

Abstract
I propose a T‐cell receptor (TcR)‐based mechanism by which immunity mediates both “genetic self” and “microbial self” thereby, connecting microbiome disease with autoimmunity. The hypothesis is based on simple principles. First, TcR are selected to avoid strong cross‐reactivity with “self,” resulting in selection for a TcR repertoire mimicking “genetic self.” Second, evolution has selected for a “microbial self” that mimics “genetic self” so as to share tolerance. In consequence, our TcR repertoire also mimics microbiome antigenicity, providing a novel mechanism for modulating tolerance to it. Also, the microbiome mimics the TcR repertoire, acting as a secondary immune system. I call this TcR‐microbiome mimicry “holoimmunity” to denote immune tolerance to the “holobiont self.” Logically, microbiome‐host mimicry means that autoimmunity directed at host antigens will also attack components of the microbiome, and conversely, an immunological attack on the microbiome may cross‐react with host antigens producing “holoautoimmunity.”
Keywords: antigenic mimicry, autoimmunity, Crohn's disease, diabetes, holobiont, immune tolerance, immunologic mimicry, microbiome, non‐self, T‐cell receptors
T‐cell receptors (TcR) and antibodies are selected to mimic “self” antigens. The microbiome avoids immunity by mimicking “self” antigens and TcR (“holoimmunity”). Pathogens, however, are complementary to “self,” TcR, and the microbiome. A pathogen that also mimics “self” can trigger autoimmunity to the host, TcR, and microbiome simultaneously (“holoautoimmunity”).
Introduction: The problem of “microbiome self”‐“genetic self” immunological interactions within holobionts
Our understanding of immunological tolerance is challenged by the discovery that most multicellular organisms are “holobionts,” consisting not only of a “genetic self” but an equally important “commensal and symbiotic self” represented by the host microbiome. Not only is most of the microbiome acquired after clonal selection or deletion have already shaped the immune system, but the microbiome can change with time, geography and diet, altering the overall antigenic make‐up of the holobiont “self” 1, 2. How can such antigenic alterations be tolerated? The conceptual difficulties are enhanced by increasing numbers of observations that autoimmune diseases are often (perhaps always) associated with significant alterations in the host microbiome, and conversely, that autoimmunity may be induced, modified or even prevented by various manipulations of the host microbiome. In Sjogren's syndrome, tear‐ and saliva‐producing cells are attacked, and patients harbor higher numbers and frequencies of Streptococcus spp., Staphylococcus spp., Lactobacillus spp., and Candida albicans, and slightly lower proportions of Fusobactera and Prevotella spp. than unaffected individuals 3. Many of these microbes, as well as Escherichia coli, produce peptides that induce autoantibodies reactive against Sjogren's syndrome autoantigens 4. Similarly, rheumatoid arthritis patients exhibit decreases in Haemophilus spp. and concomitant increases in Prevotella copri, Lactobacilli, and Porphyromonas gingivalis. These microbes exhibit antigens that mimic RA autoantigens 5, 6. Behcet's syndrome (an autoimmune disease causing inflammation of the circulatory system) is similarly characterized by specific depletion in the genera Roseburia and Subdoligranulum 7. And specific alterations in the microbiome that will be reviewed below are present in type 1 diabetes mellitus and Crohn's disease. How can such specific changes in the microbiome be related to autoimmune‐disease‐related loss of tolerance to “genetic self”?
I suggest the following hypothesis, based upon two well‐understood principles of immunology, to explain how the immune system mediates the relationship between “microbiome self” and “genetic self.”
First, I assume that clonal selection and/or deletion are carried out as generally accepted, with the result that strongly autoreactive clones are eliminated or tolerized. An unappreciated consequence of eliminating TcR and BcR that are complementary to “self” is that the remaining TcR and BcR will mimic “genetic self,” and therefore, be tolerant to it.
Second, I assume, following Damian's molecular mimicry theory 8, 9 that microbes evolve to evade the immune system by being selected to mimic the host's “genetic self.” Because the microbiome and the TcR, and BcR repertoires are all selected to mimic “genetic self,” “microbiome self” will also mirror the host TcR and BcR repertoires. This microbiome‐TcR and BcR mimicry provides a possible mechanism by which tolerance for the microbiome (like tolerance for “genetic self”) is attained. While the microbiome clearly helps shape the early immune system 10), one implication of this hypothesis is that TcR and BcR repertoires intrinsically bound or limit the possible compositions of the “microbiome self” to those microbes that can best evade an active immune response.
I call the tolerance of the immune system for the combined “microbiome‐genetic host self,” or holobiont self, “holoimmunity.” “Immunity” is used here broadly to refer not only to the ability of the system to attack and eliminate foreign antigens, but also to carry out house‐keeping activities such as monitoring “self,” maintaining tolerance, performing cellular debris sampling, and promoting healing. Thus, “holoimmunity” involves not only the elimination of non‐commensal and non‐symbiotic microbes, but also the maintenance of a healthy and host‐appropriate microbiome.
Just as immunity has its correlate in autoimmunity, the concept of “holoimmunity” implies the existence of its correlate, “holoautoimmunity.” In autoimmune diseases, the immune system loses tolerance for “genetic self.” TcR or BcR become activated against “self” antigens. Because the microbiome has evolved to mimic the host's “genetic self,” TcR and BcR that are autoreactive may also attack the components of the microbiome that mimic the targeted “genetic self” antigens. Thus, specific alterations in the microbiome repertoire should characterize each autoimmune disease. Conversely, immunization against components of the microbiome may result in concomitant autoreactivity against corresponding “genetic self” antigens. The concept of holoautoimmunity provides one possible mechanism by which autoimmune diseases produce corresponding changes in the microbiome repertoire, and helps to explain how manipulating the microbiome can both initiate and treat “autoimmune” disease (e.g. 11, 12).
Tests of the holoimmunity hypothesis
Six tests of the “holoimmunity” and “holoautoimmunity” concepts are reported below using previously published sets of TcR. I use Crohn's disease (CD) and type 1 diabetes mellitus (T1DM) as case studies. Further testable predictions are proposed in the Discussion section.
Test 1 investigates whether TcR mimicry might be due to random matches or database artifacts by comparing various types of control sets.
Test 2 investigates whether TcR mimic the “genetic self,” creating a molecular “mirror” of host antigens. TcR mimicry of the “genetic self” may provide a mechanism by which “self” tolerance is achieved.
Test 3 investigates the prediction that TcR sequences in normal human hosts will “mirror” the “commensal and symbiotic self.” Such TCR‐“commensal and symbiotic self” mimicry could provide a mechanism for host tolerance of the normal microbiome.
Test 4 evaluates the prediction that microbes unrelated to human disease or to the human microbiome will not display similarities to either human “genetic self” or to human TcR.
Tests 2–4 establish the plausibility of the hypothesis that the immune system may mediate the development of the “commensal and symbiotic self” in relation to the “genetic self” by selecting for their immunological compatibility.
Test 5 investigates whether specific deviations of the TcR mimicry repertoire from “normal” are associated with individual autoimmune diseases and might identify the triggers/targets of the autoimmune/holoautoimmune process.
Test 6 evaluates whether disease‐specific alterations in TcR mimicry repertoires produce altered tolerance for specific elements of the microbiome.
Tests 5 and 6 establish the plausibility of the concept of holoautoimmunity resulting in correlated losses of tolerance for “genetic self” and “microbiome self.”
Methods
TcR data sets
Three sets of controls were run to establish baseline distributions of TcR mimicry of the microbes listed in Tables 1 and 2. One set was derived from a convenience sample of 101 TcR CD3 V beta/D/J beta regions sequenced from nine “normal” (i.e. disease‐free) individuals 13, 14, 15, 16, 17. A second set was derived from a convenience sample of 109 CD3 V beta/D/J beta regions of TcR sequences from 13 people after uncomplicated mono‐infections with streptococcal 18, staphylococcal 19, influenza A virus 20, 21, 22, 23, HTLV‐1 24, 25, Epstein‐Barr virus 26, 27, and tuberculosis 28, 29, 30 infections. All 210 control TcR sequences were analyzed for similarity to human “self” antigens. The third set of randomized controls was derived from the second by transforming each TcR sequence into its antisense sequence 31 utilizing the following substitutions: A to R; C to T; D to L; E to L; F to I or M; G to P; H to V; I to F; K to Y; L to E or D; M to F; N to I; P to G; Q to I; R to A or S; S to R or S; T to W or C; V to H; W to T; Y to K.
Table 1.
Summary of the frequency (in percent) of TcR mimics of 42 genera of bacteria, protozoa, and yeasts characterizing the human microbiome and some of its common pathogens, as well as 11 genera of plant bacteria and fungi
| Human pathogen | # Of taxons listed in UniProt | UniProtKB entries × 1,000 | Anti TCR% N = 101 | NOR TCR% N = 114 | CON TCR% N = 101 | CD TCR% N = 103 | T1D TCR% N = 68 | p‐value (χ 2) NOR versus CONT | p‐value (χ 2) CD versus NOR | p‐value (χ 2) CD versus CON | p‐value (χ 2) T1D versus NOR | p‐value (χ 2) T1D versus CON | p‐value (χ 2) T1D versus CD |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| B. cereus | 411 | 1,094 | 40 | 35 | 33 | 31 | 19 | ||||||
| Bacteroides | 738 | 709 | 71 | 58 | 68 | 53 | 44 | ||||||
| Bifidobacteria | 184 | 215 | 45 | 32 | 29 | 14 | 25 | ||||||
| B. pertussis | 12 | 51 | 23 | 9 | 4 | 4 | 3 | ||||||
| C. jejuni | 72 | 42 | 2 | 7 | 3 | 6 | 3 | ||||||
| C. albicans | 12 | 159 | 9 | 4 | 8 | 17 | 3 | ||||||
| Cardiobacteria | 25 | 5 | 12 | 1 | 1 | 0 | 0 | ||||||
| C. pneumoniae | 9 | 4 | 1 | 7 | 5 | 6 | 3 | ||||||
| Clostridium pathogenic | 76 | 127 | 32 | 23 | 28 | 21 | 53 | <0.0001 | <0.0001 | <0.0001 | |||
| Clostridium commensal | 498 | 796 | 64 | 40 | 66 | 47 | 11 | (<0.0001) | (<0.0001) | (<0.0001) | |||
| Coprococcus | 19 | 26 | 6 | 6 | 24 | 11 | 1 | (0.0004) | (<0.0001) | ||||
| Corynebacteria | 312 | 228 | 61 | 21 | 6 | 27 | 1 | 0.0002 | <0.0001 | (<0.0001) | (<0.0001) | ||
| C. neoformans | 29 | 127 | 19 | 0 | 0 | 22 | 3 | <0.0001 | <0.0001 | (<0.0001) | |||
| Entamoeba | 57 | 62 | 20 | 9 | 7 | 7 | 3 | ||||||
| Enterobacter | 612 | 595 | 68 | 25 | 2 | 19 | 0 | <0.0001 | <0.0001 | (<0.0001) | |||
| E. faecium | 162 | 106 | 9 | 21 | 28 | 29 | 13 | ||||||
| E. coli | 1,192 | 1,468 | 60 | 30 | 39 | 24 | 21 | ||||||
| Eubacterium | 202 | 123 | 40 | 25 | 34 | 23 | 3 | (<0.0001) | (<0.0001) | (<0.0001) | |||
| Giardia | 60 | 30 | 24 | 11 | 15 | 13 | 7 | ||||||
| H. influenzae | 43 | 41 | 5 | 5 | 7 | 6 | 1 | ||||||
| H. pyelori | 244 | 417 | 8 | 8 | 17 | 2 | 1 | (0.0003) | (<0.0001) | ||||
| K. pneumoniae | 128 | 190 | 20 | 11 | 16 | 12 | 6 | ||||||
| Lactobacilli | 673 | 819 | 66 | 50 | 54 | 27 | 34 | (0.0001) | (0.0001) | ||||
| L. pneumophila | 45 | 23 | 10 | 6 | 12 | 1 | 7 | ||||||
| Listeria | 122 | 116 | 15 | 9 | 8 | 16 | 4 | ||||||
| M. tuberculosis | 211 | 195 | 39 | 16 | 7 | 5 | 7 | ||||||
| Mycobacteria | 1,828 | 989 | 93 | 47 | 27 | 44 | 16 | 0.0008 | (<0.0001) | (<0.0001) | |||
| Mycoplasma | 329 | 74 | 11 | 16 | 7 | 11 | 3 | ||||||
| Neisseria | 318 | 143 | 24 | 7 | 9 | 7 | 1 | ||||||
| Prevotella | 500 | 240 | 51 | 49 | 60 | 44 | 10 | (<0.0001) | (<0.0001) | (<0.0001) | |||
| P. aeruginosa | 473 | 337 | 44 | 12 | 11 | 48 | 12 | <0.0001 | <0.0001 | (<0.0001) | |||
| Salmonella. | 890 | 503 | 34 | 14 | 12 | 11 | 7 | ||||||
| S. marcescens | 34 | 54 | 13 | 7 | 10 | 6 | 3 | ||||||
| S. dysenteriae | 18 | 36 | 5 | 6 | 1 | 6 | 4 | ||||||
| Staphylococcus | 834 | 238 | 28 | 14 | 16 | 15 | 9 | ||||||
| Streptococcus group A | 19 | 90 | 48 | 17 | 9 | 5 | 7 | ||||||
| Streptococcus (all) | 1,334 | 950 | 88 | 41 | 36 | 30 | 13 | (0.0001) | (0.0002) | ||||
| T. gondii | 70 | 96 | 53 | 26 | 29 | 25 | 21 | ||||||
| T. pallidum | 16 | 6 | 0 | 3 | 2 | 0 | 0 | ||||||
| T. vaginalis | 11 | 50 | 33 | 17 | 15 | 21 | 7 | ||||||
| T. cruzi | 15 | 59 | 31 | 14 | 15 | 17 | 8 | ||||||
| Plant bacteria | |||||||||||||
| Agrobacterium | 1,731 | 537 | 71 | 6 | 13 | ||||||||
| A. radiobacter and tumefaciens | 2 | 12 | 24 | 3 | 6 | ||||||||
| Fusarium | 1,922 | 460 | 74 | 44 | 64 | ||||||||
| F. oxysporum | 263 | 301 | 54 | 32 | 45 | ||||||||
| Janthinobacteria | 29 | 42 | 35 | 20 | 31 | ||||||||
| J. lividum | 1 | 11 | 4 | 12 | 10 | ||||||||
| Leuconostoc | 71 | 37 | 12 | 4 | 7 | ||||||||
| L. mesenteroides and ventriculitis | 2 | 2 | 4 | 2 | 4 | ||||||||
| Pantoea | 132 | 113 | 54 | 8 | 13 | ||||||||
| P. anthophila, ananatis, dispersa | 3 | 8 | 16 | 5 | 7 | ||||||||
| Pectobacterium | 68 | 77 | 21 | 4 | 9 | ||||||||
| Phytoplasma | 25 | 14 | 2 | 3 | 2 | ||||||||
| Thielavia | 28 | 19 | 36 | 17 | 16 | ||||||||
| T. terrestris | 2 | 19 | 24 | 15 | 12 | ||||||||
| Ustilago | 52 | 15 | 44 | 28 | 26 | ||||||||
| U. maydis | 8 | 7 | 10 | 10 | 17 | ||||||||
| Xanthomonas | 610 | 394 | 71 | 37 | 48 | ||||||||
| X. campestris | 139 | 65 | 9 | 25 | 14 | ||||||||
| Xylella | 24 | 24 | 9 | 4 | 3 | ||||||||
Plant bacterial and fungal species that are commensal or pathogenic for human beings are shaded. The number (N) of TCR screened for each group (Antisense, Normals, Controls, Crohn's Disease [CD], and type 1 diabetes mellitus [T1DM]) are provided. Statistically significant differences between groups are provided in the columns to the right: the p values of those that are significantly greater (by χ2 analysis) than the comparison group are in bold, underlined; those that are significantly less, are shown within parentheses. If no p‐value is provided, then any differences were found to have a p‐value greater than 0.0012 (the cutoff value for significance after a Bonferroni correction for the 42 bacterial categories tested). No significant correlations (by linear regression) were found for the number of TcR similarities and either the number of taxons or the number of proteins entries in the UniProtKB database (see text for details). Shaded boxes indicate plant bacteria and fungi species that have become human commensal organisms or are known to cause human disease. The unshaded plant bacteria and fungi data are the sum of the “humanized” and non‐humanized rates of mimicry, so that the rate of non‐humanized mimicry can be calculated by subtracting out the “humanized” rate.
Table 2.
Summary of the frequency (in percent) of TcR mimics of 38 viruses capable of infecting human beings as well as eight common classes of plant viruses
| Human pathogen | # Of taxons listed in UniProt | UniProtKB entries × 1,000 | Anti TCR% N = 101 | NOR TCR% N = 114 | CON TCR% N = 101 | CD TCR% N = 103 | T1D TCR% N = 68 | p‐value (χ2) NOR versus CON | p‐value (χ 2) CD versus NOR | p‐value (χ 2) CD versus CON | p‐value (χ 2) T1D versus NOR | p‐value (χ 2) T1D versus CON | p‐value (χ 2) T1D versus CD |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Adenovirus | 199 | 12 | 19 | 16 | 13 | 6 | 15 | ||||||
| Astrovirus | 107 | 2.5 | 3 | 2 | 2 | 0 | 0 | ||||||
| Bocavirus | 41 | 3 | 2 | 0 | 0 | 0 | 1 | ||||||
| Cardiovirus | 29 | 0.8 | 3 | 0 | 0 | 0 | 1 | ||||||
| Coronavirus | 195 | 3 | 6 | 3 | 16 | 0 | 1 | (<0.001) | (<0.001) | ||||
| Coxsackie A | 79 | 17 | 4 | 5 | 8 | 3 | 5 | ||||||
| Coxsackie B | 28 | 17 | 0 | 3 | 7 | 0 | 34 | <0.0001 | <0.0001 | <0.0001 | |||
| CMV | 117 | 18 | 19 | 21 | 21 | 22 | 26 | ||||||
| Echoviruses | 60 | 10 | 3 | 10 | 10 | 6 | 3 | ||||||
| Enteroviruses | 542 | 21 | 4 | 10 | 11 | 5 | 3 | ||||||
| EBV | 27 | 65 | 11 | 6 | 8 | 9 | 10 | ||||||
| HAV | 59 | 230 | 0 | 1 | 1 | 1 | 0 | ||||||
| HBV | 119 | 167 | 33 | 8 | 8 | 2 | 10 | ||||||
| HCV | 204 | 238 | 44 | 18 | 29 | 15 | 24 | ||||||
| HEV | 40 | 137 | 22 | 1 | 4 | 3 | 3 | ||||||
| HHV1 | 17 | 12 | 25 | 5 | 8 | 1 | 4 | ||||||
| HHV2 | 10 | 6 | 11 | 3 | 4 | 2 | 9 | ||||||
| HHV6 | 5 | 3 | 7 | 6 | 7 | 1 | 1 | ||||||
| HHV8 | 4 | 2 | 13 | 5 | 6 | 1 | 3 | ||||||
| HTLV | 14 | 3 | 11 | 1 | 10 | 0 | 0 | ||||||
| Infl A Virus | 266 | 583 | 34 | 24 | 24 | 11 | 24 | ||||||
| Infl B virus | 56 | 368 | 7 | 1 | 2 | 1 | 1 | ||||||
| Infl C virus | 21 | 356 | 2 | 0 | 2 | 0 | 1 | ||||||
| Jap enc virus | 25 | 6 | 1 | 2 | 2 | 1 | 1 | ||||||
| Measles virus | 15 | 12 | 14 | 6 | 5 | 0 | 0 | ||||||
| Mumps virus | 36 | 1 | 6 | 1 | 1 | 0 | 0 | ||||||
| Norovirus | 3 | 32 | 5 | 5 | 6 | 4 | 6 | ||||||
| Papilloma virus | 217 | 12 | 43 | 15 | 21 | 14 | 12 | ||||||
| Parainfluenza | 48 | 2 | 4 | 1 | 2 | 0 | 0 | ||||||
| Polio virus | 25 | 9 | 0 | 0 | 1 | 1 | 0 | ||||||
| Polyoma virus | 3 | 7 | 1 | 2 | 3 | 2 | 1 | ||||||
| Reovirus | 126 | 5 | 3 | 3 | 0 | 5 | 1 | ||||||
| RSV | 56 | 0.06 | 7 | 0 | 1 | 1 | 0 | ||||||
| Rhinovirus | 292 | 8 | 3 | 3 | 4 | 3 | 6 | ||||||
| Rotaviruses | 508 | 28 | 19 | 9 | 8 | 4 | 4 | ||||||
| Rubella | 41 | 31 | 6 | 2 | 0 | 0 | 3 | ||||||
| Varicella zoster | 2 | 4 | 8 | 3 | 10 | 1 | 3 | ||||||
| Plant viruses | |||||||||||||
| Curl | 730 | 13 | 25 | 0 | 0 | ||||||||
| Mosaic | 873 | 29 | 70 | 0 | 1 | ||||||||
| Mottle | 175 | 4 | 51 | 0 | 2 | ||||||||
| Rattle | 7 | 0.3 | 3 | 3 | 1 | ||||||||
| Spot | 86 | 6 | 33 | 3 | 6 | ||||||||
| Stunt | 54 | 3 | 15 | 0 | 1 | ||||||||
| Wilt | 22 | 3 | 9 | 4 | 3 | ||||||||
| Woodiness | 5 | 0.05 | 0 | 0 | 0 | ||||||||
The number (N) of TCR screened for each group (Antisense, Normals, Controls, Crohn's Disease [CD], and type 1 diabetes mellitus [T1DM]) are provided. Statistically significant differences between groups are provided in the columns to the right: the p values of those that are significantly greater (by χ2 analysis) than the comparison group are in bold, underlined; those that are significantly less, are shown within parentheses. If no p‐value is provided, then any differences were found to have a p‐value greater than 0.0013 (the cutoff value for significance after a Bonferroni correction for the 38 virus categories utilized in the study). No significant correlations (by linear regression) were found for the number of TcR similarities, and either the number of taxons nor the number of protein entries in the UniProtKB database (see bold, underlined and shaded). CMV, cytomegalovirus; EBV, Epstein–Barr virus; HAV, hepatitis A virus; HBV, hepatitis B virus; HCV, hepatitis C virus; HEV, hepatitis E virus; HHV, human herpes virus; HTLV, human T cell lymphoma viruses; Inf, influenza; Jap Enc, Japanese encephalitis virus; RSV, respiratory syncytial virus.
The first two sets of controls were compared with 103 CD3 V beta/D/J beta TCR sequences from six people with CD 32, 33. The controls were also compared with 68 CD3 V beta TCR sequences from eight people with T1DM 34, 35, 36, 37, 38, 39.
All TCR sequences are provided in the Supplementary Material.
Proteonomics
The TcR sequences were used as search strings in BLAST2.0 (http://www.expasy.org) for comparison with the entire UniProtKB database and screened for bacterial, fungal, yeast, and protozoal similarities by keyword search. A separate search was made of the UniProtKB virus database and yet another separate search on the Homo sapiens database. For microbe searches, the BLAST parameters were set on an E threshold (1,000), number of best scoring sequences to show (1,000), and number of best alignments to show (1,000); gapped similarities were permitted and the low complexity filter was turned off. The PAM‐30 Matrix was employed because of the short length of the TcR sequences. An example of the type of data generated is shown in Fig. 1.
Figure 1.

Example of sequence similarities between a Crohn's disease patient T‐cell receptor CD3 V beta/D/J beta region, human proteins, and microbial proteins. (BLAST 2.0 search of the UniProtKB database using the PAM‐30 Matrix with E‐value set to 100 for human proteins and 1,000 for microbial proteins). Data such as these are the basis for Tables 1 and 2.
For the Homo sapiens search, the BLAST parameters were set on an E threshold of 100, number of best sequences to show 100, and number of best alignments to show 100. The rest of the parameters were the same as in the microbe searches.
The resulting BLAST results were screened by the names of the microbes listed in Tables 1 and 2. In all, 41 specific genera of human‐associated bacteria, protozoa, and yeast were included in the survey and 38 types of pathogenic viruses. As negative controls for whether microbiome‐associated microbes are specifically mimicked by human TcR, 11 genera of plant‐specific bacteria and fungi, and eight classes of plant‐specific viruses were screened as well (Tables 1 and 2). This exercise turned out to be more complicated than expected as a number of plant bacteria and fungi have become human commensal organisms (listed in the shaded boxes at the bottom of Table 1). Such “humanized” plant microbes are predicted to mimic human TcR and were analyzed separately from “non‐humanized” species. A PubMed search was conducted on each species of microbe to determine whether it was a human commensal, symbiont or pathogen, or had no known human relationship. Distinctions between pathogenic and non‐pathogenic (or opportunistic) forms of Clostridioforme bacteria were made on the basis of published criteria 40, and Streptococcal species were divided into group A Streptococci and viridans groupings according to recent convention (http://viridans.emlsa.net/).
The Homo sapiens search data were screened for TcR‐related sequences, which were discarded as being irrelevant to the current study, since the V‐ and J‐beta subunits are widely shared.
Only sequence similarities in which at least six amino acids were identical in a 10‐amino acid sequence were considered to be significant similarities. Using the T1DM TcR used here, and a subset of the control TCR 41, I have previously demonstrated that the number‐of‐identities criterion fits experimental results involving antigenic cross‐reactivity better than measures such as the Waterman‐Eggert score or E value from the BLAST search 41, 42, 43, 44, 45. In the event, the vast majority of the similarities found during this study exceeded this cut‐off (Figs. 1, 2, 3).
Figure 2.

Examples of sequence similarities between T‐cell receptor CD3 V beta/D/J beta regions derived from Crohn's disease patients demonstrating simultaneous similarities between the TcR sequences, human sequences, and microbial sequences. Many additional similarities to individual human and microbial antigens that do not cluster also exist for these TcR, but are not shown (see Fig. 1 for a more complete example).
Figure 3.

Examples of sequence similarities between T‐cell receptor CD3 V beta/D/J beta regions derived from type 1 diabetes mellitus patients demonstrating simultaneous similarities between the TcR sequences, human sequences, and microbial sequences. Many additional similarities to individual human and microbial antigens that do not cluster also exist for these TcR, but are not shown (see Fig. 1 for a more complete example).
Statistics
Linear regression (R 2) values (http://vassarstats.net/index.html) were calculated to determine whether the number of taxons or the number of protein entries in the UniProtKB database significantly skewed the results of the similarity searches.
A chi squared test (http://www.quantpsy.org/chisq/chisq.htm) was used to compare the observed frequencies of the microbial similarities to TcR between the control and the CD groups; the control and the T1DM groups; and the CD and T1DM groups. Because multiple chi squared tests were run on the same sets of data, a Bonferonni correction was employed (http://www.winsteps.com/winman/bonferroni.htm).
Kolmogorov‐Smirnov (http://www.wessa.net/rwasp_Reddy-Moores%20K-S%20Test.wasp) and Mann‐Whitney (http://vassarstats.net/index.html) tests were used to determine whether the patient control TcR and the antisense patient control TcR derived from them showed significantly different distributions of microbial similarities.
Results
Test 1: The distribution of microbial mimics of TcR sequences is not random or a database artifact
Linear regression (R 2) values were calculated to examine whether the number of taxons or the number of protein entries associated with each microbe is a factor in determining the number of similarities found. They are not: Bacteria normal controls versus taxons, R 2 = 0.12; Bacteria normal controls versus UniProtKB entries, R 2 = 0.37; Bacterial patient controls versus taxons, R 2 = 0.21; Bacterial patient controls versus UniProtKB entries, R 2 = 0.34; Virus normal controls versus taxons, R 2 = 0.02; Virus normal controls versus UniProtKB entries, R 2 = 0.11; Virus patient controls versus taxons R 2 = 0.02; Virus patient controls versus UniProtKB entries R 2 = 0.06.
The distribution of microbial mimics of the antisense TcR controls was more significantly related to number of taxons and protein entries: Bacteria antisense controls versus taxons, R 2 = 0.54; Bacteria antisense controls versus UniProtKB entries, R 2 = 0.52; Virus antisense controls versus taxons, R 2 = 0.32; Virus antisense controls versus UniProtKB entries, R 2 = 0.02.
In sum, antisense TcR mimic microbes on a random basis determined by taxon and microbial protein entry number; normal human TcR do not. The difference between the two sets is statistically significant according both Kolmogorov‐Smirnov and Mann‐Whitney tests: Bacterial TcR (sense vs. antisense), Kolmogorov‐Smirnov Test statistic, 0.31148, p = 0.005; Mann‐Whitney Test U A = 117.5, z = −2.56, p = 0.005. Viral TcR (sense vs. antisense) Kolmogorov‐Smirnov Test statistic 0.30769, p = 0.049; Mann‐Whitney Test U A = 1570.5, z = −2.21, p = 0.014. In contrast, no significant differences were found between the patient control TcR and the normal control TcR by the Kolmogorov‐Smirnov or Mann‐Whitney tests. Thus, antisense TcR behave significantly differently than patient or normal control TcR in terms of microbial mimicry.
Test 2: TcR mimic “genetic self” antigens
The standard view of the immune system is that it produces T‐ and B‐cell receptors, as well as antibodies, that are complementary to host, commensal, and pathogen antigens. This complementary response is weak, and controlled by tolerizing mechanisms to the host, and commensal antigens, but strong and actively promoted against pathogen antigens. One unexpected observation from the current study is that all TcR other than the randomized antisense ones, mimicked many human antigens. It is not possible to present all of the relevant data here, even in tabular or graphical form, because every human TcR mimicked at least a dozen different human proteins, yielding thousands of significant similarities. Typical examples are provided in Figs. 1, 2, 3.These data, in aggregate, demonstrate that TcR selection by the immune system creates a set of lymphocytes that mirror a significant portion (if not all of) the antigenic diversity of the host itself.
Test 3: TcR mimic human microbiome antigens
Another surprising result is that TcR frequently mimic microbiome antigens. In other words, the immune system appears either to adapt to its microbiome, by selecting TcR that mirror the microbiome itself, or selects from available microbes a microbiome that is compatible with the existing TcR repertoire. Table 1 demonstrates that, on average, the probability that any set of human TcR will mimic key constituents of the gut microbiome such as Bacteroides, Bifidobacterium, E. coli, Bifidobacteria, Coprococcus, Corynebacteriuim, Clostridia, Enterobacteria, Eubacteria, Lactobacilli, or Prevotella is 36.0%. Every TcR of every patient, without exception, mimicked more than one of these commensal microbes. In contrast, the probability that any given TcR mimicked the remaining microbes in Table 1 is much lower (13.6%) and approaches zero for infectious agents such as Bacillus pertussis, Campylobacter jejuni, Cardiobacteria, Chlamydia pneumonia, Shigella dysenteriae, and Haemophilus influenza. Pathogens, in short, do not appear to mimic host TcR at anywhere near the rate that microbes making up the microbiome do.
Table 2 similarly demonstrates that TcR mimics are relatively rare with regard to human viruses, with the exceptions of those viruses that can induce chronic, persistent infections such as adenoviruses, enteroviruses, cytomegalovirus, hepatitis B and C viruses, papillomaviruses, rotaviruses, and varicella zoster virus. On average, 13.8% of TcR mimic these chronic viruses as compared with 4.2% of TcR that mimic viruses associated with acute disease. Chronic or persistent infection may therefore, be facilitated by having an unusually high degree of TcR mimicry, resulting in antigenic tolerance.
Antigenic exposure to a microbe can be ruled out as a causative factor in TcR mimicry of microbes: Despite almost universal vaccination against B. pertussis, polio, measles, mumps, and rubella viruses, TcR rarely showed similarities to these microbes.
Overall, these results suggest that the greater the number of TcR that mimic a particular microbial genera or species, the more likely that genera or species is to be tolerated as part of the microbiome.
Test 4: Human TcR rarely mimic plant microbes but “randomized” TcR do
If TcR and BcR are selected to mimic or mirror host antigens, it follows that pathogens will be characterized by being antigenically complementary to TcR and BcR, while commensal and symbiotic microbes will camouflage themselves by looking like “genetic self.” However, there is a third possibility as well, which is that antigens may exist that are neither complementary nor similar to the host's “genetic self,” or to its TcR and BcR. Such “non‐antigens” would be unable to interact with host proteins, and therefore, have no effect on the host.
Plant‐infecting microbes might be expected to display such “non‐antigens.” Indeed, human TcR almost never mimic plant viruses (Table 2): While 13.8% of TcR mimic chronic human viruses; 4.2%, acute human viruses; only 1.5% mimic plant viruses. And while 36% of TcR mimic human microbiome bacteria, only 6.4% of TcR mimic plant bacterial or fungal genera unrelated to human disease (Table 1). Exceptions “prove the rule”: 14.9% of plant bacteria and fungi that have become “humanized” and are able to live as commensal organisms or to cause human disease mimic TcR (Table 1, shaded entries).
Antisense control TcR mimic plant microbes at far higher rates than do control TcR. An average of 41.4% of antisense TcR mimic plant bacteria and fungi; “humanized” ones account for only 18.1%. Additionally, 25.8% of antisense TcR mimic plant viruses.
Overall, the differences between control TcR and antisense TcR mimicry of plant microbes is significant by both the Kolmogorov‐Smirnov (statistic 0.35714; p = 0.05) and Mann‐Whitney (U A = 218, z = 2.84; p = 0.002) tests. No differences were found by these tests between patient control TcR and normal control TcR.
In short, TcR sequences are non‐random, such that they mimic microbes that can effectively interact with the host but these TcR do not mimic “non‐antigens” from plant microbes. Randomized antisense TcR, in contrast, mimic these “non‐antigen” plant microbes at rates commensurate with their appearance in the UniProt database (see Test 1 above).
Test 5: Evidence for disease‐specific altered TcR mimicry distributions in CD and T1DM
To test whether distributions of microbe‐TcR mimicry are modified by specific autoimmune diseases, control TcR were compared with TcR from CD and T1DM patients.
With regard to CD, Table 1 shows that TcR derived from CD patients are significantly more likely to mimic C. albicans, Corynebacterium, Enterobacteriaciae, Mycobacteria, and Pseudomonas species than are control TcR. These increases are accompanied by significant decreases in TcR mimicry of Lactobacilli among the CD TcR. No significant differences in viral mimicry were observed between control and CD TcR (Table 2).
With regard to T1DM, Table 1 shows that alterations in T1DM TCR mimicry of bacteria are significantly different from the CD or control population TcR mimicries. T1DM TcR display significantly increased mimicry only to pathogenic Clostridia, accompanied by significant decreases in mimicry to commensal Clostridia, Eubacteria, Mycobacteria, Prevotella, and Streptococcus viridans species.
No significant alterations in TcR mimicry were observed with viruses in CD compared with the control groups, but the T1DM TcR showed a significant increase in mimicry of coxsackie B viruses (Table 2).
In sum, TCR mimicry of both commensal and pathogenic microbes changes significantly as a result of autoimmune disease in ways that suggest both enhanced activation against, and enhanced tolerance to, different microbes in the host microbiome. Thus, the triggering of both CD and T1DM appear to be accompanied by disease‐specific shifts in TcR mimicry of the microbiome.
Test 6: Alterations in TcR repertoire mirror changes in microbiome constituents in T1DM and CD that may reflect their causative agents
In general, microbiome diversity tends to decrease in people with autoimmune diseases, including T1DM 46 and CD 47, 48. This decrease, sometimes statistically significant, is also apparent in the TcR mimicries reported in Tables 1 and 2. It appears that activation of peripheral blood TcR reflects the distribution of the microbiome, and that as that microbiome changes, so does the repertoire of circulating TcR.
TcR alterations in T1DM correlate with probable causative agents
Only two sets of microbial‐TcR mimicry are significantly altered in T1DM according to the data summarized in Tables 1 and 2: Coxsackie B viruses and Clostridium species. This evidence is consistent with evidence that coxsackie B viruses are triggers of T1DM (e.g. 49, 50, 51). However, coxsackieviruses have repeatedly proven unable to induce T1DM in animal models 52. While no one has yet experimentally explored the possibility that Clostridia are causative agents of T1DM, Clostridia are among the types of gut bacteria (including Bacteroides, Lactobacilli, and Bacilli such as Streptococci) that are known to be seriously disturbed during diabetes 53, 54, 55, 56, 57, 58. A BLAST search on the UniProtKB bacterial database using human insulin A and B chains – the major antigenic targets in T1DM 38, 59, 60, 61 – as the search strings found that Clostridia and Lactobacilli are the only human bacteria to appear in the top 10 matches in each case (data not shown). Whether Clostridia can cause T1DM alone, or rather work in conjunction with coxsackieviruses, has not been tested, but would be consistent with experiments demonstrating that virally induced T1DM in an animal model can be prevented or cured with antibiotics 11. Indeed, Filippi and von Herrath 62 have recently suggested that, “This could be explained by the fact that viral association with T1D will likely be multifactorial.” Perhaps T1DM is actually caused by a combined coxsackievirus‐Clostridium infection, a testable prediction of holoautoimmunity.
TcR alterations in Crohn's disease correlate with probable causative agents
As with T1DM, the cause or causes of CD are not known 63, but antigens expressed by an imbalanced microbiome have been implicated in eliciting the immune response that drives gut inflammation 64, 65. Viruses do not appear to be among these, and many have explicitly been excluded (including all hepatitis viruses, enteroviruses including coxsackie types A and B, Epstein–Barr virus, measles virus, human herpes virus types 1, 6, and 8, varicella‐zoster virus, mumps virus, rubella virus, rotavirus, norovirus, and adenovirus [reviewed in 66, 67, 68]). Notably, there is no significant change in this study of CD TcR mimicry of any virus (Table 2).
Significant increases in some bacteria‐TcR mimicries are, however, evident in Table 1, and as in T1DM, these involve microbes associated epidemiologically and pathologically with the development of CD. These include increases in Enterobacteriaceae, including E. coli, 69, 70, 71, 72; Pseudomonas species 73, 74, 75; atypical Mycobacteria 76, 77, 78; and cryptococcal infections 79, 80, 81. TcR mimics of all of these putative triggers of CD are significantly increased in the data analyzed here (Table 1). Corynebacteria mimicry of TcR also increases, but I can find no evidence that this group of bacteria is (or is not) associated with CD pathogenesis, providing a testable prediction from the current data. In addition, Salmonella flagellin has been implicated as a possible antigenic trigger of anti‐TLR5 autoimmunity in CD 82, but many other bacteria have also been found to carry antigenically similar flagellins, including some, such as E. coli, implicated above in as CD triggers 83, 84. Table 1 also suggests that rather than S. cerevisiae, which is commonly used in the diagnosis of CD 85, 86, 87, 88, the responsible agent may be C. albicans, which also produces positive results in the anti‐S. cerevisiae antibody (ASCA) test 88, 89, 90.
As with T1DM, the TcR mimicry data provided here (Table 1) support a multifactorial trigger for CD, in agreement with recent opinion 91, and the requirement for CD diagnosis that evidence of multiple bacteria and yeast infections be present 85, 86, 87, 92, 93, 94. Another testable prediction from the TcR data are therefore, that a combination of such agents should be able to trigger CD holoautoimmunity.
In sum, significant alterations in TcR mimicry from normal distributions may identify combinations of infectious agents responsible for triggering autoimmune/holoautoimmune diseases. Unlike current unifactorial approaches, the TcR data implicate specific sets of infections as the triggers of such diseases.
Discussion
The results of the six tests performed here establish the plausibility of the holoimmune hypothesis that TcR are selected to mimic the “genetic self,” thereby, constraining possible microbiome constituents to those that also mimic both “genetic self” and “self”‐defining TcRs. The concept of holoimmunity also provides a framework for understanding holoautoimmunity, in which both “genetic self” and “microbiome self” are attacked simultaneously. Specific alterations in TcR mimicry of microbiome constituents may provide evidence concerning the (multifactorial) triggers of holoautoimmune diseases. Undoubtedly other mechanisms are also at work in determining potential tolerance and antigenicity, including activation of T‐regs, MHC‐, and CD1‐antigen display 95, 96, TcR α‐β‐chain pairing 97, innate immune pathways 1, 2, 11, 98, 99, 100 and idiotype‐anti‐idiotype networks (see Figure 4). The current hypothesis is compatible with these other mechanisms and undoubtedly works in concert with them. Key tests will involve whether TcR mimicry can accurately predict tolerance and “non‐antigenicity,” and whether the putative “triggers” of holoautoimmunity can be used to set up viable animal models of disease.
Figure 4.
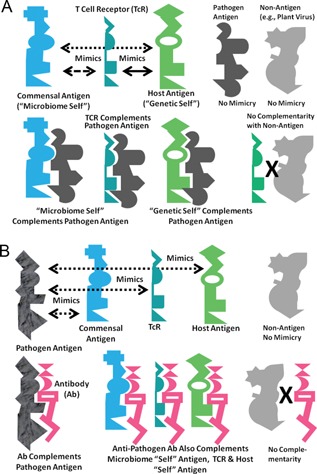
A, Top: A visual summary of the concept of holoimmunity. At the top, T‐cell receptors (TcR) are selected to mimic host antigens (the “genetic self”) and produce a molecular or antigenic “immunological mirror” of the host. Commensal and symbiotic microbes making up the microbiome of the host are tolerated by the immune system in part because they express a significant number of antigens mimicking both host antigens and the TcR “immunological mirror” (Tables 1 and 2). The result is simultaneous tolerance for a “microbiome self” and “genetic self” that overlap. Some potential antigens, such as plant viruses (upper right) neither mimic nor are complementary to human hosts, and interact with neither their cells nor their TcR. The bottom part of the figure illustrates how pathogens can be defined by their molecular complementarity to the host antigens. Only microbes able to interact with host physiological processes can become pathogenic (center). TcR mimic host antigens and prevent or counter infections by pathogens by means of molecular complementarity to antigens on the pathogen (right). Thus, agents that are not molecularly complementary to the host are of no physiological or immunological importance (in keeping with the plant bacteria, fungi, and virus data presented here [Tables 1 and 2]). B, Bottom: A visual summary of the concept of holoautoimmunity. At the top, some pathogenic microbes express antigens that mimic antigens of the microbiome and of the host, as well as host TcR (Figs. 1, 2, 3). In consequence, (below), antibodies induced against pathogenic mimics may recognize, through molecular complementarity, not only the pathogenic antigen, but also, to a greater or lesser degree, the “microbiome self” antigen, the “host self” antigen, and the TcR that the pathogenic antigen mimics. Thus, any autoimmune disease will affect not only the host, but its microbiome counterpart to produce “holoautoimmunity.” Antigens that neither mimic nor are complementary to host antigens (e.g. plant viruses) play no role in holoautoimmunity (right).
Further tests of the holoautoimmunity hypothesis
Many testable predictions follow from the hypothesis. One is that autoimmune diseases such as rheumatoid arthritis, lupus, myocarditis, idiopathic thrombocytopenia purpura, thyroiditis, etc. should display disease‐specific alterations in the microbiome that are reflected in TcR mimicry, and identify possible antigenic triggers. The TcR data should also be reflected in alterations in BcR and antibodies, so that, for example, IgA antibodies will mediate the microbiome and holoautoimmunity in ways directly analogous to those described here for TcR.
A second set of testable predictions is that the immune system has evolved only to “care about” foreign materials that can interact effectively with the host through molecularly complementary interactions. Specifically, pathogens or toxins are only of potential significance to the holobiont if they are be molecularly complementary to host antigens (e.g. cellular receptors or enzymes), and use that complementarity to recognize and interact effectively with host molecular processes. Figure 4A illustrates the concept. The hypothesis makes the testable prediction that plant viruses or most plant bacterial and fungal antigens that are neither similar nor complementary to TcR and BcR have no ability to interact effectively with human physiological functions. An expanded version of this idea could help to explain what the immune system views as “antigenic” or not.
Figure 4B illustrates how “immunological mirroring” applies to understanding holoautoimmunity. The model incorporates evidence that anti‐TcR antibodies and anti‐idiotype TcR exist in many autoimmune diseases 101, 102, 103, 104, 105. Whether such antibodies and TcR are pathogenic or are elicited as part of a Jerne‐type idiotype‐anti‐idiotype regulatory network is subject for debate 101, 102, 103, 104, 105, 106, 107, 108. A novel prediciton is that these anti‐TcR antibodies and anti‐idiotype TcR will also target the host and microbiome as well as immune system (TcR) antigens. No one has yet studied such anti‐TcR antibodies or anti‐idiotypic TcR to determine whether they also participate in recognition of host or microbiome antigens associated with autoimmune disease. Further investigation of this fascinating topic might reveal the deeper mechanisms by which the microbiome mediates, and is mediated by, the host immune system, thereby, playing a networked role in immune regulation.
A further logical consequence of “immunological mirroring” illustrated in Fig. 4A is that the “microbiome self” will be molecularly complementary to some pathogens (left), permitting the “microbiome self” to act as a secondary “immune system” for the host. Such immune‐like function would explain (beyond producing antibiotics and maintaining a stable microbiome ecosystem) how a healthy microbiome mediates against infectious disease. Some anti‐pathogen activity may reside literally in the host‐like molecular composition of the microbiome. This prediction is supported by evidence that helminths can modulate both the constituents of the bacterial microbiome and the host immune system in ways that can be beneficial 109, 110, 111.
It follows from TcR‐host‐microbiome mimicry that the microbiome will also be affected by many of the same pathogens and toxins as the host. For example, C. difficile infection causes specific alterations in the microbiome that may be responsible for the chronic nature of the infection 112, 113. Reconstituting a healthy microbiome by fecal transplant often restores the health of the holobiont as is does in cases of inflammatory bowel disease 114, 115. Thus, the holobiont may be viewed as an integrated ecology.
Because the host and its microbiome share integral elements of a common ecology, any invasive species that affects one is likely to affect the other. The risk of holoautoimmunity will therefore, be a function of how well the overall holobiont ecosystem is balanced. Any factor that can modify a key element of that ecosystem may also alter the rest. For example, recent research demonstrates that a functional microbiome is established partly at birth, as a result of exposure of the infant to its mother's microbiome while passing through the vagina. Infants born by ceasarean section do not have such exposure. Significant differences in susceptibility to allergic and autoimmune diseases have been associated with caesarean births 116, 117. Another factor is diet: Spicy foods can contain compounds with antibiotic properties, such that the ingestion of them can increase susceptibility to holoautoimmunity 118, 119. Antibiotics themselves can alter the risk of autoimmunity by altering the microbiome. And recent studies suggest that cancer therapies are mediated by the gut microbiome, implying that cancer risk may be mediated similarly 120. Such cross‐talk may help to explain the effectiveness of microbially derived adjuvants in cancer therapy and the apparent effectiveness of Coley's toxins for some cancer types. Thus, specific deviations of the microbiome from normal may precede and predict particular holoautommune diseases.
Conclusions and outlook
To summarize, my hypothesis is that “genetic self”‐“microbiome self” compatibility in mammalian holobionts is mediated by TcR and BcR mimicry of both, simultaneously (holoimmunity). The microbiome is not determined by the immune system, but is highly constrained by that to which the immune system is tolerant. Loss of tolerance to “genetic self” or “microbiome self” leads to holoautoimmunity, affecting both “selves” simultaneously. In consequence, every autoimmune disease is accompanied (and perhaps preceded) by characteristic alterations in particular portions of the microbiome corresponding, by molecular mimicry, to the host target of the disease. Conversely, what appears to be an autoimmune disease against the host may, in fact, be initiated against microbiome constituents that mimic the host antigens targeted in disease. Manipulating the host microbiome will therefore, affect host immunity and susceptibility to autoimmunity. The six tests carried out in this paper provide initial data confirming the plausibility of the hypothesis, but far more data‐intensive tests are clearly required to validate the details. Most importantly, the hypothesis provides a rationale for using alterations in TcR repertoires to identify possible multifactorial microbial triggers of holoautoimmune processes, from which new animal models might be developed, and treatment strategies (perhaps involving microbiome manipulation) devised.
The author declares that he has no conflicts of interest.
Authorship
All research, data analysis, interpretation, and writing was done by the author.
Funding
This research was carried out without funding.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supporting Data
References
- 1. Koenig JE, Spor A, Scalfone N, Fricker AD, et al. 2011. Succession of microbial consortia in the developing infant gut microbiome. Proc Natl Acad Sci USA 108: 4578–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Voreades N, Kozil A, Weir TL. 2014. Diet and the development of the human intestinal microbiome. Front Microbiol 5: 494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Almståhl A, Wikström M, Kroneld U. 2001. Microflora in oral ecosystems in primary Sjögren's syndrome. J Rheumatol 28: 1007–13. [PubMed] [Google Scholar]
- 4. Szymula A, Szczerba B, Bagavant H, Fu S‐M, et al. 2012. Oral and gut microbiota influence immune responses to Sjogren's syndrome associated antigen Ro60. Arthritis Rheum 64: 2675. [Google Scholar]
- 5. Brusca SB, Abramson SB, Scher JU. 2014. Microbiome and mucosal inflammation as extra‐articular triggers for rheumatoid arthritis and autoimmunity. Curr Opin Rheumatol 26: 101–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Zhang X, Zhang D, Jia H, Feng Q, et al. 2015. The oral and gut microbiomes are perturbed in rheumatoid arthritis and partly normalized after treatment. Nat Med 21: 895–905. [DOI] [PubMed] [Google Scholar]
- 7. Consolandi C, Turroni S, Emmi G, Severgnini M, et al. 2015. Behcet's syndrome patients exhibit specific microbiome signature. Autoimmune Rev 14: 269–76. [DOI] [PubMed] [Google Scholar]
- 8. Damian RT. 1964. Molecular mimicry: antigen sharing by parasite and host and its consequences. Am Natur 98: 129–49. [Google Scholar]
- 9. Damian RT. 1979. Molecular mimicryinbiological adaptation. In Nickol BB, ed; Host‐Parasite Interfaces. New York: Academic Press. 103–26. [Google Scholar]
- 10. Gensollen T, Iyer SS, Kasper DL, Blumberg RS. 2016. How colonoization by microbiota in early life shapes the immune system. Science 352: 539–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Hara N, Alkanani AK, Ir D, Robertson CE, et al. 2012. Prevention of virus‐induced type 1 diabetes with antibiotic therapy. J Immunol 189: 3805–14. Erratum in: J Immunol 189: 5995. [DOI] [PubMed] [Google Scholar]
- 12. Qian Q, Xiong S, Xu W. 2012. Manipulating intestinal immunity and microflora: an alternative solution to viral myocarditis? Future Microbiol 7: 1207–16. [DOI] [PubMed] [Google Scholar]
- 13.Biomedimmunotech. 2016. http://www.biomedimmunotech.com/store/technology.asp accessed February, 2016.
- 14. Forman JD, Klein JT, Silver RF, Liu MC, et al. 1994. Selective activation and accumulation of oligoclonal V beta‐specific T cells in active pulmonary sarcoidosis. J Clin Invest 94: 1533–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Probert CS, Chott A, Turner JR, Saubermann LJ, et al. 1996. Persistent clonal expansions of peripheral blood CD4+ lymphocytes in chronic inflammatory bowel disease. J Immunol 157: 3183–91. [PubMed] [Google Scholar]
- 16. Zeng W, Maciejewski JP, Chen G, Young NS. 2001. Limited heterogeneity of T cell receptor BV usage in aplastic anemia. J Clin Invest 108: 765–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Mamedov IZ, Britanova OV, Bolotin DA, Chkalina AV, et al. 2011. Quantitative tracking of T cell clones after haematopoietic stem cell transplantation. EMBO Mol Med 3: 201–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Diluvio L, Vollmer S, Besgen P, Ellwart JW, et al. 2006. Identical TCR beta‐chain rearrangements in streptococcal angina and skin lesions of patients with psoriasis vulgaris. Immunol 176: 7104–11. [DOI] [PubMed] [Google Scholar]
- 19. Quiròs Roldan E, Sottini A, Imberti L, Mattioli S, et al. 1994. Superantigen‐reactive human T cells express a biased repertoire of T‐cell receptor V beta joining regions. Res Immunol 145: 517–31. [DOI] [PubMed] [Google Scholar]
- 20. Brawley JV, Concannon P. 1999. Systematic mutagenesis of TCR complementarity‐determining region 3 residues: a single conservative substitution dramatically improves response to both multiple HLA‐DR alleles and peptide variants. J Immunol 163: 4946–52. [PubMed] [Google Scholar]
- 21. Goulmy E, Pool J, van den Elsen PJ. 1995. Interindividual conservation of T‐cell receptor beta chain variable regions by minor histocompatibility antigen‐specific HLA‐A*0201‐restricted cytotoxic T‐cell clones. Blood 85: 2478–81. [PubMed] [Google Scholar]
- 22. Bowness P, Moss PA, Rowland‐Jones S, Bell JI, et al. 1993. Conservation of T cell receptor usage by HLA B27‐restricted influenza‐specific cytotoxic T lymphocytes suggests a general pattern for antigen‐specific major histocompatibility complex class I‐restricted responses. Eur J Immunol 23: 1417–21. [DOI] [PubMed] [Google Scholar]
- 23. Moss PA, Moots RJ, Rosenberg WM, Rowland‐Jones SJ, et al. 1991. Extensive conservation of alpha and beta chains of the human T‐cell antigen receptor recognizing HLA‐A2 and influenza A matrix peptide. Proc Natl Acad Sci USA 88: 8987–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Utz U, Banks D, Jacobson S, Biddison WE. 1996. Analysis of the T‐cell receptor repertoire of human T‐cell leukemia virus type 1 (HTLV‐1) Tax‐specific CD8+ cytotoxic T lymphocytes from patients with HTLV‐1‐associated disease: evidence for oligoclonal expansion. J Virol 70: 843–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Bourcier KD, Lim D‐G, Ding Y‐H, Smith KJ, et al. Conserved CDR3 regions in T‐cell receptor (TCR) CD8+ T cells that recognize the TAX11‐19/HLA‐A*0201 complex in a subject infected with human T‐cell leukemia virus type 1: relationship of T‐cell fine specificity and major histocompatibility complex/peptide/tcr crystal structure. J Virol 75: 9836–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Levitsky V, de Campos‐Lima PO, Frisan T, Masucci MG. 1998. The clonal composition of a peptide‐specific oligoclonal CTL repertoire selected in response to persistent EBV infection is stable over time. J Immunol 161: 594–601. [PubMed] [Google Scholar]
- 27. Silins SL, Cross SM, Elliott SL, Pye SJ, et al. 1997. Selection of a diverse TCR repertoire in response to an Epstein‐Barr virus‐encoded transactivator protein BZLF1 by CD8+ cytotoxic T lymphocytes during primary and persistent infection. Int Immunol 9: 1745–55. [DOI] [PubMed] [Google Scholar]
- 28. Luo W, Su J, Zhang XB, Yang Z, et al. 2012. Limited T cell receptor repertoire diversity in tuberculosis patients correlates with clinical severity. PLoS ONE 7: e48117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Jacobsen M, Detjen AK, Mueller H, Gutschmidt A, et al. 2007. Clonal expansion of CD8+ effector T cells in childhood tuberculosis. J Immunol 179: 1331–9. [DOI] [PubMed] [Google Scholar]
- 30. Van Rhijn I, Kasmar A, de Jong A, Gras S, et al. 2013. A conserved human T cell population targets mycobacterial antigens presented by CD1b. Nat Immunol 14: 706–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Root Bernstein RS. 1982. Amino acid pairing. J Theor Biol 94: 885–94. [DOI] [PubMed] [Google Scholar]
- 32. Gulwani‐Akolkar B, Akolkar PN, Minassian A, Pergolizzi R, et al. 1996. Selective expansion of specific T cell receptors in the inflamed colon of Crohn's disease. J Clin Invest 98: 1344–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Probert CS, Aitken EW, Saubermann LJ, Balk SP, et al. 1998. T‐cell receptor usage in the intestine. Chem Immunol 71: 27–39. [DOI] [PubMed] [Google Scholar]
- 34. Durinovic‐Bellò I, Steinle A, Ziegler AG, Schendel DJ. 1994. HLA‐DQ‐restricted, islet‐specific T‐cell clones of a type I diabetic patient. T‐cell receptor sequence similarities to insulitis‐inducing T‐cells of nonobese diabetic mice. Diabetes 43: 1318–25. [DOI] [PubMed] [Google Scholar]
- 35. De Berardinis P, Ombra MN, Buono C, Toraldo R, et al. 1994. Long‐term culture and T cell receptor analysis of T cell clones isolated from a patient with adenosine deaminase deficiency and type I diabetes. Clin Immunol Immunopathol 73: 362–6. [DOI] [PubMed] [Google Scholar]
- 36. Conrad B, Weidmann E, Trucco G, Rudert WA, et al. 1994. Evidence for superantigen involvement in insulin‐dependent diabetes mellitus aetiology. Nature 371: 351–5. [DOI] [PubMed] [Google Scholar]
- 37. Naserke HE, Durinovic‐Bellò I, Seidel D, Ziegler AG. 1996. The T‐cell receptor beta chain CDR3 region of BV8S1/BJ1S5 transcripts in type 1 diabetes. Immunogenetics 45: 87–96. [DOI] [PubMed] [Google Scholar]
- 38. Kent SC, Chen Y, Bregoli L, Clemmings SM, et al. 2005. Expanded T cells from pancreatic lymph nodes of type 1 diabetic subjects recognize an insulin epitope. Nature 435: 224–8. [DOI] [PubMed] [Google Scholar]
- 39. Codina‐Busqueta E, Scholz E, Muñoz‐Torres PM, Roura‐Mir C, et al. 2011. TCR bias of in vivo expanded T cells in pancreatic islets and spleen at the onset in human type 1 diabetes. J Immunol 186: 3787–97. [DOI] [PubMed] [Google Scholar]
- 40. Ellner PD, Green SS. 1963. Serological grouping of the pathogenic Clostridia . J Bacteriol 86: 1098–100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Rudensky AYu, Preston‐Hurlburt P, Hong SC, Barlow A, et al. 1991. Sequence analysis of peptides bound to MHC class II molecules. Nature 353: 622–7. [DOI] [PubMed] [Google Scholar]
- 42. Cunningham MW, McCormack JM, Fenderson PG, Ho MK, et al. 1989. Human and murine antibodies cross‐reactive with streptococcal M protein and myosin recognize the sequence GLN‐LYS‐SER‐LYS‐GLN in M protein. J Immunol 143: 2677–83. [PubMed] [Google Scholar]
- 43. Root‐Bernstein RS. 2014. Rethinking molecular mimicry in rheumatic heart disease and autoimmune myocarditis: laminin, collagen IV, CAR, and B1AR as initial targets of disease. Front Ped Rheumatol 2: 85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Root‐Bernstein RS, Podufaly A. 2012. Autoreactive T‐cell receptor (Vbeta/D/Jbeta) sequences in diabetes recognize insulin, the insulin receptor, and each other, and are targets of insulin antibodies. Open Autoimmun J 4: 10–22. [Google Scholar]
- 45. Root‐Bernstein RS. 2009. Autoreactive T‐cell receptor (Vbeta/D/Jbeta) sequences in diabetes are homologous to insulin, glucagon, the insulin receptor, and the glucagon receptor. J Mol Recognit 22: 177–87. [DOI] [PubMed] [Google Scholar]
- 46. Kostic AD, Gevers D, Siljander H, Vatanen T, et al. 2015. The dynamics of the human infant gut microbiome in development and in the progression toward type 1 diabetes. Cell Host Microbe 17: 260–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Hofer U. 2014. Bacterial imbalance in Crohn's disease. Nat Rev Microbio 12: 312–3. [DOI] [PubMed] [Google Scholar]
- 48. Wright EK, Kamm MA, Teo SM, Inouye M, et al. 2015. Recent advances in characterizing the gastrointestinal microbiome in Crohn's disease: a systematic review. Inflamm Bowel Dis 21: 1219–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Oikarinen M, Tauriainen S, Oikarinen S, Honkanen T, et al. 2012. Type 1 diabetes is associated with enterovirus infection in gut mucosa. Diabetes 61: 687–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Hober D, Sauter P. 2010. Pathogenesis of type 1 diabetes mellitus: interplay between enterovirus and host. Nat Rev Endocrinol 6: 279–89. [DOI] [PubMed] [Google Scholar]
- 51. Richer MJ, Horwitz MS. 2009. Coxsackievirus infection as an environmental factor in the etiology of type 1 diabetes. Autoimmun Rev 8: 611–5. [DOI] [PubMed] [Google Scholar]
- 52. Zipris D. 2009. Epidemiology of type 1 diabetes and what animal models teach us about the role of viruses in disease mechanisms. Clin Immunol 131: 11–23. [DOI] [PubMed] [Google Scholar]
- 53. Goffau MC, Fuentes S, van den Bogert B, Honkanen H, et al. 2014. Aberrant gut microbiota composition at the onset of type 1 diabetes in young children. Diabetologia 57: 1569–77. [DOI] [PubMed] [Google Scholar]
- 54. Murri M, Leiva I, Gomez‐Zumaquero JM, Tinahones FJ, et al. 2013. Gut microbiota in children with type 1 diabetes differs from that in healthy children: a case‐control study. BMC Med 11: 46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Zipris D. 2013. The interplay between the gut microbiota and the immune system in the mechanism of type 1 diabetes. Curr Opin Endocrinol Diabetes Obes 20: 265–70. [DOI] [PubMed] [Google Scholar]
- 56. Davis‐Richardson AG, Triplett EW. 2015. A model for the role of gut bacteria in the development of autoimmunity for type 1 diabetes. Diabetologia 58: 1386–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Endesfelder D, zu Castell W, Ardissone A, Davis‐Richardson AG, et al. 2014. Compromised gut microbiota networks in children with anti‐islet cell autoimmunity. Diabetes 63: 2006–14. [DOI] [PubMed] [Google Scholar]
- 58. Giongo A, Gano KA, Crabb DB, Mukherjee N, et al. 2011. Toward defining the autoimmune microbiome for type 1 diabetes. ISME J 5: 82–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Griffin A, Zhao CW, Wegmann KW, Hickley WF. 1995. Experimental autoimmune insulitis: induction by T lymphocytes specific for a peptide of proinsulin. Am J Pathol 147: 845–57. [PMC free article] [PubMed] [Google Scholar]
- 60. Schloot NC, Willemen S, Duinkerken G, de Vries RR, et al. 1998. Cloned T cells from a recent onset IDDM patient reactive with in‐sulin B‐chain. J Autoimmun 11: 169–75. [DOI] [PubMed] [Google Scholar]
- 61. Narendran P, Williams AJ, Elsegood K, Leech NJ, et al. 2003. Humoral and cellular immune responses to proinsulin in adults with newly diagnosed type 1 diabetes. Diabetes Metab Res Rev 19: 52–9. [DOI] [PubMed] [Google Scholar]
- 62. Filippi C, von Herrath M. 2005. How viral infections affect the autoimmune process leading to type 1 diabetes. Cell Immunol 233: 125–32. [DOI] [PubMed] [Google Scholar]
- 63. Farrukh A, Mayberry JF. 2014. Inflammatory bowel disease in Hispanic communities: a concerted South American approach could identify the aetiology of Crohn's disease and ulcerative colitis. Arq Gastroenterol 51: 271–5. [DOI] [PubMed] [Google Scholar]
- 64. Berg D, Clemente JC, Colombel JF. 2015. Can inflammatory bowel disease be permanently treated with short‐term interventions on the microbiome? Exp Rev Gastroenterol Hepatol 10: 1–5. [DOI] [PubMed] [Google Scholar]
- 65. Haag LM, Hofmann J, Kredel LI, Holzem C, et al. 2015. Herpes simplex virus sepsis in a young woman with Crohn's disease. J Crohns Colitis 9: 1169–73. [DOI] [PubMed] [Google Scholar]
- 66. Wagner J, Sim WH, Lee KJ, Kirkwood CD. 2013. Current knowledge and systematic review of viruses associated with Crohn's disease. Rev Med Virol 23: 145–71. [DOI] [PubMed] [Google Scholar]
- 67. Masclee GM, Penders J, Pierik M, Wolffs P, et al. 2013. Enteropathogenic viruses: triggers for exacerbation in IBD? A prospective cohort study using real‐time quantitative polymerase chain reaction. Inflamm Bowel Dis 19: 124–31. [DOI] [PubMed] [Google Scholar]
- 68. Norman JM, Handley SA, Baldridge MT, Droit L, et al. 2015. Disease‐specific alterations in the enteric virome in inflammatory bowel disease. Cell 160: 447–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Seksik P, Rigottier‐Gois L, Gramet G, Sutren M, et al. 2003. Alterations of the dominant faecal bacterial groups in patients with Crohn's disease of the colon. Gut 52: 237–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Garrett WS, Gallini CA, Yatsunenko T, Michaud M, et al. 2010. Enterobacteriaceae act in concert with the gut microbiota to induce spontaneous and maternally transmitted colitis. Cell Host Microbe 8: 292–300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Harmsen HJ, Pouwels SD, Funke A, Bos NA, et al. 2012. Crohn's disease patients have more IgG‐binding fecal bacteria than controls. Clin Vaccine Immunol 19: 515–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Winter SE, Winter MG, Xavier MN, Thiennimitr P, et al. 2013. Host‐derived nitrate boosts growth of E. coli in the inflamed gut. Science 339: 708–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Liu L, Chen H, Brecher MB, Li Z, et al. 2013. Pfit is a structurally novel Crohn's disease‐associated superantigen. PLoS Pathog 9: e1003837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Wei B, Huang T, Dalwadi H, Sutton CL, et al. 2002. Pseudomonas fluorescens encodes the Crohn's disease‐associated I2 sequence and T‐Cell superantigen. Infect Immun 70: 6567–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Graham DY, Yoshimura HH, Estes MK. 1983. DNA hybridization studies of the association of Pseudomonas maltophilia with inflammatory bowel diseases. J Lab Clin Med 101: 940–54. [PubMed] [Google Scholar]
- 76. McNees AL, Markesich D, Zayyani NR, Graham DY. 2015. Mycobacterium paratuberculosis as a cause of Crohn's disease. Exp Rev Gastroenterol Hepatol 16: 1–2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Pierce ES. 2010. Ulcerative colitis and Crohn's disease: is Mycobacterium avium subspecies paratuberculosis the common villain? Gut Pathog 2: 21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Behr MA, Kapur V. 2008. The evidence for Mycobacterium paratuberculosis in Crohn's disease. Curr Opin Gastroenterol 24: 17–21. [DOI] [PubMed] [Google Scholar]
- 79. Li Q, Wang C, Tang C, He Q, et al. 2014. Dysbiosis of gut fungal microbiota is associated with mucosal inflammation in Crohn's disease. J Clin Gastroenterol 48: 513–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Sciaudone G, Pellino G, Guadagni I, Somma A, et al. 2011. Disseminated Cryptococcus neoformans infection and Crohn's disease in an immunocompetent patient. Crohns Colitis 5: 60–3. [DOI] [PubMed] [Google Scholar]
- 81. Epple HJ. 2009. Therapy‐ and non‐therapy‐dependent infectious complications in inflammatory bowel disease. Dig Dis 27: 555–9. [DOI] [PubMed] [Google Scholar]
- 82. Lunardi C, Bason C, Dolcino M, Navone R, et al. 2009. Antiflagellin antibodies recognize the autoantigens Toll‐Like Receptor 5 and Pals 1‐associated tight junction protein and induce monocytes activation and increased intestinal permeability in Crohn's disease. J Intern Med 265: 250–65. [DOI] [PubMed] [Google Scholar]
- 83. Duck LW, Walter MR, Novak J, Kelly D, et al. 2007. Isolation of flagellated bacteria implicated in Crohn's disease. Inflamm Bowel Dis 13: 1191–201. [DOI] [PubMed] [Google Scholar]
- 84. Lodes MJ, Cong Y, Elson CO, Mohamath R, et al. 2004. Bacterial flagellin is a dominant antigen in Crohn disease. J Clin Invest 113: 1296–306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Davis MK, Andres JM, Jolley CD, Novak DA, et al. 2007. Antibodies to Escherichia coli outer membrane porin C in the absence of anti‐Saccharomyces cerevisiae antibodies and anti‐neutrophil cytoplasmic antibodies are an unreliable marker of Crohn disease and ulcerative colitis. J Pediatr Gastroenterol Nutr 45: 409–13. [DOI] [PubMed] [Google Scholar]
- 86. Zholudev A, Zurakowski D, Young W, Leichtner A, et al. 2004. Serologic testing with ANCA, ASCA, and anti‐OmpC in children and young adults with Crohn's disease and ulcerative colitis: diagnostic value and correlation with disease phenotype. Am J Gastroenterol 99: 2235–41. [DOI] [PubMed] [Google Scholar]
- 87. Saibeni S, Folli C, de Franchis R, Borsi G, et al. 2003. Diagnostic role and clinical correlates of anti‐Saccharomyces cerevisiae antibodies (ASCA) and anti‐neutrophil cytoplasmic antibodies (p‐ANCA) in Italian patients with inflammatory bowel diseases. Dig Liver Dis 35: 862–8. [DOI] [PubMed] [Google Scholar]
- 88. Standaert‐Vitse A, Sendid B, Joossens M, François N, et al. 2009. Candida albicans colonization and ASCA in familial Crohn's disease. Am J Gastroenterol 104: 1745–53. [DOI] [PubMed] [Google Scholar]
- 89. Sendid B, Dotan N, Nseir S, Savaux C, et al. 2008. Antibodies against glucan, chitin, and Saccharomyces cerevisiae mannan as new biomarkers of Candida albicans infection that complement tests based on C. albicans mannan. Clin Vaccine Immunol 15: 1868–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Schaffer T, Müller S, Flogerzi B, Seibold‐Schmid B, et al. 2007. Anti‐Saccharomyces cerevisiae mannan antibodies (ASCA) of Crohn's patients crossreact with mannan from other yeast strains, and murine ASCA IgM can be experimentally induced with Candida albicans . Inflamm Bowel Dis 13: 1339–46. [DOI] [PubMed] [Google Scholar]
- 91. Pérez‐Brocal V, García‐López R, Nos P, Beltrán B, et al. 2015. Metagenomic analysis of Crohn's disease patients identifies changes in the virome and microbiome related to disease status and therapy, and detects potential interactions and biomarkers. Inflamm Bowel Dis 21: 2515–32. [DOI] [PubMed] [Google Scholar]
- 92. Kassan H, Cohavy O, Rosenbaum JT, Braun J, et al. 2005. Uveitis seroreactivity to candidate pANCA antigens: mycobacterial HupB and histone H1 (69‐171). Ocul Immunol Inflamm 13: 191–8. [DOI] [PubMed] [Google Scholar]
- 93. Eggena M, Cohavy O, Parseghian MH, Hamkalo BA, et al. 2000. Identification of histone H1 as a cognate antigen of the ulcerative colitis‐associated marker antibody pANCA. J Autoimmun 14: 83–97. [DOI] [PubMed] [Google Scholar]
- 94. Cohavy O, Harth G, Horwitz M, Eggena M, et al. 1999. Identification of a novel mycobacterial histone H1 homologue (HupB) as an antigenic target of pANCA monoclonal antibody and serum immunoglobulin A from patients with Crohn's disease. Infect Immun 67: 6510–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Kosmrlj A, Jha AK, Huseby ES, Kardar M, et al. 2008. How the thymus designs antigen‐specific and self‐tolerant T cell receptor sequences. Proc Natl Acad Sci USA 105: 16671–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Pierce BG, Vreven T, Weng Z. 2014. Modeling T cell receptor recognition of CD1‐lipid and MR1‐metabolite complexes. BMC Bioinformatics 15: 319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Han A, Glanville J, Hansmann L, Davis MM. Linking T‐cell receptor sequence to functional phenotype at the single‐cell level. Nat Biotechnol 32: 684–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Rakoff‐Nahoum S, Paglino J, Eslami‐Varzaneh F, Edberg S, et al. 2004. Recognition of commensal microflora by toll‐like receptors is required for intestinal homeostasis. Cell 118: 229–41. [DOI] [PubMed] [Google Scholar]
- 99. Man SM, Kaakoush NO, Mitchell HM. 2011. The role of bacteria and pattern‐recognition receptors in Crohn's disease. Nat Rev Gastro Hepato 8: 152–68. [DOI] [PubMed] [Google Scholar]
- 100. Schieber AM, Lee YM, Chang MW, Leblanc M, et al. 2015. Disease tolerance mediated by microbiome E. coli involves inflammasome and IGF‐1 signaling. Science 350: 558–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Acha‐Orbea H. 1993. Anti‐T‐cell receptor V beta antibodies in autoimmunity. Immunol Ser 59: 193–202. [PubMed] [Google Scholar]
- 102. Adelman MK, Schluter SF, Robey IF, Marchalonis JJ. 2007. Natural and autoantibodies to human T‐cell receptor Vbeta segments: potential roles in immunomodulation. Crit Rev Immunol 27: 221–32. [DOI] [PubMed] [Google Scholar]
- 103. Marchalonis JJ, Kaymaz H, Schluter SF, Yocum DE. 1993. Human autoantibodies to a synthetic putative T cell receptor beta‐chain regulatory idiotype: expression in autoimmunity and aging. Exp Clin Immunogenet 10: 1–5. [PubMed] [Google Scholar]
- 104. Boitard C, Sempé P, Bach JF. 1993. Anti‐TcR antibodies in autoimmune diseases. Immunol Ser 59: 167–73. [PubMed] [Google Scholar]
- 105. Schluter SF, Adelman MK, Taneja V, David C, et al. 2003. Natural autoantibodies to TCR public idiotopes: potential roles in immunomodulation. Cell Mol Biol (Noisy‐le‐grand) 49: 193–207. [PubMed] [Google Scholar]
- 106. Pernis B. 1993. Do anergic T cells induce suppressor T lymphocytes through idiotypic interactions? Int Rev Immunol 10: 327–35. [DOI] [PubMed] [Google Scholar]
- 107. Root‐Bernstein RS, Couturier J. 2006. Antigenic complementarity in the origins of autoimmunity: a general theory illustrated with a case study of idiopathic thrombocytopenia purpura. Clin Develop Immunol 13: 49–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108. Root‐Bernstein RS, Fairweather D. 2014. Rethinking autoimmune disease: a case study of autoimmune myocarditis. J Theor Biol 375: 101–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109. Zaiss MM, Harris NL. 2016. Interactions between the intestinal microbiome and helminth parasites. Parasite Immunol 38: 5–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110. Mutapi F. 2015. The gut microbiome in the helminth infected host. Trends Parasitol 31: 405–6. [DOI] [PubMed] [Google Scholar]
- 111. Glendinning L, Nausch N, Free A, Taylor DW, et al. 2014. The microbiota and helminths: sharing the same niche in the human host. Parasitology 141: 1255–71. [DOI] [PubMed] [Google Scholar]
- 112. Khoruts A, Dicksved J, Jansson JK, Sadowsky MJ. 2010. Changes in the composition of the human fecal microbiome after bacteriotherapy for recurrent Clostridium difficile‐associated diarrhea. J Clin Gastroenterol 44: 354–60. [DOI] [PubMed] [Google Scholar]
- 113. Schubert AM, Rogers MAM, Ring C, Mogle J, et al. 2014. Microbiome data distinguish patients with Clostridium difficile infection and non‐C. Difficile‐associated diarrhea from healthy controls. mBio 5: e01021–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114. Seekatz AM, Theriot CM, Molloy CT, Wozniak KL, et al. 2015. Fecal microbiota transplantation eliminates Clostridium difficile in a murine model of relapsing disease. Infect Immun 83: 3838–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115. Petrof EO, Gloor GB, Vanner SJ, Weese SJ, et al. 2013. Stool substitute transplant therapy for the eradication of Clostridium difficile infection: ‘rePOOPulating’ the gut. Microbiome 1: 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116. Biasucci G, Benenati B, Morelli L, Bessi E, et al. 2008. Cesarean delivery may affect the early biodiversity of intestinal bacteria. J Nutr 138: 1796S–800S. [DOI] [PubMed] [Google Scholar]
- 117. Neu J, Rushing J. 2011. Cesarean versus vaginal delivery: long term infant outcomes and the hygiene hypothesis. Clin Perinatol 38: 321–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118. Deng N, Huang Y, Hu Y, Zhao M, et al. 2016. Some like it hot: the emerging role of spicy food (capsaicin) in autoimmune diseases. Autoimmun Rev 15: 451–6. [DOI] [PubMed] [Google Scholar]
- 119. Cichewicz O, Thorpe RH. 1996. The antimicrobial properties of chile peppers (Capsicum species) and their uses in Mayan medicine. J Ethnopharmacol 52: 61–70. [DOI] [PubMed] [Google Scholar]
- 120. Perez‐Chanona E, Jobin C. 2014. From promotion to management: the wide impact of bacteria on cancer and its treatment. Bioessays 36: 658–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supporting Data