

Summary
Initially described as an interferon (IFN)γ‐inducing factor, interleukin (IL)‐18 is indeed involved in Th1 and NK cell activation, but also in Th2, IL‐17‐producing γδ T cells and macrophage activation. IL‐18, a member of the IL‐1 family, is similar to IL‐1β for being processed by caspase 1 to an 18 kDa‐biologically active mature form. IL‐18 binds to its specific receptor (IL‐18Rα, also known as IL‐1R7) forming a low affinity ligand chain. This is followed by recruitment of the IL‐18Rβ chain. IL‐18 then uses the same signaling pathway as IL‐1 to activate NF‐kB and induce inflammatory mediators such as adhesion molecules, chemokines and Fas ligand. IL‐18 also binds to the circulating high affinity IL‐18 binding protein (BP), such as only unbound free IL‐18 is active. IL‐18Rα may also bind IL‐37, another member of the IL‐1 family, but in association with the negative signaling chain termed IL‐1R8, which transduces an anti‐inflammatory signal. IL‐18BP also binds IL‐37 and this acts as a sink for the anti‐inflammatory properties of IL‐37. There is now ample evidence for a role of IL‐18 in various infectious, metabolic or inflammatory diseases such as influenza virus infection, atheroma, myocardial infarction, chronic obstructive pulmonary disease, or Crohn's disease. However, IL‐18 plays a very specific role in the pathogenesis of hemophagocytic syndromes (HS) also termed Macrophage Activation Syndrome. In children affected by NLRC4 gain‐of‐function mutations, IL‐18 circulates in the range of tens of nanograms/mL. HS is treated with the IL‐1 Receptor antagonist (anakinra) but also specifically with IL‐18BP. Systemic juvenile idiopathic arthritis or adult‐onset Still's disease are also characterized by high serum IL‐18 concentrations and are treated by IL‐18BP.
Keywords: hemophagocytic syndromes, inflammatory diseases, interferon γ, interleukin‐1, interleukin‐18, interleukin‐18 binding protein
This article is part of a series of reviews covering The IL‐1 cytokine and receptor family appearing in Volume 281 of Immunological Reviews.
1. INTRODUCTION
IL‐18 was first identified as “IFNγ‐inducing factor” isolated in the serum of mice after an intraperitoneal endotoxin, following pretreatment with Proprionibacterium acnes, which stimulates liver's Kupffer cells.1 With purification from mouse livers and molecular cloning of “IFNγ‐inducing factor” in 1995, the name was changed to IL‐18 which unexpectedly, appears to be related to IL‐1 family and particularly to IL‐1β in several ways.2, 3 Human IL‐18 and IL‐1β although sharing only 15% sequence homology, share a common β‐pleated sheet structure.3, 4, 5 IL‐18 similarly to IL‐1β is synthesized as an inactive precursor that lacks a signal peptide and needs caspase 1‐mediated cleavage to become biologically active. Despite binding to different receptors, IL‐1β and IL‐18 use the same signaling pathways. However, apart from these important similarities, IL‐18 and IL‐1β appear to have a different biology.4, 6, 7
2. IL‐18 BIOLOGY
2.1. Cell origin and processing
IL‐18 gene is located on chromosome 11 in humans and chromosome 9 in mice whose gene contains 7 exons with two distinct promoters on exon 1 and 2 including an interferon consensus sequence binding protein and a PU.1 binding sites.5 In contrast to other cytokine genes, IL‐18 gene has few RNA‐destabilizing elements, resulting in an unusually stable cytokine expression. Transcription of IL‐18 precursor can be induced after TLR binding of PAMPs and activation of the NF‐κB pathway. IL‐18 gene encodes for a 193 amino acids precursor, first synthesized as an inactive 24‐kDa precursor with no signal peptide, which accumulates in cell cytoplasm. In contrast with IL‐1β, the IL‐18 precursor is constitutively present in blood monocytes, macrophages, dendritic cells from healthy subjects.6, 8 Similarly to IL‐1α and IL‐33, the IL‐18 precursor is constitutively expressed in endothelial cells, keratinocytes or intestinal epithelial cells throughout the gastrointestinal tract and remains in the intracellular compartment of mesenchymal cells.
Similarly to IL‐1β, the IL‐18 precursor is processed intracellularly by caspase 1 into its mature biologically molecule of 18 kDa.9, 10 For IL‐18, the consensus is I‐N‐D at amino acid 50 but the N‐terminus generated by caspase‐1 is 14 amino acids before the consensus sequence, rather than 9 amino acids for IL‐1β. Caspase 1 can be activated by various canonical inflammasomes belonging to the Nod‐like receptors, AIM2‐like receptors or TRIM family containing either a CARD or a PYD domain.11 Among the best known inflammasomes are NLRP‐3, NLRC4, NLRP‐1 and AIM2 which sense various danger signals. Caspase 1 activation also results in a cell‐death program termed pyroptosis, which induces membrane pores and mature IL‐1β and IL‐18 release. Mature IL‐1β can also be released from the cells by lysosome exocytosis or membrane microvesicles, but it is not clear whether IL‐18 used the same pathways. Caspase 1‐independent mechanisms of IL‐18 cleavage have also been described. Notably, Fas Ligand activation of Fas‐expressing Kupffer cells or splenic macrophages from Propiobacterium acnes‐infected mice, can process active IL‐18 in a caspase 1‐independent but caspase 8‐mediated fashion.12, 13 Alternatively, caspase 3 cleaves IL‐18 precursor and mature forms in inactive fragments.14 In addition, granzyme B from cytotoxic cells, chymase from mast cells or meprin β from intestinal and kidney epithelial cells can cleave IL‐18 precursor in biologically active forms.15, 16, 17 IL‐18 can also be released in its precursor form from dying cells and processed extracellularly in an active form by neutrophil proteases such as proteinase 3.18
2.2. IL‐18 receptor and signaling pathways
IL‐18 forms a signaling complex by binding to the IL‐18 alpha chain (IL‐18Rα), which is the ligand binding chain for mature IL‐18; however, this binding is of low affinity (Figure 1).19 In cells that express the co‐receptor, termed IL‐18 receptor beta chain (IL‐18Rβ), a high affinity complex is formed, which then signals.20 The complex of IL‐18 with the IL‐18Rα and IL‐18Rβ chains is similar to that formed by other members of the IL‐1 family with the co‐receptor, the IL‐1R accessory chain IL‐1RAcP (also termed IL‐1R3). Although nearly all cells express the IL‐1R1, not all cells express IL‐1RAcP.21 Similarly, most cells express the IL‐18Rα but not all cells express the IL‐18Rβ. IL‐18Rβ is expressed on T‐cells and dendritic cells but not commonly expressed in mesenchymal cells. Following the formation of the heterodimer, the Toll‐IL‐1 receptor (TIR) domains approximate and it appears that the cascade of sequential recruitment of MyD88, the four IRAKs and TRAF‐6 followed by the degradation of IκB and release of NFκB are nearly identical as that for IL‐1.21 However, there are differences between IL‐1 and IL‐18 signaling. With few exceptions, IL‐1α or IL‐1β are active on cells in the low nanogram/mL range and often in the picogram/mL range. In contrast, IL‐18 activation of cells expressing the two IL‐18 receptor chains requires 10 to 20 ng/mL and sometime higher levels.22
Figure 1.
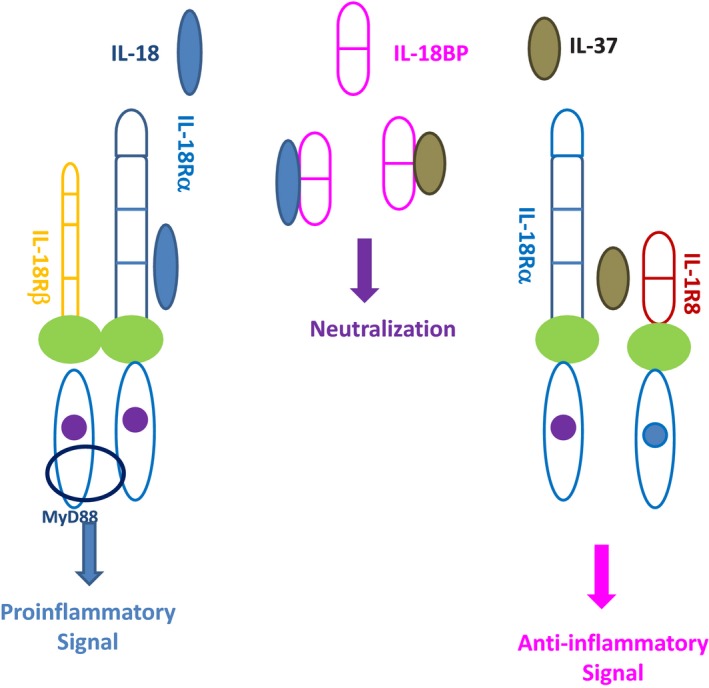
IL‐18 regulation by IL‐18 binding protein and IL‐37. IL‐18 binds the ligand receptor IL‐18Rα, inducing the recruitment of IL‐18Rβ to form a high affinity receptor. The Toll‐IL‐1R Receptor (TIR) domains approximate and allow the binding of MyD88, then inducing a pro‐inflammatory signal into the cells terminating in NF‐κB activation. IL‐18 binding protein (IL‐18BP) which is present in the extracellular compartment may bind soluble mature IL‐18 with a higher affinity than IL‐18Rα and prevents IL‐18 binding to IL‐18 receptor. IL‐18BP may also bind IL‐37, preventing its binding to IL‐18Rα. Free IL‐37 binds to IL‐18Rα inducing the recruitment of IL‐1R8 to form a high affinity receptor, which does not bind MyD88, but induces instead an anti‐inflammatory signal into the cell
In addition to NF‐kB, the IL‐18/IL‐18Rα/IL‐18Rβ complex has been shown to induce phosphorylation of STAT3 in an NK and hippocampal cell lines and the p38 MAP kinase pathway in neutrophils.23, 24, 25
2.3. IL‐37 and IL‐1R8
The tertiary structure of IL‐18 is closely related to IL‐37 and the intron‐exon borders of the IL‐18 and IL‐37 genes suggest a close association. IL‐37 is an inhibitor of the innate immune response. IL‐37 binds to the IL‐18Rα but does not recruit IL‐18Rβ.26, 27 Moreover, silencing of IL‐18Rα in mice has been shown to result in a surprising paradoxical increase in inflammation, suggesting the presence of an anti‐inflammatory ligand and of a co‐receptor that delivers an inhibitory signal.28, 29 In fact, IL‐37 binds to an orphan receptor of the IL‐1 family formerly known as SIGIRR, now designated as IL‐1R8, which forms a tripartite complex with IL‐18Rα and induces an anti‐inflammatory response (Figure 1).30, 31 The IL‐37/IL‐18Rα/IL‐1R8 activates the STAT‐3 signaling pathway, decreases NF‐κB and AP‐1 activation and reduces IFNγ production. Thus, IL‐37 and IL‐18 have opposing effects on cells, and IL‐37 may naturally modulate IL‐18 inflammatory functions.32
2.4. Biological functions in adaptive immunity
IL‐18 is a unique cytokine involved in activation and differentiation of various T cell populations (Figure 2). Together with IL‐12, IL‐18 participates in the Th1 paradigm. This property of IL‐18 is due to its ability to induce IFNγ either with IL‐12 or IL‐15, since IL‐12 or IL‐15 increases the expression of IL‐18Rα. IL‐18 in combination with IL‐12 acts on CD4, CD8 T cells and NK cells to induce IFNγ production, via the simultaneous activation of NF‐kB by IL‐18 and STAT‐4 by IL‐12.5 Interestingly, most of the effects of IL‐12 concerning IFNγ induction by T cells, appears dependent on caspase 1 and are therefore mediated via IL‐18 processing.33 IL‐18 also directly upregulates perforin‐ and FasL‐dependent cytotoxicity in NK cell and CD8 T cells.34 A population of M‐CSF‐primed macrophages expresses a membrane form of IL‐18 which after shedding, may activate NK cells.35 Macrophages can also produce IFNγ, when activated by IL‐18 and IL‐12.36
Figure 2.
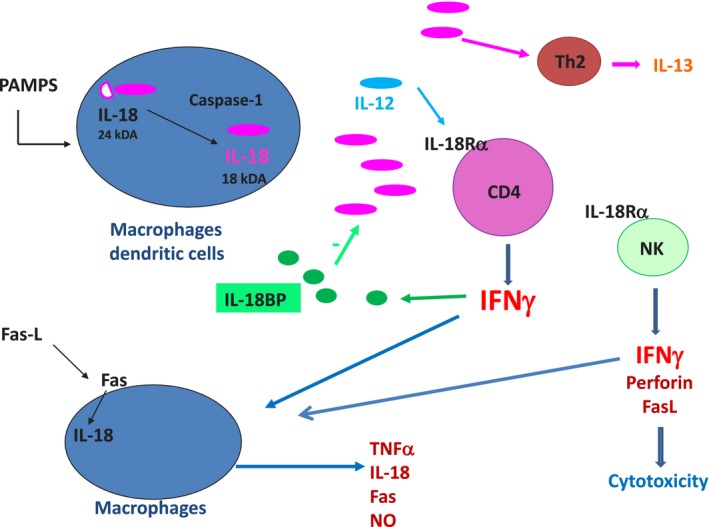
Biological functions of IL‐18. Activation of dendritic cells (DC) or macrophages may induce IL‐18 precursor transcription, but IL‐18 precursor is also constitutively present in the cells. Upon activation of NLRP3, pro‐IL‐18 is processed by caspase 1 and released in its 18 kDa‐mature form. In association with IL‐12 or IL‐15 which increase IL‐18Rβ expression on T cells, IL‐18 induces IFNγ production by CD4 T cells. IFNγ in turn, activates macrophages to produce inflammatory cytokines. IL‐18 can also activate macrophages directly to induce chemokine secretion and NK cells to induce IFNγ secretion or to stimulate perforin‐ and FasL‐mediated cytotoxicity. In macrophages, the interaction of FasL with Fas induces IL‐18 processing by caspase 8. Alternatively, in the absence of IL‐12 or IL‐15, IL‐18 activates Th2 CD4 lymphocytes to produce IL‐13 and IL‐4
Importantly, without IL‐12 or IL‐15, IL‐18 does not induce IFNγ production, but plays an important role in the differentiation of naive T cells into Th2 cells, producing IL‐13 and IL‐4.37, 38 Furthermore, basophils and mast cells, which express IL‐18Rα, produce large amounts of IL‐13 and IL‐4 in response to IL‐18 stimulation.39 In addition, whereas IL‐18 in combination with IL‐12 inhibits IgE production in an IFNγ‐dependent mechanism, IL‐18 alone induces IgE accumulation in vivo.38
It is not clear whether IL‐18 itself, similar to IL‐1β, has a role in Th17 CD4‐positive T cell differentiation, but via the induction of IFNγ by Th1 cells or macrophages, IL‐18 may in fact negatively influence Th‐17 differentiation.40 IL‐18, however, induces IL‐17 production by γδ T‐cells which express IL‐18Rα and this mechanism may be involved in diseases such as auto‐immune encephalomyelitis, systemic JIA or neonatal sepsis.41, 42, 43 In the Helicobacter pylori chronic infection, IL‐18 has been shown to play a role in FoxP3‐positive Tregs differentiation.44
2.5. Proinflammatory properties of IL‐18
Independently of IFNγ or other cytokines, IL‐18 exhibits characteristics of other proinflammatory cytokines, such as increases in cell adhesion molecules, nitric oxide synthesis, and chemokine production. IL‐18 induces ICAM‐1 expression on myeloid cells, and also VCAM‐1 expression on micro‐endothelial cells or synovial fibroblasts in vitro and in vivo via NF‐kB activation.45, 46 Blocking IL‐18 activity reduces metastasis in a mouse model of melanoma due to a reduction in IL‐18‐induced expression of VCAM‐1.47 IL‐18 also induces CXC chemokines by macrophages or synovial fibroblasts as well as angiogenic factors in rheumatoid arthritis tissues.48, 49, 50 A unique property of IL‐18 is the induction of Fas ligand (FasL), which may account for severe hepatic damages in several pathogenic conditions.12, 51 The induction of fever, a well‐studied property of IL‐1β is not a property of IL‐18 since injection of IL‐18 into mice, rabbits or humans does not produce fever.52
2.6. IL‐18 binding protein
The discovery of the IL‐18BP took place during the search for the soluble receptors for IL‐18.53, 54 IL‐18BP is a constitutively secreted protein, with an exceptionally high affinity for IL‐18 (400 pM). Present in the serum of healthy humans at a 20‐fold molar excess compared to IL‐18, IL‐18BP may contribute to a default mechanism by which a Th1 response to foreign organisms is blunted in order to reduce triggering an autoimmune responses to a routine infection.55 Although IL‐18BP is readily secreted, it falls into the functional category of being a shed soluble receptor. As shown in Figure 1, IL‐18BP contains only one IgG domain whereas the Type II IL‐1 receptor contains three domains. In this regard, the single IgG domain of IL‐18BP is similar to IL‐1R8 (SIGIRR), which also has a single IgG domain. The salient property of IL‐18BP in immune responses is in downregulating Th1 responses by binding to IL‐18 and thus reducing the induction of IFNγ. Since IL‐18 also affects Th2 responses, IL‐18BP also has properties controlling a Th2 cytokine response. Harboring a classic signal peptide, IL‐18BP is readily secreted. Serum levels in healthy subjects are in the range of 2,000‐4,000 pg/mL compared to the levels of IL‐18 in the same sera of 80‐120 pg/mL.55 Moreover, IL‐18BP binds IL‐18 with an affinity of 400 pM, an affinity significantly higher than that of IL‐18Ra. Because a single IL‐18BP molecule binds a single IL‐18 molecule, one can calculate bound vs free IL‐18 in a mixture of both molecules.55 This balance and the concentrations of free IL‐18 may be more relevant than total IL‐18 when interpreting the concentrations of IL‐18 in patients. For esample, in Wegener's granulomatosis and systemic lupus erythematosus, both IL‐18BP and IL‐18 are high, but the level of IL‐18BP is not sufficiently high enough to neutralize IL‐18 and therefore, the level of free IL‐18 is higher than in healthy subjects. The same observation has been made in situations such as sepsis, sJIA and macrophage activation syndrome.55, 56
A unique property of IL‐18BP is that the molecule also binds the anti‐inflammatory cytokine, IL‐37. Thus, in any pathological condition, the outcome is the result of the concentrations of active IL‐18, the surface level of IL‐18Rα, the presence of IL‐1R8 and the level of IL‐18BP (Figure 1).26 Hence, the anti‐inflammatory property of IL‐37 can be affected by the concentration of IL‐18BP. As the concentration of IL‐18BP increases and binds IL‐37, there is the possibility that IL‐37 becomes less available as an anti‐inflammatory cytokine. Indeed this has been observed in mice injected with IL‐18BP. At low dosing of IL‐18BP, there is reduced inflammation in a model of rheumatoid arthritis but as the doing of IL‐18BP increases, the anti‐inflammatory properties of IL‐18BP are lost.57
IL‐18BP is highly regulated at the level of gene expression and unexpectedly, IFNγ increases gene expression and synthesis of IL‐18BP.58, 59 Therefore, IFNγ driving an increase in the natural and potent inhibitor of IL‐18 falls into the category of a negative feedback loop. The concept is supported by clinical data showing that patients being treated with IFNα for hepatitis have elevated levels of IL‐18BP.60
3. IL‐18 IN PATHOLOGICAL CONDITIONS
Several autoimmune diseases are associated with elevated production of IFNγ and IL‐18. Diseases such as systemic lupus erythematosus, rheumatoid arthritis, Type‐1 diabetes, Crohn's disease, psoriasis and graft vs host disease are thought to be mediated in part, by IL‐18 and has been previously reviewed.4, 5, 6, 7 In this review, we will discuss the role of IL‐18 in the pathogenesis of inflammatory bowel diseases, metabolic syndrome and cardiovascular diseases, lung inflammatory diseases, sepsis, hemophagocytic syndromes and systemic juvenile idiopathic arthritis.
3.1. IL‐18 in inflammatory bowel diseases
Innate immunity and inflammasome activation, notably NLRP3, are implicated in the earlier phases of Crohn's disease.61 Increased total IL‐18 (in the order of 400 pg/mL) and IL‐18BP have been found in the serum of adult and pediatric patients with Crohn's disease and mature IL‐18 has been detected in the digestive tissues from patients with active Crohn's disease, but not in ulcerative colitis.62, 63, 64, 65 IL‐18 co‐localized with lamina propria cells in active lesions, whereas it appears located in epithelial cells in inactive areas.65 Furthermore patients harboring an NLRC4 mutation have both high serum IL‐18 concentrations and severe digestive tract inflammation, and IL‐18 transgenic mice have more severe colitis.66, 67 In agreement, several studies have reported the beneficial effects of IL‐18 inhibition using neutralizing anti‐IL‐18 antibodies or IL‐18BP, in DSS or TNSB‐induced models of inflammatory bowel diseases.68, 69, 70, 71 Using conditional IL‐18 and IL‐18R‐deficient mice affecting intestinal epithelial or hematopoietic cells in the DSS model, it was possible to show that IL‐18 may be produced by both cell sources, but that intestinal epithelial cells were the major targets of IL‐18 pathogenic action.72 Similarly IL‐18BP‐deficient mice demonstrated a more severe form of DSS colitis, due to decreased maturation of mucus‐producing goblet cells.72 These data involving IL‐18 in inflammatory bowel diseases pathogenesis are related to others showing the pathogenic roles of NLRP3, caspase 1, mIR223 and IL‐1β in the DSS model.73, 74, 75
Therefore, a surprising finding was the association of Crohn's disease susceptibility with NLRP3 or IL‐18 gene polymorphisms known to decrease NLRP3 and IL‐18 expression.76, 77, 78 Subsequently, several groups reported that NLRP3‐, caspase 1‐ or ASC‐deficient mice developed a more severe form of DSS colitis due to a rupture of the intestinal barrier associated with proliferation of commensal bacteria, increased chemokine production and leukocyte infiltration in the colon.79, 80 These phenomena are due to decreased IL‐18 (more than IL‐1β) production by intestinal epithelial cells and can be reversed by injection of IL‐18.79, 80 Another inflammasome, NLRP6 is highly expressed in the large and small intestine, especially in the intestinal epithelium and appears to be involved in IL‐18 production in the digestive tract.81 NLRP6 is involved in the response to viral pathogens and bacterial components of the commensal microbiome, and microbiome‐derived modulators of NLRP6 activity have been identified, such as the bile acid derivative taurine as a positive regulator of IL‐18 production, or spermin and histamine as negative regulators.82 NLRP6 activation induces protective mucus production by goblet cells, as well as IL‐18‐dependent anti‐microbial peptides production by epithelial cells and plays a role in the maintenance of a normal microbiota in the lumen. Indeed NLRP6 KO mice developed a dysbiotic microbiota, notably increased of prevotellacae.83 The dysbiotic microbiota is transferrable to wildtype mice in which it suppresses NLRP6 activation, dampens IL‐18 production, decreases antimicrobial peptide production, and increases the severity of DSS colitis.83, 84 Interestingly, this phenotype can be reversed by IL‐18 injection.82, 83
The digestive tract is a complex organ since it tolerates millions of commensal bacteria yet the intestine rapidly recognizes pathogenic microorganisms and eliminates them. This is due to the different barriers composing the intestinal mucosa. First, an anatomical barrier consisting in an epithelial cell and mucus layer associated with an antimicrobial barrier including antimicrobial peptides and immunoglobulin A, then an immunological barrier in the lamina propria containing cells of the innate and adaptive immune system constituting the gut‐associated lymphoid tissue, GALT.85, 86 In this context, IL‐18 may play a dichotomous role (Figure 3). In homeostatic conditions, IL‐18 is protective. NLRP6 and NLRP3 inflammasomes activation in epithelial cells may induce IL‐18 production, which in turn, induces the maturation of goblet cells, mucus and production of antimicrobial peptides which maintained both an intact intestinal barrier and normal microbiota in the lumen. In addition, IL‐18 may also decrease Th‐17 differentiation and modulate Treg cell function, since these cells in the lamina propria express the IL‐18R.87 In case of chronic inflammation with anatomical barrier rupture and dysbiosis on the contrary, IL‐18 is detrimental. NLRP3 and other inflammasomes may be activated in mononuclear cells from the lamina propria inducing large concentrations of pathogenic IL‐18, leukocyte recruitment and severe inflammation.88, 89 In this setting, IL‐1α constitutively present in intestinal epithelial cells is released by injured cells and has an important pro‐inflammatory role, whereas IL‐1β secreted by mononuclear cells from the lamina propria has been shown to play a rather protective healing role.90 Anti‐TNFα and anti‐IL‐12/23 are efficient treatments in inflammatory bowel diseases, likely via the decrease in IFNγ.91, 92 Since TNFα can induce IL‐18, it is possible that part of the action of anti‐TNFα may in fact be due to IL‐18 inhibition.
Figure 3.
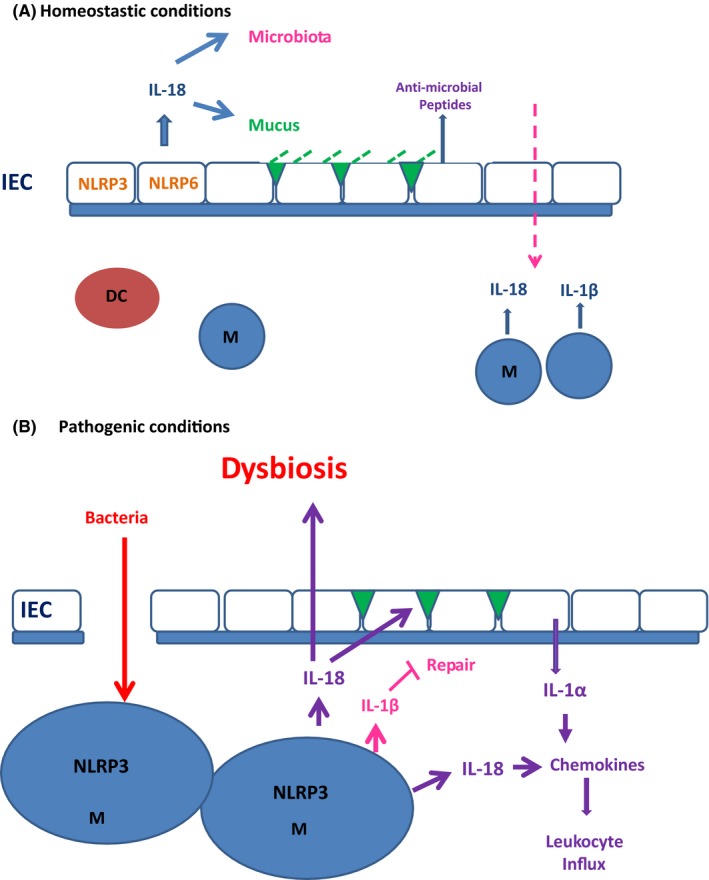
Role of IL‐18 in inflammatory bowel diseases. In homeostatic conditions (A), IL‐18 is produced by intestinal epithelial cells (IEC) after processing by NLRP3 or NLRP6 inflammasomes. IL‐18 has a protective role on intestinal barrier through mucus synthesis by goblet cells (green triangles) or secretion of anti‐microbial peptides (AMP) by IEC. In this way, IL‐18 maintains a normal microbiota in the digestive tract. In case commensal bacteria enter the mucosa (pink arrow), macrophages from the lamina propria secrete IL‐18 which controls infection and IL‐1β which has healing properties. In pathogenic conditions (B), the epithelial barrier is disrupted and bacteria enter massively in the lamina propria where they induce local macrophages to produce IL‐18 which is detrimental (purple) and IL‐1β (pink) which is protective. Disrupted IEC also release IL‐1α which is highly proinflammatory in this setting (purple) and activates macrophages. This leads to chemokine secretion and leukocyte recruitment from the peripheral blood into the lamina propria. In the meantime, IL‐18 inhibits mucus production by goblet cells and modify microbiota favoring dysbiosis
3.2. IL‐18 in metabolic syndrome and diabetes type 2
The metabolic syndrome is characterized by central obesity, acquired insulin resistance, high blood triglycerides, low HDL, blood hypertension and increases the risk of type 2 diabetes (5‐7 fold), and that of cardiovascular disease by twofold.93, 94 Obesity, type 2 diabetes and atherogenesis are characterized by low‐grade underlying inflammation. NLRP3, caspase 1 and IL‐1β play an important role in type 2 diabetes pathogenesis since IL‐1β mediates obesity‐induced inflammation, increases insulin resistance and destroys β‐cells.95 Thus high IL‐18 levels in obese patients, metabolic syndrome or type 2 diabetes is not unexpected.96, 97, 98 In addition, a polymorphism of IL‐18 gene associated with increased serum IL‐18 levels has been linked to insulin resistance and metabolic syndrome.99 The visceral rather than subcutaneous adipose tissue from obese individuals produces more IL‐18 than lean controls, and serum IL‐18 concentrations decreased after bariatric surgery.100, 101, 102
Despite these data suggesting a link between high IL‐18 concentrations and metabolic syndrome or type 2 diabetes, studies in mice reveal paradoxical observations. IL‐18 or IL‐18Rα‐deficient mice, far from being protected from metabolic syndrome, appeared to become obese and develop insulin‐resistance after 6 months of normal chow diet, due to both hyperphagia and decrease energy consumption.103, 104 Intravenous, intracerebral or intraperitoneal injection of exogenous IL‐18 corrected hyperphagia, increased catabolism (possibly via IL‐18‐induced IFNγ production) and decreased insulin‐resistance.100, 101 IL‐18 may increase insulin sensitivity via the phosphorylation of STAT3 in cells and possibly via activation of AMP‐activated kinase (AMPK) in muscles.105 Insulin resistance despite high IL‐18 levels is reminiscent of leptin resistance in obesity and type 2 diabetes, indeed PBMC of patients with obesity or type 2 diabetes were found to be resistant to the stimulating action of IL‐18 on STAT3‐mediated IFNγ production, due to decreased IL18Rα and IL‐18Rβ expression.106 Thus it appears that IL‐18, similar to leptin is an adipocytokine which controls both food intake and energy homeostasis.104 Increased IL‐18 concentrations in patients with obesity and type 2 diabetes may in fact, constitute a retrocontrol mechanism in order to compensate insulin resistance induced by other pro‐inflammatory cytokines.107, 108 In agreement with this hypothesis, treatment with anti‐IL‐18 monoclonal antibodies demonstrated no improvement in patients with type 2 diabetes.109
3.3. IL‐18 in atherosclerosis and myocardial infarction
NLRP3 inflammasome and IL‐1β play important roles in early atherogenesis.110 Cholesterol crystals which accumulate in atherosclerotic lesions activate NLRP3 and induce caspase1 activation, leading to IL‐1β and IL‐18 secretion. In addition, irradiated atherogenic‐prone LDLR‐deficient mice were protected from atherosclerosis, when reconstituted with NLRP3 or ASC‐ deficient bone marrow.110 IL‐18 and IL‐18Rα are higly expressed in macrophages from atherogenic plaque, significantly more in unstable plaques.111, 112 Alternatively, transfection with an expression‐plasmid DNA encoding IL‐18BP in atheroma‐prone ApoE KO mice prevented plaque progression.113 Injection of IL‐18 to ApoE KO mice increased the severity of the disease, an effect which was not observed in IFNγ KO mice, suggesting that the effects of IL‐18 were mediated in a large part by IFNγ in this model.114 IL‐18 may also play a long term pro‐atherogenic role, since ApoE‐deficient mice crossed with IL‐18‐deficient mice appeared protected, despite increased circulating cholesterol concentrations.115 Again, the effects of IL‐18 may be mediated by IFNγ, since IL‐18‐deficient mice had a decreased IFNγ signature. In summary, IL‐18 aggravates atherosclerosis by inducing IFNγ, which promotes the acceleration of the vascular disease.116 Despite these pro‐atherogenic properties, IL‐18 gave controversial results when used as a biomarker in cardiovascular diseases. Several studies suggest that IL‐18 is increased (in the order 70 to 300 pg/mL) in the serum of patients with coronary diseases, with a predictive value for death occurrence and a good correlation with coronary atherosclerosis evaluated by coronarography, but these results were not confirmed when carotid atheroma was evaluated by ultrasound.97, 117, 118, 119, 120
IL‐18 is, however, increased in patients at risk or with myocardial infarction as well as in patients with congestive heart failure.121, 122, 123, 124 Increased serum IL‐18 concentrations and tissue IL‐18 expression were found in a murine model of acute myocardial infarction and IV injection of both anti‐IL‐18 or mesenchymal stem cells overexpressing IL‐18BP into the coronary artery before ischemia, reduced infarct size.125, 126, 127 IL‐18 may be involved in several aspects of myocardial infarction or congestive heart failure. Stimulation of a cardiomyocyte cell line with IL‐18 induces cell hypertrophy and production of atrial natriuretic peptide, a marker of cardiac hypertrophy.128 Injection of IL‐18 to WT mice induces atrial natriuretic peptide expression in the heart, as well as myocardial hypertrophy and contractile dysfunction.129 IL‐18 has cardiodepressant effects and neutralization of IL‐18 with either anti‐IL‐18 monoclonal antibodies or IL‐18BP improved contractile function.129, 130 Importantly, IL‐18 may mediate some of the well‐known cardiodepressant effects of IL‐1β, since IL‐18 blockade with IL‐18BP has been shown to prevent the systolic dysfunction induced by IL‐1β.131 Finally, IL‐18 participates in extracellular remodeling and myocardial fibrosis. Indeed administration of IL‐18 to WT mice induced cardiac tissue fibrosis after 7 days and IL‐18 induced the synthesis of osteopontin by cardiac fibroblasts, which mediates cardiac fibrosis.132, 133
3.4. IL‐18 in lung diseases
3.4.1. Asthma
Whereas IL‐18 is a well‐known inducer of the Th1‐type immune response, as discussed earlier, several studies have shown that in absence of IL‐12, IL‐18 acts to induce Th2 functions. IL‐18 is involved in the pathogenesis of allergic asthma, which is characterized by a Th2‐type airway inflammation with eosinophils, IgE production, airway hyperresponsiveness, mucus metaplasia and cytokines such as IL‐13 and IL‐5. IL‐18 gene polymorphisms have been associated with asthma severity in adults, notably the rs5744247C>G variants, which found increase IL‐18 mRNA transcription and IL‐18 secretion by LPS‐stimulated monocytes.134 Similarly, IL‐18Rα polymorphisms have been associated with allergic asthma in three different North European populations.135 Increased IL‐18 concentrations have been transiently found in the serum of asthmatic patients, especially during asthma exacerbations with increased IL‐18 expression in epithelial and smooth muscle cells from patients airway biopsies.136, 137 IL‐18 concentrations correlate with the methacholine test, and inversely with the peak expiratory flow. In addition, in lung autopsies performed on 12 patients with fatal asthma, IL‐18 and IL‐18Rα were highly expressed and associated with increased eosinophils and CD8+ T lymphocytes.138In animal the common model of ovalbumin sensitized and challenged mice, intraperitoneal IL‐18 injection increases eosinophil recruitment into the airways but not airway hyperesponsiveness.139 More recently, ovalbumin sensitization/inhalation in IL‐18 transgenic mice overproducing IL‐18 in the lungs, demonstrated airway hyperresponsiveness and severe inflammation with leukocyte infiltration as well as IFNγ, eotaxin and IL‐13 production. In addition, treatment with anti‐CD4 decreased airway hyperesponsiveness, lymphocyte count, IL‐13 and IFNγ concentrations in broncho‐alveolar lavage fluids, whereas the same model in IL‐13‐deficient mice, the infiltrating eosinophils were less and airway hyperesponsiveness was reduced.140 In summary, due to its Th‐2 inducing functions, IL‐18 may participate in airway inflammation and hyperresponsiveness in allergic asthma and may be an interesting therapeutic target in severe asthma resistant to steroids.
3.4.2. Chronic obstructive pulmonary disease
Chronic obstructive pulmonary diseases (COPD) include lung emphysema and chronic bronchitis, both characterized by chronic inflammation, alveolar destruction, airway remodeling and fibrosis.141 The main causes of COPD are cigarette smoking (primary or second‐hand) and air pollution. Cigarette smoke contain more than 4500 chemical products, oxidants and free‐radicals that activate the innate immune system and induce lung inflammation.142 IL‐18 has been shown to be highly expressed in alveolar macrophages, CD8 T lymphocytes as well as bronchiolar and alveolar epithelial cells from patient lungs, whereas circulating 18Rα‐expressing T cells were higher in COPD patients.143, 144, 145 Serum IL‐18 concentrations are increased in COPD and smokers when compared to non‐smokers healthy controls, correlated positively with disease severity and negatively with the forced expiratory volume tests.144
In mice, chronic exposure to cigarette smoke induces emphysema, small‐airway remodeling and even pulmonary hypertension thus mimicking COPD. After 2‐week smoke exposure in this model, IL‐18 was increased in lung biopsies and the broncho‐alveolar lavage fluid, especially in alveolar macrophages, associated with increased levels of caspases 1 and 11. In IL‐18Rα KO mice, alveolar cell apoptosis, protease and chemokine production significantly decreased, reducing both inflammation and lung emphysema.144 Cigarette smoke exposure combined with poly (I:C) TLR activation to reproduce viral infection, acted synergistically to induce a more severe lung inflammation with alveolar remodeling, emphysema and airway fibrosis. IL‐18 was highly increased in alveolar macrophages associated with IFNα, followed by IL‐12 and IFNγ. Inflammation, epithelial and endothelial cell apoptosis were significantly decreased in IL‐18Rα or IFNγ‐deficient mice, demonstrating the important role of Th1‐mediated inflammation in this model.146
A second‐hand smoke model has been created in Sprague‐Dawley rats which after 2 month‐exposure, demonstrated emphysema associated with mild pulmonary hypertension, increased “foamy” macrophages, excess epithelial and endothelial cell apoptosis associated with decreased VEGFR1 and VEGFR2 expression. IL‐18 was increased while IL‐18BP was decreased, and free IL‐18 induced endothelial cell apoptosis, increased endothelial permeability and decreased VEGFR expression.147 Another interesting animal model appears to be lung‐specific IL‐18 transgenic mice which demonstrated typical COPD lesions such as emphysema, mucus metaplasia and fibrosis. Accumulation of inflammatory cells including macrophages, neutrophils, eosinophils and lymphocytes is observed In BAL fluids. All lymphocyte subpopulations are represented, notably CD4 which consists in both Th1, Th17 and Th2.148 Notably Th1 lymphocytes seem to have a rather protective role, since IFNγ‐deficient animals have more severe inflammation and emphysema, and IFNγ decreases IL‐18, IL‐17 and IL‐13. On the contrary IL‐13 KO mice demonstrate decreased inflammation, emphysema and fibrosis.149
3.4.3. Acute lung injury
NLRP3 is expressed in lung epithelial cells as well as in monocytes and macrophages and NLRP3 activation is protective in various models of lung infection, however, excessive NLRP3 stimulation has been demonstrated to be detrimental and may cause acute lung injury.150 The most severe form of acute lung injury is represented by acute respiratory distress syndrome (ARDS) with a 40%‐mortality.151 IL‐18 concentrations have been found elevated in the serum and lung of patients with ARDS (in the order of 600 pg/mL), and correlated with severity score and death.152, 153Among the most severe forms of ARDS are those induced by avian influenza virus. Seasonal influenzae RNA and M2 proteins are known to activate NLRP3 inflammasome in a protective way, since NLRP3‐ and caspase‐1‐deficient mice demonstrate more severe lung lesions as well as increased lethality.154, 155 In this setting, although controversial, IL‐18 but not IL‐12, has been shown to be protective in the early defenses against influenza by inducing NK cell cytotoxicity and INFγ production.156, 157, 158 On the contrary, avian influenzae H5N1 and H7N9 contain a PB1‐F2 protein which maintain both inhibits IFNα production and activates NLRP3 in a strong and prolonged way.159 This prolonged NLRP3 activation leads to excess IL‐18 production and induces a very detrimental IFNγ‐biased cytokine storm which appears to characterize ARDS pathogenesis.160, 161, 162 Interestingly, this pathogenesis appears common to coronavirus‐induced severe acute respiratory syndrome (SARS), another severe form of ARDS and possibly to the 1918 influenza virus.163, 164, 165 This dual role of NLRP3 in influenza infection and potentially in ARDS has been recently demonstrated by temporal inhibition of NLRP3 using MCC950, a specific NLRP3 activation inhibitor.166, 167
3.5. IL‐18 in sepsis
Sepsis is characterized by both excessive inflammation and immune suppression.168 IL‐18 and IL‐18BP concentrations are elevated in the serum of pediatric and adult patients with sepsis, are free IL‐18 remains higher than in healthy individuals or in non‐septic patients.43, 55 In postoperative sepsis patients, high IL‐18 serum levels during the 2 first days represent an early predictive factor for a lethal outcome, possibly suggesting a role for IL‐18 in the pathogenesis of sepsis.169Interestingly, caspase 1 KO mice are protected against LPS‐induced septic shock, whereas IL‐1β KO mice are not.170 In agreement, anti‐IL‐18 monoclonal antibody treatment appears to be protective in two models of E. coli or Salmonella LPS‐induced septic shock, decreasing neutrophils in the liver and lungs, and IFNγ, TNFα or chemokine serum concentrations. 170 Also, combined IL‐1β and IL‐18 inhibition has been shown to be protective in models induced by LPS injection or cecal ligation.171 IL‐18 appears to be especially important in mediating endotoxin‐induced liver and lung injuries.172, 173 The presence of free IL‐18 in sepsis serum due to an imbalance of IL‐18/IL‐18BP ratio, lead to the use of IL‐18BP in a murine model of cecal ligation. However, IL‐18BP injection 8 hours after sepsis onset appeared slightly protective in the more severe forms, but not in those with a low predicted mortality rate.174
Similar to what is observed in influenza, in the LPS‐induced model of sepsis, there is a duality role for IL‐18. Injection of low doses LPS seems to induce moderate and transient serum increase of IL‐18 and IFNγ, which enhanced antibacterial host defenses and are protective. In contrast, after the administration of high doses of LPS, high concentrations of IL‐18 and IFNγ are observed, which increased until death, associated with reduced antibacterial defenses. In the latter case, anti‐IL‐18 restores antibacterial defenses and is protective of a fatal outcome, whereas it decreased defenses in the former situation, suggesting an optimal range of IL‐18 concentration is essential for host defenses.175 These data may be in part explained by a positive loop of induction of caspase 1 mRNA transcription by IFNγ, leading to sustained IL‐18 and IFNγ levels.176 More recently, the role of IL‐18 and its capacity to induce IL‐17 in conjunction with IL‐1 has been outlined in a model of neonatal sepsis.43 Premature infants have elevated serum concentrations of IL‐18 which increases in sepsis, associated with an inverse correlation with the gestational age. In a lethal model of neonatal sepsis, IL‐18‐deficient mice were protected whereas the administration of IL‐18 increased lethality. In this model, ablation of the adaptive immune system afforded no protection, whereas treatment with anti‐IL‐17 was protective, due to the fact that IL‐18 induced IL‐17 secretion by γδ T cells from injured digestive tract or the lungs. Interestingly, in this model IL‐1R1‐ but not IL‐1β‐deficient mice were protected, suggesting a role for IL‐1α which was found highly expressed in the gut of septic neonatal mice treated with IL‐18. IL‐17 is known to play a role in neutrophil‐induced inflammation and IL‐18 is known to increase PMN functions.25, 177 Thus, despite a protective role of IL‐18 and IL‐17 in host defense against infection, their combined excessive actions during sepsis may become deleterious.
The duality of IL‐18 in sepsis and possibly in other models of cytokine storm might be explained by different inflammasome and caspase involvement. Caspase 1‐deficient mice which are resistant to endotoxic shock, are in fact deficient in both caspase 1 and caspase 11.11 Interestingly, NLRP3‐deficient mice are not protected against sepsis, despite the fact that NLRP3 controls caspase 1 activation and both IL‐1β and IL‐18 secretion, suggesting that caspase 11, rather than caspase 1 may play an essential role in the protection of caspase 1/11‐deficient mice toward sepsis.178 Caspase 11 is mainly involved in a non‐canonical inflammasome in mice. Caspase 11 oligomerizes with CARD to sensor intra‐cytoplasmic LPS, usually due to high serum concentrations of LPS, and induces pyroptosis‐mediated cell death. This mechanism dominates over that of canonical inflammasomes, which activate caspase1, such as NLRP3.179 Excess of pyroptotic cell death may induce both strong IL‐18 and DAMPS release inducing an “ecosanoid storm,” shock and death.11
3.6. IL‐18 in hemophagocytic syndromes
Hemophagocytic syndromes or hemophaphagocytic lymphohistiocytosis (HLH) are characterized by the association of clinical and biological symptoms such as fever, hepatomegaly, splenomegaly, cytopenia, hyperferritininemia, hypertriglyceridemia, intravascular coagulation and could result in multivisceral deficiency.180 The presence of hemophagocytosis in the bone marrow is not required for the diagnosis, but its presence in this clinical picture is highly suggestive of the disease. Noteworthy is the fact that hemophagocytosis may also be frequently observed in the bone marrow of sepsis patients.181
HLH can be divided in two presentations. The primary form usually occurs early in life and is due to recessive mutations affecting proteins involved in lymphocyte cytotoxicity, such as perforin, Munc 13D, syntaxin 11, Munc 18‐2, rab27 or Lyst.182 The other HLH syndrome, is also known as Secondary HLH or Macrophage Activation Syndrome, complicates infections, notably due to intracellular micro‐organisms, lymphoma or rheumatic diseases, such as systemic lupus erythematosus or systemic juvenile idiopathic arthritis (sJIA) and its adult equivalent, adult‐onset Still's disease (AOSD).183Animal models can mimick the two forms of HLH. Perforin‐deficient mice infected with lymphocyte choriomeningitis virus (LCMV) was the first published model of primary HLH, followed by models using Munc 13D‐or Rab27‐deficient mice, or murine cytomegalovirus (MCMV) infection.184, 185, 186 Another model mimicking secondary HLH is due to TLR‐9 hyperstimulation by repeated CpG injections into wildtype mice.187 More recently, other models of secondary HLH have been reported, such as LPS stimulation in mice overexpressing IL‐6, EBV, Salmonella typhimurium or murine cytomegalovirus (MCMV) infection in humanized or wildtype mice.188, 189 In humans as well as in all these animal models, excess circulating inflammatory cytokines with a rather Th1 profile, designed as a “cytokine storm,” is the primary event.184, 187, 190 Among them, IFNγ seems to play the important pathogenic role, since inhibition of IFNγ appears protective in most murine models.184, 187, 191 It is not completely clear, however, since some models depend also on TNFα or other cytokines.189, 192, 193
The role of IL‐18 in HLH is likely due to its capacity to induce IFNγ and pro‐inflammatory cytokines. Several reports have found elevated IL‐18 concentrations in the serum of patients with both primary and secondary HLH as well as in animal models and IL‐18 correlated with the biological criteria and evolution of the disease.56, 184, 187, 191, 194, 195 Usually, the concentrations of IL‐18 range from 0.6 to 3 ng/mL in HLH patients. There is the exception in two conditions, in which IL‐18 concentrations are higher than 5 ng/mL, namely XLP type 2 due to XIAP mutations and sJIA/AOSD.195, 196 In both situations, IL‐18 concentrations do not return to normal levels even after recovery of the HLH. Such elevated concentrations are unusual for an inflammatory cytokine and question its significance, especially since in the majority of these studies, the distinction between total IL‐18 and free IL‐18 was not made. In a previous study of Secondary HLH complicating lymphoma, infections and AOSD, we observed an increase in free IL‐18, IL‐18BP concentrations being similar to those in control patients without HLH, perhaps suggesting a relative resistance of IL‐18BP synthesis to IFNγ which was elevated in these patients.56 A resistance of mononuclear cells to produce IL‐18BP in response to IFNγ stimulation, was indeed reported in a patient with a primary HLH.197 This relative imbalance between IL‐18 and its natural inhibitor may favor IFNγ and TNFα excessive production. Thus, there was a study on the effect of recombinant human IL‐18BP in a model of perforin‐deficient mice infected with MCMV. Although there was no reduction in survival, IL‐18BP demonstrated a significant protective effect on liver and spleen lesions due to the decrease in IFNγ, TNFα, and FasL concentrations.198 Noteworthy is the fact that in this animal model, no anti‐cytokine strategy has been shown to increase survival probably due to the fact that concentrations of IFNγ and TNFα are found, respectively, 10‐fold and 5‐fold higher than in the LCMV‐infected perforin‐deficient model.184, 186, 199 However even in the perforin KO‐LCMV model, anti‐IL‐18 treatment did not significantly influence animal survival.184
Despite disappointing animal data, the role of IL‐18 in HLH is valide. In 2014, two independent groups reported on two pediatric patients with severe refractory HLH and digestive tract lesions without mutations affecting cytotoxicity, but bearing gain‐of‐function mutations of the NLRC4 inflammasome.66, 200 Ex vivo experiments showed that these mutations induced very high concentrations of IL‐18 (10 ng/mL), whereas IL‐1β was modestly affected. Interestingly, IL‐18 concentrations in patients with NLRC4 mutations were significantly higher than in NOMID patients, the most severe form of disease associated with NLRP3 gain‐of‐function mutations, despite the fact that both inflammasomes activate caspase 1. Pyroptosis appears to be induced by mutant NLRC4, more than by NLRP3 mutations which are not commonly associated with HLH development in humans.66, 201 Moreover, there is the successful treatment with recombinant human IL‐18BP (tadekinig) of a patient with severe and refractory HLH bearing a NLRC4 mutation. That report demonstrates that IL‐18 inhibition is likely an effective strategy in HLH, at least in this particular genetic background.202
3.7. IL‐18 in systemic juvenile idiopathic arthritis/adult onset Still's Disease
sJIA (and its adult homolog AOSD) constitutes a distinct group of patients among JIA.203 Systemic JIA is clinically characterized by high spiking fever, cutaneous rash, hepato/splenomegaly, serositis, whereas arthritis the main symptom in JIA, could be absent at least at the disease onset. Biologically, sJIA is characterized by severe generalized inflammation, neutrophilia, thrombocytosis, and hyperferritinemia.204 An unusually frequent complication of sJIA is macrophage activation syndrome (MAS), a form of secondary HLH, which occurs in 15% of the patients, whereas hemophagocytosis (“occult MAS”) may be present in the bone marrow of almost 50% of the patients with active sJIA, suggesting a special pathogenic link between these 2 diseases.205
Initial gene expression profile study discovered an IL‐1 signature in sJIA, and other studies have found upregulation of a cluster of genes involved in innate immunity and IL‐1R/TLR signaling.206, 207 Nevertheless, treatments with anakinra (IL‐1 Receptor antagonist) or canakinumab (anti‐IL‐1β monoclonal antibody) are effective in nearly 50% of the patients. Another group of patients, however, with a greater number of involved joints and lower neutrophil count may not or incompletely respond to anakinra, developing persistent synovitis.208 It is also possible to distinguish two different subgroups of sJIA patients depending on their serum cytokine profile, the group with more severe arthritis having higher concentrations of IL‐6.209 Indeed IL‐6 is markedly elevated in sJIA patients and correlates with the severity of joint involvement.210 Inhibition of IL‐6 using the IL‐6 receptor blocking antibody tocilizumab may be an effective treatment in sJIA patients not responding to IL‐1β blockade.211
IL‐18 has been found highly elevated in the serum of active sJIA in numerous studies.212, 213, 214 The concentrations of IL‐18 in sJIA range 5‐10 ng/mL, close to those found in NLRC4 mutations. Elevated IL‐18 concentrations correlate with ferritin concentrations and the occurrence of MAS, with a cut‐off of 47 ng/mL predictive of MAS occurence.209, 213, 215 All but one of these reports studied total IL‐18, however, using a new ELISA able to directly detect free IL‐18, elevation of free IL‐18 was recently confirmed in the serum of sJIA patients.216 IL‐18BP concentrations are increased in sJIA patients compared to healthy controls, but not differently than patients with other auto‐immune diseases, thus out of proportion of IL‐18 increase. The reason for this imbalance may involve epigenetic regulation. miRNA134 which inhibits IL‐18BP synthesis is overexpressed in the PBMC of AOSD patients and miRNA134 concentrations are elevated in the serum of patients with active disease.217 Correcting IL‐18/IL‐18BP imbalance is a rationale strategy, and an open‐label phase 2 trial using IL‐18BP has recently been conducted. AOSD patients were treated and normalization of both CRP and ferritin levels were observed with a dose of 160 mg subcutaneously three times a week (Gabay C, personal communication 2017).
Despite high concentrations of IL‐18, IFNγ is rarely found in patients with active JIA and numerous studies of gene expression failed to detect an IFNγ‐signature in these patients.207, 216, 218, 219 Moreover, in a recently described animal model of sJIA using complete Freund's adjuvant, IFNγ appeared rather protective, since IFNγ‐deficient mice developed a more severe phenotype which was dependent on IL‐17.220 The authors considered IFNγ‐mediated inhibition of Th17 as a possible explanation of this protective effect. It should be noted that IL‐17 has been found elevated in sJIA patients, due to increased IL‐17 positive γδ cells expressing IL‐18Rα whose proliferation is dependent on IL‐1β and IL‐18.42 Another explanation may be the long‐term recognized negative effect of IFNγ on IL‐1β production.214, 221 On the contrary, IFNγ may be found in the serum of sJIA patients complicated by MAS and a decrease IL‐18/IFNγ ratio may characterized MAS appearance.214 However, both IL‐18, sCD163 and HO‐1 concentrations are elevated in active sJIA as well as in MAS and “occult MAS,” corresponding to hemophagocytosis mediated by M2 macrophages expressing CD163. IL‐18 is present in the bone marrow of patients with active sJIA, suggesting that the underlying proliferation of hemophagocytic M2 is part of sJIA and possibly linked to increased IL‐18 concentrations.222
In addition, the links of MAS with primary HLH leads to the search for a common pathogenic pathway. NK cell cytotoxicity was reported to be abnormal in patients with active sJIA with or without MAS, consisting in decreased circulating NK numbers, perforin expression and cytotoxicity.223, 224, 225 In addition, whole exome sequencing reported that heterozygous mutations affecting Munc13.4, Munc 18.2 or Lyst were present in 35% of sJIA patients with MAS, but only in 14% of patients with sJIA without MAS.226 Furthermore, in animal models of sJIA consisting in complete Freund's adjuvant administration in IFNγ KO mice or in LPS‐stimulated mice overexpressing IL‐6, NK cell lymphopenia as well as decreased cytotoxicity was observed.220, 227 A more recent study, however, showed that after normalization of NK percentage, no cytotoxicity deficiency was observed in sJIA patients and only minor NK abnormalities such as increased surface NKp44 expression and decreased granzyme K expression were present.228 Despite these discrepancies, NK cells indeed are dysfunctional during sJIA, a surprising observation in view on the high concentrations of IL‐18 present in this disease. Two reports demonstrated that in sJIA patients NK cells are resistant to the action of IL‐18, due to an absence of phosphorylation of IL‐18Rβ, rendering NK cells incapable to produce IFNγ in response to IL‐18.228, 229 Moreover, some reports have suggested that high IL‐18 concentrations may induce lymphocyte apoptosis and NK cell death possibly explaining the observed NK lymphopenia.230, 231
4. CONCLUSION
As a member of the IL‐1 family, IL‐18 appears to share characteristics of IL‐1β with caspase‐1 processing of the inactive precursor to an active cytokine. But the IL‐18 precursor also shares characteristics with the precursor IL‐1α, as both are constitutively present in healthy mesenchymal tissues and are released following necrotic cell death. IL‐18 acts differently than IL‐1β notably due its effects on lymphocyte activation Through induction of IFNγ, IL‐18 participates in the defense against intracellular bacteria and some virus. Possibly, via IL‐13 and IgE production, it may have a role in the immune response against helminths.
In some inflammatory diseases, it remains unclear whether IL‐18 or IFNγ is causative. Unusually high serum concentrations of free IL‐18 (more than 10 ng/mL) such as NLRC4 mutations or systemic JIA/AOSD implicate a dominant role for IL‐18. Treatment with recombinant IL‐18BP or anti‐IL‐18 is likely to be effective in the treatment of HLH syndrome associated with both of these diseases. In other diseases, such as avian flu, corticosteroid‐resistant asthma, sepsis or other secondary HLH, an INFγ‐biased cytokine storm is found, an imbalance between IL‐18 and IL‐18BP has been described, and the concentrations of free IL‐18 are in the nanogram range. In these diseases, the role of pyroptosis, the levels and the role of IL‐37 should probably be studied in the future, and IL‐18‐targeted therapy possibly considered. In addition, as shown in the heart, some of the pathogenic effects of IL‐1β are mediated by IL‐18, suggesting that IL‐18 could be a target for therapy for heart diseases besides IL‐1 and NLRP3. The presence of IL‐18 in mesenchymal cells, notably keratinocytes and epithelial cells suggest an important role for IL‐18 in local barrier defenses which in some chronic inflammatory diseases may be disrupted.
CONFLICT OF INTEREST
The author declares no conflict of interest.
ACKNOWLEDGEMENTs
G. K. is the recipient of grants from Projet de Recherche Clinique National (#A0‐1109‐44) and Agence Nationale de Recherche‐Maladies Rares (#ANR‐07‐MRAR‐019).
Kaplanski G. Interleukin‐18: Biological properties and role in disease pathogenesis. Immunol Rev. 2018;281:138–153. 10.1111/imr.12616
REFERENCES
- 1. Nakamura K, Okamura H, Nagata K, Komatsu T, Tamura T. Purification of a factor which provides a costimulatory signal for gamma interferon production. Infect Immun. 1993;61:64‐70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Okamura H, Tsutsi H, Komatsu T, et al. Cloning of a new cytokine that induces IFN‐gamma production by T cells. Nature. 1995;378:88‐91. [DOI] [PubMed] [Google Scholar]
- 3. Dinarello CA. IL‐18: A TH1‐inducing, proinflammatory cytokine and new member of the IL‐1 family. J Allergy Clin Immunol. 1999;103:11‐24. [DOI] [PubMed] [Google Scholar]
- 4. Dinarello CA. Interleukin‐18. Methods. 1999;19:121‐132. [DOI] [PubMed] [Google Scholar]
- 5. Nakanishi K, Yoshimoto T, Tsutsui H, Okamura H. Interleukin‐18 regulates both Th1 and Th2 responses. Annu Rev Immunol. 2001;19:423‐474. [DOI] [PubMed] [Google Scholar]
- 6. Dinarello CA, Novick D, Kim S, Kaplanski G. Interleukin‐18 and IL‐18 binding protein. Front Immunol. 2013;4:289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Novick D, Kim S, Kaplanski G, Dinarello CA. Interleukin‐18, more than a Th1 cytokine. Semin Immunol. 2013;25:439‐448. [DOI] [PubMed] [Google Scholar]
- 8. Dinarello CA, Novick D, Puren AJ, et al. Overview of interleukin‐18: more than an interferon‐gamma inducing factor. J Leukoc Biol. 1998;63:658‐664. [PubMed] [Google Scholar]
- 9. Gu Y, Kuida K, Tsutsui H, et al. Activation of interferon‐gamma inducing factor mediated by interleukin‐1beta converting enzyme. Science. 1997;275:206‐209. [DOI] [PubMed] [Google Scholar]
- 10. Ghayur T, Banerjee S, Hugunin M, et al. Caspase‐1 processes IFN‐gamma‐inducing factor and regulates LPS‐induced IFN‐gamma production. Nature. 1997;386:619‐623. [DOI] [PubMed] [Google Scholar]
- 11. Jorgensen I, Miao EA. Pyroptotic cell death defends against intracellular pathogens. Immunol Rev. 2015;265:130‐142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Tsutsui H, Kayagaki N, Kuida K, et al. Caspase‐1‐independent, Fas/Fas ligand‐mediated IL‐18 secretion from macrophages causes acute liver injury in mice. Immunity. 1999;11:359‐367. [DOI] [PubMed] [Google Scholar]
- 13. Bossaller L, Chiang PI, Schmidt‐Lauber C, et al. Cutting edge: FAS (CD95) mediates noncanonical IL‐1β and IL‐18 maturation via caspase‐8 in an RIP3‐independent manner. J Immunol. 2012;189:5508‐5512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Akita K, Ohtsuki T, Nukada Y, et al. Involvement of caspase‐1 and caspase‐3 in the production and processing of mature human interleukin 18 in monocytic THP.1 cells. J Biol Chem. 1997;272:26595‐26603. [DOI] [PubMed] [Google Scholar]
- 15. Omoto Y, Yamanaka K, Tokime K, et al. Granzyme B is a novel interleukin‐18 converting enzyme. J Dermatol Sci. 2010;59:129‐135. [DOI] [PubMed] [Google Scholar]
- 16. Omoto Y, Tokime K, Yamanaka K, et al. Human mast cell chymase cleaves pro‐IL‐18 and generates a novel and biologically active IL‐18 fragment. J Immunol. 2006;177:8315‐8319. [DOI] [PubMed] [Google Scholar]
- 17. Banerjee S, Bond JS. Prointerleukin‐18 is activated by meprin beta in vitro and in vivo in intestinal inflammation. J Biol Chem. 2008;283:31371‐31377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Sugawara S, Uehara A, Nochi T, et al. Neutrophil proteinase 3‐mediated induction of bioactive IL‐18 secretion by human oral epithelial cells. J Immunol. 2001;167:6568‐6575. [DOI] [PubMed] [Google Scholar]
- 19. Torigoe K, Ushio S, Okura T, et al. Purification and characterization of the human interleukin‐18 receptor. J Biol Chem. 1997;272:25737‐25742. [DOI] [PubMed] [Google Scholar]
- 20. Hoshino K, Tsutsui H, Kawai T, et al. Cutting edge: Generation of IL‐18 receptor‐deficient mice: Evidence for IL‐1 receptor‐related protein as an essential IL‐18 binding receptor. J Immunol. 1999;162:5041‐5044. [PubMed] [Google Scholar]
- 21. Dinarello CA. Interleukin‐1, interleukin‐1 receptors and interleukin‐1 receptor antagonist. Int Rev Immunol. 1998;16:457‐499. [DOI] [PubMed] [Google Scholar]
- 22. Lee JK, Kim SH, Lewis EC, Azam T, Reznikov LL, Dinarello CA. Differences in signaling pathways by IL‐1beta and IL‐18. Proc Natl Acad Sci USA. 2004;101:8815‐8820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Kalina U, Kauschat D, Koyama N, et al. IL‐18 activates STAT3 in the natural killer cell line 92, augments cytotoxic activity, and mediates IFN‐gamma production by the stress kinase p38 and by the extracellular regulated kinases p44erk‐1 and p42erk‐21. J Immunol. 2000;165:1307‐1313. [DOI] [PubMed] [Google Scholar]
- 24. Alboni S, Montanari C, Benatti C, et al. Interleukin 18 activates MAPKs and STAT3 but not NF‐κB in hippocampal HT‐22 cells. Brain Behav Immun. 2014;40:85‐94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Wyman TH, Dinarello CA, Banerjee A, et al. Physiological levels of interleukin‐18 stimulate multiple neutrophil functions through p38 MAP kinase activation. J Leukoc Biol. 2002;72:401‐409. [PubMed] [Google Scholar]
- 26. Bufler P, Azam T, Gamboni‐Robertson F, et al. A complex of the IL‐1 homologue IL‐1F7b and IL‐18‐binding protein reduces IL‐18 activity. Proc Natl Acad Sci USA. 2002;99:13723‐13728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Nold MF, Nold‐Petry CA, Zepp JA, Palmer BE, Bufler P, Dinarello CA. IL‐37 is a fundamental inhibitor of innate immunity. Nat Immunol. 2010;11:1014‐1022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Lewis EC, Dinarello CA. Responses of IL‐18‐ and IL‐18 receptor‐deficient pancreatic islets with convergence of positive and negative signals for the IL‐18 receptor. Proc Natl Acad Sci USA. 2006;103:16852‐16857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Nold‐Petry CA, Nold MF, Nielsen JW, et al. Increased cytokine production in interleukin‐18 receptor alpha‐deficient cells is associated with dysregulation of suppressors of cytokine signaling. J Biol Chem. 2009;284:25900‐25911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Nold‐Petry CA, Lo CY, Rudloff I, et al. IL‐37 requires the receptors IL‐18Rα and IL‐1R8 (SIGIRR) to carry out its multifaceted anti‐inflammatory program upon innate signal transduction. Nat Immunol. 2015;16:354‐365. [DOI] [PubMed] [Google Scholar]
- 31. Li S, Neff CP, Barber K, Hong J, et al. Extracellular forms of IL‐37 inhibit innate inflammation in vitro and in vivo but require the IL‐1 family decoy receptor IL‐1R8. Proc Natl Acad Sci USA. 2015;112:2497‐2502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. McNamee EN, Masterson JC, Jedlicka P, et al. Interleukin 37 expression protects mice from colitis. Proc Natl Acad Sci USA. 2011;108:16711‐16716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Fantuzzi G, Dinarello CA. Interleukin‐18 and interleukin‐1 beta: Two cytokine substrates for ICE (caspase‐1). J Clin Immunol. 1999;19:1‐11. [DOI] [PubMed] [Google Scholar]
- 34. Tsutsui H, Nakanishi K, Matsui K, et al. IFN‐gamma‐inducing factor up‐regulates Fas ligand‐mediated cytotoxic activity of murine natural killer cell clones. J Immunol. 1996;157:3967‐3973. [PubMed] [Google Scholar]
- 35. Bellora F, Castriconi R, Doni A, et al. M‐CSF induces the expression of a membrane‐bound form of IL‐18 in a subset of human monocytes differentiating in vitro toward macrophages. Eur J Immunol. 2012;42:1618‐1626. [DOI] [PubMed] [Google Scholar]
- 36. Munder M, Mallo M, Eichmann K, Modolell M. Murine macrophages secrete interferon gamma upon combined stimulation with interleukin (IL)‐12 and IL‐18: A novel pathway of autocrine macrophage activation. J Exp Med. 1998;187:2103‐2108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Hoshino T, Wiltrout RH, Young HA. IL‐18 is a potent coinducer of IL‐13 in NK and T cells: A new potential role for IL‐18 in modulating the immune response. J Immunol. 1999;162:5070‐5077. [PubMed] [Google Scholar]
- 38. Yoshimoto T, Mizutani H, Tsutsui H, et al. IL‐18 induction of IgE: Dependence on CD4+ T cells, IL‐4 and STAT6. Nat Immunol. 2000;1:132‐137. [DOI] [PubMed] [Google Scholar]
- 39. Yoshimoto T, Tsutsui H, Tominaga K, et al. IL‐18, although antiallergic when administered with IL‐12, stimulates IL‐4 and histamine release by basophils. Proc Natl Acad Sci USA. 1999;96:13962‐13966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Hitzler I, Sayi A, Kohler E, et al. Caspase‐1 has both proinflammatory and regulatory properties in Helicobacter infections, which are differentially mediated by its substrates IL‐1β and IL‐18. J Immunol. 2012;188:3594‐3602. [DOI] [PubMed] [Google Scholar]
- 41. Sutton CE, Lalor SJ, Sweeney CM, Brereton CF, Lavelle EC, Mills KH. Interleukin‐1 and IL‐23 induce innate IL‐17 production from gammadelta T cells, amplifying Th17 responses and autoimmunity. Immunity. 2009;31:331‐341. [DOI] [PubMed] [Google Scholar]
- 42. Kessel C, Lippitz K, Weinhage T, et al. Proinflammatory cytokine environments can drive interleukin‐17 overexpression by γ/δ T cells in systemic juvenile idiopathic arthritis. Arthritis Rheumatol. 2017;69:1480‐1494. [DOI] [PubMed] [Google Scholar]
- 43. Wynn JL, Wilson CS, Hawiger J, et al. Targeting IL‐17A attenuates neonatal sepsis mortality induced by IL‐18. Proc Natl Acad Sci USA. 2016;113:E2627‐E2635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Oertli M, Sundquist M, Hitzler I, et al. DC‐derived IL‐18 drives Treg differentiation, murine Helicobacter pylori‐specific immune tolerance, and asthma protection. J Clin Invest. 2012;122:1082‐1096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Kohka H, Yoshino T, Iwagaki H, et al. Interleukin‐18/interferon‐gamma‐inducing factor, a novel cytokine, up‐regulates ICAM‐1 (CD54) expression in KG‐1 cells. J Leukoc Biol. 1998;64:519‐527. [DOI] [PubMed] [Google Scholar]
- 46. Morel JC, Park CC, Woods JM, Koch AE. A novel role for interleukin‐18 in adhesion molecule induction through NF kappa B and phosphatidylinositol (PI)3‐kinase‐dependent signal transduction pathways. J Biol Chem. 2001;276:37069‐37075. [DOI] [PubMed] [Google Scholar]
- 47. Vidal‐Vanaclocha F, Fantuzzi G, Mendoza L, et al. IL‐18 regulates IL‐1beta‐dependent hepatic melanoma metastasis via vascular cell adhesion molecule‐1. Proc Natl Acad Sci USA. 2000;97:734‐739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Puren AJ, Fantuzzi G, Gu Y, Su MS, Dinarello CA. Interleukin‐18 (IFNgamma‐inducing factor) induces IL‐8 and IL‐1beta via TNFalpha production from non‐CD14 + human blood mononuclear cells. J Clin Invest. 1998;101:711‐721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Morel JC, Park CC, Kumar P, Koch AE. Interleukin‐18 induces rheumatoid arthritis synovial fibroblast CXC chemokine production through NFkappaB activation. Lab Invest. 2001;81:1371‐1383. [DOI] [PubMed] [Google Scholar]
- 50. Amin MA, Mansfield PJ, Pakozdi A, et al. Interleukin‐18 induces angiogenic factors in rheumatoid arthritis synovial tissue fibroblasts via distinct signaling pathways. Arthritis Rheum. 2007;56:1787‐1797. [DOI] [PubMed] [Google Scholar]
- 51. Tsutsui H, Matsui K, Okamura H, Nakanishi K. Pathophysiological roles of interleukin‐18 in inflammatory liver diseases. Immunol Rev. 2000;174:192‐209. [DOI] [PubMed] [Google Scholar]
- 52. Gatti S, Beck J, Fantuzzi G, Bartfai T, Dinarello CA. Effect of interleukin‐18 on mouse core body temperature. Am J Physiol Regul Integr Comp Physiol. 2002;282:R702‐R709. [DOI] [PubMed] [Google Scholar]
- 53. Novick D, Kim SH, Fantuzzi G, Reznikov LL, Dinarello CA, Rubinstein M. Interleukin‐18 binding protein: A novel modulator of the Th1 cytokine response. Immunity. 1999;10:127‐136. [DOI] [PubMed] [Google Scholar]
- 54. Kim SH, Eisenstein M, Reznikov L, et al. Structural requirements of six naturally occurring isoforms of the IL‐18 binding protein to inhibit IL‐18. Proc Natl Acad Sci USA. 2000;97:1190‐1195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Novick D, Schwartsburd B, Pinkus R, et al. A novel IL‐18BP ELISA shows elevated serum IL‐18BP in sepsis and extensive decrease of free IL‐18. Cytokine. 2001;14:334‐342. [DOI] [PubMed] [Google Scholar]
- 56. Mazodier K, Marin V, Novick D, et al. Severe imbalance of IL‐18/IL‐18BP in patients with secondary hemophagocytic syndrome. Blood. 2005;106:3483‐3489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Banda NK, Vondracek A, Kraus D, et al. Mechanisms of inhibition of collagen‐induced arthritis by murine IL‐18 binding protein. J Immunol. 2003;170:2100‐2105. [DOI] [PubMed] [Google Scholar]
- 58. Mühl H, Kämpfer H, Bosmann M, Frank S, Radeke H, Pfeilschifter J. Interferon‐gamma mediates gene expression of IL‐18 binding protein in nonleukocytic cells. Biochem Biophys Res Commun. 2000;267:960‐963. [DOI] [PubMed] [Google Scholar]
- 59. Hurgin V, Novick D, Rubinstein M. The promoter of IL‐18 binding protein: Activation by an IFN‐gamma ‐induced complex of IFN regulatory factor 1 and CCAAT/enhancer binding protein beta. Proc Natl Acad Sci USA. 2002;99:16957‐16962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Ludwiczek O, Kaser A, Novick D, et al. Plasma levels of interleukin‐18 and interleukin‐18 binding protein are elevated in patients with chronic liver disease. J Clin Immunol. 2002;22:331‐337. [DOI] [PubMed] [Google Scholar]
- 61. Ringel‐Scaia VM, McDaniel DK, Allen IC. The goldilocks conundrum: NLR inflammasome modulation of gastrointestinal inflammation during inflammatory bowel disease. Crit Rev Immunol. 2016;36:283‐314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Naftali T, Novick D, Gabay G, Rubinstein M, Novis B. Interleukin‐18 and its binding protein in patients with inflammatory bowel disease during remission and exacerbation. Isr Med Assoc J. 2007;9:504‐508. [PubMed] [Google Scholar]
- 63. Leach ST, Messina I, Lemberg DA, Novick D, Rubenstein M, Day AS. Local and systemic interleukin‐18 and interleukin‐18‐binding protein in children with inflammatory bowel disease. Inflamm Bowel Dis. 2008;14:68‐74. [DOI] [PubMed] [Google Scholar]
- 64. Monteleone G, Trapasso F, Parrello T, et al. Bioactive IL‐18 expression is up‐regulated in Crohn's disease. J Immunol. 1999;163:143‐147. [PubMed] [Google Scholar]
- 65. Pizarro TT, Michie MH, Bentz M, et al. IL‐18, a novel immunoregulatory cytokine, is up‐regulated in Crohn's disease: Expression and localization in intestinal mucosal cells. J Immunol. 1999;162:6829‐6835. [PubMed] [Google Scholar]
- 66. Canna SW, de Jesus AA, Gouni S, et al. An activating NLRC4 inflammasome mutation causes autoinflammation with recurrent macrophage activation syndrome. Nat Genet. 2014;46:1140‐1146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Ishikura T, Kanai T, Uraushihara K, et al. Interleukin‐18 overproduction exacerbates the development of colitis with markedly infiltrated macrophages in interleukin‐18 transgenic mice. J Gastroenterol Hepatol. 2003;18:960‐969. [DOI] [PubMed] [Google Scholar]
- 68. Siegmund B, Fantuzzi G, Rieder F, et al. Neutralization of interleukin‐18 reduces severity in murine colitis and intestinal IFN‐gamma and TNF‐alpha production. Am J Physiol Regul Integr Comp Physiol. 2001;281:R1264‐R1273. [DOI] [PubMed] [Google Scholar]
- 69. Kanai T, Watanabe M, Okazawa A, et al. Macrophage‐derived IL‐18‐mediated intestinal inflammation in the murine model of Crohn's disease. Gastroenterology. 2001;121:875‐888. [DOI] [PubMed] [Google Scholar]
- 70. Ten Hove T, Corbaz A, Amitai H, et al. Blockade of endogenous IL‐18 ameliorates TNBS‐induced colitis by decreasing local TNF‐alpha production in mice. Gastroenterology. 2001;121:1372‐1379. [DOI] [PubMed] [Google Scholar]
- 71. Sivakumar PV, Westrich GM, Kanaly S, et al. Interleukin 18 is a primary mediator of the inflammation associated with dextran sulphate sodium induced colitis: Blocking interleukin 18 attenuates intestinal damage. Gut. 2002;50:812‐820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Nowarski R, Jackson R, Gagliani N, et al. Epithelial IL‐18 equilibrium controls barrier function in colitis. Cell. 2015;163:1444‐1456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Bauer C, Duewell P, Mayer C, et al. Colitis induced in mice with dextran sulfate sodium (DSS) is mediated by the NLRP3 inflammasome. Gut. 2010;59:1192‐1199. [DOI] [PubMed] [Google Scholar]
- 74. Siegmund B, Lehr HA, Fantuzzi G, Dinarello CA. IL‐1 beta ‐converting enzyme (caspase‐1) in intestinal inflammation. Proc Natl Acad Sci USA. 2001;98:13249‐13254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Neudecker V, Haneklaus M, Jensen O, et al. Myeloid‐derived miR‐223 regulates intestinal inflammation via repression of the NLRP3 inflammasome. J Exp Med. 2017;214:1737‐1752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Villani AC, Lemire M, Fortin G, et al. Common variants in the NLRP3 region contribute to Crohn's disease susceptibility. Nat Genet. 2009;41:71‐76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Takagawa T, Tamura K, Takeda N, et al. Association between IL‐18 gene promoter polymorphisms and inflammatory bowel disease in a Japanese population. Inflamm Bowel Dis. 2005;11:1038‐1043. [DOI] [PubMed] [Google Scholar]
- 78. Zhernakova A, Festen EM, Franke L, et al. Genetic analysis of innate immunity in Crohn's disease and ulcerative colitis identifies two susceptibility loci harboring CARD9 and IL18RAP. Am J Hum Genet. 2008;82:1202‐1210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Zaki MH, Boyd KL, Vogel P, Kastan MB, Lamkanfi M, Kanneganti TD. The NLRP3 inflammasome protects against loss of epithelial integrity and mortality during experimental colitis. Immunity. 2010;32:379‐391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Dupaul‐Chicoine J, Yeretssian G, Doiron K, et al. Control of intestinal homeostasis, colitis, and colitis‐associated colorectal cancer by the inflammatory caspases. Immunity. 2010;32:367‐378. [DOI] [PubMed] [Google Scholar]
- 81. Levy M, Shapiro H, Thaiss CA, Elinav E. NLRP6: A multifaceted innate immune sensor. Trends Immunol. 2017;38:248‐260. [DOI] [PubMed] [Google Scholar]
- 82. Levy M, Thaiss CA, Zeevi D, et al. Microbiota‐modulated metabolites shape the intestinal microenvironment by regulating NLRP6 inflammasome signaling. Cell. 2015;163:1428‐1443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Elinav E, Strowig T, Kau AL, et al. NLRP6 inflammasome regulates colonic microbial ecology and risk for colitis. Cell. 2011;145:745‐757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Wlodarska M, Thaiss CA, Nowarski R, et al. NLRP6 inflammasome orchestrates the colonic host‐microbial interface by regulating goblet cell mucus secretion. Cell. 2014;156:1045‐1059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Bamias G, Corridoni D, Pizarro TT, Cominelli F. New insights into the dichotomous role of innate cytokines in gut homeostasis and inflammation. Cytokine. 2012;59:451‐459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Lopetuso LR, Chowdhry S, Pizarro TT. Opposing functions of classic and novel IL‐1 family members in gut health and disease. Front Immunol. 2013;4:181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Harrison OJ, Srinivasan N, Pott J, et al. Epithelial‐derived IL‐18 regulates Th17 cell differentiation and Foxp3⁺ Treg cell function in the intestine. Mucosal Immunol. 2015;8:1226‐1236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Siegmund B. Interleukin‐18 in intestinal inflammation: Friend and foe? Immunity. 2010;32:300‐302. [DOI] [PubMed] [Google Scholar]
- 89. Hand TW. Interleukin‐18: The bouncer at the mucosal bar. Cell. 2015;163:1310‐1312. [DOI] [PubMed] [Google Scholar]
- 90. Bersudsky M, Luski L, Fishman D, et al. Non‐redundant properties of IL‐1α and IL‐1β during acute colon inflammation in mice. Gut. 2014;63:598‐609. [DOI] [PubMed] [Google Scholar]
- 91. Peyrin‐Biroulet L, Fiorino G, Buisson A, Danese S. First‐line therapy in adult Crohn's disease: Who should receive anti‐TNF agents? Nat Rev Gastroenterol Hepatol. 2013;10:345‐351. [DOI] [PubMed] [Google Scholar]
- 92. Niederreiter L, Adolph TE, Kaser A. Anti‐IL‐12/23 in Crohn's disease: Bench and bedside. Curr Drug Targets. 2013;14:1379‐1384. [DOI] [PubMed] [Google Scholar]
- 93. Eckel RH, Grundy SM, Zimmet PZ. The metabolic syndrome. Lancet. 2005;365:1415‐1428. [DOI] [PubMed] [Google Scholar]
- 94. Grundy SM, Cleeman JI, Daniels SR, et al. ; American Heart Association; National Heart, Lung, and Blood Institute . Diagnosis and management of the metabolic syndrome: An American Heart Association/National Heart, Lung, and Blood Institute Scientific Statement. Circulation. 2005;11:2735‐2752. [DOI] [PubMed] [Google Scholar]
- 95. Dinarello CA, Donath MY, Mandrup‐Poulsen T. Role of IL‐1beta in type 2 diabetes. Curr Opin Endocrinol Diabetes Obes. 2010;17:314‐321. [DOI] [PubMed] [Google Scholar]
- 96. Hung J, McQuillan BM, Chapman CM, Thompson PL, Beilby JP. Elevated interleukin‐18 levels are associated with the metabolic syndrome independent of obesity and insulin resistance. Arterioscler Thromb Vasc Biol. 2005;25:1268‐1273. [DOI] [PubMed] [Google Scholar]
- 97. Zirlik A, Abdullah SM, Gerdes N, et al. Interleukin‐18, the metabolic syndrome, and subclinical atherosclerosis: Results from the Dallas Heart Study. Arterioscler Thromb Vasc Biol. 2007;27:2043‐2049. [DOI] [PubMed] [Google Scholar]
- 98. Fischer CP, Perstrup LB, Berntsen A, Eskildsen P, Pedersen BK. Elevated plasma interleukin‐18 is a marker of insulin‐resistance in type 2 diabetic and non‐diabetic humans. Clin Immunol. 2005;117:152‐160. [DOI] [PubMed] [Google Scholar]
- 99. Presta I, Andreozzi F, Succurro E, et al. IL‐18 gene polymorphism and metabolic syndrome. Nutr Metab Cardiovasc Dis. 2009;19:e5‐e6. [DOI] [PubMed] [Google Scholar]
- 100. Bruun JM, Stallknecht B, Helge JW, Richelsen B. Interleukin‐18 in plasma and adipose tissue: Effects of obesity, insulin resistance, and weight loss. Eur J Endocrinol. 2007;157:465‐471. [DOI] [PubMed] [Google Scholar]
- 101. Skurk T, Kolb H, Müller‐Scholze S, Röhrig K, Hauner H, Herder C. The proatherogenic cytokine interleukin‐18 is secreted by human adipocytes. Eur J Endocrinol. 2005;152:863‐868. [DOI] [PubMed] [Google Scholar]
- 102. Schernthaner GH, Kopp HP, Kriwanek S, et al. Effect of massive weight loss induced by bariatric surgery on serum levels of interleukin‐18 and monocyte‐chemoattractant‐protein‐1 in morbid obesity. Obes Surg. 2006;16:709‐715. [DOI] [PubMed] [Google Scholar]
- 103. Netea MG, Joosten LA, Lewis E, et al. Deficiency of interleukin‐18 in mice leads to hyperphagia, obesity and insulin resistance. Nat Med. 2006;12:650‐656. [DOI] [PubMed] [Google Scholar]
- 104. Zorrilla EP, Sanchez‐Alavez M, Sugama S, et al. Interleukin‐18 controls energy homeostasis by suppressing appetite and feed efficiency. Proc Natl Acad Sci USA. 2007;104:11097‐11102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. Lindegaard B, Matthews VB, Brandt C, et al. Interleukin‐18 activates skeletal muscle AMPK and reduces weight gain and insulin resistance in mice. Diabetes. 2013;62:3064‐3074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106. Zilverschoon GR, Tack CJ, Joosten LA, Kullberg BJ, van der Meer JW, Netea MG. Interleukin‐18 resistance in patients with obesity and type 2 diabetes mellitus. Int J Obes (Lond). 2008;32:1407‐1414. [DOI] [PubMed] [Google Scholar]
- 107. Trøseid M, Seljeflot I, Arnesen H. The role of interleukin‐18 in the metabolic syndrome. Cardiovasc Diabetol. 2010;9:11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108. Ballak DB, Stienstra R, Tack CJ, Dinarello CA, van Diepen JA. IL‐1 family members in the pathogenesis and treatment of metabolic disease: Focus on adipose tissue inflammation and insulin resistance. Cytokine. 2015;75:280‐290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109. McKie EA, Reid JL, Mistry PC, et al. A study to investigate the efficacy and safety of an anti‐interleukin‐18 monoclonal antibody in the treatment of type 2 diabetes mellitus. PLoS ONE. 2016;11:e0150018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110. Duewell P, Kono H, Rayner KJ, et al. NLRP3 inflammasomes are required for atherogenesis and activated by cholesterol crystals. Nature. 2010;464:1357‐1361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111. Gerdes N, Sukhova GK, Libby P, Reynolds RS, Young JL, Schönbeck U. Expression of interleukin (IL)‐18 and functional IL‐18 receptor on human vascular endothelial cells, smooth muscle cells, and macrophages: Implications for atherogenesis. J Exp Med. 2002;195:245‐257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112. Mallat Z, Corbaz A, Scoazec A, et al. Expression of interleukin‐18 in human atherosclerotic plaques and relation to plaque instability. Circulation. 2001;104:1598‐1603. [DOI] [PubMed] [Google Scholar]
- 113. Mallat Z, Corbaz A, Scoazec A, et al. Interleukin‐18/interleukin‐18 binding protein signaling modulates atherosclerotic lesion development and stability. Circ Res. 2001;89:E41‐E45. [DOI] [PubMed] [Google Scholar]
- 114. Whitman SC, Ravisankar P, Daugherty A. Interleukin‐18 enhances atherosclerosis in apolipoprotein E(‐/‐) mice through release of interferon‐gamma. Circ Res. 2002;90:E34‐E38. [DOI] [PubMed] [Google Scholar]
- 115. Elhage R, Jawien J, Rudling M, et al. Reduced atherosclerosis in interleukin‐18 deficient apolipoprotein E‐knockout mice. Cardiovasc Res. 2003;59:234‐240. [DOI] [PubMed] [Google Scholar]
- 116. Badimon L. Interleukin‐18: A potent pro‐inflammatory cytokine in atherosclerosis. Cardiovasc Res. 2012;96:172‐175; discussion 176‐80. [DOI] [PubMed] [Google Scholar]
- 117. Blankenberg S, Tiret L, Bickel C, et al. Interleukin‐18 is a strong predictor of cardiovascular death in stable and unstable angina. Circulation. 2002;106:24‐30. [DOI] [PubMed] [Google Scholar]
- 118. Blankenberg S, Luc G, Ducimetière P, et al. nterleukin‐18 and the risk of coronary heart disease in European men: The Prospective Epidemiological Study of Myocardial Infarction (PRIME). Circulation. 2003;108:2453‐2459. [DOI] [PubMed] [Google Scholar]
- 119. Hulthe J, McPheat W, Samnegård A, Tornvall P, Hamsten A, Eriksson P. Plasma interleukin (IL)‐18 concentrations is elevated in patients with previous myocardial infarction and related to severity of coronary atherosclerosis independently of C‐reactive protein and IL‐6. Atherosclerosis. 2006;188:450‐454. [DOI] [PubMed] [Google Scholar]
- 120. Chapman CM, McQuillan BM, Beilby JP, Thompson PL, Hung J. Interleukin‐18 levels are not associated with subclinical carotid atherosclerosis in a community population. The Perth Carotid Ultrasound Disease Assessment Study (CUDAS). Atherosclerosis. 2006;189:414‐419. [DOI] [PubMed] [Google Scholar]
- 121. Seta Y, Kanda T, Tanaka T, et al. Interleukin 18 in acute myocardial infarction. Heart. 2000;84:668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122. Jefferis BJ, Whincup PH, Welsh P, et al. Prospective study of IL‐18 and risk of MI and stroke in men and women aged 60‐79 years: A nested case‐control study. Cytokine. 2013;61:513‐520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123. Seta Y, Kanda T, Tanaka T, et al. Interleukin‐18 in patients with congestive heart failure: Induction of atrial natriuretic peptide gene expression. Res Commun Mol Pathol Pharmacol. 2000;108:87‐95. [PubMed] [Google Scholar]
- 124. Mallat Z, Heymes C, Corbaz A, et al. Evidence for altered interleukin 18 (IL)‐18 pathway in human heart failure. FASEB J. 2004;18:1752‐1754. [DOI] [PubMed] [Google Scholar]
- 125. Woldbaek PR, Tønnessen T, Henriksen UL, et al. Increased cardiac IL‐18 mRNA, pro‐IL‐18 and plasma IL‐18 after myocardial infarction in the mouse; a potential role in cardiac dysfunction. Cardiovasc Res. 2003;59:122‐131. [DOI] [PubMed] [Google Scholar]
- 126. Venkatachalam K, Prabhu SD, Reddy VS, Boylston WH, Valente AJ, Chandrasekar B. Neutralization of interleukin‐18 ameliorates ischemia/reperfusion‐induced myocardial injury. J Biol Chem. 2009;284:7853‐7865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127. Wang M, Tan J, Wang Y, Meldrum KK, Dinarello CA, Meldrum DR. IL‐18 binding protein‐expressing mesenchymal stem cells improve myocardial protection after ischemia or infarction. Proc Natl Acad Sci USA. 2009;106:17499‐17504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128. Chandrasekar B, Mummidi S, Claycomb WC, Mestril R, Nemer M. Interleukin‐18 is a pro‐hypertrophic cytokine that acts through a phosphatidylinositol 3‐kinase‐phosphoinositide‐dependent kinase‐1‐Akt‐GATA4 signaling pathway in cardiomyocytes. J Biol Chem. 2005;280:4553‐4567. [DOI] [PubMed] [Google Scholar]
- 129. Woldbaek PR, Sande JB, Strømme TA, et al. Daily administration of interleukin‐18 causes myocardial dysfunction in healthy mice. Am J Physiol Heart Circ Physiol. 2005;289:H708‐H714. [DOI] [PubMed] [Google Scholar]
- 130. Pomerantz BJ, Reznikov LL, Harken AH, Dinarello CA. Inhibition of caspase 1 reduces human myocardial ischemic dysfunction via inhibition of IL‐18 and IL‐1beta. Proc Natl Acad Sci USA. 2001;98:2871‐2876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131. Toldo S, Mezzaroma E, O'Brien L, et al. Interleukin‐18 mediates interleukin‐1‐induced cardiac dysfunction. Am J Physiol Heart Circ Physiol. 2014;306:H1025‐H1031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132. Platis A, Yu Q, Moore D, Khojeini E, Tsau P, Larson D. The effect of daily administration of IL‐18 on cardiac structure and function. Perfusion. 2008;23:237‐242. [DOI] [PubMed] [Google Scholar]
- 133. Yu Q, Vazquez R, Khojeini EV, Patel C, Venkataramani R, Larson DF. IL‐18 induction of osteopontin mediates cardiac fibrosis and diastolic dysfunction in mice. Am J Physiol Heart Circ Physiol. 2009;297:H76‐H85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134. Harada M, Obara K, Hirota T, et al. A functional polymorphism in IL‐18 is associated with severity of bronchial asthma. Am J Respir Crit Care Med. 2009;180:1048‐1055. [DOI] [PubMed] [Google Scholar]
- 135. Zhu G, Whyte MK, Vestbo J, et al. Interleukin 18 receptor 1 gene polymorphisms are associated with asthma. Eur J Hum Genet. 2008;16:1083‐1090. [DOI] [PubMed] [Google Scholar]
- 136. Tanaka H, Miyazaki N, Oashi K, et al. IL‐18 might reflect disease activity in mild and moderate asthma exacerbation. J Allergy Clin Immunol. 2001;107:331‐336. [DOI] [PubMed] [Google Scholar]
- 137. Imaoka H, Gauvreau GM, Watson RM, et al. Interleukin‐18 and interleukin‐18 receptor‐α expression in allergic asthma. Eur Respir J. 2011;38:981‐983. [DOI] [PubMed] [Google Scholar]
- 138. Oda H, Kawayama T, Imaoka H, et al. Interleukin‐18 expression, CD8(+) T cells, and eosinophils in lungs of nonsmokers with fatal asthma. Ann Allergy Asthma Immunol. 2014;112:23‐28. [DOI] [PubMed] [Google Scholar]
- 139. Kumano K, Nakao A, Nakajima H, et al. Interleukin‐18 enhances antigen‐induced eosinophil recruitment into the mouse airways. Am J Respir Crit Care Med. 1999;160:873‐878. [DOI] [PubMed] [Google Scholar]
- 140. Sawada M, Kawayama T, Imaoka H, et al. IL‐18 induces airway hyperresponsiveness and pulmonary inflammation via CD4+ T cell and IL‐13. PLoSOne. 2013;8:e54623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141. Hogg JC, Chu F, Utokaparch S, et al. The nature of small‐airway obstruction in chronic obstructive pulmonary disease. N Engl J Med. 2004;350:2645‐2653. [DOI] [PubMed] [Google Scholar]
- 142. Sopori M. Effects of cigarette smoke on the immune system. Nat Rev Immunol. 2002;2:372‐377. [DOI] [PubMed] [Google Scholar]
- 143. Kang MJ, Homer RJ, Gallo A, et al. IL‐18 is induced and IL‐18 receptor alpha plays a critical role in the pathogenesis of cigarette smoke‐induced pulmonary emphysema and inflammation. J Immunol. 2007;178:1948‐1959. [DOI] [PubMed] [Google Scholar]
- 144. Imaoka H, Hoshino T, Takei S, et al. Interleukin‐18 production and pulmonary function in COPD. Eur Respir J. 2008;31:287‐297. [DOI] [PubMed] [Google Scholar]
- 145. Wang J, Liu X, Xie M, Xie J, Xiong W, Xu Y. Increased expression of interleukin‐18 and its receptor in peripheral blood of patients with chronic obstructive pulmonary disease. COPD. 2012;9:375‐381. [DOI] [PubMed] [Google Scholar]
- 146. Kang MJ, Lee CG, Lee JY, et al. Cigarette smoke selectively enhances viral PAMP‐ and virus‐induced pulmonary innate immune and remodeling responses in mice. J Clin Invest. 2008;118:2771‐2784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147. Kratzer A, Salys J, Nold‐Petry C, et al. Role of IL‐18 in second‐hand smoke‐induced emphysema. Am J Respir Cell Mol Biol. 2013;48:725‐732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148. Hoshino T, Kato S, Oka N, et al. Pulmonary inflammation and emphysema: Role of the cytokines IL‐18 and IL‐13. Am J Respir Crit Care Med. 2007;176:49‐62. [DOI] [PubMed] [Google Scholar]
- 149. Kang MJ, Choi JM, Kim BH, et al. IL‐18 induces emphysema and airway and vascular remodeling via IFN‐γ, IL‐17A, and IL‐13. Am J Respir Crit Care Med. 2012;185:1205‐1217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150. Pinkerton JW, Kim RY, Robertson AAB, et al. Inflammasomes in the lung. Mol Immunol. 2017;86:44‐55. [DOI] [PubMed] [Google Scholar]
- 151. Butt Y, Kurdowska A, Allen TC. Acute lung injury: A clinical and molecular review. Arch Pathol Lab Med. 2016;140:345‐350. [DOI] [PubMed] [Google Scholar]
- 152. Dolinay T, Kim YS, Howrylak J, et al. Inflammasome‐regulated cytokines are critical mediators of acute lung injury. Am J Respir Crit Care Med. 2012;185:1225‐1234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153. Makabe H, Kojika M, Takahashi G, et al. Interleukin‐18 levels reflect the long‐term prognosis of acute lung injury and acute respiratory distress syndrome. J Anesth. 2012;26:658‐663. [DOI] [PubMed] [Google Scholar]
- 154. Allen IC, Scull MA, Moore CB, et al. The NLRP3 inflammasome mediates in vivo innate immunity to influenza A virus through recognition of viral RNA. Immunity. 2009;30:556‐565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155. Thomas PG, Dash P, Aldridge JR Jr, et al. The intracellular sensor NLRP3 mediates key innate and healing responses to influenza A virus via the regulation of caspase‐1. Immunity. 2009;30:566‐575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156. Liu B, Mori I, Hossain MJ, Dong L, Takeda K, Kimura Y. Interleukin‐18 improves the early defence system against influenza virus infection by augmenting natural killer cell‐mediated cytotoxicity. J Gen Virol. 2004;85:423‐428. [DOI] [PubMed] [Google Scholar]
- 157. Van Der Sluijs KF, Van Elden LJ, Arens R, et al. Enhanced viral clearance in interleukin‐18 gene‐deficient mice after pulmonary infection with influenza A virus. Immunology. 2005;114:112‐120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158. Denton AE, Doherty PC, Turner SJ, La Gruta NL. IL‐18, but not IL‐12, is required for optimal cytokine production by influenza virus‐specific CD8+ T cells. Eur J Immunol. 2007;37:368‐375. [DOI] [PubMed] [Google Scholar]
- 159. McAuley JL, Tate MD, MacKenzie‐Kludas CJ, et al. Activation of the NLRP3 inflammasome by IAV virulence protein PB1‐F2 contributes to severe pathophysiology and disease. PLoS Pathog. 2013;9:e1003392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160. Peiris JS, Yu WC, Leung CW, et al. Re‐emergence of fatal human influenza A subtype H5N1 disease. Lancet. 2004;363:617‐619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161. Yang ZF, Mok CK, Liu XQ, et al. Clinical, virological and immunological features from patients infected with re‐emergent avian‐origin human H7N9 influenza disease of varying severity in Guangdong province. PLoS ONE. 2015;10:e0117846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162. Guo J, Huang F, Liu J, et al. The serum profile of hypercytokinemia factors identified in H7N9‐infected patients can predict fatal outcomes. Sci Rep. 2015;5:10942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163. Huang KJ, Su IJ, Theron M, et al. An interferon‐gamma‐related cytokine storm in SARS patients. J Med Virol. 2005;75:185‐194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164. Loo YM, Gale M Jr. Influenza: Fatal immunity and the 1918 virus. Nature. 2007;445:267‐268. [DOI] [PubMed] [Google Scholar]
- 165. Kobasa D, Jones SM, Shinya K, et al. Aberrant innate immune response in lethal infection of macaques with the 1918 influenza virus. Nature. 2007;445:319‐323. [DOI] [PubMed] [Google Scholar]
- 166. Tate MD, Ong JD, Dowling JK, et al. Reassessing the role of the NLRP3 inflammasome during pathogenic influenza A virus infection via temporal inhibition. Sci Rep. 2016;6:27912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167. Ong JD, Mansell A, Tate MD. Hero turned villain: NLRP3 inflammasome‐induced inflammation during influenza A virus infection. J Leukoc Biol. 2017;101:863‐874. [DOI] [PubMed] [Google Scholar]
- 168. van der Poll T, van de Veerdonk FL, Scicluna BP, Netea MG. The immunopathology of sepsis and potential therapeutic targets. Nat Rev Immunol. 2017;17:407‐420. [DOI] [PubMed] [Google Scholar]
- 169. Emmanuilidis K, Weighardt H, Matevossian E, et al. Differential regulation of systemic IL‐18 and IL‐12 release during postoperative sepsis: High serum IL‐18 as an early predictive indicator of lethal outcome. Shock. 2002;18:301‐305. [DOI] [PubMed] [Google Scholar]
- 170. Netea MG, Fantuzzi G, Kullberg BJ, et al. Neutralization of IL‐18 reduces neutrophil tissue accumulation and protects mice against lethal Escherichia coli and Salmonella typhimurium endotoxemia. J Immunol. 2000;164:2644‐2649. [DOI] [PubMed] [Google Scholar]
- 171. Vanden Berghe T, Demon D, Bogaert P, et al. Simultaneoustargeting of IL‐1 and IL‐18 is required for protection against inflammatory and septic shock. Am J Respir Crit Care Med. 2014;189:282‐291. [DOI] [PubMed] [Google Scholar]
- 172. Sakao Y, Takeda K, Tsutsui H, et al. IL‐18‐deficient mice are resistant to endotoxin‐induced liver injury but highly susceptible to endotoxin shock. Int Immunol. 1999;11:471‐480. [DOI] [PubMed] [Google Scholar]
- 173. Luo YP, Jiang L, Kang K, et al. Hemin inhibits NLRP3 inflammasome activation in sepsis‐induced acute lung injury, involving heme oxygenase‐1. Int Immunopharmacol. 2014;20:24‐32. [DOI] [PubMed] [Google Scholar]
- 174. Remick DG, Bolgos GE, Siddiqui J. Inflammatory status in sepsis alters efficacy of interleukin‐18 binding protein therapy. Crit Care Med. 2003;31:2096‐2101. [DOI] [PubMed] [Google Scholar]
- 175. Joshi VD, Kalvakolanu DV, Hasday JD, Hebel RJ, Cross AS. IL‐18 levels and the outcome of innate immune response to lipopolysaccharide: Importance of a positive feedback loop with caspase‐1 in IL‐18 expression. J Immunol. 2002;169:2536‐2544. [DOI] [PubMed] [Google Scholar]
- 176. Joshi VD, Kalvakolanu DV, Hebel JR, Hasday JD, Cross AS. Role of caspase 1 in murine antibacterial host defenses and lethal endotoxemia. Infect Immun. 2002;70:6896‐6903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177. Gelderblom M, Weymar A, Bernreuther C, et al. Neutralization of the IL‐17 axis diminishes neutrophil invasion and protects from ischemic stroke. Blood. 2012;120:3793‐3802. [DOI] [PubMed] [Google Scholar]
- 178. Kayagaki N, Warming S, Lamkanfi M, et al. Non‐canonical inflammasome activation targets caspase‐11. Nature. 2011;479:117‐121. [DOI] [PubMed] [Google Scholar]
- 179. Shi J, Zhao Y, Wang Y, et al. Inflammatory caspases are innate immune receptors for intracellular LPS. Nature. 2014;514:187‐192. [DOI] [PubMed] [Google Scholar]
- 180. Janka GE. Familial and acquired hemophagocytic lymphohistiocytosis. Annu Rev Med. 2012;63:233‐246. [DOI] [PubMed] [Google Scholar]
- 181. Strauss R, Neureiter D, Westenburger B, Wehler M, Kirchner T, Hahn EG. Multifactorial risk analysis of bone marrow histiocytic hyperplasia with hemophagocytosis in critically ill medical patients–a postmortem clinicopathologic analysis. Crit Care Med. 2004;32:1316‐1321. [DOI] [PubMed] [Google Scholar]
- 182. de Saint BG, Ménasché G, Fischer A. Molecular mechanisms of biogenesis and exocytosis of cytotoxic granules. Nat Rev Immunol. 2010;10:568‐579. [DOI] [PubMed] [Google Scholar]
- 183. Grom AA, Horne A, De Benedetti F. Macrophage activation syndrome in the era of biologic therapy. Nat Rev Rheumatol. 2016;12:259‐268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184. Jordan MB, Hildeman D, Kappler J, Marrack P. An animal model of hemophagocytic lymphohistiocytosis (HLH): CD8+ T cells and interferon gamma are essential for the disorder. Blood. 2004;104:735‐743. [DOI] [PubMed] [Google Scholar]
- 185. Jessen B, Kögl T, Sepulveda FE, de Saint BG, Aichele P, Ehl S. Graded defects in cytotoxicity determine severity of hemophagocytic lymphohistiocytosis in humans and mice. Front Immunol. 2013;4:448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186. van Dommelen SL, Sumaria N, Schreiber RD, Scalzo AA, Smyth MJ, Degli‐Esposti MA. Perforin and granzymes have distinct roles in defensive immunity and immunopathology. Immunity. 2006;25:835‐848. [DOI] [PubMed] [Google Scholar]
- 187. Behrens EM, Canna SW, Slade K, et al. Repeated TLR9 stimulation results in macrophage activation syndrome‐like disease in mice. J Clin Invest. 2011;121:2264‐2277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188. Strippoli R, Carvello F, Scianaro R, et al. Amplification of the response to Toll‐like receptor ligands by prolonged exposure to interleukin‐6 in mice: Implication for the pathogenesis of macrophage activation syndrome. Arthritis Rheum. 2012;64:1680‐1688. [DOI] [PubMed] [Google Scholar]
- 189. Brisse E, Imbrechts M, Put K, et al. Mouse cytomegalovirus infection in BALB/c mice resembles virus‐associated secondary hemophagocytic lymphohistiocytosis and shows a pathogenesis distinct from primary hemophagocytic lymphohistiocytosis. J Immunol. 2016;196:3124‐3134. [DOI] [PubMed] [Google Scholar]
- 190. Henter JI, Elinder G, Söder O, Hansson M, Andersson B, Andersson U. Hypercytokinemia in familial hemophagocytic lymphohistiocytosis. Blood. 1991;78:2918‐2922. [PubMed] [Google Scholar]
- 191. Buatois V, Chatel L, Cons L, et al. Use of a mouse model to identify a blood biomarker for IFNγ activity in pediatric secondary hemophagocytic lymphohistiocytosis. Transl Res. 2017;180:37‐52 e2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192. Canna SW, Wrobel J, Chu N, Kreiger PA, Paessler M, Behrens EM. Interferon‐γ mediates anemia but is dispensable for fulminant toll‐like receptor 9‐induced macrophage activation syndrome and hemophagocytosis in mice. Arthritis Rheum. 2013;65:1764‐1775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193. Tesi B, Sieni E, Neves C, et al. Hemophagocytic lymphohistiocytosis in 2 patients with underlying IFN‐γ receptor deficiency. J Allergy Clin Immunol. 2015;135:1638‐1641. [DOI] [PubMed] [Google Scholar]
- 194. Maeno N, Takei S, Imanaka H, et al. Increased interleukin‐18 expression in bone marrow of a patient with systemic juvenile idiopathic arthritis and unrecognized macrophage‐activation syndrome. Arthritis Rheum. 2004;50:1935‐1938. [DOI] [PubMed] [Google Scholar]
- 195. Shimizu M, Nakagishi Y, Inoue N, et al. Interleukin‐18 for predicting the development of macrophage activation syndrome in systemic juvenile idiopathic arthritis. Clin Immunol. 2015;160:277‐281. [DOI] [PubMed] [Google Scholar]
- 196. Wada T, Kanegane H, Ohta K, et al. Sustained elevation of serum interleukin‐18 and its association with hemophagocytic lymphohistiocytosis in XIAP deficiency. Cytokine. 2014;65:74‐78. [DOI] [PubMed] [Google Scholar]
- 197. Nold‐Petry CA, Lehrnbecher T, Jarisch A, et al. Failure of interferon gamma to induce the anti‐inflammatory interleukin 18 binding protein in familial hemophagocytosis. PLoS ONE. 2010;5:e8663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198. Chiossone L, Audonnet S, Chetaille B, et al. Protection from inflammatory organ damage in a murine model of hemophagocytic lymphohistiocytosis using treatment with IL‐18 binding protein. Front Immunol. 2012;3:239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199. Das R, Guan P, Sprague L, et al. Janus kinase inhibition lessens inflammation and ameliorates disease in murine models of hemophagocytic lymphohistiocytosis. Blood. 2016;127:1666‐1675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200. Romberg N, Al Moussawi K, Nelson‐Williams C, et al. Mutation of NLRC4 causes a syndrome of enterocolitis and autoinflammation. Nat Genet. 2014;46:1135‐1139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201. Raghawan AK, Sripada A, Gopinath G, et al. A disease‐associated mutant of NLRC4 shows enhanced interaction with SUG1 leading to constitutive FADD‐dependent caspase‐8 activation and cell death. J Biol Chem. 2017;292:1218‐1230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202. Canna SW, Girard C, Malle L, et al. Life‐threatening NLRC4‐associated hyperinflammation successfully treated with IL‐18 inhibition. J Allergy Clin Immunol. 2017;139:1698‐1701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203. Ombrello MJ, Arthur VL, Remmers EF, et al. Genetic architecture distinguishes systemic juvenile idiopathic arthritis from other forms of juvenile idiopathic arthritis: Clinical and therapeutic implications. Ann Rheum Dis. 2017;76:906‐913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204. Martini A. Systemic juvenile idiopathic arthritis. Autoimmun Rev. 2012;12:56‐59. [DOI] [PubMed] [Google Scholar]
- 205. Behrens EM, Beukelman T, Paessler M, Cron RQ. Occult macrophage activation syndrome in patients with systemic juvenile idiopathic arthritis. J Rheumatol. 2007;34:1133‐1138. [PubMed] [Google Scholar]
- 206. Pascual V, Allantaz F, Arce E, Punaro M, Banchereau J. Role of interleukin‐1 (IL‐1) in the pathogenesis of systemic onset juvenile idiopathic arthritis and clinical response to IL‐1 blockade. J Exp Med. 2005;201:1479‐1486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207. Fall N, Barnes M, Thornton S, Luyrink L, et al. Gene expression profiling of peripheral blood from patients with untreated new‐onset systemic juvenile idiopathic arthritis reveals molecular heterogeneity that may predict macrophage activation syndrome. Arthritis Rheum. 2007;56:3793‐3804. [DOI] [PubMed] [Google Scholar]
- 208. Gattorno M, Piccini A, Lasigliè D, et al. The pattern of response to anti‐interleukin‐1 treatment distinguishes two subsets of patients with systemic‐onset juvenile idiopathic arthritis. Arthritis Rheum. 2008;58:1505‐1515. [DOI] [PubMed] [Google Scholar]
- 209. Shimizu M, Nakagishi Y, Yachie A. Distinct subsets of patients with systemic juvenile idiopathic arthritis based on their cytokine profiles. Cytokine. 2013;61:345‐348. [DOI] [PubMed] [Google Scholar]
- 210. de Benedetti F, Massa M, Robbioni P, Ravelli A, Burgio GR, Martini A. Correlation of serum interleukin‐6 levels with joint involvement and thrombocytosis in systemic juvenile rheumatoid arthritis. Arthritis Rheum. 1991;34:1158‐1163. [DOI] [PubMed] [Google Scholar]
- 211. De Benedetti F, Brunner HI, Ruperto N, et al. Randomized trial of tocilizumab in systemic juvenile idiopathic arthritis. N Engl J Med. 2012;367:2385‐2395. Erratum in: N Engl J Med 2015;372:887. [DOI] [PubMed] [Google Scholar]
- 212. Kawashima M, Yamamura M, Taniai M, et al. Levels of interleukin‐18 and its binding inhibitors in the blood circulation of patients with adult‐onset Still's disease. Arthritis Rheum. 2001;44:550‐560. [DOI] [PubMed] [Google Scholar]
- 213. Maeno N, Takei S, Nomura Y, Imanaka H, Hokonohara M, Miyata K. Highly elevated serum levels of interleukin‐18 in systemic juvenile idiopathic arthritis but not in other juvenile idiopathic arthritis subtypes or in Kawasaki disease: Comment on the article by Kawashima et al. Arthritis Rheum. 2002;46:2539‐2541; author reply 2541‐2. [DOI] [PubMed] [Google Scholar]
- 214. Put K, Avau A, Brisse E, et al. Cytokines in systemic juvenile idiopathic arthritis and haemophagocytic lymphohistiocytosis: Tipping the balance between interleukin‐18 and interferon‐γ. Rheumatology (Oxford). 2015;54:1507‐1517. [DOI] [PubMed] [Google Scholar]
- 215. Ichida H, Kawaguchi Y, Sugiura T, et al. Clinical manifestations of Adult‐onset Still's disease presenting with erosive arthritis: Association with low levels of ferritin and Interleukin‐18. Arthritis Care Res (Hoboken). 2014;66:642‐646. [DOI] [PubMed] [Google Scholar]
- 216. Girard C, Rech J, Brown M, et al. Elevated serum levels of free interleukin‐18 in adult‐onset Still's disease. Rheumatology (Oxford). 2016;55:2237‐2247. [DOI] [PubMed] [Google Scholar]
- 217. Liao TL, Chen YM, Hsieh CW, et al. Upregulation of circulating microRNA‐134 in adult‐onset Still's disease and its use as potential biomarker. Sci Rep. 2017;7:4214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 218. Ogilvie EM, Khan A, Hubank M, Kellam P, Woo P. Specific gene expression profiles in systemic juvenile idiopathic arthritis. Arthritis Rheum. 2007;56:1954‐1965. [DOI] [PubMed] [Google Scholar]
- 219. Sikora KA, Fall N, Thornton S, Grom AA. The limited role of interferon‐γ in systemic juvenile idiopathic arthritis cannot be explained by cellular hyporesponsiveness. Arthritis Rheum. 2012;64:3799‐3808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 220. Avau A, Mitera T, Put S, et al. Systemic juvenile idiopathic arthritis‐like syndrome in mice following stimulation of the immune system with Freund's complete adjuvant: Regulation by interferon‐γ. Arthritis Rheumatol. 2014;66:1340‐1351. [DOI] [PubMed] [Google Scholar]
- 221. Ghezzi P, Dinarello CA. IL‐1 induces IL‐1. III. Specific inhibition of IL‐1 production by IFN‐gamma. J Immunol. 1988;140:4238‐4244. [PubMed] [Google Scholar]
- 222. Shimizu M, Yachie A. Compensated inflammation in systemic juvenile idiopathic arthritis: Role of alternatively activated macrophages. Cytokine. 2012;60:226‐232. [DOI] [PubMed] [Google Scholar]
- 223. Grom AA, Villanueva J, Lee S, Goldmuntz EA, Passo MH, Filipovich A. Natural killer cell dysfunction in patients with systemic‐onset juvenile rheumatoid arthritis and macrophage activation syndrome. J Pediatr. 2003;142:292‐296. [DOI] [PubMed] [Google Scholar]
- 224. Villanueva J, Lee S, Giannini EH, et al. Natural killer cell dysfunction is a distinguishing feature of systemic onset juvenile rheumatoid arthritis and macrophage activation syndrome. Arthritis Res Ther. 2005;7:R30‐R37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 225. Park JH, Kim HS, Lee JS, et al. Natural killer cell cytolytic function in Korean patients with adult‐onset Still's disease. J Rheumatol. 2012;39:2000‐2007. [DOI] [PubMed] [Google Scholar]
- 226. Kaufman KM, Linghu B, Szustakowski JD, et al. Whole‐exome sequencing reveals overlap between macrophage activation syndrome in systemic juvenile idiopathic arthritis and familial hemophagocytic lymphohistiocytosis. Arthritis Rheumatol. 2014;66:3486‐3495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 227. Cifaldi L, Prencipe G, Caiello I, et al. Inhibition of natural killer cell cytotoxicity by interleukin‐6: Implications for the pathogenesis of macrophage activation syndrome. Arthritis Rheumatol. 2015;67:3037‐3046. [DOI] [PubMed] [Google Scholar]
- 228. Put K, Vandenhaute J, Avau A, et al. Inflammatory gene expression profile and defective interferon‐γ and granzyme K in natural killer cells from systemic juvenile idiopathic arthritis patients. Arthritis Rheumatol. 2017;69:213‐224. [DOI] [PubMed] [Google Scholar]
- 229. de Jager W, Vastert SJ, Beekman JM, et al. Defective phosphorylation of interleukin‐18 receptor beta causes impaired natural killer cell function in systemic‐onset juvenile idiopathic arthritis. Arthritis Rheum. 2009;60:2782‐2793. [DOI] [PubMed] [Google Scholar]
- 230. Shibatomi K, Ida H, Yamasaki S, et al. A novel role for interleukin‐18 in human natural killer cell death: High serum levels and low natural killer cell numbers in patients with systemic autoimmune diseases. Arthritis Rheum. 2001;44:884‐892. [DOI] [PubMed] [Google Scholar]
- 231. Chen DY, Hsieh TY, Hsieh CW, Lin FJ, Lan JL. Increased apoptosis of peripheral blood lymphocytes and its association with interleukin‐18 in patients with active untreated adult‐onset Still's disease. Arthritis Rheum. 2007;57:1530‐1538. [DOI] [PubMed] [Google Scholar]