI] [PubMed] [Google Scholar]
- 1438.Theon A.P., Wilson W.D., Magdesian K.G. Long-term outcome associated with intratumoral chemotherapy with cisplatin for cutaneous tumors in equidae: 573 cases (1995–2004) J Am Vet Med Assoc. 2007;230:1506–1513. doi: 10.2460/javma.230.10.1506. [DOI] [PubMed] [Google Scholar]
- 1439.Mosunic C.B., Moore P.A., Carmichael K.P. Effects of treatment with and without adjuvant radiation therapy on recurrence of ocular and adnexal squamous cell carcinoma in horses: 157 cases (1985–2002) J Am Vet Med Assoc. 2004;225:1733–1738. doi: 10.2460/javma.2004.225.1733. [DOI] [PubMed] [Google Scholar]
- 1440.Theon A.P., Pascoe J.R., Galuppo L.D. Comparison of perioperative versus postoperative intratumoral administration of cisplatin for treatment of cutaneous sarcoids and squamous cell carcinomas in horses. J Am Vet Med Assoc. 1999;215:1655–1660. [PubMed] [Google Scholar]
- 1441.Scherrer N.M., Lassaline-Utter M., McKenna B.C. Characterization and outcome following excision of masses in the nictitating membranes of horses: 50 cases (1998–2012) J Am Vet Med Assoc. 2014;245:812–815. doi: 10.2460/javma.245.7.812. [DOI] [PubMed] [Google Scholar]
- 1442.Moore A.S., Beam S.L., Rassnick K.M. Long-term control of mucocutaneous squamous cell carcinoma and metastases in a horse using piroxicam. Equine Vet J. 2003;35:715–718. doi: 10.2746/042516403775696320. [DOI] [PubMed] [Google Scholar]
- 1443.Leach M.W., Pool R.R. Hypertrophic osteopathy in a Shetland pony attributable to pulmonary squamous cell carcinoma metastases. Equine Vet J. 1992;24:247–249. doi: 10.1111/j.2042-3306.1992.tb02825.x. [DOI] [PubMed] [Google Scholar]
- 1444.Moran J.A., Lemberger K., Cadore J.L. Small intestine adenocarcinoma in conjunction with multiple adenomas causing acute colic in a horse. J Vet Diagn Invest. 2008;20:121–124. doi: 10.1177/104063870802000128. [DOI] [PubMed] [Google Scholar]
- 1445.Kirchhof N., Steinhauer D., Fey K. Equine adenocarcinomas of the large intestine with osseous metaplasia. J Comp Pathol. 1996;114:451–456. doi: 10.1016/s0021-9975(96)80020-x. [DOI] [PubMed] [Google Scholar]
- 1446.Roy M.F., Parente E.J., Donaldson M.T. Successful treatment of a colonic adenocarcinoma in a horse. Equine Vet J. 2002;34:102–104. doi: 10.2746/042516402776181178. [DOI] [PubMed] [Google Scholar]
- 1447.Mair T.S., Davies E.V., Lucke V.M. Small colon intussusception associated with an intralumenal leiomyoma in a pony. Vet Rec. 1992;130:403–404. doi: 10.1136/vr.130.18.403. [DOI] [PubMed] [Google Scholar]
- 1448.Schaudien D., Muller J.M., Baumgartner W. Omental leiomyoma in a male adult horse. Vet Pathol. 2007;44:722–726. doi: 10.1354/vp.44-5-722. [DOI] [PubMed] [Google Scholar]
- 1449.Hanes G.E., Robertson J.T. Leiomyoma of the small intestine in a horse. J Am Vet Med Assoc. 1983;182:1398. [PubMed] [Google Scholar]
- 1450.Collier M.A., Trent A.M. Jejunal intussusception associated with leiomyoma in an aged horse. J Am Vet Med Assoc. 1983;182:819–821. [PubMed] [Google Scholar]
- 1451.Watt B.C., Trostle S.S., Cooley A.J. Intraluminal leiomyoma colon polyp in a mare. Equine Vet J. 2001;33:326–328. doi: 10.2746/042516401776249769. [DOI] [PubMed] [Google Scholar]
- 1452.Kasper C., Doran R. Duodenal leiomyoma associated with colic in a two-year-old horse. J Am Vet Med Assoc. 1993;202:769–770. [PubMed] [Google Scholar]
- 1453.Johnson P.J., Wilson D.A., Turk J.R. Disseminated peritoneal leiomyomatosis in a horse. J Am Vet Med Assoc. 1994;205:725–728. [PubMed] [Google Scholar]
- 1454.Pirie R.S., Tremaine W.H. Neoplasia of the mouth and surrounding structures. In: Robinson N.E., editor. Current Therapy in Equine Medicine. WB Saunders; Philadelphia, PA: 1997. pp. 153–155. [Google Scholar]
- 1455.Morse C.C., Saik J.E., Richardson D.W. Equine juvenile mandibular ossifying fibroma. Vet Pathol. 1988;25:415–421. doi: 10.1177/030098588802500603. [DOI] [PubMed] [Google Scholar]