

PHF20L1, a H3K27me2 reader, promotes breast tumorigenesis by cooperating with the PRC2/NuRD complex.
Abstract
TUDOR domain–containing proteins (TDRDs) are chiefly responsible for recognizing methyl-lysine/arginine residue. However, how TDRD dysregulation contributes to breast tumorigenesis is poorly understood. Here, we report that TUDOR domain–containing PHF20L1 as a H3K27me2 reader exerts transcriptional repression by recruiting polycomb repressive complex 2 (PRC2) and Mi-2/nucleosome remodeling and deacetylase (NuRD) complex, linking PRC2-mediated methylation and NuRD-mediated deacetylation of H3K27. Furthermore, PHF20L1 was found to serve as a potential MYC and hypoxia-driven oncogene, promoting glycolysis, proliferation, and metastasis of breast cancer cells by directly inhibiting tumor suppressors such as HIC1, KISS1, and BRCA1. PHF20L1 expression was also strongly correlated with higher histologic grades of breast cancer and markedly up-regulated in several cancers. Meanwhile, Phf20l1 deletion not only induces growth retardation and mammary ductal outgrowth delay but also inhibits tumorigenesis in vivo. Our data indicate that PHF20L1 promotes tumorigenesis, supporting the pursuit of PHF20L1 as a target for cancer therapy.
INTRODUCTION
A diverse array of posttranslational modifications that often occur on histone tails represents an essential means to regulate DNA-templated processes such as gene transcription (1). The methylation of histone lysine residue regulates multiple biological processes, including genome stability, gene expression, cell proliferation, and nuclear architecture (2). Histone methylation homeostasis is mediated by a series of methylase and demethylase complexes, and the recognition of methylated histones is accomplished by “readers” that usually contain plant homeodomain (PHD) finger domains, WD40 repeats, CW domains, PWWP domains, and the royal superfamily, including proteins with chromodomains, TUDOR domains, and malignant brain tumor (MBT) repeats (3). TUDOR domain–containing proteins (TDRDs) have the potential to recognize histone methylation, and the abnormal overexpression of several TDRDs has been observed in breast cancer (4). PHD finger protein 20 (PHF20) and PHF20L1 share similar domains and are homolog TDRDs. PHF20 is a component of the MOF (male absent on the first)–nonspecific lethal lysine acetyltransferase complex, which is involved in transcriptional activation (5–7). As a histone reader, PHF20 recognizes histone H3K4me2 via its PHD finger, and the H3K4me2-binding function of the PHD finger is essential for PHF20-dependent histone acetylation, target gene activation, and cancer cell growth (8). PHF20L1 was reported to recognize nonhistone methylation (9, 10). However, its roles in the recognition of histone modifications and in tumor progression remain largely unknown.
The polycomb repressive complex 2 (PRC2) is involved in repressing gene transcription through the methyltransferase activity of EZH2 for H3K27me2 and H3K27me3 writing, thus playing an important role in a number of biological processes, including embryonic development, cell fate decisions, and cancer progression (11). In mouse embryonic stem cells, H3K27me2 is the dominant modification form, reaching 70%, while H3K27me1 and H3K27me3 only occupy 7 and 4% of the total H3, respectively (12). H3K27me3 is mainly enriched within the promoters of silenced genes (13); conversely, H3K27me1 and H3K27ac accumulate on transcriptionally active genes (12, 14). Although H3K27me2 is distributed in large chromatin regions, its function remains enigmatic, and the readers that recognize H3K27 methylation modifications need to be further elucidated.
As one of the four major types of adenosine 5′-triphosphate (ATP)–dependent chromatin remodeling complexes, the nucleosome remodeling and deacetylase (NuRD) complex participates in a variety of biological processes, such as chromatin assembly, tumor progression, genomic stability, mitochondrial homeostasis, and pluripotency, through diverse assembly methods (15, 16). It has been reported that the NuRD complex promotes tumor progression via its deacetylation activity, which results in the silencing of various tumor suppressor genes (TSGs) (17). Metastasis associated 1 (MTA1) is a core factor of the NuRD complex, whose methylation is essential for the formation of the NuRD complex (18). Increasingly, key nuclear proteins such as lysine specific demethylase 1 (LSD1) have been reported to be incorporated into the NuRD complex superfamily, adding new features to this complex (19). It has been demonstrated that PRC2 and the NuRD complex can synergistically mediate H3K27 methylation and acetylation homeostasis to modulate the expression of transcriptionally poised genes in embryonic stem cells (20). However, the regulation of H3K27 modifications by PRC2 and the NuRD complex remains to be further explored in breast cancer.
The Warburg effect refers to cancer cells that exhibit aberrant metabolism characterized by high glycolysis even in the presence of abundant oxygen. This mechanism has now been widely accepted as a hallmark of cancer, which facilitates tumor growth with elevated glucose uptake and lactate production (21). Here, we report that PHF20L1 is a histone methylation reader protein, which recognizes H3K27me2 and collaborates with PRC2 and the NuRD complex in regulating H3K27 modifications to suppress a series of tumor suppressors, ultimately promoting the Warburg effect and breast tumorigenesis.
RESULTS
PHF20L1 is critical for breast cancer cell proliferation
TDRDs are often dysregulated in breast cancer (The Cancer Genome Atlas and Molecular Taxonomy of Breast Cancer International Consortium datasets) (4), and the vast majority of TUDOR domain–recognizing ligands have been reported (fig. S1A). To explore the characteristics of these TDRDs that govern breast cancer proliferation, small interfering RNA (siRNAs) targeting indicated that TDRDs were transfected into human mammary carcinoma MDA-MB-231 or Hs 578T cells to assess the state of cell growth. In these experiments, at least two independent siRNA sequences were tested for each gene (fig. S1B) and then mixed for subsequent growth curve experiments and 5-ethynyl-2′-deoxyuridine (EdU) assays. As reported (22), the knockdown (KD) of some TDRDs such as lysine demethylase 4A (KDM4A) substantially inhibited the growth of MDA-MB-231 cells. However, unexpectedly, our results showed that the depletion of PHF20L1 had a stronger inhibitory effect on the proliferation of MDA-MB-231 cells than the suppression of other TUDOR domain proteins (fig. S1C). To further consolidate our results, we transfected MDA-MB-231 and Hs 578T cells with siRNAs for 48 hours and then performed the EdU assays using a Click-iT EdU Alexa Fluor 488 imaging kit (Life Technologies). Immunofluorescence staining followed by microscopic analysis indicated that the deficiency of TDRDs, including PHF20L1, KDM4A, or ubiquitin like with PHD and ring finger domains 1 (UHRF1), could notably inhibit the proliferative activity of breast cancer cells (fig. S1, D to F). Together, these results suggest that PHF20L1 is necessary to maintain the proliferative state of breast cancer cells.
PHF20L1 regulates TSGs and glycolysis-related gene expression, participating in MYC and hypoxia signaling
To determine how PHF20L1 regulates breast cancer cell growth, we performed RNA sequencing (RNA-seq) experiments in MDA-MB-231 cells using siRNA against PHF20L1 and control oligonucleotides. Compared to levels in the control, we identified a total of 1793 up-regulated genes and 1436 down-regulated genes (fold change, >1.5; P < 0.001) in PHF20L1-deficienct cells (Fig. 1A, left). Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway analysis of the differentially expressed genes revealed that the dysregulated genes were involved in vital biological processes. Further, down-regulated genes were enriched in pathways that regulate metabolic pathways, cell cycle, and glycolysis/gluconeogenesis, whereas up-regulated genes were enriched in pathways related to cell adhesion, insulin resistance, and lysosome (Fig. 1A, right). The epigenetic silencing of TSGs is one of the crucial reasons that promote tumorigenesis (23). Considering that PHF20L1 is essential for the proliferation of breast cancer cells, by analysis of the RNA-seq results, we found that the depletion of PHF20L1 could indeed up-regulate the expression of several well-known TSGs, including HIC1, KISS1, RASSF1, FBXW7, BRCA1, PTPRG, IGFBPL1, MTUS1, FHIT, CHFR, CASP7, FOXO3, and GLI3 (Fig. 1B, top). Meanwhile, the enrichment of differentially expressed genes in the metabolic pathways and glycolysis pathways indicated that PHF20L1 may play important roles in promoting the Warburg effect. In the RNA-seq data, we also found many glycolysis-related genes (GRGs) including SIRT1, GLUT1, HK2, GPI, ALDOA, GAPDH, PGK1, PGAM1, ENO1, ENO2, PKM, and LDHA were decreased in PHF20L1-depleted cells (Fig. 1B, bottom). Five representative differentially expressed genes of both up-regulated TSGs and down-regulated GRGs were further validated by quantitative reverse transcription polymerase chain reaction (qRT-PCR) analysis using PHF20L1 KD MDA-MB-231 cells (Fig. 1, C and D). Further, the reexpressing siRNA-resistant FLAG-PHF20L1 (WTres) was found to rescue the up-regulation of TSGs and the down-regulation of GRGs in PHF20L1-deficient cells (Fig. 1E).
Fig. 1. PHF20L1 regulates TSGs and GRGs expression and participates in MYC and hypoxia signaling.
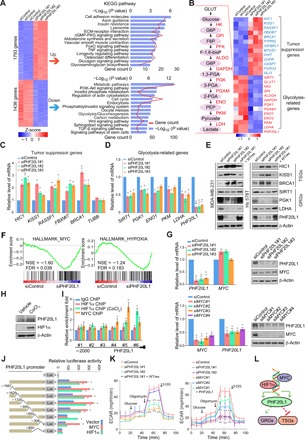
(A) Heatmap representation of differentially expressed genes (fold change, >1.5; P < 0.001) in control (siControl) and PHF20L1 KD (siPHF20L1-1, siPHF20L1-2, and siPHF20L1-3) MDA-MB-231 cells. Red, up-regulated genes; blue, down-regulated genes. The right panel shows the results of the KEGG pathway analysis of differentially expressed genes. Data were analyzed using KOBAS 3.0 software (B) Heatmap of known TSGs and GRGs identified by RNA-seq. (C and D) qRT-PCR analysis of selected TSGs and GRGs in PHF20L1 KD (siPHF20L1) MDA-MB-231 cells. TUBB (β-tublin) served as an irrelevant control gene. The mRNA levels were normalized to those of ATCB (β-actin). (E) Western blotting analysis of selected TSGs and GRGs in control, PHF20L1 KD, and PHF20L1 KD MDA-MB-231 or Hs 578T cells stably expressing short hairpin RNA (shRNA)–resistant PHF20L1 (WTres). β-Actin served as loading control. (F) Gene set enrichment analysis (GSEA) plot of MYC signal pathway (left) and hypoxia signal pathway (right). FDR, false discovery rate; NES, normalized enrichment score. (G) MYC KD significantly down-regulates the expression of PHF20L1. The expression of MYC or PHF20L1 was measured by qRT-PCR and Western blotting in MDA-MB-231 cells transfected with siRNAs as indicated. (H) Western blotting analysis of the expression of PHF20L1 and hypoxia-inducible factor 1α (HIF1α) in MDA-MB-231 cells treated with CoCl2. (I) Primer pairs including #1 to #6 were synthesized to cover the promoter region of PHF20L1 and quantitative chromatin immunoprecipitation (qChIP)–based promoter walk was performed using normal or CoCl2-treated MDA-MB-231 cells. (J) Luciferase activity of PHF20L1 promoter reporters in human embryonic kidney (HEK) 293T cells transfected with vector, MYC, or HIF1α. (K) MDA-MB-231 cells were transfected with indicated siRNAs or infected with lentiviruses as indicated. ECAR (extracellular acidification rate) was then determined separately. (L) A proposed model underlying the role of the MYC/HIF1a-PHF20L1 axis in regulating the expression of TSGs and GRGs. All error bars represent means ± SD. Two-tailed unpaired t test, *P < 0.05 and **P < 0.01 (C, D, G, I, J, and K).
To further investigate the biological significance of PHF20L1, we performed gene set enrichment analysis (GSEA) using GSEA v2.2.2 software on differentially expressed PHF20L1 target genes and found strong enrichment on the targets of MYC and hypoxia signature genes (Fig. 1F). It has been reported that MYC and hypoxia-inducible factor 1α (HIF1α) are key factors in the regulation of glycolysis in cancer cells and that their abnormal expression could promote the glycolysis process (24, 25). GSEA results suggested that PHF20L1 might participate in the MYC and hypoxia signaling pathways. To explore the crucial role of PHF20L1 in the MYC signaling pathway, we transfected PHF20L1 siRNAs into MDA-MB-231 cells and found that the KD of PHF20L1 did not influence mRNA and protein levels of MYC (Fig. 1G, top) but that mRNA and protein levels of PHF20L1 were notably decreased in MYC KD MDA-MB-231 cells (Fig. 1G, bottom), which indicated that PHF20L1 is downstream of the MYC signaling pathway. It has been reported that HIF1α expression in mammalian cells can be induced in response to hypoxic conditions (1% O2) or hypoxia activators, such as deferoxamine and cobalt chloride (CoCl2) (26). To further determine whether hypoxic conditions induce PHF20L1 expression, we examined protein levels of PHF20L1 in MDA-MB-231 cells exposed to CoCl2 treatment. We found that PHF20L1 was indeed induced by hypoxic conditions (Fig. 1H). Moreover, further quantitative chromatin immunoprecipitation (qChIP) assays using specific antibodies against MYC and HIF1α showed strong binding to PHF20L1 #5 and #6 promoter regions of MYC and HIF1α, respectively (Fig. 1I). To further test whether MYC and HIF1α could directly regulate PHF20L1 transcription, we searched up to ~2 kb of the PHF20L1 promoter regions for possible MYC- and HIF1α-binding sites. The luciferase reporter assays were performed using constructs containing the deletion mutants of each putative binding region. The results showed that overexpression of MYC and HIF1α significantly increased the reporter activity of the PHF20L1 promoter. Moreover, deletion of the −650 to −308 fragment abrogated the MYC-mediated promoter reporter activity, whereas deletion of the −308 to 0 fragment eliminated HIF1α-mediated promoter reporter activity. Meanwhile, deletion of the −650 to 0 promoter fragment almost completely abolished reporter activity (Fig. 1J). Together, these data indicate that PHF20L1 is a direct target gene of MYC/ HIF1α. To further explore the key role of PHF20L1 in altering glycolysis levels in breast cancer cells, MDA-MB-231 cells were transfected with siRNAs or infected with lentiviruses as indicated, and glycolysis levels were measured using a Seahorse XFe24 system (Seahorse Bioscience). Our experiments revealed that PHF20L1 loss of function could significantly reduce the extracellular acidification rate (ECAR), which reflects overall glycolysis levels. Meanwhile, these effects could be reversed by the reexpression of siRNA-resistant PHF20L1 (Fig. 1K, left). In addition, the reduction in glycolysis flux due to MYC depletion was also partially reversed by the reexpression of PHF20L1 (Fig. 1K, right). Together, these experiments revealed that PHF20L1, as a MYC- and HIF1α-driven gene, could repress the expression of several tumor suppressors such as HIC1, KISS1, and BRCA1 and then promotes the expression of GRGs. (Fig. 1L).
The TUDOR domain of PHF20L1 is a H3K27me2-recognizing module
PHF20L1 has MBT, TUDOR, and PHD domains; to further explore the molecular mechanisms through which PHF20L1 exerts its biological functions, we first used a modified histone peptide array containing peptide-cellulose conjugates spotted onto the planar surface of a standard microscope slide in a three-dimensional layer, carrying various histone modifications in duplicate (available on the Active Motif official website), to screen potential histone-binding sites. We found that the glutathione S-transferase (GST)–fused TUDOR domain of PHF20L1 binds strongly to the H3K27me2 peptide, whereas the MBT and PHD domains had no specific binding sites (Fig. 2, A and B). The TUDOR domain of PHF20L1 could only recognize the peptide with H3K27me2 but not the peptide with H3K27me2S28p, H3R26me2sK27me2S28p, or H3R26me2aK27me2S28p. The finding that the GST-TUDOR could not bind the peptides that contain H3S28p in addition to H3K27me2 suggested that the binding is inhibited by S28p. Biotinylated histone peptide pull-down assays with GST- or FLAG-fused PHF20L1 full-length or truncated mutants, as indicated, further confirmed the screening results of peptide array (Fig. 2C). A schematic illustration of the four different domains of PHF20L1 and GST-fused domains purified from BL21 Escherichia coli are shown in Fig. 2 (D and E). To further explore binding between the TUDOR domain and H3K27me2, we performed quantitative isothermal titration calorimetry (ITC) assays, and the results revealed an affinity dissociation constant (KD) of 73.6 μM for the PHF20L1 TUDOR domain to the H3K27me2 peptide, which was much lower than that with other modifications (Fig. 2F). Meanwhile, the results from surface plasmon resonance (SPR) assays further confirmed the specific binding between the PHF20L1 TUDOR domain and the H3K27me2 peptide (Fig. 2G). Previous studies have shown that the TUDOR domain of PHF1 recognizes the H3K36me3 peptide (27), and the results of ITC and SPR assays further revealed that the PHF20L1 TUDOR domain did not bind H3K36me3, which also indicated the specificity of PHF20L1 for H3K27me2 recognition. Moreover, we found that the PHF20L1 TUDOR domain is highly conserved among different species (Fig. 2H). To further explore the key amino acids of PHF20L1 that play important roles in recognizing H3K27me2, peptide pull-down experiments with GST-fused several TUDOR mutants were performed. The results showed that except for mutations in the glutamate-92 and threonine-98, other mutations could not abolish the interaction between H3K27me2 and the PHF20L1 TUDOR domain (Fig. 2I). Similar results were observed in peptide pull-down assays using FLAG-fusion PHF20L1 WT (wild type) and mutants (Fig. 2J). These results revealed the importance of the E92 and T98 residues of PHF20L1 TUDOR in H3K27me2 binding, probably because E92 and T98 residues are essential for the formation of hydrogen bonds between TUDOR and H3K27me2 or they are crucial for the maintenance of the structure of the TUDOR protein. Together, these experiments identified PHF20L1 as an important histone reader exhibiting high affinity and selectivity for H3K27me2 based on the TUDOR domain, which might be involved in transcriptional regulation.
Fig. 2. The TUDOR domain of PHF20L1 is a H3K27me2-recognizing module.
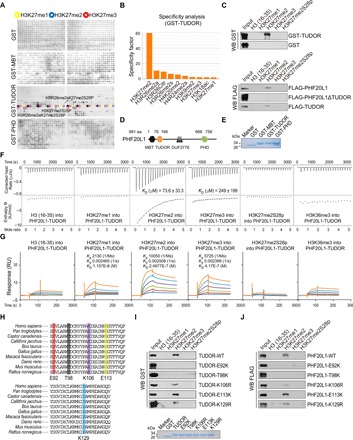
(A) Anti-GST immunoblot of the GST, GST-fused MBT, TUDOR, and PHD domains were measured on a histone peptide array. Peptides were spotted in duplicate as shown in two boxes on the same array. The positions of H3K27me1-, H3K27me2-, and H3K27me3-containing peptides are highlighted with yellow, blue, and red circles. (B) Graphical analysis of the highest binding events detected showing the binding specificity of the GST-TUDOR domain measured on a histone peptide array. (C) Western blotting analysis of histone peptide pull-down assays with GST- or FLAG-fused proteins as indicated. (D and E) Schematic illustrating the four different domains of PHF20L1 and the GST-fused domains purified from BL21 E. coli. (F) Experimental ITC titration curves of the PHF20L1 TUDOR domain to the indicated peptides. (G) SPR analysis of the interaction of PHF20L1 TUDOR with peptides as indicated. (H) Conservation of the PHF20L1 TUDOR domain among 10 species and the designated mutation amino acid sites were shown. (I and J) Western blot analysis of the peptide pull-down analysis using the GST- or FLAG-fused point mutants as indicated.
PHF20L1 exerts transcriptional repressive activity by interacting with PRC2 and the NuRD complex
H3S28p was reported to lead to gene promoter remodeling and transcriptional activation (28). Since the interaction between H3K27me2 and PHF20L1 was repelled by H3S28p, we next investigated whether PHF20L1 is involved in transcriptional repression. First, to identify PHF20L1 interacting proteins, cellular extracts from human embryonic kidney (HEK) 293T and MDA-MB-231 cells stably expressing FLAG-PHF20L1 were subjected to affinity purification using anti-FLAG beads, and the eluates were resolved using an SDS–polyacrylamide gel electrophoresis (SDS-PAGE) gel followed by silver staining to identify interacting proteins. Mass spectrometry analysis showed that PHF20L1 was indeed copurified with subunits of transcription repression–related complexes such as PRC2 and the NuRD complex, including EZH2, SUZ12, EED, Mi-2α/β, histone deacetylase 1/2 (HDAC1/2), MTA1/2, and MBD3 in both cell lines with a high abundance (Fig. 3A). The mass spectrometry details are shown in tables S1 and S2. The presence of PRC2 and NuRD subunits in the PHF20L1 interactome was confirmed by Western blotting with antibodies against the indicated components in the corresponding two cell lines (fig. S2A). Since PHF20 and PHF20L1 are homolog TDRDs with similar domains, we further performed coimmunoprecipitation (Co-IP) experiments using the FLAG antibody in MDA-MB-231 cells that stably expressed FLAG-PHF20L1 or PHF20. The results showed obvious interactions between PHF20L1 and the PRC2/NuRD complex, as well as interaction between PHF20 and MOF (Fig. 3B), as previously reported (5, 6). Unexpectedly, we also found that the PHF20 and PRC2 had some interactions. To further confirm whether their interaction was caused by the DNA fragments that might link some epigenetic regulators together, the Co-IP experiments stated before were performed in the presence or absence of deoxyribonuclease (DNase). The results showed that the interaction between PHF20 and PRC2 disappeared in the presence of DNase, indicating that this binding was indirect (Fig. 3B). Collectively, these results support the notion that PHF20L1 selectively interacts with the PRC2/NuRD complex, whereas PHF20 specifically interacts with MOF. To further confirm the interaction between PHF20L1 and the two transcriptional repressor complexes, we performed Co-IP experiments with HEK293T, MDA-MB-231, and Hs 578T cells. The results showed robust interactions between PHF20L1 and PRC2 or the NuRD complex in vivo (Fig. 3C). We next performed protein fractionation experiments with nuclear proteins by fast protein liquid chromatography (FPLC) with Superose 6 gel filtration chromatography. Western blotting analysis showed that the elution pattern of PHF20L1 largely overlapped with that of PRC2 components, including EZH2 and SUZ12, and NuRD complex proteins, including Mi-2, MTA1/2, HDAC1/2, RbAp46/48, and MBD3 (fig. S2B). These results also indicated a major peak at approximately 667 to 2000 kDa for PHF20L1, PRC2, and NuRD subunits. Furthermore, analysis of the FLAG-PHF20L1 affinity eluate by FPLC with Superose 6 gel filtration chromatography showed that FLAG-PHF20L1 exists in a multiprotein complex, containing PRC2 and NuRD subunits (fig. S2C). To define the key domains of PHF20L1 responsible for directly interacting with PRC2 and the NuRD complex in vivo, a series of PHF20L1 FLAG-tagged domain or deletion mutants were expressed in HEK293T cells. Co-IP with an anti-FLAG antibody followed by Western blotting with indicated antibodies showed that the middle part of PHF20L1, termed the PRC2-NuRD–interacting domain (PNID), was responsible for interactions with PRC2 and the NuRD complex (fig. S2D).
Fig. 3. PHF20L1 exerts transcriptional repressive activity by interacting with PRC2 and the NuRD complex.
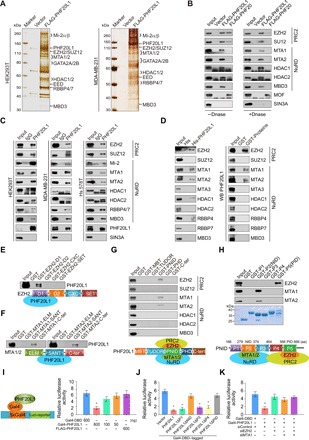
(A) Immunopurification and mass spectrometry analysis of PHF20L1-associated proteins in HEK293T and MDA-MB-231 cells. The eluates were resolved by SDS-PAGE and silver-stained, and the bands were retrieved and analyzed by mass spectrometry. (B) Cellular lysates from MDA-MB-231 cells were immunoprecipitated with antibodies against FLAG in the presence or absence of DNase. (C) Association of PHF20L1 with PRC2 and NuRD in HEK293T, MDA-MB-231, and Hs 578T cells. Whole-cell lysates were prepared, and Co-IP was performed. (D) Molecular interaction between PHF20L1 and PRC2 or NuRD subunits. GST/His pull-down assays using bacterially expressed GST/His-fused proteins and in vitro transcribed/translated proteins are shown as indicated. (E and F) GST pull-down assays with GST-fused truncated EZH2 or MTA proteins and in vitro–transcribed/translated PHF20L1. (G) Mapping the interface in PHF20L1 for the interaction between PHF20L1 and PRC2 or NuRD by GST pull-down assays with GST-fused PHF20L1 domain constructs and in vitro–transcribed/translated PRC2 and NuRD subunits. (H) Mapping the interface in PNID for the interaction between PHF20L1 and PRC2 or NuRD by GST pull-down assays with GST-fused PNID domain constructs and in vitro–transcribed/translated EZH2 and MTA1/2. (I) PHF20L1 has intrinsic transcription repressive activity. HEK293T cells were transfected with the indicated plasmids, and Gal4 luciferase reporter activity was measured. (J) Identification of the essential domains required for the transcriptional repressive activity of PHF20L1. The PHF20L1 deletions fused to the C terminus of Gal4 DNA binding domain were transfected into HEK293T cells, and Gal4 luciferase reporter activity was measured. (K) Effect of depletion of EZH2 or MTA1 on PHF20L1 repressive activity. HEK293T cells were transfected as indicated constructs along with siRNAs against EZH2 or MTA1 for 48 hours, and Gal4 luciferase reporter activity was measured. All error bars represent means ± SD of triplicate measurements that have been repeated three times with similar results. Two-tailed unpaired t test, *P < 0.05 (I to K).
To further address the role of PHF20L1 in the context of a multiprotein complex, we then performed pull-down experiments by incubating of His-fused PHF20L1 with in vitro–transcribed/translated individual components of PRC2 and the NuRD complex as indicated. These experiments indicate that PHF20L1 interacts with EZH2, MTA1, MTA2, and potentially HDAC1, but not MTA3 (Fig. 3D, left). Similarly, GST pull-down experiments with GST-fused components of PRC2/NuRD complex and in vitro–transcribed/translated PHF20L1 obtained similar results (Fig. 3D, right). Meanwhile, GST pull-down assays with GST-fused D1, D2, CXC, or the SET domain of EZH2 and in vitro–transcribed/translated PHF20L1 suggested that the D1 domain of EZH2 is responsible for the interaction between EZH2 and PHF20L1 (Fig. 3E). Similar experiments also showed that the Swi3-Ada2-N-CoR-TFIIIB (SANT) domains of MTA1/2 are responsible for the interaction between MTA1/2 and PHF20L1 (Fig. 3F). Moreover, GST pull-down assays showed that the GST-fused PHF20L1 PNID domain directly interacts with EZH2 and MTA1/2 in vitro (Fig. 3G), which is consistent with the aforementioned in vivo results. The PNID domain is a large domain with 500 amino acids. To elucidate the PRC2 and NuRD interacting region more precisely, we subdivided the PNID domain into five parts (named P1 to P5 for short). GST pull-down assays were performed with GST-fused segments, and in vitro–transcribed/translated EZH2, MTA1, and MTA2 showed that P2 and P5 are responsible for the interactions between PHF20L1 and MTA1/2 or EZH2, respectively (Fig. 3H). Therefore, P2 and P5 were named the NuRD-interacting domain (NID) and PRC2-interacting domain (PID). The results of Co-IP assays further substantiated that the NID and PID was corresponded for NuRD and PRC2 binding, respectively (fig. S2E). Collectively, these results indicate that PHF20L1 interacts with PRC2 and the NuRD complex through the PID and NID regions. The GST/His-fused proteins purified from BL21 E. coli are shown in fig. S2 (F to K).
The physical association between PHF20L1 and the PRC2-NuRD complex led us to hypothesize that PHF20L1 might be functionally involved in transcriptional repression. To verify our hypothesis, full-length PHF20L1 was fused to the C terminus of the Gal4 DNA binding domain (Gal4-PHF20L1), and the fused construct was expressed in HEK293T cells. A Gal4-driven luciferase reporter system containing five copies of the Gal4-binding sequence was used to test the transcriptional activity. The results revealed that the expression of Gal4-PHF20L1, but not FLAG-PHF20L1, leads to a significant reduction in expression of the reporter gene, in a dose-dependent manner (Fig. 3I), indicating that PHF20L1 exerts robust repressive activity. Since the PID and NID domain of PHF20L1 is responsible for interacting with PRC2 and the NuRD complex, we further investigated whether the PID or NID domain is essential for the transcriptional repression activity of PHF20L1. For this, we investigated the contribution of each PHF20L1 domain to its repressive transcriptional function. A series of Gal4 DNA-binding domain (Gal4-DBD) fused deletion constructs were generated, and the repressive activities of those constructs were monitored using Gal4 upstream activating sequence (UAS) luciferase reporter assays. Notably, deletion of the MBT, TUDOR, C-terminal, or PHD domain did not affect the repressive activity of PHF20L1, whereas the deletion of PNID resulted in a substantial reduction in the repressive transcriptional activity of PHF20L1 (fig. S2L). The results also showed that deletion of the P1, P3, or P4 region did not affect the transcriptional repressor activity of PHF20L1, whereas deletion of PID or NID resulted in a significant reduction in PHF20L1 transcriptional repressor activity (Fig. 3J). To determine whether PRC2 and NuRD activity are required for PHF20L1-mediated repression, we performed loss-of-function experiments with the Gal4 UAS luciferase reporter system. As shown, the KD of EZH2 and MTA1 led to a substantial reduction in the repressive transcriptional activity of PHF20L1 (Fig. 3K). Meanwhile, we measured reporter activity in HEK293T cells upon treatment with GSK126, a specific EZH2 inhibitor (29), and trichostatin A (TSA), a specific HDAC inhibitor (30). The results indicated that GSK126 or TSA treatment could almost completely alleviate the PHF20L1-mediated repression of reporter activity (fig. S2M), suggesting that PHF20L1-mediated repression requires the assistance of PRC2 and the NuRD complex. Together, these results suggest that PHF20L1 has intrinsic transcriptional repressor activity through coordinating with PRC2 and the NuRD complex.
PHF20L1 loss of function impairs the deposition of PRC2 and the NuRD complex, perturbing the balance of H3K27 modifications
The NuRD complex removes H3K27ac from certain target gene regions, facilitating PRC2 binding, and, subsequently, the catalysis of histone methylation on H3K27 (31). The findings that PHF20L1 is an H3K27me2 reader that interacts with PRC2 and the NuRD complex, prompted us to explore its function in chromosomal events and the underlying mechanism of transcriptional repression. First, we performed a series of ChIP sequencing (ChIP-seq) experiments, with specific antibodies against PHF20L1, EZH2, MTA1, H3K27me2, H3K27me3, and H3K27ac in normal or PHF20L1 KD MDA-MB-231 cells. We found that the enrichment of PHF20L1, EZH2, MTA1, H3K27me2, and H3K27me3 at the promoter region was substantially less than that of H3K27ac (Fig. 4A), suggesting that PHF20L1 might not share a large-scale chromatin region with H3K27ac. To further explore the relationship between PHF20L1 and two transcriptional repressor complexes in chromatin, we further analyzed ChIP-seq data. The characteristic genomic landscapes of EZH2, MTA1, H3K27me2, H3K27me3, and H3K27ac at PHF20L1-binding sites showed that these proteins were notably enriched in regions surrounding the PHF20L1 binding peaks except H3K27ac (Fig. 4, B and C), which was reported to be associated with enhanced activation of transcription (32).
Fig. 4. PHF20L1 loss of function impairs the deposition of PRC2 and the NuRD complex, perturbing the balance of H3K27 modifications.
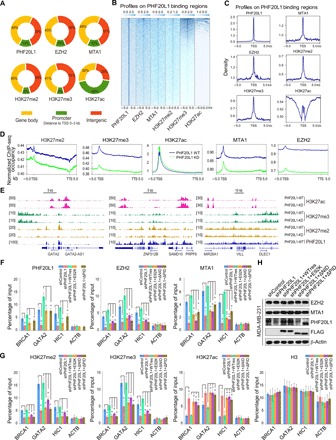
(A) Genomic distribution of PHF20L1, EZH2, MTA1, H3K27me2, H3K27me3, and H3K27ac ChIP-seq peaks. (B and C) ChIP-seq density heatmaps and profiles of EZH2, MTA1, H3K27me2, H3K27me3, and H3K27ac on PHF20L1 binding regions. TSS, transcription start site. (D) The average occupancy of EZH2, MTA1, H3K27me2, H3K27me3, and H3K27ac along the transcription unit in normal and PHF20L1 KD MDA-MB-231 cells. TTS, transcription termination site. (E) Visualized peaks at representative loci using an integrative genomics viewer. (F and G) qChIP analysis using specific antibodies against PHF20L1, EZH2, MTA1, H3K27me2, H3K27me3, H3K27ac, and H3 were performed in control, PHF20L1 KD, and PHF20L1 KD MDA-MB-231 cells stably expressing shRNA-resistant PHF20L1 (represented as WTres), PHF20L1E98K, PHF20L1ΔNID, or PHF20L1ΔPID. ACTB served as control. (H) Western blotting analysis of EZH2 and MTA1 in cells as in (F and G). Data shown are means ± SD of triplicate measurements that have been repeated three times with similar results. Two-tailed unpaired t test, *P < 0.05 and **P < 0.01 (F and G).
We next sought to confirm that PHF20L1 is required for the chromatin recruitment of PRC2 and the NuRD complex. Consistent with our expectations, the analysis of ChIP-seq data showed that PHF20L1 loss of function led to a moderate reduction in EZH2, MTA1, H3K27me2, and H3K27me3 on chromatin, whereas the average genome-wide occupancy of H3K27ac was slightly increased (Fig. 4D). Genomic distributions and peak locations in PHF20L1 KD MDA-MB-231 cells also demonstrated that decreases in H3K27me2 and H3K27me3 levels were linked to increased H3K27ac levels in PHF20L1-occupied genes (Fig. 4E).
qChIP analyses were also performed using specific antibodies against PHF20L1, EZH2, MTA1, H3K27me2, H3K27me3, and H3K27ac at selected gene regions, including BRCA1, GATA binding protein 2 (GATA2), glutathione S-transferase mu 2 (GSTM2), hypermethylated in cancer 1 (HIC1), KiSS-1 metastasis suppressor (KiSS1), stathmin 3 (STMN3), villin like (VILL), and zinc finger protein 512B (ZNF512B). Consistent with ChIP-seq results, PHF20L1 KD significantly reduced the enrichment of PHF20L1, EZH2, MTA1, H3K27me2, and H3K27me3 on PHF20L1 target genes, whereas the enrichment of H3K27ac resulted in a noteworthy increase. Moreover, the KD of EZH2 or MTA1 also resulted in a similar trend (fig. S3, A and B). qRT-PCR and Western blotting analysis confirmed that KD of PHF20L1 does not result in the down-regulation of EZH2 or MTA1 expression (fig. S3C), suggesting that the decreased recruitment was not caused by changes in overall expression levels. To further explore whether TUDOR, NID, and PID domains are essential for the recruitment of PRC2 and NuRD to the targets’ promoters, rescue experiments were performed by ectopically expressing short hairpin RNA (shRNA)–resistant WT PHF20L1 (WTres) or other mutants including E92K, PHF20L1 lacking the NID domain (ΔNID), and PHF20L1 lacking PID domain (ΔPID) in PHF20L1-depleted MDA-MB-231 cells. Then, qChIP assays were used to assess the occupancy of PHF20L1, EZH2 (representing PRC2), and MTA1 (representing the NuRD complex) at the indicated TSGs in Fig. 4F. Because of the loss of antibody recognition epitope, we could not conduct qChIP experiment stated above in MDA-MB-231 cells stably expressing shRNA-resistant PHF20L1ΔNID. The results showed that the wild-type PHF20L1 could bind stably with the target promoters, and only the wild-type PHF20L1, but not the E92K, PHF20L1ΔNID, or PHF20L1ΔPID mutant, restored the recruitments of PRC2 and the NuRD complex caused by the depletion of PHF20L1 (Fig. 4F). At the same time, the related histone modifications H3K27me2, H3K27me3, and H3K27ac are also tested with the rescue experiments with the same design using qChIP assays. The results revealed that wild-type PHF20L1, but not the E92K, PHF20L1ΔNID, or PHF20L1ΔPID mutant, could reinstate the epigenetic changes caused by the depletion of PHF20L1 (Fig. 4G). These results showed that none of the mutants fully rescues PRC2 or NuRD binding and the modification status of the genes, indicating that the whole complex could only function if all parts are present. The Western blotting assays confirmed the KD or overexpression efficiency of PHF20L1 along with the mutations or deletions; moreover, the results also demonstrated that those experimental designs did not result in the change of EZH2 or MTA1’s expression level (Fig. 4H). Together, these results suggest that the TUDOR domain, PID, and NID are critical for the transcriptional repressor activity of PHF20L1 through recognition of H3K27me2 to coordinate with PRC2 and the NuRD complex.
PHF20L1 acts in concert with its associated corepressor complexes to promote breast carcinogenesis
On the basis of the transcriptome sequencing analysis results and the role of PHF20L1 in tumor glycolytic processes, it was reasonable to postulate that PHF20L1 in association with PRC2 and NuRD plays a role in breast tumorigenesis. To this end, we first detected the protein expression profiles at different cell cycle stages synchronized using thymidine and found that PHF20L1, EZH2, and MTA1 were coexpressed in a cell cycle–dependent manner and are relatively abundant during the stages of DNA synthesis (fig. S4A, left). We further found that, compared with that in the control, the KD of PHF20L1 could notably block the cell cycle at the G1-S checkpoint (fig. S4A, right). To further explore the functional significance of PHF20L1 in breast cancer progression and metastasis, colony formation and transwell invasion assays were performed in PHF20L1-depleted MDA-MB-231 and Hs 578T cells, which were stably expressed shRNA-resistant PHF20L1 (WTres). We found that the KD of PHF20L1 notably decreased the colony number and invasive potential of MDA-MB-231 and Hs 578T cells but that the reexpression of shRNA-resistant PHF20L1 could reverse these effects (fig. S4, B to D). Together, these results indicate that PHF20L1 plays an important role in the development of breast cancer.
We demonstrated that the TUDOR, PID, and NID domains are critical for PHF20L1 to recognize H3K27me2 and recruit transcriptional repressor complexes. To explore the intrinsic function of each PHF20L1 domain, full-length PHF20L1 or MBT, TUDOR, PNID, PHD, and C-terminal deletion mutations were stably expressed in MDA-MB-231 cells, and growth curve experiments and transwell assays were performed. The results showed that deletion of the PNID or TUDOR domain could significantly reduce the ability of PHF20L1 to promote cell proliferation and invasion but that the PHD domain and C terminus of PHF20L1 were not required (fig. S4, E and F). Moreover, the rescue experiments were conducted as stated previously in Fig. 3 (F to H) for cell proliferation assays and transwell assays to further determine whether the H3K27me2 recognition function and the recruitment of PRC2 and the NuRD complex by PHF20L1 are essential for its carcinogenetic and metastatic promoting effects. We found that the expression of PHF20L1 WTres but not E92K, PHF20L1ΔNID, or PHF20L1ΔPID fully rescued the colony formation ability and invasive potential of PHF20L1 KD MDA-MB-231 and Hs 578T cells (Fig. 5, A to D), suggesting that both the recognition of H3K27me2 by the TUDOR domain and the recruitment of PRC2 and the NuRD complex by the PID and NID are important for the function of PHF20L1 in breast cancer cells. To further explore whether TUDOR, NID, and PID domains are necessary for transcriptional inhibitory activity of PHF20L1, rescue experiments were performed and confirmed that the up-regulation of TSGs and the down-regulation of GRGs caused by the depletion of PHF20L1 could be completely rescued by the ectopic expression of PHF20L1 WTres but not the E92K, PHF20L1ΔNID, or PHF20L1ΔPID mutants (Fig. 5E). These results suggested that the TUDOR, NID, and PID domains are required for PHF20L1 to function as a transcriptional repressor in breast cancer cells. To investigate the functional synergy between PRC2, NuRD, and PHF20L1, the KD of PHF20L1, together with EZH2 or MTA1 gain-of-function experiments, was performed in MDA-MB-231 cells. Colony formation assays and transwell assays results revealed that the KD of PHF20L1 notably decreased the proliferation and invasion of MDA-MB-231 cells, and this effect could hardly be rescued by the reexpression of EZH2 or MTA1 (fig. S4, G to I). These results indicate that the functions of EZH2 and MTA1 are dependent on the existence of PHF20L1. Since PHF20L1, PRC2, and the NuRD complex could act as a whole complex to exert transcriptional repression activity, we thus further investigated whether PRC2 and the NuRD complex also regulate the expression of PHF20L1 target genes. qRT-PCR and Western blotting analysis demonstrated that the KD of EZH2 or MTA1, respectively, in MDA-MB-231 cells could lead to increased expression of PHF20L1 target TSGs and the decreased expression of PHF20L1 target GRGs at the mRNA and protein level (fig. S4, J and K). These results support our arguments that PHF20L1 may play important roles in breast cancer by recruiting PRC2 and the NuRD complex to transcriptionally repress a range of TSGs including HIC1, KISS1, and BRCA1, thus synergizing the functions of PRC2 and NuRD.
Fig. 5. PHF20L1 acts in concert with its associated corepressor complexes to promote breast carcinogenesis.
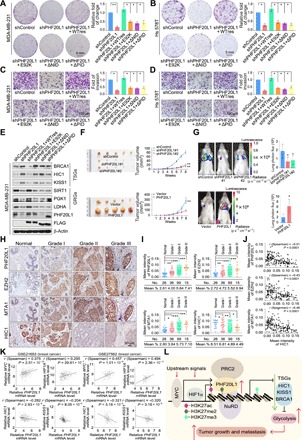
(A) Colony formation assays were performed in control, PHF20L1 KD, and PHF20L1 KD MDA-MB-231 cells stably expressing shRNA-resistant PHF20L1 (represented as WTres), PHF20L1E98K, PHF20L1ΔNID, or PHF20L1ΔPID. (B) Colony formation assays were performed in control, PHF20L1 KD, and PHF20L1 KD Hs 578T cells stably expressing shRNA-resistant PHF20L1, PHF20L1E98K, PHF20L1ΔNID, or PHF20L1ΔPID. (C) Transwell invasion assays were performed in cells as in (A). IgG, immunoglobulin G. (D) Transwell invasion assays were performed in cells as in (B). Data shown are means ± SD. Two-tailed unpaired t test, *P < 0.05 and **P < 0.01 (A to D). (E) Western blotting analysis of the TSGs and GRGs in MDA-MB-231 cells as in (A). (F) MDA-MB-231 cells infected with lentiviruses carrying shControl, shPHF20L1, or stably expressing vector PHF20L1 were inoculated orthotopically into the abdominal mammary fat pads of 6-week-old female BALB/c nude mice (n = 5), and tumor volumes were measured weekly. Data shown are means ± SD. **P < 0.01 at the final day. (G) MDA-MB-231 cells stably expressing firefly luciferase were infected as in (F) then injected intravenously through the tail veins of 6-week-old female severe combined immunodeficient (SCID) mice (n = 6). Lung metastasis was monitored using bioluminescent imaging up to 7 weeks after injection. Representative in vivo bioluminescent images are shown. Data shown are means ± SD. Two-tailed unpaired t test, *P < 0.05. (H) Immunohistochemical (IHC) staining of PHF20L1, EZH2, MTA1, and HIC1 in breast carcinoma samples (histological grades I, II, and III) paired with adjacent normal mammary tissues. Representative images (original magnification, ×200) are shown. (I) Scores of the stained sections from (H) were determined by Image-Pro Plus software and are presented with box plots. Boxes represent the 25th and 75th percentiles; lines represent the median, and whiskers show the minimum and maximum points. *P < 0.05, **P < 0.01, and ***P < 0.001 by one-way analysis of variance (ANOVA). (J) Immunohistochemistry results from (H) were used to analyze the correlation coefficient and P values as indicated. (K) Analysis of public datasets (GSE21653 and GSE27562) from breast cancer for the correlation of MYC, HIF1A, HIC1, KISS1, and PHF20L1. (L) The proposed model for the MYC/HIF1α-(PHF20L1-PRC2-NuRD)-HIC1/KISS1 axis in breast carcinogenesis. Photo credit: Yongqiang Hou, Tianjin Medical University.
To further establish the role of PHF20L1 in breast carcinogenesis in vivo, we first examined how PHF20L1 loss of function affects the growth of tumors developed from MDA-MB-231 cells in a mouse model. MDA-MB-231 cells infected with lentiviruses carrying shPHF20L1 or corresponding shControl were transplanted into the abdominal mammary fat pad of athymic BALB/c female mice (n = 5). The tumors were measured weekly to assess proliferation. As shown, PHF20L1 KD was associated with a notable decrease in the growth of primary MDA-MB-231 tumors (Fig. 5F, top). Furthermore, MDA-MB-231 cells stably expressing PHF20L1 were transplanted into the abdominal mammary fat pad of athymic BALB/c female mice (n = 5). Results showed that PHF20L1 overexpression could substantially promote breast cancer tumor growth (Fig. 5F, bottom). To assess the function of PHF20L1 in tumor metastasis, MDA-MB-231 cells stably expressing firefly luciferase were infected with lentiviruses carrying shPHF20L1, FLAG-PHF20L1, and the corresponding control; then, the cells were intravenously injected into immunocompromised severe combined immunodeficient (SCID) female mice (n = 6). Metastatic tumors were measured by quantitative bioluminescence imaging after 7 weeks using an IVIS imaging system (Xenogen). We found that PHF20L1 deficiency significantly reduced breast cancer cells lung metastasis in vivo, whereas the overexpression of PHF20L1 could promote lung metastasis (Fig. 5G). Together, these results support the notion that PHF20L1 cooperates with PRC2 and the NuRD complex to promote breast carcinogenesis.
Clinical relevance of the MYC/HIF1α–(PHF20L1-EZH2-MTA1)–HIC1/KISS1 axis in cancers
To confirm the clinicopathological relevance of the MYC/HIF1α–(PHF20L1-EZH2-MTA1)–HIC1/KISS1 axis in breast cancer, we collected 176 breast carcinoma samples and analyzed the expression profiles of PHF20L1, EZH2, MTA1, and HIC1 by immunohistochemical (IHC) staining. Notably, IHC analysis using Image-Pro Plus software showed that the expression of PHF20L1, EZH2, and MTA1 was concurrently up-regulated and appeared to be positively correlated with histological grades, whereas the expression of HIC1 was down-regulated and negatively correlated with histological grades or the expression of PHF20L1, EZH2, and MTA1 (Fig. 5, H to J). In addition, to gain a deeper understanding of the role of PHF20L1 in breast cancer progression, analysis of two published clinical datasets (GSE21653 and GSE27562) showed that the expression level of PHF20L1 is positively correlated with the expression of MYC, HIF1A, EZH2, MTA1, SIRT1, and LDHA while negatively correlated with the expression of HIC1 and KISS1 (Fig. 5K and fig. S5A). To further extend our observations on clinical relevance, we analyzed Kaplan-Meier plots based on PHF20L1, EZH2, MTA1, and HIC1 in breast cancer. As shown in fig. S5B, higher PHF20L1 expression is associated with worse overall survival for patients with breast cancer. Consistently, high expression levels of EZH2 and MTA1 were also associated with poor prognosis, whereas patients with high HIC1 expression had longer survival times. To explore whether the oncogenic effect of PHF20L1 also exists in other kind of cancers, we collected several carcinoma samples and performed tissue microarrays, followed by IHC staining to examine the expression of PHF20L1. At least six samples paired with adjacent normal tissues were used. The results showed that in addition to that in breast cancer, PHF20L1 is also progressively increased in lymphoma, cerebral cancer, esophageal cancer, prostate cancer, and pancreatic cancer (fig. S5C). In addition, the analysis of published lymphoma clinical datasets (GSE132929) and glioma datasets (GSE51062) also showed that the expression level of PHF20L1 is positively correlated with the expression of MYC, HIF1A, EZH2, MTA1, SIRT1, and LDHA while negatively correlated with the expression of HIC1 or KISS1 (fig. S5D). Together, these data support our overall hypothesis that PHF20L1 as an H3K27me2 reader could cooperate with the PRC2/NuRD complex to inhibit the expression of TSGs such as HIC1 and KISS1, participating in MYC and hypoxia signaling and leading to tumor progression (Fig. 5L).
Phf20l1 deletion induces growth retardation and mammary ductal outgrowth delay and inhibits tumorigenesis in vivo
Although our previous work herein confirmed the important role of PHF20L1 in breast cancer cells, its intrinsic role in vivo remained unknown. To further investigate the core function of PHF20L1 in vivo, we first established Phf20l1 knockout (KO) mice using CRISPR/Cas9-mediated genome editing (fig. S6A). Genotyping of offspring revealed that Phf20l1-null mice were viable; although embryonic death were observed in a small number of mice, the proportions of genotypes in newborn mice were not notably different in accordance with Mendel’s law of inheritance. However, unexpectedly, some Phf20l1 KO homozygous embryos and individuals exhibited growth retardation (Fig. 6A). We further tracked the growth and development of these mice after birth. Statistical analysis showed that Phf20l1-null mice exhibited marked growth retardation. Moreover, body weight statistics revealed that Phf20l1 KO mice of the same age weighed significantly less than wild type. With age, the weight differences gradually diminished (fig. S6B). In addition, further analysis results showed that, compared to that in wild-type mice, the reproductive age of Phf20l1-null mice was significantly delayed, and these animals exhibited lower fertility (Fig. 6B).
Fig. 6. Phf20l1 deletion induces growth retardation and mammary ductal outgrowth delay and inhibits breast cancer in vivo.
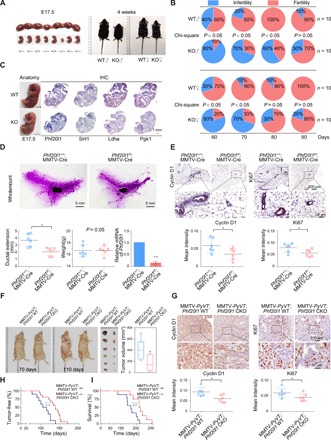
(A) Uterine tissue excised from a pregnant female at 17.5 days post coitum (left). Phf20l1 KO adults are smaller than normal at about 4 weeks old (right). E17.5, embryonic day 17.5. (B) Phf20l1-null mice has delayed reproductive age and exhibited lower fertility. (C) The expression profiles of indicated GRGs were measured using IHC staining in littermate embryos. (D) Mammary ductal developmental defects in Phf20l1 CKO mice (represented as Phf20l1f/f; MMTV-Cre) at about 6 weeks old. (E) IHC staining of cyclin D1 and Ki67 in mammary glands of 6-week-old control and Phf20l1-null mice. (F) Representative bright-field imaging of mammary adenocarcinoma from MMTV-PyVT; Phf20l1f/f; MMTV-Cre (represented as MMTV-PyVT; Phf20l1 CKO) and MMTV-PyVT; Phf20l1+/+; MMTV-Cre (represented as MMTV-PyVT; Phf20l1 WT) mice. The circles indicate surface tumors. The biggest tumors of 110-day-old mice were obtained and calculated (n = 6). (G) IHC staining of Ki67 and cyclin D1 in mammary tumors isolated from MMTV-PyVT; Phf20l1 CKO and MMTV-PyVT; Phf20l1 WT mice. Data shown are means ± SD. Two-tailed unpaired t test, *P < 0.05 and **P < 0.01 (D to G). (H) Mammary adenocarcinoma incidence in MMTV-PyVT; Phf20l1 WT (n = 10) and MMTV-PyVT; Phf20l1 CKO (n = 16) mice depicted as the percentage of tumor-free mice. Mice were considered to be tumor free until a palpable mass (>4.0 mm) persisted for longer than 4 days. Log-rank test was used. (I) Overall survival analysis of the MMTV-PyVT; Phf20l1 WT (n = 9), MMTV-PyVT; Phf20l1 CKO (n = 8) mice, log-rank test. Photo credit: Yongqiang Hou, Tianjin Medical University.
Since we have shown that PHF20L1, as a MYC/HIF1α-driven oncogene, could regulate the expression of GRGs and glycolysis process in breast cancer cells, we tested whether the expression levels of these genes were also changed in Phf20l1-null mice. First, the results of IHC staining with the littermate embryos at day 17.5 of gestation showed that the expression levels of GRGs such as Sirt1, Ldha, and Pgk1 were indeed down-regulated in Phf20l1-null mice (Fig. 6C). By detecting the mRNA levels of target genes in major organs of 4-week-old Phf20l1-null mice compared to those in wild-type mice, we found that the expression levels of GRGs such as Sirt1, Ldha, Pgk1, and Gapdh were down-regulated in the liver, spleen, and kidney (fig. S6C). Together, these results further indicate that a series of GRGs is indeed down-regulated in Phf20l1-null mice, which might contribute to growth retardation.
Next, we investigated the physiological role of PHF20L1 in mammary gland development with the Phf20l1 KO mice. The results showed that Phf20l1 deletion induced notable mammary ductal outgrowth delay. However, the KO mice were smaller than the wild-type mice (fig. S6D). To exclude the effects of differences in body weights and sizes, we generated Phf20l1 conditional knockout (CKO) mice, by crossed mice bearing floxed Phf20l1 with MMTV-Cre mice in which Cre expression was driven by the mouse mammary tumor virus promoter (MMTV-Cre) (fig. S6E). Compared to Phf20+/+; MMTV-Cre mice, virgin Phf20l1flox/flox; MMTV-Cre (Phf20l1f/f; MMTV-Cre) mice also showed a phenotype with mammary ductal outgrowth delay, whereas these animals appeared normal and did not differ from wild-type mice with respect to bodyweight. Furthermore, the results of qRT-PCR assays validated that the Phf20l1’s deletion occurs in the mammary epithelium (Fig. 6D). The observation of these small but otherwise normal mammary glands revealed that Phf20l1 deficiency suppressed mammary ductal growth during puberty. Furthermore, we confirmed that the deletion of Phf20l1 could significantly reduce the number of proliferative cells in virgin mice based on IHC staining for cyclin D1 and Ki67 (Fig. 6E). Together, our findings indicate that Phf20l1 deletion contributes to the down-regulation of GRGs and growth retardation, especially delaying mammary ductal outgrowth.
To unravel the pathological roles of PHF20L1 in breast cancer in vivo, we crossed Phf20l1f/f; MMTV-Cre (Phf20l1 CKO) or WT mice with MMTV–polyoma virus middle T (PyVT) transgenic mice, respectively. The results showed that the volumes of the tumors from MMTV-PyVT; Phf20l1f/f; MMTV-Cre (represented as MMTV-PyVT; Phf20l1 CKO) mice were notably smaller than those of MMTV-PyVT; Phf20l1+/+; MMTV-Cre (represented as MMTV-PyVT; Phf20l1 WT) control mice (Fig. 6F). Compared to those in breast cancer tissues of control mice, decreased cyclin D1 and Ki67 protein levels were observed in tumors of MMTV-PyVT; Phf20l1 CKO mice (Fig. 6G). In addition, all MMTV-PyVT; Phf20l1 WT mice spontaneously developed breast tumors at 77 to 138 days after birth. Notably, the earliest tumor lumps in MMTV-PyVT; Phf20l1 CKO mice appeared at 100 days (Fig. 6H). The survival analysis revealed that genetic ablation of Phf20l1 resulted in a markedly prolonged survival (Fig. 6I). Together, these results demonstrated that PHF20L1 inhibits tumorigenesis in vivo and is a potential oncogene for breast cancer.
DISCUSSION
Our results identify that PHF20L1 is a reader for H3K27me2, which is predominantly recognized by the TUDOR domain, and links PRC2-mediated methylation and NuRD-mediated deacetylation to repress gene expression. PHF20L1 has three classical domains, namely MBT, TUDOR, and PHD. On the basis of in vitro studies, the isolated MBT domain preferentially binds mono- or dimethylated histones but not trimethylated or unmethylated histone peptides (33). Most TDRDs recognize a variety of histone methylations. For example, the tandem-TUDOR domain of JMJD2 family proteins (JMJD2A, JMJD2B, and JMJD2C) is able to read H3K4me3 or H4K20me3 (34). Most of the PHD fingers recognize the methylation of H3K4 and the partial methylation state of H3R2 and H4R3 (35). Several studies have shown that the MBT domain of PHF20L1 binds nonhistone methylation sites instead of binding to methylated histones (9). However, we found that isolated MBT and PHD domain of PHF20L1 demonstrates no specific binding to a modified histone peptide array, while the TUDOR domain exhibited strong specificity for binding to H3K27me2 but not to other histone modifications. It is reported that the second TUDOR domain of PHF20 binds dimethylated peptides derived from the H3 and H4 histone tails, including H3K27me2; although PHF20 could bind dimethylated peptides, it exhibited a preference for peptides containing H3K36me2 than H3K27me2, H3K9me2, H4K20me2, and H3K79me2 (36). Moreover, PHF20L1 and PHF20 share analogous domains, but PHF20L1 preferentially mediates transcriptional repression, while PHF20 mediates transcriptional activation. Their functional similarity and differences as well as the potential molecular mechanisms need to be further studied. Collectively, combined with other studies, our view is that the MBT domain of PHF20L1 might be more likely to bind nonhistone binding sites, whereas the TUDOR domain allows PHF20L1 to participate in chromatin events. However, the ligands of the PHF20L1 PHD domain are still unknown, and this requires further investigation.
Our results confirmed that the TUDOR domain of PHF20L1 recognizes H3K27me2, while the H3S28p impairs the binding of PHF20L1 TUDOR to H3K27me2. It was reported that H3S28 phosphorylation blocks the deposition of PRC2 and exerts a strong transcriptional activation signal (28, 37), further supporting our notion that PHF20L1 has transcriptional inhibitory activity. In addition, ITC and SPR experiments showed that the PHF20L1 TUDOR domains also have the slight ability to bind H3K27me3 (Fig. 2, F and G), considering that the depletion of PHF20L1 reduced not only H3K27me2 but also H3K27me3 at PHF20L1-occupied genes, we conclude that PHF20L1 is essential to maintain a microenvironment of transcriptional repression at the H3K27 site.
PRC2 and the NuRD complex could coexist in specific gene regions to govern the transcription of related genes during embryonic development, and the NuRD complex is mainly responsible for removing histone acetylation, whereas PRC2 can catalyze di- or trimethylation on H3K27 (20). We showed that in breast cancer, PHF20L1 inhibits the transcription of target genes by coordinating with the PRC2/NuRD complex on H3K27me2 enrichment gene regions, bridging histone cross-talk between methylation and deacetylation at H3K27. Our series of ChIP-seq results showed that KD of PHF20L1 could cause a relatively mild but sufficiently clear change in the modifications of H3K27 site. We further validated the changes using qChIP experiments on the promoters of the target genes. The results suggested that PHF20L1 KD could notably reduce the occupancy of PRC2 and the NuRD complex at target gene promoters and lead to decreased H3K27me2/3 and increased H3K27ac levels at the corresponding regions. We suspect that, as a reader protein, PHF20L1 is not able to write or erase epigenetic modifications directly, thus regulating epigenetic markers in a quite modest manner. Moreover, our further work will focus on the intrinsic links among PHF20L1, PRC2, and NuRD during the regulation of H3K27 modifications.
TSGs refer to those for which loss of function contributes to the malignant phenotype, whereas oncogene expression promotes cancerous phenotypes (23). HIC1 is an epigenetically regulated tumor suppressor that forms a transcriptional repressive complex with SIRT1 deacetylase binding the SIRT1 promoter and repressing its transcription (38). HIC1 could participate in tumor metabolism, especially the glycolytic process, through the HIC-SIRT1-TP53 axis (39). There are also other tumor suppressors such as tumor protein p53 (TP53), phosphatase and tensin homolog (PTEN), BRCA1, KISS1 that could cause metabolic reprogramming, especially lowering glycolysis levels to inhibit tumorigenesis (40–42). Combined with ChIP-seq and RNA-seq analysis, we identified many TSGs that were inhibited by the PHF20L1/PRC2/NuRD complex, from which we selected HIC1, KISS1, and BRCA1 for further validation at the mRNA and protein level. RNA-seq analysis also showed that PHF20L1 KD causes the significant down-regulation of SIRT1, which further supports a mechanism whereby PHF20L1 directly inhibits HIC1 expression. Although the mechanism through which HIC1, KISS1, and BRCA1 inhibit the Warburg effect is relatively clear, the effect of other PHF20L1 target TSGs on glycolysis requires further clarification.
HIF1α is a key regulator of the Warburg effect and transcriptionally activates the expression of the majority of GRGs by binding hypoxia-responsive elements of glycolytic gene promoters (25). The overexpression or hyperactivation of MYC, a helix-loop-helix leucine zipper transcription factor, is one of the most common drivers of human cancer, and MYC also directly transactivates GRGs and stimulates aerobic glycolysis (24). Although the role of HIF1α and MYC in various cancers has become increasingly apparent, there are still many challenges regarding their application as drug targets in clinical practice. RNA-seq followed by GSEA analysis and subsequent experiments revealed that PHF20L1 is a downstream component of the MYC, and hypoxia signaling pathway and overexpression of PHF20L1, to a certain extent, could obviously promote glycolysis in breast cancer cells. Given the central roles of PHF20L1 in coordinating the function of the PRC2/NuRD complex and participating in the MYC/hypoxia signaling pathway, it could be a potential drug target for breast cancer.
Our results revealed that PHF20L1 is significantly up-regulated in breast cancer and that its expression appears to be positively associated with histological grades. However, the correlation between PHF20L1 and the molecular pathological subtypes including the luminal, HER2-positive, and basal-like breast cancer requires further investigation. Moreover, compared to levels in adjacent normal tissues, PHF20L1 was also found to be notably overexpressed in lymphoma, cerebrum cancer, esophageal cancer, prostate cancer, and pancreatic cancer, but significant up-regulation was not observed in some cancers such as lung cancer and cervical cancer. Therefore, we speculated that PHF20L1 might have tissue-specific expression patterns in different tumors. At present, we know little about PHF20L1, and the physiological and pathological functions of PHF20L1 in other tissues need to be further studied.
Suz12-, Eed-, or Ezh2-deficient mice are not viable and die during early implantation stages (43); meanwhile, PRC2 was reported to be essential for the development of the mammary gland (44). Further, Mta1 CKOs cause inappropriate mammary gland development (45). We showed that Phf20l1-deficient mice are viable but exhibit mammary ductal outgrowth delay. Badeaux et al. (46) reported that without Phf20, some mice died after birth, while surviving mice were notably smaller than wild-type mice, which is similar to the phenotype of Phf20l1 KO mice. PHF20 was reported to recognize histone H3K4me2 via its PHD finger and participates in transcriptional activation through interaction with MOF, while we found that PHF20L1 as a H3K27me2 reader coordinates with the PRC2/NuRD complex to mediate transcriptional inhibition. The epigenetic mechanistic difference between PHF20 and PHF20L1 has yet to be determined and needs to be studied in the future.
In summary, our findings indicate that PHF20L1, a H3K27me2 recognition protein that is characterized by its TUDOR domain, serves as a potential MYC and hypoxia-driven oncogene and plays a vital role in transcriptional repression by coordinating with PRC2 and the NuRD complex to repress several tumor suppressors such as HIC1, KISS1, and BRCA1, thus up-regulating the GRGs, leading to Warburg effect and tumor progression. Moreover, Phf20l1 deletion induces growth retardation and mammary ductal outgrowth delay and inhibits tumorigenesis in vivo. These findings support the pursuit of PHF20L1 as a potential therapeutic target of breast cancer.
MATERIALS AND METHODS
Antibodies and reagents
The sources of antibodies against the following proteins were as follows: FLAG (F1408, IP; 1:10,000 for WB), PHF20L1 (HPA028417; IP; ChIP; 1:500 for WB and 1:100 for IHC), HDAC1 (H3284; 1:10,000 for WB), HDAC2 (H3159; 1:10,000 for WB), EZH2 (AV38470; 1:1,000 for WB), RbAp46/48 (R3779; 1:1000 for WB), and actin (A1978; 1:10,000 for WB) from Sigma-Aldrich; Mi-2 (sc-11378x; 1:500 for WB), SIN3A (sc-994; 1:1000 for WB), MBD3 (sc-271521; 1:1000 for WB), KISS1 (sc-101246; 1:500 for WB), HIC1 (sc-271499; 1:500 for WB), cyclin E (sc-247; 1:1000 for WB), cyclin D1 (sc-450; 1:1000 for WB), and m-IgGk BP-HRP (horse radish peroxidase) (sc-516102; 1:5,000 for WB) from Santa Cruz Biotechnology; H3 (ab1791; ChIP; 1:10,000 for WB); MTA2 (ab50209; 1:1000 for WB), LDHA (ab101562; 1:1000 for WB), BRCA1 (9010; 1:500 for WB), PGK2 (ab38007; 1:1000 for WB), MYC (ab32072; ChIP; 1:1000 for WB), and HIF1α (ab1; ChIP; 1:500 for WB) from Abcam; SUZ12 (3737s; 1:1000 for WB); MTA1 (5647/5647s; IP; ChIP; 1:1000 for WB), and cyclin D1 (55506; 1:200 for IHC) from Cell Signaling Technology; EED (GTX628007; 1:500 for WB) from GeneTeX; H3K27me1 (07-448; ChIP), H3K27me2 (07-452; ChIP), H3K27me3 (07-449; ChIP), and H3K27ac (07-360; ChIP) were purchased from Millipore; and GST (27457701v; 1:5000 for WB) from GE Healthcare Life Sciences. The histone tail peptides were purchased from Scilight-Peptide (Beijing, China). Protein A/G, Sepharose CL-4B beads were purchased from Amersham Biosciences, and the protease inhibitor mixture cocktail was purchased from Roche Applied Science. The siRNAs and shRNAs of PHF20L1 were purchased from Sigma-Aldrich. siRNAs and shRNAs of the other genes were obtained from GenePharma (Shanghai, China).
Plasmid construction
We thank S. Pradhan (New England Biolabs) for providing the FLAG-tagged PHF20L1 plasmid, and plasmids containing complementary DNA (cDNA) of MTA1, EZH2, and PHF20 were purchased from Open Biosystems. cDNAs were cloned into pLVX-Tight-Puro (Addgene), p3 × FLAG-CMV-10 (Addgene), pCMV-Tag 2B (Addgene), pcDNA3.1-A (Addgene), pET-30a (+) (Addgene), and pGEX GST-fusion plasmids (GE Life Science). Deletion and mutation were introduced by PCR and site-directed mutagenesis using Mut Express MultiS Fast Mutagenesis Kit V2 (Vazyme). All plasmids used were confirmed by sequencing.
Cell culture and transfection
All cell lines were obtained from the American Type Culture Collection. HEK293T and Hs 578T cells were maintained in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% fetal bovine serum (FBS). Cells were maintained in a humidified incubator equilibrated with 5% CO2 at 37°C. MDA-MB-231 cells were cultured in L-15 medium supplemented with 10% FBS and without CO2. Transfections were performed using Lipofectamine 2000 or Lipofectamine RNAiMAX Reagent (Invitrogen, Carlsbad, CA) according to the manufacturer’s instructions. Each experiment was performed in triplicate and repeated at least three times. For RNA interference experiments, at least two independent siRNA sequences were tested for each gene, and the details of siRNA sequences covered in this article are available in table S3.
Real-time qRT-PCR and RNA-seq analysis
Total RNA was isolated from samples using TRIzol reagent following the manufacturer’s instructions (Invitrogen). Potential DNA contamination was removed using a ribonuclease-free DNase treatment (Promega). cDNA was prepared using the MMLV Reverse Transcriptase (Fermentas). Relative quantitation was performed using the ABI PRISM 7500 System (Applied Biosystems), which measures real-time SYBR Green fluorescence. Quantitation was then performed using the comparative Ct method (2−ΔΔCt) with the expression of ACTB (β-actin) as an internal control. The primers used are listed as the following in table S4. For RNA-seq analysis, total RNA was extracted, and three biological replicates were prepared. RNA-seq samples were sequenced using Illumina NextSeq 500. Raw reads were mapped to the human reference genome (hg19). The TopHat2 package was used to analyze the transcriptome, and htseq-count v0.6.0 was used to quantize transcript abundances. Differentially expressed genes were determined using DESeq2. Genes with a fold change of 1.5 and P value of <0.001 were selected as differential genes, and raw data are available on www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE128232.
Histone peptide array and peptide pull-downs
A modified histone peptide array (Active Motif) was used for MBT, TUDOR, or PHD domain binding detection. A modified protein domain kit and an analysis software (Active Motif) were used in accordance with the manufacturer’s instructions. Peptide pull-down assays were performed. Briefly, biotinylated peptides (20 mg) were immobilized on 10 ml of streptavidin beads (Sigma, St. Louis, MO, USA) in 200 ml of binding buffer [50 mM tris-HCl (pH 7.5), 15 mM NaCl, 1 mM EDTA, 2 mM dithiothreitol, and 0.5% NP-40] at 4°C. The next day, the beads were washed three times with binding buffer and then incubated with 25 mg of GST fusion protein or FLAG-tagged protein for 2.5 hours with rotation at 4°C. After five washes with the binding buffer, the beads were boiled in protein loading buffer, and the resulting proteins were fractionated using 10% SDS-PAGE and subjected to Western blotting analysis using an anti-GST or FLAG antibody. The modified histone peptide arrays were analyzed using Active Motif’s Array Analyze software. The software can analyze the spot intensity of the interactions.
Isothermal titration calorimetry
ITC experiments were performed using an Affinity ITC system (TA Instruments). Briefly, the synthesized peptides (>98% purity) and purified proteins were all subjected to extensive dialysis against 100 mM NaCl and 25 mM tris (pH 7.5). Protein concentration was measured using a BCA Pierce protein assay kit (Thermo Fisher Scientific). Peptides at concentrations of ∼1.0 mM were loaded into the ITC syringe, and PHF20L1 TUDOR at a concentration of ∼0.1 mM was loaded into the ITC cell. Each titration consisted of 20 successive injections at 25°C. The binding isotherm results were analyzed using NanoAnalyze Software (TA Instruments).
Surface plasmon resonance
SPR experiments were performed using a Biacore T200 (GE Healthcare). All SPR-based materials were purchased from GE Healthcare. Biotin peptides and Scilight-Peptide (Beijing, China) were diluted in HEPES buffered saline-EP (HBS-EP; GE Healthcare) and immobilized on an SA chip. Approximately 600 resonance units (RU) of the immobilized peptides were obtained. Interaction analyses were tested using HBS-EP as a running buffer. Increasing concentrations of PHF20L1 TUDOR (0.2, 0.4, 0.8, 1.6, 3.2, and 6.4 μΜ) were injected using the “Kinetics/Affinity” program. A flow cell without immobilized peptide served as a nonspecific binding control. The SA chip surface was regenerated after each cycle by injecting 10 mM NaOH for 30 s. Ka, Kd, and KD were determined using the “Kinetics” model in the Biacore T200 evaluation software version 2.0.
Immunopurification and mass spectrometry
Immunopurification assays were performed as described previously (47). Briefly, a FLAG-tagged PHF20L1 plasmid was transfected into HEK293T cells, which were harvested 48 hours later. Anti-FLAG immune affinity columns were prepared using anti-FLAG M2 affinity gel (Sigma) following the manufacturer’s suggestions. Cell lysates were obtained from about 5 × 108 cells and applied to an equilibrated FLAG column of 1-ml bed volume to allow for the adsorption of the protein complex to the column resin. After binding, the column was washed with cold BC500 buffer containing 50 mM tris, 2 mM EDTA, 500 mM KCl, 10% glycerol, and protease inhibitors. FLAG peptide (0.2 mg/ml; Sigma-Aldrich) was applied to the column to elute the FLAG protein complex, as described by the vendor. Fractions of the bed volume were collected and resolved on SDS–polyacrylamide gel, silver-stained, and subjected to liquid chromatography–tandem mass spectrometry sequencing and data analysis.
Immunoprecipitation
For immunoprecipitation assays, cells were washed with cold phosphate-buffered saline (PBS) and lysed with cold lysis buffer at 4°C for 30 min. A total of 500 μg of cellular extracts was incubated with appropriate primary antibodies or normal rabbit/mouse immunoglobin G (IgG) on a rotator at 4°C overnight, followed by the addition of protein A/G Sepharose CL-4B beads for 2 hours at 4°C. Beads were then washed five times with lysis buffer [50 mM tris-Cl (pH 7.4), 150 mM NaCl, 1 mM EDTA, 1% NP-40, 0.25% sodium deoxycholate, and a protease inhibitor mixture]. The immune complexes were subjected to SDS-PAGE followed by immunoblotting with secondary antibodies. Immunodetection was performed using enhanced chemiluminescence (ECL System, Thermo Scientific) according to the manufacturer’s instructions.
GST/His pull-down assays
GST/His-fused constructs were expressed in BL21 E. coli. In vitro transcription and translation experiments were performed using rabbit reticulocyte lysate (TNT systems, Promega) according to the manufacturer’s recommendation. In GST/His pull-down assays, about 5 μg of the appropriate GST/His fusion proteins with 30 μl of glutathione-Sepharose or Ni beads was incubated with 5 to 8 μl of the in vitro–transcribed/translated products in binding buffer [75 mM NaCl and 50 mM Hepes (pH 7.9)] at 4°C for 2 hours in the presence of the protease inhibitor mixture. The beads were washed five times with binding buffer, resuspended in 30 μl of 2× SDS-PAGE loading buffer, and detected by Western blotting.
Luciferase reporter assays
Luciferase activity was measured using a dual luciferase kit (Promega, Madison, WI) according to the manufacturer’s protocol. Each experiment was performed in triplicate and repeated at least three times.
ChIP-seq and qChIP
Normal cells or PHF20L1-depleted MDA-MB-231 cells were maintained in DMEM supplemented with 10% FBS. Approximately 5 × 107 cells were used for each ChIP-seq assay. The chromatin DNA precipitated by polyclonal antibodies against PHF20L1, EZH2, MTA1, H3K27me2, H3K27me3, or H3K27ac. The DNA was purified with a Qiagen PCR purification kit, and a Vazyme TruePrep DNA Library Prep Kit V2 for Illumina (Vazyme Biotech) was used for DNA library construction. In-depth whole genome DNA sequencing was performed by the Annoroad, Beijing. The raw sequencing image data were examined using the Illumina analysis pipeline, aligned to the unmasked human reference genome (hg19) using ELAND (Illumina), and further analyzed by MACS. Enriched binding peaks were generated after filtering through the input data. The ChIP-seq peak distribution statistics were performed using the Cis-regulatory element annotation system. All ChIP-seq data are available on www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE128231. Eluted DNA was purified using a PCR purification kit (QIAGEN), and qChIPs were performed using the TransStart Top Green qPCR Supermix (TransGen Biotech) by quantitative real-time PCR on the ABI 7500-FAST System. The qChIP PCR primers are available in table S4.
ECAR assays
The ECAR was measured using the Seahorse XF24 Extracellular Flux Analyzer (Seahorse Bioscience). Experiments were performed according to the manufacturer’s instructions. ECAR was measured using a Seahorse XF Glycolysis Stress Test Kit (Agilent Technologies).
Colony formation assays
MDA-MB-231 cells were treated as indicated, and the cells were maintained in culture media for about 14 days and then stained with crystal violet.
Cell invasion assays
Transwell chamber filters (Chemicon Incorporation) were coated with Matrigel. Cells were suspended in serum-free DMEM at a concentration of 5.0 × 105 cells/ml, and 300 μl of the cell suspension was placed in the upper chamber of the transwell. The chamber was transferred to a well containing 500 μl of media that included 10% FBS. Cells were incubated for 36 hours at 37°C. Cells in the top well were removed by wiping the top of the membrane using a cotton swab. The membranes were then stained, and the remaining cells were counted. Four high-powered fields were counted for each membrane.
Tumor xenografts
MDA-MB-231 cells were infected with indicated lentiviruses, and 5 × 106 viable cells in 100 ml PBS were injected subcutaneously into 6-week-old BALB/c nude mice (Vital River Laboratories, Beijing, China). Female nude mice (n = 5) were used in each experiment. Tumors were measured every 7 days using a vernier caliper, and the volume was calculated according to the formula: 1/2 × length × square width.
In vivo metastasis
MDA-MB-231 cells stably expressing firefly luciferase (Xenogen) were infected with indicated lentiviruses, and 2 × 106 cells were injected into the lateral tail vein of 6-week-old female SCID mice. For bioluminescence imaging, mice were injected abdominally with 200 mg/g of d-luciferin in PBS. Fifteen minutes after injection, mice were anesthetized, and bioluminescence was imaged with a charge-coupled device camera (IVIS, Xenogen). Bioluminescence images were obtained with a 15-cm field of view, a binning (resolution) factor of 8, 1/f stop, open filter, and imaging time of 30 s to 2 min. Bioluminescence from the relative optical intensity was defined manually. Photon flux was normalized to background, which was defined from a relative optical intensity drawn over a mouse not administered an injection of luciferin.
Mouse models
The Phf20l1 KO and CKO mouse models were generated by Shanghai Model Orgnaisms Center Inc. Strategies of Phf20l1 KO and CKO mouse model were illustrated in fig. S6 (A and D). To obtain MMTV-PyVT; Phf20l1flox/flox; MMTV-Cre female mice, MMTV-PyVT (mouse mammary tumor virus–polyoma virus middle T antigen) transgenic male mice were crossed with Phf20l1flox/flox; MMTV-Cre female mice, and the tail DNA was analyzed by PCR to determine the mouse genotype. All mice studies were approved by the Ethical Committee of Tianjin Medical University (permit number: SYXK 2009-0001).
Tissue specimens and IHC staining
Embryonic day 17.5 (E17.5) embryos, mouse mammary glands, or samples from adjacent normal tissues of pathological grade I, II, and III were fixed in 10% neutral-buffered formalin overnight, then processed, paraffin-embedded, sectioned, and stained with hematoxylin and eosin according to a standard protocol. For IHC staining, 6-μm sample sections were incubated with primary antibodies overnight at 4°C in a humidified chamber, followed by incubation with the HRP-conjugated secondary antibodies for 2 hours. Staining was completed by 5- to 10-min incubation with diaminobenzidine substrate, which results in a brown-colored precipitate at the antigen site.
Whole mounting staining
Mammary glands were harvested and fixed in Carnoy’s solution (6:3:1 of 100% ethanol, chloroform, and glacial acetic acid) and stained with carmine alum. The extent of ductal outgrowth was measured on whole inguinal mounts as the distance from the center of the lymph node to the leading edge of the ductal mass.
Statistical analysis
Results were reported as means ± SD for triplicate experiments unless otherwise noted. SPSS version 17.0 and two-tailed unpaired t tests were used for statistical analysis. The correlation coefficients were calculated using Cor function of the R programming software. Datasets were downloaded from www.ncbi.nlm.nih.gov/geo (Ivhsina; Gene Expression Omnibus: GSE21653, GES27562, GSE132929, and GSE51062). Data for the Kaplan-Meier survival analysis were from http://kmplot.com/analysis/index.php?p=service&cancer=breast.
Supplementary Material
Acknowledgments
Funding: This work was supported by grants from the Major State Basic Research Development Program of China (grant number 2016YFA0102400 to Y.W.) and National Natural Science Foundation of China (grant numbers 81773017 and 41931291 to Y. W.); Author contributions: Y.H. and Y.W. conceived this project. Y.H., W.L., D.S., X.Y., W.H., Yang Yang, Ying Yang, W.F., T.Z., and K.Z. mainly conducted experiments. Y.H., D.S., W.H., J.G., H.Y., X.T., R.Q., and K.Z. acquired data. Y.H., D.S., and Y.W. analyzed data. Y.H., D.S., W.H., and Y.W. wrote the manuscript. Competing interests: The authors declare that they have no competing interests. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. For the RNA-seq and ChIP-seq data, they can be found at the Gene Expression Omnibus database under accession numbers GSE128231 and GSE128232. Additional data related to this paper may be requested from the authors.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/6/16/eaaz0356/DC1
REFERENCES AND NOTES
- 1.Strahl B. D., Allis C. D., The language of covalent histone modifications. Nature 403, 41–45 (2000). [DOI] [PubMed] [Google Scholar]
- 2.Black J. C., Van Rechem C., Whetstine J. R., Histone lysine methylation dynamics: Establishment, regulation, and biological impact. Mol. Cell 48, 491–507 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Musselman C. A., Lalonde M. E., Côté J., Kutateladze T. G., Perceiving the epigenetic landscape through histone readers. Nat. Struct. Mol. Biol. 19, 1218–1227 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Jiang Y., Liu L., Shan W., Yang Z. Q., An integrated genomic analysis of Tudor domain-containing proteins identifies PHD finger protein 20-like 1 (PHF20L1) as a candidate oncogene in breast cancer. Mol. Oncol. 10, 292–302 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hoe M., Nicholas H. R., Evidence of a MOF histone acetyltransferase-containing NSL complex in C. elegans. Worm 3, e982967 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cai Y., Jin J., Swanson S. K., Cole M. D., Choi S. H., Florens L., Washburn M. P., Conaway J. W., Conaway R. C., Subunit composition and substrate specificity of a MOF-containing histone acetyltransferase distinct from the male-specific lethal (MSL) complex. J. Biol. Chem. 285, 4268–4272 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Avvakumov N., Côté J., The MYST family of histone acetyltransferases and their intimate links to cancer. Oncogene 26, 5395–5407 (2007). [DOI] [PubMed] [Google Scholar]
- 8.Klein B. J., Wang X., Cui G., Yuan C., Botuyan M. V., Lin K., Lu Y., Wang X., Zhao Y., Bruns C. J., Mer G., Shi X., Kutateladze T. G., PHF20 readers link methylation of histone H3K4 and p53 with H4K16 acetylation. Cell Rep. 17, 1158–1170 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Estève P. O., Terragni J., Deepti K., Chin H. G., Dai N., Espejo A., Corrêa I. R. Jr., Bedford M. T., Pradhan S., Methyllysine reader plant homeodomain (PHD) finger protein 20-like 1 (PHF20L1) antagonizes DNA (cytosine-5) methyltransferase 1 (DNMT1) proteasomal degradation. J. Biol. Chem. 289, 8277–8287 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Carr S. M., Munro S., Sagum C. A., Fedorov O., Bedford M. T., La Thangue N. B., Tudor-domain protein PHF20L1 reads lysine methylated retinoblastoma tumour suppressor protein. Cell Death Differ. 24, 2139–2149 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bracken A. P., Helin K., Polycomb group proteins: Navigators of lineage pathways led astray in cancer. Nat. Rev. Cancer 9, 773–784 (2009). [DOI] [PubMed] [Google Scholar]
- 12.Ferrari K. J., Scelfo A., Jammula S., Cuomo A., Barozzi I., Stützer A., Fischle W., Bonaldi T., Pasini D., Polycomb-dependent H3K27me1 and H3K27me2 regulate active transcription and enhancer fidelity. Mol. Cell 53, 49–62 (2014). [DOI] [PubMed] [Google Scholar]
- 13.Morey L., Helin K., Polycomb group protein-mediated repression of transcription. Trends Biochem. Sci. 35, 323–332 (2010). [DOI] [PubMed] [Google Scholar]
- 14.Heintzman N. D., Hon G. C., Hawkins R. D., Kheradpour P., Stark A., Harp L. F., Ye Z., Lee L. K., Stuart R. K., Ching C. W., Ching K. A., Antosiewicz-Bourget J. E., Liu H., Zhang X., Green R. D., Lobanenkov V. V., Stewart R., Thomson J. A., Crawford G. E., Kellis M., Ren B., Histone modifications at human enhancers reflect global cell-type-specific gene expression. Nature 459, 108–112 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Dos Santos R. L., Tosti L., Radzisheuskaya A., Caballero I. M., Kaji K., Hendrich B., Silva J. C. R., MBD3/NuRD facilitates induction of pluripotency in a context-dependent manner. Cell Stem Cell 15, 392 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Xue Y. T., Wong J., Moreno G. T., Young M. K., Côté J., Wang W., NURD, a novel complex with both ATP-dependent chromatin-remodeling and histone deacetylase activities. Mol. Cell 2, 851–861 (1998). [DOI] [PubMed] [Google Scholar]
- 17.Xia L. M., Huang W., Bellani M., Seidman M. M., Wu K., Fan D., Nie Y., Cai Y., Zhang Y. W., Yu L.-R., Li H., Zahnow C. A., Xie W., Yen R.-W. C., Rassool F. V., Baylin S. B., CHD4 has oncogenic functions in initiating and maintaining epigenetic suppression of multiple tumor suppressor genes. Cancer Cell 31, 653–668.e7 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Nair S. S., Li D. Q., Kumar R., A core chromatin remodeling factor instructs global chromatin signaling through multivalent reading of nucleosome codes. Mol. Cell 49, 704–718 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wang Y., Zhang H., Chen Y., Sun Y., Yang F., Yu W., Liang J., Sun L., Yang X., Shi L., Li R., Li Y., Zhang Y., Li Q., Yi X., Shang Y., LSD1 is a subunit of the NuRD complex and targets the metastasis programs in breast cancer. Cell 138, 660–672 (2009). [DOI] [PubMed] [Google Scholar]
- 20.Reynolds N., Salmon-Divon M., Dvinge H., Hynes-Allen A., Balasooriya G., Leaford D., Behrens A., Bertone P., Hendrich B., NuRD-mediated deacetylation of H3K27 facilitates recruitment of polycomb repressive complex 2 to direct gene repression. EMBO J. 31, 593–605 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Koppenol W. H., Bounds P. L., Dang C. V., Otto Warburg's contributions to current concepts of cancer metabolism. Nat. Rev. Cancer 11, 325–337 (2011). [DOI] [PubMed] [Google Scholar]
- 22.Li B. X., Zhang M. C., Luo C. L., Yang P., Li H., Xu H. M., Xu H. F., Shen Y. W., Xue A. M., Zhao Z. Q., Effects of RNA interference-mediated gene silencing of JMJD2A on human breast cancer cell line MDA-MB-231 in vitro. J. Exp. Clin. Cancer Res. 30, 90 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lee E. Y., Muller W. J., Oncogenes and tumor suppressor genes. Cold Spring Harb. Perspect. Biol. 2, a003236 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Adhikary S., Eilers M., Transcriptional regulation and transformation by Myc proteins. Nat. Rev. Mol. Cell Biol. 6, 635–645 (2005). [DOI] [PubMed] [Google Scholar]
- 25.Semenza G. L., Targeting HIF-1 for cancer therapy. Nat. Rev. Cancer 3, 721–732 (2003). [DOI] [PubMed] [Google Scholar]
- 26.Masoud G. N., Li W., HIF-1α pathway: Role, regulation and intervention for cancer therapy. Acta Pharm. Sin. B 5, 378–389 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Cai L., Rothbart S. B., Lu R., Xu B., Chen W. Y., Tripathy A., Rockowitz S., Zheng D., Patel D. J., Allis C. D., Strahl B. D., Song J., Wang G. G., An H3K36 methylation-engaging Tudor motif of polycomb-like proteins mediates PRC2 complex targeting. Mol. Cell 49, 571–582 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Gehani S. S., Agrawal-Singh S., Dietrich N., Christophersen N. S., Helin K., Hansen K., Polycomb group protein displacement and gene activation through MSK-dependent H3K27me3S28 phosphorylation. Mol. Cell 39, 886–900 (2010). [DOI] [PubMed] [Google Scholar]
- 29.McCabe M. T., Ott H. M., Ganji G., Korenchuk S., Thompson C., Van Aller G. S., Liu Y., Graves A. P., Della Pietra A. III, Diaz E., La France L. V., Mellinger M., Duquenne C., Tian X., Kruger R. G., Mc Hugh C. F., Brandt M., Miller W. H., Dhanak D., Verma S. K., Tummino P. J., Creasy C. L., EZH2 inhibition as a therapeutic strategy for lymphoma with EZH2-activating mutations. Nature 492, 108–112 (2012). [DOI] [PubMed] [Google Scholar]
- 30.Vigushin D. M., Ali S., Pace P. E., Mirsaidi N., Ito K., Adcock I., Coombes R. C., Trichostatin A is a histone deacetylase inhibitor with potent antitumor activity against breast cancer in vivo. Clin. Cancer Res. 7, 971–976 (2001). [PubMed] [Google Scholar]
- 31.Kim T. W., Kang B. H., Jang H., Kwak S., Shin J., Kim H., Lee S. E., Lee S. M., Lee J. H., Kim J. H., Kim S. Y., Cho E. J., Kim J. H., Park K. S., Che J. H., Han D. W., Kang M. J., Yi E. C., Youn H. D., Ctbp2 modulates NuRD-mediated deacetylation of H3K27 and facilitates PRC2-mediated H3K27me3 in active embryonic stem cell genes during exit from pluripotency. Stem Cells 33, 2442–2455 (2015). [DOI] [PubMed] [Google Scholar]
- 32.Creyghton M. P., Cheng A. W., Welstead G. G., Kooistra T., Carey B. W., Steine E. J., Hanna J., Lodato M. A., Frampton G. M., Sharp P. A., Boyer L. A., Young R. A., Jaenisch R., Histone H3K27ac separates active from poised enhancers and predicts developmental state. Proc. Natl. Acad. Sci. U.S.A. 107, 21931–21936 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Nady N., Krichevsky L., Zhong N., Duan S., Tempel W., Amaya M. F., Ravichandran M., Arrowsmith C. H., Histone recognition by human malignant brain tumor domains. J. Mol. Biol. 423, 702–718 (2012). [DOI] [PubMed] [Google Scholar]
- 34.Lee J., Thompson J. R., Botuyan M. V., Mer G., Distinct binding modes specify the recognition of methylated histones H3K4 and H4K20 by JMJD2A-tudor. Nat. Struct. Mol. Biol. 15, 109–111 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sanchez R., Zhou M. M., The PHD finger: A versatile epigenome reader. Trends Biochem. Sci. 36, 364–372 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Adams-Cioaba M. A., Li Z., Tempel W., Guo Y., Bian C., Li Y., Lam R., Min J., Crystal structures of the Tudor domains of human PHF20 reveal novel structural variations on the Royal Family of proteins. FEBS Lett. 586, 859–865 (2012). [DOI] [PubMed] [Google Scholar]
- 37.Drobic B., Perez-Cadahia B., Yu J., Kung S. K., Davie J. R., Promoter chromatin remodeling of immediate-early genes is mediated through H3 phosphorylation at either serine 28 or 10 by the MSK1 multi-protein complex. Nucleic Acids Res. 38, 3196–3208 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Chen W. Y., Wang D. H., Yen R. C., Luo J., Gu W., Baylin S. B., Tumor suppressor HIC1 directly regulates SIRT1 to modulate p53-dependent DNA-damage responses. Cell 123, 437–448 (2005). [DOI] [PubMed] [Google Scholar]
- 39.Rood B. R., Leprince D., Deciphering HIC1 control pathways to reveal new avenues in cancer therapeutics. Expert Opin. Ther. Targets 17, 811–827 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Levine A. J., Puzio-Kuter A. M., The control of the metabolic switch in cancers by oncogenes and tumor suppressor genes. Science 330, 1340–1344 (2010). [DOI] [PubMed] [Google Scholar]
- 41.Singh K. K., Shukla P. C., Yanagawa B., Quan A., Lovren F., Pan Y., Wagg C. S., Teoh H., Lopaschuk G. D., Verma S., Regulating cardiac energy metabolism and bioenergetics by targeting the DNA damage repair protein BRCA1. J. Thorac. Cardiovasc. Surg. 146, 702–709 (2013). [DOI] [PubMed] [Google Scholar]
- 42.Liu W., Beck B. H., Vaidya K. S., Nash K. T., Feeley K. P., Ballinger S. W., Pounds K. M., Denning W. L., Diers A. R., Landar A., Dhar A., Iwakuma T., Welch D. R., Metastasis suppressor KISS1 seems to reverse the Warburg effect by enhancing mitochondrial biogenesis. Cancer Res. 74, 954–963 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Aloia L., Di Stefano B., Di Croce L., Polycomb complexes in stem cells and embryonic development. Development 140, 2525–2534 (2013). [DOI] [PubMed] [Google Scholar]
- 44.Michalak E. M., Milevskiy M. J. G., Joyce R. M., Dekkers J. F., Jamieson P. R., Pal B., Dawson C. A., Hu Y., Orkin S. H., Alexander W. S., Lindeman G. J., Smyth G. K., Visvader J. E., Canonical PRC2 function is essential for mammary gland development and affects chromatin compaction in mammary organoids. PLOS Biol. 16, e2004986 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Singh R. R., Kumar R., MTA family of transcriptional metaregulators in mammary gland morphogenesis and breast cancer. J. Mammary Gland Biol. Neoplasia 12, 115–125 (2007). [DOI] [PubMed] [Google Scholar]
- 46.Badeaux A. I., Yang Y., Cardenas K., Vemulapalli V., Chen K., Kusewitt D., Richie E., Li W., Bedford M. T., Loss of the methyl lysine effector protein PHF20 impacts the expression of genes regulated by the lysine acetyltransferase MOF. J. Biol. Chem. 287, 429–437 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Su D., Ma S., Shan L., Wang Y., Wang Y., Cao C., Liu B., Yang C., Wang L., Tian S., Ding X., Liu X., Yu N., Song N., Liu L., Yang S., Zhang Q., Yang F., Zhang K., Shi L., Ubiquitin-specific protease 7 sustains DNA damage response and promotes cervical carcinogenesis. J. Clin. Invest. 128, 4280–4296 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/6/16/eaaz0356/DC1