

Summary
Many viruses, including coronaviruses, induce host translational shutoff, while maintaining synthesis of their own gene products. In this study we performed genome‐wide microarray analyses of the expression patterns of mouse hepatitis coronavirus (MHV)‐infected cells. At the time of MHV‐induced host translational shutoff, downregulation of numerous mRNAs, many of which encode protein translation‐related factors, was observed. This downregulation, which is reminiscent of a cellular stress response, was dependent on viral replication and caused by mRNA decay. Concomitantly, phosphorylation of the eukaryotic translation initiation factor 2α was increased in MHV‐infected cells. In addition, stress granules and processing bodies appeared, which are sites for mRNA stalling and degradation respectively. We propose that MHV replication induces host translational shutoff by triggering an integrated stress response. However, MHV replication per se does not appear to benefit from the inhibition of host protein synthesis, at least in vitro, since viral replication was not negatively affected but rather enhanced in cells with impaired translational shutoff.
Introduction
Virus infections often give rise to a shutoff of host cell translation, while synthesis of viral proteins is maintained. This cellular response provides two advantages for viral replication; it facilitates rapid viral protein synthesis and inhibits the production of antiviral proteins (Lyles, 2000). On the other hand, viruses and their hosts share the same translational machinery. Therefore, global inhibition of protein synthesis can also be regarded as part of an antiviral response of the host aimed to minimize viral replication.
Coronaviruses (CoVs) are enveloped, positive‐stranded RNA viruses and are common pathogens in man and animals. Their relevance has increased considerably with the recent emergence of new human CoVs (HCoVs), such as the severe acute respiratory syndrome (SARS)‐CoV (Drosten et al., 2003), HCoV‐NL63 (van der Hoek et al., 2004) and HCoV‐HKU1 (Woo et al., 2005). CoVs replicate entirely within the cytoplasm of their host cells, where they produce a nested set of (sub)genomic mRNAs, containing identical 5′‐leader sequences and coterminal 3′‐ends (Pasternak et al., 2006). These mRNAs, which are transcribed via a discontinuous transcription mechanism, are 5′‐capped and 3′‐polyadenylated, making them structurally equivalent to host cellular mRNAs (Sawicki and Sawicki, 2005). Therefore, CoVs must hijack the host translational machinery to produce their own proteins.
The mouse hepatitis CoV (MHV), a close relative of the SARS‐CoV (Snijder et al., 2003), and the feline infectious peritonitis virus have been shown to induce host translational shutoff in susceptible cells (Rottier et al., 1981; Siddell et al., 1981; Hilton et al., 1986; M. Raaben and C. A. M. de Haan, unpubl. results). The mechanism by which this occurs and how CoVs themselves escape from it is not known. As the leader sequence, present on all viral mRNAs, enhances translation in MHV‐infected cells, it has been suggested that the apparent downregulation of host protein synthesis is not primarily due to inhibition of translation (Tahara et al., 1994). On the other hand, the reduced host mRNA translation in MHV‐infected cells has also been associated with a decrease in the number and size of polysomes and an increase of inactive single 80S ribosomes (Hilton et al., 1986), and with cleavage of 28S ribosomal RNA (Banerjee et al., 2000). Furthermore, it has been suggested that a decline in steady‐state levels, found for some host cell mRNAs, contributes to the downregulation of host protein translation (Hilton et al., 1986; Kyuwa et al., 1994; Tahara et al., 1994). More recently, the expression of the SARS‐CoV non‐structural protein 1 (nsp1) was reported to induce mRNA degradation and inhibition of host protein synthesis (Kamitani et al., 2006).
Host shutoff is not restricted to virus infected cells, but is induced by various stress stimuli (Brostrom and Brostrom, 1998). For example, cells deprived of nutrients inhibit protein synthesis to conserve amino acids for essential metabolic processes. In addition, cells also shut down protein synthesis in response to endoplasmic reticulum (ER) stress, heat and oxidative stress. These different stress stimuli induce translational shutoff via the increased phosphorylation of eukaryotic initiation factor (eIF)2α (de Haro et al., 1996). In mammalian cells there are at least four eIF2α kinases, including GCN2, PKR, PERK and HRI, which are activated by amino acid starvation, double stranded RNA, ER stress and haem depletion, respectively (Berlanga et al., 1999; Harding et al., 1999; Kaufman, 2000; Lu et al., 2001). Upon phosphorylation of eIF2α, cytoplasmic structures containing stalled translational preinitiation complexes, frequently referred to as stress granules (SGs), are formed. The actual formation of SGs occurs upon aggregation of the RNA binding proteins TIA‐1 (T‐cell internal antigen‐1) and TIAR (TIA‐1‐related protein) (Kedersha et al., 1999). SGs are thought to recruit specific mRNA transcripts, such as ‘housekeeping’ protein encoding mRNAs, to regulate their translation in adaptation to altered conditions (Anderson and Kedersha, 2002). SGs are also referred to as triage centres because they sort mRNAs for either storage, reinitiation of translation, or degradation (Anderson and Kedersha, 2006).
Degradation of mRNA is another way to control gene expression at a post‐transcriptional level. While 3′−5′ mRNA decay occurs by a complex of exonucleases termed the exosome, 5′−3′ mRNA decay takes place in cytoplasmic foci related to SGs, called processing bodies (P bodies) (Anderson and Kedersha, 2006). P bodies contain decapping enzymes, the exonuclease Xrn1 and components of the RNA‐induced silencing complex, including GW182, which is an RNA binding protein required for microRNA‐dependent silencing (Ingelfinger et al., 2002; Eystathioy et al., 2003; Cougot et al., 2004; Liu et al., 2005). The formation of P bodies is also induced in response to different stress stimuli including glucose deprivation and osmotic stress. However, unlike SGs their assembly does not require phosphorylation of eIF2α (Kedersha et al., 2005; Teixeira et al., 2005).
To get more insight into the interference of CoVs with gene expression of their host, we investigated the global mRNA profiles in MHV‐infected cells by using genome‐wide microarray analysis. At the time of host shutoff, significant downregulation of many mRNAs, particularly of those encoding translation‐related factors, was observed. This downregulation, which is reminiscent of a cellular stress response, was shown to be dependent on viral replication and to result from mRNA decay. At the same time, increased phosphorylation of eIF2α and the assembly of SGs and P bodies were observed, suggesting that these cellular structures play an important role in the host translational shutoff and mRNA decay respectively. MHV replication was not negatively affected, but rather enhanced in mouse embryo fibroblast (MEF) cells, in which host translational shutoff and SG formation were impaired by mutation. Apparently, virus replication does not benefit, at least in vitro, from the induced translational shutoff.
Results
Kinetics of MHV‐induced host translational shutoff
Before starting with the microarray analyses, the kinetics of the MHV‐induced host translational shutoff was determined for the experimental conditions used. To this end, metabolic labelling of MHV‐infected LR7 cells was performed at different time points post infection. The results, shown in Fig. 1A, clearly demonstrated that host translation was shut down at 6, but not at 4 h post infection, while the viral spike (S), nucleocapsid (N) and membrane (M) proteins were clearly visible at 6 h and at later time points post infection. Thus, between 4 and 6 h post infection, a shift occurred from host to viral protein synthesis. This conclusion was corroborated by quantification of the radioactivity in the gel (Fig. 1B). The data show that there is a continuous rise in expression of the viral protein S, N and M, with the largest increase appearing between 4 and 6 h post infection. The amount of radioactivity in the regions between the viral proteins, representative of host protein synthesis, were also quantified, and confirmed the induction of host shutoff between 4 and 6 h post infection.
Figure 1.
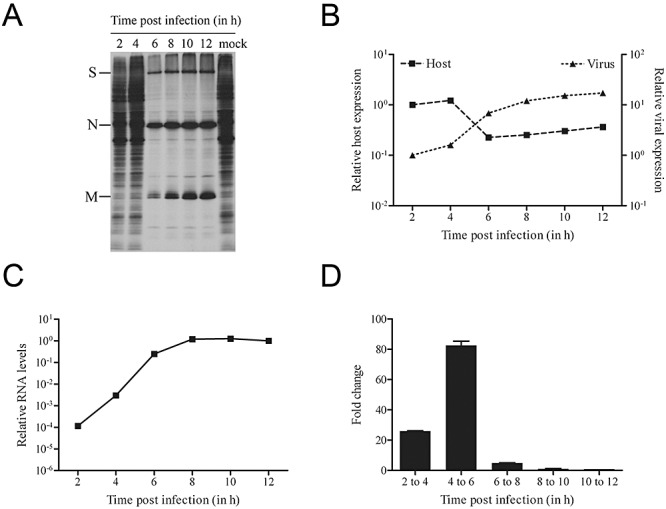
Kinetics of the MHV‐induced host translational shutoff. A. LR7 cells were infected with MHV (moi 10) and metabolically labelled for 15 min starting at the indicating time points. Cell lysates were processed and subjected to SDS‐PAGE as described in Experimental procedures. Positions of the viral proteins S, N and M are indicated. B. The amount of radioactivity in the gel was quantified with a PhosphorImager. For each time point, the amount of radioactivity in the MHV structural proteins S, N and M was combined (viral expression). For the host proteins, the amount of radioactivity in the regions between the MHV proteins was quantified (host expression). The data are presented as relative expression (2 h post infection = 1). C and D. Genomic viral RNA levels in MHV‐infected LR7 cells (moi 10) were measured by quantitative RT‐PCR at the indicated time points post infection. The data are presented as relative viral RNA levels (12 h post infection = 1) in C, or as the fold change increase relative to the previous time point in D. Error bars indicate the standard deviations (n = 3).
Next, the levels of genomic viral RNA in the MHV‐infected cells were determined. Total RNA was isolated at different time points post infection and viral RNA was quantified using quantitative reverse transcription polymerase chain reaction (RT‐PCR). The results show that viral RNA accumulated during infection, reaching a steady‐state level at approximately 8 h post infection (Fig. 1C). Consistent with the results of the metabolic labelling experiment, the largest relative increase in viral RNA levels was observed between 4 and 6 h post infection. (Fig. 1D). Altogether the results demonstrate that, under the experimental conditions used, host translational shutoff is induced in MHV‐infected LR7 cells between 4 and 6 h post infection and is accompanied by a relatively large increase in viral RNA and protein synthesis.
Microarray analysis of MHV‐infected LR7 cells reveals large‐scale mRNA degradation
To get more insight into the MHV‐induced host translational shutoff, we investigated the global mRNA profiles in infected cells by using genome‐wide microarray analysis. To this end, parallel cultures of LR7 cells were either mock‐infected or infected with MHV‐A59 and total RNA was isolated at 4 and 6 h post infection. As will be published elsewhere, at 6 h post infection viral mRNA constitutes approximately 40% of the total mRNA content in infected cells. By removing these unwanted transcripts using the oligo capture technique we reduced non‐specific hybridization by approximately 80%.
The mRNAs in the infected and uninfected samples were amplified, labelled with either Cy3 or Cy5, and hybridized to the arrays after which they were scanned and analysed. At 4 h post infection, relatively few changes in the expression profile were observed (Fig. 2A). However, at 6 h post infection, a significant number of genes appeared to be altered in their expression (Fig. 2B). Besides the apparent upregulation of several mRNAs, a large number of mRNAs (678) was significantly downregulated upon infection. This set of mRNAs is represented by the yellow dots in the different panels of Fig. 2. Actinomycin D (ActD), an inhibitor of cellular transcription but not of CoV replication (Robb and Bond, 1979; Lai et al., 1981), was used to establish whether the observed mRNA downregulation was the result of a transcriptional or a post‐transcriptional mechanism. Treatment of both infected and uninfected cells with ActD, did not affect the downregulation of mRNAs, indicating that the observed decrease is the result of post‐transcriptional mRNA degradation (Fig. 2C). To study the specificity of this virus‐induced mRNA degradation, the effect of ActD treatment was analysed in the absence of a viral infection. Treatment of cells with ActD, or with other transcriptional inhibitors, induces the downregulation of short‐lived mRNAs (Cheadle et al., 2005). Indeed, downregulation of many mRNAs was observed in ActD‐treated cells when compared with mock‐treated cells (Fig. 2D). However, the mRNA population downregulated by ActD treatment clearly differed from the mRNAs downregulated in response to MHV infection at 6 h post infection (the latter ones are represented by the yellow dots). In order to determine the role of virus replication in the mRNA decay, cells were inoculated with fusion‐competent, replication incompetent, UV‐inactivated virus. The results show, that the MHV‐induced downregulation of mRNA (shown again as yellow dots) is replication‐dependent, as it was not observed after inoculation with the UV‐inactivated virus (Fig. 2E).
Figure 2.
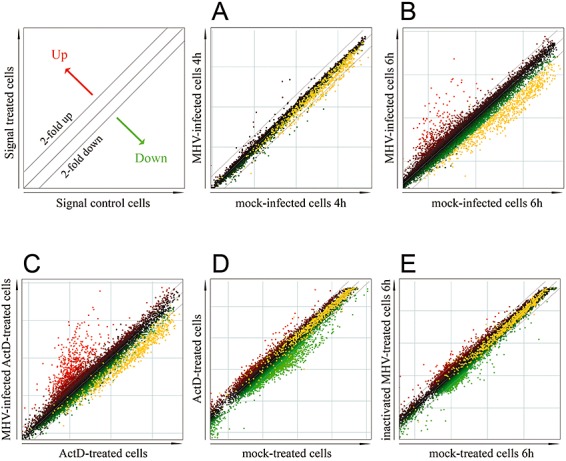
Replication‐dependent mRNA decay in MHV‐infected cells as determined by microarray analyses. LR7 cells were infected with MHV (moi 10). Total RNA was isolated from MHV‐infected and mock‐infected cells and processed for microarray analysis as described in Experimental procedures. The scatter plots display the average expression values from independent dye‐swap hybridizations for each gene present on the arrays. Red spots represent upregulated gene transcripts while green spots represent downregulated transcripts. The dashed lines indicate the twofold change cut‐off. Transcripts downregulated at 6 h post infection (in B) are represented by the yellow dots throughout the figure. Note that these transcripts are significantly changed at 6 h post infection according to SAM, applying a false‐discovery rate of 1% and a cut‐off at a twofold change (described in Experimental procedures). A. The expression profile of MHV‐infected cells compared to mock‐infected cells at 4 h post infection (average of four arrays; two independent dye‐swaps; n = 4). B. The expression profile of MHV‐infected cells compared with mock‐infected cells at 6 h post infection (n = 6). C. The expression profile of MHV‐infected cells compared with mock‐infected cells at 6 h post infection (n = 4). Both MHV‐ and mock‐infected cells were treated with ActD for 7 h (from −1 till 6 h post infection). D. The expression profile of ActD‐treated cells compared with mock‐treated cells. Non‐infected cells were treated or mock treated with ActD for 7 h (n = 1). E. The expression profile of cells treated with UV‐inactivated viral particles compared with mock‐treated cells at 6 h post inoculation (n = 1).
By using Gene Ontology (GO) databases, 240 of the 678 mRNAs downregulated at 6 h post infection could be annotated by their GO terms for biological functions/processes (see Supplementary Table S1). Strikingly, 50% of the mRNAs that could be annotated encode protein translation‐related factors (ribosome biogenesis factors, translation initiation/elongation associated factors). In summary, the results show that, at the time of virus‐induced host translational shutoff, significant downregulation of many mRNAs encoding largely translation‐related factors was observed. This downregulation was found to be dependent on viral replication and to result from mRNA decay.
Transcriptional upregulation of mRNAs
Comparison of the transcriptional profiles of MHV‐infected cells in the absence or presence of ActD (Fig. 2B and C) revealed that the mRNAs from relatively few genes were upregulated at the transcriptional level, as the majority of the upregulated mRNAs were obtained for both conditions. As approximately 20% of the viral RNA present in the LR7 cells at 6 h post infection was not removed by the oligo capture technique (M. Raaben and C. A. M. de Haan, unpubl. results), a large number of the mRNAs upregulated in the presence of ActD are likely to result from cross hybridization of the remaining viral RNA. Indeed, this was confirmed for some hits using quantitative RT‐PCR analysis (data not shown). However, we cannot exclude that some of these hits might also result from increased mRNA stability upon virus infection, a subject of further research. In order to exclude (potential) false‐positive hits, the genelists of the experiments performed in the absence or presence of ActD were compared. As a result, 243 genes were excluded from the analysis since they were upregulated under both experimental conditions, leaving 40 genes that were specifically upregulated at the transcriptional level in MHV‐infected LR7 cells at 6 h post infection. The genes identified were clustered according to their GO terms for biological processes (see Supplementary Table S2). Out of the 40 genes, we could annotate 29 genes. Examples of genes of which the expression was induced were those encoding inflammatory cytokines (Cxcl1, Cxcl2), a marker of the unfolded protein response (Herpud1), and dual specificity phosphatase 1 (Dusp1). In addition, several genes involved in regulation of transcription were differentially expressed. The expression of several genes at 4 and 6 h post infection was confirmed by quantitative RT‐PCR (see Supplementary Table S3).
Phosporylation of eIF2α, and the formation of stress granules and processing bodies
The observed downregulation of numerous mRNAs, many of which encode protein translation‐related factors, is reminiscent of a cellular stress response. Recently, SGshave been associated with the initiation of host translational shutoff during stress. A key aspect in the formation of these cytoplasmic structures, which contain stalled translation preinitiation complexes, is the phosphorylation of the translation initiation factor eIF2α at serine 51, which results in the aggregation of the mRNA binding proteins TIA‐1 and TIAR, both markers for SGs (Kedersha et al., 1999; Kedersha et al., 2005). Phosphorylation of eIF2α was analysed in MHV‐infected LR7 cells at 4 and 6 h post infection, i.e. before and at the time of host translational shutoff, respectively (Fig. 3A). Mock‐infected cells and cells treated for 30 min with 0.5 mM sodium arsenite, were taken along as controls. Sodium arsenite has been shown to induce phosphorylation of eIF2α at serine 51 (McEwen et al., 2005). The expression levels and phosphorylation status of eIF2α in all samples were visualized by immunoblotting using antibodies against total eIF2α and phosphorylated eIF2α (eIF2αP) respectively. Quantitative analysis revealed that the levels of phosphorylated eIF2α, corrected for the total amount of eIF2α, in MHV‐infected cells were reproducibly ∼2.5‐fold higher at 6 h post infection compared with mock‐infected cells, while at 4 h post infection no increase could be observed.
Figure 3.
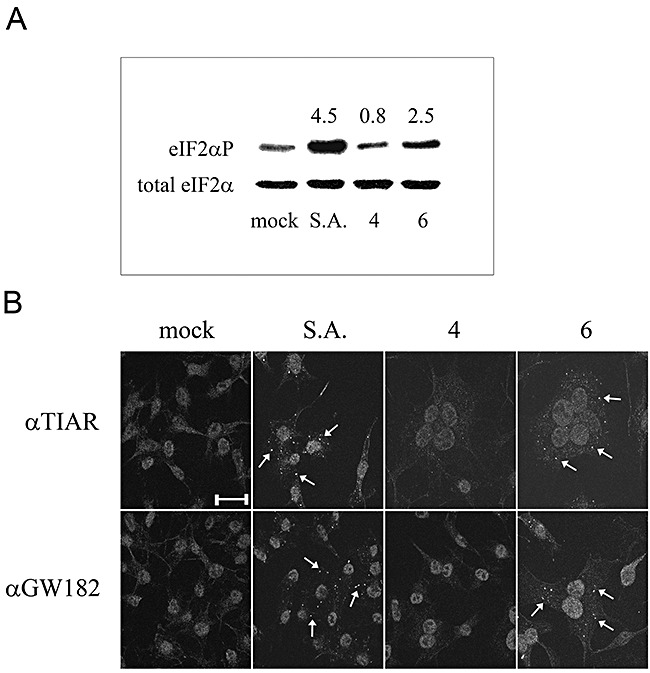
MHV induces phosphorylation of eIF2α at Ser51, and the subsequent formation of SGs and P bodies. A. The phosphorylation state of eIF2α in MHV‐infected LR7 cells at 4 and 6 h post infection (indicated by 4 and 6) or after mock infection was determined by Western blotting using eIF2αP‐specific antibodies (top) and related to total eIF2α levels by stripping and reprobing of the membrane (bottom). The fold change differences between MHV‐infected and mock‐infected cells are indicated above each lane. As a positive control, LR7 cells were treated with 0.5 mM sodium arsenite (indicated as S.A.) for 30 min. This experiment was repeated twice with similar results. B. LR7 cells were mock‐infected, treated with S.A. for 30 min, or infected with MHV. Cells were fixed and processed for immunofluorescence using TIAR (αTIAR) and GW182 (αGW182) antibodies according to Experimental procedures. MHV‐infected cells were fixed at 4 and 6 h post infection (indicated by 4 and 6). Representative images are shown for each condition. The scaled bar corresponds to 20 μM.
Subsequently, we investigated whether SGs were formed in LR7 cells upon MHV infection. As a positive control, LR7 cells were incubated with sodium arsenite as described above. The cells were fixed at the indicated time points post infection, and processed for immunofluorescence using anti‐TIAR antibodies. Indeed, cytoplasmic foci containing TIAR, representing SGs, could be detected at the time of increased eIF2α phosphorylation and host translational shutoff (at 6 h post infection), but hardly or not at 4 h post infection (Fig. 3B). SGs were observed both in single cells and in syncytia.
Next, we analysed the formation of other cytoplasmic structures, P bodies, which are sites of mRNA degradation. P bodies are related to SGs, although their formation does not require the phosphorylation of eIF2α. The GW182 antigen has recently been described as a marker for P bodies (Eystathioy et al., 2002; 2003). Consistent with the results described above, P bodies could be detected by immunofluorescence assays using antibodies against GW182 at 6 h post infection, when mRNA decay could be observed, but hardly or not at 4 h post infection (Fig. 3B). Altogether, the results show that MHV infection of LR7 cells induces the phosphorylation of eIF2α at serine 51, and the formation of SGs and P bodies, concomitant with the observed host translational shutoff and mRNA decay.
MHV replication is not negatively affected in cells with impaired host translational shutoff
To investigate to what extent the cellular stress response and the host translational shutoff affects MHV infection, we studied replication in MEFs expressing a mutant form of eIF2α (eIF2αS51A) that cannot be phosphorylated at serine 51 (Scheuner et al., 2001). Host translational shutoff is not observed in these cells during stress or alphavirus infection (Scheuner et al., 2001; McInerney et al., 2005). In addition, replication was monitored in TIA‐1–/– and TIAR–/– MEFs. These MEFs have been demonstrated to exhibit impaired SG assembly, as well as delayed host translational shutoff after virus infections (Gilks et al., 2004; McInerney et al., 2005). Unfortunately, host translational shutoff could not be studied by metabolic labelling of the cells upon infection with MHV, as only 10–20% of the MEF cells in a dish could be infected after a high multiplicity infection with MHV (data not shown). Nevertheless, viral replication in the MEF cells was monitored, not by determining the production of infectious virions, but by following the expression of a reporter gene after infection with MHV‐EFLM, which carries the firefly luciferase (FL) in an MHV‐A59 background. FL expressed by MHV has been shown to be a reliable measure for virus replication (de Haan et al., 2005). The intracellular FL levels were determined at different time points post infection and normalized for the number of infected cells, which differed slightly between the MEF cell lines. Clearly, MHV replication was enhanced in the eIF2αS51A‐expressing cells as compared with the wild‐type eIF2α‐expressing cells (Fig. 4A). Similarly, replication was also enhanced in the TIAR–/– cells, while MHV replicated to approximately the same extent in the TIA‐1–/– and wild‐type MEF cells (Fig. 4B). Overall, the results show that replication of MHV is not negatively affected but rather enhanced in cells with impaired host translational shutoff.
Figure 4.
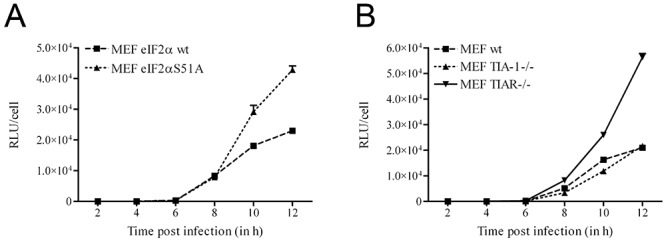
MHV replication is not negatively affected in MEF cells with impaired host translational shutoff and reduced SG assembly. Confluent monolayers of MEF cells were infected with MHV‐EFLM. Cells were lysed at the indicated time points post infection and the intracellular luciferase levels were determined (in RLU). As the number of infected cells differed to some extent between the different MEFs, the obtained RLU values for each cell line were normalized for the number of infected cells as determined by immunocytochemistry using a polyclonal anti‐MHV serum (Rottier et al., 1981). Error bars indicate the standard deviations in all graphs (n = 3). A. Luciferase expression in MEF cells expressing wild‐type eIF2α (MEF eIF2α wt) or mutant eIF2α (MEF eIF2αS51A). B. Luciferase expression in wild type (MEF wt), TIA‐1 knockout (MEF TIA‐1–/–) and TIAR knockout (MEF TIAR–/–) cells.
Discussion
A variety of strategies has been described by which viruses dominate the host‐cell protein synthetic machinery, optimize viral mRNA translation and evade the host‐cell antiviral responses that act at the translational level (Schneider and Mohr, 2003). For MHV it was shown already in the eighties that infection induces host translational shutoff, while maintaining its own protein synthesis (Rottier et al., 1981; Siddell et al., 1981; Hilton et al., 1986). However, the cellular processes underlying MHV‐induced inhibition of host protein synthesis remained largely unknown. We now show that this inhibition coincides with degradation of numerous mRNAs, many of which encode protein translation‐related factors. In addition, infection with MHV results in increased phosphorylation of eIF2α and in the formation of SGs and P bodies, which are sites of mRNA stalling and degradation respectively. Altogether these results indicate that MHV infection induces an integrated stress response, which results in increased phosphorylation of eIF2α and in the formation of SGs and P bodies, leading to shutdown of host mRNA translation and to mRNA decay (Fig. 5).
Figure 5.
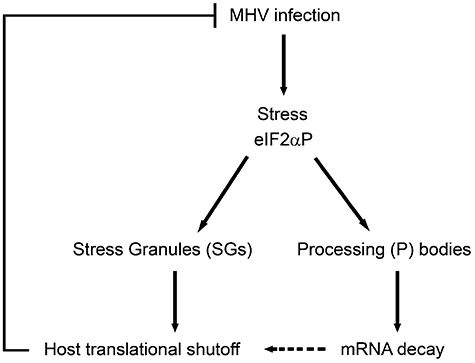
Model for MHV‐induced host translational shutoff. A cellular stress response is elicited upon infection with MHV, which results in increased phosphorylation of eIF2α (eIF2αP). Subsequently, SGs containing stalled translational preinitiation complexes are formed and host translational shutoff is induced. In addition, the stress response also induces the formation of P bodies, which are cytoplasmic sites of mRNA decay. Degradation of many mRNAs encoding protein translation‐related factors is observed, which may contribute to the MHV‐induced host translational shutoff. Overall, the initiated stress response appears to inhibit MHV replication.
Our application of microarray analysis to assess changes in cellular mRNA profiles in MHV‐infected cells revealed that mRNA levels were regulated both by transcriptional and by post‐transcriptional mechanisms. Relatively few genes (40) were identified, of which transcription was enhanced (see Supplementary data). Among these were genes encoding inflammatory cytokines (Cxcl1 and Cxcl2) and proteins associated with cellular transcription. The induction of Dusp1, which plays an important role as a protein phosphatase in the cellular response to stress (Keyse and Emslie, 1992; Ma and Hendershot, 2004) and of Herpud1, which is a marker of the unfolded protein response (Keyse and Emslie, 1992; Ma and Hendershot, 2004), suggests that cells infected with MHV elicit a stress response. Several of these upregulated gene transcripts have also been identified in a gene expression profiling study of MHV‐infected cells using a different microarray platform (Versteeg et al., 2006).
Perhaps more striking is the decline of a large number of mRNAs at the time of host translational shutoff (see Supplementary data). This reduction was shown to be dependent on viral replication and to be caused by mRNA degradation, because it was also observed in the presence of the cellular transcription inhibitor ActD. Transcriptional inhibition results in a fast degradation of short‐lived mRNAs, especially those containing AU‐rich elements (AREs) in their 3′ UTRs (Tebo et al., 2003). However, ActD treatment itself resulted in a different expression profile as compared with MHV infection, indicating that MHV infection does not simply result in increased instability of short‐lived mRNAs. Interestingly, MHV infection induced the degradation of transcripts encoding the chemokines Cxcl1 and Cxcl2 in the presence of ActD (data not shown), whereas higher levels of these transcripts were observed during infection in the absence of ActD, indicating that the expression of individual genes can be regulated both at the transcriptional and post‐transcriptional level.
Downregulation of many transcripts encoding ribosome biogenesis factors during CoV infection has also been observed after infection of Vero cells with SARS‐CoV (Leong et al., 2005). These reduced mRNA levels are reminiscent of the decline of ribosome biogenesis factor encoding mRNAs observed in yeast in response to different stress stimuli such as heat and nutrient deprivation (Gasch et al., 2000; Causton et al., 2001). In agreement with our results, mRNA decay was demonstrated to be the main cause of the observed downregulation of these mRNA levels (Grigull et al., 2004). These results indicate that CoV infection elicits a cellular response, which resembles the reaction of yeast cells upon environmental stress. Recently, expression of the SARS‐CoV nsp1 has been shown to result in degradation of several host mRNAs and in translational shutoff (Kamitani et al., 2006). A direct role for nsp1 in mRNA degradation might explain the observed effects, even though nsp1 has no sequence similarities with any known RNases, and is only poorly conserved among CoVs. Alternatively, expression of the SARS‐CoV nsp1 might induce a cellular stress response resulting in mRNA decay and in inhibition of protein synthesis. In agreement herewith, others showed that expression of SARS‐CoV nsp1 induced activation of the stress‐related transcription factor NF‐κB and chemokine upregulation (Law et al., 2007).
Phosphorylation of eIF2α is induced in response to several stress stimuli (Clemens, 2005). Indeed, a modestly increased level (∼2.5‐fold) of phosphorylated eIF2α was detected after infection with MHV. A similar increase in eIF2α phosphorylation has also been reported for some reoviruses (Smith et al., 2006). The kinase responsible for the phosphorylation of eIF2α after infection with MHV is not known. PERK, which is activated by ER stress, might be a good candidate as infection of cultured cells with SARS‐CoV (Chan et al., 2006) or MHV (Versteeg et al., 2006) appears to lead to the induction of the unfolded protein response.
The recent discovery of cellular structures involved in translational arrest (SGs) and turnover of mRNA pools (P bodies) during stress prompted us to investigate the formation of these cytoplasmic structures in MHV‐infected cells. In general, phosphorylation of eIF2α is required for the formation of SGs. Indeed, SGs were detected concomitant with increased phosphorylation of eIF2α and host translational shutoff. Similarly, the induction of SGs has recently also been demonstrated after infection with reovirus (Smith et al., 2006), Semliki Forest virus (McInerney et al., 2005) and herpes simplex virus type 1 (Esclatine et al., 2004), all of which are able to induce host translational shutoff.
To the best of our knowledge, this is the first report that describes the induction of P bodies by virus infection. P bodies were formed at the time of mRNA decay, suggesting that these structures play a role in the degradation of the host transcripts. Whether also viral mRNAs localize to P bodies and are subsequently degraded remains to be established. We cannot exclude that other RNA decay pathways may be induced upon infection with MHV as well. However, the interferon‐depended 2′,5′‐oligoadenylate synthetase pathway (Clemens, 2005) is not likely to play a major role in MHV‐infected cells, because no prominent upregulation of interferon‐mediated gene transcription was observed, which is consistent with the results of others (Banerjee et al., 2000; Versteeg et al., 2006). Whether the mRNA decay contributes to MHV‐induced host translational shutoff remains to be established.
The CoV‐induced cellular stress response, resulting in the inhibition of host protein synthesis, might be beneficial for the virus by downregulating cellular components of the antiviral response. On the other hand, as the coronaviral mRNAs are structurally equivalent to the host mRNAs, viral protein synthesis might also be impaired, even though viral protein synthesis is still robust after host translational shutoff. The results obtained with the host translational shutoff‐deficient (eIF2αS51A) MEF cells show that in the absence of phosphorylatable eIF2α MHV replication is not negatively affected. Thus, the stress response induced upon infection with MHV, which results in the phosphorylation of eIF2α and the subsequent inhibition of protein synthesis, does not appear to be beneficial for MHV replication in vitro. This is in contrast to reoviruses, replication of which has been demonstrated to be facilitated by the induced cellular stress response and eIF2α phosphorylation (Smith et al., 2006). Our conclusions are corroborated by the results obtained with the TIA‐1–/– and TIAR–/– MEFs, which exhibit impaired SG assembly and host translational shutoff, and in which MHV replication was also not negatively affected but, rather, enhanced. Similar results, using the same TIA‐1–/– and TIAR–/– MEFs, were previously obtained for the replication of vesicular stomatitis virus, Sindbis virus and herpes simplex virus type 1, but not for West Nile virus (Li et al., 2002). The latter virus was found to require TIA‐1 and TIAR binding to viral RNA for efficient replication. The importance of the CoV‐induced cellular stress response and the host translational shutoff for virus replication in vivo remains to be established.
Even though it appears that infection with MHV induced an integrated stress response resulting in host translational shutoff, the viral proteins are still efficiently made in comparison with the host proteins. Several factors are likely to play a role in the CoV ‘escape’ from translational inhibition. Previously it has been reported that the leader sequence present at the 5′‐end of all MHV encoded mRNAs mediates their preferential translation (Tahara et al., 1993; 1994). However, the precise mechanism by which this is established is still unknown. The exponential increase in the abundance of viral mRNAs during infection is also likely to contribute to a preferential translation of these mRNAs, simply as a result of increasing abundance in the competition for the limited translational machinery. We have calculated that at 6 h post infection up to 40% of all mRNAs in the cell is virus‐encoded. Furthermore, the downregulation of many mRNAs encoding protein translation‐related factors, probably via the induction of P bodies, is also likely to increase the success of the viral mRNAs in their competition with the host‐encoded mRNAs.
Experimental procedures
Cells and viruses
LR7 mouse fibroblast cells (Kuo et al., 2000), were maintained in Dulbecco's modified Eagle's medium (DMEM) (Cambrex Bio Science) containing 10% (v/v) fetal calf serum (Bodinco B.V.), 100 U ml−1 Penicillin and 100 μg ml−1 Streptomycin (referred to as complete DMEM), supplemented with Geneticin G418 (250 μg ml−1). The MEFs expressing wild type or mutant (S51A) eIF2α (Scheuner et al., 2001) as well as the MEF cell lines from wild type, TIAR knockout (TIAR–/–) and TIA‐1 knockout (TIA–/–) embryo mice (Li et al., 2002) were also maintained in complete DMEM. MHV strain A59 and MHV‐EFLM, the latter containing the firefly luciferase reporter gene (de Haan et al., 2005), were grown on LR7 cells. Inactivation of MHV‐A59 was performed by UV light (366 nm), using a Chromato‐Vue transilluminator (1 min, 6000 μW cm−2) from Ultraviolet products. The inactivated particles retained their fusion activity, as was demonstrated using a fusion‐from‐without assay on LR7 cells.
Metabolic labelling of MHV‐infected cells
LR7 cells were inoculated with MHV‐A59 at a multiplicity of infection (moi) of 10 TCID50 (50% tissue culture infectious doses) per cell, in phosphate‐buffered saline (PBS) containing 50 μg ml−1 diethylaminoethyl‐dextran (PBS‐DEAE). After 1 h, the cells were washed with and the culture medium was replaced by complete DMEM. At 2 h post infection the fusion inhibitory mHR2 peptide (1 μM) (Bosch et al., 2003) was added to the culture medium. At the indicated time points, the cells were labelled with 35S labelled amino acids (Amersham) for 15 min, after which cells were lysed and lysates were prepared for sodium dodecylsulphate‐polyacrylamide gel electrophoresis (SDS‐PAGE) and subsequent fluorography as previously described (de Haan et al., 2000). Radioactivity in protein bands was quantified in dried gels using a PhosphorImager (Molecular Dynamics).
Luciferase assay
Cell monolayers were infected with MHV‐EFLM as described above. At the indicated times, the cells were lysed using the appropriate buffer provided with the steadylite HTS reporter gene assay system (PerkinElmer). Intracellular luciferase expression was measured according to the manufacturer's instructions, and the relative light units (RLU) were determined with a Berthold Centro LB 960 plate luminometer.
Total RNA isolation
LR7 cells were either mock‐infected or infected with MHV‐A59 as described above. When indicated, the cells were incubated with ActD (20 μg ml−1) 1 h before infection, and maintained in the presence of this drug throughout the experiment. Total RNA was isolated from mock or MHV‐A59 infected cells at the indicated time post infection using the TRIzol reagent (Invitrogen). RNA was further purified using the RNeasy mini‐kit with subsequent DNaseI treatment on the column (Qiagen). Viral RNA was removed from the total RNA samples using the GlobinClear kit (Ambion) with the use of an alternative oligo capture mix, containing three 5′ biotinylated oligo's that anneal to either the 5′ leader, the nucleocapsid gene and the 3′ untranslated region of the MHV‐A59 genome, as will be reported in more detail elsewhere. All subsequent steps were performed according to the manufacturer's protocol. RNA integrity was determined by spectrometry and by a microfluidics‐based platform using a UV‐mini1240 device (Shimadzu) and a 2100 Bioanalyzer (Agilent Technologies) respectively.
cRNA synthesis, labelling and hybridization onto microarrays
mRNA was amplified from 1 μg of total RNA by cDNA synthesis with oligo(dT) double‐anchored primers, followed by in vitro transcription using a T7 RNA polymerase kit (Ambion) as described previously (Roepman et al., 2005). During transcription, 5‐(3‐aminoallyl)‐UTP was incorporated into the single stranded cRNA. Cy3 and Cy5 NHS‐esters (Amersham Biosciences) were coupled to 2 μg cRNA. RNA quality was monitored after each successive step using the methods described above. A Mouse Array‐Ready Oligo set (version 3.0) was purchased (Operon) and printed on Corning UltraGAPS slides. Mouse slides containing 35 000 spots (32 101 70‐mer oligonucleotides, and 2891 control spots) were hybridized with 1 μg of each alternatively labelled cRNA target at 42°C for 16–20 h using LifterSlips (Erie Scientific) and Corning Hybridization Chambers (van de Peppel et al., 2003). After hybridization the slides were washed extensively and scanned using the Agilent G2565AA DNA Microarray Scanner.
Microarray data analysis
Images were quantified and background corrected using Imagene 5.6 software. The data were normalized using Lowess print‐tip normalization as described previously (Yang et al., 2002). To identify the genes that were significantly different within each experiment, a one‐class Significance Analysis of Microrarrays (SAM) (Tusher et al., 2001) was performed on the average of independent dye‐swap hybridizations (unless indicated otherwise), using a false discovery rate of 1%. To increase the confidence level, a cut‐off at a twofold change in expression was applied. The resulting genelists were subjected to Genespring 7.2 software for further analysis.
ArrayExpress accession numbers
MIAME‐compliant data in MAGE‐ML format as well as complete descriptions of protocols have been submitted to the public microarray database ArrayExpress (http://www.ebi.ac.uk/arrayexpress/) with the following accession numbers: microarray layout, P‐UMCU‐8; gene expression data of MHV‐infected LR7 cells, E‐MEXP‐895; protocols for total RNA isolation and mRNA amplification, P‐MEXP‐34397; cRNA labelling, P‐MEXP‐34400 and P‐MEXP‐35534; hybridization and washing of slides, P‐MEXP‐34401; scanning of slides, P‐MEXP‐34430; data normalization, P‐MEXP‐34431.
Quantitative RT‐PCR
Altered gene expression levels of some genes, identified by microarray analysis, were verified by quantitative RT‐PCR using Assay‐On‐Demand reagents (PE Applied Biosystems), according to the manufacturer's instructions, on cDNA generated as described above. Reactions were performed using an ABI Prism 7000 sequence detection system. The comparative Ct‐method was used to determine the fold change for each individual gene. The housekeeping gene GAPDH was used as a reference in all experiments. The amounts of viral genomic RNA were determined by quantitative RT‐PCR as described before (de Haan et al., 2004).
Immunoblotting
Cells were lysed in ice‐cold lysis buffer [20 mM MOPS (pH 7.2), 5 mM EDTA, 2 mM EGTA, and 0.5% (w/v) Nonidet‐P40, containing 30 mM NaF, 40 mM β‐Glycerophosphate, 20 mM Na‐Pyrophosphate, 1 mM Na‐Orthovanadate, 1 mM PMSF, 3 mM Benzamidine, 1.5 μM Pepstatin A and 10 μM Leupeptin]. Cell lysates were cleared by centrifugation at 100 000 g at 4°C for 30 min. Proteins present in the cell lysates were separated by SDS‐PAGE and transferred to Nitrocellulose membranes (0.1 μM, Schleicher Schuell). Subsequently, the membranes were incubated over night in block buffer [PBS containing 0.05% Tween‐20, 5% (w/v) Protifar and 5% (v/v) normal goat serum]. Next, the membranes were washed three times with PBS (containing 0.05% Tween‐20) and incubated for 16 h at 4°C with a rabbit polyclonal antibody specifically recognizing eIF2α phosphorylated on serine 51 (DeGracia et al., 1997). Following extensive washing, the membranes were incubated with peroxidase labelled goat anti‐rabbit IgG (Bio‐Rad Laboratories), after which the amount of protein was visualized and quantified using the Enhanced ChemoLuminescence (ECL) plus kit, a Typhoon imager, and ImageQuant TL software (Amersham Biosciences). For the detection of the total amount of eIF2α, membranes were stripped by incubating the blot in stripping buffer (62.5 mM Tris‐HCl [pH 6.7], 100 mM β‐mercaptoethanol and 2% SDS) at 50°C for 30 min, after which the membranes were incubated with a monoclonal mouse anti‐eIF2α antibody (Scorsone et al., 1987) and peroxidase labelled goat anti‐mouse IgG (Cappel), and the protein bands were quantified as described above.
Indirect immunofluorescence
LR7 cells grown on 10‐mm cover slips were infected with MHV‐A59 as described above. At the indicated time points, cells were fixed with 1% (w/v) paraformaldehyde in phosphate buffer (48 mg l−1 KH2PO4, 3 g l−1 NaCl and 265 mg l−1 Na2HPO4) for 20 min, after which the cells were permeabilized using 0.1% Triton‐X‐100 in PBS for 10 min. After blocking with PBS containing 5% FCS, TIAR or GW182 were detected using a rabbit polyclonal antibody (Santa Cruz Biotechnologies) or a monoclonal antibody (Abcam) respectively. Subsequently, affinity‐purified goat anti‐rabbit or donkey anti‐mouse secondary antibodies conjugated to Cy3 were used for visualization. The cells were mounted with FluorSave Reagent (Calbiochem) and fluorescence was viewed with a Leica TCS SP confocal microscope.
Acknowledgements
We would like to thank the people at the Microarray Facility (UMC Utrecht, the Netherlands) for technical support, A.A.M. Thomas (Department of Developmental Biology, Utrecht University, Utrecht, the Netherlands) and G.S. Krause (Wayne State University School of Medicine, MI 48201, USA) for providing us the antibodies against total and phosphorylated eIF2α, Berend Jan Bosch for supplying us with the mHR2 peptide, P. Anderson (Division of Rheumatology and Immunology, Brigham and Women's Hospital, Boston, Massachusetts 02115, USA), R.J. Kaufman (Howard Hughes Medical Institute, University of Michigan, Ann Arbor, MI 48109, USA) and C. Koumenis (Radiation Oncology, University of Pennsylvania, Philadelphia, USA) for providing the different MEF cell lines, and Monique Oostra for stimulating discussions. This work was supported by a grant from the Netherlands Organization for Scientific Research (NWO‐VIDI‐700.54.421) to C.A.M. de Haan.
References
- Anderson, P. , and Kedersha, N. (2002) Stressful initiations. J Cell Sci 115: 3227–3234. [DOI] [PubMed] [Google Scholar]
- Anderson, P. , and Kedersha, N. (2006) RNA granules. J Cell Biol 172: 803–808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banerjee, S. , An, S. , Zhou, A. , Silverman, R.H. , and Makino, S. (2000) RNase 1‐independent specific 28S rRNA cleavage in murine coronavirus‐infected cells. J Virol 74: 8793–8802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berlanga, J.J. , Santoyo, J. , and De Haro, C. (1999) Characterization of a mammalian homolog of the GCN2 eukaryotic initiation factor 2 alpha kinase. Eur J Biochem 265: 754–762. [DOI] [PubMed] [Google Scholar]
- Bosch, B.J. , Van Der Zee, R. , De Haan, C.A. , and Rottier, P.J. (2003) The coronavirus spike protein is a class I virus fusion protein: structural and functional characterization of the fusion core complex. J Virol 77: 8801–8811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brostrom, C.O. , and Brostrom, M.A. (1998) Regulation of translational initiation during cellular responses to stress. Prog Nucleic Acid Res Mol Biol 58: 79–125. [DOI] [PubMed] [Google Scholar]
- Causton, H.C. , Ren, B. , Koh, S.S. , Harbison, C.T. , Kanin, E. , Jennings, E.G. , et al. (2001) Remodeling of yeast genome expression in response to environmental changes. Mol Biol Cell 12: 323–337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan, C.P. , Siu, K.L. , Chin, K.T. , Yuen, K.Y. , Zheng, B. , and Jin, D.Y. (2006) Modulation of the unfolded protein response by the severe acute respiratory syndrome coronavirus spike protein. J Virol 80: 9279–9287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheadle, C. , Fan, J. , Cho‐Chung, Y.S. , Werner, T. , Ray, J. , Do, L. , et al. (2005) Stability regulation of mRNA and the control of gene expression. Ann N Y Acad Sci 1058: 196–204. [DOI] [PubMed] [Google Scholar]
- Clemens, M.J. (2005) Translational control in virus‐infected cells: models for cellular stress responses. Semin Cell Dev Biol 16: 13–20. [DOI] [PubMed] [Google Scholar]
- Cougot, N. , Babajko, S. , and Seraphin, B. (2004) Cytoplasmic foci are sites of mRNA decay in human cells. J Cell Biol 165: 31–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeGracia, D.J. , Sullivan, J.M. , Neumar, R.W. , Alousi, S.S. , Hikade, K.R. , Pittman, J.E. , et al. (1997) Effect of brain ischemia and reperfusion on the localization of phosphorylated eukaryotic initiation factor 2 alpha. J Cereb Blood Flow Metab 17: 1291–1302. [DOI] [PubMed] [Google Scholar]
- Drosten, C. , Gunther, S. , Preiser, W. , Van Der Werf, S. , Brodt, H.R. , Becker, S. , et al. (2003) Identification of a novel coronavirus in patients with severe acute respiratory syndrome. N Engl J Med 348: 1967–1976. [DOI] [PubMed] [Google Scholar]
- Esclatine, A. , Taddeo, B. , and Roizman, B. (2004) Herpes simplex virus 1 induces cytoplasmic accumulation of TIA‐1/TIAR and both synthesis and cytoplasmic accumulation of tristetraprolin, two cellular proteins that bind and destabilize AU‐rich RNAs. J Virol 78: 8582–8592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eystathioy, T. , Chan, E.K. , Tenenbaum, S.A. , Keene, J.D. , Griffith, K. , and Fritzler, M.J. (2002) A phosphorylated cytoplasmic autoantigen, GW182, associates with a unique population of human mRNAs within novel cytoplasmic speckles. Mol Biol Cell 13: 1338–1351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eystathioy, T. , Jakymiw, A. , Chan, E.K. , Seraphin, B. , Cougot, N. , and Fritzler, M.J. (2003) The GW182 protein colocalizes with mRNA degradation associated proteins hDcp1 and hLSm4 in cytoplasmic GW bodies. RNA 9: 1171–1173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gasch, A.P. , Spellman, P.T. , Kao, C.M. , Carmel‐Harel, O. , Eisen, M.B. , Storz, G. , et al. (2000) Genomic expression programs in the response of yeast cells to environmental changes. Mol Biol Cell 11: 4241–4257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilks, N. , Kedersha, N. , Ayodele, M. , Shen, L. , Stoecklin, G. , Dember, L.M. , and Anderson, P. (2004) Stress granule assembly is mediated by prion‐like aggregation of TIA‐1. Mol Biol Cell 15: 5383–5398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grigull, J. , Mnaimneh, S. , Pootoolal, J. , Robinson, M.D. , and Hughes, T.R. (2004) Genome‐wide analysis of mRNA stability using transcription inhibitors and microarrays reveals posttranscriptional control of ribosome biogenesis factors. Mol Cell Biol 24: 5534–5547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Haan, C.A. , Vennema, H. , and Rottier, P.J. (2000)Assembly of the coronavirus envelope: homotypic interactions between the M proteins. J Virol 74: 4967–4978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Haan, C.A. , Stadler, K. , Godeke, G.J. , Bosch, B.J. , and Rottier, P.J. (2004) Cleavage inhibition of the murine coronavirus spike protein by a furin‐like enzyme affects cell‐cell but not virus‐cell fusion. J Virol 78: 6048–6054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Haan, C.A. , Haijema, B.J. , Boss, D. , Heuts, F.W. , and Rottier, P.J. (2005) Coronaviruses as vectors: stability of foreign gene expression. J Virol 79: 12742–12751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harding, H.P. , Zhang, Y. , and Ron, D. (1999) Protein translation and folding are coupled by an endoplasmic‐reticulum‐resident kinase. Nature 397: 271–274. [DOI] [PubMed] [Google Scholar]
- De Haro, C. , Mendez, R. , and Santoyo, J. (1996) The eIF‐2alpha kinases and the control of protein synthesis. FASEB J 10: 1378–1387. [DOI] [PubMed] [Google Scholar]
- Hilton, A. , Mizzen, L. , MacIntyre, G. , Cheley, S. , and Anderson, R. (1986) Translational control in murine hepatitis virus infection. J Gen Virol 67: 923–932. [DOI] [PubMed] [Google Scholar]
- Van Der Hoek, L. , Pyrc, K. , Jebbink, M.F. , Vermeulen‐Oost, W. , Berkhout, R.J. , Wolthers, K.C. , et al. (2004) Identification of a new human coronavirus. Nat Med 10: 368–373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ingelfinger, D. , Arndt‐Jovin, D.J. , Luhrmann, R. , and Achsel, T. (2002) The human LSm1–7 proteins colocalize with the mRNA‐degrading enzymes Dcp1/2 and Xrnl in distinct cytoplasmic foci. RNA 8: 1489–1501. [PMC free article] [PubMed] [Google Scholar]
- Kamitani, W. , Narayanan, K. , Huang, C. , Lokugamage, K. , Ikegami, T. , Ito, N. , et al. (2006) Severe acute respiratory syndrome coronavirus nsp1 protein suppresses host gene expression by promoting host mRNA degradation. Proc Natl Acad Sci USA 103: 12885–12890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaufman, R.J. (2000) The double‐stranded RNA‐activated protein kinase PKR In Translational Control of Gene Expression. Sonenberg N., Hershey J.W.B., and Mathews M.B. (eds). Cold Spring Harbor, NY: Cold Spring Harbor Laboratory Press, pp. 503–527. [Google Scholar]
- Kedersha, N.L. , Gupta, M. , Li, W. , Miller, I. , and Anderson, P. (1999) RNA‐binding proteins TIA‐1 and TIAR link the phosphorylation of eIF‐2 alpha to the assembly of mammalian stress granules. J Cell Biol 147: 1431–1442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kedersha, N. , Stoecklin, G. , Ayodele, M. , Yacono, P. , Lykke‐Andersen, J. , Fitzler, M.J. , et al. (2005) Stress granules and processing bodies are dynamically linked sites of mRNP remodeling. J Cell Biol 169: 871–884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keyse, S.M. , and Emslie, E.A. (1992) Oxidative stress and heat shock induce a human gene encoding a protein‐tyrosine phosphatase. Nature 359: 644–647. [DOI] [PubMed] [Google Scholar]
- Kuo, L. , Godeke, G.J. , Raamsman, M.J. , Masters, P.S. , and Rottier, P.J. (2000) Retargeting of coronavirus by substitution of the spike glycoprotein ectodomain: crossing the host cell species barrier. J Virol 74: 1393–1406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kyuwa, S. , Cohen, M. , Nelson, G. , Tahara, S.M. , and Stohlman, S.A. (1994) Modulation of cellular macromolecular synthesis by coronavirus: implication for pathogenesis. J Virol 68: 6815–6819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lai, M.M. , Brayton, P.R. , Armen, R.C. , Patton, C.D. , Pugh, C. , and Stohlman, S.A. (1981) Mouse hepatitis virus A59: mRNA structure and genetic localization of the sequence divergence from hepatotropic strain MHV‐3. J Virol 39: 823–834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Law, A.H. , Lee, D.C. , Cheung, B.K. , Yim, H.C. , and Lau, A.S. (2007) Role for nonstructural protein 1 of severe acute respiratory syndrome coronavirus in chemokine dysregulation. J Virol 81: 416–422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leong, W.F. , Tan, H.C. , Ooi, E.E. , Koh, D.R. , and Chow, V.T. (2005) Microarray and real‐time RT‐PCR analyses of differential human gene expression patterns induced by severe acute respiratory syndrome (SARS) coronavirus infection of Vero cells. Microbes Infect 7: 248–259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, W. , Li, Y. , Kedersha, N. , Anderson, P. , Emara, M. , Swiderek, K.M. , et al. (2002) Cell proteins TIA‐1 and TIAR interact with the 3′ stem‐loop of the West Nile virus complementary minus‐strand RNA and facilitate virus replication. J Virol 76: 11989–12000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu, J. , Rivas, F.V. , Wohlschlegel, J. , Yates, J.R. , 3rd, Parker, R. , and Hannon, G.J. (2005) A role for the P‐body component GW182 in microRNA function. Nat Cell Biol 7: 1261–1266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu, L. , Han, A.P. , and Chen, J.J. (2001) Translation initiation control by heme‐regulated eukaryotic initiation factor 2 alpha kinase in erythroid cells under cytoplasmic stresses. Mol Cell Biol 21: 7971–7980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lyles, D.S. (2000) Cytopathogenesis and inhibition of host gene expression by RNA viruses. Microbiol Mol Biol Rev 64: 709–724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma, Y. , and Hendershot, L.M. (2004) Herp is dually regulated by both the endoplasmic reticulum stress‐specific branch of the unfolded protein response and a branch that is shared with other cellular stress pathways. J Biol Chem 279: 13792–13799. [DOI] [PubMed] [Google Scholar]
- McEwen, E. , Kedersha, N. , Song, B. , Scheuner, D. , Gilks, N. , Han, A. , et al. (2005) Heme‐regulated inhibitor kinase‐mediated phosphorylation of eukaryotic translation initiation factor 2 inhibits translation, induces stress granule formation, and mediates survival upon arsenite exposure. J Biol Chem 280: 16925–16933. [DOI] [PubMed] [Google Scholar]
- McInerney, G.M. , Kedersha, N.L. , Kaufman, R.J. , Anderson, P. , and Liljestrom, P. (2005) Importance of eIF2alpha phosphorylation and stress granule assembly in alphavirus translation regulation. Mol Biol Cell 16: 3753–3763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pasternak, A.O. , Spaan, W.J. , and Snijder, E.J. (2006) Nidovirus transcription: how to make sense…? J Gen Virol 87: 1403–1421. [DOI] [PubMed] [Google Scholar]
- Van De Peppel, J. , Kemmeren, P. , Van Bakel, H. , Radonjic, M. , Van Leenen, D. , and Holstege, F.C. (2003) Monitoring global messenger RNA changes in externally controlled microarray experiments. EMBO Rep 4: 387–393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robb, J.A. , and Bond, C.W. (1979) Pathogenic murine coronaviruses. I. Characterization of biological behavior in vitro and virus‐specific intracellular RNA of strongly neurotropic JHMV and weakly neurotropic A59V viruses. Virology 94: 352–370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roepman, P. , Wessels, L.F. , Kettelarij, N. , Kemmeren, P. , Miles, A.J. , Lijnzaad, P. , et al. (2005) An expression profile for diagnosis of lymph node metastases from primary head and neck squamous cell carcinomas. Nat Genet 37: 182–186. [DOI] [PubMed] [Google Scholar]
- Rottier, P.J. , Horzinek, M.C. , and Van Der Zeijst, B.A. (1981) Viral protein synthesis in mouse hepatitis virus strain A59‐infected cells: effect of tunicamycin. J Virol 40: 350–357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sawicki, S.G. , and Sawicki, D.L. (2005) Coronavirus transcription: a perspective. Curr Top Microbiol Immunol 287: 31–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scheuner, D. , Song, B. , McEwen, E. , Liu, C. , Laybutt, R. , Gillespie, P. , et al. (2001) Translational control is required for the unfolded protein response and in vivo glucose homeostasis. Mol Cell 7: 1165–1176. [DOI] [PubMed] [Google Scholar]
- Schneider, R.J. , and Mohr, I. (2003) Translation initiation and viral tricks. Trends Biochem Sci 28: 130–136. [DOI] [PubMed] [Google Scholar]
- Scorsone, K.A. , Panniers, R. , Rowlands, A.G. , and Henshaw, E.C. (1987) Phosphorylation of eukaryotic initiation factor 2 during physiological stresses which affect protein synthesis. J Biol Chem 262: 14538–14543. [PubMed] [Google Scholar]
- Siddell, S. , Wege, H. , Barthel, A. , and Ter Meulen, V. (1981) Intracellular protein synthesis and the in vitro translation of coronavirus JHM mRNA. Adv Exp Med Biol 142: 193–207. [DOI] [PubMed] [Google Scholar]
- Smith, J.A. , Schmechel, S.C. , Raghavan, A. , Abelson, M. , Reilly, C. , Katze, M.G. , et al. (2006) Reovirus induces and benefits from an integrated cellular stress response. J Virol 80: 2019–2033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Snijder, E.J. , Bredenbeek, P.J. , Dobbe, J.C. , Thiel, V. , Ziebuhr, J. , Poon, L.L. , et al. (2003) Unique and conserved features of genome and proteome of SARS‐coronavirus, an early split‐off from the coronavirus group 2 lineage. J Mol Biol 331: 991–1004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tahara, S. , Bergmann, C. , Nelson, G. , Anthony, R. , Dietlin, T. , Kyuwa, S. , and Stohlman, S. (1993) Effects of mouse hepatitis virus infection on host cell metabolism. Adv Exp Med Biol 342: 111–116. [DOI] [PubMed] [Google Scholar]
- Tahara, S.M. , Dietlin, T.A. , Bergmann, C.C. , Nelson, G.W. , Kyuwa, S. , Anthony, R.P. , and Stohlman, S.A. (1994) Coronavirus translational regulation: leader affects mRNA efficiency. Virology 202: 621–630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tebo, J.S. , Frevel, M. , Khabar, K.S. , Williams, B.R. , and Hamilton, T.A. (2003) Heterogeneity in control of mRNA stability by AU‐rich elements. J Biol Chem 278: 12085–12093. [DOI] [PubMed] [Google Scholar]
- Teixeira, D. , Sheth, U. , Valencia‐Sanchez, M.A. , Brengues, M. , and Parker, R. (2005) Processing bodies require RNA for assembly and contain nontranslating mRNAs. RNA 11: 371–382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tusher, V.G. , Tibshirani, R. , and Chu, G. (2001) Significance analysis of microarrays applied to the ionizing radiation response. Proc Natl Acad Sci USA 98: 5116–5121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Versteeg, G.A. , Slobodskaya, O. , and Spaan, W.J. (2006) Transcriptional profiling of acute cytopathic murine hepatitis virus infection in fibroblast‐like cells. J Gen Virol 87: 1961–1975. [DOI] [PubMed] [Google Scholar]
- Woo, P.C. , Lau, S.K. , Chu, C.M. , Chan, K.H. , Tsoi, H.W. , Huang, Y. , et al. (2005) Characterization and complete genome sequence of a novel coronavirus, coronavirus HKU1, from patients with pneumonia. J Virol 79: 884–895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang, Y.H. , Dudoit, S. , Luu, P. , Lin, D.M. , Peng, V. , Ngai, J. , and Speed, T.P. (2002) Normalization for cDNA microarray data: a robust composite method addressing single and multiple slide systematic variation. Nucleic Acids Res 30: e15. [DOI] [PMC free article] [PubMed] [Google Scholar]