

Acetylation is a conserved modification used to regulate a variety of cellular pathways, such as gene expression, protein synthesis, detoxification, and virulence. Acetyltransferase enzymes transfer an acetyl moiety, usually from acetyl coenzyme A (AcCoA), onto a target substrate, thereby modulating activity or stability. Members of the GCN5-N-acetyltransferase (GNAT) protein superfamily are found in all domains of life and are characterized by a core structural domain architecture.
KEYWORDS: GCN5-related acetyltransferases, acetyltransferases, acylation, antibiotic resistance, chemical modification, histone acetylation, metabolic control, toxin-antitoxin
SUMMARY
Acetylation is a conserved modification used to regulate a variety of cellular pathways, such as gene expression, protein synthesis, detoxification, and virulence. Acetyltransferase enzymes transfer an acetyl moiety, usually from acetyl coenzyme A (AcCoA), onto a target substrate, thereby modulating activity or stability. Members of the GCN5-N-acetyltransferase (GNAT) protein superfamily are found in all domains of life and are characterized by a core structural domain architecture. These enzymes can modify primary amines of small molecules or of lysyl residues of proteins. From the initial discovery of antibiotic acetylation, GNATs have been shown to modify a myriad of small-molecule substrates, including tRNAs, polyamines, cell wall components, and other toxins. This review focuses on the literature on small-molecule substrates of GNATs in bacteria, including structural examples, to understand ligand binding and catalysis. Understanding the plethora and versatility of substrates helps frame the role of acetylation within the larger context of bacterial cellular physiology.
INTRODUCTION
Overview of Acetylation
Acetylation as a regulatory mechanism was first reported over 50 years ago, when the acetylation of histone tails was shown to regulate gene expression in eukaryotes (1). Only later did the importance of acetylation in bacteria become clear. Small-molecule acetylation by bacteria was first described in 1965 within the context of antibiotic resistance (2). Since then, our knowledge of the acetylation capabilities of bacteria has expanded to include proteins, polyamines, toxins, tRNAs, and cell wall components (3–7).
Acetylation is a ubiquitous modification, and acetyltransferase enzymes are found in all domains of life (3). The three main classes of acetyltransferases include (i) the GCN5-N-acetyltransferase (GNAT) family (Pfam 00583), (ii) the MYST family (Pfam 01853), and (iii) the p300/CBP family (Pfam 06466). Only the GNAT family of acetyltransferases is found in both eukaryotes and prokaryotes, while the MYST and p300/CBP families are found exclusively in eukaryotes (3). This review focuses on the acetylation of small molecules by bacterial GNATs. Recent reviews on bacterial protein acetylation and other posttranslational modifications can be found in references 8 to 10.
Bacterial GNATs
The GNAT superfamily is one of the largest families of proteins, containing more than 300,000 proteins (11). GNATs share little sequence homology and are instead characterized based on the core GNAT domain. As mentioned above, GNATs can acetylate a wide variety of small molecules. The largest group is the antibiotic-modifying GNATs, especially GNATs modifying aminoglycoside antibiotics. Bacterial GNATs also have roles in detoxification, synthesis, and transcription. In this review, we address each of these topics and describe the GNATs involved. An overview of the GNATs and their targets is provided in Table 1.
TABLE 1.
GNATs and their targets
| GNAT function and GNAT | Major acetylated small-molecule substrate(s) | Reference(s) |
|---|---|---|
| GNATs modifying antibiotics | ||
| AAC(1) | Apramycin, butirosin, lividomycin, and paromomycin | 30, 31 |
| AAC(3) | Gentamicin, tobramycin, and sisomicin | 27, 36, 37 |
| AAC(2′) | Broad range of aminoglycosides with 2′-amino groups (e.g., tobramycin, gentamicin) | 48, 49, 55 |
| AAC(6′) | Broad range of 4,6-disubstituted aminoglycosides (e.g., kanamycin, amikacin) and 4,5-disubstituted aminoglycosides (e.g., neomycin) | 4 |
| Hpa2 | Kanamycin and streptomycin | 70 |
| Eis | Kanamycin A, amikacin, and capreomycin | 76, 77 |
| PA3994 | Polymyxin B and polymyxin E (colistin) | 82, 83 |
| SatA | Streptothricin | 87, 90–93, 95–97 |
| MccE | Microcin C7 | 103 |
| RimL | Microcin C7 | 104 |
| YhhY | Leucine sulfamoyl adenylates | 104 |
| GNATs modifying polyamines | ||
| SSAT, SpeG | Spermidine and spermine | 107 |
| SpeG | Spermidine and spermine | 114, 119 |
| PmvE | Putrescine and spermine | 116 |
| PaiA | Spermidine and spermine | 106 |
| BltD | Spermidine and spermine | 121 |
| SnaB | Spermidine, spermine, cadaverine, and putrescine | 122 |
| GNATs modifying glutamate-mimicking substrates | ||
| PitA (PA4866), AcePitA (ACIAD1637), and MddA (STM1590) | Methionine sulfoximine and methionine sulfone | 134–136 |
| Bar, Pat | Phosphinothricin | 123, 127, 128 |
| PhoN1 | Phosphinothricin | 139 |
| PhoN2 | Methionine sulfoximine | 139 |
| Ttr | Tabtoxinine-β-lactam | 143–146 |
| Antitoxin-GNAT toxin systems | ||
| AtaT | Initiator methionine-tRNAfMet | 153, 154 |
| ItaT | Isoleucine-tRNAIle | 155 |
| TacT, TacT2, TacT3 | Amino acids of charged tRNAs, with a preference for glycine and isoleucine/leucine | 156, 157 |
| KacT | Unknown, possibly tRNAs | 161 |
| GmvT | Unknown, possibly tRNAs | 162 |
| GNATs involved in anabolism | ||
| TmcA | Wobble base (position 34) of tRNAMet | 6 |
| MshD | 1-d-myo-Inosityl-2-l-cysteinyl-amido-2-deoxy-α-d-glucopyranoside | 175 |
| CBG | Unknown, predicted involvement in clavulanic acid synthesis | 182, 183 |
| PseH | UDP-4-amino-4,6-dideoxy-β-l-AltNAc | 185 |
| WecD | dTDP-4-amino-4,6-dideoxy-α-d-galactose | 192, 193 |
| Aminoacyl GNATs | ||
| MurM | Transfers the first alanine or serine to the cross-link in S. pneumoniae | 205, 209, 210 |
| MurN | Transfers the second alanine to the cross-link in S. pneumoniae | 206, 209 |
| BppA1 | Transfers the first alanine of the cross-link in E. faecalis | 211 |
| BppA2 | Transfers the second alanine of the cross-link in E. faecalis | 211 |
| FemX (also called FmhB) | Transfers the first glycine of the cross-link in S. aureus or Weissella viridescens | 203, 204 |
| FemA | Transfers the second and third glycine residues of the cross-link in S. aureus | 213, 215 |
| FemB | Transfers the fourth and fifth glycine residues of the cross-link in S. aureus | 213, 216 |
| MprF, LysX, L-PGS | Transfers a lysine residue onto phosphatidylglycerol | 229–231, 233 |
| A-PGS | Transfers an alanine residue onto phosphatidylglycerol | 233 |
| LFT | Transfers a leucine or phenylalanine residue to the N-terminal arginyl or lysyl residue of a protein | 235 |
| VlmA | Transfers serine in the biosynthesis of valanimycin | 237 |
| PacB | Transfers alanine in the biosynthesis of pacidamycin | 238 |
| DhpH | Transfers leucine in the biosynthesis of dehydrophos | 240 |
| DhpK | Transfers glycine in the biosynthesis of dehydrophos | 240 |
| FmzI | Transfers valine in the biosynthesis of fosfazinomycin | 241 |
| Orf11 | Transfers glycine in the biosynthesis of BD-12 | 242 |
| GNATs involved in small-molecule-dependent transcription | ||
| BadL | Aminobenzoates | 247 |
| OatA | O-Acetylserine | 248 |
| CD1211 | O-Acetylserine, l-serine, l-threonine, and l-methionine | 249 |
General topology of GNATs.
The GNAT domain consists of six or seven β-strands and four α-helices (12). A cartoon representation of the GNAT topology is shown in Fig. 1A. GNATs contain four conserved motifs, with motif A being the most conserved (13). Another conserved aspect of GNATs is the binding of acetyl coenzyme A (AcCoA) via the pyrophosphate binding loop, referred to as the “P loop,” between β-4 and α-3. This conserved sequence [(R/Q)-X-X-G-X-(A/G)] allows for multiple sites of hydrogen bonding to the pyrophosphate of AcCoA to the amine backbone (14). The AcCoA molecule sits in the β-bulge, a V-shaped splaying of β-4 and β-5, where the pantothenate moiety of AcCoA interacts with the C-terminal end of β-4 (15). Neither the acetyl group nor the adenine ring of AcCoA makes many interactions with the GNAT; thus, GNATs may bind other acyl-CoAs as well as coenzyme A (CoA) nonspecifically (14). Structurally, GNATs have a conserved site for CoA binding and catalysis but display structural variations that accommodate other acyl-CoA substrates (e.g., propionyl-CoA [PrpCoA], succinly-CoA [SucCoA]) (16).
FIG 1.
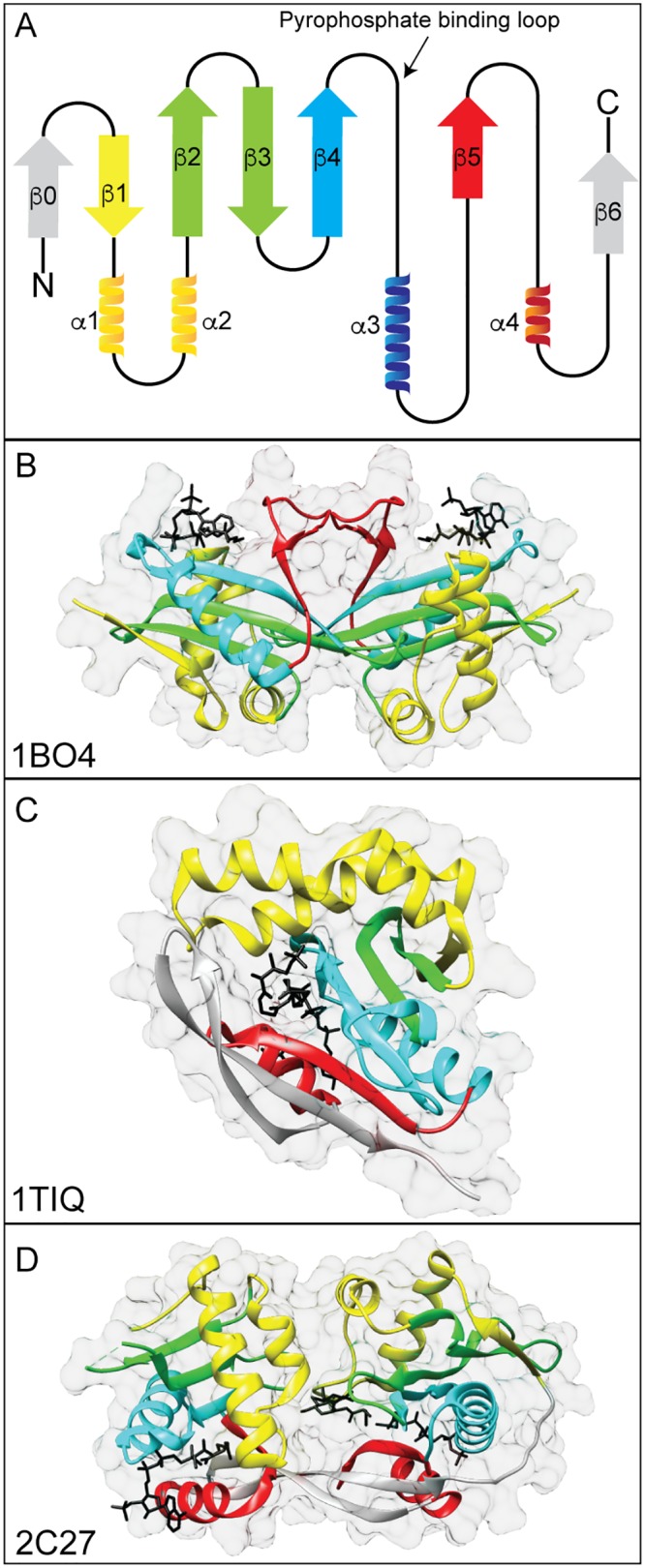
General structure of GNATs. (A) Cartoon representation of GNAT topology. GNATs contain six or seven β-sheets and four α-helices. The β-sheets are shown as arrows, and the α-helices are shown as ribbons. The four motifs are colored (yellow, motif C; green, motif D; blue, motif A; red, motif B), and this coloring applies to panels B to D. The least-conserved elements are colored gray. Examples of GNAT structures are shown with substrates in black. (B) AAC(3)-Ia dimer from Serratia marcescens in complex with AcCoA (PDB accession number 1BO4). (C) PaiA from Bacillus subtilis in complex with an oxidized CoA dimer (PDB accession number 1TIQ). (D) MshD from Mycobacterium tuberculosis in complex with AcCoA, CoA, and desacetylmycothiol (PDB accession number 2C27).
Catalytic mechanism of GNATs.
GNATs usually act as dimers and use a sequential mechanism to catalyze the transfer of an acetyl moiety from AcCoA to the substrate (Fig. 2) (15). GNATs use a general acid/base mechanism, where a catalytic glutamate or aspartate acts as a general base to abstract a proton from the amine. The nucleophilic amine attacks the thioester carbonyl carbon, resulting in a direct transfer of the acetyl group. A general acid (usually a tyrosine or a serine) reprotonates the thiolate of coenzyme A (9). A conserved tyrosine that acts as a general acid is found in 62% of characterized GNATs, while 36% have a conserved glutamate as a general base (17). In lieu of a residue acting as a general base, a water molecule(s) deprotonates before the nucleophilic attack occurs (11). GNATs may follow a random kinetic mechanism, where the binding order does not matter, or an ordered kinetic mechanism, where the binding order of the substrate and AcCoA is dependent on the other (18, 19).
FIG 2.
GNAT acetylation mechanism. A catalytic glutamate or aspartate acts as a general base to abstract a proton from an amine. The nucleophilic amine attacks the carbonyl carbon of the acetyl moiety, allowing for a direct transfer of the acetyl group. A general acid (usually a tyrosine) reprotonates the thiolate of CoA.
GNAT-DEPENDENT ANTIBIOTIC INACTIVATION
Aminoglycoside-Modifying GNATs
Aminoglycosides are hydrophilic sugars with several amino and hydroxyl moieties that vary by number and location based on the specific antibiotic (20) (Fig. 3A). These antibiotics are produced mainly by actinomycetes and inhibit protein synthesis by interacting with the 16S rRNA of the bacterial ribosome and block the A site (4, 21–24). A detailed review on the mechanism of aminoglycosides can be found in references 25 and 26. The most common means of aminoglycoside resistance arises from aminoglycoside-modifying enzymes that can phosphorylate, adenylate, or acetylate the antibiotic (20).
FIG 3.

Acetylation of aminoglycoside antibiotics. (A) Chemical structure of the aminoglycoside ribostamycin showing the sites of acetylation (1, 3, 2′, 6′). (B) Example of acetylation of kanamycin A by AAC(6′). The transferred acetyl moiety is highlighted in yellow.
Aminoglycoside acetyltransferases, called AACs, belong to the GNAT superfamily (4). They are classified based on the site of acetylation (Fig. 3A) and further subclassified based on the antibiotics that they modify (20, 27). AAC(1) and AAC(3) modify the position 1 and 3 amino groups, respectively, of the 2-deoxystreptamine ring, and AAC(2′) and AAC(6′) modify the 2′ and 6′ amino positions, respectively, of the 2,6-dideoxy-2,6-diaminoglucose ring (26, 28). An example of AAC(6′) acetylation is shown in Fig. 3B. Acetylation of the aminoglycoside impairs its ability to bind the rRNA because of unfavorable steric or electrostatic interactions, thereby reducing the cytotoxicity of the antibiotic (20, 29).
The first AAC was described by Okamoto and Suzuki in 1965, marking the first time that this family of antibiotics was reported to be inactivated in such a manner (2). Since then, more than 70 AACs have been reported (26). A brief summary of each class is discussed below.
AAC(1) family.
The AAC(1) family is comprised of only two GNATs. AAC(1)-I was originally found in Escherichia coli to monoacetylate apramycin, butirosin, lividomycin, and paromomycin and to diacetylate ribostamycin and neomycin (30). Later, AAC(1)-I was discovered in Campylobacter to be the first reported example of an AAC(1) enzyme found in a clinical isolate (4). In contrast, AAC(1)-II was identified in an Actinomycetes isolate and shown to have a broader substrate range than AAC(1)-I (31). However, these studies showed that AAC(1) modifications did not significantly reduce the antibacterial effect of the aminoglycoside.
AAC(3) family.
The AAC(3) family includes nine subclasses of GNATs (4) and includes the first aminoglycoside-modifying enzyme to be purified (gentamicin acetyltransferase) (32). Kinetic studies with AAC(3) enzymes showed that these enzymes use a sequential kinetic mechanism and prefer AcCoA as the acyl donor, helping to establish the mechanism used by most GNATs in general (33–35).
(i) Members of the AAC(3)-I subfamily confer resistance to gentamicin, sisomicin, and astromicin and are found in the integrons of Gram-negative bacteria (4, 36, 37). The first AAC-type enzyme to be crystalized, AAC(3)-Ia from Serratia marcescens (Fig. 1B), was also from this family of GNATs (38). The crystal structure of AAC(3)-Ia in complex with CoA displays the typical GNAT architecture with a V-shaped splay between β-4 and β-5 for CoA binding. Acidic residues found in motif B are important for binding the positively charged antibiotic (38). This first AAC crystal structure allowed for structure-based sequence comparisons for other GNATs as well as a means to define structurally the four GNAT motifs.
(ii) The AAC(3)-II subfamily includes three GNATs [AAC(3)-IIa to -IIc] that confer resistance to gentamicin, netilmicin, tobramycin, sisomicin, 2′-N-ethylnetilmicin, 6′-N-ethylnetilmicin, and dibekacin (4, 27). The AAC(3)-III and AAC(3)-IV GNAT subfamilies have a broader substrate range than the AAC(3)-I and AAC(3)-II enzymes (35). The AAC(3)-III subfamily includes three enzymes [AAC(3)-IIIa to -IIIc], all of which were isolated from Pseudomonas aeruginosa isolates (4, 39). There is only one example of a AAC(3)-IV enzyme, which is found in E. coli, Campylobacter jejuni, and Pseudomonas stutzeri strains, and only one example of a AAC(3)-VI enzyme, which is found in enteric bacteria (40–43).
(iii) Members of the AAC(3)-VII, AAC(3)-VIII, AAC(3)-IX, and AAC(3)-X subclasses are all chromosomally encoded in Gram-positive actinomycetes (4, 44–46). Surprisingly, the AAC(3)-X enzyme also displays AAC(3ʺ) activity, since it can acetylate the 3ʺ-amino group of arbekacin and amikacin, though only 3ʺ-N-acetylamikacin lost antimicrobial activity when acetylated (47).
AAC(2′) family.
All the aminoglycoside acetyltransferases in the AAC(2′) family are chromosomally encoded (28). AAC(2′)-Ia from Providencia stuartii was the first of this family to be described (48). This GNAT shows broad specificity for aminoglycosides with 2′-amino groups (49) and can acetylate the aminoglycoside tobramycin using an acetyl group from AcCoA, soluble fragments of peptidoglycan, or N-acetylglucosamine (50). However, kinetic and phenotypic analyses suggest that the physiological role of AAC(2′)-Ia appears to be in cell wall metabolism, such as O-acetylation of peptidoglycan (50–52).
The four other GNATs belonging to this family [AAC(2′)-Ib to -Ie] are found in the Mycobacterium species, though recently, an AAC(2′)-Ib enzyme was detected in Acinetobacter baumannii (53, 54). AAC(2′)-Ib from Mycobacterium fortuitum FC1K showed homology to AAC(2′)-Ia (38.5% identity), and expression of the aac(2′)-Ib gene in M. smegmatis provided increased resistance to gentamicin, tobramycin, dibekacin, netilmicin, and 6-N-ethylnetilmicin compared to that for the vector-only control (55). Use of the aac(2′)-Ib gene as a probe in DNA hybridization experiments indicated the universal distribution of aminoglycoside 2′-N-acetyltransferase genes throughout the Mycobacterium genus and facilitated the identification of the aac(2′)-Ic, aac(2′)-Id, and aac(2′)-Ie genes from M. tuberculosis H37Rv, M. smegmatis mc2155, and M. leprae, respectively (56). All the encoded Mycobacterium proteins share ∼60% to 80% identity, while they are only ∼30% to 40% identical to AAC(2′)-Ia from P. stuartii (56).
A deletion of the aac(2′)-Id gene in M. smegmatis results in decreased MIC values for aminoglycosides and a lack of aminoglycoside acetyltransferase activity in crude extracts, indicating a role for AAC(2′)-Id in aminoglycoside inactivation (56). Besides a sensitivity to aminoglycosides, the aac(2′)-Id-deficient strain also displayed increased sensitivity to lysozyme (56). This suggests that another physiological role of AAC(2′)-Id is involvement in the synthesis of the cell wall, similar to that of AAC(2′)-Ia. Also similar to AAC(2′)-Ia, AAC(2′)-Ic has been shown to O-acetylate kanamycin A and amikacin, suggesting that some AAC(2′) enzymes are capable of N- and O-acetylation (57, 58) and may play a role in cell wall synthesis (59).
The AAC(2′)-Ic protein has been crystalized as a dimer, displaying the typical GNAT fold with an atypical P-loop sequence (60). The C-terminal aminoglycoside-binding site is lined with acidic residues (D35, D40, E82, D152, and D179) and the carboxyl terminus of W181, all of which are conserved among AAC(2′) enzymes (60). These residues interact with the hydroxyl and amino substituents of the aminoglycoside either directly or through water molecules. The hydroxyl of Y126 is 3.6 Å away from the sulfur moiety of CoA and may act as a general acid, while residue E82 or W181 may act as a remote general base (60).
AAC(6′) family.
The AAC(6′) family is the largest family and is found in Gram-positive and Gram-negative bacteria alike (4). An example of the reaction catalyzed by this family is shown in Fig. 3B, which shows how the enzyme acetylates kanamycin A. There are two subclasses, based on the resistance profiles for amikacin and gentamicin C1. AAC(6′)-I enzymes have low levels of activity against gentamicin C1 and high levels of activity against amikacin and gentamicin C1a and C2, while AAC(6′)-II enzymes have high levels of activity against all forms of gentamicin but low levels of activity against amikacin (4). Though the AAC(6′)-Ib-cr enzyme can acetylate fluoroquinolones, it is not considered a third subclass because only two amino acid substitutions (W102R and D179Y) differentiate it from AAC(6′)-Ib (61).
The AAC(6′)-I subfamily is the largest, with over 50 unique enzymes organized into three distinct subclasses characterized by structure and the catalytic efficiencies as a function of their aminoglycoside substrates (4, 62). Subclass A contains AAC(6′)-Ig, -Ih, and -Iy as well as Acinetobacter-specific AAC(6′)-I enzymes. These proteins act as dimers and are the least catalytically efficient (62). Subclass B contains AAC(6′)-Ii, is characterized by dimerization and intermediate catalytic efficiency, and exhibits subunit cooperativity (62, 63). The final subclass, subclass C, contains AAC(6′)-Ib and -Ie, which are monomers and which have the highest known catalytic efficiency toward aminoglycosides (62).
AAC(6′)-Ib is the most clinically relevant AAC(6′) enzyme and is found in over 70% of AAC(6′)-I-positive Gram-negative bacterial clinical isolates (4, 64). Because of their medical importance, many AAC(6′)-Ib enzymes have been crystalized to identify active-site residues and understand molecular mechanisms. The crystal structure of AAC(6′)-Ib from E. coli shows that D115 acts as a general base and that Y164 may be a general acid (65). Structures also exist for a close homologue, AAC(6′)-Ib11, which has a broader range of antibiotic substrates than AAC(6′)-Ib from E. coli (66). Both structures display the conserved pyrophosphate binding loop as well as the V shape to accommodate AcCoA binding (65, 67). Unique to AAC(6′)-Ib enzymes is an extended α-helical flap that forms a lid over the antibiotic-binding pocket and that may account for the ordered kinetic mechanism where AcCoA binds first (19, 65, 67). Surprisingly, AAC(6′)-Ib acts as a monomer, though Salmonella enterica AAC(6′)-Ib11 acts as a mixture of monomers and dimers (65, 67). Comparing the structures of AAC(6′)-Ib and AAC(6′)-Ib11 reveals that the active-site residues of AAC(6′)-Ib (Q106 and L107) are replaced by leucine and serine, respectively, in AAC(6′)-Ib11. These substituted residues induce a structural change that creates a gaping of the active site to allow larger aminoglycosides to bind and explains the broader substrate range of AAC(6′)-Ib11 (66, 67). Many other AAC(6′)-Ib variants with changes in the N terminus have been described, indicating flexibility in this portion of the protein, since these variants still confer resistance (4).
In addition to the subfamilies, there is a variety of fusion proteins with the AAC(6′) portion at the N or C terminus of the protein (68). These fusion proteins can have aminoglycoside adenylylase (ANT) or phosphotransferase (APH) activity (4), highlighting how bacteria can become resistant to multiple antibiotics. A single tyrosine substitution (D179Y) of AAC(6′)-Ib-cr appears to allow for π-stacking interactions with the quinolone ring, enabling fluoroquinolone binding and acetylation (65). Some fusion proteins are a fusion of two AAC proteins, such as AAC(3)-Ib/AAC(6′)-Ib′ of P. aeruginosa (69). This enzyme has two GNAT domains, both of which are active and use an ordered sequential kinetic mechanism where AcCoA binds first (19).
Recently, the histone acetyltransferase Hpa2 of A. baumannii was reported to acetylate and inactivate kanamycin and streptomycin (70). A. baumannii Hpa2 (AbHpa2) shows a topology similar to the crystal structure of Salmonella enterica AAC(6′)-Iy, which has been shown to acetylate both histone peptides and aminoglycosides (71). A virtual substrate screen also indicated that aminoglycosides may be a possible substrate of AbHpa2 (72). Similar to AAC(6′)-Iy, apo-AbHpa2 acts as a dimer in solution (73); however, the addition of AcCoA converts the Hpa2-AcCoA complex into monomers (70). The binding of kanamycin induces structural changes in AbHpa2, and isothermal titration calorimetry experiments demonstrate that AbHpa2 binds aminoglycosides kanamycin and streptomycin well (Kd [dissociation constant], 8 μM and 79 μM, respectively) (70). Importantly, noncanonical AAC GNATs are emerging as reservoirs of aminoglycoside resistance.
The Eis GNAT.
The enhanced intracellular survival (Eis) protein is a unique aminoglycoside-acetylating enzyme that is divergent from other AACs but that is still capable of acetylating multiple amino groups (74). Eis was originally described in M. tuberculosis as a secreted protein that enhances survival in macrophages (75), and Eis homologues are found in other mycobacteria and several nonmycobacterial prokaryotes (76). Eis has been reported to acetylate many second-line antituberculosis drugs, such as kanamycin A, amikacin, and capreomycin, with this being reported as the first instance of inactivation of capreomycin via acetylation (76, 77). M. tuberculosis Eis (MtEis) can acetylate the 3-amino and 6′-amino groups of kanamycin, the 3-amino- and 4-amino-2-hydroxybutyryl groups of amikacin, and the ε-amino group of the β-lysine side chain of capreomycin (76, 77). The physiological role of Eis aminoglycoside acetylation is unclear since the Km is high for both substrates (154 μM for kanamycin and 112 μM for amikacin), and the authors of these studies suggested that Eis may acetylate host factors during M. tuberculosis infection (77). Recently, it was shown that Eis from M. tuberculosis, but not that from M. smegmatis, acetylates K55 of dual-protein phosphatase 16/mitogen-activated protein kinase phosphatase 7 (DUSP16/MKP-7), thereby suppressing cytokine production in macrophages (78). Inspecting the crystal structure of the two Eis homologues, MtEis contains a narrow channel in the active site that is absent in M. smegmatis Eis (MsEis), and this channel may act as a binding site for DUSP16/MKP-7 (78).
Eis monomers contain three structural domains and form hexamers in solution (74, 78). Surprisingly, both domain 1 (residues 1 to 150) and domain 2 (residues 151 to 291) adopt a GNAT fold, even though domain 2 shows little sequence similarity to other GNATs (78). Domain 3 (residues 292 to 402) shares homology to sterol carrier protein 2 (78). Domain 1 can bind AcCoA or CoA, but domain 2 cannot, as it lacks the R/Q residue needed in the P loop to bind the pyrophosphate of CoA, indicating that domain 2 is an inactive GNAT (74, 78). The active site is formed between the N-terminal domain 1 and central domain 2 GNAT domains, creating a negatively charged pocket for aminoglycosides of different sizes to bind (74). The substrate range and multiacetylation patterns of MtEis and MsEis differ, which is proposed to be the result of substitutions to several nonconserved residues lining the aminoglycoside-binding pocket (79). Residue H119 positions the aminoglycoside amino group for direct attack, while F402 is proposed to be a remote base via a water molecule. Mutational analysis confirmed the role of residue Y126 as a general acid, which is 3.4 Å away from the thiolate of CoA (74). A series of substituted 1,2,4- triazino-[5,6b]indole-3-thioether molecules was found to inhibit Eis acetylation (80). Structural data indicate that these inhibitor compounds bind in the kanamycin binding pocket and are competitive inhibitors that may have clinical use in the future (80).
Polymyxin-Modifying GNATs
Polymyxin antibiotics are cyclic lipodecapeptide antimicrobial molecules composed of a cyclic heptapeptide, a linear tripeptide, and a fatty acyl tail (Fig. 4). Polymyxins are basic because they contain five free amino groups, thereby facilitating interaction with the anionic lipopolysaccharide (LPS) of Gram-negative bacteria. These interactions lead to cracks in the outer membrane and the leakage of cytoplasmic contents (81). Currently, polymyxins are clinically used as a last-resort drug in multidrug-resistant cases (81), and the sources of resistance are of particular medical interest.
FIG 4.

Acetylation of the antibiotic polymyxin B. PA3994 acetylates the γ-amino group of α,γ-diaminobutryic acid proximal to the cyclic peptide. The transferred acetyl moiety is highlighted in yellow.
Pseudomonas aeruginosa PAO1 encodes a GNAT (PA3994) that acetylates polymyxin B and polymyxin E (colistin) (82). Acetylation by PA3994 occurs on a single diaminobutyric acid (residue 3) closest to the cyclic peptide, with PA3994 being reported to be the first instance of a polymyxin-modifying enzyme (Fig. 4) (83). The crystal structure displays the classic GNAT domain, which acts as a monomer in solution and in the crystal (83). The authors note that polymyxin acetylation is probably not the physiological role of PA3994, since the Km for the antibiotics are in the millimolar range, but that this function can be exploited for biotechnology applications to modify selectively structurally similar antibiotics (83). PA3994 was also found to acetylate aspartame (83), though more work must be done to understand the physiological function of that modification by PA3994.
Streptothricin-Modifying GNATs
Besides aminoglycoside antibiotics, bacteria have evolved ways to acylate and inactivate other antimicrobial agents. Streptothricin is one of the most commonly found antibiotics in the soil and is composed of a streptolidine, a gulosamine sugar, and a β-lysine that varies from 1 to 7 repeating units (Fig. 5A) (84, 85). It is produced by Streptomyces species and acts as a broad-spectrum antimicrobial that inhibits protein synthesis by interacting with the bacterial ribosome (86–89). To protect themselves, many streptothricin-producing species encode a streptothricin acetyltransferase, as shown in Streptomyces lavendulae (87, 90), Streptomyces noursei (91, 92), and Streptomyces rochei (93). These GNATs monoacetylate the β-lysine, inhibiting the cytotoxicity of the antibiotic (94). Since the initial discovery of streptothricin acetyltransferases in Streptomyces species, other Sat (streptothricin acetyltransferase) enzymes have been characterized in Campylobacter coli (95), E. coli (96), and Bacillus subtilis and Bacillus anthracis (97), suggesting a selective pressure on soil-dwelling bacteria for the acquisition of streptothricin resistance.
FIG 5.
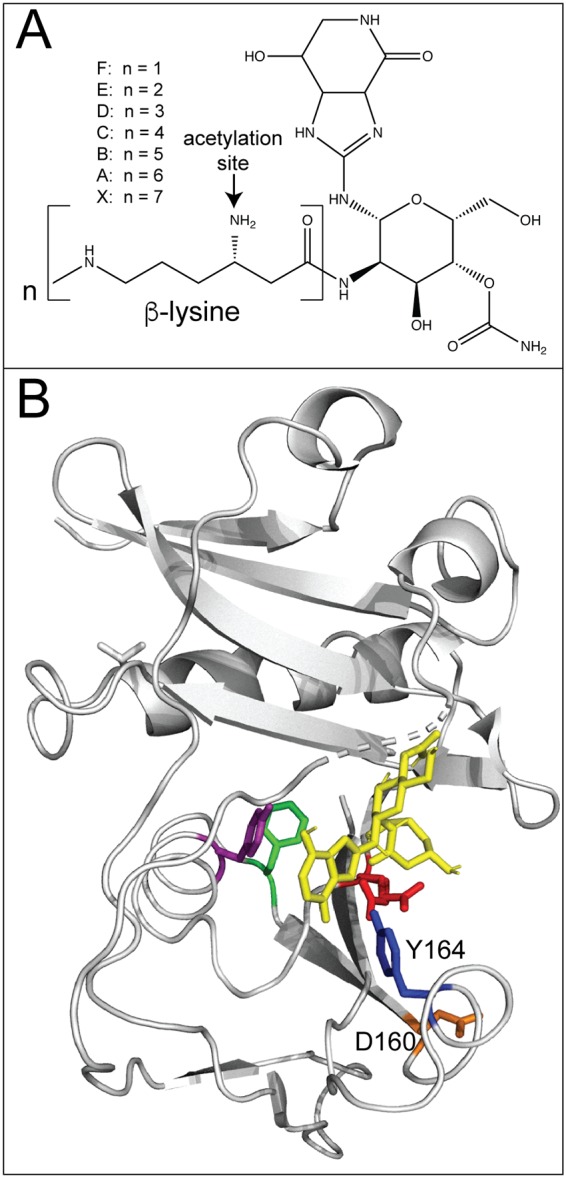
Acetylation of streptothricin. (A) Chemical structure of streptothricin showing the site of acetylation on the β-lysine. (B) Model of SatA (PDB accession number 3PP9) in complex with streptothricin (yellow). A conserved tyrosine (Y164, blue) appears to be in close proximity to a hydrogen bond with streptothricin. A conserved aspartate (D160, orange) is important for dimerization. (Adapted from reference 98.)
To gain insights into streptothricin binding to SatA, a detailed study of the B. anthracis SatA (BaSatA) enzyme was performed (98). The crystal structure of BaSatA displays a typical GNAT architecture with AcCoA situated in a V-shaped cleft. Examination of the structure indicated a putative C-terminal streptothricin-binding pocket. Through the use of a combination of site-directed mutagenesis, isothermal titration calorimetry (ITC), and docking software, the authors concluded that a conserved tyrosine (Y164) may interact with the streptothricin ligand (Fig. 5B). In addition, a conserved aspartate (D160) was found to be necessary for dimerization (98). Together, these data help shape our understanding of how, in general, GNATs recognize and bind their ligand.
McC-Modifying GNATs
Microcin C7 (McC) is a small-molecule antimicrobial produced by the Enterobacteriaceae (99). Microcin is the product of the 21-bp mccA gene (100). It is produced from a ribosomally synthesized heptapeptide with a C-terminal aspartate linked to AMP and a formylated N-terminal methionine (101). Inside sensitive cells, the nonhydrolyzable aspartyl adenylate analogue inhibits aspartyl-tRNA synthetase, thereby inhibiting translation (102). As a protection mechanism, the genomes of McC producers encode the mccE gene, which encodes a two-domain protein with an N-terminal domain homologous to decarboxylases and a C-terminal domain with homology to GNATs (103, 104). Overexpression of the C-terminal GNAT domain, but not the N-terminal domain, leads to cells with resistance to McC, albomycin, as well as other nonhydrolyzable aminoacyl adenylates with primary amines (103). Mass spectrometry analysis indicated the McC-dependent acetylation of the primary amino group of the aminoacyl moiety, rendering McC nontoxic (103).
E. coli MccE (EcMccE) was crystalized with CoA or acetyl-CoA and a variety of substrates to understand its broad substrate specificity (105). EcMccE displays typical GNAT domain folds, consisting of seven β-sheets and four α-helices, and binds the pyrophosphate and pantetheine of CoA with hydrogen bonds similar to those of other known GNATs (105). EcMccE crystalized as a monomer, which is consistent with size exclusion chromatography data. Residues S553 and E572 are implicated to serve as the general acid/base for catalysis, respectively, since replacement of both of these residues by alanine results in a protein with an ∼25-fold lower acetyltransferase activity (105). The adenine ring of the substrate predominantly determines specificity. The substrate binding pocket contains several hydrophobic residues that participate in van der Waals interactions with the adenine ring, with Try453 and Phe466 allowing for strong π-stacking interactions with the aromatic adenine ring (105).
Interestingly, RimL of E. coli was found to provide basal resistance to McC through acetylation, similar to MccE (104). Overexpression of RimL leads to McC resistance, as seen with MccE overexpression (104, 106). RimL can also acetylate other aminoacyl adenylates, like MccE (104). Another E. coli GNAT, YhhY, was shown to acetylate and detoxify other nonhydrolyzable aminoacyl adenylates, though it did not process McC (104). YhhY conferred full resistance to leucine sulfamoyl adenylates (LSA) and partial resistance to alanine sulfamoyl adenylates (ASA) and phenylalanine sulfamoyl adenylate (FSA) (104). More work is needed to understand the physiological role of the acetylation of aminoacyl adenylates by these other GNATs.
GNAT-DEPENDENT DETOXIFICATION
As in the case of antibiotics, acetylation of toxins is a common mechanism for detoxification. GNATs have evolved to recognize and inactivate a series of toxic substrates.
Spermidine/Spermine-Acetylating GNATs
Polyamines (e.g., spermine, spermidine) are low-molecular-weight molecules with more than two amino groups (Fig. 6A), making them good targets for acetylation. Spermidine and spermine are found in cells of all domains of life and play a variety of roles, including the regulation of transcription, ion channels, and enzymatic activity (107, 108). Thus, the regulation of polyamine levels is strictly regulated in cells.
FIG 6.

Acetylation of toxic compounds. (A) Acetylation of spermine or spermidine by a spermidine/spermine acetyltransferase (SSAT) signals for the N1-acetylated product’s export or degradation. (B to D) Acetylation of glutamine synthetase (GS) inhibitors blocks the toxic effect of phosphinothricin (B), methionine sulfoximine or methionine sulfone (C), or tabtoxinine-β-lactam (D).
One means of targeting polyamines for degradation is acetylation by a spermidine/spermine acetyltransferase (SSAT). These enzymes acetylate spermidine or spermine under stress conditions to promote degradation via the polyamine oxidase, thereby preventing polyamine toxicity (109–112). Acetylation also neutralizes the positive charge of polyamines to inhibit their interaction with other macromolecules (5). SSAT enzymes are induced by polyamines and help maintain proper cellular polyamine concentrations (107).
The involvement of SSATs in virulence has been documented (113). Staphylococcus aureus and Enterococcus faecalis do not synthesize polyamines and are hypersensitive to them. The virulent S. aureus USA300 strain has acquired a SSAT (speG) on a mobile element, which the authors suggest makes the strains resistant to high polyamine levels inside a human host and thus may aid in overall virulence (114). High polyamine levels can inhibit biofilm formation, but S. aureus and E. faecalis use their respective SSATs to acetylate and neutralize host polyamines, allowing for effective biofilm formation and infection (115). PmvE (polyamine metabolism and virulence of Enterococcus faecalis) acetylates putrescine and spermine, and overexpression of pmvE leads to higher virulence (116). The authors suggest that this PmvE-dependent virulence phenotype may be in part linked to a better ability to survive conditions in the macrophage, especially since pmvE appears to be essential in E. faecalis (116). In contrast, inactivation of speE in Shigella increases intracellular spermidine levels and enhances survival under oxidative stress in the macrophage (117). Besides the role of SSATs in virulence, polyamine inhibition is exploited in the development of chemotherapeutics. Since the activities of some polyamine analogues mimic SSAT activity, this leads to a decrease in polyamine levels and, thus, a decrease in cell growth of cancerous tissue (107).
Because of their role in virulence and cell viability, several SSAT enzymes have been crystalized. PaiA from Bacillus subtilis (BsPaiA) crystalized as a dimer, though it acts as a monomer in solution (106). Surprisingly, an oxidized CoA dimer was crystalized in the active site, suggesting that a long, linear molecule—such as a polyamine—would be the physiological substrate (Fig. 1C) (106). Residue Y142 was determined to act as a general acid, and a water molecule was determined to act as a general base (106). PaiA (locus tag, Ta0374) from Thermoplasma acidophilum (TaPaiA) also displays the GNAT architecture, though the loops between β-4/β-5 and β-6/β-7 are structurally different from the BsPaiA structure (118). Like BsPaiA, TaPaiA acts as a monomer with a putative polyamine-binding cleft made up of residues highly conserved in PaiA enzymes (106, 118). Cocrystallization of TaPaiA and CoA shows that the conserved Y138 is located close to the sulfur atom of CoA and may act as a general acid (118). In contrast to PaiA proteins, the spermidine N-acetyltransferase SpeG from Vibrio cholerae (VcSpeG) acts as a dodecamer, formed by a dimer of hexamers that sit on top of each other (119). A monomer from one hexamer interacts with a corresponding monomer from the other hexamer to form the classic GNAT dimer (119). This oligomerization allows for an allosteric site that binds spermidine/spermine and that results in a conformational change that allows for AcCoA and polyamine binding (120). The VcSpeG crystal shows that residue Y134 is located in close proximity to the sulfur atom of AcCoA and may act as a possible general acid (119).
Other SSATs include BltD, a SSAT that helps export polyamines via the multidrug Blt transporter (121). Bacillus subtilis BltD acetylates spermine and spermidine with a high affinity (Km, <67 μM and 200 μM, respectively). BltD can also diacetylate spermine, using N1-acetylspermine as a substrate (5). The spermidine N-acetyltransferase A (SnaA) from Corynebacterium glutamicum modifies a variety of polyamines, including spermidine, spermine, cadaverine, and putrescine (122). SnaA can use AcCoA, PrpCoA, and SucCoA, with a preference for AcCoA (122). Surprisingly, a snaA deletion did not cause a growth defect in C. glutamicum under the conditions tested, and the physiological role of polyamines in this organism is not well understood (122).
Phosphinothricin and Methionine Derivatives Inactivating GNATs
Streptomyces species produce the herbicide bialaphos, which is a tripeptide of two l-alanine residues and a glutamate mimic called phosphinothricin (PPT) (Fig. 6B) (123, 124). Once inside cells, the two alanine residues are cleaved by peptidases and release free PPT. As an analogue of glutamate, PPT inhibits glutamine synthetase and leads to ammonium toxicity in plants (125, 126). To protect themselves, Streptomyces species encode a PPT-modifying GNAT to acetylate and inactivate PPT (Fig. 6B) (123, 127, 128).
The first reported characterization of a PPT-modifying GNAT is the Bar (for bialaphos resistance) protein from Streptomyces hygroscopicus (123). Thompson et al. demonstrated that strains overexpressing the bar gene had 15-fold more acetylated PPT or demethyl-PPT than the vector control (123). The Bar protein of Streptomyces viridochromogenes, which is 73% identical to S. hygroscopicus Bar, also acetylates and detoxifies PPT (129).
The use of phosphinothricin acetyltransferases to make crops resistant to bialaphos, thereby allowing the antibiotic to be used selectively in the field, has received much attention in agriculture (128, 130). Phosphinothricin resistance was first introduced into plants (e.g., canola) in 1995, and since then, its use has been expanded to other crops (131). Recently, it was discovered that Bar can acetylate aminoadipate and tryptophan, leading to N-acetyl-aminoadipate and N-acetyl-tryptophan accumulation in plants (132). To understand its mechanism, Bar with CoA and PPT was crystalized. The crystal structure shows that E88 and Y107 act as a general base and an acid, respectively (132). The structure also highlights residues that interact with PTT, including F36, G127, and V161, which stabilize the methylphosphoryl group, as well as K78, R80, and Y83, which hydrogen bond with the two phosphoryl oxygen molecules (132). Based on structural data, Christ et al. engineered Bar variants that target only PPT, highlighting the importance of enzymological studies, needed to improve biotechnologies (132).
Besides PPT, another potent inhibitor of glutamine synthetase is methionine sulfoximine (MSX) (125, 133). As with PPT, acetylation of the amine of MXS blocks its inhibitory effects (Fig. 6C). Many GNATs annotated as PPT-modifying GNATs have been shown to acetylate MSX or the structurally related compound methionine sulfone. The Bar protein of S. hygroscopicus can also acetylate MSX, although it does so with a 3-order-of-magnitude decreased efficiency relative to that of PPT (123). The PitA protein (encoded by PA4866) of Pseudomonas aeruginosa (PaPitA), the PitA protein of Acinetobacter baylyi (AcePitA; encoded by ACIAD1637), and MddA (methionine derivative detoxifier A, encoded by the gene formerly known as yncA) of Salmonella enterica have all been shown to acetylate MSX and methionine sulfone but not PPT (134–136). The structures of PaPitA and AcePitA both showed the GNAT α/β fold of β-sheets and α-helices and a V shape for AcCoA binding (135, 137). PaPitA was cocrystalized with MSX in the active site, and R75 appears to hydrogen bond with the oxygen, while residues I30 and F77 help stabilize the methyl portion of MSX (134). In the S. hygroscopicus Pat and S. viridochromogenes Pat enzymes, I30 is replaced by an asparagine, which may be a reason for the different substrate specificity. In comparison to the apoenzyme, it appears that PaPitA undergoes a conformational change upon binding the substrate (134). All MSX- and methionine sulfone-modifying GNATs act as dimers (134–136). In an effort to predict the substrate specificity of annotated PPT-modifying GNATs, one study used a combination of in vivo, in vitro, and bioinformatics approaches to determine that phylogeny is a key criterion for substrate specificity (PPT, MSX, or methionine sulfone) of Bar homologues (138). Some organisms contain two separate GNATs, with one for PPT and one for MSX. This is the case in Pseudomonas putida, where PhoN1 is a PPT-modifying GNAT and PhoN2 is an MSX-modifying GNAT (139). The physiological reason why some organisms have different numbers of PPT-like modifying enzymes is unclear.
Tabtoxin-Inactivating GNATs
As mentioned above, inhibition of glutamine synthetase leads to cell death via ammonium toxicity. Another inhibitor of glutamine synthetase is tabtoxin (140). This toxin is a dipeptide made up of a threonine linked by a peptide bond to a monocyclic β-lactam (tabtoxinine-β-lactam [TβL]) (141) (Fig. 6D). This toxin is produced by Pseudomonas syringae, and once inside cells, the cleavage of threonine releases the active TβL moiety, which inhibits glutamine synthetase and causes wildfire disease of tobacco plants (140, 142). To protect themselves, P. syringae carries a tabtoxin resistance (ttr) gene that encodes a GNAT to monoacetylate the α-amino group, which inactivates TβL (143–146). Tobacco plants expressing ttr were resistant to TβL and infection by P. syringae pv. tabaci (143). Ttr acetylates TβL but not the dipeptide tabtoxin (146).
The crystal structure of Ttr in complex with AcCoA shows typical GNAT folding with a V shape, facilitating AcCoA binding via 10 direct hydrogen bonds and 3 water-mediated hydrogen bonds (17). Ttr crystalized as a dimer, consistent with other GNATs (17). Residue Y141 is highly conserved among other crystalized GNATs, and the crystal structure shows a hydrogen bond between Y141 and the carbonyl of AcCoA (17). TβL interacts with a water molecule stabilized by residues E92 and D130, prompting the authors to suggest that these residues may act together to promote a water-mediated general base, while Y141 may be a general acid (17).
GNATs AS TOXINS IN TA SYSTEMS
Overview of TA Systems
Toxin-antitoxin (TA) systems are a common mechanism for cells to cope with stress. TA systems are usually encoded by a two-gene operon; one gene encodes a toxin protein (∼100 amino acids), and the other one encodes the antitoxin, which is a small protein (∼80 amino acids) or regulatory antisense RNA that neutralizes the activity of the toxin, hence the name antitoxin (147). Usually the antitoxin is more labile than the toxin and degraded by specific proteases or RNases (147, 148). Thus, under stressful conditions, the antitoxin is degraded so as to let the toxin inhibit growth until conditions are more favorable. The role of TA systems in establishing a persister state has been extensively studied (148).
There are currently six types of TA systems, classified based on how the antitoxin neutralizes the toxin (148, 149). Below we discuss only type II systems because some of the described toxins contain a GNAT domain. Type II systems are comprised of a two-domain protein antitoxin in which one domain binds the cognate toxin protein, resulting in the inhibition of toxin activity, and the second domain binds DNA (148). Usually, the antitoxin acts as a repressor for the TA operon, with the toxin acting as a corepressor (147, 148). Recently, a TA system with a toxin with a GNAT fold was reported (150, 151). Since then, more GNAT toxins with a ribbon-helix-helix fold antitoxin have been reported, establishing a new subfamily of type II TA systems (149, 152).
Toxin GNATs Inhibit Translation
Toxins with a GNAT domain acetylate charged tRNAs and halt translation (152). AtaT (aminoacyl tRNA acetyltransferase; locus tag, Z4832) of E. coli O157:H7 specifically acetylates the initiator methionine-tRNAfMet, blocking the interaction with initiation factor II and thus halting translation initiation (Fig. 7A) (153, 154). ItaT (locus tag, EcHs_A0501) from E. coli acetylates isoleucine-tRNAIle specifically, blocking elongation (155). In contrast to the highly specific AtaT and ItaT, TacT (tRNA-acetylating toxin; locus tag, STM3651) from Salmonella enterica acetylates amino acids on charged tRNAs, thereby inhibiting elongation (Fig. 7B) (156). Two other GNAT toxins were found in S. enterica and were named TacT2 and TacT3 (157). All TacT-like toxins acetylate a variety of charged tRNAs with a preference for glycine and isoleucine/leucine (157). Another group reported that TacT acetylates its cognate antitoxin (TacA) as well, thereby increasing the activity of TacT and leading to decreased protein synthesis (158). The authors report that this is the first instance of a GNAT acetylating both a protein and a small molecule (158), and more work is needed to understand how the acetylation of both small-molecule and protein targets affects persistence.
FIG 7.

GNAT toxins acetylate tRNAs to halt translation. (A) AtaT acetylates the initiator tRNA, preventing binding to the initiation factor 2 (IF2), thus impairing initiation of translation. (B) TacT and ItaT acetylate charged tRNAs to prevent elongation.
The TacT2 proteins from two Salmonella enterica serovars, Salmonella enterica serovars Typhimurium and Enteritidis, show differences in toxicity and persister state formation, as well as the profile of acetylated charged tRNAs (157). Comparing the sequences, there is a single substitution (E29K) that reverses the charge of the residue at this position (157). The lysyl residue is near the active site and appears to hydrogen bond with neighboring residues F17 and Y15, helping make the active site more accessible to the substrate and thus helping make the protein more potent as a toxin (157). The TacT structure and mutational analysis demonstrate that the active site is a positively charged patch at the dimer interface, and the above-mentioned stabilization bonds are present in the TacT structure (156); this groove is necessary for binding a tRNA molecule (156). The structure of TacT3 and the modeled structure of TacT2 also reveal a groove of positively charged residues that, when substituted, resulted in a lack of toxicity (157). However, the orientation of AcCoA is different between the TacT3 structure and the TacT structure (156, 157). In TacT3, the bulky W142 prevents full engagement with the adenine base in the binding pocket, while residue G145 outside the binding pocket hydrogen bonds with the base for stabilization (157).
The TacT structure shows a conserved tyrosine, residue Y140, coordinating an oxygen molecule of AcCoA to allow for correct positioning. The Y140F variant is inactive (156). When the corresponding residue of AtaT (Y144) was replaced by a phenylalanine, this variant was also inactive (159). As with TacT-type toxins, AtaT was shown to have a positive patch formed by the dimers that interacts with methionine-tRNAfMet (160). The antitoxin AtaR binds AtaT in a heterohexameric complex (AtaT-AtaT2-AtaR2-AtaT), trapping it from its toxic form as a dimer (160). The GNAT domain-containing toxin KacT (locus tag, KPHS_05890) of Klebsiella pneumoniae is also neutralized by forming a heterohexamer with its cognate ribbon-helix-helix antitoxin, KacA (161). The transcription of kacT increased significantly when K. pneumoniae bacteria were challenged with the antibiotic meropenem (10-fold increase) or tigecycline (40-fold increase), suggesting that this TA system is used for entering into a persister state during antibiotic stress (161). The crystal structure of KacT complexed with AcCoA displays typical GNAT folding and has a basic region like TacT does, suggesting that it may bind tRNAs (161). The sulfur of CoA is within hydrogen bonding distance of residue Y145 and is hypothesized by the authors to act as a general acid (161). Though having low sequence homology (∼20% to 30%), these similarities in structure and activity suggest a common mechanism for GNATs in tRNA acetylation and TA systems in general.
The TA systems that involve a GNAT may have additional physiological roles other than triggering the persistent state. In Shigella flexneri, GmvT (GNAT maintenance of virulence toxin) halts protein synthesis in an AcCoA-dependent manner, possibly via tRNA acetylation (162). The activity of GmvT is sufficient for virulence plasmid stability at environmental temperatures (162). Bioinformatics studies have annotated other GNAT-dependent TA systems with unknown roles (163–166). In Acinetobacter baumannii, a TA system is comprised of a helix-turn-helix toxin protein and a GNAT antitoxin, thus named CheTA (switched-element toxin-antitoxin system) because it is the opposite of other TA systems with a GNAT toxin (167). Thus, validation will be needed to determine the common mechanisms and structures of these GNAT-dependent TA systems.
GNATs INVOLVED IN ANABOLISM
Besides inactivating toxic compounds, GNATs have roles in the synthesis of a variety of compounds. Acetylation of biosynthetic intermediates results in molecules central to many necessary cellular processes, including protein synthesis, antigen production, flagellar modifications, and cell wall synthesis.
TmcA-Dependent Acetylation for tRNA Synthesis
GNATs found in toxin-antitoxin (TA) systems have been shown to acetylate charged tRNAs in order to block translation. In contrast, TmcA (tRNAMet cytidine acetyltransferase) acetylates the wobble base of tRNAMet (6). First identified in 1972 by Oashi et al., N4-acetylcytidine (ac4C) is a modified nucleoside found in a variety of tRNAs and rRNAs (168). This modification of position 34 of tRNAMet has been shown to prevent misreading of the isoleucine AUA codon in favor of the correct methionine AUG codon (169).
E. coli strains lacking tmcA did not produce ac4C, though no phenotype was observed for this strain, while in Saccharomyces cerevisiae yeast, the tmcA homologue is essential (6, 170). In vitro assays show that ac4C formation is dependent on TmcA, ATP/GTP, and AcCoA (6). These data show that ATP/GTP hydrolysis is important for acetylation and that AcCoA binding stimulates hydrolysis and tRNA binding (6, 171). The structure of the E. coli TmcA shows an N-terminal ATP-binding RNA helicase-like domain and a C-terminal GNAT domain. The C terminus is composed of helices that form a positively charged groove that may facilitate tRNA binding (171). Mutational analysis demonstrates that the anticodon stem of tRNAMet is important for TmcA recognition (6). The crystal was solved to be complexed with AcCoA and ADP, with AcCoA binding occurring in the traditional V-shaped β-bulge in the middle of the structure (171). The two domains interact, though surprisingly, the two active sites are ∼30 Å apart (171). The authors suggest that the RNA helicase activity is needed to remodel the tRNA molecule to allow the C-34 amino group to make a direct nucleophilic attack since no residues corresponding to a general acid or base were observed (171). These data highlight how, in different contexts, GNATs can positively or negatively regulate translation.
MshD-Dependent Acetylation for MSH Synthesis
Mycothiol (MSH) is a redox-active molecule that is found only in actinomycetes (172). MSH appears to act analogously to glutathione; that is, MSH is needed to maintain the redox balance of the cell (173); actinomycetes do not synthesize glutathione. Mycobacteria produce high levels of MSH, and M. smegmatis strains lacking MSH become more sensitive to alkylating agents, oxidizing agents, and antibiotics (172, 174). The final step in MSH biosynthesis involves the acetylation of the amino group of cysteine present in 1-d-myo-inosityl-2-l-cysteinyl-amido-2-deoxy-α-d-glucopyranoside by the GNAT mycothiol synthase MshD (Fig. 8A) (175). An M. tuberculosis mshD strain displays decreased survival in the macrophage and higher sensitivity than the wild type to hydrogen peroxide (176, 177). These data highlight the importance of MSH for actinomycetes, potentially making MSH biosynthesis a good drug target.
FIG 8.

Acetylation involved in anabolism. (A) MshD acetylates the final step in mycothiol synthesis. (B) PseH-dependent acetylation is involved in pseudaminic acid production. (C) WecD acetylates intermediates in the enterobacterial common antigen (EAC) biosynthetic pathway. Acetyl moieties are highlighted in yellow.
To understand the mechanism of MshD, the protein from M. tuberculosis was crystalized both in complex with AcCoA and in complex with CoA and desacetylmycothiol (Fig. 1D) (178, 179). Both structures showed that MshD acts as a monomer that contains two GNAT domains linked by a random coil (178, 179). This double GNAT domain is similar to that of Staphylococcus aureus FemA and possibly resulted from gene duplication (175, 180). Each GNAT domain can bind AcCoA via the pyrophosphate binding loop between β-4 and β-5, though the authors propose that the C-terminal domain is active, while the N-terminal domain is a structural element because AcCoA is not positioned correctly to donate an acetyl group (178). Desacetylmycothiol binds in the central region between the GNAT domains and interacts with both domains, while only the C-terminal GNAT domain transfers an acetyl moiety (179). The N-terminal GNAT domain retains an AcCoA molecule, confirming its lack of catalytic ability (179). The authors suggest that residues E234 and Y294 may act as a general base and acid, respectively (179). Comparison of the binary and ternary complex shows a substantial movement of the N-terminal domain toward the C-terminal domain upon binding of the substrate (179).
CBG-Dependent Acetylation for CA Synthesis
Clavulanic acid (CA) is a secondary metabolite produced by Streptomyces clavuligerus that inhibits β-lactamases by binding irreversibly to an active-site serine (181). Because of the unique chemistry required to produce CA and its acylated versions, understanding the enzymes involved in its biosynthesis is of interest. The CA biosynthesis GNAT (CBG) is predicted to be involved in the synthesis of CA, since orf14 of S. clavuligerus produced notably less CA, though the specific role of CBG in CA synthesis is still unknown (182, 183).
The structure and gel filtration of CBG show that it is mainly a monomer containing two tandem GNAT domains linked by a β-strand, similar to the structure of the MshD and Fem proteins (178, 180, 184). While AcCoA was bound to the N-terminal GNAT domain in the crystal, the authors suggest that this may be a structural domain since the AcCoA is buried and not accessible for acetyl transfer (182). The C-terminal GNAT domain is most likely responsible for acyl transfer, containing a hydrophobic pocket that may accommodate other CoA analogs (182). Other acyl-CoAs (palmitoyl-CoA, lauroyl-CoA, myristoyl-CoA, succinyl-CoA) bind CBG, indicating that this enzyme may use other acyl-CoAs as substrates.
PseH-Dependent Acetylation for Pseudaminic Acid Synthesis
Pseudaminic acid (5,7-diacetamido-3,5,7,9-tetradeoxy-l-glycero-α-l-manno-nonulosonic acid) is used by some bacteria to modify their flagella (185). In the biosynthetic pathway, PseH acetylates UDP-4-amino-4,6-dideoxy-β-l-AltNAc to produce UDP-2,4-diacetamido-2,4,6-trideoxy-β-l-altropyranose using AcCoA as the acetyl donor (Fig. 8B) (185). Glycosylation is essential for flagellum synthesis and motility in Helicobacter pylori and Campylobacter jejuni, and pseH mutants are nonmotile (186, 187). Because pseudaminic acid production is unique to bacteria and acts as a flagellum decoration in pathogenic bacteria, this pathway is a good target for antimicrobial agents (185). The importance of glycosylation for virulence has been shown, thus providing an understanding of the mechanism of PseH that may make it a good therapeutic target (187, 188).
H. pylori PseH (HpPseH) in complex with AcCoA shows the typical GNAT domain with AcCoA binding between the splayed β-4 and β-5 sheets, though HpPseH has an additional C-terminal helix (α-5) in comparison to the structures of other GNATs (189, 190). The proximity of AcCoA to a conserved tyrosine, Y138, suggests that this residue may be a general acid, though no obvious residues appear to act as a general base (190). The acetyl group is located at the bottom of an active-site pocket made up of polar and aromatic residues (190). Modeling of UDP-4-amino-4,6-dideoxy-β-l-AltNAc in the pocket shows extensive interactions with the substrate and places the uracil ring between R30 and F52, allowing for π-stacking interactions (190). The authors suggest that a water molecule hydrogen bonded to S78 and T80 acts as a general base to deprotonate the 4-amino group for the nucleophilic attack (190). The dimer interface is composed of a β-sheet from each monomer to form a continuous β-sheet (190). In contrast, PseH from C. jejuni (CjPseH) acts as a monomer, though it also shows typical GNAT folding with a splayed AcCoA binding pocket (191). In CjPseH, residue Y128 is proposed to be the general acid because it is close to the sulfur atom of AcCoA (∼4.4 Å) and D70 may be a remote base in a mechanism similar to that described for HpPseH (191).
WecD-Dependent Acetylation for Antigen Synthesis
The outer membrane glycolipid enterobacterial common antigen (ECA) is found in many Enterobacteriaceae. The final step of synthesis involves acetylation of the dTDP-4-amino-4,6-dideoxy-α-d-galactose to TDP-4-acetamido-4,6-dideoxy-d-galactose by the TDP-fucosamine acetyltransferase WecD (Fig. 8C) (192, 193). ECA contributes to maintaining resistance to bile salts and thus plays an important role in virulence (194). Salmonella enterica wecD mutants have decreased virulence in a mouse model, highlighting the importance of the acetylation of this glycolipid (194). ECA may also be involved in biofilm formation, since an ECA-deficient strain of nontypeable Haemophilus influenzae produced smaller biofilms (195), and transcriptomics analysis of Actinobacillus pleuropneumoniae shows that wecD expression is upregulated in a biofilm (196). The production of ECA is also necessary for flagellar biosynthesis and motility in Serratia marcescens (197).
The crystal structure of E. coli WecD (EcWecD) with AcCoA has been solved (193). EcWecD acts as a dimer both in gel filtration chromatography experiments and in the crystal (193). EcWecD lacks 43 N-terminal residues compared to the sequences of other GNATs, thus lacking two α-helices and two β-strands (193). Like PseH, WecD acetylates the 4-amino group of a nucleotide-linked sugar, and the two crystals are similar, though EcWecD has an extra 70 amino acids at the N terminus compared to the sequence of HpPseH (190, 193). Residue Y208 is positioned near (∼3.0 Å) the sulfur atom of AcCoA and may act as a general acid, while residue E68 may be a remote base via a water-mediated deprotonation (193).
Aminoacyl-transferases Use Aminoacyl-tRNA as an Acyl Donor
Some GNATs can use an aminoacyl-tRNA as a donor instead of AcCoA. In these cases, the GNAT transfers the aminoacyl group to the substrate molecule. Aminoacyl-transferases are involved in the synthesis of various bacterial structures, including the cell wall, cell membranes, and secondary metabolites.
Fem aminoacyl-transferases are involved in cell wall synthesis.
Peptidoglycan is made up of repeating units of N-acetylglucosamine (GlcNAc) and N-acetyl muramic acid (MurNAc) glycans with pentapeptide stems cross-linked by peptide bridges (198–200). In some bacteria, the FemABX aminoacyl-transferase enzymes are responsible for synthesis of the peptide cross-bridges, transferring an aminoacyl group from aminoacyl-tRNA to the growing peptide cross-link (Fig. 9) (7). In bacteria with Fem proteins, the pentapeptide stem (l-Ala)-(d-Glu)-X-(d-Ala)-(d-Ala) is modified on the X residue, which can be lysine, ornithine, or meso-diaminopimelic acid (DAP) (7). These cross-links are important for cell structure and antibiotic resistance, with Fem proteins first being recognized for their importance for methicillin resistance, hence the name factors essential for the expression of methicillin resistance (200, 201). Fem proteins from different species form different peptide cross-links (7, 202). Fem proteins have been reported in a variety of organisms, including Staphylococcus, Streptococcus, and Enterococcus species (203–207). Bioinformatics studies indicate the presence of Fem proteins in diverse bacteria, including Borrelia burgdorferi, Streptomyces coelicolor, Streptomyces toyocaensis, and Clostridium perfringens, though experimental validation is needed (202, 208). Thus, the ability to aminoacylate the peptide cross-link is a conserved feature important for overall cell viability.
FIG 9.

Fem protein aminoacyl peptide cross-bridges in cell wall synthesis. Fem aminoacyl-transferase enzymes transfer an aminoacyl group from aminoacyl-tRNA to create a peptide cross-link. In Staphylococcus aureus, FemX transfers the first glycine residue, FemA transfers the second and third glycine residues, and FemB transfers the fourth and fifth glycine residues.
Streptococcus pneumoniae produces a dipeptide cross-bridge of l-Ser–l-Ala or l-Ala–l-Ala (202). MurM in S. pneumoniae transfers an alanine or a serine to the lysine of the peptide stem (205, 209, 210). MurN then adds the second residue (alanine), showing a greater than 10-fold higher catalytic efficiency for the l-Ala cross-link than for the l-Ser cross-link (206, 209). In Enterococcus faecalis, BppA1 adds the first alanine and BppA2 transfers the second alanine to form the dipeptide cross-link of l-Ala–l-Ala (207, 211).
Staphylococcus aureus produces pentaglycine cross-links that connect l-Lys (position 3) to a neighboring d-Ala (position 4) and that have been shown to be vital for cell integrity (212). FemX (also called FmhB) is essential and adds the first glycine residue of the cross-link (203). FemAB were found to add the next set of residues, with FemA adding the second and third glycine residues and FemB adding the last two (213–216). Though the essentiality of femAB in S. aureus is in question (212), femAB mutants were more susceptible to antibiotics than the wild type (214, 215, 217). The crystal structure of FemA from S. aureus is a monomer that adopts a globular fold made up of two GNAT domains with an L-shaped channel in the second GNAT domain, similar to the structure of CBG and MshD (180). The authors suggest that this channel is the binding location of the disaccharide hexapeptide substrate. Additionally, two antiparallel helices extend out, creating a coiled-coil domain similar to the coiled-coil domains found in seryl-tRNA synthetases, suggesting that this domain binds tRNA (180, 218).
Instead of the pentaglycine cross-link used by S. aureus, Weissella viridescens (also known as Lactobacillus viridescens) uses an l-Ala–l-Ser cross-link (202). W. viridescens FemX (WvFemX) was shown to transfer an amino acid (alanine, serine, or glycine) to a lysine residue of the UDP-MurNAc pentapeptide precursor (204). Kinetic analysis shows that WvFemX uses an ordered sequential binding mechanism (18). Mutational analysis suggests that D109 acts as a general base, while E320 is important for overall catalysis (18). The structure shows that WvFemX contains two GNAT domains separated by a deep cleft (∼15 Å deep and ∼20 Å wide) (184). Unlike S. aureus FemA (SaFemA), WvFemX does not contain a coil-coil region (180, 184). The UDP-MurNAc pentapeptide binds in the cleft, making contact with domain 1 mostly, with residues Lys36, Arg211, Tyr215, and Tyr256 being shown to be important for substrate binding (184, 219). The structure of WvFemX in complex with a peptidoglycan analogue and an RNA molecule mimicking Ala-tRNAAla shows that peptidyl-tRNA binding is also found in this cleft (220). The tRNA makes contact with a long channel spanning domain 2, interacting with Ile208 and Leu301 (220). Mutational analysis indicates that Lys305 is important for stabilizing the negative charge of l-Ala after the nucleophilic attack of Ala-tRNAAla, while Phe304 is important for π-stacking with the substrate (220).
Because peptide cross-links are important for cell viability and antibiotic resistance in a variety of bacteria, Fem proteins are a good target for antimicrobial agents. Studies showing the specificity of Fem proteins for their tRNA and lipid substrates have been reported (210, 221). Villet et al. demonstrate that posttranscriptional modifications and recognition of the acceptor stem of the tRNA are important criteria for the recognition by Fem proteins (222). Mutational analysis of S. pneumoniae MurM chimeras with substitutions in a 30-amino-acid region in the coil-coil domain demonstrates that this region determines residue specificity (Ala versus Ser) (223). Analogues of aminoacyl-tRNA substrates that inhibit Fem proteins have been reported (224–226). More work is needed to understand the mechanisms and specificity of Fem proteins in order for these proteins to be a possible future drug target.
Aminoacyl-phosphatidylglycerol synthases are involved in cell membrane synthesis.
Besides peptide stems, phospholipids can be aminoacylated. The addition of an amino acid reduces the overall negative charge of the bacterial membrane to help cells cope with cationic antimicrobial agents released by the immune system (227). A recent review of lipid aminoacylation can be found in reference 228. MprF (multiple-peptide resistance factor) and its homologue, LysX, transfer a lysyl residue from Lys-tRNALys onto phosphatidylglycerol (229–231). Staphylococcus aureus, Listeria monocytogenes, and Mycobacterium tuberculosis cells lacking mprF or lysX were more sensitive to antimicrobial agents than the wild type and had attenuated virulence in mouse models (230–232). These aminoacyl-phosphatidylglycerol synthases are bifunctional enzymes composed of a GNAT cytosolic C-terminal domain (to modify the lipid) and an integral membrane N-terminal domain (to flip the lipids into the periplasm) (228).
The structures of the GNAT domains of Pseudomonas aeruginosa Ala-tRNAAla-dependent alanyl-phosphatidylglycerol synthase (PaA-PGS; PA0920) and Bacillus licheniformis Lys-tRNALys-dependent lysyl-phosphatidylglycerol synthase (L-PGS; yfix) have been solved (233). The structures show that the C-terminal domain is made up of a tandem GNAT fold, similar to that of S. aureus FemA (SaFemA) and Weissella viridescens FemX (180, 184, 233). Like WvFemX, the crystal structure and mutational analysis of PaA-PGS demonstrate that residues Lys840 and Phe839 (corresponding to Lys305 and Phe304 of WvFemX) are important for binding tRNA (184, 233). The crystal structure of PaA-PGS shows that only the aminoacyl moiety is in direct contact with the main-chain atoms of the enzyme, consistent with work with MprF paralogs from Clostridium perfringens that indicates that the aminoacyl moiety is the main determinant for aminoacyl-tRNA specificity (233, 234). Docking and site-directed mutagenesis experiments show that a tunnel connected to but opposite the aminoacyl-tRNA binding pocket may act as a binding site for the lipid substrate in the membrane, with residues D765 and R768 being important for catalysis (233).
LFTs tag proteins for degradation.
The leucyl/phenylalanyl-tRNA protein transferase (LFT) also shows high homology to Fem proteins, with both enzymes transferring a residue from their cognate aminoacyl-tRNA. These proteins transfer a leucine or a phenylalanine residue to the N-terminal arginyl or lysyl residue of a protein (235). This N-end rule modification marks the protein for degradation (236). The structure of E. coli LFT shows that it contains a C-terminal GNAT domain, similar to that of Staphylococcus aureus FemA and Weissella viridescens FemX, while the N-terminal domain is not similar to a GNAT domain (180, 184, 235). The N-terminal domain appears to be important for stabilizing the aminoacyl-tRNA substrate via π-stacking with residue W49 (235). Site-directed mutagenesis demonstrates that the C-terminal GNAT domain is important for recognizing the correct aminoacyl-tRNA via a hydrophobic pocket that recognizes the aminoacyl moiety (235). The authors suggest that residues in the GNAT domain (E156 and Q188) are also important for orienting the substrate protein in close proximity of the aminoacyl-tRNA, promoting aminoacyl transfer (235).
GNAT aminoacyl-transferases are involved in antibiotic synthesis in Streptomyces.
Many Streptomyces species produce a variety of antibiotics. The synthesis of some of these antibiotics involves a tRNA-dependent GNAT aminoacyl-transferase (228). VlmA from Streptomyces viridifaciens transfers serine from Ser-tRNASer in the biosynthesis of valanimycin (237). In Streptomyces coeruleorubidus, PacB transfers alanine from Ala-tRNAAla in the production of pacidamycin, which inhibits cell wall synthesis (238). The antibiotic dehydrophos is produced by Streptomyces luridus and inhibits pyruvate dehydrogenase (239). DhpH and DhpK use Leu-tRNALeu and Gly-tRNAGly, respectively, to synthesize the tripeptide dehydrophos (240). FzmI uses Val-tRNAVal to synthesize fosfazinomycin (241). In Streptomyces luteocolor, the protein encoded by orf11 transfers glycine from Gly-tRNAGly to produce the streptothricin-like antibiotic BD-12, which has a glycine side chain instead of a β-lysine (Fig. 5A). This is the first reported instance of a tRNA-dependent peptide bond-forming enzyme capable of using an amino sugar as a substrate (242).
SMALL-MOLECULE ACETYLATION-DEPENDENT TRANSCRIPTION
Modulation of transcription is another way in which acetylation regulates bacterial cellular processes. Acetylation was first discovered on eukaryotic histones as a means to modulate transcriptionally active areas of the genome. In bacteria, direct acetylation of lysine residues of transcription factors has been shown to interfere with DNA binding. In E. coli, the GNAT Pat acetylates RcsB on residue K180 in the DNA-binding helix-turn-helix motif (243). Acetylation inhibits RcsB binding to the flhDC promoter, thereby modulating RcsB-dependent gene expression (243). Since this initial report, other acetylated bacterial transcription factors have been reported, such as RutR, RpsD, McbR, HilD, GntR, and RprY (243–246). Besides direct acetylation of transcription factors, acetylation of small molecules has been shown to modulate transcription.
BadL Acetylation of Aminobenzoates Regulates the Benzoate Degradation Operon via BadM
In Rhodopseudomonas palustris, the GNAT BadL acetylates the amino group of aminobenzoates, which bind to the repressor BadM (247). This BadM/acetylated aminobenzoate complex can no longer repress transcription of the benzoate degradation operon badDEFGAB, allowing the bacterium to catabolize benzoate (247). Acetylated aminobenzoates were also shown to be involved in the transcriptional control of light-harvesting complexes I and II in this purple nonsulfur photosynthetic bacterium (247). BadL is suggested to sense aminobenzoates in the environment to connect carbon metabolism and proton motor force generation via photosynthesis (247).
OatA Acetylation of O-Acetylserine Regulates Cysteine Biosynthesis Genes
In S. enterica, the GNAT OatA (OAS acetyltransferase A; formerly YjgM) acetylates the Nα-amino group of O-acetylserine (OAS) to produce N,O-diacetylserine (DAS) (248). OatA is a monomer that acetylates OAS and not N-acetylserine, indicating that it is an N-acetyltransferase (248). Overexpression of oatA resulted in increased cysteine biosynthesis gene expression, leading to shorter lag times in sulfate-limiting medium (248). Critically, the authors suggested that DAS, and not OAS, is the physiological signal that alters cysteine regulator CysB DNA binding to its regulon, though the addition of DAS does not alter binding in vitro (248). Possibly under physiological conditions, DAS may bind to CysB to activate the cysteine regulon, or the positive effect of DAS on the cys regulon is indirect (248). A GNAT from Clostridium difficile 630 (CD1211) has also been shown to acetylate OAS (82). Kinetic characterization of the CD1211 enzyme showed that the enzyme acetylated OAS, l-Ser, l-Thr, and l-Met with similar catalytic efficiencies and displayed positive cooperativity with AcCoA in the presence of each of the four substrates (249). The physiological role of this GNAT, possibly in cysteine biosynthesis, remains to be answered.
CONCLUSIONS
As schematized in Fig. 10, GNATs affect many vital cellular processes, ranging from detoxification and degradation to the synthesis of proteins or cell wall components. The plethora of substrates modified by the same GNAT fold displays the high versatility of this protein family.
FIG 10.

GNAT-dependent acylation modulates diverse cellular processes.
Characterization and Validation of Physiologically Relevant Substrates of GNATs
While the physiological role of many GNATs has been identified, many predicted GNATs have yet to be studied or validated to be bona fide GNATs. Currently, bioinformatic annotation is the main mechanism for the identification and classification of GNATs. There are more than 300,000 proteins that belong to this family (11), suggesting that there are many novel GNATs that are waiting to be discovered. This review focused mainly on GNATs that catalyze the transfer of acetyl moieties onto small molecules, but the transfer of other acyl moieties from acyl-CoA molecules should be explored in the future. In S. aureus, the GNAT AcuA (SaAcuA) is the first reported case of a bacterial GNAT that can succinylate a protein target (250). SaAcuA can also acetylate or propionylate, using acetyl-CoA or propionyl-CoA, respectively, suggesting that the aforementioned GNATs in this review may be capable of using other acyl donors. There is a great need for more in vitro and in vivo studies with predicted GNATs to validate their role as acetyl/acyltransferases.
With the great number of predicted GNATs, more work is also needed to elucidate the substrates of various GNATs. High-throughput studies have found possible substrates (82), but validation of each enzyme for those substrates is needed, as is a wider variety of substrates to be tested. Crystallization studies of annotated GNATs have given more structural information (249, 251), but the physiological role of these proteins must be explored in vivo.
Interestingly, many amino acids have been reported to be acetylated (82). Staphylococcus aureus SACOL1063 acetylates l-threonine and l-tryptophan, though it acetylates l-Trp with an order-of-magnitude lower catalytic efficiency than it does l-Thr (249). The Bar GNATs have been reported to acetylate tryptophan, in addition to phosphinothricin (132). However, the physiological role of amino acid acetylation by these proteins and others has not yet been determined and should be a focus of future study.
GNATs as Antimicrobial Targets
Acetylation of antibiotics is a major source of antibiotic resistance in bacteria. With the worldwide increase in the incidence of antibiotic-resistant bacteria, elucidating the molecular mechanisms of antibiotic-acetylating proteins can aid the development of new drugs and therapies. The crystal structures of medically relevant GNATs that have modified or expanded substrate specificity can help elucidate the structural reasons for the change in substrate range. The crystal structure of AAC(6′)-Ib11 highlighted how a few residue substitutions induced structural changes that led to a larger active site for larger aminoglycosides to bind (66, 67). The crystallization of other AAC(6′)-Ib N-terminus variants may lead to a better understanding of how bacterial GNATs adapt to the human use of antibiotics. These structural data may also aid in the development of GNAT inhibitors that can bind and block the active site. It is hoped that these inhibitors can be used in combination with aminoglycoside antibiotics clinically in the future (80).
Since GNATs are conserved in all domains of life, understanding the wide variety of acylated substrates can foster the identification of targets in various other organisms. Acetylation of numerous small molecules modulates many cellular processes specific to bacteria. Thus, GNATs may serve as good therapeutic targets, and recent work has focused on inhibitors of GNATs (80, 226). For example, mycobacteria exclusively use mycothiol as a redox-balancing molecule, and mycothiol synthesis is dependent on the GNAT MshD (175). An M. tuberculosis mshD strain displays decreased survival in the macrophage and higher sensitivity to hydrogen peroxide, suggesting that future work should explore exploiting MshD as a drug target (176, 177). Research understanding the wide variety of acylated substrates through genetic, biochemical, and structural means will facilitate a better comprehension and appreciation of the physiological impact of acetylation in all domains of life.
ACKNOWLEDGMENTS
We do not have a conflict of interest to declare.
This work was supported by U.S. PHS grant R35 GM130399 to J.C.E.-S.
We thank the reviewers of this work for their constructive criticism of the manuscript.
R.M.B. conceptualized the review, wrote the original draft, and edited the manuscript. J.C.E.-S. was responsible for acquiring the funding for this work, conceptualized the manuscript, and reviewed and edited all drafts.
Biographies

Rachel M. Burckhardt received her B.S. degree in biology (summa cum laude) from Alma College (2012) and her Ph.D. degree in microbiology from The University of Georgia (UGA; 2019). Her dissertation focused on the role of lysine acetylation in the detoxification of antibiotics in Gram-positive bacteria under the direction of Dr. Jorge C. Escalante-Semerena. In 2019, Dr. Burckhardt was selected by the Department of Microbiology of UGA to be the recipient of The William Jackson and Jane Payne Graduate Award, which recognizes excellence in graduate student research. Dr. Burckhardt is currently exploring employment opportunities in the private sector.

Jorge C. Escalante-Semerena received his Q.F.B. from the Universidad Nacional Autónoma de México, his M.S. and Ph.D. from The University of Illinois Urbana—Champaign, and postdoctoral training at The University of Utah. He was a Professor in the Department of Bacteriology of The University of Wisconsin—Madison until he moved to the Department of Microbiology of The University of Georgia, Athens, GA. Dr. Escalante-Semerena’s group discovered reversible lysine acetylation (RLA) in bacteria and was the first to establish the connection between this important posttranslational modification and metabolism in any organism. The widespread distribution of RLA in all domains of life reflects its importance to cell physiology. Dr. Escalante-Semerena is interested in establishing the physiological role of putative protein and small-molecule acyltransferases and deacylases in a diversity of prokaryotes. Dr. Escalante-Semerena’s group has been actively working in this area of cell physiology since 1998.
REFERENCES
- 1.Phillips DM. 1963. The presence of acetyl groups of histones. Biochem J 87:258–263. doi: 10.1042/bj0870258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Okamoto S, Suzuki Y. 1965. Chloramphenicol-, dihydrostreptomycin-, and kanamycin-inactivating enzymes from multiple drug-resistant Escherichia coli carrying episome ‘R.’ Nature 208:1301–1303. doi: 10.1038/2081301a0. [DOI] [PubMed] [Google Scholar]
- 3.Hentchel KL, Escalante-Semerena JC. 2015. Acylation of biomolecules in prokaryotes: a widespread strategy for the control of biological function and metabolic stress. Microbiol Mol Biol Rev 79:321–346. doi: 10.1128/MMBR.00020-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ramirez MS, Tolmasky ME. 2010. Aminoglycoside modifying enzymes. Drug Resist Updat 13:151–171. doi: 10.1016/j.drup.2010.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Woolridge DP, Martinez JD, Stringer DE, Gerner EW. 1999. Characterization of a novel spermidine/spermine acetyltransferase, BltD, from Bacillus subtilis. Biochem J 340(Pt 3):753–758. doi: 10.1042/0264-6021:3400753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ikeuchi Y, Kitahara K, Suzuki T. 2008. The RNA acetyltransferase driven by ATP hydrolysis synthesizes N4-acetylcytidine of tRNA anticodon. EMBO J 27:2194–2203. doi: 10.1038/emboj.2008.154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Dare K, Ibba M. 2012. Roles of tRNA in cell wall biosynthesis. Wiley Interdiscip Rev RNA 3:247–264. doi: 10.1002/wrna.1108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.VanDrisse CM, Escalante-Semerena JC. 2019. Protein acetylation in bacteria. Annu Rev Microbiol 73:111–132. doi: 10.1146/annurev-micro-020518-115526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Christensen DG, Xie X, Basisty N, Byrnes J, McSweeney S, Schilling B, Wolfe AJ. 2019. Post-translational protein acetylation: an elegant mechanism for bacteria to dynamically regulate metabolic functions. Front Microbiol 10:1604. doi: 10.3389/fmicb.2019.01604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Macek B, Forchhammer K, Hardouin J, Weber-Ban E, Grangeasse C, Mijakovic I. 2019. Protein post-translational modifications in bacteria. Nat Rev Microbiol 17:651–664. doi: 10.1038/s41579-019-0243-0. [DOI] [PubMed] [Google Scholar]
- 11.Salah Ud-Din AI, Tikhomirova A, Roujeinikova A. 2016. Structure and functional diversity of GCN5-related N-acetyltransferases (GNAT). Int J Mol Sci 17:E1018. doi: 10.3390/ijms17071018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Vetting MW, Carvalho L, Yu M, Hegde SS, Magnet S, Roderick SL, Blanchard JS. 2005. Structure and functions of the GNAT superfamily of acetyltransferases. Arch Biochem Biophys 433:212–226. doi: 10.1016/j.abb.2004.09.003. [DOI] [PubMed] [Google Scholar]
- 13.Neuwald AF, Landsman D. 1997. GCN5-related histone N-acetyltransferases belong to a diverse superfamily that includes the yeast SPT10 protein. Trends Biochem Sci 22:154–155. doi: 10.1016/s0968-0004(97)01034-7. [DOI] [PubMed] [Google Scholar]
- 14.Dyda F, Klein DC, Hickman AB. 2000. GCN5-related N-acetyltransferases: a structural overview. Annu Rev Biophys Biomol Struct 29:81–103. doi: 10.1146/annurev.biophys.29.1.81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Favrot L, Blanchard JS, Vergnolle O. 2016. Bacterial GCN5-related N-acetyltransferases: from resistance to regulation. Biochemistry 55:989–1002. doi: 10.1021/acs.biochem.5b01269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Rojas JR, Trievel RC, Zhou J, Mo Y, Li X, Berger SL, Allis CD, Marmorstein R. 1999. Structure of Tetrahymena GCN5 bound to coenzyme A and a histone H3 peptide. Nature 401:93–98. doi: 10.1038/43487. [DOI] [PubMed] [Google Scholar]
- 17.He H, Ding Y, Bartlam M, Sun F, Le Y, Qin X, Tang H, Zhang R, Joachimiak A, Liu J, Zhao N, Rao Z. 2003. Crystal structure of tabtoxin resistance protein complexed with acetyl coenzyme A reveals the mechanism for beta-lactam acetylation. J Mol Biol 325:1019–1030. doi: 10.1016/s0022-2836(02)01284-6. [DOI] [PubMed] [Google Scholar]
- 18.Hegde SS, Blanchard JS. 2003. Kinetic and mechanistic characterization of recombinant Lactobacillus viridescens FemX (UDP-N-acetylmuramoyl pentapeptide-lysine N6-alanyltransferase). J Biol Chem 278:22861–22867. doi: 10.1074/jbc.M301565200. [DOI] [PubMed] [Google Scholar]
- 19.Kim C, Villegas-Estrada A, Hesek D, Mobashery S. 2007. Mechanistic characterization of the bifunctional aminoglycoside-modifying enzyme AAC(3)-Ib/AAC(6′)-Ib′ from Pseudomonas aeruginosa. Biochemistry 46:5270–5282. doi: 10.1021/bi700111z. [DOI] [PubMed] [Google Scholar]
- 20.Kotra LP, Haddad J, Mobashery S. 2000. Aminoglycosides: perspectives on mechanisms of action and resistance and strategies to counter resistance. Antimicrob Agents Chemother 44:3249–3256. doi: 10.1128/aac.44.12.3249-3256.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Rather PN. 1998. Origins of the aminoglycoside modifying enzymes. Drug Resist Updat 1:285–291. doi: 10.1016/s1368-7646(98)80044-7. [DOI] [PubMed] [Google Scholar]
- 22.Moazed D, Noller HF. 1987. Interaction of antibiotics with functional sites in 16S ribosomal RNA. Nature 327:389–394. doi: 10.1038/327389a0. [DOI] [PubMed] [Google Scholar]
- 23.Fourmy D, Recht MI, Blanchard SC, Puglisi JD. 1996. Structure of the A site of Escherichia coli 16S ribosomal RNA complexed with an aminoglycoside antibiotic. Science 274:1367–1371. doi: 10.1126/science.274.5291.1367. [DOI] [PubMed] [Google Scholar]
- 24.Recht MI, Fourmy D, Blanchard SC, Dahlquist KD, Puglisi JD. 1996. RNA sequence determinants for aminoglycoside binding to an A-site rRNA model oligonucleotide. J Mol Biol 262:421–436. doi: 10.1006/jmbi.1996.0526. [DOI] [PubMed] [Google Scholar]
- 25.Davis BD. 1987. Mechanism of bactericidal action of aminoglycosides. Microbiol Rev 51:341–350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Krause KM, Serio AW, Kane TR, Connolly LE. 2016. Aminoglycosides: an overview. Cold Spring Harb Perspect Med 6:a027029. doi: 10.1101/cshperspect.a027029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Shaw KJ, Rather PN, Hare RS, Miller GH. 1993. Molecular genetics of aminoglycoside resistance genes and familial relationships of the aminoglycoside-modifying enzymes. Microbiol Rev 57:138–163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Magnet S, Blanchard JS. 2005. Molecular insights into aminoglycoside action and resistance. Chem Rev 105:477–498. doi: 10.1021/cr0301088. [DOI] [PubMed] [Google Scholar]
- 29.Mingeot-Leclercq MP, Glupczynski Y, Tulkens PM. 1999. Aminoglycosides: activity and resistance. Antimicrob Agents Chemother 43:727–737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lovering AM, White LO, Reeves DS. 1987. AAC(1): a new aminoglycoside-acetylating enzyme modifying the Cl aminogroup of apramycin. J Antimicrob Chemother 20:803–813. doi: 10.1093/jac/20.6.803. [DOI] [PubMed] [Google Scholar]
- 31.Sunada A, Nakajima M, Ikeda Y, Kondo S, Hotta K. 1999. Enzymatic 1-N-acetylation of paromomycin by an actinomycete strain #8 with multiple aminoglycoside resistance and paromomycin sensitivity. J Antibiot (Tokyo) 52:809–814. doi: 10.1002/chin.200007216. [DOI] [PubMed] [Google Scholar]
- 32.Williams JW, Northrop DB. 1976. Purification and properties of gentamicin acetyltransferase I. Biochemistry 15:125–131. doi: 10.1021/bi00646a019. [DOI] [PubMed] [Google Scholar]
- 33.Williams JW, Northrop DB. 1978. Substrate specificity and structure-activity relationships of gentamicin acetyltransferase I. The dependence of antibiotic resistance upon substrate Vmax/Km values. J Biol Chem 253:5908–5914. [PubMed] [Google Scholar]
- 34.Williams JW, Northrop DB. 1978. Kinetic mechanisms of gentamicin acetyltransferase I. Antibiotic-dependent shift from rapid to nonrapid equilibrium random mechanisms. J Biol Chem 253:5902–5907. [PubMed] [Google Scholar]
- 35.Magalhaes ML, Blanchard JS. 2005. The kinetic mechanism of AAC3-IV aminoglycoside acetyltransferase from Escherichia coli. Biochemistry 44:16275–16283. doi: 10.1021/bi051777d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Gionechetti F, Zucca P, Gombac F, Monti-Bragadin C, Lagatolla C, Tonin E, Edalucci E, Vitali LA, Dolzani L. 2008. Characterization of antimicrobial resistance and class 1 integrons in Enterobacteriaceae isolated from Mediterranean herring gulls (Larus cachinnans). Microb Drug Resist 14:93–99. doi: 10.1089/mdr.2008.0803. [DOI] [PubMed] [Google Scholar]
- 37.Wilson NL, Hall RM. 2010. Unusual class 1 integron configuration found in Salmonella genomic island 2 from Salmonella enterica serovar Emek. Antimicrob Agents Chemother 54:513–516. doi: 10.1128/AAC.00895-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wolf E, Vassilev A, Makino Y, Sali A, Nakatani Y, Burley SK. 1998. Crystal structure of a GCN5-related N-acetyltransferase: Serratia marcescens aminoglycoside 3-N-acetyltransferase. Cell 94:439–449. doi: 10.1016/s0092-8674(00)81585-8. [DOI] [PubMed] [Google Scholar]
- 39.Vliegenthart JS, Ketelaar-van Gaalen PA, van de Klundert JA. 1991. Nucleotide sequence of the aacC3 gene, a gentamicin resistance determinant encoding aminoglycoside-(3)-N-acetyltransferase III expressed in Pseudomonas aeruginosa but not in Escherichia coli. Antimicrob Agents Chemother 35:892–897. doi: 10.1128/aac.35.5.892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Brau B, Pilz U, Piepersberg W. 1984. Genes for gentamicin-(3)-N-acetyltransferases III and IV: I. Nucleotide sequence of the AAC(3)-IV gene and possible involvement of an IS140 element in its expression. Mol Gen Genet 193:179–187. doi: 10.1007/bf00327434. [DOI] [PubMed] [Google Scholar]
- 41.Heuer H, Krogerrecklenfort E, Wellington EM, Egan S, van Elsas JD, van Overbeek L, Collard JM, Guillaume G, Karagouni AD, Nikolakopoulou TL, Smalla K. 2002. Gentamicin resistance genes in environmental bacteria: prevalence and transfer. FEMS Microbiol Ecol 42:289–302. doi: 10.1111/j.1574-6941.2002.tb01019.x. [DOI] [PubMed] [Google Scholar]
- 42.Call DR, Singer RS, Meng D, Broschat SL, Orfe LH, Anderson JM, Herndon DR, Kappmeyer LS, Daniels JB, Besser TE. 2010. blaCMY-2-positive IncA/C plasmids from Escherichia coli and Salmonella enterica are a distinct component of a larger lineage of plasmids. Antimicrob Agents Chemother 54:590–596. doi: 10.1128/AAC.00055-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Rather PN, Mann PA, Mierzwa R, Hare RS, Miller GH, Shaw KJ. 1993. Analysis of the aac(3)-VIa gene encoding a novel 3-N-acetyltransferase. Antimicrob Agents Chemother 37:2074–2079. doi: 10.1128/aac.37.10.2074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ishikawa J, Sunada A, Oyama R, Hotta K. 2000. Identification and characterization of the point mutation which affects the transcription level of the chromosomal 3-N-acetyltransferase gene of Streptomyces griseus SS-1198. Antimicrob Agents Chemother 44:437–440. doi: 10.1128/aac.44.2.437-440.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.López-Cabrera M, Pérez-González JA, Heinzel P, Piepersberg W, Jiménez A. 1989. Isolation and nucleotide sequencing of an aminocyclitol acetyltransferase gene from Streptomyces rimosus forma paromomycinus. J Bacteriol 171:321–328. doi: 10.1128/jb.171.1.321-328.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Salauze D, Perez-Gonzalez JA, Piepersberg W, Davies J. 1991. Characterisation of aminoglycoside acetyltransferase-encoding genes of neomycin-producing Micromonospora chalcea and Streptomyces fradiae. Gene 101:143–148. doi: 10.1016/0378-1119(91)90237-6. [DOI] [PubMed] [Google Scholar]
- 47.Hotta K, Sunada A, Ishikawa J, Mizuno S, Ikeda Y, Kondo S. 1998. The novel enzymatic 3ʺ-N-acetylation of arbekacin by an aminoglycoside 3-N-acetyltransferase of Streptomyces origin and the resulting activity. J Antibiot (Tokyo) 51:735–742. doi: 10.7164/antibiotics.51.735. [DOI] [PubMed] [Google Scholar]
- 48.Chevereau M, Daniels PJ, Davies J, LeGoffic F. 1974. Aminoglycoside resistance in bacteria mediated by gentamicin acetyltransferase II, an enzyme modifying the 2′-amino group of aminoglycoside antibiotics. Biochemistry 13:598–603. doi: 10.1021/bi00700a030. [DOI] [PubMed] [Google Scholar]
- 49.Franklin K, Clarke AJ. 2001. Overexpression and characterization of the chromosomal aminoglycoside 2′-N-acetyltransferase of Providencia stuartii. Antimicrob Agents Chemother 45:2238–2244. doi: 10.1128/AAC.45.8.2238-2244.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Payie KG, Clarke AJ. 1997. Characterization of gentamicin 2′-N-acetyltransferase from Providencia stuartii: its use of peptidoglycan metabolites for acetylation of both aminoglycosides and peptidoglycan. J Bacteriol 179:4106–4114. doi: 10.1128/jb.179.13.4106-4114.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Payie KG, Rather PN, Clarke AJ. 1995. Contribution of gentamicin 2′-N-acetyltransferase to the O acetylation of peptidoglycan in Providencia stuartii. J Bacteriol 177:4303–4310. doi: 10.1128/jb.177.15.4303-4310.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Payie KG, Strating H, Clarke AJ. 1996. The role of O-acetylation in the metabolism of peptidoglycan in Providencia stuartii. Microb Drug Resist 2:135–140. doi: 10.1089/mdr.1996.2.135. [DOI] [PubMed] [Google Scholar]
- 53.Adams MD, Goglin K, Molyneaux N, Hujer KM, Lavender H, Jamison JJ, MacDonald IJ, Martin KM, Russo T, Campagnari AA, Hujer AM, Bonomo RA, Gill SR. 2008. Comparative genome sequence analysis of multidrug-resistant Acinetobacter baumannii. J Bacteriol 190:8053–8064. doi: 10.1128/JB.00834-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Lin T, Tang CG, Li QH, Ji J, Ge HY, Zhang XY, Sun HP. 2015. Identification of aac(2′)-I type b aminoglycoside-modifying enzyme genes in resistant Acinetobacter baumannii. Genet Mol Res 14:1828–1835. doi: 10.4238/2015.March.13.11. [DOI] [PubMed] [Google Scholar]
- 55.Ainsa JA, Martin C, Gicquel B, Gomez-Lus R. 1996. Characterization of the chromosomal aminoglycoside 2′-N-acetyltransferase gene from Mycobacterium fortuitum. Antimicrob Agents Chemother 40:2350–2355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Ainsa JA, Perez E, Pelicic V, Berthet FX, Gicquel B, Martin C. 1997. Aminoglycoside 2′-N-acetyltransferase genes are universally present in mycobacteria: characterization of the aac(2′)-Ic gene from Mycobacterium tuberculosis and the aac(2′)-Id gene from Mycobacterium smegmatis. Mol Microbiol 24:431–441. doi: 10.1046/j.1365-2958.1997.3471717.x. [DOI] [PubMed] [Google Scholar]
- 57.Hegde SS, Javid-Majd F, Blanchard JS. 2001. Overexpression and mechanistic analysis of chromosomally encoded aminoglycoside 2′-N-acetyltransferase (AAC(2′)-Ic) from Mycobacterium tuberculosis. J Biol Chem 276:45876–45881. doi: 10.1074/jbc.M108810200. [DOI] [PubMed] [Google Scholar]
- 58.Draker KA, Boehr DD, Elowe NH, Noga TJ, Wright GD. 2003. Functional annotation of putative aminoglycoside antibiotic modifying proteins in Mycobacterium tuberculosis H37Rv. J Antibiot (Tokyo) 56:135–142. doi: 10.7164/antibiotics.56.135. [DOI] [PubMed] [Google Scholar]
- 59.Sanz-Garcia F, Anoz-Carbonell E, Perez-Herran E, Martin C, Lucia A, Rodrigues L, Ainsa JA. 2019. Mycobacterial aminoglycoside acetyltransferases: a little of drug resistance, and a lot of other roles. Front Microbiol 10:46. doi: 10.3389/fmicb.2019.00046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Vetting MW, Hegde SS, Javid-Majd F, Blanchard JS, Roderick SL. 2002. Aminoglycoside 2′-N-acetyltransferase from Mycobacterium tuberculosis in complex with coenzyme A and aminoglycoside substrates. Nat Struct Biol 9:653–658. doi: 10.1038/nsb830. [DOI] [PubMed] [Google Scholar]
- 61.Robicsek A, Strahilevitz J, Jacoby GA, Macielag M, Abbanat D, Park CH, Bush K, Hooper DC. 2006. Fluoroquinolone-modifying enzyme: a new adaptation of a common aminoglycoside acetyltransferase. Nat Med 12:83–88. doi: 10.1038/nm1347. [DOI] [PubMed] [Google Scholar]
- 62.Stogios PJ, Kuhn ML, Evdokimova E, Law M, Courvalin P, Savchenko A. 2017. Structural and biochemical characterization of Acinetobacter spp. aminoglycoside acetyltransferases highlights functional and evolutionary variation among antibiotic resistance enzymes. ACS Infect Dis 3:132–143. doi: 10.1021/acsinfecdis.6b00058. [DOI] [PubMed] [Google Scholar]
- 63.Draker KA, Northrop DB, Wright GD. 2003. Kinetic mechanism of the GCN5-related chromosomal aminoglycoside acetyltransferase AAC(6′)-Ii from Enterococcus faecium: evidence of dimer subunit cooperativity. Biochemistry 42:6565–6574. doi: 10.1021/bi034148h. [DOI] [PubMed] [Google Scholar]
- 64.Vakulenko SB, Mobashery S. 2003. Versatility of aminoglycosides and prospects for their future. Clin Microbiol Rev 16:430–450. doi: 10.1128/cmr.16.3.430-450.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Vetting MW, Park CH, Hegde SS, Jacoby GA, Hooper DC, Blanchard JS. 2008. Mechanistic and structural analysis of aminoglycoside N-acetyltransferase AAC(6′)-Ib and its bifunctional, fluoroquinolone-active AAC(6′)-Ib-cr variant. Biochemistry 47:9825–9835. doi: 10.1021/bi800664x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Casin I, Hanau-Bercot B, Podglajen I, Vahaboglu H, Collatz E. 2003. Salmonella enterica serovar Typhimurium bla(PER-1)-carrying plasmid pSTI1 encodes an extended-spectrum aminoglycoside 6′-N-acetyltransferase of type Ib. Antimicrob Agents Chemother 47:697–703. doi: 10.1128/aac.47.2.697-703.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Maurice F, Broutin I, Podglajen I, Benas P, Collatz E, Dardel F. 2008. Enzyme structural plasticity and the emergence of broad-spectrum antibiotic resistance. EMBO Rep 9:344–349. doi: 10.1038/embor.2008.9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Zhang W, Fisher JF, Mobashery S. 2009. The bifunctional enzymes of antibiotic resistance. Curr Opin Microbiol 12:505–511. doi: 10.1016/j.mib.2009.06.013. [DOI] [PubMed] [Google Scholar]
- 69.Dubois V, Poirel L, Marie C, Arpin C, Nordmann P, Quentin C. 2002. Molecular characterization of a novel class 1 integron containing bla(GES-1) and a fused product of aac3-Ib/aac6′-Ib′ gene cassettes in Pseudomonas aeruginosa. Antimicrob Agents Chemother 46:638–645. doi: 10.1128/aac.46.3.638-645.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Tomar JS, Peddinti RK, Hosur RV. 2019. Aminoglycoside antibiotic resistance conferred by Hpa2 of MDR Acinetobacter baumannii: an unusual adaptation of a common histone acetyltransferase. Biochem J 476:795–808. doi: 10.1042/BCJ20180791. [DOI] [PubMed] [Google Scholar]
- 71.Vetting MW, Magnet S, Nieves E, Roderick SL, Blanchard JS. 2004. A bacterial acetyltransferase capable of regioselective N-acetylation of antibiotics and histones. Chem Biol 11:565–573. doi: 10.1016/j.chembiol.2004.03.017. [DOI] [PubMed] [Google Scholar]
- 72.Tomar JS, Peddinti RK. 2017. A. baumannii histone acetyl transferase HpaII: optimization of homology modeling, analysis of protein-protein interaction and virtual screening. J Biomol Struct Dyn 35:1115–1126. doi: 10.1080/07391102.2016.1172025. [DOI] [PubMed] [Google Scholar]
- 73.Magnet S, Lambert T, Courvalin P, Blanchard JS. 2001. Kinetic and mutagenic characterization of the chromosomally encoded Salmonella enterica AAC(6′)-Iy aminoglycoside N-acetyltransferase. Biochemistry 40:3700–3709. doi: 10.1021/bi002736e. [DOI] [PubMed] [Google Scholar]
- 74.Chen W, Biswas T, Porter VR, Tsodikov OV, Garneau-Tsodikova S. 2011. Unusual regioversatility of acetyltransferase Eis, a cause of drug resistance in XDR-TB. Proc Natl Acad Sci U S A 108:9804–9808. doi: 10.1073/pnas.1105379108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Wei J, Dahl JL, Moulder JW, Roberts EA, O'Gaora P, Young DB, Friedman RL. 2000. Identification of a Mycobacterium tuberculosis gene that enhances mycobacterial survival in macrophages. J Bacteriol 182:377–384. doi: 10.1128/jb.182.2.377-384.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Houghton JL, Green KD, Pricer RE, Mayhoub AS, Garneau-Tsodikova S. 2013. Unexpected N-acetylation of capreomycin by mycobacterial Eis enzymes. J Antimicrob Chemother 68:800–805. doi: 10.1093/jac/dks497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Zaunbrecher MA, Sikes RD Jr, Metchock B, Shinnick TM, Posey JE. 2009. Overexpression of the chromosomally encoded aminoglycoside acetyltransferase eis confers kanamycin resistance in Mycobacterium tuberculosis. Proc Natl Acad Sci U S A 106:20004–20009. doi: 10.1073/pnas.0907925106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Kim KH, An DR, Song J, Yoon JY, Kim HS, Yoon HJ, Im HN, Kim J, Kim DJ, Lee SJ, Kim KH, Lee HM, Kim HJ, Jo EK, Lee JY, Suh SW. 2012. Mycobacterium tuberculosis Eis protein initiates suppression of host immune responses by acetylation of DUSP16/MKP-7. Proc Natl Acad Sci U S A 109:7729–7734. doi: 10.1073/pnas.1120251109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Chen W, Green KD, Tsodikov OV, Garneau-Tsodikova S. 2012. Aminoglycoside multiacetylating activity of the enhanced intracellular survival protein from Mycobacterium smegmatis and its inhibition. Biochemistry 51:4959–4967. doi: 10.1021/bi3004473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Ngo HX, Green KD, Gajadeera CS, Willby MJ, Holbrook SYL, Hou C, Garzan A, Mayhoub AS, Posey JE, Tsodikov OV, Garneau-Tsodikova S. 2018. Potent 1,2,4-triazino[5,6 b]indole-3-thioether inhibitors of the kanamycin resistance enzyme Eis from Mycobacterium tuberculosis. ACS Infect Dis 4:1030–1040. doi: 10.1021/acsinfecdis.8b00074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Vaara M. 2019. Polymyxins and their potential next generation as therapeutic antibiotics. Front Microbiol 10:1689. doi: 10.3389/fmicb.2019.01689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Kuhn ML, Majorek KA, Minor W, Anderson WF. 2013. Broad-substrate screen as a tool to identify substrates for bacterial Gcn5-related N-acetyltransferases with unknown substrate specificity. Protein Sci 22:222–230. doi: 10.1002/pro.2199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Czub MP, Zhang B, Chiarelli MP, Majorek KA, Joe L, Porebski PJ, Revilla A, Wu W, Becker DP, Minor W, Kuhn ML. 2018. A Gcn5-related N-acetyltransferase (GNAT) capable of acetylating polymyxin B and colistin antibiotics in vitro. Biochemistry 57:7011–7020. doi: 10.1021/acs.biochem.8b00946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Baltz RH. 2007. Antimicrobials from actinomycetes: back to the future. Microbe 2:125–131. [Google Scholar]
- 85.Johnson AW, Westley JW. 1962. Streptothricin group of antibiotics. Part I. The general structural pattern. J Chem Soc 1962:1642–1655. doi: 10.1039/JR9620001642. [DOI] [Google Scholar]
- 86.Waksman SA, Woodruff HB. 1942. Streptothricin, a new selective bacteriostatic and bactericidal agent active against gram-negative bacteria. Proc Soc Exp Biol Med 49:207–210. doi: 10.3181/00379727-49-13515. [DOI] [Google Scholar]
- 87.Kobayashi T, Horinouchi S, Uozumi T, Beppu T. 1987. Purification and biochemical characterization of streptothricin acetyltransferase coded by the cloned streptothricin-resistance gene of Streptomyces lavendulae. J Antibiot (Tokyo) 40:1016–1022. doi: 10.7164/antibiotics.40.1016. [DOI] [PubMed] [Google Scholar]
- 88.Haupt I, Hubener R, Thrum H. 1978. Streptothricin F, an inhibitor of protein synthesis with miscoding activity. J Antibiot (Tokyo) 31:1137–1142. doi: 10.7164/antibiotics.31.1137. [DOI] [PubMed] [Google Scholar]
- 89.Haupt I, Jonak J, Rychlik I, Thrum H. 1980. Action of streptothricin F on ribosomal functions. J Antibiot (Tokyo) 33:636–641. doi: 10.7164/antibiotics.33.636. [DOI] [PubMed] [Google Scholar]
- 90.Kobayashi T, Uozumi T, Beppu T. 1986. Cloning and characterization of the streptothricin-resistance gene which encodes streptothricin acetyltransferase from Streptomyces lavendulae. J Antibiot (Tokyo) 39:688–693. doi: 10.7164/antibiotics.39.688. [DOI] [PubMed] [Google Scholar]
- 91.Krugel H, Fiedler G, Haupt I, Sarfert E, Simon H. 1988. Analysis of the nourseothricin-resistance gene (nat) of Streptomyces noursei. Gene 62:209–217. doi: 10.1016/0378-1119(88)90559-8. [DOI] [PubMed] [Google Scholar]
- 92.Krugel H, Fiedler G, Smith C, Baumberg S. 1993. Sequence and transcriptional analysis of the nourseothricin acetyltransferase-encoding gene nat1 from Streptomyces noursei. Gene 127:127–131. doi: 10.1016/0378-1119(93)90627-f. [DOI] [PubMed] [Google Scholar]
- 93.Fernández-Moreno MA, Vallín C, Malpartida F. 1997. Streptothricin biosynthesis is catalyzed by enzymes related to nonribosomal peptide bond formation. J Bacteriol 179:6929–6936. doi: 10.1128/jb.179.22.6929-6936.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Taniyama H, Sawada Y, Tanaka S. 1974. Chemical modification of streptothricin group antibiotics. II. Some amino derivatives in racemomycin-A and their biological activity. Chem Pharm Bull (Tokyo) 22:337–341. doi: 10.1248/cpb.22.337. [DOI] [PubMed] [Google Scholar]
- 95.Jacob J, Evers S, Bischoff K, Carlier C, Courvalin P. 1994. Characterization of the sat4 gene encoding a streptothricin acetyltransferase in Campylobacter coli BE/G4. FEMS Microbiol Lett 120:13–17. doi: 10.1111/j.1574-6968.1994.tb07000.x. [DOI] [PubMed] [Google Scholar]
- 96.Tietze E, Brevet J, Tschape H. 1987. Relationships among the streptothricin resistance transposons Tn1825 and Tn1826 and the trimethoprim resistance transposon Tn7. Plasmid 18:246–249. doi: 10.1016/0147-619x(87)90067-9. [DOI] [PubMed] [Google Scholar]
- 97.Burckhardt RM, Escalante-Semerena JC. 2017. In Bacillus subtilis, the SatA (formerly YyaR) acetyltransferase detoxifies streptothricin via lysine acetylation. Appl Environ Microbiol 83:e01590-17. doi: 10.1128/AEM.01590-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Burckhardt RM, Escalante-Semerena JC. 2019. Insights into the function of the N-acetyltransferase SatA that detoxifies streptothricin in Bacillus subtilis and Bacillus anthracis. Appl Environ Microbiol 85:e03029-18. doi: 10.1128/AEM.03029-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Moreno F, Gonzalez-Pastor JE, Baquero MR, Bravo D. 2002. The regulation of microcin B, C and J operons. Biochimie 84:521–529. doi: 10.1016/s0300-9084(02)01452-9. [DOI] [PubMed] [Google Scholar]
- 100.González-Pastor JE, San Millán JL, Moreno F. 1994. The smallest known gene. Nature 369:281. doi: 10.1038/369281a0. [DOI] [PubMed] [Google Scholar]
- 101.Garcia-Bustos JF, Pezzi N, Mendez E. 1985. Structure and mode of action of microcin 7, an antibacterial peptide produced by Escherichia coli. Antimicrob Agents Chemother 27:791–797. doi: 10.1128/aac.27.5.791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Metlitskaya A, Kazakov T, Kommer A, Pavlova O, Praetorius-Ibba M, Ibba M, Krasheninnikov I, Kolb V, Khmel I, Severinov K. 2006. Aspartyl-tRNA synthetase is the target of peptide nucleotide antibiotic microcin C. J Biol Chem 281:18033–18042. doi: 10.1074/jbc.M513174200. [DOI] [PubMed] [Google Scholar]
- 103.Novikova M, Kazakov T, Vondenhoff GH, Semenova E, Rozenski J, Metlytskaya A, Zukher I, Tikhonov A, Van Aerschot A, Severinov K. 2010. MccE provides resistance to protein synthesis inhibitor microcin C by acetylating the processed form of the antibiotic. J Biol Chem 285:12662–12669. doi: 10.1074/jbc.M109.080192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Kazakov T, Kuznedelov K, Semenova E, Mukhamedyarov D, Datsenko KA, Metlitskaya A, Vondenhoff GH, Tikhonov A, Agarwal V, Nair S, Van Aerschot A, Severinov K. 2014. The RimL transacetylase provides resistance to translation inhibitor microcin C. J Bacteriol 196:3377–3385. doi: 10.1128/JB.01584-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Agarwal V, Metlitskaya A, Severinov K, Nair SK. 2011. Structural basis for microcin C7 inactivation by the MccE acetyltransferase. J Biol Chem 286:21295–21303. doi: 10.1074/jbc.M111.226282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Forouhar F, Lee IS, Vujcic J, Vujcic S, Shen J, Vorobiev SM, Xiao R, Acton TB, Montelione GT, Porter CW, Tong L. 2005. Structural and functional evidence for Bacillus subtilis PaiA as a novel N1-spermidine/spermine acetyltransferase. J Biol Chem 280:40328–40336. doi: 10.1074/jbc.M505332200. [DOI] [PubMed] [Google Scholar]
- 107.Pegg AE. 2008. Spermidine/spermine-N(1)-acetyltransferase: a key metabolic regulator. Am J Physiol Endocrinol Metab 294:E995–E1010. doi: 10.1152/ajpendo.90217.2008. [DOI] [PubMed] [Google Scholar]
- 108.Perez-Leal O, Merali S. 2012. Regulation of polyamine metabolism by translational control. Amino Acids 42:611–617. doi: 10.1007/s00726-011-1036-6. [DOI] [PubMed] [Google Scholar]
- 109.Tabor CW. 1968. The effects of temperature on the acetylation of spermidine. Biochem Biophys Res Commun 30:339–342. doi: 10.1016/0006-291x(68)90747-x. [DOI] [PubMed] [Google Scholar]
- 110.Limsuwun K, Jones PG. 2000. Spermidine acetyltransferase is required to prevent spermidine toxicity at low temperatures in Escherichia coli. J Bacteriol 182:5373–5380. doi: 10.1128/jb.182.19.5373-5380.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Vujcic S, Liang P, Diegelman P, Kramer DL, Porter CW. 2003. Genomic identification and biochemical characterization of the mammalian polyamine oxidase involved in polyamine back-conversion. Biochem J 370:19–28. doi: 10.1042/BJ20021779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Carper SW, Willis DG, Manning KA, Gerner EW. 1991. Spermidine acetylation in response to a variety of stresses in Escherichia coli. J Biol Chem 266:12439–12441. [PubMed] [Google Scholar]
- 113.Shah P, Nanduri B, Swiatlo E, Ma Y, Pendarvis K. 2011. Polyamine biosynthesis and transport mechanisms are crucial for fitness and pathogenesis of Streptococcus pneumoniae. Microbiology 157:504–515. doi: 10.1099/mic.0.042564-0. [DOI] [PubMed] [Google Scholar]
- 114.Joshi GS, Spontak JS, Klapper DG, Richardson AR. 2011. Arginine catabolic mobile element encoded speG abrogates the unique hypersensitivity of Staphylococcus aureus to exogenous polyamines. Mol Microbiol 82:9–20. doi: 10.1111/j.1365-2958.2011.07809.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Li B, Maezato Y, Kim SH, Kurihara S, Liang J, Michael AJ. 2019. Polyamine-independent growth and biofilm formation, and functional spermidine/spermine N-acetyltransferases in Staphylococcus aureus and Enterococcus faecalis. Mol Microbiol 111:159–175. doi: 10.1111/mmi.14145. [DOI] [PubMed] [Google Scholar]
- 116.Martini C, Michaux C, Bugli F, Arcovito A, Iavarone F, Cacaci M, Paroni Sterbini F, Hartke A, Sauvageot N, Sanguinetti M, Posteraro B, Giard JC. 2015. The polyamine N-acetyltransferase-like enzyme PmvE plays a role in the virulence of Enterococcus faecalis. Infect Immun 83:364–371. doi: 10.1128/IAI.02585-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Barbagallo M, Di Martino ML, Marcocci L, Pietrangeli P, De Carolis E, Casalino M, Colonna B, Prosseda G. 2011. A new piece of the Shigella pathogenicity puzzle: spermidine accumulation by silencing of the speG gene [corrected]. PLoS One 6:e27226. doi: 10.1371/journal.pone.0027226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Filippova EV, Shuvalova L, Minasov G, Kiryukhina O, Zhang Y, Clancy S, Radhakrishnan I, Joachimiak A, Anderson WF. 2011. Crystal structure of the novel PaiA N-acetyltransferase from Thermoplasma acidophilum involved in the negative control of sporulation and degradative enzyme production. Proteins 79:2566–2577. doi: 10.1002/prot.23062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Filippova EV, Kuhn ML, Osipiuk J, Kiryukhina O, Joachimiak A, Ballicora MA, Anderson WF. 2015. A novel polyamine allosteric site of SpeG from Vibrio cholerae is revealed by its dodecameric structure. J Mol Biol 427:1316–1334. doi: 10.1016/j.jmb.2015.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Filippova EV, Weigand S, Osipiuk J, Kiryukhina O, Joachimiak A, Anderson WF. 2015. Substrate-induced allosteric change in the quaternary structure of the spermidine N-acetyltransferase SpeG. J Mol Biol 427:3538–3553. doi: 10.1016/j.jmb.2015.09.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Woolridge DP, Vazquez-Laslop N, Markham PN, Chevalier MS, Gerner EW, Neyfakh AA. 1997. Efflux of the natural polyamine spermidine facilitated by the Bacillus subtilis multidrug transporter Blt. J Biol Chem 272:8864–8866. doi: 10.1074/jbc.272.14.8864. [DOI] [PubMed] [Google Scholar]
- 122.Nguyen AQ, Schneider J, Wendisch VF. 2015. Elimination of polyamine N-acetylation and regulatory engineering improved putrescine production by Corynebacterium glutamicum. J Biotechnol 201:75–85. doi: 10.1016/j.jbiotec.2014.10.035. [DOI] [PubMed] [Google Scholar]
- 123.Thompson CJ, Movva NR, Tizard R, Crameri R, Davies JE, Lauwereys M, Botterman J. 1987. Characterization of the herbicide-resistance gene bar from Streptomyces hygroscopicus. EMBO J 6:2519–2523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Bayer E, Gugel KH, Hagele K, Hagenmaier H, Jessipow S, Konig WA, Zahner H. 1972. Metabolic products of microorganisms. 98. Phosphinothricin and phosphinothricyl-alanyl-analine. Helv Chim Acta 55:224–239. (In German.) doi: 10.1002/hlca.19720550126. [DOI] [PubMed] [Google Scholar]
- 125.Gill HS, Eisenberg D. 2001. The crystal structure of phosphinothricin in the active site of glutamine synthetase illuminates the mechanism of enzymatic inhibition. Biochemistry 40:1903–1912. doi: 10.1021/bi002438h. [DOI] [PubMed] [Google Scholar]
- 126.Tachibana K, Watanabe T, Sekizawa Y, Takematsu T. 1986. Accumulation of ammonia in plants treated with bialaphos. J Pesticide Sci 11:33–37. doi: 10.1584/jpestics.11.33. [DOI] [Google Scholar]
- 127.Murakami T, Anzai H, Imai S, Satoh A, Nagaoka K, Thompson CJ. 1986. The bialaphos biosynthetic genes of Streptomyces hygroscopicus: molecular cloning and characterization of the gene cluster. Mol Gen Genet 205:42–50. doi: 10.1007/BF02428031. [DOI] [Google Scholar]
- 128.Wohlleben W, Arnold W, Broer I, Hillemann D, Strauch E, Puhler A. 1988. Nucleotide sequence of the phosphinothricin N-acetyltransferase gene from Streptomyces viridochromogenes Tu494 and its expression in Nicotiana tabacum. Gene 70:25–37. doi: 10.1016/0378-1119(88)90101-1. [DOI] [PubMed] [Google Scholar]
- 129.Strauch E, Wohlleben W, Puhler A. 1988. Cloning of a phosphinothricin N-acetyltransferase gene from Streptomyces viridochromogenes Tu494 and its expression in Streptomyces lividans and Escherichia coli. Gene 63:65–74. doi: 10.1016/0378-1119(88)90546-x. [DOI] [PubMed] [Google Scholar]
- 130.Block MD, Botterman J, Vandewiele M, Dockx J, Thoen C, Gossele V, Movva NR, Thompson C, Montagu MV, Leemans J. 1987. Engineering herbicide resistance in plants by expression of a detoxifying enzyme. EMBO J 6:2513–2518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.O’Connor SE. 2017. Raising the BAR of specificity. Nat Plants 3:924–925. doi: 10.1038/s41477-017-0074-9. [DOI] [PubMed] [Google Scholar]
- 132.Christ B, Hochstrasser R, Guyer L, Francisco R, Aubry S, Hortensteiner S, Weng JK. 2017. Non-specific activities of the major herbicide-resistance gene BAR. Nat Plants 3:937–945. doi: 10.1038/s41477-017-0061-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Manning JM, Moore S, Rowe WB, Meister A. 1969. Identification of l-methionine S-sulfoximine as the diastereoisomer of l-methionine SR-sulfoximine that inhibits glutamine synthetase. Biochemistry 8:2681–2685. doi: 10.1021/bi00834a066. [DOI] [PubMed] [Google Scholar]
- 134.Davies AM, Tata R, Beavil RL, Sutton BJ, Brown PR. 2007. l-Methionine sulfoximine, but not phosphinothricin, is a substrate for an acetyltransferase (gene PA4866) from Pseudomonas aeruginosa: structural and functional studies. Biochemistry 46:1829–1839. doi: 10.1021/bi0615238. [DOI] [PubMed] [Google Scholar]
- 135.Davies AM, Tata R, Snape A, Sutton BJ, Brown PR. 2009. Structure and substrate specificity of acetyltransferase ACIAD1637 from Acinetobacter baylyi ADP1. Biochimie 91:484–489. doi: 10.1016/j.biochi.2008.12.003. [DOI] [PubMed] [Google Scholar]
- 136.Hentchel KL, Escalante-Semerena JC. 2015. In Salmonella enterica, the Gcn5-related acetyltransferase MddA (formerly YncA) acetylates methionine sulfoximine and methionine sulfone, blocking their toxic effects. J Bacteriol 197:314–325. doi: 10.1128/JB.02311-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Davies AM, Tata R, Agha R, Sutton BJ, Brown PR. 2005. Crystal structure of a putative phosphinothricin acetyltransferase (PA4866) from Pseudomonas aeruginosa PAC1. Proteins 61:677–679. doi: 10.1002/prot.20603. [DOI] [PubMed] [Google Scholar]
- 138.VanDrisse CM, Hentchel KL, Escalante-Semerena JC. 2016. Phosphinothricin acetyltransferases identified using in vivo, in vitro, and bioinformatic analyses. Appl Environ Microbiol 82:7041–7051. doi: 10.1128/AEM.02604-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Paez-Espino AD, Chavarria M, de Lorenzo V. 2015. The two paralogue phoN (phosphinothricin acetyl transferase) genes of Pseudomonas putida encode functionally different proteins. Environ Microbiol 17:3330–3340. doi: 10.1111/1462-2920.12798. [DOI] [PubMed] [Google Scholar]
- 140.Bender CL, Alarcón-Chaidez F, Gross DC. 1999. Pseudomonas syringae phytotoxins: mode of action, regulation, and biosynthesis by peptide and polyketide synthetases. Microbiol Mol Biol Rev 63:266–292. doi: 10.1128/MMBR.63.2.266-292.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Stewart WW. 1971. Isolation and proof of structure of wildfire toxin. Nature 229:174–178. doi: 10.1038/229174a0. [DOI] [PubMed] [Google Scholar]
- 142.Thomas MD, Langston-Unkefer PJ, Uchytil TF, Durbin RD. 1983. Inhibition of glutamine synthetase from pea by tabtoxinine-beta-lactam. Plant Physiol 71:912–915. doi: 10.1104/pp.71.4.912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Anzai H, Yoneyama K, Yamaguchi I. 1989. Transgenic tobacco resistant to a bacterial disease by the detoxification of a pathogenic toxin. Mol Gen Genet 219:492–494. doi: 10.1007/BF00259626. [DOI] [Google Scholar]
- 144.Anzai H, Yoneyama K, Yamaguchi I. 1990. The nucleotide sequence of tabtoxin resistance gene (ttr) of Pseudomonas syringae pv. tabaci. Nucleic Acids Res 18:1890. doi: 10.1093/nar/18.7.1890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Liu J, Le Y, Ye B, Zhen Y, Zhu C, Shen J, Zhang R. 2002. Tabtoxin-resistant protein: overexpression, purification, and characterization. Protein Expr Purif 24:439–444. doi: 10.1006/prep.2001.1586. [DOI] [PubMed] [Google Scholar]
- 146.Wencewicz TA, Walsh CT. 2012. Pseudomonas syringae self-protection from tabtoxinine-beta-lactam by ligase TblF and acetylase Ttr. Biochemistry 51:7712–7725. doi: 10.1021/bi3011384. [DOI] [PubMed] [Google Scholar]
- 147.Buts L, Lah J, Dao-Thi MH, Wyns L, Loris R. 2005. Toxin-antitoxin modules as bacterial metabolic stress managers. Trends Biochem Sci 30:672–679. doi: 10.1016/j.tibs.2005.10.004. [DOI] [PubMed] [Google Scholar]
- 148.Page R, Peti W. 2016. Toxin-antitoxin systems in bacterial growth arrest and persistence. Nat Chem Biol 12:208–214. doi: 10.1038/nchembio.2044. [DOI] [PubMed] [Google Scholar]
- 149.Jurėnas D, Garcia-Pino A, Van Melderen L. 2017. Novel toxins from type II toxin-antitoxin systems with acetyltransferase activity. Plasmid 93:30–35. doi: 10.1016/j.plasmid.2017.08.005. [DOI] [PubMed] [Google Scholar]
- 150.Iqbal N, Guerout AM, Krin E, Le Roux F, Mazel D. 2015. Comprehensive functional analysis of the 18 Vibrio cholerae N16961 toxin-antitoxin systems substantiates their role in stabilizing the superintegron. J Bacteriol 197:2150–2159. doi: 10.1128/JB.00108-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Van Acker H, Sass A, Dhondt I, Nelis HJ, Coenye T. 2014. Involvement of toxin-antitoxin modules in Burkholderia cenocepacia biofilm persistence. Pathog Dis 71:326–335. doi: 10.1111/2049-632X.12177. [DOI] [PubMed] [Google Scholar]
- 152.Yeo CC. 2018. GNAT toxins of bacterial toxin-antitoxin systems: acetylation of charged tRNAs to inhibit translation. Mol Microbiol 108:331–335. doi: 10.1111/mmi.13958. [DOI] [PubMed] [Google Scholar]
- 153.Jurėnas D, Chatterjee S, Konijnenberg A, Sobott F, Droogmans L, Garcia-Pino A, Van Melderen L. 2017. AtaT blocks translation initiation by N-acetylation of the initiator tRNA(fMet). Nat Chem Biol 13:640–646. doi: 10.1038/nchembio.2346. [DOI] [PubMed] [Google Scholar]
- 154.Van Melderen L, Jurenas D, Garcia-Pino A. 2018. Messing up translation from the start: how AtaT inhibits translation initiation in E. coli. RNA Biol 15:303–307. doi: 10.1080/15476286.2017.1391439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Wilcox B, Osterman I, Serebryakova M, Lukyanov D, Komarova E, Gollan B, Morozova N, Wolf YI, Makarova KS, Helaine S, Sergiev P, Dubiley S, Borukhov S, Severinov K. 2018. Escherichia coli ItaT is a type II toxin that inhibits translation by acetylating isoleucyl-tRNAIle. Nucleic Acids Res 46:7873–7885. doi: 10.1093/nar/gky560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Cheverton AM, Gollan B, Przydacz M, Wong CT, Mylona A, Hare SA, Helaine S. 2016. A Salmonella toxin promotes persister formation through acetylation of tRNA. Mol Cell 63:86–96. doi: 10.1016/j.molcel.2016.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Rycroft JA, Gollan B, Grabe GJ, Hall A, Cheverton AM, Larrouy-Maumus G, Hare SA, Helaine S. 2018. Activity of acetyltransferase toxins involved in Salmonella persister formation during macrophage infection. Nat Commun 9:1993. doi: 10.1038/s41467-018-04472-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.VanDrisse CM, Parks AR, Escalante-Semerena JC. 2017. A toxin involved in Salmonella persistence regulates its activity by acetylating its cognate antitoxin, a modification reversed by CobB sirtuin deacetylase. mBio 8:e00708-17. doi: 10.1128/mBio.00708-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Jurėnas D, Van Melderen L, Garcia-Pino A. 2018. Crystallization and X-ray analysis of all of the players in the autoregulation of the ataRT toxin-antitoxin system. Acta Crystallogr F Struct Biol Commun 74:391–401. doi: 10.1107/S2053230X18007914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Jurėnas D, Van Melderen L, Garcia-Pino A. 2019. Mechanism of regulation and neutralization of the AtaR-AtaT toxin-antitoxin system. Nat Chem Biol 15:285–294. doi: 10.1038/s41589-018-0216-z. [DOI] [PubMed] [Google Scholar]
- 161.Qian H, Yao Q, Tai C, Deng Z, Gan J, Ou HY. 2018. Identification and characterization of acetyltransferase-type toxin-antitoxin locus in Klebsiella pneumoniae. Mol Microbiol 108:336–349. doi: 10.1111/mmi.13934. [DOI] [PubMed] [Google Scholar]
- 162.McVicker G, Tang CM. 2016. Deletion of toxin-antitoxin systems in the evolution of Shigella sonnei as a host-adapted pathogen. Nat Microbiol 2:16204. doi: 10.1038/nmicrobiol.2016.204. [DOI] [PubMed] [Google Scholar]
- 163.Makarova KS, Wolf YI, Koonin EV. 2009. Comprehensive comparative-genomic analysis of type 2 toxin-antitoxin systems and related mobile stress response systems in prokaryotes. Biol Direct 4:19. doi: 10.1186/1745-6150-4-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Wei YQ, Bi DX, Wei DQ, Ou HY. 2016. Prediction of type II toxin-antitoxin loci in Klebsiella pneumoniae genome sequences. Interdiscip Sci 8:143–149. doi: 10.1007/s12539-015-0135-6. [DOI] [PubMed] [Google Scholar]
- 165.Habib G, Zhu Q, Sun B. 2018. Bioinformatics and functional assessment of toxin-antitoxin systems in Staphylococcus aureus. Toxins (Basel) 10:E473. doi: 10.3390/toxins10110473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Xie Y, Wei Y, Shen Y, Li X, Zhou H, Tai C, Deng Z, Ou HY. 2018. TADB 2.0: an updated database of bacterial type II toxin-antitoxin loci. Nucleic Acids Res 46:D749–D753. doi: 10.1093/nar/gkx1033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Jurenaite M, Markuckas A, Suziedeliene E. 2013. Identification and characterization of type II toxin-antitoxin systems in the opportunistic pathogen Acinetobacter baumannii. J Bacteriol 195:3165–3172. doi: 10.1128/JB.00237-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Oashi Z, Murao K, Yahagi T, Von Minden DL, McCloskey JA, Nishimura S. 1972. Characterization of C+ located in the first position of the anticodon of Escherichia coli tRNAMet as N4-acetylcytidine. Biochim Biophys Acta 262:209–213. doi: 10.1016/0005-2787(72)90234-1. [DOI] [PubMed] [Google Scholar]
- 169.Stern L, Schulman LH. 1978. The role of the minor base N4-acetylcytidine in the function of the Escherichia coli noninitiator methionine transfer RNA. J Biol Chem 253:6132–6139. [PubMed] [Google Scholar]
- 170.Giaever G, Chu AM, Ni L, Connelly C, Riles L, Véronneau S, Dow S, Lucau-Danila A, Anderson K, André B, Arkin AP, Astromoff A, El-Bakkoury M, Bangham R, Benito R, Brachat S, Campanaro S, Curtiss M, Davis K, Deutschbauer A, Entian K-D, Flaherty P, Foury F, Garfinkel DJ, Gerstein M, Gotte D, Güldener U, Hegemann JH, Hempel S, Herman Z, Jaramillo DF, Kelly DE, Kelly SL, Kötter P, LaBonte D, Lamb DC, Lan N, Liang H, Liao H, Liu L, Luo C, Lussier M, Mao R, Menard P, Ooi SL, Revuelta JL, Roberts CJ, Rose M, Ross-Macdonald P, Scherens B, et al. 2002. Functional profiling of the Saccharomyces cerevisiae genome. Nature 418:387–391. doi: 10.1038/nature00935. [DOI] [PubMed] [Google Scholar]
- 171.Chimnaronk S, Suzuki T, Manita T, Ikeuchi Y, Yao M, Suzuki T, Tanaka I. 2009. RNA helicase module in an acetyltransferase that modifies a specific tRNA anticodon. EMBO J 28:1362–1373. doi: 10.1038/emboj.2009.69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Newton GL, Arnold K, Price MS, Sherrill C, Delcardayre SB, Aharonowitz Y, Cohen G, Davies J, Fahey RC, Davis C. 1996. Distribution of thiols in microorganisms: mycothiol is a major thiol in most actinomycetes. J Bacteriol 178:1990–1995. doi: 10.1128/jb.178.7.1990-1995.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Fan F, Vetting MW, Frantom PA, Blanchard JS. 2009. Structures and mechanisms of the mycothiol biosynthetic enzymes. Curr Opin Chem Biol 13:451–459. doi: 10.1016/j.cbpa.2009.07.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Rawat M, Newton GL, Ko M, Martinez GJ, Fahey RC, Av-Gay Y. 2002. Mycothiol-deficient Mycobacterium smegmatis mutants are hypersensitive to alkylating agents, free radicals, and antibiotics. Antimicrob Agents Chemother 46:3348–3355. doi: 10.1128/aac.46.11.3348-3355.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Koledin T, Newton GL, Fahey RC. 2002. Identification of the mycothiol synthase gene (mshD) encoding the acetyltransferase producing mycothiol in actinomycetes. Arch Microbiol 178:331–337. doi: 10.1007/s00203-002-0462-y. [DOI] [PubMed] [Google Scholar]
- 176.Rengarajan J, Bloom BR, Rubin EJ. 2005. Genome-wide requirements for Mycobacterium tuberculosis adaptation and survival in macrophages. Proc Natl Acad Sci U S A 102:8327–8332. doi: 10.1073/pnas.0503272102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Buchmeier NA, Newton GL, Fahey RC. 2006. A mycothiol synthase mutant of Mycobacterium tuberculosis has an altered thiol-disulfide content and limited tolerance to stress. J Bacteriol 188:6245–6252. doi: 10.1128/JB.00393-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Vetting MW, Roderick SL, Yu M, Blanchard JS. 2003. Crystal structure of mycothiol synthase (Rv0819) from Mycobacterium tuberculosis shows structural homology to the GNAT family of N-acetyltransferases. Protein Sci 12:1954–1959. doi: 10.1110/ps.03153703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Vetting MW, Yu M, Rendle PM, Blanchard JS. 2006. The substrate-induced conformational change of Mycobacterium tuberculosis mycothiol synthase. J Biol Chem 281:2795–2802. doi: 10.1074/jbc.M510798200. [DOI] [PubMed] [Google Scholar]
- 180.Benson TE, Prince DB, Mutchler VT, Curry KA, Ho AM, Sarver RW, Hagadorn JC, Choi GH, Garlick RL. 2002. X-ray crystal structure of Staphylococcus aureus FemA. Structure 10:1107–1115. doi: 10.1016/s0969-2126(02)00807-9. [DOI] [PubMed] [Google Scholar]
- 181.Liras P, Rodríguez-García A. 2000. Clavulanic acid, a beta-lactamase inhibitor: biosynthesis and molecular genetics. Appl Microbiol Biotechnol 54:467–475. doi: 10.1007/s002530000420. [DOI] [PubMed] [Google Scholar]
- 182.Iqbal A, Arunlanantham H, Brown T Jr, Chowdhury R, Clifton IJ, Kershaw NJ, Hewitson KS, McDonough MA, Schofield CJ. 2010. Crystallographic and mass spectrometric analyses of a tandem GNAT protein from the clavulanic acid biosynthesis pathway. Proteins 78:1398–1407. doi: 10.1002/prot.22653. [DOI] [PubMed] [Google Scholar]
- 183.Mellado E, Lorenzana LM, Rodrı Guez-Sáiz M, Dı Ez B, Liras P, Barredo JL. 2002. The clavulanic acid biosynthetic cluster of Streptomyces clavuligerus: genetic organization of the region upstream of the car gene. Microbiology 148:1427–1438. doi: 10.1099/00221287-148-5-1427. [DOI] [PubMed] [Google Scholar]
- 184.Biarrotte-Sorin S, Maillard AP, Delettre J, Sougakoff W, Arthur M, Mayer C. 2004. Crystal structures of Weissella viridescens FemX and its complex with UDP-MurNAc-pentapeptide: insights into FemABX family substrates recognition. Structure 12:257–267. doi: 10.1016/j.str.2004.01.006. [DOI] [PubMed] [Google Scholar]
- 185.Schoenhofen IC, McNally DJ, Brisson JR, Logan SM. 2006. Elucidation of the CMP-pseudaminic acid pathway in Helicobacter pylori: synthesis from UDP-N-acetylglucosamine by a single enzymatic reaction. Glycobiology 16:8c–14c. doi: 10.1093/glycob/cwl010. [DOI] [PubMed] [Google Scholar]
- 186.Josenhans C, Vossebein L, Friedrich S, Suerbaum S. 2002. The neuA/flmD gene cluster of Helicobacter pylori is involved in flagellar biosynthesis and flagellin glycosylation. FEMS Microbiol Lett 210:165–172. doi: 10.1111/j.1574-6968.2002.tb11176.x. [DOI] [PubMed] [Google Scholar]
- 187.Guerry P, Ewing CP, Schirm M, Lorenzo M, Kelly J, Pattarini D, Majam G, Thibault P, Logan S. 2006. Changes in flagellin glycosylation affect Campylobacter autoagglutination and virulence. Mol Microbiol 60:299–311. doi: 10.1111/j.1365-2958.2006.05100.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Friedrich V, Janesch B, Windwarder M, Maresch D, Braun ML, Megson ZA, Vinogradov E, Goneau MF, Sharma A, Altmann F, Messner P, Schoenhofen IC, Schaffer C. 2017. Tannerella forsythia strains display different cell-surface nonulosonic acids: biosynthetic pathway characterization and first insight into biological implications. Glycobiology 27:342–357. doi: 10.1093/glycob/cww129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Liu YC, Ud-Din AI, Roujeinikova A. 2014. Cloning, purification and preliminary crystallographic analysis of the Helicobacter pylori pseudaminic acid biosynthesis N-acetyltransferase PseH. Acta Crystallogr F Struct Biol Commun 70:1276–1279. doi: 10.1107/S2053230X14015398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Ud-Din AI, Liu YC, Roujeinikova A. 2015. Crystal structure of Helicobacter pylori pseudaminic acid biosynthesis N-acetyltransferase PseH: implications for substrate specificity and catalysis. PLoS One 10:e0115634. doi: 10.1371/journal.pone.0115634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Song WS, Nam MS, Namgung B, Yoon SI. 2015. Structural analysis of PseH, the Campylobacter jejuni N-acetyltransferase involved in bacterial O-linked glycosylation. Biochem Biophys Res Commun 458:843–848. doi: 10.1016/j.bbrc.2015.02.041. [DOI] [PubMed] [Google Scholar]
- 192.Meier-Dieter U, Starman R, Barr K, Mayer H, Rick PD. 1990. Biosynthesis of enterobacterial common antigen in Escherichia coli. Biochemical characterization of Tn10 insertion mutants defective in enterobacterial common antigen synthesis. J Biol Chem 265:13490–13497. [PubMed] [Google Scholar]
- 193.Hung MN, Rangarajan E, Munger C, Nadeau G, Sulea T, Matte A. 2006. Crystal structure of TDP-fucosamine acetyltransferase (WecD) from Escherichia coli, an enzyme required for enterobacterial common antigen synthesis. J Bacteriol 188:5606–5617. doi: 10.1128/JB.00306-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Ramos-Morales F, Prieto AI, Beuzón CR, Holden DW, Casadesús J. 2003. Role for Salmonella enterica enterobacterial common antigen in bile resistance and virulence. J Bacteriol 185:5328–5332. doi: 10.1128/jb.185.17.5328-5332.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Jurcisek J, Greiner L, Watanabe H, Zaleski A, Apicella MA, Bakaletz LO. 2005. Role of sialic acid and complex carbohydrate biosynthesis in biofilm formation by nontypeable Haemophilus influenzae in the chinchilla middle ear. Infect Immun 73:3210–3218. doi: 10.1128/IAI.73.6.3210-3218.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Tremblay YD, Deslandes V, Jacques M. 2013. Actinobacillus pleuropneumoniae genes expression in biofilms cultured under static conditions and in a drip-flow apparatus. BMC Genomics 14:364. doi: 10.1186/1471-2164-14-364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197.Castelli ME, Fedrigo GV, Clementín AL, Ielmini MV, Feldman MF, García Véscovi E. 2008. Enterobacterial common antigen integrity is a checkpoint for flagellar biogenesis in Serratia marcescens. J Bacteriol 190:213–220. doi: 10.1128/JB.01348-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198.Vollmer W, Blanot D, de Pedro MA. 2008. Peptidoglycan structure and architecture. FEMS Microbiol Rev 32:149–167. doi: 10.1111/j.1574-6976.2007.00094.x. [DOI] [PubMed] [Google Scholar]
- 199.Lovering AL, Safadi SS, Strynadka NC. 2012. Structural perspective of peptidoglycan biosynthesis and assembly. Annu Rev Biochem 81:451–478. doi: 10.1146/annurev-biochem-061809-112742. [DOI] [PubMed] [Google Scholar]
- 200.Schleifer KH, Kandler O. 1972. Peptidoglycan types of bacterial cell walls and their taxonomic implications. Bacteriol Rev 36:407–477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201.Berger-Bächi B, Barberis-Maino L, Strässle A, Kayser FH. 1989. FemA, a host-mediated factor essential for methicillin resistance in Staphylococcus aureus: molecular cloning and characterization. Mol Gen Genet 219:263–269. doi: 10.1007/bf00261186. [DOI] [PubMed] [Google Scholar]
- 202.Rohrer S, Berger-Bächi B. 2003. FemABX peptidyl transferases: a link between branched-chain cell wall peptide formation and beta-lactam resistance in gram-positive cocci. Antimicrob Agents Chemother 47:837–846. doi: 10.1128/aac.47.3.837-846.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203.Rohrer S, Ehlert K, Tschierske M, Labischinski H, Berger-Bächi B. 1999. The essential Staphylococcus aureus gene fmhB is involved in the first step of peptidoglycan pentaglycine interpeptide formation. Proc Natl Acad Sci U S A 96:9351–9356. doi: 10.1073/pnas.96.16.9351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204.Hegde SS, Shrader TE. 2001. FemABX family members are novel nonribosomal peptidyltransferases and important pathogen-specific drug targets. J Biol Chem 276:6998–7003. doi: 10.1074/jbc.M008591200. [DOI] [PubMed] [Google Scholar]
- 205.Filipe SR, Pinho MG, Tomasz A. 2000. Characterization of the murMN operon involved in the synthesis of branched peptidoglycan peptides in Streptococcus pneumoniae. J Biol Chem 275:27768–27774. doi: 10.1074/jbc.M004675200. [DOI] [PubMed] [Google Scholar]
- 206.De Pascale G, Lloyd AJ, Schouten JA, Gilbey AM, Roper DI, Dowson CG, Bugg TD. 2008. Kinetic characterization of lipid II-Ala:alanyl-tRNA ligase (MurN) from Streptococcus pneumoniae using semisynthetic aminoacyl-lipid II substrates. J Biol Chem 283:34571–34579. doi: 10.1074/jbc.M805807200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207.Bouhss A, Josseaume N, Allanic D, Crouvoisier M, Gutmann L, Mainardi JL, Mengin-Lecreulx D, van Heijenoort J, Arthur M. 2001. Identification of the UDP-MurNAc-pentapeptide:l-alanine ligase for synthesis of branched peptidoglycan precursors in Enterococcus faecalis. J Bacteriol 183:5122–5127. doi: 10.1128/jb.183.17.5122-5127.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208.Pootoolal J, Thomas MG, Marshall CG, Neu JM, Hubbard BK, Walsh CT, Wright GD. 2002. Assembling the glycopeptide antibiotic scaffold: the biosynthesis of A47934 from Streptomyces toyocaensis NRRL15009. Proc Natl Acad Sci U S A 99:8962–8967. doi: 10.1073/pnas.102285099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209.Filipe SR, Tomasz A. 2000. Inhibition of the expression of penicillin resistance in Streptococcus pneumoniae by inactivation of cell wall muropeptide branching genes. Proc Natl Acad Sci U S A 97:4891–4896. doi: 10.1073/pnas.080067697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210.Lloyd AJ, Gilbey AM, Blewett AM, De Pascale G, El Zoeiby A, Levesque RC, Catherwood AC, Tomasz A, Bugg TD, Roper DI, Dowson CG. 2008. Characterization of tRNA-dependent peptide bond formation by MurM in the synthesis of Streptococcus pneumoniae peptidoglycan. J Biol Chem 283:6402–6417. doi: 10.1074/jbc.M708105200. [DOI] [PubMed] [Google Scholar]
- 211.Bouhss A, Josseaume N, Severin A, Tabei K, Hugonnet JE, Shlaes D, Mengin-Lecreulx D, Van Heijenoort J, Arthur M. 2002. Synthesis of the l-alanyl-l-alanine cross-bridge of Enterococcus faecalis peptidoglycan. J Biol Chem 277:45935–45941. doi: 10.1074/jbc.M207449200. [DOI] [PubMed] [Google Scholar]
- 212.Monteiro JM, Covas G, Rausch D, Filipe SR, Schneider T, Sahl HG, Pinho MG. 2019. The pentaglycine bridges of Staphylococcus aureus peptidoglycan are essential for cell integrity. Sci Rep 9:5010. doi: 10.1038/s41598-019-41461-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 213.Ehlert K, Schroder W, Labischinski H. 1997. Specificities of FemA and FemB for different glycine residues: FemB cannot substitute for FemA in staphylococcal peptidoglycan pentaglycine side chain formation. J Bacteriol 179:7573–7576. doi: 10.1128/jb.179.23.7573-7576.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 214.Strandén AM, Ehlert K, Labischinski H, Berger-Bächi B. 1997. Cell wall monoglycine cross-bridges and methicillin hypersusceptibility in a femAB null mutant of methicillin-resistant Staphylococcus aureus. J Bacteriol 179:9–16. doi: 10.1128/jb.179.1.9-16.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 215.Maidhof H, Reinicke B, Blümel P, Berger-Bächi B, Labischinski H. 1991. femA, which encodes a factor essential for expression of methicillin resistance, affects glycine content of peptidoglycan in methicillin-resistant and methicillin-susceptible Staphylococcus aureus strains. J Bacteriol 173:3507–3513. doi: 10.1128/jb.173.11.3507-3513.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 216.Henze U, Sidow T, Wecke J, Labischinski H, Berger-Bächi B. 1993. Influence of femB on methicillin resistance and peptidoglycan metabolism in Staphylococcus aureus. J Bacteriol 175:1612–1620. doi: 10.1128/jb.175.6.1612-1620.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 217.Ling B, Berger-Bächi B. 1998. Increased overall antibiotic susceptibility in Staphylococcus aureus femAB null mutants. Antimicrob Agents Chemother 42:936–938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 218.Cusack S, Berthet-Colominas C, Hartlein M, Nassar N, Leberman R. 1990. A second class of synthetase structure revealed by X-ray analysis of Escherichia coli seryl-tRNA synthetase at 2.5 Å. Nature 347:249–255. doi: 10.1038/347249a0. [DOI] [PubMed] [Google Scholar]
- 219.Maillard AP, Biarrotte-Sorin S, Villet R, Mesnage S, Bouhss A, Sougakoff W, Mayer C, Arthur M. 2005. Structure-based site-directed mutagenesis of the UDP-MurNAc-pentapeptide-binding cavity of the FemX alanyl transferase from Weissella viridescens. J Bacteriol 187:3833–3838. doi: 10.1128/JB.187.11.3833-3838.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 220.Fonvielle M, Li de La Sierra-Gallay I, El-Sagheer AH, Lecerf M, Patin D, Mellal D, Mayer C, Blanot D, Gale N, Brown T, van Tilbeurgh H, Ethève-Quelquejeu M, Arthur M. 2013. The structure of FemX(Wv) in complex with a peptidyl-RNA conjugate: mechanism of aminoacyl transfer from Ala-tRNA(Ala) to peptidoglycan precursors. Angew Chem Int Ed Engl 52:7278–7281. doi: 10.1002/anie.201301411. [DOI] [PubMed] [Google Scholar]
- 221.Fonvielle M, Bouhss A, Hoareau C, Patin D, Mengin-Lecreulx D, Iannazzo L, Sakkas N, El Sagheer A, Brown T, Ethève-Quelquejeu M, Arthur M. 2018. Synthesis of lipid-carbohydrate-peptidyl-RNA conjugates to explore the limits imposed by the substrate specificity of cell wall enzymes on the acquisition of drug resistance. Chemistry 24:14911–14915. doi: 10.1002/chem.201802360. [DOI] [PubMed] [Google Scholar]
- 222.Villet R, Fonvielle M, Busca P, Chemama M, Maillard AP, Hugonnet JE, Dubost L, Marie A, Josseaume N, Mesnage S, Mayer C, Valery JM, Etheve-Quelquejeu M, Arthur M. 2007. Idiosyncratic features in tRNAs participating in bacterial cell wall synthesis. Nucleic Acids Res 35:6870–6883. doi: 10.1093/nar/gkm778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 223.Filipe SR, Severina E, Tomasz A. 2001. Functional analysis of Streptococcus pneumoniae MurM reveals the region responsible for its specificity in the synthesis of branched cell wall peptides. J Biol Chem 276:39618–39628. doi: 10.1074/jbc.M106425200. [DOI] [PubMed] [Google Scholar]
- 224.Chemama M, Fonvielle M, Villet R, Arthur M, Valery JM, Etheve-Quelquejeu M. 2007. Stable analogues of aminoacyl-tRNA for inhibition of an essential step of bacterial cell-wall synthesis. J Am Chem Soc 129:12642–12643. doi: 10.1021/ja0749946. [DOI] [PubMed] [Google Scholar]
- 225.Fonvielle M, Mellal D, Patin D, Lecerf M, Blanot D, Bouhss A, Santarem M, Mengin-Lecreulx D, Sollogoub M, Arthur M, Ethève-Quelquejeu M. 2013. Efficient access to peptidyl-RNA conjugates for picomolar inhibition of non-ribosomal FemX(Wv) aminoacyl transferase. Chemistry 19:1357–1363. doi: 10.1002/chem.201201999. [DOI] [PubMed] [Google Scholar]
- 226.Fonvielle M, Sakkas N, Iannazzo L, Le Fournis C, Patin D, Mengin-Lecreulx D, El-Sagheer A, Braud E, Cardon S, Brown T, Arthur M, Etheve-Quelquejeu M. 2016. Electrophilic RNA for peptidyl-RNA synthesis and site-specific cross-linking with tRNA-binding enzymes. Angew Chem Int Ed Engl 55:13553–13557. doi: 10.1002/anie.201606843. [DOI] [PubMed] [Google Scholar]
- 227.Arendt W, Hebecker S, Jager S, Nimtz M, Moser J. 2012. Resistance phenotypes mediated by aminoacyl-phosphatidylglycerol synthases. J Bacteriol 194:1401–1416. doi: 10.1128/JB.06576-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 228.Fields RN, Roy H. 2018. Deciphering the tRNA-dependent lipid aminoacylation systems in bacteria: novel components and structural advances. RNA Biol 15:480–491. doi: 10.1080/15476286.2017.1356980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 229.Lennarz WJ, Bonsen PP, van Deenen LL. 1967. Substrate specificity of O-l-lysylphosphatidylglycerol synthetase. Enzymatic studies on the structure of O-l-lysylphosphatidylglycerol. Biochemistry 6:2307–2312. doi: 10.1021/bi00860a005. [DOI] [PubMed] [Google Scholar]
- 230.Peschel A, Jack RW, Otto M, Collins LV, Staubitz P, Nicholson G, Kalbacher H, Nieuwenhuizen WF, Jung G, Tarkowski A, van Kessel KP, van Strijp JA. 2001. Staphylococcus aureus resistance to human defensins and evasion of neutrophil killing via the novel virulence factor MprF is based on modification of membrane lipids with l-lysine. J Exp Med 193:1067–1076. doi: 10.1084/jem.193.9.1067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 231.Maloney E, Stankowska D, Zhang J, Fol M, Cheng QJ, Lun S, Bishai WR, Rajagopalan M, Chatterjee D, Madiraju MV. 2009. The two-domain LysX protein of Mycobacterium tuberculosis is required for production of lysinylated phosphatidylglycerol and resistance to cationic antimicrobial peptides. PLoS Pathog 5:e1000534. doi: 10.1371/journal.ppat.1000534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 232.Thedieck K, Hain T, Mohamed W, Tindall BJ, Nimtz M, Chakraborty T, Wehland J, Jansch L. 2006. The MprF protein is required for lysinylation of phospholipids in listerial membranes and confers resistance to cationic antimicrobial peptides (CAMPs) on Listeria monocytogenes. Mol Microbiol 62:1325–1339. doi: 10.1111/j.1365-2958.2006.05452.x. [DOI] [PubMed] [Google Scholar]
- 233.Hebecker S, Krausze J, Hasenkampf T, Schneider J, Groenewold M, Reichelt J, Jahn D, Heinz DW, Moser J. 2015. Structures of two bacterial resistance factors mediating tRNA-dependent aminoacylation of phosphatidylglycerol with lysine or alanine. Proc Natl Acad Sci U S A 112:10691–10696. doi: 10.1073/pnas.1511167112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 234.Roy H, Ibba M. 2008. RNA-dependent lipid remodeling by bacterial multiple peptide resistance factors. Proc Natl Acad Sci U S A 105:4667–4672. doi: 10.1073/pnas.0800006105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 235.Suto K, Shimizu Y, Watanabe K, Ueda T, Fukai S, Nureki O, Tomita K. 2006. Crystal structures of leucyl/phenylalanyl-tRNA-protein transferase and its complex with an aminoacyl-tRNA analog. EMBO J 25:5942–5950. doi: 10.1038/sj.emboj.7601433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 236.Tobias JW, Shrader TE, Rocap G, Varshavsky A. 1991. The N-end rule in bacteria. Science 254:1374–1377. doi: 10.1126/science.1962196. [DOI] [PubMed] [Google Scholar]
- 237.Garg RP, Qian XL, Alemany LB, Moran S, Parry RJ. 2008. Investigations of valanimycin biosynthesis: elucidation of the role of seryl-tRNA. Proc Natl Acad Sci U S A 105:6543–6547. doi: 10.1073/pnas.0708957105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 238.Zhang W, Ntai I, Kelleher NL, Walsh CT. 2011. tRNA-dependent peptide bond formation by the transferase PacB in biosynthesis of the pacidamycin group of pentapeptidyl nucleoside antibiotics. Proc Natl Acad Sci U S A 108:12249–12253. doi: 10.1073/pnas.1109539108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 239.O'Brien TA, Kluger R, Pike DC, Gennis RB. 1980. Phosphonate analogues of pyruvate. Probes of substrate binding to pyruvate oxidase and other thiamin pyrophosphate-dependent decarboxylases. Biochim Biophys Acta 613:10–17. doi: 10.1016/0005-2744(80)90186-2. [DOI] [PubMed] [Google Scholar]
- 240.Bougioukou DJ, Mukherjee S, van der Donk WA. 2013. Revisiting the biosynthesis of dehydrophos reveals a tRNA-dependent pathway. Proc Natl Acad Sci U S A 110:10952–10957. doi: 10.1073/pnas.1303568110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 241.Huang Z, Wang KA, van der Donk WA. 2016. New insights into the biosynthesis of fosfazinomycin. Chem Sci 7:5219–5223. doi: 10.1039/C6SC01389A. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 242.Maruyama C, Niikura H, Izumikawa M, Hashimoto J, Shin-Ya K, Komatsu M, Ikeda H, Kuroda M, Sekizuka T, Ishikawa J, Hamano Y. 2016. tRNA-dependent aminoacylation of an amino sugar intermediate in the biosynthesis of a streptothricin-related antibiotic. Appl Environ Microbiol 82:3640–3648. doi: 10.1128/AEM.00725-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 243.Thao S, Chen CS, Zhu H, Escalante-Semerena JC. 2010. N(epsilon)-Lysine acetylation of a bacterial transcription factor inhibits its DNA-binding activity. PLoS One 5:e15123. doi: 10.1371/journal.pone.0015123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 244.Sang Y, Ren J, Ni JJ, Tao J, Lu J, Yao YF. 2016. Protein acetylation is involved in Salmonella enterica serovar Typhimurium virulence. J Infect Dis 213:1836–1845. doi: 10.1093/infdis/jiw028. [DOI] [PubMed] [Google Scholar]
- 245.Christensen DG, Meyer JG, Baumgartner JT, D’Souza AK, Nelson WC, Payne SH, Kuhn ML, Schilling B, Wolfe AJ. 2018. Identification of novel protein lysine acetyltransferases in Escherichia coli. mBio 9:e01905-18. doi: 10.1128/mBio.01905-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 246.Li Y, Krishnan K, Duncan MJ. 2018. Post-translational regulation of a Porphyromonas gingivalis regulator. J Oral Microbiol 10:1487743. doi: 10.1080/20002297.2018.1487743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 247.VanDrisse CM, Escalante-Semerena JC. 2018. Small-molecule acetylation controls the degradation of benzoate and photosynthesis in Rhodopseudomonas palustris. mBio 9:e01895-18. doi: 10.1128/mBio.01895-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 248.VanDrisse CM, Escalante-Semerena JC. 2018. In Salmonella enterica, OatA (formerly YjgM) uses O-acetyl-serine and acetyl-CoA to synthesize N,O-diacetylserine, which upregulates cysteine biosynthesis. Front Microbiol 9:2838. doi: 10.3389/fmicb.2018.02838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 249.Majorek KA, Osinski T, Tran DT, Revilla A, Anderson WF, Minor W, Kuhn ML. 2017. Insight into the 3D structure and substrate specificity of previously uncharacterized GNAT superfamily acetyltransferases from pathogenic bacteria. Biochim Biophys Acta Proteins Proteom 1865:55–64. doi: 10.1016/j.bbapap.2016.10.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 250.Burckhardt RM, Buckner BA, Escalante-Semerena JC. 2019. Staphylococcus aureus modulates the activity of acetyl-coenzyme A synthetase (Acs) by sirtuin-dependent reversible lysine acetylation. Mol Microbiol 112:588–604. doi: 10.1111/mmi.14276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 251.Srivastava R, Kaur A, Sharma C, Karthikeyan S. 2018. Structural characterization of ribT from Bacillus subtilis reveals it as a GCN5-related N-acetyltransferase. J Struct Biol 202:70–81. doi: 10.1016/j.jsb.2017.12.006. [DOI] [PubMed] [Google Scholar]