

Summary
Bacteria have evolved diverse mechanisms to survive environments with antibiotics. Temperature is both a key factor that affects the survival of bacteria in the presence of antibiotics and an environmental trait that is drastically increasing due to climate change. Therefore, it is timely and important to understand links between temperature changes and selection of antibiotic resistance. This review examines these links by synthesizing results from laboratories, hospitals, and environmental studies. First, we describe the transient physiological responses to temperature that alter cellular behavior and lead to antibiotic tolerance and persistence. Second, we focus on the link between thermal stress and the evolution and maintenance of antibiotic resistance mutations. Finally, we explore how local and global changes in temperature are associated with increases in antibiotic resistance and its spread. We suggest that a multidisciplinary, multiscale approach is critical to fully understand how temperature changes are contributing to the antibiotic crisis.
Subject Areas: Global Change, Microbiology
Graphical Abstract
Global Change; Microbiology
Introduction
We are rapidly losing options to treat bacterial infections. This is due primarily to pathogens evolving resistance to antibiotics. For example, some strains of Acinetobacter baumannii, Pseudomonas aeruginosa, and Klebsiella pneumoniae have evolved resistance to all known antimicrobial drugs (Souli et al., 2008). This resistance has dire consequences such as drug-resistant tuberculosis leading to over 200,000 deaths globally per year with more than 2,000 deaths caused by extensively drug-resistant tuberculosis (XDR-TB; World Health Organization, 2019). Overall, multidrug-resistant bacterial pathogens cause at least 700,000 deaths globally per year. Deaths due to drug-resistance are projected to increase to 10 million globally per year by 2050 (O’Neill, 2014, Interagency Coordination Group on Antimicrobial Resistance, 2019).
Antimicrobial resistance commonly occurs in hospitals, communities where people live, and agricultural settings. In farms, industrial agriculture, and aquaculture, the misuse and overuse of antibiotics is selecting for antibiotic-resistant bacteria in both animal and plant hosts (Van Boeckel et al., 2017). For instance, antibiotics used in industrial agriculture are not typically used to treat bacterial infections but to promote faster growth of animals. This “antibiotic misuse” promotes the evolution of drug-resistant bacteria (Van Boeckel et al., 2015). Furthermore, small doses of antibiotics are being released into the environment—through rivers, lakes, soils—in the form of urine, feces, manure, and pharmaceuticals waste. These sublethal doses only reduce bacterial growth compared with growth in the absence of antibiotics (Andersson and Hughes, 2014), whereas higher concentrations of antibiotics either completely arrest growth or kill bacteria.
Bacteria have evolved three primary mechanisms to survive and grow in the presence of antibiotics (Brauner et al., 2016, Balaban et al., 2019). First, a population can transiently survive antibiotics through physiological changes that slow down growth—a phenomenon known as tolerance (Handwerger and Tomasz, 1985, Kester and Fortune, 2014). By comparison, persistence is when only a subpopulation of cells is in a slowly growing or non-growing state that is able to transiently survive antibiotics (Balaban et al., 2004, Wakamoto et al., 2013). Finally, bacteria can evolve genetic modifications that make them survive higher concentrations of antibiotics for longer periods, resulting in resistance. Environmental factors such as temperature, pH, and nutrient availability modulate these mechanisms and thus the survival chances of bacteria in the presence of antibiotics.
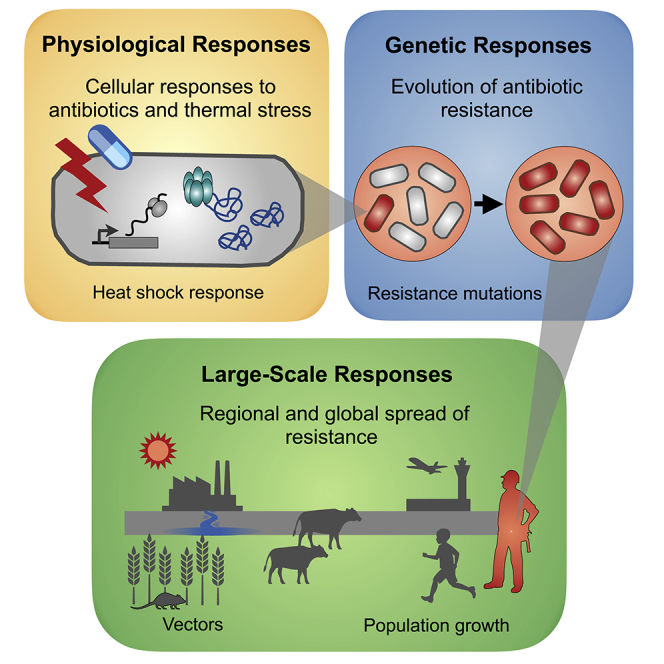
In recent years it has become evident that temperature plays a key role in cellular, physiological, ecological, and evolutionary processes that affect the survival of bacteria. In this review we synthesize recent studies of antibiotic-temperature links, dissecting them by three types of responses: physiological, genetic, and large-scale responses. These responses manifest at different levels of biological organization and at different spatiotemporal scales (Figure 1). First, we focus on the transient physiological responses to temperature that alter cellular behavior and lead to antibiotic tolerance and persistence. Second, we synthesize observations that link thermal stress with the appearance and maintenance of antibiotic resistance mutations in populations (i.e., genetic responses). Third, we explore how local and global changes in temperature are associated with increases in antibiotic resistance and its spread (i.e., large-scale responses). Overall, we believe this is a critical time to synthesize these observations, especially considering the alarming global rises in both temperature and antibiotic resistance.
Figure 1.

Temperature and Antibiotics Can Affect Bacterial Survival at Three Temporal and Spatial Scales
Left: Physiological responses to antibiotics and thermal stress (e.g., heat shock response) are local. That is, they occur at a microscale and mostly affect individual cells. Cells may be exposed to antibiotics and stressful temperatures simultaneously or may encounter these stresses sequentially. In either case, these events are typically short (0.5–48 h) and affect cells over their lifetime or possibly a handful of subsequent generations. Center: When antibiotics and/or stressful temperatures persist for days, resistant bacteria (i.e., individuals carrying heritable genetic mutations that confer stress resistance) take over the population, displacing susceptible bacteria. Right: Finally, resistance spreads across communities (i.e., across different species). Local and global temperatures affect processes such as population growth and the spread of pathogens and vectors that modulate the transmission of antibiotic resistance.
Physiological Responses to Temperature and Antibiotic Stressors
Temperature fluctuations have been present since the very beginning of life, and substantial changes in temperature are associated with major biological epochs such as ice ages or the existence of giant insects. As such, living organisms have developed mechanisms to deal with the physiological effects of temperature fluctuations to improve their chances of survival. In this section we review the literature that shows that changes in temperature can harm cellular processes in ways similar to harm caused by certain kinds of antibiotics. Moreover, we note evidence that the heat- and cold shock stress responses are elicited in response to antibiotics. These observations suggest that these general physiological stress responses to temperature could be involved in and partly co-opted for the stress responses to antibiotics and the evolution of antibiotic resistance.
The Heat- and Cold Shock Stress Responses
Any environmental condition—such as a non-optimal temperature or sublethal concentrations of antibiotics—that reduces the growth of an organism can be considered a stressor. High-temperature damages the cell by causing cellular proteins to unfold and produce aggregates that harm cellular functioning. Living organisms combat these effects through the activation of a highly conserved cellular stress response known as the heat shock response (Lindquist, 1986, Richter et al., 2010, Yura et al., 1984). The cell first detects increased temperatures through specific sensory biomolecules that can be either nucleic acids (DNA or RNA) or proteins (Roncarati and Scarlato, 2017). The heat shock response then induces increased expression of two main sets of proteins: (1) chaperones to prevent and reverse the aggregation of misfolded proteins and (2) proteases to degrade misfolded protein aggregates. Besides protein misfolding, heat stress also causes increased membrane fluidity and damage to DNA and RNA (Richter et al., 2010). In addition to chaperones, a genome-wide screen for genes essential for growth at high temperature (47°C) in Escherichia coli (Murata et al., 2011) found genes involved in energy metabolism, outer membrane stabilization, membrane transport, DNA repair, tRNA modification, translation control, and cell division. In eukaryotes, heat shock also damages the cytoskeleton and intracellular transport mechanisms (Richter et al., 2010).
RNA polymerase and elongation factors, important components of the cellular machinery involved in transcription and translation, have been observed to have decreased stability in E. coli after a shift from 30°C to 42°C (Mogk et al., 1999). This suggests that gene expression is slowed down during heat stress. The E. coli transcription and translation machinery is also regulated at a transcriptional level (Caspeta et al., 2009): expression of rpoA and tufB decreases after temperature shifts from 30°C to 42°C. For a milder temperature shift (from 30°C to 38°C), expression of these genes also decreases under fast heating rates (0.6–8°C/min), but not for slow heating (0.4°C/min).
In E. coli the main regulator of the heat shock response is the sigma factor σ32, transcribed by the rpoH gene (Arsène et al., 2000). High heat increases the concentration of free intracellular σ32. In turn, σ32 binds to the RNA polymerase and initiates the transcription of heat shock genes such as numerous global transcriptional regulators and genes involved in maintaining membrane functionality and homeostasis (Grossman et al., 1987, Nonaka et al., 2006, Straus et al., 1987, Yura et al., 1984, Zhao et al., 2005). Many chaperones, including DnaK, have been shown to participate in the negative regulation of the heat shock response by binding σ32 and preventing it from activating the expression of heat shock genes (Arsène et al., 2000, Roncarati and Scarlato, 2017). DnaK also binds to unfolded proteins. As such, many other stresses that cause protein unfolding can compete for DnaK, freeing σ32 and activating the heat shock response. Other organisms have different mechanisms to regulate the heat shock response, but its involvement in other stresses—especially those that cause protein misfolding—seems to be a common theme. In fact, the heat shock response has been shown to be involved in responding to stresses as diverse as cadmium in Drosophila spp. (Courgeon et al., 1984), heavy metals in Oncorhynchus tshawytscha (Heikkila et al., 1982), and ethanol in Zymomonas mobilis (Michel and Starka, 1986) and E. coli (Chaudhuri et al., 2006).
The cold shock response has not been as extensively studied as the heat shock response and seems to be less conserved across organisms (Yamanaka, 1999). Under cold stress, DNA and RNA secondary structures are over-stabilized. This leads to a reduced efficiency of transcription and translation due to impaired movement of DNA polymerase and the ribosome, respectively. For instance, exposure of E. coli to cold initially stops growth and severely inhibits the synthesis of proteins except those that constitute the cold shock response (Phadtare and Inouye, 2008). These cold shock proteins mitigate the damaging physiological effects of low temperatures, which include translational block, reduced membrane fluidity, slow protein folding, and increased negative DNA supercoiling (Barria et al., 2013, Phadtare and Inouye, 2008, Yamanaka, 1999). The cold shock response also modifies the metabolism of the cell to produce the sugar trehalose, which has a protective effect at low temperatures (Kandror et al., 2002). Importantly, cold shock proteins have been shown to be involved in responding to various stresses besides low temperature such as oxidative stress (Loepfe et al., 2010), osmotic shock (Schmid et al., 2009), acid stress, and ethanol (Derman et al., 2015).
Similarity between the Physiological Effects of Antibiotics and Temperature
Extreme temperatures and antibiotics harm cells through many different pathways and mechanisms of action. Some of the cellular processes damaged by antibiotics overlap with those affected by changes in temperature. For example, aminoglycosides are a class of antibiotics that induce physiological effects in the cell that are qualitatively similar to those of heat stress. Aminoglycosides harm the cell through two main mechanisms: (1) they bind to the ribosome and introduce errors in protein translation, producing aggregates of misfolded proteins, and (2) they inhibit protein synthesis (Greulich et al., 2015, Ramirez and Tolmasky, 2010). Given that aminoglycosides increase misfolded proteins in cells, they also induce the heat shock machinery. Indeed, DnaK and GroEL, chaperones involved in the heat shock response, have been shown to be induced in response to streptomycin (an aminoglycoside) in A. baumannii (Cardoso et al., 2010) and E. coli (Cruz-Loya et al., 2019). Overexpression of both the DnaK/DnaJ/GrpE and GroEL/GroES chaperone systems have a protective effect against sublethal concentrations of gentamicin (Goltermann et al., 2013). Overexpression of the GroEL/GroES chaperones resulted in both increased survival to gentamicin and rescued cell growth, whereas the DnaK/DnaJ/GrpE chaperones increased survival but did not rescue growth.
Although aminoglycosides provide a compelling example, similarity to temperature-induced damage is by no means an exclusive feature of this antibiotic class. In a study exploring the effects of several antibiotics that bind to the ribosome in E. coli, it was observed that the protein expression profiles of bacteria exposed to many non-aminoglycoside protein synthesis inhibitors (macrolides, fusidic acid, and tetracycline) is similar to that induced under cold shock (VanBogelen and Neidhardt, 1990).
Another line of evidence supporting the similarity between physiological effects caused by antibiotics and temperature stress comes from genetic manipulations in Staphylococcus aureus. Notably, deletion of cspB—a major cold shock gene—was found to modify antibiotic susceptibility. Specifically, it resulted in increased resistance to aminoglycosides (≈80-fold for gentamicin) and trimethoprim (>16-fold) as well as increased sensitivity to daptomycin (≈20-fold) and teicoplanin (≈4-fold), as measured by changes in the minimum inhibitory concentration (MIC; Duval et al., 2010).
Finally, similarities between the physiological effects of antibiotics and temperature can be learned from interactions between stressors. Interactions between stressors can be defined in terms of the effect on the growth of the organism when they are present simultaneously. Stressors can interact synergistically, having a greater effect in reducing the growth of an organism than would be expected given the effect of the individual stressors. Alternatively, they can interact antagonistically when the combination of the stressors is less effective than expected, or additively when the effects of the stressors on growth are independent and the combination is as effective as expected. Stressors that harm the same cellular components tend to interact similarly with other stressors when used in combination (Segrè et al., 2005, Yeh et al., 2006). Because of this, drugs and temperatures that induce similar physiological damage to the cell tend to be synergistic or antagonistic to the same set of stressors.
A recent study by some of us measured the interactions (synergy or antagonism) between the effects of twelve antibiotics and six non-optimal growth temperatures on the growth of E. coli (Cruz-Loya et al., 2019). By grouping antibiotics and temperatures with similar interaction profiles, antibiotics were classified into groups with specific growth temperatures based on similar physiological effects in the cell. This provides an expanded look at the relationship between temperature-induced and antibiotic-induced damage to the cell by including previously unexplored antibiotic classes and multiple temperatures that correspond to different levels of heat or cold stress. The similarity to temperature stress of the specific antibiotics evaluated in our study (Cruz-Loya et al., 2019) is summarized in Table 1 and compared with the results of a previous study that explored the similarity between the protein expression profile induced under temperature stress and various ribosome-targeting antibiotics (macrolides, fusidic acid, tetracycline, and aminoglycosides; VanBogelen and Neidhardt, 1990).
Table 1.
Classification of Antibiotics by Similarity to Temperature Stress according to the Induced Protein Expression Profile (VanBogelen and Neidhardt, 1990) or Interactions with Other Stressors (Cruz-Loya et al., 2019) in E. coli
| Antibiotic Class: Antibiotic Tested (Abbreviation) |
Cellular Process (Effect) | Protein Expression Similarity | Interaction Similarity |
|---|---|---|---|
| Chloramphenicol: | Protein synthesis (inhibition) | Cold shock | Not measured |
| Chloramphenicol (CHL) | |||
| Macrolides: | Protein synthesis (inhibition) | Cold shock (SPR, ERY) | Cold (22°C–25°C) (ERY, CLI) |
| Clindamycin (CLI) | |||
| Erythromycin (ERY) | |||
| Spiramycin (SPR) | |||
| Fusidanes: | Protein synthesis (inhibition) | Cold shock | Not measured |
| Fusidic acid (FUS) | |||
| Tetracyclines: | Protein synthesis (inhibition) | Cold shock | Cold (22°C–37°C) |
| Tetracycline (TET) | |||
| Fluoroquinolones: | DNA supercoiling | Not measured | Cold (22°C–37°C) |
| Ciprofloxacin (CPR) | |||
| Levofloxacin (LVX) | |||
| Folic acid synthesis inhibitors: | DNA synthesis (Reduction) | Not measured | Hot (44°C) |
| Trimethoprim (TMP) | |||
| Nitrofurans: | Multiple, including damage to DNA | Not measured | Hot (44°C) |
| Nitrofurantoin (NTR) | |||
| Aminoglycosides: | Protein synthesis (misfolding and aggregation) | Heat shock (KAN, STR, PUR) | Very hot (46°C) (GEN, TOB, STR) |
| Gentamicin (GEN) | |||
| Kanamycin (KAN) | |||
| Puromycin (PUR) | |||
| Streptomycin (STR) | |||
| Tobramycin (TOB) | |||
| Beta-lactams: | Cell wall synthesis | Not measured | None |
| Ampicillin (AMP) | |||
| Cefoxitin (FOX) |
The specific antibiotics from each class evaluated in each study are indicated in parenthesis, except when an antibiotic class was only explored in one study (in which case we mark the other study as “not measured”). The specific temperatures or temperature ranges with similar interaction profiles to the class from Cruz-Loya et al. (2019) are indicated in the right.
Antibiotic Classes with Similar Physiological Effects to Heat
Aminoglycosides—gentamicin, tobramycin, and streptomycin—were found to interact similarly in E. coli to 46°C, the highest temperature evaluated in Cruz-Loya et al. (2019) (Table 1). The protein-expression profile induced by kanamycin, streptomycin, and puromycin in E. coli has also been found to be similar to that induced under heat shock (VanBogelen and Neidhardt, 1990). These observations are likely due to the similarity between the effects of temperature-induced protein unfolding and aminoglycoside-induced misfolded proteins due to translational misreading (Kohanski et al., 2008). The presence of misfolded proteins under aminoglycosides may induce the expression of chaperones involved the heat shock response, as has been shown for DnaK and GroEL in A. baumannii (Goltermann et al., 2013). Overexpression of the heat shock chaperones DnaK and GroEL has also been shown to confer protection against sublethal concentrations of gentamicin in E. coli (Goltermann et al., 2013).
Other antibiotics were also found to have effects similar to heat. In particular, nitrofurantoin and trimethoprim—antibiotics that either damage DNA or prevent its synthesis—were found to have interactions similar to those of a milder heat stress (44°C) in E. coli (Table 1). Interestingly, DNA repair mechanisms have also been linked to heat stress. A genome-wide screen searching for thermotolerant proteins in E. coli (Murata et al., 2011) discovered that various proteins involved in DNA repair (dnaQ, holC, priA, ruvA, and ruvC) are indispensable for growth at 47°C. It has also been reported that Hsp70, a heat shock protein, protects a human-derived cell line from DNA damage (Niu et al., 2006). This raises the possibility that this interaction similarity could perhaps be due to the involvement of DNA-protecting and -repairing enzymes. However, further study is needed to clarify the extent to which DNA damage occurs during high-temperature stress and the mechanistic details of the involvement of DNA-protecting and -repairing enzymes in responding to high-temperature stress and antibiotics. More research is also needed to elucidate if there are any differences between the DNA repair systems involved in responding to heat-similar DNA-damaging antibiotics (nitrofurantoin and trimethoprim) and fluoroquinolones, cold-similar antibiotics that inhibit DNA gyrase (see Table 1 and below).
Antibiotic Classes with Similar Physiological Effects to Cold
All the non-aminoglycoside protein synthesis inhibitors studied in VanBogelen and Neidhardt (1990) and Cruz-Loya et al. (2019) were found to either induce a cold-shock-like protein expression pattern or interact with other stressors in a way similar to cold temperatures in E. coli (Table 1). Since translational block is one of the main effects of cold stress (Yamanaka, 1999), it seems possible that the cellular machinery that responds to these antibiotics might overlap with that activated under cold shock. One important qualitative difference exists between macrolides and tetracycline. Tetracycline was found to have similar interactions to cold stress across a wide range of low temperatures (22–37°C), whereas macrolides were found to be similar to a narrower temperature range (22°C–25°C) corresponding to the lowest temperatures evaluated (Cruz-Loya et al., 2019).
Another class of antibiotics that was found to interact in a similar way as cold temperatures (22°C –37°C) with other stressors in E. coli is fluoroquinolones (Table 1). This class of antibiotics inhibits DNA gyrase, thereby reducing the negative supercoiling of the bacterial chromosome (Aldred et al., 2014). Notably, DNA gyrase is induced in the cold shock response, where it has an important role in regulating the supercoiling of the bacterial chromosome (Yamanaka, 1999). It seems plausible that some of the cellular machinery that regulates DNA supercoiling could be used for dealing with both stressors, which might account for their similar interaction profiles.
Sequential Exposure to Antibiotics and Temperature
Thus far, we have focused on the physiological effects of antibiotic and temperature stress on bacterial cells, both when encountered individually and when combined simultaneously. In this section we explore the implications when they are exposed sequentially. Previous studies have explored the effects of sequential exposure of both different antibiotics (Lenhard et al., 2015, MacGowan and Bowker, 1998, Miller et al., 1996) and antibiotics and temperature (Andrade-Linares et al., 2016, Hilker et al., 2016, Manrique et al., 2016, Rangel, 2011). Even transient exposure to a stressor can modify how an organism responds to subsequent stress. This can result in a primed response to a second stressor (termed the triggering stressor), where the organism has better survivability compared with the unprimed response (Hilker et al., 2016).
A recent meta-analysis (Andrade-Linares et al., 2016) evaluated more than thirty studies of sequential exposure to stressors in various microbes, including gram-negative bacteria (Salmonella typhimurium, Vibrio harveyi, Vibrio cholerae, E. coli, and Pseudomonas putida), gram-positive bacteria (Listeria monocytogenes, Lactobacillus lactis, and S. aureus), and fungi (Saccharomyces cerevisiae, Schizosaccharomyces pombe, Candida albicans, Aspergillus nidulans, Metarhizium anisopliae, and Neurospora crassa). The authors found that, on average, the survival of microorganisms to a stressor is about 10-fold higher after priming. The stressors evaluated in the individual studies were categorized by the authors into temperature, pH, osmotic, oxidative, growth, and physiological stress (a detailed list can be found in Supplementary Table 3 of Andrade-Linares et al., 2016). The greatest cross-protection to a second stressor (as measured by increased survival) was found when temperature, osmotic, and physiological stress were the priming stressors.
Antibiotics are sometimes used in sequential exposure and combinations in part to help prevent tolerant bacterial populations. Tolerant bacteria can survive temporary exposure to a stressor by transiently stopping growth but lack the ability to grow or reproduce while exposed to the stressor (Oz et al., 2014). When bacteria are exposed to different stressors sequentially there is the possibility of cross-tolerance arising. This phenomenon occurs when previous exposure to a stressor renders tolerance to a different kind of stress (Wiuff et al., 2005). Persister cells exhibit multidrug tolerance, which has been proposed to arise through metabolic quiescence by the inactivation of the cellular processes targeted by antibiotics (Lewis, 2007). However, a more recent study testing cross-tolerance between various antibiotics (ciprofloxacin, ampicillin, and gentamicin) in E. coli and S. saprophycus has suggested that the mechanisms for tolerance may be shared between antibiotics that target similar processes rather than through generalized cell dormancy (Goneau et al., 2014, Wiuff et al., 2005). We expect these shared tolerance mechanisms might also extend to different kinds of stressors (such as temperature, pressure, or pH) when the cellular processes targeted by an antibiotic and the stressor overlap. In general, we hypothesize that cross-tolerance is likely to be stronger in contexts in which the physiological damage to the cell is similar, so that the stress response elements induced by a priming stressor are effective at mitigating the damage by the triggering stressor.
Indeed, pre-exposing cells to temperature, antibiotics, or other stressors has been shown to provide protective effects against subsequent stressful temperature or antibiotics (Andrade-Linares et al., 2016, Hilker et al., 2016, Rangel, 2011). Cardoso et al. (2010) offer an example of cross-tolerance involving temperature and antibiotic stress in A. baumannii. In this study, bacteria pretreated with high temperature (45°C for 30 min) and then exposed to an even higher temperature (50°C for 15 min) had a greater survival rate (≈80% versus ≈50% in the absence of pretreatment). The cells pretreated with heat shock also had higher rates of survival (≈75% versus ≈50%) when exposed to streptomycin, an aminoglycoside. Consistent with the temperature classification above (Table 1), the authors suggest that molecular chaperones can play important roles for both thermotolerance and antibiotic tolerance (Cardoso et al., 2010). Further evidence for the role of chaperones in mitigating aminoglycoside-induced damage is given by a study (Goltermann et al., 2013) that shows that overexpression of the chaperone GroEL protects against the bactericidal effect of gentamicin in E. coli.
However, in contrast to these studies, susceptibility to amikacin (as measured by changes in MIC) was found to increase by at least 4-fold in E. coli and S. enterica and between 2- and 4-fold in S. aureus after incubating at high temperature (45°C) until stationary phase (McMahon et al., 2007). Increased susceptibility to amikacin remained even upon the removal of the high temperature. This discrepancy on the protective effect of high-temperature exposure to aminoglycosides across studies may be related to differences in the experimental conditions (for example, short- versus long-term high-temperature exposure or cells in exponential versus stationary phase) or metrics for antibiotic susceptibility (percent survival versus MIC determination). This example highlights that more research on sequential exposure to temperature and antibiotics is needed to fully understand the mechanisms of cross-tolerance.
Persister Cells
Antibiotic persistence can occur either spontaneously or can be triggered (Balaban et al., 2004, Balaban et al., 2019, Cabral et al., 2018, Wood et al., 2013). In the first case, a subpopulation of cells randomly switches into the persister phenotype under normal/unstressed growth conditions. In contrast, triggered persister cells form after exposure to a previous stressor and may remain persisters for a substantial amount of time after removal of the stressor (Balaban et al., 2004, Balaban et al., 2019). The exact mechanisms of the persistence response are variable and still up for debate (Levin et al., 2014, Michiels et al., 2016, Radzikowski et al., 2017). However, persister cells have been shown to appear with many different types of stressors, including nutrient limitation in E.coli, S. aureus, and Mycobacterium smegmatis (Gutierrez et al., 2017); high cell density in E. coli (Vega et al., 2012); and exposure to antibiotics in E. coli (Dörr et al., 2010) and S. aureus (Johnson and Levin, 2013).
Although it has not been directly shown that heat shock can lead to persister cells, it is known that heat shock can induce phenotypic heterogeneity (Bruhn-Olszewska et al., 2018). Bruhn-Olszewska and colleagues showed that two distinct subpopulations form after an E. coli culture is exposed to heat shock at 50°C. Upon transfer to LB medium, a high-density subpopulation resumed growth immediately. However, the growth of a low-density subpopulation was considerably postponed. This subpopulation was determined to primarily contain viable but non-culturable cells by quantifying the number of live cells through fluorescence microscopy and comparing with colony-forming unit (CFU) counts. The low-density subpopulation exhibited a higher tolerance (i.e., increased rate of survival) to sublethal concentrations of antibiotics and hydrogen peroxide.
Arguably, this low-density population should be considered persister cells, since it meets all requirements of the definition. That is, it is a slow-growing subpopulation of cells that acquired an enhanced tolerance to all antibiotics tested (ampicillin, kanamycin, rifampicin, trimethoprim, and nalidixic acid). Especially striking results were found for low sublethal concentrations of ampicillin and kanamycin. The low-density fraction exhibited near 100% survival compared with approximately 20% (ampicillin) and 40% (kanamycin) survival for the high-density fraction. Moreover, the acquired tolerance was not greatly correlated to the concentration of the antibiotic (Bruhn-Olszewska et al., 2018), which is one of the hallmarks of persister cells (Balaban et al., 2019). The fact that previous exposure to heat increases tolerance to some antibiotics is further support for similarities between both the physiological effects and the protective responses to antibiotic and temperature stress.
Genetic Adaptations to Temperature and Antibiotic Stressors
Previously, we discussed how temperature modifies the susceptibility to antibiotics through changes in growth and related gene expression. These effects are transient, affecting individuals (i.e., plastic changes). Here we discuss temperature effects on long-term heritable effects conferred by genetic modifications that can truly be termed the evolution of antibiotic resistance. Antibiotic resistance can evolve through spontaneous mutations or can be acquired through horizontal gene transfer (e.g., plasmids, bacteriophages). Horizontal gene transfer is an important evolutionary force for spreading resistance, but its relationship with temperature is not well studied (but see Walsh et al., 2011 showing that horizontal gene transfer is more common at 30°C than at 25°C). Thus, we focus on reviewing spontaneous mutations that confer resistance to antibiotics—antibiotic resistance mutations—and their relationship with temperature. We first focus on how temperature stress selects for de novo evolution of antibiotic resistance and then review how it maintains resistance mutations in the population.
De Novo Evolution of Antibiotic Resistance
The use of antibiotics is the main selective pressure favoring the evolution of antibiotic resistance. For example, in clinics and farms the use of antibiotics selects for antibiotic resistance mutations (Van Boeckel et al., 2017). Antibiotic resistance mutations confer the ability to survive high concentrations of antibiotics, and therefore, individuals that carry them have a selective advantage over their susceptible counterparts. Resistant individuals outcompete susceptible ones and, over time, take over the population. But, the use of antibiotics is not the only selective pressure favoring the evolution of antibiotic resistance. Temperature also selects for resistance mutations. As previously discussed, both antibiotics and temperature act on similar cellular functions. Consequently, mutations conferring resistance to temperature stress can confer resistance to antibiotics and vice versa (Rodríguez-Verdugo et al., 2013), a phenomenon known as collateral resistance or cross-protection (Dragosits et al., 2013).
One example of cross-protection was observed during an in vitro evolution experiment in which E.coli was exposed to high-temperature stress for 2,000 generations (Rodríguez-Verdugo et al., 2013). After hundreds of generations, 12 of 115 experimental lines acquired de novo mutations conferring rifampicin resistance, despite the fact that bacteria were never exposed to antibiotics during the evolution experiment. The selective pressure was a high temperature of 42°C, within the range of both temperatures found in humans (Box 1) and in soils in the environment. Cross-resistance evolved because temperature and rifampicin acted on the same target of selection—RNA polymerase—that transcribes DNA into RNA and controls gene expression. Mutations in the active site of the RNA polymerase led to changes in gene expression that were adaptive under high-temperature stress (Rodríguez-Verdugo et al., 2016). The same amino acid substitutions that alter gene expression also prevent the binding of rifampicin to the active site of the RNA polymerase. Given that rifampicin could not bind to the RNA polymerase it could not exert its inhibitory action. Therefore, rifampicin resistance evolved as a collateral effect of heat-stress adaptation. Interestingly, some of the high-temperature adapted strains showed increased resistance to two antibiotics (trimethoprim and nitrofurantoin) and increased sensitivity to two other antibiotics (erythromycin and clindamycin; Cruz-Loya et al., 2019). This resistance pattern was different between rpoB single-mutants and high-temperature adapted strains suggesting that additional mutations contributing to thermal stress adaptation contribute to antibiotic resistance besides rpoB mutations. Further studies are needed to elucidate the molecular mechanisms underlying the acquired sensitivity and resistance to these antibiotics.
Box 1. The Case of Fever in Animals and Antibiotic Resistance.
We have reviewed several studies in which stressful high temperatures, ranging between 40°C and 45°C, influence how bacteria respond to antibiotics. In humans, a high fever can occur around 40°C and sometimes body temperature may rise above 41°C (e.g., hyperthermia; Simon, 1993, El-Radhi et al., 2009). It could be clinically useful to consider how fevers influence antibiotic treatment (Clint and Fessler, 2016).
On the one hand, fever could speed evolution of resistance or select for de novo antibiotic resistance mutations (e.g., rpoB mutations). One caveat for this hypothesis is that fever is transient. After a few hours of fever, body temperature returns to around 37°C, a temperature at which rpoB mutations (advantageous at 40°C–42°C) are deleterious. So, even if a fever is long enough for resistance mutations to appear, a “normal” body temperature would purge costly resistance mutations. Nevertheless, a recent study shows that rpoB mutations are not necessarily costly at 37°C in physiological conditions similar to the human body (Maharjan and Ferenci, 2017).
On the other hand, fever could occur after an antibiotic treatment. Therefore, resistance mutations may appear because of the antibiotic treatment and fever could maintain resistance mutation even after an antibiotic treatment stops. A fever could alleviate the cost of resistance and could provide time for compensatory mutations to appear. We acknowledge our speculations are based on a handful of studies conducted in E. coli exposed to one antibiotic (rifampicin). Nevertheless, rifampicin is a clinically relevant antibiotic prescribed to treat Mycobacterium tuberculosis infections (i.e., tuberculosis) with treatments extending up to 3, months (World Health Organization, 2019). Alarmingly, in 2018 there were half a million new cases of tuberculosis testing positive for rifampicin resistance (World Health Organization, 2019). A common mechanism of rifampicin resistance in clinical isolates involves rpoB mutations (Gagneux et al., 2006). Therefore, it is worth considering the potential effects that fever could have on patients with tuberculosis and treated with rifampicin.
Another mechanism for indirectly acquiring antibiotic resistance is to transition from a free-living to a surface-attached lifestyle (i.e., biofilms; Stewart, 2002). Even if antibiotics can freely penetrate biofilms, many cells inside biofilms are in a dormant state and therefore are protected against bactericidal antibiotics (Stewart and Franklin, 2008). In addition, increased temperature may select for increased biofilm formation (Kent et al., 2018). Kent et al. (2018) observed that a marine bacterium, Roseovarius sp., formed more biofilms than its ancestral form after evolving for 500 generations at a high temperature of 33°C (25°C being its optimal temperature). Increased biofilm formation was caused by mutations in a quorum-sensing regulator, luxRI, and a small RNA-binding regulation protein, hfq, that modulates changes in gene expression under stress. Interestingly, increased biofilm formation was not an adaptation to thermal stress per se but was an adaptation to reduced oxygen availability caused by the increase in temperature. Therefore, temperature increases modify several environmental factors that in turn select for alternative lifestyles (e.g., biofilms). These alternative lifestyles may result in a higher tolerance to antibiotics that might have broader implications for marine bacteria that are experiencing warmer seawater temperatures caused by climate change.
Temperature not only selects for antibiotic resistance mutations but can also increase the likelihood of their appearance in the first place. Temperature can potentiate adaptive evolution by increasing the phenotypic and genotypic variation in a population (i.e., evolvability; Payne and Wagner, 2019). Previously we discussed how temperature may induce phenotypic variation by either inducing persistence or by triggering cellular “memory.” In both cases, when exposed to antibiotics some cells have a higher survival compared with cells that were actively growing or were primed with a previous stress. Although these physiological responses are short and transient, they may provide time for an antibiotic resistance mutation to appear and fix in the population (Bódi et al., 2017). This phenomenon was previously observed in E.coli exposed to ampicillin pulses (Levin-Reisman et al., 2017). Levin-Reisman et al. (2017) observed that tolerance to ampicillin, conferred by a long lag phase, allowed cells to survive ampicillin until antibiotic resistance mutations spontaneously appeared and fixed in the population. In principle, a similar phenomenon could occur with pulses of heat stress and antibiotics.
Another mechanism potentiating adaptive evolution is to increase the spontaneous mutation rate and, consequently, increase the probability of evolving adaptive mutations. Increased mutagenesis occurs during the heat shock response and the general stress response (Foster, 2007). These responses enhance the activity of error-prone DNA polymerases (e.g., Pol IV) and down-regulate error-correcting enzymes (e.g., MutS and MutH). Both the heat shock response and the general stress response can be induced by shifts in temperature (Rodríguez-Verdugo et al., 2016, Weber et al., 2005, Nonaka et al., 2006). Thus, temperature may potentiate adaptive evolution by increasing mutagenesis and providing a large mutational supply.
Interactions between Temperature Effects and Fitness Effects of Antibiotic Resistance Mutations
After resistance mutations appear, they can either sweep to fixation or be purged from the population. The fate of a mutation depends on its fitness effect in a given environment (MacLean et al., 2010). Naturally, resistance mutations are essentially always beneficial in the presence of antibiotics. But when antibiotics are removed, most resistance mutations become deleterious owing to their fitness costs. These costs occur because enzymes that perform essential cellular functions—such as DNA replication, transcription, protein and cell wall synthesis—work sub-optimally when harboring resistance mutations that result in lower growth rate.
Intriguingly, temperature can alleviate these fitness costs while preserving antibiotic resistance. In this way, they serve a similar genetic role as compensatory mutations that can also ameliorate these fitness costs (Andersson and Hughes, 2010, Knies et al., 2017). A handful of studies have shown that resistance mutations are not necessarily costly or deleterious under conditions of thermal stress with no antibiotic present (Trindade et al., 2012, Rodríguez-Verdugo et al., 2013). At least five mutations in the RNA polymerase beta subunit (rpoB) that confer resistance to rifampicin in E.coli are beneficial at a high temperature of 40°C (Trindade et al., 2012) and 42°C (Rodríguez-Verdugo et al., 2013). The exact mechanisms underlying this advantage are not fully known, but rpoB mutations confer large fitness advantages at 42°C because they reset gene expression patterns from a stressed state toward an unstressed state (Rodríguez-Verdugo et al., 2016).
Antibiotic resistance mutations are not necessarily costly in antibiotic-free environments, especially if these environments are warm (Trindade et al., 2012, Rodríguez-Verdugo et al., 2013). That said, in some cases antibiotic resistance mutations are more detrimental at high temperatures (Gifford et al., 2016). Gifford et al. (2016) found that three rpoB mutations in P. aeruginosa are more costly at 42°C than at 37°C. Why resistance mutations are advantageous in some cases and deleterious in others is still poorly understood and a ripe area for future investigation.
Ecological Shifts to Temperature and Antibiotic Stressors
Up until now, we have primarily discussed how temperature changes affect bacteria at the cellular and genetic level—within a single bacterium and within single populations of bacteria. Now, we expand our discussion to examine the effects of temperature across a larger spatial scale at the global level. Over the last several decades, there has been a growing awareness that climate change is affecting not only humans and other megafauna and flora but also the world's microbial organisms. Here we first discuss how changing temperatures impact infectious disease in terms of geographic ranges and intensities, along with global microbial distributions. We also briefly review how changing climate affects the functioning of microbes within ecosystems. Then, we examine the literature on how global changes in temperatures are changing the presence and prevalence of antibiotic resistance. Critically, when we talk about changing temperatures, we mean not only changes in the mean temperature but also changes in the extreme maximum and minimum temperatures as well as changes in the distribution of warmer and cooler days because all of these can impact the types and frequencies of infectious diseases.
Climate Change Influences Distribution and Incidence of Pathogens
For humans, one of the most striking and consequential effects of a global change in temperature is the change in the occurrence, intensity, and distribution of infectious diseases (Boxall et al., 2009, Cohen, 2000, Epstein, 2001, Semenza and Menne, 2009). Numerous reviews in recent years have focused on climate change and its effects on infectious disease (e.g., Blum and Hotez, 2018, Brooks and Boeger, 2019, Campbell-Lendrum et al., 2015, Cavicchioli et al., 2019, Hinz et al., 2019, Liang and Gong, 2017, Levy et al., 2016, Watts et al., 2017). Much of the focus has been on the effects of changing climate patterns on the vectors of the disease, rather than the pathogen causing the disease. For example, in the early 1990s an increase in rainfall increased plant growth and in turn caused the rodent population to take off. This led to increased interactions between humans and rodents that resulted in the hantavirus outbreak in the southwestern U.S. states (Hjelle and Glass, 2000). The same is true for Lyme disease, where increasing temperatures affect tick phenology and are predicted to cause earlier seasonal outbreaks of Lyme disease (Monaghan et al., 2015, Ogden and Lindsay, 2016, Ostfeld and Brunner, 2015). For malaria, increasing temperature as little as 0.5°C could result in a 30%–100% increase in mosquito abundance (Caminade et al., 2014, Gething et al., 2010, Pascual et al., 2006, Rogers and Randolph, 2000).
More generally, for malaria, dengue fever, Ross River virus, Zika, and other mosquito-borne diseases, increasing temperatures affect mosquito survival, reproduction, and biting rates (Campbell et al., 2015, Colón-González et al., 2018, Messina et al., 2019, Mordecai et al., 2019). By mechanistically modeling mosquito species, pathogens, and their thermal responses, more nuanced predictions can be made. In particular, differences among tropical and temperate pathogens—with some increasing and some decreasing—can be predicted based on future temperature means and ranges (Mordecai et al., 2019).
Such studies that include how temperature affects microbial pathogens—as opposed to only their vectors or carriers—are quite rare and only now appearing, even though temperature directly affects pathogens on a global scale. Most studies connecting increasing temperatures to microbes have measured microbial rates of reproduction (Frey et al., 2013, Ratkowsky et al., 1982, Zwietering et al., 1991) Increasingly, however, there is an interest in understanding large-scale effects of temperature as well. For example, one recent study found that increasing temperatures would likely result in a substantial shift in microbial communities, particularly an increase in the biodiversity of microbes across much of North America and the Tibetan Plateau (Ladau et al., 2018). A recent review presented a consensus statement—substantial evidence that climate change is affecting microorganisms—by some of the leading climate change microbiologists in the world (Cavicchioli et al., 2019). For example, ocean warming, acidification, and eutrophication result in increases in macroalgae and harm corals and the hundreds of thousands of microbial species associated with corals (Bourne et al., 2016, Cavicchioli et al., 2019, Quigley et al., 2018, Torda et al., 2017, Webster and Reusch, 2017, Ziegler et al., 2017).
In turn microorganisms feedback to affect climate change through their ecosystem functioning capabilities, such as sequestering carbon and contributing to greenhouse gas emissions (Atwood et al., 2015, Behrenfeld et al., 2001, Behrenfeld, 2014, Boetius et al., 2013, Bonan, 2008, Cavicchioli et al., 2019, Field et al., 1998, Pan et al., 2011). Although the mechanisms and details may be different in marine and terrestrial biomes, the overarching conclusion is the same: warming temperatures have and will continue to have effects on the range, distribution, community composition, and physiological and ecosystem functioning of microorganisms (Cavicchioli et al., 2019, García et al., 2018).
Novel Studies Associating Climate Change with Antibiotic Resistance
Given that there is substantial research showing how climate change affects the global distribution of bacteria and their physiological functioning, it should not be surprising that climate change will affect antibiotic resistance at the global scale. Yet it has only been in the last couple of years and only in four papers that there are details of how temperature affects the level of antibiotic resistance on a large geographic scale (MacFadden et al., 2018, McGough et al., 2018, Parnanen et al., 2019, Casadevall et al., 2019). These papers are particularly informative because they not only detail how levels of antibiotic resistance have changed with increasing temperature, but also lay out the foundations for how we can make predictions about the spread of antibiotic resistance on a global scale.
In a recent tour de force, MacFadden et al. (2018) set out to examine the relationship between temperature and antibiotic resistance across the entire United States (MacFadden et al., 2018). They collected temperature data from 1980 to 2010, using minimum and mean temperatures from throughout the country. The researchers quantified percentages of antibiotic-resistant strains by collecting data from hundreds of hospitals and laboratories across 41 states, ultimately ending up with over 1.6 million bacterial pathogens in their dataset. They focused on resistance in three widespread pathogens of concern: gram-negative E. coli and K. pneumoniae and gram-positive S. aureus. MacFadden and colleagues found that, at the population level, for every 10°C increase in minimum temperature there was a significant increase in the percentage of resistant strains (4.2%, 2.2%, and 2.7%, respectively, for the three pathogens they studied). To our knowledge this is the first study that explicitly connects the increase in temperature expected from climate change to increased antibiotic resistance in pathogens.
Based on these empirical findings, statistical extrapolations of these data into the future could provide guidelines for what we should expect in terms of the scope of the problem of antibiotic-resistant pathogens in the future. Furthermore, by combining these and similar data with climate predictions and more evolutionary and physiologically grounded models—that include mutation rates, standing variance, or effective population size and thermal responses—more targeted predictions by region and in time could be made.
An even more recent follow-up study by McGough et al. (2018) examined temperature effects of resistance across 28 countries in Europe, from 2000 to 2016 (McGough et al., 2018). Consistent with MacFadden et al. (2018), the researchers found that increases in temperature increased the percentage of antibiotic-resistant bacteria. This study went a step further. The researchers were not only able to correlate warmer temperatures with an increase in overall presence of resistant bacteria but they also found a correlation between warmer temperatures and increased rates of change for levels of the same resistant bacteria. The careful and meticulous measurements across time in this study over the last two decades allowed a temporal analysis of resistance throughout most of Europe. Importantly, the rapid rate of resistance was observed even after controlling for known factors—such as overall antibiotic consumption and human population density—that affect resistance.
This same pattern of increased percentage of both antibiotic-resistant bacteria and antibiotic-resistant genes in warmer temperatures has also been found in a large study of wastewater treatment plants across Europe (Parnanen et al., 2019). Given that sewage systems are a major point of transfer of human bacteria to the environment, the researchers focused on twelve wastewater treatment systems in seven European countries. By examining the influent and effluent for antibiotic-resistant bacteria and genes, the authors found that temperature is a critical driver of antibiotic resistance as measured by the persistence of resistance and the spread in the environment (Parnanen et al., 2019). There appear to be numerous ways higher temperatures can affect resistance, including through a faster rate of mutations with potential for resistance, through effects on which bacteria persist and consequently shift community composition, and through the speed of dispersal across spatial scales.
Another recent paper by Casadevall et al. (2019) hypothesized that drug-resistant fungi Candida auris emerged as a new human pathogen because of increasing temperatures caused by global climate change (Casadevall et al., 2019). It has been suggested that mammals have relatively few fungal diseases compared with abundance of fungal species and ones that are pathogenic in ectotherms. This rarity is hypothesized to be because of the “mammalian thermal barrier”—the temperature difference between the ambient environmental temperature at which fungi can survive and the higher internal temperature of mammals. It has been hypothesized that this thermal barrier evolved during the cretaceous period, with fungal diseases serving as a selective agent for mammals (Casadevall, 2012). Thus, mammals that survived were more likely to be resistant to fungal diseases (Casadevall, 2012).
In contrast, many other species that comprise substantial parts of the planet's biodiversity and ecosystem services are being devastated by fungal diseases. Specifically, chytrid fungus is estimated to have caused the extinction of hundreds of species and endangered nearly a third of amphibian species globally, making it one of the deadliest pathogens and invasive species in existence. It has been argued that the spread and deadliness of chytrid is linked to climate change and temperature increases, with substantial work still being done to parse out the various contributing factors (O’Hanlon et al., 2018, Rohr et al., 2008, Murray et al., 2011, Rohr and Raffel, 2010).
With increasing temperatures there will likely be an increase in fungal adaptations to heat in general (Garcia-Solache and Casadevall, 2010). Adaptations to warmer environments can already be seen in experimental studies (de Crecy et al., 2009), studies of culture collections (Robert et al., 2015), and studies comparing fungal growth in cooler rural habitats versus warmer urban habitats (McLean et al., 2005).
C. auris likely started out as a plant saprophyte in wetlands. Subsequent changes in environmental temperatures contributed to both increased presence of C. auris (via changes in ecological niches of plant hosts) and beneficial mutations that arose owing to greater UV exposure and higher temperatures. Seabirds could have served as an intermediate host between plants and humans, since fungi that can grow at 40°C–42°C can infect avian hosts. Although definitive pathways to the emergence of pathogens are difficult to discern, the arguments set forth are persuasive that C. auris could be the first species discovered that has arisen as a human pathogen as a result of warming temperatures (Casadevall et al., 2019).
Conclusions
In summary, changes in temperatures are likely to lead to thermal adaptation in microbes that experience rises and declines in temperature—either inside a host or in the external environment. These thermal adaptations may have collateral effects such as higher antibiotic resistance. Even if most microbes are not pathogens, the overall community of microbes serves as a reservoir of antibiotic-resistant genes that can be transmitted to pathogens. Therefore, we suggest that it is critical to study the emergence and persistence of antibiotic resistance both in clinically relevant pathogens as well as non-pathogenic environmental bacteria. In covering all the bases above that we think are needed for this, we invoked processes across many biological scales—from genomic to cellular to physiological to ecological—that also correspond to enormous ranges in spatial and temporal scales—from sub-cellular compartments to global dispersion and from nearly immediate response to stimuli to evolutionary times over eons. We also described work that transcends disciplines by bringing together clinical concerns, drug studies, genomics, mathematical theory, physiology, computation, ecology, statistics, evolution, data analysis, biochemistry, protein dynamics, climate predictions, and even thermal physics. Consequently, to fully understand the factors promoting the current and growing antibiotic resistance crisis, we will need a multidisciplinary, multiscale approach that fully captures and considers effects of temperature.
Acknowledgments
We thank Nicholas Ida and two anonymous reviewers for helpful comments that improved the manuscript. We are grateful for funding from the Hellman Foundation (P.Y.), a KL2 Fellowship (P.Y.) through the NIH/National Center for Advancing Translational Sciences (NCATS) UCLA CTSI Grant Number UL1TR001881, and a James F. McDonnell Foundation Complex Systems Scholar Award (V.S.).
Author Contributions
A.R.-V. and P.Y. outlined the manuscript. A.R.-V. prepared the graphical abstract and figure. All authors wrote, edited, and provided feedback on the manuscript.
Declaration of Interests
The authors declare no competing interests.
Published: April 24, 2020
References
- Aldred K.J., Kerns R.J., Osheroff N. Mechanism of quinolone action and resistance. Biochemistry. 2014;53:1565–1574. doi: 10.1021/bi5000564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andersson D.I., Hughes D. Antibiotic resistance and its cost: is it possible to reverse resistance. Nat. Rev. Microbiol. 2010;8:260–271. doi: 10.1038/nrmicro2319. [DOI] [PubMed] [Google Scholar]
- Andersson D.I., Hughes D. Microbiological effects of sublethal levels of antibiotics. Nat. Rev. Microbiol. 2014;12:465–478. doi: 10.1038/nrmicro3270. [DOI] [PubMed] [Google Scholar]
- Andrade-Linares D.R., Lehmann A., Rillig M.C. Microbial stress priming: a meta-analysis. Environ. Microbiol. 2016;18:1277–1288. doi: 10.1111/1462-2920.13223. [DOI] [PubMed] [Google Scholar]
- Arsène F., Tomoyasu T., Bukau B. The heat shock response of Escherichia coli. Int. J. Food Microbiol. 2000;55:3–9. doi: 10.1016/s0168-1605(00)00206-3. [DOI] [PubMed] [Google Scholar]
- Atwood T.B., Connolly R.M., Ritchie E.G., Lovelock C.E., Hwithaus M.R., Hays G.C., Fourqurean J.W., Macreadie P.I. Predators help protect carbon stock in blue carbon ecosystems. Nat. Clim. Change. 2015;5:1038–1045. [Google Scholar]
- Balaban N.Q., Helaine S., Lewis K., Ackermann M., Aldridge B., Andersson D.I., Brynildsen M.P., Bumann D., Camilli A., Collins J.J. Definitions and guidelines for research on antibiotic persistence. Nat. Rev. Microbiol. 2019;17:441–448. doi: 10.1038/s41579-019-0196-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balaban N.Q., Merrin J., Chait R., Kowalik L., Leibler S. Bacterial persistence as a phenotypic switch. Science. 2004;305:1622–1625. doi: 10.1126/science.1099390. [DOI] [PubMed] [Google Scholar]
- Barria C., Malecki M., Arraiano C.M. Bacterial adaptation to cold. Microbiology. 2013;159:2437–2443. doi: 10.1099/mic.0.052209-0. [DOI] [PubMed] [Google Scholar]
- Behrenfeld M.J. Climate-mediated dance of the plankton. Nat. Clim. Change. 2014;4:880–887. [Google Scholar]
- Behrenfeld M.J., Randerson J.T., McClain C.R., Feldman G.C., Los S.O., Tucker C.J., Falkowski P.G., Field C.B., Frouin R., Esaias W.E. Biospheric primary production during an ENSO transition. Science. 2001;291:2594–2597. doi: 10.1126/science.1055071. [DOI] [PubMed] [Google Scholar]
- Blum A.J., Hotez P.J. Global “worming”: climate change and its projected general impact on human helminth infections. PLoS Negl. Trop. Dis. 2018;12:e0006370. doi: 10.1371/journal.pntd.0006370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bódi Z., Farkas Z., Nevozhay D., Kalapis D., Lázár V., Csörgő B., Nyerges Á., Szamecz B., Fekete G., Papp B. Phenotypic heterogeneity promotes adaptive evolution. PLoS Biol. 2017;15:e2000644. doi: 10.1371/journal.pbio.2000644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boetius A., Albrecht S., Bakker K., Bienhold C., Felden J., Fernández-Méndez M., Hendricks S., Katlein C., Lalande C., Krumpen T. Export of algal biomass from the melting Arctic sea ice. Science. 2013;339:1430–1432. doi: 10.1126/science.1231346. [DOI] [PubMed] [Google Scholar]
- Bonan G.B. Forests and climate change: forcings, feedbacks, and the climate benefits of forests. Science. 2008;320:1444–1449. doi: 10.1126/science.1155121. [DOI] [PubMed] [Google Scholar]
- Bourne D.G., Morrow K.M., Webster N.S. Insights into the coral microbiome: underpinning the health and resilience of reef ecosystems. Annu. Rev. Microbiol. 2016;70:317–340. doi: 10.1146/annurev-micro-102215-095440. [DOI] [PubMed] [Google Scholar]
- Boxall A.B., Hardy A., Beulke S., Boucard T., Burgin L., Falloon P.D., Haygarth P.M., Hutchinson T., Kovats R.S., Leonardi G. Impacts of climate change on indirect human exposure to pathogens and chemicals from agriculture. Environ. Health Perspect. 2009;117:508–514. doi: 10.1289/ehp.0800084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brauner A., Fridman O., Gefen O., Balaban N.Q. Distinguishing between resistance, tolerance and persistence to antibiotic treatment. Nat. Rev. Microbiol. 2016;14:320–330. doi: 10.1038/nrmicro.2016.34. [DOI] [PubMed] [Google Scholar]
- Brooks D.R., Boeger W.A. Climate change and emerging infectious diseases: evolutionary complexity in action. Curr. Opin. Syst. Biol. 2019;13:75–81. [Google Scholar]
- Bruhn-Olszewska B., Szczepaniak P., Matuszewska E., Kuczyńska-Wiśnik D., Stojowska-Swędrzyńska K., Moruno Algara M., Laskowska E. Physiologically distinct subpopulations formed in Escherichia coli cultures in response to heat shock. Microbiol. Res. 2018;209:33–42. doi: 10.1016/j.micres.2018.02.002. [DOI] [PubMed] [Google Scholar]
- Cabral D.J., Wurster J.I., Belenky P. Antibiotic persistence as a metabolic adaptation: stress, metabolism, the host, and new directions. Pharmaceuticals. 2018;11:14. doi: 10.3390/ph11010014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caminade C., Kovats S., Rocklov J., Tompkins A.M., Morse A.P., Colón-González F.J., Stenlund H., Martens P., Lloyd S.J. Impact of climate change on global malaria distribution. Proc. Natl. Acad. Sci. U S A. 2014;111:3286–3291. doi: 10.1073/pnas.1302089111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campbell-Lendrum D., Manga L., Bagayoko M., Sommerfeld J. Climate change and vector-borne diseases: what are the implications for public health research and policy. Philos. Trans. R Soc. Lond. B Biol. Sci. 2015;370:20130552. doi: 10.1098/rstb.2013.0552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campbell L.P., Luther C., Moo-Llanes D., Ramsey J.M., Danis-Lozano R., Peterson A.T. Climate change influences on global distributions of dengue and chikungunya virus vectors. Philos. Trans. R Soc. Lond. B Biol. Sci. 2015;370:20140135. doi: 10.1098/rstb.2014.0135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cardoso K., Gandra R.F., Wisniewski E.S., Osaku C.A., Kadowaki M.K., Felipach-Neto V., Haus L.F., Simão R.C. DnaK and GroEL are induced in response to antibiotic and heat shock in Acinetobacter baumannii. J. Med. Microbiol. 2010;59:1061–1068. doi: 10.1099/jmm.0.020339-0. [DOI] [PubMed] [Google Scholar]
- Casadevall A. Fungi and the rise of mammals. PLoS Pathog. 2012;8:e1002808. doi: 10.1371/journal.ppat.1002808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Casadevall A., Kontoyiannis D.P., Robert V. On the emergence of Candida auris: climate change, azoles, swamps, and birds. MBio. 2019;10 doi: 10.1128/mBio.01397-19. e01397–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caspeta L., Flores N., Perez N.O., Bolivar F., Ramirez O.T. The effect of heating rate on Escherichia coli metabolism, physiological stress, transcriptional response, and production of temperature-induced recombinant protein: a scale-down study. Biotechnol. Bioeng. 2009;102:468–482. doi: 10.1002/bit.22084. [DOI] [PubMed] [Google Scholar]
- Cavicchioli R., Ripple W.J., Timmis K.N., Azam F., Bakken L.R., Baylis M., Behrenfeld M.J., Boetius A., Boyd P.W., Classen A.T. Scientists’ warning to humanity: microorganisms and climate change. Nat. Rev. Microbiol. 2019;17:569–586. doi: 10.1038/s41579-019-0222-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chaudhuri S., Jana B., Basu T. Why does ethanol induce cellular heat-shock response? Cell Biol. Toxicol. 2006;22:29–37. doi: 10.1007/s10565-006-0029-9. [DOI] [PubMed] [Google Scholar]
- Clint E., Fessler D.M. Insurmountable heat: the evolution and persistence of defensive hyperthermia. Q. Rev. Biol. 2016;91:25–46. doi: 10.1086/685302. [DOI] [PubMed] [Google Scholar]
- Cohen M.L. Changing patterns of infectious disease. Nature. 2000;406:762–767. doi: 10.1038/35021206. [DOI] [PubMed] [Google Scholar]
- Colón-González F.J., Harris I., Osborn T.J., Steiner São Bernardo C., Peres C.A., Hunter P.R., Lake I.R. Limiting global-mean temperature increase to 1.5-2 °C could reduce the incidence and spatial spread of dengue fever in Latin America. Proc. Natl. Acad. Sci. U S A. 2018;115:6243–6248. doi: 10.1073/pnas.1718945115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Courgeon A.M., Maisonhaute C., Best-Belpomme M. Heat shock proteins are induced by cadmium in Drosophila cells. Exp. Cell Res. 1984;153:515–521. doi: 10.1016/0014-4827(84)90618-9. [DOI] [PubMed] [Google Scholar]
- Cruz-Loya M., Kang T.M., Lozano N.A., Watanabe R., Tekin E., Damoiseaux R., Savage V.M., Yeh P.J. Stressor interaction networks suggest antibiotic resistance co-opted from stress responses to temperature. ISME J. 2019;13:12–23. doi: 10.1038/s41396-018-0241-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Crecy E., Jaronski S., Lyons B., Lyons T.J., Keyhani N.O. Directed evolution of a filamentous fungus for thermotolerance. BMC Biotechnol. 2009;9:74. doi: 10.1186/1472-6750-9-74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Derman Y., Söderholm H., Lindström M., Korkeala H. Role of csp genes in NaCl, pH, and ethanol stress response and motility in Clostridium botulinum ATCC 3502. Food Microbiol. 2015;46:463–470. doi: 10.1016/j.fm.2014.09.004. [DOI] [PubMed] [Google Scholar]
- Dörr T., Vulić M., Lewis K. Ciprofloxacin causes persister formation by inducing the TisB toxin in Escherichia coli. PLoS Biol. 2010;8:e1000317. doi: 10.1371/journal.pbio.1000317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dragosits M., Mozhayskiy V., Quinones-Soto S., Park J., Tagkopoulos I. Evolutionary potential, cross-stress behavior and the genetic basis of acquired stress resistance in Escherichia coli. Mol. Syst. Biol. 2013;9:643. doi: 10.1038/msb.2012.76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duval B.D., Mathew A., Satola S.W., Shafer W.M. Altered growth, pigmentation, and antimicrobial susceptibility properties of Staphylococcus aureus due to loss of the major cold shock gene cspB. Antimicrob. Agents Chemother. 2010;54:2283–2290. doi: 10.1128/AAC.01786-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- El-Radhi A.S., Carroll J., Klein N., editors. Clinical Manual of Fever in Children. Springer; 2009. [DOI] [Google Scholar]
- Epstein P.R. Climate change and emerging infectious diseases. Microbes Infect. 2001;3:747–754. doi: 10.1016/s1286-4579(01)01429-0. [DOI] [PubMed] [Google Scholar]
- Field C.B., Behrenfeld M.J., Randerson J.T., Falkowski P. Primary production of the biosphere: integrating terrestrial and oceanic components. Science. 1998;281:237–240. doi: 10.1126/science.281.5374.237. [DOI] [PubMed] [Google Scholar]
- Foster P.L. Stress-induced mutagenesis in bacteria. Crit. Rev. Biochem. Mol. Biol. 2007;42:373–397. doi: 10.1080/10409230701648494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frey S.D., Lee J., Melillo J.M., Six J. The temperature response of soil microbial efficiency and its feedback to climate. Nat. Clim. Chang. 2013;3:395–398. [Google Scholar]
- Gagneux S., Long C.D., Small P.M., Van T., Schoolnik G.K., Bohannan B.J. The competitive cost of antibiotic resistance in Mycobacterium tuberculosis. Science. 2006;312:1944–1946. doi: 10.1126/science.1124410. [DOI] [PubMed] [Google Scholar]
- Garcia-Solache M.A., Casadevall A. Global warming will bring new fungal diseases for mammals. MBio. 2010;1 doi: 10.1128/mBio.00061-10. e00061-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- García F.C., Bestion E., Warfield R., Yvon-Durocher G. Changes in temperature alter the relationship between biodiversity and ecosystem functioning. Proc. Natl. Acad. Sci. U S A. 2018;115:10989–10994. doi: 10.1073/pnas.1805518115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gething P.W., Smith D.L., Patil A.P., Tatem A.J., Snow R.W., Hay S.I. Climate change and the global malaria recession. Nature. 2010;465:342–345. doi: 10.1038/nature09098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gifford D.R., Moss E., MacLean R.C. Environmental variation alters the fitness effects of rifampicin resistance mutations in Pseudomonas aeruginosa. Evolution. 2016;70:725–730. doi: 10.1111/evo.12880. [DOI] [PubMed] [Google Scholar]
- Goltermann L., Good L., Bentin T. Chaperonins fight aminoglycoside-induced protein misfolding and promote short-term tolerance in Escherichia coli. J. Biol. Chem. 2013;288:10483–10489. doi: 10.1074/jbc.M112.420380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goneau L.W., Yeoh N.S., MacDonald K.W., Cadieux P.A., Burton J.P., Razvi H., Reid G. Selective target inactivation rather than global metabolic dormancy causes antibiotic tolerance in uropathogens. Antimicrob. Agents Chemother. 2014;58:2089–2097. doi: 10.1128/AAC.02552-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greulich P., Scott M., Evans M.R., Allen R.J. Growth-dependent bacterial susceptibility to ribosome-targeting antibiotics. Mol. Syst. Biol. 2015;11:796. doi: 10.15252/msb.20145949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grossman A.D., Straus D.B., Walter W.A., Gross C.A. Sigma 32 synthesis can regulate the synthesis of heat shock proteins in Escherichia coli. Genes Dev. 1987;1:179–184. doi: 10.1101/gad.1.2.179. [DOI] [PubMed] [Google Scholar]
- Gutierrez A., Jain S., Bhargava P., Hamblin M., Lobritz M.A., Collins J.J. Understanding and sensitizing density-dependent persistence to quinolone antibiotics. Mol. Cell. 2017;68:1147–1154.e3. doi: 10.1016/j.molcel.2017.11.012. [DOI] [PubMed] [Google Scholar]
- Handwerger S., Tomasz A. Antibiotic tolerance among clinical isolates of bacteria. Annu. Rev. Pharmacol. Toxicol. 1985;25:349–380. doi: 10.1146/annurev.pa.25.040185.002025. [DOI] [PubMed] [Google Scholar]
- Heikkila J.J., Schultz G.A., Iatrou K., Gedamu L. Expression of a set of fish genes following heat or metal ion exposure. J. Biol. Chem. 1982;257:12000–12005. [PubMed] [Google Scholar]
- Hilker M., Schwachtje J., Baier M., Balazadeh S., Bäurle I., Geiselhardt S., Hincha D.K., Kunze R., Mueller-Roeber B., Rillig M.C. Priming and memory of stress responses in organisms lacking a nervous system. Biol. Rev. Camb. Philos. Soc. 2016;91:1118–1133. doi: 10.1111/brv.12215. [DOI] [PubMed] [Google Scholar]
- Hinz R., Frickmann H., Krüger A. International climate protection. In: Palocz-Andresen M., Szalay D., Gosztom A., Sipos L., Taligas T., editors. Chapter: Climate Change and Infectious Diseases. Springer; 2019. [Google Scholar]
- Hjelle B., Glass G.E. Outbreak of hantavirus infection in the Four Corners region of the United States in the wake of the 1997-1998 El Nino-southern oscillation. J. Infect. Dis. 2000;181:1569–1573. doi: 10.1086/315467. [DOI] [PubMed] [Google Scholar]
- Interagency Coordination Group on Antimicrobial Resistance (2019). No time to wait: securing the future from drug-resistant infections. Report to the Secretary-General of the United Nations. April 2019.
- Johnson P.J., Levin B.R. Pharmacodynamics, population dynamics, and the evolution of persistence in Staphylococcus aureus. PLoS Genet. 2013;9:e1003123. doi: 10.1371/journal.pgen.1003123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kandror O., DeLeon A., Goldberg A.L. Trehalose synthesis is induced upon exposure of Escherichia coli to cold and is essential for viability at low temperatures. Proc. Natl. Acad. Sci. U S A. 2002;99:9727–9732. doi: 10.1073/pnas.142314099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kent A.G., Garcia C.A., Martiny A.C. Increased biofilm formation due to high-temperature adaptation in marine Roseobacter. Nat. Microbiol. 2018;3:989–995. doi: 10.1038/s41564-018-0213-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kester J.C., Fortune S.M. Persisters and beyond: mechanisms of phenotypic drug resistance and drug tolerance in bacteria. Crit. Rev. Biochem. Mol. Biol. 2014;49:91–101. doi: 10.3109/10409238.2013.869543. [DOI] [PubMed] [Google Scholar]
- Knies J.L., Cai F., Weinreich D.M. Enzyme efficiency but not thermostability drives cefotaxime resistance evolution in TEM-1 ß-lactamase. Mol. Biol. Evol. 2017;34:1040–1054. doi: 10.1093/molbev/msx053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kohanski M.A., Dwyer D.J., Wierzbowski J., Cottarel G., Collins J.J. Mistranslation of membrane proteins and two-component system activation trigger antibiotic-mediated cell death. Cell. 2008;135:679–690. doi: 10.1016/j.cell.2008.09.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ladau J., Shi Y., Jing X., He J.S., Chen L., Lin X., Fierer N., Gilbert J.A., Pollard K.S., Chu H. Existing climate change will lead to pronounced shifts in the diversity of soil prokaryotes. mSystems. 2018;3 doi: 10.1128/mSystems.00167-18. e00167–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lenhard J.R., Brown T., Rybak M.J., Meaney C.J., Norgard N.B., Bulman Z.P., Brazeau D.A., Gill S.R., Tsuji B.T. Sequential evolution of vancomycin-intermediate resistance alters virulence in Staphylococcus aureus: pharmacokinetic/pharmacodynamic targets for vancomycin exposure. Antimicrob. Agents Chemother. 2015;60:1584–1591. doi: 10.1128/AAC.02657-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levin-Reisman I., Ronin I., Gefen O., Braniss I., Shoresh N., Balaban N.Q. Antibiotic tolerance facilitates the evolution of resistance. Science. 2017;355:826–830. doi: 10.1126/science.aaj2191. [DOI] [PubMed] [Google Scholar]
- Levin B.R., Concepción-Acevedo J., Udekwu K.I. Persistence: a copacetic and parsimonious hypothesis for the existence of non-inherited resistance to antibiotics. Curr. Opin. Microbiol. 2014;21:18–21. doi: 10.1016/j.mib.2014.06.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levy K., Woster A.P., Goldstein R.S., Carlton E.J. Untangling the impacts of climate change on waterborne diseases: a systematic review of relationships between diarrheal diseases and temperature, rainfall, flooding, and drought. Environ. Sci. Technol. 2016;50:4905–4922. doi: 10.1021/acs.est.5b06186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewis K. Persister cells, dormancy and infectious disease. Nat. Rev. Microbiol. 2007;5:48–56. doi: 10.1038/nrmicro1557. [DOI] [PubMed] [Google Scholar]
- Liang L., Gong P. Climate change and human infectious diseases: a synthesis of research findings from global and spatio-temporal perspectives. Environ. Int. 2017;103:99–108. doi: 10.1016/j.envint.2017.03.011. [DOI] [PubMed] [Google Scholar]
- Lindquist S. The heat-shock response. Annu. Rev. Biochem. 1986;55:1151–1191. doi: 10.1146/annurev.bi.55.070186.005443. [DOI] [PubMed] [Google Scholar]
- Loepfe C., Raimann E., Stephan R., Tasara T. Reduced host cell invasiveness and oxidative stress tolerance in double and triple csp gene family deletion mutants of Listeria monocytogenes. Foodborne Pathog. Dis. 2010;7:775–783. doi: 10.1089/fpd.2009.0458. [DOI] [PubMed] [Google Scholar]
- MacFadden D.R., McGough S.F., Fisman D., Santillana M., Brownstein J.S. Antibiotic resistance increases with local temperature. Nat. Clim. Chang. 2018;8:510–514. doi: 10.1038/s41558-018-0161-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacGowan A.P., Bowker K.E. Sequential antimicrobial therapy: pharmacokinetic and pharmacodynamic considerations in sequential therapy. J. Infect. 1998;37(Suppl 1):30–36. doi: 10.1016/s0163-4453(98)92721-9. [DOI] [PubMed] [Google Scholar]
- MacLean R.C., Hall A.R., Perron G.G., Buckling A. The population genetics of antibiotic resistance: integrating molecular mechanisms and treatment contexts. Nat. Rev. Genet. 2010;11:405–414. doi: 10.1038/nrg2778. [DOI] [PubMed] [Google Scholar]
- Maharjan R., Ferenci T. The fitness costs and benefits of antibiotic resistance in drug-free microenvironments encountered in the human body. Environ. Microbiol. Rep. 2017;9:635–641. doi: 10.1111/1758-2229.12564. [DOI] [PubMed] [Google Scholar]
- Manrique Y., Suriyarak S., Gibis M., Schmidt H., Weiss J. Survival of spoilage bacteria subjected to sequential eugenol and temperature treatments. Int. J. Food Microbiol. 2016;218:6–16. doi: 10.1016/j.ijfoodmicro.2015.10.027. [DOI] [PubMed] [Google Scholar]
- McGough S.F., MacFadden D.R., Hattab M.W., Molback K., Santillana M. Rates of increase of antibiotic resistance and ambient temperature in Europe: a cross-national analysis of 28 countries between 2000-2016. Biorxiv. 2018 doi: 10.1101/414920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McLean M.A., Angilletta M.J., Williams K.S. If you can’t stand the heat, stay out of the city: thermal reaction norms of chitinolytic fungi in an urban heat island. J. Therm. Biol. 2005;30:384–391. [Google Scholar]
- McMahon M.A., Xu J., Moore J.E., Blair I.S., McDowell D.A. Environmental stress and antibiotic resistance in food-related pathogens. Appl. Environ. Microbiol. 2007;73:211–217. doi: 10.1128/AEM.00578-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Messina J.P., Brady O.J., Golding N., Kraemer M.U.G., Wint G.R.W., Ray S.E., Pigott D.M., Shearer F.M., Johnson K., Earl L. The current and future global distribution and population at risk of dengue. Nat. Microbiol. 2019;4:1508–1515. doi: 10.1038/s41564-019-0476-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michel G.P., Starka J. Effect of ethanol and heat stresses on the protein pattern of Zymomonas mobilis. J. Bacteriol. 1986;165:1040–1042. doi: 10.1128/jb.165.3.1040-1042.1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michiels J.E., Van den Bergh B., Verstraeten N., Michiels J. Molecular mechanisms and clinical implications of bacterial persistence. Drug Resist. Updat. 2016;29:76–89. doi: 10.1016/j.drup.2016.10.002. [DOI] [PubMed] [Google Scholar]
- Miller Y.W., Eady E.A., Lacey R.W., Cove J.H., Joanes D.N., Cunliffe W.J. Sequential antibiotic therapy for acne promotes the carriage of resistant staphylococci on the skin of contacts. J. Antimicrob. Chemother. 1996;38:829–837. doi: 10.1093/jac/38.5.829. [DOI] [PubMed] [Google Scholar]
- Mogk A., Tomoyasu T., Goloubinoff P., Rüdiger S., Röder D., Langen H., Bukau B. Identification of thermolabile Escherichia coli proteins: prevention and reversion of aggregation by DnaK and ClpB. EMBO J. 1999;18:6934–6949. doi: 10.1093/emboj/18.24.6934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monaghan A.J., Moore S.M., Sampson K.M., Beard C.B., Eisen R.J. Climate change influences on the annual onset of Lyme disease in the United States. Ticks Tick Borne Dis. 2015;6:615–622. doi: 10.1016/j.ttbdis.2015.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mordecai E.A., Caldwell J.M., Grossman M.K., Lippi C.A., Johnson L.R., Neira M., Rohr J.R., Ryan S.J., Savage V., Shocket M.S. Thermal biology of mosquito-borne disease. Ecol. Lett. 2019;22:1690–1708. doi: 10.1111/ele.13335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murata M., Fujimoto H., Nishimura K., Charoensuk, Nagamitsu H., Raina S., Kosaka T., Oshima T., Ogasawara N., Yamada M. Molecular strategy for survival at a critical high temperature in Escherichia coli. PLoS One. 2011;6:e20063. doi: 10.1371/journal.pone.0020063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murray K.A., Retallick R.W.R., Puschendorf R., Skerratt L.F., Rosauer D., McCallum H., Berger L., Speare R., VanDerWal J. Assessing spatial patterns of disease risk to biodiversity: implications for the management of the amphibian pathogen, Batrachochytrium dendrobatidis. J. Appl. Ecol. 2011;48:163–173. [Google Scholar]
- Niu P., Liu L., Gong Z., Tan H., Wang F., Yuan J., Feng Y., Wei Q., Tanguay R.M., Wu T. Overexpressed heat shock protein 70 protects cells against DNA damage caused by ultraviolet C in a dose-dependent manner. Cell Stress Chaperon. 2006;11:162–169. doi: 10.1379/CSC-175R.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nonaka G., Blankschien M., Herman C., Gross C.A., Rhodius V.A. Regulon and promoter analysis of the E. coli heat-shock factor, sigma32, reveals a multifaceted cellular response to heat stress. Genes Dev. 2006;20:1776–1789. doi: 10.1101/gad.1428206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Hanlon S.J., Rieux A., Farrer R.A., Rosa G.M., Waldman B., Bataille A., Kosch T.A., Murray K.A., Brankovics B., Fumagalli M. Recent Asian origin of chytrid fungi causing global amphibian declines. Science. 2018;360:621–627. doi: 10.1126/science.aar1965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Neill (2014). Antimicrobial Resistance: Tackling a Crisis for the Health and Wealth of nations. Review on Antimicrobial Resistance, 2014.
- Ogden N.H., Lindsay L.R. Effects of climate and climate change on vectors and vector-borne diseases: ticks are different. Trends Parasitol. 2016;32:646–656. doi: 10.1016/j.pt.2016.04.015. [DOI] [PubMed] [Google Scholar]
- Ostfeld R.S., Brunner J.L. Climate change and Ixodes tick-borne diseases of humans. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2015;370:20140051. doi: 10.1098/rstb.2014.0051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oz T., Guvenek A., Yildiz S., Karaboga E., Tamer Y.T., Mumcuyan N., Ozan V.B., Senturk G.H., Cokol M., Yeh P. Strength of selection pressure is an important parameter contributing to the complexity of antibiotic resistance evolution. Mol. Biol. Evol. 2014;31:2387–2401. doi: 10.1093/molbev/msu191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan Y., Birdsey R.A., Fang J., Houghton R., Kauppi P.E., Kurz W.A., Phillips O.L., Shvidenko A., Lewis S.L., Canadell J.G. A large and persistent carbon sink in the world’s forests. Science. 2011;333:988–993. doi: 10.1126/science.1201609. [DOI] [PubMed] [Google Scholar]
- Parnanen K.M.M., Narciso-da-Rocha C., Kneis D., Berendonk T.U., Cacace D., Do T.T., Elpers C., Fatta-Kassinos D., Henriques I., Jaeger T. Antibiotic resistance in European wastewater treatment plants mirrors the pattern of clinical antibiotic resistance prevalence. Sci. Adv. 2019;5:eaau9124. doi: 10.1126/sciadv.aau9124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pascual M., Ahumada J.A., Chaves L.F., Rodó X., Bouma M. Malaria resurgence in the East African highlands: temperature trends revisited. Proc. Natl. Acad. Sci. U S A. 2006;103:5829–5834. doi: 10.1073/pnas.0508929103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Payne J.L., Wagner A. The causes of evolvability and their evolution. Nat. Rev. Genet. 2019;20:24–38. doi: 10.1038/s41576-018-0069-z. [DOI] [PubMed] [Google Scholar]
- Phadtare S., Inouye M. The cold shock response. EcoSal Plus. 2008;3 doi: 10.1128/ecosalplus.5.4.2. [DOI] [PubMed] [Google Scholar]
- Quigley K.M., Baker A.C., Coffroth M.A., Willis B.L., Van Oppen M.J.H. Coral Bleaching: patterns, processes, causes and consequences. In: van Oppen M.J.H., Lough J.M., editors. Chapter: Bleaching Resistance and the Role of Algal Endosymbionts. Springer; 2018. [Google Scholar]
- Radzikowski J.L., Schramke H., Heinemann M. Bacterial persistence from a system-level perspective. Curr. Opin. Biotechnol. 2017;46:98–105. doi: 10.1016/j.copbio.2017.02.012. [DOI] [PubMed] [Google Scholar]
- Ramirez M.S., Tolmasky M.E. Aminoglycoside modifying enzymes. Drug Resist. Updat. 2010;13:151–171. doi: 10.1016/j.drup.2010.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rangel D.E. Stress induced cross-protection against environmental challenges on prokaryotic and eukaryotic microbes. World J. Microbiol. Biotechnol. 2011;27:1281–1296. doi: 10.1007/s11274-010-0584-3. [DOI] [PubMed] [Google Scholar]
- Ratkowsky D.A., Olley J., McMeekin T.A., Ball A. Relationship between temperature and growth rate of bacterial cultures. J. Bacteriol. 1982;149:1–5. doi: 10.1128/jb.149.1.1-5.1982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richter K., Haslbeck M., Buchner J. The heat shock response: life on the verge of death. Mol. Cell. 2010;40:253–266. doi: 10.1016/j.molcel.2010.10.006. [DOI] [PubMed] [Google Scholar]
- Robert V., Cardinali G., Casadevall A. Distribution and impact of yeast thermal tolerance permissive for mammalian infection. BMC Biol. 2015;13:18. doi: 10.1186/s12915-015-0127-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodríguez-Verdugo A., Gaut B.S., Tenaillon O. Evolution of Escherichia coli rifampicin resistance in an antibiotic-free environment during thermal stress. BMC Evol. Biol. 2013;13:50. doi: 10.1186/1471-2148-13-50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodríguez-Verdugo A., Tenaillon O., Gaut B.S. First-step mutations during adaptation restore the expression of hundreds of genes. Mol. Biol. Evol. 2016;33:25–39. doi: 10.1093/molbev/msv228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rogers D.J., Randolph S.E. The global spread of malaria in a future, warmer world. Science. 2000;289:1763–1766. doi: 10.1126/science.289.5485.1763. [DOI] [PubMed] [Google Scholar]
- Rohr J.R., Raffel T.R. Linking global climate and temperature variability to widespread amphibian declines putatively caused by disease. Proc. Natl. Acad. Sci. U S A. 2010;107:8269–8274. doi: 10.1073/pnas.0912883107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rohr J.R., Raffel T.R., Romansic J.M., McCallum H., Hudson P.J. Evaluating the links between climate, disease spread, and amphibian declines. Proc. Natl. Acad. Sci. U S A. 2008;105:17436–17441. doi: 10.1073/pnas.0806368105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roncarati D., Scarlato V. Regulation of heat-shock genes in bacteria: from signal sensing to gene expression output. FEMS Microbiol. Rev. 2017;41:549–574. doi: 10.1093/femsre/fux015. [DOI] [PubMed] [Google Scholar]
- Schmid B., Klumpp J., Raimann E., Loessner M.J., Stephan R., Tasara T. Role of cold shock proteins in growth of Listeria monocytogenes under cold and osmotic stress conditions. Appl. Environ. Microbiol. 2009;75:1621–1627. doi: 10.1128/AEM.02154-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Segrè D., Deluna A., Church G.M., Kishony R. Modular epistasis in yeast metabolism. Nat. Genet. 2005;37:77–83. doi: 10.1038/ng1489. [DOI] [PubMed] [Google Scholar]
- Semenza J.C., Menne B. Climate change and infectious diseases in Europe. Lancet Infect. Dis. 2009;9:365–375. doi: 10.1016/S1473-3099(09)70104-5. [DOI] [PubMed] [Google Scholar]
- Simon H.B. Hyperthermia. N. Engl. J. Med. 1993;329:483–487. doi: 10.1056/NEJM199308123290708. [DOI] [PubMed] [Google Scholar]
- Souli M., Galani I., Giamarellou H. Emergence of extensively drug-resistant and pandrug-resistant Gram-negative bacilli in Europe. Euro Surveill. 2008;13 [PubMed] [Google Scholar]
- Stewart P.S. Mechanisms of antibiotic resistance in bacterial biofilms. Int. J. Med. Microbiol. 2002;292:107–113. doi: 10.1078/1438-4221-00196. [DOI] [PubMed] [Google Scholar]
- Stewart P.S., Franklin M.J. Physiological heterogeneity in biofilms. Nat. Rev. Microbiol. 2008;6:199–210. doi: 10.1038/nrmicro1838. [DOI] [PubMed] [Google Scholar]
- Straus D.B., Walter W.A., Gross C.A. The heat shock response of E. coli is regulated by changes in the concentration of sigma 32. Nature. 1987;329:348–351. doi: 10.1038/329348a0. [DOI] [PubMed] [Google Scholar]
- Torda G., Donelson J.M., Aranda M., Barshis D.J., Bay L., Berumen M.L., Bourne D.G., Cantin N., Foret S., Matz M. Rapid adaptive responses to climate change in corals. Nat. Clim. Chang. 2017;7:627–636. [Google Scholar]
- Trindade S., Sousa A., Gordo I. Antibiotic resistance and stress in the light of Fisher’s model. Evolution. 2012;66:3815–3824. doi: 10.1111/j.1558-5646.2012.01722.x. [DOI] [PubMed] [Google Scholar]
- Van Boeckel T.P., Brower C., Gilbert M., Grenfell B.T., Levin S.A., Robinson T.P., Teillant A., Laxminarayan R. Global trends in antimicrobial use in food animals. Proc. Natl. Acad. Sci. U S A. 2015;112:5649–5654. doi: 10.1073/pnas.1503141112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Boeckel T.P., Glennon E.E., Chen D., Gilbert M., Robinson T.P., Grenfell B.T., Levin S.A., Bonhoeffer S., Laxminarayan R. Reducing antimicrobial use in food animals. Science. 2017;357:1350–1352. doi: 10.1126/science.aao1495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- VanBogelen R.A., Neidhardt F.C. Ribosomes as sensors of heat and cold shock in Escherichia coli. Proc. Natl. Acad. Sci. U S A. 1990;87:5589–5593. doi: 10.1073/pnas.87.15.5589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vega N.M., Allison K.R., Khalil A.S., Collins J.J. Signaling-mediated bacterial persister formation. Nat. Chem. Biol. 2012;8:431–433. doi: 10.1038/nchembio.915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wakamoto Y., Dhar N., Chait R., Schneider K., Signorino-Gelo F., Leibler S., McKinney J.D. Dynamic persistence of antibiotic-stressed Mycobacteria. Science. 2013;339:91–95. doi: 10.1126/science.1229858. [DOI] [PubMed] [Google Scholar]
- Walsh T.R., Weeks J., Livermore D.M., Toleman M.A. Dissemination of NDM-1 positive bacteria in the New Delhi environment and its implications for human health: an environmental point prevalence study. Lancet Infect. Dis. 2011;11:355–362. doi: 10.1016/S1473-3099(11)70059-7. [DOI] [PubMed] [Google Scholar]
- Watts N., Adger W.N., Ayeb-Karlsson S., Bai Y., Byass P., Campbell-Lendrum D., Colbourn T., Cox P., Davies M., Depledge M. The Lancet Countdown: tracking progress on health and climate change. Lancet. 2017;389:1151–1164. doi: 10.1016/S0140-6736(16)32124-9. [DOI] [PubMed] [Google Scholar]
- Weber H., Polen T., Heuveling J., Wendisch V.F., Hengge R. Genome-wide analysis of the general stress response network in Escherichia coli: sigmaS-dependent genes, promoters, and sigma factor selectivity. J. Bacteriol. 2005;187:1591–1603. doi: 10.1128/JB.187.5.1591-1603.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Webster N.S., Reusch T.B.H. Microbial contributions to the persistence of coral reefs. ISME J. 2017;11:2167–2174. doi: 10.1038/ismej.2017.66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiuff C., Zappala R.M., Regoes R.R., Garner K.N., Baquero F., Levin B.R. Phenotypic tolerance: antibiotic enrichment of noninherited resistance in bacterial populations. Antimicrob. Agents Chemother. 2005;49:1483–1494. doi: 10.1128/AAC.49.4.1483-1494.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wood T.K., Knabel S.J., Kwan B.W. Bacterial persister cell formation and dormancy. Appl. Environ. Microbiol. 2013;79:7116–7121. doi: 10.1128/AEM.02636-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- World Health Organization (2019). Global Tuberculosis Report 2019. ISBN 978-92-4-156571-4.
- Yamanaka K. Cold shock response in Escherichia coli. J. Mol. Microbiol. Biotechnol. 1999;1:193–202. [PubMed] [Google Scholar]
- Yeh P., Tschumi A.I., Kishony R. Functional classification of drugs by properties of their pairwise interactions. Nat. Genet. 2006;38:489–494. doi: 10.1038/ng1755. [DOI] [PubMed] [Google Scholar]
- Yura T., Tobe T., Ito K., Osawa T. Heat shock regulatory gene (htpR) of Escherichia coli is required for growth at high temperature but is dispensable at low temperature. Proc. Natl. Acad. Sci. U S A. 1984;81:6803–6807. doi: 10.1073/pnas.81.21.6803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao K., Liu M., Burgess R.R. The global transcriptional response of Escherichia coli to induced sigma 32 protein involves sigma 32 regulon activation followed by inactivation and degradation of sigma 32 in vivo. J. Biol. Chem. 2005;280:17758–17768. doi: 10.1074/jbc.M500393200. [DOI] [PubMed] [Google Scholar]
- Ziegler M., Seneca F.O., Yum L.K., Palumbi S.R., Voolstra C.R. Bacterial community dynamics are linked to patterns of coral heat tolerance. Nat. Commun. 2017;8:14213. doi: 10.1038/ncomms14213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zwietering M.H., de Koos J.T., Hasenack B.E., de Witt J.C., van’t Riet K. Modeling of bacterial growth as a function of temperature. Appl. Environ. Microbiol. 1991;57:1094–1101. doi: 10.1128/aem.57.4.1094-1101.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]