

ABSTRACT
We have utilized a de novo designed two‐stranded α‐helical coiled‐coil template to display conserved α‐helical epitopes from the stem region of hemagglutinin (HA) glycoproteins of influenza A. The immunogens have all the surface‐exposed residues of the native α‐helix in the native HA protein of interest displayed on the surface of the two‐stranded α‐helical coiled‐coil template. This template when used as an immunogen elicits polyclonal antibodies which bind to the α‐helix in the native protein. We investigated the highly conserved sequence region 421–476 of HA by inserting 21 or 28 residue sequences from this region into our template. The cross‐reactivity of the resulting rabbit polyclonal antibodies prepared to these immunogens was determined using a series of HA proteins from H1N1, H2N2, H3N2, H5N1, H7N7, and H7N9 virus strains which are representative of Group 1 and Group 2 virus subtypes of influenza A. Antibodies from region 449–476 were Group 1 specific. Antibodies to region 421–448 showed the greatest degree of cross‐reactivity to Group 1 and Group 2 and suggested that this region has a great potential as a “universal” synthetic peptide vaccine for influenza A. © 2016 Wiley Periodicals, Inc. Biopolymers (Pept Sci) 106: 144–159, 2016.
Keywords: helical epitopes, synthetic peptide vaccines, coiled‐coil immunogens, hemagglutinin, influenza A viruses
INTRODUCTION
Influenza A viruses spread rapidly, causing widespread seasonal epidemics of respiratory disease worldwide, which results in more than a billion cases and 500,000 deaths annually.1 Annually in the USA, there are 25–50 million cases of influenza, 225,000 hospitalizations and about 36,000 deaths.2 A new type of influenza vaccine that can provide long‐lasting protection against all strains of influenza A virus is urgently needed.2 Although influenza vaccines and several influenza drugs are available, the burden of influenza on society remains enormous. Natural influenza virus infection and current influenza vaccines elicit antibodies that block binding of the head of the viral hemagglutinin (HA) glycoprotein to its cell surface receptor, but the virus readily evades these antibodies by mutations in the antibody‐binding region (Figure 1).5 In contrast, several groups have recently identified conserved regions in the stem domain of HA that elicit rare human neutralizing antibodies that can protect against multiple influenza HA subtypes.6, 7, 8, 9, 10, 11, 12 These human monoclonal antibodies are candidate therapeutics for influenza, but because of the complexity of the three‐dimensional binding sites of the monoclonal antibodies on the stem region of HA, it is extremely difficult to design an immunogen to mimic these epitopes to create immunogens that will elicit these neutralizing antibodies.
Figure 1.
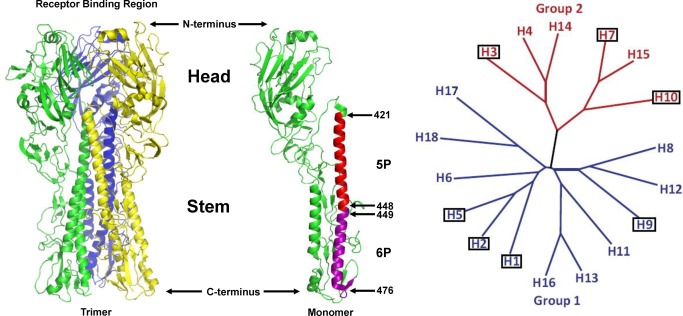
Hemagglutinin protein (HA) of Influenza A virus (A/South Carolina/1/1918(H1N1), PDB ID: 1RUZ). The left panel shows the three‐dimensional structure of the trimeric HA protein expressed in recombinant vaccinia virus‐infected CV1 cells.3 The right panel shows the structure of a single monomer where the regions of interest 421–448 (5P) and 449–476 (6P) are colored red and purple, respectively. The head region is involved in receptor binding. The stem region is involved in membrane fusion and virus entry. The models were created with PyMOL (version 0.99) program. Phylogenetic tree of the hemagglutinin genes of all known influenza A virus subtypes. Adapted from Wu et al., 2014.4 The categorized HA molecules can be grouped into two groups, group 1 (colored blue) and group 2 (colored red). The subtypes that have been confirmed in humans are boxed.
The sequence of different influenza HA subtypes is more conserved in the stem region across different HA subtypes compared to the globular head.13 Many attempts to develop a vaccine based on the stem domain (HA2 subunit) have been unsuccessful. Recently, results suggest that HA2‐based immunogens can provide cross protection against several strains within a subtype.14, 15 However, these vaccines are still large in size and require C‐terminal trimerization motifs connected by flexible linkers to properly fold the trimeric structure of the headless stem domain.15 It still remains to be shown that cross‐reactive protection against many different subtypes can be achieved with stem domain constructs by in vivo challenge experiments in animals with heterologous subtypes. Existing influenza vaccines display limited efficacy for several reasons.16 The head region of HA rapidly mutates and antibodies in the human population to previous influenza strains select for antigenic variants so that new vaccine strains that target these variants must be developed every year, based on predictions of viral strains that may circulate the next winter.17 Errors in predicting antigenic types and long vaccine development times compromise vaccination programs. Eighteen HA subtypes of influenza A have been identified in humans, animals and birds (Figure 1),4 including H1, H2 and H3 which have caused human pandemics. H1 and H3 subtypes of influenza A are continually undergoing immune selection (antigenic drift) in humans, so that antibody‐resistant strains continue to emerge and cause new human pandemics. Subtypes H5, H7, and H9 are recognized by the World Health Organization as “potential pandemic viruses” and humans have no pre‐existing immunity to these subtypes.18 Avian H5N1 virus is highly pathogenic in humans with a mortality rate of 60% in humans, but fortunately there is not efficient human‐to‐human transmission.19 Mutations in virus genes that alter antigenicity or host range, or increase virus virulence or transmissibility can occur spontaneously or can be genetically engineered to create biothreat strains. Therefore, a new strategy is needed for development of a universal influenza vaccine that could protect against all HA types and their antigenic variants, be manufactured quickly and inexpensively, be very stable with a long self‐life, and induce long‐lasting immunity.
We are developing a novel synthetic peptide vaccine platform technology20, 21, 22 that targets α‐helical regions in proteins. We have chosen the stem region of Class 1 Viral Fusion Proteins such as Influenza A, Severe Acute Respiratory Syndrome Coronavirus (SARS‐CoV) spike glycoprotein and Middle East Respiratory Syndrome Coronavirus (MERS‐CoV) spike glycoprotein, Respiratory Syncytial Virus (RSV) F protein and Ebola glycoprotein23, 24 because the α‐helical regions in the stem are highly conserved and ideal candidates for vaccine development. The stem domains of the fusion glycoproteins of these diverse viruses share a common coiled‐coil structure that must be triggered to undergo a massive conformational change in order to induce fusion of the virus envelope with host cell membranes and initiate virus infection. Blocking the conformational change in the stem region of the viral fusion protein with antibodies can prevent virus infection in vitro and in vivo. Thus, if one can elicit antibodies to functionally significant and/or highly conserved regions of these glycoproteins, the ability to provide a broadly protective vaccine is dramatically enhanced. Synthetic peptide vaccines have many advantages over traditional vaccines25 if they can mimic the structure of the epitope found in the native protein target: (1) The peptide vaccine can be synthesized soon after virus emergence when the key regions of the viral immunogen fusion protein have been deduced from the viral nucleic acid sequence; (2) The peptide immunogen, in our case, is small containing a B‐cell epitope of 28 amino acid residues in length and can be rapidly and inexpensively manufactured in very large quantities; (3) The synthesized peptide immunogen does not have to be purified away from cellular components or irrelevant viral proteins; (4) Traditional vaccines with intact proteins may contain additional epitopes that elicit undesired B‐cell or T‐cell responses, thus, there is improved safety for synthetic peptide vaccines; (5) The amino acid sequences of the coiled‐coil regions of the viral fusion proteins are highly conserved among the many strains of emerging viruses, so that a single peptide immunogen has the potential to protect against multiple subtypes and host range mutants of an emerging virus, making the vaccine useful not only for humans, but also the animal reservoirs of the virus; (6) Our unique, conformation‐stabilized templated, two‐stranded synthetic peptide immunogens are highly stable to temperature, facilitating storage and stockpiling of the vaccine.
The Technology
We are developing an extremely simple, innovative and robust technology to present α‐helical epitopes from native proteins to the immune system such that the resulting conformation‐specific antibodies bind to the native protein target. This procedure involves a process called templating where the helical sequence of interest is inserted into a parallel two‐stranded α‐helical coiled‐coil and disulfide‐bridged template which maintains the native helical conformation of the epitope, presents all the surface‐exposed residues of the α‐helix in the native protein and optimizes stability of the immunogen (Figure 2). To optimize the conformational stability of the immunogen, two synthetic peptides are stabilized in a two‐stranded, coiled‐coil configuration by substituting at each of the hydrophobic residues in the a and d positions of the native α‐helix with isoleucine and leucine, respectively; and also linking the two templated peptides with a disulfide bond. It is well known that residues at the a and d positions define the hydrophobic interface between the helices and are critical for determining the oligomerization state and stability of coiled‐coils. For example, the β‐branched hydrophobic residues Ile and Val provide significantly greater stability to the coiled‐coil than that of the similarly sized hydrophobic residue, Leu, when placed at position a,26, 27, 28 whereas Leu appeared to provide more stability at position d.28 Harbury et al. using synthetic analogs of GCN4, have shown that subtle changes in the type of hydrophobic core residues can result in selective formation of dimer, trimer and tetramer structures.29 Hodges and co‐workers carried out the complete biophysical characterization of a 38‐residue model coiled‐coil substituted by 20 different amino acid residues at position a and position d to determine their effect on protein stability.30, 31, 32 Woolfson and Alber developed an algorithm to predict the oligomerization states of coiled‐coils.33
Figure 2.
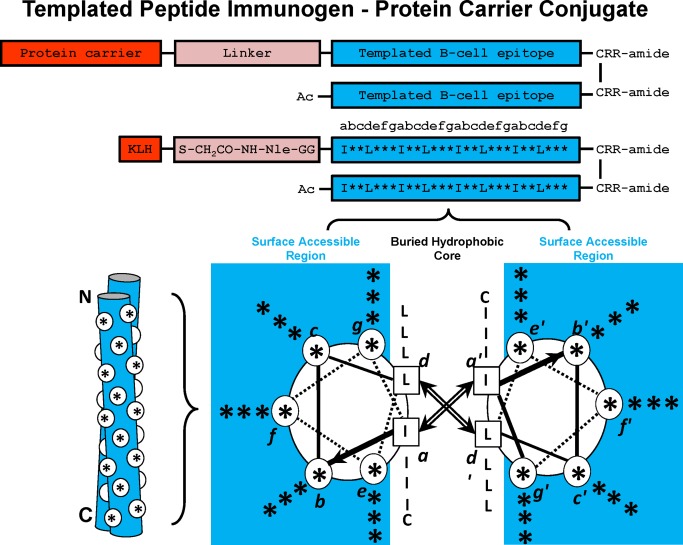
Templated α–Helical B‐cell epitopes. The top panel shows the disulfide‐bridged homo‐two‐stranded α‐helical coiled‐coil templated B‐cell epitopes. The residue positions b, c, e, f, and g, which can be substituted with the native B‐cell epitope, are indicated with an asterisk (*). The relative position of the * residues when in a two‐stranded coiled‐coil structure are shown in a cross‐sectional view (lower right) and as a cartoon (lower left). In the cross‐sectional view, the direction of the helices is into the page from the NH2 to the COOH terminus, with the polypeptide chains parallel and in‐register. Heptad positions are labeled a–g, with the prime indicating corresponding positions on the opposing helix. Arrows depict the hydrophobic interactions that occur between residues a and a′, d and d′. In the cartoon model, the white circles denote the b, c, e, f, and g positions exposed from the front view of the two‐stranded coiled‐coil. Diagram of templated peptide immunogens used in this study. Templated B‐cell epitope is conjugated to the protein carrier, Keyhole limpet hemocyanin (KLH) via a linker consisting of a GG spacer, norleucine (for quantitation of moles of peptide per mole protein carrier) and an iodoacetyl group used to couple the peptide to SH groups on KLH. The disulfide bond at the C‐terminus of the template is at an a position and is used to further stabilize the two‐stranded α–helical coiled‐coil. The RR residues at the C‐terminus are used to enhance solubility of the immunogen, if required.
In the native α‐helix the hydrophobic residues in positions a and d are buried while each of the b, c, e, f, and g residues are exposed on the surface. Similarly, in the two‐stranded α‐helical coiled‐coil immunogen, each of the b, c, e, f, and g residues are also exposed on the surface and are identical to the corresponding residues displayed on the surface of the selected native α‐helix in the stem domain (denoted by asterisks Figure 2 which creates two helical surfaces (blue), each which generate antibodies that bind the helix of interest in the native protein. In the native trimeric spike protein the helix may be a single α‐helix either stabilized by surrounding protein regions or stabilized in a trimeric coiled‐coil in the stem domain of the spike protein. What is unique with our technology is that the resulting antibodies from our immunogens bind to a single α‐helical surface and are dramatically different from the binding sites of monoclonal antibodies that bind to the stem region of hemagglutinin. These monoclonal antibodies binding sites involve complex conformational epitopes rather than a single helix.6, 7, 8, 12
MATERIALS AND METHODS
Peptide Synthesis
The peptides were synthesized on 4‐methylbenzhydrylamine (MBHA) hydrochloride salt polystyrene resin (substitution: 0.4 mmole g−1 resin, bead size 100–200 mesh, 1% crosslinked with divinylbenzene) using the general procedures for solid‐phase peptide synthesis on a Beckman peptide synthesizer, model 990. Amino acids were coupled as their Nα‐tert‐butoxycarbonyl (Boc) derivatives (fivefold molar excess), and reactive side‐chains were protected as follows: Arg, tosyl (Tos); Asn, 9‐H‐xanthen‐9‐yl (Xan); Asp, cyclohexyl (OcHx); Cys, 4‐methylbenzyl (MeBzl); Gln, 9‐H‐xanthen‐9‐yl (Xan); Glu, cyclohexyl (OcHx); His, benzyloxymethyl (Bom); Lys, 2‐chlorobenzyloxycarbonyl (2‐Cl‐Z); Ser, benzyl (Bzl); Thr, benzyl (Bzl); Trp, formyl (For). Coupling reactions were performed using 5 equivalents of the mixture Boc‐amino acid:BOP (benzotriazol‐l‐yloxy‐tris (dimethylamino) phosphonium, hexafluorophosphate): HOBt (hydroxybenzotriazole): DIEA (diisoproylethylamine) (1:1:1:2) in DMF (N,N‐dimethylformamide), with a 3‐5 min pre‐activation step before coupling. Cys and His were coupled without pre‐activation to prevent racemization. Coupling reactions were monitored at each step by using the ninhydrin (Kaiser) test. Couplings that were incomplete after 3 h were recoupled. Removal of N α‐Boc protection at each step was accomplished by treatment for 30 min with 35% trifluoroacetic acid/CH2Cl2 (v/v) containing indole (1 mg ml−1). The NH2‐terminal Boc‐group was removed from the completed protected peptides before HF cleavage. Deprotection of Ni‐formyl‐tryptophan was carried out using piperidine in DMF (1.5 h; 0°C) before HF‐cleavage. Side‐chain deprotection and peptide cleavage from the resin was carried out using a cleavage cocktail consisting of HF:anisol:DMS (dimethylsulfide):EDT (1,2‐ethanedithiol):p‐thiocresol (8.5:0.5:0.5:0.3:0.2, 1.5 h; 0°C). After removal of HF under vacuum, the crude free sulfhydryl peptides were precipitated with ether, filtered, and dissolved in glacial acetic acid. Lyophilization gave the crude peptides as white fluffy powders which were purified using reversed‐phase high‐performance liquid chromatography (RP‐HPLC).
Peptide Purification by Reversed‐phase HPLC
Preparative reversed‐phase HPLC (RP‐HPLC) was carried out on a Luna C18 column from Phenomenex (10‐µm particle size, 100Å pore size; 250 × 21.2 mm2 I.D.). The column was equilibrated in 80% eluent A/20% eluent B, where eluent A is 0.2% aq. trifluoracetic acid (TFA) and eluent B is 0.15% TFA in CH3CN. The sample (800 mg) was dissolved in 20% aq. CH3CN containing 0.2% TFA to a concentration of 5 mg ml−1, then loaded onto the column at a flow‐rate of 5 ml min−1. The column was then eluted with 80% eluent A/20% eluent B for 30 min at 10 ml min−1, followed by a shallow linear AB gradient of 0.1% B/min up to 50% CH3CN. Fractions were collected every 2 min. Subsequent fraction analysis was carried out on a Luna C18 RP‐HPLC column (5‐µm particle size, 100Å pore size; 250 × 3 mm I.D.) using a linear AB gradient (1% B/min) from starting conditions of 80% A/20% B. Purity of fractions was also verified by LC/MS using a HALO Peptide ES‐C18 2.0 µm, 2.1 mm I.D × 100 mm column from Advanced Materials Technology, USA. Pooled fractions containing purified material were subsequently lyophilized.
Formation of Disulfide‐bridged Two‐stranded Peptides
The disulfide‐bridged two‐stranded peptides were prepared as follows: 2,2′‐dithiodipyridine (DTDP) was dissolved in DMF to make a final concentration of 100 μg μL−1. The N‐terminal acetylated peptide was dissolved in 3:1 (v/v) acetic acid/H2O to make a final concentration of 0.8 mM. DTDP solution was added to the peptide solution with a molar ratio of 1:10 and stirred for 2 h with monitoring of the mass increase by LC/MS. Distilled water was added and the peptide with the thiopyridine derivative (TP) of cysteine was purified with RP‐HPLC. The TP‐derived peptide (first strand) and the peptide with a free cysteine (second strand) were dissolved in 40 mM ammonium acetate buffer, pH 5.5, (molar ratio 1:1) and stirred for 2 h, monitoring the mass change with LC/MS (Figure 3). Once the disulfide bridge formation was complete the disulfide‐bridged two‐stranded peptide was purified by RP‐HPLC and lyophilized.
Figure 3.
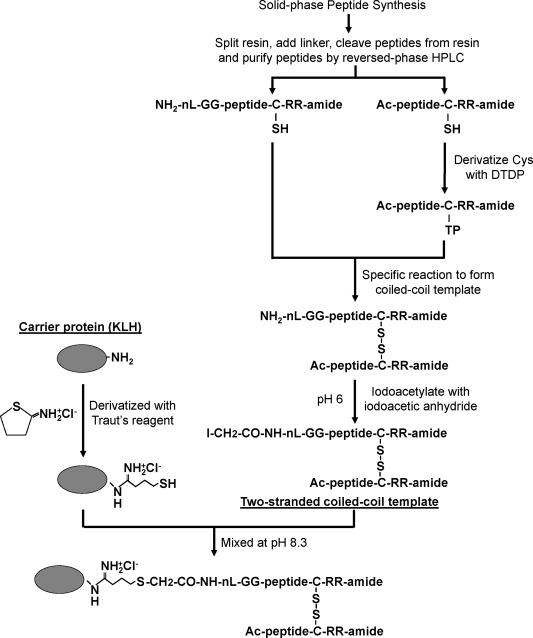
Flowchart for the generation of the two‐stranded α‐helical coiled‐coil template and conjugation of the template to the protein carrier (see Methods). nL denotes norleucine; DTDP, 2,2'‐dithiodipyridine; KLH, keyhole limpet hemocyanin; Traut's reagent, 2‐iminothiolane hydrochloride.
Purification of the Disulfide‐bridged Peptides
The disulfide‐bridged two‐stranded peptides were purified on a Luna C18 reversed‐phase column from Phenomenex (10 µm particle size, 100 Å pore size; 250 × 10 mm2 ID.). The column was equilibrated in 80% eluent A/20% eluent B, where eluent A is 0.2% aq. TFA and eluent B is 0.15% TFA in CH3CN. The sample (10–20 mg, depending on the yield of crude sample) was dissolved in 20% aq. CH3CN containing 0.2% TFA to a concentration of 5 mg ml−1, then loaded onto the column at a flow‐rate of 1 ml mm−1, followed by a rapid gradient to 30% aq. CH3CN in 5 min and a final shallow gradient of 0.2% B/min up to 50% CH3CN at a flow rate of 2 ml min−1. Fractions (1 min) were collected and subsequently analyzed as described in Peptide Purification by Reversed‐phase HPLC section. Pooled fractions containing purified material were subsequently lyophilized.
Conjugation of Peptide Immunogen to Keyhole Limpet Hemocyanin (KLH) or Bovine Serum Albumin (BSA)
Peptide immunogens conjugated to keyhole limpet hemocyanin (KLH) and peptide immunogens conjugated to bovine serum albumin (BSA) used in enzyme‐linked immunosorbent assays (ELISA) were prepared as follows: The single free α‐amino group on the two‐stranded peptide immunogen was iodoacetylated using iodoacetic anhydride. Iodoacetic anhydride was dissolved in 1,4‐dioxane at concentration of 100 mM. The disulfide‐bridged two‐stranded peptide was dissolved in 100 mM 2‐(N‐morpholine) ethanesulfonic acid (MES), pH 6.0 in 60% acetonitrile/H2O at a concentration of 0.15 mM. The iodoacetic anhydride solution was slowly added into the peptide solution to a molar ratio of 1.2:1, incubated at room temperature for 10 min and the mass change was monitored with LC/MS. The iodoacetyl‐two‐stranded disulfide‐bridged peptide was purified by RP‐HPLC and lyophilized. The protein carriers (KLH and BSA) were derivatized with Traut's reagent (2‐iminothiolane) as follows: dissolve KLH or BSA in PBS (pH 8.9 with 8M urea and 5 mM EDTA) at a concentration of 5 mg ml−1. Add Traut's reagent dissolved in water (4 mg ml−1) to the protein solution with a molar ratio of 40:1, and then incubate the mixture at room temperature for 1 h in a dark room. The unused Traut's reagent was removed by dialysis. The Traut's reagent modified KLH or BSA then was lyophilized. Iodoacetyl‐two‐stranded disulfide‐bridged peptide was dissolved in 0.5 mM PBS, 8M urea at a concentration of 4 mgml−1. The modified KLH or BSA was dissolved in 0.2 mM PBS, 8M urea the above solutions were mixed at a molar ratio of peptide:KLH(6:1) or peptide:BSA(5:1) and then incubated at room temperature overnight. Iodoacetamide (28 mM) in water was added to the reaction mixture and incubate at room temperature for 30 min. Free peptides were removed by dialysis in PBS/8M urea, and then 60% acetonitrile in water with 0.2% TFA. KLH or BSA‐conjugated peptide was purified by RP‐HPLC and lyophilized (Figure 3).
Circular Dichroism Spectroscopy
Circular dichroism (CD) spectra were recorded on a Jasco J‐815 spectropolarimeter (Jasco Easton, MD) at 5°C under benign (non‐denaturing) conditions (50 mM NaH2PO4/Na2HPO4/100 mM KCl, pH 7.0), as well as in the presence of an α‐helix inducing solvent, 2,2,2‐trifluoroethanol (TFE), (50 mM NaH2PO4/Na2HPO4/100 mM KC1, pH 7.0 buffer/50% TFE). Peptide ellipticity was scanned from 195 to 250 nm in a 1 nm path length cell. Temperature denaturation was measured by following the change in molar ellipicity at 222 nm from 5 to 95°C and a temperature increase rate of 1°C min−1.
Animal Immunization Protocol
The animal work was carried out at New England Peptide in accordance with established protocols on file. Briefly, for each immunogen, two New Zealand white rabbits were immunized using at least four subcutaneous sites. Primary immunization (day 1) contained 400 μg of the KLH‐conjugated peptides emulsified with complete Freund's adjuvant (CFA). Secondary and tertiary immunizations at day 15 and day 44 also contained 200 μg of KLH‐conjugated peptides that were emulsified with incomplete Freund's adjuvant (IFA). After exsanguination at day 78, serum was collected and stored at −20°C.
Purification of IgG
Polyclonal antibodies were precipitated from sera using ammonium sulfate. Nearly 10–15 ml sera were diluted 1:1 into PBS and then crystalline ammonium sulfate was added to 45% saturation (0.277 g ml−1) while stirring in an ice bath to precipitate the immunoglobulins for 2 h. Centrifugation at 12,000g at 4°C for 30 min was used to collect the precipitated antibodies. The pellet was resuspended in PBS using the same volume as the starting serum and dialyzed against PBS, pH 7.4. The dialysis solution was changed at least 3 times during the total dialysis. The antibody solution was then further purified on a protein G affinity column (1.6 cm I.D. × 2.5 cm, 5 ml, protein G–sepharose 4 Fast Flow, GE Healthcare, Piscataway, NJ). The bound antibody was eluted from the column using 0.5M ammonium acetate, pH 3 buffer after which the solution was immediately adjusted to pH 7–8 with ammonium hydroxide and dialyzed against PBS overnight. Subsequently, the antibody solution was concentrated to ∼10 mg ml−1 in an Amicon concentration unit using YM30 ultrafiltration discs (Millipore, Bedford, MA). The concentration of each antibody solution was determined by OD280 using a concentration standard IgG sample as a reference. Finally, the antibody solution was stored at −20°C until use. Purification of IgG was critical to ensure identical concentrations of IgG for comparison of antibody cross‐reactivity.
Enzyme‐Linked Immnosorbent Assays (ELISA) Using Peptides, BSA‐conjugated Peptides or Hemagglutinin Proteins
High‐binding, 96‐well polystyrene microplates were coated with diluted peptide antigens (0.002 mg ml−1 using 100 mM carbonate, pH 9.6) or hemagglutinin protein antigens (0.002 mg mL−1 in PBS) for 1 h at 37°C. After removing the coating solution and washing three times with PBS, each well was blocked with 100 μl 5% BSA in PBS for 1 h at 37°C. The sera or Protein G purified antibodies (IgG) were diluted in 2% BSA in PBS and added to each well with a serial dilution and incubated at 37°C for 1 h. The sample solution was removed and washed three times with PBS containing 0.2% Tween20. Next, 100 μl horseradish peroxidase (HRP) conjugated anti‐rabbit IgG (goat) (diluted 1:5000) was added to each well and incubated for 1 h at 37°C. Sample solution was then removed and the wells were washed three times with PBS containing 0.2% Tween 20. A 100 μl solution of 2,2′‐azino‐bis‐3‐ethylbenzothiazoline‐6‐sulfonic acid (0.6 mg ml−1) in 10 mM citrate, pH 4.2 containing 0.1% H2O2 was added to each well, the ELISA plates were shaken for 15 min and then the plates were read at OD450nm.
Hemagglutinin (HA) Proteins
The hemagglutinin proteins were obtained through BEI Resources, NIAID, NIH as follows: H1 hemagglutinin protein from influenza virus, A/Solomon Islands/3/2006 (H1N1), is a full‐length glycosylated recombinant protein that was produced in Sf9 insect cells using a baculovirus expression vector system, NR‐15170; H1 hemagglutinin protein with C‐terminal histidine tag from Influenza Virus, A/Puerto Rico/8/1934 (H1N1), recombinant from Baculovirus produced in Sf9 insect cells, NR‐19240; H1 Hemagglutinin Protein with N‐Terminal Histidine Tag from Influenza Virus, A/Toulon/1173/2011 (H1N1)pdm09, recombinant from Baculovirus produced in Trichoplusia ni insect larvae, NR‐34587; H1 Hemagglutinin Protein with C‐Terminal Histidine Tag from Influenza Virus, A/Czech Republic/32/2011 (H1N1)pdm09, recombinant from Baculovirus produced in Trichoplusia ni insect larvae, NR‐42486; H1 Hemagglutinin Protein with C‐Terminal Histidine Tag from Influenza Virus, A/Christchurch/16/2011 (H1N1)pdm09, recombinant from Baculovirus produced in Trichoplusia ni insect larvae, NR‐42487; H1 Hemagglutinin Protein with N‐Terminal Histidine Tag from Influenza Virus, A/Brisbane/59/2007 (H1N1), recombinant from Baculovirus produced in Sf9 insect cells, NR‐13411; H2 Hemagglutinin Protein from Influenza Virus, A/Singapore/1/1957 (H2N2), recombinant from Baculovirus produced in Sf9 insect cells, NR‐2668; H5 Hemagglutinin Protein from Influenza Virus, A/duck/Laos/3295/2006 (H5N1), is a full‐length glycosylated recombinant protein that was produced in Sf9 insect cells using a baculovirus expression vector system, NR‐13509; H3 Hemagglutinin Protein from Influenza Virus, A/Uruguay/716/2007 (H3N2), is a full‐length glycosylated recombinant protein that was produced in Sf9 insect cells using a baculovirus expression vector system, NR‐15168; H7 Hemagglutinin Protein from Influenza Virus, A/Netherlands/219/2003 (H7N7), is a full‐length glycosylated recombinant protein that was produced in Sf9 insect cells using a baculovirus expression vector system, NR‐2633; H7 Hemagglutinin Protein from Influenza Virus, A/Anhui/1/2013 (H7N9), is a full‐length glycosylated recombinant protein that was produced in expresSF+® insect cells using a baculovirus expression vector system, NR‐45118.
The concentrations of HA proteins were provided by the supplier based on bicinchoninic acid colorimetric assay which determines the total protein concentration in solution. We wanted to verify the concentrations of the HA proteins by an alternative method since the degree of cross‐reactivity to HA proteins is so dependent on concentration. Because we have the sequence of each HA protein we can carry out quantitative amino acid analysis to determine concentration. Both methods gave identical results.
Amino acid analysis was performed according to the method described by Cohen and Michaud.34 Briefly, 10 µl of each protein sample was aliquoted into glass tubes and lyophilized. To these tubes, 300 µl of 6M HCl w/0.1% phenol was added and the resulting solution was heated to 110°C for 48 h in order to hydrolyze the peptide bonds in the sample. Each sample tube was allowed to come to room temperature and then vacuum‐dried to remove the HCl. Each sample was then re‐suspended in 10 mM HCl and 20 µl of the sample was added to 60 µl of 0.2M sodium borate buffer, pH 8.8. To this mixture, 20 µl of 6‐aminoquinoyl‐N‐hydroxysuccinimidyl carbamate in acetonitrile was added to derivatize the amino acids present in the sample. After this addition, the derivatized sample was heated to 55°C for 15 min to convert Tyr byproducts to one form. High‐Performance Liquid Chromatography using an Agilent 1260 series instrument and a Waters AccQ Tag 3.9 I.D. × 150 mm column was used to separate and quantify the derivatized amino acids present in each sample. Quantification was accomplished by UV absorbance at 254 nm.
Passive Immunization to Protect Mice against Lethal Infection with Influenza Virus a PR8/34
Female 6‐ to 8‐week‐old BALB/c mice were divided into 8 experimental groups of 10 mice each. One group received no virus and no IgG. For the other seven groups, the following reagents were inoculated intraperitoneally on one day before, one day after and 3 days after Avertin anesthesia followed by intranasal challenge with 10 LD50 (105 TCID50) of influenza virus A PR8/34, phosphate buffered saline (PBS), 1 mg/inoculation with IgG purified with Protein G from rabbit pre‐immunization serum (Pre‐immune), or IgG similarly purified from rabbits immunized with KLH‐coupled homo‐2‐stranded influenza A immunogens 1A, 3A, 4A, 5A, or 6A. Mice were weighed daily and observed for signs of illness for 14 days. According to our IACUC protocol, any animals that lost more than 15% of body weight or appeared moribund were immediately humanely euthanized.
Acquisition and Analysis of Hemagglutinin Sequences from H1N1, H5N1 and H3N2
All sequences were obtained from http://fludb.org using the protein search utility. Searches were run for virus type A HA (“classical” proteins: 4 HA) protein sequences (Data to Return: Proteins) from all hosts and any date range without specifying geographic groupings or countries. Proteins pH1N1 were included in the search, along with partial sequences. This resulted in 29,848 H1N1 sequences; 5708 H5N1 sequences; and 25,361 H3N2 sequences. A filter was applied to the sequence names to obtain the date of origin, and although we did not filter by date in the analyses presented, we did filter out sequences with ambiguous dates, along with chimeric sequences. The downloaded sequences are available in the files fluH1n1.fasta, fluH5n1.fasta, and fluH3N2.fasta. To obtain a complete analysis of the variability in the full sets of applicable sequences, we wrote a short program in perl, called “flucheck.pl”; this will be made available on our laboratory web site (http://www.evolutionarygenomics.com). The aim of the analysis is to show the variability of short sequence fragments (kmers) by position. This is an alignment‐free analysis, so the kmer positions are relative to one another. The program parameters can be adjusted through a control file, but the most relevant parameters are the minimum kmer count considered, and also the minimum transition count considered to obtain the relative positions. For the H1N1 and H3N2 datasets, these were set to 200, and for the H5N1 dataset (with far fewer sequences) they were set to 100. We note that the highest kmer counts vary along the sequences primarily because not all sequences are full‐length. The kmer length in these analyses is seven (i.e., a 7mer).
Rapid Selection of Potential α‐helical B‐cell Epitopes for Vaccine Development
To avoid a long and costly process of trial and error to the selection of α‐helical B‐cell epitopes for vaccine development, a method was needed to rapidly identify from the deduced amino acid sequences key α‐helical B‐cell epitopes of 28–35 residues, which would be optimal vaccine candidates. We developed a novel computational technology called StableCoil to select the most stable amphipathic α‐helices in any protein that have the potential to form stable coiled‐coil structures. StableCoil is based entirely on experimentally determined physiochemical parameters. These parameters include stability coefficients derived from model coiled‐coils30, 31, 32 and α‐helical propensity values derived from synthetic amphipathic α‐helical peptides.35 When considering a protein sequence of interest, there are seven possible reading frames in which a repeating heptad sequence (abcdefg)n can be found. StableCoil considers all seven possible reading frames and generates predicted stability data for each one. As a result, StableCoil avoids the need for screening large numbers of vaccine constructs by defining the reading frame(s) that contain predicted coiled‐coil sequences and indicating which residues belong to each position in the heptad repeat (abcdefg)n. StableCoil rapidly identifies the region(s) of interest, thus reducing the number of B‐cell epitopes that must be studied experimentally. StableCoil cannot predict which of these amphipathic α‐helices will produce neutralizing antibodies but the importance of greatly reducing the number of helical epitopes as potential vaccine candidates cannot be over emphasized.
We first reported on StableCoil36 and used this algorithm to identify two heptad repeat regions, HR‐N and HR‐C, in the 1255‐residue spike glycoprotein of SARS‐coronavirus37 and then use our templated two‐stranded coiled‐coil technology to elicit neutralizing antibodies to the SARS‐coronavirus.20 We are presently updating StableCoil and will make it available worldwide when it is complete. It turns out, that for emerging and re‐emerging viruses that enter cells using Class 1 Viral Fusion Proteins, the most stable amphipathic α‐helices capable of forming coiled‐coil structures are the α‐helices involved in the major conformational change to induce fusion of the virus envelope with host cell membranes and initiate virus entry.
RESULTS AND DISCUSSION
The initial goal of this project was to show that we could take helical amino acid sequences from the trimeric hemagglutinin (HA) protein, insert them into a conformation‐stabilized two‐stranded α‐helical coiled‐coil template and use these immunogens to generate polyclonal antibodies in rabbits that bound to native HA proteins. The two‐stranded disulfide‐bridged immunogen is conjugated to a carrier protein (keyhole limpet hemocyanin, KLH) for immunization of rabbits and to bovine serum albumin (BSA) for screening sera for antibody production to the peptide immunogen (Figure 3).
As shown in Figure 1, the helical regions in the stem of HA that we investigated were denoted 5P (421‐448) and 6P (449‐476) colored red and purple, respectively. Figure 4 shows the results of antibodies generated to a 21‐residue sequence of HA 457–477 from WSN/1933 (H1N1) denoted 6A and a 28‐residue sequence of HA 448‐475 from Puerto Rico/8/1934/Mount Sinai (H1N1) denoted 6P inserted into our disulfide‐bridged two‐stranded α‐helical coiled‐coil template. In the case of sequence 6A the disulfide‐bridge was located at the N‐terminal and in the case of sequence 6P the disulfide‐bridge was located at the C‐terminus of the sequence of interest.
Figure 4.
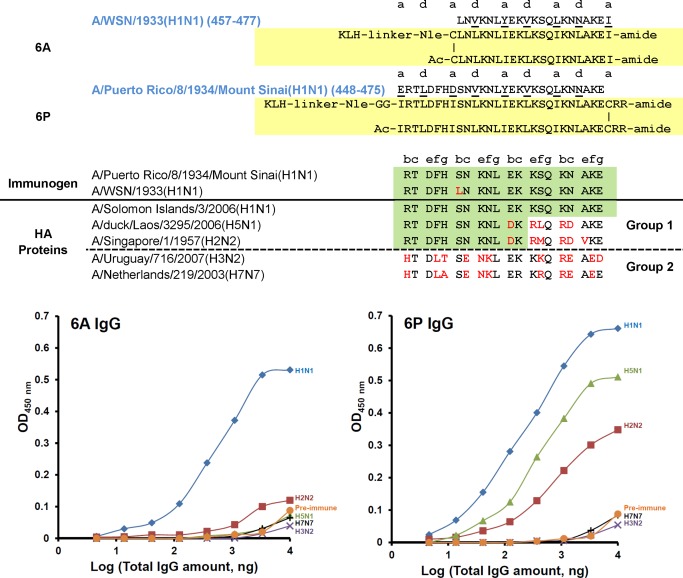
6A and 6P KLH‐conjugated templated immunogens. Top panel: sequence alignment and peptide immunogen constructs (yellow shaded) of 6A and 6P. Middle panel: sequence alignment of H1, H2, H5, H3, and H7 in the region of 6A and 6P, red denotes nonconservative substitutions and green shaded area denotes the highly conserved region between different HA proteins to explain antibody cross‐reactivity. Bottom panel: ELISA of cross‐reactivity of anti‐6A (bottom left panel) or anti‐6P (bottom right panel) IgG against five different HA proteins. Coated HA proteins are from influenza A viruses: H1N1, A/Solomon islands/3/06 (NR‐15170); H2N2, A/Singapore/1/57 (NR‐2668); H5N1, A/Laos/3295/06 (NR‐13509); H3N2, A/Uruguary/716/07 (NR‐15168); H7N7, A/Netherlands/219/03 (NR‐2633). Negative control was the IgG isolated from pre‐immune sera (H1N1 HA protein coated on the plate).
The coiled‐coil template replaces the native hydrophobic core residues at positions a and d with Ile at position a and Leu at position d. These changes not only stabilize the resulting coiled‐coil structure, but ensure that the coiled‐coil is two‐stranded.26, 27, 28, 29, 30, 31, 32, 33 A disulfide‐bridge between the two strands can further stabilize the two‐stranded coiled‐coil immunogen. We previously demonstrated that the contribution of disulfide bonds to protein stability was dependent on whether the disulfide bond was at position a or d in the hydrophobic core of the coiled‐coil. A disulfide bond at any position d (near‐optimal geometry) makes the largest contribution to coiled‐coil stability.38 The only a position that contributes significantly to coiled‐coil stability is a disulfide bond inserted at the N‐terminal where the increase in stability is similar to a d position at the C‐terminal end of the coiled‐coil.38 We also showed that the hydrophobic interactions at position a and d are less important at the ends of coiled‐coil and the ends of coiled‐coil are more flexible.27 Thus, a disulfide‐bond at the ends of coiled‐coil prevents end‐fraying, stabilizes the coiled‐coil structure and the helical conformation at the ends of the two‐stranded coiled‐coil better mimicking the helical‐structure of the native protein.
The immunogen is coupled to the protein carrier keyhole limpet hemocyanin (KLH) and used to immunize rabbits (Figure 3). The resulting IgG purified from rabbit sera was evaluated for binding and cross‐reactivity to HA proteins from five Influenza A virus subtypes H1N1, H5N1, and H2N2 from Group 1 and H3N2 and H7N7 from Group 2. In the case of immunogen 6A, the antibodies were very specific for H1N1 with no significant binding to HA of H2N2, H5N1, H3N2, and H7N7 (Figure 4) The results show that the extension of the sequence from 21 to 28 residues for 6P enhanced the cross‐reactivity of the antibodies to Group 1 sequences represented by H1, H5 and H2. Sequence alignment clearly shows why the 6P antibodies do not cross‐react with Group 2 sequences represented by H3 and H7 (Figure 4). The sequence differences between Group 1 and Group 2 are dramatic in this region. These results also show that sequence identity for two heptads (14‐residues) is enough to generate cross‐reactive antibodies to H1, H5, and H2 HA‐proteins indicated by the green highlight (Figure 4).
Figure 5 shows the results of antibody cross‐reactivity from two immunogens in the region 421–448. Antibodies generated to immunogen 5A covering 23‐residues of the native sequence in this region demonstrated cross‐reactivity to both Group 1 and Group 2 HA‐proteins (H1N1, H2N2, and H5N1 from Group 1 and H3N2 from Group 2). These 5A antibodies showed no cross‐reactivity to H7 HA protein from Group 2. Extension of the sequence from 5A to 5P resulted in a 28‐residue sequence with greater sequence identity as indicated by the green shaded area (Figure 5). The results show that only 14‐residues of sequence identity in the immunogen between Group 1 and Group 2 are enough to generate antibody cross‐reactivity to Group 2 HA proteins (represented by H3N2 and H7N7 subtypes). These results demonstrate that one 28‐residue immunogen can generate cross‐reactive antibodies that bind Group 1 and Group 2 HA‐proteins thus showing the potential of a “Universal” synthetic peptide vaccine. It is interesting that we were able to add two additional Arg residues at the C‐terminus of the immunogen sequence 5P and 6P as solubility enhancing residues without preventing the generation of cross‐reactive antibodies to the native HA proteins.
Figure 5.
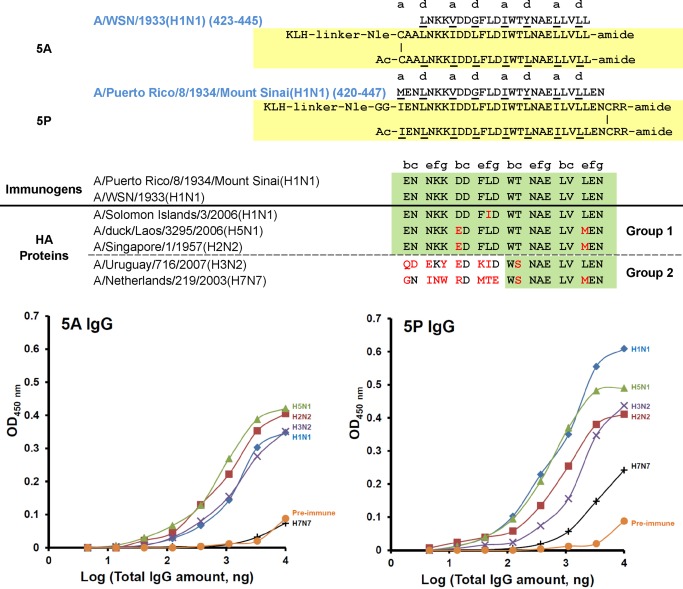
5A and 5P KLH‐conjugated templated immunogens. Top panel: sequence alignment and peptide immunogen constructs (yellow shaded) of 5A and 5P. Middle panel: sequence alignment of H1, H2, H5, H3, and H7 in the region of 5A and 5P, red denotes non‐conservative substitutions and green shaded area denotes the highly conserved region between different HA proteins to explain antibody cross‐reactivity. Bottom panel: ELISA of cross‐reactivity of anti‐5A (bottom left panel) or anti‐5P (bottom right panel) IgG against five different HA proteins. Coated HA proteins are from influenza A viruses: H1N1, A/Solomon islands/3/06 (NR‐15170); H2N2, A/Singapore/1/57 (NR‐2668); H5N1, A/Laos/3295/06 (NR‐13509); H3N2, A/Uruguary/716/07 (NR‐15168); H7N7, A/Netherlands/219/03 (NR‐2633). Negative control was the IgG isolated from pre‐immune sera (H1N1 HA protein coated on the plate).
In our antibody binding studies to HA proteins, we wanted to know if glycosylation of the HA proteins would affect antibody binding. To address this concern we have used H1 hemagglutinin from influenza virus A/Solomon Island/3/2006 (H1N1) which is a full length glycosylated recombinant protein that was produced in insect cells using a baculovirus expression vector system.39, 40 As shown in Figures 4 and 5, our antibodies bind to the fully glycosylated H1 protein from Solomon Islands. The N‐glycosylation sites in H1N1 influenza viruses have been determined.41 In the stem region, residue 304 and 498 are glycosylated and highly conserved but distant from our antibody binding region 421–448. The analysis of HA sequences of human H1N1 virus isolates from 1918 to 2010 has revealed a gradual increase in glycosylation over time and the specific glycosylation sites responsible for virulence and antigenic properties have been identified.42 Fortunately, glycosylation seems to leave the region of interest for our vaccine untouched probably because glycosylation would prevent this region from undergoing the conformational change necessary for membrane fusion and virus entry.
Because we wanted to learn whether antibody binding to HA proteins would also protect mice from virus challenge, we carried out passive immunization of mice with the rabbit antibody to immunogen 5A shown in Figure 5. The results in Figure 6 show that passive immunization with antibody to 5A protected 60% of the mice from intranasal challenge with 10 LD50 of influenza virus H1N1 PR 8/34. Taken together, the ELISA cross‐reactivity and mouse protection data suggest that our vaccine would likely provide broad cross‐protection against multiple influenza strains with antigenic variants and different subtypes of HA.
Figure 6.
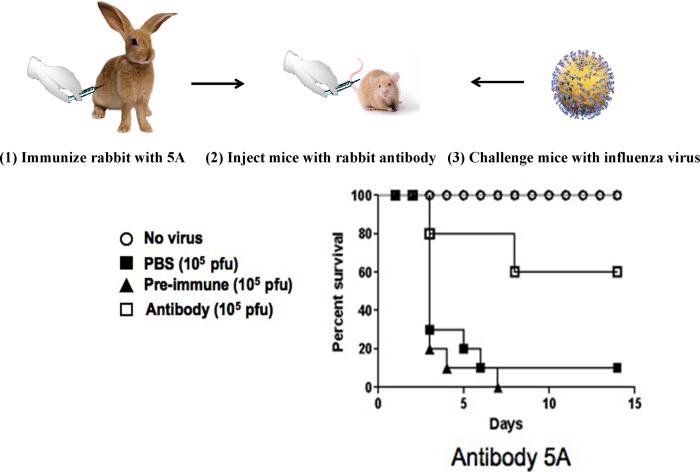
Rabbits were immunized three times with the 5A peptide‐KLH conjugate using Freund's adjuvant. Rabbit IgGs prepared from pre‐ and post‐immune sera, were used to passively immunize Balb/c mice on days −1, +1 and +3 (1 mg/IP inoculation). Mice were challenged intranasally with 10 LD50 PR 8/34 virus on day 0.
We carried out a detailed analysis of all hemagglutinin sequences from H1N1 (29,845), H5N1 (5,708) and H3N2 (25,356). Figure 7 shows the plots of Kmer variant count versus sequence position in the stem domain 300–550 region. The location of sequences of 5P and 6P are colored green and brown and the frequency occurrence of the sequences identified in this analysis are shown in Figure 8. In the 5P region, for H3 one sequence (13,847) has been identified which represents 98% of all sequences identified with occurrences greater than 100; for H5 in the 5P region, only one sequence was identified with a frequency of occurrence of 4,689; for H1 in the 5P region, ten sequences were observed with a frequency of occurrence >100 and with one sequence occurring 14,315 times representing 74% of occurrence. Region 6P is slightly more variable than 5P with 3 major sequences in H3 with a frequency of occurrence at 9019; 2157; and 1,307 representing 88% of occurrences. For H5 in the 6P region, one sequence (3994) represented 86% of occurrences. For H1 in the 6P region, three sequences represented 94% of all occurrences (10,579; 3682; and 2809).
Figure 7.
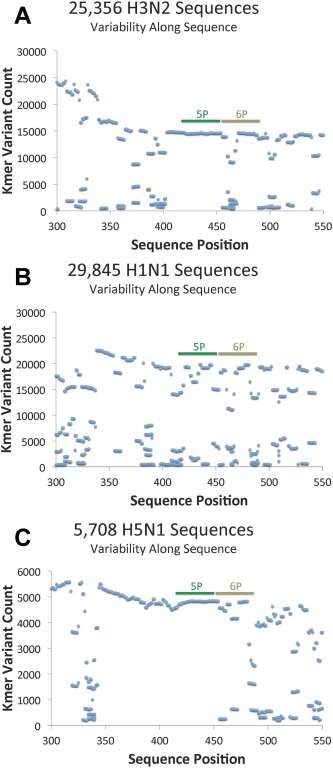
Acquisition and analysis of hemagglutinin sequence from H3N2 (25,356), H1N1 (29,845), and H5N1 (5708) sequences in the stem region residues 300‐550. The results in panel A, B, and C show the variability of short sequence fragments (Kmers) by sequence position. The Kmer length in these analyses is seven (i.e., a 7 mer). See method section (Acquisition and Analysis of Hemagglutinin Sequences from H1N1, H5N1 and H3N2) for more detail. The two regions studied for synthetic peptide vaccine development are 5P (421–448) and 6P (449–476) denoted in green and brown, respectively.
Figure 8.

Alignment of hemagglutinin sequences identified from the analysis shown in Figure 7 for regions 5P and 6P for H3N2, H1N1, and H5N1. We have compared all identified sequences to the sequence with highest occurrence in the 5P and 6P regions. The occurrence of each sequence is indicated. The blue shading in the sequence denotes conservative substitutions (e.g., K/R, I/M, L/I, I/V, Y/F, N/S). The red shading denotes non‐conservative substitutions (e.g., S/F, S/L, K/N). We have not included any sequences with an occurrence <100.
The H1 sequences have remained highly conserved from 1918 (Spanish flu pandemic) to present with very subtle substitutions of conserved residues (e.g., M/I, L/I, V/I, F/Y, and R/K). The H5 sequences are essentially identical from 1959 to present. Similarly, the H3 sequences are extremely conserved since 1968 to present and the major changes observed are K to R or R to K substitutions. Based on this sequence information, it is easy to understand antibody cross‐reactivity from a single peptide immunogen. Because these helices are involved in the major conformational change necessary for membrane fusion and virus entry it is clear why the 5P and 6P sequence remains so highly conserved over time. The existing trivalent vaccine consists of three HA proteins, two from influenza A (H1N1 and H3N2) and one from influenza B. Antigenic variants of these three proteins have been in global circulation since 1977. It is our goal to select the best sequence from this region that provides an immune response in humans where the antibodies protect against all viral strains from Group 1 and Group 2.
The templated disulfide‐bridged two‐stranded peptide immunogens are completely helical in aqueous conditions and no significant α‐helical structure is induced by the addition of the helix inducing solvent, trifluoroethanol (Figure 9 A). Temperature denaturation from 5 to 85°C shows a temperature midpoint of unfolding of 70.6°C demonstrating the exceptional stability of the immunogen, 5P (Figure 9 B).
Figure 9.
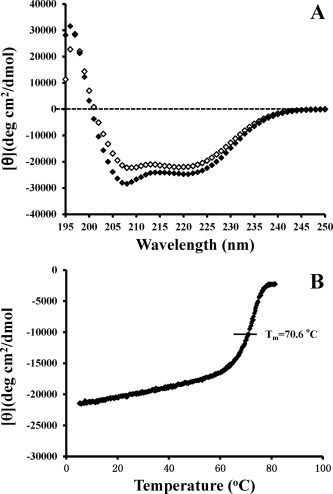
Circular dichroism spectroscopy (CD) results of templated disulfide‐bridged peptide immunogens. Panel A shows the CD spectra of the peptide immunogen, 5P in aqueous benign buffer, pH 7 (50 mM sodium phosphate buffer, 100 mM KCl, pH 7.0) (open symbols) and in the above buffer containing the helix‐inducing solvent 50% trifluoroethanol (closed symbols). Panel B shows the temperature denaturation curve as the temperature is increased from 5 to 85°C for the template two‐stranded α‐helical coiled‐coil with a disulfide‐bridge.
CONCLUSIONS
The templated peptide immunogen technology provides a highly structured and stable peptide immunogen in the α‐helical conformation that can tolerate a wide variety of sequences. This technology can elicit antibodies that are cross‐reactive with HA protein subtypes in the α‐helical regions of the stem domain that are involved in membrane fusion and virus entry. The template was designed for maximum stability by creating a hydrophobic core consisting of Ile at position a and Leu at position d and an interchain disulfide‐bridge at position a at the end of the coiled‐coil. These changes also lock the immunogen into a two‐stranded coiled‐coil structure. It is interesting that the α‐helices on the two‐stranded structure generate antibodies that bind to the α‐helices in the native hemagglutinin three‐stranded coiled‐coil in the stem domain. Our design greatly simplifies the immunogen structure. This technology leaves five out of every seven residues surface‐exposed and mimics the native amphipathic α‐helix in the native protein. Our research suggests that 28‐residue B‐cell epitopes are ideal to generate conformation‐specific antibodies. We also discovered that subtle changes in the design of the peptide immunogens can have dramatic effects on the cross‐reactivity of the resulting antibodies. For example, as shown with immunogens 6A and 6P (Figure 4) we extended the sequence of immunogen 6A by 7‐residues at the N‐terminus and repositioned the disulfide bond from the N‐terminus to the C‐terminus for immunogen 6P. These subtle changes in sequence changed antibody cross‐reactivity from H1N1 specific (Figure 4(6A)) to antibodies that were cross‐reactive to three group 1 HA proteins, including H1N1, H5N1 and H2N2 (Figure 4(6P)). Similarly, when comparing immunogens 5A and 5P (Figure 5) we extended this sequence at the C‐terminal by 2 residues and repositioned the disulfide bond from the N‐terminal of immunogen 5A to the C‐terminus of 5P. The immunogen 5P shows the best cross‐reactivity against Group1 and Group 2 HA proteins and this change improved the cross‐reactivity to H7 hemagglutinin.
The authors are very grateful for the Hemagglutinin (HA) proteins obtained through BEI Resources, NIAID, NIH, which are listed in the Methods section.
This article was originally published online as an accepted preprint. The “Published Online” date corresponds to the preprint version. You can request a copy of any preprints from the past two calendar years by emailing the Biopolymers editorial office at biopolymers@wiley.com.
Author Contributions: Robert S. Hodges planned and organized the study including peptide design and was responsible for the manuscript writing. Ziqing Jiang was involved in peptide synthesis using Fmoc‐chemistry, peptide purification, ELISA evaluation of antibody cross‐reactivity to HA proteins and biophysical characterization of immunogens. Lajos Gera was involved in large scale synthesis of peptide immunogens using Boc‐chemistry. Colin Mant was responsible for the large scale reversed‐phase HPLC purification of peptide immunogens. Brooke Hirsch was involved in preliminary studies on epitope selection which involved small scale peptide synthesis, purification, biophysical characterization and antibody evaluation by ELISA. Zhe Yan developed the methodology for conjugation of peptide immunogens to protein carriers, KLH and BSA, purification of IgG from rabbit polyclonal sera and ELISA evaluation of purified antibodies. Zhaohui Qian and Kathryn Holmes were responsible for the initial in vivo study in mice to demonstrate protection against lethal influenza virus infection by passive immunization with rabbit IgG to one of our peptide immunogens. Jonathan Shortt and David Pollock provided the analysis of all hemagglutinin sequences from H1N1, H5N1 and H3N2.
REFERENCES
- 1.WHO. Available at: http://www.who.int/topics/influenza/en
- 2. Lambert, L. C. ; Fauci, A. S. N Engl J Med 2010, 363, 2036–2044. [DOI] [PubMed] [Google Scholar]
- 3. Gamblin, S. J. ; Haire, L. F. ; Russell, R. J. ; Stevens, D. J. ; Xiao, B. ; Ha, Y. ; Vasisht, N. ; Steinhauer, D. A. ; Daniels, R. S. ; Elliot, A. ; Wiley, D. C. ; Skehel, J. J Science 2004, 303, 1838–1842. [DOI] [PubMed] [Google Scholar]
- 4. Wu, Y. ; Wu, Y. ; Tefsen, B. ; Shi, Y. ; Gao, G. F. Trends Microbiol 2014, 22, 183–191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Nabel, G. J. ; Fauci, A. S. Nat Med 2010, 16, 1389–1391. [DOI] [PubMed] [Google Scholar]
- 6. Ekiert, D. C. ; Bhabha, G. ; Elsliger, M. A. ; Friesen, R. H. ; Jongeneelen, M. ; Throsby, M. ; Goudsmit, J. ; Wilson, I. A. Science 2009, 324, 246–251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Ekiert, D. C. ; Friesen, R. H. ; Bhabha, G. ; Kwaks, T. ; Jongeneelen, M. ; Yu, W. ; Ophorst, C. ; Cox, F. ; Korse, H. J. ; Brandenburg, B. ; Vogels, R. ; Brakenhoff, J. P. ; Kompier, R. ; Koldijk, M. H. ; Cornelissen, L. A. ; Poon, L. L. ; Peiris, M. ; Koudstaal, W. ; Wilson, I. A. ; Goudsmit, J. Science 2011, 333, 843–850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Sui, J. ; Hwang, W. C. ; Perez, S. ; Wei, G. ; Aird, D. ; Chen, L. M. ; Santelli, E. ; Stec, B. ; Cadwell, G. ; Ali, M. ; Wan, H. ; Murakami, A. ; Yammanuru, A. ; Han, T. ; Cox, N. J. ; Bankston, L. A. ; Donis, R. O. ; Liddington, R. C. ; Marasco, W. A. Nat Struct Mol Biol 2009, 16, 265–273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Sui, J. ; Sheehan, J. ; Hwang, W. C. ; Bankston, L. A. ; Burchett, S. K. ; Huang, C. Y. ; Liddington, R. C. ; Beigel, J. H. ; Marasco, W. A. Clin Infect Dis, Infect Dis Soc Am 2011, 52, 1003–1009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Throsby, M. ; Geuijen, C. ; Goudsmit, J. ; Bakker, A. Q. ; Korimbocus, J. ; Kramer, R. A. ; Clijsters‐van der Horst, M. ; de Jong, M. ; Jongeneelen, M. ; Thijsse, S. ; Smit, R. ; Visser, T. J. ; Bijl, N. ; Marissen, W. E. ; Loeb, M. ; Kelvin, D. J. ; Preiser, W. ; ter Meulen, J. ; de Kruif, J. J Virol 2006, 80, 6982–6992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Wrammert, J. ; Koutsonanos, D. ; Li, G. M. ; Edupuganti, S. ; Sui, J. ; Morrissey, M. ; McCausland, M. ; Skountzou, I. ; Hornig, M. ; Lipkin, W. I. ; Mehta, A. ; Razavi, B. ; Del Rio, C. ; Zheng, N. Y. ; Lee, J. H. ; Huang, M. ; Ali, Z. ; Kaur, K. ; Andrews, S. ; Amara, R. R. ; Wang, Y. ; Das, S. R. ; O'Donnell, C. D. ; Yewdell, J. W. ; Subbarao, K. ; Marasco, W. A. ; Mulligan, M. J. ; Compans, R. ; Ahmed, R. ; Wilson, P. C. J Exp Med 2011, 208, 181–193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Julien, J. P. ; Lee, P. S. ; Wilson, I. A. Immunol Rev 2012, 250, 180–198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Krammer, F. ; Pica, N. ; Hai, R. ; Margine, I. ; Palese, P. J Virol 2013, 87, 6542–6550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Bommakanti, G. ; Lu, X. ; Citron, M. P. ; Najar, T. A. ; Heidecker, G. J. ; ter Meulen, J. ; Varadarajan, R. ; Liang, X. J Virol 2012, 86, 13434–13444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Mallajosyula, V. V. ; Citron, M. ; Ferrara, F. ; Lu, X. ; Callahan, C. ; Heidecker, G. J. ; Sarma, S. P. ; Flynn, J. A. ; Temperton, N. J. ; Liang, X. ; Varadarajan, R. Proc Natl Acad Sci USA 2014, 111, E2514–E2523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Osterholm, M. T. ; Kelley, N. S. ; Sommer, A. ; Belongia, E. A. Lancet Infect Dis 2012, 12, 36–44. [DOI] [PubMed] [Google Scholar]
- 17. Kaur, K. ; Sullivan, M. ; Wilson, P. C. Trends Immunol 2011, 32, 524–531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Fiore, A. ; Bridges, C. ; Cox, N. In Vaccines for Pandemic Influenza; Compans R. W.; Orenstein W. A., Eds.; Springer: Berlin, Heidelberg, 2009; pp 43–82. [Google Scholar]
- 19.CDC. Available at: http://www.cdc.gov/flu/avianflu/h5n1-people.htm
- 20. Tripet, B. ; Kao, D. J. ; Jeffers, S. A. ; Holmes, K. V. ; Hodges, R. S. J Struct Biol 2006, 155, 176–194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Yan, Z. ; Hartsock, W. J. ; Qian, Z. ; Holmes, K. V. ; Hodges, R. S. Small Wonders: Peptides for Disease Control; Rajasekaran, Kanniah, et al.; American Chemical Society, Washington, DC, 2012; pp 93–136. [Google Scholar]
- 22. Jiang, Z. ; Gera, L. ; Hartsock, W. J. ; Yan, Z. ; Hirsch, B. ; Mant, C. T. ; Qian, Z. ; Holmes, K. V. ; Kirwan, J. P. ; Hodges, R. S. In Peptides Across the Pacific: Proceedings of the 23rd American Peptide Symposium and the 6th International Peptide Symposium; Lebl M., Ed.; Prompt Scientific Publishing, San Diego, CA, 2013; pp 132– 133. [Google Scholar]
- 23. Bosch, B. J. ; van der Zee, R. ; de Haan, C. A. ; Rottier, P. J. J Virol 2003, 77, 8801–8811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. White, J. M. ; Delos, S. E. ; Brecher, M. ; Schornberg, K. Crit Rev Biochem Mol Biol 2008, 43, 189–219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Milich, D. R. Semin Immunol 1990, 2, 307–315. [PubMed] [Google Scholar]
- 26. Zhou, N. E. ; Kay, C. M. ; Hodges, R. S. Biochemistry 1992, 31, 5739–5746. [DOI] [PubMed] [Google Scholar]
- 27. Zhou, N. E. ; Kay, C. M. ; Hodges, R. S. J Biol Chem 1992, 267, 2664–2670. [PubMed] [Google Scholar]
- 28. Zhu, B. Y. ; Zhou, N. E. ; Kay, C. M. ; Hodges, R. S. Protein Sci 1993, 2, 383–394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Harbury, P. B. ; Zhang, T. ; Kim, P. S. ; Alber, T. Science 1993, 262, 1401–1407. [DOI] [PubMed] [Google Scholar]
- 30. Wagschal, K. ; Tripet, B. ; Hodges, R. S. J Mol Biol 1999, 285, 785–803. [DOI] [PubMed] [Google Scholar]
- 31. Wagschal, K. ; Tripet, B. ; Lavigne, P. ; Mant, C. T. ; Hodges, R. S. Protein Sci 1999, 8, 2312–2329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Tripet, B. ; Wagschal, K. ; Lavigne, P. ; Mant, C. T. ; Hodges, R. S. J Mol Biol 2000, 300, 377–402. [DOI] [PubMed] [Google Scholar]
- 33. Woolfson, D. N. ; Alber, T. Protein Sci 1995, 4, 1596–1607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Cohen, S. A. ; Michaud, D. P. Anal Biochem 1993, 211, 279–287. [DOI] [PubMed] [Google Scholar]
- 35. Zhou, N. E. ; Monera, O. D. ; Kay, C. M. ; Hodges, R. S. Protein Peptide Lett 1994, 1, 114–119. [Google Scholar]
- 36. Tripet, B. ; Hodges, R. S. In Peptides: The Wave of the Future; Proceedings of the 17th American Peptide Symposium; Lebl M.; Houghten R., Eds.; Springer: Netherlands, 2001; pp 365–366. [Google Scholar]
- 37. Tripet, B. ; Howard, M. W. ; Jobling, M. ; Holmes, R. K. ; Holmes, K. V. ; Hodges, R. S. J Biol Chem 2004, 279, 20836–20849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Zhou, N. E. ; Kay, C. M. ; Hodges, R. S. Biochemistry 1993, 32, 3178–3187. [DOI] [PubMed] [Google Scholar]
- 39. Smith, G. E. ; Foellmer, H. G. ; Knell, J. ; DeBartolomeis, J. ; Voznesensky, A. I. 2000. US Patent 6103526.
- 40. Smith, G. E. ; Volvovitz, F. ; Wilkinson, B. E. ; Hackett, C. S. 1998. US Patent 5762939.
- 41. Sun, S. ; Wang, Q. ; Zhao, F. ; Chen, W. ; Li, Z. PloS One 2011, 6, e22844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Sun, X. ; Jayaraman, A. ; Maniprasad, P. ; Raman, R. ; Houser, K. V. ; Pappas, C. ; Zeng, H. ; Sasisekharan, R. ; Katz, J. M. ; Tumpey, T. M. J Virol 2013, 87, 8756–8766. [DOI] [PMC free article] [PubMed] [Google Scholar]