

Abstract
Readily available acrylamide naphthoquinones can be converted into the corresponding aza-anthraquinones using 6π-photoelectrocyclization reactions. Not only do these reactions not proceed thermally but, as demonstrated here, they can also be used to generate a range of aza-anthraquinone and aza-tetracycline derivatives including the natural products griffithazanone A and marcanine A. Several of the aza-anthraquinones generated in this work showed antibacterial activity.
Graphical Abstract
INTRODUCTION
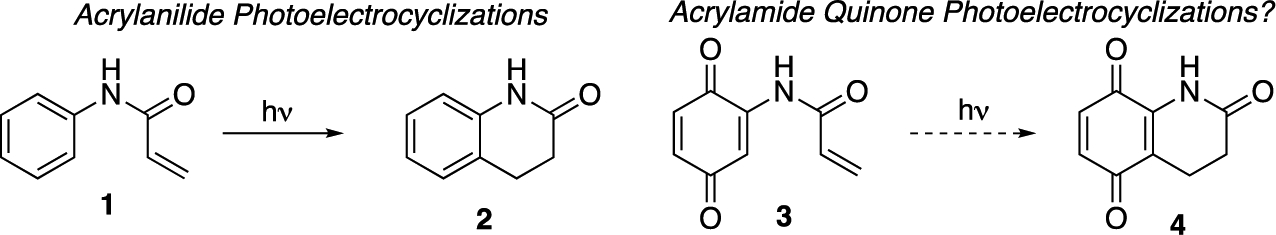
In addition to being ubiquitous in biologically active natural and non-natural products, quinones have demonstrated impressive properties in both synthetic and biological chemistries.1 In spite of this, there are a very few examples of the use of quinones in either thermal or photochemical electrocyclization reactions (3–4, Scheme 1).2 This comes in spite of their wide use in [2 + 2] photocycloaddition reactions,3 the use of quinone methides and derivatives in 6π electrocyclizations,4 and the rich photochemical and electron transport properties of quinones.5 In contrast to the dearth of examples of quinone reactivity, the photoelectrocyclization chemistry of the corresponding acrylanilides has received a significant amount of attention (1–2, Scheme 1).6 These transformations were first reported by Chapman in 1967 and have subsequently been shown to be a valuable means of synthesizing substituted quinolines.7,8 From an interest in using the reactions in the synthesis of complex targets, we set out to determine whether the photoelectrocyclization reactions of quinones have simply been overlooked or whether they might have inherent problems. Outlined here are experiments that address these questions and that demonstrate that acrylamide-containing naphthoquinones undergo photoelectrocyclization reactions. In addition, also included in this work are the first total synthesis of the aza-anthraquinone natural product griffithazanone A,9 the total synthesis of the quinolone natural product marcanine A,10 and preliminary antibacterial activity of the substrates generated as a consequence of these investigations.
Scheme 1.
Photoelectrocyclizations to Lactams
RESULTS AND DISCUSSION
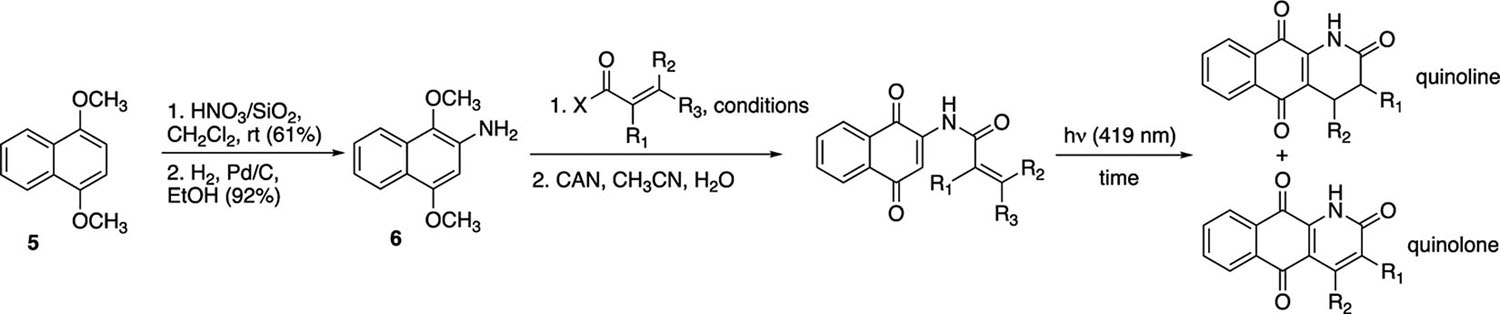
For the synthesis of the requisite acrylamide and cinnamide cyclization precursors, we initially explored the coupling of 2-aminonaphthoquinone with acrylic and cinnamic acid derivatives. As has been reported by other laboratories,11 this reaction proved to be capricious in our hands. To date, a much more reliable route to the cyclization precursors has involved a two-step process that has coupled acrylamide or cinnamide formation from the reaction of 2-amino-1,4-dimethoxynaphthalene 6 and unsaturated acids or acid chlorides with oxidation of the resulting hydroquinone to the corresponding quinone (Table 1).12 In turn, aminohydroquinone 6 resulted from the nitration and reduction of 1,4-dimethylhydroquinone 5.13
Table 1.
Generation of Aza-Anthraquinones via Photoelectrocyclization Reactions
 | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| entry | X | Conditions | Coupling Yield | Oxidation Yield | aminoquinone | time (min.)b | quinoline | yield | quinolone | yield |
| 1 | CI | A | 76% | 75% | 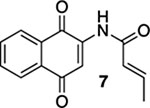 |
255 | 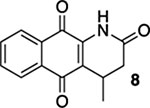 |
47% | 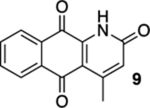 |
50% |
| 2 | OH | B | 56% | 80% | 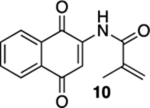 |
75 | 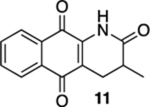 |
76% | 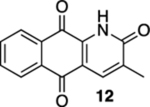 |
21% |
| 3 | OH | B | 81% | 95% | 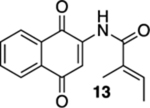 |
25 | 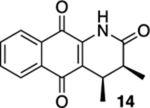 |
90% (cis:trans = 15:1) | 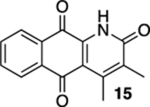 |
--d |
| 4 | CI | A | 78% | 70% | 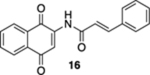 |
50 | 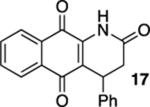 |
33% |  |
--e |
| 5 | OH | B | — | 52%c | 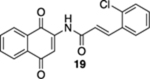 |
60 | 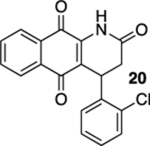 |
20% |  |
--e |
| 6 | OH | B | — | 45%c | 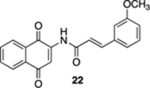 |
60 | 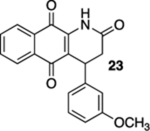 |
20% | 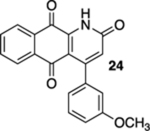 |
--e |
Conditions: A: Et3N, CH2Cl2; B: EDC·HCl, 4-DMAP, and CH2Cl2.
Time required for the disappearance of the starting material by thin-layer chromatography (TLC).
Yield for two steps.
A trace amount of 15 was formed if the reaction was allowed to proceed for more than 60 min.
An intractable mixture of what we presume as oxidized byproducts was obtained.
With the precursors in hand, we explored their photoelectrocyclization reactions using a 419 nm light source (Table 1). While we were pleased that the reactions proceeded and that they gave the requisite aza-anthraquinones, we were surprised that the substitution pattern on the acrylamide starting materials was important in the efficacy of the reactions. For example, when β-methyl acrylamide 7 was exposed to light, we isolated a 1:1 mixture of quinoline 8 and the corresponding oxidized product, quinolone 9, in an overall yield of 97% (entry 1). In contrast, the reaction of the isomeric α-methyl acrylamide 10 not only gave much less of the oxidized product (12) but the reaction also required less time to go to completion (entry 2). Our presumption was that 9 arose from the oxidation of 8 by O2 in spite of our efforts to deoxygenate the reaction mixture by bubbling N2 through it immediately prior to subjecting the mixture to light.14 It was interesting and ultimately implying that this deoxygenation protocol was used in the reaction of the α-methyl analogue 10 without the same oxidation issues. In addition to the cyclization and the oxidation, another interesting feature of this reaction includes the fact that 9 is the natural product marcanine A.10 Marcanine A was originally isolated from the bark of the Thai flowering plant Goniothalamus marcanii, and although it was not our initial focus, it is interesting in its own right as it has shown impressive activity against a number of cancer cell lines and malaria including a drug-resistant strain of Plasmodium falciparum.15 The use of photochemistry was critical here, and our attempts to carry out the electrocyclization reaction of 7 and 10 using thermal conditions were unsuccessful, resulting in the recovery of the starting material.16
In a reaction that was more similar to the reaction of 10 than that of 7, 2,3-dimethyl acrylamide 13 gave exclusively cisdimethyl quinoline 14 (see Table 1, entry 3)17 unless the reaction was allowed to proceed for more than 1 h after which we began observing the formation of a byproduct that we presume as oxidized quinolone 15 (entry 3). Assuming a photochemically allowed conrotatory electrocyclization, the relative stereochemistry of 14 would be consistent with a mechanism involving the tautomerization of the electrocyclization product 27 and not a [1,5]-hydride shift mechanism (Scheme 2).18 This result was particularly interesting to us in light of the fact that the related acrylanilide reactions have not only shown lower levels of selectivity but they also provide a predominance of the trans-product in the absence of a proton source.7
Scheme 2.
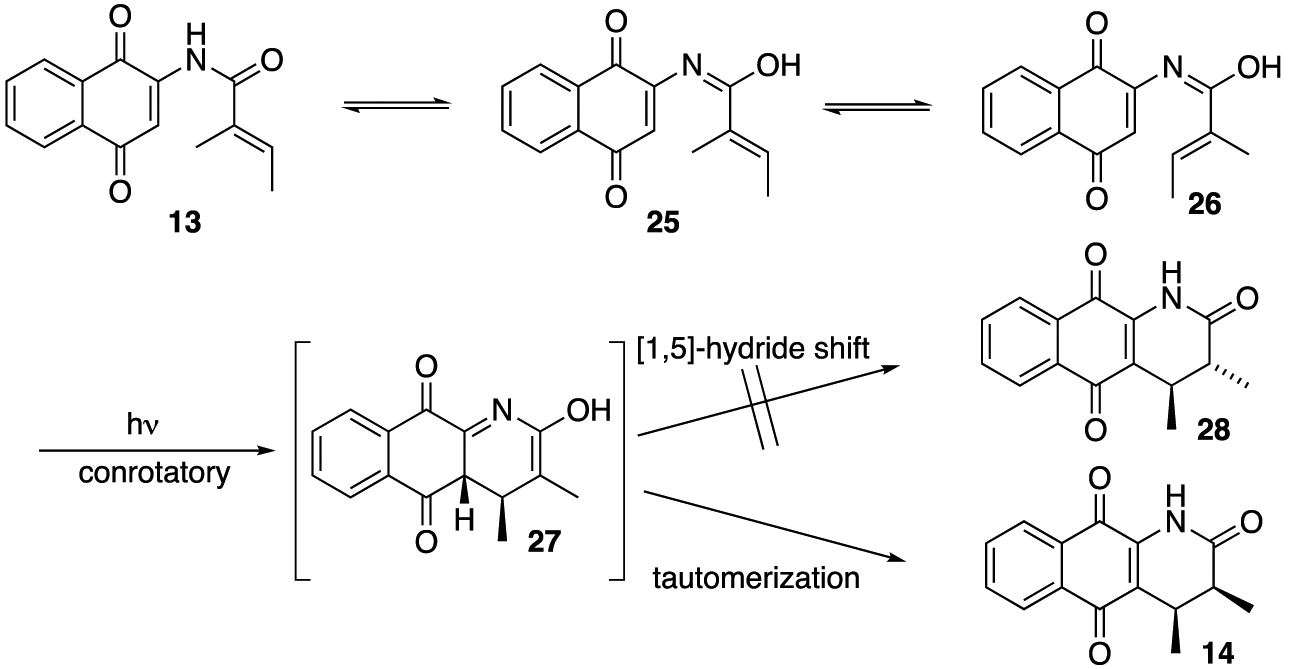
Proposed Mechanism for the Photoelectrocyclization of Naphthoquinone Acrylamides
In contrast to those of the acrylamide substrates, the photoelectrocyclization reactions of cinnamide derivatives 16, 19, and 22 were problematic; while the corresponding quinolines 17, 20, and 23 were isolated, the yields were low (Table 1, entries 4–6). These reactions resulted in the formation of a number of intractable byproducts that we presume to arise from the oxidation of the electrocyclization starting materials, intermediates, or products.
As mentioned above, we assumed that the oxidation problems that were outlined arose from the presence of residual O2 in the reaction mixture. In an attempt to overcome this, we explored the use of a more rigorous deoxygenation protocol by subjecting a solution of 7 to three freeze–pump–thaw cycles prior to subjecting it to 419 nm light.19 This resulted in the generation of aza-anthraquinone 8 in 88% yield without any noticeable quinolone 9 (Scheme 3). Unfortunately, this solution to the oxidation problem was not general; when we applied the freeze–pump–thaw technique to the cinnamide derivatives 16, 19, and 22, we continued to isolate a low yield of the corresponding quinolines along with a predominance of what appeared to be the same intractable oxidized material that we had observed previously.
Scheme 3.
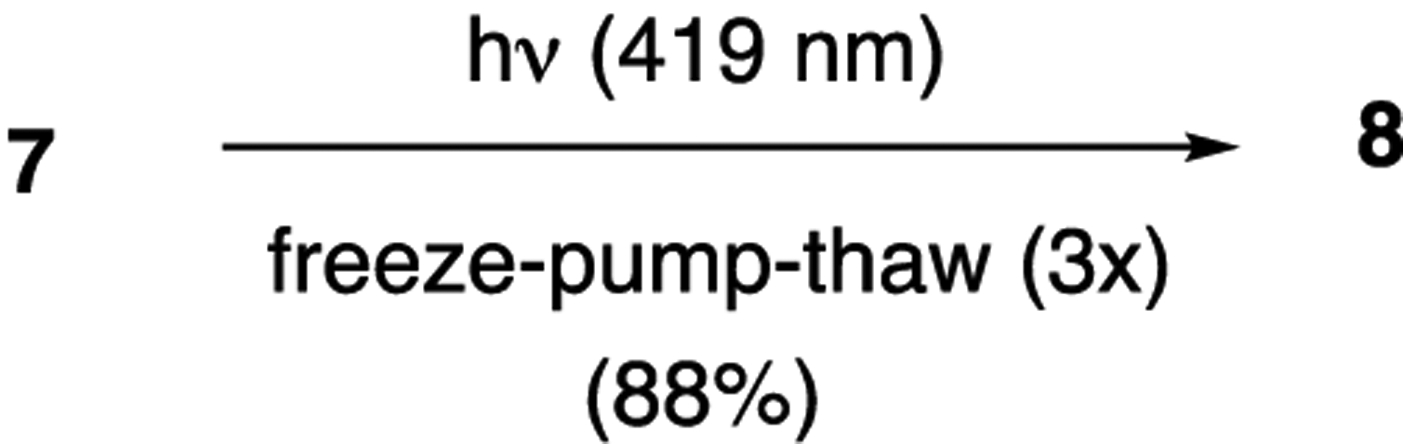
Detrimental Role of O2 in Acrylamide Naphthoquinone Photoelectrocyclizations
Having established that the photoelectrocyclization reactions of acrylamide and cinnamide naphthoquinones can be utilized to generate relatively simple aza-anthraquinones, we set out to examine the scope of the reactions by exploring more elaborate substrates. Along these lines, the presence of spirocyclic quinones in antibacterial and antivirulence natural products inspired us to examine geminal disubstituted substrates 29, 31, 33, 35, and 37.20 These cyclization precursors were generated in a similar fashion to that of those examined previously, namely, from the coupling of aminonaphthalene 6 with the corresponding acrylic acid or acid chlorides followed by oxidation (Table 2). We were ultimately pleased to find that geminal disubstituted acrylamides could be converted into the corresponding quaternary substituted aza-anthraquinones in moderate (45–60%) to high (92%) yields when they were exposed to 419 nm light. With tetrasubstituted acrylamide 31 as the exception, it was not surprising that these substrates generally required much longer reaction times to go to completion when compared to the nongeminal disubstituted substrates.21 With regard to the effect of pre-existing stereocenters on the stereochemical outcome of the reactions, we were encouraged that the photoelectrocyclization of unsymmetrical acrylamide 37 resulted in the generation of spirocycle 38 in 92% yield as a single diastereomer by 1H NMR.
Table 2.
Photoelectrocyclizations to Quaternary Substituted Aza-Anthraquinones
 | ||||||||
|---|---|---|---|---|---|---|---|---|
| entry | X | Conditionsa | Coupling Yield | Oxidation Yield | Amide | time | quinoline | yield |
| 1 | CI | A | 93% | 87% |  |
16 h | 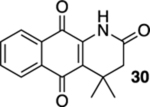 |
56%b |
| 2 | OH | B | 54% | 75% | 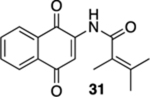 |
40 min | 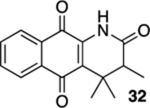 |
91% |
| 3 | OH | B | 63% | 92% | 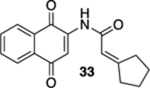 |
26 h | 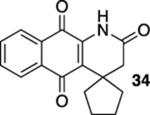 |
48% |
| 4 | OH | B | 66% | 90% | 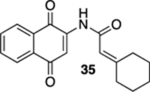 |
10 h | 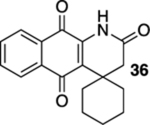 |
82% |
| 5 | OH | B | 42% | 81% | 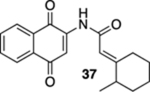 |
10 h | 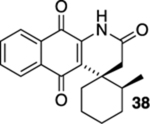 |
92% (d.r. >20:1)c |
Conditions: A: Et3N, CH2Cl2; B: (COCl)2, dimethylformamide (DMF), and CH2Cl2; NEt3.
13% of the starting amide 29 was recovered.
The ratio was determined by 1H NMR of the crude reaction mixture.
As a final substrate class for these studies, we examined the formation of angularly fused aza-tetracyclic quinones. Related substrates have shown a number of interesting properties including anticancer and antibiotic activities.22 As illustrated in Table 3, the cyclization precursors for these studies were accessed in the same fashion as for those that were described previously. We were pleased to find that the photoelectrocyclization was general; exposing acrylamides 39 and 41 to 419 nm light efficiently resulted in the formation of 40, 42, and 43 (entries 1 and 3).23 Of note was that no oxidized material was observed and that these reactions gave exclusively the cis-fused products.24 As was mentioned earlier, it was interesting that the analogous acrylanilide cyclizations were both less selective and gave a predominance of the transfused products unless the reaction was run in the presence of a proton source.8 In contrast to our results with 37, the presence of the multiple stereocenters in shikimic acid cyclization precursor 41 complicated the reaction, giving the two cis-isomers 42 and 43 in essentially equal quantities.
Table 3.
Angularly Fused Aza-Tetracycles
 | ||||||||
|---|---|---|---|---|---|---|---|---|
| entry | X | Coupling Yield | Oxidation Yield | Amide | time | additive | Yield | quinoline |
| 1 | OH | 66% | 80% | 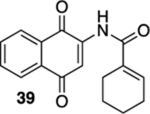 |
45 min | none | 85% (cis:trans >20:1) | 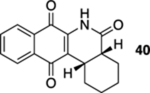 |
| 2 | OH | 66% | 80% | 39 | 60 min |  |
11% (cis:trans >20:1) | 40 |
| 3 | OH | 97% | 86% | 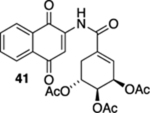 |
10.5 h | none | 81% (42:43 = 1.2:1) | 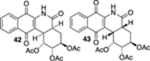 |
In an effort to better understand the nature of the excited state, we carried out the photoreaction of 39 in the presence of 5 equiv. of tetramethylbutadiene as a triplet quencher (Table 3, entry 2). That the yield of the cyclized product was much lower in the presence of the quencher than it was in its absence is suggestive of a triplet excited state.25
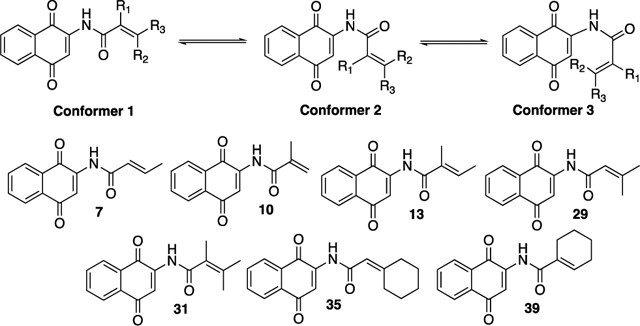
Assuming that the nonequilibration of excited state rotomers (NEER) principle was operative here,26 we carried out density functional theory (DFT) calculations on seven representative substrates in an effort to better understand their relative reaction rates.27,28 We focused on three relevant conformations, the s-trans conformation (Conformer 1) and the two scis conformations (Conformers 2 and 3, Table 4). In all instances, the s-trans conformation (Conformer 1) was the lowest in energy and therefore was arbitrarily assigned a value of 0 kcal/mol. We then compared the differences in the energies between Conformers 2 and 3 with the amount of time required for the starting materials to be consumed. It was interesting that the rate of each reaction correlated reasonably well with the differences in energy between the two conformers. The substrates that decomposed the fastest had substitution at the α-position (8, 9, 31, and 40) and either had a product-forming conformation (Conformer 3) that was lower in energy or one that was relatively close in energy to their nonproduct-forming conformation (Conformer 2). In contrast, Conformer 2 was much more stable for those substrates that lacked the α-substituent. These substrates also required much longer reaction times (see 7, 29, and 35).
Table 4.
Effects of Relative Energetics of Acrylamide Naphthoquinone Conformations on Their Photoelectrocyclization Rates
| compound | 7 | 10 | 13 | 29 | 31 | 35 | 39 |
|---|---|---|---|---|---|---|---|
| Conformer 3 (kcal/mol) | 8.51 | 7.06 | 6.60 | 8.39 | 4.95 | 8.22 | 6.46 |
| Conformer 2 (kcal/mol) | 5.47 | 7.90 | 7.24 | 4.91 | 4.14 | 5.03 | 7.24 |
| ΔE (3–2) | 3.04 | −0.84 | −0.64 | 3.39 | 0.81 | 3.19 | −0.78 |
| reaction timea | 255 min | 75 min | 25 min | 960 min | 40 min | 600 min | 45 min |
Reaction mixtures were removed from the photochamber when the starting material had disappeared by TLC.
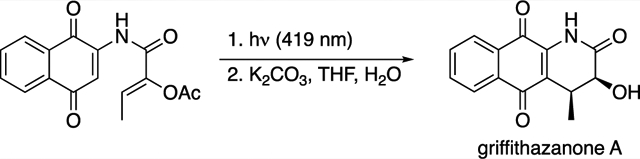
Having established the photoelectrocyclization reaction of quinones as a powerful means of generating aza-anthraquinones, we set out to utilize the reaction in the synthesis of the natural product griffithazanone A (Scheme 4). Griffithazanone A was originally isolated from the roots of the tropical plant Goniothalamus griffithii, a member of the Annonaceae family.29 Griffithazanone A has demonstrated cytotoxicity against the colon cancer cell line HCT-116, with an IC50 value of 2.39 μM (doxorubicin had an IC50 value of 9.74 μM in the same assay) and to the best of our knowledge has not been targeted in total synthesis studies.30 Our initial efforts for its synthesis focused on the direct oxidation of 8. These experiments resulted in either the recovery of the starting material or its decomposition to an unrecognizable material.31 We then turned to the incorporation of a heteroatom into the cyclization precursor and examined the photochemistry of α-bromoacrylamide 44 and α-acetoxyacrylamide 45. The photoelectrocyclizations of these substrates resulted in the formation of 46 and 47, respectively. While these reactions led to the cis-isomers as the major products, in contrast to the reactions of 13, 39, and 41, they gave a measurable quantity of the corresponding trans-products. It was not surprising that the α-bromo analogue 46 proved to be unstable to isolation, chromatographic purification, and the attempted displacement with nucleophiles. In contrast, α-acetoxy aza-anthraquinone 47 was relatively stable and underwent hydrolysis when exposed to K2CO3 and H2O to give griffithazanone A.
Scheme 4.
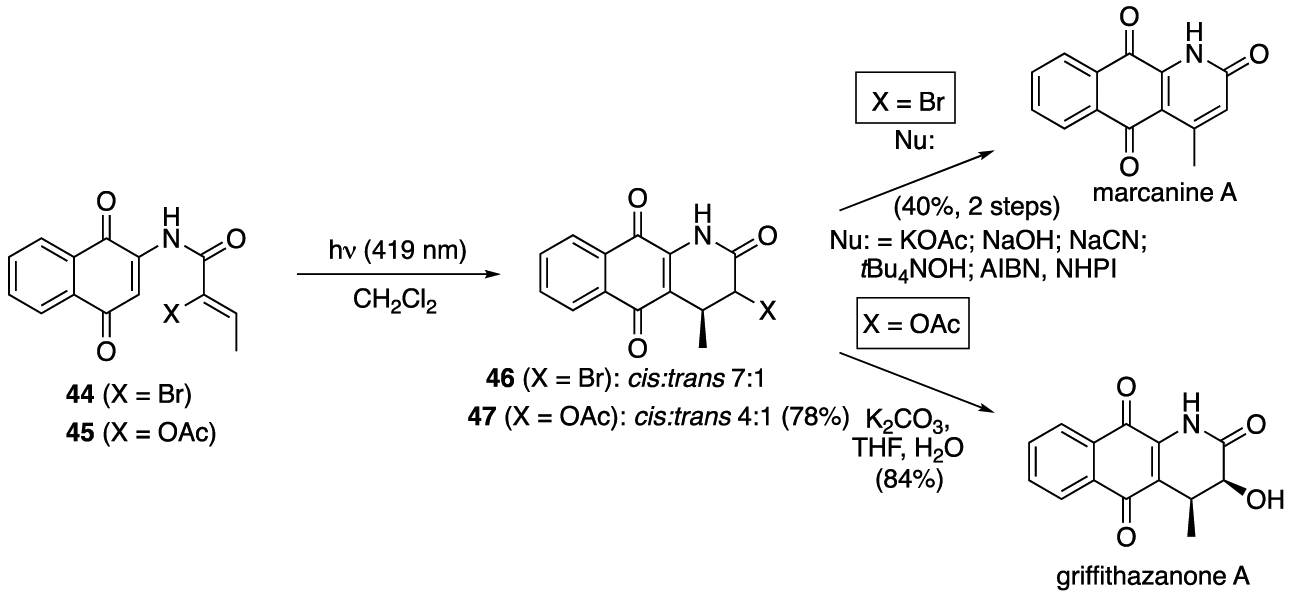
Syntheses of Marcanine A and Griffithazanone A
A selection of the above quinolines and quinolones were tested for their ability to inhibit bacterial growth (Table 5). The minimum inhibitory concentration (MIC) for these compounds was determined according to the Clinical and Laboratory Standards Institute guidelines.32 While the compounds did not show inhibitory activity against Escherichia coli (ATCC-25922) or Enterococcus faecalis (ATCC-19433), several aza-anthraquinones and aza-angucyclinones did show promising activity against Staphylococcus aureus (ATCC-12600). Interestingly, of the compounds tested, marcanine A was the most active with an MIC of 1 μg/mL.
Table 5.
Activity of Photoproducts against S. aureus
| compound | MIC (S. aureus) | compound | MIC, μg/mL (S. aureus) |
|---|---|---|---|
| 8 | 32 μg/mL | 32 | 2 |
| marcanine A | 1 μg/mL | 34 | 8 |
| 11 | 4 μg/mL | 36 | 64 |
| 12 | 32 μg/mL | 38 | >64 |
| 14 | 32 μg/mL | 40 | 8 |
| 17 | 16 μg/mL | 42 | 64 |
| 20 | 32 μg/mL | 43 | 64 |
| 23 | 16 μg/mL | 47 | 32 |
| 30 | N.D. | (±)-Griffithazanone A | 16 |
In conclusion, the work described here shows that photoelectrocyclization reactions of acrylamide naphthoquinones are an effective means of generating aza-anthraquinones and angularly fused aza-angucyclinone derivatives. In addition to demonstrating the breadth of substrates that undergo this transformation, we have also shown that this class of targets has interesting and in some cases impressive activity against S. aureus. In our future studies, we will continue to examine the use of photoelectrocyclization reactions to synthesize complex small molecules while also exploring the antibiotic properties of aza-anthraquinones.
EXPERIMENTAL INFORMATION
General Information.
Glassware was dried in an oven at 130 °C or flame-dried and cooled under a N2 atmosphere prior to use. All reactions were performed using common dry, inert atmosphere techniques. Solvents were purified according to the guidelines in Purification of Common Laboratory Chemicals (Perrin, Armarego, and Perrin: Oxford, 1966). Dichloromethane was distilled from CaH2.
All photoreactions were conducted in 10 mm pyrex tubes using a Rayonet-100 Photoreactor. Pyrex tubes were placed in the center of the photoreactor. The distances from the pyrex tubes to the light source were ca. 9 cm. The light source used in the photoreactions was manufactured by Southern New England Ultraviolet Company (RPR-4190A) with a peak intensity of 419 nm. The emission spectrum of this light source ranged from 373 to 460 nm with an intensity at 419 nm of 1.20 × 10−5 W/cm2; see the Supporting Information for the emission spectra. Reactions were monitored by TLC and visualized by a dual short-wave/long-wave UV lamp and stained with an ethanolic solution of potassium permanganate or p-anisaldehyde. Column flash chromatography was performed using hexanes and ethyl acetate as eluents with SilicaFlash F60-grade silica gel, 230–400-mesh, from Silicycle. NMR spectra were recorded on a Varian Inova-500 or a Varian Inova-400 spectrometer. Chemical shifts for 1H NMR were reported as δ, parts per million (ppm), relative to the tetramethylsilane signal at 0 ppm or for the CHCl3 signal at 7.26 ppm. Mass spectra were recorded at the Mass Spectrometry Facility in the Department of Chemistry at the University of Utah on a Finnigan MAT 95 double focusing high-resolution mass spectrometer. Crystal structures were obtained at the X-ray crystallography center in the Department of Chemistry at the University of Utah.
1,4-Dimethoxynaphthalene (5).
To a solution of 1,4-naphthoqui-none (20.0 g, 126 mmol) in tetrahydrofuran (THF, 500 mL) at room temperature (rt) was added 10% Pd/C (1 g). The resulting mixture was charged with H2 gas (1 atm) and allowed to stir for 4 h after which the reaction mixture was cooled to 0 °C. To this was slowly added NaH (60% dispersion in mineral oil, 15.2 g, 379 mmol). This resulted in the reaction mixture undergoing a color change to green. Dimethyl sulfate (60.0 mL, 632.3 mmol) was then added dropwise to the mixture. The reaction mixture was allowed to stir for 1 h and then warmed to rt and stirred for an additional 16 h at that temperature. The mixture was passed through a short pad of Celite using THF (50 mL) as the eluent. Concentration gave a brown paste that was dissolved in EtOAc (200 mL) and washed sequentially with NH4OH (33% aq., 2 × 40 mL), H2O (2 × 50 mL), and brine (50 mL). The organic phase was dried (Na2SO4) and concentrated. Flash chromatography (hexanes/ethyl acetate = 20:1) gave 21.4 g of 5 (90%) as a white solid.
Mp 86–87 °C; 1H NMR (500 MHz, CDCl3) δ 8.30–8.28 (m, 2H), 7.38–7.56 (m, 2H), 6.72 (s, 2H), 3.99 (s, 6H); 13C{1H} (126 MHz, CDCl3) δ 149.6, 126.5, 125.9, 121.9, 103.3, 55.8; IR (neat) 3070, 3011, 2953, 2920, 2841, 1630, 1593, 1463, 1445, 1384, 1270, 1239, 1159, 1086, 1023, 955, 803, 764, 720 cm−1; HRMS (ESI) m/z: [M + H]+ calcd for C12H13O2 189.0916; found 189.0915.
1,4-Dimethoxy-2-nitronaphthalene (S1).
Preparation of nitric acid-impregnated silica gel: To a beaker charged with 50 g of silica gel (GFS chemical, 50–200 mesh) was carefully added 60 mL of concentrated nitric acid (aq.). The mixture was sonicated at rt for 2 h and then allowed to sit for an additional 24 h. The mixture was filtered, and the solid was air-dried at rt. The nitric acid content of SiO2 was determined to be 2.3 mmol/g through its titration with 0.1 N NaOH (aq.) using phenolphthalein as the indicator.
To a solution of 5 (11.0 g, 58.4 mmol) in CH2Cl2 (500 mL) at rt was slowly added silica-gel-supported nitric acid (28 g, 64 mmol HNO3) in three equal portions. The resulting reaction mixture was stirred for 15 min and then filtered. Concentration and flash column chromatography (hexanes/ethyl acetate = 20:1) gave 8.30 g of S1 (61%) as a yellow solid.
Mp 95–96 °C; 1H NMR (500 MHz, CDCl3) δ 8.28–8.24 (m, 2H), 7.66–7.65 (m, 2H), 7.21 (s, 1H), 4.08 (s, 3H), 4.03 (s, 3H); 13C{1H} NMR (126 MHz, CDCl3) δ 152.0, 145.8, 138.8, 129.1, 129.0, 128.9, 128.3, 124.2, 122.8, 98.6, 63.7, 56.2; IR (neat) 2936, 2844, 1587, 1526, 1503, 1462, 1417, 1329, 1265, 1212, 1110, 1088, 992, 967, 750, 720 cm−1; HRMS (ESI) m/z: [M + H]+ calcd for C12H12NO4 234.0766; found 234.0771.
1,4-Dimethoxynaphthalen-2-amine (6).
To a solution of S1 (6.00 g, 25.7 mmol) in anhydrous EtOH (100 mL) at rt was added 10% Pd/C (0.6 g). The resulting mixture was charged with H2 gas (1 atm) and allowed to stir for 8 h after which it was quickly passed through a short pad of Celite. Concentration under reduced pressure followed by flash column chromatography (hexanes/ethyl acetate = 3:1) using a positive pressure of N2 to avoid oxidation provided 4.8 g of 6 (92%) as an air-sensitive white solid.
Mp 102–103 °C; 1H NMR (500 MHz, CDCl3) δ 8.11 (d, J = 8.4 Hz, 1H), 7.85 (d, J = 8.4 Hz, 1H), 7.45 (td, J = 7.6, 1.1 Hz, 1H), 7.24 (td, J = 8.0, 0.9 Hz, 1H), 6.31 (s, 1H), 3.93 (s, 2H), 3.88 (s, 3H), 3.84 (s, 3H); 13C{1H} NMR (126 MHz, CDCl3) δ 152.8, 135.4, 133.1, 129.2, 127.0, 122.4, 121.8, 120.7, 119.8, 97.4, 60.0, 55.7; IR (neat) 3458, 3362, 2935, 2834, 1628, 1602, 1462, 1447, 1398, 1380, 1261, 1222, 1156, 1081, 999, 827, 764 cm−1; HRMS (ESI) m/z: [M + H]+ calcd for C12H14NO2 204.1025; found 204.1026.
(E)-N-(1,4-Dimethoxynaphthalen-2-yl)but-2-enamide (S2).
To a solution of 6 (203 mg, 1.00 mmol) in CH2Cl2 (5 mL) at rt was added crotonoyl chloride (105 μL, 1.10 mmol). After the resulting reaction mixture was stirred for 15 min, a solution of Et3N (139 μL, 1.00 mmol) in CH2Cl2 (5 mL) was added dropwise over 10 min. The reaction mixture was stirred for an additional 20 min and then concentrated. The residue was dissolved in EtOAc (20 mL) and then washed sequentially with sat. NH4Cl (aq., 10 mL), H2O (2 × 10 mL), and brine (aq., 10 mL). The organic layer was dried (Na2SO4) and concentrated. Flash column chromatography (hexanes/ethyl acetate = 5:1) gave 206 mg of S2 (76%) as a white solid.
Mp 135–137 °C; 1H NMR (500 MHz, CDCl3) δ 8.20 (d, J = 8.4 Hz, 1H), 8.11 (broad s, 1H), 7.94–7.93 (m, 2H), 7.51 (t, J = 7.4 Hz, 1H), 7.39 (t, J = 7.6 Hz, 1H), 7.05 (dq, J = 13.7, 6.7 Hz, 1H), 6.07 (d, J = 15.0 Hz, 1H), 4.01 (s, 3H), 3.88 (s, 3H), 1.93 (d, J = 6.8 Hz, 3H); 13C{1H} NMR (126 MHz, CDCl3) δ 164.2, 152.2, 141.8, 136.7, 128.1,127.5, 126.9, 125.7, 124.3, 123.2, 122.7, 120.9, 98.9, 61.6, 55.8, 17.9; IR (neat) 3277, 2937, 2850, 1674, 1627, 1601, 1522, 1496, 1457, 1371, 1216, 1093, 975, 927, 830, 764 cm−1; HRMS (ESI) m/z: [M + H]+ calcd for C16H18NO3 272.1287; found 272.1287.
(E)-N-(1,4-Dioxo-1,4-dihydronaphthalen-2-yl)but-2-enamide (7).
To a solution of S2 (272 mg, 1.00 mmol) in CH3CN (16 mL) at rt was added a solution of ceric ammonium nitrate (1.21 g, 2.20 mmol) in a mixture of CH3CN (4 mL) and H2O (4 mL). The resulting reaction mixture was stirred for 15 min and then poured into H2O (20 mL). The aqueous phase was extracted with EtOAc (3 × 40 mL), and the combined organic extracts were washed with brine, dried (Na2SO4), and concentrated. Flash column chromatography (hexanes/ethyl acetate = 4:1) gave 181 mg of 7 (75%) as a yellow solid.
Mp 218–222 °C; 1H NMR (500 MHz, CDCl3) δ 8.34 (broad s, 1H), 8.11 (d, J = 7.7 Hz, 2H), 7.92 (s, 1H), 7.78 (t, J = 7.6 Hz, 1H), 7.72 (t, J = 7.6 Hz, 1H), 7.07 (dq, J = 13.9, 6.9 Hz, 1H), 6.08–6.06 (m, 1H), 1.97 (dd, J = 6.9, 1.4 Hz, 3H); 13C{1H} NMR (126 MHz, CDCl3) δ 185.3, 181.2, 164.6, 144.8, 140.3, 135.1, 133.3, 132.4, 130.1, 126.8, 126.6, 124.7, 117.3, 18.3; IR (neat) 3313, 1662, 1642, 1614, 1592, 1507, 1341, 1307, 1194, 1098, 736 cm−1; HRMS (ESI) m/z: [M + Na]+ calcd for C14H11NO3Na 264.0637; found 263.0640.
4-Methyl-3,4-dihydrobenzo[g]quinoline-2,5,10(1H)-trione (8) and 4-Methylbenzo[g]quinoline-2,5,10(1H)-trione (Marcanine A, 9).
A solution of 7 (12.1 mg, 0.0500 mmol) in CHCl3 (20 mL) was placed in two 10 mm NMR tubes and purged with Ar for 10 min. The solution was then placed in the center of a photoreactor and irradiated for 4.25 h using 419 nm light (spectral window: 373–460 nm). The reaction mixture was concentrated and the resulting residue was subjected to flash column chromatography (gradient from hexanes/ethyl acetate = 3:1 to hexanes/ethyl acetate = 1:1) to give 5.6 mg of 8 (47%) and 6.1 mg of 9 (marcanine A, 50%), both as yellow solids.
8:
Mp 186–189 °C; 1H NMR (500 MHz, CDCl3) δ 8.13 (d, J = 7.7 Hz, 1H), 8.11 (d, J = 7.5 Hz, 1H), 8.11 (partially obscured s, 1H), 7.77 (td, J = 7.5, 1.2 Hz, 1H), 7.71 (td, J = 7.6, 1.2 Hz, 1H), 3.50 (pd, J = 7.2, 1.1 Hz, 1H), 2.76 (dd, J = 16.7, 7.3 Hz, 1H), 2.56 (d, J = 16.8 Hz, 1H), 1.20 (d, J = 7.2 Hz, 3H); 13C{1H} NMR (126 MHz, CDCl3) δ 182.2, 179.3, 169.2, 137.8, 135.0, 133.5, 132.4, 130.4, 126.8, 126.5, 125.0, 37.1, 25.2, 19.0; IR (neat) 3299, 3218, 2962, 2923, 1691, 1674, 1645, 1631, 1593, 1465, 1445, 1358, 1332, 1297, 1239, 1209, 722 cm−1; HRMS (ESI) m/z: [M + H]+ calcd for C14H12NO3 242.0817; found 242.0818.
9 (marcanine A):
Mp 264–289 °C, decomposed; 1H NMR (500 MHz, CDCl3) δ 9.68 (broad s, 1H), 8.24 (dt, J = 7.7, 0.7 Hz, 1H), 8.19 (dt, J = 7.7, 0.7 Hz, 1H), 7.87 (td, J = 7.6, 0.7 Hz, 1H), 7.78 (td, J = 7.6, 0.7 Hz, 1H), 6.69 (q, J = 0.7 Hz, 1H), 2.71 (d, J = 0.7 Hz, 3H); 13C{1H} NMR (126 MHz, CDCl3) δ 181.5, 178.1, 160.4, 152.3, 139.9, 136.0, 133.9, 133.3, 130.1, 127.8, 127.7, 126.8, 116.2, 23.0; IR (neat) 2926, 2856, 1646, 1591, 1465, 1400, 1374, 1339, 1295, 1165 cm−1; HRMS (ESI) m/z: calcd for C14H10NO3 [M + H]+ 240.0661; found 240.0664.
N-(1,4-Dimethoxynaphthalen-2-yl)methacrylamide (S3).
To a solution of 6 (87.4 mg, 0.430 mmol) in CH2Cl2 (5 mL) at rt were added methacrylic acid (55 mg, 0.65 mmol), EDC·HCl (125 mg, 0.650 mmol), and 4-DMAP (10 mg, 0.090 mmol). The reaction mixture was stirred overnight and then concentrated. The resulting residue was dissolved in EtOAc (5 mL) and washed sequentially with sat. NH4Cl (aq., 5 mL), H2O (2 × 2 mL), and brine (2 mL). The organic layer was dried (Na2SO4), concentrated, and purified using flash column chromatography (hexanes/ethyl acetate = 5:1) to provide 65.1 mg of S3 (56%) as white solid and as a mixture of amide rotomers.
Mp 96–108 °C. 1H NMR (500 MHz, CDCl3) δ 8.36 (bs, 1H), 8.21 (d, J = 8.4 Hz, 1H), 8.10 (s, 1H), 7.92 (d, J = 8.3 Hz, 1H), 7.49 (dd, J = 8.0, 6.9 Hz, 1H), 7.38 (dd, J = 8.1, 7.0 Hz, 1H), 5.91 (s, 1H), 5.50 (s, 1H), 4.01 (s, 3H), 3.87 (s, 1.5H), 3.86 (s, 1.5H), 2.12 (s 3H). 13C{1H} NMR (126 MHz, CDCl3) δ 166.2, 152.2, 140.6, 136.7, 127.8, 127.4, 126.9, 124.2, 123.1, 122.6, 120.7, 120.4, 98.4, 61.4, 55.7, 18.6; IR (neat) 3424, 3071, 2936, 2850, 1680, 1627, 1601, 1520, 1495, 1457, 1419, 1372, 1291, 1253, 1216,1162, 1122, 1091, 1032, 997, 974 cm−1; HRMS (ESI) m/z: [M + H]+ calcd for C16H18NO3 272.1287; found 272.1293.
N-(1,4-Dioxo-1,4-dihydronaphthalen-2-yl)methacrylamide (10).
To a solution of S3 from above in CH3CN (4 mL) at rt was added a mixture of ceric ammonium nitrate (290 mg, 0.53 mmol) in CH3CN (1 mL) and H2O (1 mL). The resulting reaction mixture was stirred for 15 min and then poured into H2O (5 mL). The aqueous phase was extracted with EtOAc (3 × 5 mL), and the combined organic extracts were washed with brine (15 mL), dried (Na2SO4), and concentrated. Flash column chromatography (hexanes/ethyl acetate = 4:1) gave 46.7 mg of 10 (80%) as a yellow solid.
Mp 116–118 °C; 1H NMR (500 MHz, CDCl3) δ 8.84 (broad s, 1H), 8.10–8.09 (m, 2H), 7.87 (s, 1H), 7.77 (t, J = 7.5 Hz, 1H), 7.71 (t, J = 7.4 Hz, 1H), 5.93 (s, 1H), 5.62 (s, 1H), 2.08 (s, 3H); 13C{1H} NMR (126 MHz, CDCl3) δ 185.2, 181.2, 166.8, 140.1, 139.8, 135.1, 133.4, 132.3, 130.1, 126.8, 126.5, 122.5, 117.2, 18.5; IR (neat) 3373, 1699, 1666, 1650, 1620, 1595, 1505, 1458, 1337, 1298, 1198, 1107, 723 cm−1; HRMS (ESI) m/z: [M + H]+ calcd for C14H12NO3 242.0817; found 242.0821.
3-Methyl-3,4-dihydrobenzo[g]quinoline-2,5,10(1H)-trione (11) and 3-Methylbenzo[g]quinoline-2,5,10(1H)-trione (12).
A solution of 10 (12.1 mg, 0.05 mmol) in CHCl3 (20 mL) in two 10 mm NMR tubes was purged with Ar for 10 min. The resulting solution was placed in the center of a photoreactor and irradiated with 419 nm light (spectral window: 373–460 nm) for 75 min after which the reaction mixture was concentrated. The resulting residue was subjected to flash column chromatography (gradient from hexanes/ethyl acetate = 2:1 to hexanes/ethyl acetate = 1:1) to give 9.2 mg of 11 (76%) along with 2.5 mg of 12 (21%), both as yellow solids.
11:
Mp 251–252 °C; 1H NMR (500 MHz, CDCl3) δ 8.13 (dd, J = 7.6, 1.0 Hz, 1H), 8.00 (dd, J = 7.6, 1.3 Hz, 1H), 7.99 (broad s, 1H), 7.77 (td, J = 7.5, 1.4 Hz, 1H), 7.71 (td, J = 7.5, 1.4 Hz, 1H), 3.21 (dd, J = 17.7, 6.9 Hz, 1H), 2.72–2.67 (m, 1H), 2.58 (dd, J = 17.7, 11.7 Hz, 1H), 1.35 (d, J = 6.9 Hz, 3H); 13C{1H} NMR (126 MHz, CDCl3) δ 182.7, 178.9, 172.5, 139.0, 135.0, 133.4, 132.4, 130.5, 126.8, 126.6, 120.1, 34.2, 26.5, 15.7; IR (neat) 3226, 2923, 2850, 1687, 1674, 1644, 1632, 1593, 1461, 1448, 1370, 1330, 1301, 1221, 791, 719 cm−1; HRMS (ESI) m/z: [M + H]+ calcd for C14H12NO3 242.0817; found 242.0824.
12:
Mp 240–280 °C, decomposed, 1H NMR (500 MHz, CDCl3) δ 9.46 (broad s, 1H), 8.25 (d, J = 7.5 Hz, 1H), 8.20 (d, J = 7.5 Hz, 1H), 8.00 (s, 1H), 7.85 (t, J = 7.5 Hz, 1H), 7.79 (t, J = 7.5 Hz, 1H), 2.31 (s, 3H); 13C{1H} NMR (126 MHz, CDCl3) δ 180.4, 177.9, 161.9, 138.8, 137.2, 135.5, 134.1, 132.8, 132.3, 131.0, 127.5, 127.1, 117.0, 17.6; IR (neat) 2919, 2849, 1681, 1661, 1638, 1594, 1564, 1486, 1393, 1378, 1336, 1245 cm−1; HRMS (ESI) m/z: [M + H]+ calcd for C14H10NO3 240.0661; found 240.0665.
(E)-N-(1,4-Dimethoxynaphthalen-2-yl)-2-methylbut-2-enamide (S4).
To a solution of 6 (203 mg, 1.00 mmol) in CH2Cl2 (10 mL) at rt were added tiglic acid (150 mg, 1.50 mmol), EDC·HCl (290 mg, 1.5 mmol), and 4-DMAP (24.4 mg, 0.200 mmol). The reaction mixture was stirred overnight and then concentrated. The resulting residue was dissolved in EtOAc (10 mL) and washed with saturated NH4Cl (aq., 10 mL), H2O (2 × 5 mL), and brine (5 mL). The organic layer was dried (Na2SO4) and concentrated. Flash column chromatography (hexanes/ethyl acetate = 8:1) gave 232 mg of S4 (81%) as a white solid.
Mp 94–97 °C; 1H NMR (500 MHz, CDCl3) δ 8.28 (broad s, 1H), 8.21 (d, J = 8.4 Hz, 1H), 8.12 (s, 1H), 7.94 (d, J = 8.4 Hz, 1H), 7.51 (t, J = 7.6 Hz, 1H), 7.39 (td, J = 7.6, 1.1 Hz, 1H), 6.68–6.64 (m, 1H), 4.02 (s, 3H), 3.90 (s, 3H), 2.02 (d, J = 0.8 Hz, 3H), 1.87 (d, J = 6.9 Hz, 3H); 13C{1H} NMR (126 MHz, CDCl3) δ 167.4, 152.4, 136.7, 132.8, 132.1, 128.4, 127.6, 127.0, 124.2, 123.1, 122.7, 120.9, 98.7, 61.6, 55.9, 14.4, 12.6; IR (neat) 3430, 2937, 1676, 1629, 1602, 1520, 1495, 1457, 1375, 1266, 1214, 1095, 997, 767 cm−1; HRMS (ESI) m/z: [M + H]+ calcd for C17H20NO3 286.1443; found 286.1449.
(E)-N-(1,4-Dioxo-1,4-dihydronaphthalen-2-yl)-2-methylbut-2-enamide (13).
To a solution of S4 (57.1 mg, 0.200 mmol) in CH3CN (4 mL) at rt was added a mixture of ceric ammonium nitrate (241 mg, 0.440 mmol) in CH3CN (1 mL) and H2O (1 mL). The reaction mixture was stirred for 15 min and then poured into H2O (5 mL). The aqueous phase was extracted with EtOAc (3 × 5 mL), and the combined organic extracts were washed with brine (15 mL), dried (Na2SO4), and concentrated. Flash column chromatography (toluene/ethyl acetate = 20:1) gave 48.5 mg of 13 (95%) as a yellow solid.
Mp 126–128 °C; 1H NMR (500 MHz, CDCl3) δ 1:1 mixture of two conformers, 8.78 (broad s, 1H), 8.10 (d, J = 7.7 Hz, 2H), 7.88 (s, 1H), 7.78 (td, J = 7.5, 1.3 Hz, 1H), 7.71 (td, J = 7.5, 1.4 Hz, 1H), 6.69–6.65 (m, 1H), 1.970 (s, 1.5H), 1.968 (s, 1.5H), 1.880 (d, J = 6.9 Hz, 1.5H), 1.866 (d, J = 6.9 Hz, 1.5H); 13C{1H} NMR (126 MHz, CDCl3) δ 185.3, 181.5, 167.7, 140.4, 135.1, 134.5, 133.3, 132.4, 132.2, 130.2, 126.7, 126.5, 116.9, 14.5, 12.3; IR (neat) 3376, 1666, 1649, 1502, 1334, 1299, 1228, 1197, 1103, 885, 783, 716 cm−1; HRMS (ESI) m/z: [M + H]+ calcd for C15H14NO3 256.0974; found 255.0975.
3,4-Dimethyl-3,4-dihydrobenzo[g]quinoline-2,5,10(1H)-trione (14).
A solution of 13 (12.8 mg, 0.0500 mmol) in CHCl3 (20 mL) was placed in two 10 mm NMR tubes and then purged with Ar for 10 min. The resulting solution was placed in the center of a photoreactor and irradiated with 419 nm light (spectral window: 373–460 nm) for 25 min after which the reaction mixture was concentrated. The resulting residue was subjected to flash column chromatography (hexanes/ethyl acetate = 5:1) to give 11.5 mg of 14 (90%, cis/trans = 15.2:1) as a yellow solid.
Mp 230–233 °C; 1H NMR (500 MHz, CDCl3) δ 8.12 (d, J = 7.7 Hz, 1H), 8.08 (d, J = 7.6 Hz, 1H), 8.02 (broad s, 1H), 7.76 (td, J = 7.5, 1.0 Hz, 1H), 7.70 (td, J = 7.5, 1.0 Hz, 1H), 3.37 (dq, J = 6.9, 6.9 Hz, 1H), 2.28 (qd, J = 6.8, 6.8 Hz, 1H), 1.32 (d, J = 7.0 Hz, 3H), 1.05 (d, J = 7.2 Hz, 3H); 13C{1H} NMR (126 MHz, CDCl3) δ 182.1, 179.4, 172.3, 138.0, 134.9, 133.3, 132.4, 130.5, 126.7, 126.5, 126.2, 38.8, 30.4, 12.4, 11.1; IR (neat) 3309, 2924, 2852, 1708, 1670, 1632, 1594, 1468, 1454, 1330, 1301, 1213, 722 cm−1; HRMS (ESI) m/z: [M + H]+ calcd for C15H14NO3 256.0974; found 256.0976.
N-(1,4-Dimethoxynaphthalen-2-yl)cinnamamide (S5).
To a solution of 6 (203 mg, 1.00 mmol) in CH2Cl2 (5 mL) at rt was added cinnamoyl chloride (183 mg, 1.10 mmol). After the resulting reaction mixture was stirred for 15 min, a solution of Et3N (139 μL, 1.00 mmol) in CH2Cl2 (5 mL) was added dropwise over a period of 10 min. The reaction mixture was allowed to stir for an additional 2 h and then concentrated. The resulting residue was dissolved in EtOAc (20 mL) and then washed with sat. NH4Cl (aq., 10 mL), H2O (2 × 10 mL), and brine (10 mL). The organic phase was dried (Na2SO4) and concentrated. Flash column chromatography (hexanes/ethyl acetate = 5:1) gave 260 mg of S5 (78%) as a pale-yellow solid.
Mp 170–172 °C; 1H NMR (500 MHz, CDCl3) δ 8.23 (d, J = 8.4 Hz, 1H), 8.19 (broad s, 1H), 8.09 (broad s, 1H), 7.96 (d, J = 8.4 Hz, 1H), 7.82 (d, J = 15.5 Hz, 1H), 7.60–7.58 (m, 2H), 7.53 (t, J = 7.1, 7.1 Hz, 1H), 7.43–7.39 (m, 4H), 6.67 (d, J = 15.5 Hz, 1H), 4.04 (s, 3H), 3.93 (s, 3H); 13C{1H} NMR (126 MHz, CDCl3) δ 164.2, 152.3, 142.7, 136.8, 134.7, 130.2, 129.0, 128.2, 127.6, 127.1, 124.4, 123.4, 122.8, 121.1, 121.0, 98.8, 61.8, 55.9; IR (neat) 3242, 3004, 2933, 2841, 1665, 1623, 1600, 1588, 1522, 1496, 1457, 1448, 1416, 1371, 1332, 1275, 1259, 1217, 1162, 1124, 1092, 1035, 993, 976, 824, 763, 734, 717 cm−1; HRMS (ESI) m/z: [M + H]+ calcd for C21H20NO3 334.1443; found 334.1446.
N-(1,4-Dioxo-1,4-dihydronaphthalen-2-yl)cinnamamide (16).
To a solution of S5 (123 mg, 0.370 mmol) in CH3CN (5 mL) at rt was added a mixture of ceric ammonium nitrate (446 mg, 0.810 mmol) in CH3CN (1.5 mL) and H2O (1.5 mL). The resulting reaction mixture was stirred for 15 min and then poured into H2O (8 mL). The aqueous phase was extracted with EtOAc (3 × 15 mL), and the combined organic extracts were washed with brine (45 mL), dried (Na2SO4), and concentrated. Flash column chromatography (toluene/ethyl acetate = 20:1) gave 79 mg of 16 (70%) as a yellow solid.
Mp 250–252 °C; 1H NMR (500 MHz, CDCl3) δ 8.52 (broad s, 1H), 8.15 (ddd, J = 7.3, 5.9, 1.0 Hz, 2H), 7.99 (s, 1H), 7.81 (d, J = 15.5 Hz, 1H), 7.80 (td, J = 7.5, 1.3 Hz, 1H), 7.74 (td, J = 7.6, 1.3 Hz, 1H), 7.60–7.58 (m, 2H), 7.44–7.42 (m, 3H), 6.64 (d, J = 15.6 Hz, 1H); 13C{1H} NMR (126 MHz, CDCl3) δ 185.3, 181.3, 164.8, 144.9, 140.4, 135.2, 134.2, 133.4, 132.5, 130.9, 130.2, 129.2, 128.5, 126.9, 126.6, 119.7, 117.6; IR (neat) 3309, 1669, 1637, 1593, 1579, 1506, 1450, 1336, 1303, 1197, 1155, 1112, 984, 721 cm−1; HRMS (ESI) m/z: [M + Na]+ calcd for C19H13NO3Na 326.0793; found 326.0788.
4-Phenyl-3,4-dihydrobenzo[g]quinoline-2,5,10(1H)-trione (17).
A solution of 16 (15.0 mg, 0.0500 mmol) in CHCl3 (20 mL) that had been purged with N2 gas for 10 min was placed in a photoreactor and irradiated with 419 nm light (spectral window of lamps: 373–460 nm). After TLC indicated that the starting material had been consumed (ca. 6 h), the reaction mixture was concentrated. Flash column chromatography (hexanes/ethyl acetate = 3:1) afforded 4.9 mg of 17 (33%) as a yellow solid.
Mp 197–200 °C; 1H NMR (500 MHz, CDCl3) δ 8.15 (broad s, 1H), 8.11 (partially obscured dd, J = 6.9, 0.8 Hz, 1H), 8.09 (partially obscured dd, J = 6.9, 0.8 Hz, 1H), 7.77 (td, J = 7.5, 1.4 Hz, 1H), 7.72 (td, J = 7.5, 1.4 Hz, 1H), 7.29–7.27 (m, 4H), 7.24–7.21 (m, 1H), 4.67 (dd, J = 8.2, 1.3 Hz, 1H), 3.05 (dd, J = 17.1, 8.2 Hz, 1H), 2.95 (d, J = 17.0 Hz, 1H); 13C{1H} NMR (126 MHz, CDCl3) δ 182.2, 179.4, 168.8, 140.6, 138.4, 135.1, 133.6, 132.3, 130.4, 129.3, 127.8, 127.0, 127.0, 126.6, 122.6, 36.9, 35.4; IR (neat) 3296, 2922, 2851, 1708, 1672, 1635, 1594, 1579, 1464, 1452, 1350, 1330, 1301, 1222, 1159, 1145, 980, 936, 721, 707 cm−1; HRMS (ESI) m/z: [M + Na]+ calcd for C19H13NO3Na 326.0793; found 326.0801.
(E)-3-(2-Chlorophenyl)-N-(1,4-dioxo-1,4-dihydronaphthalen-2-yl)acrylamide (19).
A mixture of 6 (203 mg, 1.00 mmol), 2-chlorocinnamic acid (274 mg, 1.50 mmol), EDC·HCl (288 mg, 1.50 mmol), and 4-DMAP (24.4 mg, 0.200 mmol) in CH2Cl2 (10 mL) at rt was allowed to stir overnight and then concentrated. The resulting residue was dissolved in EtOAc (10 mL) and washed sequentially with sat. NH4Cl (aq., 10 mL), H2O (2 × 5 mL), and brine (5 mL). The organic layer was dried (Na2SO4) and concentrated. The resulting hydroquinone (E)-3-(2-chlorophenyl)-N-(1,4-dimethoxynaphthalen-2-yl)acrylamide was used without additional purification.
To hydroquinone (E)-3-(2-chlorophenyl)-N-(1,4-dimethoxynaphthalen-2-yl)acrylamide from above in CH3CN (5 mL) at rt was added a mixture of ceric ammonium nitrate (1.2 g, 2.2 mmol) in CH3CN (7.5 mL) and H2O (7.5 mL). The reaction mixture was stirred for 15 min and then poured into H2O (15 mL). The aqueous phase was extracted with EtOAc (3 × 40 mL), and the combined organic extracts were washed with brine (120 mL), dried (Na2SO4), and concentrated. Flash column chromatography (toluene/ethyl acetate = 20:1) gave 176 mg of 19 (52%, 2 steps) as a yellow solid.
Mp 261–263 °C; 1H NMR (500 MHz, CDCl3) δ 8.56 (broad s, 1H), 8.20 (d, J = 15.6 Hz, 1H), 8.15 (d, J = 6.6 Hz, 1H), 8.14 (d, J = 7.0 Hz, 1H), 8.00 (s, 1H), 7.81 (td, J = 7.5, 1.1 Hz, 1H), 7.74 (td, J = 7.6, 1.1 Hz, 1H), 7.67 (dd, J = 7.4, 1.8 Hz, 1H), 7.46 (dd, J = 8.8, 1.1 Hz, 1H), 7.37–7.31 (m, 2H), 6.65 (d, J = 15.6 Hz, 1H); 13C{1H} NMR (126 MHz, DMSO-d6) 185.1, 180.4, 165.4, 141.8, 137.3, 134.9, 133.8, 133.7, 132.2, 131.8, 131.4, 130.3, 130.2, 127.9, 127.8, 126.4, 125.5, 124.5, 116.7; IR (neat) 3313, 3066, 2920, 1694, 1664, 1642, 1594, 1498, 1351, 1334, 1301, 1255, 1190, 1157, 1108, 981, 737, 721, 666 cm−1; HRMS (ESI) m/z: [M + Na]+ calcd for C19H12NO3Na35Cl 360.0403; found 360.0403, calcd for C19H12NO3Na37Cl 362.0374; found 362.0396.
4-(2-Chlorophenyl)-3,4-dihydrobenzo[g]quinoline-2,5,10(1H)-trione (20).
A solution of 19 (15 mg, 0.050 mmol) in CHCl3 (20 mL) that had been purged with N2 gas for 10 min was placed in a photoreactor and irradiated at 419 nm (spectral window of lamps: 373–460 nm). After TLC indicated that the starting material had been consumed (ca. 7 h), the reaction mixture was removed from the photoreactor and concentrated. Flash column chromatography (hexanes/ethyl acetate = 5:1) afforded 3 mg of 20 (20%) as a yellow solid.
Mp 241–243 °C; 1H NMR (400 MHz, CDCl3) 8.20 (broad s, 1H), 8.17 (dd, J = 7.4, 1.3 Hz, 1H), 8.07 (dd, J = 7.3, 1.3 Hz, 1H), 7.78–7.76 (m, 2H), 7.44 (dd, J = 7.9, 1.3 Hz, 1H), 7.19 (td, J = 7.5, 1.7, Hz, 1H), 7.11 (td, J = 7.6, 1.4 Hz, 1H), 7.00 (dd, J = 7.7, 1.6 Hz, 1H), 5.12 (dd, J = 8.8, 1.6 Hz, 1H), 3.05 (dd, J = 17.2, 8.9 Hz, 1H), 2.90 (ddd, J = 17.3, 1.7, 1.0 Hz, 1H); 13C{1H} NMR (126 MHz, CDCl3) δ 181.7, 179.2, 168.0, 140.2, 137.2, 135.3, 133.6, 133.4, 132.3, 130.8, 130.5, 129.2, 127.6, 127.1, 127.0, 126.7, 121.3, 36.5, 33.1; IR (neat) 3200, 3100, 2921, 2852, 1719, 1672, 1651, 1633, 1593, 1467, 1355, 1333, 1301, 1288, 1222, 1037, 750, 716 cm−1; HRMS (ESI) m/z: [M + Na]+ calcd for C19H12NO3Na35Cl 360.0403; found 360.0404, calcd for C19H12NO3Na37Cl 362.0374; found 360.0385.
(E)-N-(1,4-Dioxo-1,4-dihydronaphthalen-2-yl)-3-(3-methoxyphenyl)acrylamide (22).
A solution containing 6 (203 mg, 1.00 mmol), 3-methoxycinnamic acid (267 mg, 1.50 mmol), EDC· HCl (288 mg, 1.50 mmol), and 4-DMAP (24.4 mg, 0.200 mmol) in CH2Cl2 (10 mL) at rt was allowed to stir overnight. After the reaction mixture was concentrated, the resulting residue was dissolved in EtOAc (10 mL) and washed sequentially with sat. NH4Cl (aq., 10 mL), H2O (2 × 5 mL), and brine (5 mL). The organic layer was dried (Na2SO4) and concentrated. The resulting hydroquinone (E)-N-(1,4-dimethoxynaphthalen-2-yl)-3-(3-methoxyphenyl)acrylamide was used in the subsequent oxidation without additional purification.
To a solution of the hydroquinone (E)-N-(1,4-dimethoxynaphthalen-2-yl)-3-(3-methoxyphenyl)acrylamide from above in CH3CN (15 mL) at rt was added a mixture of ceric ammonium nitrate (1.2 g, 2.2 mmol) in CH3CN (4 mL) and H2O (4 mL). The reaction mixture was stirred for 15 min and then poured into H2O (15 mL). The aqueous phase was extracted with EtOAc (3 × 40 mL), and the combined organic extracts were washed with brine (120 mL), dried (Na2SO4), and concentrated. Flash column chromatography (toluene/ethyl acetate = 20:1) gave 150 mg of 22 (45%, 2 steps) as a yellow solid.
Mp 197–199 °C; 1H NMR (500 MHz, CDCl3) δ 8.52 (broad s, 1H), 8.13 (t, J = 7.1 Hz, 2H), 7.98 (s, 1H), 7.79 (t, J = 7.6 Hz, 1H), 7.76 (d, J = 15.5 Hz, 1H), 7.73 (partially obscured t, J = 7.3 Hz, 1H), 7.33 (dd, J = 8.0, 7.9 Hz, 1H), 7.18 (d, J = 7.7 Hz, 1H), 7.09 (broad s, 1H), 6.97 (dd, J = 8.2, 2.2 Hz, 1H), 6.62 (d, J = 15.5 Hz, 1H), 3.86 (s, 3H); 13C{1H} NMR (126 MHz, CDCl3) δ 185.3, 181.3, 164.7, 160.2, 144.8, 140.3, 135.5, 135.2, 133.4, 132.5, 130.2, 130.2, 126.9, 126.6, 121.1, 119.9, 117.5, 116.8, 113.4, 55.5; IR (neat) 3285, 3098, 2918, 1670, 1633, 1593, 1527, 1353, 1304, 1232, 1201, 1163, 1109, 974, 783, 732 cm−1; HRMS (ESI) m/z: [M + Na]+ calcd for C20H15NO4Na 356.0899; found 356.0897.
4-(3-Methoxyphenyl)-3,4-dihydrobenzo[g]quinoline-2,5,10(1H)-trione (23).
A solution of 22 (34.0 mg, 0.100 mmol) in CHCl3 (40 mL) was purged with N2 gas for 10 min, placed in a photoreactor, and irradiated at 419 nm (spectral window of lamps: 373–460 nm). After TLC indicated that the starting material had been consumed (ca. 12 h), the reaction mixture was concentrated. Flash column chromatography (hexanes/ethyl acetate = 5:1) afforded 6.9 mg of 23 (20%) as a yellow solid.
Mp 225–228 °C; 1H NMR (500 MHz, CDCl3) δ 8.15 (broad s, 1H), 8.11 (td, J = 7.1, 7.1, 0.9 Hz, 1H), 8.10 (d, J = 6.5 Hz, 1H), 7.76 (td, J = 7.4, 1.4 Hz, 1H), 7.71 (td, J = 7.4, 1.5 Hz, 1H), 7.20 (t, J = 8.0 Hz, 1H), 6.86 (d, J = 7.8 Hz, 1H), 6.84 (t J = 1.8 Hz, 1H), 6.76 (dd, J = 7.5, 1.8 Hz, 1H), 4.64 (d, J = 7.9 Hz, 1H), 3.76 (s, 3H), 3.03 (dd, J = 17.0, 8.2 Hz, 1H), 2.93 (d, J = 16.7 Hz, 1H); 13C{1H} NMR (126 MHz, CDCl3) δ 182.2, 179.3, 168.7, 160.1, 142.1, 138.5, 135.1, 133.5, 132.3, 130.4, 130.3, 127.0, 126.6, 122.4, 119.1, 113.3, 112.7, 55.4, 37.0, 35.4; IR (neat) 3296, 2924, 2854, 1706, 1672, 1633, 1594, 1489, 1463, 1452, 1331, 1299, 1219, 1140, 1050, 733 cm−1; HRMS (ESI) m/z: [M + Na]+ calcd for C20H15NO4Na 356.0899; found 356.0894.
N-(1,4-Dimethoxynaphthalen-2-yl)-3-methylbut-2-enamide (S6).
To a solution of 6 (203 mg, 1.00 mmol) in CH2Cl2 (5 mL) at rt was added 3,3-dimethylacryloyl chloride (122 μL, 1.10 mmol). After the reaction mixture was stirred for 15 min, a solution of Et3N (139 μL, 1.00 mmol) in CH2Cl2 (5 mL) was added to it dropwise over 10 min. After being stirred for an additional 2 h, the reaction mixture was concentrated. The resulting residue was dissolved in EtOAc (20 mL) and washed sequentially with sat. NH4Cl (aq., 10 mL), H2O (2 × 10 mL), and brine (10 mL). The organic phase was then dried (Na2SO4) and concentrated. Flash column chromatography (hexanes/ethyl acetate = 9:1) gave 265 mg of S6 (93%) as a white solid and a 1:1 mixture of two conformational isomers.
Mp 122–123 °C; 1H NMR (500 MHz, CDCl3) δ, 8.20 (d, J = 8.4 Hz, 1H), 8.12 (broad s, 1H), 7.93 (d, J = 8.4 Hz, 1H), 7.84 (broad s, 1H), 7.50 (dd, J = 7.8, 7.0 Hz, 1H), 7.38 (dd, J = 7.8, 7.1 Hz, 1H), 5.82 (s, 0.5H), 5.821 (s, 0.5H), 4.03 (s, 3H), 3.887 (s, 1.5H), 3.886 (s, 1.5H), 2.28 (s, 3H), 1.93 (s, 3H); 13C{1H} NMR (126 MHz, CDCl3) δ 165.2, 153.8, 152.2, 136.3, 128.5, 127.6, 126.9, 124.0, 123.0, 122.7, 120.9, 119.0, 98.7, 61.6, 55.9, 27.5, 20.1; IR (neat) 3313, 2934, 2850, 1669, 1626, 1599, 1491, 1454, 1416, 1368, 1279, 1255, 1211, 1158, 1140, 1095, 1030, 992, 976 cm−1; HRMS (ESI) m/z: [M + H]+ calcd for C17H20NO3 286.1443; found 286.1445.
N-(1,4-Dioxo-1,4-dihydronaphthalen-2-yl)-3-methylbut-2-en-amide (29).
To a solution of S6 (143 mg, 0.500 mmol) in CH3CN (8 mL) at rt was added a mixture of ceric ammonium nitrate (603 mg, 1.10 mmol) in CH3CN (2 mL) and H2O (2 mL). The resulting reaction mixture was stirred for 15 min and then poured into H2O (10 mL). The aqueous phase was extracted with EtOAc (3 × 20 mL), and the combined organic extracts were washed with brine (60 mL), dried (Na2SO4), and concentrated. Flash column chromatography (hexanes/ethyl acetate = 5:1) gave 111 mg of 29 (87%) as a yellow solid.
Mp 193–195 °C; 1H NMR (500 MHz, CDCl3) δ 8.25 (broad s, 1H), 8.09 (d, J = 7.5 Hz, 2H), 7.90 (s, 1H), 7.76 (t, J = 7.2 Hz, 1H), 7.70 (t, J = 7.4 Hz, 1H), 5.83 (s, 1H), 2.24 (s, 3H), 1.96 (s, 3H); 13C{1H} NMR (126 MHz, CDCl3) δ 185.3, 181.3, 165.3, 158.0, 140.6, 135.0, 133.2, 132.4, 130.2, 126.8, 126.5, 117.9, 116.5, 27.8, 20.5; IR (neat) 3308, 3079, 1667, 1639, 1612, 1594, 1493, 1338, 1308, 1218, 1200, 1146, 1105,1084 cm−1; HRMS (ESI) m/z: [M + H]+ calcd for C15H14NO3 256.0974; found 256.0975.
4,4-Dimethyl-3,4-dihydrobenzo[g]quinoline-2,5,10(1H)-trione (30).
A solution of 29 (12.8 mg, 0.0500 mmol) in CHCl3 (20 mL) was placed in two 10 mm NMR tubes and then purged with Ar for 10 min. The resulting solution was placed in the center of a photoreactor and irradiated with 419 nm light (spectral window: 373–460 nm) for 16 h after which the reaction mixture was concentrated. The resulting residue was subjected to flash column chromatography (hexanes/ethyl acetate = 2:1) to give 7.2 mg of 30 (56%) as a yellow solid.
Mp 198–201 °C; 1H NMR (500 MHz, CDCl3) δ 8.26 (broad s, 1H), 8.08 (dd, J = 7.7, 1.4 Hz, 1H), 8.06 (dd, J = 7.7, 1.3 Hz, 1H), 7.77 (td, J = 7.4, 1.3 Hz, 1H), 7.69 (td, J = 7.6, 1.3 Hz, 1H), 2.52 (s, 2H), 1.48 (s, 6H); 13C{1H} NMR (126 MHz, CDCl3) δ 182.8, 179.5, 168.8, 138.1, 135.1, 133.5, 133.1, 129.8, 127.2, 126.8, 126.2, 46.7, 35.3, 27.0; IR (neat) 3208, 3132, 2962, 2921, 1717, 1689, 1674, 1645, 1611, 1594, 1465, 1327, 1296, 1210 cm−1; HRMS (ESI) m/z: [M + H]+ calcd for C15H14NO3 256.0974; found 256.0977.
N-(1,4-Dimethoxynaphthalen-2-yl)-2,3-dimethylbut-2-enamide (S7).
To a solution of 2,3-dimethylbut-2-enoic acid (28.5 mg, 0.250 mmol) in CH2Cl2 (2 mL) were added oxalyl chloride (20 μL, 0.25 mmol) and a drop of DMF. The resulting reaction mixture was stirred at rt for 1 h after which a solution of 6 (55.9 mg, 0.280 mmol) in CH2Cl2 was added. After being stirred for an additional 10 min, Et3N (35 μL, 0.25 mmol) was added to the reaction mixture. After being stirred for an additional 1 h, the reaction mixture was concentrated. The residue was dissolved in EtOAc (5 mL) and washed with sat. NH4Cl (aq., 2 mL), H2O (2 × 2 mL), and brine (2 mL). The organic phase was dried (Na2SO4) and concentrated. Flash column chromatography (hexanes/ethyl acetate = 5:1) gave 40.7 mg (54%) of S7 as a colorless viscous oil.
1H NMR (500 MHz, CDCl3) δ 8.22 (d, J = 8.4 Hz, 1H), 8.14 (s, 1H), 7.93 (d, J = 8.4 Hz, 1H), 7.89 (broad s, 1H), 7.51 (dd, J = 7.9, 7.0 Hz, 1H), 7.40 (t, J = 7.6 Hz, 1H), 4.04 (s, 3H), 3.89 (s, 3H), 1.99 (s, 3H), 1.96 (s, 3H), 1.81 (s, 3H); 13C{1H} NMR (126 MHz, CDCl3) δ 170.7, 152.4, 136.5, 134.1, 128.2, 127.9, 127.6, 127.0, 124.2, 123.2, 122.8, 120.9, 98.6, 61.9, 56.0, 22.5, 20.6, 16.3; IR (neat) 3418, 2936, 2853, 1673, 1628, 1601, 1589, 1491, 1456, 1417, 1372, 1291, 1217, 1110, 1088, 996, 767 cm−1; HRMS (ESI) m/z: [M + Na]+ calcd for C18H21NO3Na 322.1419; found 322.1423.
N-(1,4-Dioxo-1,4-dihydronaphthalen-2-yl)-2,3-dimethylbut-2-enamide (31).
To a solution of S7 (21.1 mg, 0.0700 mmol) in CH3CN (2 mL) at rt was added a mixture of CAN (85.0 mg, 0.155 mmol) in CH3CN (1 mL) and H2O (1 mL). The resulting reaction mixture was stirred for 15 min and then poured into H2O (2 mL). The aqueous phase was extracted with EtOAc (3 × 5 mL), and the combined organic extracts were washed with brine (15 mL), dried (Na2SO4), and concentrated. Flash column chromatography (hexanes/ethyl acetate = 5:1) gave 14.8 mg (75%) of 31 as a yellow solid.
Mp 106–108 °C; 1H NMR (500 MHz, CDCl3) δ 8.39 (broad s, 1H), 8.11–8.09 (m, 2H), 7.91 (s, 1H), 7.78 (td, J = 7.5, 1.3 Hz, 1H), 7.71 (td, J = 7.5, 1.3 Hz, 1H), 1.94 (m, 3H), 1.92 (m, 3H), 1.81–1.80 (m, 3H); 13C{1H} NMR (126 MHz, CDCl3) δ 185.4, 181.4, 170.9, 140.2, 138.2, 135.1, 133.3, 132.3, 130.2, 126.8, 126.5, 126.2, 117.0, 22.6, 21.2, 16.1; IR (neat) 3376, 2919, 1666, 1649, 1616, 1594, 1495, 1334, 1300, 1198, 1083, 721 cm−1; HRMS (ESI) m/z: [M + Na]+ calcd for C16H15NO3Na 292.0950; found 292.0948.
3,4,4-Trimethyl-3,4-dihydrobenzo[g]quinoline-2,5,10(1H)-trione (32).
A solution of 31 (13.5 mg, 0.0500 mmol) in CHCl3 (20 mL) was placed in two 10 mm NMR tubes and purged with Ar for 10 min. The resulting solution was placed in the center of a photoreactor and irradiated with 419 nm light (spectral window of lamps: 373–460 nm) for 40 min after which the reaction mixture was concentrated. The resulting residue was subjected to flash column chromatography (hexanes/ethyl acetate = 5:1) to give 12.3 mg of 32 (91%) as a yellow solid.
Mp 172–174 °C; 1H NMR (500 MHz, CDCl3) δ 8.18 (broad s, 1H), 8.08 (dd, J = 7.2, 1.1 Hz, 1H), 8.06 (dd, J = 7.2, 1.1 Hz, 1H), 7.77 (td, J = 7.6, 1.2 Hz, 1H), 7.69 (td, J = 7.6, 1.2 Hz, 1H), 2.38 (q, J = 7.2 Hz, 1H), 1.43 (s, 3H), 1.41 (s, 3H), 1.22 (d, J = 7.2 Hz, 3H); 13C{1H} NMR (126 MHz, CDCl3) δ 183.2, 179.4, 172.7, 137.8, 135.1, 133.8, 133.0, 129.7, 126.8, 126.6, 126.1, 48.1, 38.1, 26.3, 21.9, 11.4; IR (neat) 3309, 2972, 1709, 1671, 1646, 1611, 1464, 1450, 1329, 1297 cm−1; HRMS (ESI) m/z: [M + Na]+ calcd for C16H15NO3Na 292.0950; found 292.0954.
2-Cyclopentylidene-N-(1,4-dimethoxynaphthalen-2-yl)-acetamide (S8).
To a solution of 2-cyclopentylideneacetic acid (121 mg, 0.960 mmol) in CH2Cl2 (10 mL) at rt were added oxalyl chloride (82 μL, 0.96 mmol) and one drop of DMF. After being stirred for 1 h, amine 6 (162 mg, 0.800 mmol) was added to the reaction mixture. After an additional 10 min, Et3N (134 μL, 0.96 mmol) was added dropwise and the reaction mixture was stirred for another 1 h after which it was concentrated. The resulting residue was dissolved in EtOAc (20 mL) and washed sequentially with sat. NH4Cl (aq., 10 mL), H2O (2 × 10 mL), and brine (10 mL). The organic phase was then dried (Na2SO4) and concentrated. Flash column chromatography (hexanes/ethyl acetate = 5:1) gave 157 mg of S8 (63%) as a white solid.
Mp 136–137 °C; 1H NMR (500 MHz, CDCl3) δ 8.20 (d, J = 8.4 Hz, 1H), 8.15 (broad s, 1H), 7.92 (d, J = 8.4 Hz, 1H), 7.83 (broad s, 1H), 7.51 (td, J = 6.8, 1.0 Hz, 1H), 7.38 (td, J = 7.6, 1.0 Hz, 1H), 5.98 (t, J = 2.1 Hz, 1H), 4.03 (s, 3H), 3.88 (s, 3H), 2.94 (t, J = 7.3 Hz, 2H), 2.51 (t, J = 7.1 Hz, 2H), 1.81 (p, J = 6.9 Hz, 2H), 1.70 (p, J = 6.8 Hz, 2H); 13C{1H} NMR (126 MHz, CDCl3) δ 166.6, 165.2, 152.2, 136.1, 128.7, 127.6, 126.9, 124.0, 122.9, 122.7, 120.8, 114.6, 98.6, 61.6, 56.0, 36.2, 32.5, 26.7, 25.5; IR (neat) 3314, 2954, 2926, 2854, 1669, 1627, 1603, 1520, 1495, 1456, 1417, 1377, 1255, 1216, 1190, 1118, 1094 cm−1; HRMS (ESI) m/z: [M + H]+ calcd for C19H22NO3 312.1600; found 312.1603.
2-Cyclopentylidene-N-(1,4-dioxo-1,4-dihydronaphthalen-2-yl)-acetamide (33).
To a solution of S8 (141 mg, 0.500 mmol) in CH3CN (7 mL) at rt was added a mixture of ceric ammonium nitrate (603 mg, 1.10 mmol) in CH3CN (2 mL) and H2O (2 mL). The reaction mixture was stirred for 15 min and then poured into H2O (10 mL). The aqueous phase was extracted with EtOAc (3 × 20 mL), and the combined organic extracts were washed with brine (60 mL), dried (Na2SO4), and concentrated. Flash column chromatography (hexanes/ethyl acetate = 5:1) gave 115 mg of 33 (92%) as a yellow solid.
Mp 166–167 °C; 1H NMR (500 MHz, CDCl3) δ 8.28 (broad s, 1H), 8.08 (d, J = 7.3 Hz, 2H), 7.91 (s, 1H), 7.76 (td, J = 7.5, 1.3 Hz, 1H), 7.70 (td, J = 7.6, 1.3 Hz, 1H), 5.99 (p, J = 2.1 Hz, 1H), 2.87 (t, J = 7.3 Hz, 2H), 2.50 (t, J = 7.1 Hz, 2H), 1.79 (p, J = 6.9 Hz, 2H), 1.69 (p, J = 7.0 Hz, 2H); 13C{1H} NMR (126 MHz, CDCl3) δ 185.3, 181.4, 170.9, 165.3, 140.6, 135.0, 133.2, 132.4, 130.1, 126.7, 126.4, 116.2, 113.5, 36.5, 33.0, 26.6, 25.5; IR (neat) 3274, 2947, 1668, 1634, 1611, 1593, 1512, 1340, 1302, 1201, 1106 cm−1; HRMS (ESI) m/z: [M + H]+ calcd for C17H16NO3 282.1130; found 282.1131.
1H-Spiro[benzo[g]quinoline-4,1′-cyclopentane]-2,5,10(3H)-trione (34).
A solution of 33 (14.1 mg, 0.0500 mmol) in CHCl3 (20 mL) was placed in two 10 mm NMR tubes and then purged with Ar for 10 min. The resulting solution was placed in the center of a photoreactor and irradiated with 419 nm light (spectral window: 373–460 nm) for 26 h after which the reaction mixture was concentrated. The resulting residue was subjected to flash column chromatography (hexanes/ethyl acetate = 5:1) to give 6.8 mg of 34 (48%) as a yellow solid.
Mp 196–198 °C; 1H NMR (500 MHz, CDCl3) δ 8.21 (broad s, 1H), 8.07 (d, J = 7.8 Hz, 2H), 7.77 (td, J = 7.5, 0.8 Hz, 1H), 7.69 (t, J = 7.6 Hz, 1H), 2.57 (s, 2H), 2.30 (dt, J = 12.5, 7.4 Hz, 2H), 2.04–1.96 (m, 2H), 1.81–1.73 (m, 2H), 1.61 (dt, J = 12.1, 6.0 Hz, 2H); 13C{1H} NMR (126 MHz, CDCl3) δ 182.7, 179.3, 168.6, 138.6, 135.1, 133.5, 133.1, 129.8, 127.7, 126.8, 126.2, 44.7, 44.6, 38.4, 26.2; IR (neat) 3010, 2956, 2868, 1716, 1667, 1642, 1608, 1594, 1578, 1465, 1329, 1299, 1288, 1207, 1147, 998, 714 cm−1; HRMS (ESI) m/z: [M + H]+ calcd for C17H16NO3 282.1130; found 282.1131.
2-Cyclohexylidene-N-(1,4-dimethoxynaphthalen-2-yl)acetamide (S9).
To a solution of 2-cyclohexylideneacetic acid (168 mg, 1.20 mmol) in CH2Cl2 (10 mL) at rt were added oxalyl chloride (103 μL, 1.20 mmol) and one drop of DMF. After being stirred for 1 h, a solution of 6 (203 mg, 1.00 mmol) in CH2Cl2 (2 mL) was added to the reaction mixture. After 10 min, Et3N (167 μL, 1.20 mmol) was added dropwise and the reaction mixture was stirred for an additional 1 h after which it was concentrated. The resulting residue was dissolved in EtOAc (20 mL) and washed sequentially with sat. NH4Cl (aq., 10 mL), H2O (2 × 10 mL), and brine (10 mL). The organic phase was then dried (Na2SO4) and concentrated. Flash column chromatography (hexanes/ethyl acetate = 10:1) gave 215 mg of S9 (66%) as a white solid.
Mp 132–134 °C; 1H NMR (500 MHz, CDCl3) δ 8.20 (d, J = 8.4 Hz, 1H), 8.14 (broad s, 1H), 7.93 (d, J = 8.4 Hz, 1H), 7.86 (broad s, 1H), 7.51 (t, J = 7.2 Hz, 1H), 7.39 (t, J = 7.4 Hz, 1H), 5.76 (s 1H), 4.03 (s, 3H), 3.89 (s, 3H), 2.96–2.94 (m, 2H), 2.27–2.24 (m, 2H), 1.73–1.67 (m, 4H), 1.65–1.61 (m, 2H); 13C{1H} NMR (126 MHz, CDCl3) δ 165.4, 161.0, 152.3, 136.2, 128.5, 127.6, 127.0, 124.1, 123.0, 122.7, 120.9, 116.3, 98.6, 61.7, 56.0, 38.2, 29.9, 28.8, 28.1, 26.5; IR (neat) 3316, 2924, 2852, 1665, 1627, 1601, 1519, 1493, 1455, 1417, 1370, 1255, 1217, 1163, 1124, 1092, 995, 978, 847, 765 cm−1; HRMS (ESI) m/z: [M + H]+ calcd for C20H24NO3 326.1756; found 326.1756.
2-Cyclohexylidene-N-(1,4-dioxo-1,4-dihydronaphthalen-2-yl)-acetamide (35).
To a solution of S9 (163 mg, 0.500 mmol) in CH3CN (7 mL) at rt was added a mixture of ceric ammonium nitrate (603 mg, 1.10 mmol) in CH3CN (2 mL) and H2O (2 mL). The reaction mixture was stirred for 15 min and then poured into H2O (10 mL). The aqueous phase was extracted with EtOAc (3 × 20 mL), and the combined organic extracts were washed with brine (60 mL), dried (Na2SO4), and concentrated. Flash column chromatography (hexanes/ethyl acetate = 10:1) gave 132 mg of 35 (90%) as a yellow solid.
Mp 182–184 °C; 1H NMR (500 MHz, CDCl3) δ 8.29 (broad s, 1H), 8.08 (d, J = 7.7 Hz, 2H), 7.89 (s, 1H), 7.76 (td, J = 7.4, 1.3 Hz, 1H), 7.69 (td, J = 7.6, 1.3 Hz, 1H), 5.75 (s, 1H), 2.88 (t, J = 5.5 Hz, 2H), 2.24 (t, J = 5.5 Hz, 2H), 1.69–1.61 (m, 6H); 13C{1H} NMR (126 MHz, CDCl3) δ 185.3, 181.3, 165.5, 165.0, 140.5, 135.0, 133.2, 132.4, 130.1, 126.7, 126.4, 116.5, 115.0, 38.3, 30.0, 28.9, 28.0, 26.3; IR (neat) 3330, 2927, 2854, 1664, 1643, 1609, 1594, 1500, 1339, 1307, 1192, 1151, 1101 cm−1; HRMS (ESI) m/z: [M + Na]+ calcd for C18H17NO3Na 318.1106; found 318.1111.
1H-Spiro[benzo[g]quinoline-4,1′-cyclohexane]-2,5,10(3H)-trione (36).
A solution of 35 (14.8 mg, 0.0500 mmol) in CHCl3 (20 mL) was placed in two 10 mm NMR tubes and then purged with Ar for 10 min. The resulting solution was placed in the center of a photoreactor and irradiated with 419 nm light (spectral window: 373–460 nm) for 10 h after which the reaction mixture was concentrated. The resulting residue was subjected to flash column chromatography (hexanes/ethyl acetate = 5:1) to give 12.1 mg of 36 (82%) as a yellow solid.
Mp 227–229 °C; 1H NMR (500 MHz, CDCl3) δ 8.32 (broad s, 1H), 8.05 (d, J = 7.7 Hz, 1H), 8.04 (d, J = 7.3 Hz, 1H), 7.76 (td, J = 7.6, 1.0 Hz, 1H), 7.67 (td, J = 7.6, 0.8 Hz, 1H), 2.79 (s, 2H), 2.55 (t, J = 3.5 Hz, 1H), 2.54 (t, J = 4.3 Hz, 1H), 1.76–1.74 (m, 1H), 1.66–1.63 (m, 2H), 1.56–1.47 (m, 4H), 1.43–1.34 (m, 1H); 13C{1H} NMR (126 MHz, CDCl3) δ 183.1, 179.5, 168.7, 138.6, 135.1, 133.9, 133.0, 129.6, 127.3, 126.9, 126.0, 39.4, 39.1, 32.5, 25.3, 21.1; IR (neat) 3230, 3124, 2923, 2852, 1715, 1666, 1641, 1604, 1464, 1367, 1296, 712 cm−1; HRMS (ESI) m/z: [M + H]+ calcd for C18H18NO3 296.1287; found 296.1291.
(E)-N-(1, 4-Dimethoxynaphthalen-2-yl)-2-(2-methylcyclohexylidene)acetamide (S10).
To a solution of (E)-2-(2-methylcyclohexylidene)acetic acid (254 mg, 1.65 mmol) in CH2Cl2 (30 mL) at rt were added oxalyl chloride (154 μL, 1.80 mmol) and one drop of DMF. After being stirred for 1 h, a solution of 6 (305 mg, 1.5 mmol) in CH2Cl2 (10 mL) was added to the reaction mixture. This was followed by the dropwise addition of NEt3 (209 μL, 1.50 mmol) after 10 min. The resulting reaction mixture was stirred for another 1 h and then concentrated. The resulting residue was dissolved in EtOAc (30 mL) and washed sequentially with sat. NH4Cl (aq., 20 mL), H2O (2 × 20 mL), and brine (20 mL). The organic phase was dried (Na2SO4) and concentrated. Flash column chromatography (hexanes/ethyl acetate = 10:1) gave 214 mg of S10 (42%) as a white solid as its E isomer.
Mp 167–169 °C; 1H NMR (500 MHz, CDCl3) δ 8.21 (d, J = 8.4 Hz, 1H), 8.15 (broad s, 1H), 7.94 (d, J = 8.4 Hz, 1H), 7.88 (broad s, 1H), 7.51 (t, J = 7.3 Hz, 1H), 7.39 (t, J = 7.5 Hz, 1H), 5.72 (s, 1H), 4.03 (s, 3H), 3.90 (s, 3H), 3.55–3.52 (m, 1H), 2.35–2.27 (m, 2H), 1.92–1.76 (m, 3H), 1.60–1.51 (m, 2H), 1.36–1.30 (m, 1H), 1.14 (d, J = 6.7 Hz, 3H); 13C{1H} NMR (126 MHz, CDCl3) δ 166.0, 164.0, 152.3, 136.3, 128.6, 127.6, 126.9, 124.1, 123.1, 122.7, 120.9, 114.2, 98.7, 61.7, 56.0, 39.9, 37.0, 29.4, 28.6, 25.2, 18.7; IR (neat) 2930, 2852, 1663, 1626, 1601, 1520, 1494, 1456, 1418, 1371, 1216, 1170, 1094, 994, 849, 767 cm−1; HRMS (ESI) m/z: [M + H]+ calcd for C21H26NO3 340.1913; found 340.1912.
(E)-N-(1,4-Dioxo-1,4-dihydronaphthalen-2-yl)-2-(2-methylcyclohexylidene)acetamide (37).
To a solution of S10 (163 mg, 0.410 mmol) in CH3CN (16 mL) at rt was added a mixture of ceric ammonium nitrate (499 mg, 0.910 mmol) in CH3CN (2 mL) and H2O (2 mL). The reaction mixture was stirred for 15 min and then poured into H2O (10 mL). The aqueous phase was extracted with EtOAc (3 × 30 mL), and the combined organic extracts were washed with brine (60 mL), dried (Na2SO4), and concentrated. Flash column chromatography (hexanes/ethyl acetate = 10:1) gave 99 mg of 37 (81%) as a yellow solid.
Mp 140–142 °C; 1H NMR (500 MHz, CDCl3) δ 8.32 (broad s, 1H), 8.08 (dd, J = 7.5, 1.1 Hz, 1H), 8.07 (dd, J = 7.5, 1.1 Hz, 1H), 7.89 (s, 1H), 7.75 (td, J = 7.5, 1.3 Hz, 1H), 7.68 (td, J = 7.6, 1.3 Hz, 1H), 5.70 (s, 1H), 3.54–3.50 (m, 1H), 2.31–2.19 (m, 2H), 1.90–1.73 (m, 3H), 1.55–1.47 (m, 2H), 1.31–1.24 (m, 1H), 1.10 (d, J = 6.7 Hz, 3H); 13C{1H} NMR (126 MHz, CDCl3) δ 185.2, 181.3, 168.4, 166.0, 140.6, 135.0, 133.2, 132.4, 130.1, 126.7, 126.4, 116.5, 112.9, 40.2, 37.2, 29.6, 28.6, 25.1, 18.5; IR (neat) 3332, 2928, 2854, 1696, 1667, 1644, 1612, 1594, 1496, 1335, 1301, 1230, 1198, 1151, 1102, 1082, 886, 783 cm−1; HRMS (ESI) m/z: [M + Na]+ calcd for C19H19NO3Na 332.1263, found 332.1262.
2′-Methyl-1H-spiro[benzo[g]quinoline-4,1′-cyclohexane]-2,5,10(3H)-trione (38).
A solution of 37 (14.8 mg, 0.0500 mmol) in CHCl3 (20 mL) was placed in two 10 mm NMR tubes and then purged with Ar for 10 min. The resulting solution was placed in the center of a photoreactor and irradiated with 419 nm light (spectral window of lamps: 373–460 nm) for 10 h after which the reaction mixture was concentrated. The resulting residue was subjected to flash column chromatography (hexanes/ethyl acetate = 5:1) to give 13.6 mg of 38 (92%, d.r. > 20:1) as a yellow solid.
Mp 158–160 °C; 1H NMR (500 MHz, CDCl3) δ 8.22 (broad s, 1H), 8.07 (ddd, J = 7.9, 6.8, 1.2 Hz, 2H), 7.77 (td, J = 7.5, 7.5, 1.4 Hz, 1H), 7.68 (td, J = 7.6, 7.6, 1.3 Hz, 1H), 2.99 (ddq, J = 10.7, 6.9, 3.9 Hz, 1H), 2.79 (d, J = 16.9 Hz, 1H), 2.63 (d, J = 16.9 Hz, 1H), 2.34 (td, J = 13.8, 13.8, 5.0 Hz, 1H), 1.78–1.74 (m, 1H), 1.67–1.44 (m, 5H), 1.32–1.23 (m, 1H), 0.79 (d, J = 6.9 Hz, 3H); 13C{1H} NMR (126 MHz, CDCl3) δ 183.1, 179.3, 169.2, 139.7, 135.1, 133.8, 133.0, 129.7, 127.0, 126.1, 126.0, 44.1, 35.4, 34.9, 34.3, 29.3, 25.9, 20.8, 18.0; IR (neat) 3307, 2927, 2856, 1705, 1669, 1645, 1611, 1595, 1579, 1497, 1463, 1452, 1334, 1292, 1231, 1200, 1152, 1103, 721 cm−1; HRMS (ESI) m/z: [M + Na]+ calcd for C19H19NO3Na 332.1263, found 332.1263.
N-(1,4-Dimethoxynaphthalen-2-yl)cyclohex-1-ene-1-carboxamide (S11).
To a solution of 6 (203 mg, 1.00 mmol) in CH2Cl2 (10 mL) at rt were added 1-cyclohexene-1-carboxylic acid (189 mg, 1.50 mmol), EDC·HCl (288 mg, 1.50 mmol), and 4-DMAP (24.4 mg, 0.200 mmol). The reaction mixture was stirred overnight and then concentrated. The resulting residue was dissolved in EtOAc (10 mL) and washed sequentially with saturated NH4Cl (aq., 10 mL), H2O (2 × 5 mL), and brine (5 mL). The organic layer was dried (Na2SO4) and concentrated. The residue was purified using flash column chromatography (hexanes/ethyl acetate = 10:1) to provide 207 mg of S11 (66%) as a purple foam.
1H NMR (500 MHz, CDCl3): δ 8.26 (bs, 1H), 8.21 (d, J = 8.4 Hz, 1H), 8.13 (s, 1H), 7.94 (d, J = 8.4 Hz, 1H), 7.51 (ddd, J = 8.1, 6.8, 1.2 Hz, 1H), 7.39 (ddd, J = 8.2, 6.8, 1.2 Hz, 1H), 6.87 (septet, J = 1.9 Hz, 1H), 4.02 (s, 3H), 3.91 (s, 3H), 2.46–2.42 (m, 2H), 2.30–2.26 (m, 2H), 1.81–1.77 (m, 2H), 1.71–1.66 (m, 2H). 13C{1H} NMR (126 MHz, CDCl3): δ 166.6, 152.4, 136.7, 135.1, 134.0, 128.4, 127.6, 127.0, 124.2, 123.1, 122.7, 120.9, 98.8, 61.6, 55.9, 25.8, 24.5, 22.3, 21.6; IR (neat) 3423, 2999, 2934, 2857, 1672, 1627, 1600, 1588, 1520, 1493, 1455, 1417, 1372, 1271, 1259, 1221, 1161, 1098, 1081, 995, 970 cm−1; HRMS (ESI) m/z: [M + H]+ calcd for C19H22NO3 312. 1600; found 312.1603.
N-(1,4-Dioxo-1,4-dihydronaphthalen-2-yl)cyclohex-1-ene-1-carboxamide (39).
To a solution of S11 (207 mg, 0.736 mmol) in CH3CN (6 mL) at rt was added a mixture of ceric ammonium nitrate (800 mg, 1.46 mmol) in CH3CN (1 mL) and H2O (1 mL). The resulting reaction mixture was stirred for 15 min and then poured into H2O (5 mL). The aqueous phase was extracted with EtOAc (3 × 8 mL), and the combined organic extracts were washed with brine (15 mL), dried (Na2SO4), and concentrated. Flash column chromatography (toluene/ethyl acetate = 20:1) gave 148 mg of 39 (80%) as a yellow solid.
Mp 177–180 °C; 1H NMR (500 MHz, CDCl3) δ 8.77 (s, 1H), 8.12–8.10 (m, 2H), 7.90 (s, 1H), 7.78 (td, J = 7.5, 1.4 Hz, 1H), 7.72 (td, J = 7.6, 1.3 Hz, 1H), 6.88 (ddd, J = 5.6, 3.8, 1.6 Hz, 1H), 2.40–2.37 (m, 2H), 2.30–2.26 (m, 2H), 1.78–1.73 (m, 2H), 1.68–1.64 (m, 2H); 13C{1H} NMR (126 MHz, CDCl3) δ 185.2, 181.4, 166.8, 140.2, 137.4, 135.0, 133.4, 133.2, 132.4, 130.1, 126.7, 126.4, 116.8, 25.8, 24.1, 22.0, 21.4; IR (neat) 3371, 2933, 1666, 1648, 1500, 1334, 1298, 1213, 1081 cm−1; HRMS (ESI) m/z: [M + H]+ calcd for C17H16NO3 282.1130; found 282.1126.
2,3,4,4a,6,12b-Hexahydrobenzo[b]phenanthridine-5,7,12(1H)-trione (40).
A solution of 39 (14.1 mg, 0.05 mmol) in CHCl3 (20 mL) was placed in two 10 mm NMR tubes that had been purged with Ar for 10 min. The resulting solution was placed in the center of a photoreactor and irradiated with 419 nm light (spectral window: 373–460 nm) for 45 min after which the reaction mixture was concentrated. The resulting residue was subjected to flash column chromatography (hexanes/ethyl acetate = 5:1) to give 12 mg of 40 (85%, cis/trans ≥ 20:1) as a yellow solid.
Mp 255–257 °C; 1H NMR (500 MHz, CDCl3) δ 8.11 (d, J = 7.6 Hz, 1H), 8.07 (d, J = 7.6 Hz, 1H), 8.03 (broad s, 1H), 7.76 (t, J = 7.5 Hz, 1H), 7.69 (t, J = 7.5 Hz, 1H), 3.31 (ddd, J = 12.7, 6.1, 4.0 Hz, 1H), 2.78 (broad s, 1H), 2.55 (broad d, J = 13.4 Hz, 1H), 1.81–1.76 (m, 2H), 1.68–1.65 (m, 1H), 1.55–1.33 (m, 3H), 1.25–1.17 (m, 1H); 13C{1H} NMR (126 MHz, CDCl3) δ 182.2, 179.3, 171.4, 137.8, 134.9, 133.3, 132.5, 130.5, 126.7, 126.5, 124.8, 39.8, 32.9, 27.5, 26.0, 24.0, 22.3; IR (neat) 3298, 2926, 2843, 1709, 1666, 1632, 1593, 1468, 1453, 1366, 1333, 1307, 1210, 718 cm−1; HRMS (ESI) m/z: [M + H]+ calcd for C17H16NO3 282.1130; found 282.1128.
(1R,2S,3R)-5-((1,4-Dimethoxynaphthalen-2-yl)carbamoyl)-cyclohex-4-ene-1,2,3-triyl Triacetate (S12).
To a solution of 6 (203 mg, 1.00 mmol) in CH2Cl2 (20 mL) at rt were added (3R,4S,5R)-3,4,5-triacetoxycyclohex-1-ene-1-carboxylic acid (901 mg, 3.00 mmol), EDC·HCl (575 mg, 3.00 mmol), and 4-DMAP (24.4 mg, 0.200 mmol). The reaction mixture was stirred overnight and then concentrated. The resulting residue was dissolved in EtOAc (20 mL) and washed sequentially with sat. NH4Cl (aq., 10 mL), H2O (2 × 10 mL), and brine (10 mL). The organic layer was dried (Na2SO4) and concentrated. Purification using flash column chromatography (hexanes/ethyl acetate = 3:1) provided 471 mg of S12 (97%) as a white solid.
Mp 84–86 °C; 1H NMR (500 MHz, CDCl3) δ 1:1 mixture of two conformers, 8.28 (broad s, 1H), 8.19 (d, J = 8.3 Hz, 1H), 7.94 (s, 1H), 7.92 (partially obscured d, J = 8.5 Hz, 1H), 7.50 (ddd, J = 8.2, 6.8, 1.2 Hz, 1H), 7.39 (ddd, J = 8.3, 6.8, 1.2 Hz, 1H), 6.51 (dt, J = 4.1, 2.1 Hz, 1H), 5.78 (t, J = 4.0, 4.0 Hz, 1H), 5.42–5.36 (m, 1H), 5.32–5.26 (m, 1H), 3.98 (s, 1.5H), 3.97 (s, 1.5H), 3.89 (s, 3H), 3.17–3.08 (m, 1H), 2.59–2.51 (m, 1H), 2.10 (s, 3H), 2.09 (s, 3H), 2.07 (s, 3H); 13C{1H} NMR (126 MHz, CDCl3) δ 170.1, 170.0, 169.9, 164.2, 152.2, 137.0, 136.2, 127.4, 127.3, 127.1, 124.6, 123.4, 122.7, 120.9, 98.3, 68.0, 66.7, 66.1, 61.8, 55.8, 29.0, 21.0, 20.9, 20.8; IR (neat) 3422, 2940, 1748, 1680, 1603, 1499, 1373, 1248, 1226, 1043 cm−1; HRMS (ESI) m/z: [M + Na]+ calcd for C25H27NO9Na 508.1584; found 508.1590.
(1R,2S,3R)-5-((1,4-Dioxo-1,4-dihydronaphthalen-2-yl)-carbamoyl)cyclohex-4-ene-1,2,3-triyl Triacetate (41).
To a solution of S12 (336 mg, 0.690 mmol) in CH3CN (9 mL) at rt was added a mixture of ceric ammonium nitrate (883 mg, 1.52 mmol) in CH3CN (2.5 mL) and H2O (2.5 mL). The resulting reaction mixture was stirred for 15 min and then poured into H2O (20 mL). The aqueous phase was extracted with EtOAc (3 × 20 mL), and the combined organic extracts were washed with brine (60 mL), dried (Na2SO4), and concentrated. Flash column chromatography (hexanes/ethyl acetate = 2:1) gave 272 mg of 41 (86%) as a yellow solid.
Mp 146–148 °C; 1H NMR (500 MHz, CDCl3) δ 8.80 (s, 1H), 8.13 (d, J = 7.5 Hz, 2H), 7.87 (s, 1H), 7.81 (td, J = 7.6, 1.2 Hz, 1H), 7.74 (td, J = 7.5, 1.2 Hz, 1H), 6.55–6.54 (m, 1H), 5.81 (broad s, 1H), 5.36 (ddd, J = 7.7, 5.1, 5.1 Hz, 1H), 5.32 (dd, J = 7.7, 3.9 Hz, 1H), 3.04–3.00 (m, 1H), 2.58–2.54 (m, 1H), 2.12 (s, 3H), 2.10 (s, 3H), 2.08 (s, 3H); 13C{1H} NMR (126 MHz, CDCl3) δ 185.1, 181.1, 170.0, 169.9, 169.9, 164.7, 139.7, 135.3, 134.8, 133.5, 132.3, 129.9, 129.6, 126.9, 126.6, 117.7, 67.4, 66.6, 66.0, 28.3, 21.1, 20.9, 20.9; IR (neat) 3364, 2940, 1748, 1669, 1653, 1506, 1370, 1336, 1301, 1225, 1044, 719 cm−1; HRMS (ESI) m/z: [M + Na]+ calcd for C23H21NO9Na 478.1114; found 478.1105.
(1R,2S,3R,4aS,12bR)-5,7,12-Trioxo-1,2,3,4,4a,5,6,7,12,12bdecahydrobenzo[b]phenanthridine-1,2,3-triyl Triacetate (42, 43).
A solution of 41 (22.8 mg, 0.0500 mmol) in CHCl3 (20 mL) was placed in two 10 mm NMR tubes and then purged with Ar for 10 min. The resulting solution was placed in the center of a photoreactor and irradiated with 419 nm light (spectral window: 373–460 nm) for 10.5 h after which the reaction mixture was concentrated. The resulting residue was subjected to flash column chromatography (hexanes/ethyl acetate = 2:1) to give 18.5 mg of 42 and 43 (81%, 42:43 = 1.2:1) as yellow solids.
42:
Mp 252–254 °C; 1H NMR (500 MHz, CDCl3) δ 1:1 mixture of two conformers, 8.16 (broad s, 1H), 8.11 (dd, J = 8.0, 0.5 Hz, 2H), 7.79 (td, J = 7.4, 1.0 Hz, 1H), 7.73 (td, J = 7.4, 1.1 Hz, 1H), 5.34 (dd, J = 11.6, 2.6 Hz, 1H), 5.27 (t, J = 3.4 Hz, 1H), 5.03 (dd, J = 6.4, 2.8 Hz, 1H), 4.00 (dd, J = 11.6, 6.7 Hz, 1H), 2.95–2.89 (m, 2H), 2.225 (s, 1.5H), 2.224 (s, 1.5H), 2.049 (s, 1.5H), 2.048 (s, 1.5H), 2.00 (ddd, J = 15.2, 5.9, 2.9 Hz, 1H), 1.807 (s, 1.5H), 1.806 (s, 1.5H); 13C{1H} NMR (126 MHz, CDCl3) δ 182.0, 178.6, 170.3, 169.7, 169.7, 169.6, 139.0, 135.2, 133.6, 132.1, 130.3, 126.8, 126.6, 119.8, 68.6, 68.1, 67.7, 37.6, 30.9, 21.8, 21.3, 21.1, 20.7; IR (neat) 3289, 2840, 1745, 1675, 1639, 1467, 1368, 1329, 1302, 1221, 1053, 724 cm−1; HRMS (ESI) m/z: [M + Na]+ calcd C23H21NO9Na 478.1114; found 478.1101.
43:
Mp 286–288 °C; 1H NMR (500 MHz, CDCl3) δ 8.13 (d, J = 7.8 Hz, 1H), 8.08 (d, J = 7.5 Hz, 1H), 7.95 (broad s, 1H), 7.79 (t, J = 7.5 Hz, 1H), 7.72 (t, J = 7.4 Hz, 1H), 5.56 (t, J = 2.4 Hz, 1H), 5.38 (td, J = 11.0, 5.2 Hz, 1H), 5.11 (dd, J = 10.3, 2.5 Hz, 1H), 3.71 (dd, J = 7.6, 2.1 Hz, 1H), 3.07 (ddd, J = 13.4, 5.0, 1.8 Hz, 1H), 2.99 (t, J = 5.8 Hz, 1H), 2.06 (s, 3H), 1.96 (s, 3H), 1.93 (s, 3H), 1.76 (ddd, J = 13.0, 13.0, 5.7 Hz, 1H); 13C{1H} NMR (126 MHz, CDCl3) δ 181.4, 178.9, 170.0, 169.9, 169.8, 169.0, 140.4, 135.4, 133.7, 132.0, 130.2, 127.1, 126.7, 117.7, 73.3, 71.9, 66.5, 37.0, 35.5, 27.2, 21.1, 20.8, 20.7; IR (neat) 3282, 2930, 1742, 1673, 1635, 1594, 1467, 1454, 1370, 1330, 1299, 1210, 1161, 1046, 960, 722 cm−1; LRMS (ESI) m/z: [M + Na]+ calcd C23H21NO9Na 478.1114; found 478.1110.
(Z)-1-((1,4-Dimethoxynaphthalen-2-yl)amino)-1-oxobut-2-en-2-yl Acetate (S13).
To a solution of 6 (203 mg, 1.00 mmol) in CH2Cl2 (20 mL) at rt were added carboxylic acid (Z)-2-acetoxybut-2-enoic acid (216 mg, 1.50 mmol), EDC·HCl (288 mg, 1.50 mmol), and 4-DMAP (24.4 mg, 0.200 mmol). The resulting reaction mixture was stirred overnight and then concentrated. The resulting residue was dissolved in EtOAc (20 mL) and washed sequentially with sat. NH4Cl (aq., 10 mL), H2O (2 × 10 mL), and brine (10 mL). The organic layer was dried (Na2SO4), concentrated, and purified using flash column chromatography (hexanes/ethyl acetate = 5:1) to provide 233 mg of S13 (71%, isolated as the Z isomer) as a white solid.
Mp 102–104 °C; 1H NMR (500 MHz, CDCl3) δ 8.47 (broad s, 1H), 8.21 (d, J = 8.4 Hz, 1H), 8.02 (s, 1H), 7.92 (d, J = 8.4 Hz, 1H), 7.51 (td, J = 7.6, 1.2 Hz, 1H), 7.40 (td, J = 7.7, 1.2 Hz, 1H), 6.73 (q, J = 7.2 Hz, 1H), 4.01 (s, 3H), 3.87 (s, 3H), 2.39 (s, 3H), 1.76 (d, J = 7.2 Hz, 3H); 13C{1H} NMR (126 MHz, CDCl3) δ 167.7, 159.8, 152.4, 141.5, 136.9, 127.4, 127.3, 127.1, 124.5, 124.1, 123.4, 122.7, 120.8, 98.4, 61.7, 55.9, 20.5, 12.1; IR (neat) 3415, 3072, 2940, 2853, 1769, 1691, 1662, 1628, 1603, 0589, 1522, 1500, 1458, 1421, 1374, 1266, 1213, 1187, 1095, 1065 cm−1; HRMS (ESI) m/z: [M + H]+ calcd for C18H20NO5 330.1341; found 330.1348.
(Z)-1-((1,4-Dioxo-1,4-dihydronaphthalen-2-yl)amino)-1-oxobut-2-en-2-yl Acetate (45).
To a solution of S13 (157 mg, 0.477 mmol) in CH3CN (7 mL) at rt was added a mixture of CAN (575 mg, 1.05 mmol) in CH3CN (1 mL) and H2O (1 mL). The reaction mixture was stirred for 15 min and then poured into H2O (5 mL). The aqueous phase was extracted with EtOAc (3 × 10 mL), and the combined organic extracts were washed with brine (20 mL), dried (Na2SO4), and concentrated. Flash column chromatography (hexanes/ethyl acetate = 5:1) gave 96 mg of 45 (67%) as a yellow solid.
Mp 168–170 °C; 1H NMR (500 MHz, CDCl3) δ 8.93 (broad s, 1H), 8.09 (dd, J = 7.5, 1.2 Hz, 1H), 8.08 (dd, J = 8.4, 1.2 Hz, 1H), 7.84 (s, 1H), 7.78 (td, J = 7.6, 1.3 Hz, 1H), 7.72 (td, J = 7.6, 1.3 Hz, 1H), 6.74 (q, J = 7.2 Hz, 1H), 2.41 (s, 3H), 1.76 (d, J = 7.2 Hz, 3H); 13C{1H} NMR (126 MHz, CDCl3) δ 185.1, 181.0, 167.7, 160.5, 140.5, 139.7, 135.2, 133.5, 132.3, 130.0, 126.8, 126.6, 126.6, 117.7, 20.6, 12.3; IR (neat) 3363, 3073, 2923, 1771, 1706, 1650, 1620, 1592, 1500, 1342, 1297, 1226, 1181, 1018 cm−1; HRMS (ESI) m/z: [M + H]+ calcd for C16H14NO5 300.0872; found 300.0878.
4-Methyl-2,5,10-trioxo-1,2,3,4,5,10-hexahydrobenzo[g]quinolin-3-yl Acetate (47).
A solution of 45 (30.0 mg, 0.100 mmol) in CHCl3 (40 mL) in a glass tube was placed in the center of a photoreactor and irradiated at 419 nm (spectral window of lamps: 373–460 nm) for 6 h after which the reaction mixture was concentrated. The resulting residue was subjected to flash column chromatography (hexanes/ethyl acetate = 5:1) to give 21.4 mg of 47 (78%) as a yellow solid.
Mp 223–280 °C, decomposed; 1H NMR (500 MHz, CDCl3) δ 8.14 (d, J = 7.4, Hz, 1H), 8.11 (d, J = 7.6 Hz, 1H), 8.03 (broad s, 1H), 7.80 (t, J = 7.5 Hz, 1H), 7.74 (t, J = 7.5 Hz, 1H), 5.65 (d, J = 6.7 Hz, 1H), 3.67 (p, J = 7.0 Hz, 1H), 2.26 (s, 3H), 1.21 (d, J = 7.1 Hz, 3H); 13C{1H} NMR (126 MHz, CDCl3) δ 181.6, 178.7, 169.8, 166.0, 137.3, 135.2, 133.7, 132.3, 130.3, 127.0, 126.7, 123.9, 69.9, 29.7, 20.8, 12.6; IR (neat) 3277, 2921, 2853, 1746, 1720, 1671, 1637, 1621, 1596, 1578, 1465, 1452, 1329, 1304, 1259, 1215, 1167, 1082, 941 cm−1; HRMS (ESI) m/z: [M + Na]+ calcd for C16H13NO5Na 320.0691; found 322.0700.
3-Hydroxy-4-methyl-3,4-dihydrobenzo[g]quinoline-2,5,10(1H)-trione [(±)-Griffithazanone A].
To a solution of acetate 47 (36.1 mg, 0.120 mmol) in THF (6 mL) and MeOH (3 mL) at rt was added K2CO3 (83 mg, 0.60 mmol). The reaction mixture was stirred for 2 h, and then, the reaction was quenched with sat. NH4Cl (aq., 5 mL) and extracted with EtOAc (3 × 10 mL). The organic extracts were combined, washed with brine (10 mL), dried (Na2SO4), and concentrated. Flash column chromatography (hexanes/ethyl acetate = 2:1) gave 26.1 mg of griffithazanone (84%) as a yellow solid.
Mp 240–280 °C, decomposed; 1H NMR (500 MHz, CDCl3) δ 8.15 (d, J = 7.6 Hz, 1H), 8.10 (d, J = 7.6 Hz, 1H), 8.04 (bs, 1H), 7.80 (td, J = 7.5, 1.1 Hz, 1H), 7.74 (td, J = 7.5, 1.1 Hz, 1H), 4.48 (dd, J = 7.0, 1.8 Hz, 1H), 3.71 (d, J = 7.1 Hz, 1H), 3.43 (d, J = 1.8 Hz, 1H), 1.13 (d, J = 7.1 Hz, 3H); 13C{1H} NMR (126 MHz, CDCl3) δ 181.9, 178.9, 171.3, 137.1, 135.2, 133.7, 132.3, 130.3, 127.0, 126.6, 125.6, 69.4, 30.7, 11.4; IR (neat) 3471, 3298, 2922, 2849, 1706, 1663, 1629, 1592, 1468, 1451, 1334, 1302, 1213, 1158, 1103 cm−1; HRMS (ESI) m/z: [M + Na]+ calcd for C14H11NO4Na 280.0586; found 280.0594.
Supplementary Material
ACKNOWLEDGMENTS
We are grateful for financial support from the National Science Foundation (CHE 1465113) and the National Institutes of Health (GM132531-01). We would also like to thank Dr. Peter Flynn (University of Utah), Dr. Hsiaonung Chen (University of Utah), and Dr. Ryan Vanderlinden (University of Utah) for help with NMR, mass spectrometry, and X-ray structure acquisition, respectively.
Footnotes
Supporting Information
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.joc.9b03417.
Experimental protocols and spectral data for all compounds; spectral distribution and intensity of RPR-4190A; Methods of XRD sample preparations and XRD determination (PDF)
Crystal structures were deposited to the CCDC with deposition numbers 1967034 (14) (CIF)
Crystal structures were deposited to the CCDC with deposition numbers 1967030 (40) (CIF)
Complete contact information is available at: https://pubs.acs.org/10.1021/acs.joc.9b03417
The authors declare no competing financial interest.
REFERENCES
- (1).Qiu H-Y; Wang P-F; Lin H-Y; Tang C-Y; Zhu H-L; Yang Y-H Napthoquinones: A continuing source for discovery of therapeutic antineoplastic agents. Chem. Biol. Drug Des 2018, 91, 681–690. [DOI] [PubMed] [Google Scholar]
- (2).Maruyama K; Otsuki T; Tai S Photoinduced Electron-Transfer-Initiated Aromatic Cyclization. J. Org. Chem 1985, 50, 52–60. [Google Scholar]
- (3).(a) Crimmins MT; Reinhold TL Enone olefin [2+2] photochemical cycloadditions. Org. React 1993, 297. [Google Scholar]; (b) Naito T; Makita Y; Yazaki S; Kaneko C Cycloadditions in Syntheses. XXVI. I,2-Dihydrocyclobuta[b]-naphthalene-3,8-diones: Synthesis by Photo-chemical Means and Their Reactions via 2,3-Dimethylene-1,4-dioxo-1,2,3,4-tetrahydronaphthalenes. Chem. Pharm. Bull 1986, 34, 1505–1517. [Google Scholar]; (c) Bryce-Smith D; Evans EH; Gilbert A; McNeill HS Photoaddition of Ethenes to 1,4-Naphthoquinone: Factors Influencing the Site of Reaction. J. Chem. Soc. Perkin Trans 1 1992, 485–489. [Google Scholar]; (d) Nielsen LB; Wege D The enantioselective synthesis of elecanacin through an intramolecular naphthoquinone-vinyl ether photochemical cycloaddition. Org. Biomol. Chem 2006, 4, 868–876. [DOI] [PubMed] [Google Scholar]
- (4).Creed D; Werbin H; Daniel T The Mechanism of Photooxidation of the Menaquinones. Tetrahedron Lett 1981, 22, 2039–2042. [Google Scholar]
- (5).(a) Rajendran M Quinones as photosensitizers for photo-dynamic therapy: ROS generation, mechanism and detection methods. Photodiagn. Photodyn. Ther 2016, 13, 175–187. [DOI] [PubMed] [Google Scholar]; (b) Nowicka B; Kruk J Occurrence, biosynthesis, and function of isoprenoid quinones. Biochim. Biophys. Acta, Bioenerg 2010, 1797, 1587–1605. [DOI] [PubMed] [Google Scholar]
- (6).Mallory FB; Mallory CW Photocyclization of stilbenes and related molecules. Org. React 1984, 1–456. [Google Scholar]
- (7).Cleveland PG; Chapman OL Non-oxidative Photo-cyclizations of Alkyl-substituted Acrylic Acid Anilides to Dihydrocarbostyrils. Chem. Commun 1967, 1064–1065. [Google Scholar]
- (8).Bach T; Grosch B; Strassner T; Herdtweck E Enantioselective [6p]-Photoelectrocyclization Reaction of an Acrylanilide Mediated by a Chiral Host. Interplay between Enantioselective Ring Closure and Enantioselective Protonation. J. Org. Chem 2003, 68, 1107–1116. [DOI] [PubMed] [Google Scholar]
- (9).(a) Zhang Y-J; Kong M; Chen R-Y; Yu D-Q Alkaloids from the Roots of Goniothalamus griffithii. J. Nat. Prod 1999, 62, 1050–1052. [DOI] [PubMed] [Google Scholar]; (b) Tip-pyang S; Limpipatwattana Y; Khumkratok S; Siripong P; Sichaem J A new cytotoxic 1-azaanthraquinone from the stems of Goniothalamus laoticus. Fitoterapia 2010, 81, 894–896. [DOI] [PubMed] [Google Scholar]; (c) Jaidee W; Andersen RJ; Patrick BO; Pyne SG; Muanprasat C; Borwornpinyo S; Laphookhieo S Alkaloids and styryllactones from Goniothalamus cheliensis. Phytochemistry 2019, 157, 8–20. [DOI] [PubMed] [Google Scholar]
- (10).Soonthornchareonnon N; Suwanborirux K; Bavovada R; Patarapanich C; Cassady JM New Cytotoxic 1-Azaanthraquinones and 3-Aminonaphthoquinone from the Stem Bark of Goniothalamus marcanii. J. Nat. Prod 1999, 62, 1390–1394. [DOI] [PubMed] [Google Scholar]
- (11).Corey EJ; Clark. Studies on the Total Synthesis of Rifamycin. A Method for the Closure of the Macrocyclic Unit. Tetrahedron Lett 1980, 21, 2045–2048. [Google Scholar]
- (12).Feng Y; Liu J; Carrasco YP; MacMillan JB; De Brander JK Rifamycin Biosynthetic Congeners: Isolation and Total Synthesis of Rifsaliniketal and Total Synthesis of Salinisporamycin and Saliniketals A and B. J. Am. Chem. Soc 2016, 138, 7130–7142. [DOI] [PubMed] [Google Scholar]
- (13).Tapia R; Torres G; Valderrama JA Nitric Acid on Silica Gel: A Useful Nitrating Reagent for Activated Aromatic Compounds. Synth. Commun 2006, 39, 681–687. [Google Scholar]
- (14).(a) Gandy MN; Piggott MJ Synthesis of Kalasinamide, a Putative Plant Defense Phototoxin. J. Nat. Prod 2008, 71, 866–868. [DOI] [PubMed] [Google Scholar]; (b) Siewart B; Stuppner H The photoactivity of natural products – An overlooked potential of phytomedicines. Phytomedicine 2019, 60, No. 152985. [DOI] [PubMed] [Google Scholar]
- (15).Ichino C; Soonthornchareonnon N; Chuakul W; Kiyohara H; Ishiyama A; Sekiguchi H; Namatame M; Otoguro K; Omura S; Yamada H Screening of Thai Medicinal Plant Extracts and Their Active Consituents for In Vitro Antimalarial Activity. Phytother. Res 2006, 20, 307–309. [DOI] [PubMed] [Google Scholar]
- (16).Acrylamides 7 and 10 were recovered after subjecting them to 180 °C for 5 h in the dark.
- (17). See Figure S2 for the X-ray structure analysis of 14.
- (18).(a) Woodward RB; Hoffmann R The Conservation of Orbital Symmetry; Verlag Chemie: Weinheim, 1970. [Google Scholar]; (b) Woodward RB; Hoffmann R Angew. Chem., Int. Ed 1969, 8, 781. [Google Scholar]
- (19).Shriver DF; Drezdn MA The Manipulation of Air Sensitive Compounds, 2nd ed.; Wiley & Sons: New York, NY, 1986. [Google Scholar]
- (20).Tohma H; Harayama Y; Hashizume M; Iwata M; Kiyono Y; Egi M; Kita Y The First Total Synthesis of Discorhabdin A. J. Am. Chem. Soc 2003, 125, 11235–11240. [DOI] [PubMed] [Google Scholar]
- (21).Bach T; Grosch B; Strassner T; Herdtweck E Enantioselective [6π]-Photocyclization Reaction of an Acrylanilide Mediated by a Chiral Host. Interplay between Enantioselective Ring Closure and Enantioselective Protonation. J. Org. Chem 2003, 68, 1107–1116. [DOI] [PubMed] [Google Scholar]
- (22).Kharel MK; Pahari P; Shepherd MD; Tibrewal N; Nybo SE; Shaaban KA; Rohr J Angucyclines: Biosynthesis, mode-of-action, new natural products, and synthesis. Nat. Prod. Rep 2012, 29, 264–325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Acrylamide 39 was recovered after subjecting it to 180 °C for 24 h in the dark.
- (24). See Figure S3 for the X-ray crystal structure analysis of 40.
- (25).Modha SG; Pöthig A; Dreuw A; Bach T [6π] Photocyclization to cis-Hexahydrocarbazol-4-ones: Substrate Modification, Mechanism, and Scope. J. Org. Chem 2019, 84, 1139–1153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).(a) Jackobs HJC; Havinga E Photochemistry of vitamin D and its isomers and of simple trienes. Adv. Photochem 1979, 11, 305–373. [Google Scholar]; (b) Mazzucator U; Momicchioli F Rotational Isomerism in trans-1,2-Diarylethylenes. Chem. Rev 1991, 91, 1679–1719. [Google Scholar]
- (27).Ground state geometries and energies were obtained from DFT calculations performed with the Gaussian09, version D.01, using the B3LYP functional and 6–311G+g(d) basis set.
- (28).Frisch MJ; Trucks GW; Schlegel HB; Scuseria GE; Robb MA; Cheeseman JR; Scalmani G; Barone V; Mennucci B; Petersson GA; Nakatsuji H; Caricato M; Li X; Hratchian HP; Izmaylov AF; Bloino J; Zheng G; Sonnenberg JL; Hada M; Ehara M; Toyota K; Fukuda R; Hasegawa J; Ishida M; Nakajima T; Honda Y; Kitao O; Nakai H; Vreven T; Montgomery JA Jr.; Peralta JE; Ogliaro F; Bearpark M; Heyd JJ; Brothers E; Kudin KN; Staroverov VN; Keith T; Kobayashi R; Normand J; Raghavachari K; Rendell A; Burant JC; Iyengar SS; Tomasi J; Cossi M; Rega N; Millam JM; Klene M; Knox JE; Cross JB; Bakken V; Adamo C; Jaramillo J; Gomperts R; Stratmann RE; Yazyev O; Austin AJ; Cammi R; Pomelli C; Ochterski JW; Martin RL; Morokuma K; Zakrzewski VG; Voth GA; Salvador P; Dannenberg JJ; Dapprich S; Daniels AD; Farkas O; Foresman JB; Ortiz JV; Cioslowski J; Fox DJ Gaussian 09, Revision D.01; Gaussian, Inc.: Wallingford CT, 2013. [Google Scholar]
- (29).Zhang YJ; Kong M; Chen RY; Yu DQ Alkaloids from the roots of Goniothalamus griffithii. J. Nat. Prod 1999, 62, 1050–1052. [DOI] [PubMed] [Google Scholar]
- (30).Jaidee W; Andersen RJ; Patrick BO; Pyne SG; Muanprasat C; Borwornpinyo S; Laphookhieo S Alkaloids and styryllactones from Goniothalamus cheliensis. Phytochemistry 2019, 157, 8–20. [DOI] [PubMed] [Google Scholar]
- (31).We explored the oxidation of the N,O-ketene acetal from 8 (or the N-methyl lactam) with O2, Davis’ oxaziridine, and m-CPBA. We also examined the use of a free-radical oxidation using NHPI.
- (32).Clinical and Laboratory Standards Institute. Methods for Dilution Antimicrobial Susceptibility Tests for Bacteria That Grow Aerobically, Approved Standard—Ninth ed.; CLSI. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.