

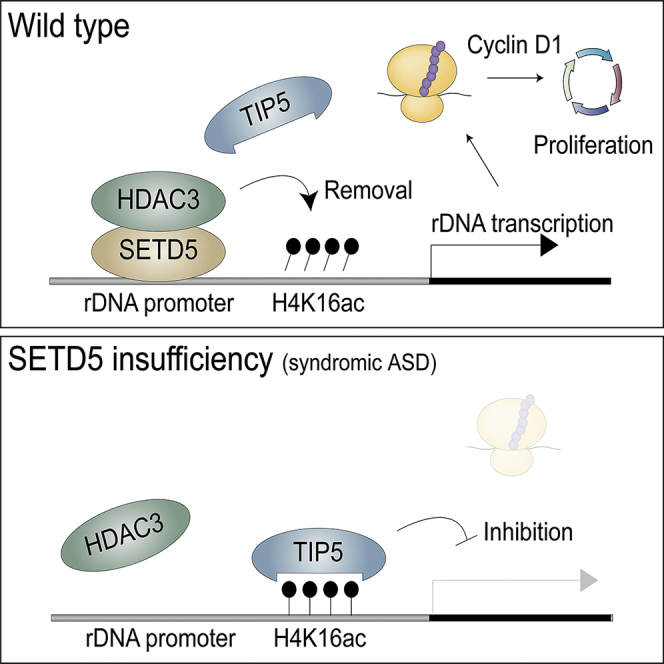
Summary
Haploinsufficiency of SETD5 is implicated in syndromic autism spectrum disorder (ASD), but the molecular mechanism underlying the pathological role of this protein has remained unclear. We have now shown that Setd5+/– mice manifest ASD-related behavioral phenotypes and that the expression of ribosomal protein genes and rDNA is disturbed in the brain of these mice. SETD5 recruited the HDAC3 complex to the rDNA promoter, resulting in removal of the histone mark H4K16ac and its reader protein TIP5, a repressor of rDNA expression. Depletion of SETD5 attenuated rDNA expression, translational activity, and neural cell proliferation, whereas ablation of TIP5 in SETD5-deficient cells rescued these effects. Translation of cyclin D1 mRNA was specifically down-regulated in SETD5-insufficient cells. Our results thus suggest that SETD5 positively regulates rDNA expression via an HDAC3-mediated epigenetic mechanism and that such regulation is essential for translation of cyclin D1 mRNA and neural cell proliferation.
Subject Areas: Behavior Genetics, Molecular Genetics, Behavioral Neuroscience, Molecular Neuroscience
Graphical Abstract
Highlights
-
•
Setd5+/– mice manifest syndromic autism-related phenotypes
-
•
SETD5 recruits the HDAC3 complex to the rDNA promoter
-
•
SETD5 deficiency reduces rRNA abundance and attenuates translational activity
-
•
SETD5 deficiency inhibits cyclin D1 mRNA translation and neural cell proliferation
Behavior Genetics; Molecular Genetics; Behavioral Neuroscience; Molecular Neuroscience
Introduction
Autism spectrum disorder (ASD) is a neurodevelopmental disorder that affects nearly 1% of the human population and is characterized by impairment of social communication and restricted and repetitive behaviors (de la Torre-Ubieta et al., 2016). ASD has been classified into syndromic and nonsyndromic types, with patients with syndromic type manifesting additional phenotypes such as intellectual disability (ID), attention-deficit/hyperactivity disorder, epilepsy, and craniofacial dysmorphology. Although most cases of nonsyndromic ASD are thought to be polygenic in etiology (Sztainberg and Zoghbi, 2016), most cases of syndromic ASD appear to be caused by chromosomal abnormalities, submicroscopic copy number variations, or single-gene mutations, thus providing an opportunity to investigate the molecular mechanisms underlying ASD pathophysiology with the use of targeted gene modification in cellular and animal models. Indeed, ASD-related mutations have been found to impair a variety of cellular processes including neurogenesis, neurite growth, and synaptic plasticity (Gilbert and Man, 2017). Accumulating evidence indicates that three major cellular activities are commonly affected by ASD-related gene mutations: protein translation, WNT signaling, and synaptic signaling (de la Torre-Ubieta et al., 2016).
De novo heterozygous deletion of the gene for SET domain-containing 5 (SETD5) has recently been identified in patients with ID and ASD phenotypes (Fernandes et al., 2018, Green et al., 2017, Grozeva et al., 2014, Kuechler et al., 2015, Pinto et al., 2014, Powis et al., 2018, Stur et al., 2017, Szczaluba et al., 2016). SETD5 is a ubiquitously expressed protein (Osipovich et al., 2016) that belongs to the SET domain-containing protein family (Herz et al., 2013). The SET domains of most proteins catalyze protein lysine methylation, but those of SETD5 and its paralog MLL5 lack methyltransferase activity as a result of amino acid substitutions at several critical positions (Deliu et al., 2018, Madan et al., 2009, Mas et al., 2016). Instead, SETD5 was recently shown to bind to two chromatin-regulating complexes—the polymerase-associated factor 1 (PAF1) and histone deacetylase 3 (HDAC3) complexes (Deliu et al., 2018, Osipovich et al., 2016, Yu et al., 2017)—suggesting that SETD5 contributes to epigenetic regulation and control of gene expression through its association with these complexes. Importantly, heterozygous loss-of-function mutations in genes encoding several components of the HDAC3 complex have been identified in individuals with ASD or ID (O'Roak et al., 2012, Pons et al., 2015, Sirmaci et al., 2011), suggestive of a functional link between SETD5 and the HDAC3 complex in the pathogenesis of ASD and ID. However, whether or how SETD5 regulates gene expression related to ASD and ID has remained unclear.
We have now subjected Setd5+/– mice to comprehensive behavioral analysis and thereby shown that these animals manifest phenotypes similar to those of patients with ASD and ID. Transcriptomics analysis of the brain of Setd5+/– mice revealed impairment of the expression of rDNA as well as of ribosomal protein genes. Given that rRNA, which is encoded by rDNA, is also a key structural component of ribosomes, we analyzed the regulation of rDNA expression by SETD5 with the use of neuroblastoma cell lines. We found that SETD5 binds to the rDNA promoter and thereby recruits HDAC3, which reduces the amount of the Lys16-acetylated form of histone 4 (H4K16ac) and thereby triggers the dissociation of TIP5 and promotes rDNA expression. We also found that depletion of rRNA induced by SETD5 deficiency results in attenuation of cell proliferation in cultured neuroblastoma cells, and a reduced extent of cell proliferation was also detected in the brain of Setd5+/– mouse embryos in vivo as well as in adult neural stem cells of these mice in vitro. In addition, translation of cyclin D1 mRNA was found to be specifically down-regulated in SETD5-deficient cells. Together, our findings reveal that SETD5 plays a key role in neural cell proliferation through regulation of rDNA expression at the epigenetic level.
Results
Setd5+/– Mice Manifest Syndromic ASD Phenotypes
To examine the consequences of heterozygous deletion of Setd5 in vivo, we obtained Setd5+/– mice generated by the International Mouse Phenotyping Consortium (Figure S1) and determined whether these animals manifest phenotypes similar to those of humans with syndromic ASD. Consistent with a previous study (Deliu et al., 2018), Setd5+/– mice were smaller than their wild-type (WT) littermates (Figure S2A). In contrast, we did not observe any significant difference between Setd5+/– mice and WT littermates with regard to body temperature (Figure S2B), grip strength and balance in the wire-hang test (Figure S2C), or performance in the light-dark transition test (Figure S2D), the open-field test (Figure S2E), the elevated plus-maze test (Figure S2F), the Porsolt forced-swim test (Figure S2G), and the tail-suspension test (Figure S2H), revealing that Setd5+/– mice do not manifest anxiety- or depression-related behaviors.
We then performed social behavioral tests with Setd5+/– mice. In the neonatal ultrasonic vocalization test, which assesses early communicative behavior of pups toward the mother (Scattoni et al., 2009), Setd5+/– mice emitted substantially fewer ultrasonic vocalizations than did WT littermates (Figure 1A). In the social-interaction test in a novel environment, there was no difference between genotypes in the total number of contacts (47 ± 2 and 53 ± 4 events for WT and Setd5+/– mice, respectively, p = 0.271), but the mean duration per contact was significantly increased and the total duration of contacts tended to be increased (p = 0.054) in Setd5+/– mice compared with WT littermates (Figure 1B). Setd5+/– mice and WT mice performed similarly in three-chamber sociability and social preference tests (Figure S3).
Figure 1.

Behavioral Abnormalities of Setd5+/– Mice
(A) Social communication of neonatal Setd5+/+ or Setd5+/– mice assessed by the ultrasonic vocalization test at postnatal day 6. Call number over 5 min was determined. Data are means ± SEM (n = 30, Setd5+/+; n = 25, Setd5+/–). ∗p < 0.05 (rank-sum test).
(B) Social interaction of adult mice assessed in a novel environment. Two mice of the same genotype and sex that had been housed in different cages were placed together in a box. Mean duration time per contact and total duration time of contacts were determined. Data are means ± SEM (n = 8 for each genotype). ∗p < 0.05 by one-way analysis of variance (ANOVA).
(C) Association memory formation in adult mice assessed by the cued fear-conditioning test. A conditioned stimulus of sound (55-dB white noise for 30 s) and aversive unconditioned stimulus of foot shock (0.3 mA for 2 s) were given during the indicated periods. The percentage time for freezing behavior in consecutive blocks of 1 min during the conditioning phase (left panel) and cued testing on the following day (right panel) was determined. Data are means ± SEM (n = 17 for each genotype). ∗p < 0.05, ∗∗p < 0.01 (two-way repeated-measures ANOVA).
(D) Association memory retention in adult mice assessed by the cued fear-conditioning test. Cued testing as in (C) was performed 1 month after the last conditioning. Data are means ± SEM (n = 17 for each genotype). ∗p < 0.05, ∗∗p < 0.01 (two-way repeated-measures ANOVA).
(E) Activity level of adult mice assessed by monitoring of the home cage over 24 h. Two mice of the same genotype and sex that had been housed in different cages were placed together in a box, and their activity was continuously monitored over 24 h. Data for 7 days are shown compressed and are means ± SEM (n = 8 per genotype). ∗p < 0.05, ∗∗p < 0.01 (two-way repeated-measures ANOVA).
See also Figures S1–S4 as well as Table S1.
To assess memory performance, we subjected mice to a cued fear-conditioning test. During the training phase, Setd5+/– mice showed reduced freezing behavior compared with WT littermates (Figure 1C), and this difference was maintained in the subsequent test phase (Figure 1D). In the Barnes maze test, Setd5+/– mice took more time to reach the target hole than did the control mice (Figures S4A and S4D). The number of errors committed in reaching the target hole as well as memory accuracy did not differ significantly between Setd5+/– and WT littermates (Figures S4B, S4C, S4E, and S4F). Setd5+/– mice also made similar numbers of correct responses as did WT littermates in the T-maze spontaneous alternation task (Figure S4G and S4H). Finally, we monitored animals for 24 h in their home cage and found that Setd5+/– mice were hyperactive in both light and dark periods compared with control mice (Figure 1E). Together, these data revealed that Setd5+/– mice manifest phenotypes similar to those of syndromic ASD (Table S1).
Impaired Expression of Ribosome-Related Genes in the Setd5+/– Mouse Brain
We performed RNA-sequencing (RNA-seq) analysis of the brain of Setd5+/– and WT mice to examine the effects of reduced abundance of SETD5 on the transcriptome. Of a total of 12,333 protein-coding genes with an FPKM (fragments per kilobase of exon model per million mapped fragments) value of >1 in the WT brain, the expression of 10 genes (Pmch, Cox5b, Rpl30, Ndufs5, Tfap2d, Cox8b, Chrna6, Slc6a3, Gm7541, Rps8) was found to be up-regulated, whereas that of 20 genes (Slc6a4, Atp5l, Tph2, Tuba1c, Rpl17, Rpl36, Rps10, Rpl23a, Amd2, Rps29, Slc18a3, Rpl18, Rpl37a, Cyr61, Rln3, Crabp1, Eno1, Prph, Npas4, Rps7) was down-regulated in the Setd5+/– brain with a cutoff of a 2-fold change (Figure 2A, Table S2). We then conducted Gene Ontology (GO) analysis of differentially expressed genes with the use of the PANTHER overrepresentation test (Mi et al., 2013), which revealed that genes related to transcription were enriched among those whose expression was increased in the Setd5+/– mouse brain (Figure S5A), whereas genes for ribosomal proteins were enriched among those whose expression was decreased (Figure 2B). The differences in expression of ribosomal protein genes between the two genotypes were validated by reverse transcription (RT) and quantitative polymerase chain reaction (qPCR) analysis (Figure S5B). Given that ribosomal protein expression and ribosome assembly are strongly correlated with rRNA levels (Klinge and Woolford, 2019, Xiao and Grove, 2009), we examined the abundance of rRNA, which was excluded from the RNA-seq analysis as a result of its lack of a poly(A) tail. With the use of primer sets for the primary transcript of rDNA (47S rRNA) and a processed product (18S rRNA) (Chang et al., 2009, Corsini et al., 2018), we detected a significant reduction in the amounts of both rRNA species in the Setd5+/– mouse brain compared with the WT brain (Figure 2C), implicating SETD5 in the transcription of rDNA.
Figure 2.

Impaired Ribosome-Related Gene Expression in the Brain of Setd5+/– Mice
(A) Scatterplot of gene expression levels in the brain of adult Setd5+/+ and Setd5+/– mice (n = 1 for each genotype) determined by RNA-seq analysis. Only genes with an FPKM value >1 in the Setd5+/+ mouse brain (12,333 genes) are shown and were subjected to GO analysis.
(B) GO analysis of molecular function and cellular component for the 617 (5%) genes showing the highest level of down-regulation in the Setd5+/– mouse brain as performed with the PANTHER overrepresentation test (Mi et al., 2013). FDR, false discovery rate.
(C) Schematic representation of rRNA maturation (ETS, external transcribed spacer) as well as RT-qPCR analysis of 47S and 18S rRNA abundance in adult mouse brain. Data are means ± SEM (n = 3 for each genotype). ∗p < 0.05, ∗∗p < 0.01 (Student's t test).
SETD5 Binds to HDAC3 and PAF1 Complexes
To elucidate the molecular mechanism by which SETD5 might regulate the expression of ribosomal protein genes and rDNA, we investigated which proteins associate with SETD5. We thus expressed 3×FLAG-tagged human SETD5 in HEK293T cells, subjected cell lysates to immunoprecipitation with antibodies to FLAG, and analyzed the precipitates by liquid chromatography and tandem mass spectrometry (LC-MS/MS). Consistent with previous findings (Deliu et al., 2018, Osipovich et al., 2016, Yu et al., 2017), we detected components of the HDAC3 complex (NCoR1/2, KDM4A, MTA2, HDAC3, TBL1) and of the PAF1 complex (CDC73, WDR61) in the 3×FLAG-SETD5 immunoprecipitates (Figures 3A and S6A, and Table S3). LC-MS/MS analysis of similar immunoprecipitates prepared from SH-SY5Y cells expressing 3×FLAG-SETD5 confirmed the coprecipitation of components of the HDAC3 (NCoR1/2, TBL1, HDAC3) and PAF1 (CDC73) complexes (Figures 3B and S6B, and Table S3). Gel filtration of cell lysates prepared from SH-SY5Y cells expressing 3×FLAG-SETD5 revealed that the recombinant protein coeluted with endogenous HDAC3 and PAF1 in fractions corresponding to a molecular size of >669 kDa (Figure 3C), indicating that most 3×FLAG-SETD5 was likely present in HDAC3 and PAF1 complexes. Immunoprecipitation analysis with a series of deletion mutants of SETD5 showed that HDAC3 bound to the NH2-terminal portion of SETD5 comprising amino acids 1 to 972, but not to one consisting of residues 1 to 767, suggesting that the region of SETD5 spanning amino acids 768 to 972 is responsible for binding to the HDAC3 complex (Figure 3D). PAF1 bound to full-length SETD5, but not to any of the deletion mutants tested, suggesting that PAF1 binds to the extreme COOH-terminal region of SETD5 (amino acids 1286–1442) (Figure 3D). Importantly, the NH2-terminal fragments of SETD5 examined in this immunoprecipitation analysis were selected on the basis of SETD5 mutations—including nonsense (R445X, R768X, S973X) and frameshift (S1286Lfs∗84) mutations—identified in patients with ASD or ID. Patients harboring R445X or R768X nonsense mutations were reported to exhibit severe syndromic ASD phenotypes (Grozeva et al., 2014, Kuechler et al., 2015), whereas those with the S973X nonsense mutation or the S1286Lfs∗84 frameshift mutation were reported to show only mild motor defects and ID without ASD or other comorbidities (Stur et al., 2017, Szczaluba et al., 2016), suggesting that loss of HDAC3 binding is critical for the pathogenesis of syndromic ASD caused by SETD5 mutations.
Figure 3.

Association of SETD5 with HDAC3 and PAF1 Complex Components
(A) Silver staining of an SDS-PAGE gel loaded with an immunoprecipitate of 3×FLAG-tagged human SETD5 expressed in HEK293T cells. An immunoprecipitate prepared from cells transfected with the corresponding empty vector served as a control. Proteins identified by LC-MS/MS analysis are indicated.
(B) Silver staining of an SDS-PAGE gel loaded with an immunoprecipitate of 3×FLAG-SETD5 expressed in SH-SY5Y cells with the use of the doxycyline-inducible system. An immunoprecipitate prepared from corresponding cells not treated with doxycycline served as a control. Proteins identified by LC-MS/MS analysis are indicated.
(C) Immunoblot (IB) analysis of the indicated proteins in fractions obtained by gel filtration of lysates of SH-SY5Y cells expressing 3×FLAG-SETD5.
(D) Lysates of HEK293T cells expressing full-length 3×FLAG-SETD5 or the indicated deletion mutants thereof (or transfected with the corresponding empty vector) were subjected to immunoprecipitation (IP) with antibodies to FLAG, and the resulting precipitates as well as the original cell lysates were subjected to immunoblot analysis with antibodies to the indicated proteins.
Recruitment of HDAC3 to the rDNA Promoter by SETD5
We next generated Neuro2a mouse neuroblastoma cells that lack SETD5 with the use of the CRISPR/Cas9 system (Figures 4A and S7A). Consistent with data obtained with the Setd5+/– mouse brain, loss of SETD5 resulted in down-regulation of the expression of rDNA (Figure 4B). With the use of antibodies to SETD5 whose specificity was validated with the Setd5 knockout (KO) cells (Figure S7B), we detected the apparent presence of SETD5 in the nucleolus (Figure 4C), the site of rDNA transcription (Boisvert et al., 2007). To examine whether SETD5 binds to rDNA, we engineered the Setd5 KO cells to express hemagglutinin (HA)-epitope-tagged full-length (FL) or N767 mutant (amino acids 1–767) forms of SETD5 (Figure 4D) and then subjected the cells to chromatin immunoprecipitation (ChIP) with antibodies to HA followed by qPCR analysis with primers targeted to the rDNA gene body or its promoter region. Of note, expression of SETD5(FL) rescued the expression of rDNA in the Setd5 KO cells (Figure 4E), excluding the possibility of an off-target effect of the KO procedure on the expression of rDNA. We detected the binding of SETD5(FL) to the rDNA promoter (Figure 4F), but not to the gene body (Figure S7C). In contrast, SETD5(N767) did not show any binding to these genomic regions (Figures 4F and S7C) and also did not restore the expression of rDNA in the Setd5 KO cells (Figure 4E), indicating that the association of SETD5 with the rDNA promoter is mediated by the COOH-terminal portion of the protein comprising amino acids 768 to 1442 and results in the production of rRNA. To examine whether HDAC3 also binds to rDNA, we introduced the DNA sequence encoding the 3×FLAG tag into the endogenous Hdac3 locus (HDAC3-3×FLAG KI) in both control and Setd5 KO Neuro2a cells (Figure 4G) and performed ChIP with antibodies to FLAG followed by qPCR analysis. HDAC3 was found to bind to the rDNA promoter (Figure 4H), but not to the gene body (Figure S7D), in the control (SETD5-expressing) cells. However, this binding was not detected in the Setd5 KO cells (Figure 4H), indicating that SETD5 recruits HDAC3 to the rDNA promoter.
Figure 4.

Recruitment of HDAC3 to the rDNA Promoter by SETD5
(A) Immunoblot analysis of SETD5 and DDB1 (loading control) in control (WT) and Setd5 KO Neuro2a cells. Asterisks indicate nonspecific signals.
(B) RT-qPCR analysis of 47S and 18S rRNA in WT and Setd5 KO Neuro2a cells. Data are means ± SEM (n = 3 independent experiments). ∗p < 0.05, ∗∗p < 0.01 (Student's t test).
(C) Immunofluorescence analysis of SETD5 and UBF (nucleolar marker) in Neuro2a cells. Nuclei were stained with 4′,6-diamidino-2-phenylindole (DAPI). The boxed regions in the upper panels are shown at higher magnification in the lower panels. Scale bar, 10 μm.
(D) Immunoblot analysis of WT or Setd5 KO Neuro2a cells as well as of KO cells transfected with expression vectors for HA-tagged FL or N767 mutant forms of SETD5 (or with the empty vector, Vec). Tubulin served as a loading control.
(E) RT-qPCR analysis of 47S rRNA in Setd5 KO Neuro2a cells complemented (Comp) with FL or N767 mutant forms of SETD5 as in (D). Data are means ± SEM (n = 4 independent experiments). ∗p < 0.05 (one-way ANOVA followed by Tukey's test).
(F) ChIP-qPCR analysis of HA-SETD5 binding to the rDNA promoter in Setd5 KO Neuro2a cells complemented with FL or N767 forms of HA-SETD5 as in (D). ChIP was performed with antibodies to HA and with control immunoglobulin G (IgG). Data are means ± SEM (n = 4 independent experiments). ∗∗p < 0.01 (one-way ANOVA followed by Tukey's test).
(G) Immunoblot analysis of WT or Setd5 KO Neuro2a cells as well as of WT or KO cells modified by knockin of the DNA sequence encoding the 3×FLAG tag into the Hdac3 locus (HDAC-3×FLAG KI).
(H) ChIP-qPCR analysis of endogenous HDAC3-3×FLAG binding to the rDNA promoter in WT or Setd5 KO Neuro2a cells engineered as in (G). ChIP was performed with antibodies to FLAG and with control IgG. Data are means ± SEM (n = 3 independent experiments). ∗p < 0.05 (one-way ANOVA followed by Tukey's test).
See also Figures S7 and S10.
TIP5 Mediates Repression of rDNA in Setd5 KO Cells
ChIP-qPCR analysis revealed that the level of the HDAC3 substrate H4K16ac (Johnson et al., 2002) at the rDNA promoter, but not that at the gene body, was increased in Setd5 KO cells compared with control cells (Figures 5A and S8A). TTF-I-interacting protein 5 (TIP5) was previously shown to recognize H4K16ac and thereby to recruit transcriptional repressor proteins to silence rDNA (Sharifi and Bierhoff, 2018). Consistent with these previous findings, we found that the amount of TIP5 bound to the rDNA promoter was also increased in the Setd5 KO cells (Figure 5B). The amount of TIP5 bound to the 5′ external transcribed spacer of rDNA, but not that bound at downstream regions, was also increased in the KO cells (Figure S8B); this increase might be mediated by a mechanism independent of H4K16ac, possibly involving transcription termination factor-I (TTF-I) and long noncoding RNA (Sharifi and Bierhoff, 2018). Transcription of rDNA is silenced by the TIP5-containing nucleolar remodeling complex by establishment of heterochromatin-like features—including DNA methylation, hypoacetylation of histone 4 (mediated by HDAC1), and methylation (me) of H3K9—at the rDNA promoter (Santoro et al., 2002, Zhou et al., 2002). Consistent with these previous findings, we detected increased levels of DNA methylation (Figure S8C) and H3K9me3 (Figure S8D) at the rDNA promoter in the SETD5-deficient cells. In contrast, we found that the level of pan-acetylated histone 4 (K5+K8+K12) was increased, as was that of H4K16ac, whereas the level of pan-acetylated histone 3 (K9+K14 + K18 + K23 + K27) was reduced, in Setd5 KO cells (Figure S8E). We speculate that TIP5 bound to H4K16ac in the Setd5 KO cells might interfere with deacetylation of histone 4, but not with that of histone 3, or that deacetylation of histone 4 is specifically catalyzed by HDAC3 recruited by SETD5 rather than by HDAC1 bound to TIP5. SETD5 was recently found to possess intrinsic histone methyltransferase activity for H3K36 (Sessa et al., 2019). However, we did not detect a reduction in the level of H3K36me3 at the rDNA promoter in Setd5 KO cells (Figure S8D). Given that KDM4A, which we found to be associated with SETD5 (Figure 3A), possesses H3K36 demethylase activity (Whetstine et al., 2006), it is possible that loss of SETD5 results in the simultaneous loss of KDM4A from the rDNA promoter and the consequent inability to remove associated H3K36me3.
Figure 5.

Essential Role of TIP5 in Repression of rDNA in Setd5 KO Neuro2a Cells
(A–C) ChIP-qPCR analysis of H4K16ac (A), TIP5 (B), or RPA194 (C) at the rDNA promoter in WT or Setd5 KO Neuro2a cells. Data are means ± SEM (n = 4 independent experiments). ∗p < 0.05, ∗∗p < 0.01 (one-way ANOVA followed by Tukey's test).
(D) Immunoblot analysis of TIP5 in Setd5 KO Neuro2a cells with (KO1 and KO2) or without (WT) additional KO of TIP5.
(E) RT-qPCR analysis of 47S and 18S rRNA in Setd5 KO Neuro2a cells with or without TIP5 KO. Data are means ± SEM (n = 4 independent experiments). ∗∗p < 0.01 (one-way ANOVA followed by Tukey's test).
(F and G) Immunoblot analysis of translational activity on the basis of puromycin incorporation into newly synthesized proteins in WT or Setd5 KO Neuro2a cells (F) or in Setd5 KO Neuro2a cells with or without TIP5 KO (G).
(H and I) Immunofluorescence analysis (left) of bromodeoxyuridine (BrdU) incorporation in WT or Setd5 KO Neuro2a cells (H) or in Setd5 KO Neuro2a cells with or without TIP5 KO (I). Nuclei were stained with DAPI. Scale bars, 20 μm. The percentage of BrdU-positive cells was also determined for each condition (right). Data are means ± SEM (n = 3 independent experiments). ∗∗p < 0.01 by Student's t test (H) or one-way ANOVA followed by Tukey's test (I).
See also Figures S8–S10.
To examine further the molecular mechanism of rDNA repression in Setd5 KO cells, we performed ChIP-qPCR analysis with antibodies to RPA194 to measure the binding of RNA polymerase I (Pol I). The amount of Pol I binding throughout the promoter and gene body of rDNA was reduced in the absence of SETD5 (Figures 5C and S8F), suggesting that SETD5 regulates rDNA transcription by promoting the recruitment of Pol I, rather than by an effect on Pol I translocation at the rDNA promoter.
To validate the role of TIP5 in repression of rDNA in Setd5 KO cells, we knocked out Tip5 (also known as Baz2a) in Setd5 KO Neuro2a cells with the use of the CRISPR/Cas9 system (Figures 5D and S9A). Such KO of TIP5 restored the expression of rDNA in the Setd5 KO cells (Figure 5E).
The reduction in the amount of rRNA we found to be associated with SETD5 deficiency might have been expected to impair translational activity. Consistent with this notion, we found that the extent of puromycin incorporation into newly synthesized peptides, a measure of translational activity (Schmidt et al., 2009), was reduced in Setd5 KO cells (Figures 5F and S9B) and that this effect was reversed by ablation of TIP5 (Figures 5G and S9C). In addition, the proliferation rate of Setd5 KO cells was reduced compared with that of WT cells (Figure 5H), and again this effect was reversed by TIP5 KO (Figure 5I). Of note, Tip5 KO cells on the WT background (Figures S9D and S9E) did not exhibit any increase in rDNA expression (Figure S9F) or in cell proliferation (Figure S9G), indicating that repression of rDNA expression by TIP5 is minimal in WT cells. Together, these results revealed that SETD5 recruits HDAC3 to the rDNA promoter and thereby reduces the levels of H4K16ac and its reader TIP5, resulting in up-regulation of rRNA abundance, translational activity, and cell proliferation, in Neuro2a cells.
Depletion of SETD5 Impairs rDNA Expression, Translational Activity, and Cell Proliferation in SH-SY5Y Cells
To confirm that the observed effects of SETD5 loss were not restricted to mouse cells, we depleted SH-SY5Y human neuroblastoma cells of SETD5 by RNA interference (Figure S10A). The expression of rDNA (Figure S10B), translational activity (Figure S10C), and cell proliferation (Figure S10D) were all attenuated in SH-SY5Y cells depleted of SETD5.
SETD5 Haploinsufficiency Impairs Proliferation of Neural Stem/Progenitor Cells
We next investigated the effect of SETD5 haploinsufficiency on neural cell proliferation in vivo. Immunoblot analysis revealed that the abundance of SETD5 was markedly greater in the embryonic mouse brain than in the neonatal brain (Figure 6A). Immunofluorescence staining of the brain at embryonic day (E) 15 showed the presence of SETD5 in the nucleus of SOX2-positive neural stem/progenitor cells as well as in that of more differentiated cells located outside of the ventricular zone (Figure 6B), indicating that the expression of SETD5 begins early in the differentiation process of neural stem/progenitor cells and continues in differentiated neuronal cells. Immunostaining of the E15 brain with antibodies to Ki67, a marker of cell proliferation, revealed that the proportion of Ki67-positive cells was reduced in Setd5+/– mice compared with WT littermates (Figure 6C), indicative of impairment of cell proliferation by SETD5 haploinsufficiency. To determine whether SETD5 also regulates proliferation of adult neural stem cells, we performed a neurosphere formation assay with cells derived from the adult Setd5+/– mouse brain. The brain of Setd5+/– mice contained fewer neurosphere-initiating cells than did the WT brain (Figure 6D). Furthermore, the extent of rDNA expression was significantly reduced in neurospheres formed by Setd5+/– cells compared with those formed by WT cells (Figure 6E). Together, these results thus indicated that SETD5 positively regulates the proliferation of neural stem/progenitor cells in both the embryonic and adult mouse brain.
Figure 6.

Impaired Proliferation of Neural Stem/Progenitor Cells in the Embryonic and Adult Brain of Setd5+/– Mice
(A) Immunoblot analysis of SETD5 in mouse brain from embryonic day (E) 10 to postnatal day (P) 3.
(B) Immunohistofluorescence staining of SOX2 and SETD5 in a cortical section of the E15 mouse brain. Nuclei were stained with DAPI. The boxed region in the merged image is shown at higher magnification on the right. Scale bar, 20 μm. CP, cortical plate; IZ, intermediate zone; SVZ, subventricular zone; VZ, ventricular zone.
(C) Immuohistofluorescence staining of Ki67 in the ventricular zone of the Setd5+/+ or Setd5+/– mouse brain at E15. Scale bar, 10 μm. The numbers of Ki67-positive cells were determined as mean ± SEM values (n = 5 for each genotype). ∗p < 0.05 (Student's t test).
(D) Phase-contrast microscopic images of neurospheres derived from the brain of adult Setd5+/+ or Setd5+/– mice. Scale bar, 100 μm. The numbers of neurospheres were also determined as means ± SEM (n = 5 for each genotype). ∗p < 0.05 (Student's t test).
(E) RT-qPCR analysis of 47S rRNA in neurospheres derived from the brain of adult Setd5+/+ or Setd5+/– mice. Data are means ± SEM (n = 3 for each genotype). ∗∗p < 0.01 (Student's t test).
Translation of Cyclin D1 mRNA Is Down-regulated in Setd5 KO Cells
Finally, we explored the molecular mechanism by which SETD5 haploinsufficiency impairs cell proliferation. Analysis of cell cycle profile by flow cytometric analysis after staining with 5-bromo-2′-deoxyuridine and propidium iodide showed that the proportion of cells in G1 phase was increased and that in S phase was reduced for Setd5 KO cells compared with WT cells, indicative of G1 arrest in the former cells (Figure 7A). Given that expression of G1-S cyclins is required for G1 to S progression (Liu et al., 2019), we examined the translation of mRNAs encoding such proteins that have been shown to be subject to translational regulation: cyclin D1, cyclin E1, and cyclin E2 (Tarn and Lai, 2011). RT-qPCR analysis of mRNAs associated with polysome and monosome fractions revealed a shift of cyclin D1 mRNA, but not of cyclin E1 and cyclin E2 mRNAs, from polysomes to monosomes in Setd5 KO cells compared with WT cells, suggesting that translation of cyclin D1 mRNA is specifically down-regulated as a result of SETD5 deficiency (Figures 7B–7F). Consistent with these findings, the amount of cyclin D1 protein, but not that of the corresponding mRNA, was reduced, whereas those of cyclin E1 and cyclin E2 proteins and mRNAs were not affected, in Setd5 KO cells (Figures 7G and 7H). We also detected a reduction in the amount of cyclin D1 protein in the E15.5 brain of Setd5+/– mice compared with that of WT mice (Figure 7I). Together, these results indicated that SETD5 insufficiency attenuates the translation of cyclin D1 mRNA and thereby restrains cell proliferation. Given that cyclin D1 KO mice manifest neurological phenotypes (Sicinski et al., 1995) and that cyclin D1 is implicated in ASD-related protein-protein interaction networks (Neale et al., 2012), we hypothesize that down-regulation of cyclin D1 and the consequent impairment of neural cell proliferation are responsible at least in part for the ASD-like phenotypes of Setd5+/– mice.
Figure 7.

Down-regulation of Cyclin D1 mRNA Translation in Setd5 KO Neuro2a Cells
(A) Flow cytometric analysis of the cell cycle in WT and Setd5 KO Neuro2a cells stained with bromodeoxyuridine (BrdU) and propidium iodide (PI). Representative profiles as well as mean ± SEM values for the proportion of cells in each phase of the cell cycle (n = 6 independent experiments) are shown. ∗p < 0.05 (Student's t test).
(B) Ribosome fractionation by sucrose gradient centrifugation for WT and Setd5 KO Neuro2a cells as revealed by measurement of absorbance at 254 nm (A254). Fractions 9 and 10 were assayed as 80S monosomes, and fractions 11 to 17, as polysomes.
(C–E) Representative RT-qPCR analysis of mRNAs for cyclin D1 (C), cyclin E1 (D), and cyclin E2 (E) in ribosome fractions of WT and Setd5 KO Neuro2a cells.
(F) The polysome/monosome ratio for cyclin D1, cyclin E1, and cyclin E2 mRNAs determined as in (C–E). Data are means ± SEM (n = 2 independent experiments).
(G) RT-qPCR analysis of mRNAs for cyclin D1, cyclin E1, and cyclin E2 in WT and Setd5 KO Neuro2a cells. Data are means ± SEM (n = 3 independent experiments).
(H) Immunoblot analysis of cyclin D1, cyclin E1, and cyclin E2 in WT and Setd5 KO Neuro2a cells. DDB1 served as a loading control. Representative blots as well as quantitative data presented as means ± SEM (n = 3 independent experiments) are shown. ∗∗p < 0.01 (Student's t test).
(I) Immunoblot analysis of cyclin D1 in the E15.5 brain of Setd5+/+ and Setd5+/– mice. Representative blots as well as quantitative data presented as means ± SEM (n = 4, Setd5+/+; n = 2, Setd5+/–) are shown.
Discussion
Translation is one of the three major cellular activities shown to be disrupted in patients with ASD (de la Torre-Ubieta et al., 2016). However, whether or how rDNA expression, which is required for ribosome production, is related to syndromic ASD has been unexplored. We have now shown that SETD5 regulates the expression of rDNA at the epigenetic level and that SETD5 haploinsufficiency gives rise to syndromic-ASD-like phenotypes in mice. SETD5 was found to recruit the HDAC3 complex to the rDNA promoter, resulting in the removal of both H4K16ac and its reader TIP5 and a consequent increase in rDNA transcription by Pol I. A reduction in the amount of SETD5 thus resulted in attenuation of rRNA production, translational activity, and cell proliferation.
During the preparation of our manuscript, several studies of Setd5+/– mice, including behavioral analyses, were published (Deliu et al., 2018, Moore et al., 2019, Sessa et al., 2019). Our results are mostly consistent with those of these previous studies showing ASD-related behavioral phenotypes of the mutant mice including impaired social behavior and cognitive function as well as increased locomotor activity (Table S1), although we did not detect increased anxiety-like behavior in the elevated plus-maze test (Figure S2F). Our results on the effect of SETD5 deficiency on cell proliferation also differ from those reported in these previous studies (Deliu et al., 2018, Moore et al., 2019, Sessa et al., 2019). These apparent discrepancies might be due to differences in genetic background of the mice studied, in the methods adopted for disruption of Setd5, in animal housing conditions, in the age of embryos examined, or in the methods applied for measurement of cell proliferation. Further analyses are required to elucidate the reasons for the discrepant findings.
In general, HDACs are thought to function as repressors of transcription (Seto and Yoshida, 2014). In the case of rDNA, HDAC1 has been shown to down-regulate its expression (Sharifi and Bierhoff, 2018). In contrast, our data now indicate that HDAC3—a member of the class I HDAC family that includes HDAC1, HDAC2, and HDAC8—up-regulates rDNA expression. Of note, HDAC3 was previously shown to activate transcription controlled by retinoic acid–responsive elements (Jepsen et al., 2000). In addition, cells derived from HDAC3 KO mice manifest not only up-regulation but also down-regulation of gene expression (Bhaskara et al., 2008). These observations suggest that transcriptional activation by HDAC3 is not limited to rDNA. It will be of interest to determine the genomic contexts in which HDAC3 functions as either a transcriptional activator or a transcriptional repressor. In this regard, histone deacetylation at the rDNA promoter in cells might provide a model system to identify relevant histone substrates of HDAC3 and other class I HDAC family members, given that the attempted identification of such substrates with purified proteins has yielded conflicting and inconsistent results in vitro (Seto and Yoshida, 2014). Of particular interest is our finding that the level of histone 3 acetylation is decreased, whereas that of histone 4 acetylation is increased in SETD5-deficient Neuro2a cells (Figure S8E).
We focused on impaired cell proliferation as a cellular phenotype induced by deficiency of SETD5, given that this phenotype was readily detectable by microscopy during cell culture. Knockdown of SETD5 was recently shown to attenuate the proliferation of a human prostate carcinoma cell line (Zhang et al., 2019), indicating that regulation of cell proliferation by SETD5 might not be specific to neural cells. Our polysome analysis revealed that the translation of cyclin D1 mRNA was down-regulated in SETD5-insufficient cells. It remains unclear how the translation of cyclin D1 mRNA, but not that of cyclin E1 and cyclin E2 mRNAs, is sensitive to the loss of SETD5. Given that mechanistic target of rapamycin complex 1 (mTORC1) (Frost et al., 2009, Holmes et al., 2016) and heterogeneous nuclear ribonucleoprotein A1 (hnRNPA1) (Gao et al., 2017) are both implicated in the regulation of cyclin D1 mRNA translation, it is likely that several mechanisms, which might be regulated by SETD5 independently of translation, may play a role in this selectivity. The findings that cyclin D1 KO mice manifest neurological phenotypes (Sicinski et al., 1995) and that cyclin D1 is implicated in ASD-related protein-protein interaction networks (Neale et al., 2012) suggest the possibility that down-regulation of cyclin D1 and the consequent disruption of neural cell proliferation are at least partially responsible for the ASD phenotypes of Setd5+/– mice.
In addition to cell proliferation, however, SETD5-mediated regulation of translation might be linked to other processes with relevance to ASD, such as synaptic function. In support of this notion, neurons in the brain of Setd5+/– mice were recently found to manifest abnormal synaptic activity (Deliu et al., 2018) and connectivity (Moore et al., 2019). It will be of particular interest to determine the extent to which synaptic dysfunction in Setd5+/– mice is due to impairment of translation.
We found that TIP5 ablation rescued rDNA expression, translational activity, and cell proliferation in Setd5 KO cells. These data suggest the possibility that either a drug that interferes with the binding of TIP5 to H4K16ac at the rDNA promoter or depletion of TIP5 might ameliorate phenotypes associated with SETD5 haploinsufficiency.
Limitations of the Study
Results of this study utilizing cultured neuronal cells might not exactly reflect the in vivo functions of neurons.
Methods
All methods can be found in the accompanying Transparent Methods supplemental file.
Acknowledgments
We thank the Wellcome Trust Sanger Institute Mouse Genetics Project (Sanger MGP) and its funders for providing the mutant mouse line (allele: Setd5tm1a(EUCOMM)Wtsi). Funding and associated primary phenotypic information may be found at www.sanger.ac.uk/mouseportal. We thank T. Kitamura for pMX-puro and Plat-E cells, T. Sato and laboratory members for discussion, and the Biomedical Research Core of Tohoku University Graduate School of Medicine for technical support. This work was funded by Japan Society for the Promotion of Science (JSPS) KAKENHI grants 15K18365, 17K14955, and 19K07837 (to T.N.) and 17H04035 and JP16H06276 (to K.N.); a Grant-in-Aid for Scientific Research on Innovative Areas–Platform for Advanced Technologies and Research Resources (16H06276 to T.M.); and in part by the Joint Usage/Research Center for Genes, Brain, and Behavior supported by the Ministry of Education, Culture, Sports, Science and Technology of Japan.
Author Contributions
T.N. designed and performed most of the experiments and cowrote the manuscript. K.N. directed and coordinated the study, oversaw the results, and cowrote the manuscript. M.N. and Y.N. assisted with experiments. S.H. coordinated and assisted with most of the mouse behavioral analyses. R.N. performed polysome analysis. R.K. performed ultrasonic vocalization tests. M.M. performed LC-MS/MS analysis. R.F. performed transcriptomics analysis. T.I., T.M., N.O., and K.I.N. provided advice. All authors discussed the results and commented on the manuscript.
Declaration of Interests
The authors declare no competing interests.
Published: April 24, 2020
Footnotes
Supplemental Information can be found online at https://doi.org/10.1016/j.isci.2020.101030.
Supplemental Information
Differences in mouse origin, mutations introduced into the Setd5 gene, mouse genetic background, and behavioral data between the present study and the studies of Deliu et al., 2018, Moore et al., 2019, and Sessa et al. (2019) are compared.
FPKM values were calculated from the RNA-seq data with the use of Cufflinks.
Peptides derived from the band slices shown in Figure S6 were identified with the use of the MASCOT data set. 293T, HEK293T cells; SH, SH-SY5Y cells. 293T-n denotes band n of the HEK293T sample, and SH-n indicates band n of the SH-SY5Y sample.
References
- Bhaskara S., Chyla B.J., Amann J.M., Knutson S.K., Cortez D., Sun Z.W., Hiebert S.W. Deletion of histone deacetylase 3 reveals critical roles in S phase progression and DNA damage control. Mol. Cell. 2008;30:61–72. doi: 10.1016/j.molcel.2008.02.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boisvert F.M., van Koningsbruggen S., Navascues J., Lamond A.I. The multifunctional nucleolus. Nat. Rev. Mol. Cell Biol. 2007;8:574–585. doi: 10.1038/nrm2184. [DOI] [PubMed] [Google Scholar]
- Chang T.C., Zeitels L.R., Hwang H.W., Chivukula R.R., Wentzel E.A., Dews M., Jung J., Gao P., Dang C.V., Beer M.A. Lin-28B transactivation is necessary for Myc-mediated let-7 repression and proliferation. Proc. Natl. Acad. Sci. U S A. 2009;106:3384–3389. doi: 10.1073/pnas.0808300106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corsini N.S., Peer A.M., Moeseneder P., Roiuk M., Burkard T.R., Theussl H.C., Moll I., Knoblich J.A. Coordinated control of mRNA and rRNA processing controls embryonic stem cell pluripotency and differentiation. Cell Stem Cell. 2018;22:543–558.e12. doi: 10.1016/j.stem.2018.03.002. [DOI] [PubMed] [Google Scholar]
- de la Torre-Ubieta L., Won H., Stein J.L., Geschwind D.H. Advancing the understanding of autism disease mechanisms through genetics. Nat. Med. 2016;22:345–361. doi: 10.1038/nm.4071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deliu E., Arecco N., Morandell J., Dotter C.P., Contreras X., Girardot C., Kasper E.L., Kozlova A., Kishi K., Chiaradia I. Haploinsufficiency of the intellectual disability gene SETD5 disturbs developmental gene expression and cognition. Nat. Neurosci. 2018;21:1717–1727. doi: 10.1038/s41593-018-0266-2. [DOI] [PubMed] [Google Scholar]
- Fernandes I.R., Cruz A.C.P., Ferrasa A., Phan D., Herai R.H., Muotri A.R. Genetic variations on SETD5 underlying autistic conditions. Dev. Neurobiol. 2018;78:500–518. doi: 10.1002/dneu.22584. [DOI] [PubMed] [Google Scholar]
- Frost P., Shi Y., Hoang B., Gera J., Lichtenstein A. Regulation of D-cyclin translation inhibition in myeloma cells treated with mammalian target of rapamycin inhibitors: rationale for combined treatment with extracellular signal-regulated kinase inhibitors and rapamycin. Mol. Cancer Res. 2009;8:83–93. doi: 10.1158/1535-7163.MCT-08-0254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao G., Dhar S., Bedford M.T. PRMT5 regulates IRES-dependent translation via methylation of hnRNP A1. Nucleic Acids Res. 2017;45:4359–4369. doi: 10.1093/nar/gkw1367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilbert J., Man H.Y. Fundamental elements in autism: from neurogenesis and neurite growth to synaptic plasticity. Front. Cell. Neurosci. 2017;11:359. doi: 10.3389/fncel.2017.00359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Green C., Willoughby J., Study D.D.D., Balasubramanian M. De novo SETD5 loss-of-function variant as a cause for intellectual disability in a 10-year old boy with an aberrant blind ending bronchus. Am. J. Med. Genet. A. 2017;173:3165–3171. doi: 10.1002/ajmg.a.38461. [DOI] [PubMed] [Google Scholar]
- Grozeva D., Carss K., Spasic-Boskovic O., Parker M.J., Archer H., Firth H.V., Park S.M., Canham N., Holder S.E., Wilson M. De novo loss-of-function mutations in SETD5, encoding a methyltransferase in a 3p25 microdeletion syndrome critical region, cause intellectual disability. Am. J. Hum. Genet. 2014;94:618–624. doi: 10.1016/j.ajhg.2014.03.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herz H.M., Garruss A., Shilatifard A. SET for life: biochemical activities and biological functions of SET domain-containing proteins. Trends Biochem. Sci. 2013;38:621–639. doi: 10.1016/j.tibs.2013.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holmes B., Lee J., Landon K.A., Benavides-Serrato A., Bashir T., Jung M.E., Lichtenstein A., Gera J. Mechanistic target of rapamycin (mTOR) inhibition synergizes with reduced internal ribosome entry site (IRES)-mediated translation of cyclin D1 and c-MYC mRNAs to treat glioblastoma. J. Biol. Chem. 2016;291:14146–14159. doi: 10.1074/jbc.M116.726927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jepsen K., Hermanson O., Onami T.M., Gleiberman A.S., Lunyak V., McEvilly R.J., Kurokawa R., Kumar V., Liu F., Seto E. Combinatorial roles of the nuclear receptor corepressor in transcription and development. Cell. 2000;102:753–763. doi: 10.1016/s0092-8674(00)00064-7. [DOI] [PubMed] [Google Scholar]
- Johnson C.A., White D.A., Lavender J.S., O’Neill L.P., Turner B.M. Human class I histone deacetylase complexes show enhanced catalytic activity in the presence of ATP and co-immunoprecipitate with the ATP-dependent chaperone protein Hsp70. J. Biol. Chem. 2002;277:9590–9597. doi: 10.1074/jbc.M107942200. [DOI] [PubMed] [Google Scholar]
- Klinge S., Woolford J.L., Jr. Ribosome assembly coming into focus. Nat. Rev. Mol. Cell Biol. 2019;20:116–131. doi: 10.1038/s41580-018-0078-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuechler A., Zink A.M., Wieland T., Ludecke H.J., Cremer K., Salviati L., Magini P., Najafi K., Zweier C., Czeschik J.C. Loss-of-function variants of SETD5 cause intellectual disability and the core phenotype of microdeletion 3p25.3 syndrome. Eur. J. Hum. Genet. 2015;23:753–760. doi: 10.1038/ejhg.2014.165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu L., Michowski W., Kolodziejczyk A., Sicinski P. The cell cycle in stem cell proliferation, pluripotency and differentiation. Nat. Cell Biol. 2019;21:1060–1067. doi: 10.1038/s41556-019-0384-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madan V., Madan B., Brykczynska U., Zilbermann F., Hogeveen K., Dohner K., Dohner H., Weber O., Blum C., Rodewald H.R. Impaired function of primitive hematopoietic cells in mice lacking the Mixed-Lineage-Leukemia homolog MLL5. Blood. 2009;113:1444–1454. doi: 10.1182/blood-2008-02-142638. [DOI] [PubMed] [Google Scholar]
- Mas Y.M.S., Barbon M., Teyssier C., Demene H., Carvalho J.E., Bird L.E., Lebedev A., Fattori J., Schubert M., Dumas C. The human mixed lineage leukemia 5 (MLL5), a sequentially and structurally divergent SET domain-containing protein with no intrinsic catalytic activity. PLoS One. 2016;11:e0165139. doi: 10.1371/journal.pone.0165139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mi H., Muruganujan A., Thomas P.D. PANTHER in 2013: modeling the evolution of gene function, and other gene attributes, in the context of phylogenetic trees. Nucleic Acids Res. 2013;41:D377–D386. doi: 10.1093/nar/gks1118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moore S.M., Seidman J.S., Ellegood J., Gao R., Savchenko A., Troutman T.D., Abe Y., Stender J., Lee D., Wang S. Setd5 haploinsufficiency alters neuronal network connectivity and leads to autistic-like behaviors in mice. Transl. Psychiatry. 2019;9:24. doi: 10.1038/s41398-018-0344-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neale B.M., Kou Y., Liu L., Ma’ayan A., Samocha K.E., Sabo A., Lin C.F., Stevens C., Wang L.S., Makarov V. Patterns and rates of exonic de novo mutations in autism spectrum disorders. Nature. 2012;485:242–245. doi: 10.1038/nature11011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Roak B.J., Vives L., Fu W., Egertson J.D., Stanaway I.B., Phelps I.G., Carvill G., Kumar A., Lee C., Ankenman K. Multiplex targeted sequencing identifies recurrently mutated genes in autism spectrum disorders. Science. 2012;338:1619–1622. doi: 10.1126/science.1227764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Osipovich A.B., Gangula R., Vianna P.G., Magnuson M.A. Setd5 is essential for mammalian development and the co-transcriptional regulation of histone acetylation. Development. 2016;143:4595–4607. doi: 10.1242/dev.141465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pinto D., Delaby E., Merico D., Barbosa M., Merikangas A., Klei L., Thiruvahindrapuram B., Xu X., Ziman R., Wang Z. Convergence of genes and cellular pathways dysregulated in autism spectrum disorders. Am. J. Hum. Genet. 2014;94:677–694. doi: 10.1016/j.ajhg.2014.03.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pons L., Cordier M.P., Labalme A., Till M., Louvrier C., Schluth-Bolard C., Lesca G., Edery P., Sanlaville D. A new syndrome of intellectual disability with dysmorphism due to TBL1XR1 deletion. Am. J. Med. Genet. A. 2015;167A:164–168. doi: 10.1002/ajmg.a.36759. [DOI] [PubMed] [Google Scholar]
- Powis Z., Farwell Hagman K.D., Mroske C., McWalter K., Cohen J.S., Colombo R., Serretti A., Fatemi A., David K.L., Reynolds J. Expansion and further delineation of the SETD5 phenotype leading to global developmental delay, variable dysmorphic features, and reduced penetrance. Clin. Genet. 2018;93:752–761. doi: 10.1111/cge.13132. [DOI] [PubMed] [Google Scholar]
- Santoro R., Li J., Grummt I. The nucleolar remodeling complex NoRC mediates heterochromatin formation and silencing of ribosomal gene transcription. Nat. Genet. 2002;32:393–396. doi: 10.1038/ng1010. [DOI] [PubMed] [Google Scholar]
- Scattoni M.L., Crawley J., Ricceri L. Ultrasonic vocalizations: a tool for behavioural phenotyping of mouse models of neurodevelopmental disorders. Neurosci. Biobehav. Rev. 2009;33:508–515. doi: 10.1016/j.neubiorev.2008.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt E.K., Clavarino G., Ceppi M., Pierre P. SUnSET, a nonradioactive method to monitor protein synthesis. Nat. Methods. 2009;6:275–277. doi: 10.1038/nmeth.1314. [DOI] [PubMed] [Google Scholar]
- Sessa A., Fagnocchi L., Mastrototaro G., Massimino L., Zaghi M., Indrigo M., Cattaneo S., Martini D., Gabellini C., Pucci C. SETD5 regulates chromatin methylation state and preserves global transcriptional fidelity during brain development and neuronal wiring. Neuron. 2019;104:271–289. doi: 10.1016/j.neuron.2019.07.013. [DOI] [PubMed] [Google Scholar]
- Seto E., Yoshida M. Erasers of histone acetylation: the histone deacetylase enzymes. Cold Spring Harb. Perspect. Biol. 2014;6:a018713. doi: 10.1101/cshperspect.a018713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharifi S., Bierhoff H. Regulation of RNA polymerase I transcription in development, disease, and aging. Annu. Rev. Biochem. 2018;87:51–73. doi: 10.1146/annurev-biochem-062917-012612. [DOI] [PubMed] [Google Scholar]
- Sicinski P., Donaher J.L., Parker S.B., Li T., Fazeli A., Gardner H., Haslam S.Z., Bronson R.T., Elledge S.J., Weinberg R.A. Cyclin D1 provides a link between development and oncogenesis in the retina and breast. Cell. 1995;82:621–630. doi: 10.1016/0092-8674(95)90034-9. [DOI] [PubMed] [Google Scholar]
- Sirmaci A., Spiliopoulos M., Brancati F., Powell E., Duman D., Abrams A., Bademci G., Agolini E., Guo S., Konuk B. Mutations in ANKRD11 cause KBG syndrome, characterized by intellectual disability, skeletal malformations, and macrodontia. Am. J. Hum. Genet. 2011;89:289–294. doi: 10.1016/j.ajhg.2011.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stur E., Soares L.A., Louro I.D. SETD5 gene variant associated with mild intellectual disability—a case report. Genet. Mol. Res. 2017;16:2. doi: 10.4238/gmr16029615. [DOI] [PubMed] [Google Scholar]
- Szczaluba K., Brzezinska M., Kot J., Rydzanicz M., Walczak A., Stawinski P., Werner B., Ploski R. SETD5 loss-of-function mutation as a likely cause of a familial syndromic intellectual disability with variable phenotypic expression. Am. J. Med. Genet. A. 2016;170:2322–2327. doi: 10.1002/ajmg.a.37832. [DOI] [PubMed] [Google Scholar]
- Sztainberg Y., Zoghbi H.Y. Lessons learned from studying syndromic autism spectrum disorders. Nat. Neurosci. 2016;19:1408–1417. doi: 10.1038/nn.4420. [DOI] [PubMed] [Google Scholar]
- Tarn W., Lai M. Translational control of cyclins. Cell Div. 2011;6:5. doi: 10.1186/1747-1028-6-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whetstine J.R., Nottke A., Lan F., Huarte M., Smolikov S., Chen Z., Spooner E., Li E., Zhang G., Colaiacovo M. Reversal of histone lysine trimethylation by the JMJD2 family of histone demethylases. Cell. 2006;125:467–481. doi: 10.1016/j.cell.2006.03.028. [DOI] [PubMed] [Google Scholar]
- Xiao L., Grove A. Coordination of ribosomal protein and ribosomal RNA gene expression in response to TOR signaling. Curr. Genomics. 2009;10:198–205. doi: 10.2174/138920209788185261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu S.E., Kim M.S., Park S.H., Yoo B.C., Kim K.H., Jang Y.K. SET domain-containing protein 5 is required for expression of primordial germ cell specification-associated genes in murine embryonic stem cells. Cell Biochem. Funct. 2017;35:247–253. doi: 10.1002/cbf.3269. [DOI] [PubMed] [Google Scholar]
- Zhang Y., Yan L., Yao W., Chen K., Xu H., Ye Z. Integrated analysis of genetic abnormalities of the histone lysine methyltransferases in prostate cancer. Med. Sci. Monit. 2019;25:193–239. doi: 10.12659/MSM.912294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Y., Santoro R., Grummt I. The chromatin remodeling complex NoRC targets HDAC1 to the ribosomal gene promoter and represses RNA polymerase I transcription. EMBO J. 2002;21:4632–4640. doi: 10.1093/emboj/cdf460. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Differences in mouse origin, mutations introduced into the Setd5 gene, mouse genetic background, and behavioral data between the present study and the studies of Deliu et al., 2018, Moore et al., 2019, and Sessa et al. (2019) are compared.
FPKM values were calculated from the RNA-seq data with the use of Cufflinks.
Peptides derived from the band slices shown in Figure S6 were identified with the use of the MASCOT data set. 293T, HEK293T cells; SH, SH-SY5Y cells. 293T-n denotes band n of the HEK293T sample, and SH-n indicates band n of the SH-SY5Y sample.