

Abstract
Summary: The main functions of memory T cells are to provide protection upon re‐exposure to a pathogen and to prevent the re‐emergence of low‐grade persistent pathogens. Memory T cells achieve these functions through their high frequency and elevated activation state, which lead to rapid responses upon antigenic challenge. The significance and characteristics of memory CD8+ T cells in viral infections have been studied extensively. In many of these studies of T‐cell memory, experimental viral immunologists go to great lengths to assure that their animal colonies are free of endogenous pathogens in order to design reproducible experiments. These experimental results are then thought to provide the basis for our understanding of human immune responses to viruses. Although these findings can be enlightening, humans are not immunologically naïve, and they often have memory T‐cell populations that can cross‐react with and respond to a new infectious agent or cross‐react with allo‐antigens and influence the success of tissue transplantation. These cross‐reactive T cells can become activated and modulate the immune response and outcome of subsequent heterologous infections, a phenomenon we have termed heterologous immunity. These large memory populations are also accommodated into a finite immune system, requiring that the host makes room for each new population of memory cell. It appears that memory cells are part of a continually evolving interactive network, where with each new infection there is an alteration in the frequencies, distributions, and activities of memory cells generated in response to previous infections and allo‐antigens.
Cross‐reactivity and T‐cell receptor repertoires
T‐cell cross‐reactivity
The antigen‐specific memory T‐cell populations that are important in protection against subsequent infection 1, 2, 3 have diverse T‐cell receptor (TCR) repertoires (4, 5) but also are intrinsically cross‐reactive. The TCR of a CD8+ T cell discriminates peptides of usually eight to 10 amino acids that are embedded in major histocompatibility complex class I (MHC‐I) molecules (6). Crystal structural studies revealed that only a few TCR contact residues on the peptide presented by MHC molecules are required for activation 7, 8, 9, 10, 11, although a TCR can tolerate certain amino acid substitutions in the peptide sequence and still become activated. For example, amino acid substitutions for a human leukocyte antigen (HLA) B8‐restricted Epstein – Barr virus (EBV) peptide at positions 1, 2, and 8 were tolerated, while substitutions at positions 4, 6, and 7 were crucial for cytotoxic T‐lymphocyte recognition (12). Thus, ‘molecular mimicry’, whereby a peptide ligand differing from the original ligand retains sites that are necessary for interaction with the TCR (13), is one of several paths to cross‐reactive T‐cell responses. However, an alternative explanation for this flexibility in TCR binding may be explained by thermodynamic studies of TCR – peptide – MHC interactions 14, 15, 16, 17. The TCR undergoes conformational changes of the TCR complementarity determining region 3 (CDR3) for proper accommodation to cognate antigen, as seen with human T cells binding flu‐M158‐66/HLA‐A2.1 and EBV‐EBNA3339‐347/HLA‐B8 epitopes (15, 16) and mouse T cells binding a cytochrome C epitope MCC88‐103/H2‐Ek (14). Wu et al. (17) have suggested an ‘induced fit’ model, where an αβ‐TCR with low conformational complementarity to a peptide‐MHC first contacts the MHC molecule via the CDR1 and CDR2 loops, followed by readjustment of the flexible CDR3 loops to the peptide – MHC complex. This flexibility in the CDR3 molecule leads to an enhanced ability of a single TCR to accommodate structurally diverse peptides. It is possible that this kinetic flexibility in the TCR may also lead to different regions of the same TCR interacting with different sites on two separate ligands, termed as alternative recognition (18, 19). Another mechanism for T‐cell cross‐reactivity is the ability of T cells to express two different TCRs, due to incomplete allelic exclusion of the TCRα chain (20). Mathematical calculations suggest that a single TCR should be able to react against 106 different nonamer peptides (21). Taken together, these mechanisms make cross‐reactivity very difficult to predict and a fairly common event.
Cross‐reactivity may be valuable to the host, considering the large number of potential pathogenic antigens to which one is exposed over a lifetime. Cross‐reactivity could compensate for a situation with a limited TCR repertoire and still allow a normal immune response. For instance, mice deficient for the enzyme terminal deoxynucleotidyl transferase (TdT) have impaired CDR3 diversification with a TCR repertoire which is only 5–10% of that calculated for wildtype mice (22), but upon lymphocytic choriomeningitis virus (LCMV) or Sendai virus infection, these mice have a relatively normal immune response and recover from infection (23). These TdT‐deficient mice are reported to have T cells with a highly cross‐reactive profile when compared to wildtype mice (24), and thus, they may compensate for the less diverse TCR repertoire. Additionally, many TCR transgenic mice, which have limited TCR diversity, are capable of responding to many antigens and resisting viral infections (25, 26).
Reports of pathogen‐specific memory CD8+ T cells recognizing cross‐reactive epitopes on different proteins of the same pathogen or proteins from closely related or totally unrelated pathogens are increasing (reviewed in 27). Perhaps it is not surprising that cross‐reactive T‐cell responses can be seen directed at evolutionarily conserved sites within virus groups, such as different strains of influenza virus 28, 29, 30 or dengue virus (31, 32), or conserved sites between different members of the same virus group, such as hantaviruses (33), arenaviruses (34), and flaviviruses (35). However, examples of cross‐reactive T‐cell responses involving completely unrelated viruses such as LCMV and vaccinia virus (VV) (36), influenza virus and hepatitis C virus (HCV) (37), influenza virus and EBV (38), influenza virus and human immunodeficiency virus (HIV) (39), and human papillomavirus and coronavirus (40), have now been shown. These cross‐reactive T‐cell responses are more frequently observed once memory T‐cell populations have been generated, due the increased frequency and higher activation state of memory T cells 41, 42, 43. When cross‐reactive immune responses are present, they can alter T‐cell dynamics and have considerable consequences on the pathogenesis of infection and either inhibit or enhance the replication of a newly encountered heterologous virus 44, 45, 46, 47. They can also have a significant impact on allospecific T‐cell activity prior to and following transplantation (48, 49). In addition, autoimmunity has been associated with viral infections (50), and it is likely that an individual's history of virus infections and the unique composition of the cross‐reactive memory T‐cell pool may either initiate or reactivate T cells with auto‐immune potential.
Cross‐reactivity, immunodominance, and TCR diversity
Although hundreds of peptides in any viral infection have appropriate sequences to bind MHC‐I, usually only a small number of epitopes stimulate immunogenic responses. The CD8+ T‐cell memory pool created after a virus infection has a distinct hierarchy of epitope‐specific responses in a naïve host. Some viral epitopes are dominant, stimulating high frequency T‐cell responses, while others are subdominant, stimulating weaker or barely detectable T‐cell responses (51). This immunodominance hierarchy is regulated by various parameters, including the efficiency of processing and presentation of the peptide, the affinity between peptide and the MHC‐I, the availability of T cells with TCRs that recognize the peptide‐MHC complex, and the competition between T cells for domains on the antigen‐presenting cell (APC) (52).
Public versus private specificity
Many epitope‐specific responses have distinct TCR CDR3 amino acid motifs that are maintained between clonotypes and between different individuals. For example, in the human HLA‐A2‐restricted influenza A M1‐58 Vβ17 response, the amino acid motif IRSS is common (5), and in the H2‐Kd‐restricted HLA‐CW3 Vb10 response in DBA/2 mice, SxG in the first three positions of the CDR3 region was a common motif (53). Other CDR3 binding motifs have been identified, including the murine H2‐Db‐restricted nucleoprotein (NP)396‐specific Vβ8.1 response (GxxN) in LCMV infection (54) and the HLA‐B14‐restricted HIV Env EL9 response (GQG) (55). Conservation of CDR3 amino acid motifs suggests that these sites are required for the TCR to bind to the MHC – ligand structure. These similarities in Vβ usage and amino acid motifs, as well as conservation of immunodominance hierarchies, can be thought of as the ‘public specificities’ of epitope‐specific T‐cell responses that are similar between individuals.
Despite the public specificities in T‐cell responses, there can be tremendous diversity in the TCR repertoire between individuals. The TCR usage per epitope differs between individual hosts, even though there might be general similarities in preferred TCR Vβ usage or specific CDR3 amino acid motifs (53, 56, 57, 58). Thus, the TCRs on the antigen‐specific T‐cell clones are unique to the individual, and these unique regions have been referred to as the ‘private specificity’ for that epitope‐specific response. This variation is probably a consequence of the random stochastic process of TCR rearrangement in the thymus, which results in variations in the naïve peripheral TCR repertoire, and of the random stochastic process whereby a T‐cell encounters an APC presenting its cognate ligand (59). T‐cell clones that are stimulated early may dominate the response by interfering with the stimulation of other T cells (52).
Private specificity of cross‐reactive memory alters subsequent immune repertoires
When a memory CD8+ T‐cell has specificity for an epitope of an unrelated pathogen, the high frequency and activation state of memory cells gives it an advantage over naïve T cells, which can lead to preferential expansion of the cross‐reactive CD8+ T‐cell population. This cross‐reactive expansion can alter the hierarchy of T‐cell responses; we saw this in sequential heterologous virus infections in mice with two distantly related arenaviruses, LCMV and Pichinde virus (PV) (34). These viruses encode epitopes in NP205 with six of eight amino acids in common. This epitope is normally subdominant for either virus in a naïve host. However, due to a selective expansion of NP205‐specific cross‐reactive memory CD8+ T cells, this NP205‐specific T‐cell response became dominant when LCMV‐immune mice were infected with PV or when PV‐immune mice were infected with LCMV (34). These data support the concept that expansions of cross‐reactive T‐cell populations substantially contribute to the immune hierarchies of T‐cell responses. This study may explain some of the variability in immunodominant hierarchies observed in human viral infections, where the host has been exposed to numerous infections throughout life. For instance, patients with identical haplotypes infected with HIV or HCV show high variability in their T‐cell immune hierarchies (60, 61). Interestingly, cross‐reactive CD8+ T‐cell responses to other pathogens have been documented for both of these viruses (37, 62).
Our studies with VV infection of LCMV‐immune mice showed that VV sometimes elicited the expansion of T cells with one LCMV epitope specificity but other times elicited the expansion of T cells with a different specificity. This outcome could be explained by the stochastic nature of clonal dominance and by the private specificities in antigen‐specific TCR repertoires in each immune host. Thus, the proportion of epitope‐specific memory T cells cross‐reactive with another antigen would differ from host to host. This variation is indeed what we have observed in LCMV‐immune hosts challenged with VV (36, 63). In our study to predict which LCMV‐encoded epitopes might be driving cross‐reactive responses to VV, substantial variability was noted when individual mice were tested. In 50% of mice, the VV infection stimulated strong expansion of T cells specific to the LCMV NP205‐212 epitope, in 23% of mice they were specific to either the glycoprotein (GP)33 or GP34 overlapping epitopes, and in 15% of mice specific to the GP118‐125 epitope ( Fig. 1A ). Often there was expansion of T cells specific to only one LCMV epitope, but sometimes T cells specific to more than one epitope were expanded. The question was whether these variations in expansion represent random stochastic events in an LCMV‐immune mouse challenged with VV, where only a limited number of the cross‐reactive T cells actually engage antigen, or whether each mouse had a unique T‐cell repertoire in regards to its potential cross‐reactivity with VV. To address this point, carboxyfluorescein succinimidyl ester (CSFE)‐labeled splenocytes from different donor LCMV‐immune mice were adoptively transferred into three recipients, which were each then challenged with VV. The pattern of epitope‐specific T‐cell expansion was virtually identical among the recipients of a single donor but differed in recipients from different donors. This result showed that these variations in T‐cell responses were reflections of the private specificities of the individual immune host.
Figure 1.

(A ) Hierarchy of lymphocytic choriomeningitis virus (LCMV) epitope‐specific memory populations which expand upon vaccinia virus (VV) infection. Determined by private specificity of the T‐cell receptor (TCR) repertoire of each individual mouse, NP205(50% of mice) > GP34(23%) > GP118(15%). (B) Structural modeling demonstrates similar structures evident in areas important for TCR interaction between VV a11r198 and LCMV GP118, LCMV GP34, LCMV NP205, and VV e7r130. Using the concept of molecular mimicry, LCMV epitopes were identified that induced cross‐reactive CD8+ T‐cell responses recognizing the VV a11r198 epitope (shown in green). The arrows mark positions 4, 7 (white), and 6 (yellow), which are important for TCR interaction. For modeling 3‐D structures of putative peptide‐H‐2Kb complexes, we used X‐ray coordinates of the crystallized TCR(2C)–dEV8–H‐2Kb (PDB access number 2CKB). Using Swiss‐PDB Viewer software (GlaxoSmithKline R & D, Geneva, Switzerland), we mutated dEV8 to simulate the peptides whose sequences are shown in (B). The modified variants with minimal rotamer penalties were subjected to an energy minimization protocol. For graphical presentation of the simulated peptide‐H‐2Kb complexes, PyMol software (DeLano scientific LLC, San Carlos, California) was utilized. The H‐2Kb‐restricted ovalbumin epitope, SIINFEKL, is shown as the actual crystal structure adapted from PDB access number 1VAC (185).
Although there is a high level of T‐cell cross‐reactivity involving the two LCMV and PV NP205 epitopes in regards to peptide‐induced interferon‐γ (IFNγ) production, a heterologous virus challenge selects for a very small subset of the cross‐reactive T cells, leading to a substantial narrowing of the TCR repertoire (64). This narrowing of the repertoire had different patterns between individuals, and adoptive transfer studies indicated that this variation was a reflection of the private specificities of the immune system that developed after the primary infection. Most studies, with few exceptions (65), have linked narrow TCR repertoires to poor viral clearance and an enhanced probability of selecting for epitope escape variants 66, 67, 68, 69. Thus, private specificities of memory TCR repertoires adds another level of complexity in understanding cross‐reactive T‐cell responses and further explain the variability in disease outcome upon infection with the same pathogen (70).
Cross‐reactivity and heterologous immunity: balancing protection and pathology
Cross‐reactivity and protective immunity
Memory T cells cross‐reactive with a heterologous virus can provide partial protective immunity and, in experimental models, can provide the difference between life and death in the infected host (44, 45, 71) ( Table 1 ). For example, LCMV‐immune mice control PV infection, presumably due to the cross‐reactivity of T cells specific for the subdominant NP205 epitope (34, 63). LCMV‐immune mice also manifest strong protective immunity against infections with the large DNA poxvirus VV compared to naïve mice (44). In a respiratory model of infection, this heterologous immunity prevented mortality to an otherwise lethal dose of VV (45). Adoptive transfer studies demonstrated that CD4+ and CD8+ T cells from LCMV‐immune mice were required to transfer protective immunity to naïve mice challenged with PV or VV (44). Because of the tenuous balance between T‐cell immunodominance, protective immunity, and immunopathology, heterologous immunity is not always beneficial. Although immunity to LCMV protected against respiratory VV infection, it inhibited the clearance of respiratory syncytial virus (RSV) (45, 47). Similarly, our work demonstrated that a history of influenza A infection protected against VV but inhibited clearance of murine cytomegalovirus (MCMV) and LCMV (46) ( Table 1 ).
Table 1.
Heterologous immunity: changes in viral titer as a consequence of prior infection
| Immunizing virus | Challenge virus | change in titer versus control |
|---|---|---|
| A. Systemic (i.p) infection | ||
| Altered viral titers in the spleen or liver | ||
| LCMV | PV | ↓93% |
| LCMV | MCMV | ↓60% |
| LCMV | VV | ↓97% |
| PV | LCMV | ↓60% |
| PV | MCMV | ↓75% |
| PV | VV | ↓98% |
| MCMV | LCMV | ↓84% |
| MCMV | PV | ↓69% |
| MCMV | VV | ↓97% |
| VV | LCMV | ↓50% |
| VV | PV | ↓21% |
| VV | MCMV | −0% |
| B. Mucosal (i.n) infection altered viral titers in the lung | ||
| LCMV | VV | ↓87% |
| MCMV | VV | ↓68% |
| INFLUENZA | VV | ↓75% |
| INFLUENZA | LCMV | ↑400% |
| INFLUENZA | MCMV | ↑500% |
LCMV, lymphocytic choriomeningitis virus; PV, Pichinde virus; MCMV, murine cytomegalovirus; VV, vaccinia virus.
The numbers above represent the percentage reduction of PFU titer in (A) the spleen or liver or (B) lung.
Lack of reciprocity
Although LCMV immunity protects against VV, immunity to VV does not protect against LCMV (44). Correspondingly, although VV elicits the proliferation of a subset of CSFE‐labeled adoptively transferred LCMV memory cells, LCMV stimulates very little proliferation of a VV‐immune population (63). Since VV encodes over 200 proteins and perhaps thousands of potential epitopes, it is probably much more likely to encode an epitope that would activate some cells from an LCMV‐immune T‐cell population, whereas LCMV, which encodes only four proteins and a far more limited number of epitopes, may be less likely to encounter a VV‐immune T‐cell to stimulate. This finding could explain why so many large DNA viruses have evolved to encode gene products that interfere with MHC‐I antigen presentation (72). Other factors may also be involved. For instance, VV might be a better inducer of interleukin‐12 (IL‐12) than LCMV and could lead to enhanced IFNγ production by cross‐reactive T cells (45).
Matrix of cross‐reactivity
The selective expansion of LCMV‐specific memory CD8+ T cells upon VV infection suggested the possibility of cross‐reactive CD8+ T‐cell responses between these two viruses (45, 63). Based on the concept of molecular mimicry, we have identified cross‐reactive epitopes by searching for VV sequence homology with the LCMV NP205 epitope ( Fig. 1B ). An epitope, VVa11r189, generated CD8+ T‐cell responses that were highly cross‐reactive with three LCMV‐specific memory CD8+ T‐cell responses, NP205, GP34, and GP118 (which, incidentally, showed no cross‐reactivity with each other), with one PV‐specific response, NP205, and another VV epitope‐specific response, e7r130 (36, unpublished data). Hence, a whole matrix of cross‐reactivity was revealed, where one VV epitope could activate five different CD8+ memory populations and impact heterologous immunity. Of significance, however, was when a11r198‐stimulated cell lines were derived from individual LCMV‐immune donors, each line had different patterns of cross‐reactivity, with some high for one epitope and others high for a different epitope, again reflecting the private specificity of cross‐reactivity (unpublished data).
Cross‐reactivity and altered pathology
T cells mediate not only protective immunity but also substantial immunopathology 73, 74, 75, 76, 77, 78. In LCMV infection, T cells clear the virus but can also mediate a severe leptomeningitis (73, 74). The T‐cell‐mediated pathology during acute infection likely results from the inflammatory conditions induced by the presence of high numbers of T cells lysing infected tissues, producing pro‐inflammatory cytokines and chemokines which recruit other cells. Disease outcome is ultimately based on a fine balance between the number of memory T cells recruited to sites of viral replication, the efficiency and length of time these T cells are present, and amplifying pro‐inflammatory responses, before virus is cleared. The efficiency of the activated T cells in viral clearance is at least partially dependent on the avidity of the TCR interaction with its ligand. Studies with altered peptide ligands (APLs), which represent an in vitro model for cross‐reactive responses, show that high‐ and low‐potency ligands differ in the length of time the TCR interacts with MHC/ligand, i.e. TCR ‘avidity’, and modify the functional potential of individual clones (7, 79). These studies help explain why a lower avidity cross‐reactive response may not protect as well against a heterologous virus or why heterologous immunity may not be as effective at mediating protective immunity as homologous immunity. It is easy to imagine how an enhanced early activation of a lower avidity cross‐reactive clone might be inefficient at protection but could lead to different types of immunopathology.
Heterologous immunity and altered pathology in mice
An example of the balance between T‐cell immunity and pathology is seen in VV infection of LCMV‐immune mice. LCMV‐immune mice challenged with VV developed panniculitis presenting as necrosis of visceral fat (44), which is analogous to human erythema nodosum. In a respiratory infection model, reduced mortality of LCMV‐immune mice infected with VV was accompanied by altered lung pathology (45). The lungs were infiltrated by LCMV‐specific T cells, which contributed to obstruction of bronchioles by fibrin and inflammatory cells (bronchiolitis obliterans). In humans, erythema nodosum and bronchiolitis obliterans are of unknown etiology but can be seen in some viral and bacterial infections and are also associated with autoimmune diseases 80, 81, 82. Erythema nodosum has been observed after vaccination for smallpox or hepatitis B. The development of bronchiolitis obliterans in lung allografts is associated with transplant rejection (82). Altered pathology also occurred in the influenza‐immune mice with augmented viral replication upon LCMV or MCMV infection. Instead of the usual mild mononuclear infiltrate observed in acute MCMV infection of naïve mice, influenza‐immune mice infected with MCMV developed a severe consolidating mononuclear pneumonia with evidence of bronchiolization. During bronchiolization, alveolar epithelium is replaced by bronchiolar‐like cells, which is an indicator of lung repair (83).
Heterologous immunity and altered pathology in humans
Manifestations of heterologous immunity may therefore explain variations in human disease pathogenesis thought previously to be only affected by genetic differences, the physiological condition of the patient, or the inoculation route and dose. The individual's history of infections may shape the T‐cell memory pool in ways that contribute to this variability. Heterologous immunity and cross‐reactive T‐cell responses are reminiscent of the phenomenon of ‘original antigenic sin’, which was first described for B‐cell responses against influenza virus subtypes (84). Different strains and variants of influenza virus are commonly cross‐reactive at the T‐cell level in mice and humans, leading to speculations that these cross‐reactive cells may be involved in the pathogenesis of influenza virus infections 28, 29, 30. Perhaps the most recognized human examples of heterologous immunity come from dengue virus infections. Infection with a dengue virus serotype generated CD8+ T cells with a higher avidity to a second and presumably previously encountered dengue virus serotype, suggesting that cross‐reactive memory CD8+ T cells had preferentially expanded over T cells with greater avidity to the serotype causing infection (31, 85). These lower avidity cross‐reactive T cells may mediate a more severe disease outcome, including hemorrhagic fever, which has been observed in subsequent infections with different dengue virus serotypes.
EBV and acute infectious mononucleosis
The difference between a clinical and an asymptomatic acute EBV infection is the magnitude of the T‐cell response not the viral load (86). Symptomatic disease in the form of acute infectious mononucleosis syndrome is less likely in young children than in teenagers and young adults, who have a longer history of infections and presumably a more complex pool of memory cells than young children (87). Other infections follow a similar pattern including mumps, chicken pox, polio, and measles. A subset of T cells directed against a major HLA‐A2.1‐restricted immunodominant EBV epitope, BMLF‐1280, can cross‐react with the invariant HLA‐A2.1‐restricted influenza A virus epitope M158, even though they share only three of nine amino acids (38). Our recent studies have shown activation of these cross‐reactive T cells in some but not all acute mononucleosis patients, perhaps again reflecting private specificities in the host response (38). Due to the large size of its genome, EBV likely presents an extensive pool of potential CD8+ T‐cell epitopes that could activate other cross‐reactive memory CD8+ T cells of different specificities. In fact, we have been able to define a matrix of cross‐reactive EBV and influenza virus epitopes, analogous to the VV/LCMV a11r130 matrix, which centers on the EBV BMLF1280 epitope (unpublished data) ( Fig. 2 ).
Figure 2.
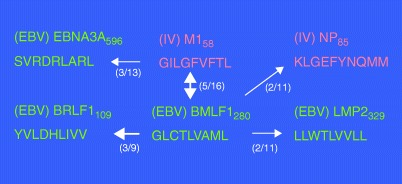
Cross‐reactive CD8+ T‐cell matrix. The Epstein – Barr virus BMLF1280 peptides induce CD8+ T‐cell responses that cross‐react with four other CD8+ T‐cell populations. The influenza M158 peptide can induce CD8+ T‐cell responses that cross‐react with at least three other CD8+ T‐cell populations. Which cross‐reactive pattern any individual displays depends on the private specificity of each individual's T‐cell receptor repertoire. The numbers in the figure indicate how many individuals of the total tested demonstrated the predominant cross‐ reactive response indicated (the thickness of arrows correlates with the frequency).
Analyses of the M158 TCR repertoire from two individuals experiencing EBV‐associated acute infectious mononucleosis, however, revealed a substantially different hierarchy of Jβ usage than in healthy influenza A immune donors, suggesting that a skewed subset of the M158‐specific TCR repertoire, probably those cross‐reactive with EBV, were being stimulated to proliferate. Interestingly, these cross‐reactive T cells, as in APL studies, behaved differently in their functional responses to each ligand. Some cross‐reactive cells bound both tetramers and produced tumor necrosis factor α (TNFα), IFNγ, and macrophage inflammatory protein 1β (MIP1β) to both ligands, some bound only one tetramer but produced TNFα, IFNγ, and MIP1β to the alternate ligand, while some bound only one tetramer but were able to produce only MIP1β to the alternate ligand. It would appear that how a cross‐reactive T‐cell interacts with its alternative ligand is highly variable and that functional patterns of T‐cell cross‐reactivity are indeed heterogeneous. Hence, multiple techniques are required to detect T‐cell cross‐reactivity, including tetramer staining and different functional assays. A potentially important factor in TCR interaction with its ligand is TCR avidity. The interaction between a cross‐reactive T‐cell and its alternative ligand could be too weak to stably bind tetramer but could be sufficient to induce a distinct hierarchy of cytokine production. This is analogous to the observation that influenza M1‐specific clones were unable to bind M1‐specific tetramers but produced IFNγ following M1 peptide stimulation (88).
HCV and fulminant hepatitis
There is extreme variability in the pathogenesis of HCV in humans, ranging from asymptomatic to fulminant and from sterilizing to persistent infections (89). Heterologous immunity may play a role in this variability (90, 91), as HCV encodes an HLA‐A2‐restricted epitope, NS31073‐1081, that shares six of eight amino acids with the influenza epitope NA231‐239, and T cells from influenza‐immune individuals with no evidence of a past HCV infection can often respond to the HCV epitope in vitro (37). Hence, the human population may be partially immune to HCV as a consequence of this cross‐reactivity. A pronounced cross‐reactive T‐cell response between influenza and HCV was noted in some patients with HCV infection, with resultant narrowing of the repertoire to this single epitope (90). These same patients experienced a fulminant necrotizing hepatitis far more severe than patients who mounted a more diverse T‐cell response. These patients would likely be immune to the ubiquitous influenza virus suggesting that private specificities may have dictated the altered immune responses.
Heterologous immunity and immune deviation
Immunity to previously encountered viruses can alter the cytokine response to subsequent virus encounters. Prior immunity to LCMV induces much higher levels of IFNγ and lower levels of IL‐6 upon VV infection (45, 46). Much of the enhanced IFNγ production comes directly from the LCMV epitope‐specific T cells activated by the VV infection (45, 46). Immunity to influenza protects mice from severe disease caused by infection with RSV after an RSV G‐protein vaccination (92) by shifting the usual T‐helper 2 (Th2) response into a Th1 response and preventing severe eosinophilic infiltrates in the lung. Changes in Th1/Th2 responses due to infections or vaccinations early in childhood might explain why some children are more likely to develop allergies than others. Several studies reported beneficial effects of early infections in childhood on the development of asthma, which may be the consequence of an early skewing of the memory pool toward a Th1 phenotype that would inhibit Th2‐based allergic responses (93).
Heterologous immunity and allografts
The impact of heterologous immunity extends to the field of transplantation, as viral infections can influence the generation of cellular immune responses to allo‐antigens and have been associated with the rejection of foreign tissue transplants (48, 49, 94, 95, 96, 97, 98, 99). The contribution of heterologous immunity to the induction of allospecific immune responses is supported by evidence that virus‐specific T cells cross‐react with allo‐antigens (12, 48, 99, 100, 101, 102, 103, 104). This cross‐reactivity provides a mechanism for the observed activation of allospecific T cells following infections and for the detection of memory allospecific T cells within individuals that have not been previously exposed to allo‐antigens (105). The activation of allospecific T cells by viral infections presents a unique obstacle for the transplant of foreign tissues and for the induction of transplant tolerance.
Detection of allospecific T cells
The naïve T‐cell repertoire contains a large number of allospecific T cells, some of which may be recruited into virus‐specific T‐cell responses. The frequency of naïve allospecific T cells has been estimated to range between 0.1 and 10% of the total T cells, using limiting dilution and cell outgrowth techniques 106, 107, 108, 109. However, these protocols are not optimal for quantification directly ex vivo, as they require lengthy periods of in vitro culture and may not accurately reflect the frequency of naïve allospecific T cells in vivo. More recently, intracellular cytokine assays have been utilized to detect allospecific T cells more efficiently directly ex vivo (105, 110, 111, 112). We have adopted this cytokine assay to specifically identify naïve T cells. Naïve T cells had previously been thought to acquire cytokine‐producing effector functions only after undergoing a differentiation process, but we demonstrated that naïve T cells rapidly produce TNFα but not IFNγ following TCR engagement in vitro (113). Using TNFα production as a marker, naïve allospecific T cells were readily detected directly ex vivo at frequencies ranging between 0.5 and 0.7% of the total T cells. Naïve allospecific T cells were distinguishable from effector/memory allospecific T cells by the differential production of TNFα and IFNγ, as effector/memory cells produced both cytokines (unpublished data).
Cross‐reactivity and activation of allospecific T cells by viral infection
The cross‐reactive nature of antigen recognition by T cells suggests that allospecific T cells may be activated by viral infections. Initial results from our laboratory demonstrated that acute infection of B6 mice with LCMV stimulated allospecific cytotoxicity against a broad range of foreign haplotypes (94, 95). The in vitro killing of allogeneic target cells detected seven days after infection was primarily mediated by CD8+ T cells (94, 99). Allospecific CD8+ T cells that were activated by infection with LCMV were also detected by the production of IFNγ in response to allogeneic targets (48, 49). Infection of mice with other viruses, including PV, VV, and MCMV, also stimulated allospecific T‐cell cytotoxicity, suggesting that this was a generalized phenomenon following infection (95). Cytotoxic allospecific CD8+ T cells were also found in humans infected with EBV during acute infectious mononucleosis (100, 114, 115).
Heterologous immunity is an important mechanism for the activation of allospecific T cells following infection, and CD8+ T‐cell cross‐reactivity between viruses and alloantigens has been demonstrated in numerous infection models, with T‐cell clones derived from mice infected with LCMV, influenza virus, or VSV and from humans infected with EBV or herpes simplex virus (12, 99, 100, 101, 102, 103, 104). Studies examining cross‐reactivity between LCMV‐specific CD8+ T cells and alloantigens showed that this cross‐reactivity was broad‐based, as a portion of T cells specific for each of the four LCMV epitopes examined (GP33, NP205, GP276, and NP396) cross‐reacted with H2d antigens, as determined by the production of IFNγ (48). However, while this cross‐reactivity with H2d antigens was broad, it was also distinctive, with different proportions of each epitope‐specific population recognizing the allogeneic targets. This distinctive nature of cross‐reactivity was clearly evident between LCMV‐specific T cells and H2k antigens, which was still broad‐based but was also more selective, as two of the four LCMV‐epitopes examined (GP33 and NP205) were cross‐reactive. These results suggest that the viral infections can dramatically alter the T‐cell repertoire in an unexpected manner by the activation of allospecific T cells and that infections may account for the presence of allospecific T cells with a memory phenotype in individuals that have not been previously exposed to alloantigens (105, 110, 111, 112). Indeed, after the acute LCMV infection resolved, memory phenotype allospecific CD8+ T cells remained present in the resting immune state (48, 49, 99).
Viral induced rejection of allogeneic implants in vivo
The activation of allospecific CD8+ T cells by viral infections may have important consequences for the transplantation of foreign tissues and for the maintenance of graft function. Viral infections have been demonstrated to rapidly induce a CD8+ T‐cell‐mediated rejection of allogeneic splenocyte implants in mice (116). In these studies, the fate of allogeneic cells implanted into virus‐infected mice was monitored using in vivo cytotoxicity assays. This assay allows T‐cell‐mediated rejection of CFSE‐labeled allogeneic splenocytes to be examined in vivo within 20 h after transfer. The in vivo cytotoxicity assay will also detect natural killer (NK) cell‐mediated rejection of allogeneic splenocytes, and to specifically examine T‐cell‐dependent mechanisms, NK cells need to be depleted before transfer of the target splenocytes (117). Infection of B6 mice with either LCMV or PV induced a CD8+ T‐cell‐dependent mechanism for the rejection of implanted allogeneic splenocytes that was first detectable one day after infection and reached peak levels by day three postinfection (116). This virus‐induced CD8+ T‐cell‐dependent rejection was surprising, in that it occurred at an early time point after infection, when NK cell functions are peaking. This outcome could either be due to allospecific T cells cross‐reacting with viral antigens or be due to a vigorous primary T‐cell response to the implanted allogeneic cells in the wake of the rich cytokine environment created during acute infection.
Viral infections abrogate the induction of allospecific tolerance
Traditional methods to prolong survival of allogeneic tissue grafts have involved the use of chronic immunosuppression to prevent the generation of donor‐specific immune responses 118, 119, 120. The use of costimulation blockade to tolerize T cells against specific donor tissues has proven to be a promising alternative approach to achieve long‐term survival of transplanted tissues without the need for generalized immunosuppression 121, 122, 123, 124, 125, 126. However, recent studies have demonstrated that acute viral infections can seriously diminish the efficacy of tolerance‐inducing protocols. Acute infection of mice with viruses such as LCMV or PV within 1–15 days post‐transplant or by persistent infection with LCMV interfered with the induction of tolerance to alloantigens and resulted in rapid rejection of allogeneic skin grafts 96, 97, 98. Depletion of CD8+ cells from LCMV‐infected recipient mice significantly delayed rejection of the skin allografts, supporting a role for CD8+ T cells in mediating the rejection. The inability of poly(I:C), VV, or MCMV to abrogate tolerance under these conditions suggests that the production of cytokines alone is not sufficient to induce rejection (97), and the mechanisms mediating this virus‐induced abrogation of transplant tolerance still remain to be clarified, though they could involve cross‐reactivity.
Although a cytokine‐mediated disruption of tolerance was not evident at time points after transplantation, mice treated with Toll‐like receptor (TLR) agonists, such as poly(I:C) and lipopolysaccharide, prior to the transplant of allogeneic tissues were not effectively tolerized, suggesting that the very early stages of tolerance induction are sensitive to a cytokine‐dependent mechanism (127). These findings suggest that ongoing viral infections will be an impediment to the use of costimulation blockade for the induction of tolerance in humans.
Heterologous immunity abrogates the induction of allospecific tolerance
The presence of memory allospecific T cells has been associated with accelerated rejection of allogeneic tissues in transplant recipients (105, 110, 111, 112). Memory allospecific T cells generated by exposure to alloantigens are also refractory to the effects of costimulation blockade, because memory T cells are less reliant on costimulatory signals for activation (49, 128, 129). Memory allospecific T cells generated by infections are also refractory to costimulation blockade (48, 49, 130). Mice previously infected with LCMV and possessing memory phenotype allospecific T cells were not sufficiently tolerized by costimulation blockade to accept allogeneic skin grafts (48, 49). Moreover, sequential infection of mice with heterologous viruses generated higher frequencies of allospecific memory T cells and increased resistance to costimulation blockade (48, 49). These results suggest that an individual's history of infections will influence their responsiveness to allo‐antigens, and this history will be an important factor for the design of novel methodologies to achieve optimal graft survival in humans due to their large complex memory populations.
Heterologous immunity and modulation of prior memory
Heterologous immunity between unrelated pathogens has a great impact on the stability of the memory T‐cell pool. The homeostasis of T cells is a tightly regulated phenomenon with, in the resting state, relatively fixed proportions of CD4+ and CD8+ T cells and somewhat fixed proportions of memory to naïve cells. As the individual ages, the thymus, the source of naïve T cells, involutes, and there can be a proportional increase of memory phenotype over naïve phenotype T cells (131). The distinction between bona fide memory cells and other cells possessing a memory‐like antigenic phenotype is an important one. The mouse memory marker of CD44 can be expressed on naïve T cells after they receive a foreign antigen signal and go through programmed proliferation and contraction and become bona fide memory cells, or CD44 can be expressed on naïve T cells when they are introduced into a lymphopenic environment (131, 132). Such an environment stimulates naïve T cells to respond to self‐antigens or perhaps to microbial flora to acquire a memory CD44 phenotype and fill up the available space, but they do not go through the same differentiation scheme as bona fide memory cells. These cells are thus the product of homeostatic proliferation and are referred to as ‘HP’ cells. Other types of memory phenotype cells that are not bona fide memory cells include classical CD1‐restricted NKT cells bearing an invariant TCRα chain (133) and CD1‐nonrestricted NKT cells, such as CD8+ T cells coexpressing Ly49 markers (134, 135). The net result is that cells expressing memory phenotypic markers may have been derived from different sources.
Under resting non‐lymphopenic conditions, both bona fide memory and HP cells undergo a steady state low or basal level division, which maintains T‐cell number and keeps T cells of different specificities in their same proportional frequencies 136, 137, 138. This is likely because the cells all have similar division rates. The number of cells remains the same either because of the limitation of protective niches in the lymphoid organs or because each division gives rise to one surviving and one dead daughter. This question has not been resolved. This steady state low‐level proliferation, which also is referred to as homeostatic proliferation, is not driven by antigen and does not require an MHC‐containing environment. It is at least partially dependent on signaling by IL‐15 or IL‐7 139, 140, 141.
Because of this steady‐state homeostasis, the proportion of bona fide memory T cells in a resting immunological environment can be very stable for a long period of time, and studies in mice have shown that CD8+ T‐cell memory is very stable (142, 143). Some reports suggest that CD4+ T‐cell memory gradually wanes (144, 145), but this point is controversial and is not supported by other studies 146, 147, 148. For some time there was a debate over whether antigen was needed for the preservation of T‐cell memory. That concept was refuted by adoptive transfer studies showing that both CD8+ and CD4+ memory T cells could be maintained in environments lacking expression of MHC antigens (146, 149). These experiments were performed with fully differentiated memory cells, and some evidence suggests that an inadequate priming of T cells may lead to less stable memory cells 150, 151, 152. Nevertheless, in humans, VV‐specific T cells can be found several decades after immunization (153), and recent quantitative longitudinal studies have shown moderately declining but still impressive T‐ and B‐cell memory to VV over a similar period (154, 155). There are suggestions of apparently better protective memory induced by attenuated rather than inactivated viral vaccines, but it is unclear whether the benefit of the attenuated vaccines is from generating a stronger immune response in the first place because of replication of the antigen, generating a response more biased to CD8+ T cells, or having persistent antigen continue to stimulate responses over time. Our data have shown that heterologous viral infections can cause a substantial reduction of memory to previously encountered pathogens (34, 142, 156, 157). In nature, where individuals are constantly challenged with infections by different pathogens, the persistence of antigen may counter balance the loss of memory that would occur by such infections.
Apoptosis in T‐cell responses
T‐cell numbers are regulated by the ratio of cell division to cell death, which usually occurs by apoptosis. It is important to distinguish the concepts of cell division from cell proliferation, the latter meaning an increase in cell number. Division may occur without an increase in cell number, as is the case in steady‐state homeostasis. Dividing cells may become sensitized to various types of proapoptotic stimuli, and division must overcome the apoptosis to result in an increase in number. The dynamics of cell number changes during LCMV infection of mice are displayed in Fig. 3 . There initially is a significant loss in T‐cell number during the early stage of LCMV and many other viral infections in mice and humans (156, 158, 159, 160, 161). An expansion phase of virus‐specific T cells follows, where division outpaces apoptosis. Next, there is a contraction or silencing phase, where T‐cell number in the spleen and lymph nodes declines through apoptosis (162) and migration into peripheral tissue (45, 163, 164, 165). The parameters of these responses are similar in mouse and human.
Figure 3.
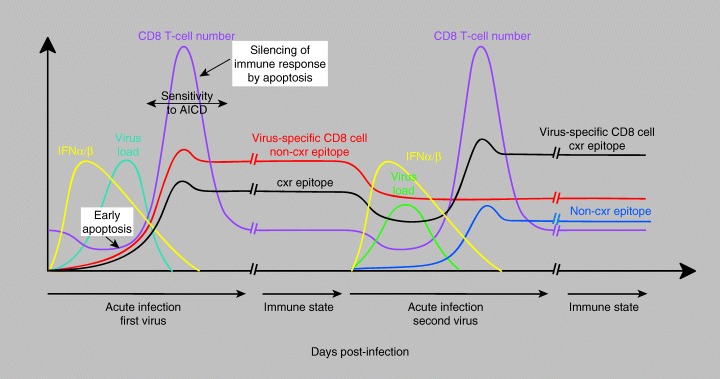
Dynamics of the CD8+ T‐cell response during sequential heterologous virus infections. This figure demonstrates the kinetics of virus growth, interferon synthesis, virus‐induced lymphopenia, T‐cell expansion and then apoptotic decline, and stability of CD8+ memory in mice infected with lymphocytic choriomeningitis virus. It shows the enhancement of T cells specific to cross‐reactive (cxr) and the attrition against non‐cxr epitopes after challenge with a heterologous virus, such as Pichinde virus. Reprinted, with permission, from the Annual Reviews of Immunology, Volume 22 ©2004 by Annual Reviews http://www.annualreviews.org.
Virus‐induced lymphopenia
Many viral infections in mouse and human induce a substantial loss in lymphocyte and leukocyte numbers in the early stages of infections (166). Undoubtedly, there may be many mechanisms for this loss in cell number, but a common mediator may be type 1 IFN, which is induced at high levels in the early stages of infections in direct response to viral replication. A substantial loss in CD8+ and CD4+ T‐cell number during the first five days of LCMV infection parallels the levels of type 1 IFN production, does not occur in type 1 IFN receptor knockout mice (159), is blocked by antibody to type 1 IFN (167), and is mimicked by IFN‐inducing TLR agonists, such as poly(I:C) (159). The cells suffering the highest levels of apoptosis are CD44hi CD8+ T cells. These cells activate caspases and express phosphatidyl serine on their membranes, which interacts with annexin V (158, 159). The apoptotic loss of either memory or, to a lesser extent, naïve CD8+ T cells occurs prior to cell division, as shown by bromodeoxyuridine uptake studies in vivo (159) or by implanting CSFE‐labeled T cells into mice and looking for loss of CSFE staining as an indicator of cell division versus apoptosis (168). This early apoptosis of T cells is not inhibited by stimulation of the T cells with their ligand. Injection of mice with enough LCMV epitope GP33‐43 to cause activation of GP33‐specific transgenic or memory CD8+ T cells in vivo in regards to up‐regulation of CD69 did not inhibit LCMV‐ or poly(I:C)‐induced apoptosis of the cells. This finding is of interest, because some models of T‐cell activation have indicated that exposure of T cells to ligand, especially in the context of proper costimulation, causes an upregulation of antiapoptotic factors such as Bcl‐XL to protect the dividing T cells from apoptosis (169). Our computer modeling, however, indicates that without the apoptosis of memory T cells at the early stages of infection, cross‐reactive T cells might dominate the response to any infection (168, 170). By selectively reducing the frequency of memory cells at the beginning of an immune response, there may be a more highly diverse T‐cell response to a new pathogen, and this high diversity may be beneficial for pathogen control.
Type 1 IFN appears to play a number of other roles in the early stages of T‐cell responses. It has a major effect on the differentiation of antigen‐presenting dendritic cells, acting, somewhat surprisingly, through a signal transducer and activator of transcription 2 (STAT2)‐dependent pathway (171). Recent work on the ‘growth inhibitory’ function of type 1 IFN, which might be linked to its pro‐apoptotic properties, has implicated the importance of the STAT1 signaling pathway (172). Of interest is that CD8+ T cells downregulate STAT1 after activation and then become resistant to the growth inhibitory effects of IFN. Type 1 IFN can also induce the division of memory T cells through its ability to induce IL‐15 (173). Most memory T cells express receptors for IL‐15, which is also a mediator of homeostatic division (139, 140). Together, it seems that type 1 IFN first causes apoptosis of memory T cells and a generalized lymphopenic state. Then IL‐15 stimulates them to divide until the lymphopenic state is corrected. They do not ‘overshoot’ in number, unless antigen is there to drive a new immune response.
Contraction or silencing phase
At the peak of the T‐cell response, the activated T cells in the spleen and lymph nodes express elevated levels of Fas and Fas ligand (FasL) (54), become susceptible to antigen‐driven activation‐induced cell death (AICD) (174), express a preapoptotic phenotype, as indicated by staining with annexin V, but maintain functionality, such as cytotoxicity or cytokine production (175). There has been controversy concerning whether staining with annexin V really means impending apoptosis or whether it might just be a marker for T‐cell activation. Our studies showed that the fate of LCMV‐specific annexin V+ CD8+ T cells, when introduced into culture in vitro, was to fragment their DNA and complete the apoptotic processes. When transferred into mice in vivo, the annexin V+ cell populations gradually decreased in number and failed to mediate a recall response, whereas similarly transferred annexin V– cells could mediate a recall response (175). Some correlations were made between cells that were annexin V– and expression of receptors for IL‐7 (175). IL‐7R is expressed on cells bearing high concentrations of antiapoptotic markers such as Bcl‐2, and these are precursors for fully functional memory cells (176). We thus feel that annexin V is indeed a harbinger of apoptosis, though the apoptosis does not necessarily happen immediately.
Part of the silencing phase is associated with the dissemination of T cells from the spleen and lymph nodes into the peripheral organs. It is commonly stated that 95% of the activated T cells die by apoptosis, but that number is based on examining T‐cell frequencies in lymphoid tissue. One study in the influenza system indicated that there was a much smaller reduction when all the cells in the body were added up (165). A feature of LCMV‐specific CD8+ cells in the periphery, meaning lung, visceral fat, or peritoneal cavity, is that they have a different apoptotic phenotype than those present in the spleen and lymph nodes at the same time. These peripheral T cells react less with annexin V, express more IL‐7R, stain less with antibodies to Fas or FasL, and are relatively resistant to ligand‐triggered AICD (54, 175). In the LCMV system, part of the resistance of peritoneal cells to AICD was linked to the presence of transforming growth factor‐β (54). In the lung of influenza‐infected mice, the resistance to apoptosis was linked to collagen in the lung signaling through α1β1 integrin expressed on the T cells (177). Signaling through certain integrins and adhesion molecules has long been known to render T cells resistant to apoptosis (178), and changes in the tissue environment of T cells may well alter their apoptotic properties. The significance of this finding may relate to the fact that effector memory T cells may linger in peripheral tissue long after clearance of a virus, providing a potential fast acting line of defense to pathogens.
An intriguing aspect about the apoptosis of T cells is that there are epitope‐specific preferences regarding the degree of apoptosis (175). This can easily be seen in the LCMV infection of C57BL/6 mice, where T cells specific for the Db‐restricted NP396‐404 epitope express higher annexin V reactivity, higher Fas, lower IL‐7R (175), and higher mitochondrial membrane potential (179) than do T cells specific for the Db‐restricted GP33‐41 epitope. This finding might be explained by the fact that the T cells specific to this epitope may be triggered more often than by the GP33 epitope, as the LCMV NP is expressed at much higher levels than the GP during the LCMV infection of cells. However, VV recombinants expressing NP versus G‐proteins under similar promoters stimulate T cells with the same apoptotic properties (175). Nevertheless, the NP396 epitope is a very high affinity binder to H2Db and may still be expressed better than GP33. What is most intriguing, however, is that this differential in apoptosis between NP396 and GP33 is also seen on secondary immunizations, where there would be less antigen presented, and in the resting immune state, where there is no antigen at all. The apoptotic levels are lower in the resting immune state, but the apoptotic differential between NP396‐specific versus GP33‐specific T cells remains. It seems that the apoptotic properties of epitope‐specific T cells are an intrinsic property of the epitope, presumably learned during the early phases of infection and imprinted permanently in those T cells. One might expect that the ratio of NP396‐ to GP33‐specific T cells should change in the resting memory state, and indeed there is a moderate shift to GP33 dominance over time within this otherwise moderately stable memory population (175).
Loss of memory after viral infections
With the caveat of some subtle shifting of the hierarchies of T cells having different apoptotic properties, our studies of infections with LCMV, PV, VV, MCMV, and VSV have indicated that CD8+ T‐cell memory is generally quite stable for well over a year but that it is significantly reduced after subsequent infections with heterologous viruses (34, 142, 157). Very little information is available concerning the stability of CD8+ T‐cell memory in humans, because most studies are done with persistent virus infections, such as HIV, EBV, CMV, and HCV, or with repeatedly encountered pathogens like influenza virus. Recent studies of individuals immunized with VV, the small pox vaccine, have indicated relatively stable memory populations over several decades, though the memory frequencies did decline over 10‐fold (155). It will be necessary to determine whether the VV‐specific T‐cell repertoires remain stable over time to assess how stable this memory is. That is because our studies in the mouse have suggested that VV may be highly cross‐reactive with other viruses, inviting the possibility that some of the VV‐specific T cells may be maintained through cross‐reactivity (36, 71)( Fig. 3 ).
A loss of pre‐existing memory after subsequent infections has also not been clarified during human infections. With a ‘larger’ immune system, humans may be able to accommodate large pools of new memory cells without excluding others. The somewhat stable VV‐immune frequencies, assuming that the T cells are not maintained through cross‐reactivity, might argue that the loss of memory is not a major issue in human infections. However, the infections that drive memory loss in mice are major systemic infections that cause a substantial lymphopenia and then splenomegaly, so one might expect substantial memory loss to occur more frequently in children rather than adults, who do not have many diseases of this nature.
Cause of the loss in memory
We have proposed two models to explain the loss of pre‐existing memory occurring as a consequence of a heterologous viral infection: the Active Attrition Model, whereby something directly kills off the memory cells, and the Passive Attrition Model, where the newly formed memory cells compete with the previously formed memory cells for protective niches in the spleen and lymph nodes as the immune response contracts (27, 71, 170). Although both these mechanisms might influence the survival of memory, our data support the active model, occurring during the early cytokine‐induced lymphopenia phase of infection (156). By quantifying pre‐existing antigen‐specific CD8+ memory cells and following their frequencies through the course of infection, it is clear that their numbers go down very early during the virus‐induced lymphopenia and that their recovery is rather modest. There may still be some accommodation issues in competing for protective niches later in infection, but its relative importance seems less than the active attrition in these models of infection. Because virus‐induced lymphopenia is a commonly described event occurring during many human infections, including influenza, West Nile, Ebola, HIV, severe acute respiratory syndrome corona, and even human LCMV (166, 168), we suspect that this will be a factor driving attrition in memory in humans. There has been little analysis of the effects of human virus‐induced lymphopenia on CD8+ memory to other antigens, though it is known that patients receiving IFN therapy at least initially have a loss in memory phenotype CD8+ T cells (180). These types of issues are now being examined in controlled vaccination studies, but the vaccines may not drive the level of lymphopenia necessary to cause a loss in memory. Otherwise, the vaccinees would get sick. This issue may be best addressed in those many volunteers who get bad reactions to the small pox vaccine!
Memory cell attrition during persistent infections
T‐cell dynamics are greatly altered during persistent infections, which not only induce proapoptotic cytokines but also drive T cells into apoptotic deletion by AICD or functional exhaustion (181). Our studies have shown that memory T cells specific to heterologous viruses are greatly deleted after the initiation of infection with a high dose of LCMV clone 13, which will cause a persistent infection, and those T cells do not rebound in frequency (156). In addition, when CSFE‐labeled memory T cells are transferred into mice persistently infected with the LCMV clone 13, a few attempt modest levels of homeostatic division, but most are rapidly eliminated. If similar mechanisms occur in humans, it would suggest that persistent infections such as HIV or HCV should vigorously delete memory to everything else that does not receive re‐stimulation by its own antigen or by cross‐reactive antigen. During persistent human infections, could residual memory T cells specific to nonpersistent antigens be cross‐reactive with the pathogen driving the infection?
Failure of depleted memory cell populations to recover
The residual populations of memory cells remaining after the early cytokine‐induced deletion during infections are ineffective in restoring their population frequencies (156). This ineffectiveness is for two reasons, the first being that they have to compete with antigen‐specific T cells responding to the ongoing infection. Antigen‐driven proliferation at three to four divisions per day occurs at a much higher rate than homeostatic division (43, 182), and the pre‐existing memory cells cannot compete. The second factor is that bona fide memory cells are not very effective at homeostatic proliferation in the first place, even in the absence of an infection (183). When CSFE‐labeled LCMV‐immune splenocytes were transferred into T‐cell‐deficient mice or mice rendered lymphopenic by irradiation or poly(I:C) inoculation, the proportion of LCMV‐specific T cells in the reconstituting population of cells became, in comparison with its starting frequency, substantially reduced as the cells proliferated to fill up the lymphopenic environment. Thus, the memory cells compete poorly with the HP cells that respond to self‐antigens or microbial flora (131, 183, 184). We predict that conditions in humans that cause lymphopenia other than infections, such as irradiation or immunosuppressive drug treatment, should induce a memory cell loss, which needs to be tested.
Conclusions
In humans as well as mice, the immune system has evolved such that multiple diverse antigen‐specific memory TCR repertoires accumulate over a lifetime. With each new infection, memory T cells must compete for limited space. This competition is at least partially accomplished by the preferential loss of prior memory populations by active cytokine‐dependent mechanisms. Also, memory T cells specific to previously encountered pathogens but also cross‐reactive with the newly encountered pathogen are preferentially maintained or expanded, such that the T‐cell repertoire specific to the previous pathogen becomes permanently altered ( Fig. 3 ). These activated cross‐reactive memory T cells play a role in heterologous immunity by modulating the T‐cell immune hierarchy and the private specificity of individual antigen‐specific TCR repertoires, leading to an alteration in the balance between protective immunity and immunopathology. Virus‐specific T‐cell responses cross‐reactive with allo‐antigens can also alter the memory allospecific T‐cell pool and influence graft rejection and tolerance induction. Thus, getting the wrong infection in a host with a particular MHC and at the wrong time in a sequence of other infections might have significant consequences for the host. To understand the fine balance memory T cells can play in the induction of protective immunity versus pathology, we need to learn more about the consequences of heterologous immunity and cross‐reactive T‐cell responses.
Acknowledgements
This work was supported by NIH research grants AI17672, AI46629, AI46578, AI49320, AR35506, and DK32520, and DFG fellowship CO310/1–1.
References
- 1. Ahmed R, Gray D. Immunological memory and protective immunity: understanding their relation. Science 1996;272: 54–60. [DOI] [PubMed] [Google Scholar]
- 2. Dutton RW, Bradley LM, Swain SL. T cell memory. Annu Rev Immunol 1998;16: 201–223. [DOI] [PubMed] [Google Scholar]
- 3. Zinkernagel RM. On differences between immunity and immunological memory. Curr Opin Immunol 2002;14: 523–536. [DOI] [PubMed] [Google Scholar]
- 4. Pewe LL, Netland JM, Heard SB, Perlman S. Very diverse CD8 T cell clonotypic responses after virus infections. J Immunol 2004;172: 3151–3156. [DOI] [PubMed] [Google Scholar]
- 5. Naumov YN, Naumova EN, Hogan KT, Selin LK, Gorski J. A fractal clonotype distribution in the CD8+ memory T cell repertoire could optimize potential for immune responses. J Immunol 2003;170: 3994–4001. [DOI] [PubMed] [Google Scholar]
- 6. Falk K, Rotzschke O, Stevanovic S, Jung G, Rammensee H. Allele‐specific motifs revealed by sequencing of self‐peptides eluted from MHC molecules. Nature 1991;351: 290–296. [DOI] [PubMed] [Google Scholar]
- 7. Ding Y‐H, Baker BM, Garboczi DN, Biddison WE, Wiley DC. Four A6‐TCR/peptide/HLA‐A2 structures that generate very different T cell signals are nearly identical. Immunity 1999;11: 45–56. [DOI] [PubMed] [Google Scholar]
- 8. Kjer‐Nielsen L, et al. A structural basis for the selection of dominant alphabeta T cell receptors in antiviral immunity. Immunity 2003;18: 53–64. [DOI] [PubMed] [Google Scholar]
- 9. Rudolph MG, Wilson IA. The specificity of TCR/pMHC interaction. Curr Opin Immunol 2002;14: 52–65. [DOI] [PubMed] [Google Scholar]
- 10. Reiser JB, et al. CDR3 loop flexibility contributes to the degeneracy of TCR recognition. Nat Immunol 2003;4: 241–247. [DOI] [PubMed] [Google Scholar]
- 11. Reiser JB, et al. A T cell receptor CDR3beta loop undergoes conformational changes of unprecedented magnitude upon binding to a peptide/MHC class I complex. Immunity 2002;16: 345–354. [DOI] [PubMed] [Google Scholar]
- 12. Burrows SR. Cross‐reactive recognition of viral and self‐peptides by a ‘public’ T cell receptor expressed by cytotoxic T lymphocytes expanded in multiple unrelated individuals. Immunol Lett 2004;93: 7–9. [DOI] [PubMed] [Google Scholar]
- 13. Barnett LA, Fujinami RS. Molecular mimicry: a mechanism for autoimmune injury. FASEB J 1992;6: 840–844. [DOI] [PubMed] [Google Scholar]
- 14. Boniface J, Reich Z, Lyons DS, Davis MM. Thermodynamics of T cell receptor binding to peptide‐MHC: evidence for a general mechanism of molecular scanning. Proc Natl Acad Sci USA 1999;96: 11446–11451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Willcox B, et al. TCR binding to peptide‐MHC stabilizes a flexible recognition interface. Immunity 1999;10: 357–365. [DOI] [PubMed] [Google Scholar]
- 16. Borg N, et al. The CDR3 regions of an immunodominant T cell receptor dictate the ‘energetic landscape’ of peptide‐MHC recognition. Nat Immunol 2005;6: 171–180. [DOI] [PubMed] [Google Scholar]
- 17. Wu L, Tuot D, Lyons D, Garcia K, Davis MM. Two‐step binding mechanism for T‐cell receptor recognition of peptide MHC. Nature 2002;418: 552–556. [DOI] [PubMed] [Google Scholar]
- 18. Daniel C, Horvath S, Allen PM. A basis for alloreactivity: MHC helical residues broaden peptide recognition by the TCR. Immunity 1998;8: 543–552. [DOI] [PubMed] [Google Scholar]
- 19. Speir JA, et al. Structural basis of 2C TCR allorecognition of H‐2Ld peptide complexes. Immunity 1998;8: 553–562. [DOI] [PubMed] [Google Scholar]
- 20. Alam SM, Gascoigne NR. Posttranslational regulation of TCR Valpha allelic exclusion during T cell differentiation. J Immunol 1998;160: 3883–3890. [PubMed] [Google Scholar]
- 21. Mason D. A very high level of crossreactivity is an essential feature of the T cell repertoire. Immunol Today 1998;19: 395–404. [DOI] [PubMed] [Google Scholar]
- 22. Cabaniols JP, Fazilleau N, Casrouge A, Kourilsky P, Kanellopoulos JM. Most alpha/beta T cell receptor diversity is due to terminal deoxynucleotidyl transferase. J Exp Med 2001;194: 1385–1390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Gilfillan S, et al. Efficient immune responses in mice lacking N‐region diversity. Eur J Immunol 1995;25: 3115–3122. [DOI] [PubMed] [Google Scholar]
- 24. Gavin MA, Bevan MJ. Increased peptide promiscuity provides a rationale for the lack of N regions in the neonatal T cell repertoire. Immunity 1995;3: 793–800. [DOI] [PubMed] [Google Scholar]
- 25. Zarozinski CC, Welsh RM. Minimal bystander activation of CD8 T cells during the virus‐ induced polyclonal T cell response. J Exp Med 1997;185: 1629–1639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Daly K, Nguyen P, Woodland DL, Blackman MA. Immunodominance of major histocompatibility complex class‐I‐restricted influenza virus epitopes can be influenced by the T‐cell receptor repertoire. J Virol 1995;69: 7416–7422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Welsh RM, Selin LK, Szomolanyi‐Tsuda E. Immunological memory to viral infections. Annu Rev Immunol 2004;22: 711–743. [DOI] [PubMed] [Google Scholar]
- 28. Haanen JBAG, Wolkers MC, Kruisbeek AM, Schumacher TNM. Selective expansion of cross‐reactive CD8+ memory T cells by viral variants. J Exp Med 1999;190: 1319–1328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Effros RB, Doherty PC, Gerhard W, Bennick JR. Generation of both cross‐reactive and virus‐specific T‐cell populations after immunization with serologically distinct influenza A viruses. J Exp Med 1977;145: 557–568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Boon AC, et al. Recognition of homo‐ and heterosubtypic variants of influenza A viruses by human CD8+ T lymphocytes. J Immunol 2004;172: 2453–2460. [DOI] [PubMed] [Google Scholar]
- 31. Mongkolsapaya J, et al. Original antigenic sin and apoptosis in the pathogenesis of dengue hemorrhagic fever. Nat Med 2003;9: 921–927. [DOI] [PubMed] [Google Scholar]
- 32. Zivny J, et al. Partial agonist effect influences the CTL response to a heterologous dengue virus serotype. J Immunol 1999;163: 2754–2760. [PubMed] [Google Scholar]
- 33. Maeda K, Toyosaki‐Maeda T, Rothman AL, Ennis FA. Identification and analysis for cross‐reactivity among hantaviruses of the H‐2b‐restrictied cytotoxic T‐lymphocyte epitopes in Sin Nombre virus nucleocapsid protein. J Gen Virol 2004;85: 1909–1919. [DOI] [PubMed] [Google Scholar]
- 34. Brehm MA, Pinto AK, Daniels KA, Schneck JP, Welsh RM, Selin LK. T cell immunodominance and maintenance of memory regulated by unexpectedly cross‐reactive pathogens. Nat Immunol 2002;3: 627–634. [DOI] [PubMed] [Google Scholar]
- 35. Spaulding AC, Kurane I, Ennis FA, Rothman AL. Analysis of murine CD8(+) T‐cell clones specific for the Dengue virus NS3 protein: flavivirus cross‐reactivity and influence of infecting serotype. J Virol 1999;73: 398–403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Kim SK, Cornberg M, Wang XZ, Chen HD, Selin LK, Welsh RM. Private specificities of CD8 T cell responses control patterns of heterologous immunity. J Exp Med 2005;201: 523–533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Wedemeyer H, Mizukoshi E, Davis AR, Bennink JR, Rehermann B. Cross‐reactivity between hepatitis C virus and influenza A virus determinant‐specific cytotoxic T cells. J Virol 2001;75: 11392–11400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Clute SC, et al. Cross‐reactive influenza virus‐specific CD8 T cells contribute to the lymphoproliferation in Epstein – Barr virus‐associated infectious mononucleosis. J Clin Invest 2005;115: 3602–3612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Acierno PM, Newton DA, Brown EA, Maes LA, Baatz JE, Gattoni‐Celli S. Cross‐reactivity between HLA‐A2‐restricted FLU‐M1:58–66 and HIV p17 GAG:77–85 epitopes in HIV‐infected and uninfected individuals. J Transl Med 2003;1: 3–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Nilges K, et al. Human papillomavirus type 16, E7 peptide‐directed CD8+ T cells from patients with cervical cancer are cross‐reactive with the coronavirus NS2 protein. J Virol 2003;75: 5464–5474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Veiga‐Fernandes H, Walter U, Bourgeois C, McLean A, Rocha B. Response of naive and memory CD8 T cells to antigen stimulation in vivo. Nat Immunol 2000;1: 47–53. [DOI] [PubMed] [Google Scholar]
- 42. Kaech SM, Ahmed R. Memory CD8+ T cell differentiation: initial antigen encounter triggers a developmental program in naive cells. Nat Immunol 2001;2: 415–422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Van Stipdonk MJ, Lemmens EE, Schoenberger SP. Naive CTLs require a single brief period of antigenic stimulation for clonal expansion and differentiation. Nat Immunol 2001;2: 423–429. [DOI] [PubMed] [Google Scholar]
- 44. Selin LK, Varga SM, Wong IC, Welsh RM. Protective heterologous antiviral immunity and enhanced immunopathogenesis mediated by memory T cell populations. J Exp Med 1998;188: 1705–1715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Chen HD, Fraire AE, Joris I, Brehm MA, Welsh RM, Selin LK. Memory CD8+ T cells in heterologous antiviral immunity and immunopathology in the lung. Nat Immunol 2001;2: 1067–1076. [DOI] [PubMed] [Google Scholar]
- 46. Chen HD, Fraire A, Joris I, Welsh R, Selin LK. Specific history of heterologous virus infections determines antiviral immunity and immunopathology in the lung. Am J Pathol 2003;188: 1341–1355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Ostler T, Pircher H, Ehl S. ‘Bystander’ recruitment of systemic memory T cells delays the immune response to respiratory virus infection. Eur J Immunol 2003;33: 1839–1848. [DOI] [PubMed] [Google Scholar]
- 48. Brehm MA, Markees TG, Daniels KA, Greiner DL, Rossini AA, Welsh RM. Direct visualization of cross‐reactive effector and memory allo‐specific CD8 T cells generated in response to viral infections. J Immunol 2003;170: 4077–4086. [DOI] [PubMed] [Google Scholar]
- 49. Adams AB, et al. Heterologous immunity provides a potent barrier to transplantation tolerance. J Clin Invest 2003;111: 1887–1895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Zhao Z‐S, Granucci F, Yeh L, Schaffer PA, Cantor H. Molecular mimicry by herpes simplex vrus‐type 1: autoimmune disease after viral infection. Science 1998;279: 1344–1347. [DOI] [PubMed] [Google Scholar]
- 51. Chen W, Anton LC, Bennink JR, Yewdell JW. Dissecting the multifactorial causes of immunodominace in class I‐restricted T cell responses to viruses. Immunity 2000;12: 83–93. [DOI] [PubMed] [Google Scholar]
- 52. Yewdell JW, Bennink JR. Immunodominance in major histocompatibility complex class I‐restricted T lymphocyte responses. Annu Rev Immunol 1999;17: 51–88. [DOI] [PubMed] [Google Scholar]
- 53. Maryanski JL, Attuil V, Bucher P, Walker PR. A quantitative, single‐cell PCR analysis of an antigen‐specific TCR repertoire selected during an in vivo CD8 response: direct evidence for a wide range of clone sizes with uniform tissue distribution. Mol Immunol 1999;36: 745–753. [DOI] [PubMed] [Google Scholar]
- 54. Wang XZ, Stepp SE, Brehm MA, Chen HD, Selin LK, Welsh RM. Virus‐specific CD8 T cells in peripheral tissues are more resistant to apoptosis than those in lymphoid organs. Immunity 2003;18: 631–642. [DOI] [PubMed] [Google Scholar]
- 55. Cohen GB, et al. Clonotype tracking of TCR repertoires during chronic virus infections. Virology 2002;304: 474–484. [DOI] [PubMed] [Google Scholar]
- 56. Lin MY, Welsh RM. Stability and diversity of T cell receptor (TCR) repertoire usage during lymphocytic choriomeningitis virus infection of mice. J Exp Med 1998;188: 1993–2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Blattman JN, Sourdive DJ, Murali‐Krishna K, Ahmed R, Altman JD. Evolution of the T cell repertoire during primary, memory, and recall responses to viral infection. J Immunol 2000;165: 6081–6090. [DOI] [PubMed] [Google Scholar]
- 58. Camereon TO, Cohen GB, Islam SA, Stern LJ. Examination of the highly diverse CD4 T‐cell repertoire directed against an influenza peptide: a step toward TCR proteomics. Immunogenetics 2002;54: 611–620. [DOI] [PubMed] [Google Scholar]
- 59. Bousso P, et al. Individual variations in the murine T cell response to a specific peptide reflect variability in naive repertoire. Immunity 1998;9: 169–178. [DOI] [PubMed] [Google Scholar]
- 60. Betts MR, et al. Putative immunodominant human immunodeficiency virus‐specific CD8(+) T cell responses cannot be predicted by major histocompatibility complex class I haplotype. J Virol 2000;74: 9144–9151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Koziel MJ, Walker BD. Characteristics of the intrahepatic cytotoxic T lymphocyte response in chronic hepatitis C virus infection. Springer Semin Immunopathol 1997;19: 69–83. [DOI] [PubMed] [Google Scholar]
- 62. Hohn H, et al. Longitudinal analysis of Mycobacterium tuberculosis 19‐kDa antigen‐specific T cells in patients with pulmonary tuberculosis: association with disease activity and cross‐reactivity to a peptide from HIVenv gp120. Eur J Immunol 2003;33: 1613–1623. [DOI] [PubMed] [Google Scholar]
- 63. Kim SK, Brehm MA, Welsh RM, Selin LK. Dynamics of memory T cell proliferation under conditions of heterologous immunity and bystander stimulation. J Immunol 2002;169: 90–98. [DOI] [PubMed] [Google Scholar]
- 64. Cornberg M, et al. Narrowed T cell receptor repertoire and viral escape as a consequence of heterologous immunity. J Clin Invest 2006;116: (in press). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Dong T, et al. HIV‐specific cytotoxic T cells from long‐term survivors select a unique T cell receptor. J Exp Med 2004;200: 1547–1757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Wilson JDK, et al. Oligoclonal expansions of CD8+ T cells in chronic HIV infection are antigen specific. J Exp Med 1998;188: 785–790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Pantaleo G, et al. Major expansion of CD8+ T cells with a predominant V beta usage during the primary immune response to HIV. Nature 1994;370: 463–467. [DOI] [PubMed] [Google Scholar]
- 68. Meyer‐Olson D, et al. Limited T cell receptor diversity of HCV‐specific T cell responses is associated with CTL escape. J Exp Med 2004;200: 307–319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Borrow P, et al. Antiviral pressure exerted by HIV‐1‐specific cytotoxic T lymphocytes (CTLs) during primary infection demonstrated by rapid selection of CTL escape virus. Nat Med 1997;3: 205–211. [DOI] [PubMed] [Google Scholar]
- 70. Welsh RM, Kim SK, Cornberg M, Clute SC, Selin LK, Naumov YN. The privacy of T cell memory to viruses. Curr Top Microbiol Immunol (in press). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Selin LK, Welsh RM. Plasticity of T cell memory responses to viruses. Immunity 2004;20: 5–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Ploegh HL. Viral strategies of immune evasion. Science 1998;280: 248–253. [DOI] [PubMed] [Google Scholar]
- 73. Doherty PC, Zinkernagel RM. T‐cell‐mediated immunopathology in viral infections. Transplant Rev 1974;19: 89–120. [DOI] [PubMed] [Google Scholar]
- 74. Cole GA, Nathanson N, Prendergast RA. Requirement for θ‐bearing cells in lymphocytic choriomeningitis virus‐induced central nervous system disease. Nature 1972;238: 335–337. [DOI] [PubMed] [Google Scholar]
- 75. Kapikian AZ, Mitchell RH, Chanock RM, Shvedoff RA, Stewart CE. An epidemiological study of altered clinical reactivity to respiratory syncitial (RS) virus infection in children previously vaccinated with an inactivated RS virus vaccine. Am J Epidemiol 1969;89: 405–421. [DOI] [PubMed] [Google Scholar]
- 76. Cannon MJ, Openshaw PJM, Askonas BA. Cytotoxic T cells clear virus but augment lung pathology in mice infected with respiratory syncytial virus. J Exp Med 1988;168: 1163–1168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Graham BS, Bunton LA, Wright PF, Karzon DT. Role of T lymphocyte subsets in the pathogenesis of primary infection and rechallenge with respiratory syncytial virus in mice. J Clin Invest 1991;88: 1026–1033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Moskophidis D, Kioussis D. Contribution of virus‐specific CD8+ cytotoxic T cells to virus clearance or pathologic manifestations of influenza virus infection in a T cell receptor transgenic mouse model. J Exp Med 1998;188: 223–232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Sloan‐Lancaster J, Allen PM. Altered peptide ligand‐induced partial T cell activation: molecular mechanisms and role in T cell biology. Annu Rev Immunol 1996;14: 1–27. [DOI] [PubMed] [Google Scholar]
- 80. Smoller BR, Weishar M, Gray MH. An unusual cutaneous manifestation in Crohn's disease. Arch Pathol Lab Med 1990;6: 609–610. [PubMed] [Google Scholar]
- 81. Requena L, Requena C. Erythema nodosum. Dermatol Online 2002;8: 4. [PubMed] [Google Scholar]
- 82. Schlesinger C, Meyer CA, Veeraraghavan S, Koss MN. Constrictive (obliterative) bronchiolitis: diagnosis, etiology, and a critical review of the literature. Ann Diagn Pathol 1998;2: 321–334. [DOI] [PubMed] [Google Scholar]
- 83. Nettesheim P, Szakal AK. Morphogenesis of alveolar bronchiolization. Lab Invest 1972;26: 210–219. [PubMed] [Google Scholar]
- 84. De St. Groth SF, Webster RG. Disquisitions on original antigenic sin. II. Proof in lower creatures. J Exp Med 1966;124: 347–361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Welsh RM, Rothman AL. Dengue immune response: low affinity, high febrility. Nat Med 2003;9: 820–822. [DOI] [PubMed] [Google Scholar]
- 86. Silins SL, et al. Asymptomatic primary Epstein – Barr virus infection occurs in the absence of blood T‐cell repertoire perturbations despite high levels of systemic virus load. Blood 2001;98: 3739–3744. [DOI] [PubMed] [Google Scholar]
- 87. Rickinson AB, Kieff E. Epstein – Barr virus In: Fields BN, et al., eds. Virology, Vol. 2 Philadelphia: Lippincott‐Raven Publishers, 1996. :2397–2446. [Google Scholar]
- 88. Lawson TM, et al. Functional differences between influenza A‐specific cytotoxic T lymphocyte clones expressing dominant and subdominant TCR. Int Immunol 2001;13: 1383–1390. [DOI] [PubMed] [Google Scholar]
- 89. Farci P, et al. Hepatitis C virus‐associated fulminant hepatic failure. N Engl J Med 1996;335: 631–634. [DOI] [PubMed] [Google Scholar]
- 90. Urbani S, et al. Heterologous T cell immunity in severe hepatitis C virus infection. J Exp Med 2005;201: 675–680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Rehermann B, Shin EC. Private aspects of heterologous immunity. J Exp Med 2005;201: 667–670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Walzl G, Tafuro S, Moss P, Openshaw PJ, Hussell T. Influenza virus lung infection protects from respiratory syncitial virus‐induced immunopathology. J Exp Med 2000;192: 1317–1326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Douglass JA, O'Hehir RE. What determines asthma phenotype? Respiratory infections and asthma. Am J Respir Crit Care Med 2000;161: S211–S214. [DOI] [PubMed] [Google Scholar]
- 94. Yang H, Welsh RM. Induction of alloreactive cytotoxic T cells by acute virus infection of mice. J Immunol 1986;136: 1186–1193. [PubMed] [Google Scholar]
- 95. Yang H, Dundon PL, Nahill SR, Welsh RM. Virus‐induced polyclonal cytotoxic T lymphocyte stimulation. J Immunol 1989;142: 1710–1718. [PubMed] [Google Scholar]
- 96. Williams MA, et al. Characterization of virus‐mediated inhibition of mixed chimerism and allospecific tolerance. J Immunol 2001;167: 4987–4995. [DOI] [PubMed] [Google Scholar]
- 97. Welsh RM, et al. Virus‐induced abrogation of transplantation tolerance induced by donor‐specific transfusion and anti‐CD154 antibody. J Virol 2000;74: 2210–2218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Williams MA, et al. Cutting edge: persistent viral infection prevents tolerance induction and escapes immune control following CD28/CD40 blockade‐based regimen. J Immunol;169: 5387–5391. [DOI] [PubMed] [Google Scholar]
- 99. Nahill SR, Welsh RM. High frequency of cross‐reactive cytotoxic T lymphocytes elicited during the virus‐induced polyclonal cytotoxic T lymphocyte response. J Exp Med 1993;177: 317–327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Burrows SR, Khanna R, Burrows JM, Moss DJ. An alloresponse in humans is dominated by cytotoxic T lymphocytes (CTL) cross‐reactive with a single Epstein – Barr Virus CTL epitope: implications for graft‐versus‐host disease. J Exp Med 1994;179: 1155–1161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Braciale TJ, Andrew ME, Braciale VL. Simultaneous expression of H‐2 restricted and alloreactive recognition by a cloned line of influenza virus‐specific cytotoxic T lymphocytes. J Exp Med 1981;153: 1371–1376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Jennings SR. Cross‐reactive recognition of mouse cells expressing the bm3 and bm11 mutations within H‐2Kb by H‐2Kb‐restricted herpes simplex virus‐specific cytotoxic T lymphocytes. J Immunol 1985;135: 3530–3536. [PubMed] [Google Scholar]
- 103. Sheil JM, Bevan MJ, Lefrancois L. Characterization of dual‐reactive H‐2Kb‐restricted anti‐ vesicular stomatitus virus and alloreactive cytotoxic T cells. J Immunol 1987;138: 3654–3660. [PubMed] [Google Scholar]
- 104. Gamadia LE, et al. Cross‐reactivity of cytomegalovirus‐specific CD8+ T cells to allo‐major histocompatibility complex class I molecules. Transplantation 2004;77: 1879–1885. [DOI] [PubMed] [Google Scholar]
- 105. Heeger PS, et al. Pretransplant frequency of donor‐specific, IFN‐gamma‐producing lymphocytes is a manifestation of immunologic memory and correlates with the risk of posttransplant rejection episodes. J Immunol 1999;163: 2267–2275. [PubMed] [Google Scholar]
- 106. Suchin EJ, Langmuir PB, Palmer E, Sayegh MH, Wells AD, Turka LA. Quantifying the frequency of alloreactive T cells in vivo: new answers to an old question. J Immunol 2001;166: 973–981. [DOI] [PubMed] [Google Scholar]
- 107. Lindahl KF, Wilson DB. Histocompatibility antigen‐activated cytotoxic T lymphocytes. II. Estimates of the frequency and specificity of precursors. J Exp Med 1977;145: 508–522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108. Ford WL, Simmonds SJ, Atkikn RC. Early cellular events in a systemic graft‐vs.‐host reaction. II. Autoradiographic estimates of donor lymphocytes which response to Ag‐B‐determined antigenic complexes. J Exp Med 1975;141: 681–696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109. Heeger PS. T‐cell allorecognition and transplant rejection: a summary and update. Am J Transplant 2003;3: 525–533. [DOI] [PubMed] [Google Scholar]
- 110. Bendjelloul F, Desin TS, Shoker AS. Donor non‐specific IFN‐gamma production by primed alloreactive cells as a potential screening test to predict the alloimmune response. Transpl Immunol 2004;12: 167–176. [DOI] [PubMed] [Google Scholar]
- 111. Nickel P, et al. Enzyme‐linked immunosorbent spot assay for donor‐reactive interferon‐gamma‐producing cells identifies T‐cell presensitization and correlates with graft function at 6 and 12 months in renal‐transplant recipients. Transplantation 2004;78: 1640–1646. [DOI] [PubMed] [Google Scholar]
- 112. Sester U, et al. Rapid identification of preformed alloreactive T cells for use in a clinical setting. Transplantation 2004;78: 607–614. [DOI] [PubMed] [Google Scholar]
- 113. Brehm MA, Daniels KA, Welsh RM. Rapid production of TNF‐α following TCR‐engagement of naïve CD8 T cells. J Immunol 2005;175: 5043–5049. [DOI] [PubMed] [Google Scholar]
- 114. Tomkinson BE, Maziarz R, Sullivan JL. Characterization of the T cell‐mediated cellular cytotoxicity during infectious mononucleosis. J Immunol 1989;143: 660–670. [PubMed] [Google Scholar]
- 115. Strang G, Rickinson AB. Multiple HLA class I‐dependent cytotoxicities constitute the ‘non‐HLA‐restricted’ response in infectious mononucleosis. Eur J Immunol 1987;17: 1007–1013. [DOI] [PubMed] [Google Scholar]
- 116. Brehm MA, Daniels KA, Ortaldo JA, Welsh RM. Rapid conversion of effector mechanisms from NK to T cells during virus‐induced lysis of allogeneic implants in vivo. J Immunol 2005;174: 6663–6671. [DOI] [PubMed] [Google Scholar]
- 117. Oberg L, et al. Loss or mismatch of MHC class I is sufficient to trigger NK cell‐mediated rejection of resting lymphocytes in vivo – role of KARAP/DAP12‐dependent and – independent pathways. Eur J Immunol 2004;34: 1646–1653. [DOI] [PubMed] [Google Scholar]
- 118. Chen G, Dong JH. Individualized immunosuppression: new strategies from pharmacokinetics, pharmacodynamics and pharmacogenomics. Hepatobiliary Pancreat Dis Int 2005;4: 332–338. [PubMed] [Google Scholar]
- 119. Odorico JS, Sollinger HW. Technical and immunosuppressive advances in transplantation for insulin‐dependent diabetes mellitus. World J Surg 2002;26: 194–211. [DOI] [PubMed] [Google Scholar]
- 120. Shapiro R, Young JB, Milford EL, Trotter JF, Bustami RT, Leichtman AB. Immunosuppression: evolution in practice and trends, 1993–2003. Am J Transplant 2005;5: 874–886. [DOI] [PubMed] [Google Scholar]
- 121. Kenyon NS, et al. Long‐term survival and function of intrahepatic islet allografts in baboons treated with humanized anti‐CD154. Diabetes 1999;48: 1473–1481. [DOI] [PubMed] [Google Scholar]
- 122. Kirk AD, et al. CTLA4‐Ig and anti‐CD40 ligand prevent renal allograft rejection in primates. Proc Natl Acad Sci USA 1997;94: 8789–8794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123. Larsen CP, et al. Long‐term acceptance of skin and cardiac allografts after blocking CD40 and CD28 pathways. Nature 1996;381: 434–438. [DOI] [PubMed] [Google Scholar]
- 124. Markees TG, et al. Prolonged survival of skin allografts in recipients treated with donor splenocytes and antibody to CD40 ligand. Transplantation 1997;64: 329–335. [DOI] [PubMed] [Google Scholar]
- 125. Parker DC, et al. Survival of mouse pancreatic islet allografts in recipients treated with allogeneic small lymphocytes and antibody to CD40 ligand. Proc Natl Acad Sci USA 1995;92: 9560–9564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126. Hancock WW, Sayegh MH, Zheng XG, Peach R, Linsley PS, Turka LA. Costimulatory function and expression of CD40 ligand, CD80, and CD86 in vascularized murine cardiac allograft rejection. Proc Natl Acad Sci USA 1996;93: 13967–13972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127. Thornley TB, et al. Toll‐like receptor agonists abrogate costimulation blockade‐induced transplantation tolerance. J Immunol 2006;176: 1561–1570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128. Chen Y, Heeger PS, Valujskikh A. In vivo helper functions of alloreactive memory CD4+ T cells remain intact despite donor‐specific transfusion and anti‐CD40 ligand therapy. J Immunol 2004;172: 5456–5466. [DOI] [PubMed] [Google Scholar]
- 129. Zhai Y, Meng L, Gao F, Busuttil RW, Kupiec‐Weglinski JW. Allograft rejection by primed/memory CD8 (+) T cells is CD154 blockade resistant: therapeutic implications for sensitized transplant recipients. J Immunol 2002;169: 4667–4673. [DOI] [PubMed] [Google Scholar]
- 130. Pantenburg B, Heinzel F, Das L, Heeger PS, Valujskikh A. T cells primed by Leishmania major infection cross‐react with alloantigens and alter the course of allograft rejection. J Immunol 2002;169: 3686–3693. [DOI] [PubMed] [Google Scholar]
- 131. Budd RC, et al. Distinction of virgin and memory T lymphocytes. Stable acquisition of the Pgp‐1 glycoprotein concomitant with antigenic stimulation. J Immunol 1987;138: 3120–3129. [PubMed] [Google Scholar]
- 132. Kieper WC, Jameson SC. Homeostatic expansion and phenotypic conversion of naive T cells in response to self peptide/MHC ligands. Proc Natl Acad Sci USA 1999;96: 13306–13311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133. Bendelac A, Rivera MN, Park SH, Roark JH. Mouse CD1‐specific NK1 T cells: development, specificity, and function. Annu Rev Immunol 1997;15: 535–562. [DOI] [PubMed] [Google Scholar]
- 134. McMahon CW, et al. Viral and bacterial infections induce expression of multiple NK cell receptors in responding CD8(+) T cells. J Immunol 2002;169: 1444–1452. [DOI] [PubMed] [Google Scholar]
- 135. Peacock CD, Welsh RM. Origin and fate of lymphocytic choriomeningitis virus‐specific CD8+ T cells coexpressing the inhibitory NK cell receptor Ly49G2. J Immunol 2004;173: 478–484. [DOI] [PubMed] [Google Scholar]
- 136. Razvi ES, Welsh RM, McFarland HI. In vivo state of antiviral CTL precursors: characterization of a cycling population containing CTL precursors in immune mice. J Immunol 1995;154: 620–632. [PubMed] [Google Scholar]
- 137. Selin LK, Welsh RM. Cytolytically active memory CTL present in lymphocytic choriomeningitis virus (LCMV) ‐immune mice after clearance of virus infection. J Immunol 1997;158: 5366–5373. [PubMed] [Google Scholar]
- 138. Zimmermann C, Brduscha‐Riem K, Blaser C, Zinkernagel RM, Pircher H. Visualization, characterization, and turnover of CD8+ memory T cells in virus‐infected hosts. J Exp Med 1996;183: 1367–1375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139. Prlic M, Lefrancois L, Jameson SC. Multiple choices. regulation of memory CD8 T cell generation and homeostasis by interleukin (IL) ‐7 and IL‐15. J Exp Med 2002;195: F49–F52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140. Tan JT, Ernst B, Kieper WC, LeRoy E, Sprent J, Surh CD. Interleukin (IL) ‐15 and IL‐7 jointly regulate homeostatic proliferation of memory phenotype CD8+ cells but are not required for memory phenotype CD4+ cells. J Exp Med 2002;195: 1523–1532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141. Kieper WC, et al. Overexpression of interleukin (IL) ‐7 leads to IL‐15‐independent generation of memory phenotype CD8+ T cells. J Exp Med 2002;195: 1533–1539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142. Selin LK, Vergilis K, Welsh RM, Nahill SR. Reduction of otherwise remarkably stable virus‐specific cytotoxic T lymphocyte memory by heterologous viral infections. J Exp Med 1996;183: 2489–2499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143. Lau LL, Jamieson BD, Somasundaram T, Ahmed R. Cytotoxic T‐cell memory without antigen. Nature 1994;369: 648–652. [PubMed] [Google Scholar]
- 144. Homann D, Teyton L, Oldstone MB. Differential regulation of antiviral T‐cell immunity results in stable CD8+ but declining CD4+ memory. Nat Med 2001;7: 892–893. [DOI] [PubMed] [Google Scholar]
- 145. Varga SM, Welsh RM. High frequency of virus‐specific interleukin‐2‐producing CD4+ T cells and Th1 dominance during lymphocytic choriomeningitis virus infection. J Virol 2000;74: 4429–4432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146. Swain SL, Hu H, Huston G. Class II‐independent generation of CD4 memory T cells from effectors. Science 1999;286: 1381–1383. [DOI] [PubMed] [Google Scholar]
- 147. Varga SM, Welsh RM. Stability of virus‐specific CD4+ T cell frequencies from acute infection into long term memory. J Immunol 1998;161: 367–374. [PubMed] [Google Scholar]
- 148. Varga SM, Selin LK, Welsh RM. Independent regulation of lymphocytic choriomeningitis virus‐specific T cell memory pools: relative stability of CD4 memory under conditions of CD8 memory T cell loss. J Immunol 2001;166: 1554–1561. [DOI] [PubMed] [Google Scholar]
- 149. Murali‐Krishna K, Lau LL, Sambhara S, Lemonnier F, Altman J, Ahmed R. Persistence of memory CD8 T cells in MHC class I‐deficient mice. Science 1999;286: 1377–1381. [DOI] [PubMed] [Google Scholar]
- 150. Bourgeois C, Rocha B, Tanchot C. A role for CD40 expression on CD8+ T cells in the generation of CD8+ T cell memory. Science 2002;297: 2060–2063. [DOI] [PubMed] [Google Scholar]
- 151. Sun JC, Bevan MJ. Defective CD8 T cell memory following acute infection without CD4 T cell help. Science 2003;300: 339–342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152. Janssen EM, Lemmens EE, Wolfe T, Christen U, Von Herrath MG, Schoenberger SP. CD4+ T cells are required for secondary expansion and memory in CD8+ T lymphocytes. Nature 2003;421: 852–856. [DOI] [PubMed] [Google Scholar]
- 153. Demkowicz WE, Littaua RA, Wang J, Ennis FA. Human cytotoxic T‐cell memory: long‐lived responses to vaccinia virus. J Virol 1996;70: 2627–2631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154. Crotty S, Felgner P, Davies H, Glidewell J, Villarreal L, Ahmed R. Cutting edge: long‐term B cell memory in humans after smallpox vaccination. J Immunol 2003;171: 4969–4973. [DOI] [PubMed] [Google Scholar]
- 155. Hammarlund E, et al. Duration of antiviral immunity after smallpox vaccination. Nat Med 2003;9: 1131–1137. [DOI] [PubMed] [Google Scholar]
- 156. Kim SK, Welsh RM. Comprehensive early and lasting loss of memory CD8 T cells and functional memory during acute and persistent viral infections. J Immunol 2004;172: 3139–3150. [DOI] [PubMed] [Google Scholar]
- 157. Selin LK, et al. Attrition of T cell memory: selective loss of lymphocytic choriomeningitis virus (LCMV) epitope‐specific memory CD8 T cells following infections with heterologous viruses. Immunity 1999;11: 733–742. [DOI] [PubMed] [Google Scholar]
- 158. Jiang J, Lau L, Shen H. Selective depletion of nonspecific T cells during the early stage of immune responses to infection. J Immunol 2003;171: 4352–4358. [DOI] [PubMed] [Google Scholar]
- 159. McNally JM, Zarozinski CC, Lin MY, Brehm MA, Chen HD, Welsh RM. Attrition of bystander CD8 T cells during virus‐induced T cell and interferon responses. J Virol 2001;75: 5965–5976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160. Wong R, et al. Haematological manifestations in patients with severe acute respiratory syndrome: retrospective analysis. BMJ 2003;326: 1358–1362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161. Nabeshima S, Murata M, Kikuchi K, Ikematsu H, Kashiwagi S, Hayashi J. A reduction in the number of peripheral CD28+CD8+T cells in the acute phase of influenza. Clin Exp Immunol 2002;128: 339–346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162. Razvi ES, Jiang Z, Woda BA, Welsh RM. Lymphocyte apoptosis during the silencing of the immune response to acute viral infections in normal, lpr and Bcl‐2‐transgenic mice. Am J Pathol 1995;147: 79–91. [PMC free article] [PubMed] [Google Scholar]
- 163. Masopust D, Vezys V, Marzo AL, Lefrancois L. Preferential localization of effector memory cells in nonlymphoid tissue. Science 2001;291: 2413–2417. [DOI] [PubMed] [Google Scholar]
- 164. Hogan RJ, et al. Activated antigen‐specific CD8+ T cells persist in the lungs following recovery from respiratory virus infections. J Immunol 2001;166: 1813–1822. [DOI] [PubMed] [Google Scholar]
- 165. Marshall DR, et al. Measuring the diaspora for virus‐specific CD8+ T cells. Proc Natl Acad Sci USA 2001;98: 6313–6318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166. Welsh RM, Bahl K, Wang XZ. Apoptosis and loss of virus‐specific CD8(+) T‐cell memory. Curr Opin Immunol 2004;16: 271–276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167. Jiang J, Gross D, Nogusa S, Elbaum P, Murasko DM. Depletion of T cells by type I interferon: differences between young and aged mice. J Immunol 2005;175: 1820–1826. [DOI] [PubMed] [Google Scholar]
- 168. Bahl K, et al. Interferon‐induced attrition of CD8 T cells in the presence or absence of cognate antigen during the early stages of viral infections. J Immunol 2006;176: 4284–4295. [DOI] [PubMed] [Google Scholar]
- 169. Rathmell JC, Thompson CB. Pathways of apoptosis in lymphocyte development, homeostasis, and disease. Cell 2002;109: S97–S107. [DOI] [PubMed] [Google Scholar]
- 170. Selin LK, et al. CD8 memory T cells: cross‐reactivity and heterologous immunity. Semin Immunol 2004;16: 335–347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171. Hahm B, Trifilo MJ, Zuniga EI, Oldstone MB. Viruses evade the immune system through type I interferon‐mediated STAT2‐dependent, but STAT1‐independent, signaling. Immunity 2005;22: 247–257. [DOI] [PubMed] [Google Scholar]
- 172. Gil MP, Salomon R, Louten J, Biron CA. Modulation of STAT1 protein levels: a mechanism shaping CD8 t cell responses in vivo. Blood 2006;107: 987–993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173. Zhang X, Sun S, Hwang I, Tough DF, Sprent J. Potent and selective stimulation of memory‐phenotype CD8+ T cells in vivo by IL‐15. Immunity 1998;8: 591–599. [DOI] [PubMed] [Google Scholar]
- 174. Razvi ES, Welsh RM. Programmed cell death of T lymphocytes during acute viral infection: a mechanism for virus‐induced immune deficiency. J Virol 1993;67: 5754–5765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175. Wang XZ, Brehm MA, Welsh RM. Preapoptotic phenotype of viral epitope‐specific CD8 T cells precludes memory development and is an intrinsic property of the epitope. J Immunol 2004;173: 5138–5147. [DOI] [PubMed] [Google Scholar]
- 176. Kaech SM, Tan JT, Wherry EJ, Konieczny BT, Surh CD, Ahmed R. Selective expression of the interleukin 7 receptor identifies effector CD8 T cells that give rise to long‐lived memory cells. Nat Immunol 2003;4: 1191–1198. [DOI] [PubMed] [Google Scholar]
- 177. Ray SJ, et al. The collagen binding alpa1beta1 integrin VLA‐1 regulates CD8 T cell‐mediated immune protection against heterologous influenza infection. Immunity 2004;20: 167–179. [DOI] [PubMed] [Google Scholar]
- 178. Ayroldi E, Cannarile L, Migliorati G, Bartoli A, Nicoletti I, Riccardi C. CD44 (Pgp‐1) inhibits CD3 and dexamethasone‐induced apoptosis. Blood 1995;86: 2672–2678. [PubMed] [Google Scholar]
- 179. Grayson JM, Laniewski NG, Lanier JG, Ahmed R. Mitochondrial potential and reactive oxygen intermediates in antigen‐specific CD8+ T cells during viral infection. J Immunol 2003;170: 4745–4751. [DOI] [PubMed] [Google Scholar]
- 180. Lauer GM, et al. Full‐breadth analysis of CD8+ T‐cell responses in acute hepatitis C virus infection and early therapy. J Virol 2005;79: 12979–12988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181. Zhou S, Ou R, Huang L, Moskophidis D. Critical role for perforin‐, Fas/FasL‐, and TNFR1‐mediated cytotoxic pathways in down‐regulation of antigen‐specific T cells during persistent viral infection. J Virol 2002;76: 829–840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182. Selin LK, Nahill SR, Welsh RM. Cross‐reactivities in memory cytotoxic T lymphocyte recognition of heterologous viruses. J Exp Med 1994;179: 1933–1943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183. Peacock C, Kim S, Welsh RM. Attrition of virus‐specific memory CD8(+) T cells during reconstitution of lymphopenic environments. J Immunol 2003;171: 655–663. [DOI] [PubMed] [Google Scholar]
- 184. Kieper WC, Burghardt JT, Surh CD. A role for TCR affinity in regulating naive T cell homeostasis. J Immunol 2004;172: 40–44. [DOI] [PubMed] [Google Scholar]
- 185. Fremont DH, Stura EA, Matsumura M, Peterson PA, Wilson IA. Crystal structure of an H‐2Kb‐ovalbumin peptide complex reveals the interplay of primary and secondary anchor positions in the major histocompatibility complex binding groove. Proc Natl Acad Sci USA 1995;92: 2479–2483. [DOI] [PMC free article] [PubMed] [Google Scholar]