

Nuclear actin promotes the formation of inducible transcription factory, as a general mechanism of the rapid response to stimuli.
Abstract
Gene expression in response to stimuli underlies many fundamental processes. However, how transcription is regulated under these scenarios is largely unknown. Here, we find a previously unknown role of nuclear actin in transcriptional regulation. The RNA-seq data reveal that nuclear actin is required for the serum-induced transcriptional program. Using super-resolution imaging, we found a remarkable enhancement of RNA polymerase II (Pol II) clustering upon serum stimulation, and this enhancement requires nuclear actin. Pol II clusters colocalized with the serum-response genes and nuclear actin filaments upon serum stimulation. Furthermore, N-WASP is required for serum-enhanced Pol II clustering. N-WASP phase-separated with Pol II and nuclear actin. In addition to serum stimulation, nuclear actin also enhanced Pol II clustering upon interferon-γ treatment. Together, our work unveils that nuclear actin promotes the formation of transcription factory on inducible genes, acting as a general mechanism underlying the rapid response to environmental cues.
INTRODUCTION
Response to stimuli is a hallmark of life. Gene regulation in response to stimuli determines the fundamental decision a living cell makes on proliferation, differentiation, motility, and cell death. At tissue and organism levels, precise control of genetic response has substantial impacts on organ development, immune responses, and neuronal plasticity and may contribute to tumor progression upon its dysregulation (1–3). Unexpectedly, little is known about how the transcription machinery is regulated in these inducible transcription processes.
Nuclear actin is implicated in gene regulation through various mechanisms. Nuclear actin is present in chromatin remodeling complexes (4); it interacts with all three classes of RNA polymerases for their full transcriptional activity (5–7); it modulates the localization and activity of megakaryocytic acute leukemia (MAL), a coactivator of serum response factor (SRF) (8); it associates with nascent transcripts through specific heterogeneous nuclear ribonucleoproteins (hnRNPs) (9, 10); and it is involved in long-range chromatin organization (11–13) and DNA damage repair (14–16). Despite recent progress, the mechanisms whereby nuclear actin regulates gene expression remain unclear.
Recent studies have revealed that serum stimulation induces dynamic assembly of nuclear actin filaments, which regulates the SRF-MAL signaling pathway, a key pathway involved in inducible gene expression (17). Noteworthily, we and others reported that serum stimulation also markedly changes the dynamics of “transcription factories” (18, 19), which are discrete RNA polymerase II (Pol II)–clustered foci implicated in gene regulation (20–24). Thus, it would be intriguing to determine whether these two serum stimulation–dependent phenomena are physiologically connected. Here, we formulate a hypothesis that nuclear actin regulates inducible transcription by modulating the formation of transcription factories.
We test this hypothesis with a combination of next-generation transcriptome sequencing and super-resolution microscopy. We find that nuclear actin contributes to the establishment of the serum-induced transcriptional program by scaffolding larger, more active, and more long-lasting Pol II clusters upon serum stimulation. Similar observation is also obtained upon interferon-γ (IFN-γ) treatment. We demonstrate that nuclear actin promotes Pol II cluster formation through de novo nuclear actin polymerization induced by N-WASP/Arp2/3. Using an optoDroplet system, we observe that N-WASP phase-separates with Pol II and nuclear actin. Together, our work unveils a novel role of nuclear actin in transcriptional modulation of inducible genes by promoting enhanced-level Pol II clustering, acting as a general mechanism underlying the rapid response to environmental cues.
RESULTS
Nuclear actin is required for the establishment of the serum-induced transcriptional program
To investigate the role of nuclear actin in inducible transcription activity, we interrogated whether the disruption of nuclear actin results in any change in the transcriptome upon serum stimulation. Commonly used pharmaceutical agents targeting actin, such as cytochalasin D and latrunculin B, lack the specificity to disrupt actin polymerization only in the nucleus. To overcome this difficulty and achieve high specificity toward nuclear actin, we fused actin mutant G13R, a dominant-negative mutant that cannot polymerize (25), with a nuclear localization signal (NLS) sequence and overexpressed the recombinant protein NLS-actin G13R (G13R) to specifically perturb the polymerization state of nuclear actin. We performed next-generation transcriptome sequencing on cells overexpressing G13R and those expressing green fluorescent protein (GFP) as the control, under two conditions: normal growth and serum stimulation. Serum stimulation modulates the transcription of specific genes called serum-response genes (1, 2). To identify differentially expressed genes in response to serum, we defined a parameter termed “fold change” as the ratio of the transcription level under serum stimulation condition to that under normal growth condition. We found that, in general, these serum-response genes exhibited a lower fold change in cells overexpressing G13R than in control cells overexpressing GFP (Fig. 1A). Furthermore, most of the serum-response genes in GFP-control cells were not present in cells overexpressing G13R (Fig. 1B). In contrast, for basal-level transcription under normal growth condition, cells overexpressing G13R had a highly similar transcriptional profile to those expressing GFP, with only 38 differentially expressed genes. To confirm the RNA sequencing (RNA-seq) results, we selected nine canonical serum-response genes and two housekeeping genes, RPLP0 and GAPDH, for quantitative reverse transcription polymerase chain reaction (qRT-PCR) (GAPDH as the internal control). The qRT-PCR results were highly consistent with the RNA-seq data (Fig. 1C). Noteworthily, most canonical serum-response genes had a lower fold change in G13R-overexpressing cells than in GFP-control cells (Fig. 1C, left). Next, we sorted the up-regulated genes by the level of transcriptional change caused by G13R overexpression (Materials and Methods and table S1) and conducted functional annotation analysis of the top ~30% ranked genes (Fig. 1D). These results indicate that nuclear actin is required for the serum-induced expression of specific transcriptional regulators. Together, these results demonstrate that nuclear actin is required for the establishment of the serum-induced transcriptional profile.
Fig. 1. Nuclear actin is required for the establishment of the serum-induced transcriptional program.
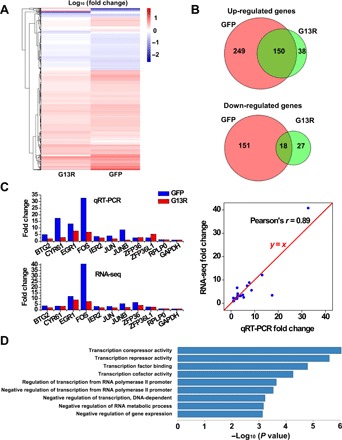
(A) Heatmap showing the fold change for differentially expressed genes upon serum stimulation in cells overexpressing G13R and cells overexpressing GFP. (B) Venn diagrams showing the numbers of differentially expressed genes. (C) qRT-PCR conformation for nine canonical serum-response genes and two housekeeping genes. (D) The functional annotations of the top-ranked genes after sorting by the level of transcriptional change caused by actin mutant overexpression.
Serum-response genes are localized within Pol II clusters upon serum stimulation for transcription
To probe the underlying mechanism for nuclear actin–dependent inducible transcription, we first investigate the transcriptional state of serum-responsive genes by imaging their localization relative to Pol II molecules via ImmunoFISH. Specifically, we found that Pol II molecules form clusters on two canonical serum-response genes, FOS and JUNB (1), upon serum stimulation (fig. S1, A and B). On the contrary, for the control gene PLA2G2E, the percentage of alleles that colocalized with Pol II clusters remained virtually the same under both conditions (fig. S1, A and B). These results suggest that, upon serum stimulation, inducible transcription of serum-response genes takes place in clusters of Pol II molecules.
To confirm that Pol II clusters are active transcription hubs, we investigated the spatial relationship between Pol II clusters and nascent RNA using two-color super-resolution imaging (26, 27). To visualize Pol II, we constructed a human osteosarcoma cell line (U2OS) that stably expresses the Dendra2-fused catalytic subunit of Pol II (RPB1), replacing endogenous RPB1 as previously reported (18, 19). We labeled nascent RNA by pulse incorporation of 5-ethynyl uridine (EU) and Click Chemistry–based conjugation of dyes to EU (Materials and Methods). We quantified the colocalization between Pol II clusters and nascent RNA by pair cross-correlation analysis (28) and found that the colocalization was evidently higher than that of pixel-permutated images (fig. S1, C and D). Together, we reason that Pol II clusters form on specific serum-response genes for active transcription upon serum stimulation.
Serum stimulation enhances the spatiotemporal dynamics of Pol II clustering
We previously reported that Pol II forms clusters in the nucleus and that these clusters appeared and disappeared in an asynchronous manner (19). To quantify the spatiotemporal dynamics of Pol II clusters at higher resolution, we recorded live-cell photoactivated localization microscopy (PALM) (26) images of this stable cell line (Fig. 2A). Pair-correlation analysis (28) confirmed Pol II clustering, with a correction for the photophysical properties of Dendra2 (fig. S2A). We then used SR-Tesseler, a segmentation-based cluster analysis method (29), to identify clusters and extract cluster characteristics (Fig. 2, B and C). For the temporal dynamics of Pol II clusters, we applied the time-correlated PALM (tcPALM) analysis as previously described (18). In tcPALM, assembly of a Pol II cluster is represented by an abrupt increase of single-molecule localizations detected in a cluster, termed a “burst” (Materials and Methods and fig. S2B). Using these analyses, we quantitatively compared Pol II clustering under three conditions: serum stimulation, serum deprivation, and normal growth. We found that cluster density, cluster area, and the number of localizations per cluster were higher in serum-stimulated cells than under normal growth condition (Fig. 2D and fig. S2C), in accord with our previous results (19). Similarly, the number of localizations in a burst (burst size), the duration of a burst (burst lifetime), and the number of bursts per cluster obtained by tcPALM analysis were also larger in serum-stimulated cells (Fig. 2E and fig. S2D), consistent with a previous report (18). In contrast, all of these cluster parameters in cells under serum deprivation condition did not show significant difference from those under normal growth condition (Fig. 2, D and E, and fig. S2, C and D), indicating that the serum-induced changes in Pol II clustering were due to serum stimulation rather than serum deprivation that preceded stimulation (Materials and Methods). These results suggest that more Pol II clusters form de novo upon serum stimulation, which accords with the on-demand model of transcription factory formation (18, 19, 23), and that Pol II clusters become more active, grow larger, and contain more Pol II molecules upon serum stimulation.
Fig. 2. Enhanced Pol II clustering upon serum stimulation.
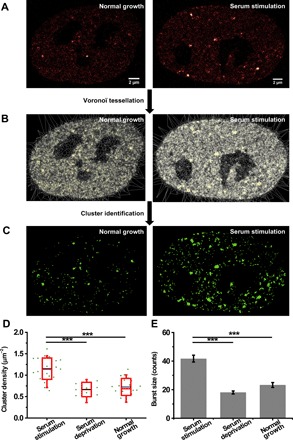
(A) Representative live-cell PALM images of Pol II under normal growth condition (n = 35 cells) and under serum stimulation condition (n = 39 cells). (B) Voronoϊ tessellation of the images in (A) by SR-Tesseler. (C) Images of Pol II clusters identified by SR-Tesseler. (D) Cluster density under serum stimulation (n = 23 cells), serum deprivation (n = 10 cells), and normal growth conditions (n = 20 cells). Each green dot is the mean for all Pol II clusters from a single nucleus. The blue line is the mean for the whole population of nuclei under the indicated condition. The red box shows the SD around the mean. The red line within the red box is the median. The whiskers show 5 and 95%. (E) Burst size under serum stimulation (n = 829 bursts of 104 clusters from 23 cells), serum deprivation (n = 174 bursts of 61 clusters from 10 cells), and normal growth conditions (n = 306 bursts of 111 clusters from 20 cells). Data are shown as means ± SEM. Statistical significance was determined by one-way analysis of variance (ANOVA) with Tukey-Kramer test. ***P < 0.001.
Nuclear actin is required for serum-enhanced Pol II clustering
As serum stimulation drastically enhanced Pol II clustering dynamics, we investigated whether nuclear actin was required for this enhanced-level Pol II clustering. To address this question, we overexpressed NLS-actin mutants (G13R, R62D, and S14C) (25) to perturb the polymerization state of nuclear actin. G13R and R62D are two dominant-negative mutants that cannot polymerize, while S14C is a mutant that is more likely to polymerize than wild-type actin. Overexpression of S14C promoted a massive assembly of nuclear actin filaments (fig. S3, A and B), while overexpression of G13R and R62D reduced nuclear F-actin significantly compared to the control (fig. S3C). With overexpression of nuclear actin mutants, serum stimulation was no longer able to promote the formation of Pol II clusters, which reduced to a level similar to that in cells under normal growth condition (Fig. 3). Note that without serum stimulation, there was no significant difference in any of the cluster parameters in cells overexpressing these actin mutants compared to those under normal growth condition (fig. S4). These results suggest that dynamic polymerization and depolymerization of nuclear actin is required for enhanced-level Pol II clustering upon serum stimulation but is dispensable for basal-level Pol II clustering under normal growth condition.
Fig. 3. Overexpression of actin mutants abolishes serum-enhanced Pol II clustering.
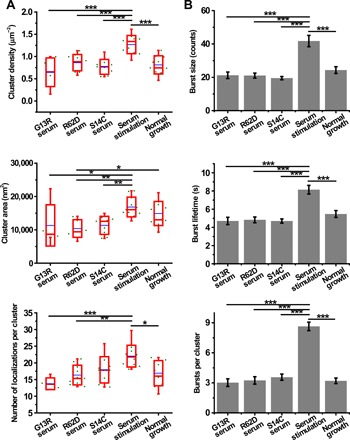
(A) Cluster density, cluster area, and the number of localizations per cluster in cells overexpressing different actin mutants under serum stimulation condition (n = 5, 10, and 9 cells, respectively, from left to right), cells under serum stimulation condition (n = 11 cells), and cells under normal growth condition (n = 10 cells). Each green dot is the mean for all Pol II clusters from a single nucleus. The blue line is the mean for the whole population of nuclei under the indicated condition. The red box shows the SD around the mean. The red line within the red box is the median. The whiskers show 5 and 95%. (B) Burst size, burst lifetime, and the number of bursts per cluster in cells overexpressing different actin mutants under serum stimulation condition (n = 163 bursts of 54 clusters from 5 cells, 230 bursts of 71 clusters from 10 cells, and 271 bursts of 76 clusters from 9 cells, respectively, from left to right), cells under serum stimulation condition (n = 432 bursts of 50 clusters from 11 cells), and cells under normal growth condition (n = 192 bursts of 60 clusters from 10 cells). Data are shown as means ± SEM. Statistical significance was determined by one-way ANOVA with Tukey-Kramer test. *P < 0.05, **P < 0.01, and ***P < 0.001.
In addition to actin mutants, we also carried out spatial knockdown of actin in the nucleus by overexpressing exportin 6 (XPO6) and studied its effects on Pol II clustering. XPO6 specifically exports actin-profilin complexes into the cytoplasm (30). As expected, the fluorescent levels of nuclear actin were markedly lower in XPO6-overexpressing cells than in cells that did not overexpress XPO6 (fig. S5, A to C). Upon depletion of nuclear actin by overexpressing XPO6, we found that serum stimulation was no longer able to promote the formation of Pol II clusters, which reduced to a level similar to that in cells under normal growth condition (fig. S5, D and E). Note that without serum stimulation, there was no significant difference in any of the cluster parameters in XPO6-overexpressing cells compared to the control cells under normal growth condition (fig. S5, D and E). These results confirm that nuclear actin is required for enhanced-level Pol II clustering but dispensable for basal-level Pol II clustering.
To eliminate the possibility that the transfection procedure interfered with the enhanced-level Pol II clustering upon serum stimulation, we overexpressed GFP as the control to investigate its effect on Pol II clustering. We found that GFP overexpression hardly affected either the spatial organization or the temporal dynamics of Pol II clusters under both serum stimulation and normal growth conditions (fig. S6).
Nuclear actin filaments form in adjacent to Pol II clusters upon serum stimulation
To investigate how nuclear actin regulates Pol II clustering upon serum stimulation, we studied the spatial relationship between nuclear actin and Pol II clusters using two-color super-resolution imaging. Imaging nuclear actin has long been a challenge because the signal of cytoplasmic actin overwhelms that of nuclear actin due to its relatively low abundance (31). Moreover, the currently available live-cell imaging probes for nuclear actin interfere with actin dynamics and result in artifacts (32–34). To overcome these challenges, we used an extraction procedure to remove most of the cytoplasmic substances and then labeled actin by dye-conjugated phalloidin, a well-established F-actin probe, in fixed cells (Materials and Methods). Nuclear actin appeared as dense clumps in the nucleoplasm (Fig. 4A) (14, 16, 32). These clumps probably consisted of short actin filaments with more than seven actin subunits, allowing the binding of phalloidin. Nuclear actin was also enriched in nucleoli and nuclear membrane (Fig. 4A). The enrichment of nuclear actin in nucleoli is consistent with its role in Pol I transcription (7, 35, 36). Nuclear membrane–associated cytoplasmic actin is tethered to the nuclear lamina via the linker of nucleoskeleton and cytoskeleton complex (37). Regarding the spatial relationship between nuclear actin and Pol II clusters, both visual inspection (Fig. 4A) and quantitative analysis (Fig. 4B) indicate that nuclear actin clumps evidently colocalized with Pol II clusters at a much higher level under serum stimulation condition than that under normal growth condition. In addition, this serum-induced colocalization was significantly higher than the random level (fig. S7). These results suggest that nuclear actin filaments scaffold Pol II clusters through protein-protein interactions upon serum stimulation.
Fig. 4. Nuclear actin filaments colocalize with Pol II clusters upon serum stimulation.
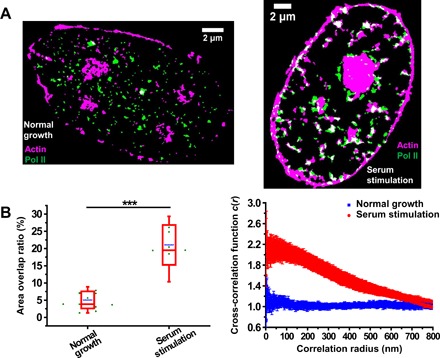
(A) Representative cluster images obtained by SR-Tesseler showing actin (magenta) and Pol II (green) in cells under normal growth (n = 12 cells) and serum stimulation (n = 8 cells) conditions. (B) Quantification of colocalization by calculating the area overlap ratio (left) and analyzing the pair cross-correlation (right). Left: Each green dot is the area overlap ratio for a single nucleus. The blue line is the mean for the whole population of nuclei under the indicated condition. The red box shows the SD around the mean. The red line within the red box is the median. The whiskers show 5 and 95%. Statistical significance was determined by two-tailed t test. ***P < 0.001. Right: Data are shown as means ± SEM (n = 12 cells under normal growth condition, n = 8 cells under serum stimulation condition).
N-WASP–induced nuclear actin polymerization is required for serum-enhanced Pol II clustering
Previous studies have found that both the actin nucleator Arp2/3 and its activating protein N-WASP localized in the nucleus and play a crucial role in Pol II–dependent transcription (38, 39). N-WASP is found to physically associate with Pol II through its interaction with NonO, which binds the C-terminal domain (CTD) of Pol II. It is also demonstrated that the regulation of Pol II–dependent transcription by N-WASP depends on de novo formation of actin filaments induced by N-WASP, rather than the preexisting actin filaments (38). A following work confirms that N-WASP–induced nuclear actin polymerization is mediated by the Arp2/3 complex, which also physically associates with Pol II (39). To investigate whether the nuclear actin filaments induced by N-WASP and Arp2/3 are responsible for serum-enhanced Pol II clustering, we constructed a nuclear-localized dominant-negative mutant of N-WASP (NLS-CA) (38) and studied the effect of N-WASP on the temporal dynamics of Pol II clusters. We found that upon overexpression of NLS-CA, serum stimulation was no longer able to promote the formation of Pol II clusters. The burst size, burst lifetime, and the number of bursts per cluster all reduced to a level similar to that in cells under normal growth condition (Fig. 5) and also similar to that in cells overexpressing actin mutants (Fig. 3B) or XPO6 (fig. S5E). These results indicate that de novo nuclear actin polymerization through the N-WASP–Arp2/3 pathway is required for enhanced Pol II clustering upon serum stimulation.
Fig. 5. Overexpression of a dominant-negative mutant of N-WASP (NLS-CA) interferes with the temporal dynamics of Pol II clusters.
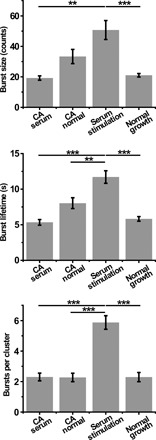
Burst size, burst lifetime, and the number of bursts per cluster in cells overexpressing NLS-CA under serum stimulation (n = 115 bursts of 50 clusters from 10 cells) and normal growth (n = 152 bursts of 67 clusters from 13 cells) conditions, cells under serum stimulation condition (n = 323 bursts of 55 clusters from 11 cells), and cells under normal growth condition (n = 149 bursts of 65 clusters from 13 cells). Data are shown as means ± SEM. Statistical significance was determined by one-way ANOVA with Tukey-Kramer test. **P < 0.01 and ***P < 0.001.
As both N-WASP and Pol II contain low-complexity domains for liquid-liquid phase separation (40–43), we hypothesized that N-WASP–induced nuclear actin scaffolds Pol II clusters through a phase separation mechanism. To test this hypothesis, we constructed an optoDroplet system (44) by fusing N-WASP to Cry2 for light-activated control of intracellular phase separation. Upon light activation, N-WASP phase-separated with the CTD of RPB1 and nuclear actin into liquid droplets in the nucleus (fig. S8). These results suggest a phase separation mechanism by which N-WASP–induced nuclear actin scaffolds Pol II clusters.
Nuclear actin is also required for enhanced-level Pol II clustering upon IFN-γ treatment
To test whether nuclear actin–enhanced Pol II clustering acts as a general mechanism for cells in response to environmental cues, we investigated the effect of nuclear actin in another inducible transcription system. IFN-γ is a crucial cytokine for innate and adaptive immunity (45). Treatment of IFNs was previously found to cause the recruitment of nuclear actin to the promoter regions of IFN-inducible genes (6). For IFN-γ–induced gene expression, we found that nuclear actin promoted enhanced-level Pol II clustering upon IFN-γ treatment in a similar manner to serum stimulation (fig. S9), suggesting the general role of nuclear actin in regulating Pol II clustering and gene expression in response to external stimuli.
DISCUSSION
Both nuclear actin and transcription factories have been under fierce debate (31, 46). In this study, we propose a mechanism in which nuclear actin regulates inducible transcription by promoting enhanced-level Pol II clustering in response to stimuli. Specifically, we investigated the effects of nuclear actin on Pol II clustering upon serum stimulation and IFN-γ treatment. Under normal growth condition, single Pol II molecules assemble into clusters for basal-level transcription, which is independent of nuclear actin dynamics (fig. S4). In contrast, under stimulation condition, Pol II molecules assemble into larger, more dynamic, and more long-lasting clusters, which requires dynamic polymerization and depolymerization of nuclear actin (Figs. 3 and 6). This work connects nuclear actin dynamics with Pol II transcription factories by unveiling the mechanism underlying rapid modulation of gene expression in response to environmental cues, adding insights on actin-related signaling pathways.
Fig. 6. Model that nuclear actin dynamics regulate Pol II clustering in response to serum.
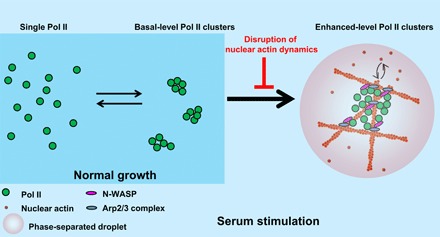
Pol II molecules assemble into basal-level clusters for transcription under normal growth condition, which is independent of nuclear actin dynamics. Upon serum stimulation, Pol II molecules aggregate into enhanced-level clusters for serum-induced transcription, which requires dynamic polymerization and depolymerization of nuclear actin.
The regulation of Pol II clustering by nuclear actin may promote the formation of the preinitiation complex and transcription initiation in response to stimuli. This conclusion is based on previous studies by others and us reporting that inhibitors of transcription elongation do not reduce the dynamics or density of Pol II clusters (18, 19), suggesting that Pol II clustering takes place in the transcription initiation stage. Thus, our model is compatible and complementary to previous models that monomeric actin contributes to chromatin remodeling by mediating the binding of the chromatin remodeling complex INO80 to chromatin (47), and nuclear actin facilitates transcription elongation by assembling at the Ser2-phosphorylated Pol II CTD with hnRNP U to recruit the histone acetyltransferase PCAF (5, 10, 48). Besides, a recent study investigated the role of nuclear actin in basal-level transcription and Pol II clustering (49), complementing our work in inducible transcription under external stimuli.
Our data suggest that nuclear actin may work as a dynamic scaffold to foster the formation of enhanced-level Pol II clusters through a phase separation mechanism (Fig. 6 and fig. S8). The phase separation of N-WASP through its proline-rich motif has been shown to facilitate Arp2/3-branched actin polymerization (42). The phase separation of Pol II through the CTD of the RPB1 subunit is regulated by CTD phosphorylation and plays a role in promoter escape and transcription elongation (40, 41, 43). N-WASP and Arp2/3 physically associate with Pol II, and this association is involved in Pol II transcription (38, 39). Using an optoDroplet system, we demonstrated that N-WASP phase-separated with the CTD of Pol II and nuclear actin upon light activation, mimicking the inducible formation of enhanced-level Pol II clusters upon serum stimulation (fig. S8). Our model that enhanced-level Pol II clusters phase-separate upon serum stimulation (Fig. 6) complements a recent report showing transient interactions between transcription factors and Pol II without detectable phase separation under normal growth condition (50).
The precise molecular mechanisms of how nuclear actin scaffold Pol II clusters on specific genes in response to stimuli still remain to be elucidated. First, it is intriguing to understand how nuclear actin scaffolding, co-phase–separating with Pol II clusters, can function as a general mechanism to promote transcription factory formation in response to different stimuli, in this work, serum and IFN-γ, respectively. Several studies have suggested a transcription factor–dependent transcription factory formation model in which genes regulated by the same transcription factor and cofactors tend to cluster in the same transcription factory (22, 51). We speculate that following the transcription factor–dependent clustering of genes and Pol II molecules, Pol II can recruit N-WASP and Arp2/3, which subsequently activate nuclear actin polymerization and enhance Pol II clustering in a positive feedback manner through multivalency interaction and phase separation (42). Detailed investigation of this speculation and the roles of other factors such as NonO and PSF (38, 39) and phosphorylation of the CTD of Pol II (40, 41, 43) would be of great importance for understanding the precise mechanisms. Second, technological advancements, especially in imaging, would greatly benefit this investigation (52, 53). Recent studies, together with our unpublished data, indicate that various live-cell imaging probes for nuclear actin induce artifacts (32–34). The development of methods that allow artifact-free, high-contrast, and live-cell imaging of nuclear actin would aid in dissecting the details of gene regulation by nuclear actin.
MATERIALS AND METHODS
Cells and plasmids
The original U2OS cell line was purchased from the Cell Bank of Chinese Academy of Sciences. A stable cell line was constructed to express Dendra2-RPB1, replacing endogenous RPB1 as previously described (18, 19). The cell line was cultured in low-glucose Dulbecco’s modified Eagle’s medium (DMEM) (Life Technologies, 10567-014) supplemented with 10% fetal bovine serum (FBS) (Life Technologies, 10099-141) and 1% penicillin/streptomycin (Life Technologies, 15140-122). Cells were tested for the absence of mycoplasma using the LookOut Mycoplasma PCR Detection Kit (Sigma-Aldrich, MP0035). No cell lines used in this study were listed in the database of commonly misidentified cell lines maintained by International Cell Line Authentication Committee and National Center for Biotechnology Information BioSample. We did not attempt to authenticate the cell lines. Transfections were conducted using FuGENE 6 (Promega, E2691) for imaging experiments or Amaxa Cell Line Nucleofector Kit V (Lonza Amaxa, VCA-1003) for RNA-seq experiments, according to the supplier’s protocols.
The plasmids coexpressing actin mutants and GFP were constructed as follows. Actin mutant fragments were PCR-amplified from wild-type β-actin with mutagenic primers. Thereafter, GFP, P2A, NLS, and actin mutant fragments were fused together by overlap PCR. Then, the GFP-P2A-NLS-actin mutant fragments were inserted into the pcDNA 3.1(+) vector. The plasmid coexpressing wild-type actin and GFP was constructed in the same way. The plasmid coding for XPO6-mCerulean was constructed as follows: The XPO6 gene was PCR-amplified from human complementary DNA (cDNA), fused with mCerulean by overlap PCR, and inserted into the pcDNA 3.1(+) vector. The plasmid expressing yellow fluorescent protein (YFP)–actin was constructed by inserting the PCR-amplified β-actin gene sequence into the pYFP-C1 vector. The plasmid coexpressing NLS-CA (a nuclear-localized dominant-negative mutant of N-WASP) and GFP was constructed as follows: the CA sequence [the C-terminal portion of the N-WASP verprolin-homology, cofilin-homology, acidic (VCA) domain] was PCR-amplified from the glutathione S-transferase (GST)–CA plasmid (a gift from W. Guo); GFP, P2A, NLS, and CA fragments were fused together by overlap PCR; and then, the GFP-P2A-NLS-CA fragment was inserted into the pcDNA 3.1(+) vector.
Serum deprivation and stimulation experiments
Cells under serum deprivation condition were obtained by replacing the normal culture medium with serum-free DMEM containing 1% penicillin/streptomycin and culturing overnight. Serum-free L-15 medium (Life Technologies, 21083-027) was used for imaging under serum deprivation condition. Cells under serum stimulation condition were cultured in the serum-free medium overnight and then stimulated with L-15 medium containing 20% FBS for 30 min before imaging. As a control, cells under normal growth condition were grown in the normal culture medium and then transferred to L-15 medium containing 10% FBS right before imaging. Transfections were performed 1 day before culturing cells in the serum-free medium overnight.
IFN-γ treatment
Human IFN-γ (Sino Biological Inc., 11725-HNAS) was a gift from Z. Jiang laboratory (Peking University). Cells were left untreated or treated with IFN-γ (500 IU/ml) for 16 hours before imaging. Transfections were performed 1 day before the treatment.
Phase separation using an optoDroplet system
N-WASP was fused with Cry2-mCherry, which can form clusters containing several molecules upon blue light illumination. This process can be strengthened when Cry2-mCherry was fused with proteins containing intrinsically disorder domain. Cells were transfected with plasmids encoding Cry2–mCherry–N-WASP and EGFP-NLS-RPB1CTD. After 24 hours, cells were illuminated by a light-emitting diode (LED) light source for 20 min. Then, cells were fixed using 4% paraformaldehyde for 15 min. During the fixation process, the blue light was kept illuminating on cells to avoid the disassembly of phase-separated droplets. Cells were washed twice with prewarmed phosphate-buffered saline (PBS) (pH 7.4). Then, cells were permeabilized with 0.1% Triton X-100 in PBS for 15 min. Cells were stained in PBS solution containing 165 nM Alexa Fluor 647 Phalloidin (Molecular Probes, A22287) and 1% bovine serum albumin (BSA) for 30 min. Cells were stained with 4′,6-diamidino-2-phenylindole (DAPI) for 5 min and washed three times with PBS. Images were taken under a spinning disc confocal microscope.
Fluorescence recovery after photobleaching assay
Cells were illuminated with a blue LED light for 10 min to induce the droplet formation. FRAP (fluorescence recovery after photobleaching) assay was then performed on a Zeiss LSM 880 confocal microscope equipped with live-cell incubation system. mCherry signal was bleached using a 561-nm laser beam, using 10 iterations with an acousto-optic tunable filter of the 561-nm laser line set to 100% transmission. A 2-μm-diameter circular region that can cover a phase separation droplet was applied. After five prebleach frames, 200 postbleach frames with a time interval of 1 s were recorded.
Characterization of actin mutants
Cells cultured on glass-bottom dishes were left untreated or transfected with the plasmids encoding actin mutants. After 24 hours, the cells were washed twice with PBS, fixed with 4% paraformaldehyde in PBS for 20 min, washed twice with PBS, and permeabilized with 0.5% Triton X-100 in PBS for 10 min. The cells were then washed twice with PBS and stained with 0.5 μM Alexa Fluor 647 Phalloidin in PBS for 30 min. The cells were washed twice with PBS and stained with DAPI (500 ng/ml) for 2 min. Thereafter, the cells were washed three times with PBS and immediately mounted for epifluorescence imaging.
RNA sequencing
After cells in a six-well plate were rinsed with Dulbecco’s PBS (Gibco, 14190-250), 1 ml of TRIzol LS Reagent (Ambion, 10296010) was added to one well. The cells were resuspended by pipetting and transferred into a 1.5-ml nuclease-free tube. The cell suspension was vigorously vortexed for 30 s and then maintained at room temperature for 5 min. Thereafter, 200 μl of chloroform-isoamyl alcohol (24:1; Sigma-Aldrich, C0549-1PT) was added to the cell lysate and mixed by pipetting. The mixture was maintained at room temperature for 5 min and then centrifuged at 12,000g for 15 min at 4°C. The top aqueous phase containing the total RNA was transferred to a new 1.5-ml tube. An equal volume of isopropanol (Beijing Chemical Works) was added, vortexed, and then maintained at room temperature for 10 min. The tube was centrifuged at 12,000g for 10 min at 4°C to obtain the pellet, which was then washed with 1 ml of 80% ethanol (Sigma-Aldrich, 24102) diluted with diethyl pyrocarbonate–treated water (Ambion, AM9915G). After the pellet was dried, the total RNA was dissolved in 20 μl of ribonuclease (RNase)–free water. Approximately 500 ng of total RNA (quantified by NanoDrop 2000) was used as the starting material for mRNA separation [NEBNext Poly(A) mRNA Magnetic Isolation Module, New England Biolabs (NEB), E7490L] and library preparation (NEBNext Ultra RNA Library Prep Kit for Illumina, NEB, E7530L). All steps were performed following the supplier’s protocols, except that the volume of each solution was half of the recommended volume for each reaction. The libraries were quality-tested for the appropriate fragment distribution and then sequenced on Illumina platform with 150–base pair (bp) pair-end reads.
Bioinformatic analysis of RNA-seq
Whole-transcriptome reads were aligned using the TopHat (v2.0.13) (54) with the Ensembl Homo_sapiens_Ensembl_GRCh37.gtf as a reference (http://www.ensembl.org). In total, we sequenced 10 and 13.7 Mb of 150-bp read pairs for duplicates of cells overexpressing actin mutant G13R under normal growth condition, 10.9 and 13.9 Mb of 150-bp read pairs for duplicates of cells overexpressing actin mutant G13R under serum stimulation condition, 10.2 and 16 Mb of 150-bp read pairs for duplicates of cells overexpressing GFP under normal growth condition, and 14 and 10 Mb of 150-bp read pairs for duplicates of cells overexpressing GFP under serum stimulation condition. The transcriptional analysis was performed using Cufflinks (v2.2.1) (54). The RPKM (reads per kilobase of transcript per million mapped sequence reads) method (55) was used for normalizing gene counts. We calculated the fold change and P value (t test) of RPKM between the samples under serum stimulation condition and the samples under normal growth condition. Differentially expressed genes were screened using a fold change >2 and an absolute difference of FPKM (fragments per kilobase of transcript per million mapped reads) >5 between the samples under serum stimulation condition and the samples under normal growth condition (table S1). To determine the level of transcriptional change caused by actin mutant overexpression, we defined the fold change ratio of a gene as the ratio of its fold change in GFP-control cells to its fold change in cells overexpressing G13R. All the up-regulated genes in either GFP-control cells or cells overexpressing G13R (437 genes in total) were sorted by descending fold change ratio (table S1). The up-regulated genes were then divided into three groups: high–fold change ratio group (140 genes), medium–fold change ratio group (140 genes), and low–fold change ratio group (157 genes). Functional annotation analysis was conducted for these three gene groups using DAVID (https://david.ncifcrf.gov) (56). The first annotation cluster of each group was presented in table S1. For the high–fold change ratio group, the functional annotations in the first annotation cluster with a P value below 0.001 were presented in Fig. 1D. The R density plot package (R Core Team, 2015. R: A Language and Environment for Statistical Computing. R Foundation for Statistical Computing, Vienna, Austria; https://www.R-project.org/) was used to generate the heatmaps, scatterplots, and Venn diagrams. Principal components analysis was conducted using the princomp function in the R graphics package.
Quantitative reverse transcription polymerase chain reaction
Isolation of total RNA was performed as the procedures in RNA-seq. Approximately 200 ng of total RNA (quantified by NanoDrop 2000) was used for reverse transcription in a volume of 19 μl with oligo-d(T) primers using SuperScript III reverse transcriptase (Invitrogen, 18080-044). After reverse transcription at 50°C for 1 hour and 70°C for 15 min to inactivate the enzyme, 1 μl of RNase H (Invitrogen, 18021-071) was added into each reaction tube to digest RNA. Thereafter, 180 μl of nuclease-free water was added to dilute the cDNA to 1 ng/μl. Of this diluted DNA, 1.5 μl was used for each 10 μl of quantitative PCR. The quantitative PCR was run on an ABI 7500 system using SYBR Green (Roche, 13396700) with the following protocol: 95°C for 10 min and 40 cycles at 95°C for 5 s and 60°C for 30 s. Besides the nine serum-response genes we selected from the RNA-seq data, we also selected two housekeeping genes, GAPDH and RPLP0, which served as controls. By normalizing the Ct values of each target gene to that of GAPDH for each sample, we calculated the fold change of a gene as dividing the ∆Ct of the gene in cells under serum stimulation condition by that under normal growth condition.
Probe design and synthesis for ImmunoFISH
Probe design and synthesis was conducted as previously described (57). We designed human genomic libraries of PCR primer pairs delimiting amplicons of 200 to 220 nucleotides (nt) in length for three genes: 44 pairs of primers for FOS, 19 pairs of primers for JUNB, and 21 pairs of primers for PLA2G2E (table S2). For each probe, forward and reverse primers were synthesized by Sangon Biotech Company. Forward and reverse primer pairs were diluted to 100 μM in nuclease-free water. We carried out PCRs by mixing the following reagents: 5 μl of 10× rTaq buffer, 4 μl of 10× deoxyribonucleoside triphosphate (dNTP) mix, 0.25 μl of rTaq enzyme, 0.2 μl each of the 100 μM forward and reverse primers, 100 ng of human genomic DNA, and nuclease-free water to a final volume of 50 μl. For each PCR, 30 cycles were performed with Ta = 55°C. After PCR, the contents of all the wells corresponding to a given probe were purified by agarose gel electrophoresis, and gel recovery was carried out using the HiPure Gel Pure DNA Mini Kit (Magen). Each of the amplicons was mixed together to generate an amplicon pool. One microgram of the 200- to 220-nt amplicon pool was used for subsequent ethanol precipitation.
The FOS/JUNB/PLA2G2E PCR-based DNA fluorescence in situ hybridization (FISH) probes comprised 44/19/21 different 200- to 220-nt amplicon pools. For gene labeling, a volume corresponding to 1 μg from each 20- to 220-nt amplicon pool was precipitated by ethanol and then labeled with Alexa Fluor 647 using the Ulysis Nucleic Acid Labeling Kit (Invitrogen, U21660), according to the supplier’s protocol. Unbound dyes were removed by Micro Bio-Spin Columns, P-30 Tris (Bio-Rad, 732-6223). The labeled probes were stored at −20°C.
Sample preparation for ImmunoFISH
The sample was prepared for ImmunoFISH as previously described (58), with minor modifications. For each hybridization, we used the DNA probe (2 ng/μl) in hybridization solution (namely, 100 ng of the FISH probe was precipitated and dissolved in 50 μl of the hybridization buffer). The hybridization buffer contained 50% formamide, 10% dextran sulfate, 2× SSC, and 1% Tween 20. Unlabeled competitor DNA (Cot-1 DNA and salmon sperm DNA) was added to the probes to inhibit nonspecific hybridization. The concentration of the competitor DNA was approximately 10 to 50 times that of the probe DNA. Therefore, we added 100 ng of the labeled probes, 10 μg of the unlabeled salmon sperm DNA, 1 μg of the unlabeled human Cot-1 DNA, and nuclease-free water to a final volume of 100 μl. Thereafter, 10 μl of 3 M sodium acetate and 275 μl of ice-cold 100% ethanol were added to the mixture. The labeled probe mix was precipitated at −80°C for at least 1 hour. Efficient precipitation of the labeled probe mix was obtained by centrifuging at 14,800 rpm for 30 min. A second washing with 70% ethanol was then performed, and the probe mix was centrifuged at 14,800 rpm for 5 min. The pellet was resuspended with hybridization mix consisting of 50% formamide, 10% dextran sulfate, 2× SSC, and 1% Tween 20. The hybridization mix was first denatured at 86°C in a heat block for 2 min and immediately immersed on ice.
The cells were fixed with 4% paraformaldehyde in PBS for 10 min at room temperature. The cells were then washed twice with PBST (0.1% Tween 20 in PBS) and quenched with freshly prepared 20 mM glycine dissolved in PBS for 10 min. The cells were washed twice with PBST for 5 min each, permeabilized with 0.5% Triton X-100 and 0.5% saponin in PBS for 10 min, and then washed twice with PBST for 5 min each. The cells were incubated with 20% glycerol in PBS for at least 1 hour at room temperature and then submerged into a liquid N2-filled container using fine forceps for approximately 6 s. This freezing-thawing step was repeated twice to improve the penetration and accessibility of the probes to the target DNA sequences. The cells were washed twice with PBS for 2 min each. Thereafter, 0.1 M HCl was applied to the cells for 10 min at room temperature. The cells were washed twice with PBS for 2 min each and then washed twice with 2× SSC for 2 min each. The cells were incubated in 50% formamide and 2× SSC at 4°C overnight. On the following day, the cells were predenatured in a water bath set at 73°C for 10 min and then immediately placed on ice for 2 min. The cells were dehydrated through a series of 70, 85, and 100% ethanol baths for 1 min each and dried for 2 min. The predenatured probe hybridization mix was added to the dried cells. The cells were co-denatured with the probe on a heating block set at 86°C for 3 min and then immediately placed on ice for 5 min. Last, the samples were incubated in a hybridization oven set at 37°C for at least 2 days. After hybridization, the samples were washed as follows: three times for 5 min each with 0.1% Tween 20 in 2× SSC at 37°C on a shaking platform, three times for 5 min each with 0.3% Tween 20 in 0.1× SSC preheated to 63°C under moderate shaking, two times for 5 min each with 0.1% Tween 20 in 4× SSC at 37°C under moderate shaking, and two times for 5 min each with 2× SSC under moderate shaking.
The cells were incubated in PBST blocking buffer (2% BSA, 0.5% fish skin gelatin, and 0.02% Tween 20 in PBS) for 1 hour at room temperature. The primary antibody (mouse Pol II 8WG16; Abcam, ab817) was diluted 1:200 in PBST. The cells were incubated with the primary antibody in a dark humidified chamber for 3 hours. After incubation, the cells were washed four times with PBST. The secondary antibody (donkey anti-mouse, Cy3B) was diluted 1:20 in PBST. The samples were incubated with the secondary antibody in a dark humidified chamber for 45 min. After incubation, the cells were washed four times with PBST and then postfixed in 4% paraformaldehyde in PBS for 10 min at room temperature. After postfixation, the cells were washed four times with PBST and then mounted for imaging.
Sample preparation for two-color PALM/STORM imaging of nascent RNA and Pol II
The samples were prepared using Click-iT Plus Alexa Fluor 647 Picolyl Azide Toolkit (Molecular Probes, C10643), according to the supplier’s protocol. A 2 mM working solution of EU (Molecular Probes, E10345) was prepared from the 100 mM stock solution in prewarmed complete medium. Half of the media within the 35-mm glass-bottom dish was replaced with fresh media containing 2 mM of EU. The cells were incubated under normal cell culture conditions for 10 min. The media were then aspirated, and the cells were fixed with 1 ml of 4% formaldehyde in PBS for 15 min at room temperature. Thereafter, the fixative was removed, and the cells were washed twice with 3% BSA in PBS, followed by permeabilization with 1 ml of 0.5% Triton X-100 in PBS for 20 min at room temperature. A 1× working solution of Click-iT reaction buffer additive was prepared by diluting the 10× solution 1:10 in deionized water. The CuSO4 and copper protectant premix was prepared by mixing 5 μl of CuSO4 with 5 μl of copper protectant. The Click-iT Plus reaction cocktail was prepared by mixing 435 μl of 1× Click-iT Plus reaction buffer, 5 μl of 500 μM Alexa Fluor 647 picolyl azide solution, 10 μl of CuSO4-copper protectant premix, and 50 μl of 1× Click-iT reaction buffer additive. The cells were washed twice with 3% BSA in PBS. Five hundred microliters of Click-iT Plus reaction cocktail was added to the cells. The dish was wrapped with aluminum foil and incubated for 30 min at room temperature. Thereafter, the sample was washed once with 3% BSA in PBS and immediately mounted for imaging.
Spinning disc confocal microscopy
Spinning disc confocal microscopy was conducted on an UltraVIEW VoX spinning disc microscope (PerkinElmer) with an electron-multiplying charge-coupled device (EMCCD) (Hamamatsu C9100-13). Single z-slice images were acquired at an exposure time of 100 ms for both 514- and 440-nm laser lines. Image analysis was performed using Volocity software.
Sample preparation for two-color PALM/STORM imaging of nuclear actin and Pol II
Cells cultured on a 35-mm glass-bottom dish were briefly washed twice with PBS and then permeabilized with 1 ml of hypotonic buffer [10 mM tris-HCl (pH 7.3), 10 mM KCl, and 1.5 mM MgCl2] for 3 min. Thereafter, the cells were washed with 500 μl of 0.5% PBST (0.5% Triton X-100 in PBS) for 45 s, followed by a second washing with PBST for 30 s and a third washing with PBS. The cells were fixed with 1 ml of 4% paraformaldehyde [electron microscopy (EM) grade, Electron Microscopy Sciences] in cytoskeleton buffer [10 mM MES (pH 6.1), 150 mM NaCl, 5 mM EGTA, 5 mM glucose, and 5 mM MgCl2] for 10 min. The cells were then washed with PBS three times for 10 min each. To label each dish of sample, the staining solution was prepared by diluting 30 μl of the Alexa Fluor 647 Phalloidin (Molecular Probes, A22287) stock solution (~6.6 μM in methanol) into 400 μl of PBS (a final concentration of ~0.5 μM). The dishes were wrapped with aluminum foil and incubated at 4°C overnight on a shaker. Thereafter, the sample was briefly washed once with PBS and immediately mounted for imaging.
Epifluorescence, tcPALM, and two-color PALM/STORM imaging
Epifluorescence, tcPALM, and two-color PALM/STORM imaging were conducted using an Olympus inverted microscope equipped with a 100× oil immersion objective [Olympus; numerical aperture (NA), 1.49] and oblique incidence excitation. A 488-nm laser (MPB Communications, 500 mW) was used to excite the fluorophore Dendra2 for conventional epifluorescence imaging. A 405-nm laser (Coherent, Cube 405-100C) was used for the photoconversion of Dendra2 from the green emission state to the red emission state or for the activation of Alexa Fluor 647 from the dark state to the fluorescent state. A 561-nm laser (MPB Communications, 2 W) was used to illuminate Dendra2 at the red emission state. A 647-nm laser (MPB Communications, 1.5 W) was used to excite Alexa Fluor 647. The laser lines were combined with a custom dichroic (Chroma, zt405/488/561/647/752RPC). The emission was filtered with a custom dichroic (Chroma, zet405/488/561/647-656/752M). The emission was separated with a QuadView (Photometrics, QV2) using the dichroics T560lpxr, T650lpxr, and 750dcxxr (Chroma) and the emission filters ET525/50m, ET605/70m, ET700/75m, and HQ795/50m (Chroma) and imaged with an EMCCD (Andor-897). The z-position was maintained during acquisition by detecting a separate infrared beam reflected from the coverslip using a quadrant photodiode, which reads the position of the coverslip and feeds back the position to a piezo-controlled objective.
For epifluorescence imaging, images were acquired at an exposure time of 200 ms and an EM gain of 30 for five consecutive frames (the five frames were averaged for image analysis). The laser power density on the sample was 0.2 W/cm2 (488 nm).
For tcPALM imaging, images were acquired at exposure time of 50 ms and an EM gain of 90 for 10,000 frames under continuous illumination for both 405- and 561-nm lasers. The laser power densities on the sample were 0.15 W/cm2 (405 nm) and 2.0 kW/cm2 (561 nm).
For two-color PALM/STORM imaging, images of Alexa Fluor 647 were acquired at an exposure time of 20 ms and an EM gain of 30 for ~20,000 frames under continuous illumination for a single 647-nm laser; then, images of Dendra2 were acquired at an exposure time of 50 ms and an EM gain of 90 for ~10,000 frames under continuous illumination for both 405- and 561-nm lasers. Fiducial beads for drift correction and chromatic correction were simultaneously tracked during image acquisition. The laser power densities on the sample were 1.5 kW/cm2 (647 nm) and 2.0 kW/cm2 (561 nm). The 405-nm laser intensity on the sample was ramped from 0.1 to 30 W/cm2 to maintain an almost constant rate of photoswitching. The cells were imaged in STORM imaging buffer. The STORM imaging buffer was prepared by mixing 2 ml of 20 mM NaCl, 100 mM tris (pH 8.0), and 10% (w/v) glucose with 20 μl of Glucose Oxidase Oxygen Scavenger solution and 20 μl of β-mercaptoethanol (Sigma-Aldrich, 63689-25ML-F). The Glucose Oxidase Oxygen Scavenger solution contained glucose oxidase (60 mg/ml) (Sigma-Aldrich, G6125-10KU), catalase (6 mg/ml) from bovine liver (Sigma-Aldrich, C100-50MG), and 40% glycerol in PBS.
Image analysis was conducted using Insight3 software (provided by B. Huang from University of California, San Francisco) for fluorophore localization and custom-written MATLAB scripts for drift correction, chromatic correction, and image reconstruction.
Structured illumination microscopy imaging for ImmunoFISH
Structured illumination microscopy (SIM) imaging was performed under an N-SIM microscope (Nikon) equipped with a 100× oil immersion objective (Nikon, NA 1.49) and an EMCCD (Andor, iXon3 DU-897E). A z-stack of seven layers with a step size of 0.12 μm was acquired at an exposure time of 20 to 200 ms, an EM gain of 300, and a conversion gain of 2.4× for 640- and 561-nm laser lines. The exposure time and laser intensities were adjusted to achieve a good image quality. SIM images were reconstructed by the three-dimensional (3D) Reconstruct Stack program in the N-SIM module. The reconstructed Z-stack image was projected into a 2D image by maximum intensity projection. The cells were imaged in STORM imaging buffer without β-mercaptoethanol.
Cluster analysis
Clusters were identified using open-source SR-Tesseler software (29). Clusters smaller than 40 nm in diameter (1256.637 nm2 in area) were excluded from our analysis. Pair autocorrelation analysis and tcPALM analysis were performed as previously described (18, 28) using custom-written MATLAB scripts. For tcPALM analysis, localizations separated by no more than 30 dark frames were considered part of the same burst; bursts with fewer than 10 localizations were excluded from our analysis; hence, a burst might arise from the blinking of a single Dendra2 molecule.
tcPALM is a method to capture and quantify transient clustering dynamics in living cells. For a Pol II cluster in living cells, single-molecule signals appeared clustered in the detection profile and exhibited large steps in the cumulative count (fig. S2B, left). This large step represented an abrupt increase in local Pol II concentration, indicating the assembly of a Pol II cluster. This assembly event was termed a burst. The number of localizations in a burst was termed burst size, and the duration of a burst was termed burst lifetime (fig. S2B, left). On the contrary, for a Pol II cluster in fixed cells, single-molecule signals concentrated at the start of the acquisition and showed a monotonic slope gradually followed by a plateau (fig. S2B, right). This curve represented the gradual photobleaching of immobile Dendra2-Pol II in fixed cells.
Colocalization analysis
Colocalization of two channels in two-color PALM/STORM imaging was analyzed by calculating the percentage of cluster area overlap (area overlap ratio) and analyzing the pair cross-correlation. For the calculation of the area overlap ratio, the clusters in the two channels were identified by SR-Tesseler (29). The two cluster images were exported from SR-Tesseler and then opened in ImageJ software (National Institutes of Health, USA). The colocalization analysis was conducted on a cluster-to-cluster basis for the area of nucleoplasm (the regions of nucleoli and nuclear membrane were excluded from the analysis). The total areas of Pol II clusters, actin clusters, and overlap between these two clusters were calculated by the particle analyzer in ImageJ. The area overlap ratio was calculated as follows
Pair cross-correlation analysis was performed as previously described (28) using custom-written MATLAB scripts.
Statistics and reproducibility
Statistical analyses were conducted using OriginPro 8 software. Statistical significance was determined by one-way analysis of variance (ANOVA) with Tukey-Kramer test, two-tailed t test, pair-sample two-tailed t test, and chi-square test as indicated in the corresponding figure legends. All experiments presented in the manuscript were repeated at least in two independent experiments/biological replicates.
Supplementary Material
Acknowledgments
We thank X. Zhuang (Harvard University), C. Wu (Peking University), and K. Xu (University of California, Berkeley) for discussion; H. Babcock (Harvard University) for help with optical setup of the STORM microscopy; B. Huang (University of California, San Francisco) for Insight3 software and discussion; I. I. Cisse (Massachusetts Institute of Technology) for help with tcPALM experiments; F. Levet (University of Bordeaux) for help with SR-Tesseler analysis; W. Du (Tsinghua University) for help with cell biology experiments; N. Wang (University of California, Los Angeles) for help with bioinformatic analysis; W. Guo (University of Pennsylvania) for the gift of the GST-CA plasmid; and Z. Jiang (Peking University) for the gift of IFN-γ. We thank H. Lv, C. Shan, and the Core Facilities at School of Life Sciences, Peking University for imaging support. Funding: This work was supported by grants from the National Key R&D Program of China, No. 2017YFA0505300 and the National Science Foundation of China 21825401 to Y.S. Author contributions: Y.S. and M.W. conceived the project and designed the experiments. M.W. performed the imaging experiments and data analysis with the help of M.D. X.F. performed RNA-seq and qRT-PCR experiments. R.L. conducted bioinformatic analysis. Y.H., M.W., and S.M. prepared the samples for ImmunoFISH. S.S. performed the optoDroplet assay. M.D. assisted with plasmid construction. M.W. and Y.S. wrote the manuscript, with inputs from X.F., R.L., Y.H., S.S., and M.D. All authors participated in discussion and editing of the manuscript. Competing interests: The authors declare that they have no competing interests. Data and materials availability: The RNA-Seq data have been deposited at Gene Expression Omnibus (GEO) under the accession code GSE82320. All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. Additional data related to this paper may be requested from the authors.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/6/16/eaay6515/DC1
REFERENCES AND NOTES
- 1.Herschman H. R., Primary response genes induced by growth factors and tumor promoters. Annu. Rev. Biochem. 60, 281–319 (1991). [DOI] [PubMed] [Google Scholar]
- 2.Weake V. M., Workman J. L., Inducible gene expression: Diverse regulatory mechanisms. Nat. Rev. Genet. 11, 426–437 (2010). [DOI] [PubMed] [Google Scholar]
- 3.de Nadal E., Ammerer G., Posas F., Controlling gene expression in response to stress. Nat. Rev. Genet. 12, 833–845 (2011). [DOI] [PubMed] [Google Scholar]
- 4.Kapoor P., Shen X., Mechanisms of nuclear actin in chromatin-remodeling complexes. Trends Cell Biol. 24, 238–246 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Percipalle P., Co-transcriptional nuclear actin dynamics. Nucleus 4, 43–52 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hofmann W. A., Stojiljkovic L., Fuchsova B., Vargas G. M., Mavrommatis E., Philimonenko V., Kysela K., Goodrich J. A., Lessard J. L., Hope T. J., Hozak P., de Lanerolle P., Actin is part of pre-initiation complexes and is necessary for transcription by RNA polymerase II. Nat. Cell Biol. 6, 1094–1101 (2004). [DOI] [PubMed] [Google Scholar]
- 7.Philimonenko V. V., Zhao J., Iben S., Dingová H., Kyselá K., Kahle M., Zentgraf H., Hofmann W. A., de Lanerolle P., Hozák P., Grummt I., Nuclear actin and myosin I are required for RNA polymerase I transcription. Nat. Cell Biol. 6, 1165–1172 (2004). [DOI] [PubMed] [Google Scholar]
- 8.Vartiainen M. K., Guettler S., Larijani B., Treisman R., Nuclear actin regulates dynamic subcellular localization and activity of the SRF cofactor MAL. Science 316S, 1749–1752 (2007). [DOI] [PubMed] [Google Scholar]
- 9.Percipalle P., Zhao J., Pope B., Weeds A., Lindberg U., Daneholt B., Actin bound to the heterogeneous nuclear ribonucleoprotein hrp36 is associated with Balbiani ring mRNA from the gene to polysomes. J. Cell Biol. 153, 229–236 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kukalev A., Nord Y., Palmberg C., Bergman T., Percipalle P., Actin and hnRNP U cooperate for productive transcription by RNA polymerase II. Nat. Struct. Mol. Biol. 12, 238–244 (2005). [DOI] [PubMed] [Google Scholar]
- 11.Chuang C.-H., Carpenter A. E., Fuchsova B., Johnson T., de Lanerolle P., Belmont A. S., Long-range directional movement of an interphase chromosome site. Curr. Biol. 16, 825–831 (2006). [DOI] [PubMed] [Google Scholar]
- 12.Dundr M., Ospina J. K., Sung M.-H., John S., Upender M., Ried T., Hager G. L., Matera A. G., Actin-dependent intranuclear repositioning of an active gene locus in vivo. J. Cell Biol. 179, 1095–1103 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Baarlink C., Plessner M., Sherrard A., Morita K., Misu S., Virant D., Kleinschnitz E.-M., Harniman R., Alibhai D., Baumeister S., Miyamoto K., Endesfelder U., Kaidi A., Grosse R., A transient pool of nuclear F-actin at mitotic exit controls chromatin organization. Nat. Cell Biol. 19, 1389–1399 (2017). [DOI] [PubMed] [Google Scholar]
- 14.Schrank B. R., Aparicio T., Li Y., Chang W., Chait B. T., Gundersen G. G., Gottesman M. E., Gautier J., Nuclear ARP2/3 drives DNA break clustering for homology-directed repair. Nature 559, 61–66 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Caridi C. P., D’Agostino C., Ryu T., Zapotoczny G., Delabaere L., Li X., Khodaverdian V. Y., Amaral N., Lin E., Rau A. R., Chiolo I., Nuclear F-actin and myosins drive relocalization of heterochromatic breaks. Nature 559, 54–60 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Belin B. J., Lee T., Mullins R. D., DNA damage induces nuclear actin filament assembly by Formin-2 and Spire-1/2 that promotes efficient DNA repair. eLife 4, e07735 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Baarlink C., Wang H., Grosse R., Nuclear actin network assembly by formins regulates the SRF coactivator MAL. Science 340, 864–867 (2013). [DOI] [PubMed] [Google Scholar]
- 18.Cisse I. I., Izeddin I., Causse S. Z., Boudarene L., Senecal A., Muresan L., Dugast-Darzacq C., Hajj B., Dahan M., Darzacq X., Real-time dynamics of RNA polymerase II clustering in live human cells. Science 341, 664–667 (2013). [DOI] [PubMed] [Google Scholar]
- 19.Chen X., Wei M., Zheng M. M., Zhao J., Hao H., Chang L., Xi P., Sun Y., Study of RNA polymerase II clustering inside live-cell nuclei using bayesian nanoscopy. ACS Nano 10, 2447–2454 (2016). [DOI] [PubMed] [Google Scholar]
- 20.Li G., Ruan X., Auerbach R. K., Sandhu K. S., Zheng M., Wang P., Poh H. M., Goh Y., Lim J., Zhang J., Sim H. S., Peh S. Q., Mulawadi F. H., Ong C. T., Orlov Y. L., Hong S., Zhang Z., Landt S., Raha D., Euskirchen G., Wei C. L., Ge W., Wang H., Davis C., Fisher-Aylor K. I., Mortazavi A., Gerstein M., Gingeras T., Wold B., Sun Y., Fullwood M. J., Cheung E., Liu E., Sung W.-K., Snyder M., Ruan Y., Extensive promoter-centered chromatin interactions provide a topological basis for transcription regulation. Cell 148, 84–98 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Tang Z., Luo O. J., Li X., Zheng M., Zhu J. J., Szalaj P., Trzaskoma P., Magalska A., Wlodarczyk J., Ruszczycki B., Michalski P., Piecuch E., Wang P., Wang D., Tian S. Z., Penrad-Mobayed M., Sachs L. M., Ruan X., Wei C.-L., Liu E. T., Wilczynski G. M., Plewczynski D., Li G., Ruan Y., CTCF-mediated human 3D genome architecture reveals chromatin topology for transcription. Cell 163, 1611–1627 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Schoenfelder S., Sexton T., Chakalova L., Cope N. F., Horton A., Andrews S., Kurukuti S., Mitchell J. A., Umlauf D., Dimitrova D. S., Eskiw C. H., Luo Y., Wei C.-L., Ruan Y., Bieker J. J., Fraser P., Preferential associations between co-regulated genes reveal a transcriptional interactome in erythroid cells. Nat. Genet. 42, 53–61 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Fanucchi S., Shibayama Y., Burd S., Weinberg M. S., Mhlanga M. M., Chromosomal contact permits transcription between coregulated genes. Cell 155, 606–620 (2013). [DOI] [PubMed] [Google Scholar]
- 24.Park S.-K., Xiang Y., Feng X., Garrard W. T., Pronounced cohabitation of active immunoglobulin genes from three different chromosomes in transcription factories during maximal antibody synthesis. Genes Dev. 28, 1159–1164 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Posern G., Sotiropoulos A., Treisman R., Mutant actins demonstrate a role for unpolymerized actin in control of transcription by serum response factor. Mol. Biol. Cell 13, 4167–4178 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Betzig E., Patterson G. H., Sougrat R., Lindwasser O. W., Olenych S., Bonifacino J. S., Davidson M. W., Lippincott-Schwartz J., Hess H. F., Imaging intracellular fluorescent proteins at nanometer resolution. Science 313, 1642–1645 (2006). [DOI] [PubMed] [Google Scholar]
- 27.Rust M. J., Bates M., Zhuang X., Sub-diffraction-limit imaging by stochastic optical reconstruction microscopy (STORM). Nat. Methods 3, 793–796 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sengupta P., Jovanovic-Talisman T., Skoko D., Renz M., Veatch S. L., Lippincott-Schwartz J., Probing protein heterogeneity in the plasma membrane using PALM and pair correlation analysis. Nat. Methods 8, 969–975 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Levet F., Hosy E., Kechkar A., Butler C., Beghin A., Choquet D., Sibarita J.-B., SR-Tesseler: A method to segment and quantify localization-based super-resolution microscopy data. Nat. Methods 12, 1065–1071 (2015). [DOI] [PubMed] [Google Scholar]
- 30.Stüven T., Hartmann E., Görlich D., Exportin 6: A novel nuclear export receptor that is specific for profilin·actin complexes. EMBO J. 22, 5928–5940 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Grosse R., Vartiainen M. K., To be or not to be assembled: Progressing into nuclear actin filaments. Nat. Rev. Mol. Cell Biol. 14, 693–697 (2013). [DOI] [PubMed] [Google Scholar]
- 32.Belin B. J., Cimini B. A., Blackburn E. H., Mullins R. D., Visualization of actin filaments and monomers in somatic cell nuclei. Mol. Biol. Cell 24, 982–994 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Du J., Fan Y.-L., Chen T.-L., Feng X.-Q., Lifeact and Utr230 induce distinct actin assemblies in cell nuclei. Cytoskeleton 72, 570–575 (2015). [DOI] [PubMed] [Google Scholar]
- 34.Courtemanche N., Pollard T. D., Chen Q., Avoiding artefacts when counting polymerized actin in live cells with LifeAct fused to fluorescent proteins. Nat. Cell Biol. 18, 676–683 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ye J., Zhao J., Hoffmann-Rohrer U., Grummt I., Nuclear myosin I acts in concert with polymeric actin to drive RNA polymerase I transcription. Genes Dev. 22, 322–330 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Sarshad A., Sadeghifar F., Louvet E., Mori R., Böhm S., Al-Muzzaini B., Vintermist A., Fomproix N., Östlund A.-K., Percipalle P., Nuclear myosin 1c facilitates the chromatin modifications required to activate rRNA gene transcription and cell cycle progression. PLOS Genet. 9, e1003397 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Crisp M., Liu Q., Roux K., Rattner J. B., Shanahan C., Burke B., Stahl P. D., Hodzic D., Coupling of the nucleus and cytoplasm: Role of the LINC complex. J. Cell Biol. 172, 41–53 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wu X., Yoo Y., Okuhama N. N., Tucker P. W., Liu G., Guan J.-L., Regulation of RNA-polymerase-II-dependent transcription by N-WASP and its nuclear-binding partners. Nat. Cell Biol. 8, 756–763 (2006). [DOI] [PubMed] [Google Scholar]
- 39.Yoo Y., Wu X., Guan J.-L., A novel role of the actin-nucleating Arp2/3 complex in the regulation of RNA polymerase II-dependent transcription. J. Biol. Chem. 282, 7616–7623 (2007). [DOI] [PubMed] [Google Scholar]
- 40.Boehning M., Dugast-Darzacq C., Rankovic M., Hansen A. S., Yu T., Marie-Nelly H., McSwiggen D. T., Kokic G., Dailey G. M., Cramer P., Darzacq X., Zweckstetter M., RNA polymerase II clustering through carboxy-terminal domain phase separation. Nat. Struct. Mol. Biol. 25, 833–840 (2018). [DOI] [PubMed] [Google Scholar]
- 41.Cho W.-K., Spille J.-H., Hecht M., Lee C., Li C., Grube V., Cisse I. I., Mediator and RNA polymerase II clusters associate in transcription-dependent condensates. Science 361, 412–415 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Li P., Banjade S., Cheng H.-C., Kim S., Chen B., Guo L., Llaguno M., Hollingsworth J. V., King D. S., Banani S. F., Russo P. S., Jiang Q.-X., Nixon B. T., Rosen M. K., Phase transitions in the assembly of multivalent signalling proteins. Nature 483, 336–340 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lu H., Yu D., Hansen A. S., Ganguly S., Liu R., Heckert A., Darzacq X., Zhou Q., Phase-separation mechanism for C-terminal hyperphosphorylation of RNA polymerase II. Nature 558, 318–323 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Shin Y., Berry J., Pannucci N., Haataja M. P., Toettcher J. E., Brangwynne C. P., Spatiotemporal control of intracellular phase transitions using light-activated optoDroplets. Cell 168, 159–171.e14 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Stark G. R., Kerr I. M., Williams B. R., Silverman R. H., Schreiber R. D., How cells respond to interferons. Annu. Rev. Biochem. 67, 227–264 (1998). [DOI] [PubMed] [Google Scholar]
- 46.Zhao Z. W., Roy R., Gebhardt J. C. M., Suter D. M., Chapman A. R., Xie X. S., Spatial organization of RNA polymerase II inside a mammalian cell nucleus revealed by reflected light-sheet superresolution microscopy. Proc. Natl. Acad. Sci. U.S.A. 111, 681–686 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kapoor P., Chen M., Winkler D. D., Luger K., Shen X., Evidence for monomeric actin function in INO80 chromatin remodeling. Nat. Struct. Mol. Biol. 20, 426–432 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Obrdlik A., Kukalev A., Louvet E., Östlund Farrants A.-K., Caputo L., Percipalle P., The histone acetyltransferase PCAF associates with actin and hnRNP U for RNA polymerase II transcription. Mol. Cell. Biol. 28, 6342–6357 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Serebryannyy L. A., Parilla M., Annibale P., Cruz C. M., Laster K., Gratton E., Kudryashov D., Kosak S. T., Gottardi C. J., de Lanerolle P., Persistent nuclear actin filaments inhibit transcription by RNA polymerase II. J. Cell Sci. 129, 3412–3425 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Chong S., Dugast-Darzacq C., Liu Z., Dong P., Dailey G. M., Cattoglio C., Heckert A., Banala S., Lavis L., Darzacq X., Tjian R., Imaging dynamic and selective low-complexity domain interactions that control gene transcription. Science 361, eaar2555 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Papantonis A., Kohro T., Baboo S., Larkin J. D., Deng B., Short P., Tsutsumi S., Taylor S., Kanki Y., Kobayashi M., Li G., Poh H.-M., Ruan X., Aburatani H., Ruan Y., Kodama T., Wada Y., Cook P. R., TNFα signals through specialized factories where responsive coding and miRNA genes are transcribed. EMBO J. 31, 4404–4414 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Levine M., Cattoglio C., Tjian R., Looping back to leap forward: Transcription enters a new era. Cell 157, 13–25 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Liu Z., Lavis L. D., Betzig E., Imaging live-cell dynamics and structure at the single-molecule level. Mol. Cell 58, 644–659 (2015). [DOI] [PubMed] [Google Scholar]
- 54.Trapnell C., Roberts A., Goff L., Pertea G., Kim D., Kelley D. R., Pimentel H., Salzberg S. L., Rinn J. L., Pachter L., Differential gene and transcript expression analysis of RNA-seq experiments with TopHat and Cufflinks. Nat. Protoc. 7, 562–578 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Mortazavi A., Williams B. A., McCue K., Schaeffer L., Wold B., Mapping and quantifying mammalian transcriptomes by RNA-Seq. Nat. Methods 5, 621–628 (2008). [DOI] [PubMed] [Google Scholar]
- 56.da Huang W., Sherman B. T., Lempicki R. A., Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat. Protoc. 4, 44–57 (2009). [DOI] [PubMed] [Google Scholar]
- 57.Bienko M., Crosetto N., Teytelman L., Klemm S., Itzkovitz S., van Oudenaarden A., A versatile genome-scale PCR-based pipeline for high-definition DNA FISH. Nat. Methods 10, 122–124 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Markaki Y., Smeets D., Cremer M., Schermelleh L., Fluorescence in situ hybridization applications for super-resolution 3D structured illumination microscopy. Methods Mol. Biol. 950, 43–64 (2013). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/6/16/eaay6515/DC1