

Abstract
Identifying single amino acid variants (SAAVs) in cancer is critical for precision oncology. Several advanced algorithms are now available to identify SAAVs, but attempts to combine different algorithms and optimize them on large data sets to achieve a more comprehensive coverage of SAAVs have not been implemented. Herein, we report an expanded detection of SAAVs in the PANC-1 cell line using three different strategies, which results in the identification of 540 SAAVs in the mass spectrometry data. Among the set of 540 SAAVs, 79 are evaluated as deleterious SAAVs based on analysis using the novel AssVar software in which one of the driver mutations found in each protein of KRAS, TP53, and SLC37A4 is further validated using independent selected reaction monitoring (SRM) analysis. Our study represents the most comprehensive discovery of SAAVs to date and the first large-scale detection of deleterious SAAVs in the PANC-1 cell line. This work may serve as the basis for future research in pancreatic cancer and personal immunotherapy and treatment.
Keywords: single amino acid variant, deleterious mutation, LC-MS/MS, LC-SRM, PRISM-SRM, PANC-1 cell line, KRAS, TP53
Graphical Abstract
INTRODUCTION
Cancer development is caused by the loss of control of cell proliferation due to the accumulation of gene mutations. Thousands of mutations in a single tissue sample and in large cohorts have been detected at the nucleic acid level with next-generation sequencing.1,2 These findings have been achieved due to the advances in polymerase chain reaction (PCR) methods for DNA or RNA sequencing. However, not all mutations at the nucleic acid level affect the final protein sequence due to the existence of DNA and RNA repair or because they are not translated.3−5 Proteins perform most of the work in cells and are required for the structure, function, and regulation of the biology, which provide more biologically relevant information on the current state of the phenotype.6 In addition, the correlation between mRNA and protein levels is insufficient to predict protein abundance levels based on mRNA expression levels.7 In comparison with mutations detected at the nucleic acid level, the direct detection of single amino acid variants (SAAVs) at the proteome level will be extremely important to understand tumorigenesis and progression and eventually could provide potential targets for immunotherapy for personalized medicine.
Various efforts have contributed to the systematic discovery of SAAVs based on advances in state-of-the-art mass spectrometry. Zhang et al. identified 796 SAAVs among 86 human colon and rectal cancers for which RNA-seq data were available.7 Su et al. identified and quantified 154 SAAVs in a human brain proteome using mass spectrometry data and a de novo sequencing algorithm instead of depending on general single-nucleotide polymorphism (SNP) databases.8 Lichti et al. identified SAAVs in glioma stem-cell-derived chromosome 19 and validated three SAAVs at the protein level by selected reaction monitoring (SRM).9 Alternative to the genomic variant database or transcript data for customized database construction, an error-tolerant peptide search engine such as BICEPS for identifying SAAVs based on the standard UniProt database was developed by Giese et al. This approach has the advantage that the search space is not limited to known SAAVs.10 Using the error-tolerant search strategy, global quantification of the SAAVs in hepatocellular carcinoma (HCC) was obtained by integrating the stable isotope dimethyl labeling with a variant-associated database where 282 unique SAAVs sites were quantified between HCC and normal liver tissues.11 Work has been done on the detection of SAAVs in MCF-7 breast cancer cell line subpopulations and also quantitative analysis of SAAVs associated with pancreatic cancer in serum.12,13 In addition, in recent work, 79 SAAVs were detected from as few as nine PANC-1 cells using sample fractionation, TMT multiplexing, and a carrier/reference strategy.14
Most of the current strategies for the detection of SAAVs rely on customized database construction and novel database-searching algorithms.15 An important aspect in applying these strategies for SAAV discovery is filtering out the false positives from the potential SAAV results.16 In recent work, SAAVs were filtered for quality control using the SAVControl method, which detects and removes false positives to reduce the false discovery rate (FDR) for variant peptide identifications and SAAV sites with unrestrictive mass shift relocalization.14,17
There has to date been no effort to compare different strategies for SAAV detection and the overlap of the SAAVs detected in the different strategies. In addition, some studies focus on the identification of driver mutations based on large-scale exome sequencing data such as the Cancer Genome Atlas (TCGA) project and the International Cancer Genome Consortium (ICGC).18,19 However, for large-scale SAAV detection, the prediction of the impact on cancer development due to proteins with SAAVs has also not been investigated. It is critical to confirm the existence of deleterious SAAVs in cancer patients to aid in the selection of treatment for cancer patients. As an example, the drugs panitumumab and cetuximab are used to treat advanced colorectal cancers, but cancer patients that have KRAS with SAAVs will not benefit from these two drugs.20−22
There are two biological classes of somatic mutations that occur in all dividing cells: “driver” mutations and “passenger” mutations.23 Driver mutations confer a growth advantage for cancer cells during tissue invasion and metastasis, angiogenesis, and evasion of apoptosis. Driver mutations, therefore, are positively selected during the evolution of cancer. By definition, these mutant genes are cancer genes. Conversely, passenger mutations are biologically neutral and, therefore, these mutations do not contribute to the growth advantage where passenger mutations have not been subjected to selection.24 A current challenge involves the comprehensive identification of mutations in conjunction with an accurate classification of the mutations into driver mutations and passenger mutations.23
In this work, we have completed the comprehensive detection of SAAVs on a PANC-1 cell line by three complementary strategies. The full set of 540 detected SAAVs was then used to predict the impact on cancer development for the combined set of SAAVs using the AssVar server. Of these, 70 SAAVs were found to overlap among the three detection strategies applied to PANC-1 cells. Of critical importance, 79 SAAVs were predicted as deleterious mutations using the AssVar online server and one of the driver mutations found in each protein KRAS, TP53, and SLC37A4 was further validated using independent SRM analysis. The current comprehensive detection of SAAVs in the PANC-1 cell line; the prediction of a large number of deleterious SAAVs; and the validation of SAAVs in KRAS, TP53, and other proteins will be essential in diagnosing cancer, monitoring the effects of treatment for some clinical anticancer drugs, early detection and prognosis, and the development of new drugs for specific targeted SAAVs sites.
MATERIALS AND METHODS
Cell Culture
The PANC-1 cell line was purchased from the American Type Culture Collection (ATCC). The cell culture was described in a previous study with a minor adjustment.14 Briefly, the PANC-1 cells were cultured in a dish (100 mm × 20 mm) with Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% (v/v) fetal bovine serum (FBS) and 1% (v/v) antibiotic–antimycotic. PANC-1 cells were cultured in a humidified atmosphere at 37 °C with 5% CO2. The cells were harvested using trypsin while in the exponential growth phase. Harvested cells were collected in a 15 mL tube and centrifuged at 100g for 5 min. After removing the supernatant, the cell pellets were washed with 10 mL of 1× phosphate-buffered saline (PBS) prior to centrifugation at 100g for 5 min. The washing step was repeated five times, and the resulting cell pellets were stored at −80 °C until used for proteomic analysis.
Protein Extraction and Sample Fractionation
Different numbers of cells, ranging from 9 cells to 1 × 106 cells, were subjected to protein extraction and digestion. For samples with a small number of cells, lysis was performed by sonication, as described in our previous work,14 while for a bulk cell scale, lysis was accomplished by incubation with 200 μL of radioimmunoprecipitation assay buffer (RIPA) with protease inhibitors on ice for 15 min with periodic pipetting. To improve the protein yield, the lysate was sonicated for 30 s at 50% pulse on ice. The cell lysate was then centrifuged at 13 000g for 5 min at 4 °C, and the supernatant was collected into an Eppendorf tube. Buffer exchange was conducted using a 3 K MWCO centrifugal filter (EMD Millipore) with triethylammonium bicarbonate (TEAB) buffer. The lysate was then aliquoted and stored at −80 °C for further analysis.
Protein reduction was conducted with 0.5 mM tris(2-carboxyethyl)phosphine (TCEP) in 100 mM TEAB (pH 8.5) solution for 1 h, followed by alkylation in 2 mM iodoacetamide for 30 min under dark conditions. Sequencing-grade trypsin (Promega) was added into sample solution [1:30 trypsin-to-protein ratio (w/w)] with 0.05% ProteaseMax (Promega) to improve the protein digestion efficiency at 37 °C for 12 h. After the digestion, the samples were dried in a SpeedVac and then labeled with TMT 11-plex reagent following the manufacturer’s instructions. The advantage of using an isobaric labeling strategy is not only to achieve high-resolution tandem mass spectra (MS/MS) to improve the accuracy of SAAV identification but also to detect different samples at the same time. After quenching the remaining TMT reagent using ammonium hydroxide solution, the same sample set was pooled and dried using a SpeedVac.
The sample mixture was fractionated using two different methods. Half of the sample was fractionated with the Pierce high-pH reversed-phase peptide fractionation kit (Thermo Fisher Scientific) into 10 fractions for analysis, as previously described in detail.14 Then, another one-fourth of the sample was fractionated using high-pH reversed-phase liquid chromatography (RPLC) and 11 fractions were obtained. The last one-fourth of the sample was subjected to low-pH nano-RPLC for fractionation. An EASY nanoLC 1200 (Thermo Fisher Scientific) equipped with a capillary column (75 μm i.d. × 50 cm length, C18, 2 μm bead, 100 Å pore, Thermo Fisher Scientific) was used for fractionation. Mobile phase A contained 2% (v/v) acetonitrile (ACN), 98% (v/v) H2O, and 0.1% (v/v) formic acid (FA). Mobile phase B contained 80% (v/v) ACN, 20% (v/v) H2O, and 0.1% (v/v) FA. The flow rate was set at 200 nL/min. The gradient was set as follows: from 8 to 30% (v/v) B in 40 min, from 30 to 50% (v/v) B in 30 min, from 50 to 80% (v/v) B in 10 min and keeping at 80% (v/v) B for 10 min. The first fraction collection started from the sample loading and continued for 15 min from the initiation of the gradient. The rest of the fraction was collected every 4 min, and the last fraction was collected from 75 min to the end of the gradient. In total, 17 fractions were collected. The eluates of each fraction were deposited into an Eppendorf tube (0.6 mL) containing 2 μL of 50 mM NH4HCO3 (pH 8.5). The final volume of each fraction was around 3 μL, and the final pH was about 8.0. The first and last fractions were then lyophilized and re-suspended with 3 μL of 50 mM NH4HCO3 (pH 8.0). After desalting with C18 ZipTip (Thermo Fisher Scientific), the samples were analyzed by the proteomics workflow.
Mass Spectrometry Analysis
LC-MS/MS data were acquired on three different instruments. These include an Orbitrap Q-Exactive HF mass spectrometer coupled with high-performance liquid chromatography (HPLC) or capillary zone electrophoresis (CZE) for the sample fractionated by low-pH nano-RPLC and an Orbitrap Fusion mass spectrometer coupled with HPLC for the sample fractionated by the Pierce high-pH reversed-phase peptide fractionation kit (Thermo Fisher Scientific).
For the 17 fractions collected using low-pH nano-RPLC, each fraction was then analyzed by CZE-MS/MS, as previously described in detail.25 Briefly, the sample injection vials of CZE were treated with a BSA solution to reduce nonspecific adsorption of peptides on the inner wall of the vials. A 100 cm linear polyacrylamide (LPA)-coated capillary (50/360 μm i.d./o.d.) with an etched end by hydrofluoric acid was used for CZE separation.26 The commercialized electrokinetically pumped sheath flow CE–MS interface (CMP scientific, Brooklyn, NY) was used to couple CZE to MS.27 An ECE-001 autosampler (CMP scientific) was used for the automated CZE operation. The background electrolyte (BGE) was 5% (v/v) acetic acid. For sample loading, 5 psi was first applied for 90 s so that approximately 500 nL of the sample was injected for CZE-MS/MS. The capillary distal end was then moved into BGE, and 30 kV was applied for 120 min for CZE separation. During the last 5 min of separation, 15 psi was applied to rinse the capillary.
The 11 fractions collected using high-pH HPLC were analyzed by low-pH nano-RPLC-MS/MS. An EASY nanoLC 1200 (Thermo Fisher Scientific) equipped with a capillary column (75 μm i.d. × 50 cm length, C18, 2 μm bead, 100 Å pore, Thermo Fisher Scientific) was coupled with an Orbitrap Q-Exactive HF (Thermo Fisher Scientific) for low-pH nano-RPLC-MS/MS analysis. Mobile phase A contained 2% (v/v) acetonitrile (ACN), 98% (v/v) H2O, and 0.1% (v/v) formic acid (FA). Mobile phase B contained 80% (v/v) ACN, 20% (v/v) H2O, and 0.1% (v/v) FA. The flow rate was set at 200 nL/min. The gradient for separation was set as follows: from 8 to 30% (v/v) B in 55 min, from 30 to 50% (v/v) B in 45 min, from 50 to 80% (v/v) B in 15 min, and keeping at 80% (v/v) B for 5 min.
For CZE-ESI-MS/MS and RPLC-MS/MS analysis on the Q-Exactive, the parameters were set according to a previous study with some modifications.25 The data acquisition was performed in data-dependent mode using the software Xcalibur v2.3 (Thermo Fisher Scientific). Full MS scans were acquired in the Orbitrap mass analyzer over m/z 400–1500 range with a resolution of 60 K at m/z 200. The AGC target was set at 3e6. The quadrupole isolation window was set at 4 m/z. The top 10 most intense peaks with charge state ≥2 were fragmented in the HCD collision cell with a normalized collision energy of 30%. The tandem mass spectra were recorded in the Orbitrap mass analyzer with a resolution of 35 K at m/z 200. The AGC target was set at 1e5. The maximum ion injection time was set at 50 ms for full MS scans and 200 ms for MS/MS mass spectra. The ion selection intensity threshold was set at 1e4, and the dynamic exclusion was set at 30 s.
For the samples fractionated by the high-pH reversed-phase peptide fractionation kit, an Orbitrap Fusion mass spectrometer was used, coupled with an EASY 1000 nano UHPLC system (Thermo Fisher Scientific) and an Acclaim PepMap 100, 75 μm × 2 cm trap with an Acclaim PepMap RSLC, 75 μm × 25 cm column (Dionex). The samples were analyzed using a 90 min gradient from 4 to 30% acetonitrile with 0.1% FA. The mass spectrometer was operated in the data-dependent mode to acquire the mass spectral data using the software Xcalibur v2.3 (Thermo Fisher Scientific). The electrospray ionization (ESI) spray voltage was set as positive ion mode at 2500 V. A full mass scan (m/z 350–1600) was performed, and the most intense ions in the full scan were chosen for MS/MS analysis. The normalized collision energy was set at 40% for higher-energy collision-induced dissociation (HCD) fragmentation. The maximum injection times were set at 100 and 250 ms for MS1 and MS/MS, respectively. The Orbitrap resolutions were set at 120 and 60 K for MS1 and MS2 spectra, respectively. For filter dynamic exclusion, the repeat count was set to 1 and the exclusion duration was set to 60 s. Both low and high mass tolerances were set at 10 ppm. The mass spectrometry proteomics data have been deposited at the public repository ProteomeXchange Consortium28 using the PRIDE29 partner repository with the data set identifier PXD017449.
Database Searching on Proteome Discoverer 1.4
All RAW data were analyzed by database searching using the SEQUEST HT algorithm on Proteome Discoverer 1.4 (PD 1.4). The Swiss-CanSAAVs database that contains 87 733 amino acid variant sequences from 73,910 UniProtKB/Swiss-Prot canonical proteins was used.30 The carbamidomethylation of cysteine was set as a fixed modification, and N-terminal TMT, TMT of lysine, and methionine oxidation were set as variable modifications. The tolerances for precursor ions and fragment ions were set as 10 ppm and 0.05 Da, respectively. A maximum of two missed cleavages and the shortest peptide length set at six amino acids were allowed. The identified results were filtered using high confidence at 1% peptide-level false discovery rate (FDR). Protein identification is based on at least two confidently identified peptides. The resulting peptide data were exported into the XML-formatted file from PD 1.4, and all potential peptides with SAAVs derived from the same group sample were combined. A large percentage of false-positive SAAV peptides are often observed due to the existence of missed cleavage sites surrounding the site of variation during database construction. A manual check of each SAAV site was further performed to remove false SAAVs. Only the shorter peptide sequence with the SAAV sites was selected due to up to two tryptic missed cleavages in the database search setting.
Customized Database Construction Based on RNA-seq and Database Searching
The PANC-1 customized database was derived from RNA-seq data from GEO SRX5053565. This data was processed by proteogenomics software named Spritz (https://github.com/smith-chem-wisc/Spritz). This software analyzes the RNA-seq data from beginning to end, starting using fasterq-dump31 to download the RNA-Seq data, skewer32 to trim and filter the reads, STAR33 to align the RNA sequences to the reference genome, and GATK34−36 to call variants from the alignments. Then, a custom version of SnpEff37 is used to annotate variants and produce a proteogenomic database containing SAAVs that is annotated with posttranslational modifications (PTMs) from UniProt to allow detection of both amino acid variations and PTMs. MetaMorpheus38 (https://github.com/smith-chemwisc/MetaMorpheus) was used to search this database.
SAVControl Database Searching
The database searching based on SAVControl has been described in previous studies.14,39 The CanProVar 2.0 database was downloaded from http://canprovar2.zhang-lab.org/, which includes 65 963 distinct human cancer protein variants and 825 106 coding SNPs from dbSNP.40 Corresponding nonvariant proteins are from the Ensembl database (Homo sapiens, v53). A decoy database derived from the reversed sequences of the same size was mapped into the protein sequence database for false discovery rate (FDR) estimation. The RAW files were searched using the Mascot algorithm with the following parameter settings: (1) carbamidomethylation (+57.021 Da) at cysteine and TMT reaction with the peptide N-terminus and ε-amino group of lysine (+229.163 Da) were set as static modifications; (2) oxidation (+15.995 Da) at methionine was set as a dynamic modification; (3) precursor ion mass tolerance was set to 10 ppm, and fragment ion mass tolerance was set to 0.05 Da; (4) trypsin was set as the enzyme, and the maximum missed trypsin cleavage sites was set as 2; (5) charge status was set as + 2, + 3, and + 4; (6) the minimum tryptic peptide length was set at five amino acids, and unrestricted peptide-level FDR was enabled. The unrestricted peptide-level FDR setting results in extremely exaggerated high coverage of peptide identification, which includes not only genuine SAAVs but also false-positive SAAVs. False-positive peptides with SAAVs were removed by further quality control via SAVControl.
Prediction of the SAAV Effect on Protein Function Using the Online AssVar Server
AssVar has been developed to quantitatively assess the impact of SAAVs on tumor development (https://zhanglab.ccmb.med.umich.edu/AssVar/), which has shown an advantage over other similar pipelines with a higher Matthews correlation coefficient.41 A total of 540 SAAVs were submitted for analysis. The default cutoff impact score was set at 0.57, which means that if the value of the impact score for SAAVs is greater than 0.57, the variant will be assigned as a driver mutation. Otherwise, the SAAVs are assigned as passenger mutations without a deleterious feature in the protein that is related to cancer development.
Validation of Three SAAVs by the SRM Assay
Three SAAVs derived from proteins KRAS, TP53, and SLC37A4 were selected for validation by the SRM assay. The peptide pairs were LVVVGAGGVGK (canonical peptide) and LVVVGADGVGK (variant peptide G12D) for KRAS, FVSGVLSGQMSAR (canonical peptide) and FVSGVLSD QMSAR (variant peptide G88D) for SLC37A4, and NSFEVRVCACPGR (canonical peptide) and NSFEVHVCACPGR (variant peptide R273H) for TP53. Heavy-isotope-labeled variant peptides and one canonical peptide LVVVGAGGVGK with the 13C/15N labeling on the C-terminal K or R were synthesized by New England Peptide (Gardner, MA). For each peptide, three or four best transitions with a higher SRM response were selected for reliable detection and quantification. The standard peptides were spiked into the tryptic digests derived from the PANC-1 cell lysate with final concentrations at 2 fmol/μL for the standard and 1 μg/μL for the sample.
A tiered approach was used for sensitive SRM quantification of the three variant peptides and one canonical peptide. Regular LC-SRM was first used for the simultaneous quantification of all of the four peptides. For the peptide FVSGVLSDQMSAR from SLC37A4, which cannot be reliably detected and quantified by LC-SRM, PRISM-SRM was employed for sensitive targeted quantification because it can provide ~100-fold higher detection sensitivity.42
Regular LC-SRM.
The LC-SRM analysis was performed using a nanoACQUITY UPLC (Waters, Milford, MA) coupled to a TSQ Vantage triple quadrupole mass spectrometer (Thermo Scientific, San Jose, CA). The nanoACQUITY UPLC ethylene bridged hybrid (BEH) 1.7 μm C18 column (75 μm i.d. × 20 cm) was connected to a chemically etched 20 μm inner diameter fused silica electrospray emitter via a stainless steel union. Mobile phase A (0.1% FA) and mobile phase B (90% acetonitrile in 0.1% FA) were used with a linear gradient: 5–20% B for 26 min, 20–25% B for 10 min, 25–40% B for 8 min, 40–95% B for 1 min, and at 95% B for 7 min. The flow rate was set as 350 nL/min. The analytical column was reconditioned at 99.5% mobile phase A for 8 min. Approximately 1 μL of the peptide sample was directly loaded onto the BEH C18 column from the vial (Waters, Milford, MA) without using a trapping column.
The TSQ Vantage mass spectrometer was operated with the following parameters: ion spray voltages (2400 ± 100 V), capillary offset voltage (35 V), skimmer offset voltage (−5 V), and capillary inlet temperature (220 °C). The tube lens offset voltage was obtained from automatic tuning and calibration without any further optimization. A scan width of 0.002 m/z and a dwell time of 20 ms were set for all SRM transitions.
PRISM-SRM.
The PRISM-SRM approach has been previously described for the quantification of low-abundance proteins in human plasma or serum.42,43 Briefly, high-resolution reversed-phase capillary LC with pH 9 mobile phase was used as the first dimensional separation of peptides from trypsin-digested PANC-1 proteins. Following separation, the column eluent was automatically collected every minute into a 96-well plate during a ~100 min LC run, while on-line SRM monitoring of heavy internal standard peptides was performed on a small split stream of the flow. Intelligent selection (termed iSelection) of target peptide fractions was achieved based on the on-line SRM signal of internal standard peptides. Prior to peptide fraction collection, 27 μL of water was added to each well to minimize the excessive loss of peptides and to dilute the peptide fractions (~1:10) for LCSRM analysis. Following iSelection, the target peptide-containing fractions were subjected to LC-SRM measurements. A total of 15 μL of individual peptide fractions (total volume 30 μL) following PRISM was injected for LC separations followed by SRM analysis. The nanoACQUITY UPLC and TSQ Vantage were operated in the same manner as described above for regular LC-SRM analysis.
SRM Data Analysis.
Skyline software was employed for SRM data analysis.42,44 For each peptide, the best transition without matrix interference from co-eluting peptides was used for precise quantification. Two criteria were applied to determine the peak detection and integration: (1) the same retention time; (2) approximately the same relative SRM peak intensity ratios among multiple transitions between heavy peptide internal standards and endogenous (light) peptides.45 All data were manually inspected to confirm the correct peak detection and accurate integration. All RAW files generated on the TSQ Vantage mass spectrometer were imported into Skyline software, and the graphs of extracted ion chromatograms (XICs) to multiple transitions of the target peptides monitored were displayed.45
RESULTS AND DISCUSSION
Overview of the Workflow
We have established an integrated workflow for high-confidence global SAAV analysis that can minimize false positives and predict deleterious SAAVs and incorporate validation of SAAVs using SRM. The workflow is summarized in Figure 1. Two methods were applied in parallel for sample fractionation. The first was the low-pH nano-RPLC method, where a total of 60 HPLC fractions were collected and then combined into 11 fractions for LC-MS/MS and CZE-MS/MS analyses. The second was the high-pH RP fractionation method, where a total of 10 fractions were collected for LC-MS/MS analysis. The fractionated samples were separated using CZE or UHPLC prior to tandem mass spectrometry analysis. The sets of SAAVs derived from the same sample sets using different separation methods and CZE or UHPLC are presented in Supporting Information Table S1. In general, the HPLC method produced a greater depth of coverage than the CZE method for the same sample. A total of 13 sample sets were measured, which resulted in 139 RAW files for SAAV detection. Data were searched using three different algorithms, SEQUEST, Mascot incorporated with SAVControl, and MetaMorpheus,38 against three different databases, CanProVar 2.0, Swiss-CanSAAVs, and RNA-seq databases, respectively, as shown in Figure 1C. After data refinement including removal of false-positive SAAVs, all SAAVs with high confidence were evaluated by an online server AssVar to assess the impact on tumor development. One of the deleterious SAAVs found in each protein of KRAS, TP53, and SLC37A4 was validated by SRM.
Figure 1.
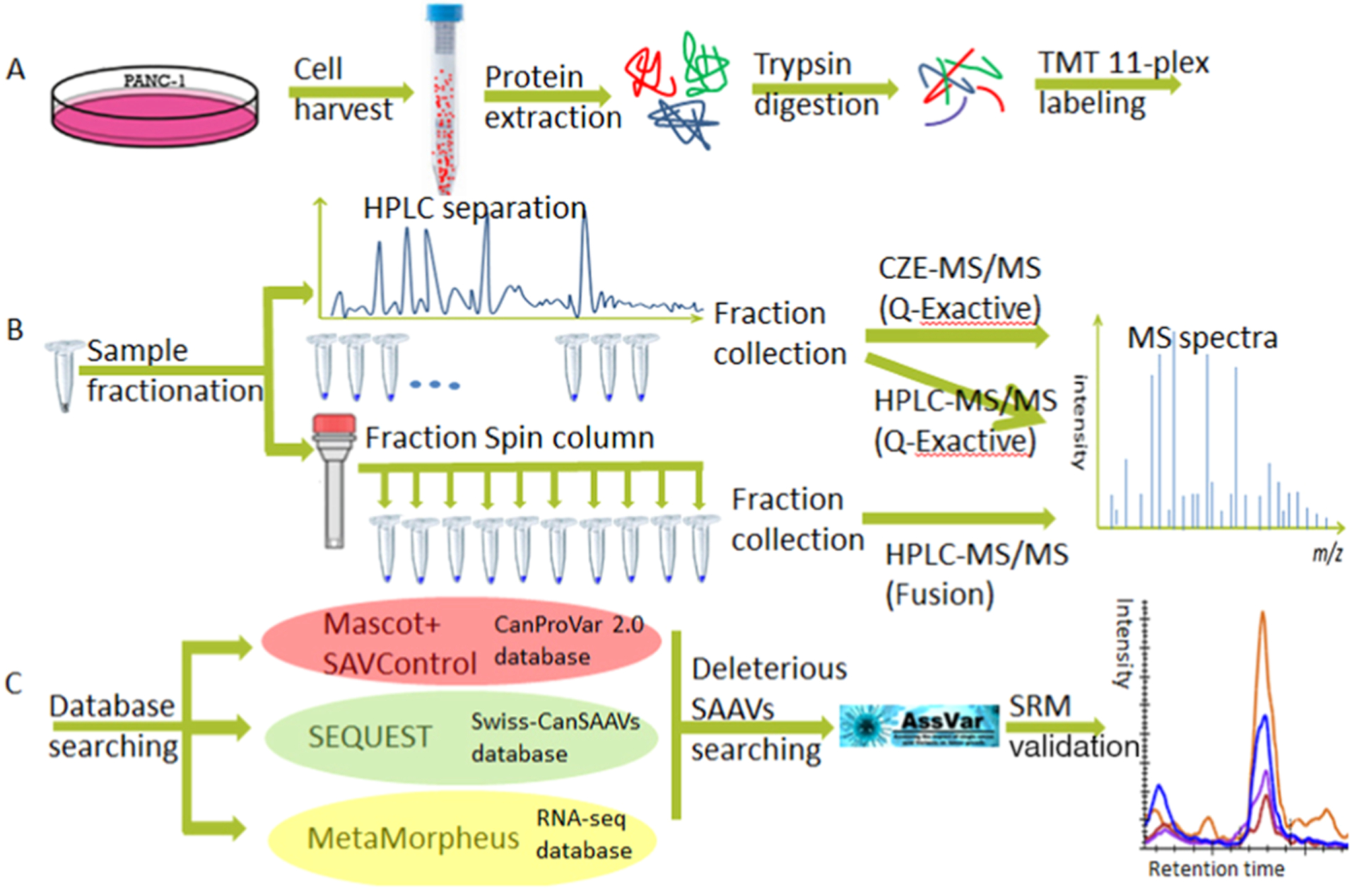
Overview of comprehensive detection of SAAVs in the PANC-1 cell line including three major steps of sample preparation: (A) sample fractionation, (B) MS/MS analysis, and (C) SAAV identification and validation.
SAAVs Derived from Different Strategies
To increase the coverage of SAAV detection, we sampled the tryptic digests of PANC-1 cells into 13 sets, which were subjected to fractionation and MS/MS analysis using different strategies. The sample information is shown in Table 1. Two sample sets were fractionated using low-pH nano-RPLC, while the remaining 11 sets were fractionated using the Pierce high-pH reversed-phase peptide fractionation kit. A total of 11 or 17 fractions were obtained from the low-pH nano-RPLC method and 10 fractions from the high-pH RP fractionation method. Fractions from the low-pH RP method were analyzed on the Oribtrap Q-Exactive HF, while fractions from the high-pH RP method were analyzed on the Orbitrap Fusion mass spectrometer. Due to the high workload for sample preparation and MS analysis, we performed the entire workflow at three different time points for 5, 2, and 6 sample sets. A total of 133 mass spectral runs were performed, and the related RAW files were acquired for subsequent database searching.
Table 1.
Sample Fractionation and Mass Spectrometry Analysis
| sample set | fractionation method | number of fractions | MS/MS analysis | number of runs per set |
|---|---|---|---|---|
| sets 1–5 | Pierce high-pH reversed-phase peptide fractionation kit | 9 | LC-MS/MS | 9 |
| sets 6–7 | low-pH nano-RPLC | 11 | CZE-MS/MS | 11 |
| 17 | LC-MS/MS | 17 | ||
| sets 8–13 | Pierce high-pH reversed-phase peptide fractionation kit | 10 | LC-MS/MS | 10 |
Three different search algorithms were employed for MS/MS database searching to identify SAAVs, i.e., SEQUEST algorithm in software Proteome Discoverer (PD) 1.4 against Swiss-CanSAAVs database, MetaMorpheus against the customized RNA-seq database, and Mascot algorithm coupled with SAVControl quality control. The results derived from the sample sets that were processed at the same time point were combined. The numbers of proteins and peptides containing SAAVs identified using the three different searching algorithms are listed in Table 2. A total of 418 SAAVs from 380 proteins were identified from all of the sample sets using the SEQUEST algorithm in Proteome Discoverer (PD) 1.4 software (Table 2). A total of 196 proteins and 221 peptides with SAAVs were identified using MetaMorpheus against the customized RNA-seq database. The number of proteins and peptides with SAAVs by this strategy is approximately half that obtained by the strategy using the SEQUEST algorithm in PD 1.4. The total number of proteins and peptides with SAAVs was 165 and 181, respectively, when using the Mascot algorithm coupled with SAVControl quality control. Among the three algorithms, the lowest number of proteins and peptides with SAAVs were identified based on the Mascot algorithm with SAVControl. Especially in sets 6–7, only 14 and 15 proteins and peptides with SAAVs were identified, respectively. When combining the results from the three searching algorithms, 540 variant peptides from 483 proteins were identified in total, which is the most comprehensive coverage of SAAVs to date.
Table 2.
Number of SAAVs Identified Using Three Different Searching Algorithmsa
| SEQUEST algorithm against Swiss-CanSAAVs database | MetaMorpheus against RNA-seq database | Mascot algorithm and SAVControl | ||||
|---|---|---|---|---|---|---|
| samples | proteins with SAAVs | peptides with SAAVs | proteins with SAAVs | peptides with SAAVs | proteins with SAAVs | peptides with SAAVs |
| sets 1–5 | 293 | 326 | 171 | 190 | 158 | 172 |
| sets 6–7 | 118 | 123 | 58 | 64 | 14 | 15 |
| sets 8–13 | 113 | 123 | 71 | 77 | 50 | 53 |
| total | 382 | 197 | 165 | |||
| percentage of all SAAVs (%) | 78.1 | 40.9 | 34.2 | |||
SEQUEST algorithm in software Proteome Discoverer (PD) 1.4 against Swiss-CanSAAVs database, MetaMorpheus against the customized RNA-seq database, and Mascot algorithm coupled with SAVControl quality control.
The SAAVs derived from low-pH RPLC fractionation followed by two separation methods for MS/MS analysis, i.e., CZE or UHPLC, are presented in Supporting Information Table S1. In general, the HPLC method produced a greater depth of coverage than the CZE method.
The numbers of proteins and peptides with SAAVs detected in three separate strategies are different. The largest number of SAAVs was detected in the PD strategy, while the lowest number of SAAVs was detected in the SAVControl strategy. A total of 382 of 483 (78.1%) SAAVs were detected in the PD strategy, 197 of 483 (40.9%) SAAVs in the RNA-seq strategy, and 181 of 483 (33.5%) in the SAVControl strategy (Table 2). There is an overlap of 70 SAAVs identified across the three search algorithms. We have manually checked the spectra from many of the SAAVs to confirm the accuracy of the SAAVs in different strategies (Supporting Information, Figure S1). As shown in Supporting Information Figure S1, the variant peptide DSMFGITVK from protein ITGA6 (A419) was detected in all the three different strategies, while the variant peptides ATEEQLK from protein ALB (K565E), AYLEGTCVEWLR from protein HLA-A (D185E), and AGLLIFASK from protein ARL5A (N125S) were detected in the SEQUEST strategy, the MetaMorpheus strategy, and the SAVContol strategy, respectively. There are some potential reasons why only a small percentage of the 540 SAAVs was identified from all three platforms. One reason is that the databases used for searching are different. This may be the main reason for such differences where we have found from our prior work on SAAVs searching that the use of RNA databases may result in different SAAVs depending on the source from which they were derived. The second reason for these differences is that the number of SAAVs derived from SEQUEST is the largest but may contain more false positives than the other two strategies. False-positive SAAVs may still exist, although we reduce the false positives using multiple optimized methods and manual checking. The third reason is that the search algorithm is different in the three strategies and yields complementary but different results. In addition, the canonical peptide sequence is predominant compared to variant peptides in most cases, so the SAAVs are difficult to detect and assign based on the algorithm.
SAAVs Filtering in Different Strategies
The SAAVs obtained were filtered in each of the different strategies. Among these strategies, the highest duplication or false positives existed in the PD strategy, while the lowest duplication and false-positive SAAVs were detected in the SAVControl strategy. Many more duplications and false-positive SAAVs were removed from the PD strategy where more SAAVs escaped from quality control based on FDR during the database searching, resulting in potential false positives. The smallest number of SAAVs was detected in three processing steps (original searching results, combination and duplication removal, and manual check) in the SAVControl strategy compared to the other two strategies, which showed that the database searching is stringent and the highest quality control works in this strategy (Figure 2). There is a competition that exists between deeper coverage and highly rigid quality control. More false-positive SAAVs resulted from loose quality control, while less positive SAAVs were lost in this strategy. Likewise, the highest rigid quality control results in a few false-positive SAAVs where more positive SAAVs resulted. A much higher quality result for single amino acid polymorphisms (SAPs) derived from a customized database compared to those identified using a larger aggregate database has also been shown by Sheynkman et al. using a Jurkat cell line.46
Figure 2.

Filtering of SAAVs in different strategies. (A) Number of potential SAAVs in SAVControl strategy; (B) number of potential SAAVs in PD strategy; and (C) number of potential SAAVs in the RNA-seq strategy.
Overlap between Different Database Searching
The SAAVs identified from three different strategies shared a varying overlap (Figure 3). A total of 70 SAAVs were detected that are common to the three strategies, which makes them the most reliable variants detected in the PANC-1 cell line. The SAVcontrol and RNA-seq strategies shared 86 SAAVs, the SAVControl and PD strategies shared 111 SAAVs, and 119 SAAVs were shared by the RNA-seq and PD strategies. Compared to that of the PD strategy, a smaller number of SAAVs was detected in the RNA-seq strategy. Nevertheless, the customized protein sequence database could significantly improve the sensitivity of SAAV detection and reduce ambiguity in peptide identification. In addition, multiple modifications were considered in the MetaMorpheus strategy, which also increase the accuracy of the SAAV searching. A total of 83 novel SAAVs were identified in the RNA-seq strategy compared to SAAVs detected in the PD strategy. Customized databases derived from RNA-seq data can improve the efficiency and accuracy of identifying splice variants. Also, for the comparison between the PD and SAVControl strategies, a smaller number of SAAVs, i.e., 70 novel SAAVs, was detected in the SAVControl strategy. The integrated transfer FDR control, unrestricted mass shift relocation, and introduction of alternative interpretations could also significantly increase the sensitivity of SAAV detection and reduce ambiguity in peptide identification.39 Compared to that of the SEQUEST strategy, a high FDR in global searching versus a strict FDR in variant peptide assignment in the SAVControl strategy showed its advantage for SAAV detection.
Figure 3.
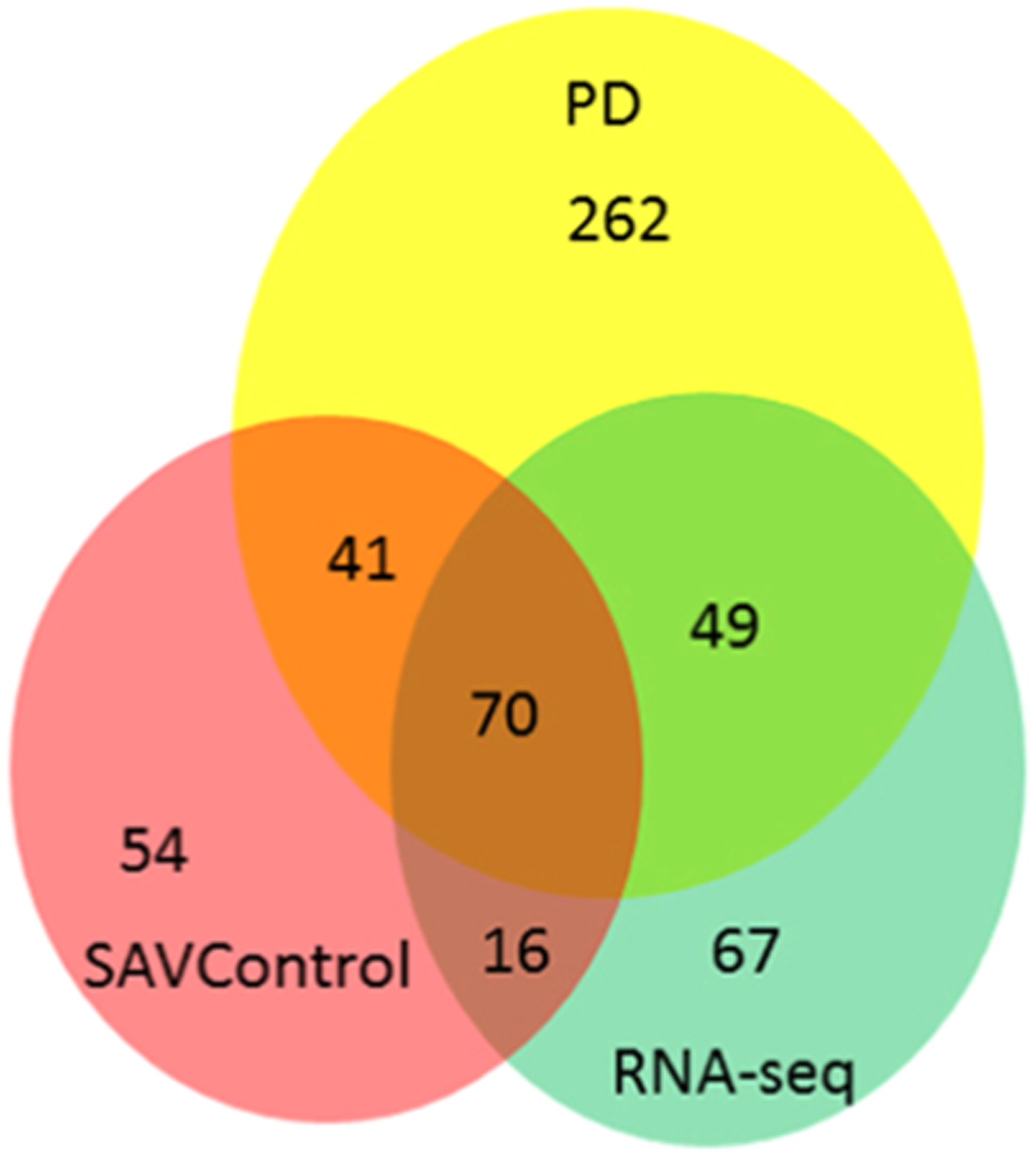
Venn diagram picture showing the SAAV identification and overlap among the three strategies. The yellow circle represents the Proteome Discoverer database searching. The green circle represents the RNA-seq database searching, and the red circle represents the SAVControl strategy.
N-Glycosylation Site Change
We also checked for the potential loss and gain of glycosylation sites due to SAAVs. There were nine potential N-glycosylation sites detected that change due to SAAVs; three peptides with SAAVs gained potential N-glycosylation sites, while six peptides lost the potential N-glycosylation sites (Table 3). Among these proteins with the gain or loss of N-glycosylation, CEND1 participates in cell cycle control.47 Intracellular and extracellular ANXA1 plays a role in stimulating pancreatic cancer cell migration and invasion.48 The mutation of ROCK2 leads to increased motility and adhesion in cancer cells.49 A specific gain or loss of glycosylation sites in human glycosylation has been associated with diseases.50 Mazumder et al. revealed that there are 259 unique variations with the loss of N-glycoslation by mapping the variation data to the UniprotKB human proteome.51 In our current study, we detected a small number of N-glycosylation sites with gain or loss compared to the study of Mazumder.
Table 3.
Nine Potential N-Glycosylation Sites Change due to SAAVsa
| peptide | protein name | accession number | gene name | variant | N-glycosylation change |
|---|---|---|---|---|---|
| ADPALLNDHSNLKPAPTVPSSPDATPEPK | cell cycle exit and neuronal differentiation protein 1 | Q8N111 | CEND1 | N74D | − |
| ASSSILINESEPTTNIQIR | NSFL1 cofactor p47 | Q9UNZ2 | NSFL1C | D290N | + |
| DIDLSCGSGSSK | GC-rich sequence DNA-binding factor 2 | P16383 | GCFC2 | N249S | − |
| EILQIMDK | polyribonucleotide nucleotidyltransferase 1, mitochondrial | Q8TCS8 | PNPT1 | N590D | − |
| ENDSIQSR | Rho-associated protein kinase 2 | O75116 | ROCK2 | T431N | + |
| GAPMDPNESPAAPEAALPK | cyclic GMP-AMP synthase | Q8N884 | MB21D1 | T35N | + |
| GGPGSAVSPYPTFDPSSDVAALH | annexin A1 | P04083 | ANXA1 | N43D | − |
| IIGELSK | GTP-binding protein 10 | A4D1E9 | GTPBP10 | N110S | − |
| VIDDTDITR | keratin, type I cytoskeletal 18 | P05783 | KRT18 | N193D | − |
+ represents gain, while − represents loss.
Prediction of Deleterious SAAVs
A total of 540 SAAVs were searched using the AssVar server to predict the impact of the SAAVs detected in each protein. If the SAAV was a driver mutation, the SAAV was considered as a deleterious mutation. Mutations can contribute to cancer by activating the protein function. We used the AssVar method to ascertain the presence of 79 deleterious SAAVs (selected deleterious SAAVs are shown in Table 4, and all 79 deleterious SAAVs are shown in Supporting Information Table S2). Among these 79 deleterious SAAVs, most proteins were detected with a single deleterious site, except for KRAS with three deleterious sites and FLNA with two deleterious sites. In addition to the oncogenic mutations of KRAS, another RAS family protein, HRAS, which was the first oncogene identified in human tumors, was also detected in PANC-1 cells. The oncogenic mutation of RAS family genes changes the balance for the normal outcome of some signaling pathways, resulting in tumor development.52 The RAS family has been detected in approximately 90% of pancreatic cancer patients.53 Also, around 30% of other cancers have a mutation in the protein KRAS.54 In the MAPK signaling pathway, the pancreatic cell relies on a process known as autophagy to create energy by disrupting the pathway derived from KRAS.55 Thus, the development of anti-KRAS therapies is one of the most elusive targets for cancer research.56 Savoy et al. revealed that FLNA is involved in cancer with dual roles. In the cytoplasm, overexpression of FLNA has a tumor-promoting effect, while FLNS acts as an inhibitor for tumor growth when FLNA undergoes proteolysis in the nucleus.57 In a study of more than 7664 tumors from 29 different cancer types from TCGA, Martincorena et al. confirmed the average number of driver genes for cancer as 1–10 driver mutations.58
Table 4.
Selected Deleterious SAAVs from Predicted 79 Deleterious SAAVs Based on the AssVar Servera
| protein name | gene name | accession number | mutation position | wild-type AA | mutant-type AA |
impact score | prediction results |
|---|---|---|---|---|---|---|---|
| glucose-6-phosphate exchanger SLC37A4 | SLC37A4 | O43826 | 88 | G | D | 0.91 | 1b |
| GTPase Hras | HRAS | P01112 | 61 | Q | E | 0.88 | 1 |
| GTPase Kras | KRAS | P01116 | 22 | Q | E | 0.87 | 1 |
| GTPase Kras | KRAS | P01116 | 12 | G | D | 0.95 | 1 |
| GTPase Kras | KRAS | P01116 | 12 | G | R | 0.94 | 1 |
| cellular tumor antigen p53 | TP53 | P04637 | 273 | R | H | 0.87 | 1 |
| aldehyde dehydrogenase, mitochondrial | ALDH2 | P05091 | 287 | G | A | 0.91 | 1 |
| insulin receptor | INSR | P06213 | 1055 | A | V | 0.86 | 1 |
| annexin A11 | ANXA11 | P50995 | 230 | R | C | 0.69 | 1c |
| neuroblast differentiation-associated protein AHNAK | AHNAK | Q09666 | 4090 | D | G | 0.78 | 1 |
| activating signal cointegrator 1 complex subunit 3 | ASCC3 | Q8N3C0 | 1995 | S | C | 0.63 | 1c |
| nuclear pore membrane glycoprotein 210 | NUP210 | Q8TEM1 | 1052 | G | S | 0.89 | 1 |
| HEAT repeat-containing protein 1 | HEATR1 | Q9H583 | 2017 | E | G | 0.68 | 1c |
| NSFL1 cofactor p47 | NSFL1C | Q9UNZ2 | 290 | D | N | 0.58 | 1c |
| ADP-ribosylation factor-like protein 5 A | ARL5A | Q9Y689 | 125 | N | S | 0.86 | 1 |
| filamin-A | FLNA | P21333 | 207 | P | L | 0.74 | 1 |
| filamin-A | FLNA | P21333 | 605 | D | N | 0.62 | 1 |
| integrin α−6 | ITGA6 | P23229 | 419 | A | T | 0.58 | 1c |
All 79 deleterious SAAVs are shown in Supporting Information Table S2.
Prediction results “1” refer to the deleterious SAAV.
Superscript “c” means it is common to three different database searching methods. A total of five deleterious SAAVs were shared by three strategies.
Three SAAVs were identified in protein TP53, while only one SAAV (R273H) was predicted as deleterious. As a tumor suppressor protein, TP53 plays a key role in some cell activities such as inducing cell cycle arrest, senescence, DNA repair, and cell apoptosis. As the most frequently mutated protein in the Pan-Cancer cohort, it is found in almost every type of cancer at rates varying from 10% in hematopoietic malignancies to 95% in high-grade serous carcinoma of the ovary.59,60 TP53 mutations including the hotspot R273H result in a gain of oncogenic function in tumor progression, invasion, and metastasis.61,62 Two proteins mitochondrial aldehyde dehydrogenase 2 (ALDH2) and SLC37A4 were also detected with a single SAAV. ALDH2 plays an essential role for alcohol detoxification to remove acetaldehyde in the pathway of alcohol metabolism.63 It is reported that more than 500 million people worldwide, mostly in East Asia, have inherited an inactive ALDH2 at residue 487 (E487K) with symptoms.64 It is the first time that the deleterious variant for ALDH2 at residue 287 (G287A) has been predicted. SLC37A4 is also known as glucose-6-phosphate translocase (G6PT) or SPX4, which belongs to the multicomponent glucose-6-phosphatase system (G6Pase-system) family.65 SLC37A4 contains 10 transmembrane helices and plays a role in maintaining blood glucose homeostasis in the liver and kidney and also maintains neutrophil and macrophage functions.66 A genetic defect of SLC37A4 was found to associate with inflammatory bowel disease (IBD)-like immunopathology for glycogen storage disease type 1b.67 It is the first time that the deleterious variants for ALDH2 and SLC37A4 at residues 287 (G287A) and 88 (G88D) were predicted, respectively.
All cancer cells carry somatic mutations including driver mutations and passenger mutations. Driver mutations are defined where the mutation confers a selective growth advantage and is causally implicated in cancer development, whereas the remainder are passenger mutations, which usually do not contribute to cancer development.24,68 Driver mutations are deleterious, and accumulated passenger mutations could also impact cancer progression.69 Ongoing research aims to identify driver mutations for all cancer types and identify therapies that can target tumors with these alterations. Accumulation of knowledge on deleterious SAAVs of cancer is a crucial step in successfully implementing precision oncology.
Validation of SAAV Sites for Variant Peptides Derived from KRAS, SLC37A4, and TP53 Using SRM
SRM was used to validate the presence of selected variants. One of these involves the KRAS (G12D) variant, which is an important driver mutation in pancreatic cancer. Both endogenous canonical peptide LVVVGAGGVGK and variant peptide LVVVGADGVGK (variant peptide G12D, italic D) from the PANC-1 cell line were detected using SRM (Figure 4A,B). They have the same retention time and SRM peak patterns with their corresponding heavy internal standards. Interestingly, the abundance of the variant peptide is slightly higher than that of the canonical peptide either by the endogenous peptide abundance alone or by the abundance ratio of endogenous over internal standard peptides (Figure 4A,B). Similarly, two other variant peptides FVSGVLSDQM SAR (G88D, italic D) and NSFEVHVCACPGR (R273H, italic H) derived from SLC37A4 and TP53 were also validated based on SRM (Figure 4C,D). For the variant peptide FVSGVLSDQ MSAR from SLC37A4, regular LC-SRM cannot provide sufficient sensitivity for confident detection with low SRM signal (Supporting Information Figure S2). Mostovenko et al. also found that not all identified variant peptides were appropriate for mass spectrometric quantification by the SRM assay after they identified approximate 400 SAAVs in glioma stem cells based on custom database searching.70 We further applied ultrasensitive PRISM-SRM to confirm its expression in the PANC-1 cell line (Figure 4C). TP53, a tumor suppressor protein, binds to DNA and regulates gene expression to prevent mutations of the genome.71 The mutant TP53 (R273H) has been found to be involved in inducing massive cell apoptosis and enhancing cancer cell malignancy.72−74
Figure 4.

Validation of SAAV sites for variant peptides derived from KRAS, SLC37A4, and TP53 using SRM. (A) Variant peptide LVVVGADGVGK (variant peptide G12D) from KRAS. (B) Canonical peptide LVVVGAGGVGK from KRAS. (C) Variant peptide FVSGVLSDQMSAR from SLC37A4. (D) Variant peptide NSFEVHVCACPGR (variant peptide R273H) from TP53. The variant peptide from SLC37A4 was detected by PRISM-SRM. The other three peptides were detected by regular LC-SRM. The endogenous peptides were confirmed by matching their corresponding heavy internal standards in the retention time and the SRM peak patterns. The top panel shows the SRM signal for endogenous peptides; the bottom panel shows the SRM signal for heavy internal standards (13C6, 15N2 on the C-terminal K or R). IS, internal standard.
CONCLUSIONS
We have detected SAAVs in the PANC-1 cell line based on mass spectrometry analysis. To improve the depth of coverage of SAAV peptide detection, we prepared samples by fractionation into approximately 10 fractionations for each sample prior to analysis by HPLC-MS/MS and CZE-MS/MS. In total, 133 mass spectrometry runs evaluating the PANC-1 cell line were conducted in this study. Different algorithms for database searching and different databases were applied. Potential SAAV spectra were evaluated manually to remove duplication and false-positive SAAVs. 70 SAAVs were identified using all three strategies indicating complementarity in the MS/MS as well as the informatics approaches. The 70 SAAVs are believed to be high-quality SAAV identifications. The 79 deleterious SAAVs predicted by the AssVar server included five SAAVs identified by all three strategies. The consistency of finding these deleterious SAAVs indicates they are high-quality identifications and are prime candidates for further study of drivers for pancreatic cancer (Table 4). In summary, the comprehensive detection of SAAVs was achieved in this study by integrating multiple strategies in both data acquisition and data analysis. A total of 79 deleterious SAAVs were distinguished from 461 passenger mutations in PANC-1 cells, and one of the driver mutations found in each protein of KRAS, TP53, and SLC37A4 was validated by SRM. Our study provides a blueprint for mutation research and potential targeted sites for anti-immunotherapy drug design for pancreatic cancer.
Supplementary Material
ACKNOWLEDGMENTS
We thank Dr. Song Nie from Regeneron Pharmaceuticals, Inc. for a critical reading of the manuscript. We acknowledge partial support of this work under NIH R01GM49500 (DML) and R01CA160254 (DML) and R21CA223715 (TS). Also, the assistance of the Wayne State University Proteomics Core, which is supported through NIH grants P30 ES020957, P30 CA022453, and S10 OD010700, is acknowledged. The support of Computation and Informatics in Biology and Medicine training grant comes from the NIH award number 5T15LM007359 (A.J.C.) and the support by NIH grant 5R21CA223887-02 (M.R.S.) from the National Cancer Institute. We also acknowledge partial support of this work under NIH R01GM125991 (L.S.) and the NSF CAREER Award DBI-1846913 (L.S.).
Footnotes
Supporting Information
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.jproteome.9b00840.
Four representative peptides with SAAVs manually confirmed by checking spectra where they are detected in different strategies (Figure S1) (PDF)
Validation of one SAAV site in protein SLC37A4 using regular LC-SRM analysis (Figure S2) (PDF)
SAAVs detected in HPLC and CZE in different strategies (Table S1) (XLSX)
79 deleterious SAAVs predicted by the AssVar server (Table S2) (PDF)
The authors declare no competing financial interest.
Contributor Information
Anthony J. Cesnik, Department of Genetics, Stanford University, Stanford, California 94305, United States
Xinpei Yi, Lester and Sue Smith Breast Center, Baylor College of Medicine, Houston, Texas 77030, United States.
Xiaohu Hao, Shanghai Institutes for Biological Science, Chinese Academy of Science, Shanghai 200031, China.
Michael R. Shortreed, Department of Chemistry, University of Wisconsin-Madison, Madison, Wisconsin 53706, United States
Tujin Shi, Integrative Omics Group, Biological Sciences Division, Pacific Northwest National Laboratory, Richland, Washington 99352, United States;.
David M. Lubman, Department of Surgery, The University of Michigan, Ann Arbor, Michigan 48109, United States;.
REFERENCES
- (1).Altshuler DM; Durbin RM; Abecasis GR; et al. An integrated map of genetic variation from 1,092 human genomes. Nature 2012, 491, 56–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).Cole C; Krampis K; Karagiannis K; Almeida JS; Faison WJ; Motwani M; Wan Q; Golikov A; Pan Y; Simonyan V; Mazumder R Non-synonymous variations in cancer and their effects on the human proteome: workflow for NGS data biocuration and proteome-wide analysis of TCGA data. BMC Bioinf. 2014, 15, 28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Bellacosa A; Moss EG RNA repair: damage control. Curr. Biol 2003, 13, R482–R484. [DOI] [PubMed] [Google Scholar]
- (4).Branzei D; Foiani M Regulation of DNA repair throughout the cell cycle. Nat. Rev. Mol. Cell Biol 2008, 9, 297–308. [DOI] [PubMed] [Google Scholar]
- (5).Li M; Wang IX; Li Y; Bruzel A; Richards AL; Toung JM; Cheung VG Widespread RNA and DNA sequence differences in the human transcriptome. Science 2011, 333, 53–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Boonen K; Hens K; Menschaert G; Baggerman G; Valkenborg D; Ertaylan G Beyond genes: Re-identifiability of proteomic data and its implications for personalized medicine. Genes 2019, 10, 682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Zhang B; Wang J; Wang X; Zhu J; Liu Q; Shi Z; Chambers MC; Zimmerman LJ; Shaddox KF; Kim S; Davies SR; Wang S; Wang P; Kinsinger CR; Rivers RC; Rodriguez H; Townsend RR; Ellis MJ; Carr SA; Tabb DL; Coffey RJ; Slebos RJ; Liebler DC Proteogenomic characterization of human colon and rectal cancer. Nature 2014, 513, 382–387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Su ZD; Sheng QH; Li QR; Chi H; Jiang X; Yan Z; Fu N; He SM; Khaitovich P; Wu JR; Zeng R De novo identification and quantification of single amino-acid variants in human brain. J. Mol. Cell Biol 2014, 6, 421–433. [DOI] [PubMed] [Google Scholar]
- (9).Lichti CF; Mostovenko E; Wadsworth PA; Lynch GC; Pettitt BM; Sulman EP; Wang Q; Lang FF; Rezeli M; Marko-Varga G; Vegvari A; Nilsson CL Systematic identification of single amino acid variants in glioma stem-cell-derived chromosome 19 proteins. J. Proteome Res 2015, 14, 778–786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Giese SH; Zickmann F; Renard BY Detection of unknown amino acid substitutions using error-tolerant database search. Methods Mol. Biol 2016, 1362, 247–264. [DOI] [PubMed] [Google Scholar]
- (11).Song C; Wang F; Cheng K; Wei X; Bian Y; Wang K; Tan Y; Wang H; Ye M; Zou H Large-scale quantification of single amino-acid variations by a variation-associated database search strategy. J. Proteome Res 2014, 13, 241–248. [DOI] [PubMed] [Google Scholar]
- (12).Tan ZJ; Nie S; McDermott SP; Wicha MS; Lubman DM Single amino acid variant profiles of subpopulations in the MCF-7 breast cancer cell line. J. Proteome Res 2017, 16, 842–851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Nie S; Yin HD; Tan ZJ; Anderson MA; Ruffin MT; Simeone DM; Lubman DM Quantitative analysis of single amino acid variant peptides associated with pancreatic cancer in serum by an isobaric labeling quantitative method. J. Proteome Res 2014, 13, 6058–6066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Tan Z; Yi X; Carruthers NJ; Stemmer PM; Lubman DM Single amino acid variant discovery in small numbers of cells. J. Proteome Res 2019, 18, 417–425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Sheynkman GM; Shortreed MR; Cesnik AJ; Smith LM Proteogenomics: Integrating next-generation sequencing and mass spectrometry to characterize human proteomic variation. Annu. Rev. Anal. Chem 2016, 9, 521–545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Nesvizhskii AI Proteogenomics: concepts, applications and computational strategies. Nat. Methods 2014, 11, 1114–1125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Yi XP; Wang B; An ZW; Gong FZ; Li J; Fu Y Quality control of single amino acid variations detected by tandem mass spectrometry. J. Proteomics 2018, 187, 144–151. [DOI] [PubMed] [Google Scholar]
- (18).Bailey MH; Tokheim C; Porta-Pardo E; et al. Comprehensive characterization of cancer driver genes and mutations. Cell 2018, 174, 1034–1035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Raphael BJ; Dobson JR; Oesper L; Vandin F Identifying driver mutations in sequenced cancer genomes: computational approaches to enable precision medicine. Genome Med. 2014, 6, 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Peeters M; Oliner KS; Price TJ; et al. Analysis of KRAS/NRAS mutations in a phase III study of panitumumab with FOLFIRI compared with FOLFIRI alone as second-line treatment for metastatic colorectal cancer. Clin. Cancer Res 2015, 21, 5469–5479. [DOI] [PubMed] [Google Scholar]
- (21).Fakih M; Vincent M Adverse events associated with anti-EGFR therapies for the treatment of metastatic colorectal cancer. Curr. Oncol 2010, 17, S18–S30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Hocking CM; Price TJ Panitumumab in the management of patients with KRAS wild-type metastatic colorectal cancer. Ther. Adv. Gastroenterol 2014, 7, 20–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Greenman C; Stephens P; Smith R; Stratton MR; et al. Patterns of somatic mutation in human cancer genomes. Nature 2007, 446, 153–158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Stratton MR; Campbell PJ; Futreal PA The cancer genome. Nature 2009, 458, 719–724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Yang ZC; Shen XJ; Chen DY; Sun LL Improved nanoflow RPLC-CZE-MS/MS system with high peak capacity and sensitivity for nanogram bottom-up proteomics. J. Proteome Res 2019, 18, 4046–4054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).McCool EN; Lubeckyr R; Shen XJ; Kou Q; Liu XW; Sun LL Large-scale top-down proteomics using capillary zone electrophoresis tandem mass spectrometry. J. Visualized Exp 2018, 140, No. e58644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Yang Z; Shen X; Chen D; Sun L Microscale reversed-phase liquid chromatography/ capillary zone electrophoresis-tandem mass spectrometry for deep and highly sensitive bottom-up proteomics: identification of 7500 proteins with five micrograms of an MCF7 proteome digest. Anal. Chem 2018, 90, 10479–10486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Deutsch EW; Csordas A; Sun Z; Jarnuczak A; Perez-Riverol Y; Ternent T; Campbell DS; Bernal-Llinares M; Okuda S; Kawano S; Moritz RL; Carver JJ; Wang MX; Ishihama Y; Bandeira N; Hermjakob H; Vizcaino JA The ProteomeXchange consortium in 2017: supporting the cultural change in proteomics public data deposition. Nucleic Acids Res. 2017, 45, D1100–D1106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Vizcaíno JA; Csordas A; del-Toro N; Dianes JA; Griss J; Lavidas I; Mayer G; Perez-Riverol Y; Reisinger F; Ternent T; Xu QW; Wang R; Hermjakob H 2016 update of the PRIDE database and its related tools. Nucleic Acids Res. 2016, 44, D447–D456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Song CX; Wang FJ; Cheng K; Wei XL; Bian YY; Wang KY; Tan YX; Wang HY; Ye ML; Zou HF Large-scale quantification of single amino-acid variations by a variation-associated database search strategy. J. Proteome Res 2014, 13, 241–248. [DOI] [PubMed] [Google Scholar]
- (31).Leinonen R; Sugawara H; Shumway M The sequence read archive. Nucleic Acids Res. 2011, 39, D19–D21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).Jiang H; Lei R; Ding SW; Zhu SF Skewer: a fast and accurate adapter trimmer for next-generation sequencing paired-end reads. BMC Bioinf. 2014, 15, 182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Dobin A; Davis CA; Schlesinger F; Drenkow J; Zaleski C; Jha S; Batut P; Chaisson M; Gingeras TR STAR: ultrafast universal RNA-seq aligner. Bioinformatics 2013, 29, 15–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).McKenna A; Hanna M; Banks E; Sivachenko A; Cibulskis K; Kernytsky A; Garimella K; Altshuler D; Gabriel S; Daly M; DePristo MA The genome analysis toolkit: A MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010, 20, 1297–1303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35).DePristo MA; Banks E; Poplin R; et al. A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nat. Genet 2011, 43, 491–498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (36).Poplin R; Chang PC; Alexander D; Schwartz S; Colthurst T; Ku A; Newburger D; Dijamco J; Nguyen N; Afshar PT; Gross SS; Dorfman L; McLean CY; DePristo MA A universal SNP and small-indel variant caller using deep neural networks. Nat. Biotechnol 2018, 36, 983–987. [DOI] [PubMed] [Google Scholar]
- (37).Cingolani P; Platts A; Wang LL; Coon M; Nguyen T; Wang L; Land SJ; Lu XY; Ruden DM A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff: SNPs in the genome of Drosophila melanogaster strain w(1118); iso-2; iso-3. Fly 2012, 6, 80–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (38).Solntsev SK; Shortreed MR; Frey BL; Smith LM Enhanced global post-translational modification discovery with MetaMorpheus. J. Proteome Res 2018, 17, 1844–1851. [DOI] [PubMed] [Google Scholar]
- (39).Yi X; Wang B; An Z; Gong F; Li J; Fu Y Quality control of single amino acid variations detected by tandem mass spectrometry. J. Protomics 2018, 187, 144–151. [DOI] [PubMed] [Google Scholar]
- (40).Zhang MH; Wang B; Xu J; Wang XJ; Xie L; Zhang B; Li YX; Li J CanProVar 2.0: An updated database of human cancer proteome variation. J. Proteome Res 2017, 16, 421–432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (41).Hao XH; Tan ZJ; Li Y; Zhang CX; Zheng W; Lubman DM; Zhang GJ; Zhang Y AssVar: A computational method for as-sessing impact of single amino acid variants in tumor, 2019. (in preparation).
- (42).Shi TJ; Fillmore TL; Sun XF; Zhao R; Schepmoes AA; Hossain M; Xie F; Wu S; Kim JS; Jones N; Moore RJ; Pasa-Tolic L; Kagan J; Rodland KD; Liu T; Tang KQ; Camp DG; Smith RD; Qian WJ Antibody-free, targeted mass-spectrometric approach for quantification of proteins at low picogram per milliliter levels in human plasma/serum. Proc. Natl. Acad. Sci. U.S.A 2012, 109, 15395–15400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (43).Shi TJ; Sun XF; Gao YQ; Fillmore TL; Schepmoes AA; Zhao R; He JT; Moore RJ; Kagan J; Rodland KD; Liu T; Liu AY; Smith RD; Tang KQ; Camp DG; Qian WJ Targeted quantification of low ng/mL level proteins in human serum without immunoaffinity depletion. J. Proteome Res 2013, 12, 3353–3361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (44).MacLean B; Tomazela DM; Shulman N; Chambers M; Finney GL; Frewen B; Kern R; Tabb DL; Liebler DC; MacCoss MJ Skyline: an open source document editor for creating and analyzing targeted proteomics experiments. Bioinformatics 2010, 26, 966–968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (45).Shi T; Gaffrey MJ; Fillmore TL; Nicora CD; Yi L; Zhang PF; Shukla AK; Wiley HS; Rodland KD; Liu T; Smith RD; Qian WJ Facile carrier-assisted targeted mass spectrometric approach for proteomic analysis of low numbers of mammalian cells. Commun. Biol 2018, 1, No. 103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (46).Sheynkman GM; Shortreed MR; Frey BL; Scalf M; Smith LM Large-scale mass spectrometric detection of variant peptides resulting from nonsynonymous nucleotide differences. J. Proteome Res 2014, 13, 228–240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (47).Katsimpardi L; Gaitanou M; Malnou CE; Lledo PM; Charneau P; Matsas R; Thomaidou D BM88/Cend1 expression levels are critical for proliferation and differentiation of subventricular zone-derived neural precursor cells. Stem Cells 2008, 26, 1796–1807. [DOI] [PubMed] [Google Scholar]
- (48).Belvedere R; Bizzarro V; Popolo A; Dal Piaz F; Vasaturo M; Picardi P; Parente L; Petrella A Role of intracellular and extracellular annexin A1 in migration and invasion of human pancreatic carcinoma cells. BMC Cancer 2014, 14, No. 961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (49).Lochhead PA; Wickman G; Mezna M; Olson MF Activating ROCK1 somatic mutations in human cancer. Oncogene 2010, 29, 2591–2598. [DOI] [PubMed] [Google Scholar]
- (50).Fan Y; Hu Y; Yan C; Goldman R; Pan Y; Mazumder R; Dingerdissen HM Loss and gain of N-linked glycosylation sequons due to single-nucleotide variation in cancer. Sci. Rep 2018, 8, No. 4322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (51).Mazumder R; Morampudi KS; Motwani M; Vasudevan S; Goldman R Proteome-wide analysis of single-nucleotide variations in the N-glycosylation sequon of human genes. PLoS One 2012, 7, No. e36212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (52).Fernandez-Medárde A; Santos E Ras in cancer and developmental diseases. Genes Cancer 2011, 2, 344–358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (53).Ryan DP; Hong TS; Bardeesy N Pancreatic adenocarcinoma. N. Engl. J. Med 2014, 371, 1039–1049. [DOI] [PubMed] [Google Scholar]
- (54).Waters AM; Der CJ KRAS: The critical driver and therapeutic target for pancreatic cancer. Cold Spring Harbor Perspect. Med 2018, 8, No. a031435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (55).Kinsey CG; Camolotto SA; Boespflug AM; et al. Protective autophagy elicited by RAF → MEK → ERK inhibition suggests a treatment strategy for RAS-driven cancers. Nat. Med 2019, 25, 620–627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (56).McCormick F Progress in targeting RAS with small molecule drugs. Biochem. J 2019, 476, 365–374. [DOI] [PubMed] [Google Scholar]
- (57).Savoy RM; Ghosh PM The dual role of filamin A in cancer: can’t live with (too much of) it, can’t live without it. Endocr.-Relat. Cancer 2013, 20, R341–R356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (58).Martincorena I; Raine KM; Gerstung M; Dawson KJ; Haase K; Van Loo P; Davies H; Stratton MR; Campbell PJ Universal patterns of selection in cancer and somatic tissues. Cell 2018, 173, 1823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (59).Kandoth C; McLellan MD; Vandin F; Ye K; Niu B; Lu C; Xie M; Zhang Q; McMichael JF; Wyczalkowski MA; Leiserson MDM; Miller CA; Welch JS; Walter MJ; Wendl MC; Ley TJ; Wilson RK; Raphael BJ; Ding L Mutational landscape and significance across 12 major cancer types. Nature 2013, 502, 333–339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (60).Rivlin N; Brosh R; Oren M; Rotter V Mutations in the p53 tumor suppressor gene: Important milestones at the various steps of tumorigenesis. Genes Cancer 2011, 2, 466–474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (61).Olive KP; Tuveson DA; Ruhe ZC; Yin B; Willis NA; Bronson RT; Crowley D; Jacks T Mutant p53 gain of function in two mouse models of Li-Fraumeni syndrome. Cell 2004, 119, 847–860. [DOI] [PubMed] [Google Scholar]
- (62).Sigal A; Rotter V Oncogenic mutations of the p53 tumor suppressor: the demons of the guardian of the genome. Cancer Res. 2000, 60, 6788–6793. [PubMed] [Google Scholar]
- (63).Zakhari S Overview: How is alcohol metabolized by the body? Alcohol Res. Health 2006, 29, 245–254. [PMC free article] [PubMed] [Google Scholar]
- (64).Jin SF; Chen J; Chen LZ; Histen G; Lin ZZ; Gross S; Hixon J; Chen Y; Kung C; Chen YW; Fu YF; Lu YX; Lin H; Cai XJ; Yang H; Cairns RA; Dorsch M; Su SM; Biller SS; Mak TW; Cang Y ALDH2(E487K) mutation increases protein turnover and promotes murine hepatocarcinogenesis. Proc. Natl. Acad. Sci. U.S.A 2015, 112, 9088–9093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (65).Cappello AR; Curcio R; Lappano R; Maggiolini M; Dolce V The physiopathological role of the exchangers belonging to the SLC37 family. Front. Chem 2018, 6, 122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (66).Chou JY; Jun HS; Mansfield BC The SLC37 family of phosphate-linked sugar phosphate antiporters. Mol. Aspects Med 2013, 34, 601–611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (67).Uhlig HH Monogenic diseases associated with intestinal inflammation: implications for the understanding of inflammatory bowel disease. Gut 2013, 62, 1795–1805. [DOI] [PubMed] [Google Scholar]
- (68).Pleasance ED; Cheetham RK; Stephens PJ; et al. A comprehensive catalogue of somatic mutations from a human cancer genome. Nature 2010, 463, 191–196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (69).McFarland CD; Korolev KS; Kryukov GV; Sunyaev SR; Mirny LA Impact of deleterious passenger mutations on cancer progression. Proc. Natl. Acad. Sci. U.S.A 2013, 110, 2910–2915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (70).Mostovenko E; Vegvari A; Rezeli M; Lichti CF; Fenyo D; Wang Q; Lang FF; Sulman EP; Sahlin KB; Marko-Varga G; Nilsson CL Large scale identification of variant proteins in glioma stem cells. ACS Chem. Neurosci 2018, 9, 73–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (71).Kern SE; Kinzler KW; Bruskin A; Jarosz D; Friedman P; Prives C; Vogelstein B Identification of P53 as a sequence-specific DNA-binding protein. Science 1991, 252, 1708–1711. [DOI] [PubMed] [Google Scholar]
- (72).Li J; Yang LX; Gaur S; Zhang KQ; Wu XW; Yuan YC; Li HZ; Hu SY; Weng YG; Yen Y Mutants TP53 p.R273H and p.R273C but not p.R273G enhance cancer cell malignancy. Hum. Mutat 2014, 35, 575–584. [DOI] [PubMed] [Google Scholar]
- (73).Kang N; Wang Y; Guo SC; Ou YW; Wang GC; Chen J; Li D; Zhan QM Mutant TP53 G245C and R273H promote cellular malignancy in esophageal squamous cell carcinoma. BMC Cell Biol. 2018, 19, 16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (74).Tan BS; Tiong KH; Choo HL; Chung FF; Hii LW; Tan SH; Yap IK; Pani S; Khor NT; Wong SF; Rosli R; Cheong SK; Leong CO Mutant p53-R273H mediates cancer cell survival and anoikis resistance through AKT-dependent suppression of BCL2-modifying factor (BMF). Cell Death Dis. 2015, 6, No. e1826. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.