

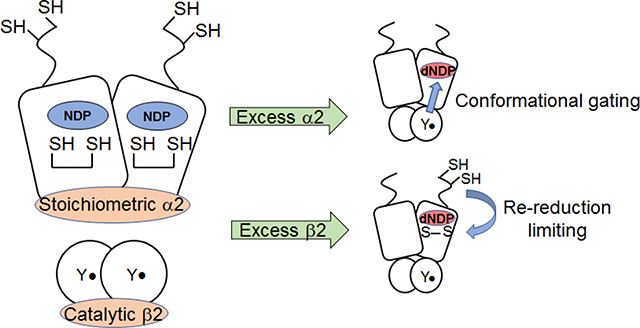
Abstract
Ribonucleotide reductases (RNRs) catalyze the conversion of nucleotides (NDP) to deoxynucleotides (dNDP), in part controlling the ratios and quantities of dNTPs available for DNA replication and repair. The active form of E. coli class Ia RNR is an asymmetric α2β2 complex in which α2 contains the active site and β2 contains the stable diferric-tyrosyl radical cofactor responsible for initiating the reduction chemistry. Each dNDP is accompanied by disulfide bond formation. We now report that β2 can act catalytically when assaying RNR in either the absence or presence of an external reductant. In the absence of reductant, rapid chemical quench analysis of a reaction of α2 (10 μM), substrate, and effector with variable amounts of β2 (0.1, 1 or 10 μM) yields 3 dCDP/α2 at all ratios of α2:β2 with a rate constant of 8–9 s−1, associated with a rate limiting conformational change(s). Stopped-flow fluorescence spectroscopy with a fluorophore-labeled β reveals that the rate constants for subunit association (163 ± 7 μM−1 s−1) and dissociation (75 ± 10 s−1) are fast relative to turnover, consistent with catalytic β2. When assaying in the presence of an external reducing system, the turnover number is dictated by the ratio of α2:β2, their concentrations, and the concentration and nature of the reducing system; the rate-limiting step can change from the conformational gating, to a step or steps involving disulfide re-reduction, dissociation of the inhibited α4β4 state or both. The issues encountered in the E. coli RNR assays are likely of importance in all class I RNR assays and are central to understand in developing screening assays for inhibitors of these enzymes.
Graphical Abstract
INTRODUCTION
E. coli class Ia ribonucleotide reductase (RNR) catalyzes the conversion of nucleoside 5′-diphosphates (CDP, UDP, GDP, ADP) to their corresponding deoxynucleotides (dNDPs), providing the essential building blocks for DNA replication and repair.1,2 The E. coli Ia enzyme, a paradigm for class I RNRs, consists of two homodimeric subunits, α2 and β2, that are active in an asymmetric, tetrameric α2β2 complex.3–6 The diferric-tyrosyl radical (Y122•) located in β2 generates a transient thiyl radical in α2 (C439•) subsequent to its binding NDP and effector, which then initiates NDP reduction. This reversible oxidation process occurs over a distance of ~35 Å and involves a pathway of conserved aromatic amino acid residues and a cysteine.6,7
The central role of RNRs in nucleic acid metabolism has made the human RNR the target of five clinically used therapeutics that inhibit distinct steps in the reduction process.8–10 The limiting factor in identifying new types of RNR inhibitors has been the development of rapid in vitro and in vivo screens for enzyme activity. In a recent study with an RNR containing a site-specifically incorporated unnatural amino acid, we identified the ability of a mutant β2 to act catalytically in α2 turnover.11 We now report that wt β2 can also act catalytically in pre-steady state and steady state assays. In support of this result, the kinetics of α/β subunit interactions are fast relative to enzymatic turnover. Consideration of the dynamics of these inter-subunit interactions are important in designing assays for RNR activity and screening for RNR inhibitors.
In early studies to purify E. coli RNR,12 the two subunits separated and required recombination for activity measurement. The Kd for subunit interaction was subsequently established to be 0.4 μM in the apo form and to increase to 0.2 μM on binding of NDP and its matched allosteric effector.13 Since that time, RNR subunits have typically been assayed independently, using a physiological concentration of one subunit (0.1–0.5 μM) and a 5 to 10-fold excess of the second subunit to ensure “active” α2β2 complex formation. However, the activity of α2 is always less than that of β2,14 suggesting that the amount of α2β2 complex alone does not govern activity.
RNR assays are typically carried out under two sets of conditions in the absence and presence of endogenous reductant. Our recent model for dNDP formation in the absence of endogenous reducing system (steps A-G) and in its presence (step H) is shown in Scheme 1.15 Many distinct perturbative biochemical studies and recent structural studies of an active α2β2 double mutant (F3Y122/E52Q-β2/α2/GDP/TTP) establish asymmetry within the “active” complex.15 We propose a model in which α2, loaded with substrate (CDP) and allosteric effector (ATP), binds to β2 and that one dCDP is formed in an α/β pair before triggering formation of a second dCDP in the adjacent α/β pair. In the wt enzyme, 2 dCDPs are generated at ~5–10 s−1.14 In all the perturbative systems examined, the asymmetry becomes apparent as the switching between the two α/β pairs is altered. For the wt system, each dCDP is accompanied by the oxidation of a C225,C462 pair to a disulfide. The C-terminal tail of each α monomer contains two additional conserved cysteines (C754, C759) that are involved in re-reduction of the active site disulfide in their own monomer.16 While the details of the re-reduction are not established, the structure suggests that the subunits likely dissociate or may undergo a slow conformational reorganization so that two more dCDPs can be generated (step G, Scheme 1) at a rate constant of ~ 0.1 s−1. While a maximum of 4 dCDPs/α2 can be generated, only 2.5–3 dCDPs are routinely measured. We attribute this observation to α2’s sensitivity to oxidation during its purification and/or handling. Under typical assay conditions for E. coli RNR, in the presence of endogenous reductants thioredoxin (TR), thioredoxin reductase (TRR) and NADPH, α2 generates many dCDPs (step H) with a rate constant of 1–2 s−1, that is likely associated with the complexity of C-terminal tail mediated re-reduction and the role of the thioredoxin.17,18
Scheme 1.

Updated model for kinetics of dNDP formation in the absence of endogenous reductant at 5–10 s−1 (steps A-G), and in the presence of endogenous reductant and multiple turnovers (step H) at 1–2 s−1.
Unlike α2, the Y• in β2 is regenerated immediately following dCDP formation.11 This scenario makes it feasible that each β2 can service multiple α2s and that fast subunit dissociation and re-association to α2β2 followed by rapid chemistry, could preclude observation of these dynamics. We hypothesize that catalytic β2 might account for the differences in the turnover number of α2 and β2.
In the current study, we describe experiments to examine the ability of β2 to act catalytically in the absence and in the presence of an external reducing system. In the absence of reductant and at ratios of α:β from 1:1, 10:1 and 100:1, ~3 dCDP/α2 are generated with a rate constant of 8 s−1 relative to Y•. When the α:β ratio is 100:1, 200 turnovers occur. Rate constants for association (163 ± 7 μM−1 s−1) and dissociation (75 ± 10 s−1) of α2 and β2, measured by stopped-flow fluorescence spectroscopy using β2 modified site-specifically with an environmentally sensitive fluorophore, support a catalytic role for β2. In the presence of endogenous reductant, we also establish that β2 can function catalytically. However, under the conditions of limiting α2 or at high concentrations of the subunits (1 – 10 μM), the rate-limiting step switches from the conformational gating of radical transfer (RT) chemistry to re-reduction of α2, likely involving conformational change(s)15 or interconversion of active and inactive quaternary structures of RNR (α2β2 and α4β4 in E. coli)19 or both.
The observations that (i) β2 can function catalytically, (ii) the discoveries of the complexity of α’s quaternary structures and their dependence on concentration of protein and (deoxy)nucleotides, (iii) incompletely loaded metallocofactor in β, and (iv) the instability of α to oxidation and its propensity to aggregate require consideration when developing assays for both old and new RNRs. These issues lead us to suggest a standard way to assay RNRs that is distinct from the one that has been used for the past few decades.
MATERIALS AND METHODS
Materials
His6-α2, specific activity (SA) of 2500 nmol(min-mg)−1 and wt-β2 with 1.2 Y•/β2, SA of 6000 nmol(min-mg)−1 were expressed from the pET28a-nrdA and pTB2-nrdB plasmids and purified as previously described.20,21 The diferric-tyrosyl radical in β2 is always generated by self-assembly,22 and by our method23 gives 1.2 Y•s that we believe are unequally distributed between each β2. Sixty percent of β2 is active and 40% contains a diferric-center with no radical, is inactive and termed met-β2. α2 was pre-reduced prior to use with dithiothreitol (DTT) and treated with hydroxyurea (HU) to reduce the tyrosyl radical in the small amount of β2 that always co-purifies with α2.24 E. coli thioredoxin, TR (40 U/mg) and thioredoxin reductase, TRR (1400 U/mg) were purified using established protocols.25,26 6-Bromoacetyl-2-dimethylaminonaphthalene (BADAN) was purchased from Molecular Probes (Eugene, OR). C268S/C305S/V365C-β2 was isolated and labeled with BADAN to make dansyl-β2 as previously described.27 For stopped flow (SF) fluorescence spectroscopy experiments, Y122• of wt-β2 or (dansyl-β2) was reduced by treatment of β2 with HU. [5-3H]-CDP was obtained from Vitrax (Placentia, CA). Calf alkaline phosphatase was purchased from Roche. Assay buffer is 50 mM HEPES, 15 mM MgSO4 and 1 mM EDTA (pH 7.6).
dCDP formation kinetics as a function of subunit ratio with no endogenous reductant
Rapid Chemical Quench (RCQ) experiments were performed on a Kintek RQF-3 instrument attached to an external circulating water bath set at 25 °C. To assess the effect of [β2] on kinetics, wt-α2 (20 μM) and ATP (6 mM) in assay buffer in syringe A was rapidly mixed with wt-β2 (0.2, 2 or 20 μM and [5-3H]-CDP (1 mM, 20,000 cpm/nmol) in an equal volume from syringe B. The reaction was quenched in the instrument (5 ms – 100 s) or by hand (> 100 s) in 2% HClO4 and samples were dephosphorylated and worked up as previously described.14,28 To assess the effect of [α4β4] on dCDP formation kinetics, a control reaction of wt-α2 (1 μM), wt-β2 (1 μM), [5-3H]-CDP (0.5 mM) and ATP (3 mM) was also monitored by RCQ. Data were fit to Eq. 1 (for 1:1 and 10:1 α2:β2), and Eq. 2 (for 100:1 α2:β2) to obtain kcat from the sum of A1k1 + A2k2.
| (1) |
| (2) |
Association and dissociation rate constants by SF fluorescence spectroscopy
kassoc and kdissoc for the α2β2 complex in the presence and absence of CDP, ATP, and dATP were determined by SF fluorescence spectroscopy using an Applied Photophysics DX.17MV instrument equipped with the Pro-Data upgrade. The temperature was maintained at 22.0 ± 0.1 °C with a Lauda RE106 circulating water bath and all samples were incubated for 3 min at this temperature prior to measurement. Total fluorescence at λ > 420 nm was detected by a PMT with a long pass filter, using an excitation bandwidth of 40 nm centered at 390 nm. All solutions were prepared in assay buffer. Association rate constants were determined by rapid mixing of 1–6 μM of α2, ± CDP (0.4 mM), and ± ATP (1.2 mM) in one syringe with an equal volume of 0.16 μM dansyl-β2 in a second syringe. The reaction was iterated to obtain ~50 traces that were then averaged and fit to Eq. 2 to obtain kobs. Plotting kobs as a function of [α2] provided kassoc from the slope of the linear fit to Eq. 3 derived from a one-step reversible binding model for pseudo-first order conditions. This process was repeated three times and reported error reported accounts for error between replicates as well as error associated with the goodness of fit.
| (3) |
As determination of kdissoc by extrapolation of Eq. 3 to [α2] = 0 gives significant error, kdissoc was measured directly by mixing equal volumes of dansyl-β2 (0.2 μM), α2 (0.5 μM), ± CDP (0.5 mM) and ± ATP (1.2 mM) or ± dATP (100 μM) in one syringe with a large excess of met-β2 (100 μM) in a second syringe with the same CDP/ATP. A minimum of 15 traces was averaged per trial and 3–6 independent trials were conducted for each condition (in the presence and absence of CDP and/or ATP). With the exception of dissociation rate constants measured in the presence of dATP, all SF fluorescence traces were well fit to a monoexponential function (Eq. 2) over 1–75 ms. In the presence of dATP, dissociation kinetics were biphasic and fit to Eq. 1 over 0.001–8 s. All fitting was performed using OriginPro 8.0 software (OriginLab). Acceptability of fitting was determined on the basis of qualitative symmetry of residuals about zero amplitude and the R2 factor.
dCDP formation kinetics as a function of subunit ratio in the presence of TR/TRR/NADPH
RCQ experiments were performed identically to those described above in the absence of reductant with the following minor modifications. Syringe A included TR (80 μM), TRR (1.6 μM) in addition to wt-α2 (20 μM) and ATP (6 mM), while syringe B contained wt-β2 (0.2 or 20 μM), [5-3H]-CDP (2 mM, 20,000 cpm/nmol) and NADPH (2 mM). Data were fit to Eq. 4 (for 1:1 and 10:1 α2:β2), and Eq. 5 (for 100:1 α2:β2) where A and kburst are the amplitude and observed rate constant for the burst phase, respectively, and klinear is the steady-state turnover number (kcat).
| (4) |
| (5) |
Steady-state turnover kinetics
Assays contained a reaction mixture of 170 μL: wt-α2 (1–10 μM), wt-β2 (0.1–10 μM), [5-3H]-CDP (8,000 cpm/nmol, 0.5 mM), ATP (3 mM), TR (40 μM), TRR (0.8 μM) and NADPH (1 mM). Aliquots (40 μL) were quenched at 30 s, 1, 2 and 3 min in 25 μL of 2% HClO4. Product separation was performed as described by Steeper and Steuart.28
Kinetics of dimanganese tyrosyl radical RNR (NrdF2-NrdE) with NrdH exogenous reductant
NrdI was purified to homogeneity and used to assemble the dimanganese-tyrosyl radical cofactor in NrdF2. Typically, 0.7 Y•/β were obtained for cluster assembled in NrdF2. In our hands, NrdF1 was always in inclusion bodies. NrdE was purified under a variety of conditions and in all cases its solubility was limited (~10 μM). Full length NrdE, with no tag, was used in these assays. Purification of thioredoxin equivalents (TrxA, TrxB1, Trxc and NrdH) as well as thioredoxin reductase were carried out and examined for activity. NrdH was established to be the physiological reductant.
RESULTS
Effect of catalytic β2 on inactive site-directed mutants of RNR
Since RNR is essential for E. coli viability, over-expression of either mutant α2 or β2 always results in small amounts of co-purifying endogenous subunits.29 For example, Y356F-β2, a variant incapable of dNDP formation,30 typically contains between 0.1–5% contaminating wt-β2. When product formation is examined for a reaction mixture containing 0.34 nmol each of Y356F-β2 and wt-α2, CDP, and ATP in the absence of a reducing system, increasing amounts of dCDP are observed with time such that 1.5 dCDP/α2 are produced within 15 min (Fig. S1). If β2 is not catalytic with respect to α2, one would predict that turnover in the absence of a reducing system would result in 0.003–0.15 dCDP/α2. The observation of a 10–500-fold enhancement over the expected number of dCDPs suggests that one β2 can service multiple α2s. In contrast to Y356F-β2, when similar experiments are performed with another catalytically inactive mutant, Y731F-α2,31 only 0.05–0.09 dCDP/α2 are generated with no visible time dependence. This observation is consistent with 2–3% contaminating wt-α2 and a non-catalytic role for α2 with respect to β2 turnover. These observations together with the observation that α2 in the presence of excess β2 vs β2 in the presence of excess α2 (Fig. S2) give different turnover numbers provided the impetus for examining the reaction of wt RNR at different α:β ratios.
dCDP formation kinetics as a function of subunit ratio in the absence of reductant
To test the hypothesis that β2 is catalytic, dCDP was measured at α:β = 1:1, 10:1 or 100:1 (Fig. 1) with 10 μM α2, and β2 varied (0.1, 1 or 10 μM). The total amount of dCDP was normalized per α2 or Y• and the rate constants calculated from the sum of A1k1 + A2k2 in Eqs. 1 and 2, where An and kn represent the amplitude and rate constant of each kinetic phase (Table 1). As seen in Fig. 1, 2.5–3 dCDP/α2 are produced at all α:β ratios. At α:β = 1:1 and 10:1, total dCDP production occurs over two kinetic phases: 1.3–1.4 dCDP/α2 are generated in the first phase and 1.1–1.6 dCDP/α2 in the second phase. As we previously proposed,14 the first phase reports on the generation of the first two dCDPs by each α monomer, and the second phase reports on re-reduction of the active site disulfide by the C-terminal dithiols of α2 (Scheme 1). Support for this interpretation is provided by the ability to generate 1.4–1.7 dCDP/α2 in a single kinetic phase in a mutant α2 containing serines in place of the C-terminal cysteines.14 At α:β = 100:1, a single kinetic phase producing 2.5 dCDP/α2 is observed.
Figure 1.

Product formation kinetics measured by RCQ in the absence of reductant. Final reaction mixtures contained 10 μM α2, 0.5 mM CDP, 3 mM ATP and (a) 10 μM, (b) 1 μM, or (c) 0.1 μMβ2 as indicated. Circles and error bars represent the mean and average error of 2 trials, respectively. Black traces represent fits to Eq. 1 (a and b) or Eq. 2 (c) and produced the rate constants and amplitudes listed in Table 1.
Table 1.
dCDP formation kinetics at varying ratios of α:β in the absence of reductant.
| α2 (μM) | β2 (μM) | First phase | Second phase | kcatb (s−1) | ||||
|---|---|---|---|---|---|---|---|---|
| k1 (s−1) | dCDP/ α2 | dCDP/Y•a | k2 (s−1) | dCDP/α2 | dCDP/Y•a | |||
| 10 | 10 | 6 (1) | 1.3 (2) | 1.1 (1) | 0.51 (8) | 1.6 (2) | 1.3 (1) | 8 (1) |
| 10 | 1 | 0.8 (2) | 1.4 (1) | 10 (1) | 0.030 (7) | 1.1 (1) | 11 (1) | 8 (2) |
| 10 | 0.1 | 0.046 (2) | 2.5 (1) | 207 (3) | – | – | – | 9.5 (4) |
| 1 | 1 | 6 (1) | 1.5 (2) | 1.2 (1) | 0.9 (1) | 1.5 (2) | 1.2 (1) | 8 (1) |
Y• amount used in the dCDP/ Y• calculation was obtained from the moles of β2 (87,000 g/mol) in the reaction mixture and the amount of radical per dimer (1.2).
kcat = A1k1 + A2k2, where An refers to the amplitude with respect to Y•.
In contrast, normalizing product formation relative to equivalents of Y• reveals that a total of 1.1, 10, and 207 dCDP/Y• are formed at α:β = 1:1, 10:1, and 100:1, respectively (Table 1). Recalling that ~60% of β2 is active and contains 2Y•, the remaining 40% is inactive with only a diferric cluster.11 Thus, a single active β2 performs many turnovers during the same time α2 makes 2.5–3 dCDP/α2 (Fig. 1, Table 1). Calculating the turnover number relative to Y• reveals that product formation occurs at 8–9 s−1 in all cases (Table 1). This rate constant is within the 5–10 s−1 range reported in previous studies14 and is associated with the protein conformational gating that occurs prior to initiation of the reaction. As the ratio of α:β increases, the second phase (re-reduction of the α2 active site) becomes unencumbered by the issues associated by re-reduction of the active site disulfide (Table 1, Scheme 1).
Our previous studies on wt E. coli RNR showed that the activity of each subunit in the presence of a 5-fold excess of the second subunit increased with concentration up to 0.5 μM (Fig. S2), but that by 2 μM it decreased by 50%.14 We recently reported, using size exclusion chromatography analysis, that α2β2 can be converted in part into an inactive α4β4 state at concentrations >10 μM or in the presence of the negative effector dATP.27 To investigate the potential effects of the α4β4 state on our pre-steady state kinetics measurements, we performed the 1:1 α2:β2 experiment at low (1 μM), and high (10 μM) subunit concentrations (Fig. S3, Table 1). No differences were observed in dCDP formation. This result suggests that if α4β4 forms under these experimental conditions that it rapidly reverts back to α2β2.
Measurement of α2β2 subunit association (kassoc) and dissociation (kdissoc) rate constants by stopped-flow fluorescence spectroscopy
For β2 to function catalytically, dissociation and re-association of the active α2β2 complex must be fast relative to the rate limiting processes. To study this possibility, we prepared β2 that contains an environmentally sensitive fluorophore site-specifically attached to a cysteine at position 365 within its C-terminal tail that is largely responsible for the binding affinity to α2.13,30 This dansyl-β2 exhibits increased fluorescence intensity when the probe is situated in a hydrophobic environment compared to its fluorescence intensity free in buffered solution, providing a method to explore subunit oligomerization.19,27
To measure the rate constant for kassoc for α2β2, stopped-flow (SF) fluorescence experiments were performed at varying concentrations of α2 in > 10-fold excess relative to dansyl-β2 (80 nM) for a range of [α2] (Fig. 2a). The subunits were rapidly mixed and the increases in fluorescence, shown in Fig. 2b, were fit to Eq. 2 for each [α2]. Plotting the resulting kobs versus [α2] (Fig. 2c) provided kassoc from a linear fit to Eq. 3. These experiments were performed in the presence and absence of CDP/ATP and yielded kassoc that were similar and range from 150–200 μM−1 s−1 (Table 2).
Figure 2.

Diagram describing SF fluorescence experiment to measure (a) kassoc, where green indicates the highly fluorescent state. (b) To extract kassoc, the fluorescence intensity was measured under pseudo-first order conditions for a range of [α2], and fit to Eq. 2 (black traces, 1 set of 3 data sets shown). (c) kobs (points) obtained in this way were then plotted versus [α2], and fit (lines) to Eq. 3 to obtain kassoc from the slope of fitted lines.
Table 2.
Association and dissociation rate constants as a function of CDP and effector measured by SF fluorescence spectroscopy.
| Substrate/Effector | kassoc (μM−1 s−1) | kdissoc (s−1) |
|---|---|---|
| CDP/ATP | 163 (7) | 75 (10) |
| CDP | 193 (7) | 93 (3) |
| ATP | 156 (14) | 86 (10) |
| None | 179 (11) | 109 (6) |
| dATPa | 0.46 (4), 7 (2) |
50 μM, sufficient to saturate both the specificity- and activity-allosteric binding sites.
Dissociation rate constants (kdissoc) were determined by rapid mixing of α2(dansyl-β2) complex (or α4(dansyl-β2)2 in the case with 50 μM dATP) with a large excess of wt-β2 (Fig. 3a). In the absence and presence of CDP and ATP, the loss in fluorescence intensity was fit to a monoexponential decay, Eq. 2, to provide kdissoc values of 75–100 s−1 (Fig. 3b, Table 2). kdissoc of the active α2β2 complex is ~8–12-fold faster than the conformational change(s) (8–9 s−1), supporting the proposal that a single β2 can service multiple α2s without attenuating turnover kinetics. In contrast, when the experiment is performed in the presence of the inhibitory effector dATP, dissociation kinetics are slow and biphasic (Fig. 3c).
Figure 3.

Scheme for SF fluorescence experiment to determine kdissoc (a), where green indicates the highly fluorescent state and n = 1 in (b) and 2 in (c). Fluorescence decay kinetics were measured in the presence of 0.5 mM CDP and 1.5 mM ATP (b), and 50 μM dATP (c). Black circles represent averages of 15 SF traces, and lines are fits to Eq. 2 (b) or 1 (c). The data shown represent 1 of 3 independent trials, the averages and sd of which are presented in Table 2.
The oligomeric state (α2β2 vs α4β4) of E. coli RNR can be controlled by protein concentration and dATP concentrations.3,19,32,33 We therefore also determined the subunit dissociation rate constant for α4β4 generated in the presence of 50 μM dATP. At these concentrations of dATP, both the specificity and activity sites of α2 are saturated,34,35 resulting in a shift toward the inactive α4β4 state.3,36 Fitting these data to a biexponential decay (Eq. 1), each phase represents ~50% of the total amplitude change and the rate constants obtained are 10- and 100-fold slower than the corresponding dissociation rate constants from the α2β2 complex (Table 2). This is consistent with previous reports of enhanced subunit affinity in the α4β4 state.19,32 As stated previously, α4β4 formation that may result from high protein concentrations (10 μM of each subunit) does not affect turnover in the absence of TR/TRR/NADPH. However, the slower dissociation kinetics of α4β4 could explain the range of values observed for steady state turnover in the regime where α2 becomes limiting (Table 3).
Table 3.
Steady-state turnover numbers for different α:β ratios.
| β2 (μM) | α2 (μM) | α2β2a (% β2 in complex) | kcat b (s−1) |
|---|---|---|---|
| 0.1 | 10 | 98 | 9.0 (3) |
| 1 | 10 | 98 | 7.8 (1) |
| 10 | 10 | 96 | 1.45 (4) |
| 1 | 1 | 64 | 4.3 (2) |
| 0.2c | 1 | 81 | 6.8 (3) |
Assuming a Kd for α2β2 of 0.2 μM (ref 13).
Turnover number in the presence of TR/TRR/NADPH. kcat has not been scaled for the percentage of active complex.
These are our standard assay conditions to assess β2 activity.
Steady state turnover kinetics
Studies by Ge et al. revealed that turnover number of RNR in the presence of TR/TRR/NADPH declines with increasing protein concentration (Fig. S2).14 With these data, we proposed that the conformational change(s) prior to turnover are rate-limiting under physiological subunit concentrations (0.1–0.5 μM) while some aspect of the re-reduction process governs turnover under higher protein concentrations (1–10 μM). To determine if β acts catalytically in the presence of reductant, standard RNR assays were performed at different concentrations and ratios of α2 and β2 (Table 3). In contrast to turnover in the absence of reductant (8–9 s−1), the steady-state turnover varied from 1.4 to 9 s−1. Maximal α2β2 activity with reductant is realized when [α2] > [β2]. These data combined with our previous studies14 provide further support for the catalytic role of β2 during turnover. Under conditions of α:β = 1:1 and subsequent to complete oxidation of α2, additional turnovers can only occur upon re-reduction by the TR/TRR/NADPH system. However, in the presence of excess α2, a single β2 can “rapidly” dissociate and re-associate with multiple α2s prior to requirement for re-reduction. Catalytic β2 could explain the discrepancy between α2 and β2 activities as measured by standard assays at physiological concentrations. These data further suggest that re-reduction is not only rate-limiting during steady-state turnover at high protein concentrations, but also under physiological concentrations when α2 is limiting.
dCDP kinetics as a function of subunit ratio in the presence of TR/TRR/NADPH
Our previous studies monitoring dCDP formation in the presence of the TR/TRR system with 15 μM α2 and 32 μM β2, reported 1.4–1.7 dCDP/α2 are generated in a burst phase at ~6 s−1, followed by a linear phase reporting on steady-state turnover at 0.9 s−1 (Table 4 (top row) and Fig. S2).14 These experiments provided evidence that re-reduction of α2 by TR/TRR/NADPH is rate-determining under steady-state turnover at high protein concentrations. Given these results, and the studies above, we predicted that altering the concentration of β2 in the reaction mixture would not alter the amplitude of the burst phase with respect to α2, but that it would change when calculated relative to Y•.
Table 4.
dCDP formation kinetics at varying ratios of α:β in the presence of TR/TRR/NADPH.
| [α2] (μM) | [β2] (μM) | Burst phase | Linear phase | |||
|---|---|---|---|---|---|---|
| kburst (s−1) | dCDP/α2 | dCDP/Y• | kcat/α2 (s−1) | kcat/Y• (s−1) | ||
| 15 | 32a | 6.1 | 1.4 | 0.7 | 0.9 | – |
| 10 | 10b | 9 ± 2 | 1.9 ± 0.1 | 1.5 ± 0.1 | 1.85 ± 0.05 | 1.45 ± 0.04 |
| 10 | 0.1b | – | – | – | – | 6.8 ± 0.08 |
Adapted from reference 14, 0.9 Y•/β2.
Current work, 1.2 Y•/β2.
RCQ experiments were therefore carried out in the presence of α:β = 1:1 ([α2] = [β2] = 10 μM) with the prediction that the burst phase should remain constant with respect to α2 but minimally double with respect to Y•. We found that 1.9 ± 0.1 dCDP/α2 and 1.5 ± 0.1 dCDP/ Y• (relative to 0.7, see Table 4 top two rows in middle column) are generated in the burst phase at 9 ± 2 s−1 (Fig. 4a,). This result is similar to product formation kinetics and stoichiometry in the absence of TR/TRR where β2 performs several turnovers, servicing all the α2s prior to re-reduction of the active site disulfide.
Figure 4.

dCDP formation kinetics measured by RCQ in the presence of TR/TRR/NADPH. Reaction mixtures contained α2 (10 μM), [5-3H]-CDP (1 mM), ATP (3 mM), TR (40 μM), TRR (0.8 μM), NADPH (1 mM), and β2 at either 10 μM, (a), or 0.1 μM, (b). Data points represent the averages of two trials. Black traces represent fits to Eq. 4 (a) or 5 (b).
Finally, based on our current model, the 8–9 s−1 observed rate constant (Table 1) reports on the protein conformational change(s) that gates radical propagation. To support this hypothesis, we measured turnover kinetics in the presence of TR/TRR/NADPH at α:β = 100:1. As shown in Fig. 4b, no burst phase is observed under these conditions. Instead, a linear phase for formation of product with a rate constant of 6.8 ± 0.1 s−1 is observed. These results are consistent with the rate-determining step occurring prior to the first turnover.
DISCUSSION
Our studies in 1999 on the adenosylcoblamain (AdoCbl)-dependent Lactobacillus leichamannii ribonucleoside triphosphate reductase (RTPR) showing that AdoCbl can act catalytically,37 were in part, the inspiration for thinking about whether β2 could function in a similar capacity. The RTPR system lent itself to careful kinetic analysis under two sets of conditions: [AdoCbl] >> [RTPR] and [RTPR] >> [AdoCbl] in the presence of the TR/TRR reducing system. Our studies in the former case revealed that carbon-cobalt bond reformation and AdoCbl dissociation from AdoCbl•RTPR into solution can accompany every turnover, that is the radical chain length of the RTPR catalyzed dNTP reaction is ~1. In that case, the rate-limiting step is disulfide re-reduction by TR/TRR at 2 s−1. Under conditions where [RTPR] >> [AdoCbl], kcat is 15–20 s−1 and is governed by the rate constant for AdoCbl dissociation. In this situation, AdoCbl services more than one RTPR, that is, it functions catalytically. The ability of RTPR to release AdoCbl after each turnover can have important implications as previous physiological studies in L leichamannii suggested that cells produce RTPR in molar excess of AdoCbl molecules taken into cells. This strategy would thus increase the effective concentration of holo enzyme and enable a higher rate of DNA biosynthesis; elevated levels of RTPR can optimize use of scarce nutrients such as AdoCbl. The turnover number of 2 s−1 is very similar to the turnover number of many class I RNRs (1–2 s−1) when re-reduction occurs by endogenous proteins. Thus, catalytic AdoCbl in the L leichmannii class II RNR set a precedent for catalytic β2 in E. coli Ia RNR.
In most cases, the intracellular concentrations of the α and β subunits of class I RNRs have not been measured, although with bacterial systems, where their genes reside in the same operon, it is likely that the α/β ratio is ~1:1. However, as in the case of RTPR, in the class Ia E. coli RNR, the availability of the active form of the metallocofactor is carefully controlled based on our studies in which the amount of β2 was varied genetically and the amount of diferric cluster and tyrosyl radical was measured quantitatively.38 Future studies will be required to determine not only the amounts of RNR subunits, but the amounts of active, diferric-tyrosyl radical loaded β2. If future studies reveal that substoichiometric amounts of Y• relative to α2 is possible, catalytic β2 could play a physiological role. We note, however, that regardless of the physiological role of catalytic β2, the ability of E. coli RNR to use β2 catalytically provides an explanation for the differences in activity of α2 and β2 under standard assay conditions in which each subunit is assayed in the presence of an excess of the second subunit to form the “same” active complex.
Based on studies of class Ia E. coli and human RNRs and class Ib S. sanguinis, B. subtilis, M. tuberculosis, and E. coli RNRs, there are important lessons to be learned for performing reliable assays.
(1) Both α and β subunits must be carefully characterized as well as their complexes:
α subunit characterization
α in general is very air sensitive and its oxidation is often irreversible within a few hours if a reductant such as dithiothreitol (DTT) is not always present during purification and protein storage. α is now known to reside in many distinct quaternary structures (monomer, dimer, non-canonical dimer, tetramer, hexamer, fibrils) and the basis for these changes reside, in part in its N-terminal, ATP cone domain or partial cone domains.15 Purification of α is often facilitated by appendage of an N-terminal (His)6-tag. An altered N-terminally tagged α must thus be used with caution. The recent availability of (His)6-SUMO-tags, which allows purification by Ni affinity column, followed by complete removal of the tag with the SUMO protease is recommended.39 The C-terminal tail of all αs is always disordered and houses the two essential cysteines required for the re-reduction of the active site disulfide (Scheme 1). Thus, a tag should never be placed at the C-terminal end. Anion exchange purification has worked well for all class I RNRs that we have studied. Purification of α in the presence of small molecule reductant and under anaerobic conditions without a tag, might well avoid many problems.
β subunit characterization
β2 is a homodimer in all class I RNRs and thus quaternary structure is not an issue. The problems routinely encountered with β2s are the stability of the Y• in the metallocofactor and the quantity of metallocofactor inserted during recombinant expression. First, the tyrosyl radicals of the dimetallo-Y• cofactors have widely varying stability. While the E. coli Ia Y• has a half-life of 4 days at 4 °C, the human enzyme has a half-life of 30 min at 37 °C. Since β is almost always isolated from recombinant sources at an elevated level of expression, it almost never has a full complement of cofactor.40 Thus, the cofactor must be assembled and characterized with respect to the metal content and Y• per β. The loading of β with 0.5 to 1 Y•/β, from the self-assembly process in vitro22 is distinct from in vivo studies38,41 thus far reported; more basic biology and biochemical studies are required to understand how the cofactors are assembled in cells. In the optimized assembly of the E. coli β2 cofactor, while there are 3.6 irons/β2, there are typically 1.2 Y•/β2.42 Sixty percent of the cluster is the active Fe3+2-Y• cofactor and 40% is inactive, tyrosyl radical reduced, Fe3+2-YOH. Thus, in most assays that are 1:1 α/β, β2 always acts catalytically! The cofactor assembly has been optimized for the E. coli RNR, but each β2 is distinct. Since the Y• is a key indicator of activity, optimizing cluster assembly for assays is essential.
α/β complex characterization
Although the α and β subunits in all class I RNRs are structurally homologous, the quaternary structures of α and the α/β complex are unique and appear to be protein concentration- and nucleotide-dependent. While α alone has been found as noted above in many states, it also forms inactive complexes with β (α4β4) in E. coli19 and in a helical α/β fibril in B. subtilis.43 Some have proposed that α6/β2 forms exist in S. cerevisiae and human RNRs,44 but in our opinion, the activity of this state is in question. Not much is known about the equilibria of these species and how other factors in cells affect them. Thus, the conditions used to study RNRs must be carefully described.
Kd for subunit interactions
The Kd for subunit interaction is also essential to determine. If the Kd is weak, (0.2 μM) and the protein is diluted during purification or in crude lysates, activity can be lost because the active complex does not form or the subunits dissociate and the active complex cannot reform. An important issue to consider is that KdS are challenging to determine, if one does not understand the quaternary structure of each subunit.
(2) Redox reductants to cycle RNRs for multiple turnovers must be identified:
Given the requirement for re-reduction of the disulfide in α accompanying formation of each dNDP, the endogenous reductant in each organism needs to be determined. DTT is often used to reduce previously unidentified αs because the protein reductant(s) are initially unknown. For this case, the efficiency varies widely and is dependent on the concentration of reductant.45 As almost all organisms have multiple candidates for α reduction including TR/TRR, glutaredoxin/glutaredoxin reductase/glutathione, NrdH, etc., identification of the most efficient system is very important to obtain the most robust assay for inhibitor(s) discovery.
(3) Assay conditions musts have optimized α:β and reductant ratios:
Knowledge about the RNR and the organism from which it is obtained is a pre-requisite for the best experimental design. Our experiences with class Ia RNRs (E. coli, human, and S. cerevisiae) and class Ib RNRs (S. sanquinis, B. subtilis, and M. tuberculosis) have suggested the following general protocol. First, assume that the ratio of α:β is 1:1 and that α2β2 is the active form. We believe this is a reasonable assumption based on the recent structures of an active E. coli Ia RNR15 and structures of B. subtilis43 and S. typhimurium46 Ib RNRs. Second, determine the concentration of RNR in the organism of interest and identify the endogenous reductant. Third, measure the activity of each RNR over the protein concentration range from 0.5 to 5× estimated physiological levels. The results from some recent studies on S. sanquinis,47 B. subtilis39,48 and M. tuberculosis dimanganese-tyrosyl radical RNRs using this approach are shown in Fig. S4. The apparent Km values obtained from these analyses are lower (Km of 6.4 nM, 0.025 μM, and 0.16 μM, respectively) than a similar experiment with E. coli and human Ia RNRs (apparent Km of 0.2 μM) suggesting “tighter” α/β affinity. However, efforts to isolate active α/β complexes of these Ib RNRs using several biophysical methods (size exclusion chromatography and analytical ultracentrifugation) all revealed that the subunit behavior, as with Ia E. coli RNR, is dynamic. Our studies on M. tb RNR have shown that it uses a dimanganese-tyrosyl radical cofactor, possesses high activity and behaves similarly to the B. subtilis and S. sanguinis enzymes, in contrast with the recent report of Fe-loaded NrdF2.49 In the case of the B. subtilis RNR, the assay approach described above appears to avoid the known complexities associated with complex quaternary structure43 in this system.
(4) The rate limiting steps for single vs multiple turnovers of RNR must be considered:
Studies on E. coli Ia RNR have shown that the rate limiting steps for single turnover is described by a rate constant of 8–9 s−1 whereas multiple turnovers of RNR yields a rate constant of 1–2 s−1. Thus, experiments on RNR are typically performed under two general conditions: one in the steady state with endogenous reductant and the other under “single” turnover in the absence of a reducing system. In the former case, low protein amounts are sufficient and the protocol described in (3) is recommended in search of small molecule RNR inhibitors. Once an inhibitor is identified, the target of the inhibition (tyrosyl radical reduction in β, active site modification in α, altered quaternary structure of α or α/β, etc.) can be identified using the “single” turnover approach. In this case, elevated levels of protein are required so that the method of analysis can detect a single equivalent or less of intermediate or product.
CONCLUDING REMARKS
We highlight here that studies on newly discovered RNRs must identify the system and describe the assay conditions carefully. Heroic studies are now turning to examining RNR in vivo owing to the importance of inhibiting the enzyme for development of antibacterial and anticancer therapies. Considering the complexity of in vitro RNR assays, as shown herein for E. coli class I RNR, the design of in vivo assays50 for such studies will be extremely challenging. Understanding RNR’s properties is important for uncovering its role in nucleotide metabolism in general, and key for development of assays for new RNR inhibitors in vitro and in vivo.
Supplementary Material
ACKNOWLEDGEMENT
The authors gratefully the NIH for funding, DGN (GM 47274) and JS (GM 29595) and LO acknowledges the NSF for a graduate fellowship.
ABBREVIATIONS
- RNR
class Ia ribonucleotide reductase from E. coli
- SF
stopped flow
- RCQ
rapid chemical quench
- TR
thioredoxin
- TRR
thioredoxin reductase
Footnotes
UniProt NUMBERS
E.coli nrdA – P00452
E. coli nrdB – P69924
B. subtilis nrdE – P50620
B. subtilis nrdF – P50621
S. Sanguinis nrdE – A3CLZ6
S. sanguinis nrdF – A3CLZ4
M. tb nrdE – P9WH75
M. tb nrdF – P9WH71
Human RRM1 – P23921
Human RRM2 – P31350
ASSOCIATED CONTENT
Observed dCDP production in the presence of pathway-blocked variants Y356F-β2, and Y731F-α2, previously reported turnover kinetics14 in the presence of TR/TRR/NADPH, pre-steady state single turnover kinetics of dCDP formation, RNR assays in the presence of optimized endogenous reductant with α:β (1:1) at increasing concentrations. This material is available free of charge via the Internet at http://pubs.acs.org.
REFERENCES
- (1).Stubbe J, and van der Donk WA (1998) Protein radicals in enzyme catalysis. Chem. Rev 98, 705–762. [DOI] [PubMed] [Google Scholar]
- (2).Jordan A, and Reichard P (1998) Ribonucleotide reductases. Annu. Rev. Biochem 67, 71–98. [DOI] [PubMed] [Google Scholar]
- (3).Brown NC, and Reichard P (1969) Ribonucleoside diphosphate reductase. Formation of active and inactive complexes of proteins B1 and B2. J. Mol. Biol 46, 25–38. [DOI] [PubMed] [Google Scholar]
- (4).Thelander L (1973) Physicochemical characterization of ribonucleoside diphosphate reductase from Escherichia coli. J. Biol. Chem 248, 4591–4601. [PubMed] [Google Scholar]
- (5).Minnihan EC, Ando N, Brignole EJ, Olshansky L, Chittuluru J, Asturias FJ, Drennan CL, Nocera DG, and Stubbe J (2013) Generation of a stable, aminotyrosyl radical-induced α2β2 complex of Escherichia coli class Ia ribonucleotide reductase. Proc. Natl. Acad. Sci. U.S.A 110, 3835–3840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Uhlin U, and Eklund H (1994) Structure of ribonucleotide reductase protein R1. Nature 370, 533–539. [DOI] [PubMed] [Google Scholar]
- (7).Stubbe J, Nocera DG, Yee CS, and Chang MCY (2003) Radical initiation in the class I ribonucleotide reductase: long-range proton-coupled electron transfer? Chem. Rev 103, 2167–2201. [DOI] [PubMed] [Google Scholar]
- (8).Shao J, Zhou B, Chu B, and Yen Y (2006) Ribonucleotide reductase inhibitors and future drug design. Curr. Cancer Drug Targets 6, 409–431. [DOI] [PubMed] [Google Scholar]
- (9).Cerqueira NMFSA, Pereira S, Fernandes PA, and Ramos MJ (2005) Overview of ribonucleotide reductase inhibitors: an appealing target in anti-tumour therapy. Curr. Med. Chem 12, 1283–1294. [DOI] [PubMed] [Google Scholar]
- (10).Wnuk SF, and Robins MJ (2006) Ribonucleotide reductase inhibitors as anti-herpes agents. Antiviral Res. 71, 122–126. [DOI] [PubMed] [Google Scholar]
- (11).Ravichandran KR, Minnihan EC, Wei Y, and Stubbe J (2015) Reverse electron transfer completes the catalytic cycle in a 2,3,5-trifluorotyrosine-substituted ribonucleotide reductase. J. Am. Chem. Soc 137, 14387–14395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Brown NC, Canellakis ZN, Lundin B, Reichard P, and Thelander L (1969) Ribonucleoside diphosphate reductase. Purification of the two subunits, proteins B1 and B2. Eur. J. Biochem 9, 561–573. [DOI] [PubMed] [Google Scholar]
- (13).Climent I, Sjöberg B-M, and Huang CY (1991) Carboxyl-terminal peptides as probes for Escherichia coli ribonucleotide reductase subunit interaction: kinetic analysis of inhibition studies. Biochemistry 30, 5164–5171. [DOI] [PubMed] [Google Scholar]
- (14).Ge J, Yu G, Ator MA, and Stubbe J (2003) Pre-steady-state and steady-state kinetic analysis of E. coli class I ribonucleotide reductase. Biochemistry 42, 10071–10083. [DOI] [PubMed] [Google Scholar]
- (15).Greene BL, Kang G, Cui C, Bennati M, Nocera DG, Drennan CL, and Stubbe J (2020) Ribonucleotide reductases (RNRs): structure, chemistry, and metabolism suggest new therapeutic targets. Annu. Rev. Biochem 89, in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Mao SS, Holler TP, Yu GX, Bollinger JM, Booker S, Johnston MI, and Stubbe J (1992) A model for the role of multiple cysteine residues involved in ribonucleotide reduction: amazing and still confusing. Biochemistry 31, 9733–9743. [DOI] [PubMed] [Google Scholar]
- (17).Kasrayan A, Birgander PL, Pappalardo L, Regnström K, Westman M, Slaby A, Gordon E, and Sjöberg BM (2004) Enhancement by effectors and substrate nucleotides of R1-R2 interactions in Escherichia coli class Ia ribonucleotide reductase. J. Biol. Chem 279, 31050-–31057. [DOI] [PubMed] [Google Scholar]
- (18).Lou M, Liu Q, Ren G, Zeng J, Xiang X, Ding Y, Lin Q, Zhong T, Liu X, Zhu L, Qi H, Shen J, Li H, and Shao J (2017) Physical interaction between human ribonucleotide reductase large subunit and thioredoxin increases colorectal cancer malignancy. J. Biol. Chem 292, 9136–9149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Ando N, Brignole EJ, Zimanyi CM, Funk MA, Yokoyama K, Asturias FJ, Stubbe J, and Drennan CL (2011) Structural interconversions modulate activity of Escherichia coli ribonucleotide reductase. Proc. Natl. Acad. Sci. U.S.A 108, 21046–21051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Minnihan EC, Seyedsayamdost MR, Uhlin U, and Stubbe J (2011) Kinetics of radical intermediate formation and deoxynucleotide production in 3-aminotyrosine-substituted Escherichia coli ribonucleotide reductases. J. Am. Chem. Soc 133, 9430–9440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Yokoyama K, Uhlin U, and Stubbe J (2010) Site-specific incorporation of 3-nitrotyrosine as a probe of pKa perturbation of redox-active tyrosines in ribonucleotide reductase. J. Am. Chem. Soc. 132, 8385–8397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Atkin CL, Thelander L, Reichard P, and Lang G (1973) Iron and free radical in ribonucleotide reductase. Exchange of iron and Mössbauer spectroscopy of the protein B2 subunit of the Escherichia coli enzyme. J. Biol. Chem 248, 7464–7472. [PubMed] [Google Scholar]
- (23).Bollinger JM Jr, Tong WH, Ravi N, Huynh BH, Edmonson DE, and Stubbe J (1995) Use of rapid kinetics methods to study the assembly of the diferric-tyrosyl radical cofactor of E. coli ribonucleotide reductase. Meth Enzymol 258, 278–303. [DOI] [PubMed] [Google Scholar]
- (24).Seyedsayamdost MR, Xie J, Chan CTY, Schultz PG, and Stubbe J (2007) Site-specific insertion of 3-aminotyrosine into subunit α2 of E. coli ribonucleotide reductase: Direct evidence for involvement of Y730 and Y731 in radical propagation. J. Am. Chem. Soc 129, 15060–15071. [DOI] [PubMed] [Google Scholar]
- (25).Chivers PT, Prehoda KE, Volkman BF, Kim BM, Markley JL, and Raines RT (1997) Microscopic pKa values of Escherichia coli thioredoxin. Biochemistry 36, 14985–14991. [DOI] [PubMed] [Google Scholar]
- (26).Russel M, and Model P (1985) Direct cloning of the trxB gene that encodes thioredoxin reductase. J. Bacteriol 163, 238–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Hassan AQ, Wang Y, Plate L, and Stubbe J (2008) Methodology to probe subunit interactions in ribonucleotide reductases. Biochemistry 47, 13046–13055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Steeper JR, and Steuart CD (1970) A rapid assay for CDP reductase activity in mammalian cell extracts. Anal. Biochem. 34, 123–130. [DOI] [PubMed] [Google Scholar]
- (29).Aberg A, Hahne S, Karlsson M, Larsson A, Ormo M, Ahgren A, and Sjöberg BM (1989) Evidence for two different classes of redox-active cysteines in ribonucleotide reductase of Escherichia coli. J. Biol. Chem 264, 12249–12252. [PubMed] [Google Scholar]
- (30).Climent I, Sjöberg B-M, and Huang CY (1992) Site-directed mutagenesis and deletion of the carboxyl terminus of Escherichia coli ribonucleotide reductase protein R2. Effects on catalytic activity and subunit interaction. Biochemistry 31, 4801–4807. [DOI] [PubMed] [Google Scholar]
- (31).Ekberg M, Sahlin M, Eriksson M, and Sjöberg BM (1996) Two conserved tyrosine residues in protein R1 participate in an intramolecular electron transfer in ribonucleotide reductase. J. Biol. Chem 271, 20655–20659. [DOI] [PubMed] [Google Scholar]
- (32).Rofougaran R, Crona M, Vodnala M, Sjöberg B-M, and Hofer A (2008) Oligomerization status directs overall activity regulation of the Escherichia coli class Ia ribonucleotide reductase. J. Biol. Chem 283, 35310–35318. [DOI] [PubMed] [Google Scholar]
- (33).Funk MA, Brignole EJ, and Drennan CL Disruption of an oligomeric interface prevents allosteric inhibition of Escherichia coli class Ia ribonucleotide reductase. (2018) J. Biol. Chem. 293, 10404–10412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Ormö M, and Sjöberg B-M (1990) An ultrafiltration assay for nucleotide binding to ribonucleotide reductase. Anal. Biochem 189, 138–141. [DOI] [PubMed] [Google Scholar]
- (35).Von Döbeln U, and Reichard P (1976) Binding of substrates to Escherichia coli ribonucleotide reductase. J. Biol. Chem 251, 3616–3622. [PubMed] [Google Scholar]
- (36).Zimanyi CM, Ando N, Brignole EJ, Asturias FJ, Stubbe J, and Drennan CL (2012) Tangled up in knots: structures of inactivated forms of E. coli class Ia ribonucleotide reductase. Structure 20, 1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (37).Licht SS, Lawrence C, C. L., and Stubbe J (1999) Class II ribonucleotide reductases catalyze carbon-cobalt bond reformation on every turnover. J. Am. Chem. Soc 121, 7463–7468. [Google Scholar]
- (38).Hristova D, Wu CH, Jiang W, Krebs C, and Stubbe J (2008) Importance of the maintenance pathway in the regulation of the activity of Escherichia coli ribonucleotide reductase. Biochemistry 47, 3989–3999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (39).Parker MJ, Ailiena, Maggiolo AO, Thomas WC, Kim A, Meisburger SP, Ando N, Boal A,K, and Stubbe J (2018) An endogenous dAMP ligand in Bacillus subtilis class Ib RNR promotes assembly of a noncanonical dimer for regulation by dATP. Proc. Natl. Acad. Sci. U.S.A 115, E4594–E4603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (40).Aye Y, and Stubbe J Clofarabine 5′-di and -triphosphates inhibit human ribonucleotide reductase by altering the quaternary structure of its large subunit. Proc. Natl. Acad. Sci. U.S.A 108, 9815–9820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (41).Hristova D, Perlstein DL, Zhang Z, Huang M, and Stubbe J (2006) Determination of the in vivo stoichiometry of tyrosyl radical per beta betá in Saccharomyces cerevisiae ribonucleotide reductase. Biochemistry 45, 12282–12294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (42).Bollinger JM (1983) On the chemical mechanism of assembly of the tyrosyl radical-dinulcear iron cluster cofactor of E. coli ribonucleotide reductase. Ph.D. Thesis Massachusetts Institute of Technology, https://dspace.mit.edu/handle/1721.1/12581
- (43).Thomas WC, Brooks FP, Burnim AA, Bacik J-P, Stubbe J, Kaelber JT, Chen JZ, and Ando N (2019) Convergent allostery in ribonucleotide reductase, Nat. Commun 10, 2653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (44).Rofougaran R, Vodnala M, and Hofer A (2006) Enzymatically active mammalian ribonucleotide reductase exists primarily as an alpha(6)beta(2) J. Biol. Chem 281, 27705–27711. [DOI] [PubMed] [Google Scholar]
- (45).Mao SS, Johnston M. I. Bollinger, J. M., and Stubbe J (1989) Mechanism-based inhibition of a mutant Escherichia coli ribonucleotide reductase (cysteine-225----serine) by its substrate CDP. Proc. Natl. Acad. Sci. U.S.A 86, 1485–1489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (46).Uppsten M, Färnegårdh M, Domkin V and Uhlin U (2006) The first holocomplex structure of ribonucleotide reductase gives new insight into its mechanism of action. J. Mol. Biol 359, 365–377. [DOI] [PubMed] [Google Scholar]
- (47).Makhlynets O, Boal AK, Rhodes DV, Kitten D, Rosenzweig AC and Stubbe J (2014) Streptococcus sanguinis Class Ib Ribonucleotide reductase: High activity with both iron and manganese cofactors and structural insights. J. Biol. Chem 289, 6259–6272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (48).Parker MJ, Zhu X, and Stubbe J (2014) Bacillus subtilis class Ib ribonucleotide reductase: High activity and dynamic subunit interactions. Biochemistry 53, 766–776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (49).Hammerstad M, Røhr AK, Andersen NH, Gräslund A, Högbom M, and Andersson KK (2014) The class Ib ribonucleotide reductase from Mycobacterium tuberculosis has two active R2F subunits. J. Biol. Inorg. Chem 19, 893–902. [DOI] [PubMed] [Google Scholar]
- (50).Tholander F, Sjöberg BM. (2012) Discovery of antimicrobial ribonucleotide reductase inhibitors by screening in microwell format. Proc. Natl. Acad. Sci. U.S.A 109, 9798–9803. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.