

ABSTRACT
Peptide‐based antiviral therapeutics has gradually paved their way into mainstream drug discovery research. Experimental determination of peptides' antiviral activity as expressed by their IC50 values involves a lot of effort. Therefore, we have developed “AVP‐IC50Pred,” a regression‐based algorithm to predict the antiviral activity in terms of IC50 values (μM). A total of 759 non‐redundant peptides from AVPdb and HIPdb were divided into a training/test set having 683 peptides (T683) and a validation set with 76 independent peptides (V76) for evaluation. We utilized important peptide sequence features like amino‐acid compositions, binary profile of N8‐C8 residues, physicochemical properties and their hybrids. Four different machine learning techniques (MLTs) namely Support vector machine, Random Forest, Instance‐based classifier, and K‐Star were employed. During 10‐fold cross validation, we achieved maximum Pearson correlation coefficients (PCCs) of 0.66, 0.64, 0.56, 0.55, respectively, for the above MLTs using the best combination of feature sets. All the predictive models also performed well on the independent validation dataset and achieved maximum PCCs of 0.74, 0.68, 0.59, 0.57, respectively, on the best combination of feature sets. The AVP‐IC50Pred web server is anticipated to assist the researchers working on antiviral therapeutics by enabling them to computationally screen many compounds and focus experimental validation on the most promising set of peptides, thus reducing cost and time efforts. The server is available at http://crdd.osdd.net/servers/ic50avp. © 2015 Wiley Periodicals, Inc. Biopolymers (Pept Sci) 104: 753–763, 2015.
Keywords: antiviral, peptide, IC50, prediction, machine learning
Abbreviations
- HIV
Human immunodeficiency virus
- HCV
Hepatitis C virus
INTRODUCTION
Antiviral peptides (AVPs) have recently emerged as an alternative strategy to fight disease causing viruses.1 The peptide Enfuvirtide (T20) is the first AVP approved by the FDA against HIV.2 Similarly, Sifuvirtide (SFT) peptide has shown potent anti‐HIV activity and pharmacokinetic profiles and is under phase‐II clinical trial.3 The peptide CIGB‐228 has shown potency against Human papilloma virus (HPV) infections and is under Phase II clinical trial.4 A peptide‐based HLA‐A2‐restricted CTL epitope capable of inducing both cellular and humoral responses is in phase I clinical trials against Hepatitis C virus (HCV).5 Another synthetic peptide (SPC3) was reported to prevent the HIV infection and is being evaluated for its antiviral properties in phase II clinical trials.6
Since peptides are involved in a number of cellular processes, they have a considerable potential to act as drugs in treating human diseases. Many peptide‐based drugs are already generating billions of dollars in annual sales.7 Peptide‐based drugs like penicillin and insulin has been widely used as therapeutics.8 Antimicrobial peptides are produced by living organisms as a means of defense against invading microbes including bacteria, protozoa, fungi and viruses, for example, cathelicidins, defensins, histatins, and so forth.9 Lately, antimicrobial peptides have been used to control different types of pathogens, particularly viruses1, 10 which are a major cause of malaise and death in the world because of their high genetic variation, different routes of transmission, efficient replication, and the capability to persist in the host cells.11 Although there are several traditional antiviral nucleoside and non‐nucleoside analogues against few viruses, many of these drugs have undesirable toxic effects.12 Whereas, AVPs with natural amino acids have lesser toxicity and are easily eliminated from the body.13
The AVPs block the viruses via different strategies including inhibition of virus fusion, signaling, replication, and so forth.14 They can interact with various glycosaminoglycans on the cell surface and thus compete with the virus for attachment sites. They may obstruct viral entry by binding virus fusion protein or cellular receptors needed for virus internalization. They can also hinder viral gene expression or translation via inhibition of essential viral proteins like polymerase, reverse transcriptase, and so forth.15
The earliest reports of AVPs were described for herpes simplex virus (HSV).16 Daher reported α‐defensin as an AVP against HSV.17 Melittin peptide has been noted to have inhibitory activity against HIV18 and Junin virus (JV).10 Similarly alloferon peptides have been demonstrated to inhibit influenza virus.19 Peptides derived from human lactoferricin were shown to possess potent antiviral activity against a variety of viruses.20 Generally, the efficacy of AVPs is measured as the half maximal inhibitory concentration (IC50) which is a quantitative measure to signify the concentration of a molecule or drug required to block a given biological process by half (50%).21 For example, “FluPep” blocks Influenza virus entry into the cells with an IC50 of 0.10 μM.22 Also, peptides derived from RhoA protein restrict Respiratory syncytial virus (RSV) replication and the best performing peptide accomplished an IC50 of 1.23 μM.23 Similarly, Pinon et al.24 were able to inhibit Human T‐cell leukemia virus (HTLV) protease using AVPs with a minimum IC50 of 0.28 μM. Also, Ray et al.25 demonstrated the ability of small peptides in inhibiting HCV translation as well as replication by disrupting the interaction between NS3 protease and HCV IRES with an IC50 value of 5 μM.
Although, for AVPs two specialized databases, “HIPdb” for HIV15a and “AVPdb” for more than 60 viruses14 are available. Additionally, AVP prediction algorithm “AVPpred,”26 which classifies a given peptide sequence as effective or non‐effective also exists. However a peptide antiviral activity predictor in terms of IC50 value is lacking. Therefore, in this study we have developed a regression‐based algorithm, “AVP‐IC50Pred” using experimentally proven datasets by employing multiple machine learning algorithms.
METHODS
Algorithm Development
Data Sets
From combined 3040 peptides available in recently published specialized AVP databases, AVPdb14 and HIPdb,15a we selected 1061 peptides having quantitative IC50 values against 42 viruses. After removal of redundant sequences, 759 peptides were left which were divided into datasets of 683 peptides for training/testing (T683) and 76 peptides for independent validation (V76). The peptide length ranged from 8 to 38 (average 22) amino acid residues. Peptides in the training/testing dataset belong to 39 diverse viruses and their quantitative IC50 values range from 0.001 to 442 μM. The validation dataset belongs to 18 viruses and their quantitative IC50 values range from 0.002 to 333 μM. Full description of the training/testing and validation dataset is available on the web server as well as in the Supporting Information Tables S1 and S2.
Peptide Features
We have used the different features like amino acid composition, binary profiles, physicochemical properties, solvent accessibility, secondary structure, and their hybrids for model development. In addition, we have also used database scanning technique to display earlier reported sequences matching with user provided peptides.
Amino Acid Composition
Amino acid composition has been widely used in a number of existing prediction methods.27 It is the fraction of each amino acid in a peptide. The fraction of all 20 natural amino acids was calculated using the following equation:
The models developed using mono and di amino acid compositions are termed as “Mono” and “Di,” respectively.
Binary Profile
Many researchers have used binary method for predicting proteins and peptides belonging to different classes.28 Binary profiles were generated for the peptides with each amino acid being represented by a vector of 20 dimensions (e.g., Ala by 1,0,0,0, 0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0 and Cys by 0,1,0,0,0,0,0,0,0,0,0,0,0,0, 0,0,0,0,0,0, etc.) to incorporate positional information of amino acids in a peptide. A pattern of window length “w” was represented by a vector of dimensions 20*w. The models created employing the binary pattern of eight carboxy and eight amino terminal amino acids are referred to as “C‐8 Bin” and “N‐8 Bin,” respectively. We have generated binary pattern of eight amino acid residues since it is the minimum length of peptides in our dataset.
Physicochemical Properties
A number of workers have demonstrated the importance of physicochemical properties in the development of different types of prediction models.26, 29 We have used the numerical values of 15 best performing physicochemical properties (Supporting Information Table S3) from AAindex, a database of indices defining various physico and biochemical properties of amino acids and pairs of amino acids.30 The model developed using the above physicochemical features is denoted as “Physico.”
Solvent Accessibility
Solvent accessibility of a peptide determines the extent to which it interacts with the solvent. It is proportional to the surface area of the exposed peptide.31 Solvent accessibility of the peptides was predicted using the method of ASAview.32 The model developed using the solvent accessibility features is denoted as “SA.”
Secondary Structure
The secondary structure of a peptide depicts the hydrogen‐bonding pattern (α‐helix, β‐sheet, or random coil) of its backbone.33 The secondary structure of the peptides was calculated using prediction PSSpred module (http://zhanglab.ccmb.med.umich.edu/PSSpred/) of I‐TASSER platform.34 The model developed using the secondary structural features is denoted as “SS.”
Database Scanning
Since sequences having high resemblance can mimic each other in structure and function, similarity search may be used to identify AVPs. This approach has been frequently used in the development of protein and peptide‐based prediction methods.3, 35 In this method, each query sequence is matched against two newly released AVP databases viz., AVPdb14 and HIPdb15a using BLAST.36
Machine Learning Techniques
Support Vector Machines
SVM has been used to develop number of bioinformatics algorithms.37 The SVMlight software package (available at http://svmlight.joachims.org/) was used to construct SVM models. In this study, we used the radial basis function kernel:
where x and y are two data vectors, and γ is a training parameter.
Random Forest
Random forests (RFs) are an ensemble learning method for classification and regression.38 The randomForest package version 4.6–7 in R (available at http://stat-www.berkeley.edu/users/breiman/RandomForests) was used.
IBk
IBk is a K‐nearest neighbors classifier available in Weka package (accessible at http://www.cs.waikato.ac.nz/ml/weka/). It can select appropriate value of K based on cross‐validation and distance weighting.
KStar
KStar is an instance‐based classifier (IBk) in Weka package that is the value of a test instance is based upon the values of those training instances similar to it, as determined by similarity function.
Evaluation
In order to evaluate performance of our models, we used Pearson's correlation coefficient (PCC). Models were evaluated using 10‐fold and leave one out cross validation (LOOCV) technique.
where n is the size of test set, and is the predicted and actual IC50, respectively.
In LOOCV each peptide in the dataset is used for testing iteratively and rest of the peptides are used to train the respective prediction models. In addition to LOOCV, we have also used Leave one virus out cross validation (LOVOCV) method. In this technique, AVPs from each virus are iteratively excluded and the classifier is trained on the remaining virus AVPs followed by testing on the excluded AVPs of that individual virus.
RESULTS
Performance Evaluation During 10‐Fold Cross Validation
AVP‐IC50Pred models have been developed using various peptide sequence features including mono and di‐amino acid composition, binary pattern of eight C‐terminal and N‐terminal amino acids, physicochemical properties, and so forth. During 10‐fold cross validation using SVM, we achieved a maximum correlation of 0.59, 0.61, 0.56, 0.51, 0.59, 0.22, 0.18 on mono, di, C8‐binary, N8‐binary and physico, solvent accessibility (sa), secondary structure (ss) models, respectively, on the T683 training/testing dataset. Using hybrid models of the above mentioned features, the performance was improved to a maximum of 0.66 in case of mono‐di‐physico‐sa‐ss composite features as shown in Table 1. The SVM and RF parameters used to develop prediction models are shown in Supporting Information Table S4. Furthermore, we calculated the P‐value for different models and found that P‐value of most of the models is statistically significant and <0.001 (For model no. 6 and 7 the P‐value is < 0.05).
Table 1.
Performance Evaluation During 10‐Fold Cross Validation
| S. No. | Feature | No. of Features | PCC | |||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Training/Testing, T683 (10×) | Validation, V76 | |||||||||
| SVM | RF | IBk | K* | SVM | RF | IBk | K* | |||
| 1 | Amino acid composition (Mono) | 20 | 0.59 | 0.61 | 0.44 | 0.41 | 0.64 | 0.64 | 0.42 | 0.41 |
| 2 | Di‐peptide composition (Di) | 400 | 0.61 | 0.60 | 0.47 | 0.43 | 0.66 | 0.62 | 0.47 | 0.45 |
| 3 | C8 Binary profile (C8 Bin) | 160 | 0.56 | 0.57 | 0.45 | 0.42 | 0.59 | 0.60 | 0.43 | 0.41 |
| 4 | N8 Binary profile (N8 Bin) | 160 | 0.51 | 0.54 | 0.45 | 0.43 | 0.48 | 0.60 | 0.45 | 0.43 |
| 5 | Physicochemical properties (Physico) | 315 | 0.59 | 0.54 | 0.46 | 0.44 | 0.63 | 0.68 | 0.46 | 0.45 |
| 6 | Solvent accessibility (SA) | 21 | 0.22 | 0.20 | 0.18 | 0.19 | 0.21 | 0.18 | 0.15 | 0.16 |
| 7 | Secondary structure (SS) | 3 | 0.18 | 0.18 | 0.16 | 0.17 | 0.19 | 0.16 | 0.17 | 0.18 |
| 8 | 1 + 2 | 420 | 0.60 | 0.61 | 0.47 | 0.45 | 0.67 | 0.62 | 0.48 | 0.48 |
| 9 | 3 + 4 | 320 | 0.59 | 0.62 | 0.51 | 0.48 | 0.62 | 0.65 | 0.52 | 0.50 |
| 10 | 1 + 2+5 | 735 | 0.63 | 0.61 | 0.52 | 0.51 | 0.70 | 0.64 | 0.54 | 0.51 |
| 11 | 3 + 4+5 | 635 | 0.63 | 0.60 | 0.51 | 0.50 | 0.72 | 0.67 | 0.52 | 0.50 |
| 12 | 1 + 2+3 + 4 | 740 | 0.61 | 0.62 | 0.51 | 0.49 | 0.67 | 0.63 | 0.51 | 0.50 |
| 13 | 1 + 2+3 + 4+5 | 1055 | 0.62 | 0.61 | 0.50 | 0.51 | 0.66 | 0.64 | 0.54 | 0.53 |
| 14 | 6 + 7 | 23 | 0.22 | 0.20 | 0.18 | 0.21 | 0.23 | 0.19 | 0.20 | 0.18 |
| 15 | 1 + 2+5 + 6+7 | 758 | 0.66 | 0.63 | 0.55 | 0.54 | 0.74 | 0.68 | 0.59 | 0.57 |
| 16 | 3 + 4+5 + 6+7 | 658 | 0.65 | 0.64 | 0.56 | 0.55 | 0.73 | 0.70 | 0.58 | 0.56 |
10‐Fold cross validation performance of predictive models on AVP dataset of 683 sequences (T683) and evaluation of performance of predictive models on validation dataset of 76 peptides (V76) using SVM, RF, IBk, and K* MLTs.
Abbreviations: SVM: support vector machine; RF: random forest; IBk: instance‐based classifier (Weka); K*: KStar (Weka); T685: Training dataset of 683 AVPs; 10×: 10‐fold cross validation; V76: independent dataset of 76 AVPs.
We also used other machine learning algorithms like RF, IBk, and K* to check their performance on the T683 training/testing data using the above mentioned features. RF performed similar to SVM with best PCC of 0.64 on N8/C8‐physico‐sa‐ss hybrid model while IBk and K* showed best performance of 0.56 and 0.55 on hybrid N8/C8‐physico‐sa‐ss features (Table 1). However, SVM performed better than other machine learning algorithms.
Performance Evaluation on Independent Data Set
Besides 10‐fold cross validation, we also checked the performance of our models on independent dataset of 76 peptides (V76) not used during training/testing. Here, we achieved a maximum correlation of 0.64, 0.66, 0.59, 0.48, 0.63, 0.21, 0.19 on mono, di, C8‐binary, N8‐binary, physico, solvent accessibility (sa), and secondary structure (ss) models, respectively, using SVM. As expected, the hybrid models gave a better correlation with a maximum PCC value of 0.74 on hybrid mono‐di‐physico‐sa‐ss model and 0.73 on N8/C8‐bin‐physico‐sa‐ss model using SVM.
Other machine learning techniques (MLTs) performed in a similar trend but their correlation was less as compared to SVM. Their best correlations on the hybrid mono‐di‐physico‐sa‐ss model were 0.68 for RF, 0.59 for IBk and 0.57 for K* (Table 1). The correlation between actual and predicted IC50 values of the independent dataset using SVM and RF is also graphically depicted in Figure 1.
Figure 1.
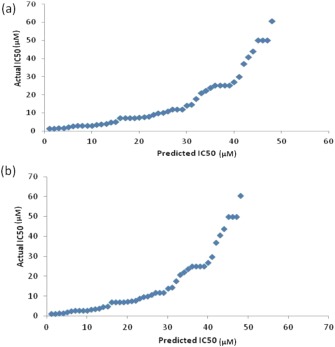
Correlation between actual and predicted IC50 values of the independent dataset using (a) SVM and (b) RF.
Performance Evaluation During LOVOCV
To further check the predictive performance for each virus in the 759 AVP dataset, we used LOVOCV method (Table 2). Overall, the training dataset performance during 10‐fold cross validation ranged from PCC value of a minimum 0.55 to a maximum 0.68 with an average 0.60. Simultaneously, the validation performance ranged from PCC 0.13 to 0.80 with an average 0.43. The method showed good correlation for 38 out of 42 viruses. However, for few viruses like HIV, RSV, INFV A, and HSV the performance was not satisfactory. Since, enough AVPs for RSV, INFV A, and HSV were not available; therefore, we made a combined dataset of these viruses to develop a prediction model with a best PCC of 0.59 using LOOCV. This model performed well with a PCC of 0.54 for independent dataset of these three viruses.
Table 2.
Performance of the SVM Model for Each Virus in the 759 AVP Dataset Using LOVOCV Method
| S. No. | Virus | Abbreviation | No. of Peptides | PCC | ||
|---|---|---|---|---|---|---|
| Training | Validation | Training | Validation | |||
| 1 | Hepatitis C virus | HCV | 635 | 124 | 0.55 | 0.8 |
| 2 | SARS coronavirus | SARS‐CoV | 733 | 26 | 0.58 | 0.53 |
| 3 | Porcine reproductive and respiratory syndrome Virus | PRRSV | 746 | 13 | 0.58 | 0.53 |
| 4 | Hepatitis B virus | HBV | 747 | 12 | 0.58 | 0.53 |
| 5 | Dengue 2 virus | DENV 2 | 752 | 7 | 0.58 | 0.53 |
| 6 | Newcastle disease virus | NDV | 752 | 7 | 0.58 | 0.53 |
| 7 | Transmissible gastroenteritis virus | TGEV | 756 | 3 | 0.64 | 0.53 |
| 8 | West Nile virus | WNV | 756 | 3 | 0.63 | 0.53 |
| 9 | Human papillomavirus | HPV | 753 | 6 | 0.59 | 0.52 |
| 10 | Human metapneumovirus | hMPV | 754 | 5 | 0.68 | 0.52 |
| 11 | Human parainfluenza virus type 3 | HPIV 3 | 734 | 25 | 0.57 | 0.51 |
| 12 | HSV 2 | HSV 2 | 754 | 5 | 0.62 | 0.51 |
| 13 | Hendra Virus | HeV | 755 | 4 | 0.63 | 0.51 |
| 14 | Human cytomegalovirus | HCMV | 755 | 4 | 0.65 | 0.5 |
| 15 | Marek's disease virus | MDV | 754 | 5 | 0.61 | 0.49 |
| 16 | Dengue 1 virus | DENV 1 | 756 | 3 | 0.61 | 0.49 |
| 17 | Feline immunodeficiency virus | FIV | 730 | 29 | 0.55 | 0.46 |
| 18 | Measles virus | MV | 739 | 20 | 0.57 | 0.45 |
| 19 | Human T‐cell leukemia virus 1 | HTLV 1 | 753 | 6 | 0.65 | 0.41 |
| 20 | HSV 1 | HSV 1 | 729 | 30 | 0.58 | 0.2 |
| 21 | Influenza A virus | INFV A | 720 | 39 | 0.59 | 0.15 |
| 22 | Human immunodeficiency virus | HIV | 464 | 295 | 0.6 | 0.13 |
| 23 | Respiratory syncytial virus | RSV | 694 | 65 | 0.58 | 0.02 |
| 24 | Othersa | Oth | 736 | 23 | 0.65 | 0.51 |
Other viruses include: ASLV‐A, JV, SeV, VACV, AIV, AMV, ASFV, BKV, BoHV 1, BRV, DENV 4, EBoV, HPIV 2, INFV B, JEV, LCMV, MHV, NiV, and SNV.
Performance Evaluation During LOOCV
We have also developed virus specific models for HIV and HCV where reasonable numbers of peptides are available. We also checked the performance of our method using LOOCV technique by employing earlier mentioned peptide sequence features (Table 3). In this method we used specific AVP datasets belonging to HIV (295 AVPs) and HCV (124 AVPs) and individually divided them into training and validation datasets, keeping 10% of the data for validation in each case. Here also the performance was increased while using hybrid mono‐di‐physico and mono‐di‐C8/N8 bin features (PCC 0.60–0.65) as compared to individual feature (PCC 0.50–0.55).
Table 3.
Performance of the SVM Models Using LOOCV Method on Virus Specific Datasets
| S. No. | Feature | No. of features | PCC | |||
|---|---|---|---|---|---|---|
| Training/Testing | Validation | |||||
| HIV | HCV | HIV | HCV | |||
| 1 | Amino acid composition (Mono) | 20 | 0.54 | 0.56 | 0.51 | 0.52 |
| 2 | Di‐peptide composition (Di) | 400 | 0.56 | 0.58 | 0.51 | 0.53 |
| 3 | C8/N8 Binary profile (C8/N8 Bin) | 160 | 0.57 | 0.57 | 0.54 | 0.55 |
| 4 | Physicochemical properties (Physico) | 315 | 0.55 | 0.55 | 0.51 | 0.51 |
| 5 | 1 + 2+3 | 735 | 0.58 | 0.64 | 0.54 | 0.61 |
| 6 | 1 + 2+4 | 635 | 0.60 | 0.67 | 0.54 | 0.58 |
| 7 | 1 + 2+3 + 4 | 740 | 0.60 | 0.65 | 0.53 | 0.63 |
Two Sample Logo
Two sample logos are used to graphically depict the differences between two sets of sequence alignments.39 The web‐based tool is freely available via the url: http://www.twosamplelogo.org. Highly and least effective AVPs were chosen to generate two sample logos using both 8 N‐terminal and 8 C‐terminal residues. The IC50 values of highly and least effective AVPs were below 1 µM and above 100 µM, respectively. In the N terminal region, we found that large acidic amino acids like Met, Thr, Asp, and Glu were enriched in highly effective AVPs while small basic amino acids like Ala, Gly, and Lys were more common in least effective AVPs (Figure 2 a). Similarly in the C‐terminal region, large polar and non‐polar amino acids like Leu, Trp, and Asn were frequent in the highly effective AVPs while small positively charged amino acids like Gly, Lys, and Arg were more common in the least effective AVPs (Figure 2 b). Two sample logos of the above sequences with different amino acids colored as per their charge, hydrophobicity, surface exposure, flexibility, and disorder are available in Supporting Information Figure S1.
Figure 2.

Two sample logo (TSL) comparison. TSLs Two sample logos of a) 8‐N terminal and b) 8‐C terminal residues of 97 highly effective peptides (IC50 < 1 µM) and an equal number of least effective peptides (IC50 > 100 µM).
Box Plots
The box plot is a convenient way to denote the summary statistics and the distribution of numerical data. It not only allows the depiction of the maximum, minimum, and median of a data set but also the visualization of lower and upper quartiles.40 We selected 97 highly effective (IC50 < 1 µM) and an equal number of least effective AVPs (IC50 > 100 µM) and calculated the 15 best performing physicochemical properties (Supporting Information Table S3a) predicted by SVM. Box plots for the individual properties were drawn using BoxPlotR (available at http://boxplot.tyerslab.com). The plots are shown in Supporting Information Figure S2(i and ii). It was observed that some properties like helix initiation parameter (c), frequency of C‐terminal non beta region (d), free energy in beta‐strand region (e, m) and frequency of helix (f, k) were more discriminative compared to others. Similar results were reported by Chang and Yang12 and Polanco et al.41 while analyzing the physicochemical properties of AVPs.
Web Server
AVP‐IC50Pred web server is freely accessible via the URL http://crdd.osdd.net/servers/ic50avp. A flowchart depicting the workflow of AVP‐IC50Pred web server is shown in Supporting Information Figure S3.
Input
On the submit page user may paste single or multiple peptide sequence(s) in FASTA format in the provided text‐box or upload a FASTA file from the system. An “Example link” has been provided to load a default set of sequences. User can select the desired model and MLT to run the prediction (Supporting Information Figure S4).
Output
The prediction output is shown in tabular form with 10 columns. The first, second, and third columns consist of the sequence identifier for the input FASTA sequence, the sequence itself and the sequence length, respectively. The fourth column gives the action buttons for database scanning in order to check the presence of similar sequences in the existing antiviral databases HIPdb and AVPdb and also in the latest UniProt release using BLAST. Rest of the columns (5–8) show the output of different MLTs used in this study (SVM, RF, IBk, K*). The predicted IC50 value for the peptide sequence is displayed in μM units. The analysis column displays the calculated physicochemical properties for the peptides in question. All the columns of the table have been provided with a sorting functionality. Search option is also provided to filter the results (Figure 3).
Figure 3.
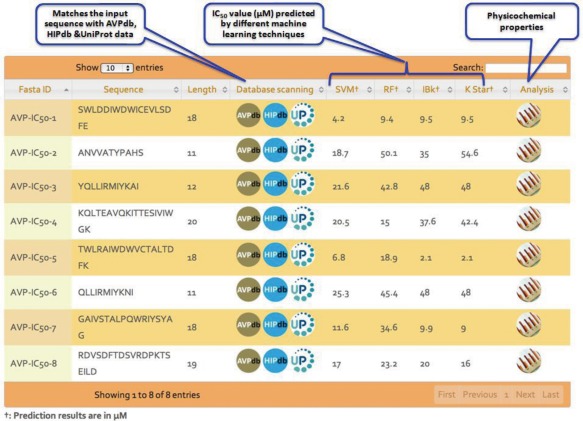
AVP‐IC50 Pred result output.
Analysis Tools
Mutation Analyzer
This tool allows the user to generate all possible combinations of amino acid mutations in a given peptide sequence and predict the IC50 of the mutant peptides using the best performing model. However, the mutated peptides are only computationally predicted and need experimental validation. Worked examples have been provided in Table 4.
Table 4.
Mutational Analysis
| Peptide | Length | Mutation Position | IC50 (µM) | Fold Change | PubMed ID |
|---|---|---|---|---|---|
| YTSLIHSLIEESQNQQEKNEQELLELDKWASLWNWF (Enfuvirtide/T‐20) | 36 | No Mutation | 7.57 | – | 19949052 |
| YTSLIHSLIAESQNQQEKNEQELLELDKWASLWNWF | 36 | E10A | 0.01 | 757.0 | |
| YTSLIHSLIEASQNQQEKNEQELLELDKWASLWNWF | 36 | E11A | 0.02 | 378.5 | |
| YTSLIHSLIEESQNQQVKNEQELLELDKWASLWNWF | 36 | E17V | 0.03 | 252.3 | |
| YTSLIHSLIEESQNQQEKNDQELLELDKWASLWNWF | 36 | E20D | 0.03 | 252.3 | |
| YTSLIHSLIEESQNQQEKNEQELLELDKWASLWGWF | 36 | N34G | 0.03 | 252.3 | |
| KVINPEPIVEPFMSKPFALF (Scr alpha1‐antitrypsin peptide) | 20 | No Mutation | 100.00 | – | 17448989 |
| KVINPEPIVEPFMSKPFLLF | 20 | A18L | 5.24 | 19.1 | |
| LVINPEPIVEPFMSKPFALF | 20 | K1L | 7.38 | 13.6 | |
| KVINPEPIVEPFMSKPFVLF | 20 | A18V | 7.46 | 13.4 | |
| KVINPEPIVEPFMSLPFALF | 20 | K15L | 7.95 | 12.6 | |
| KVILPEPIVEPFMSKPFALF | 20 | M4L | 14.55 | 6.9 | |
| GLQLLGFILAFLGWIGAI (CL58.1 peptide) | 18 | No Mutation | 25.00 | – | 22378192 |
| GLQLLYFILAFLGWIGAI | 18 | G6Y | 2.71 | 9.2 | |
| GLQLLGFILAYLGWIGAI | 18 | F11Y | 2.99 | 8.4 | |
| GLQLLGFILAFLGWIGAY | 18 | I18Y | 2.99 | 8.4 | |
| GLQLLGFILAFLRWIGAI | 18 | G13R | 3.25 | 7.7 | |
| GLQLLGFILAFLGWIYAI | 18 | G16Y | 3.5 | 7.1 | |
| MANAGLQLLGFILAFLGWIG (peptide CL58‐2) | 20 | No Mutation | 17.8 | – | 22378192 |
| MANAGLQLLGFILAFLGWIV | 20 | I20V | 0.46 | 38.7 | |
| MANAGLQLLGFILAFLGWIR | 20 | I20R | 0.51 | 34.9 | |
| MANAGLQLLRFILAFLGWIG | 20 | G10R | 0.9 | 19.8 | |
| MANAGLQLLGFILAFLRWIG | 20 | G17R | 0.99 | 18.0 | |
| MANAGLQLLGFILAFLVWIG | 20 | G17V | 1.07 | 16.6 |
Mutational analysis of different AVPs showing the top five best performing mutations and their predicted IC50 values from each peptide.
AVP‐IC50Pred‐BLAST
Users may BLAST their peptide for similarity against HIPdb and AVPdb. The result shows distribution of hits, their score, E‐value and alignment with sequences having significant similarity.
AVP‐IC50Pred‐Map
The MAP tool is used to fetch the perfectly matching peptides available in the existing AVP databases which map against a user provided protein sequence. The result output displays a list of earlier reported AVPs that match with specific portions of the user provided sequence.
Physicochemical Properties Calculator
The properties tool allows the user to visually examine some important peptide features like amino acid composition, hydrophobicity, preference for β‐strands and frequency of α‐helix in a given peptide sequence.
Motif Search
AVP‐IC50Pred Motif Scan allows to search possible AVP motifs in user provided protein/peptide sequences. This tool is based on MEME/MAST software.42
Protein Fragmentor
This tool generates peptide fragments of desired length and overlapping residues from a protein sequence.
AVP‐IC50 Conserve
Checks the conservation of user provided peptide sequence in human, viral, and antiviral proteins.
Application
Using AVP‐IC50Pred mutation analyzer, users can generate all possible combinations of amino acid mutations in a given peptide sequence and predict the IC50 of the mutant peptides. This tool also enables the users to sort the mutant peptides as per their predicted IC50 value to select the highly effective peptides. As an example we generated the mutants of Enfuvirtide/T‐20, an HIV fusion inhibitor using this tool. We found that mutations like E10A and E11A improved the predicted IC50 over hundreds of folds. Likewise, mutations of I20V and I20R in peptide CL58‐2 altered the IC50 by about 40 fold. Similar analysis of some more peptides was also carried out as provided in Table 4.
DISCUSSION
Due to the limited availability of drugs and vaccines for many viruses, there is a demand to develop more effective antiviral therapeutics.37, 43 Thus, apart from drugs and vaccines, AVPs are a potential alternative to control viral pathogenesis.26, 44 Lower toxicity and broad range AVPs such as EB peptide against HSV,45 α‐defensin against cytomegalovirus (CMV),15b enfuvirtide2 and SFT against HIV,3 Human neutrophil peptide 1 against VSV,1 and so forth have shown promising results with high specificity and relatively few off‐target side‐effects. The AVPs act via variety of routes including inhibition of viral entry into the host cell, suppression of viral gene expression or translation, immunopotentiation, and so forth.46
Antiviral activity of a peptide is often determined experimentally as its half maximal inhibitory concentration (IC50) value. Usually AVPs with < 1 µM IC50 values are considered very effective while > 100 μM are least effective in repressing a viral process or function. Although there are many antimicrobial peptide prediction servers like CAMP,47 APD2,48 AntiBP2,49 Wang et al.,3 and so forth and one AVP predictor AVPpred26 but they are all based on classification mode of machine learning, that is, they only classify a peptide sequence as effective or ineffective. In addition, none of them quantitatively predicts the antimicrobial or antiviral activity of a peptide in terms of IC50.
To develop such predictor we have extracted comprehensive non‐redundant 759 AVPs with quantitative IC50 values from specialized resources HIPdb15a and AVPdb.14 These AVPs were randomly divided into three training/testing (T683) and validation (V76) datasets that belong to as many as 42 medically important viruses. We also checked the performance of SVM models based on these three training and validation dataset and selected one set with better performance for algorithm development as shown in Supporting Information Table S5.
Peptide‐based antivirals can act in many different ways however generally the AVPs act via interference of protein–protein interactions by mimicking the properties of one of the interfaces, thus acting as competitive inhibitors by preventing interaction of the binding protein partners. The various sequence features of a peptide play an important role in their bioactivity. These features include amino acid composition, N/C terminal residues and their physicochemical properties like hydrophobicity, secondary structure, and so forth. These have also been reported previously for prediction of other important peptides viz., ABCpred,50 AntiCP,51 QSPpred,52 and so forth. Evans et al.53 have also described HIV coreceptor tropism prediction on the basis of the amino‐acid sites and physiochemical characteristics of the V3 loop sequence of the HIV envelope.
In our results, however, we found that SVM and RF comparatively performed better than IBk and K* MLTs (MLTs). In SVM by choosing appropriate kernel generalization grade, SVMs gain flexibility and robustness and deliver a unique solution in their prediction.54 RF computes proximities between pairs of cases that can be used in clustering and locating outliers. RF is a fast and highly accurate learning algorithm and can handle thousands of input variables without variable deletion.38 IBk and K* are simple MLTs that work well on basic recognition problems. One of the shortcomings of the these two algorithms is that they are lazy learners, that is, they do not learn anything from the training data but simply use the training data itself for prediction because of which they are not robust for predicting noisy data.55 Due to this reason SVM and RF performed better than these MLTs in our study.
AVP‐IC50Pred is a regression‐based algorithm employing hybrid models integrating different peptide features like amino acid composition, binary profiles, physicochemical properties, and so forth. We claim this method to be the first ever attempt to predict a numerical antiviral efficacy value for a given peptide. We had used SVM at the first place to predict the peptide IC50 values for which we achieved a maximum PCC of 0.66 during 10‐fold cross validation and a PCC of 0.74 on the independent dataset.
Since AVPs are heterogeneous in terms of their target viruses, one of the important questions remains whether this algorithm is applicable for the general viruses. It has been found by earlier researchers that a given class of peptides follow a certain pattern as is the case with antimicrobial peptide prediction algorithms like APD2,48 CAMP,47 AntiBP2,49 ClassAMP,56 AMPA,57 and so forth. To further address this issue, we have used LOVOCV strategy37 in which AVPs from each virus are iteratively excluded and the classifier is trained on AVPs of the remaining viruses followed by testing on the excluded AVPs. This approach worked for 38 out of 42 viruses. Predictive performance for IC50 of each virus was satisfactory for most of the viruses despite the fact that their data was not included during training. It shows that AVP‐IC50Pred can act as a general AVP prediction algorithm, which may be applied to other viruses as well. However, there are a few exceptions like HIV, RSV, INFV A and HSV for which algorithm did not work. It can be due to the fact that some viruses differ in their manner of infection/life cycle and hence mechanism or mode of action of AVPs may differ in some cases. Therefore we have provided virus specific models for the above viruses and also integrated them on the web server. In the future, we are interested in making target specific algorithms once appropriate and enough data are available.
It is reported in the literature that the prediction methods developed in regression mode show a lower correlation for heterogeneous datasets than that for homogenous datasets. For example, in similar studies of siRNAs, it was found that the PCC ranged from 0.4 to 0.6 on heterogeneous datasets while it increased to 0.7–0.8 on homogenous datasets.37, 58 The SVM models in this study were developed using a highly diverse dataset taken from 125 different studies tested in 64 cell lines against 42 viruses. So the PCC of 0.66 in regression mode is reasonable. This performance can be improved further if a large homogeneous dataset is available in future. However, to put more confidence in our present prediction, we used three more MLTs in addition to SVM namely, RF, IBk and K* which performed in a similar fashion. This will also encourage developing newer regression‐based algorithms besides the existing classification‐based methods in the area of peptide‐based therapeutics.
There are limited reports of experimentally improving the IC50 value of AVPs or to overcome their virus resistance by amino acid substitutions. Izumi et al.59 designed an enfuvirtide variant containing the S138A substitution and showed that it is a potent inhibitor of both enfuvirtide ‐sensitive and enfuvirtide ‐resistant viruses. Also it was shown that mutation of four C‐terminal amino acids (WNWF to ANAA) of the enfuvirtide peptide made it inactive.60 Dwyer et al.61 found that the mutations, which increased peptide helical structure, were more effective in blocking the virus. All such improvements in the peptide efficacy were achieved after considerable efforts in terms of time and cost. Had there been an algorithm which generates the possible mutants of a potential AVP and predict its efficacy, these results might have been achieved in lesser time. Simultaneously, AVPs with improved efficacy might be useful against mutated viruses. To assist the researchers in this direction, AVP‐IC50Pred mutation analyzer tool will be useful in designing AVPs with enhanced antiviral potential as illustrated in the “Application” section. Further, prediction and analysis of IC50 of all single mutants of the AVPs in our dataset have been provided on our web server. Besides, AVP‐IC50Pred web server has also been equipped with some important tools such as physicochemical properties calculator, Motif scan, BLAST, Conservation, Map, and fragment tool for further analysis of the AVPs.
CONCLUSIONS
AVP‐IC50Pred is the first regression‐based algorithm developed using experimentally validated data sets for prediction of peptide antiviral activity in terms of IC50. Multiple MLTs were used to build comprehensive prediction models exploiting important peptide sequence features such as amino acid and dipeptide compositions, binary profile of N8‐C8 residues as well as selected physicochemical properties. AVP‐IC50Pred web server is hoped to assist the researchers working on AVP therapeutics by decreasing the cost and time efforts involved in experimental validation.
MK conceived the approach, helped in analysis and interpretation of data, gave overall supervision to the project. MK, AQ wrote the manuscript. AQ, HT collected the data, implemented machine learning software and developed the web server. All of the authors read and approved the final manuscript. The authors declare that they have no competing interests.
Supporting information
Supporting Information
This article was originally published online as an accepted preprint. The “Published Online” date corresponds to the preprint version. You can request a copy of any preprints from the past two calendar years by emailing the Biopolymers editorial office at biopolymers@wiley.com.
REFERENCES
- 1. Gwyer Findlay, E. ; Currie, S. M. ; Davidson, D. J. BioDrugs 2013, 27, 479‐493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Ashkenazi, A. ; Wexler‐Cohen, Y. ; Shai, Y. Biochim Biophys Acta 2011, 1808, 2352‐2358. [DOI] [PubMed] [Google Scholar]
- 3. Wang, P. ; Hu, L. ; Liu, G. ; Jiang, N. ; Chen, X. ; Xu, J. ; Zheng, W. ; Li, L. ; Tan, M. ; Chen, Z. ; Song, H. ; Cai, Y. D. ; Chou, K. C. PLoS One 2011, 6, e18476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Solares, A. M. ; #Baladron, I. ; Ramos, T. ; Valenzuela, C. ; Borbon, Z. ; Fanjull, S. ; Gonzalez, L. ; Castillo, D. ; Esmir, J. ; Granadillo, M. ; Batte, A. ; Cintado, A. ; Ale, M. ; Fernandez de Cossio, M. E. ; #Ferrer, A. ; Torrens, I. ; Lopez‐Saura, P. ISRN Obstet Gynecol 2011, 2011, 292951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Yutani, S. ; Komatsu, N. ; Shichijo, S. ; Yoshida, K. ; Takedatsu, H. ; Itou, M. ; Kuromatu, R. ; Ide, T. ; Tanaka, M. ; Sata, M. ; Yamada, A. ; Itoh, K. Cancer Sci 2009, 100, 1935‐1942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Carlier, E. ; Mabrouk, K. ; Moulard, M. ; Fajloun, Z. ; Rochat, H. ; De Waard, M. ; Sabatier, J. M. J Pept Res 2000, 56, 427‐437. [DOI] [PubMed] [Google Scholar]
- 7.(a) Craik, D. J. ; Fairlie, D. P. ; Liras, S. ; Price, D. Chem Biol Drug Des 2013, 81, 136‐147; [DOI] [PubMed] [Google Scholar]; (b) Mooney, C. ; Haslam, N. J. ; Holton, T. A. ; Pollastri, G. ; Shields, D. C. Bioinformatics 2013, 29, 1120‐1126. [DOI] [PubMed] [Google Scholar]
- 8. Uhlig, T. ; Kyprianou, T. ; Martinelli, F. G. ; Oppici, C. A. ; Heiligers, D. ; Hills, D. ; Calvo, X. R. ; Verhaert, P. EuPA Open Proteomics 2014, 4, 58‐69. [Google Scholar]
- 9.(a) Alba, A. ; Lopez‐Abarrategui, C. ; Otero‐Gonzalez, A. J. Biopolymers 2012, 98, 251‐267; [DOI] [PubMed] [Google Scholar]; (b) Fernandes, F. C. ; Rigden, D. J. ; Franco, O. L. Biopolymers 2012, 98, 280‐287; [DOI] [PubMed] [Google Scholar]; (c) Peters, B. M. ; Shirtliff, M. E. ; Jabra‐Rizk, M. A. PLoS Pathog 2010, 6, e1001067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Albiol Matanic, V. C. ; Castilla, V. Int J Antimicrob Agents 2004, 23, 382‐389. [DOI] [PubMed] [Google Scholar]
- 11.(a) Nichol, S. T. ; Arikawa, J. ; Kawaoka, Y. Proc Natl Acad Sci USA 2000, 97, 12411‐12412; [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Domingo, E. Vet Res 2010, 41, 38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Chang, K. Y. ; Yang, J. R. PLoS One 2013, 8, e70166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Castel, G. ; Chteoui, M. ; Heyd, B. ; Tordo, N. Molecules 2011, 16, 3499‐3518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Qureshi, A. ; Thakur, N. ; Tandon, H. ; Kumar, M. Nucleic Acids Res 2014, 42, D1147‐D1153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.(a) Qureshi, A. ; Thakur, N. ; Kumar, M. PLoS One 2013, 8, e54908; [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Mulder, K. C. ; Lima, L. A. ; Miranda, V. J. ; Dias, S. C. ; Franco, O. L. Front Microbiol 2013, 4, 321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Imanishi, J. ; Oku, T. ; Cho, Y. ; Inagawa, S. ; Tanaka, A. ; Kuwayama, W. C R Seances Soc Biol Fil 1985, 179, 414‐419. [PubMed] [Google Scholar]
- 17. Daher, K. A. ; Selsted, M. E. ; Lehrer, R. I. J Virol 1986, 60, 1068‐1074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Wachinger, M. ; Kleinschmidt, A. ; Winder, D. ; von Pechmann, N. ; Ludvigsen, A. ; Neumann, M. ; Holle, R. ; Salmons, B. ; Erfle, V. ; Brack‐Werner, R. J Gen Virol 1998, 79(Pt 4), 731‐740. [DOI] [PubMed] [Google Scholar]
- 19. Chernysh, S. ; Kim, S. I. ; Bekker, G. ; Pleskach, V. A. ; Filatova, N. A. ; Anikin, V. B. ; Platonov, V. G. ; Bulet, P. Proc Natl Acad Sci USA 2002, 99, 12628‐12632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.(a) Jenssen, H. ; Hamill, P. ; Hancock, R. E. Clin Microbiol Rev 2006, 19, 491‐511; [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) van der Strate, B. W. ; Beljaars, L. ; Molema, G. ; Harmsen, M. C. ; Meijer, D. K. Antiviral Res 2001, 52, 225‐239. [DOI] [PubMed] [Google Scholar]
- 21.(a) Patankar, S. J. ; Jurs, P. C. J Chem Inf Comput Sci 2000, 40, 706‐723; [DOI] [PubMed] [Google Scholar]; (b) Riddick, G. ; Song, H. ; Ahn, S. ; Walling, J. ; Borges‐Rivera, D. ; Zhang, W. ; Fine, H. A. Bioinformatics 2011, 27, 220‐224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Nicol, M. Q. ; Ligertwood, Y. ; Bacon, M. N. ; Dutia, B. M. ; Nash, A. A. J Gen Virol 2012, 93 (Pt 5), 980‐986. [DOI] [PubMed] [Google Scholar]
- 23. Budge, P. J. ; Graham, B. S. J Antimicrob Chemother 2004, 54, 299‐302. [DOI] [PubMed] [Google Scholar]
- 24. Pinon, J. D. ; Kelly, S. M. ; Price, N. C. ; Flanagan, J. U. ; Brighty, D. W. J Virol 2003, 77, 3281‐3290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Ray, U. ; Roy, C. L. ; Kumar, A. ; Mani, P. ; Joseph, A. P. ; Sudha, G. ; Sarkar, D. P. ; Srinivasan, N. ; Das, S. Mol Ther 2013, 21, 57‐67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Thakur, N. ; Qureshi, A. ; Kumar, M. Nucleic Acids Res 2012, 40 (Web Server issue), W199‐W204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.(a) Lata, S. ; Sharma, B. K. ; Raghava, G. P . BMC Bioinformatics 2007, 8, 263; [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Garg, A. ; Bhasin, M. ; Raghava, G. P. J Biol Chem 2005, 280, 14427‐14432; [DOI] [PubMed] [Google Scholar]; (c) Holton, T. A. ; Pollastri, G. ; Shields, D. C. ; Mooney, C. Bioinformatics 2013, 29, 3094‐3096. [DOI] [PubMed] [Google Scholar]
- 28.(a) Xiao, X. ; Wang, P. ; Chou, K. C. J Comput Chem 2009, 30, 1414‐23; [DOI] [PubMed] [Google Scholar]; (b) Gautam, A. ; Chaudhary, K. ; Kumar, R. ; Sharma, A. ; Kapoor, P. ; Tyagi, A. ; Raghava, G. P. J Transl Med 2013, 11, 74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Sanders, W. S. ; Johnston, C. I. ; Bridges, S. M. ; Burgess, S. C. ; Willeford, K. O. PLoS Comput Biol 2011, 7, e1002101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Kawashima, S. ; Kanehisa, M. Nucleic Acids Res 2000, 28, 374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Durham, E. ; Dorr, B. ; Woetzel, N. ; Staritzbichler, R. ; Meiler, J. J Mol Model 2009, 15, 1093‐1108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Ahmad, S. ; Gromiha, M. ; Fawareh, H. ; Sarai, A. BMC Bioinformatics 2004, 5, 51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Heringa, J. Curr Protein Pept Sci 2000, 1, 273‐301. [DOI] [PubMed] [Google Scholar]
- 34. Roy, A. ; Kucukural, A. ; Zhang, Y. Nat Protoc 2010, 5, 725‐738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Frank, K. ; Sippl, M. J. Bioinformatics 2008, 24, 2172‐2176. [DOI] [PubMed] [Google Scholar]
- 36. Altschul, S. F. ; Gish, W. ; Miller, W. ; Myers, E. W. ; Lipman, D. J. J Mol Biol 1990, 215, 403‐410. [DOI] [PubMed] [Google Scholar]
- 37. Qureshi, A. ; Thakur, N. ; Kumar, M. J Transl Med 2013, 11, 305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Svetnik, V. ; Liaw, A. ; Tong, C. ; Culberson, J. C. ; Sheridan, R. P. ; Feuston, B. P. J Chem Inf Comput Sci 2003, 43, 1947‐1958. [DOI] [PubMed] [Google Scholar]
- 39. Vacic, V. ; Iakoucheva, L. M. ; Radivojac, P. Bioinformatics 2006, 22, 1536‐1537. [DOI] [PubMed] [Google Scholar]
- 40. Spitzer, M. ; Wildenhain, J. ; Rappsilber, J. ; Tyers, M. Nat Methods 2014, 11, 121‐122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Polanco, C. ; Samaniego, J. L. ; Castanon‐Gonzalez, J. A. ; Buhse, T. Cell Biochem Biophys 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Bailey, T. L. ; Boden, M. ; Buske, F. A. ; Frith, M. ; Grant, C. E. ; Clementi, L. ; Ren, J. ; Li, W. W. ; Noble, W. S. Nucleic Acids Res 2009, 37 (Web Server issue), W202‐W208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.(a) Sadanand, S. Yale J Biol Med 2011, 84, 353‐359; [PMC free article] [PubMed] [Google Scholar]; (b) Duffy, S. ; Shackelton, L. A. ; Holmes, E. C. Nat Rev Genet 2008, 9, 267‐276. [DOI] [PubMed] [Google Scholar]
- 44. Lopez‐Martinez, R. ; Ramirez‐Salinas, G. L. ; Correa‐Basurto, J. ; Barron, B. L. PLoS One 2013, 8, e76876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Altmann, S. E. ; Brandt, C. R. ; Jahrling, P. B. ; Blaney, J. E. Virol J 2012, 9, 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Real, E. ; Rain, J. C. ; Battaglia, V. ; Jallet, C. ; Perrin, P. ; Tordo, N. ; Chrisment, P. ; D'Alayer, J. ; Legrain, P. ; Jacob, Y. J Virol 2004, 78, 7410‐7417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Waghu, F. H. ; Gopi, L. ; Barai, R. S. ; Ramteke, P. ; Nizami, B. ; Idicula‐Thomas, S. Nucleic Acids Res 2014, 42 (Database issue), D1154‐D1158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Wang, G. ; Li, X. ; Wang, Z. Nucleic Acids Res 2009, 37 (Database issue), D933‐D937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Lata, S. ; Mishra, N. K. ; Raghava, G. P. BMC Bioinformatics 2010, 11 (Suppl 1), S19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Saha, S. ; Raghava, G. P. Proteins 2006, 65, 40‐48. [DOI] [PubMed] [Google Scholar]
- 51. Tyagi, A. ; Kapoor, P. ; Kumar, R. ; Chaudhary, K. ; Gautam, A. ; Raghava, G. P. Sci Rep 2013, 3, 2984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Rajput, A. ; Gupta, A. K. ; Kumar, M. PLoS One 2015, 10, e0120066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Evans, M. C. ; Paquet, A. C. ; Huang, W. ; Napolitano, L. ; Frantzell, A. ; Toma, J. ; Stawiski, E. W. ; Goetz, M. B. ; Petropoulos, C. J. ; Whitcomb, J. ; Coakley, E. ; Haddad, M. J Bioinform Comput Biol 2013, 11, 1350006. [DOI] [PubMed] [Google Scholar]
- 54. Muller, K. ; Mika, S. ; Ratsch, G. ; Tsuda, K. ; Scholkopf, B. IEEE Trans Neural Netw 2001, 12, 181‐201. [DOI] [PubMed] [Google Scholar]
- 55. Bhavsar, H. ; Ganatra, A. , IJSCE 2012, 2, 2231‐2307. [Google Scholar]
- 56. Joseph, S. ; Karnik, S. ; Nilawe, P. ; Jayaraman, V. K. ; Idicula‐Thomas, S. IEEE/ACM Trans Comput Biol Bioinform 2012, 9, 1535‐1538. [DOI] [PubMed] [Google Scholar]
- 57. Torrent, M. ; Di Tommaso, P. ; Pulido, D. ; Nogues, M. V. ; Notredame, C. ; Boix, E. ; Andreu, D. Bioinformatics 2012, 28, 130‐131. [DOI] [PubMed] [Google Scholar]
- 58. Peek, A. S. BMC Bioinformatics 2007, 8, 182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Izumi, K. ; Kodama, E. ; Shimura, K. ; Sakagami, Y. ; Watanabe, K. ; Ito, S. ; Watabe, T. ; Terakawa, Y. ; Nishikawa, H. ; Sarafianos, S. G. ; Kitaura, K. ; Oishi, S. ; Fujii, N. ; Matsuoka, M. J Biol Chem 2009, 284, 4914‐4920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Hildinger, M. ; Dittmar, M. T. ; Schult‐Dietrich, P. ; Fehse, B. ; Schnierle, B. S. ; Thaler, S. ; Stiegler, G. ; Welker, R. ; von Laer, D. J Virol 2001, 75, 3038‐3042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Dwyer, J. J. ; Wilson, K. L. ; Davison, D. K. ; Freel, S. A. ; Seedorff, J. E. ; Wring, S. A. ; Tvermoes, N. A. ; Matthews, T. J. ; Greenberg, M. L. ; Delmedico, M. K. Proc Natl Acad Sci USA 2007, 104, 12772‐12777. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting Information