

Abstract
The study of viral molecular genetics has produced a considerable body of research into the sequences and phylogenetic relationships of human and animal viruses. A review of this literature suggests that humans have been afflicted by viruses throughout their evolutionary history, although the number and types have changed. Some viruses show evidence of long‐standing intimate relationship and cospeciation with hominids, while others are more recently acquired from other species, including African monkeys and apes while our line was evolving in that continent, and domesticated animals and rodents since the Neolithic. Viral selection for specific resistance polymorphisms is unlikely, but in conjunction with other parasites, viruses have probably contributed to selection pressure maintaining major histocompatibility complex (MHC) diversity and a strong immune response. They may also have played a role in the loss in our lineage of N‐glycolylneuraminic acid (Neu5Gc), a cell‐surface receptor for many infectious agents. Shared viruses could have affected hominid species diversity both by promoting divergence and by weeding out less resistant host populations, while viruses carried by humans and other animals migrating out of Africa may have contributed to declines in other populations. Endogenous retroviral insertions since the divergence between humans and chimpanzees were capable of directly affecting hominid evolution through changes in gene expression and development. Yrbk Phys Anthropol 46:14–46, 2003. © 2003 Wiley‐Liss, Inc.
Keywords: infectious disease, hominids, paleoepidemiology
Glossary
Acute life strategy: A strategy (similar to r‐selection) in which a virus increases fitness by increasing its reproductive rate (offspring produced per parent generation). Often disease‐associated, it is characteristic of viruses with high mutation rates and the ability to infect multiple host species. Viral transmission tends to be horizontal and is more dependent on host population structure and density than it is for viruses using a persistent life strategy (Villarreal et al., 2000).
Apparent competition: The process by which the sharing of a common enemy (a predator or pathogen) can lead to consequences similar to those of more conventional forms of interspecies competition for limiting resources (Holt and Lawton, 1994).
Coevolution: Reciprocal evolutionary change in interacting species (e.g., host/parasite, predator/prey, plant/herbivore, or mutualism), with changes in the two species caused by mutual selective pressure.
Cospeciation: Parallel speciation of two organisms with a close ecological association (e.g., host and parasite), such that cladograms of the two are congruent.
Endogenous retroviruses: DNA sequences, related to those of infectious retroviruses, that are integrated into host genomes and have lost the ability to cause active infection; thought to be remnants of ancient germ‐cell infections, they have proliferated and evolved by retrotransposition.
Error‐catastrophe threshold: The level of critical‐copying fidelity below which (i.e., the base substitution rate above which) viral genetic information can no longer be maintained and nucleotide sequences would become essentially random (Domingo and Holland, 1994).
Gene capture: De novo gene acquisition via recombination of a viral genome with that of the host or another virus.
Host‐linked evolution: A form of cospeciation, in which well‐adapted viruses living in single hosts, through ancient association with them, have diverged and speciated along with their hosts. Isolation of virus populations and selection pressure exerted by the host are important factors in this process, which results in a correlation between virus and host phylogenies (Chan et al., 1997).
Hyperdisease: An extremely lethal disease introduced into a region by a carrier species that is relatively unaffected by it; at the same time, it is capable of infecting a variety of other species with high mortality rates and has the potential for causing their extinction (MacPhee and Marx, 1997).
Modular evolution: Acquisition of new active genes by recombination of specific nucleic acid sequences or functional domains within genes; common in viral evolution, with recombination occurring both between and within viral taxa.
Parasite‐mediated competition: A form of apparent competition, by which the sharing of a pathogen by two species can result in population decline, even extinction, of one of the hosts, even in the absence of resource competition (Holt and Pickering, 1985).
Persistent life strategy: A strategy in which a virus increases fitness by increasing the survival time of its offspring, thus enhancing persistence in an individual host and the probability of transmission over time. Characteristic of genetically stable viruses that show cospeciation with their hosts, this strategy is often associated with vertical or sexual transmission. Latent infection, with the capacity of the virus to reactivate, is a frequent result (Villarreal et al., 2000).
Quasispecies: A virus population of closely related but distinct genetic variants resulting from an error‐prone replication process (typical of RNA viruses, in particular); a self‐sustaining population of sequences that reproduce themselves imperfectly but well enough to retain a collective identity over time, but which also contain the potential to produce more virulent strains (Eigen, 1993).
Zoonosis: An infection or infectious disease transmissible under natural conditions from other vertebrate animals to humans; most zoonoses are dead‐end infections with little or no transmission among human hosts.
INTRODUCTION
The study of human evolution has concentrated on humans and their hominid1 ancestors, without as much attention to other organisms also evolving in the same environments. But a population must constantly interact with and adapt to these other organisms if it is to survive and reproduce. Food species, predators, and agents of infectious disease have all played a role in human evolution, and among the latter, viruses were probably particularly important. As significant causes of morbidity and mortality, and in their capacity to act as “molecular genetic parasites” (Luria, 1959), viruses are in a strong position to influence the evolution of their hosts (May, 1995; Villarreal, 1999; Balter, 2000). While this is well‐recognized in studies of the evolution of other organisms (especially plants, e.g., Thompson and Burdon, 1992; Simms, 1996), the impact of infectious disease on human evolution has not received the attention it deserves (Swedlund, 2000). Yet viral parasites have probably played an important role in human evolution (de Souza Leal and Zanotto, 2000). The purpose of this paper is to review recent work in the molecular genetics of human viruses and to assess the extent to which viruses were significant parasites of earlier hominids (and thus in a position to have affected human evolution before the Neolithic). It also speculates on the roles they may have played.
Haldane (1949) was among the first to suggest that the struggle against infectious disease was an important evolutionary process, and he described some possible effects. A disease that kills or lowers fertility is a likely selective agent. It can be an advantage or a disadvantage to a species in competition with others, even contributing to extinction. Most species contain considerable genetic diversity in their resistance against disease, and considering the speed with which new pathogens evolve (in general much faster than the host), it is in the best interests of the host to be genetically diverse and highly mutable in the loci concerned with disease resistance. In the view of Haldane (1949), disease may even favor speciation, by coupling this diversity and mutability with geographic isolation. In addition, transmission requirements may have contributed to host population size and structure and behavioral characteristics such as negative reaction to fecal odors.
As the scientific community became more aware of “surprising biochemical diversity” (Haldane, 1949, p. 329) in virtually every animal investigated, geneticists sought to explain it. Estimates of the proportion of polymorphic loci ranged from 20–40% in vertebrates and about 30% in humans (Selander et al., 1970). Some suggested that interactions between parasites and hosts played an important role in maintaining this degree of polymorphism (Clarke, 1976). Hamilton et al. (1990) suggested that the need for genetic diversity in a “host‐pathogen arms race” even contributed to the evolution of sexual reproduction. We now know that this level of polymorphism (30%) is probably an underestimate, based on incomplete sampling of rare mutations. Given the frequency of DNA polymorphism (about one in every 500 nucleotides), nearly all genes may be polymorphic (Cavalli‐Sforza et al., 1994). The human genome is now estimated to contain between two and three million single‐nucleotide polymorphisms (SNPs), and even though less than 1% are estimated to result in variation in proteins, this still leaves enough to involve many more than 30% of the estimated 20,000–30,000 human genes (International Human Genome Sequencing Consortium, 2003; Venter et al., 2003).
Disease‐driven selective pressure is now considered a likely reason for the astonishing degree of polymorphism in the major histocompatibility complex (MHC), which codes for membrane glycoproteins [also known as human leukocyte antigens (HLA) in humans] that play an important role in the immune system by binding fragments of infectious origin and presenting them to T‐cells (Zinkernagel et al., 1985; Howard, 1991). The importance of maintaining this first line of defense against invading pathogens is emphasized by the discovery that some polymorphic alleles in the MHC system predate the divergence between chimpanzees and humans, and have been transmitted through speciation bottlenecks via “trans‐species selection” (Takahata, 1990). While such positive selection is still believed to have maintained many MHC allelic lineages, the sharing of MHC polymorphisms by different mammalian orders and even some within the primates (e.g., between humans and New World monkeys) may be the result of convergent evolution (Yeager and Hughes, 1999; Kriener et al., 2000).
Zoologists now recognize that infectious diseases may mediate “apparent competition” between species and augment the danger of extinction (Hudson and Greenman, 1998; Daszak et al., 2000). Some have even suggested a “hyperdisease” scenario, involving infections carried by migrating humans or their dogs, to explain Pleistocene megafaunal extinctions (MacPhee and Marx, 1997).
Yet another way in which pathogens can affect the evolution of their hosts is through direct interaction with host DNA. By virtue of their simplicity and their need to use host‐cell replication and transcription machinery, viruses act as “molecular genetic parasites” (Luria, 1959) and may alter host genomes through such mechanisms as recombination, retrotransposition, and gene conversion. While lasting effects are more frequent in unicellular hosts and plants, ancient germ‐cell infections by retroviruses have left their mark on human and other primate genomes (Sverdlov, 2000).
Before one can judge the likelihood of any of these processes contributing to human evolution, one must determine whether our hominid ancestors suffered significantly from infections, whether they could have been significant causes of morbidity or mortality (and thus agents of selection), and what kinds of parasites may have been involved.
The study of human disease history has tended to assume an epidemic disease model and a focus on human ecology and population history, with most infectious diseases considered to have evolved since the increased size and concentration of human populations in the Neolithic (Cockburn, 1967b; Burnet and White, 1972; Armelagos and Dewey, 1978). The impact of parasitism before that time was not considered very important, as it appeared to be restricted to chronic latent infections unlikely to cause serious disease and the occasional zoonosis (disease of another animal species). Meanwhile, the influence of disease on human affairs since the Neolithic was acknowledged to have been considerable (McNeill, 1976). Humans were assumed to have had few viral diseases before the Neolithic (Burnet and White, 1972), but studies of antibodies in isolated South American tribes suggested that small isolated groups had their share of infections (Neel et al., 1964, 1968; Black, 1975), and Cockburn (1967a) recognized the presence of many viruses in wild primates and thus, probably, in our ancestors. Perhaps infectious agents, and viruses in particular, were more important than we thought.
Recent advances in molecular virology have provided a gold mine of information about the evolutionary history of the most important human viruses, and this allows one to decipher when they might have entered our line (Zimmer, 2001). Deep phylogenetic branches, high genetic diversity, global distributions, and trees showing cospeciation with primate hosts all suggest ancient association of many viruses with humans, while close relationships with viruses infecting other species (especially rodents and domesticates) suggest more recent acquisition of others. The ease with which our species acquires emerging infections from other primates points to the importance of these zoonoses as a source of human disease, both now and in the past. When mapping the evolutionary relationships of human and animal viruses, one obtains a different picture depending on whether the viruses in question are DNA‐based, RNA‐based, or replicated using reverse transcriptase, so these types are discussed separately.
THE EVOLUTION OF HUMAN VIRUSES
Animal viruses are grouped into a number of families, based on the nature of the viral genome (DNA or RNA, single‐ or double‐stranded, positive or negative strand) and how it is replicated (Table 1). These factors determine many of their basic properties, including modes and rates of change, which in turn influence their histories of association with hominids and possible effects on human evolution.
Table 1.
Animal virus families
| Family | Description | Evolution |
|---|---|---|
| Adenoviridae | Medium‐sized DNA viruses that cause respiratory and enteric infections in birds and mammals; numerous subtypes in humans and other primates. | Evolved along with warm‐blooded animals; subtypes diverged during evolution of primates (Song et al., 1996). |
| Arenaviridae | RNA viruses, mostly zoonotic and maintained in rodent reservoirs; spread via contact with infected rodent secretions, e.g., Lassa fever and lymphocytic choriomeningitis viruses. | Long‐term coevolution with rodent hosts; hominids unlikely to have encountered them before agriculture and permanent houses (Bowen et al., 1997). |
| Astroviridae | RNA viruses with worldwide distribution in humans and other animals; water‐ and food‐borne; significant cause of diarrheal disease in developing countries. | Not much known about these yet. |
| Bunyaviridae | Among larger RNA viruses, these are zoonotic and emerging; mostly arthropod‐borne (e.g., Rift Valley fever, California encephalitis, Crimean‐Congo hemorrhagic fever), except for hantavirus. | Originated in insects and coevolved with them; hantavirus phylogeny shows coevolution with rodents and host‐switching (Zhao and Hay, 1997; Vapalahti et al., 1999). |
| Caliciviridae | RNA viruses with worldwide distribution in humans and other vertebrates; common cause of diarrhea in children; transmitted via contaminated food and water or uncooked shellfish. | Sequences show geographic similarities that transcend host relationships; readily move across species barriers; passed back and forth between humans and domesticated animals (van der Poel et al., 2000). |
| Coronaviridae | Largest RNA viruses, infecting humans, cattle, pigs, rodents, cats, dogs, and chickens; common cause of colds in humans and a variety of conditions in other animals, primarily enteric and respiratory; CV‐like particles found in stools of adult chimps, macaques, baboons, and marmosets. | Antigenic drift and recombination constantly produce new strains; interspecies spread leads to episodic evolution (Lai, 1995). |
| Filoviridae | RNA viruses that infect only vertebrates; contains two species (Ebola and Marburg) that cause acute fatal disease in humans and other primates; reservoirs unknown. | Two species estimated to have diverged 7–8 kya, and Ebola subtypes diversified 1–2 kya (Suzuki and Gojobori, 1997). |
| Flaviviridae | RNA viruses, including numerous arboviruses (e.g., yellow fever, dengue, West Nile, tick‐borne encephalitis), bovine diarrhea, hog cholera, and hepatitis C‐like viruses (HCV, GBV‐A, ‐B, and ‐C); GB‐viruses widely distributed in simians. | Mosquito‐borne viruses originated in Africa, have affected primates for a long time. HCV long‐term presence in Africa as well, but infects only humans; GB‐viruses show cospeciation with primates; GBV‐C had ancient association with humans (Gaunt et al., 2001; Robertson, 2001). |
| Hepadnaviridae | Hepatitis B viruses found in primates, New World rodents, and birds; among smallest viruses; contain partially double‐stranded circular DNA replicated via RNA intermediate and reverse transcriptase. | May have cospeciated with primates; human strains result of ancient interspecies transmission in Africa; now found in isolated human populations in both hemispheres (Simmonds, 2001). |
| Herpesviridae | Large DNA viruses; most vertebrate species have at least one. Three subfamilies alpha‐ (herpes simplex, varicella), beta‐ (cytomegalovirus), and gammaherpesvirinae (Epstein‐Barr, Kaposi's sarcoma). All contain many strains found in other animals, including primates. | Ancient divergence of subfamilies with tissue specificity acquired early, at least 200 mya; have since coevolved in close association with hosts (McGeoch et al., 2000; McGeoch, 2001). |
| Orthomyxoviridae | Influenza subtypes A, B, and C; RNA viruses with segmented genomes that readily reassort, producing pandemic influenza A; wild A strains, maintained in avian hosts, also infect pigs, horses, and other mammals; subtypes B and C infect humans only. | Subtype A appears to be ancient parasite of aquatic birds; avian virus strains are in “evolutionary stasis;” recent and explosive evolution in pigs and humans (Webster, 1997). |
| Papovaviridae | Widespread and numerous viruses with very small circular DNA; include papillomaviruses that cause cutaneous or genital lesions (e.g., warts, genital papilloma) and polyomaviruses (JC and BK viruses, simian virus‐40) that infect kidney cells; highly species‐specific; all mammal, bird, and reptile species so far studied carry species‐specific PVs, with most infected by several types; HPV and JC found in nearly all human populations. | Papilloma species and type diversity suggests a well‐adapted parasite intimately associated with hosts for a long time and cospeciating with them; also coevolved with humans; can be used to trace migrations (Ong et al., 1993; Van Ranst et al., 1995). JC is ubiquitous human pathogen whose genotypes diverged when human populations did (Hatwell and Sharp, 2000). |
| Paramyxoviridae | RNA viruses that include parainfluenza, mumps, morbilliviruses (e.g., measles, distemper, rinderpest), and many other viruses of humans and other animals; include emerging viruses Nipah and Hendra; transmission primarily by airborne droplet. | Animal morbillivirus phylogeny matches that of host species, but human viruses probably resulted from recent interspecies transmission from domesticated animals (Norrby et al., 1992). |
| Parvoviridae | Among smallest, simplest viruses known, with single‐stranded DNA genomes; widespread in many vertebrate and invertebrate hosts; tend to infect rapidly dividing tissues (mostly fetal, intestinal epithelial, and bone marrow). | Very species‐specific; viral evolution tightly linked to that of host (Shadan and Villarreal, 1993), but not much known about human parvoviruses. |
| Picornaviridae | Several genera of RNA viruses: Aphthovirus (foot‐and‐mouth disease), Cardiovirus (encephalomyocarditis), Enterovirus (Coxsackie, echo, polio), Hepatovirus (hepatitis A), Parechovirus, and Rhinovirus (colds); last four genera infect mainly humans, with domesticated animal viruses probably derived from human strains. | Diverse and numerous simian viruses are all from Old World primates and are closest known relatives of human enteroviruses; some picornaviruses may have been long associated with humans (Gromeier et al., 1999). |
| Poxviridae | DNA genome; largest and most complex viruses known; vertebrate subfamily includes large number of poxviruses infecting many animals (e.g., variola, vaccinia, cowpox, monkeypox, camelpox, fowlpox, sheep‐and‐goatpox, swinepox) plus molluscum contagiosum virus of humans. Many (e.g., cowpox, monkeypox) are maintained in rodent reservoirs. | Origins of variola (smallpox) unclear; may be ancient hominid virus (like molluscum contagiosum) that recently became more virulent, or may be more recently evolved from African rodent virus (Fenner et al., 1988; Tucker, 2001). |
| Reoviridae | “Respiratory enteric orphan” viruses, with double‐stranded RNA genomes; ubiquitous in nature, implying wide cell tropism, ubiquitous receptor; infect vertebrates (mammals, birds, reptiles, fish), invertebrates (insects, molluscs), and plants; found in all mammals except whales; human varieties include rotaviruses, common cause of severe diarrhea worldwide; major cause of mortality in young of many species; include many emerging viruses that cause hemorrhagic fever, encephalitis, Colorado tick fever. | Rapid evolvers; members of various genera among most evolutionarily divergent RNA viruses (Duncan, 1999); frequent interspecies transmission and reassortment of segmented genome give different trees for different genes and close relationship between human and nonhuman rotaviruses (Cunliffe et al., 1997). |
| Retroviridae | RNA viruses that reproduce using reverse transcriptase and a DNA intermediate; subgroups include lentiviruses (e.g., HIV, SIV); primate T‐cell lymphotropic viruses (PTLV); endogenous retroviruses (ERV); in wide variety of vertebrates, and can jump host species, sometimes across wide phylogenetic distances | Great antiquity; retroviruses and retroid elements found in all eukaryotes; infectious retroviruses found only in vertebrates and may have evolved from retrotransposons; extremely fast mutation rates, but can also be very stable (e.g., as integrated provirus); some SIVs show evidence of cospeciation with hosts, while others are result of recent interspecies transmission (HIV‐1 from SIVcpz and HIV‐2 from SIVsm); HTLV‐I and ‐II likewise from STLVs of Asian macaques and bonobos, respectively (Holmes, 2001; Salemi et al., 2000). |
| Rhabdoviridae | RNA viruses that include genus Lyssavirus (rabies) and vesicular stomatitis virus; there are no specifically human rhabdoviruses, but rabies is one of most dangerous zoonoses. | Very ancient family; contains members that infect animals and others that infect plants; rabies appears derived from lyssaviruses of African bats (Amengual et al., 1997). |
| Togaviridae | RNA viruses, mostly mosquito‐borne and in genus Alphavirus, including a number of zoonotic viruses maintained in rodent and bird reservoirs (e.g., Eastern and Western equine encephalitis, Semliki Forest virus); also genus Rubivirus (rubella), which is not vector‐borne and infects only humans. | Evolved in insects; contain segments from plant viruses via recombination in insect hosts (Lai, 1995). Origin of rubella not known. |
DNA viruses
DNA viruses include both the largest (poxvirus) and smallest (hepatitis B) animal viruses, and there is some variability in the characteristics of this group. All except the Parvoviridae have double‐stranded DNA genomes, like the cells they infect. This similarity in structure and replication gives DNA viruses a very intimate molecular relationship with their hosts, especially viruses that replicate in the nucleus, which are capable of recombination with host genomes and horizontal gene transfer (Morse, 1994; Villarreal, 1999). DNA viruses tend to be more species‐specific, with narrower host ranges than RNA viruses. Mutation rates are slower than those of RNA viruses, on the order of only 10–100 times as fast as host rates. Many have adopted a “persistent life strategy” whereby the virus increases fitness by increasing the survival time of its offspring, thus increasing probability of transmission. This strategy results in chronic, latent infections with long periods of infectivity and the ability to persist in small host populations (although “persistent” refers to persistence in an individual host; Villarreal, 1999). DNA viruses thus tend to be stabler and older than RNA viruses, and their phylogenies often show a pattern of cospeciation with their hosts.
A word of caution, however: one must be careful applying molecular clocks to viral phylogenies; good molecular clocks cannot be assumed (Gibbs, 1987; Simmonds, 2001). Viral evolutionary rates are not constant, and even if they were, high mutation rates lead to underestimation of genetic distances due to homoplasies, reversions, and multiple substitutions at a single site (Brown, 1994). This is particularly problematic for rapidly mutating RNA viruses (more below), but must also be a concern when estimating timescales for the deeper branches on DNA viral trees (McGeoch and Davison, 1995; McGeoch et al., 1995). But in general, DNA viruses, with their slower mutation rates, yield clearer evolutionary relationships extending further back in time.
Early hominids most likely carried several kinds of DNA viruses, from the families Herpesviridae, Papovaviridae, Adenoviridae, and Parvoviridae. Most animals carry members of these groups, and our ancestors would have been no exception.
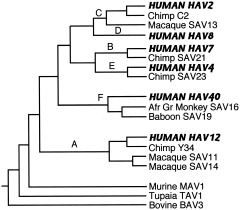
The mammalian herpesviruses have been especially well‐studied, and we now have a detailed picture of their evolution (McGeoch and Davison, 1999a; McGeoch et al., 2000; McGeoch, 2001; Davison, 2002). These large, complex DNA viruses have mutation rates comparable to those of cellular DNA. Observed mutation rates for alphaherpesviruses are compatible with estimated rates based on cospeciation with their hosts, so assumption of a fairly constant molecular clock appears warranted for this group (McGeoch and Davison, 1995). A robust phylogeny of 48 viruses has been constructed, using amino‐acid sequences of eight viral genes and maximum‐likelihood evaluation of sets of candidate trees to create composite trees (for details of the method, see McGeoch et al., 2000). Figure 1 shows part of this phylogeny, concentrating especially on the primate herpesviruses.
Figure 1.
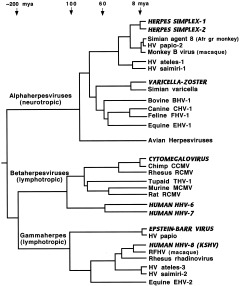
Phylogeny of mammalian herpesviruses. Composite tree based on amino‐acid sequences from eight genes. Viruses that regularly infect humans are indicated by bold amd italicized capitals. KSHV, Kaposi's sarcoma herpesvirus; RFHV, retroperitoneal fibromatosis herpesvirus of macaques; Afr gr, African green. Sources: McGeoch and Davison, 1999a; McGeoch et al., 2000; McGeoch, 2001.
It is an ancient group, with representatives in virtually all vertebrates (and at least one invertebrate). The family split into three branches with different tissue tropisms (alpha‐, beta‐, and gammaherpesviruses) around the time that mammals diverged from reptiles. Each branch further subdivided into at least two major sublineages before the mammalian radiation of 60–80 mya and subsequently cospeciated with their hosts (McGeoch et al., 1995). Alphaherpesviruses show a tight correlation with phylogenies of primates, ungulates, and carnivores, while betaherpesviruses cospeciated with primates and rodents, although few sequences and hosts are as yet known for the group containing HHV‐6 and HHV‐7 (McGeoch et al., 2000). Similarly, we need more data on other primate viruses in the gamma‐1 sublineage containing Epstein‐Barr virus, but the gamma‐2 sublineage contains many viruses that have cospeciated with primates (McGeoch, 2001). Members of the HHV‐8 subgroup have been found in two species of macaques, African green monkeys, chimpanzees, and gorillas, and these viruses show the expected host‐linked phylogenetic relationships (Greensill et al., 2000a, b; Lacoste et al., 2000). Thus it appears that hominids, and perhaps all anthropoids, have been affected by viruses in each of these sublineages throughout their evolutionary history.
An interesting detail of herpesvirus evolution is the differentiation of herpes simplex virus (HSV) into two distinct types, oral and genital (HSV‐1 and HSV‐2, respectively), about 8 mya (McGeoch et al., 1995). As the type 1 alphaherpesviruses of nonhuman primates infect both oral and genital tissues without any differentiation into separate strains, this suggests microbiological isolation of the two body areas in the hominid line. Some have proposed changes in sexual behavior as an explanation, with continual female sexual attractiveness and adoption of face‐to‐face mating in the line leading to hominids (Gentry et al., 1988). It might reflect increased distance between these two areas with the adoption of bipedalism, although 8 mya is a bit too early, even considering the recent discovery of a 6–7‐million‐year‐old putative hominid (Brunet et al., 2002), and 8 mya for the divergence of the two HSV types is likely to be an underestimate. Like several other viruses able to persist in small populations, HSV‐1 is most commonly acquired in early childhood, from mother to infant via infected saliva. HSV‐2 is usually acquired in adulthood through sexual contact, and both viruses can be transmitted by the oral‐genital or oral‐anal route (Benenson, 1995). Perhaps there were changes in social behavior and gestures that resulted in less frequent oral‐genital and oral‐anal contact, especially between mother and infant. Related viruses in African apes remain to be discovered, and further reflection on this point should probably wait until we know whether they indeed lack different oral and genital strains.
Sequence variation within human herpesvirus strains uncovers relationships between viral genotypes and ethnic groups reflecting Paleolithic migrations (HSV‐1, Umene and Sakaoka, 1999; HHV‐8, Hayward, 1999). HHV‐8 is particularly interesting, as highly divergent sequences at either end of one strain's genome suggest recombination about 100 kya with some unknown primate virus (XXX in Fig. 2), and a second more recent recombination about 35 kya in Europe or the Mideast (Hayward, 1999; McGeoch and Davison, 1999b; McGeoch, 2001). The timing and location of this second event are suggestive, inviting speculation that the source of the presumed ancestral MMM genotype may have been a Neandertal (Hayward, 1999). Perhaps more likely is that this ancestral virus was carried by one of many lineages of modern humans that died out.
Figure 2.
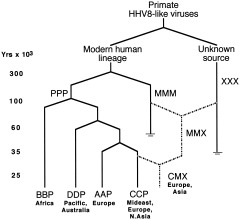
Recombination in human herpesvirus‐8. Capital letters (e.g., PPP, BBP) refer to alleles (ORF‐K1, ORF‐75, ORF‐K15) carried by viral subtypes. PPP, original intact form of HHV‐8. MMM, hypothesized older variant carrying M‐associated ORF‐75 allele. XXX, unknown exotic HHV8‐like virus with highly diverged ORF‐K15 allele. AAP, BBP, CCP, and DDP represent variants with different alleles of ORF‐K1. Dashed lines indicate possible recombination events. Reprinted from Hayward (1999), Fig. 5, with permission from Elsevier.
The Papovaviridae present a phylogeny similar to that of the herpesviruses, with ancient branching points and evidence of cospeciation (Ong et al., 1993; Shadan and Villareal, 1993; Van Ranst et al., 1995; Chan et al., 1997). This family of very small DNA viruses makes maximal use of minimal genomes by using overlapping reading frames on both DNA strands, thus putting a selective constraint on its ability to mutate and resulting in slow rates of evolution, on the same order as cellular DNA (Chan et al., 1992; Hatwell and Sharp, 2000). It contains two genera, Papillomavirus and Polyomavirus, which infect a wide variety of vertebrates and are highly species‐specific. Figure 3 shows the relationships of a few of the many viruses in this family. Again one sees deep branches with different tissue tropisms and long‐term cospeciation with hosts. While nonhuman primate viruses cluster with human viruses according to tissue tropism rather than phylogeny of the host, within these tropic subgroups, viral relationships mirror host phylogeny (Van Ranst et al., 1995). The high degree of species and type diversity within this family suggests a well‐adapted pathogen that has been closely associated with its hosts for a long time (Chan et al., 1992). Molecular clock estimates are not as robust for these viruses, however, as most have not been as intensively studied as the herpesviruses.
Figure 3.
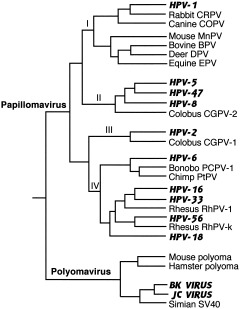
Phylogeny of mammalian Papovaviridae. Composite tree constructed from phylogenies in Ong et al. (1993), Van Ranst et al. (1995), Chan et al. (1997), and Shadan and Villareal (1993). Viruses that regularly infect humans are indicated by bold amd italicized capitals. Papilloma subtypes I and II infect cutaneous tissue and include viruses that cause warts and other skin conditions; subtype IV infects mucosal tissue and includes malignant genital papillomaviruses HPV‐16 and ‐18; subtype III contains viruses that infect both types of tissue. Polyoma viruses infect kidney cells.
Like herpesviruses, papovaviruses produce inapparent chronic infections that have allowed them to persist in small populations and migrate with them. Both papilloma and polyoma genotypes reflect human geographic and ethnic groups (Fig. 4; Ho et al., 1993; Ong et al., 1993; Van Ranst et al., 1995). JC virus has proven particularly useful for tracing modern population relationships, as it is a ubiquitous human pathogen causing lifelong asymptomatic infection in a large fraction of the population and is generally transmitted through close contact of mother to child. Thus it is a population marker almost as definitive as mtDNA and can be used to reconstruct prehistoric human migrations (Jobes et al., 1998; Hatwell and Sharp, 2000; Stoner et al., 2000; Sugimoto et al., 2002). In addition, analysis of 12 human papillomavirus types found limited diversity, reflecting a severe pruning of the HPV tree and suggesting an evolutionary bottleneck in both virus and host approximately 100 kya (Stewart et al., 1996).
Figure 4.
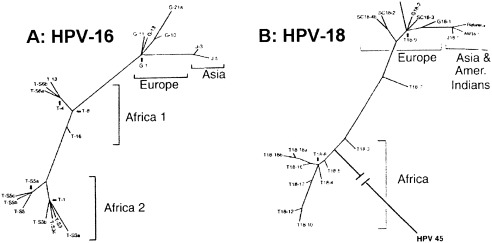
Intratype diversity in papillomaviruses 16 (A) and 18 (B). Phylogenetic trees of HPV16 and HPV18 variants from patients from Africa (Tanzania), Europe (Germany and Scotland), East Asia (Japan), and America (Mundurucu Indian, Brazil), based on analysis of 364‐bp genomic segments of HPV16 and 321‐bp segments of HPV18. HPV18 reference clone was found in both East Asia and South America. HPV45 is included in HPV18 tree to indicate probable African root. Reprinted from Ong et al. (1993), Fig. 3, with permission from American Society for Microbiology and the authors.
Other DNA viruses that have evolved along with primates include the adenoviruses (Fig. 5), which cause prolonged latent infections in birds and mammals (Bailey and Mautner, 1994; Kidd et al., 1995; and Song et al., 1996). Interpretation of adenovirus relationships is complicated, however, by evidence of recombination in its “virus‐associated RNA” (VA RNA), i.e., small, regulatory RNAs abundant in the cytoplasm of infected cells. Human and chimp strains in subgenera B through E make two classes, RNA‐I and RNA‐II, indicating a duplication of VA RNA genes in a hominoid ancestor (at what point is not clear; little is known about the adenoviruses of other apes), with subsequent diversification of these subgenera (Kidd et al., 1995). Subgenus C, however, has a monkey‐like RNA‐I and chimp‐like RNA‐II, indicating probable recombination between a human adenovirus and the macaque virus SAV13.
Figure 5.
Phylogeny of primate adenoviruses. Composite tree derived from phylogenies based on analysis of VA RNA and DNA polymerase genes (Kidd et al., 1995; Song et al., 1996). A–F are subgenera of primate adenoviruses; branch points between them were derived from comparison of trees from all regions of genome (Bailey and Mautner, 1994). Viruses that regularly infect humans are indicated by bold and italicized capitals. Genetic distances are not to scale.
There is only one known human parvovirus, B19, about which little is understood because it is difficult to grow in culture. Two lines of evidence suggest that it is an old and persistent hominid virus. First, 70–90% of most human populations are seropositive for B19 (Gamble, 1997). Parvoviruses infect a wide variety of vertebrates including primates, but the relationships between B19 and nonhuman primate viruses are not close enough to suggest recent interspecies transmission (Parrish and Truyen, 1999). Study of other species' parvoviruses indicates high species specificity and viral evolution tightly linked to that of the host (Shadan and Villarreal, 1993).
It is probably safe to conclude that early hominids carried viruses from these four families, and that these viruses diversified and migrated along with human populations. Viral phylogenies reflect evolutionary relationships of their hosts, and relatively slow mutation rates enabled them to evolve along with the primates. Production of latent and subclinical infections allowed these pathogens to persist in the small populations that were probably typical of earlier hominids. Viral genotypes reflect migrations of anatomically modern humans, indicating close acquaintance predating our species' African exodus. All the viruses discussed so far replicate in the cell nucleus and use the same DNA‐replicating machinery as the host, thus promoting an intimate evolutionary relationship.
Two other DNA virus families remain to be mentioned: Poxviridae and Hepadnaviridae. While poxviruses appear to be an ancient family that evolved along with its nonhuman hosts (Fenner and Kerr, 1994), smallpox virus, or variola (genus Orthopoxvirus), was probably acquired recently by humans, perhaps during the Neolithic, as it follows an acute strategy in humans (short‐lived, virulent infection in virtually all individuals it infects). It infects only humans, however, so its source is unclear. Its closest relatives are camelpox, gerbilpox, and monkeypox, the latter maintained in a rodent reservoir, in spite of its name (Douglas and Dumbell, 1996). Figure 6 shows some likely relationships among members of this family, but these ultimately depend on where in the genome one looks (Blasco, 1995). The world's remaining stocks of variola have been restricted since the 1970s to two research centers in the US and Russia, so the phylogeny of this virus is not as well‐studied, although researchers at the Centers for Disease Control and Prevention (CDC) sequenced 10 strains and found that these sequences were highly conserved (Stone, 2002). Because camelpox and variola are very species‐specific and incapable of causing infection in the other host, and because the genome of monkeypox is quite different from that of variola, Fenner et al. (1988) did not believe either of these was the source of the human virus, but that it descended from a “protovariola” that followed a more persistent strategy in earlier hominids and became more virulent when human populations increased. On the other hand, the close relationship to gerbilpox suggests another possibility: a zoonotic rodent virus in Africa that evolved into variola only after human populations became large enough to support it (Tucker, 2001). Another poxvirus, molluscum contagiosum (genus Molluscipoxvirus), appears to be an ancient, persistent hominid infection (Senkevich et al., 1997).
Figure 6.
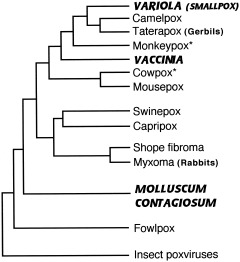
Phylogeny of mammalian poxviruses. Approximate relationships as presented in Blasco (1995), Douglas and Dumbell (1996), and Senkevich et al. (1997). Viruses specifically adapted to humans are indicated by bold and italicized capitals. *Maintained in nature in wild rodents.
The origin of vaccinia virus, the strain used in live vaccines, is also unclear. It was assumed to be derived from the cowpox virus used for vaccination in 1796 by Edward Jenner and propagated arm‐to‐arm for more than 100 years, but modern strains are distinct from cowpox and, in some areas of the genome, more closely related to variola and monkeypox (Blasco, 1995; de Souza Trindade et al., 2003). Now that the remaining stocks of smallpox have been spared destruction (for the time being) and fears of its use in bioterrorism have focused attention on it, we may get more insight into the mysteries of these viruses' evolution.
Hepadnaviruses replicate their DNA genomes via RNA intermediates and a reverse transcriptase, which makes them more similar to their cousins, the retroviruses. Discussion of this group is thus postponed until we meet up with the retroviruses later.
RNA viruses
Most animal viruses (about 70%; Domingo and Holland, 1994) carry their genomes in the form of RNA, and these are quite different from their DNA‐based counterparts. Their replication is more error‐prone, with high base substitution rates by viral enzymes that typically lack proof‐reading ability, on the order of 104–106 times the rates for DNA viruses and cellular genomes (Simmonds, 2001). Mutation rates are even greater for retroviruses, whose reverse transcriptases operate close to the error catastrophe threshold, the level of critical‐copying fidelity below which information can no longer be maintained and the nucleotide sequences of viral RNA would become essentially random (Domingo and Holland, 1994). Fast mutation rates help keep RNA viruses fairly small, as larger genomes could not sustain these rates of change and still remain capable of replicating. Many RNA viruses also undergo frequent recombination via “copy‐choice” (in which the RNA polymerase switches templates during replication), and those with segmented genomes (e.g., influenza) can reassort (Morse, 1994). All this results in a sort of “modular” evolution for these viruses and much faster rates of evolution than those of DNA viruses (Holland, 1993; Domingo and Holland, 1994) (recombination also occurs in DNA viruses, but via “breakage and reunion” as in host‐cell genomes, and it is less frequent than among RNA viruses). RNA viruses thus have a frightening ability to evolve and adapt to new hosts, and also to increase in virulence. They are less species‐specific and are readily transmitted between species; almost all viral zoonoses and “emerging” viruses are RNA viruses (Morse, 1993; Woolhouse et al., 2001).
Another consequence of this intense capacity to evolve is that genome populations within infected individuals are highly variable and known as “quasispecies” (Eigen, 1993; Domingo and Holland, 1994; Domingo et al., 1985, 1995, 1999). It is estimated that every possible single nucleotide polymorphism can be found in an HIV‐infected individual (Wain‐Hobson, 1994; Overbaugh and Bangham, 2001). Considering that RNA viruses may have been among the earliest organisms to evolve (Gorbalenya, 1995), they have explored enormous evolutionary spaces since then. At the other extreme, stabilizing selection in a well‐adapted and long‐standing natural host can result in persistence of an average or consensus genome sequence through time, with “persistence” here meaning maintenance in nature by the continuous infection of susceptible host organisms (Morse, 1994; Domingo et al., 1998). But freed from this stabilizing selection, or when single mutant genomes escape to a new host, these same RNA viruses are capable of rapid evolution and often increased virulence as well (Morse, 1994; Kilbourne, 1994; Domingo et al., 1998). While some can cause persistent inapparent infections in natural reservoir hosts, most follow an “acute life strategy” (rather like r‐selection), in which the virus increases fitness by increasing its reproductive rate (Villarreal, 1999). The ability to infect multiple species helps, as do means of transmission that do not require host fitness, such as water‐ and vector‐borne transmission (Ewald, 1994; Woolhouse et al., 2001).
The capacity for RNA viruses to evolve extremely rapidly under some circumstances, but hardly at all in others, and to undergo recombination and reassortment, makes the interpretation of their phylogenetic relationships much more difficult than for DNA viruses. Here, especially, one cannot assume constant molecular clocks (Gibbs, 1987; Simmonds, 2001), and trees tend to greatly underestimate genetic distances (Brown, 1994). The quasispecies nature of RNA virus populations means that trees compare consensus or average sequences instead of actual “species” or “strains,” and recombination or “modular evolution” results in different trees when different parts of the genome are compared. In spite of these difficulties, however, virologists still construct phylogenetic trees for RNA viruses, but they should be very careful in their interpretations.
Given their potential for interspecies transmission and rapid evolution, many human RNA viruses were acquired from other species, especially domesticated animals and commensal rodents, in the Neolithic and since, although one must be careful concluding this, as interspecies transmission has often been in the reverse direction.
Caliciviruses, distributed worldwide in humans and other vertebrates, are important causes of gastroenteritis and infant diarrhea, and show extensive sharing of strains between humans and domesticated animals (van der Poel et al., 2000). Figure 7A illustrates some phylogenetic relationships for this group. Close relationships with viruses of cattle and pigs suggest frequent interspecies transmissions both from and to these animals.
Figure 7.
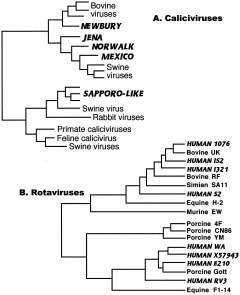
Common agents of infant diarrhea. A: Caliciviruses. Calicivirus phylogeny derived from sequence analysis of capsid gene of Sapporo‐like viruses (Jiang et al., 1997) and RNA polymerase gene of Norwalk‐like viruses (van der Poel et al., 2000). B: Rotaviruses. Rotavirus phylogeny based on analysis of VP6 capsid protein (reprinted from Tang et al., 1997, Fig. 2, with permission from Elsevier). Human viruses are indicated in bold and italicized capitals.
Figure 7B shows the phylogeny of another group of diarrheal disease agents, the rotaviruses (family Reoviridae), the most common agents of diarrhea worldwide and a major cause of infant mortality in the developing world (and in the young of many mammalian and avian species); virtually all children are seropositive (Woods et al., 1992). Rotaviruses are ubiquitous in nature and are found in both animals and plants. They are probably old viruses and might have afflicted some of our hominid ancestors. Evidence of this, however, has been overwritten by the tendency of these viruses to be shared by multiple host species. In addition, rotaviruses contain segmented genomes that frequently reassort when an individual is coinfected by multiple strains, making tree interpretation very difficult, and different genes give different trees (Joklik and Roner, 1995). Analysis of a number of structural and nonstructural proteins all show close relationships among human, bovine, and swine rotaviruses, with evidence of frequent interspecies exchanges (Goral et al., 1996; Kojima et al., 1996; Cunliffe et al., 1997; Tang et al., 1997). Figure 7B shows one of these trees, for the major capsid protein (Tang et al., 1997). While earlier hominids may have suffered from some form of rotaviral infection, they were probably sporadic and zoonotic. Today's human strains have acquired many of their genes from viruses of domesticated animals, and rotaviruses are now considered to have coevolved with nonhuman hosts (Ito et al., 2001).
Coronaviruses, a common cause of colds in humans and enteric and respiratory infections in a variety of animals, suggest a similar pattern of interspecies spread, with close relationships among viruses of humans and domesticated animals (Fig. 8A; Stephenson et al., 1999). Recombination in this viral family again results in somewhat different trees for different genes, but all show this intermingling of human and other animal strains. While most of these viruses are today very host‐specific, cross‐species transmission can still be a source of new human disease, as witnessed by the recent emergence of sudden acute respiratory syndrome (SARS). While bearing some similarities to group II and III coronaviruses, this hitherto unknown virus is sufficiently different to be placed in its own group (Marra et al., 2003; Rota et al., 2003). The SARS virus appears to have crossed over into humans in Guangdong Province, China, from some as yet undetermined zoonotic source, perhaps civet cats for sale in a local food market (Enserink, 2003), and it spread quickly from there. Fortunately, the epidemic was controlled by rapid epidemiological response, including aggressive use of quarantine, but SARS implies that there are many potential human pathogens we have yet to discover and that are increasingly likely to emerge as human numbers continue to grow, especially in parts of the world where dense populations of people come into contact with wild animals.
Figure 8.
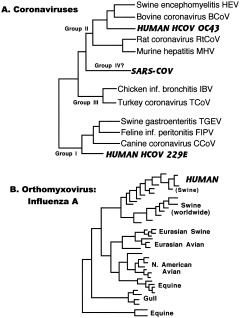
Respiratory viruses. A: Coronaviruses. Coronavirus phylogenetic relationships derived from analysis of polymerase gene (Stephenson et al., 1999; Marra et al., 2003; Rota et al., 2003). B: Orthomyxovirus: influenza A. Influenza A phylogeny based on nucleotide sequence of nonstructural (NS) protein (Kawaoka et al., 1998). Viruses that infect humans are indicated in bold and italicized capitals.
The best‐known members of the family Orthomyxoviridae are the influenza viruses types A, B, and C. Type A infects a wide range of animals, while B and C are found almost exclusively in humans. Types B and C are estimated to have diverged from type A approximately 4,000 and 8,000 years ago, respectively (Suzuki and Nei, 2002). Annual flu epidemics are caused by types A and B; serious pandemics result from novel strains of type A. Given the importance of influenza A in causing serious disease, it has been especially well‐studied, and its behavior illustrates both stable selection equilibrium in natural reservoir hosts (aquatic birds) and rapid divergent evolution in humans and pigs (Fig. 8B).
There are at least 15 strains of influenza A virus that circulate in wild aquatic birds (especially ducks) and are maintained in these populations by fecal‐oral transmission. These are the ultimate source of epidemic and pandemic influenza in humans, with pigs being an intermediate host (Webster, 1997, 1998; Kawaoka et al.. 1998). The production of many new human strains has been traced to pig‐duck‐fish farming in Asia, where the use of duck fecal material as fish food leads to transmission of avian influenza to pigs, and transmission to humans follows via the respiratory route (Scholtissek and Naylor, 1988). Like rotaviruses, influenza A has segmented RNA that can reassort, and interspecies transmission is frequently associated with reassortment among avian, swine, and human viral genes (Webster, 1997, 1998), a process known as “antigenic shift.” This creates new viruses sufficiently different to overcome immunity gained by past experience with influenza virus, and results in a strain with pandemic potential. Once in a new host species (unlike in the avian reservoir), the virus is under intense selection pressure to outwit the host immune response through mutations resulting in amino‐acid substitutions, or “antigenic drift.” Selection pressure is especially focused on the viral hemagglutinin (HA), a surface protein that binds to sialic acids on the new host's cell surfaces, and is often the target of immune surveillance (Bush et al., 1999). Drift probably occurs in other viral infections of respiratory or enteric mucous surfaces in long‐lived vertebrates, because the presence of antibodies on those surfaces means strong selection for even minor antigenic novelty (Fenner et al., 1974, p. 627–630).
The dual nature of influenza virus is evidenced by its behavior in different hosts. Sequence analysis of avian strains suggests a long history of association and evolutionary stasis, while the virus shows a completely different pattern in humans and pigs (Webster, 1997). In these species, viral mutation rates are higher (Kawaoka et al., 1998; Reid et al., 1999), but intense competition results in only a single dominant variant surviving an epidemic period to become precursor to the next set of strains (Fitch et al., 1997; Bush et al., 1999). This yields an unusual phylogenetic tree, especially for viral surface antigens such as the HA antigen, with only one of many strains persisting to give rise to the next year's crop of flu viruses (see Fig. 1 in Bush et al., 1999).
Paramyxoviruses include measles (MeV) and mumps (MuV) viruses; other human pathogens in this family of mostly respiratory agents include the parainfluenza viruses (PIV). These probably had recent separate origins, and at least some are derived from domesticated animals. Measles (MeV), canine distemper (CDV), and rinderpest (RPV) viruses are in the same genus Morbillivirus (Fig. 9), but MeV is more closely related to RPV than to CDV (although the two groups are still close enough that measles vaccine protects dogs from distemper). Acute crowd infections like measles require urban densities of at least 500,000 people, so its origin is probably recent interspecies transmission from a bovine source (Norrby et al., 1992).
Figure 9.
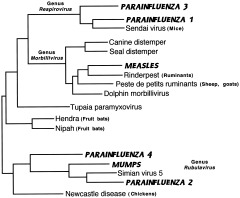
Paramyxoviruses. Phylogenetic analysis of N open reading frame (Chua et al., 2000). Human viruses are indicated in bold and italicized capitals.
Morbilliviruses are notorious for their ability to exploit new species (Baskin, 2000); examples include a rinderpest panzootic of the late 19th century that swept Africa, killing huge numbers of wildebeest, buffalo, other wild grazers, and cattle (Daszak et al., 2000), and canine distemper that today threatens both seals (Stone, 2000) and African lions (Roelke‐Parker et al., 1996). Measles now threatens mountain gorillas (Baskin, 2000). Emerging viruses closely related to this genus (but now put into their own genus) are Hendra (that killed horses and a trainer in Australia in 1995) and Nipah (fatal in humans and pigs in Malaysia and Singapore in 1999), both suspected to be maintained in nature in fruit bats (Enserink, 2000).
On the other hand, paramyxoviruses have fairly slow rates of evolution compared with other RNA viruses; measles virus is relatively stable, and there is no convincing evidence of recombination (Rima et al., 1997; Yamaguchi, 1997); this is also true for parainfluenza (Hetherington et al., 1994). The phylogeny of nonhuman morbilliviruses matches that of the host species (Fig. 9), as cetaceans are now known to be closely related to ungulates, and seals to other carnivores (Blixenkrone‐Möller et al., 1994). Not enough is known about MuV and the PIVs to make firm conclusions about their evolution; they may have been derived from other viruses such as bovine PIV, or vice versa (Kawano et al., 1990).
RNA viruses are complicated, however, and not all that afflict humans are recent acquisitions. Because of their capacity to evolve either very rapidly or hardly at all, depending on their circumstances, some have been long‐standing parasites of primates. Viruses whose replication involves overlapping reading frames or multigene products with strict secondary structures required for processing suffer constraints on their ability to evolve. Picornaviruses and hepatitis viruses are examples. Other viruses can integrate a DNA provirus into the germ‐cell genome and be passed on as Mendelian traits. Retroviruses illustrate this phenomenon. This potential for slowed evolution can result in RNA viruses that show host‐linked phylogenies, as do DNA viruses.
The Picornaviridae, which include enteroviruses (e.g., polio), rhinoviruses, and several other genera, are an interesting case because on the one hand they can evolve very quickly, close to the maximum rate compatible with maintaining genetic information (Domingo et al., 1995), and with frequent recombination (Lai, 1995), and yet their genomes are remarkably stable when grown under unchanging conditions (Gromeier et al., 1999). The viral RNA is translated into a single polyprotein that is later cleaved into several structural and other proteins, including an RNA‐dependent RNA polymerase. The polypeptide backbone of the polyprotein is highly conserved in all known picornaviruses, and thus mutations are mostly synonymous; capsid and polymerase proteins are also somewhat conserved (Gromeier et al., 1999).
Most of the genera in this family infect primarily humans and Old World anthropoids (Fig. 10), with nonprimate strains appearing to derive from their human counterparts (Rodrigo and Dopazo, 1995; Gromeier et al., 1999). The clade containing foot‐and‐mouth disease and encephalomyocarditis is considered the mostly nonhuman animal side of the tree (Doherty et al., 1999). Simian enteroviruses (isolated so far from macaques, guenons, and baboons) are as diverse and probably as numerous as human enteroviruses. Many of these are the closest known relatives of human enteroviruses (with the exception of viruses derived from human strains, e.g., swine vesicular disease virus, thought to have originated from coxsackie B; Poyry et al., 1999), while other simian viruses cluster together and probably represent a previously unknown picornavirus genus (Oberste et al., 2002). There are more than 60 human enteroviral serotypes, including 3 polio, 23 Coxsackie A, 6 Coxsackie B, and 30 echoviruses. They are common in humans, and most of them cause no apparent infection. A high proportion (>99%) of polio infections are subclinical as well, and the virus was endemic in developing countries of Asia, Africa, and the Americas until recently (Rico‐Hesse et al., 1987; polio has now been largely eradicated everywhere except sub‐Saharan Africa and India, thanks to WHO's Global Polio Eradication Initiative; World Health Organization, 2001). The neuropathogenicity of wild polio is low and requires spread of the virus from the intestinal tract to motor neurons (Crainic and Kew, 1993). Poliovirus may persist in the organism and continue to be shed after acute infection (Domingo et al., 1998), allowing it to be maintained in small isolated populations, although Black (1990) thought that poliovirus was introduced into South American Indians. Wild polio type 1 strains (the most common cause of polio) correlate better with geography than with time of isolation, meaning that viruses isolated at different times in a single geographic region are more closely related to one another than are polioviruses from several regions isolated in the same year (as would be found in the case of pandemic influenza, for example). This argues for the relative stability of this virus, with strains from Africa and the Mideast having the deepest roots (Rico‐Hesse et al., 1987). It further suggests that enteroviruses, including poliovirus, have long been associated with hominids, either coevolving with us or possibly entering our line from other primates sometime during our evolution in Africa.
Figure 10.
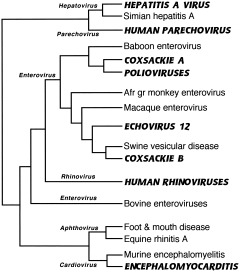
Picornaviruses. Phylogeny of polymerase protein, composite of trees from Rodrigo and Dopazo (1995), Doherty et al. (1999), and Poyry et al. (1999). Human viruses are indicated in bold and italicized capitals. Six most common genera of picornaviruses are indicated next to branches leading to members of each genus. This nomenclature may not reflect genetic relationships, however; note that genus Rhinovirus maps within Enterovirus phylogeny.
Other viruses that may have cospeciated with primates include the various hepatitis viruses (Robertson, 2001; Simmonds, 2001). As causative agents of chronic disease and latent infections, these viruses could have persisted in wild primates and small hominid groups. These include hepatitis A virus (HAV), associated with infectious hepatitis spread by food and water but also capable of causing subclinical infections (Gust, 1980); hepatitis B (HBV), the cause of serum hepatitis, transmitted by blood and blood products, and also asymptomatic in the majority of cases following perinatal or vertical transmission (Tiollais and Buendia, 1991); hepatitis C (HCV), cause of the non‐A, non‐B hepatitis of recent epidemic spread but also capable of causing prolonged, inapparent infection (Simmonds, 2001); and the closely related GB virus‐C (GBV‐C) or hepatitis G (HGV). The origins of HCV are not well‐understood, but evidence of natural infections by relatives of the other hepatitis viruses in wild primates now suggests that these viruses have evolved along with their primate hosts (Robertson, 2001).
Hepatitis A virus (HAV) is a picornavirus like polio but in its own genus Hepatovirus, along with a number of simian hepatitis A viruses (SAV) in Old World monkeys and probably chimpanzees as well (Robertson, 2001). As for poliovirus, there is a high degree of antigenic conservation. SAVs from wild‐caught monkeys group into three genotypes, while human strains fall into 4–6 others that correlate with geographic regions (Fig. 11A; Robertson et al., 1992). Antibodies have been found in virtually every population tested, with largely subclinical infections in developing countries (Gust, 1980). Continued study is necessary, especially of chimpanzee SAV, before firm conclusions can be drawn, but HAV may be an ancient associate of humans or acquired from cattarhine primates sometime during human evolution.
Figure 11.
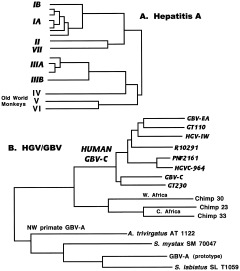
Hepatitis viruses. A: Phylogeny of human and primate hepatitis A viruses, based on a 170 base sequence from VP1/P2A junction. Genotypes IA, IB, II, IIIA, IIIB, and VII cause human hepatitis A infection. IA and IB, endemic African strains; II, single isolate from France; IIIA, endemic in many parts of Asia; IIIB, from Japan; VII, single isolate from Sierra Leone. Genotypes IV, V, and VI are strains found in Old World monkeys in Philippines, Kenya, and Indonesia, respectively. For details, see Robertson et al. (1992) and Robertson (2001). B: Phylogenetic analysis of NS5 region of HCV‐like viruses. From Simmonds (2001), Fig. 5, with permission from Society for General Microbiology. Human viruses are indicated in bold italics.
The GB viruses (“hepatitis G”) were discovered when serum from a surgeon (GB) with hepatitis was inoculated into tamarins, who also developed the disease. Serial passage of their sera in other tamarins resulted in the isolation of several GB agents that are not HAV, HBV, or HCV, although they are closely related to HCV (Robertson, 2001). HCV and the GB‐agents are members of the Flaviviridae, a family whose better known members include mosquito‐ and tick‐borne viruses like dengue, yellow fever, West Nile, and miscellaneous encephalitis agents. HCV and the GB‐agents are in their own genus, Hepacivirus, only distantly related to the arthropod‐borne members of this family. GBV‐A has since been found in a wide range of New World monkeys (tamarins, owl monkeys, and marmosets, each species having its own variant), and causes chronic infection with no obvious disease (Fig. 11B; Robertson, 2001). GBV‐B may also be a natural infection in New World primates.
Several viruses found in wild chimpanzees (both Pan troglodytes verus in West Africa and P.t. troglodytes in Central Africa) are even closer to human HGV/GBV‐C than GBV‐A is. There is more sequence diversity between the two chimp subspecies than among human variants (Adams et al., 1998). Relationships among the human GBV‐C subtypes imply an ancient association, before migration of anatomically modern humans out of Africa, with the sub‐Saharan African genotype having the greatest sequence diversity (Robertson, 2001; Simmonds, 2001). Whether HGV evolved from a chimpanzee virus or it represents an older virus that cospeciated with both apes and hominids cannot be resolved at this time, but it appears to have originated in Africa and has since diversified along with human populations as they migrated from that continent (Pavesi, 2001).
The apparent paradox of such slow evolution in viruses otherwise known for their rapid mutation rates can be explained in this case by the peculiar nature of the HGV RNA genome, which forms a complex and extensive secondary structure through internal base‐pairing. Base substitutions in any of the multiple sites that interact to form this structure require matching substitutions elsewhere, such that specific stem‐loops are conserved, thus constraining evolution in this virus (Simmonds, 2001).
As for the rest of this family, the zoonotic flaviviruses show phylogenetic relationships that are closely linked to their mosquito or tick vectors. The mosquito‐borne group separates into two clades: one is neurotropic (in humans and livestock) and associated with avian reservoirs and Culex mosquitoes; this clade includes West Nile, Japanese encephalitis, St. Louis encephalitis, and others. The other mosquito‐borne clade is hemorrhagic and associated with primates and Aedes mosquitoes (e.g., yellow fever and dengue). All mosquito‐borne flaviviruses appear to have originated in Africa, where both yellow fever and dengue still occur in sylvatic cycles in wild monkeys (Gaunt et al., 2001). The Culex and Aedes species that preferentially bite humans have coevolved with us; the ancestor of both was present by the Eocene, and primates since then (including hominids) have probably suffered from arboviral infections. Aedes appears to have diverged from the Culex line in the Oligocene, which may coincide with the origin of sylvatic yellow fever (Capasso, 1993).
Viruses that use reverse transcription
One more common hepatitis virus remains to be discussed. Although its viral capsids contain a DNA genome, hepatitis B virus (HBV) replicates via an RNA intermediate and a reverse transcriptase, giving it the same error‐prone transcription and capacity for rapid evolution as the retroviruses, to which it is related (McClure, 1995). Among the smallest of all viral genomes, it uses overlapping reading frames that generate severe evolutionary constraints and highly variable mutation rates at different sites, (Yang et al., 1995; Simmonds, 2001), and in spite of much data on nonhuman primate HBVs, interpretation of this family's phylogeny is a challenge (Fig. 12).
Figure 12.
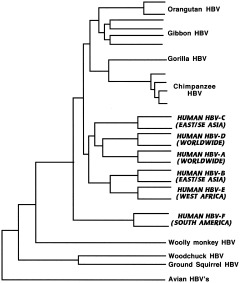
Hepatitis B viruses. Phylogenetic analysis of representative human and nonhuman HBV full genome sequences (Norder et al., 1996; Verschoor et al., 2001). Human viruses are indicated in bold and italicized capitals.
Until the discovery of wild strains in free‐living hominoids, the woolly monkey virus appeared to be ancestral to human strains, as it and the human serotypes found in indigenous South Americans (HBV‐F in Fig. 12) seemed to be the deepest roots on the tree (Lanford et al., 1998). Discovery of the virus in relatively isolated aboriginal human populations throughout Asia and in Africa presented problems for this interpretation, however (Bollyky and Holmes, 1999). Sequencing of viruses from chimpanzees (Takahashi et al., 2000; Hu et al., 2000; MacDonald et al., 2000) suggests that the infection is relatively common among chimps in West Africa, and that chimp HBV circulates in nature; up to 50% of adult chimps are seropositive (Robertson, 2001), and different subspecies of Pan troglodytes harbor distinct chimp HBV genetic variants (Hu et al., 2001). Similar viruses have been found in wild gorillas, orangutans (Verschoor et al., 2001), and gibbons (Norder et al., 1996), indicating widespread natural infection in all the apes.
The ape viruses cluster together on a branch separate from the human viruses (Fig. 12), with African and Asian ape viruses forming two subclusters, and the Asian subcluster further subdividing into orangutan and gibbon clades, suggesting cospeciation (Verschoor et al., 2001; Robertson and Margolis, 2002). The woolly monkey virus is still closest to the South American human type F; that and the existence of the only other known mammalian hepadnaviruses in American rodents (e.g., woodchucks, squirrels) still suggest to some an American origin by cross‐species transmission. Others have proposed that HBV coevolved with humans and diversified as they migrated from Africa during the last 100,000 years (Magnius and Norder, 1995). But the viral phylogeny does not parallel genetic relationships between human and nonhuman primate hosts, which presents a problem for any coevolutionary model. A third scenario involves multiple zoonotic transmissions, analogous to what some believe has happened with HIV (see below), with donor species remaining unidentified (MacDonald et al., 2000). Recent zoonotic transmission is not supported by the separation of human and ape clusters, however, as one would expect in that case to see a mosaic branching pattern of human and ape strains (Verschoor et al., 2001). The latest and perhaps best explanation of this confusing picture is that primate HBVs cospeciated with their hosts (both woolly monkey and apes) over the last 10–35 my (which leads to a very low estimate of mutation rate, by the way, but similar to that obtained if cospeciation is assumed for rodent and woolly monkey viruses), with human strains being of ancient zoonotic origin (sometime before leaving Africa), perhaps via processing of primates for food (Simmonds, 2001). Since that time, dispersal from Africa and rapid population growth (providing more opportunities for horizontal transmission) led to faster mutation rates and diversification of the human virus (Robertson and Margolis, 2002). This last hypothesis fails to account for the close relationship between South American HBV and the woolly monkey virus, however, unless one assumes multiple zoonotic origins. The mystery is as yet unresolved.
Retroviruses use an RNA‐dependent DNA polymerase, or reverse transcriptase, to generate a DNA copy of the RNA genome; this cDNA is transcribed by another enzyme into viral RNA that is packaged into infectious particles. The DNA copy, or provirus, can also be inserted into the cellular genome, where it is replicated by the host‐cell DNA polymerase with its much lower error rate. These integrated viruses also escape immune detection and the resulting pressure to evolve, making them much more stable than exogenous viruses. Retroviruses that tend to integrate, like human T‐cell lymphotropic virus (HTLV), are thus characterized by slower rates of change and less sequence diversity, while viruses that produce continual rounds of replication using the viral polymerase, like human immunodeficiency virus (HIV), have more genetically diverse populations and faster rates of evolution (Overbaugh and Bangham, 2001). “Endogenous retroviruses,” presumed remnants of ancient germ‐cell infections, are integrated proviruses that have lost the ability to produce infectious particles. In spite of this, their DNA sequences have multiplied and inserted at numerous sites through retrotransposition, and are estimated to make up about 8% of the human genome (International Human Genome Sequencing Consortium, 2001). Given their ability to insert in regions contiguous to genetic loci, these human endogenous retroviruses (HERVs) may have directly affected host gene expression and contributed to hominid evolution (see Discussion).
Simian and human immunodeficiency viruses (SIV/HIV) belong to the lentivirus family of retroviruses; SIVs have been found in a wide range of mostly African anthropoids (26 different species; Hahn et al., 2000). Most of these viruses are naturally occurring and do not appear to cause disease in their reservoir hosts, while cross‐species transmission results in pathology (Sharp et al., 1999). That the phylogeny of these viruses as a whole does not correlate with that of their hosts suggests a long history of interspecies transmission and recombination, but there is also evidence of host‐dependent evolution within some primate clades. Examples of both can be seen in Figure 13. The SIVs of the “African green monkeys” (vervets, grivets, sabaeus, and tantalus monkeys) reflect the evolutionary relationships of their hosts, and indicate a longstanding association (Fomsgaard et al., 1997). In contrast, the close relationship between macaque and sooty mangabey SIVs probably reflects interspecies transmission, as SIVmac (see Fig. 13 for explanation of subscripts) has been found only in captive animals (Sharp et al., 2001).
Figure 13.
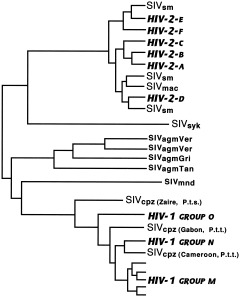
Primate immunodeficiency viruses. Schematic diagram shows phylogenetic relationships and cross‐species transmission of primate lentiviruses, from a number of analyses (Sharp et al., 1999, 2001; Hahn et al., 2000). HIV, human immunodeficiency virus; SIV, simian immunodeficiency virus; SIVsm, sooty mangabey; SIVmac, macaque; SIVsyk, Sykes' monkey; SIVagmVer, vervet; SIVagmGri, grivet; SIVagmTan, tantalus monkey; SIVmnd, mandrill; SIVcpz, chimpanzee; P.t.s., Pan troglodytes schweinfurthii; P.t.t., P. t. troglodytes.
SIVs are common subclinical infections in monkeys, but are not as prevalent in other primates. The chimpanzee virus, SIVcpz, is the only nonmonkey SIV; similar viruses are not found in gorillas or bonobos. Recent work suggests that SIVcpz is a recombinant of the SIVs of red‐capped mangabeys and greater spot‐nosed monkeys (Bailes et al., 2003). It was likely acquired by chimpanzees through hunting and eating infected monkeys, perhaps at some point before the divergence of Pan troglodytes troglodytes and P.t. schweinfurthii, as it is found in these subspecies but not in P.t. verus (Prince et al., 2002). There appear to have been numerous other cross‐species transmissions, both simian‐to‐simian and simian‐to‐human. The human immunodeficiency viruses most likely originated through recent interspecies transmissions, from chimpanzees in the case of HIV‐1 (Gao et al., 1999; Corbet et al., 2000), and from sooty mangabeys for HIV‐2 (Chen et al., 1997; for reviews, see Holmes, 2001; Sharp et al., 2001). It is now clear that HIV‐2, a less pathogenic virus endemic in West Africa, emerged in that region where mangabeys are both hunted and kept as pets, and at least four separate interspecies transmission events gave rise to six human subtypes (Chen et al., 1997).
The origins of HIV‐1 are less clear, but there appear to have been multiple cross‐species transmissions of SIVcpz to humans in west central Africa (Gao et al., 1999; Sharp et al., 1999; Hahn et al., 2000). This conclusion is still somewhat tentative, however, as it is based on only six captive chimps (Zimmer, 2001). SIV prevalence in chimpanzees, both wild and captive, is low (Corbet et al., 2000; Santiago et al., 2002). In contrast, both African green monkeys and sooty mangabeys are infected by their respective viruses at high frequencies (Hahn et al., 2000). It should be noted, however, that wild P.t. troglodytes, the suspected source of HIV‐1, has not yet been adequately sampled. SIVcpz from P.t. schweinfurthii is quite divergent from HIV (Santiago et al., 2002), and it was not found at all in 387 P.t. verus from West Africa (Prince et al., 2002). The only case of SIV infection in Nigerian P.t. vellerosus was in an animal caged with an infected P.t. troglodytes (Corbet et al., 2000), so SIVcpz of P.t. troglodytes seems the most likely HIV‐1 ancestor. That chimp and human viruses are interspersed on the tree shown in Figure 13 suggests that there have been at least three cross‐species transmissions, most likely related to the practice of hunting and field‐dressing chimpanzees for “bush meat” (Hahn et al., 2000). Indeed, there is a high potential for acquiring primate viruses in this manner, as recent sampling showed a substantial proportion (∼20%) of wild monkeys hunted or kept as pets in Cameroon to be SIV‐infected (Peeters et al., 2002).
Three different cross‐species transmissions of SIVcpz resulted in three groups of HIV‐1 (M, N, and O). Molecular clock estimates date the last common ancestor of the M group, the group responsible for the majority of HIV infections worldwide, prior to 1940 (Sharp et al., 2001), perhaps as early as 1931 (Korber et al., 2000), and the jump from chimpanzee to human may have occurred even earlier. The virus apparently remained undetected for 50 years or more in low‐risk populations before spreading to high‐risk groups, through cultural practices affecting contact with infected blood or sexual frequency which increased the potential for transmission (Armelagos et al., 1990). Increased sexual frequency may have selected for greater virulence as well (Ewald, 1994). The highly divergent O group, found in western equatorial Africa (Cameroon and neighboring countries), and the N group, a small number of viruses from Cameroon, have not diversified or spread to the same extent as the M group has; coalescence dates for these groups have not yet been reported.
An alternative hypothesis (that HIV originated from contaminated oral polio vaccine allegedly produced in chimpanzee kidney cell cultures; Hooper, 2000) has been effectively rejected (Hahn et al., 2000; Blancou et al., 2001; Sharp et al., 2001). All three groups of HIV‐1 are more closely related to SIVcpz from P.t. troglodytes, found in west central Africa, the presumed place of origin of the HIV pandemic. The chimpanzees purported to have been the source of tissue for the polio vaccine were from northeast Congo, an area inhabited by P.t. schweinfurthi. In addition, recent estimates of the time frame for the initial interspecies transmission of SIVcpz leading to the HIV‐1 M group put it some time before the use of oral polio vaccines in the late 1950s.
There is considerable discrepancy (several orders of magnitude) between molecular clock estimates based on observed mutation rates in HIV‐1 and divergence dates that assume cospeciation in SIVs and primates (Sharp et al., 2000). In spite of the retroviral capacity to rapidly evolve, stable consensus sequences can persist in natural hosts, in part because of lower immune response in nonhuman primates, and therefore less selection pressure to evade it. On the other hand, transmission to a new species leads to rapid evolution. Thus SIVagm and other primate viruses shown in Figure 13 display long branch lengths and host‐linked evolution, while HIV is evolving rapidly.
The human T‐cell lymphotropic viruses have somewhat different histories. Both HTLV‐I and ‐II are older retroviruses than HIV, and have evolved more slowly because of their greater tendency to integrate into the cellular genome and consequent potential for vertical transmission. This leads to mostly inapparent lifelong infections like those caused by DNA viruses. On the other hand, the tendency of HTLV‐I to jump species barriers can lead to rapid viral evolution, as in other retroviruses. Because of this difference, HTLV‐I and HTLV‐II have contrasting evolutionary histories (Fig. 14; Slattery et al., 1999; Salemi et al., 2000). HTLV‐I appears to have originated in Asia between 50–100 kya, moved to Africa where it explosively spread through Central African anthropoids (including humans) with many occasions of interspecies transmission, and eventually spread as a “cosmopolitan” variant to the rest of the world (Vandamme et al., 1998; Van Dooren et al., 2001). Geographic variants of HTLV‐I thus reflect recent migrations of anatomically modern humans (Slattery et al., 1999). HTLV‐II, on the other hand, is an older virus with an African origin ∼400 kya; its closest relative is the simian T‐cell lymphotropic virus (STLV) of Pan paniscus (Vandamme et al., 2000). Today there are two types of distribution for both HTLV‐I and ‐II: a cosmopolitan epidemic associated with transmission via contaminated blood or needles, and an endemic pattern produced by mother‐to‐child transmission (via breast milk), with substrains reflecting human migrations (Black, 1997), but both types I and II originated through interspecies transmission of STLVs to human ancestors (Salemi et al., 2000).
Figure 14.
Primate T‐cell lymphotropic viruses (PTLV). Phylogeny of HTLV/STLV types I and II, constructed from data in Slattery et al. (1999), Vandamme et al. (2000), and Van Dooren et al. (2001). STLVs isolated from nonhuman primates are indicated by bold italics; all others including unlabeled lines are HTLV strains isolated from humans. Note interspersal of Central African HTLV‐I subtypes among branches of STLV‐I.
Human and simian viruses in this group are now collectively referred to as “primate T‐cell lymphotropic viruses” (PTLVs) because African HTLVs and STLVs cannot be separated into distinct phylogenetic lineages according to their species of origin, but are instead related according to the geographic origin of their host (Vandamme et al., 1998). There are six PTLV‐I subtypes: subtype A is the “cosmopolitan” one, recently derived and estimated to have radiated at least 13 kya; it contains exclusively human strains; B, D, E, and F are Central African subtypes found in humans (including pygmies in Cameroon, Gabon, and Congo) and nonhuman primates; and subtype C is the Australo‐Melanesian subtype, found in Papua New Guineans, Australian Aboriginals, and Indonesian macaques (and perhaps orangutans; Verschoor et al., 1998). STLV‐I and HTLV‐I strains from the same geographic region show closer phylogenetic relationship than do HTLV‐I/STLV‐I strains from the same primate species (Van Dooren et al., 2001), i.e., the relationships between HTLV‐Is and STLV‐Is show evidence of horizontal interspecies transmission instead of cospeciation (Koralnik et al., 1994; Mahieux et al., 1998; Salemi et al., 1998).
The moderate stability and low mutation rates of these viruses have allowed investigators to make molecular clock estimates for this group (Salemi et al., 2000; Vandamme et al., 2000; Van Dooren et al., 2001). The ancestor of all PTLVs probably evolved in Africa, with one branch (the ancestor of PTLV‐I) carried to Asia by one million years ago, while viruses remaining in Africa evolved into PTLV‐II and STLV‐L (Salemi et al., 2000). Salemi et al. (2000) suggested that the PTLV‐I ancestor spread to Asia in migrating macaques at 2.2 mya, but the macaque radiation probably occurred much earlier, around 5.5 mya (Morales and Melnick, 1998). That HTLV‐I evolved in Asia is suggested by the clear separation between African and Asian/Melanesian strains (Van Dooren et al., 2001). HTLV‐I subtype C and STLV‐I from macaques and orangutans have higher levels of genetic diversity and are thought to be older than African subtypes (Giri et al., 1997; Verschoor et al., 1998), with Asian HTLV‐I estimated to have originated 50–100 kya (Salemi et al., 2000; Van Dooren et al., 2001). It appears to have reached Africa by at least 27.3 kya and explosively spread through primates. Whether this was the result of a simian or a human migration is not yet certain.
PTLV‐II is much less likely to jump species, so it is less diverse (Slattery et al., 1999). The only known STLV‐II is found in bonobos (Vandamme et al., 2000). HTLV‐II isolates from African pygmies have more sequence diversity than those from Amerinds, which supports the conclusion that HTLV‐II originated in Africa, probably via an ancient interspecies transmission of the bonobo virus ∼400 kya (Giri et al., 1997; Salemi et al., 2000; Vandamme et al., 2000). HTLV‐II is endemic in both native North and South Americans and Central African pygmies (with epidemic spread in IV drug users); subtype D, the most divergent, is found in Efe pygmies living not far from bonobos in the Congolese rain forest (Vandamme et al., 2000). HTLV‐II heterogeneity in the Americas is greater than in HTLV‐I and comparable to HTLV‐I/STLV‐I in the Old World, suggesting that type II has been in the Western Hemisphere longer and was brought over by the original migrants, while HTLV‐I was introduced more recently (Dube et al., 1993; Vandamme et al., 2000). Molecular clock estimates suggest that HTLV‐II was carried out of Africa at least 58 kya and the American subtypes diverged between 12–38 kya, after the first human migrations to the New World.
DISCUSSION
The role of viruses in human evolution
What can we conclude from this survey of the molecular phylogenies of viruses, and what does it imply about the role of viruses in human evolution?
-
1
Most of the slowly evolving DNA viruses are ancient and have coevolved in close association with their hosts. Families such as the Herpesviridae, Papovaviridae, and Adenoviridae have cospeciated with vertebrates, and even the earliest hominids undoubtedly had their share. The life‐long, mostly asymptomatic infections caused by these viruses are unlikely to have had a significant selective effect, however, unless introduced into a new host.
-
2
Many RNA viruses, with their enormous capacity for change, were more recently acquired during the Neolithic, when humans and domesticated animals entered into intimate contact, and agricultural surpluses and permanent housing attracted disease‐carrying rodents. This process was facilitated by RNA viruses' high mutation rates, propensity for interspecies transmission, and ability to recombine or reassort when related viruses coinfect the same host (e.g., mammalian and avian influenza viruses in pigs). Post‐Neolithic viruses include the diarrheal calici‐ and rotaviruses, the coronaviruses of common colds, and ortho‐ (influenza) and paramyxoviruses that cause a variety of other respiratory diseases. The DNA‐based smallpox virus may also be in this category. The generally more acute infections caused by these newer viruses are unlikely to have plagued earlier hominids. At the same time, the existence of RNA viruses in wild primates suggests that hominids had some of these, but the transient nature of these continually drifting viral genomes means that their sequences have been totally overwritten, and their propensity to switch hosts means their descendants may be anywhere. The trail on these is quite cold.
-
3
On the other hand, the capacity of some RNA viruses to be evolutionarily stable in long‐standing reservoir hosts, and other constraints on their ability to evolve, suggest that some of these may be very old human parasites. Hepatitis viruses, picornaviruses, and HTLV became endemic in hominids some time before modern humans left Africa; some of these may have infected the earliest hominids. But like the DNA viruses, these are unlikely to have acted as important agents of selection, given their association with mostly chronic and latent infections, although this may not have been the case in the past. When these viruses were first acquired they were probably more virulent, and different hominid species may have had different susceptibilities. Considering the array of retroviruses found in African monkeys and apes today and the ease with which they cross species barriers, ancient humans may have encountered more than just HTLV. Endogenous retroviruses, moreover, with their ability to retrotranspose into different parts of the host genome, may have played a more direct role in human evolution.
-
4
Zoonoses, or infectious diseases whose natural hosts are other animals, must have been important causes of disease throughout human evolution (Fenner et al., 1974, p. 632). Because viruses that must be able to replicate efficiently in more than one host are more genetically stable (Kilbourne, 1994; Scott et al., 1994), arboviruses such as yellow fever, dengue, and Rift Valley fever have probably been present in Africa during much of our evolution, and mosquitoes capable of transmitting them evolved during the Pliocene (Capasso, 1993). Bat lyssaviruses, from which rabies is derived, also have a long history in Africa (Bourhy et al., 1993). Given the diversity of tropical ecosystems, African environments are bound to have contained numerous zoonotic viruses capable of infecting hominids. Scavenging or hunting other species, including primates, would have presented constant opportunities for the transmission of viruses.
In many respects this agrees with earlier reconstructions of hominid disease patterns inferred from disease transmission requirements (Cockburn, 1967b; Armelagos and Dewey, 1978; Armelagos, 1990) or studies of isolated populations (Black, 1975, 1990). This analysis suggests, however, that viruses were even more important pathogens during earlier stages of human evolution than we realized. What roles might they have played?
As causes of morbidity and mortality they may have acted as agents of selection, especially new viruses entering a population for the first time or during apparent competition between species with differing susceptibilities. As molecular genetic parasites in intimate association with host cell DNA, they perhaps played a role in molding the human genome, especially retroviruses and DNA viruses that replicate in the cell nucleus and can integrate into germ‐cell DNA.
Viruses as agents of selection
Viral selection could operate at several levels, from the cellular level to the level of competition between species. Among the kinds of viral‐mediated selection likely to be important factors in the evolution of a species are selection among individuals within a population and apparent competition between populations or species. In cases where genetic diversity is useful (e.g., the MHC system), intrapopulation selection will be balancing and presumably result in polymorphism and maintenance of an adequate defensive response. In other cases (e.g., selection for or against cell‐surface antigens recognized by viruses), selection will be directional and favor certain alleles over others. Virus‐mediated selection between populations or species might give one a competitive edge and thus contribute to decline in the other, or it may promote divergence and habitat partitioning. But were the requirements for these kinds of selection satisfied during human evolution?
Intrapopulation selection
Some have proposed that genetic interactions between pathogens and hosts have been important in maintaining balanced polymorphisms within host populations (Clarke, 1976). For this to occur, however, several conditions must be fulfilled (Clarke, 1976; Svanborg‐Eden and Levin, 1990):
-
1
There is genetic variation in disease susceptibility and mortality within the host population;
-
2
This inherited variation is the result of only one or a few genes;
-
3
There is phenotypic variation in the parasite's ability to attack its hosts;
-
4
There is a relationship between these two kinds of variation (i.e., host and pathogen evolve in response to each other, also known as gene‐for‐gene coevolution);
-
5
There is a relationship between numbers of parasites and numbers of hosts;
-
6
Host and pathogen encounter each other continuously or at least frequently; and
-
7
The parasite causes great enough mortality (or reduction in fertility) prior to the termination of reproduction (during ages of high reproductive value) for it to be an evolutionary force.
To what extent were these conditions fulfilled during human evolution? As for the first condition, genetic variation in host susceptibility to infection appears to be the norm in most organisms (Clarke, 1976); our ancestors were unlikely to have been atypical in this regard. That even the most virulent of parasites (e.g., the plague bacillus or Ebola virus) do not kill all infected individuals suggests some innate variation in human response (but other differences—environmental, developmental, social, or behavioral—may also play a role). As another example, most children exposed to varicella zoster virus develop clinical symptoms of chickenpox, but many infected individuals show no symptoms (Benenson, 1995, p. 87).
Is this variation the result of simple inherited variation, i.e., polymorphisms at a single locus or only a few loci (condition 2)? There is substantial histocompatibility polymorphism in humans (Bodmer, 1972; Bodmer and Bodmer, 1978; Howard, 1991), but whether this is the result of selection by infectious disease is not certain (Svanborg‐Eden and Levin, 1990). Diversity in this region does appear to correlate with a population's susceptibility to infectious disease (Black, 1990), and studies now link different HLA antigens with resistance to viruses, e.g. influencing the time of onset of AIDS after HIV infection (Kaslow et al., 1996) and clearance of hepatitis B virus (Thursz et al., 1995).
The ABO blood system is another possible example of polymorphism related to disease susceptibility. Associations between infectious agents and ABO allele frequencies suggest differential susceptibilities to certain viruses and bacteria. For example, decreased A allele frequencies following smallpox epidemics suggest viral selection against that allele (Vogel, 1975), but discrepancies in the literature and a lack of hard evidence concerning the underlying biological mechanism preclude any firm conclusions (Greenwell, 1997). Other viral diseases implicated in ABO selection include hepatitis, chickenpox, poliomyelitis, influenza A, and adenovirus infection, but the evidence is ambiguous and may simply reflect a generalized effect of ABO alleles on the immune response (Vogel, 1975).
Several other polymorphisms are known to confer disease resistance, such as hemoglobin S and favism vs. falciparum malaria, but these are probably fairly recent adaptations. The only well‐studied examples are associated with malaria and plague (Stephens et al., 1998; Tishkoff et al., 2001), both unlikely to have affected humans much before the Neolithic.
The requirement for variation in parasite virulence (condition 3) was undoubtedly met, especially in the case of RNA viral quasispecies. Even in the most stable viral populations, one finds a multitude of variants that might be favored in another host, so most viruses probably meet this criterion.
Condition 4 (gene‐for‐gene coevolution) was demonstrated for a large number of plant/parasite systems, including viruses (Thompson and Burdon, 1992), but in plants, somatic variation can be inherited, producing rapid rates of evolution (although this may be an artifact of plant breeding and not as common in wild plants, which tend to have higher levels of genetic variation for pathogen resistance; Simms, 1996). In spite of the lower probability of inheritable change in sexually reproducing animals, there are now several examples of host‐pathogen coevolution in animals, probably due to selection acting on preexisting variation. A well‐studied example is the introduction of myxoma virus (a natural parasite of American rabbits) into Australian rabbits in the 1950s. The initially very high mortality rates declined over several years, and investigations showed that both rabbits and virus had changed. The same strain of virus that killed 90% of wild‐caught rabbits during the first year killed only 30% seven years later, indicating selection for resistance in the host. Meanwhile, virus isolated from wild rabbits showed reduced virulence over time (Fenner and Kerr, 1994). It is possible that such a scenario might have played itself out during hominid evolution, providing a new pathogen was sufficiently lethal yet capable of being transmitted to enough new susceptibles to be able to persist in the population (but these are a lot of “ifs”!).
Clarke (1976) suggested that this condition obtains whenever the enhanced ability to resist one pathogen reduces a host's ability to resist another, i.e., the resistant host will still be susceptible to infection with a virus that has mutated to overcome the resistance factor. Changes in cell surface receptor sites may facilitate the binding of viruses with altered coat proteins. Viral proteins may mimic host antigens and thereby escape immune surveillance in most individuals; hosts with rare antigenic types would have an advantage. Many viruses wrap themselves in a host cell membrane on their way out of the infected cell. Upon attacking another individual of a different antigenic type, the new host will respond with antibodies against the antigens picked up from the previous host. Under these conditions, a population with greater heterogeneity in cell surface antigens makes it harder for enveloped viruses to find a tolerant host, and this may have been important in promoting MHC diversity (Bodmer, 1972). Enveloped viruses include most of the RNA viruses (among the DNA viruses, herpesviruses pick up bits of nuclear membrane, and poxviruses snatch proteins from Golgi bodies).
The final three conditions are related; host and pathogen numbers are dependent on each other when the host species is the natural one and thus encounters the parasite regularly, and the pathogen has no other important natural reservoir. This means that zoonoses, in spite of their potential for dramatic consequences, are unlikely to lead to lasting selective effects; their role was probably limited to keeping hominids from exploiting certain microenvironments. And while some viruses encountered by hominids may have been fatal, the ones for which they were a frequent host could not have been too serious (or they would not have persisted in small populations), certainly not to the extent required for strong selection pressure.
Possible exceptions may have been when new viruses like the HTLVs or hepatitis B entered the human line (sometime before modern humans left Africa), or when migrating hominids encountered unfamiliar RNA viruses with their tendency to cross species barriers. These situations were probably few; individual viruses were not likely to have been important factors selecting for balanced polymorphisms in our line much before the Neolithic, if even then. The relative dearth of known resistance factors to any kind of pathogen reinforces this conclusion. The best‐known example of a specific parasite‐polymorphism relationship is falciparum malaria and the various alleles associated with resistance to it, including sickle‐cell and other hemoglobins and glucose‐6‐phosphate dehydrogenase (G6PD) variants. These polymorphisms appear to have evolved recently, however. Emergence of the ancestral strain of Plasmodium falciparum has been estimated at 3,200–7,700 years ago, since the introduction of agriculture in Africa (Volkman et al., 2001), and two of the most common G6PD polymorphisms are of about the same age (Tishkoff et al., 2001).
It is more likely that selection due to a number of pathogens collectively helped maintain general disease resistance and a strong immune response. Considering the range of environments Pliocene hominids probably exploited (Potts, 1998), they were likely to encounter a wide variety of viruses with the potential for interspecies transmission. The tendency for multiple infectious agents to share receptor sites (Baranowski et al., 2001) means that viruses may also have acted in concert with other pathogens to influence the evolution of cell surface properties. In this regard, the recent discovery that humans lack N‐glycolylneuraminic acid (Neu5Gc), a sialic acid receptor used by several viruses and other pathogens, is interesting (Varki, 2001; the possible inhibiting effect of Neu5Gc on brain development may also have contributed to selection against this receptor in hominids). Viruses that bind to Neu5Gc include some corona‐, rota‐, and influenza viruses, which explains why some strains (e.g., avian influenza; Webster, 1997) are not infectious in humans. Neu5Gc is not found in Neandertal fossils; the enzyme that produces it was inactivated in the human line by an Alu insertion estimated to have occurred 2.7–2.8 mya (Chou et al., 2002). It is tempting to link this with the behavioral shift to more meat‐eating in hominids, which would have increased their exposure to the viral families we now share with our domesticates and which are just the highly mutable agents that bind to sialic acids.
Another example of a cell‐surface mutation related to disease resistance is a 32‐base‐pair deletion in the CC chemokine receptor‐5 gene (CCR‐5), associated with resistance to HIV‐1 and possibly the plague bacillus. Homozygous individuals have nearly complete resistance to HIV infection, while heterozygotes experience delayed onset of symptoms (Stephens et al., 1998). The chemokine receptor CCR‐5 is a coreceptor (along with the CD4 protein) for HIV and related viruses in their entry into macrophages and initiation of infection. The mutant allele is present at high frequencies in some European populations, but is absent in Africans, Native Americans, and East Asians (Liu et al., 1996; Samson et al., 1996). With the use of coalescence theory to analyze the linkage disequilibrium between it and two microsatellite loci, the delta32 mutation has been dated to around 700 years ago, about the time of the 14th century Black Death in Europe, suggesting that the mutant allele may also confer some resistance to the plague (Stephens et al., 1998). If this is true, it indicates the levels of mortality (estimated to have been 25–33%) required for selection for such a polymorphism.
Along with other infectious diseases, viral infections would have helped maintain a strong immune response and promoted diversity in the major histocompatibility complex (MHC), T‐cell receptors, and B‐cells involved in antibody production, as well as in other loci that influence the quality of immune response (Zinkernagel et al., 1985; Hill and Motulsky, 1999). There would have been strong selective pressure against any reduction in the ability to resist infection (Svanborg‐Eden and Levin, 1990). The MHC, also known in humans as the human leukocyte antigen (HLA) complex, is the most highly polymorphic genetic system in mammals. These antigens are present on cell surfaces, and bind foreign antigen fragments for presentation to T‐lymphocyte receptors. MHC diversity results from meiotic recombination facilitated by high levels of heterozygosity; diversity in B‐cells and in T‐cell receptors is generated by somatic recombination, clonal selection, and higher than normal mutation rates, mechanisms thought to have evolved under pressure from parasites (Sarkar, 1992; Knapp, 2002). Balancing selection in the form of immune reactions between mother and fetus and negative assortative mating may be involved in maintaining MHC heterozygosity (Black and Hedrick, 1997; Ober, 1999; Penn and Potts, 1999; Wedekind and Penn, 2000); viral infection may also contribute. In mice, individuals infected with mouse hepatitis virus produce even more MHC heterozygotes (Rulicke et al., 1998). Descendants of Dutch colonists in Surinam who survived 19th century epidemics of typhoid and yellow fevers (with combined mortality ∼60%) have significantly higher heterogeneity at an MHC locus and others involved in control of the immune response than did a Dutch control sample (deVries et al., 1979). Individuals with high MHC heterozygosity have enhanced resistance to multiple parasites, while variation among individuals (favored by the presence of more alleles in a population) limits transmission of rapidly evolving viruses (Black, 1992). MHC mating preferences may produce a “moving target” against rapidly evolving parasites that would otherwise escape immune recognition (Penn and Potts, 1999). A clear correlation between MHC polymorphism and selection by infectious disease is difficult to prove, however, and other explanations of MHC‐dependent mate preferences include the need to avoid inbreeding (Penn and Potts, 1999; Wedekind and Penn, 2000).
The disassortative mating required to maintain high levels of MHC heterozygosity suggests that parasite‐mediated selection also affected behavior (Penn and Potts, 1999), although such nonrandom mating is not universal in animals (Wedekind and Penn, 2000). Some studies suggest that it may be mediated by mate choice in response to odor in mice (Howard, 1991; Potts et al., 1991) and in humans (Wedekind et al., 1995; Wedekind and Füri, 1997; Ober, 1999; Jacob et al., 2002). Negative assortative mating for MHC loci was not found among South American Indians, however (Hedrick and Black, 1997). In addition, South American Indians have fewer MHC alleles than other human populations (Black, 1990; Black and Hedrick, 1997), in part a legacy of founder effect and genetic drift associated with the peopling of the Americas, but which also correlates with their lower pre‐Columbian exposure to viral diseases (Van Blerkom, 1997).
Another behavior that might have evolved in response to disease is illustrated by less frequent intergroup transfer among rainforest primates with higher parasite loads (Freeland, 1979). Savanna baboons have a higher rate of exchange of individuals between groups, but also fewer species of intestinal protozoa (viruses were not examined in this study). Forest‐dwelling mangabeys, blue monkeys, and red‐tail monkeys, on the other hand, had more protozoan species, much lower rates of transfer, and greater intergroup differences in their parasites. Apparently in conditions of higher pathogen loads there has been disease‐related selection against traits that lead to high rates of transfer. Group members may also discriminate among potential new members in terms of probable disease risk. This phenomenon may be only one form of a group of avoidance behaviors that have evolved in animals in response to selection by infectious diseases; human examples might include negative reactions to the smell of feces and rotting flesh and an aversion to maggots (Curtis and Biran, 2001).
Interpopulation selection
In theory, infectious disease‐driven selection may not only maintain diversity within populations, but also promote divergence between populations, perhaps even to the point of speciation (Haldane, 1949; Clarke, 1976). If two populations share an infectious agent, it is advantageous for them to diverge from the more susceptible genotype. Shared infections can also lead to habitat partitioning, as a way of reducing interspecies transmission (Holt and Pickering, 1985).
Viruses may have affected hominid species diversity both by promoting such divergence and by weeding out less resistant host populations. Haldane (1949) was the first to propose that disease could confer a competitive advantage on a population that had some resistance. Such “parasite‐mediated competition” (a form of “apparent competition”) can influence community structure and eventually eliminate species, even in the absence of competition for resources (Holt and Pickering, 1985). Apparent competition due to disease has been demonstrated in a number of natural circumstances and often occurs when one host species invades the habitat of another. Evidence that introduced viruses can contribute to population decline in mammals includes the effects of parapoxvirus (carried by grey squirrels introduced from North America) on red squirrels in the UK (Sainsbury et al., 1997) and rinderpest in African ungulates (Dobson and Hudson, 1986). Canine distemper has been implicated in local extinctions of African wild dogs and black‐footed ferret populations, while canine parvovirus threatens gray wolves (Daszak et al., 2000), and both of these plus rabies are blamed for declines in Ethiopian wolves (Hudson and Greenman, 1998).
Mathematical models of apparent competition suggest that shared pathogens are capable of limiting host species diversity, provided certain criteria are met. Requirements for parasite‐mediated exclusion of one host species by another include the following (Holt and Pickering, 1985; Holt and Lawton, 1994):
-
1
Host species with shared pathogen(s);
-
2
Both within‐ and cross‐species transmission;
-
3
Differences in host reproductive rates of (uninfected individuals); and
-
4
Differences in host susceptibility to infection or tolerance to the disease (birth, death, and recovery rates of infected individuals).
Given the kinds of viruses likely to have been present, could they have affected hominid species diversity during the Plio‐Pleistocene? The first requirement (shared viruses) was more than likely met. The frequency with which retroviruses, for example, have crossed species lines among catarrhine primates makes it not improbable that viruses were shared by hominids in the past. The potential for shared viruses is more pronounced in closely related species, as susceptibility to available pathogens increases with phylogenetic relatedness (Holt and Pickering, 1985). Once hominids began exploiting more open environments, the sharing of parasites may have been even easier, as a survey of African cercopithecoids found that groups of savanna baboons were more likely to share protozoal parasites than were forest monkeys (Freeland, 1979).
Host population declines are more likely when the shared pathogen is not limited by resources other than susceptible hosts and there is a strong numerical response to host density (Holt and Lawton, 1994; Hudson and Greenman, 1998). Generalist pathogens (widely present in the environment or in many other species) are usually not as powerful to depress or exclude host species as are more specific ones (i.e., specific to the host species in apparent competition). Viruses implicated in apparent competition today [canine distemper, rinderpest (both paramyxoviruses), squirrel parapox (a poxvirus), canine parvovirus, and rabies] are, with the exception of rabies, all fairly host‐specific, confining their range mostly to within a particular mammalian order. Candidate viruses that specialized in infecting primates during the Plio‐Pleistocene probably included herpes‐, papilloma‐, adeno‐, parvo‐, and picornaviruses, plus PTLVs and various hepatitis viruses.
The effects of shared parasites on population structure are a function of the frequencies of transmission within and between species in apparent competition (condition 2; Holt and Pickering, 1985), with exclusion being favored by greater interspecific communication. This would appear to require frequent interspecies contact, at least for contact infections. This is generally rare but could be more common when individuals are evenly spread within territories and the territories of different species can overlap (Hudson and Greenman, 1998). The likelihood of between‐species transmission is also increased when pathogens have long‐lived free‐living stages, cause chronic carrier states in a reservoir host, or are vector‐ or water‐borne. Behaviors such as hunting or scavenging also increase the probability of interspecies transmission. Indeed, when hominids became serious meat‐eaters, they would have been exposed more frequently to other species' pathogens (McMichael, 2001), including those of other hominids if such carcasses were scavenged. Considering the zeal with which chimpanzees hunt colobus monkeys and some Africans enjoy bushmeat, the discovery that 20% of wild monkeys in Cameroon are SIV‐infected suggests a considerable potential for transmission of infection among hominids and other primates in the past (Peeters et al., 2002).
Intraspecies transmission is also required. Some populations may be highly susceptible to infection but incapable of supporting transmission within the population, in which case serious disease‐mediated consequences are unlikely. Many zoonotic infections (such as rabies) produce dead‐end infections in humans, and the chain of transmission is broken; these are unlikely to have affected hominid diversity. Any viruses involved in parasite‐mediated competition must have been able to spread in those hosts.
Conditions 3 and 4 relate the probability of exclusion to relative susceptibility and resistance of host populations, and to the birth and death rates of both infected and uninfected host individuals. If all else is equal, the host with the higher intrinsic growth rate when uninfected will be dominant and exclude the other; populations with lower growth rates are more vulnerable to disease‐mediated decline (Holt and Pickering, 1985). Reproductive rates can be low because of long generation length, low clutch size, or scarcity of resources (due to living at the edge of one's niche or close to its geographical limits, specializing in scarce resources, or engaging in interspecific competition; Holt and Lawton, 1994). Hominids were no doubt characterized by several of these conditions, giving species with a reproductive “edge” an advantage for disease‐mediated competition as well.
Exclusion is also favored if the other host has a lower susceptibility to infection or higher tolerance to the disease (i.e., faster recovery, lower death rate, or higher reproductive rate when infected). The host that by itself can sustain a higher density of infected individuals can depress or exclude an alternate host (Holt and Pickering, 1985), e.g., by being a reservoir (Holt and Lawton, 1994; Dobson and Hudson, 1986). Viruses frequently affect inexperienced populations more seriously than they do their usual hosts, so the introduction of a new pathogen by an in‐migrating host can have severe consequences for native species. Isolation increases the potential for die‐off upon first contact, although secondary transmission is less likely among animals living singly or in small family groups than among those living in large communities (Haldane, 1949). In addition, genetically more diverse populations are generally more resistant (May, 1983; Hudson and Greenman, 1998).
This implies parasites with considerable virulence, which often results when viruses of one species are introduced into another. Interspecies transmission of herpesviruses, for example, can be deadly for the new host. The rhesus strain of monkey B virus is lethal in nonmacaques (Smith et al., 1998), and human herpesvirus type 1 (HHV‐1) is fatal in owl monkeys and occasionally in other primates (gibbons, lemurs) (Skinner et al., 2001; Huemer et al., 2002). An African elephant herpesvirus kills young Asian elephants in zoos, and an Asian virus is suspected in the death of an infant African elephant (Ferber, 1999). Many RNA viruses (e.g., SIVs and hepatitis viruses) cause inapparent infections in reservoir hosts, but produce fatal consequences in other species.
Thus virus‐mediated apparent competition could have played a role in human evolution. Such competition among hominid populations in the Pliocene may have helped prune some of the branches of our phylogenetic tree (with emphasis on helped, as disease is likely to have been only one factor), but it may also have promoted habitat partitioning and speciation. Scavenging and/or hunting would have increased the potential for transmission of viruses between species. One can only speculate about the other kinds of interspecies contact that may have taken place. Infectious disease may have played an important role in shaping hominid species diversity during the Pliocene.
Later hominids may have carried viruses out of Africa and introduced them into inexperienced hominid populations in Asia and Europe (e.g., Neandertals and other archaic forms). MacPhee and Marx (1997) proposed this as a factor in late Quaternary faunal extinctions, whereby “hyperdisease” carried by migrating humans or associated fauna (especially dogs) contributed to a number of extinctions in the New World and elsewhere over the last 40,000 years. Viruses carried by domesticated dogs have been implicated in population declines and local extinctions of a number of wild carnivores (Pain, 1997; Daszak et al., 2000), and dogs carry many diseases transmissible to humans as well. The time at which humans acquired dogs is not yet settled, however. An mtDNA study of wolves and dogs suggested it was well before the earliest osteological evidence of roughly 14 kya (Vilà et al., 1999), but more recent work locates the earliest domestic dogs somewhere around 15 (or possibly 40) kya in East Asia (Savolainen, 2002), probably too late to have been a major source of disease for Neandertals but within the realm of possibility for New World faunal extinctions. Rabies virus meets all the hyperdisease requirements proposed by MacPhee and Marx (1997) (reservoir species with stable carrier state, epizootic potential, mortality rates in excess of 75%, and ability to infect multiple species without seriously threatening humans); indeed, rabies is one of the most threatening diseases for wildlife when introduced into a new area (Hudson and Greenman, 1998). Rabies may be a good candidate for mediating competition among carnivores, but its lack of intraspecific transmission in humans limits its potential for affecting population structure in hominids.
Migrating modern humans probably carried HTLVs and herpesviruses, if not others that produced chronic, inapparent infections in their long‐standing host but that might have proven dangerous to other hominids, e.g., hepatitis, genital papillomas, and picornaviruses such as enteroviruses and rhinoviruses. Any such virus should now be worldwide in distribution as a consequence of its spread during the human diaspora (MacPhee and Marx, 1997). In connection with rhinoviruses, it is interesting to note that differences in nasal aerodynamics have been correlated with susceptibility to these agents (Uliyanov, 1998). In particular, individuals with nasal airflow directed more toward the inferior rather than the medial nasal passages are not as efficient at filtering or warming inspired air, and are thus more likely to catch colds. Recent work on Neandertal nasal anatomy suggests that they may have had this airflow pattern (Churchill et al., 1999).
Other animal viruses may have inadvertently spread at the same time as humans; people were not the only thing expanding out of Africa. Migrating fauna would have brought many of their parasites with them. The rabies viruses of European bats and carnivores appear to be derived from lyssaviruses of African bats (Amengual et al., 1997). Arthropod vector distributions expanded as climates changed, as did reservoir hosts; this may have been when mosquito‐borne flaviviruses began spreading out of Africa.
Pathogens are more effective as regulatory agents among newly introduced species or when the parasites are introduced into new regions (May, 1983). They can cause both catastrophic population decline plus chronic population depression if threshold host density for transmission is low enough. In the end, it is unlikely that disease alone causes extinction (remember the rabbits in Australia), but it can cause sufficient population depression to render populations more susceptible to it, especially in combination with other stressors. Only a small demographic advantage is required to result in extinction. A mortality differential of only 2% between Neanderthals and modern humans could have led to extinction in roughly 30 generations, or 1,000 years (Zubrow, 1989).
Molecular genetic parasites
Viruses may also have affected human evolution as agents of genetic change via recombination and gene conversion. With their close association with cellular DNA, viruses have been in a position to directly affect the genomes of their hosts. One way this has happened is through recombination with cellular DNA. Most of this would have occurred in very early stages of organismal evolution, however, and it is now more likely in plants; it requires germ‐cell infection to produce inherited variation in higher animals. Similarities between viral and cellular DNA polymerases suggest frequent recombination and “gene capture” at some far distant era in the past, however (McGeoch, 1989; Villareal, 1999). Viruses may have been “gene‐shufflers” during early stages in the evolution of life, and as such may have played an active role in molding early genomes (Balter, 2000).
One type of virus has continued to be such an agent during primate and hominid evolution, however. The capacity of retroviruses to insert their genetic material into cellular DNA and to retrotranspose to new locations gives them a significant capacity to modify host genomes. Reflecting the great antiquity of retroviruses and retroid elements, these endogenous retroviruses are found in all eukaryotes, and reverse transcriptase may have been involved in the earliest phases of life (McClure, 1995). They appear to have been especially active during primate evolution. Human endogenous retroviruses (HERVs), remnants of ancient germ‐cell infections occurring 5–50 mya, make up about 1% of the human genome (Wilkinson et al., 1994; Löwer et al., 1996; Johnson and Coffin, 1999; Sverdlov, 1998). A number of HERV families are estimated to have integrated at different times and from different exogenous viruses (Table 2). They are not as common in platyrrhines as in catarrhines, and therefore most are estimated to be ∼30–40 million years old (Costas and Naveira, 2000). Highly expressed in placental tissue and certain tumors, some of these sequences result in pathological conditions such as autoimmune disease, leukemia, and other cancers (Wilkinson et al., 1994; Weiss et al., 1999), while others are involved in the regulation of host gene expression, placental formation, and immunosuppression during pregnancy (Table 2). An especially well‐studied example of HERV genetic effects is the tissue‐specific expression of salivary and pancreatic amylases, mediated by insertion of an endogenous retrovirus into the amylase gene (Fig. 15; Samuelson et al., 1990; Ting et al., 1992).
Table 2.
Human endogenous retrovirus families1
| Copy number | Also found in | Biological roles | |
|---|---|---|---|
| Class I | |||
| HERV‐E | 35–50 | Apes and Old World monkeys | Salivary amylase gene expression2 |
| HERV‐H | 50–100 intact, ∼900 deleted | Apes and Old World monkeys | LTRs involved in gene regulation in placenta3 |
| HERV‐I | ∼25 | Apes and Old World monkeys | LTR may help regulate cytochrome c1 gene4 |
| HERV‐P | 10–20 each of three types | Apes, OW and NW monkeys | Not known |
| HERV‐R | 1 | Apes and Old World monkeys | Immunosuppression in placenta (env)5 |
| HERV‐W | Multiple copies | Apes and Old World monkeys? | env gene product involved in placental morphogenesis6 |
| ERV‐9 | 30–40; at least 4,000 LTRs | Apes and Old World monkeys | LTRs may be involved in gene regulation7 |
| Class II | |||
| HERV‐K | 30–50 intact, ∼ 25,000 solitary LTRs | Apes and OW monkeys; some are human‐specific | LTRs may be involved in gene regulation; expressed in placenta8 |
Class I, more closely related to murine leukemia virus; class II, related to mouse mammary tumor virus. Families defined by type of tRNA that binds to 5′ LTR primer binding site. General references: Shih et al., 1991; Wilkinson et al., 1994; Löwer et al., 1996. NW, New World; OW, Old World.
Goodchild et al., 1992.
Suzuki et al., 1990.
Venables et al., 1995.
Costas and Naveira, 2000.
Figure 15.
Tissue‐specific gene expression mediated by HERV insertion: salivary amylase. Enhancer sequences in ERVA1 LTR inserted into 5′ flanking region of pancreatic amylase gene (AMY2) activate cryptic promoter in adjoining γ‐actin pseudogene and result in expression of salivary amylase in parotid gland (AMY1). Insertion estimated at 39 mya in an anthropoid ancestor (Samuelson et al., 1990; Ting et al., 1992).
Particularly interesting is the HERV‐K family, a transpositionally active group found in Old World monkeys and apes and which includes a number of sequences inserted into the human genome since the divergence of humans and chimpanzees (Barbulescu et al., 1999; Costas, 2001). These uniquely human proviruses and solitary long terminal repeats (LTRs) may have played a role in hominid evolution (Lebedev et al., 2000). HERV‐K LTRs are capable of affecting the expression of nearby genes (Löwer et al., 1996; Baust et al., 2000). They contain potential hormone response elements, enhancers, promoters, polyadenylation signals, and transcription factor binding sites that could be involved in the regulation of adjacent genes if inserted in an appropriate context (Sverdlov, 2000). HERV‐K LTRs can bind host‐cell nuclear proteins and have the potential to activate neighboring genes (Akopov et al., 1998). They can act as switches for alternative splicing (e.g., alternate forms of the leptin receptor) and therefore be involved in transcriptional and posttranscriptional regulation of gene expression (Kapitonov and Jurka, 1999). Some HERV‐K proviral genes are expressed in humans (Barbulescu et al., 1999).
These HERVs resemble no known human exogenous viruses; the ancestral viruses appear to have been eliminated (Wilkinson et al., 1994). A possible selective advantage is reduced susceptibility to infection with exogenous retroviruses (Löwer et al., 1996). That HERV proviral genes are expressed only in embryonic or placental tissue suggests a potential role in early development (and perhaps important species differences).
CONCLUSIONS
Our ancestors were liable to infection with a number of viruses throughout our evolutionary history. While our most ancient and coevolving DNA viruses were well‐adapted to hominids and unlikely to cause more than persistent mild infections, a variety of emerging RNA viruses undoubtedly affected hominids, some as isolated zoonotic infections incapable of being transmitted directly to other hominids, and others as new acquisitions via interspecies transmission. Some of these adapted to their new host and perhaps were shared with other species.
Even the earliest hominids carried endogenous retroviruses capable of acting as insertional mutagens. The existence of unique human HERV‐K sequences suggests that retrotranspositional events since the human‐ape divergence may have helped mold the hominid genome. The tendency for proviral genes to be expressed in placental and embryonic tissues suggests the possibility of changes in gene expression affecting development.
Until human and associated animal populations increased in size and density in the Neolithic, specific viruses were unlikely to have selected for balanced polymorphisms like those conferring resistance to falciparum malaria. In combination with other infectious agents, however, viruses must have contributed to selection pressure maintaining a strong immune response (including MHC diversity) and favoring changes in cell surface receptors. Given the range of environments Pliocene hominids exploited and the likelihood they engaged in scavenging or hunting, they were probably exposed to a wide variety of zoonotic and emerging parasites.
The late Miocene/early Pliocene epochs saw a radiation of hominids, and there was undoubtedly competition among some of them. Viruses and other parasites may have contributed to both this proliferation of species and the subsequent pruning of the hominid family tree through apparent competition mediated by parasites. Herpes‐, papilloma‐, adeno‐, picorna‐ and hepatitis viruses were all probably present in African anthropoids and could have been involved. At some point before the exodus of anatomically modern humans out of Africa, the human lineage acquired, in addition, HTLV‐I and ‐II, poliovirus, and perhaps smallpox. Hominids probably met up with RNA viruses that are readily transmitted across species lines and cause diarrheal and respiratory conditions (e.g., rotaviruses, caliciviruses, and coronaviruses).
When hominids began migrating out of Africa, they would have encountered a new cast of viruses. They would also have carried with them many of the old. This “pathogen pollution” may have contributed to faunal extinctions following first contact, and in the case of anatomically modern humans, other hominids they encountered might have been vulnerable as well. Parasite‐mediated competition might have resulted from viruses carried by humans (herpes simplex, HTLV, hepatitis, polio, and rhinoviruses), and differential susceptibility to zoonoses emergent in an environment both populations were using (e.g., rabies), or agents carried along in comigrating animals (dogs?) and arthropods (e.g., mosquito‐borne flaviviruses) could hasten the demise of already foundering populations. Infectious disease, while not likely to cause epidemic spread in small scattered Pleistocene populations, may have added to the effects of climate change and nutritional stress to tip the balance toward local extinction in Neandertals and other archaic human populations competing with anatomically modern humans.
Humans acquired many more viruses during the Neolithic, primarily from domestic and commensal animals, but by this time biological evolution in our species had become relatively unimportant. Homo sapiens was the only surviving lineage, and parasite‐mediated competition became a way for more immunologically sophisticated populations to overrun those less so, as in the cases of Native Americans and Australians confronted with European invaders. That the two sides were of the same species helped keep the conquered populations from total extinction (although not the Tasmanians or some American tribes); interbreeding may have helped save the outcompeted populations. Neandertals may have had no such safety net, or if they did, it was not enough to save them.
This study has only begun to consider the effects of disease on human evolution. Much more needs to be done. In virology, we need more work deciphering the evolutionary histories of the most common human viruses and determining just what role the unique human HERV‐K insertions might play in the human genome. In the area of ancient DNA, it might be possible to use molecular probes to search for viral nucleic acids in well‐preserved fossil remains, although this would be useful in studying only more recent events. For example, researchers working on late Pleistocene extinctions have recovered and sequenced endogenous retroviruses of mammoths by this means (Greenwood et al., 2001).
Sophisticated mathematical models of parasite‐mediated competition have been derived (Holt and Pickering, 1985; Holt and Lawton, 1994), and these might generate testable hypotheses concerning the possibility of such competition during human evolution. And finally, this analysis could be extended to bacterial, protozoal, and other human infections (e.g., recent work on trypanosomes, malaria plasmodia, tapeworms, and bacteria that cause tuberculosis, dysentery, and stomach ulcers suggests that many human diseases are surprisingly old; Zimmer, 2001), although these agents, given their larger genomes, are not as easy to study as viruses. With growing interest in the coevolution of humans and their disease, this topic is not likely to be ignored.
Acknowledgements
I thank Alan Swedlund for his encouragement and review of an earlier manuscript, and Todd Disotell, Paul Leslie, and Sara Stinson for their thoughtful reviews and helpful editorial suggestions. I also gratefully acknowledge Chris Stringer for critical discussion and advice during the gestation of this study, and Stan Ambrose for his initial skepticism, which motivated me to overcome it.
This article is a US Government work and, as such, is in the public domain in the United States of America.
Footnotes
This paper uses traditional taxonomic nomenclature, by which the term “hominid” refers to humans and their bipedal relatives, while apes and humans are included in the hominoids.
LITERATURE CITED
- Adams NJ, Prescott LE, Jarvis LM, Lewis JCM, McClure MO, Smith DB, Simmonds P. 1998. Detection in chimpanzees of a novel flavivirus related to GB virus‐C/hepatitis G virus. J Gen Virol 79: 1871–1877. [DOI] [PubMed] [Google Scholar]
- Akopov SB, Nikolaev LG, Khil PP, Lebedev YB, Sverdlov ED. 1998. Long terminal repeats of human endogenous retrovirus K family (HERV‐K) specifically bind host cell nuclear proteins. FEBS Lett 421: 229–233. [DOI] [PubMed] [Google Scholar]
- Amengual B, Whitby JE, King A, Cobo JS, Bourhy H. 1997. Evolution of European bat lyssaviruses. J Gen Virol 78: 2319–2328. [DOI] [PubMed] [Google Scholar]
- Armelagos GJ. 1990. Health and disease in prehistoric populations in transition In: Swedlund AC, Armelagos GJ, editors. Disease in populations in transition. New York: Bergin & Garvey; p 127–144. [Google Scholar]
- Armelagos GJ, Dewey JR. 1978. Evolutionary response to human infectious diseases In: Logan MB, Hunt EE, Sr, editors. Health and the human condition: perspectives in medical anthropology. Belmont, CA: Wadsworth Publishing Co. p 101–107. [Google Scholar]
- Armelagos GJ, Ryan M, Leatherman T. 1990. Evolution of infectious disease: a biocultural analysis of AIDS. Am J Hum Biol 2: 353–363. [DOI] [PubMed] [Google Scholar]
- Bailes E, Gao F, Bibollet‐Ruche F, Courgnaud V, Peeters M, Marx PA, Hahn BH, Sharp PM. 2003. Hybrid origin of SIV in chimpanzees. Science 300: 1713. [DOI] [PubMed] [Google Scholar]
- Bailey A, Mautner V. 1994. Phylogenetic relationships among adenovirus serotypes. Virology 205: 438–452. [DOI] [PubMed] [Google Scholar]
- Balter M. 2000. Evolution on life's fringes. Science 289: 1866–1867. [DOI] [PubMed] [Google Scholar]
- Baranowski E, Ruiz‐Jarabo CM, Domingo E. 2001. Evolution of cell recognition by viruses. Science 292: 1102–1105. [DOI] [PubMed] [Google Scholar]
- Barbulescu M, Turner G, Seaman MI, Deinard AS, Kidd KK, Lenz J. 1999. Many human endogenous retrovirus K (HERV‐K) proviruses are unique to humans. Curr Biol 9: 861–868. [DOI] [PubMed] [Google Scholar]
- Baskin Y. 2000. A sickening situation: emerging pathogens pose a threat to wildlife. Nat Hist 2000; 109: 24–27. [Google Scholar]
- Baust C, Seifarth W, Germaier H, Hehlmann R, Leib‐Mosch C. 2000. HERV‐K‐T47D‐related long terminal repeats mediate polyadenylation of cellular transcripts. Genomics 66: 98–103. [DOI] [PubMed] [Google Scholar]
- Benenson AS, editor. 1995. Control of communicable diseases manual, 16th ed. Washington, DC: American Public Health Association. [Google Scholar]
- Black FL. 1975. Infectious diseases in primitive societies. Science 187: 515–518. [DOI] [PubMed] [Google Scholar]
- Black FL. 1990. Infectious disease and evolution of human populations: the example of South American forest tribes In: Swedlund AC, Armelagos GJ, editors. Disease in populations in transition. New York: Bergin & Garvey; p 55–74. [Google Scholar]
- Black FL. 1992. Why did they die? Science 258: 1739–1740. [DOI] [PubMed] [Google Scholar]
- Black FL. 1997. Tracing prehistoric migrations by the viruses they carry: human T‐cell lymphotropic viruses as markers of ethnic relationships. Hum Biol 69: 467–482. [PubMed] [Google Scholar]
- Black FL, Hedrick PW. 1997. Strong balancing selection at HLA loci: evidence from segregation in South Amerindian families. Proc Natl Acad Sci USA 94: 12452–12456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blancou P, Vartanian J‐P, Christopherson C, Chenciner N, Basilico C, Kwok S, Wain‐Hobson S. 2001. Polio vaccine samples not linked to AIDS. Nature 410: 1045–1046. [DOI] [PubMed] [Google Scholar]
- Blasco R. 1995. Evolution of poxviruses and African swine fever virus In: Gibbs AJ, Calisher CH, Garcia‐Arenal F, editors. Molecular basis of virus evolution. Cambridge: Cambridge University Press; p 255–269. [Google Scholar]
- Blixenkrone‐Möller M, Bolt G, Gottschalck E, Kenter M. 1994. Comparative analysis of the gene encoding the nucleocapsid protein of dolphin morbillivirus reveals its distant evolutionary relationship to measles virus and ruminant morbilliviruses. J Gen Virol 75: 2829–2834. [DOI] [PubMed] [Google Scholar]
- Blond J‐L, Besème F, Duret L, Bouton O, Bedin F, Perron H, Mandrand B, Mallet F. 1999. Molecular characterization and placental expression of HERV‐W, a new human endogenous retrovirus family. J Virol 73: 1175–1185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bodmer WF. 1972. Evolutionary significance of the HL‐A system. Nature 237: 139–145, 183. [DOI] [PubMed] [Google Scholar]
- Bodmer WF, Bodmer JG. 1978. Evolution and function of the HLA system. Br Med Bull 34: 309–316. [DOI] [PubMed] [Google Scholar]
- Bollyky PL, Holmes EC. 1999. Reconstructing the complex evolutionary history of hepatitis B virus. J Mol Evol 49: 130–141. [DOI] [PubMed] [Google Scholar]
- Bourhy H, Kissi B, Tordo N. 1993. Molecular diversity of the Lyssavirus genus. Virology 194: 70–81. [DOI] [PubMed] [Google Scholar]
- Brown AJL. 1994. Methods of evolutionary analysis of viral sequences In: Morse SS, editor. The evolutionary biology of viruses. New York: Raven Press; p 75–84. [Google Scholar]
- Brunet M, Guy F, Pilbeam D, Mackaye HT, Likius A, Ahounta D, Beauvilain A, Blondel C, Bocherens H, Boisserie J‐R, De Bonis L, Coppens Y, Dejax J, Denys C, Duringer P, Elsenmann V, Fanone G, Fronty P, Geraads D, Lehmann T, Lihoreau F, Louchart A, Mahamat A, Merceron G, Mouchelin G, Otero O, Campomanes PP, Ponce De Leon M, Rage J‐C, Sepanet M, Schuster M, Sudre J, Tassy P, Valentin X, Vignaud P, Viriot L, Zazzo A, Zollikofer C. 2002. A new hominid from the upper Miocene of Chad, Central Africa. Nature 418: 145–151. [DOI] [PubMed] [Google Scholar]
- Burnet M, White DO. 1972. Natural history of infectious disease, 4th ed. Cambridge: Cambridge University Press. [Google Scholar]
- Bush RM, Bender CA, Subbarao K, Cox NJ, Fitch WM. 1999. Predicting the evolution of human influenza A. Science 286: 1921–1925. [DOI] [PubMed] [Google Scholar]
- Capasso L. 1993. Fossil mosquitoes and the spread of infectious diseases in man's ancestors. J Paleopathol 3: 171–201. [Google Scholar]
- Cavalli‐Sforza LL, Menozzi P, Piazza A. 1994. The history and geography of human genes. Princeton: Princeton University Press. [Google Scholar]
- Chan S‐Y, Bernard H‐U, Ong C‐K, Chan S‐P, Hofmann B, Delius H. 1992. Phylogenetic analysis of 48 papillomavirus types and 28 subtypes and variants: a showcase for the molecular evolution of DNA viruses. J Virol 66: 5714–5725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan S‐Y, Bernard H‐U, Ratterree M, Birkebak TA, Faras AJ, Ostrow RS. 1997. Genomic diversity and evolution of papillomaviruses in rhesus monkeys. J Virol 71: 4938–4943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Z, Luckay A, Sodora DL, Telfer P, Reed P, Gettie A, Kanu JM, Sadek RF, Yee J, Ho DD, Zhang L, Marx PA. 1997. Human immunodeficiency virus type 2 (HIV‐2) seroprevalence and characterization of a distinct HIV‐2 genetic subtype from the natural range of simian immunodeficiency virus‐infected sooty mangabeys. J Virol 71: 3953–3960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chou H‐H, Hayakawa T, Diaz S, Krings M, Indriati E, Leakey M, Pääbo S, Satta Y, Takahata N, Varki A. 2002. Inactivation of CMP‐N‐acetylneuraminic acid hydroxylase occurred prior to brain expansion during human evolution. Proc Natl Acad Sci USA 99: 11736–11741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chua KB, Bellini WJ, Rota PA, Harcourt BH, Tamin A, Lam SK, Ksiazek TG, Rollin PE, Zaki SR, Shieh W‐J, Goldsmith CS, Gubler DJ, Roehrig JT, Eaton B, Gould AR, Olson J, Field H, Daniels P, Ling AE, Peters CJ, Anderson LJ, Mahy BWJ. 2000. Nipah virus: a recently emergent deadly paramyxovirus. Science 288: 1432–1435. [DOI] [PubMed] [Google Scholar]
- Churchill SE, Shackelford LL, Georgi JN, Black MT. 1999. Airflow dynamics in the Neandertal nose. J Hum Evol 36: A5 [abstract]. [Google Scholar]
- Clarke B. 1976. The ecological genetics of host‐parasite relationships In: Taylor AER, Muller R, editors. Genetic aspects of host‐parasite relationships Symposia of the British Society for Parasitology, volume 14 Oxford: Blackwell Scientific; p 87–103. [Google Scholar]
- Cockburn A. 1967a. Infections of the order primates In: Cockburn A, editor. Infectious diseases: their evolution and eradication. Springfield, IL: Charles C. Thomas; p 38–49. [Google Scholar]
- Cockburn A. 1967b. The evolution of human infectious diseases In: Cockburn A, editor. Infectious diseases: their evolution and eradication. Springfield, IL: Charles C. Thomas; p 84–107. [Google Scholar]
- Corbet S, Müller‐Trutwin MC, Versmisse P, Delarue S, Ayouba A, Lewis J, Brunak S, Martin P, Brun‐Vezinet F, Simon F, Barre‐Sinoussi F, Mauclere P. 2000. env sequences of simian immunodeficiency viruses from chimpanzees in Cameroon are strongly related to those of human immunodeficiency virus group N from the same geographic area. J Virol 74: 529–534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Costas J. 2001. Evolutionary dynamics of the human endogenous retrovirus family HERV‐K inferred from full‐length proviral genomes. J Mol Evol 53: 237–243. [DOI] [PubMed] [Google Scholar]
- Costas J, Naveira H. 2000. Evolutionary history of the human endogenous retrovirus family ERV9. Mol Biol Evol 17: 320–330. [DOI] [PubMed] [Google Scholar]
- Crainic R, Kew O. 1993. Evolution and polymorphism of poliovirus genomes. Biologicals 21: 379–384. [DOI] [PubMed] [Google Scholar]
- Cunliffe NA, Woods PA, Leite JPG, Das BK, Ramachandran M, Bhan MK, Hart CA, Glass RI, Gentsch JR. 1997. Sequence analysis of NSP4 gene of human rotavirus allows classification into two main genetic groups. J Med Virol 53: 41–50. [PubMed] [Google Scholar]
- Curtis V, Biran A. 2001. Dirt, disgust, and disease. Is hygiene in our genes? Perspect Biol Med 44: 17–31. [DOI] [PubMed] [Google Scholar]
- Daszak P, Cunningham AA, Hyatt AD. 2000. Emerging infectious diseases of wildlife—threats to biodiversity and human health. Science 287: 443–449. [DOI] [PubMed] [Google Scholar]
- Davison AJ. 2002. Evolution of the herpesviruses. Vet Microbiol 86: 69–88. [DOI] [PubMed] [Google Scholar]
- de Souza Leal É, Zanotto PMA. 2000. Viral diseases and human evolution. Mem Inst Oswaldo Cruz [Suppl] 95: 193–200. [DOI] [PubMed] [Google Scholar]
- de Souza Trindade G, da Fonseca FG, Marques JT, Nogueira ML, Mendes LCN, Borges AS, Peiró JR, Pituco EM, Bonjardim CA, Ferreira PCP, Kroon EG. 2003. Araçatuba virus: a vaccinialike virus associated with infection in humans and cattle. Emerg Infect Dis [serial online] 2003; 9 [cited February 10, 2003]. Available from URL: http://www.cdc.gov/ncidod/EID/vol9no2/02-0244.htm. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Vries RRP, MeeraKhan P, Bernini LF, van Loghem E, van Rood JJ. 1979. Genetic control of survival in epidemics. J Immunogenet 6: 271–287. [DOI] [PubMed] [Google Scholar]
- Dobson AP, Hudson PJ. 1986. Parasites, disease and the structure of ecological communities. Trends Ecol Evol 1: 11–14. [DOI] [PubMed] [Google Scholar]
- Doherty M, Todd D, McFerran N, Hoey EM. 1999. Sequence analysis of a porcine enterovirus serotype 1 isolate: relationships with other picornaviruses. J Gen Virol 80: 1929–1941. [DOI] [PubMed] [Google Scholar]
- Domingo E, Holland J. 1994. Mutation rates and rapid evolution of RNA viruses In: Morse SS, editor. The evolutionary biology of viruses. New York: Raven Press; p 161–184. [Google Scholar]
- Domingo E, Martínez‐Salas E, Sobrino F, de la Torre JC, Portela A, Ortin J, López‐Galindez C, Pérez‐Breña P, Villanueva N, Nájera R, VandePol S, Steinhauer D, DePolo N, Holland J. 1985. The quasispecies (extremely heterogeneous) nature of viral RNA genome populations: biological relevance—a review. Gene 40: 1–8. [DOI] [PubMed] [Google Scholar]
- Domingo E, Holland J, Biebricher C, Eigen M. 1995. Quasi‐species: the concept and the word In: Gibbs AJ, Calisher CH, García‐Arenal F, editors. Molecular basis of virus evolution. Cambridge: Cambridge University Press; p 181–191. [Google Scholar]
- Domingo E, Baranowski E, Ruiz‐Jarabo CM, Martin‐Hernández AM, Sáiz JC, Escarmís C. 1998. Quasispecies structure and persistence of RNA viruses. Emerg Infect Dis [serial online] 1998; 4 [cited July 2,1999]. Available from URL: http://www.cdc.gov/ncidod/eid/vol4no4/domingo.htm. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Domingo E, Escarmís C, Menéndez‐Arias L, Holland JJ. 1999. Viral quasispecies and fitness variations In: Domingo E, Webster R, Holland J, editors. Origin and evolution of viruses. London: Academic Press; p 141–162. [Google Scholar]
- Douglas NJ, Dumbell KR. 1996. DNA sequence variation as a clue to the phylogenesis of orthopoxviruses. J Gen Virol 77: 947–951. [DOI] [PubMed] [Google Scholar]
- Dube DK, Sherman MP, Saksena NK, Bryz‐Gornia V, Mendelson J, Love J, Arnold CB, Spicer T, Dube S, Glaser JB, Williams AE, Nishimura M, Jacobsen S, Ferrer JF, del Pino N, Quiruelas S, Poiesz BJ. 1993. Genetic heterogeneity in human T‐cell leukemia/lymphoma virus type II. J Virol 67: 1175–1184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duncan R. 1999. Extensive sequence divergence and phylogenetic relationships between the fusogenic and nonfusogenic orthoreoviruses: a species proposal. Virology 260: 316–328. [DOI] [PubMed] [Google Scholar]
- Eigen M. 1993. Viral quasispecies. Sci Am 1993; 269: 42–49. [DOI] [PubMed] [Google Scholar]
- Enserink M. 2000. Malaysian researchers trace Nipah virus outbreak to bats. Science 289: 518–519. [DOI] [PubMed] [Google Scholar]
- Enserink M. 2003. Clues to the animal origins of SARS. Science 300: 1351. [DOI] [PubMed] [Google Scholar]
- Ewald PW. 1994. Evolution of infectious disease. New York: Oxford University Press. [Google Scholar]
- Fenner F, Kerr PJ. 1994. Evolution of the poxviruses, including the coevolution of virus and host in myxomatosis In: Morse SS, editor. The evolutionary biology of viruses. New York: Raven Press; p 273–292. [Google Scholar]
- Fenner F, McAuslan BR, Mims CA, Sambrook J, White DO. 1974. The biology of animal viruses, 2nd ed. New York: Academic Press. [Google Scholar]
- Fenner F, Henderson DA, Arita I, Jezek Z, Ladnyi ID. 1988. Smallpox and its eradication. Geneva: World Health Organization. [Google Scholar]
- Ferber D. 1999. Virus suspect identified in elephant deaths. Science 283: 1093–1094. [DOI] [PubMed] [Google Scholar]
- Fitch WM, Bush RM, Bender CA, Cox NJ. 1997. Long term trends in the evolution of H(3) HA1 human influenza type A. Proc Natl Acad Sci USA 94: 7712–7718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fomsgaard S, Müller‐Trutwin MC, Diop O, Hansen J, Mathiot C, Corbet S, Barré‐Sinoussi F, Allan JS. 1997. Relation between phylogeny of African green monkey CD4 genes and their respective simian immunodeficiency virus genes. J Med Primatol 26: 120–128. [DOI] [PubMed] [Google Scholar]
- Freeland WJ. 1979. Primate social groups as biological islands. Ecology 60: 719–728. [Google Scholar]
- Gamble ER. 1997. Family: Parvoviridae. Available at URL: http://www.greenapple.com/~rgamble/parvo.htm [cited July 29, 1999].
- Gao F, Bailes E, Robertson DL, Chen Y, Rodenburg CM, Michael SF, Cummins LB, Arthur LO, Peeters M, Shaw GM, Sharp PM, Hahn BH. 1999. Origin of HIV‐1 in the chimpanzee Pan troglodytes troglodytes . Nature 397: 436–441. [DOI] [PubMed] [Google Scholar]
- Gaunt MW, Sall AA, de Lamballerie X, Falconar AKI, Dzhivanian TI, Gould EA. 2001. Phylogenetic relationships of flaviviruses correlate with their epidemiology, disease association and biogeography. J Gen Virol 82: 1867–1876. [DOI] [PubMed] [Google Scholar]
- Gentry GA, Lowe M, Alford G, Nevins R. 1988. Sequence analyses of herpesviral enzymes suggest an ancient origin for human sexual behavior. Proc Natl Acad Sci USA 85: 2658–2661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gibbs A. 1987. Molecular evolution of viruses; “trees”, “clocks”and “modules”. J Cell Sci [Suppl] 7: 319–337. [DOI] [PubMed] [Google Scholar]
- Giri A, Slattery JP, Heneine W, Gessain A, Rivadeneira E, Desrosiers RC, Rosen L, Anthony R, Pamungkas J, Iskandriati D, Richards AL, Herve V, McClure H, O'Brien SJ, Franchini G. 1997. The tax gene sequences form two divergent monophyletic lineages corresponding to types I and II of simian and human T‐cell leukemia/lymphotropic viruses. Virology 231: 96–104. [DOI] [PubMed] [Google Scholar]
- Goodchild NL, Wilkinson DA, Mager DL. 1992. A human endogenous long terminal repeat provides a polyadenylation signal to a novel, alternatively spliced transcript in normal placenta. Gene 121: 287–294. [DOI] [PubMed] [Google Scholar]
- Goral MI, Mochow‐Grundy M, Dermody TS. 1996. Sequence diversity within the reovirus S3 gene: reoviruses evolve independently of host species, geographic locale, and date of isolation. Virology 216: 265–271. [DOI] [PubMed] [Google Scholar]
- Gorbalenya AE. 1995. Origin of RNA viral genomes: approaching the problem by comparative sequence analysis In: Gibbs AJ, Calisher CH, García‐Arenal F, editors. Molecular basis of virus evolution. Cambridge: Cambridge University Press; p 49–66. [Google Scholar]
- Greensill J, Sheldon JA, Murthy KK, Bessonette JS, Beer BE, Schulz TF. 2000a. A chimpanzee rhadinovirus sequence related to Kaposi's sarcoma‐associated herpesvirus/human herpesvirus 8: increased detection after HIV‐1 infection in the absence of disease. AIDS 14: 129–135. [DOI] [PubMed] [Google Scholar]
- Greensill J, Sheldon JA, Renwick NM, Beer BE, Norley S, Goudsmit J, Schulz TF. 2000b. Two distinct gamma‐2 herpesviruses in African green monkeys: a second gamma‐2 herpesvirus lineage among Old World primates? J Virol 74: 1572–1577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greenwell P. 1997. Blood group antigens: molecules seeking a function? Glycoconj J 14: 159–173. [DOI] [PubMed] [Google Scholar]
- Greenwood AD, Lee F, Capelli C, DeSalle R, Tikhonov A, Marx PA, MacPhee RDE. 2001. Evolution of endogenous retrovirus‐like elements of the woolly mammoth (Mammuthus primigenius) and its relatives. Mol Biol Evol 18: 840–847. [DOI] [PubMed] [Google Scholar]
- Gromeier M, Wimmer E, Gorbalenya AE. 1999. Genetics, pathogenesis and evolution of picornaviruses In: Domingo E, Webster R, Holland J, editors. Origin and evolution of viruses. London: Academic Press; p 287–343. [Google Scholar]
- Gust I. 1980. Acute viral hepatitis In: Stanley NF, Joske RA, editors. Changing disease patterns and human behaviour. London: Academic Press; p 85–113. [Google Scholar]
- Hahn BH, Shaw GM, De Cock KM, Sharp PM. 2000. AIDS as a zoonosis: scientific and public health implications. Science 287: 607–614. [DOI] [PubMed] [Google Scholar]
- Haldane JBS. 1949. Disease and evolution. Ric Sci [Suppl] 19: 2–11. [Google Scholar]
- Hamilton WD, Axelrod R, Tanese R. 1990. Sexual reproduction as an adaptation to resist parasites: a review. Proc Natl Acad Sci USA 87: 3566–3573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hatwell JN, Sharp PM. 2000. Evolution of human polyomavirus JC. J Gen Virol 81: 1191–1200. [DOI] [PubMed] [Google Scholar]
- Hayward GS. 1999. KSHV strains: the origins and global spread of the virus. Cancer Biol 9: 187–199. [DOI] [PubMed] [Google Scholar]
- Hedrick PW, Black FL. 1997. HLA and mate selection: no evidence in South Amerindians. Am J Hum Genet 61: 505–511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hetherington SV, Watson AS, Scroggs RA, Portner A. 1994. Human parainfluenza virus type 1 evolution combines cocirculation of strains and development of geographically restricted lineages. J Infect Dis 169: 248–252. [DOI] [PubMed] [Google Scholar]
- Hill AVS, Motulsky AG. 1999. Genetic variation and human disease: the role of natural selection In: Stearns SC, editor. Evolution in health and disease. Oxford: Oxford University Press; p 50–61. [Google Scholar]
- Ho L, Chan S‐Y, Burk RD, Das BC, Fujinaga K, Icenogle JP, Kahn T, Kiviat N, Lancaster W, Mavromara‐Nazos P, Labropoulou V, Mitrani‐Rosenbaum S, Norrild B, Pillai MR, Stoerker J, Syrjaenen K, Syrjaenen S, Tay S‐K, Villa LL, Wheeler CM, Williamson A‐L, Bernard H‐U. 1993. The genetic drift of human papillomavirus type 16 is a means of reconstructing prehistoric viral spread and the movement of ancient human populations. J Virol 67: 6413–6423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holland J. 1993. Replication error, quasispecies populations, and extreme evolution rates of RNA viruses In: Morse SS, editor. Emerging viruses. New York: Oxford University Press; p 203–218. [Google Scholar]
- Holmes EC. 2001. On the origin and evolution of the human immunodeficiency virus (HIV). Biol Rev 76: 239–254. [DOI] [PubMed] [Google Scholar]
- Holt RD, Lawton JH. 1994. The ecological consequences of shared natural enemies. Annu Rev Ecol Syst 25: 495–520. [Google Scholar]
- Holt RD, Pickering J. 1985. Infectious disease and species coexistence:a model of Lotka‐Volterra form. Am Nat 126: 196–211. [Google Scholar]
- Hooper E. 2000. The river: a journey back to the source of HIV and AIDS, revised edition. London: Penguin. [Google Scholar]
- Howard JC. 1991. Disease and evolution. Nature 352: 565–567. [DOI] [PubMed] [Google Scholar]
- Hu X‐L, Margolis HS, Purcell RH, Ebert J, Robertson BH. 2000. Identification of hepatitis B virus indigenous to chimpanzees. Proc Natl Acad Sci USA 97: 1661–1664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu X‐L, Javadian A, Gagneux P, Robertson BH. 2001. Paired chimpanzee hepatitis B virus (ChHBV) and mtDNA sequences suggest different ChHBV genetic variants are found in geographically distinct chimpanzee subspecies. Virus Res 79: 103–108. [DOI] [PubMed] [Google Scholar]
- Hudson PJ, Greenman J. 1998. Competition mediated by parasites: biological and theoretical progress. Trends Ecol Evol 13: 387–390. [DOI] [PubMed] [Google Scholar]
- Huemer HP, Larcher C, Czedik‐Eysenberg T, Nowotny N, Reifinger M. 2002. Fatal infection of a pet monkey with human herpesvirus 1 . Emerg Infect Dis [serial online] 2002; 8 [cited May 28, 2002]. Available from URL: http://www.cdc.gov/ncidod/EID/vol8no6/01-0341.htm. [DOI] [PMC free article] [PubMed] [Google Scholar]
- International Human Genome Sequencing Consortium . 2001. Initial sequencing and analysis of the human genome. Nature 409: 860–921. [DOI] [PubMed] [Google Scholar]
- Ito H, Sugiyama M, Masubuchi K, Mori Y, Minamoto N. 2001. Complete nucleotide sequence of a group A avian rotavirus genome and a comparison with its counterparts of mammalian rotaviruses. Virus Res 75: 123–138. [DOI] [PubMed] [Google Scholar]
- Jacob S, McClintock MK, Zelano B, Ober C. 2002. Paternally inherited HLA alleles are associated with women's choice of male odor. Nat Genet 30: 175–179. [DOI] [PubMed] [Google Scholar]
- Jiang X, Cubitt WD, Berke T, Zhong W, Dai X, Nakata S, Pickering LK, Matson DO. 1997. Sapporo‐like human caliciviruses are genetically and antigenically diverse. Arch Virol 142: 1813–1827. [DOI] [PubMed] [Google Scholar]
- Jobes DV, Chima SC, Ryschkewitsch CF, Stoner GL. 1998. Phylogenetic analysis of 22 complete genomes of the human polyomavirus JC virus. J Gen Virol 79: 2491–2498. [DOI] [PubMed] [Google Scholar]
- Johnson WE, Coffin JM. 1999. Constructing primate phylogenies from ancient retrovirus sequences. Proc Natl Acad Sci USA 96: 10254–10260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joklik WK, Roner MR. 1995. The evolution of the Reoviridae In: Gibbs AJ, Calisher CH, Garcia‐Arenal F, editors. Molecular basis of virus evolution. Cambridge: Cambridge University Press; p 369–384. [Google Scholar]
- Kapitonov VV, Jurka J. 1999. The long terminal repeat of an endogenous retrovirus induces alternative splicing and encodes an additional carboxy‐terminal sequence in the human leptin receptor. J Mol Evol 48: 248–251. [DOI] [PubMed] [Google Scholar]
- Kaslow RA, Carrington M, Apple R, Park L, Muñoz A, Saah AJ, Goedert JJ, Winkler C, O'Brien SJ, Rinaldo C, Detels R, Blattner W, Phair J, Erlich H, Mann DL. 1996. Influence of combinations of human major histocompatibility complex genes on the course of HIV‐1 infection. Nat Med 2: 405–411. [DOI] [PubMed] [Google Scholar]
- Kawano M, Bando H, Yuasa T, Kondo K, Tsurudome M, Komada H, Nishio M, Ito Y. 1990. Sequence determination of the hemagglutinin‐neuraminidase (HN) gene of human parainfluenza type 2 virus and the construction of a phylogenetic tree for HN proteins of all the paramyxoviruses that are infectious to humans. Virology 174: 308–313. [DOI] [PubMed] [Google Scholar]
- Kawaoka Y, Gorman OT, Ito T, Wells K, Donis RO, Castrucci MR, Donatelli I, Webster RG. 1998. Influence of host species on the evolution of the nonstructural (NS) gene of influenza A viruses. Virus Res 55: 143–156. [DOI] [PubMed] [Google Scholar]
- Kidd AH, Garwicz D, Oberg M. 1995. Human and simian adenoviruses: phylogenetic inferences from analysis of VA RNA genes. Virology 207: 32–45. [DOI] [PubMed] [Google Scholar]
- Kilbourne ED. 1994. Host determination of viral evolution: a variable tautology In: Morse SS, editor. The evolutionary biology of viruses. New York: Raven Press; p 253–271. [Google Scholar]
- Knapp LA. 2002. Evolution and immunology. Evol Anthropol [Suppl] 11: 140–144. [Google Scholar]
- Kojima K, Taniguchi K, Urasawa T, Urasawa S. 1996. Sequence analysis of normal and rearranged NSP5 genes from human rotavirus strains isolated in nature: implications for the occurrence of the rearrangement at the step of plus strand synthesis. Virology 224: 446–452. [DOI] [PubMed] [Google Scholar]
- Koralnik IJ, Boeri E, Saxinger WC, Monico AL, Fullen J, Gessain A, Guo H‐G, Gallo RC, Markham P, Kalyanaraman V, Hirsch V, Allan J, Murthy K, Alford P, Slattery JP, O'Brien SJ, Franchini G. 1994. Phylogenetic associations of human and simian T‐cell leukemia/lymphotropic virus type I strains: evidence for interspecies transmission. J Virol 68: 2693–2707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korber B, Muldoon M, Theiler J, Gao F, Gupta R, Lapedes A, Hahn BH, Wolinsky S, Bhattacharya T. 2000. Timing the ancestor of the HIV‐1 pandemic strains. Science 288: 1789–1796. [DOI] [PubMed] [Google Scholar]
- Kriener K, O'hUigin C, Tichy H, Klein J. 2000. Convergent evolution of major histocompatibility complex molecules in humans and New World monkeys. Immunogenetics 51: 169–178. [DOI] [PubMed] [Google Scholar]
- Lacoste V, Mauclère P, Dubreuil G, Lewis J, Georges‐Courbot M‐C, Gessain A. 2000. KSHV‐like herpesviruses in chimps and gorillas. Nature 407: 151–152. [DOI] [PubMed] [Google Scholar]
- Lai MMC. 1995. Recombination and its evolutionary effect on viruses with RNA genomes In: Gibbs AJ, Calisher CH, Garcia‐Arenal F, editors. Molecular basis of virus evolution. Cambridge: Cambridge University Press; p 119–132. [Google Scholar]
- Lanford RE, Chavez D, Brasky KM, Burns RB III, Rico‐Hesse R. 1998. Isolation of a hepadnavirus from the woolly monkey, a New World primate. Proc Natl Acad Sci USA 95: 5757–5761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lebedev YB, Belonovitch OS, Zybrova NV, Khil PP, Kurdyukov SG, Vinogradova TV, Hunsmann G, Sverdlov ED. 2000. Differences in HERV‐K LTR insertions in orthologous loci of humans and great apes. Gene 247: 265‐–277. [DOI] [PubMed] [Google Scholar]
- Liu R, Paxton WA, Choe S, Ceradini D, Martin SR, Horuk R, MacDonald ME, Stuhlmann H, Koup RA, Landau NR. 1996. Homozygous defect in HIV‐1 coreceptor accounts for resistance of some multiply‐exposed individuals to HIV‐1 infection. Cell 86: 367–377. [DOI] [PubMed] [Google Scholar]
- Löwer R, Löwer J, Kurth R. 1996. The viruses in all of us: characteristics and biological significance of human endogenous retrovirus sequences. Proc Natl Acad Sci USA 93: 5177–5184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luria SE. 1959. Viruses: a survey of some current problems In: Isaacs A, Lacey BW, editors. Virus growth and variation. Princeton, NJ: Cambridge University Press; p 1–10. [Google Scholar]
- MacDonald DM, Holmes EC, Lewis JCM, Simmonds P. 2000. Detection of hepatitis B virus infection in wild‐born chimpanzees (Pan troglodytes verus): phylogenetic relationships with human and other primate genotypes. J Virol 74: 4253–4257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacPhee RDE, Marx PA. 1997. The 40,000‐year plague: humans, hyperdisease, and first‐contact extinctions In: Goodman SM, Patterson BD, editors. Natural change and human impact in Madagascar. Washington, DC: Smithsonian Institution; p 169–217. [Google Scholar]
- Magnius LO, Norder H. 1995. Subtypes, genotypes and molecular epidemiology of the hepatitis B virus as reflected by sequence variability of the S‐gene. Intervirology 38: 24–34. [DOI] [PubMed] [Google Scholar]
- Mahieux R, Chappey C, Georges‐Courbot M‐C, Dubreuil G, Mauclere P, Georges A, Gessain A. 1998. Simian T‐cell lymphotropic virus type 1 from Mandrillus sphinx as a simian counterpart of human T‐cell lymphotropic virus type 1 subtype D. J Virol 72: 10316–10322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marra MA, Jones SJN, Astell CR, Holt RA, Brooks‐Wilson A, Butterfield YSN, Khattra J, Asano JK, Barber SA, Chan SY, Cloutier A, Coughlin SM, Freeman D, Girn N, Griffith OL, Leach SR, Mayo M, McDonald H, Montgomery SB, Pandoh PK, Petrescu AS, Robertson AG, Schein JE, Siddiqui A, Smailus DE, Stott JM, Yang GS, Plummer F, Andonov A, Artsob H, Bastien N, Bernard K, Booth TF, Bowness D, Czub M, Drebot M, Fernando L, Flick R, Garbutt M, Gray M, Grolla A, Jones S, Feldmann H, Meyers A, Kabani A, Li Y, Normand S, Stroher U, Tipples GA, Tyler S, Vogrig R, Ward D, Watson B, Brunham RC, Krajden M, Petric M, Skowronski DM, Upton C, Roper RL. 2003. The genome sequence of the SARS‐associated coronavirus. Science 300: 1399–1404. [DOI] [PubMed] [Google Scholar]
- May RM. 1983. Parasitic infections as regulators of animal populations. Am Sci 71: 36–45. [PubMed] [Google Scholar]
- May RM. 1995. The co‐evolutionary dynamics of viruses and their hosts In: Gibbs AJ, Calisher CH, Garcia‐Arenal F, editors. Molecular basis of virus evolution. Cambridge: Cambridge University Press; p 192–212. [Google Scholar]
- McClure MA. 1995. Molecular evolution of the retroid family In: Gibbs AJ, Calisher CH, Garcia‐Arenal F, editors. Molecular basis of virus evolution. Cambridge: Cambridge University Press; p 404–415. [Google Scholar]
- McGeoch DJ. 1989. The genomes of the human herpesviruses: contents, relationships, and evolution. Annu Rev Microbiol 43: 235–265. [DOI] [PubMed] [Google Scholar]
- McGeoch DJ. 2001. Molecular evolution of the g‐Herpesvirinae . Philos Trans R Soc Lond [Biol] 356: 421–435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGeoch DJ, Davison AJ. 1995. Origins of DNA viruses In: Gibbs AJ, Calisher CH, Garcia‐Arenal F, editors. Molecular basis of virus evolution. Cambridge: Cambridge University Press; p 67–75. [Google Scholar]
- McGeoch DJ, Davison AJ. 1999a. The molecular evolutionary history of the herpesviruses In: Domingo E, Webster R, Holland J, editors. Origin and evolution of viruses. London: Academic Press; p 441–465. [Google Scholar]
- McGeoch DJ, Davison AJ. 1999b. The descent of human herpesvirus 8. Cancer Biol 9: 201–209. [DOI] [PubMed] [Google Scholar]
- McGeoch DJ, Cook S, Dolan A, Jamieson FE, Telford EAR. 1995. Molecular phylogeny and evolutionary timescale for the family of mammalian herpesviruses. J Mol Biol 246: 443–458. [DOI] [PubMed] [Google Scholar]
- McGeoch DJ, Dolan A, Ralph AC. 2000. Toward a comprehensive phylogeny for mammalian and avian herpesviruses. J Virol 74: 10401–10406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McMichael T. 2001. Human frontiers, environments and disease: past patterns, uncertain futures. Cambridge: Cambridge University Press. [Google Scholar]
- McNeill WH. 1976. Plagues and peoples. Garden City, NY: Anchor Books. [Google Scholar]
- Mi S, Lee X, Li X‐P, Veldman GM, Finnerty H, Racie L, LaVallie E, Tang X‐Y, Edouard P, Howes S, Keith JC Jr, McCoy JM. 2000. Syncytin is a captive retroviral envelope protein involved in human placental morphogenesis. Nature 403: 785–789. [DOI] [PubMed] [Google Scholar]
- Morales JC, Melnick DJ. 1998. Phylogenetic relationships of the macaques (Cercopithecidae: Macaca), as revealed by high resolution restriction site mapping of mitochondrial ribosomal genes. J Hum Evol 34: 1–23. [DOI] [PubMed] [Google Scholar]
- Morse SS. 1993. Examining the origins of emerging viruses In: Morse SS, editor. Emerging viruses. New York: Oxford University Press; p 10–28. [Google Scholar]
- Morse SS. 1994. Toward an evolutionary biology of viruses In: Morse SS, editor. The evolutionary biology of viruses. New York: Raven Press; p 1–28. [Google Scholar]
- Neel JV, Salzano FM, Junqueira PC, Keiter F, Mayberry‐Lewis D. 1964. Studies on the Xavante Indians of the Brazilian Mato Grosso. Am J Hum Genet 16: 52–140. [PMC free article] [PubMed] [Google Scholar]
- Neel JV, Andrade AHP, Brown GE, Eveland WE, Goobar J, Sodeman WA Jr, Stollerman GH, Weinstein ED, Wheeler AH. 1968. Further studies of the Xavante Indians, IX. Immunologic status with respect to various diseases and organisms. Am J Trop Med Hyg 17: 486–498. [PubMed] [Google Scholar]
- Norder H, Ebert JW, Fields HA, Mushahwar IK, Magnius LO. 1996. Complete sequencing of a gibbon hepatitis B virus genome reveals a unique genotype distantly related to the chimpanzee hepatitis B virus. Virology 218: 214–223. [DOI] [PubMed] [Google Scholar]
- Norrby E, Kövamees J, Blixenkrone‐Möller M, Sharma B, Örvell C. 1992. Humanized animal viruses with special reference to the primate adaptation of morbillivirus. Vet Microbiol 33: 275–286. [DOI] [PubMed] [Google Scholar]
- Ober C. 1999. Studies of HLA, fertility and mate choice in a human isolate. Hum Reprod Update 5: 103–107. [DOI] [PubMed] [Google Scholar]
- Oberste MS, Maher K, Pallansch MA. 2002. Molecular phylogeny and proposed classification of the simian picornaviruses. J Virol 76: 1244–1251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ong C‐K, Chan S‐Y, Campo MS, Fujinaga K, Mavromara‐Nazos P, Labropoulou V, Pfister H, Tay S‐K, ter Meulen J, Villa LL, Bernard H‐U. 1993. Evolution of human papillomavirus type 18: an ancient phylogenetic root in Africa and intratype diversity reflect coevolution with human ethnic groups. J Virol 67: 6424–6431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Overbaugh J, Bangham CRM. 2001. Selection forces and constraints on retroviral sequence variation. Science 292: 1106–1109. [DOI] [PubMed] [Google Scholar]
- Pain S. 1997. Plague dogs. New Sci 154: 32–37. [Google Scholar]
- Parrish CR, Truyen U. 1999. Parvovirus variation and evolution In: Domingo E, Webster R, Holland J, editors. Origin and evolution of viruses. London: Academic Press; p 421–439. [Google Scholar]
- Pavesi A. 2001. Origin and evolution of GBV‐C/hepatitis G virus and relationships with ancient human migrations. J Mol Evol 53: 104–113. [DOI] [PubMed] [Google Scholar]
- Peeters M, Courgnaud V, Abela B, Auzel P, Pourrut X, Bibollet‐Ruche F, Loul S, Liegeois F, Butel C, Koulagna D, Mpoudi‐Ngole E, Shaw GM, Hahn BH, Delaporte E. 2002. Risk to human health from a plethora of simian immunodeficiency viruses in primate bushmeat. Emerg Infect Dis [serial online] 2002; 8 [cited April 26, 2002]. Available from URL: http://www.cdc.gov/ncidod/EID/vol8no5/01-0522.htm. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Penn DJ, Potts WK. 1999. The evolution of mating preferences and major histocompatibility complex genes. Am Nat 153: 145–164. [DOI] [PubMed] [Google Scholar]
- Potts R. 1998. Environmental hypotheses of hominin evolution. Yrbk Phys Anthropol 41: 93–136. [DOI] [PubMed] [Google Scholar]
- Potts WK, Manning CJ, Wakeland EK. 1991. Mating pattterns in seminatural populations of mice influenced by MHC genotype. Nature 352: 619–621. [DOI] [PubMed] [Google Scholar]
- Poyry T, Kinnunen L, Hovi T, Hyypiä T. 1999. Relationships between simian and human enteroviruses. J Gen Virol 80: 635–638. [DOI] [PubMed] [Google Scholar]
- Prince AM, Brotman B, Lee DH, Andrus L, Valinsky J, Marx P. 2002. Lack of evidence for HIV type 1‐related SIVcpz infection in captive and wild chimpanzees (Pan troglodytes verus) in West Africa. AIDS Res Hum Retroviruses 18: 657–660. [DOI] [PubMed] [Google Scholar]
- Reed KE. 1997. Early hominid evolution and ecological change through the African Plio‐Pleistocene. J Hum Evol 32: 289–322. [DOI] [PubMed] [Google Scholar]
- Reid AH, Fanning TG, Hultin JV, Taubenberger JK. 1999. Origin and evolution of the 1918 “Spanish” influenza virus hemagglutinin gene. Proc Natl Acad Sci USA 96: 1651–1656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rico‐Hesse R, Pallansch MA, Nottay BK, Kew OM. 1987. Geographic distribution of wild poliovirus type 1 genotypes. Virology 160: 311–322. [DOI] [PubMed] [Google Scholar]
- Rima BK, Earle JAP, Baczko K, ter Meulen V, Liebert UG, Carstens C, Carabaña J, Caballero M, Celma ML, Fernandez‐Muñoz R. 1997. Sequence divergence of measles virus haemagglutinin during natural evolution and adaptation to cell culture. J Gen Virol 78: 97–106. [DOI] [PubMed] [Google Scholar]
- Robertson BH. 2001. Viral hepatitis and primates: historical and molecular analysis of human and nonhuman primate hepatitis A, B, and the GB‐related viruses. J Viral Hepat 8: 233–242. [DOI] [PubMed] [Google Scholar]
- Robertson BH, Margolis HS. 2002. Primate hepatitis B viruses—genetic diversity, geography and evolution. Rev Med Virol 12: 133–141. [DOI] [PubMed] [Google Scholar]
- Robertson BH, Jansen RW, Khanna B, Totsuka A, Nainan OV, Siegl G, Widell A, Margolis HS, Isomura S, Ito K, Ishizu T, Moritsugu Y, Lemon SM. 1992. Genetic relatedness of hepatitis A virus strains recovered from different geographical regions. J Gen Virol 73: 1365–1377. [DOI] [PubMed] [Google Scholar]
- Rodrigo MJ, Dopazo J. 1995. Evolutionary analysis of the picornavirus family. J Mol Evol 40: 362–371. [DOI] [PubMed] [Google Scholar]
- Roelke‐Parker ME, Munson L, Packer C, Kock R, Cleaveland S, Carpenter M, O'Brien SJ, Pospischil A, Hofmann‐Lehmann R, Lutz H, Mwamengele GLM, Mgasa MN, Machange GA, Summers BA, Appel MJG. 1996. A canine distemper virus epidemic in Serengeti lions (Panthera leo). Nature 379: 441–445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rota PA, Oberste MS, Monroe SS, Nix WA, Canpagnoli R, Icenogle JP, Peñaranda S, Bankamp B, Maher K, Chen M, Tong S, Tamin A, Lowe L, Frace M, DeRisi JL, Chen Q, Wang D, Erdman DD, Peret TCT, Burns C, Ksiazek TG, Rolin PE, Sanchez A, Liffick S, Holloway B, Limor J, McCaustland K, Olsen‐Rasmussen M, Fouchier R, Günther S, Osterhaus ADME, Drosten C, Pallansch MA, Anderson LJ, Bellini WJ. 2003. Characterization of a novel coronavirus associated with severe acute respiratory syndrome. Science 300: 1394–1399. [DOI] [PubMed] [Google Scholar]
- Rulicke T, Chapuisat M, Homberger FR, Macas E, Wedekind C. 1998. MHC‐genotype of progeny influenced by parental infection. Proc R Soc Lond [Biol] 265: 711–716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sainsbury AW, Nettleton P, Gurnell J. 1997. In: Furnell J, Lurz P, editors. The conservation of red squirrels, Sciurus vulgaris L. London: People's Trust for Endangered Species; p 105–108. [Google Scholar]
- Salemi M, Van Dooren S, Audenaert E, Delaporte E, Goubau P, Desmyter J, Vandamme A‐M. 1998. Two new human T‐lymphotropic virus type I phylogenetic subtypes in seroindeterminates, a Mbuti pygmy and a Gabonese, have closest relatives among African STLV‐I strains. Virology 246: 277–287. [DOI] [PubMed] [Google Scholar]
- Salemi M, Desmyter J, Vandamme A‐M. 2000. Tempo and mode of human and simian T‐lymphotropic virus (HTLV/STLV) evolution revealed by analyses of full‐genome sequences. Mol Biol Evol 17: 374–386. [DOI] [PubMed] [Google Scholar]
- Samson M, Libert F, Doranz BJ, Rucker J, Liesnard C, Farber C‐M, Saragosti S, Lapouméroulie C, Cognauz J, Forceille C, Muyidermans G, Verhofstede C, Burtonboy G, Georges M, Imai T, Rana S, Yi Y, Smyth RJ, Collman RG, Doms RW, Vassart G, Parmentier M. 1996. Resistance to HIV‐1 infection in Caucasian individuals bearing mutant alleles of the CCR‐5 chemokine receptor gene. Nature 382: 722–725. [DOI] [PubMed] [Google Scholar]
- Samuelson LC, Wiebauer K, Snow CM, Meisler MH. 1990. Retroviral and pseudogene insertion sites reveal the lineage of human salivary and pancreatic amylase genes from a single gene during primate evolution. Mol Cell Biol 10: 2513–2520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santiago ML, Rodenburg CM, Kamenya S, Bibollet‐Ruche F, Gao F, Bailes E, Meleth S, Soong S‐J, Kilby JM, Moldoveanu Z, Fahey B, Muller MN, Ayouba A, Nerrienet E, McClure HM, Heeney JL, Pusey AE, Collins DA, Boesch C, Wrangham RW, Goodall J, Sharp PM, Shaw GM, Hahn BH. 2002. SIVcpz in wild chimpanzees. Science 295: 465. [DOI] [PubMed] [Google Scholar]
- Sarkar S. 1992. Sex, disease, and evolution—variations on a theme from J.B.S. Haldane. Bioscience 42: 448–454. [Google Scholar]
- Savolainen P, Zhang Y‐P, Luo J, Lundeberg J, Leitner T. 2002. Genetic evidence for an East Asian origin of domestic dogs. Science 298: 1610–1613. [DOI] [PubMed] [Google Scholar]
- Scholtissek C, Naylor E. 1988. Fish farming and influenza pandemics. Nature 331: 215. [DOI] [PubMed] [Google Scholar]
- Scott TW, Weaver SC, Mallampalli VL. 1994. Evolution of mosquito‐borne viruses In: Morse SS, editor. The evolutionary biology of viruses. New York: Raven Press; p 293–324. [Google Scholar]
- Selander RK, Yang SY, Lewontin RC, Johnson WE. 1970. Genetic variation in the horseshoe crab (Limulus polyphemus), a phylogenetic “relic.” Evolution 24: 402–414. [DOI] [PubMed] [Google Scholar]
- Senkevich TG, Koonin EV, Bugert JJ, Darai G, Moss B. 1997. The genome of Molluscum contagiosum virus: analysis and comparison with other poxviruses. Virology 233: 19–42. [DOI] [PubMed] [Google Scholar]
- Shadan FF, Villarreal LP. 1993. Coevolution of persistently infecting small DNA viruses and their hosts linked to host‐interactive regulatory domains. Proc Natl Acad Sci USA 90: 4117–4121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharp PM, Bailes E, Robertson DL, Gao F, Hahn BH. 1999. Origins and evolution of AIDS viruses. Biol Bull Woods Hole 196: 338–342. [DOI] [PubMed] [Google Scholar]
- Sharp PM, Bailes E, Gao F, Beer BE, Hirsch VM, Hahn BH. 2000. Origins and evolution of AIDS viruses: estimating the time‐scale. Biochem Soc Trans 28: 275–282. [DOI] [PubMed] [Google Scholar]
- Sharp PM, Bailes E, Chaudhuri RR, Rodenburg CM, Santiago MO, Hahn BH. 2001. The origins of acquired immune deficiency syndrome viruses: where and when? Philos Trans R Soc Lond [Biol] 356: 867–876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shih A, Coutavas EE, Rush MG. 1991. Evolutionary implications of primate endogenous retroviruses. Virology 182: 495–502. [DOI] [PubMed] [Google Scholar]
- Simmonds P. 2001. The origin and evolution of hepatitis viruses in humans. J Gen Virol 82: 693–712. [DOI] [PubMed] [Google Scholar]
- Simms EL. 1996. The evolutionary genetics of plant‐pathogen systems. Bioscience 46: 136–145. [Google Scholar]
- Skinner GRB, Ahmad A, Davies JA. 2001. The infrequency of transmission of herpesviruses between humans and animals; postulation of an unrecognised protective host mechanism. Comp Immunol Microbiol Infect Dis 24: 255–269. [DOI] [PubMed] [Google Scholar]
- Slattery JP, Franchini G, Gessain A. 1999. Genomic evolution, patterns of global dissemination, and interspecies transmission of human and simian T‐cell Leukemia/lymphotropic viruses. Genome Res 9: 525–540. [PubMed] [Google Scholar]
- Smith AL, Black DH, Eberle R. 1998. Molecular evidence for distinct genotypes of monkey B virus (Herpesvirus simiae) which are related to the macaque host species. J Virol 72: 9224–9232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song B, Hu S‐L, Darai G, Spindler KR, Young CSH. 1996. Conservation of DNA sequence in the predicted major late promoter regions of selected mastadenoviruses. Virology 220: 390–401. [DOI] [PubMed] [Google Scholar]
- Stephens JC, Reich DE, Goldstein DB, Shin HD, Smith MW, Carrington M, Winkler C, Huttley GA, Allikmets R, Schriml L, Gerrard B, Malasky M, Ramos MD, Morlot S, Tzetis M, Oddoux C, de Giovine FS, Nasioulas G, Chandler D, Aseev M, Hanson M, Kalaydjieva L, Glavac D, Gasparini P, Kanavakis E, Claustres M, Kambouris M, Ostrer H, Duff G, Baranov V, Sibul H, Metspalu A, Goldman D, Martin N, Duffy D, Schmidtke J, Estivell X, O'Brien SJ, Dean M. 1998. Dating the origin of the CCR5‐D32 AIDS‐resistance allele by the coalescence of haplotypes. Am J Hum Genet 62: 1507–1515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stephenson CB, Casebolt DB, Gangopadhyay NN. 1999. Phylogenetic analysis of a highly conserved region of the polymerase gene from 11 coronaviruses and development of a consensus polymerase chain reaction assay. Virus Res 60: 181–189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stewart A‐CM, Eriksson AM, Manos MM, Muñoz N, Bosch FX, Peto J, Wheeler CM. 1996. Intratype variation in 12 human papillomavirus types: a worldwide perspective. J Virol 70: 3127–3136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stone R. 2000. Canine virus blamed in Caspian seal deaths. Science 289: 2017–2018. [DOI] [PubMed] [Google Scholar]
- Stone R. 2002. WHO puts off destruction of US, Russian caches. Science 295: 598–599. [DOI] [PubMed] [Google Scholar]
- Stoner GL, Jobes DV, Cobo MF, Agostini HT, Chima SC, Ryschkewitsch CF. 2000. JC virus as a marker of human migration to the Americas. Microbes Infect 2: 1905–1911. [DOI] [PubMed] [Google Scholar]
- Sugimoto C, Hasegawa M, Kato A, Zheng H‐Y, Ebihara H, Taguchi F, Kitamura T, Yogo Y. 2002. Evolution of human polyomavirus JC: implications for the population history of humans. J Mol Evol 54: 285–297. [DOI] [PubMed] [Google Scholar]
- Suzuki H, Hosokawa Y, Toda H, Nishikimi M, Ozawa T. 1990. Common protein‐binding sites in the 5′‐flanking regions of human genes for cytochrome c1 and ubiquinone‐binding protein. J Biol Chem 265: 8159–8163. [PubMed] [Google Scholar]
- Suzuki Y, Gojobori T. 1997. The origin and evolution of Ebola and Marburg viruses. Mol Biol Evol 14: 800–806. [DOI] [PubMed] [Google Scholar]
- Suzuki Y, Nei M. 2002. Origin and evolution of influenza virus hemagglutinin genes. Mol Biol Evol 19: 501–509. [DOI] [PubMed] [Google Scholar]
- Svanborg‐Eden C, Levin BR. 1990. Infectious disease and natural selection in human populations: a critical reexamination In: Swedlund AC, Armelagos GJ, editors. Disease in populations in transition. New York: Bergin & Garvey; p 31–46. [Google Scholar]
- Sverdlov ED. 1998. Perpetually mobile footprints of ancient infections in human genome. FEBS Lett 428: 1–6. [DOI] [PubMed] [Google Scholar]
- Sverdlov ED. 2000. Retroviruses and primate evolution. Bioessays 22: 161–171. [DOI] [PubMed] [Google Scholar]
- Swedlund AC. 2000. A view on the science: physical anthropology at the millenium. Am J Phys Anthropol 113: 1–4. [DOI] [PubMed] [Google Scholar]
- Takahashi K, Brotman B, Usuda S, Mishiro S, Prince AM. 2000. Full‐genome sequence analyses of hepatitis B virus (HBV) strains recovered from chimpanzees infected in the wild: implications for an origin of HBV. Virology 267: 58–64. [DOI] [PubMed] [Google Scholar]
- Takahata N. 1990. A simple geneaological structure of strongly balanced allelic lines and trans‐species evolution of polymorphism. Proc Natl Acad Sci USA 87: 2419–2423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang B, Gilbert JM, Matsui SM, Greenberg HB. 1997. Comparison of the rotavirus gene 6 from different species by sequence analysis and localization of subgroup‐specific epitopes using site‐directed mutagenesis. Virology 237: 89–96. [DOI] [PubMed] [Google Scholar]
- Thompson JN, Burdon JJ. 1992. Gene‐for‐gene coevolution between plants and parasites. Nature 360: 121–125. [Google Scholar]
- Thursz MR, Kwiatkowski D, Allsopp CEM, Greenwood BM, Thomas HC, Hill AVS. 1995. Association between an MHC class II allele and clearance of hepatitis B virus in the Gambia. N Engl J Med 332: 1065–1069. [DOI] [PubMed] [Google Scholar]
- Ting CN, Rosenberg MP, Snow CM, Samuelson LC, Meisler MH. 1992. Endogenous retroviral sequences are required for tissue‐specific expression of a human salivary amylase gene. Genes Dev 6: 1457–1465. [DOI] [PubMed] [Google Scholar]
- Tiollais P, Buendia M‐A. 1991. Hepatitis B virus. Sci Am 1991; 264: 116–123. [DOI] [PubMed] [Google Scholar]
- Tishkoff SA, Varkony R, Cahinhinan N, Abbes S, Argyropoulos G, Destro‐Bisol G, Drousiotou A, Dangerfield B, Lefranc G, Loiselet J, Piro A, Stoneking M, Tagarelli A, Tagarelli G, Touma EH, Williams SM, Clark AG. 2001. Haplotype diversity and linkage disequilibrium at human G6PD: recent origin of alleles that confer malarial resistance. Science 293: 455–462. [DOI] [PubMed] [Google Scholar]
- Tucker JB. 2001. Scourge: the once and future threat of smallpox. New York: Atlantic Monthly Press. [Google Scholar]
- Uliyanov YP. 1998. Clinical manifestations of the variants of nasal aerodynamics. Otolaryngol Head Neck Surg 119: 152–153. Available from URL: http://www.airsilver.net/ch25.html [cited February 7, 2003]. [Google Scholar]
- Umene K, Sakaoka H. 1999. Evolution of herpes simplex virus type 1 under herpesviral evolutionary processes. Arch Virol 144: 637–656. [DOI] [PubMed] [Google Scholar]
- Van Blerkom LM. 1997. Zoonoses and the origins of Old and New World viral diseases: a reappraisal In: Romanucci‐Ross L, Moerman DE, Tancredi LR, editors. The anthropology of medicine, 3rd ed. New York: Bergin and Garvey; p 143–168. [Google Scholar]
- Vandamme A‐M, Salemi M, Desmyter J. 1998. The simian origins of the pathogenic human T‐cell lymphotropic virus type I. Trends Microbiol 6: 477–483. [DOI] [PubMed] [Google Scholar]
- Vandamme A‐M, Bertazzoni U, Salemi M. 2000. Evolutionary strategies of human T‐cell lymphotropic virus type II. Gene 261: 171–180. [DOI] [PubMed] [Google Scholar]
- van der Poel WHM, Vinjé J, van der Heide R, Herrera M‐I, Vivo A, Koopmans MPG. 2000. Norwalk‐like calicivirus genes in farm animals. Emerg Infect Dis [serial online] 2000; 6 [cited February 11, 2000]. Available from URL: http://www.cdc.gov/ncidod/eid/vol6no1/van_der_poel.htm [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Dooren S, Salemi M, Vandamme A‐M. 2001. Dating the origin of the African human T‐cell lymphotropic virus (HTLV‐I) subtypes. Mol Biol Evol 18: 661–671. [DOI] [PubMed] [Google Scholar]
- Van Ranst M, Kaplan JB, Sundberg JP, Burk RD. 1995. Molecular evolution of papillomaviruses In: Gibbs AJ, Calisher CH, Garcia‐Arenal F, editors. Molecular basis of virus evolution. Cambridge: Cambridge University Press; p 455–476. [Google Scholar]
- Vapalahti O, Lundkvist A, Fedorov V, Conroy CJ, Hirvonen S, Plyusnina A, Nemirov K, Fredga K, Cook JA, Niemimaa J, Kaikusalo A, Henttonen H, Vaheri A, Plyusnin A. 1999. Isolation and characterization of a hantavirus from Lemmus sibiricus: evidence for host switch during hantavirus evolution. J Virol 73: 5586–5592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varki A. 2001. Loss of N‐glycolylneuraminic acid in humans: mechanisms, consequences, and implications for hominid evolution. Yrbk Phys Anthropol 44: 54–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Venables PJW, Brookes SM, Griffiths D, Weiss RA, Boyd MT. 1995. Abundance of an endogenous retroviral envelope protein in placental trophoblasts suggests a biological function. Virology 211: 589–592. [DOI] [PubMed] [Google Scholar]
- Venter JC, Adams MD, Myers EW, Li PW, Mural, RJ , Sutton GG, et al. 2003. The sequence of the human genome. Science 291: 1304–1351. [DOI] [PubMed] [Google Scholar]
- Verschoor EJ, Warren KS, Niphuis H, Heriyanto, Swan RA, Heeney JL. 1998. Characterization of a simian T‐lymphotropic virus from a wild‐caught orang‐utan (Pongo pygmaeus) from Kalimantan, Indonesia. J Gen Virol 79: 51–55. [DOI] [PubMed] [Google Scholar]
- Verschoor EJ, Warren KS, Langenhuijzen S, Heriyanto, Swan RA, Heeney JL. 2001. Analysis of two genomic variants of orang‐utan hepadnavirus and their relationship to other primate hepatitis B‐like viruses. J Gen Virol 82: 893–897. [DOI] [PubMed] [Google Scholar]
- Vilà C, Maldonado JE, Wayne RK. 1999. Phylogenetic relationships, evolution, and genetic diversity of the domestic dog. J Hered 90: 71–77. [DOI] [PubMed] [Google Scholar]
- Villarreal LP. 1999. DNA virus contribution to host evolution In: Domingo E, Webster R, Holland J, editors. Origin and evolution of viruses. London: Academic Press; p 391–420. [Google Scholar]
- Villarreal LP, Defilippis VR, Gottlieb KA. 2000. Acute and persistent viral life strategies and their relationship to emerging diseases. Virology 272: 1–6. [DOI] [PubMed] [Google Scholar]
- Vogel F. 1975. ABO blood groups, the HL‐A system and diseases In: Salzano FM, editor. The role of natural selection in human evolution. Amsterdam: North‐Holland Publishing Co. p 247–269. [Google Scholar]
- Volkman SK, Barry AE, Lyons EJ, Nielsen KM, Thomas SM, Choi M, Thakore SS, Day KP, Wirth DF, Harti DL. 2001. Recent origin of Plasmodium falciparum from a single progenitor. Science 293: 482–484. [DOI] [PubMed] [Google Scholar]
- Wain‐Hobson S. 1994. Is antigenic variation in HIV important for AIDS and what might be expected in the future? In: Morse SS, editor. The evolutionary biology of viruses. New York: Raven Press; p 185–209. [Google Scholar]
- Webster RG. 1997. Influenza virus: transmission between species and relevance to emergence of the next human pandemic. Arch Virol [Suppl] 13: 105–113. [DOI] [PubMed] [Google Scholar]
- Webster RG. 1998. Influenza: an emerging disease. Emerg Infect Dis [serial online] 1998; 4 [cited February 10, 2000]. Available from URL: http://www.cdc.gov/ncidod/eid/vol4no3/webster.htm [DOI] [PMC free article] [PubMed] [Google Scholar]
- Webster RG, Bean WJ, Gorman OT. 1995. Evolution of influenza viruses: rapid evolution and stasis In: Gibbs AJ, Calisher CH, García‐Arenal F, editors. Molecular basis of virus evolution. Cambridge: Cambridge University Press; p 531–543. [Google Scholar]
- Wedekind C, Füri S. 1997. Body odour preferences in men and women: do they aim for specific MHC combinations or simply heterozygosity? Proc R Soc Lond [Biol] 264: 1471–1479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wedekind C, Penn D. 2000. MHC genes, body odours, and odour preferences. Nephrol Dial Transplant 15: 1269–1271. [DOI] [PubMed] [Google Scholar]
- Wedekind C, Seebeck T, Bettens F, Paepke AJ. 1995. MHC‐dependent mate preferences in humans. Proc Biol Sci 260: 245–249. [DOI] [PubMed] [Google Scholar]
- Weiss RA, Griffiths D, Takeuchi Y, Patience C, Venables PJW. 1999. Retroviruses: ancient and modern. Arch Virol [Suppl] 15: 171–177. [DOI] [PubMed] [Google Scholar]
- Wilkinson DA, Mager DL, Leong J‐AC. 1994. Endogenous human retroviruses In: Levy JA, editor. The Retroviridae, volume 3 New York: Plenum Press; p 465–535. [Google Scholar]
- Woods PA, Gentsch J, Gouvea V, Mata L, Simhon A, Santosham M, Bai Z‐S, Urasawa S, Glass RI. 1992. Distribution of serotypes of human rotavirus in different populations. J Clin Microbiol 30: 781–785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woolhouse MEJ, Taylor LH, Haydon DT. 2001. Population biology of multihost pathogens. Science 292: 1109–1112. [DOI] [PubMed] [Google Scholar]
- World Health Organization . 2001. Poliomyelitis. WHO information fact sheets. Available from URL: http://www.who.int/inf-fs/en/fact114.html. Revised April 2001 [cited February 1, 2002].
- Yamaguchi S. 1997. Identification of three lineages of wild measles virus by nucleotide sequence analysis of N, P, M, F, and L genes in Japan. J Med Virol 52: 113–120. [DOI] [PubMed] [Google Scholar]
- Yang Z, Lauder IJ, Lin HJ. 1995. Molecular evolution of the hepatitis B virus genome. J Mol Evol 41: 587–596. [DOI] [PubMed] [Google Scholar]
- Yeager M, Hughes AL. 1999. Evolution of the mammalian MHC: natural selection, recombination, and convergent evolution. Immunol Rev 167: 45–58. [DOI] [PubMed] [Google Scholar]
- Zhao X, Hay J. 1997. The evolution of hantaviruses. Immunol Invest 26: 191–197. [DOI] [PubMed] [Google Scholar]
- Zimmer C. 2001. Genetic trees reveal disease origins. Science 292: 1090–1093. [DOI] [PubMed] [Google Scholar]
- Zinkernagel RM, Hengartner H, Stitz L. 1985. On the role of viruses in the evolution of immune responses. Br Med Bull 41: 92–97. [DOI] [PubMed] [Google Scholar]
- Zubrow E. 1989. The demographic modeling of Neanderthal extinction In: Mellars P, Stringer C, editors. The human revolution: behavioural and biological perspectives on the origins of modern humans. Princeton, NJ: Princeton University Press; p 212–231. [Google Scholar]