

Abstract
A novel 5‐membered iminocyclitol derivative was found to be a potent and selective inhibitor of the glycoprotein‐processing α‐glucosidase with a K i value of 53 nm. This compound was further derivatized to antiviral agents against Japanese encephalitis virus, dengue virus serotype 2 (DEN‐2), human SARS coronavirus, and human β‐hexosaminidase (K i =2.6 nm), a new target for the development of osteoarthritis therapeutics.
Keywords: antiviral agents, glycoproteins, iminocyclitols, inhibitors, osteoarthritis
Five against five: A novel 5‐membered iminocyclitol derivative (1) was found to be a potent and selective inhibitor of the glycoprotein‐processing α‐glucosidase with a K i value of 53 nm. It was further developed into antiviral agents such as 2 (against Japanese encephalitis virus, dengue virus serotype 2, human SARS coronavirus, and human β‐hexosaminidase (K i=2.6 nm), a new target for development of osteoarthritis therapeutics.
Introduction
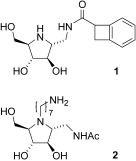
Glycosidases are involved in the biosynthesis and processing of the oligosaccharide chains of N‐linked glycoproteins in the endoplasmic reticulum (ER; Figure 1).1 Inhibition of these glycosidases, especially α‐glucosidases, has a profound effect on the glycan structure and, consequently, affects the maturation, transport, secretion, and function of glycoproteins, and could therefore alter cell–cell or cell–virus recognition processes.2–4 The neuraminidase inhibitor Tamiflu, for example, has been prescribed for the treatment of influenza virus infection.5 The digestive α‐glucosidase inhibitor, N‐hydroxyethyldeoxynojirimycin (1, also called Glyset or Miglitol, Scheme 1), has been used for the treatment of non‐insulin‐dependent diabetes.6, 7 Recent studies also showed that deoxynojirimycin (DNJ, 2) and its derivatives (especially N‐butyl DNJ) were potentially useful for treating several human diseases such as cancers,8 viruses (AIDS,9, 10 hepatitis B,11, 12 hepatitis C,13, 14 and dengue[15, 16]), and glycosphingolipid storage disorders.17, 18 The efficacy of iminocyclitols is attributed to their mimicry of the transition state of enzymatic glycosidic cleavage.19
Figure 1.
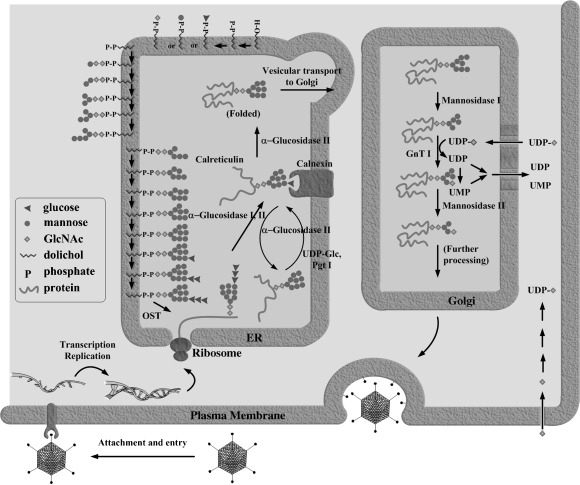
The assembly of viral envelope glycoproteins involves the use of host enzymes such as glycosidases and glycosyltransferases. Inhibition of glycosidases could disrupt the process and inhibit viral replication.
Scheme 1.
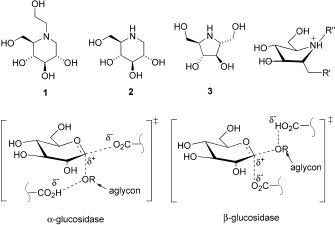
Structures of iminocylitols 1 to 3 and representative transition states of α‐ and β‐glucosidase‐catalyzed reactions showing the aglycon R. The five‐membered iminocyclitol shown top right is used for derivatization at R′ and R′′ to probe the aglycon binding sites.
The iminocyclitol 2,5‐dideoxy‐2,5‐imino‐d‐mannitol (3), a natural product,20 is a powerful inhibitor of a large range of α‐ and β‐glucosidases, even surpassing the activity of DNJ, which is frequently used as a standard.19, 21 The five‐membered iminocyclitols that carry hydroxyl groups of a specific orientation can mimic the shape and charge of the reacting sugar moiety of the transition state.19, 22, 24 Since enzyme inhibition could be significantly enhanced with slight modifications at the aglycon moiety,25–27 iminocyclitols could be used as common cores for the development of selective glycosidase inhibitors through identification of an additional group to occupy the aglycon space. To quickly find such a group for attachment to the 5‐membered iminocyclitol, we decided to conduct a combinatorial modification of compound 4 (Scheme 2) at the amine group in microtiter plates followed by screening in situ, a strategy successfully applied to other enzymes.27, 28 We initially screened the library for inhibitors of a panel of glycosidases in vitro, the potent α‐glucosidase inhibitors identified in the screen were then tested in cell‐based assays against Japanese encephalitis virus (JEV), dengue virus serotype 2 (DEN‐2), as well as severe acute respiratory syndrome coronavirus (SARS‐CoV).
Scheme 2.
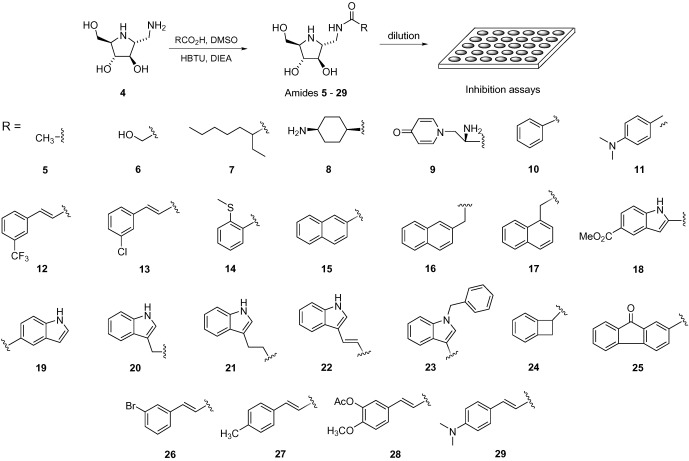
The reaction of iminocylitol 4 with a library of carboxylic acids for the subsequent high‐throughput in situ screening of glycosidase.
In addition, we discovered a new potent inhibitor of human N‐acetyl β‐hexosaminidase, which is the dominant glycosyaminoglycan‐degrading glycosidase released by the chrondrocytes into the extracellular compartment, and is the dominant glycosidase in synovial fluid from patients with osteoarthritis.29 Inhibitors of this enzyme could be used in the treatment of osteoarthritis.30
Results and Discussion
Rapid combinatorial synthesis and high‐throughput screening
The preliminary inhibitory assays of core 4 against a panel of glycosidases from different sources showed a broad spectrum of inhibitory activities, except for α‐, β‐galactosidase and β‐mannosidase (see Supporting Information). Because core 4 had only moderate inhibitory activities against α‐mannosidase and α‐fucosidase, the library generated from 4 was screened against α‐, β‐glucosidase and N‐acetyl‐β‐hexosaminidase in this study.
A mixture of iminocyclitol 4, a carboxylic acid (1 equiv, Scheme 2), diisopropyl ethylamine (DIEA, 2.2 equiv), and (1H‐benzotriazole‐1‐yl)‐1,1,3,3‐(tetramethyl)uronium hexafluorophosphate (HBTU, 1.1 equiv) in DMSO was shaken in 96‐well microtiter plates for 5 h. Crude reaction products were randomly selected (30 %) and analyzed by ESI‐MS to ensure the presence of the desired products. The reaction mixture was diluted with appropriate buffer solutions and transferred to another 96‐well microtiter plate for screening directly without any purification. Based on the IC50 values of 4 (see Supporting Information), the concentrations of reaction products were set at 20 μm for α‐, β‐glucosidase and 30 μm for β‐hexosaminidase. Assuming that amide formation is complete, the percentage of inhibition relative to the control was calculated on the basis of absorbance at 405 nm for p‐nitrophenol released from their corresponding substrates.
Of the 144 compounds generated from the amide‐forming reaction, several inhibitors were found (see Supporting Information). Good inhibitors for each enzyme were selected, resynthesized as pure compounds, and reevaluated for determination of IC50 or K i values (Table 1, Scheme 2). In the screening for α‐glucosidase (bakers’ yeast) inhibition, about two‐thirds of reaction products were found to be more potent than 4 (see Supporting Information for overall library inhibitory activities). Briefly, α‐glucosidase was inhibited to a greater extent when the compound contained a fused‐aromatic ring (e.g. 17 and 25) and a heteroaromatic component (e.g. 19–21). It is worth noting that the enzyme was more interactive with a bicyclic than a tri‐ or mono‐cyclic aglycon (e.g. 16 vs. 25 vs. 10). The α‐glucosidase from Bacillus stearothermophilus was also investigated with the same library. The results were similar to those with bakers’ yeast. The most potent one against yeast α‐glucosidase was compound 24, with a K i value of 53 nm and a 600‐fold selectivity for α‐ over β‐glucosidase. Interestingly, the IC50 values of compounds 15–17 for α‐glucosidase were about two‐to‐three orders of magnitude different. These results suggest that, while a lipophilic binding pocket exists that allows a bicyclic ring such as indole or naphthalene to fit, the specific orientation of these bicyclic rings is also very important.
Table 1.
The publisher did not receive permission from the copyright owner to include this object in this version of this product. Please refer either to the publisher's own online version of this product or the printed product where one exists.
In the screening of β‐glucosidase (almonds), about one‐sixth of the reaction products were found to be comparable to or more potent than 4. C‐1‐modified α‐iminocyclitols showed weaker effects for β‐glucosidase than for α‐glucosidase. A dimethylamino group attached to the aromatic ring could enhance the inhibitory activity (e.g. 11 and 29), as documented previously.26 In particular, structures with a trans‐cinnamic moiety were shown to significantly inhibit this enzyme, especially those with meta‐halogen substituents (e.g. 12, 13, and 26). Because the trans‐cinnamic moiety has a board range of biological properties, including hepatoprotective,31 antimalarial,32 and antioxidant,33 our finding suggests a new application of trans‐cinnamic acid and its derivatives. The most inhibitory was reaction product 28 (up to 67 % inhibition), with a K i value of 1.2 μm. In general, derivatives of 4 are more selective α‐ than β‐glucosidase inhibitors.
Inhibition of jack bean N‐acetyl‐β‐hexosaminidase seems to be greater when there are hydrophilic substituents in the C‐1 position (e.g. 6, 8, and 9). However, none of the substituents is more potent than the acetamido group; this indicates its crucial role in the enzyme active site. This study also showed that potent inhibitors of β‐hexosaminidase can be found from derivatives of 4, though relatively weaker inhibitors of β‐glucosidase were observed.
Overall, combinatorial synthesis followed by rapid screening in situ as described here provided a platform to identify selective glycosidase inhibitors through modification of a common transition‐state core at the aglycon side chain.
Antiviral activities of 5‐membered iminocyclitols
Endoplasmic reticulum (ER) α‐glucosidase inhibitors, which block the trimming step of N‐linked glycosylation, have been shown to eliminate the production of several ER‐budding viruses.34 Previously, we reported the antivirus effects of an N‐n‐nonyl DNJ (NN‐DNJ) on flavivirus infections.16 However, five‐membered iminocyclitols had not been tested for their antiviral activities. With the potent α‐glucosidase inhibitors in hand, we tested their potential antiviral effect based on our previous assay system for JEV and DEN‐2. Details of the antivirus assay are given in the Experimental Section.
Compounds 7, 17, 20, 22, and 24 exhibited no inhibition at 50 or 100 μm (data not shown). The peracetylated compound 30, which is believed to increase the cellular uptake and is then converted to compound 24 by cellular esterases,35 was subjected to the cell assay. However, it also showed no inhibition at 50 μm (data not shown). We then turned our attention to modifying the ring nitrogen on the most potent α‐glucosidase inhibitor 24. Side‐chain modification by alkylation of DNJ has previously been reported to enhance both the ability to inhibit glycan processing and virus production.13, 36–38
To study the influence of the alkyl‐chain length on the cell‐based assay, a series of compounds with alkyl chains ranging from C4 to C12 attached to compound 24 was synthesized by using reductive amination with appropriate aldehydes to give rise to compounds 31–38 (Scheme 3). The inhibition activities of N‐alkylated derivatives against α‐ and β‐glucosidase were also investigated (see Supporting Information). The results showed that these molecules still have α‐glucosidase selectivity, albeit with low activity compared to the parent compound 24. This result was also observed in the in vitro anti‐α‐glucosidase activity of DNJ and NN‐DNJ. The cell‐based sodium 3,3′‐{1,[(phennylamino)carbonyl]‐3,4‐tetrazolium}‐bis(4‐methoxy‐6‐nitro)benzene sulfonic acid hydrate (XTT) assay, which measured cell proliferation, indicated that most of the compounds were not toxic at 10 μm (Figure 2 A). The exception was compound 38, which showed a low level of cytotoxicity. In general, the alkylated iminocylitols were more active against DEN‐2 than JEV infection (Figure 2 B). Relative to the virus titer derived from cells treated with NN‐DNJ, these newly synthesized molecules were less potent against DEN‐2 infection (Figure 2 C). However, in anti‐JEV infection, compounds 36–38 were more potent than NN‐DNJ. Compound 37 (Figure 3) appeared to be less cytotoxic and more potent with IC50=4.7 μm and IC90=9.2 μm for DEN‐2 (Table 2). In the present studies, compounds 36–38 with chain lengths of nine, ten, and twelve carbons, respectively, were found to be optimal for the antiviral activity. A similar effect was also noticed against varicella zoster virus.39 Longer alkyl chains, such as, the decyl chain in compound 38, provided a modest increase in potency in cell‐based assays, but also resulted in an increase in cytotoxicity, presumably due to the disruption of the lipid bilayer.36
Scheme 3.
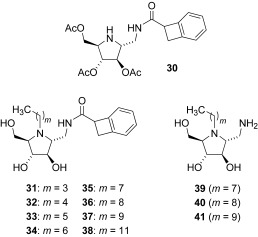
Compounds 30–41 for antivirus assays.
Figure 2.
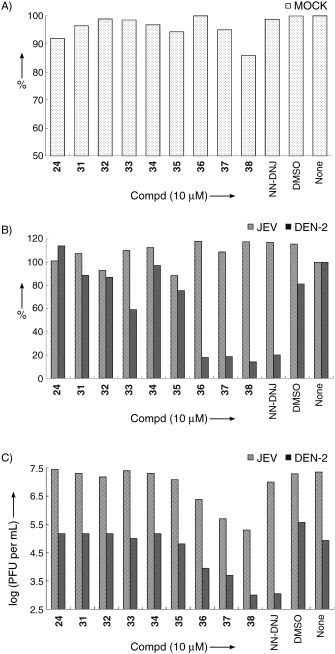
Cell‐based assays of the anti‐JEV and anti‐DEN effects of compounds 24, 31–38 and NN‐DNJ (10 μm). A) Cell survival was measured by XTT assays. Data are shown as the percentage versus the mock‐infected BHK‐21 cells without inhibitor treatment (none, 100 %). B) The viral protein expressions of IFA were read by fluorescence microplate reader (Molecular Device) with an excitation wavelength of 355 nm and an emission wavelength of 488 nm. Data are shown as the percentage versus untreated inhibitor 1. C) The culture supernatants were collected for viral titration by plaque‐forming assay. The virus titers are shown as PFU (plaque forming unit) per milliliter. Representative results from two independent experiments are shown here.
Figure 3.
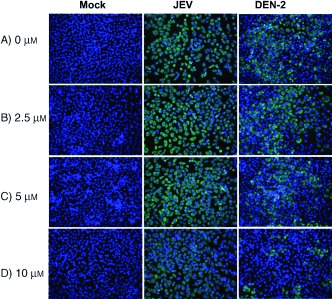
Morphology of favivirus‐infected BHK‐21 cells. Cells infected with mock, JEV (M.O.I. of 0.1) and DEN‐2 (MOI of 0.1) were treated with various dose of inhibitor 37 as indicated. Two days postinfection, cells were fixed and stained with anti‐JEV and anti‐DEN NS3 MAb and a FITC‐conjugated secondary antibody (green). Cell nuclei were stained by DAPI (blue). Pictures were taken by using an inverted fluorescent microscope (Leica) with filters for FITC and DAPI, and the pictures were superimposed in the same fields.
Table 2.
The publisher did not receive permission from the copyright owner to include this object in this version of this product. Please refer either to the publisher's own online version of this product or the printed product where one exists.
From a structural perspective, the alkylated iminocyclitol can be viewed as consisting of two distinct molecular elements: 1) an imino sugar head group and 2) an N‐alkyl side chain. The head group is recognized by the ER‐α‐glucosidase. The role of the tail, as shown for NN‐DNJ, is unclear but it might be able to insert into the membrane to increase its local concentration near the membrane‐associated ER glucosidase.40 In recent studies, N‐nonyldeoxygalactonojirimycin (NN‐DGJ), a galactose‐type iminocyclitol, was found to still have anti‐HBV12 and anti‐BVDV13 activities, although it lacks the ability to inhibit α‐glucosidase. These observations suggest that NN‐DNJ might possess a different antiviral mechanism. Nevertheless, we found that NN‐DGJ did not inhibit either JEV or DEN‐2 in our cell‐based assay system.16 The weaker α‐glucosidase inhibitors 39–41, derived from core 4 with eight to ten carbons (Scheme 3), were then evaluated, and we found that 39–41 at 10 or 50 μm did not inhibit either JEV or DEN‐2 infection (data not shown) in the cell‐based assay.
Inhibitors 36–38 were also screened against the infection of SARS‐CoV by following our previously established procedure.41 The IC50 for compound 37 was around 3.3–10 μm. The antiviral effects of compounds 36–38 on JEV, DEN‐2, and SARS‐CoV shown in the present study are likely mediated by inhibition of the ER α‐glucosidase; however the possibility of other mechanisms besides α‐glucosidase inhibition cannot be rigorously excluded.16
Discovery of potent human β‐hexosaminidase inhibitors
In our previous study, compound 5 and its N‐methyl derivative 42 ((2R,3R,4R,5R)‐N‐methyl‐2‐(acetamidomethyl)‐3,4‐dihydroxy‐5‐(hydroxymethyl)pyrrolidine) were found to be potent inhibitors of human N‐acetyl‐β‐hexosaminidases, with K i≈24 nm.30 In particular, incubation of human chondrosarcoma cells with iminocyclitol 42 resulted in an accumulation of glycosaminoglycans (GAGs) in the cell‐associated fraction and a decrease in the release of GAGs into the culture supernatant. The discovery of iminocyclitols as potential chondroprotective agents suggests a new avenue for the development of drugs to treat osteoarthritis.
In order to further improve the potency of 42, considerable effort has been directed toward modification of the ring nitrogen and the C1 nitrogen of iminocyclitol 4.42 However, none of the synthesized inhibitors was more potent than compound 42. The structure–activity relationship of iminocyclitols revealed that the acetamido group at the C1 position is crucial, and that the active site pocket of β‐hexosaminidase does not tolerate larger substituents. The methyl group at the ring nitrogen enhances the inhibition activity, whereas aromatic ring substituents cause a decrease in inhibition activity. Our alternative approach to increasing potency is to probe a distant aglycon‐binding site of β‐hexosaminidase. We therefore decided to attach a long‐chain alkyl group with a terminal amine to the ring nitrogen through reductive animation. The resulting primary amine is easy to diversify by amide‐bond formation to generate libraries as mentioned above.
Our strategy began with reductive amination of compound 5 with aldehydes of different lengths to give compounds 43–49 (Scheme 4). These intermediates were either deprotected under acidic conditions or hydrogenolyzed to give primary amines 50–56. In our first attempt to generate a library from compound 50 by amide‐bond formation as mentioned above, the high‐throughput screening showed no compounds with significantly enhanced inhibitory activities (data not shown). Thus, inhibition studies of compounds 51–56 against human placenta N‐acetyl‐β‐hexosaminidase were carried out. 4‐Methylumbelliferyl N‐acetyl‐β‐d‐glucosaminide (4‐MU‐GNAc) was used as the substrate. The apparent K m and V max values for each substrate were calculated from the Lineweaver–Burke double reciprocal plot of [1/v]/[1/S]. The K i values were determined from a replot of the K m app versus the inhibitor concentration. Compound 54 is the most potent competitive inhibitor with a K i value 2.6 nm (Figure 4). Interestingly, in varying the linkage from N‐propyl to N‐octyl of compounds 50–55, we observed a trend in human β‐hexosaminidase inhibition. Compound 54, with a heptamine moiety, was the strongest inhibitor. In an attempt to further optimize the inhibitor, the linker was replaced by an ethylene glycol chain (hydrophilic linker) to give compound 56 (K i=60 nm) and by moderate N‐alkyl chains to give compounds such as 57, 58, and 59 (K i=180, 250, and 160 nm, respectively). However, these modifications had a negative effect; this indicated the important role of the amino group and the lipophilic chain in compound 54. As the crystal structure of human β‐hexosaminidase B is now available,43 its complex with 54 was modeled to reveal a narrow hydrophobic cleft in the active site, which is mainly enclosed by the side‐chain groups of residues W424, Y450, A447, and L453 and the backbone of residues K425 and D426 (Figure 5). The amino group at the terminal site is expected to be largely protonated when binding to the enzyme. It presumably forms a salt bridge with the secondary aglycon binding site of the enzyme.44 It appears that the alkyl linkage is long enough to bring the amine end near enough to have hydrogen‐bond interactions with the backbone carbonyl of K425 and A447 and a possible ionic interaction with the carboxyl group of D426.
Scheme 4.
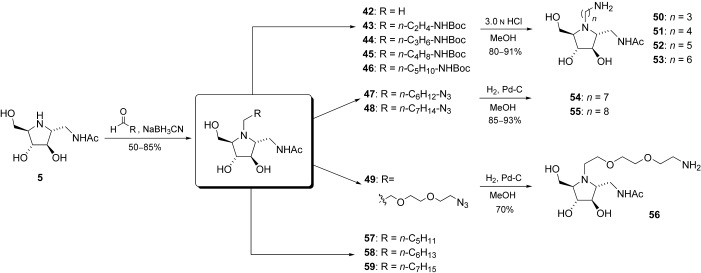
Synthesis of hexosaminidase inhibitors 50–58.
Figure 4.
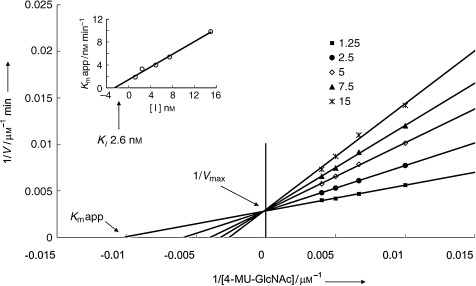
Lineweaver–Burk double reciprocal plots of compound 54 were carried out to obtained K m app and V max values. Moreover, K i was obtained from a replot of the K m app and inhibitor concentrations. The K i value of each molecule is shown on the y axis.
Figure 5.
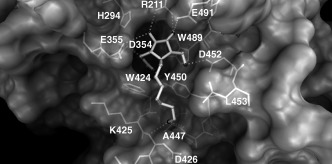
A modeling complex of human β‐hexosaminidase with 54 (light gray at central). The hydroxyl groups of the inhibitor could form hydrogen‐bond interactions with residues R211, D355, D452, and E491. The C1 N‐acetyl group of 54 hydrogen bonds with D354, and its carbonyl group hydrogen bonds with the side‐chain hydroxyl group of Y450. The computed molecular surface exhibits a narrow hydrophobic cleft near the binding site of the iminocyclitol ring. Additional binding affinity from the long alkyl chain could result from hydrogen‐bond interactions of the end amino group with the backbone carbonyl of K425 and A447 or possible induced ionic interactions with the carboxyl group of D426. Figure produced with MGLTOOLS.
Conclusion
Based on the structure of 1‐amino‐1,2,5‐dideoxy‐2,5‐imnio‐d‐mannitol (4), we applied a combinatorial synthesis in microtiter plates for in situ screening. This method is a powerful procedure to rapidly identify significant binding‐site differences among similar enzymes and develop potent and selective inhibitors. For the inhibitors of α‐glucosidase, structures with bicyclic rings such as indole and naphthalene gave the best inhibitory potency, as illustrated by 24 with a K i value of 53 nm and 100‐fold increase in activity compared to parent core 4. The N‐alkylated derivatives of compound 24 were also tested for antiviral activity, and 36–38, which contain lipophilic alkyl groups, were the most active, with an IC50 of about 5–10 μm against JEV, DEN‐2, and SARS‐CoV infection.
Given its important role in osteoarthritis, the inhibition of N‐acetyl‐β‐hexosaminidase was investigated with regard to the substituent effects on the C1 nitrogen and ring nitrogen of core 4. The results showed that the acetamido group at the C1 position was crucial, with modification at the ring nitrogen with aromatic groups causing loss of inhibition. However, extending the alkyl chain at the ring nitrogen gave the most potent human β‐hexosaminidase inhibitor known to date, that is, compound 54, with a K i value of 2.6 nm. Modeling indicated strong binding of compound 54 with β‐hexosaminidase in its lipophilic cleft and an ionic interaction with the secondary binding site. Altogether, this work clearly demonstrates the effectiveness of our simple combinatorial approach strategy for the rapid discovery of potent inhibitors as potential candidates for medical applications.
Experimental Section
Materials: The sources of enzymes were as follows: α‐glucosidase (EC 3.2.1.20) from bakers’ yeast and Bacillus stearothermophilus; β‐glucosidase (EC 3.2.1.21) from almonds; α‐galactosidase (EC 3.2.1.22) from Aspergillus niger; α‐mannosidase (EC 3.2.1.24) from jack bean; β‐mannosidase (EC 3.2.1.25) from snail acetone powder; N‐acetyl‐β‐hexosaminidase (EC 3.2.1.52) from jack beans and human placenta. All of the above enzymes were purchased from Sigma (St. Louis, MO). β‐Glucosidase from sweet almonds and β‐galactosidase (EC 3.2.1.23) from Escherichia coli were purchased from Toyobo Co., Ltd. (Osaka, Japan). Human α‐fucosidase (EC 3.2.1.51), a recombinant protein, was a gift from Professor Chun‐Hung Lin at Academia Sinica (Taipei, Taiwan).
General method for chemical synthesis: All nonaqueous reactions were run in oven‐dried and vacuum‐cooled glassware under nitrogen atmosphere. Reactions were monitored by thin‐layer chromatography (Merck, silica gel 60F‐254) by utilizing ninhydrin, p‐anisaldehyde, or cerium molybdate as the stain reagent. Silica gel used for flash column chromatography was Mallinckrodt type 60 (230–400 mesh). Unless otherwise noted, reagents and materials were obtained from commercial sources and used as provided without further purification. 1H and 13C NMR spectra were recorded on a Bruker AV‐400 or AV‐500 spectrometer and referenced to residual solvent peaks (CDCl3: 1H δ=7.24, 13C δ=77.0; [D4]MeOH: 1H δ=3.30, 13C δ=49).
General procedure for coupling reactions and in situ screening: Carboxylic acid (10 μL from a 10 mm stock solution in DMSO, 0.1 μmol), HBTU (10 μL from a 11 mm stock solution in DMSO, 1.1 equiv, 0.11 μmol), and DIEA (10 μL from a 22 mm stock solution in DMSO, 2.2 equiv, 0.22 μmol) were added to DMSO (10 μL) in each well of a 96‐well microtiter plate. The reaction was initiated by adding compound 4 (10 μL from a 10 mm stock solution, 0.1 μmol) to each well. The reaction mixtures were shaken at room temperature for 5 h and were monitored for completion based on the disappearance of 4 by TLC with a mobile phase of CHCl3/MeOH/NH4OH (1:3:1, R f=0.33). Then an aliquot (5 μL) was withdrawn from the previous reaction mixture and mixed with phosphate buffer (95 μL, pH 7.0) to reach a 20‐fold dilution. The same procedure was repeated to give the desired dilution (i.e. a final concentration of 20 μm product in each well of a microtiter plate). In each well of another plate, the α‐glucosidase from baker’s yeast (10 μL, 0.1 U mL−1) and p‐nitrophenyl‐α‐glucopyranoside (50 μL, 2 mm) were mixed with an aliquot (50 μL) of the aforementioned mixture and 90 μL buffer (to give ∼20 μm inhibitor) for the enzyme inhibition assay.
Enzyme assay. General procedure for the assay with various glycosidases: The initial velocities of hydrolysis at 30 °C were measured spectrophotometrically at various concentrations of p‐nitrophenylglycopyranoside (4 mm, 2 mm, 1 mm, 0.5 mm, 0.25 mm, 0.125 mm, 0.0625 mm) at 405 nm by using Bio Assay Reader (Perkin–Elmer HTS 7000 Plus). The obtained data were fitted into the Michaelis–Menten equation by using the Kaleida Graph program to determine the apparent K m values. The substrate concentrations were used at three‐ to fivefold K m values for evaluation of the inhibitory effect against various glycosidases. To give an ideal progress curve, an appropriate enzyme concentration (0.01–0.2 units per mL) and inhibitor concentration (from 1 nm to 500 μm) were used. The 50 % inhibitory concentration (IC50) was determined as the concentration at which the velocity of the hydrolysis was reduced to 50 % as compared to the untreated control. The assays performed in wells of the microtiter plate contained either sodium phosphate buffer (50 mm, pH 7, for α‐ and β‐glucosidase, α‐ and β‐galactosidase, α‐ and β‐mannosidase, α‐fucosidase), or McIlvaine’s buffer (25 mm, pH 6, N‐acetyl‐β‐hexosaminidase, jack bean).
The K i values of the inhibitors were determined by the double reciprocal plot (1/V vs. 1/[S]) to give apparent K m (the K m in the presence of inhibitors). The secondary plot was generated by plotting the apparent K m values as a function of inhibitor concentrations. K i was calculated from the negative value of the x‐intercept of this plot.
Kinetic analysis of human placenta β‐hexosaminidase. Incubations were performed in a total volume of 200 μL. Unless otherwise stated, reaction mixtures contained citrate buffer (100 mm, pH 4.5), various amount of 4‐methylumbelliferyl N‐acetyl‐β‐d‐glucosaminide, and various amounts of inhibitors with 0.02 mU per well of β‐hexosaminidase. After incubation for 15 min at 30 °C, the reaction was terminated by the addition of sodium glycine buffer (0.5 m), pH 10.5. Enzyme activity was measured by the release of 4‐methylumbelliferone with an excitation wavelength of 360 nm and an emission wavelength of 460 nm.
Cell lines, viruses, and virus infection: BHK‐21 cells were cultured in RPMI 1640 medium containing 5 % fetal bovine serum (FBS) and l‐glutamine (2 mm). JEV strain RP‐9 45 and the Taiwanese DEN‐2 strain PL046 46 were used in this study. Virus propagation was carried out in C6/36 cells by using RPMI 1640 medium containing 5 % FBS. Inhibitors were dissolved in DMSO. For infection with JEV or DEN‐2, monolayers of BHK‐21 cells in six‐ or 12‐well plates were adsorbed with virus for 1 h at 37 °C. After adsorption, unbound viruses were removed by gentle washing with serum‐free medium, followed by addition of fresh medium containing various amounts of inhibitors for further incubation at 37 °C. To determine virus titers, culture media were harvested for plaque‐forming assays. Various virus dilutions were added to 80 % confluent BHK‐21 cells and incubated at 37 °C for 1 h. After adsorption, cells were washed and overlaid with 1 % agarose (SeaPlaque; FMC BioProducts, Cambrix Bioscience, Rockland, ME, USA) containing RPMI 1640 with 1 % FBS. After incubation for 4 days for JEV and 7 days for DEN‐2, cells were fixed with 10 % formaldehyde and stained with 0.5 % crystal violet. The inhibitors concentrations required to inhibit virus production by 50 % (IC50) and 90 % (IC90) were determined.
Indirect immunofluorescence assay (IFA): Cells were fixed in acetone/methanol (1:1) for 3 min and then treated with a monoclonal antibody (MAb) against JEV NS3 47 or DEN‐2 NS3 .46 After being washed with phosphate‐buffered saline (PBS), cells were further stained with a goat anti‐mouse fluorescein isothiocyanate (FITC)‐conjugated secondary antibody (Jackson ImmunoResearch, West Grove, PA, USA), and the resulting cells were examined under a Leica fluorescent microscope. The viral protein expressions by IFA were read by fluorescence microplate reader (Molecular Devices) with an excitation wavelength of 355 nm and an emission wavelength of 488 nm. Data are shown as percentage infected BHK‐21 cells versus those without inhibitor treatment (none, 100 %). Cell nuclei were visualized by 4′,6′‐diamidino‐2‐phenylindole (DAPI) staining in 0.9 % sodium chloride at room temperature for 5 min.
XTT assay: To determine cell viability, a colorimetric XTT‐based assay was performed (Cell Proliferation Kit II; Roche). BHK‐21 cells in a 96‐well plate were incubated with various concentrations of inhibitors for 2 days before the XTT labeling reagent was added to the culture medium. Cells were incubated at 37 °C for about 30 min and then read by an ELISA reader at 450 nm (Molecular Devices).
Primary screening for anti‐SARS‐CoV activity: Vero E6 cells (2×104 per well) were cultured in a 96‐well plate in Dulbecco's Modified Eagle Medium (DMEM) supplemented with 10 % FBS. The culture medium was removed after 1 day's incubation, when the cells reached 80–90 % confluence. A solution of DMEM (100 μL), with 2 % FBS containing the compound to be tested was placed in three wells. Cells were incubated in a CO2 incubator at 37 °C for 2 h and inoculated with SARS‐CoV (H.K. strain) at a dose of 100 TCID50 per well; the cytopathic morphology of the cells was examined by using an inverted microscope 72 h after infection.
Computer modeling. Docking experiments were conducted by using Autodock 3.0.5 with a Lamarckian genetic algorithm (LGA).48 The crystal structure of a human β‐hexosaminidase B complex with a transition‐state‐analogue inhibitor, 2‐acetamido‐2‐deoxy‐d‐glucono‐1,5‐lactone (δ‐lactone),44 was downloaded from the RCSB Protein Data Bank (PDB code 1o7a). Chain A of the structure was extracted and utilized in docking simulation. The structure models of inhibitors were built in CAChe (Fujitsu, Japan) and refined by performing an optimized geometry calculation in Mechanics by using augmented MM3 parameters and stored in PDB format. MGLTOOLS (Molecular Graphics Lab, Scripps Research Institute)49 was used for protein‐structure preparation and parameter creation to meet the input requirements of Autodock. Briefly, essential hydrogen atoms were added to the structure model of hexosaminidase followed by assigning Kollman united atom charges and solvation parameters. Compound molecules were assigned Gasteiger–Marsili charges, nonpolar H atoms were merge, and torsions were defined. Autogrid tool in Autodock 3.0.5 was applied to produce energy grids (50×50×50 in xyz directions with 0.375 Å spacing) of various types of compound atoms. These grip maps were centered at the active site where the δ‐lactone bound. During docking experiments, each compound was kept flexible and the protein was kept rigid. Solis & Wets' local search method with LGA was applied to generate available conformations of compound structures within the active site. The conformational search was conducted by utilizing 0.2 Å quaternion and 2° torsion steps. For each compound structure, a maximum of 5×106 energy units were evaluated, and 50 poses were selected from 2.7×105 generations per run. Plausible docking modes were selected from the most abundant cluster (RMSD=2.0 Å), which had the strongest affinity energy. Pictures of the final simulated complex were generated in MGLTOOLS.
Supporting information
Supporting information for this article is available on the WWW under http://www.wiley-vch.de/contents/jc_2268/2006/f500321_s.pdf or from the author.
Acknowledgements
We thank Dr. Jia‐Tsrong Jan at the National Defense University (Taipei, Taiwan) for the anti‐SARS assay and the Academia Sinica for financial support.
Contributor Information
Yi‐Ling Lin, Email: yll@ibms.sin
Chi‐Huey Wong, Email: wong@scripps.edu
References
- 1. Ellgaard L., Helenius A., Nat. Rev. Mol. Cell. Biol. 2003, 4, 181–191. [DOI] [PubMed] [Google Scholar]
- 2. Asano N., Glycobiology 2003, 13, 93R–140R. [DOI] [PubMed] [Google Scholar]
- 3. Dwek R. A., Butters T. D., Platt F. M., Zitzmann N., Nat. Rev. Drug Discovery 2002, 1, 65–75. [DOI] [PubMed] [Google Scholar]
- 4. Jacob G. S., Curr. Opin. Struct. Biol. 1995, 5, 605–611. [DOI] [PubMed] [Google Scholar]
- 5. Lew W., Chen X., Kim C. U., Curr. Med. Chem. 2000, 7, 663–672. [DOI] [PubMed] [Google Scholar]
- 6. Mitrakou A., Tountas A. E., Raptis R. J., Bauer H., Schulz S. A., Raptis. Diabet. Med. 1998, 15, 657–660. [DOI] [PubMed] [Google Scholar]
- 7. Scott L. J., Spencer C. M., Drugs 2000, 59, 521–549. [DOI] [PubMed] [Google Scholar]
- 8. Goss P. E., Baptiste J., Fernandes B., Baker M., Dennis J. W., Cancer Res. 1994, 54, 1450–1457. [PubMed] [Google Scholar]
- 9. Gruters R. A., Neefjes J. J., Tersmette M., Goede R. E. de, Tulp A., Huisman H. G., Miedema F., Ploegh H. L., Nature 1987, 330, 74–77. [DOI] [PubMed] [Google Scholar]
- 10. Walker B. D., Kowalski M., Goh W. C., Kozarsky K., Krieger M., Rosen C., Rohrschneider L., Haseltine W. A., Sodroski J., Proc. Natl. Acad. Sci. USA 1987, 84, 8120–8124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Block T. M., Lu X., Platt F. M., Foster G. R., Gerlich W. H., Blumberg B. S., Dwek R. A., Proc. Natl. Acad. Sci. USA 1994, 91, 2235–2239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Mehta A., Carrouee S., Conyers B., Jordan R., Butters T., Dwek R. A., Block T. M., Hepatology 2001, 33, 1488–1495. [DOI] [PubMed] [Google Scholar]
- 13. Durantel D., Branza‐Nichita N., Carrouee‐Durantel S., Butters T. D., Dwek R. A., Zitzmann N., J. Virol. 2001, 75, 8987–8998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Durantel D., Carrouee‐Durantel S., Branza‐Nichita N., Dwek R. A., Zitzmann N., Antimicrob. Agents Chemother. 2004, 48, 497–504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Courageot M. P., Frenkiel M. P., Santos C. D. Dos, Deubel V., Despres P., J. Virol. 2000, 74, 564–572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Wu S.‐F., Lee C.‐J., Liao C.‐L., Dwek R. A., Zitzmann N., Lin Y.‐L., J. Virol. 2002, 76, 3596–3604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Butters T. D., Dwek R. A., Platt F. M., Chem. Rev. 2000. 100, 4683–4696. [DOI] [PubMed] [Google Scholar]
- 18. Fan J. Q., Trends Pharmacol. Sci. 2003. 24, 355–360. [DOI] [PubMed] [Google Scholar]
- 19. Stütz A. E., Iminosugars as Glycosidase Inhibitors—Norjirimycin and Beyond, Wiley‐VCH, Weinheim, 1999. [Google Scholar]
- 20. Welter A., Dardene G., Marlier M., Casimir J., Phytochemistry 1976, 15, 747–749. [Google Scholar]
- 21. Card P. J., Hitz W. D., J. Org. Chem. 1985, 50, 891–893. [Google Scholar]
- 22. Andersen S. M., Ebner M., Ekhart C. W., Gradnig G., Legler G., Lundt I., Stütz A. E., Withers S. G., Wrodnigg T., Carbohydr. Res. 1997, 301, 155–166. [Google Scholar]
- 23. Look G. C., Fotsch C. H., Wong C.‐H., Acc. Chem. Res. 1993, 26, 182–190. [Google Scholar]
- 24. Saotome C., Wong C.‐H., Kanie O., Chem. Biol. 2001, 8, 1061–1070. [DOI] [PubMed] [Google Scholar]
- 25. Takebayashi M., Iranuma S., Kanie Y., Kajimoto T., Kanie O., Wong C.‐H., J. Org. Chem. 1999, 64, 5280–5291. [DOI] [PubMed] [Google Scholar]
- 26. Wrodigg T. M., Diness F., Gruber C., Häusler H., Lundt I., Rupitz K., Steiner A. J., Stütz A. E., Tarling C. A., Withers S. G., Wölfler H., Bioorg. Med. Chem. 2004, 12, 3458–3495. [DOI] [PubMed] [Google Scholar]
- 27. Chang C.‐F., Ho C.‐W., Wu C.‐Y., Chao T.‐A., Wong C.‐H., Lin C.‐H., Chem. Biol. 2004, 11, 1301–1306. [DOI] [PubMed] [Google Scholar]
- 28. Brik A., Lin Y. C., Elder J., Wong C.‐H., Chem. Biol. 2002, 9, 891–896. [DOI] [PubMed] [Google Scholar]
- 29. Shikhman A. R., Brinson D. C., Lotz M., Arthritis Rheum. 2000, 43, 1307–1314. [DOI] [PubMed] [Google Scholar]
- 30. Liu J., Shikhman A. R., Lotz M. K., Wong C.‐H., Chem. Biol. 2001, 8, 701–711. [DOI] [PubMed] [Google Scholar]
- 31. Perez‐Alvarez V., Bobadilla R. A., Muriel P., J. Appl. Toxicol. 2001, 21, 527–531. [DOI] [PubMed] [Google Scholar]
- 32. Wiesner J., Mitsch A., Wißner P., Jomaa H., Schlitzer M., Bioorg. Med. Chem. Lett. 2001, 11, 423–424. [DOI] [PubMed] [Google Scholar]
- 33. Natella F., Nardini M., Felice M. Di, Scaccini C., J. Agric. Food Chem. 1999, 47, 1453–1459. [DOI] [PubMed] [Google Scholar]
- 34. Tan A., Broek L. van den, Boeckel S. van, Ploegh H., Bolscher J., J. Biol. Chem. 1991, 266, 14504–15410. [PubMed] [Google Scholar]
- 35. Jacobs C. L., Yarema K. Y., Mahal L. K., Nauman D. A., Charters N. W., Bertozzi C. R., Methods. Enzymol. 2000, 327, 260–275. [DOI] [PubMed] [Google Scholar]
- 36. Mellor H. R., Platt F. M., Dwek R. A., Butters T. D., Biochem. J. 2003, 374, 307–314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Mehta A., Ouzounov S., Jordan R., Simsek E., Lu X., Moriarty R. M., Jacob G., Dwek R. A., Block T. M., Antiviral Chem. Chemother. 2002, 13, 299–304. [DOI] [PubMed] [Google Scholar]
- 38. Block T. M., Lu X., Mehta A., Blumberg B., Tennant B., Ebling M., Korba B., Lansky D. M., Jacob G. S., Dwek R. A., Nat. Med. 1998, 4, 610–614. [DOI] [PubMed] [Google Scholar]
- 39. McGuigan C., Yarnold C. J., Jones G., Velazquez S., Barucki H., Brancale A., Andrei G., Snoeck R., Clercq E. De, Balzarini J., J. Med. Chem. 1999, 42, 4479–4484. [DOI] [PubMed] [Google Scholar]
- 40. Zitzmann N., Mehta A. S., Carrouee S., Butters T. D., Platt F. M., McCauley J., Blumberg B. S., Dwek R. A., Block T. M., Proc. Natl. Acad. Sci. USA 1999, 96, 11878–11882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Wu C.‐Y., Jan J.‐T., Ma S.‐H., Kuo C.‐J., Juan H.‐F., Cheng Y.‐S. E., Hsu H.‐H., Huang H.‐C., Wu D., Brik A., Liang F.‐S., Liu R.‐S., Fang J.‐M., Chen S.‐T., Liang P.‐H., Wong C.‐H., Proc. Natl. Acad. Sci. USA 2004, 101, 10012–10017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Liu J., Numa M. M. D., Liu H., Huang S.‐J., Sears P., Shikhman A. R., Wong C.‐H., J. Org. Chem. 2004, 69, 6273–6283. [DOI] [PubMed] [Google Scholar]
- 43. Maier T., Strater N., Schuette C. G., Klingenstein R., Sandhoff K., Saenger W., J. Mol. Biol. 2003, 328, 669–681. [DOI] [PubMed] [Google Scholar]
- 44. Bulow A., Plesner I. W., Bols M., J. Am. Chem. Soc. 2000, 122, 8567–8568. [Google Scholar]
- 45. Chen L. K., Lin Y. L., Liao C. L., Lin C. G., Huang Y. L., Yeh C. T., Lai S. C., Jan J. T., Chin C., Virology 1996, 223, 79–88. [DOI] [PubMed] [Google Scholar]
- 46. Lin Y. L., Liao C. L., Chen L. K., Yeh C. T., Liu C. I., Ma S. H., Huang Y. Y., Huang Y. L., Kao C. L., King C. C., J. Virol. 1998, 72, 9729–9737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Chen L. K., Liao C. L., Lin C. G., Lai S. C., Liu C. I., Ma S. H., Huang Y. Y., Lin Y. L., Virology 1996, 217, 220–229. [DOI] [PubMed] [Google Scholar]
- 48. Morris G. M., Goodsell D. S., Halliday R. S., Huey R., Hart W. E., Belew R. K., Olson A. J., J. Comput. Chem. 1998, 19, 1639–1662. [Google Scholar]
- 49.MGLTOOLS: http://www.scripps.edu/∼sanner/python Molecular Graphics Lab (MGL) of the Scripps Research Institute.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting information for this article is available on the WWW under http://www.wiley-vch.de/contents/jc_2268/2006/f500321_s.pdf or from the author.