

Abstract
Bats and rodents are recognized to host a great diversity of viruses and several important viral zoonoses, but how this viral diversity is structured and how viruses are connected, shared and distributed among host networks is not well understood. To address this gap in knowledge, we compared the associative capacity of the host–virus networks in rodents and bats with the identification of those viruses with zoonotic potential. A virus database, detected by molecular methods, was constructed in the two taxonomic groups. We compiled 5,484 records: 825 in rodents and 4,659 in bats. We identified a total of 173 and 166 viruses, of which 53 and 40 are zoonotic viruses, in rodents and bats, respectively. Based on a network theory, a non‐directed bipartite host–virus network was built for each group. Subsequently, the networks were collapsed to represent the connections among hosts and viruses. We identified both discrete and connected communities. We observed a greater degree of connectivity in bat viruses and more discrete communities in rodents. The Coronaviridae recorded in bats have the highest values of degree, betweenness and closeness centralities. In rodents, higher degree positions were distributed homogeneously between viruses and hosts. At least in our database, a higher proportion of rodent viruses were zoonotic. Rodents should thus not be underestimated as important reservoirs of zoonotic disease. We found that viruses were more frequently shared among bats than in rodents. Network theory can reveal some macroecological patterns and identify risks that were previously unrecognized. For example, we found that parvovirus in megabats and Gbagroube virus in rodents may represent a zoonotic risk due to the proximity to humans and other zoonotic viruses. We propose that epidemiological surveillance programmes should consider the connectivity of network actors as a measure of the risks of dispersion and transmission.
Keywords: disease ecology, host–parasite network, viral diversity, zoonoses
Impacts.
The analysis of virus and host networks (rodents and bats) allows us to measure the potential risk of zoonotic diseases.
Measuring network connectivity can be a useful tool for identifying hosts and viruses of potential importance in the transmission dynamic of zoonotic diseases.
Bats presented twice as many connections between virus and host as rodents, indicating a higher zoonotic potential transmission.
1. INTRODUCTION
Bats and rodents are hosts of a significant proportion of zoonoses, higher than any other mammalian order. Over 200 viruses belonging to 27 viral families have been isolated or detected in bats; however, bat–human transmission has only been observed for 11 viruses, belonging to four different viral families: Rhabdoviridae, Filoviridae, Coronaviridae and Paramyxoviridae (Allocati et al., 2016). Some examples of those viruses are as follows: SARS‐related coronavirus, Sosuga rubulavirus, Ebola virus and Marburg virus, rabies lyssavirus, Nipah henipavirus and Hendra henipavirus (Allocati et al., 2016; Calisher, Childs, Field, Holmes, & Schountz, 2006; Hayman, 2016; O'Shea et al., 2014; Plowright et al., 2015).
Rodents have similar zoonotic potential to bats and are associated with a large number of zoonotic viruses, such as Sin Nombre virus, Puumala virus, Crimean‐Congo hemorrhagic fever virus, Kyasanur forest virus, tick‐borne encephalitis virus, Lassa fever virus and Venezuelan equine encephalitis virus, among others. All of the aforementioned bat‐ and rodent‐associated viruses have a large impact on public health. However, it is important to take into count that not all of these viruses are obligate pathogens; some are generally commensal.
Previous studies have explored viral associations on relatively restricted spatial or phylogenetic scales. For example, Hayman (2016) propose maps of viral distributions according to the distribution of hosts' families, Streicker et al. (2010) explored rabies viruses using a phylogenetic approach, and Cui, Tachedjian, and Wang (2015) compared retrovirus associations between bats and rodents. Anthony et al. (2017) explored coronavirus networks at the level of host family, and Bordes, Caron, Blasdell, Garine‐Wichatitsky, and Morand (2017) analyse the relationships among zoonotic diseases in Southeast Asia. Luis et al. (2015) analyse viral networks between rodents and bats at global scale identifying several ecology factors to explain virus–host associations. Recently, works explored the specificity and frequency of sharing DNA and RNA viruses among Carnivores and bats (Wells, Morand, Wardeh, & Baylis, 2018) and the importance of the phylogeny to explain the viral richness associated with bats and rodents (Guy, Thiagavel, Mideo, & Ratcliffe, 2019). However, there are currently no studies at a global level that incorporate the human influence in the viral networks.
While some authors consider bats and rodents to belong to a similar category of high zoonotic risk potential (Han, Schmidt, Bowden, & Drake, 2015), other work examines the differences between bats and rodents (Luis et al., 2015). Several different distinctive features of bats have been hypothesized to explain their particularly high viral richness, such as their ability to fly, long migrations, high trophic diversity and social structure (Brook & Dobson, 2015; Moratelli & Calisher, 2015). However, currently the viral diversity and connectivity among different species of bats are not well understood (Moratelli & Calisher, 2015; O'Shea et al., 2014) making it difficult to evaluate the implications of those relationships for emerging and re‐emerging zoonoses. While the literature does explain why bats harbour a particularly high number of viruses, it does not describe viral associations at the level of host species or describe direct relationships (only associations), does not address the whole viral species complex and does not describe the statistics or metrics that characterize the associations.
Bats and rodents are similar in that both are highly diverse, are basal taxa within the mammal phylogeny and have similar life history characteristics (Luis et al., 2013). Rodents, like bats, have been recognized as reservoirs for several zoonotic viruses (Han et al., 2015), such as virus of hantaviridae (Schmaljohn & Hjelle, 1997) and arenaviridae families (Charrel & de Lamballerie, 2010). However, there are differences in rodent–virus associations that impact their zoonotic potential compared with bats.
In disease ecology, analytical tools have been used to holistically explain the dynamics of infections and provide novel hypothesis to explain macroecological patterns (Johnson, Roode, & Fenton, 2016). One of the theories that helps to predict dynamic changes in host–pathogen systems is graph theory, also known as network theory (Bordes et al., 2017; Johnson et al., 2016). This approach can be used to gain better understanding of how interactions take place within pathogen communities, how hosts are connected with pathogens, their preferred association and patterns of pathogen transmission (Godfrey, 2013; White, Forester, & Craft, 2017).
The graphs, better known as networks, focus on the interactions between entities (Newman, 2014), and they have the potential to infer relationships within a larger framework (Hossain & Feng, 2016; Luke & Stamatakis, 2012). A network is capable of emphasizing the preferred union as a process (Hartonen & Annila, 2011) and capturing both the individual elements in a system as well as their relevant interconnections (Kolaczyk & Csardi, 2014). In disease ecology, this kind of analysis could be applied to describe viral diversity associated with different hosts and detect hosts and viruses that share associations, and therefore identify groups that share similar characteristics (White et al., 2017).
In network theory, centrality and dispersion metrics quantify the importance of each component member (Martínez‐López, Perez, & Sánchez‐Vizcaíno, 2009; Newman, 2014; Opsahl, Agneessens, & Skvoretz, 2010). The parameter “betweenness” can be used to recognize dispersing hosts and key viruses in the evolution or viral transmission (Opsahl et al., 2010; White et al., 2017), while “closeness” can indicate hosts and viruses that may have little direct connectivity but are surrounded by important highly connected nodes (Opsahl et al., 2010; White et al., 2017). In terms of disease ecology, we can employ these and other parameters to explore the host–host, virus–host and virus–virus interactions by collapsing the networks and identifying communities. Network analysis thus offers the opportunity to recognize highly diverse viruses and hosts based on a high degree of connectedness. Bats and rodents are excellent taxa in which to implement this tool because they harbour a large number of highly adaptable viruses and hosts with high resistance.
Therefore, in this study we aimed to compare and recognize the differences in the associative capacity of the host–virus networks in rodents and bats worldwide, as well as to identify the viruses that may shift across species, including humans, suggesting zoonotic potential.
2. METHODS
2.1. Database
Data were compiled from several sources. For bats, we retrieved data from the DBatVir database (http://www.mgc.ac.cn/DBatVir/). For rodents, data on viruses in rodents were searched in Web of Science (https://login.webofknowledge.com), Elsevier (https://www.elsevier.com/advanced-search) and World Wide Science (https://worldwidescience.org/).
In each of the aforementioned databases, we searched the keywords: rodent, virus, PCR, wild and zoonotic. We then constructed two separate large databases of viruses isolated from rodents and bats. When the taxonomic classification of the virus was not clear, we searched the ICTV database (https://talk.ictvonline.org/) to confirm. In the case of Coronaviridae and Paramyxoviridae families, we assigned as bat coronavirus and bat Paramyxovirus all unclassified coronaviruses and paramyxoviruses. Because bat astrovirus does not exist in the ICTV classification, we assigned all reports from Astroviridae family as astrovirus. To increase the certainty of identification of the viruses, only studies that used molecular methods to detect viruses were included in the database we compiled. Subsequently, each of the viruses identified in rodents or bats was classified as direct zoonotic or non‐zoonotic pathogens (Allocati et al., 2016; Calisher et al., 2006; Han et al., 2015).
2.2. Overall networks analyses
An independent undirected bipartite network was built for each of the orders of Rodentia and Chiroptera. That is to say, each network included two types of nodes—viruses and hosts. Viral nodes were connected to host nodes when the virus indicated by the node has been detected in the species indicated by the host node. We included a human host node, which was connected to viruses that have been classified as zoonotic. This helped us to group and identify zoonotic viruses and viruses close to them (which could have zoonotic potential).
2.3. Collapsed networks
Then, host‐to‐host and virus‐to‐virus networks were constructed in order to explore these networks in different dimensions. The “bipartite.projection” function in the igraph package implemented in r software version 3.4.2 (R Core Team, 2017) was used to collapse the bipartite network. Basically, in the collapsed networks a host was connected to another host when they shared a common virus and a virus to a virus when they shared a common host (Figure 1).
Figure 1.
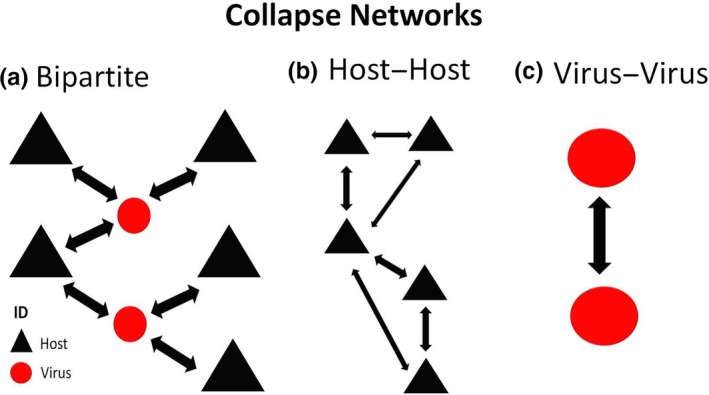
Collapsed networks. (a) Bipartite network, (b) Collapsed host–host network and (c) Collapsed virus–virus network [Colour figure can be viewed at http://www.wileyonlinelibrary.com]
In each host–host collapsed network, the host nodes were conserved and the virus nodes were transformed using the weight of corresponding links in order to illustrate the relationships among different hosts (Figure 1a,b). In each virus–virus network (one for bats and one for rodents), the virus–virus relationship was highlighted by collapsing the host nodes into the weighted links (Figure 1c).
2.4. Network measurements
We measured the networks on two levels: individual node and the entire network. At the node level, we measured different centrality values including: degree (number of links that a node has), betweenness (number of times a node is an intermediary to connect each possible pair of nodes) and closeness (the degrees of average separation in relation to other nodes) (Martínez‐López et al., 2009; Newman, 2014). At the network level, we measured density and diameter. Network density is the proportion of links that are actually observed in the network divided by those that could possibly occur and network diameter is the length of the longest geodesic distance (Newman, 2014).
Network level measurements were useful for summarizing the “big picture” of the network and identify the key nodes that are closely related to humans. Network level measurements were generated using the algorithms provided in the packages “igraph” (Csárdi & Nepusz, 2006) and “network” (Butts, 2008; Table 1), and plots were produced using the packages “igraph” and “ggplot2” (Gómez‐Rubio, 2017) in r (R Development Core Team, 2011).
Table 1.
Formulas applied to calculate networks parameters
| Measure | Formula | Reference | |
|---|---|---|---|
| Degree centrality (D) |
|
Freeman (1978) | |
| Betweenness centrality (B) |
|
Freeman (1978) | |
| Closeness centrality (C) |
|
Freeman (1978) | |
| Density (ρ) |
|
Martínez‐López et al. (2009) | |
| Diameter (Ø) | maxik d(i, k) | West Douglas (2001) |
This article is being made freely available through PubMed Central as part of the COVID-19 public health emergency response. It can be used for unrestricted research re-use and analysis in any form or by any means with acknowledgement of the original source, for the duration of the public health emergency.
2.5. Community detection
Communities were detected using the Maximization of Modularity method (Newman, 2016), which recognize nodes with dense and weak connections between groups. We used the function “cluster_edge_betweenness” (Girvan, Girvan, Newman, & Newman, 2002; Newman & Girvan, 2003) in the “igraph” package (Csárdi & Nepusz, 2006) to identify the nodes with dense connections with humans, which is based on the following equation:
| (1) |
where m denotes the total number of links in the network, Aij refers to the actual number of links between nodes i and node j, γ is a parameter calculated by the algorithm; K, degree of i; δgigj is a randomized number of links between a pair of nodes. Community detection facilitates the recognition of groups of hosts that share viruses and viruses that share hosts, and which therefore may continue to enter in contact with each other because they share similar characteristics.
2.6. Subnetwork
A subnetwork was built by choosing communities with more than four host–virus pairs, which is above the minimum number accepted in statistical normality samples (n = 3) (Hammer, Harper, & Ryan, 2001; Royston, 1982). These subnetworks were illustrated to gain better community visualization and recognize the most relevant communities for the detection of potentially zoonotic viruses.
2.7. Subcommunities
From the subnetwork, the most important communities were chosen using to the measures of the members (top five nodes) and the number of zoonotic viruses (80%) as selection criteria. Then, a sociogram representing the preferential unions and the complex interaction on the largest communities was constructed using the package “visnetwork” (Almende, Thieurmel, & Robert, 2016) in r. This choice of subnetworks helps us to focus and observe in more detail the interactions within these important communities.
3. RESULTS
3.1. Database
The rodent database contained 825 records including 172 rodent species and 123 viruses, of which 53 are zoonotic viruses. The bat database contained 4,659 records, consisting of 220 bats species associated with 166 viruses, of which 40 viruses were classified as zoonotic. Both databases are detailed in Appendix S1.
3.2. Rodentia network
3.2.1. Overall network analyses
The bipartite Rodentia network contained 269 nodes (172 rodents, 123 viruses, human node) and 323 links (Figure 2a). The diameter of the network was nine, and the density was 0.0044. Mus musculus was the host with the highest degree and betweenness values, at 17 and 2,496, respectively. The top five nodes based on centrality values are shown in Table 2, and centrality values are in Appendix S2. 78.06% of the nodes had a degree value of 1 or 2, making them uninformative in terms of epidemiological information, though they may be involved in co‐evolutionary processes.
Figure 2.
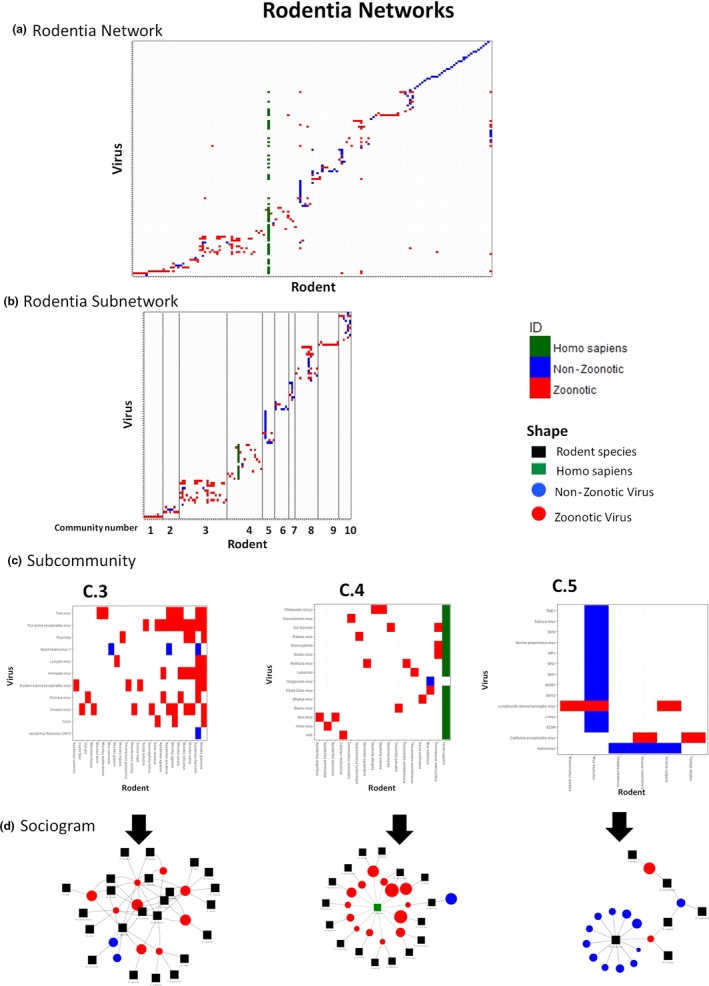
Rodentia networks. (a) Whole Rodentia network. (b) Subnetwork, with 10 selected communities, renamed with a consecutive number. Lines were added to separate the communities. (c) Subcommunities selected to show the host–species interactions; (d) Sociogram to facilitate the visualization of the interactions [Colour figure can be viewed at http://www.wileyonlinelibrary.com]
Table 2.
Rodent network. The top five nodes with the highest centrality values
| Node | Centrality values | ||
|---|---|---|---|
| Degree | Betweenness | Closeness | |
| Homo sapiens | 53 | 18,475.0 | 6.4 × 10−5 |
| Mus musculus | 17 | 2,496.5 | 6.3 × 10−5 |
| Andes virus | 13 | 2003.5 | 6.3 × 10−5 |
| Rattus norvegicus | 13 | 1,362.6 | 6.3 × 10−5 |
| Cowpox virus | 11 | 1869.2 | 6.3 × 10−5 |
This article is being made freely available through PubMed Central as part of the COVID-19 public health emergency response. It can be used for unrestricted research re-use and analysis in any form or by any means with acknowledgement of the original source, for the duration of the public health emergency.
3.2.2. Community detection
Thirty‐nine different communities were detected. Sixteen communities included only two members, while the largest group consisted of 32 members. This particular group included humans, as shown in Appendix S2.
3.2.3. Subnetwork
Ten communities with at least eight members were selected in the subnetwork (Figure 2b). We excluded communities 1 and 9 despite fulfilling the inclusion criteria because they are linear, with a single virus that influences the whole.
3.2.4. Subcommunities
Communities 3, 4 and 5 satisfied our selection criteria. Community 3 included 23 hosts and 11 viruses. This community was considered a dense network because the number of links greatly exceeds the number of nodes (34 nodes and 55 links). In this community, only two viruses are non‐zoonotic: herpesvirus 1 and cytomegalovirus (CMV3). Figure 2c3 shows the gradient representing the number of viruses associated with each host within that community. It was noteworthy that Myodes glareolus is linked with the highest number of associated viruses (eight), and all of which are zoonotic. In the fourth community, 32 nodes and 33 links were observed. Within this subcommunity, 16 rodent species were recognized along with 16 viruses, and of which, only one virus was not zoonotic, Gbagroube virus, proposed as Arenaviridae virus (Coulibaly‐N'Golo et al., 2011). Figure 2c4 highlights the proximity of Peromyscus maniculatus to the human node, indicating a large number of shared viruses. On the other hand, community 5 consisted of 20 nodes and 19 links, representing six rodent species and 14 viruses. Only two viruses in this community were zoonotic: lymphocytic choriomeningitis virus (LCV) and California encephalitis virus. In this subcommunity, most viruses linked only with M. musculus.
3.3. Host–host Rodentia network
This network contained 147 nodes of rodent species with 502 links. The diameter of the network was 5, and the density was 0.0627 (Figure 3a). The top five nodes in terms of degree, betweenness and closeness are shown in Table 2, and the remaining values are in Appendix S3. Thirty‐five different communities were detected, and the largest of which contained 43 members, followed by a group of 21 members (Appendix S3).
Figure 3.
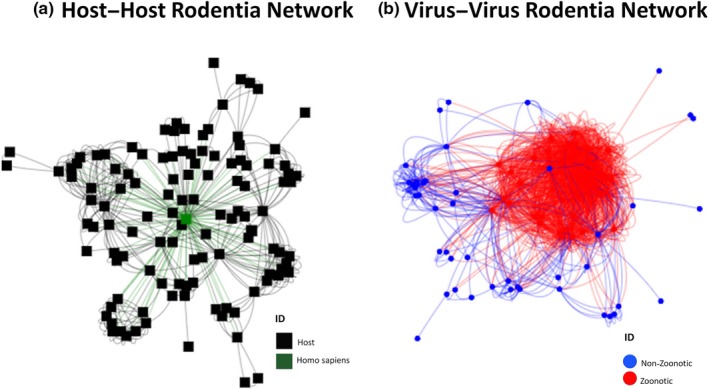
Collapsed networks (a) host–host rodentia network; (b) virus–virus rodentia network [Colour figure can be viewed at http://www.wileyonlinelibrary.com]
3.4. Virus–virus Rodentia network
In virus–virus network, there were 122 nodes and 1,661 links. The diameter and density were 4 and 0.3494, respectively (Figure 3b). The five viruses with the highest centrality values are all zoonoses (Table 3). Fifty‐three different communities were detected, the largest of which contained 52 members, and the second largest had only 12 members. Centrality values and community detection are given in Appendix S3.
Table 3.
Rodent collapsed network. Top five nodes with the highest centrality values
| Network | Node | Centrality values | ||
|---|---|---|---|---|
| Degree | Betweenness | Closeness | ||
| Host–host | Homo sapiens | 105 | 4,932.8 | 2.1 × 10−4 |
| Rattus rattus | 35 | 443.7 | 2.1 × 10−4 | |
| Rattus norvegicus | 30 | 293.6 | 2.1 × 10−4 | |
| Myodes glareolus | 26 | 111.8 | 2.1 × 10−4 | |
| Apodemus sylvaticus | 21 | 75.3 | 2.1 × 10−4 | |
| Virus–virus | Venezuelan equine encephalitis Virus | 76 | 516.6 | 3.0 × 10−4 |
| Encephalomyocarditis virus | 70 | 181.2 | 3.0 × 10−4 | |
| Severe Fever With thrombocytopenia syndrome | 69 | 203.1 | 3.0 × 10−4 | |
| Eastern equine encephalitis virus | 68 | 202.8 | 3.0 × 10−4 | |
| Lymphocytic choriomeningitis virus | 65 | 171.5 | 3.0 × 10−4 | |
This article is being made freely available through PubMed Central as part of the COVID-19 public health emergency response. It can be used for unrestricted research re-use and analysis in any form or by any means with acknowledgement of the original source, for the duration of the public health emergency.
3.5. Chiroptera network
3.5.1. Overall networks analyses
A total of 387 nodes (220 bat species, 166 viruses, human node) and 736 links were contained in the bipartite bat network. The network diameter was 10, and the density was 0.0049 (Figure 4a). Three zoonotic viruses had the highest degree and betweenness values; these were bat coronavirus, rabies lyssavirus and bat paramyxovirus (Table 4). 65.71% of the nodes had degrees of 1 or 2, so they do not provide much information to the network but they may be involved in co‐evolutionary processes. All centrality values are given in Appendix S2.
Figure 4.
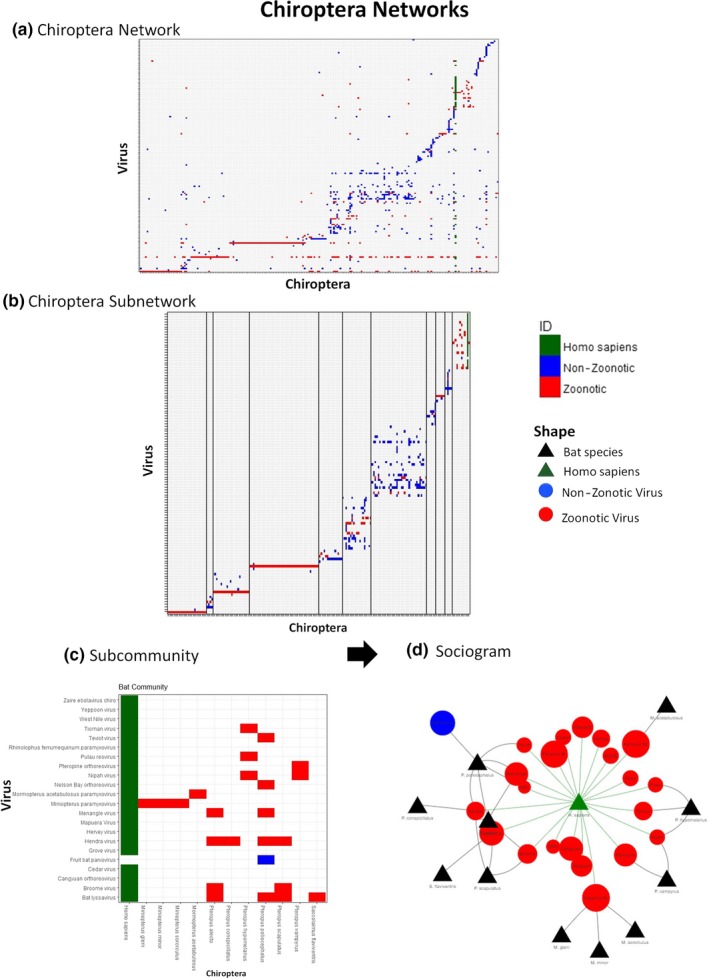
Chiroptera networks (a) Chiroptera host–virus network, (b) Subnetwork, with 11 communities selected. Lines were added to separate the different communities to observe their composition and detect the relevant communities; (c) Human subcommunity selected to show the host–species interactions; (d) Sociogram to facilitate the visualization of the interactions [Colour figure can be viewed at http://www.wileyonlinelibrary.com]
Table 4.
Top five nodes of Chiroptera network with the highest centrality network
| Node | Centrality values | ||
|---|---|---|---|
| Degree | Betweenness | Closeness | |
| Bat coronavirus | 80 | 26,930.5 | 2.8 × 10−4 |
| Rabies | 56 | 15,539.7 | 2.6 × 10−4 |
| Bat paramyxovirus | 55 | 13,920.7 | 2.7 × 10−4 |
| Homo sapiens | 39 | 13,233.5 | 2.7 × 10−4 |
| Astrovirus | 31 | 4,857.4 | 2.6 × 10−4 |
This article is being made freely available through PubMed Central as part of the COVID-19 public health emergency response. It can be used for unrestricted research re-use and analysis in any form or by any means with acknowledgement of the original source, for the duration of the public health emergency.
3.5.2. Community detection
Twenty‐nine different communities were detected; four of those had only two members, and the largest community contained 38 members (Appendix S2).
3.5.3. Subnetwork
Eleven communities contained eight or more members (Figure 4b). Communities 1, 3 and 4 were linear and possessed simple edges, with a virus that influenced the whole community. Community 7 was a homogeneous community with rich ecological interactions, but which was not highly related to zoonotic viruses. The last community was associated with humans had the highest number of zoonotic viruses involved (Figure 4c,d, details below).
3.5.4. Subcommunities
A low number of zoonotic viruses were found in homogeneous bat network communities. We focused only on the community that included the human node. In Figure 4a, three red lines representing the three highly connected viruses in the network followed by a homogeneous community and later in green the human node connections.
The community that included humans (Figure 4c,d) has 13 host nodes (including human) and 23 virus nodes with 45 links. Fruit bat parvovirus was the only non‐zoonotic virus in that community. Miniopterus, Mormopterus and Saccolaimus were the only bat genera that directly shared viruses with humans.
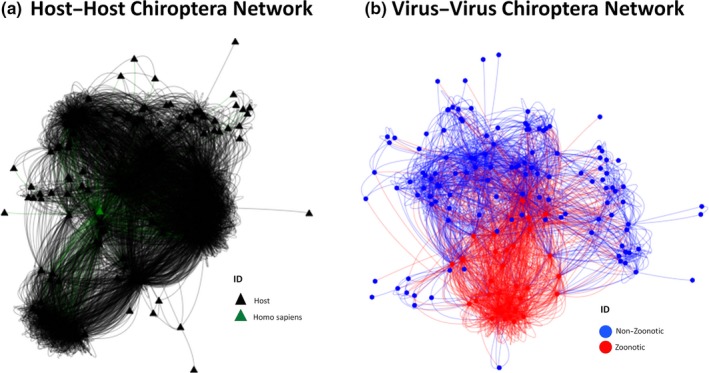
3.5.5. Host–host Chiroptera network
The host–host Chiroptera network contained 221 nodes and 6,949 links. The diameter and density of the network were 5 and 0.2911, respectively (Figure 5a). The five nodes with the highest centrality values are shown in Table 5, and detailed results are in Appendix S4. Sixty‐seven different communities were detected; the largest had 69 members. The second largest community consisted of 56 members including humans (Appendix S4).
Figure 5.
Collapsed networks (a) host–host rodentia network; (b) virus–virus rodentia network [Colour figure can be viewed at http://www.wileyonlinelibrary.com]
Table 5.
Collapsed Chiroptera network. Top five nodes with the highest centrality values
| Network | Node | Centrality values | ||
|---|---|---|---|---|
| Degree | Betweenness | Closeness | ||
| Host–host | Homo sapiens | 181 | 1556.6 | 8.2 × 10−4 |
| Myotis daubentonii | 174 | 767.5 | 8.2 × 10−4 | |
| Glossophaga soricina | 155 | 610.8 | 8.3 × 10−4 | |
| Rhinolophus ferrumequinum | 146 | 484.6 | 8.0 × 10−4 | |
| Desmodus rotundus | 138 | 873.4 | 8. 2 × 10−4 | |
| Virus–virus | Bat coronavirus | 138 | 2,402.0 | 1.3 × 10−3 |
| Bat paramyxovirus | 110 | 917.9 | 1.2 × 10−3 | |
| European bat lyssavirus | 90 | 376.8 | 1.2 × 10−3 | |
| Betacoronavirus | 85 | 581.9 | 1.2 × 10−3 | |
| Alphacoronavirus | 77 | 358.7 | 1.2 × 10−3 | |
This article is being made freely available through PubMed Central as part of the COVID-19 public health emergency response. It can be used for unrestricted research re-use and analysis in any form or by any means with acknowledgement of the original source, for the duration of the public health emergency.
3.5.6. Virus–virus Chiroptera network
The virus–virus network contained 164 nodes and 2,132 links. The diameter of the network was 4, and the density was 0.1655 (Figure 5b). The three highest centrality viruses were coronaviruses (Bat coronavirus, alphacoronavirus and betacoronavirus), and all of which are zoonotic (Appendix S4). Ninety‐seven communities were detected, the largest of which contained 35 viruses, and all of the zoonotic (Appendix S4).
4. DISCUSSION
Bats are well‐known as excellent reservoirs for zoonotic viruses that usually result in high public health impact (Gay et al., 2014; Luis et al., 2013, 2015; Plowright et al., 2015). Nevertheless, in our database, 53 of 123 (43%) rodent viruses are zoonotic, nearly twice the proportion of bat viruses, 40 of 166 (24%). Rodents should thus not be overlooked as potential hosts of zoonotic viruses. However, bats are more linked to more cosmopolitan viruses with broad distributions of their hosts.
In the bipartite network of bats, the main actors (top values of degree, betweenness and closeness) are all viruses, including bat coronavirus, rabies lyssavirus, bat paramyxovirus and astrovirus (Table 4). In the rodent bipartite network, in contrast, the main actors are two host species (M. musculus and Rattus norvegicus) and two viruses (Andes virus and Cowpox virus) (Table 2). Hence, the bat viruses have a higher degree of connectivity with a large number of bat hosts.
The ratio of nodes to edges in the bat–virus network was 1:1.9 and in rodents was 1:1.2. In other words, each bat species on average interacts with nearly two viruses and vice versa, while in rodents, on average there is a near one‐to‐one host–virus interaction, leading to the rodent network having more divisions. This is similar to results found by Luis et al. (2015) when building viral association networks in Rodentia and Chiroptera, who then used the centrality metrics as a response variable in a generalized linear model to determine the phylogenetic, functional and ecological characteristics that are responsible for this high connectivity in bats. This pattern of higher host diversity among bat viruses than rodent viruses has been observed in viral metacommunities using analyses at different spatial scales (Nieto‐Rabiela, Suzán, Wiratsudakul, & Rico‐Chávez, 2018), and it has been suggested that this is due to their higher dispersal ability compared with rodents (Wang, Walker, & Poon, 2011). However, since studies on viruses detected in others mammals have not yet been carried out at a similarly large scale, it is not clear whether bats' level of host–virus connectivity is atypically high among mammals; we can only conclude that it is higher than in rodents.
In collapsed host–host networks in both bats and rodents, the human node is closely linked with the species that host zoonotic viruses. The largest community including the human node is surrounded by three to four smaller groups in both networks. We can highlight that in the largest community where the human is embedded, the bats and rodent hosts have a high adaptability, wide distribution and low phylogenetic distinction. Also, the closest hosts to the human node are the main reservoirs of viral diseases with worldwide distribution like hantaviridae viruses, rabies‐related virus and dengue virus (Tables 3 and 5). However, the clusters in the bat network are four times denser compared with rodents; this makes it easier to continue sharing the viruses. In addition, they can act as virus mixers, allowing the viruses to acquire characteristics that allow them to infect other host species, including humans. In addition, the human node is more closely connected to bats. It shows that viruses are shared to a greater degree among bats as discussed earlier (Figures 3a and 5a). One plausible explanation is that many species of bats live in high‐density populations, with many individuals in close proximity to each other, such as in caves and roosts sites. Indeed, there are always a larger number of bat species than rodent species in a given area (Kerth, Perony, & Schweitzer, 2011).
The difference in connectivity between the bat and rodent host–virus networks has implications for the zoonotic potential of each taxon. High connectivity facilitates viral transmission within and between species, and so, bats are expected to have higher zoonotic potential than rodents. For example, the nectarivorous bat Glossophaga soricina had the highest closeness value in the host–host network despite the fact that only three viruses have been isolated in this species: bat coronavirus, bat paramyxovirus and rabies lyssavirus. This is because these three viruses have a high viral diversity and when the network is collapsed G. soricina connects directly with 155 hosts; it is the central node, even more central than humans. This suggests that this species is prone to harbour several viruses. However, we most considered the biological characteristic of the host that could prevent an efficient transmission to the human. In both the bat and rodent virus–virus networks, there are some large groups that include all of the zoonotic viruses. The grouping of zoonotic viruses suggests a high capacity for mutation and adaptability to different hosts. For that reason, the viruses are shared among different hosts and thus intricate communities are presented (Woolhouse, 2001). In bats, these clusters are disordered and close together resulting in a broad viral exchange among bats. In contrast, a closer relationship was observed in rodent viral–viral network indicating smaller groups around zoonotic viruses. It is difficult though to compare our virus–virus network with other works, since most studies focus on hosts. Moreover, we faced a lack of knowledge on the organization of the viral communities, assembly rules, co‐occurrences or even cross‐antigenicity. These issues directly affect our network architectures. For that reason, deeper comprehension is required to unravel this entanglement.
One of the objectives covered in this work was to recognize non‐zoonotic viruses that may be strongly connected with humans and therefore have zoonotic potential. The bat–virus community that contained humans was composed of bats distributed in Africa and Australia may be explained by high rates of human‐bat contact (Allocati et al., 2016; Rupprecht, 2009).
One non‐zoonotic virus that was included in the bat–virus community that included humans is fruit bat parvovirus. The parvoviridae family were transmitted from bats to other mammals by a viral ancestor suggesting their zoonotic capacity, and groups of genes in their genome denote this potential (Canuti et al., 2011). Even though the virus currently infects only bats (Canuti et al., 2011), it is firmly connected with the human node in our network by Pteropus poliocephalus, a species endemic to Eastern Australia (Lunney, Richards, & Dickman, 2008). Future studies are recommended to elucidate its potential for zoonosis.
Andes viruses (rodents), cowpox (rodents) and rabies lyssavirus (bats) were defined as main actors (high values of degree, betweenness and closeness) in the bipartite networks. However, their importance disappeared when the network was collapsed to virus–virus interactions, likely because their geographical restriction may limit their viral connectivity. Andes virus is only distributed in South America (Martinez et al., 2005), cowpox in Europe (Vorou, Papavassiliou, & Pierroutsakos, 2008) and rabies lyssavirus in America (Moratelli & Calisher, 2015). Therefore, these viruses were less important in the virus–virus networks compared with the worldwide distributed ones, so subsequent at geographical scales are important.
Coronaviruses in bats stood out throughout our study in both bipartite and collapsed networks in terms of connectivity, with high values of degree, betweenness and closeness. In this case, the three most prominent coronaviruses are bat coronavirus, alphacoronavirus and betacoronavirus. Also, each virus is a protagonist in their own community. These are RNA viruses with high mutation rates, and the viruses possess great plasticity allowing them to horizontally transfer accessory genes which facilitate new host and niche establishment (de Groot et al., 2011; Guy et al., 2019).
In the rodent community selected, community 3 has members with predominantly European distribution. Further, two non‐zoonotic viruses are included among the nine zoonotic viruses, but we do not think they are likely to have zoonotic potential because they do not have direct contact with the human node. Therefore, in this community we do not find viruses with zoonotic potential.
In the rodent community 5, M. musculus has high values of connectivity but in the sociogram (Figure 2d), it is evident that the connectivity is with non‐zoonotic viruses. This host species thus is likely less important in public health terms, but highly relevant for disease ecology. In addition, two zoonotic viruses are included in the community 5, and while they can transfer their zoonotic potential to other non‐zoonotic viruses using M. musculus as a virus mixer, we consider this unlikely because the proportion of zoonotic viruses is low, adding the specificity of rodents' viruses and the associative characteristics founding in the rodents. Thus, the one‐to‐one virus–host species relationship suggested by the node‐to‐edge ratio suggests that spillover is unlikely, though not impossible.
In the human–rodent community, Gbagroube virus is noteworthy because it is the only non‐zoonotic virus found in the community. However, the genetics of Gbagroube virus is similar to Lassa virus which is deadly in humans (Coulibaly‐N'Golo et al., 2011). Gbagroube virus could potentially adapt to infect humans because it is genetically similar to Lassa virus (Coulibaly‐N'Golo et al., 2011) and has strong connections with other zoonotic viruses and with humans. Gbagroube virus should be closely monitored along with its host, Mus setulosus, found in Central Africa (Granjon, 2016).
In our study, the human is the most relevant and largest node connected in both groups. The relevance of the human in the network is explained by several factors. First, humans' enormous population and globalization push human populations to nearly everywhere on earth and greatly increases the probability of contact with innumerable organisms, resulting in the emergence of zoonotic diseases (Kock, 2014). Secondly, because zoonotic diseases have clear social implications, once detected in one species, they are much more likely to be tested for, and thus detected, in others (Oliver‐Morales & Abarca García, 2016). It is therefore possible that the high apparent importance of humans in the networks is more due to the over‐representation of zoonotic viruses in the literature than to humans actually being particularly highly.
In the database, we do not have Ebola virus reports because in the database the DBATVIR database did not identify the host species from which the virus was isolated. Similarly, we found that 78.06% of rodent nodes and 65.71% of bats nodes were poorly connected (1–2 degree). Surely, bats have associations that we do not recognize. In addition, there are associations that could occur but do not, but these cannot be identified because cases in which viruses were tested for and not detected are not often reported. We therefore think it is important to report negative samples and the number of animals tested in meticulous reports.
A future study may complement and compare our study with models where the influence of humans is omitted. We must take into account that the human node influences the network structure, and the ecological relationships must be analysed without this influence. Surely, laxer networks will be observed when the connective force of humans is removed. However, our study does need to include both to identify potentially zoonotic viruses.
It is pertinent that, in future investigations, different characteristics of the viruses must be considered simultaneously and not only by their connectivity in the network such as gene sequence, type of transmission and virulence. In the present study, we focus only on viral host capability, not on the symbionts and their associative nature. Spatial analysis may help to further explain how our findings apply among different regions of the world.
Graph theory, beyond allowing the visualization of complex interactions, allows the quantification of many aspects of connectivity and structure.
Rodents should be taken into account as important reservoirs for zoonotic viruses, since in our database, a greater proportion of the total viruses reported were zoonotic viruses in rodents than in bats. Fruit bat parvovirus in bats and Gbagroube virus in rodents should be monitored to elucidate their zoonotic potential. In the present study, we only assessed their network proximity to humans and other zoonotic viruses, and molecular genetic approaches may help to confirm our results. Counting the number of zoonotic symbionts associated with each order is not a conclusive estimate of their zoonotic potential. Our findings reveal that viruses were more frequently shared among bats than rodents. For that reason, bats have more zoonotic potential that the rodents. However, potential emerging zoonotic diseases may arise from both taxonomic groups.
CONFLICT OF INTEREST
We declare that we have no conflict of interest.
Supporting information
ACKNOWLEDGEMENTS
We are very grateful to PAPIIT (Project IA206416), Programa de Apoyo a los Estudios de Posgrado, UNAM, CONACYT and Laboratorio de Ecología de Enfermedades y Una Salud, FMVZ, UNAM, especially to Maribel López Santana and Daniel Mendizabal Castillo for their contribution in the construction of the databases.
Nieto‐Rabiela F, Wiratsudakul A, Suzán G, Rico‐Chávez O. Viral networks and detection of potential zoonotic viruses in bats and rodents: A worldwide analysis. Zoonoses Public Health. 2019;66:655–666. 10.1111/zph.12618
REFERENCES
- Allocati, N. , Petrucci, A. , Giovanni, P. D. , Masulli, M. , Di Ilio, C. , & De Laurenzi, V. (2016). Bat–man disease transmission: Zoonotic pathogens from wildlife reservoirs to human populations. Cell Death Discovery, 2(1), 16048 10.1038/cddiscovery.2016.48 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Almende, B. V. , Thieurmel, B. , & Robert, T. (2016). visNetwork: Network visualization using ‘vis. js’ Library. R package version 2.0.1. https://CRAN.R-project.org/package=visNetwork
- Anthony, S. J. , Johnson, C. K. , Greig, D. J. , Kramer, S. , Che, X. , Wells, H. , … Goldstein, T. (2017). Global patterns in coronavirus diversity. Virus Evolution, 3(1), 1–15. 10.1093/ve/vex012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bordes, F. , Caron, A. , Blasdell, K. , de Garine‐Wichatitsky, M. , & Morand, S. (2017). Forecasting potential emergence of zoonotic diseases in South‐East Asia: Network analysis identifies key rodent hosts. Journal of Applied Ecology, 54(3), 691–700. 10.1111/1365-2664.12804 [DOI] [Google Scholar]
- Brook, C. E. , & Dobson, A. P. (2015). Bats as “special” reservoirs for emerging zoonotic pathogens. Trends in Microbiology, 23(3), 172–180. 10.1016/j.tim.2014.12.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Butts, C. T. (2008). network: A package for managing relational data in R. Journal of Statistical Software, 24(2), 1–36. 10.18637/jss.v024.i02 18612375 [DOI] [Google Scholar]
- Calisher, C. H. , Childs, J. E. , Field, H. E. , Holmes, K. V. , & Schountz, T. (2006). Bats: Important reservoir hosts of emerging viruses. Clinical Microbiology Reviews, 19(3), 531–545. 10.1128/CMR.00017-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Canuti, M. , Eis‐Huebinger, A. M. , Deijs, M. , de Vries, M. , Drexler, J. F. , Oppong, S. K. , … van der Hoek, L. (2011). Two novel parvoviruses in frugivorous new and old world bats. PLoS ONE, 6(12), 1–9. 10.1371/journal.pone.0029140 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Charrel, R. N. , & de Lamballerie, X. (2010). Zoonotic aspects of arenavirus infections. Veterinary Microbiology, 140(3–4), 213–220. 10.1016/j.vetmic.2009.08.027 [DOI] [PubMed] [Google Scholar]
- Coulibaly‐N’Golo, D. , Allali, B. , Kouassi, S. K. , Fichet‐Calvet, E. , Becker‐Ziaja, B. , Rieger, T. , … Günther, S. (2011). Novel arenavirus sequences in Hylomyscus sp. and Mus (Nannomys) setulosus from Cote d’Ivoire: Implications for evolution of arenaviruses in Africa. PLoS ONE, 6(6), e20893 10.1371/journal.pone.0020893 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Csárdi, G. , & Nepusz, T. (2006). The igraph software package for complex network research. InterJournal Complex Systems, 1695, 1695. [Google Scholar]
- Cui, J. , Tachedjian, G. , & Wang, L. F. (2015). Bats and rodents shape mammalian retroviral phylogeny. Scientific Reports, 5, 1–7. 10.1038/srep16561 [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Groot, R. J. , Baker, S. C. , Baric, R. , Enjuanes, L. , Gorbalenya, A. E. , Holmes, K. V. , Ziebuhr, J. (2011). Coronaviridae. Retrieved from ICTV 9th Report (2011). [Google Scholar]
- Freeman, L. C. (1978). Centrality in social networks. Conceptual clarification. Social Networks, 1, 215–239. [Google Scholar]
- Gay, N. , Olival, K. J. , Bumrungsri, S. , Siriaroonrat, B. , Bourgarel, M. , & Morand, S. (2014). Parasite and viral species richness of Southeast Asian bats: Fragmentation of area distribution matters. International Journal for Parasitology: Parasites and Wildlife, 3(2), 161–170. 10.1016/j.ijppaw.2014.06.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Girvan, M. , Girvan, M. , Newman, M. E. J. , & Newman, M. E. J. (2002). Community structure in social and biological networks. Proceedings of the National Academy of Sciences of the United States of America, 99(12), 7821–7826. 10.1073/pnas.122653799 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Godfrey, S. S. (2013). Networks and the ecology of parasite transmission: A framework for wildlife parasitology. International Journal for Parasitology: Parasites and Wildlife, 2(1), 235–245. 10.1016/j.ijppaw.2013.09.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gómez‐Rubio, V. (2017). ggplot2 – Elegant graphics for data analysis (2nd edition). Journal of Statistical Software, 77(Book Review 2), 2–5. 10.18637/jss.v077.b02 [DOI] [Google Scholar]
- Granjon, L. (2016). Mus setulosus. The IUCN Red List of Threatened Species 2016: e.T13980A22406874. 10.2305/IUCN.UK.2016-3.RLTS.T13980A22406874.en [DOI]
- Guy, C. , Thiagavel, J. , Mideo, N. , & Ratcliffe, J. M. (2019). Phylogeny matters: Revisiting "a comparation of bats and rodents as reservoirs of zoonotic viruses". Royal Society Open Science, 6, 181182 10.1098/rsos.181182 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hammer, Ø. , Harper, D. A. T. , & Ryan, P. D. (2001). Past: Paleontological statistics software package for education and data analysis. Palaeontologia Electronica, 4(1), art. 4: 9 pp., 178kb. http://palaeo-electronica.org/2001_1/past/issue1_01.htm [Google Scholar]
- Han, B. A. , Schmidt, J. P. , Bowden, S. E. , & Drake, J. M. (2015). Rodent reservoirs of future zoonotic diseases. Proceedings of the National Academy of Sciences, 112(22), 7039–7044. 10.1073/pnas.1501598112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartonen, T. , & Annila, A. (2011). Natural networks. ArXiv Preprint ArXiv:1106.4127, 1–10. [Google Scholar]
- Hayman, D. T. S. (2016). Bats as viral reservoirs. Annual Review of Virology, 3(1), 77–99. 10.1146/annurev-virology-110615-042203 [DOI] [PubMed] [Google Scholar]
- Hossain, L. , & Feng, S. (2016). Disaster network science: Research and applications. Frontiers in Communication, 1, 1–7. 10.3389/fcomm.2016.00001 [DOI] [Google Scholar]
- Johnson, P. T. J. , De Roode, J. C. , & Fenton, A. (2016). Why infectious disease research needs community ecology. Science, 349(6252), 1–20. 10.1126/science.1259504 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kerth, G. , Perony, N. , & Schweitzer, F. (2011). Bats are able to maintain long‐term social relationships despite the high fission‐fusion dynamics of their groups. Proceedings of the Royal Society B: Biological Sciences, 278(1719), 2761–2767. 10.1098/rspb.2010.2718 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kock, R. (2014). Drivers of disease emergence and spread: Is wildlife to blame? Onderstepoort Journal of Veterinary Research, 81, 4–7. 10.4102/ojvr.v81i2.739 [DOI] [PubMed] [Google Scholar]
- Kolaczyk, E. D. , & Csardi, G. (2014). Statistical analysis of network data with R. New York, NY: Springer‐Verlag; ISBN 978‐1‐4939‐0983‐4. 207 pp. GBP 39.99 (P). http://www.springer.com/book/9781493909827 [Google Scholar]
- Luis, A. D. , Hayman, D. T. S. , O'Shea, T. J. , Cryan, P. M. , Gilbert, A. T. , Pulliam, J. R. C. , … Webb, C. T. (2013). A comparison of bats and rodents as reservoirs of zoonotic viruses: Are bats special? Proceedings of the Royal Society B: Biological Sciences, 280(1756), 20122753 10.1098/rspb.2012.2753 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luis, A. D. , O'Shea, T. J. , Hayman, D. T. S. , Wood, J. L. N. , Cunningham, A. A. , Gilbert, A. T. , … Webb, C. T. (2015). Network analysis of host‐virus communities in bats and rodents reveals determinants of cross‐species transmission. Ecology Letters, 18(11), 1153–1162. 10.1111/ele.12491 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luke, D. A. , & Stamatakis, K. A. (2012). System science methods in public health: Dynamics, networks, and agents. Annual Review of Public Health, 33(1), 357–376. 10.1146/annurev-publhealth-031210-101222.Systems [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lunney, D. , Richards, G. , & Dickman, C. (2008). Pteropus poliocephalus. The IUCN Red List of Threatened Species 2008: e.T18751A8554062. 10.2305/IUCN.UK.2008.RLTS.T18751A8554062.en [DOI]
- Martinez, V. P. , Bellomo, C. , San Juan, J. , Pinna, D. , Forlenza, R. , Elder, M. , & Padula, P. J. (2005). Person‐to‐person transmission of Andes virus. Emerging Infectious Diseases, 11(12), 1848–1853. 10.3201/eid1112.050501 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martínez‐López, B. , Perez, A. M. , & Sánchez‐Vizcaíno, J. M. (2009). Social network analysis. Review of general concepts and use in preventive veterinary medicine. Transboundary and Emerging Diseases, 56(4), 109–120. 10.1111/j.1865-1682.2009.01073.x [DOI] [PubMed] [Google Scholar]
- Moratelli, R. , & Calisher, C. H. (2015). Bats and zoonotic viruses: Can we confidently link bats with emerging deadly viruses? Memorias do Instituto Oswaldo Cruz, 110(1), 1–22. 10.1590/0074-02760150048 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newman, M. E. J. (2014). Networks: An introduction. Oxford, UK: Oxford University. [Google Scholar]
- Newman, M. E. J. (2016). Equivalence between modularity optimization and maximum likelihood methods for community detection. Physical Review E, 94(5). 10.1103/physreve.94.052315 [DOI] [PubMed] [Google Scholar]
- Newman, M. E. J. , & Girvan, M. (2003). Finding and evaluating community structure in networks. Physical Review E, 69(2), 1–16. 10.1103/PhysRevE.69.026113 [DOI] [PubMed] [Google Scholar]
- Nieto‐Rabiela, F. , Suzán, G. , Wiratsudakul, A. , & Rico‐Chávez, O. (2018). Viral metacommunities associated to bats and rodents at different spatial scales. Community Ecology, 19(2), 168–175. 10.1556/168.2018.19.2.9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Shea, T. J. , Cryan, P. M. , Cunningham, A. A. , Fooks, A. R. , Hayman, D. T. S. , Luis, A. D. , … Wood, J. L. N. (2014). Bat flight and zoonotic viruses. Emerging Infectious Diseases, 20(5), 741–745. 10.3201/eid2005.130539 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oliver‐Morales, C. , & Abarca García, C. (2016). El juego de los reflejos: Evolución biológica y cultural In Laguna G., Marcelín R., Patrick G. A., & Vásquez G. (Eds.), Complejidad y sistemas comlejos: Un acercamiento multidimensional (pp. 127–139). Ed. CopIt‐arXives & EditoraC3. ISBN: 978‐1‐938128‐10‐3 ebook [Google Scholar]
- Opsahl, T. , Agneessens, F. , & Skvoretz, J. (2010). Node centrality in weighted networks: Generalizing degree and shortest paths. Social Networks, 32(3), 245–251. 10.1016/j.socnet.2010.03.006 [DOI] [Google Scholar]
- Plowright, R. K. , Eby, P. , Hudson, P. J. , Smith, I. L. , Westcott, D. , Bryden, W. L. , … McCallum, H. (2015). Ecological dynamics of emerging bat virus spillover. Proceedings of the Royal Society B: Biological Sciences, 282(1798), 20142124 10.1098/rspb.2014.2124 [DOI] [PMC free article] [PubMed] [Google Scholar]
- R Core Team (2017). R: A language and environment for statistical computing. Vienna, Austria: R Foundation for Statistical Computing; Retrieved from https://www.R-project.org/ [Google Scholar]
- R Development Core Team (2011). R language definition. Web, 0, 62. Vienna, Austria: R Foundation for Statistical Computing; 10.1016/0164-1212(87)90019-7 [DOI] [Google Scholar]
- Royston, J. P. (1982). An extension of Shapiro and Wilk’s W test for normality to large samples. Applied Statistics, 31, 115–124. 10.2307/2347973 [DOI] [Google Scholar]
- Rupprecht, C. E. (2009). Bats, emerging diseases, and the human interface. PLOS Neglected Tropical Diseases, 3(7), e451 10.1371/journal.pntd.0000451 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmaljohn, C. , & Hjelle, B. (1997). Hantaviruses: A global disease problem. Emerging Infectious Diseases, 3(2), 95–104. 10.3201/eid0302.970202 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Streicker, D. G. , Turmelle, A. S. , Vonhof, M. J. , Kuzmin, I. V. , McCracken, G. F. , & Rupprecht, C. E. (2010). Host phylogeny constrains cross‐species emergence and establishment of rabies virus in bats. Science, 329(5992), 676–679. 10.1126/science.1188836 [DOI] [PubMed] [Google Scholar]
- Vorou, R. M. , Papavassiliou, V. G. , & Pierroutsakos, I. N. (2008). Cowpox virus infection: An emerging health threat. Current Opinion in Infectious Diseases, 21(2), 153–156. 10.1097/QCO.0b013e3282f44c74 [DOI] [PubMed] [Google Scholar]
- Wang, L. F. , Walker, P. J. , & Poon, L. L. M. (2011). Mass extinctions, biodiversity and mitochondrial function: Are bats “special” as reservoirs for emerging viruses? Current Opinion in Virology, 1(6), 649–657. 10.1016/j.coviro.2011.10.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wells, K. , Morand, S. , Wardeh, M. , & Baylis, M. . (2018). Distinct spread of DNA and RNA viruses among mammals amid prominent role of domestic species. bioRxiv, 421511 10.1101/421511 [DOI] [PMC free article] [PubMed] [Google Scholar]
- West Douglas, B. (2001). Introduction to graph theory (2nd ed.). Prentice Hall. University of Illinois. ISBN 0‐13‐014400‐2. [Google Scholar]
- White, L. A. , Forester, J. D. , & Craft, M. E. (2017). Using contact networks to explore mechanisms of parasite transmission in wildlife. Biological Reviews, 92(1), 389–409. 10.1111/brv.12236 [DOI] [PubMed] [Google Scholar]
- Woolhouse, M. E. J. (2001). Population biology of multihost pathogens. Science, 292(5519), 1109–1112. 10.1126/science.1059026 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials