

Abstract
Introduction:
ctDNA offers a promising, non-invasive approach to monitor therapeutic efficacy in real-time. We explored whether the quantitative percent change in ctDNA early after therapy initiation can predict treatment response and PFS in metastatic GI cancer patients.
Methods:
138 patients with metastatic GI cancers and tumor profiling by NGS had serial blood draws pre-treatment and at scheduled intervals during therapy. ctDNA was assessed using individualized droplet digital PCR measuring the mutant allele fraction in plasma of mutations identified in tumor biopsies. ctDNA changes were correlated with tumor makers and radiographic response.
Results:
138 patients enrolled. 101 were evaluable for ctDNA and 68 for tumor markers at four weeks. Percent change of ctDNA by four weeks predicted partial response (PR, p<0.0001) and clinical benefit (CB: PR and stable disease (SD), p<0.0001). ctDNA decreased by 98% (median) and >30% for all PR patients. ctDNA change at eight-weeks, but not two-weeks, also predicted CB (p<0.0001). Four-week change in tumor markers also predicted response (p=0.0026) and CB (p=0.022). However, at a clinically relevant specificity threshold of 90%, four-week ctDNA change more effectively predicted CB versus tumor makers, with a sensitivity of 60% vs. 24%, respectively (p=0.0109). Patients whose four-week ctDNA decreased beyond this threshold (≥30% decrease) had a median PFS of 175 days versus 59.5 days (Hazard Ratio 3.29; 95% CI 1.55-7.00; p<0.0001).
Conclusions:
serial ctDNA monitoring may provide early indication of response to systemic therapy in metastatic GI cancer patients prior to radiographic assessments and may outperform standard tumor markers, warranting further evaluation.
Introduction
Analysis of circulating tumor DNA (ctDNA), commonly referred to as “liquid biopsy,” is a non-invasive way to detect and measure cancer-specific molecular alterations in the blood.1–5 The use of ctDNA is emerging as a useful tool in several settings, including detection of post-surgical residual disease and identifying mechanisms of drug resistance. 6–16 Recent data suggests that ctDNA levels within an individual patient correlate with tumor burden over time and that serial assessment of ctDNA may represent a promising approach for monitoring treatment response, with early decreases in ctDNA serving as a predictor of response.17–25 However, further clinical evaluation of ctDNA monitoring as a means of tracking therapeutic response is needed.
Currently, radiographic imaging remains the gold-standard for evaluating treatment response. However, imaging is typically performed several months into therapy, and more frequent radiographic assessment may not be practical or informative. Serum tumor markers (i.e. CEA, CA19-9) have also been used as a means of minimally invasive monitoring of treatment response, but the longer half-lives of these markers and lack of tumor-specificity can limit their performance.26,27 A more accurate means for early prediction of therapeutic response could be beneficial to distinguish patients most likely to benefit from continued therapy from patients unlikely to benefit, in whom an earlier switch to an alternative therapy may spare toxicity and provide clinical benefit. In this regard, ctDNA represents a promising approach to monitor treatment response and help with early prediction of therapeutic efficacy. ctDNA has the advantages of having a short half-life (~one hour), high tumor specificity, and can be performed non-invasively at more frequent intervals than imaging.28 However, the utility of serial ctDNA monitoring to predict therapeutic response has not been well characterized.
In the current study, we sought to perform a proof-of-concept analysis evaluating the use of serial ctDNA monitoring to predict treatment response in patients with metastatic GI cancer receiving systemic therapy. In this prospective cohort, we evaluated whether an early change in ctDNA levels can predict radiographic response to treatment across metastatic gastrointestinal cancer patients and compared how ctDNA performed relative to standard tumor markers.
Materials and Methods
Patients and sample collection
Between 2014 and 2018, we enrolled 138 patients with metastatic gastrointestinal cancers. All patients provided informed written consent, and specimens were collected at the Massachusetts General Hospital (MGH) Cancer Center according to Institutional Review Board-approved protocols in accordance with the Declaration of Helsinki. Patients were followed during standard-of-care cytotoxic chemotherapy or targeted therapy. Targeted therapies included EGFR, BRAF, HER2, FGFR, or MET (Table 1) directed therapy. Blood and tumor specimens were obtained. Tumor mutational profiling was performed at MGH as part of routine clinical care through a standard clinical institutional NGS panel for 104 known cancer genes. Blood was drawn prior to the start of therapy and after initiation of therapy at two weeks, four weeks, eight weeks, and then every eight weeks until progression (Figure 1a). Cell-free DNA was extracted from plasma using QIAamp Circulating Nucleic Acid Kit (QIAGEN) and assessed by digital droplet PCR using probes for tumor-specific point mutations (see Supplementary Methods for full ddPCR methods). To improve accuracy, one or more tumor-specific alterations likely to be clonal based on clinical sequencing were identified and used to evaluate ctDNA longitudinally in available plasma specimens (Supplementary Table S1). For patients with multiple assessable mutations, the percent change in mutant allele fraction of up to 3 mutations ctDNA were averaged. Informative tumor markers, if available during the same timepoints, were also analyzed and the more dynamic tumor marker was chosen if multiple tumor markers were informative. Response Evaluation Criteria in Solid Tumors (RECIST) 1.1 measurements determined by independent radiological review were used to compare baseline CT scans with first restaging scans during treatment. If unavailable, progression was determined clinically by the investigators.29 Progression-free survival (PFS) on treatment was determined by investigator review.
Table 1.
Baseline Patient and Disease Characteristics
| Characteristic | N=138 |
|---|---|
| Median age, years (range) | 61 (21-87) |
| Sex (M) | 84 (61%) |
| Race | |
| White | 122 (88%) |
| Asian | 6 (4.3%) |
| Black | 2 (1.4%) |
| Unknown | 8 (5.8%) |
| Primary tumor location | |
| Colorectal | 69 (50%) |
| Pancreas | 26 (19%) |
| Biliary | 18 (13%) |
| Esophagogastric | 17 (12%) |
| Other | 8 (5.8%) |
| Therapy types | |
| Cytotoxic therapy only | 97 (70%) |
| Targeted therapy only | 23 (17%) |
| Targeted + cytotoxic therapy | 18 (13%) |
| Metastatic at Diagnosis | 83 (60%) |
| Median time from metastatic diagnosis to treatment start, months (range) | 2.1 (0-205) |
| Lines of prior metastatic therapy, median (range) | 0 (0-6) |
| 1st line therapy | 78 (57%) |
| 2nd line therapy | 31 (22%) |
| 3rd line therapy or later | 29 (21%) |
Figure 1.
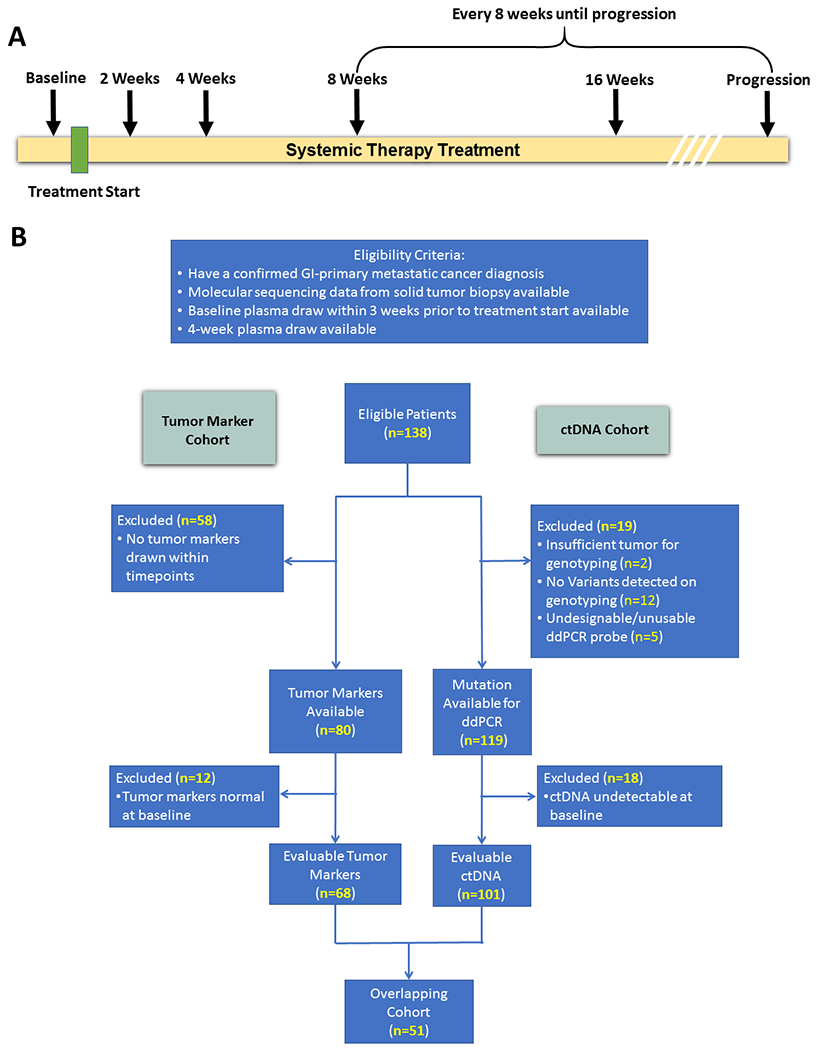
Draw Schedule (a) and Consort Diagram (b)
Eligible patients had a histologically confirmed metastatic cancer diagnosis from a GI primary, received only systemic therapy before first scan, tumor tissue which was genotyped, a baseline plasma draw within three-weeks prior to treatment start and a four-week plasma draw (between 20-45 days). We evaluated how change in ctDNA levels or change in tumor markers predicted response to treatment and clinical benefit (defined as patients who had a partial response or stable disease).
Statistical Analyses
We compared the distributions of percent change in ctDNA and in tumor markers at four-weeks between response categories using Mann-Whitney U tests and Kruskal-Wallis tests. For both ctDNA and tumor markers, we identified the cut-off for percent change at four-weeks yielding approximately 90% specificity (i.e. 90% of patients with clinical benefit classified correctly) and compared the sensitivity and positive predictive value associated with the cut-off between ctDNA and tumor markers using Fisher’s exact test. Progression-free survival (PFS) on treatment was defined as time from treatment start to progression or death (event) or most recent treatment date (censored) and summarized using the Kaplan-Meier method. We compared PFS between patients who did and did not exceed the week four percent change cut-off for ctDNA/tumor markers using the log-rank test and computed the log-rank hazard ratio. We evaluated whether clinical characteristics including cancer type, treatment type, and number of prior lines of treatment were confounders in the relationship between outcomes and percent change of ctDNA at 4 weeks using univariate and multivariate logistic (PR and CB outcomes) and Cox proportional hazards (PFS outcome) regression models. We assessed the impact of covariate adjustment on the statistical significance and effect estimate for ctDNA percent change between the univariate and multivariate models for each outcome. As exploratory analyses, we compared the distributions of percent change in ctDNA at two-weeks and eight-weeks between response categories as described above and performed subgroup analyses by tumor type and treatment type. All analyses were conducted among patients with available data for the specified variable and time point. The two-sided significance level was 0.05 for all comparisons.
Results
Overall, 138 patients met eligibility criteria and were enrolled—50% colorectal cancer (CRC), 29% pancreatic cancer, 13% biliary cancers, 12% esophagogastric cancer, and 6% other gastrointestinal primaries (Table 1). 70% were treated with cytotoxic chemotherapy, 17% with targeted therapy, and 13% with targeted therapy in combination with cytotoxic chemotherapy (Table 1, Supplementary Table S2). In 101 patients we identified at least one mutation that could be tracked in ctDNA that was detectable at baseline (Supplementary Table S3). A subset of 68 patients had evaluable tumor markers within the specified timepoints (Supplementary Table S4). 51 patients had both tumor markers and ctDNA that were evaluable at four weeks (Figure 1b). Of the patients with evaluable ctDNA, 27 patients had two-week draws and 85 patients had eight-week draws. For ctDNA, the average time from treatment start until the four-week blood draw for ctDNA analysis was 29.9 days +/− 4.8 (Standard Deviation) while the average time to first re-staging scan was 55.4 days +/− 19.8 (Standard Deviation).
We observed that the percent change in ctDNA mutant allele fraction (MAF) at four weeks predicted radiographic PR and clinical benefit (PR or SD). Patients achieving PR had a median ctDNA decrease at four weeks of 98.0% compared to patients with progressive disease (PD) who had a median decrease of 49.0% (p<0.0001) (Figure 2a). Notably, all patients with PR had a decrease of ctDNA of >30%. ctDNA change was also predictive of clinical benefit, with a median decrease of 95.5% observed in these patients (Figure 2b). Patients with PR or SD also had a significantly greater decrease of standard tumor markers (median −57.50% and median −7.00%, respectively) compared to those with PD (median 21.0%, p=0.0026 for PR vs. PD) (Figure 2c). The change in tumor markers was also predictive of clinical benefit (p=0.022) (Figure 2d).
Figure 2.
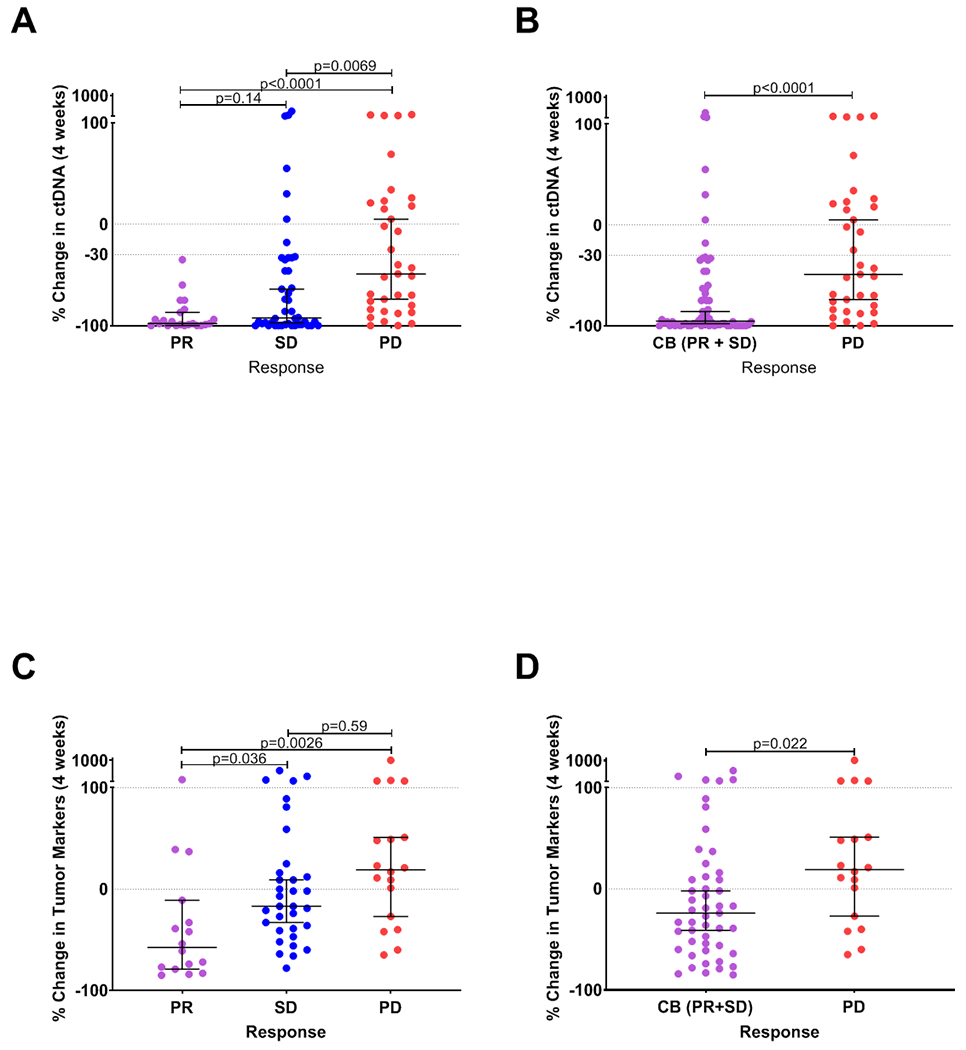
Change in ctDNA (panels a, b) and tumor markers (panels c, d) at 4 weeks are shown for patients grouped by radiographic response by RECIST1.1 criteria. Each data point represents the percent change in ctDNA or tumor markers at 4 weeks relative to baseline for a single patient. Horizontal bars represent the median and error bars indicate 95% Confidence Interval. PR: partial response; SD: stable disease; PD: progressive disease; CB: clinical benefit.
We next assessed the sensitivity and positive predictive value (PPV) for predicting clinical benefit of ctDNA and tumor markers at a clinically-relevant specificity threshold of ~90%, at which no more than one out of every 10 patients who would achieve clinical benefit from therapy would fail to be identified with each respective assay. Interestingly, all patients achieving PR exhibited ctDNA decreases beyond this threshold, which equated to a ctDNA decrease of 30% or greater. Of 28 patients who did not have a ctDNA decrease reaching this threshold, all but 2 (26 pts, 93%) developed progressive disease and discontinued treatment within 4 months of starting therapy. At this threshold, the sensitivity of ctDNA for predicting clinical benefit was 60% and PPV was 75%. By contrast, a change in tumor markers at a similar threshold yielding ~90% specificity for clinical benefit had a sensitivity of only 24% with a PPV of 44%. The difference between sensitivity of ctDNA vs. tumor markers was statistically significant (0.0109) (Supplementary Table S5.)
Furthermore, we performed additional exploratory analyses of ctDNA at this 90% specificity threshold. A >30% decrease in ctDNA also predicted PFS (HR: 3.29; 95% CI 1.55 to 7.00; p<0.0001). Patients whose ctDNA decreased by >30% had a median survival of 175 days, while patients whose ctDNA did not had a median survival of 59.5 days (Figure 3a). Similarly, patients with a ctDNA decrease of >30% after eight-weeks of therapy also showed improved PFS (HR: 4.34; 95% CI 1.69 to 11.11; p<0.0001; median survival 183 days v. 64 days, respectively) (Figure 3b).
Figure 3.
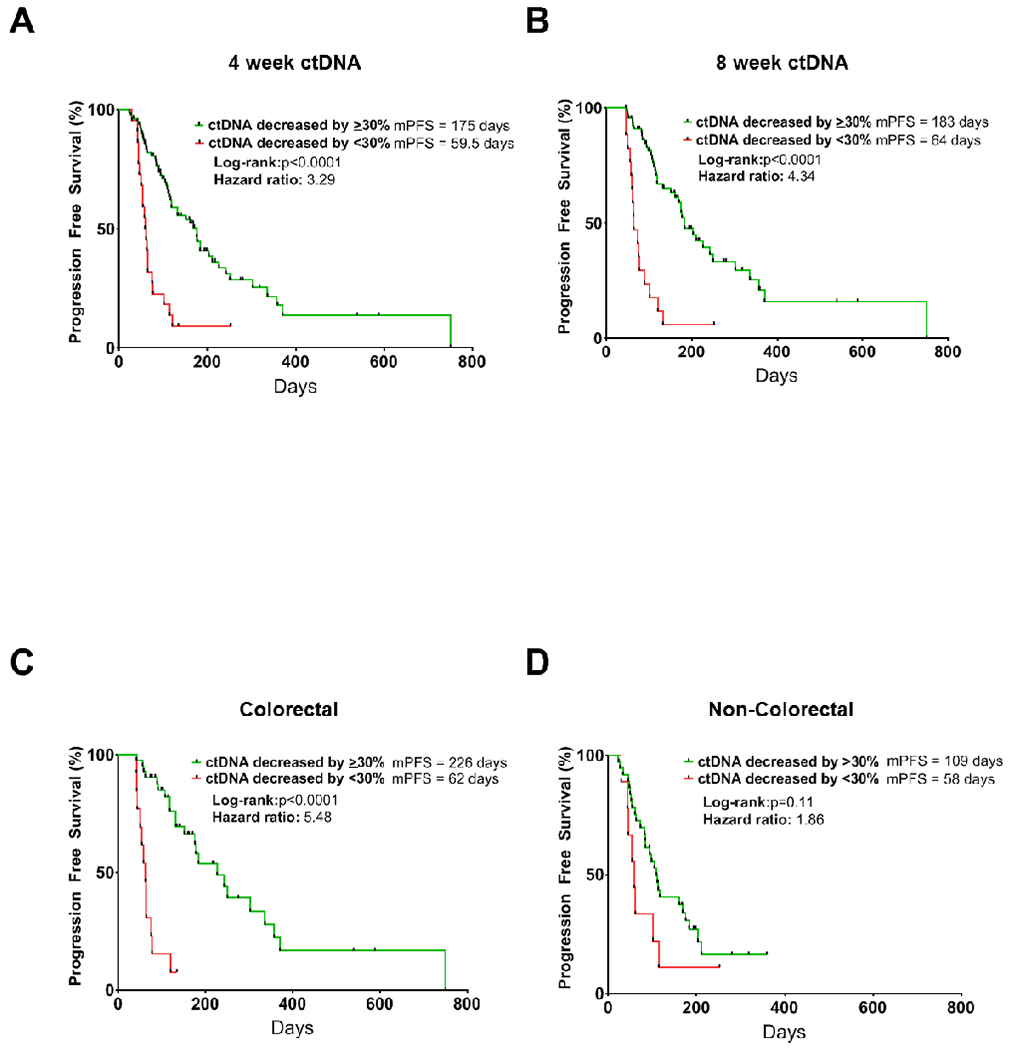
Kaplan Meier curves showing progression-free survival by percent change in ctDNA for all patients at 4 weeks (panel a), 8 weeks (panel b) from treatment initiation, or for patients with CRC only (panel c) or non-CRC at 4 weeks (panel d). mPFS: median progression free survival.
Interestingly, we observed possible differences in the association of ctDNA changes with PFS based on tumor type. Most notably, ctDNA change showed a striking association with PFS in CRC (n=55), where patients whose ctDNA decreased by >30% had a median survival of 226 days on treatment, compared to just 62 days (HR: 5.484; 95% CI 1.69 to 17.78; p<0.0001). Conversely, while a clear trend was noted, the association of ctDNA change with PFS did not reach statistical significance in non-CRC patients (n=46; p=0.11) (Figures 3c, d). Similarly, while ctDNA change predicted response (PR vs. PD) as well as clinical benefit in CRC patients (p<0.0001), this association did not reach statistical significance in non-CRC patients (p=0.085, p=0.086, respectively), though a similar trend was observed (Supplementary Figure S1a–d).
However, we observed that ctDNA change did predict PFS across tumor types in patients receiving targeted therapy with or without chemotherapy (HR: 3.49; 95% CI 1.19 to 10.30; p=0.0002) and in patients receiving chemotherapy alone (HR: 2.95; 95% CI 1.06 to 8.22; p=0.0017) (Supplementary Figure S2a,b). Similarly, ctDNA was predictive of clinical benefit for patients receiving targeted therapy (p<0.0001) and chemotherapy alone (p=0.043), although ctDNA was better able to predict radiographic response in patients receiving targeted therapy (n=34) versus cytotoxic chemotherapy alone (n=67) (p=0.0003 vs p=0.17, respectively) (Supplementary Figure S2b–f). We also evaluated patients based on line of therapy and found that ctDNA change was predictive of clinical benefit across different lines of therapy (Supplementary Figure S3a–c).
Given the heterogeneity of tumor types, treatment types, and lines of therapy, we performed a multivariate analysis adjusting for these variables and found that a ctDNA change of at least 30% remained significantly associated with both clinical benefit (OR=6.9; 95% CI 2.304 to 20.732; p=0.006) and progression-free survival (HR=0.324; 95% CI 0.183 to 0.571; p=0.0001) (Table 2). Additionally, the unadjusted and adjusted ORs and HRs were similar in magnitude, providing further evidence that our unadjusted results are not confounded by the heterogeneity in clinical characteristics.
Table 2.
Unadjusted and adjusted associations between 30% decrease in ctDNA at 4 weeks and clinical outcomes.
| Outcome | Unadjusted (univariate) | Adjusted (multivariate)1 | ||
|---|---|---|---|---|
| OR/HR2 (95% CI) | P-value | OR/HR2 95% CI | P-value | |
| Partial response | -3 | - | -3 | - |
| Clinical benefit | OR = 6.321 (2.256, 17.717) | 0.0005 | OR = 6.912 (2.304, 20.732) | 0.0006 |
| Progression-free survival | HR = 0.277 (0.161, 0.479) | <0.0001 | HR = 0.324 (0.183, 0.571) | 0.0001 |
Adjusted for diagnosis, treatment type, and number of prior lines of therapy
Odd ratios (ORs)/hazard ratios (HRs) represent odds/risk of outcome for decrease ≥ 30% in week 4 ctDNA compared to change > −30%
Cannot be estimated because all patients with PR had ≥ 30% decrease in week 4 ctDNA
Finally, we performed an exploratory analysis assessing the optimal timing for ctDNA assessment for prediction of clinical benefit. While earlier prediction of therapeutic response would certainly have advantages, we observed that at two-weeks, change in ctDNA did not show a statistically significant correlation with treatment response or clinical benefit, but at eight-weeks a similar degree of statistical significance observed at four weeks remained between patients achieving PR (median −100%) vs PD (median −46.0%) (p<0.0001) as well as SD (median −99.0%) and PD (p=0.0090) (Figure 4 a, b, c). At eight weeks, ctDNA change also predicted clinical benefit (p<0.0001) (Figure 4d). Even when the analysis was restricted the to the 27 patients who had 2-week draws (all had 4-week draws, and all but 5 patients who progressed prior to 8 weeks had 8-weeks draws), we found that 4-week and 8-week ctDNA change remained a statistically-significant predictor of clinical benefit, whereas 2-week ctDNA change did not show a statistically-significant association. (Supplementary Figure S4a–d). We observed continued evolution of ctDNA levels from two weeks to four weeks in many patients (Figure 4e). For patients achieving PR at the first set of scans, ctDNA decline was remarkably consistent (Figure 4e). Most patients with PR had benefit beyond 6 months, with only 6 patients with PR progressing within 6 months. Interestingly, for patients achieving SD, over time a rise in ctDNA levels was seen in many patients who developed progressive disease within 6 months (Figure 4e, blue), whereas ctDNA levels remained suppressed in most patients remaining on therapy for more than 6 months prior to developing progressive disease (Figure 4d, orange). The changes highlight the ability of ctDNA detection to predict clinical benefit longitudinally and the ability to detect a dynamic increase in ctDNA levels at progression (Supplementary Figure S5, Supplementary Table S4).
Figure 4.
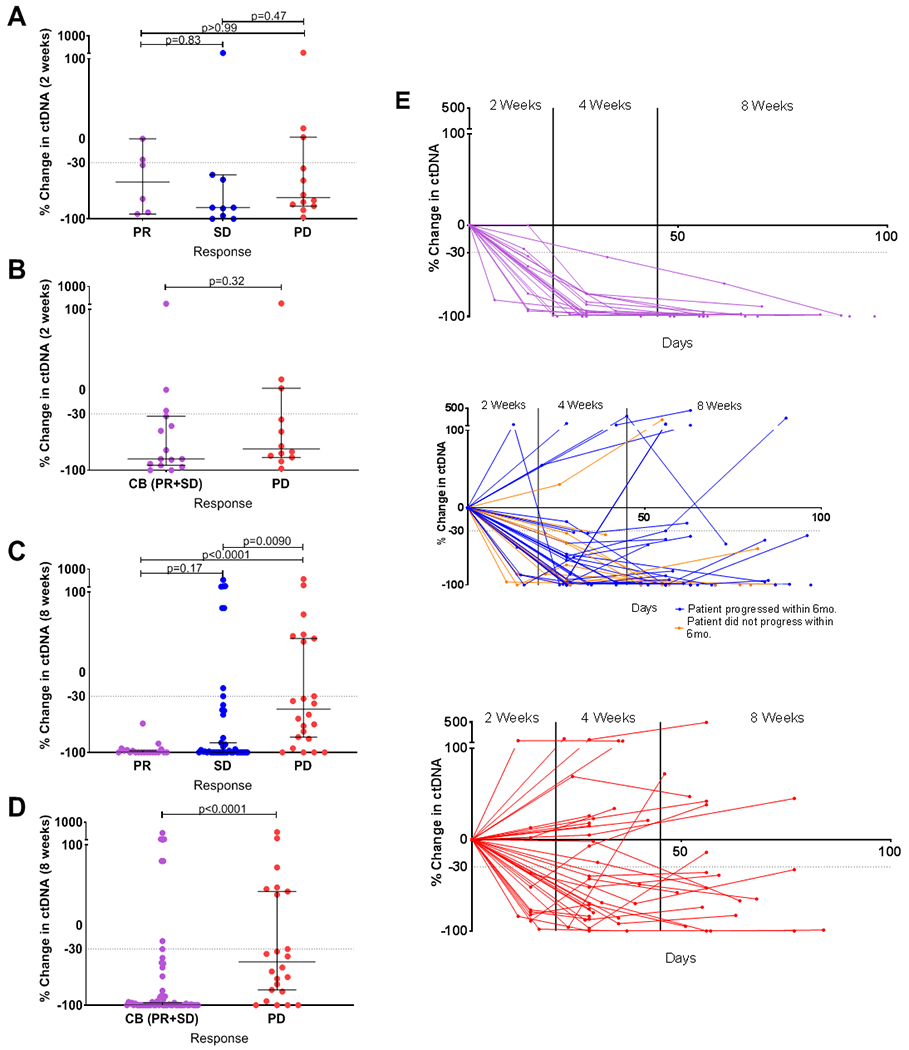
Panels a-d: Changes in ctDNA at 2 weeks (panels a, b) or 8 weeks (panels c, d) of treatment by RECIST1.1 criteria in patients achieving partial response (PR), stable disease (SD) and progressive disease (PD) (panels a, c) or clinical benefit (CB) and PD (panels b, d). Panel e: Longitudinal ctDNA changes during the first 100 days of therapy in patients with PR (upper panel, purple lines), SD (middle panel, blue and orange lines) and PD cases (lower panel, red lines). For SD patients, blue lines represent patients with PFS <6 months and orange lines represent patients with PFS > 6 months. Horizontal bars represent the median and error bars indicate 95% Confidence Interval.
Discussion
This proof-of-concept study suggests that serial ctDNA monitoring may provide an early and reliable predictor of treatment response and clinical benefit to systemic therapy. We observed a rapid and consistent decline in ctDNA levels during the first four weeks of systemic therapy in patients achieving partial response or clinical benefit. All patients who achieved a partial response had a decrease in ctDNA levels by four weeks of treatment of at least 30%, with most exhibiting a near complete decline (median 98% decrease). Importantly, the decrease in ctDNA levels by four weeks in patients achieving PR or clinical benefit was significantly greater than in patients with PD. Notably, all patients achieving PR had decreases in ctDNA beyond 30% and of the 28 patients who did not have a ctDNA decrease reaching this threshold, 93% of patients developed progressive disease and discontinued treatment within 4 months of starting therapy (Supplementary Figure 5). ctDNA decrease of 30% of more also predicted a stark difference in PFS, median PFS of 175 days versus 59.5 days (HR: 3.29; 95% CI 1.55 to 7.00; p<0.0001).
These data suggest that further evaluation of ctDNA monitoring and its potential for early prediction of response or lack of therapeutic benefit is warranted and that serial ctDNA monitoring could offer early insight into whether a patient is responding to a given therapy and should therefore continue that therapy, or whether a patient is unlikely to respond. Early identification of patients who are not responding to therapy would allow a switch to an alternative therapy sooner, increasing the chance of potential benefit and reducing unnecessary toxicity from an ineffective therapy. Thus, serial ctDNA monitoring could increase the efficiency of personalized therapeutic decisions for individual patients and allow for adaptive clinical trials where therapy might be modulated based on ctDNA.
Notably, change in standard serum tumor markers—CEA and CA19-9—by four weeks also exhibited an association with response and clinical benefit, although with more limited statistical confidence than for ctDNA (p=0.021 tumor markers vs. p<0.0001 for ctDNA). Therefore, we conducted an exploratory analysis comparing the effectiveness of ctDNA versus tumor markers in predicting clinical benefit. We assessed the predictive power of ctDNA or tumor makers at a specificity threshold of 90%. This threshold was selected as a potential clinically relevant specificity cutoff, such that if a treatment were to be discontinued or changed due to lack of a sufficient decrease in either marker, then no more than one of ten patients who would go on to derive some clinical benefit would have treatment discontinued based on this result. At this 90% specificity threshold, the sensitivity for predicting clinical benefit was 60% for ctDNA versus only 24% for tumor markers (p=0.0109). These results suggest that ctDNA monitoring may potentially outperform standard tumor markers at this early time point and ctDNA monitoring may offer advantages over the current standard of care. It is possible that the shorter half-life and increased tumor specificity of ctDNA may provide advantages over standard tumor markers for monitoring of treatment response. 30,31
We also explored how early after treatment initiation ctDNA could predict response. An earlier ability to predict whether a patient is benefiting, may allow an earlier switch of a non-responding patient to a more effective therapy, increasing the chance of benefit and limiting unnecessary exposure. However, our initial analysis suggests that if ctDNA is assessed too early after the initiation of therapy, its predictive power is more limited. At two-weeks after the start of therapy, we did not observe a significant association between change in ctDNA levels and response or clinical benefit, as we did at four and eight-weeks, though the number of patients with available two-week plasma samples was limited. One potential explanation may be due to the kinetics of ctDNA release during therapy. ctDNA levels may increase acutely after initiation of therapy, due to release of tumor DNA as a result of tumor cell death, before decreasing in parallel with a reduction in tumor burden.32 Indeed, additional evolution of ctDNA levels was observed between two weeks and four weeks (Figure 4e), and specifically, further decreases in ctDNA were observed by four weeks in patients achieving PR. These data suggest that further optimization of the timing of ctDNA assessment following therapeutic initiation will be critical.
While we observed a highly consistent decrease in ctDNA in patients achieving PR, the change in ctDNA levels by four weeks in patients with SD or PD was more variable. For SD patients, this may reflect the fact that SD, as defined by RECIST 1.1, includes patients who achieve some degree of tumor shrinkage not reaching criteria for PR, as well as patients whose tumors increase in size but not by enough to meet criteria for PD. In SD patients achieving a PFS of >6 months, a far more consistent decrease in ctDNA levels by four weeks was observed (Figure 4e). Conversely, SD patients with PFS <6 months typically exhibited a rise or rebound in ctDNA by eight weeks. Similarly, PD patients exhibiting an initial reduction in ctDNA levels by four weeks, also showed a rise or rebound in ctDNA levels by eight weeks. Likewise, in patients with PR or prolonged SD, a consistent rebound in ctDNA levels was observed as patients developed eventual disease progression (Supplementary Figure S3). These findings support the potential importance of serial monitoring of ctDNA to gain further insight into the evolution of a patient’s response over time.
This study does have several limitations. First, while over 100 patients were evaluated, the overall sample size is still limited, and not all cases had both serial tumor marker and ctDNA assessments. Second, we were unable to evaluate ctDNA in several patients (Figure 1b). In some cases, this was because no mutations were detected upon clinical tumor sequencing. This issue could be overcome by performing more broad-based tumor sequencing facilitating the identification of trackable DNA mutations in more patients. In other cases, a customized ddPCR probe could not be designed for specific mutations or patients did not have detectable baseline levels of ctDNA. While the proportion of patients with unevaluable ctDNA was similar to patients whose baseline tumor markers were in the normal range and thus also unevaluable, it is possible that different ctDNA technologies, including larger NGS panels, customized multiplexed mutation assays, multiple mutation tracking or including methylation markers could be more effective for tracking ctDNA in more patients.33–36 In this study, individualized ddPCR was utilized as a means of establishing clinical proof-of-concept for serial ctDNA monitoring, and is not necessarily the optimal approach. Third, several patients had very low levels at baseline meaning small fluctuations in ctDNA levels over time could lead to large calculated percent changes, potentially affecting the accuracy of response prediction. Indeed, outlier values were often observed in patients with very low baseline levels of ctDNA (Supplementary Table S3), and lower levels of baseline ctDNA observed in non-CRC patients (Supplementary Figure S1e,f) may be one explanation for why ctDNA change was less effective in predicting response and PFS in non-colorectal versus CRC patients (Figure 3 c,d; Supplementary Figure S1a–d). In future studies, the use of more sensitive technologies or determination of a minimum basal level of ctDNA for accurate interpretation may be important to overcome this potential issue and to define effective thresholds for clinical decision-making.
In summary, these data suggest that serial monitoring of ctDNA has the potential to provide an early indication of treatment response and clinical benefit across a range of GI cancers receiving an array of systemic cytotoxic and/or targeted therapies. While larger and more comprehensive studies are needed to define the optimal timing of ctDNA assessments, to determine the most accurate thresholds for response prediction, and to evaluate the most suitable and cost-effective technologies for ctDNA measurement, serial ctDNA monitoring has the potential to help guide clinicians in making more personalized treatment decisions and to facilitate early adaptation of therapy to limit the cost and toxicity from ineffective therapies and to allow a more rapid switch to a potentially more effective therapy. Moreover, serial monitoring of ctDNA could be used as an early marker of efficacy or lack of efficacy to facilitate adaptive clinical trial strategies. Thus, further prospective assessment of serial ctDNA monitoring as a means of predicting therapeutic response is warranted.
Supplementary Material
Translational Relevance.
While prior studies suggest that a directional change in ctDNA levels correlates generally with therapeutic response, it is unclear whether the quantitative ctDNA change might provide an early predictor of response with sufficient accuracy to guide treatment decisions. This proof-of-concept study in metastatic GI cancer patients suggests that the quantitative measure of ctDNA reduction by 4 weeks of therapy provides an accurate prediction of eventual radiographic response and progression-free survival, with favorable performance relative to standard tumor markers. Our study also provides key insights into the optimal timing of ctDNA assessment and the degree of ctDNA reduction corresponding to clinical benefit. While further evaluation in larger studies is needed, serial ctDNA monitoring could facilitate adaptive clinical trial design and help clinicians make more personalized treatment decisions for early adaptation of therapy, limiting the cost and toxicity from ineffective therapies and allowing a more rapid switch to potentially more effective therapies.
Acknowledgements
The work is partially supported by NIH/NCI Gastrointestinal Cancer SPORE P50 CA127003, R01CA208437, U54CA224068, a Damon Runyon Clinical Investigator Award, a Stand Up To Cancer Colorectal Dream Team Translational Research Grant (grant number SU2C-AACR-DT22-17; to R.B. Corcoran and supporting A.R.Parikh), and the ASCO Conquer Cancer Foundation Career Development Award to A.R.Parikh. Stand Up To Cancer is a division of the Entertainment Industry Foundation. Research grants are administered by the American Association for Cancer Research, the scientific partner of SU2C.
Disclosure of Potential Conflicts of Interest
A.R.P is a consultant/advisory board member for Puretech and Foundation Medicine. Institutional research funding from Eli Lilly, Array, Plexxikon, Guardant, BMS, Novartis, Tesaro, Macrogenics and Tolero. Research support from SU2C-AACR-DT22-17 Colorectal Dream Team: Targeting Genomic, Metabolic, and Immunological Vulnerabilities of Colorectal Cancer, P50CA127003 DF/HCC SPORE in Gastrointestinal Cancer, Conquer Cancer Foundation ASCO CDA and Gateway Foundation for Cancer Research. D.P.R. serves on advisory board for MPM Capital, Gritstone Oncology, Oncorus, Maverick Therapeutics, 28/7 Therapeutics; has equity in MPM Capital and Acworth Pharmaceuticals; serves as author for Johns Hopkins University Press, UpToDate, McGraw Hill. L.G. is a consultant or advisory board member for Debiopharm, H3 Biomedicine, Agios Pharmaceuticals, Taiho Pharmaceuticals, Klus Pharmaceuticals, QED, Alentis Pharmaceuticals, and Pieris Pharmaceuticals. E.R. is advisory board member/consultant for Helsinn, Heron, BASF, American Imaging Management, Napo, Imuneering and Vector Oncology. C.W. is a consultant for Celgene. A.X.Z is a consultant/ advisor for AstraZeneca, Bayer, Bristol-Myers Squibb, Eisai, Eli Lilly, Exelixis, Merck, Novartis, and Roche/Genentech; research funding from Bayer, Bristol-Myers Squibb, Eli Lilly, Merck, and Novartis. A.J.I is a consultant for DebioPharm, Chugai, and Roche; research support from Sanofi and has equity in ArcherDx.
T.S.H. is consultant/advisory board member for Merck, EMD Serono, and Synthetic Biologics; research support from Taiho, Astra Zeneca, Bristol Myers Squibb, Mobetron, and Ipsen. R.B.C. is a consultant/advisory board member for Amgen, Array Biopharma, Astex Pharmaceuticals, Avidity Biosciences, BMS, C4 Therapeutics, Chugai, Elicio, Fog Pharma, Fount Therapeutics, Genentech, LOXO, Merrimack, N-of-one, Novartis, nRichDx, Revolution Medicines, Roche, Roivant, Shionogi, Shire, Spectrum Pharmaceuticals, Symphogen, Taiho, and Warp Drive Bio; holds equity in Avidity Biosciences, C4 Therapeutics, Fount Therapeutics, nRichDx, and Revolution Medicines; and has received research funding from Asana, AstraZeneca, and Sanofi. A.M., J.L.S., K.K, E.V.S, I.F., A.T., M.G.F., B.T., B.N., H.S., J.N.A., L.B., B.G., R.D.N., J.Y.W., D.D.S, J.K.L., G.S., N.H., and J.C., have no disclosures.
References
- 1.Fujii T, Barzi A, Sartore-Bianchi A, et al. Mutation-Enrichment Next-Generation Sequencing for Quantitative Detection of KRAS Mutations in Urine Cell-Free DNA from Patients with Advanced Cancers. Clinical cancer research : an official journal of the American Association for Cancer Research. 2017;23(14):3657–3666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.De Mattos-Arruda L, Mayor R, Ng CKY, et al. Cerebrospinal fluid-derived circulating tumour DNA better represents the genomic alterations of brain tumours than plasma. Nature communications. 2015;6:8839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Miller AM, Shah RH, Pentsova EI, et al. Tracking tumour evolution in glioma through liquid biopsies of cerebrospinal fluid. Nature. 2019;565(7741):654–658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Siravegna G, Geuna E, Mussolin B, et al. Genotyping tumour DNA in cerebrospinal fluid and plasma of a HER2-positive breast cancer patient with brain metastases. ESMO Open. 2017;2(4):e000253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Vidal J, Muinelo L, Dalmases A, et al. Plasma ctDNA RAS mutation analysis for the diagnosis and treatment monitoring of metastatic colorectal cancer patients. Annals of oncology : official journal of the European Society for Medical Oncology. 2017;28(6):1325–1332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Tie J, Wang Y, Tomasetti C, et al. Circulating tumor DNA analysis detects minimal residual disease and predicts recurrence in patients with stage II colon cancer. Science translational medicine. 2016;8(346):346ra392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Corcoran RB, Chabner BA. Cell-free DNA Analysis in Cancer. The New England journal of medicine. 2019;380(5):501–502. [DOI] [PubMed] [Google Scholar]
- 8.Parikh AR, Leshchiner I, Elagina L, et al. Liquid versus tissue biopsy for detecting acquired resistance and tumor heterogeneity in gastrointestinal cancers. Nature medicine. 2019;25(9):1415–1421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Strickler JH, Loree JM, Ahronian LG, et al. Genomic Landscape of Cell-Free DNA in Patients with Colorectal Cancer. Cancer Discov. 2018;8(2):164–173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Abbosh C, Swanton C, Birkbak NJ. Circulating tumour DNA analyses reveal novel resistance mechanisms to CDK inhibition in metastatic breast cancer. Annals of oncology : official journal of the European Society for Medical Oncology. 2018;29(3):535–537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Fribbens C, Garcia Murillas I, Beaney M, et al. Tracking evolution of aromatase inhibitor resistance with circulating tumour DNA analysis in metastatic breast cancer. Annals of oncology : official journal of the European Society for Medical Oncology. 2018;29(1):145–153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Tsui DWY, Murtaza M, Wong ASC, et al. Dynamics of multiple resistance mechanisms in plasma DNA during EGFR-targeted therapies in non-small cell lung cancer. EMBO Mol Med. 2018;10(6). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Tarazona N, Gimeno-Valiente F, Gambardella V, et al. Targeted next-generation sequencing of circulating-tumor DNA for tracking minimal residual disease in localized colon cancer. Annals of oncology : official journal of the European Society for Medical Oncology. 2019. [DOI] [PubMed] [Google Scholar]
- 14.De Mattos-Arruda L, Weigelt B, Cortes J, et al. Capturing intra-tumor genetic heterogeneity by de novo mutation profiling of circulating cell-free tumor DNA: a proof-of-principle. Annals of oncology : official journal of the European Society for Medical Oncology. 2014;25(9):1729–1735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Abbosh C, Birkbak NJ, Wilson GA, et al. Phylogenetic ctDNA analysis depicts early-stage lung cancer evolution. Nature. 2017;545(7655):446–451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gale D, Lawson ARJ, Howarth K, et al. Development of a highly sensitive liquid biopsy platform to detect clinically-relevant cancer mutations at low allele fractions in cell-free DNA. PloS one. 2018;13(3):e0194630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Corcoran R, André T, Yoshino T, Bendell J, Atreya C, Schellens J, Ducreux M, McRee A, Siena S, Middleton G, et al. Efficacy and circulating tumor DNA (ctDNA) analysis of the BRAF inhibitor dabrafenib (D), MEK inhibitor trametinib (T), and anti-EGFR antibody panitumumab (P) in patients (pts) with BRAF V600E–mutated (BRAFm) metastatic colorectal cancer (mCRC) Ann Oncol. 2016;27:4550. [Google Scholar]
- 18.Siravegna G, Sartore-Bianchi A, Mussolin B, et al. Tracking a CAD-ALK gene rearrangement in urine and blood of a colorectal cancer patient treated with an ALK inhibitor. Annals of oncology : official journal of the European Society for Medical Oncology. 2017;28(6):1302–1308. [DOI] [PubMed] [Google Scholar]
- 19.Siravegna G, Marsoni S, Siena S, Bardelli A. Integrating liquid biopsies into the management of cancer. Nature reviews Clinical oncology. 2017;14(9):531–548. [DOI] [PubMed] [Google Scholar]
- 20.Kurtz DM, Scherer F, Jin MC, et al. Circulating Tumor DNA Measurements As Early Outcome Predictors in Diffuse Large B-Cell Lymphoma. Journal of clinical oncology : official journal of the American Society of Clinical Oncology. 2018:Jco2018785246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Mouliere F, Chandrananda D, Piskorz AM, et al. Enhanced detection of circulating tumor DNA by fragment size analysis. Science translational medicine. 2018;10(466). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Tie J, Kinde I, Wang Y, et al. Circulating tumor DNA as an early marker of therapeutic response in patients with metastatic colorectal cancer. Annals of oncology : official journal of the European Society for Medical Oncology. 2015;26(8):1715–1722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Garlan F, Laurent-Puig P, Sefrioui D, et al. Early Evaluation of Circulating Tumor DNA as Marker of Therapeutic Efficacy in Metastatic Colorectal Cancer Patients (PLACOL Study). Clinical cancer research : an official journal of the American Association for Cancer Research. 2017;23(18):5416–5425. [DOI] [PubMed] [Google Scholar]
- 24.Frenel JS, Carreira S, Goodall J, et al. Serial Next-Generation Sequencing of Circulating Cell-Free DNA Evaluating Tumor Clone Response To Molecularly Targeted Drug Administration. Clinical cancer research : an official journal of the American Association for Cancer Research. 2015;21(20):4586–4596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hsu HC, Lapke N, Wang CW, et al. Targeted Sequencing of Circulating Tumor DNA to Monitor Genetic Variants and Therapeutic Response in Metastatic Colorectal Cancer. Mol Cancer Ther. 2018;17(10):2238–2247. [DOI] [PubMed] [Google Scholar]
- 26.Shinkins B, Nicholson BD, Primrose J, et al. The diagnostic accuracy of a single CEA blood test in detecting colorectal cancer recurrence: Results from the FACS trial. PloS one. 2017;12(3):e0171810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Nicholson BD, Shinkins B, Pathiraja I, et al. Blood CEA levels for detecting recurrent colorectal cancer. The Cochrane database of systematic reviews. 2015(12):Cd011134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Thierry AR, Mouliere F, Gongora C, et al. Origin and quantification of circulating DNA in mice with human colorectal cancer xenografts. Nucleic acids research. 2010;38(18):6159–6175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Eisenhauer EA, Therasse P, Bogaerts J, et al. New response evaluation criteria in solid tumours: revised RECIST guideline (version 1.1). European journal of cancer (Oxford, England : 1990). 2009;45(2):228–247. [DOI] [PubMed] [Google Scholar]
- 30.Bidart J- M, Thuillier F, Augereau C, et al. Kinetics of Serum Tumor Marker Concentrations and Usefulness in Clinical Monitoring. Clinical Chemistry. 1999;45(10):1695–1707. [PubMed] [Google Scholar]
- 31.Ballehaninna UK, Chamberlain RS. The clinical utility of serum CA 19–9 in the diagnosis, prognosis and management of pancreatic adenocarcinoma: An evidence based appraisal. J Gastrointest Oncol. 2012;3(2):105–119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Husain H, Melnikova VO, Kosco K, et al. Monitoring Daily Dynamics of Early Tumor Response to Targeted Therapy by Detecting Circulating Tumor DNA in Urine. Clinical cancer research : an official journal of the American Association for Cancer Research. 2017;23(16):4716–4723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Reinert T, Henriksen TV, Christensen E, et al. Analysis of Plasma Cell-Free DNA by Ultradeep Sequencing in Patients With Stages I to III Colorectal Cancer. JAMA oncology. 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.CAPP-Seq: an ultrasensitive quantitative assay of ctDNA. Nature Reviews Clinical Oncology. 2014;11(6):301–301. [Google Scholar]
- 35.Newman AM, Bratman SV, To J, et al. An ultrasensitive method for quantitating circulating tumor DNA with broad patient coverage. Nature medicine. 2014;20(5):548–554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ooki A, Maleki Z, Tsay JJ, et al. A Panel of Novel Detection and Prognostic Methylated DNA Markers in Primary Non-Small Cell Lung Cancer and Serum DNA. Clinical cancer research : an official journal of the American Association for Cancer Research. 2017;23(22):7141–7152. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.