

Abstract
Viral infections are associated with coagulation disorders. All aspects of the coagulation cascade, primary hemostasis, coagulation, and fibrinolysis, can be affected. As a consequence, thrombosis and disseminated intravascular coagulation, hemorrhage, or both, may occur. Investigation of coagulation disorders as a consequence of different viral infections have not been performed uniformly. Common pathways are therefore not fully elucidated. In many severe viral infections there is no treatment other than supportive measures. A better understanding of the pathophysiology behind the association of viral infections and coagulation disorders is crucial for developing therapeutic strategies. This is of special importance in case of severe complications, such as those seen in hemorrhagic viral infections, the incidence of which is increasing worldwide. To date, only a few promising targets have been discovered, meaning the implementation in a clinical context is still hampered. This review discusses non‐hemorrhagic and hemorrhagic viruses for which sufficient data on the association with hemostasis and related clinical features is available. This will enable clinicians to interpret research data and place them into a perspective. J. Med. Virol. 84:1680–1696, 2012. © 2012 Wiley Periodicals, Inc.
Keywords: virus infection, coagulation, platelets, influenza, HIV, hemorrhagic virus, herpesvirus, hantavirus, cytomegalovirus, hepatitis, parvovirus B19, Epstein‐Barr virus, thrombosis
INTRODUCTION
An increasing body of evidence suggests the existence of an extensive correlation between inflammation and coagulation, whereby inflammation not only leads to coagulation, but coagulation also affects inflammatory activity [Keller et al., 2003; Opal, 2003; Esmon, 2004; Levi et al., 2004; Van der Poll and Levi, 2012]. Inflammation impacts the initiation, propagation and inhibitory phases of blood coagulation [Opal, 2003]. In viral and bacterial infections, this can actually lead to both thrombotic as well as hemorrhagic complications. Pathogens, as well as inflammatory cells and mediators, can induce the expression of tissue factor on monocytes and endothelial cell surfaces. Tissue factor is a major activator of coagulation [Van der Poll et al., 2011]. Direct or indirect activation of the endothelium by viruses or other pathogens may result in alterations in the coagulation and the fibrinolytic systems [van Gorp et al., 1999]. Normally, coagulation is a balance between procoagulant and (natural) anticoagulant mechanisms. A regulated activation of coagulation is part of the host's defence against infectious agents [Opal, 2003]. Inflammation may lead to altered coagulation, resulting in an imbalance between the pro‐ and anticoagulant state. The clinical picture of altered coagulation in several viral infections manifests itself in bleeding (hemorrhage), thrombosis, or both. An exaggerated response may even lead to disseminated intravascular coagulation with the formation of microvascular thrombi in various organs [Levi, 2007]. Disseminated intravascular coagulation contributes to multiple organ failure and is associated with high mortality in both bacterial and nonbacterial diseases [Levi et al., 2004; Levi, 2007]. Other syndromes associated with bacterial or viral infections are hemolytic uraemic syndrome, idiopathic thrombocytopenic purpura and thrombotic thrombocytopenic purpura [van Gorp et al., 1999]. It is not yet clear why some viruses cause hemorrhaging (e.g., Ebola), others are associated with thrombosis (e.g., cytomegalovirus) and yet others show both complications (e.g., varicella zoster virus) [Miller and Stephan, 1993; Uthman and Gharavi, 2002; Geisbert and Jahrling, 2004; Squizzato et al., 2005]. In addition to this, the bleeding complications of hemorrhagic viruses vary in severity, such as the minor bleeding complications in some forms of dengue and more severe bleeding in Ebola and Marburg. For many viral infections, targeted therapy is not available, and only supportive care can be provided. In many mild cases, treatment may not even be necessary. However, to improve therapy and supportive care for complicated viral infections, a better understanding is needed of the pathogenesis of bleeding and thrombotic complications due to viral infections. This review briefly outlines the coagulation cascade in general, as well as the interaction between the coagulation cascade and cytokines released during viral infections. Subsequently, the clinical picture of coagulation alterations seen in viral disease is reviewed. Finally, an explanation is given of the presumed mechanism of how these viruses influence hemostasis. The influence of viral infections on atherosclerosis and atherothrombosis is not discussed.
METHODS
This review includes viruses for which sufficient data on the association between hemostasis and related clinical pictures are documented. pubmed/MEDLINE was searched for articles that document the relationship between viruses and hemostatic parameters and thrombotic complications. All possible combinations between the virus and coagulation groups as stated in Appendix 1 were used.
Principles of Hemostasis, Coagulation, and Fibrinolysis: General Aspects
The formation of a blood clot is a well‐regulated process comprising three elements: (1) primary hemostasis, (2) secondary hemostasis/coagulation, and (3) fibrinolysis (Fig. 1) [Dahlback, 2005]. Primary hemostasis is characterized by the adhesion, activation, and aggregation of platelets to form a hemostatic plug. Von Willebrand factor mediates platelet adhesion to exposed subendothelium. P‐selectin, a cell adhesion molecule localized on platelets and endothelial cells, supports initial tethering of leukocytes to activated endothelial cells and activated platelets [Othman et al., 2007]. The activation of coagulation leads to the formation of fibrin strands, secondary coagulation, which stabilize the platelet plug. Coagulation results from a series of linked coagulation protease–zymogen reactions, ultimately ensuing in the formation of fibrin. Tissue factor is the main initiator of the coagulation cascade, which is localized in the subendothelium, but also on non‐circulating leukocytes and possibly on platelets. Thrombin generation is induced by the assembly of the tissue factor–factor VIIa complex. Thrombin is able to convert fibrinogen into (insoluble) fibrin. Coagulation is regulated by different inhibitory mechanisms. A first mechanism is made up of the circulating inhibitors of blood coagulation: antithrombin and heparin cofactor II (both inhibitors of thrombin), and tissue factor pathway inhibitor. Two other circulating inhibitors of blood coagulation are protein C and protein S (the latter of which is a cofactor for the proper functioning of activated protein C). A second inhibitory mechanism consists of the endothelium‐bound modulators heparin sulfate and thrombomodulin, which facilitate the inhibitory activity of antithrombin and the activation of protein C, respectively. The third element, the fibrinolytic system, is necessary to degrade the formed fibrin strands. This system is initiated by tissue plasminogen activator and urokinase after their synthesis by, and release from, endothelial cells. These activators initiate the conversion of plasminogen to plasmin, which hydrolyses polymerized fibrin strands into soluble fibrin degradation products, thus degrading the fibrin clot. The activity of the fibrinolytic system is, among other things, regulated by plasminogen activator inhibitor type I, of which may greatly increase during acute phase reactions. Thrombin‐activatable fibrinolysis inhibitor is also an inhibitor of fibrinolysis and is activated by thrombin. Fibrinolysis may be activated primarily—and thus independently of the activation of the coagulation cascade—or secondarily, in response to fibrin formation.
Figure 1.
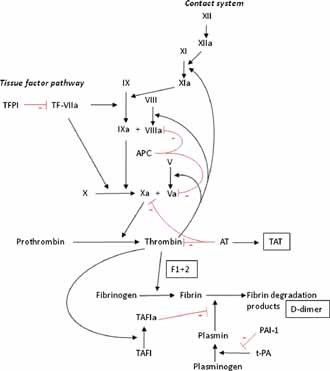
Simplified model of the coagulation cascade. Coagulation proteins are given in roman numerals. Products of coagulation activation that can be measured in blood samples are shown in squares: F1 + 2, prothrombin fragment 1 + 2; D‐dimer, fibrin degradation product; TAT, thrombin–antithrombin complex. Natural inhibitors of coagulation shown: TFPI, tissue factor pathway inhibitor; AT, antithrombin; APC, activated protein C (which cleaves activated factors V and VIII). TAFI, thrombin‐activatable fibrinolysis inhibitor. t‐PA, tissue plasminogen activator; PAI‐1, plasminogen activator inhibitor‐1. Adapted from [van Gorp et al., 1999].
Procoagulant/prothrombotic changes
Procoagulant and prothrombotic are two terms used in literature to indicate the same thing; a tendency to develop thrombosis. In this review the term procoagulant is used to refer to a change in coagulation markers determined in the laboratory, while a prothrombotic state indicates a clinical risk situation. Generally speaking, in viral infections several procoagulant markers are elevated, indicating that the coagulation system is activated. A procoagulant state can be defined by a number of alterations in the blood. Firstly, there will be an increased level of clotting factors (e.g., factor VIII, factor XI), soluble tissue factor and von Willebrand factor. Secondly, the regulatory system can be deficient, which is indicated by decreased levels of the natural anticoagulants protein C, protein S, antithrombin, and tissue factor pathway inhibitor. Furthermore, markers of thrombin generation (prothrombin fragment 1 + 2 and thrombin–antithrombin complexes), platelet activation, fibrin degradation and fibrinolysis (e.g., d‐dimer and plasmin–α2‐antiplasmin complexes) can also be increased.
The effect of inflammation on coagulation
Many studies have been carried out on the influence of inflammation, and the (pro)inflammatory cytokines on the coagulation system. The activation of the coagulation cascade during inflammation is the result of the stimulation of coagulant synthesis, the decreased synthesis of anti‐coagulants and the suppression of fibrinolysis [Lipinski et al., 2011]. This is the net result of an increased expression of tissue factor leading to the activation of the extrinsic coagulation pathway, together with the downregulation of activated protein C and the inhibition of fibrinolysis [Petaja, 2011]. Pro‐inflammatory cytokines like IL‐6, IL‐1, IL‐12, and TNF‐α are known to increase the production of von Willebrand factor, which may result in platelet activation. They upregulate tissue factor expression via the activation of protease‐activated receptors, on monocytes, microparticles, and endothelial cells [Opal, 2003; Levi et al., 2004; Schouten et al., 2008]. Furthermore, the generation of activated protein C is also decreased, due to a lowered expression of thrombomodulin on the surface of endothelial cells, which acts as a cofactor in the thrombin‐mediated activation of protein C. While on the one hand anticoagulants are decreased, on the other hand there is an upregulation of plasminogen activator inhibitor‐1, which is able to block the activation of plasminogen, thus creating a decrease in the breakdown of fibrin clots [Keller et al., 2003; Opal, 2003; Schouten et al., 2008]. The generation of thrombin is generally increased due to inflammation, all of which results in a procoagulant state of the clotting cascade [Levi et al., 2012].
Clinical Aspects of Hemostasis in Viral Infections
As previously described, the clinical picture of the vascular complications of viral infections consists of thrombosis and/or hemorrhage. Table I shows an overview of the clinical pathology and laboratory abnormalities seen in altered hemostasis due to viral infections. Common viruses known to cause alterations in hemostasis are discussed. The main focus is on respiratory viruses, HIV, herpes viruses, and hemorrhagic fever viruses. Respiratory tract infections increase the risk of deep venous thrombosis and possibly pulmonary embolism too [Smeeth et al., 2006]. Patients infected with the influenza A virus have been known to suffer disseminated intravascular coagulation and pulmonary microembolism [Davison et al., 1973; Harms et al., 2010]. In the recent outbreak of H1N1 influenza (“swine flu”), both thrombotic and hemorrhagic complications were reported, such as deep venous thrombosis, pulmonary embolism, and pulmonary hemorrhage with hemoptysis, hematemesis, petechial rash, and one case of disseminated petechial brain hemorrhage [Agarwal et al., 2009; Soto‐Abraham et al., 2009; Adalja, 2010; Gilbert et al., 2010; Harms et al., 2010; Mauad et al., 2010; Mukhopadhyay et al., 2010; Shachor‐Meyouhas and Kassis, 2010; Venkata et al., 2010; Bunce et al., 2011; Calore et al., 2011]. Frequently, a significant co‐morbidity was present, and in some reported complications a direct effect of H1N1 was uncertain. In several influenza cases intrarenal fibrin deposition was found, which lead to renal failure. H5N1 highly pathogenic avian influenza infections resulted in disseminated intravascular coagulation, pulmonary hemorrhage, and thrombocytopenia in several patients [Wiwanitkit, 2008]. One study reported a number of children who developed hematemesis during influenza A infection [Armstrong et al., 1991]. How frequently influenza infection leads to clinically relevant thrombotic disease has yet to be established. RSV and adenovirus are not known to cause clinically relevant vascular complications. A relatively recent outbreak of SARS, a novel coronavirus, showed significant morbidity and mortality. The clinical picture pertaining to coagulation consisted of vascular endothelial damage in both small‐ and mid‐sized pulmonary vessels, disseminated intravascular coagulation, deep venous thrombosis and pulmonary thromboemboli resulting in pulmonary infarction [Lee et al., 2003; Chong et al., 2004; Hwang et al., 2005]. An association between CMV infection and thromboembolic disease has been established in animals [Persoons et al., 1998] and in humans, but mostly in immunocompromised patients [Maslo et al., 1997; Kazory et al., 2004; Sengul et al., 2006; Lijfering et al., 2008], with some reports being made of thrombotic events during CMV infection in immunocompetent subjects as well [Abgueguen et al., 2003; Youd et al., 2003; Squizzato et al., 2005; Delbos et al., 2007]. In several cases an additional procoagulant risk factor was present, such as a protein C deficiency, factor V Leiden mutation, or a heterozygous prothrombin G20210A mutation [Bauduer et al., 2003; Fridlender et al., 2007; Lijfering et al., 2007]. Hepatitis C infections (HCV) have been associated with thrombosis. HCV was observed more frequently in patients suffering from liver cirrhosis with splanchnic venous thrombosis or thrombophlebitis than those without thrombosis, indicating that HCV infection may contribute to venous thrombosis [Squizzato and Gerdes, 2012; Violi et al., 1995].
Table I.
Clinical Signs of Altered Hemostasis Seen in Viral Infection
| Virus | Vascular complications | References |
|---|---|---|
| Non‐hemorrhagic viruses | ||
| Respiratory viruses in general | Elevated risk of DVT and PE |
Smeeth et al. [2006 ] |
| Influenza (general) | Thrombocytopenia, DIC (relapse of), ITP, intrarenal fibrin deposition, consumption coagulopathy (intrapulmonary), hemorrhage, hematemesis |
Davison et al. [1973 ], Armstrong et al. [1991 ], Rice and Resar [1998 ], and Kaneko et al. [2004 ] |
| Avian influenza (H5N1) | DIC, lung hemorrhage, thrombocytopenia, multiple organ hemorrhage |
Claas et al. [1998 ], Kuiken et al. [2003 ], Xu et al. [2006 ], Muramoto et al. [2006 ], Wiwanitkit [2008 ], and Korteweg and Gu [2008 ] |
| Swine flu (H1N1) | Thrombocytopenia, DVT, portal vein thrombosis, PE, pulmonary hemorrhage, hemoptysis, hematemesis, disseminated petechial brain hemorrhage, petechial rash |
Soto‐Abraham et al. [2009 ], Agarwal et al. [2009 ], Mukhopadhyay et al. [2010 ], Shachor‐Meyouhas and Kassis [2010 ], Mauad et al. [2010 ], Harms et al. [2010 ], Gilbert et al. [2010 ], Adalja [2010 ], Venkata et al. [2010 ], Bunce et al. [2011 ], and Calore et al. [2011 ] |
| SARS | Thrombocytopenia, reactive thrombocytosis, DIC, DVT, PE |
Wong et al. [2003 ], Lee et al. [2003 ], Chong et al. [2004 ], and Hwang et al. [2005 ] |
| Parvovirus B19 | Pulmonary embolism |
Cioc et al. [2002 ], Magro et al. [2002 ], and Asano et al. [2006 ] |
| HSV | DIC, thrombocytopenia |
Whitaker et al. [1974 ] Phinney et al. [1982 ] and McSorley et al. [1974 ] |
| CMV | Thrombotic microangiopathy, TTP, HUS, DIC, DVT, PE, thrombosis of subclavicular vein, internal jugular vein, cerebral vein, mesenteric vein, portal vein, antiphospholipid syndrome |
Caton et al. [1993 ], Inacio et al. [1997 ], Neau et al. [1997 ], Maslo et al. [1997 ], Persoons et al. [1998 ], Humblot et al. [2001 ], Uthman and Gharavi [2002 ], Belet et al. [2003 ], Bauduer et al. [2003 ], Abgueguen et al. [2003 ], Youd et al. [2003 ], Kazory et al. [2004 ], Cervera and Asherson [2005 ], Niewold and Bundrick [2006 ], Sengul et al. [2006 ], Squizzato et al. [2007 ], Lijfering et al. [2007 ], Lijfering et al. [2008 ] Delbos et al. [2007 ], and Fridlender et al. [2007 ] |
| EBV | Thrombocytopenia, DIC, HUS, ITP, infarction of the spleen, calf DVT, PE |
Yamazaki et al. [1991 ], Lee et al. [1998], Rand and Wright [1998 ], van Steijn et al. [2000 ], and van Hal et al. [2005 ] |
| VZV | Thrombocytopenia, cutaneous bleeding, febrile purpura, post‐infectious purpura, Henoch–Schönlein purpura, purpura fulminans, ITP, hemorrhage, DIC, DVT |
Miller et al. [1993], Rand and Wright [1998 ], Uthman and Gharavi [2002 ], Manco‐Johnson et al. [1996 ] and Cervera and Asherson [2005 ] |
| Hepatitis A | Thrombocytopenia, ITP, hepatic vein thrombosis |
Cohen et al. [1993 ], Ertem et al. [2001 ], Tanir et al. [2005 ], and Sainokami et al. [2005 ] |
| Hepatitis C | Splanchnic vein thrombosis, thrombocytopenia, renal thrombotic microangiopathy |
Violi et al. [1995 ], Prieto et al. [1996 ], and Uthman and Gharavi [2002 ] |
| HIV | DVT, PE, TTP, thrombocytopenia, HUS |
Becker et al. [2004 ], Klein et al. [2005 ], and Passos et al. [2010 ] |
| Hemorrhagic viruses | ||
| Crimean‐Congo hemorrhagic fever | Ecchymosis, visceral bleeding, DIC, thrombocytopenia, hematemesis, melena |
Geisbert and Jahrling [2004 ], Bray [2005 ], and Sonmez et al. [2007 ] Cevik et al. [2008 ] |
| Dengue | Increased vascular permeability, skin bleeding, epistaxis, gingival bleeding, gastro‐intestinal bleeding, hematuria, menorrhaegia, thrombocytopenia, thrombocytopathy, DIC |
Huang et al. [2001 ], Mairuhu et al. [2003 ], Geisbert and Jahrling [2004 ], Bray [2005 ], and Sosothikul et al. [2007 ] |
| Marburg + Ebola | DIC, conjunctival hemorrhage, mucosal hemorrhage, ecchymosis, petechiae, easy bruising, uncontrolled bleeding from venapuncture sites, hematuria, gastro‐intestinal bleeding |
Mahanty and Bray [2004 ], Geisbert and Jahrling [2004 ], Hensley and Geisbert [2005 ], and Bray [2005 ] |
| Hantavirus | Thrombocytopenia, mucosal bleeding, petechiae, gastro‐intestinal bleeding, epistaxis, hematuria, conjunctival bleeding |
Lee [1987 ], Dunst et al. [1998 ], Khaiboullina et al. [2005 ], and Laine et al. [2010 ] |
| SFTS | Thrombocytopenia |
Zhang et al. [2012 ] |
Most of the clinical pictures are based on limited case reports. CMV, cytomegalovirus; DIC, disseminated intravascular coagulation; DVT, deep venous; thrombosis; EBV, Epstein‐Barr Virus; HSV, herpes simplex virus; HUS, hemolytic uremic; syndrome; ITP, idiopathic thrombocytopenic purpura; PE, pulmonary embolism; SARS, severe acute respiratory syndrome; TTP, thrombotic thrombocytopenic purpura; VZV, varicella zoster virus.
Thrombocytopenia is a common hematological complication observed during HIV‐infection, documented numbers range from 10% to 50%. The severity of the thrombocytopenia correlates with the CD4 count. While bleeding complications are rare in HIV associated thrombocytopenia, the platelet count can remain low for a long time [Passos et al., 2010]. Thrombotic thrombocytopenic purpura in HIV‐infected patients was relatively common before the introduction of effective antiretroviral therapy. Since the introduction of this treatment, thrombotic thrombocytopenic purpura has been a rare condition and is associated with advanced HIV disease [Becker et al., 2004]. Cardiovascular complications are reported in both acute and chronic HIV infections [Friis‐Moller et al., 2007]. The overall risk of venous thrombotic disease in HIV‐infected patients may be between two and ten times as high as it is in healthy individuals [Klein et al., 2005; Crum‐Cianflone et al., 2008]. The use of protease inhibitors is linked to venous as well as arterial thrombotic events [Klein et al., 2005; Friis‐Moller et al., 2007]. There are strong indications that the incidence rates of vascular complications in the antiretroviral era are increasing [Klein et al., 2005].
Whereas HIV and respiratory viruses have more thrombotic clinical complications many acute viral infections may result in bleeding. Often these pathogens are classified as hemorrhagic fever viruses with the most devastating being members of the filoviridae (Ebola and Marburg) and the arenaviruses (Lassa fever and South American hemorrhagic fever). Bleeding often occurs from various mucous membranes together with easy bruising and persistent bleeding after venapuncture. Massive bleeding may occur in the gastro‐intestinal tract and/or intra‐cerebrally [Kortepeter et al., 2011]. These bleeding complications are most frequent in severe forms of infection and they correlate with the case fatality rate. Bleeding is thought to be the consequence of an imbalanced coagulation cascade, sometimes resulting in disseminated intravascular coagulation, as seen in hantavirus, Ebola, Marburg, Crimean‐Congo hemorrhagic fever, and Dengue infections [Geisbert and Jahrling, 2004; Bray, 2005; Laine et al., 2010]. Bleeding manifestations are most prominent in Ebola, hantavirus, and Crimean‐Congo hemorrhagic fever [Geisbert and Jahrling, 2004]. Thrombocytopenia is consistently found among viral hemorrhagic fever infections, as well as a decreased platelet function (thrombocytopathia) [Geisbert and Jahrling, 2004]. Known vascular complications in arenaviral infection, in addition to mucosal bleedings, are severe pleural‐ and pericardial effusion. The latter is also a consequence of vascular dysfunction. Disseminated intravascular coagulation does not seem to play a role in arenaviral infection [Peters et al., 1989; Richmond and Baglole, 2003].
Hantaviruses circulating in Europe and Asia are associated with hemorrhagic fever and renal syndrome (HFRS) or the milder nephropathia epidemica (NE) variant [Khaiboullina et al., 2005a; Jonsson et al., 2010]. Hallmark symptoms of HFRS are acute kidney failure and alterations in hemostasis, ranging from mild thrombocytopenia to disseminated intravascular coagulation [Sundberg et al., 2011]. At first, less severe bleeding complications like epistaxis, conjunctival bleeding, hematuria, petechiae and mucosal bleeding occur, while in a later phase gastrointestinal, intra‐cerebral and pleural bleedings are reported in severe HFRS. Less common hemorrhagic events are right atrial hemorrhage [Chun and Godfrey, 1984], spleen hemorrhage [Alexeyev et al., 1994] and pituitary gland hemorrhage resulting in endocrinal disturbance or even panhypopituitarism [Suh et al., 1995; Pekic et al., 2005]. Hantaviruses circulating in North and South America can cause the hantavirus cardiopulmonary syndrome, resulting in acute respiratory distress and thrombocytopenia. However, bleeding disorders play a less significant role in the pathology of this disease. Dengue virus, one of the most widespread mosquito‐borne viruses worldwide and with an annual infection rate around 50–100 million, used to be subdivided into dengue fever, dengue hemorrhagic fever and dengue shock syndrome [WHO, 1997]. The most recent classification breaks down dengue patients into non‐severe dengue cases, with or without warning signs, and severe dengue cases. The latter are patients presenting with either shock, respiratory distress, severe organ impairment or severe bleeding [van de Weg et al., 2012]. The occurrence of severe dengue with bleeding or signs of shock differs all over the world. Therefore, it is assumed that the interaction between host, virus, vector, and environment defines the clinical presentation and outcome. Although thrombocytopenia has been well documented in dengue infection [Sosothikul et al., 2007], no association has been established between the presence of active bleeding and the degree of thrombocytopenia on admission. Bleeding symptoms in dengue cases often start with petechiae, bleeding from the mucous membranes and epistaxis while in severe cases typically large gastrointestinal bleedings may occur. Recently, an outbreak of a novel bunyavirus caused a clinical syndrome described as severe fever with thrombocytopenia syndrome. Patients rapidly developed thrombocytopenia, accompanied with multiple organ dysfunction and gastrointestinal complaints. The case fatality rate was estimated around 16% [Zhang et al., 2012].
Pathophysiological Mechanisms of Hemostasis in Viral Infection
Data from experimental and human studies have revealed the complexity of the interaction between infectious pathogens, cytokines, effector cells, and the coagulation system. This review discusses the existing evidence of the pathogenesis of abnormal hemostasis in viral infections with regard to primary hemostasis, secondary hemostasis and fibrinolysis. Antiphospholipid antibodies are discussed separately.
Primary Hemostasis
Platelets
Platelets are the key players in primary hemostasis; the formation of the platelet plug. Reduced platelet function, as well as diminished production or destruction of platelets, is a well‐documented phenomenon in several viral infections. Thrombocytopenia often occurs in both hemorrhagic and non‐hemorrhagic viral infections. In most cases, thrombocytopenia is caused by autoimmune antibodies against platelets. Other proposed mechanisms include the increased adherence and activation of platelets, which leads to the consumption of platelets, and the infection of bone marrow directly affecting megakaryocytic, and thus the production of platelets. Thrombocytopenia caused by autoantibodies has been described in SARS, influenza, chronic parvovirus B19, herpes virus, CMV, VZV, Epstein Barr virus, HIV, and hepatitis A virus and hepatitis C virus infections [Whitaker et al., 1974; Kahane et al., 1981; Kazatchkine et al., 1984; Mayer and Beardsley, 1996; Neau et al., 1997; Rand and Wright, 1998; Scheurlen et al., 2001; Sainokami et al., 2005; Yang et al., 2005; Panzer et al., 2006; Passos et al., 2010]. Autoantibodies have been identified in several viral infections. The target is usually one of the surface glycoproteins such as GPIIb/IIIa, GPIb/IX, or GPV, with a possible cross reactivity between antibodies against the virus and the platelets [Mayer and Beardsley, 1996; Tanaka et al., 2003], as in HIV where platelets are bound via the platelet glycoprotein IIIa integrin [Passos et al., 2010]. Whether platelet destruction in viral hepatitis is caused by platelet‐specific glycoprotein antibodies, by immune complexes bound to the platelet surface or by a combination of the above, remains uncertain [Doi et al., 2002; Weksler, 2007]. In secondary dengue virus infection, platelet apoptosis seems to be enhanced, which results in increased platelet clearance [Alonzo et al., 2012]. This is probably present in any viral infection that leads to a systemic inflammatory response. Platelets may serve as carriers of viral infections, which are then damaged and partially destroyed [Terada et al., 1966]. Platelet consumption due to other mechanisms has been documented for influenza, SARS, hantavirus, and adenovirus infections [Lee, 1987; Rand and Wright, 1998; Yang et al., 2005; Othman et al., 2007]. Hantavirus‐infected endothelial cells seem to bind to quiescent platelets via an Alpha‐v beta‐3 integrin dependent mechanism, which is the cellular receptor for the hantavirus [Gavrilovskaya et al., 2002, 2010]. This may not only result in a decreased number of platelets; that is, thrombocytopenia, but also a decreased function and an increased vascular permeability. Furthermore, in hemorrhagic fever renal syndrome patients in China, the intensity of this receptor, also known as CD61—determined by flow cytometry—on platelets correlates with the severity of the disease [Liu et al., 2008].
Platelets derived from patients infected with Lassa fever had depressed capacities for platelet aggregation [Cummins et al., 1989; Roberts et al., 1989; Richmond and Baglole, 2003]. Acute‐phase plasma from Lassa fever patients showed inhibition of ADP‐induced platelet aggregation. This inhibition was found in 80% of Lassa fever patients with hemorrhage but in only 16% of those without hemorrhage. When plasma samples from Lassa fever patients were mixed 1:1 with control, platelet‐rich plasma, a marked inhibition of ADP‐induced aggregation was observed. These findings indicate that platelet dysfunction may play an important role in the bleeding characteristics of this disease [Cummins et al., 1989; Roberts et al., 1989]. However a decreased fibrinogen concentration may also play a role explaining this phenomenon. Furthermore, influenza, rhinovirus and other viruses stimulate IL‐6 production [Bouwman et al., 2002]. There is a complex platelet–endothelial–leukocyte interplay [Othman et al., 2007]. Viruses bind to platelets, which are then activated, leading to rapid exposure of P‐selectin on the platelet surface, which, in turn, triggers the formation of platelet–leukocyte aggregates. Subsequently, endothelial cells are activated, which is demonstrated by increases in vascular cell adhesion of molecule 1, von Willebrand factor and endothelial cell‐derived microparticles. A reduced production of platelets was observed in SARS, HCV, HIV, hantavirus, and Junin hemorrhagic fever infections. This can be caused by the direct infection of hemapoietic stem/progenitor cells and megakaryocytes [Carballal et al., 1981; Li et al., 1999; Yang et al., 2005; Lutteke et al., 2010; Passos et al., 2010]. For example, pathogenic hantaviruses invade and subsequently replicate in megakaryocytes, leading to upregulation of human leukocyte antigen (HLA) class 1 molecules, the target structures of cytotoxic CD8 T‐cells in vitro. These T‐cells kill the infected megakaryocyte, hypothetically leading to a decreased platelet production. This could also explain why corticosteroid treatment increases platelet count by inhibiting the cellular immune response in hantavirus‐infected patients [Dunst et al., 1998; Seitsonen et al., 2006]. In addition to this, impaired thrombopoietin production, the primary cytokine governing megakaryocyte maturation and platelet formation [Kaushansky, 1998], is present in HCV if there is hepatocellular damage [Weksler, 2007]. Reduced platelet production is not always caused by the infection itself but sometimes by the medical therapy. Well‐known examples are the pegylated interferon (peg‐IFN) treatment of HCV and antiretroviral therapy for HIV [Kowdley, 2005; Passos et al., 2010]. Finally, hypersplenism (with the sequestration of platelets) due to portal hypertension can be present in chronic hepatitis [Weksler, 2007].
Endothelial Cells, Von Willebrand Factor, Tissue Factor and the Connection to Secondary Hemostasis
Endothelial cells are key regulators of coagulation, both producing and presenting anticoagulant markers, thrombomodulin and antithrombin as well as procoagulant factors like tissue factor and plasminogen activator inhibitor‐1. The infection of endothelial cells can result in the activation of these cells and, consequently, the activation of coagulation. Endothelial cell activation is mainly marked by an increase in von Willebrand factor secretion, which can bind platelets after vessel wall damage. In vitro and in vivo studies have shown that a variety of prothrombotic viruses are able to infect endothelial cells. These viruses include influenza A and B, parainfluenza‐1, RSV, adenovirus, CMV, parvovirus B19, HIV, and the hepatitis B virus [Mason et al., 1993; Poland et al., 1995; Visseren et al., 2000; Magro et al., 2002; Arnold and Konig, 2005; Squizzato et al., 2005; Gavrilovskaya et al., 2010]. Endothelial cell activation plays a crucial role in altering coagulation and is an etiological factor for vascular complications in HIV infection. Endothelial cell activation is caused by the virus itself and by HIV‐induced cytokines [Chi et al., 2000]. HIV‐specific gag and env gene sequences have been successfully amplified by polymerase chain reaction from human microvascular endothelial cells [Poland et al., 1995]. Markedly increased levels of soluble vascular and soluble intercellular adhesion molecules and elevated von Willebrand factor, which are indicative of endothelial cell activation, have been shown in HIV‐infected patients before the start of antiretroviral therapy (ART) with only partial recovery after the start of ART [Wolf et al., 2002]. Herpes viruses are known to convert vascular endothelial cells from an anticoagulant to a procoagulant phenotype [Dam‐Mieras et al., 1992; Pryzdial and Wright, 1994; Nicholson and Hajjar, 1999; Visseren et al., 2000]. The following four mechanisms may be involved.
-
(1)
The inhibition of anticoagulant/antithrombotic properties by reducing both the heparin sulfate proteoglycan synthesis and the expression of thrombomodulin by endothelial cells, with a consequently reduced activation of protein C [Nicholson and Hajjar, 1999].
-
(2)
The induction of procoagulant properties of the endothelium by changing the phospholipid exposure. Enhanced thrombin generation and secretion of von Willebrand factor by endothelial cells result in an increase of platelet binding to HSV‐ or CMV‐infected endothelium [Nicholson and Hajjar, 1999].
-
(3)
HSV1 and HSV2 and CMV can initiate the generation of thrombin directly on their surface envelopes through the incorporation of host‐cell‐derived tissue factor and procoagulant phospholipids. Furthermore, the virus can use this generated thrombin to enhance infection through protease activated receptor‐1 stimulation of target cells [Sutherland et al., 2012].
-
(4)
An increase in binding sites for inflammatory cells, such as granulocytes and platelets, can lead to a further shift of the endothelial cell surface from thromboresistance to a prothrombotic condition. These inflammatory cells produce procoagulant cytokines, which further induce the expression of prothrombotic endothelial cell proteins [Visser et al., 1988; Nicholson and Hajjar, 1998, 1999; Sutherland et al., 2007].
The infection of endothelial cells has also been demonstrated for hemorrhagic fever viruses like dengue, Marburg, Ebola, Crimean‐Congo, hantavirus, yellow fever, and Lassa fever [van Gorp et al., 1999; Geimonen et al., 2002; Schnittler and Feldmann, 2003; Khaiboullina et al., 2005b; Kunz, 2009]. Although some of these viruses can productively replicate in endothelial cells, much of the disease pathology, including the impairment of the vascular system, is thought to result from the release of mediators from the infected cells. These mediators alter the vascular function and trigger coagulation disorders [Marty et al., 2006]. For example, hantavirus is able to infect endothelial cells directly and induce the production of chemokines and cell adhesion molecules like IL‐8, IL‐6, GRO‐β, and ICAM [Song et al., 1999; Geimonen et al., 2002; Han et al., 2008]. Levels of von Willebrand factor and soluble tissue factor are increased in patients with severe dengue infection [Sosothikul et al., 2007], while upregulation of tissue factor transcription has also been reported [Huerta‐Zepeda et al., 2008]. However, evidence for the activation of the tissue factor pathway in dengue infections is both limited and conflicting [Mairuhu et al., 2003]. Furthermore, abnormal von Willebrand factor multimers were seen in dengue hemorrhagic fever, with a shift from high molecular‐weight to lower molecular‐weight multimers [Sosothikul et al., 2007]. There might be a role for the von Willebrand factor cleavage protease: “ADAMTS13.” Known to degrade von Willebrand multimers, ADAMTS13 serves as an anticoagulant protein, and increased von Willebrand factor degradation is associated with bleeding. Decreased ADAMTS13 activity could lead to the formation of large von Willebrand factor multimers and increased platelet activation. Lowered ADAMTS13 levels have been found in acute influenza and hantavirus (Puumala) infections [Akiyama et al., 2011; Laine et al., 2011]. In recent years, it has been demonstrated that the influenza virus and other respiratory viruses can modulate inflammation and activate coagulation in vitro [Visseren et al., 2000; Bouwman et al., 2002]. The tissue factor expression on endothelial cell surfaces after infection, which leads to a reduced clotting time, may be a direct virus effect but it may also be triggered by cytokines, such as IL‐6 [Visseren et al., 2000; Bouwman et al., 2002; Marsden, 2006].
In cases of human avian influenza infection, tissue factor gene expression was upegulated [Muramoto et al., 2006]. Elevated levels of von Willebrand factor were found in SARS‐infected humans, although soluble tissue factor levels were not elevated [Wu et al., 2006]. In viral hepatitis, activated endothelial cells and macrophages express distinct cell‐surface procoagulants, which are important for both the initiation and localization of fibrin deposition in virally induced liver disease. Fgl2/fibroleukin has the ability to cleave prothrombin to thrombin directly, and the increased expression of fibrinogen‐like protein 2/fibroleukin on infected endothelial cells and macrophages has been found in hepatitis B patients [Levy et al., 2000; Marsden et al., 2003]. Via this mechanism, it can bypass the tissue factor/factor VII pathway (extrinsic pathway). In fibrinogen‐like protein 2/fibroleukin deficient mice infected with murine hepatitis virus strain‐3, fibrin deposition and liver necrosis were markedly reduced, compared to controls [Marsden et al., 2003]. An experimental therapeutic study in Ebola‐infected primates has provided evidence that the tissue factor pathway is an important pathophysiological component of this hemorrhagic fever [Geisbert et al., 2003b]. After blocking the tissue factor pathway by recombinant nematode anticoagulant protein c2 (rNAPc2), the coagulation response was attenuated. Endothelial cells are probably not an early target of the Ebola virus [Geisbert et al., 2003a]. The infection of endothelial cells occurs after the onset of disseminated intravascular coagulation, indicating that the primary coagulation abnormalities in Ebola virus infection are not the result of endothelial cell infection [Geisbert et al., 2003c]. The coagulopathy seen in Ebola fever is probably caused by several factors. Data suggests that tissue factor expression and release from infected monocytes/macrophages, and the release of tissue factor‐bearing microparticles into the circulation, are the key inducers of coagulation abnormalities [Bray, 2005; Geisbert et al., 2003a; Hensley and Geisbert, 2005; Ruf, 2004]. Together with fibrin deposition this may lead to severe disseminated intravascular coagulation.
Coagulation proteins and markers of an activated coagulation system
Viral infections can alter the levels of a variety of coagulation proteins and may consequently lead to a prothrombotic state that could result in a thrombotic event. A procoagulant state may be present through increased levels of coagulation proteins like fibrinogen [Horan et al., 2001] or factor VII [Woodhouse et al., 1994]. The presence of elevated levels of factor VIII is a risk factor found in chronic HBV and HCV patients [Papatheodoridis et al., 2003]. Laboratory studies have shown that herpes viruses can facilitate factor Xa generation from the inactive precursor factor X, but only when factor VII/VIIa and calcium ions are present [Sutherland et al., 1997]. Raised coagulation markers have also been found in animal and human tissue. Increased numbers of intravascular thrombi and fibrin deposition in lungs were found in cases of influenza, avian influenza, and SARS infection, and these may well be the result of disseminated intravascular coagulation and microthrombosis [Hwang et al., 2005; Keller et al., 2006; Muramoto et al., 2006]. In SARS infections, pulmonary infarcts were observed [Hwang et al., 2005]. Fibrin depositions were also found in tissue from primates infected with the Ebola virus [Geisbert et al., 2003a, b]. In contrast to the activated coagulation system described above, activities of factors II, V, VII, VIII, IX, and X and factor XII were decreased in dengue while in hantavirus infections factors II, V, VIII, IX, and X seemed to be decreased [Lee, 1987]. However, results in dengue research are inconclusive [Mairuhu et al., 2003]. Decreased levels of these coagulation factors are associated with hemorrhage and might be due to the consumption or loss of these factors. During the convalescent phase of DHF, factor VIII levels and factor VIIa levels were increased, when compared to dengue fever (DF) [Sosothikul et al., 2007]. Elevated levels of factor XIa‐C1‐inhibitor complexes were found in patients with DHF, indicating the activation of coagulation [van Gorp et al., 2001]. Macaques infected with the Ebola virus showed decreased levels of factor VIIa [Geisbert et al., 2003b].
Prolonged clotting times have been reported in several viral infections and in non‐hemorrhagic and hemorrhagic viral infections, such as hantavirus and VZV infections [Canpolat and Bakir, 2002; Kurugol et al., 2000; Laine et al., 2010]. The activation of the coagulation system results in elevated levels of several systemic coagulation markers. Increased levels of D‐dimer, prothrombin fragment 1 + 2, thrombin–antithrombin complexes, and/or plasmin–alpha‐2‐antiplasmin complexes have been found in respiratory tract infections, influenza, SARS, HIV, HCV, VZV, and the hanta‐, Ebola‐, and dengue‐hemorrhagic viruses [Bray et al., 2001; Canpolat and Bakir, 2002; Crum‐Cianflone et al., 2008; Feffer et al., 1995; Geisbert et al., 2003a, b; Keller et al., 2006, 2007; Kurugol et al., 2000; Laine et al., 2010; Lee et al., 2003; Mairuhu et al., 2003; Schouten et al., 2010; van Gorp et al., 2001; Violi et al., 1995; Wu et al., 2003]. Patients with HCV infection had levels of prothrombin fragment 1 + 2 that were elevated significantly [Violi et al., 1995]. Increased thrombin–antithrombin to plasmin–alpha‐2‐antiplasmin ratios, indicating a balance shifted to a procoagulant state, were found in dengue patients and associated with an adverse clinical outcome [van Gorp et al., 2002]. Fatal cases of Ebola infection showed higher levels of d‐dimer and fibrin degradation products during the acute phase of the disease [Bray et al., 2001; Rollin et al., 2007]. The level of these coagulation products correlate with death, and disseminated intravascular coagulation may therefore be an early and important component of Ebola infection [Bray et al., 2001; Rollin et al., 2007].
From the above it can be concluded that, generally speaking, hemorrhagic viruses may lead to the activation of the coagulation system during the acute phase and ultimately lead to the consumption or loss of clotting factors, microthrombi causing organ failure, endothelial cell dysfunction and bleeding phenotype. However, several issues about the relationship between viral infections and the coagulation system remain unknown. For example, there are gaps in the knowledge of the effect that viral hemorrhagic fevers have on coagulation markers; most data come from studies on dengue virus infections. Furthermore, limited data exists on the duration of a procoagulant state during or after viral infections. For example, the elevated levels of procoagulant proteins in viral respiratory tract infections returned to baseline after two weeks [Keller et al., 2007]. In a study of children with dengue shock syndrome (DSS), fibrinogen levels decreased significantly two days after hospital admission and had returned to normal within a month [Wills et al., 2002].
Natural anticoagulant proteins: protein C, protein S, antithrombin, thrombomodulin, and heparin cofactor II
Several viral infections lead to deficiencies in the natural anticoagulants protein C, protein S, antithrombin, and heparin cofactor II. It is well known that these deficiencies are associated with an increased risk of thrombosis. Such deficiencies have been reported in respiratory tract infections in general, VZV, HIV, HBV, HCV, hantavirus, DSS, and Ebola virus infections [Bissuel et al., 1992; Canpolat and Bakir, 2002; Crum‐Cianflone et al., 2008; Erbe et al., 2003; Geisbert et al., 2003b; Kaba et al., 2003; Kurugol et al., 2000; Laine et al., 2010; Papatheodoridis et al., 2003; Wills et al., 2002]. Probable deficiency mechanisms include autoantibodies against protein C, protein S and antithrombin, as described in VZV infections [Josephson et al., 2001; van Ommen et al., 2002], as well as the leakage of these proteins through the endothelium of capillaries, such as in dengue shock [Wills et al., 2002], and the consumption of these natural anticoagulants [Esmon, 2004; Sosothikul et al., 2007; Laine et al., 2010]. Another underlying mechanism could be the decreased synthesis of clotting factors by the liver. In chronic viral hepatitis the degree of antithrombin and protein C deficiency was found to be strongly associated with advanced fibrosis of the liver [Papatheodoridis et al., 2003]. An increased (local) production of thrombomodulin may have an anticoagulant effect [Dahlback, 2005]. Thrombomodulin expression is increased on the surface of sinusoidal endothelial cells in both chronic hepatitis B and C patients [Zeniya et al., 1995]. Soluble thrombomodulin levels were also increased in patients with SARS and severe dengue infection [Wills et al., 2002; Liu et al., 2005; Sosothikul et al., 2007]. Furthermore, dengue virus promotes the expression of thrombomodulin in cultured endothelial cells [Jiang et al., 2007]. Circulating soluble thrombomodulin may reflect endothelial cell activation, however thrombomodulin is most active when it is bound to endothelium [Schouten et al., 2008].
Fibrinolysis
Impaired fibrinolysis (or a hypofibrinolytic state), and thus an elevated risk of thrombosis, has been reported in several viral infections. There are two mechanisms to consider here. Firstly, increased levels of plasminogen activator inhibitor‐1 have been found in influenza, SARS, VZV, CMV, dengue, and HIV infections [Woodroffe and Kuan, 1998; Kurugol et al., 2000; Koppel et al., 2002; Wills et al., 2002; Klein et al., 2005; Keller et al., 2006; Wu et al., 2006; Sosothikul et al., 2007; Schouten et al., 2010]. In HIV infection, elevated plasminogen activator inhibitor‐1 levels have been shown to be related to the metabolic syndrome and the use of protease inhibitors as part of the antiretroviral therapy [Koppel et al., 2002]. Secondly, plasminogen deficiency is present in patients with chronic viral hepatitis [Papatheodoridis et al., 2003]. Dengue virus infection is associated with a hyperfibrinolytic state due to an increase in the levels and activity of tissue‐plasminogen activator, which results in an increased breakdown of fibrin strands, and thus an elevated risk of hemorrhage. However, increased levels of plasminogen activator inhibitor‐1 have also been found in dengue virus infections and these correlate with disease severity [Huang et al., 2001; van Gorp et al., 2001, 2002; Wills et al., 2002; Mairuhu et al., 2003; Jiang et al., 2007; Sosothikul et al., 2007]. In hantavirus infections, enhanced fibrinolysis could compensate for the increased coagulation activity and contribute to clinical recovery, but this does not explain the bleeding complications seen in hantavirus cases [Laine et al., 2010]. In addition to this, increased plasma concentrations of tissue‐plasminogen activator and soluble thrombomodulin were found in SARS infections [Liu et al., 2005]. However assays quantifying tissue‐plasminogen activator and plasminogen activator inhibitor‐1 used to measure both the circulating proteins as the protein complexes. Which makes it hard to identify actual plasma levels. In patients infected with Crimean‐Congo hemorrhagic fever and dengue virus, thrombin‐activatable fibrinolysis inhibitor activity was decreased, which may have contributed to an imbalance in fibrinolysis [Mairuhu et al., 2003; Sonmez et al., 2007; Sosothikul et al., 2007]. The decreased thrombin‐activatable fibrinolysis inhibitor activity may have been due to liver dysfunction during the infection [Sonmez et al., 2007], but it may also suggest the consumption of this inhibitor by excessive thrombin formation [Mairuhu et al., 2003].
Antiphospholipid Antibodies
The relationship between antiphospholipid antibodies, thrombosis and infection is not fully clear. Antibodies against phospholipids during acute infection are a diverse group of autoantibodies against proteins bound to phospholipids. They only remain in the plasma for a short period and most of them are not related to clinical thrombosis [de Groot and Urbanus, 2012]. However the Antiphospholipid antibodies, directed against the plasma protein β2‐glycoprotein I, are associated with arterial or venous thrombosis and pregnancy complications [Ruiz‐Irastorza et al., 2002; Asherson et al., 2008; Espinosa et al., 2008]. Antiphospholipid antibodies have been associated with infections of parvovirus B19, several herpes viruses, such as CMV, EBV and VZV, HAV, HBV, HCV, and HIV [Yamazaki et al., 1991; Prieto et al., 1996; Violi et al., 1997; Kurugol et al., 2000; 2001; Ertem et al., 2001; Uthman and Gharavi, 2002; Youd et al., 2003; Yuste and Prieto, 2003; Cervera and Asherson, 2005; van Hal et al., 2005; Crum‐Cianflone et al., 2008; Sene et al., 2008]. Antiphospholipid antibodies that are frequently found in patients with chronic HCV infection have not only been implicated in HCV‐associated thrombosis, but also in thrombocytopenia [Prieto et al., 1996]. It has been suggested that antiphospholipid antibodies might participate in the process of fibrosis by promoting thrombosis in small intrahepatic vessels. Whether these antibodies contribute to clinically important thrombotic events during infection remains controversial [Prieto et al., 1996; Mangia et al., 1999; Harada et al., 2000; Josephson et al., 2001; Cervera and Asherson, 2005; Sene et al., 2008]. Gharavi et al. [2002] have shown that immunization with peptides derived from cytomegalovirus induced lupus anticoagulant activity and resulted in thrombotic complications. While the presence of anti‐β2GPI antibodies has been reported in CMV patients presenting with thrombosis [Delbos et al., 2007; de Groot and Urbanus, 2012].
DISCUSSION AND CLINICAL IMPLICATIONS
After reviewing the available literature, it has become clear that although much is known about the pathophysiological mechanisms behind the association between viral infections and alterations of the coagulation cascade, many questions still remain. It is not clear, for example, why some viruses have a strong influence on coagulation and are associated with thrombotic complications or bleeding, while in other viral infections this effect is limited. The complex interplay between the host, the virus (virulence), the vector and the environment (infection pressure in the community) defines the clinical presentation and outcome. This might explain the divergent clinical presentations of viral infections in different parts of the world. Furthermore, differences in clinical presentation could also be explained by the diverse tropism of viruses, such as for monocytes or endothelial cells for example. It is not always clear whether a virus exerts its effect through the direct infection of the target cell, through virus‐specific antibodies, or via inflammatory mediators. In addition to this, inherited host factors also play a role in disease severity.
For implementing specific therapeutic interventions, it is crucial to know how the hemostatic balance of an individual is affected during the course of the infection. This is extremely difficult to determine, particularly in acute infections, and this is the main reason that theoretically promising interventions (anti‐TNF, activated protein C) were less successful, disappointing even, in clinical practice. Coagulation disorders vary among the viral hemorrhagic fevers. Both coagulation and fibrinolysis are activated, but the degree of activation of the coagulation system is influenced by the ability of the host to effectively balance the counteracting effects [Geisbert and Jahrling, 2004]. It is quite possible that there are major similarities in mechanisms in which different viral infections interact with the coagulation pathway. Even though studies on the interaction of several viral infections with coagulation have been performed with different methods, and these were focused on various elements of the coagulation system, it now seems evident that these viruses interact with coagulation both in a common way as with specific features related to the specific virus. Furthermore, more research on the alteration of coagulation has been performed on some viruses, such as CMV for example, than on others. It is conceivable that other, less prevalent, viral infections exert the same effect on the coagulation cascade, but studies are lacking. In the case of filoviruses, little research has been performed, partly because of the hazards and logistical difficulties associated with collecting and processing blood samples in the remote regions of Africa where outbreaks usually occur. Furthermore, most data come from in vitro studies, which may not accurately reflect the situation in actual infections in humans. In vivo data are also not clear [Squizzato et al., 2005]. For example, Ebola infections have been induced in non‐human primates, although useful to determine pathophysiological mechanisms these cannot be considered representative of human infections because the disease in monkeys appears to develop more rapidly and takes a more severe clinical course [Rollin et al., 2007]. For some other viruses there are not yet any adequate animal models. The effect of viral infections on coagulation, and the resulting clinical picture, could be exacerbated by other factors, such as thrombophilia as a host factor for example, and the environment (i.e., infection pressure). Several studies have shown that in patients with thrombotic complications during CMV infection, additional thrombophilic factors were present, such as the factor V Leiden mutation or the prothrombin G20210A mutation [Fridlender et al., 2007; Lijfering et al., 2007]. It is possible that those patients would not have developed thrombosis without the co‐existing thrombophilia. In the case of respiratory viruses, seasonal variation in several hemostatic proteins may contribute to a more pronounced effect on coagulation and therefore the development of thrombosis. It has been documented that during winter, platelet count, fibrinogen, factor VII, and plasminogen activator inhibitor‐1 are elevated [Woodhouse et al., 1994; Frohlich et al., 1997; Crawford et al., 2003]. Naturally occurring seasonal variations may exist independently of viral infections. Because respiratory viruses are more common during winter, it has been suggested that the combination of a seasonal variation and a viral infection has a more pronounced effect on the coagulation system, resulting in a higher risk of thrombosis. It was found that the risk of thrombosis is elevated during the first two weeks of a respiratory tract infection [Smeeth et al., 2006]. Bleeding may result from a multifactorial process resulting from a combination of thrombocytopenia due to autoantibodies or the consumption of platelets, the consumption of clotting factors (local), fibrinolysis, and vascular damage or leakage. For most hemorrhagic viruses, infection leads to increased anticoagulant activity and hyperfibrinolysis, partly by activated endothelial cells. For example, data on dengue virus infections showed increased endothelial cell damage [Sosothikul et al., 2007], but there is confliction in the data [Martina et al., 2009].
Despite increasing data on the association of viral infections and the coagulation cascade, the pathophysiological mechanisms behind this association have not yet been elucidated fully for most viruses. Knowledge of the underlying mechanisms leading to thrombosis or bleeding is fundamental for the development of therapeutic strategies for the treatment of thrombohemorrhagic complications. Given the potential role that tissue factor may play in some of the thrombohemorrhagic complications of viral disease, therapeutic intervention at the tissue factor level, for example, or aimed at one of the critical cytokines that mediate its cellular expression, may alter the clinical course of these infections favourably. For example, in patients infected with dengue, pharmacological agents that block tissue factor may represent an important therapeutic approach [Huerta‐Zepeda et al., 2008]. In a study in rhesus monkeys infected with the Ebola virus, treatment with a recombinant inhibitor of factor VIIa/tissue factor showed a prolonged survival time and attenuation of the coagulation and proinflammatory response [Geisbert et al., 2003a]. In another study in rhesus monkeys infected with the Ebola virus, treatment with a recombinant human activated protein C improved the chances of survival significantly [Hensley et al., 2007]. Therapy reduced coagulopathy and decreased inflammation and viral replication. These examples indicate that therapeutic strategies targeted at the coagulation cascade seem promising. However, none of them are ready for phase 2 trials. Because many issues remain unanswered, there is an urgent need for more clinical and experimental studies. Furthermore, additional studies are needed to investigate further in several areas. These include the efficacy of prophylactic LMWH, the need to test for viral pathogens in patients with thrombosis in irregular places (such as the portal vein), prothrombotic intervention in patients with hemorrhagic viral infection and the proper treatment of patients with viral DIC. In summary, during the past decade the world has been confronted with outbreaks of old and new viral infections. These have often been accompanied by the activation of coagulation at different levels of the coagulation cascade, resulting in venous thrombosis, DIC with microvascular thrombosis and bleeding. Although direct interaction between the virus and the coagulation system occurs, coagulation activation is also indirectly influenced by cytokines. An imbalance of the coagulation cascade may be the result, either procoagulant or profibrinolytic. Generally speaking, chronic viral infections seem to be associated with thrombotic complications, while acute viral infections are associated with either thrombotic or hemorrhagic complications. However, therapeutic antiviral options are still limited and vaccines are often not available, which makes supportive treatment crucial in the clinical management of these often life‐threatening infections. Although evidence is still limited, patients with unexplained thrombosis in places other than in the extremities or the lungs (the portal vein, for example), as described in HAV, HCV, and CMV infections (see Table I), should be tested for those viral pathogens. Furthermore, because there is a strong association between several viral infections and antiphospholipid antibodies, as was discussed above, patients with newly found antiphospholipid antibodies should also be screened for viral pathogens, taking into account the local epidemiology and travel history. In patients admitted to hospital with severe viral infection, thrombosis prophylaxis with low molecular‐weight heparin should be started, unless there is an increased risk of bleeding. Furthermore, patients who are bedbound but not admitted to hospital should also be considered for treatment with low molecular‐weight heparin. In mild thrombocytopenia (with a platelet count >50 × 109/L) low molecular‐weight heparin is not contraindicated. Insufficient studies have been made in this field to support the recommendation. The guidelines of the American College of Chest Physicians, ACCP, can be consulted [Geerts et al., 2008; Eikelboom et al., 2012]. Patients with bleeding complications, with or without systemic inflammatory response syndrome, or sepsis due to viral infection, should receive prompt standard supportive care (such as fluid resuscitation or ventilatory support, for example), as stated in the treatment guidelines for severe sepsis or septic shock [Dellinger et al., 2008].
In severe bleeding complications the administration of plasma products and platelets may be necessary. Interventions with antithrombin have not been investigated sufficiently, but they might be useful in individual cases. Recombinant activated protein C proved to be useful in certain cases but larger trials did not show a beneficial effect and this treatment is no longer available. Furthermore the “novel” anticoagulants blocking thrombin or factor Xa might be of high potential, however the occurrence of bleeding complications can be a problem. Studies to investigate the value of these therapeutic agents are required, especially in dengue virus infections, given that this is the most prevalent virus causing hemorrhagic fever. Therapeutic intervention of the tissue factor pathway seems promising, but supporting evidence is still limited and not yet near large human tirals. Research should focus first on the development of antiviral agents and vaccines. A better understanding of the pathogenesis of coagulation disorders during infection is urgently needed to improve supportive care.
APPENDIX 1: COMPLETE SEARCH STRATEGY
We searched PubMed for articles regarding the relationship between viruses (alternative search strategy between brackets) and hemostatic parameters and trombotic complications. All possible combinations were used between the virus and coagulation groups:
| Virus |
| Avian influenza (H5N1) |
| Adenovirus |
| Beta 2 glycoprotein I (B2GPI) |
| Coronavirus (corona virus) |
| Crimean‐congo |
| Cytomegalovirus (CMV) |
| Dengue virus/dengue hemorrahic fever/dengue schock syndrome |
| Ebola |
| Epstein Barr (EBV) |
| Hantavirus |
| Hemorrhagic fever (hemorragic fever) |
| Hepatitis A/B/C (HAV, HBV, HCV) |
| Herpesvirus/herpes virus |
| Herpes simplex virus (HSV) |
| Human immunodeficiency virus (HIV) |
| Influenza |
| Junin (junin hemorrhagic fever) |
| Lassa/lassa fever |
| Marburg |
| Mexican flu (H1N1) |
| Parvovirus (B19) |
| Respiratory tract infections |
| Respiratory syncytial virus (RSV) |
| Rhinovirus |
| Severe acute respiratory syndrome (SARS) |
| Varicella zoster (VZV) |
| Viral hepatitis |
| Yellow fever |
| Coagulation |
| Activated protein C (APC) |
| Antiphospholipid antibodies/anticardiolipin antibodies/lupus anticoagulant |
| Antithrombin (AT) |
| Bleeding/hemorrhage/hemorrhage/bleeding complication |
| Coagulation |
| Coagulation factor/clotting factor |
| d‐dimer |
| Endothelial cell protein C receptor (EPCR) |
| Factor XI |
| Factor VIII |
| Fibrin/fibrinogen |
| Fibrinolysis |
| Hemorrhagic/hemorrhagic complications |
| Hemostasis/hemostasis |
| Heparin cofactor II |
| Heparan sulfate |
| Plasmin–α2‐antiplasmin (PAP) |
| Plasminogen activator inhibitor type I (PAI‐1) |
| Platelets |
| Protein C/protein S |
| Prothrombin fragment 1 + 2 (F1 + 2) |
| P‐selectin |
| Soluble intercellular adhesion molecule (sICAM) |
| Soluble vascular adhesion molecules (sVCAM) |
| Tissue factor (TF) |
| Tissue factor pathway inhibitor (TFPI) |
| TF‐factor VIIa |
| Thrombin–antithrombin (TAT) |
| Thrombin‐activatable fibrinolysis inhibitor (TAFI) |
| Thrombomodulin (TM) |
| Thrombosis/thrombotic complication |
| Tissue plasminogen activator (t‐PA) |
| von Willebrand factor (VWF) |
| Urokinase |
M. Goeijenbier and M. van Wissen contributed equally to the manuscript.
REFERENCES
- Abgueguen P, Delbos V, Chennebault JM, Payan C, Pichard E. 2003. Vascular thrombosis and acute cytomegalovirus infection in immunocompetent patients: Report of 2 cases and literature review. Clin Infect Dis 36:E134–E139. [DOI] [PubMed] [Google Scholar]
- Adalja AA. 2010. Hematemesis in a 2009 H1N1 influenza patient. Am J Emerg Med 28:846–854. [DOI] [PubMed] [Google Scholar]
- Agarwal PP, Cinti S, Kazerooni EA. 2009. Chest radiographic and CT findings in novel swine‐origin influenza A (H1N1) virus (S‐OIV) infection. AJR Am J Roentgenol 193:1488–1493. [DOI] [PubMed] [Google Scholar]
- Akiyama R, Komori I, Hiramoto R, Isonishi A, Matsumoto M, Fujimura Y. 2011. H1N1 influenza (swine flu)‐associated thrombotic microangiopathy with a markedly high plasma ratio of von Willebrand factor to ADAMTS13. Intern Med 50:643–647. [DOI] [PubMed] [Google Scholar]
- Alexeyev OA, Morozov VG, Efremov AG, Settergren B. 1994. A case of haemorrhagic fever with renal syndrome complicated by spleen haemorrhage. Scand J Infect Dis 26:491–492. [DOI] [PubMed] [Google Scholar]
- Alonzo MT, Lacuesta TL, Dimaano EM, Kurosu T, Suarez LA, Mapua CA, Akeda Y, Matias RR, Kuter DJ, Nagata S, Natividad FF, Oishi K. 2012. Platelet apoptosis and apoptotic platelet clearance by macrophages in secondary dengue virus infections. J Infect Dis 205:1321–1329. [DOI] [PubMed] [Google Scholar]
- Armstrong KL, Fraser DK, Faoagali JL. 1991. Gastrointestinal bleeding with influenza virus. Med J Aust 154:180–182. [DOI] [PubMed] [Google Scholar]
- Arnold R, Konig W. 2005. Respiratory syncytial virus infection of human lung endothelial cells enhances selectively intercellular adhesion molecule‐1 expression. J Immunol 174:7359–7367. [DOI] [PubMed] [Google Scholar]
- Asano Y, Sarukawa M, Idezuki T, Harada S, Kaji K, Nakasu I, Igarashi A. 2006. Multiple small pulmonary emboli associated with transient antiphospholipid syndrome in human parvovirus B19 infection. Clin Rheumatol 25:585–587. [DOI] [PubMed] [Google Scholar]
- Asherson RA, Cervera R, Merrill JT, Erkan D. 2008. Antiphospholipid antibodies and the antiphospholipid syndrome: Clinical significance and treatment. Semin Thromb Hemost 34:256–266. [DOI] [PubMed] [Google Scholar]
- Bauduer F, Blanc A, Cordon B. 2003. Deep vein thrombosis and acute cytomegalovirus infection: Case report and review of the literature. Blood Coagul Fibrinolysis 14:489–491. [DOI] [PubMed] [Google Scholar]
- Becker S, Fusco G, Fusco J, Balu R, Gangjee S, Brennan C, Feinberg J. 2004. HIV‐associated thrombotic microangiopathy in the era of highly active antiretroviral therapy: An observational study. Clin Infect Dis 39:S267–S275. [DOI] [PubMed] [Google Scholar]
- Belet N, AsIlioglu N, Kucukoduk S. 2003. Two cases of congenital cytomegalovirus infection associated with disseminated intravascular coagulation. Pediatr Int 45:593–594. [DOI] [PubMed] [Google Scholar]
- Bissuel F, Berruyer M, Causse X, Dechavanne M, Trepo C. 1992. Acquired protein S deficiency: Correlation with advanced disease in HIV‐1‐infected patients. J Acquir Immune Defic Syndr 5:484–489. [PubMed] [Google Scholar]
- Bouwman JJ, Visseren FL, Bosch MC, Bouter KP, Diepersloot RJ. 2002. Procoagulant and inflammatory response of virus‐infected monocytes. Eur J Clin Invest 32:759–766. [DOI] [PubMed] [Google Scholar]
- Bray M. 2005. Pathogenesis of viral hemorrhagic fever. Curr Opin Immunol 17:399–403. [DOI] [PubMed] [Google Scholar]
- Bray M, Hatfill S, Hensley L, Huggins JW. 2001. Haematological, biochemical and coagulation changes in mice, guinea‐pigs and monkeys infected with a mouse‐adapted variant of Ebola Zaire virus. J Comp Pathol 125:243–253. [DOI] [PubMed] [Google Scholar]
- Bunce PE, High SM, Nadjafi M, Stanley K, Liles WC, Christian MD. 2011. Pandemic H1N1 influenza infection and vascular thrombosis. Clin Infect Dis 52:e14–e17. [DOI] [PubMed] [Google Scholar]
- Calore EE, Uip DE, Perez NM. 2011. Pathology of the swine‐origin influenza A (H1N1) flu. Pathol Res Pract 207:86–90. [DOI] [PubMed] [Google Scholar]
- Canpolat C, Bakir M. 2002. A case of purpura fulminans secondary to transient protein C deficiency as a complication of chickenpox infection. Turk J Pediatr 44:148–151. [PubMed] [Google Scholar]
- Carballal G, Cossio PM, Laguens RP, Ponzinibbio C, Oubina JR, Meckert PC, Rabinovich A, Arana RM. 1981. Junin virus infection of guinea pigs: Immunohistochemical and ultrastructural studies of hemopoietic tissue. J Infect Dis 143:7–14. [DOI] [PubMed] [Google Scholar]
- Caton B, Diaz dO, Aldamiz‐Echebarria M, Viguri A. 1993. Haemolytic‐uraemic syndrome with thrombotic microangiopathy of the retina following cytomegalovirus infection: Postmortem findings. Postgrad Med J 69:653–655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cervera R, Asherson RA. 2005. Antiphospholipid syndrome associated with infections: Clinical and microbiological characteristics. Immunobiology 210:735–741. [DOI] [PubMed] [Google Scholar]
- Cevik MA, Erbay A, Bodur H, Gulderen E, Bastug A, Kubar A, Akinci E. 2008. Clinical and laboratory features of Crimean‐Congo hemorrhagic fever: Predictors of fatality. Int J Infect Dis 12:374–379. [DOI] [PubMed] [Google Scholar]
- Chi D, Henry J, Kelley J, Thorpe R, Smith JK, Krishnaswamy G. 2000. The effects of HIV infection on endothelial function. Endothelium 7:223–242. [DOI] [PubMed] [Google Scholar]
- Chong PY, Chui P, Ling AE, Franks TJ, Tai DY, Leo YS, Kaw GJ, Wansaicheong G, Chan KP, Ean Oon LL, Teo ES, Tan KB, Nakajima N, Sata T, Travis WD. 2004. Analysis of deaths during the severe acute respiratory syndrome (SARS) epidemic in Singapore: Challenges in determining a SARS diagnosis. Arch Pathol Lab Med 128:195–204. [DOI] [PubMed] [Google Scholar]
- Chun PK, Godfrey LJ. 1984. Unique selective right atrial hemorrhage with epidemic (Korean) hemorrhagic fever Am. Heart J. 108:410–412. [DOI] [PubMed] [Google Scholar]
- Cioc AM, Sedmak DD, Nuovo GJ, Dawood MR, Smart G, Magro CM. 2002. Parvovirus B19 associated adult Henoch Schonlein purpura. J Cutan Pathol 29:602–607. [DOI] [PubMed] [Google Scholar]
- Claas EC, Osterhaus AD, van Beek R, De Jong JC, Rimmelzwaan GF, Senne DA, Krauss S, Shortridge KF, Webster RG. 1998. Human influenza A H5N1 virus related to a highly pathogenic avian influenza virus. Lancet 351:472–477. [DOI] [PubMed] [Google Scholar]
- Cohen O, Mevorach D, Ackerman Z, Oren R. 1993. Thrombocytopenic purpura as a manifestation of acute hepatitis A. J Clin Gastroenterol 17:166–167. [DOI] [PubMed] [Google Scholar]
- Crawford VL, McNerlan SE, Stout RW. 2003. Seasonal changes in platelets, fibrinogen and factor VII in elderly people. Age Ageing 32:661–665. [DOI] [PubMed] [Google Scholar]
- Crum‐Cianflone NF, Weekes J, Bavaro M. 2008. Review: Thromboses among HIV‐infected patients during the highly active antiretroviral therapy era. AIDS Patient Care STDS 22:771–778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cummins D, Fisher‐Hoch SP, Walshe KJ, Mackie IJ, McCormick JB, Bennett D, Perez G, Farrar B, Machin SJ. 1989. A plasma inhibitor of platelet aggregation in patients with Lassa fever. Br J Haematol 72:543–548. [DOI] [PubMed] [Google Scholar]
- Dahlback B. 2005. Blood coagulation and its regulation by anticoagulant pathways: Genetic pathogenesis of bleeding and thrombotic diseases. J Intern Med 257:209–223. [DOI] [PubMed] [Google Scholar]
- Dam‐Mieras MC, Muller AD, van Hinsbergh VW, Mullers WJ, Bomans PH, Bruggeman CA. 1992. The procoagulant response of cytomegalovirus infected endothelial cells. Thromb Haemost 68:364–370. [PubMed] [Google Scholar]
- Davison AM, Thomson D, Robson JS. 1973. Intravascular coagulation complicating influenza A virus infection. Br Med J 1:654–655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delbos V, Abgueguen P, Chennebault JM, Fanello S, Pichard E. 2007. Acute cytomegalovirus infection and venous thrombosis: Role of antiphospholipid antibodies. J Infect 54:e47–e50. [DOI] [PubMed] [Google Scholar]
- Dellinger RP, Levy MM, Carlet JM, Bion J, Parker MM, Jaeschke R, Reinhart K, Angus DC, Brun‐Buisson C, Beale R, Calandra T, Dhainaut JF, Gerlach H, Harvey M, Marini JJ, Marshall J, Ranieri M, Ramsay G, Sevransky J, Thompson BT, Townsend S, Vender JS, Zimmerman JL, Vincent JL. 2008. Surviving sepsis campaign: International guidelines for management of severe sepsis and septic shock 2008. Intensive Care Med 34:17–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doi T, Homma H, Mezawa S, Kato J, Kogawa K, Sakamaki S, Niitsu Y. 2002. Mechanisms for increment of platelet associated IgG and platelet surface IgG and their implications in immune thrombocytopenia associated with chronic viral liver disease. Hepatol Res 24:23. [DOI] [PubMed] [Google Scholar]
- Dunst R, Mettang T, Kuhlmann U. 1998. Severe thrombocytopenia and response to corticosteroids in a case of nephropathia epidemica. Am J Kidney Dis 31:116–120. [DOI] [PubMed] [Google Scholar]
- Eikelboom JW, Hirsh J, Spencer FA, Baglin TP, Weitz JI. 2012. Antiplatelet drugs: Antithrombotic Therapy and Prevention of Thrombosis, 9th ed: American College of Chest Physicians Evidence‐Based Clinical Practice Guidelines. Chest. 14:e89S–119S. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Erbe M, Rickerts V, Bauersachs RM, Lindhoff‐Last E. 2003. Acquired protein C and protein S deficiency in HIV‐infected patients. Clin Appl Thromb Hemost 9:325–331. [DOI] [PubMed] [Google Scholar]
- Ertem D, Acar Y, Pehlivanoglu E. 2001. Autoimmune complications associated with hepatitis A virus infection in children. Pediatr Infect Dis J 20:809–811. [DOI] [PubMed] [Google Scholar]
- Esmon CT. 2004. The impact of the inflammatory response on coagulation. Thromb Res 114:321–327. [DOI] [PubMed] [Google Scholar]
- Espinosa G, Bucciarelli S, Asherson RA, Cervera R. 2008. Morbidity and mortality in the catastrophic antiphospholipid syndrome: Pathophysiology, causes of death, and prognostic factors. Semin Thromb Hemost 34:290–294. [DOI] [PubMed] [Google Scholar]
- Feffer SE, Fox RL, Orsen MM, Harjai KJ, Glatt AE. 1995. Thrombotic tendencies and correlation with clinical status in patients infected with HIV. South Med J 88:1126–1130. [DOI] [PubMed] [Google Scholar]
- Fridlender ZG, Khamaisi M, Leitersdorf E. 2007. Association between cytomegalovirus infection and venous thromboembolism. Am J Med Sci 334:111–114. [DOI] [PubMed] [Google Scholar]
- Friis‐Moller N, Reiss P, Sabin CA, Weber R, Monforte A, El Sadr W, Thiebaut R, De Wit S, Kirk O, Fontas E, Law MG, Phillips A, Lundgren J.D., 2007. Class of antiretroviral drugs and the risk of myocardial infarction. N Engl J Med 356:1723–1735. [DOI] [PubMed] [Google Scholar]
- Frohlich M, Sund M, Russ S, Hoffmeister A, Fischer HG, Hombach V, Koenig W. 1997. Seasonal variations of rheological and hemostatic parameters and acute‐phase reactants in young, healthy subjects. Arterioscler Thromb Vasc Biol 17:2692–2697. [DOI] [PubMed] [Google Scholar]
- Gavrilovskaya IN, Gorbunova EE, Mackow ER. 2010. Pathogenic hantaviruses direct the adherence of quiescent platelets to infected endothelial cells. J Virol 84:4832–4839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gavrilovskaya IN, Peresleni T, Geimonen E, Mackow ER. 2002. Pathogenic hantaviruses selectively inhibit beta3 integrin directed endothelial cell migration Arch. Virol. 147:1913–1931. [DOI] [PubMed] [Google Scholar]
- Geerts WH, Bergqvist D, Pineo GF, Heit JA, Samama CM, Lassen MR, Colwell CW. 2008. Prevention of venous thromboembolism: American College of Chest Physicians Evidence‐Based Clinical Practice Guidelines (8th edition). Chest 133:381S–453S. [DOI] [PubMed] [Google Scholar]
- Geimonen E, Neff S, Raymond T, Kocer SS, Gavrilovskaya IN, Mackow ER. 2002. Pathogenic and nonpathogenic hantaviruses differentially regulate endothelial cell responses. Proc Natl Acad Sci USA 99:13837–13842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geisbert TW, Jahrling PB. 2004. Exotic emerging viral diseases: Progress and challenges. Nat Med 10:S110–S121. [DOI] [PubMed] [Google Scholar]
- Geisbert TW, Hensley LE, Jahrling PB, Larsen T, Geisbert JB, Paragas J, Young HA, Fredeking TM, Rote WE, Vlasuk GP. 2003a. Treatment of Ebola virus infection with a recombinant inhibitor of factor VIIa/tissue factor: A study in rhesus monkeys. Lancet 362:1953–1958. [DOI] [PubMed] [Google Scholar]
- Geisbert TW, Young HA, Jahrling PB, Davis KJ, Kagan E, Hensley LE. 2003b. Mechanisms underlying coagulation abnormalities in ebola hemorrhagic fever: Overexpression of tissue factor in primate monocytes/macrophages is a key event. J Infect Dis 188:1618–1629. [DOI] [PubMed] [Google Scholar]
- Geisbert TW, Young HA, Jahrling PB, Davis KJ, Larsen T, Kagan E, Hensley LE. 2003c. Pathogenesis of Ebola hemorrhagic fever in primate models: evidence that hemorrhage is not a direct effect of virus‐induced cytolysis of endothelial cells. Am J Pathol 163:2371–2382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gharavi AE, Pierangeli SS, Espinola RG, Liu X, Colden‐Stanfield M, Harris EN. 2002. Antiphospholipid antibodies induced in mice by immunization with a cytomegalovirus‐derived peptide cause thrombosis and activation of endothelial cells in vivo. Arthritis Rheum 46:545–552. [DOI] [PubMed] [Google Scholar]
- Gilbert CR, Vipul K, Baram M. 2010. Novel H1N1 influenza A viral infection complicated by alveolar hemorrhage. Respir Care 55:623–625. [PubMed] [Google Scholar]
- de Groot PG, Urbanus RT. 2012. The significance of auto‐antibodies against beta2‐Glycoprotein I. Blood, 120:266–274. [DOI] [PubMed] [Google Scholar]
- Han Q, Zhang L, Liu Z, Kang W, Lou S, Qiu J, Li Z, Zhang G, Wang Y, Li M, Li N. 2008. Elevated sICAM‐1 levels in patients with hemorrhagic fever with renal syndrome caused by Hantaan virus. Eur J Clin Microbiol Infect Dis 29:1507–1511. [DOI] [PubMed] [Google Scholar]
- Harada M, Fujisawa Y, Sakisaka S, Kawaguchi T, Taniguchi E, Sakamoto M, Sumie S, Sasatomi K, Koga H, Torimura T, Ueno T, Gondo K, Yoshida H, Tanikawa K, Sata M. 2000. High prevalence of anticardiolipin antibodies in hepatitis C virus infection: Lack of effects on thrombocytopenia and thrombotic complications. J Gastroenterol 35:272–277. [DOI] [PubMed] [Google Scholar]
- Harms PW, Schmidt LA, Smith LB, Newton DW, Pletneva MA, Walters LL, Tomlins SA, Fisher‐Hubbard A, Napolitano LM, Park PK, Blaivas M, Fantone J, Myers JL, Jentzen JM. 2010. Autopsy findings in eight patients with fatal H1N1 influenza. Am J Clin Pathol 134:27–35. [DOI] [PubMed] [Google Scholar]
- Hensley LE, Geisbert TW. 2005. The contribution of the endothelium to the development of coagulation disorders that characterize Ebola hemorrhagic fever in primates. Thromb Haemost 94:254–261. [DOI] [PubMed] [Google Scholar]
- Hensley LE, Stevens EL, Yan SB, Geisbert JB, Macias WL 2007. Recombinant human activated protein C for the postexposure treatment of Ebola hemorrhagic fever J. Infect. Dis. 196 Suppl 2:S390–S399. [DOI] [PubMed] [Google Scholar]
- Horan JT, Francis CW, Falsey AR, Kolassa J, Smith BH, Hall WJ. 2001. Prothrombotic changes in hemostatic parameters and C‐reactive protein in the elderly with winter acute respiratory tract infections. Thromb Haemost 85:245–249. [PubMed] [Google Scholar]
- Huang YH, Liu CC, Wang ST, Lei HY, Liu HL, Lin YS, Wu HL, Yeh TM. 2001. Activation of coagulation and fibrinolysis during dengue virus infection. J Med Virol 63:247–251. [DOI] [PubMed] [Google Scholar]
- Huerta‐Zepeda A, Cabello‐Gutierrez C, Cime‐Castillo J, Monroy‐Martinez V, Manjarrez‐Zavala ME, Gutierrez‐Rodriguez M, Izaguirre R, Ruiz‐Ordaz BH. 2008. Crosstalk between coagulation and inflammation during Dengue virus infection. Thromb Haemost 99:936–943. [DOI] [PubMed] [Google Scholar]
- Humblot S, Martin T, Pasquali JL, Korganow AS. 2001. Blood coagulation disorders during primary cytomegalovirus infection. Arch Intern Med 161:2149–2150. [DOI] [PubMed] [Google Scholar]
- Hwang DM, Chamberlain DW, Poutanen SM, Low DE, Asa SL, Butany J. 2005. Pulmonary pathology of severe acute respiratory syndrome in Toronto. Mod Pathol 18:1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inacio C, Hillaire S, Valla D, Denninger MH, Casadevall N, Erlinger S. 1997. Case report: Cytomegalovirus infection as a cause of acute portal vein thrombosis. J Gastroenterol Hepatol 12:287–288. [DOI] [PubMed] [Google Scholar]
- Jiang Z, Tang X, Xiao R, Jiang L, Chen X. 2007. Dengue virus regulates the expression of hemostasis‐related molecules in human vein endothelial cells. J Infect 55:e23–e28. [DOI] [PubMed] [Google Scholar]
- Jonsson CB, Figueiredo LT, Vapalahti O. 2010. A global perspective on hantavirus ecology, epidemiology, and disease. Clin Microbiol Rev 23:412–441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Josephson C, Nuss R, Jacobson L, Hacker MR, Murphy J, Weinberg A, Manco‐Johnson MJ. 2001. The varicella‐autoantibody syndrome. Pediatr Res 50:345–352. [DOI] [PubMed] [Google Scholar]
- Kaba NK, Francis CW, Hall WJ, Falsey AR, Smith BH. 2003. Protein S declines during winter respiratory infections. J Thromb Haemost 1:729–734. [DOI] [PubMed] [Google Scholar]
- Kahane S, Dvilansky A, Estok L, Nathan I, Zolotov Z, Sarov I. 1981. Detection of anti‐platelet antibodies in patients with idiopathic thrombocytopenic purpura (ITP) and in patients with rubella and herpes group viral infections. Clin Exp Immunol 44:49–56. [PMC free article] [PubMed] [Google Scholar]
- Kaneko H, Ohkawara Y, Nomura K, Horiike S, Taniwaki M. 2004. Relapse of idiopathic thrombocytopenic purpura caused by influenza A virus infection: A case report. J Infect Chemother 10:364–366. [DOI] [PubMed] [Google Scholar]
- Kaushansky K. 1998. Thrombopoietin. N Engl J Med 339:746–754. [DOI] [PubMed] [Google Scholar]
- Kazatchkine MD, Lambre CR, Kieffer N, Maillet F, Nurden AT. 1984. Membrane‐bound hemagglutinin mediates antibody and complement‐dependent lysis of influenza virus‐treated human platelets in autologous serum. J Clin Invest 74:976–984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kazory A, Ducloux D, Chalopin JM. 2004. Cytomegalovirus‐associated bleeding diathesis in renal transplant recipients. Transpl Infect Dis 6:81–83. [DOI] [PubMed] [Google Scholar]
- Keller TT, Mairuhu AT, de Kruif MD, Klein SK, Gerdes VE, ten Cate H, Brandjes DP, Levi M, van Gorp EC. 2003. Infections and endothelial cells. Cardiovasc Res 60:40–48. [DOI] [PubMed] [Google Scholar]
- Keller TT, van der Sluijs KF, de Kruif MD, Gerdes VE, Meijers JC, Florquin S, van der PT, van Gorp EC, Brandjes DP, Buller HR, Levi M. 2006. Effects on coagulation and fibrinolysis induced by influenza in mice with a reduced capacity to generate activated protein C and a deficiency in plasminogen activator inhibitor type 1. Circ Res 99:1261–1269. [DOI] [PubMed] [Google Scholar]
- Keller TT, van Wissen M, Mairuhu AT, van Doornum GJ, Brandjes DP. 2007. Acute respiratory tract infections in elderly patients increase systemic levels of hemostatic proteins. J Thromb Haemost 5:1567–1569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khaiboullina SF, Morzunov SP, St Jeor SC. 2005a. Hantaviruses: Molecular biology, evolution and pathogenesis. Curr Mol Med 5:773–790. [DOI] [PubMed] [Google Scholar]
- Khaiboullina SF, Rizvanov AA, Holbrook MR, St Jeor S. 2005b. Yellow fever virus strains Asibi and 17D‐204 infect human umbilical cord endothelial cells and induce novel changes in gene expression. Virology 342:167–176. [DOI] [PubMed] [Google Scholar]
- Klein SK, Slim EJ, de Kruif MD, Keller TT, ten Cate H, van Gorp EC, Brandjes DP. 2005. Is chronic HIV infection associated with venous thrombotic disease? A systematic review. Neth J Med 63:129–136. [PubMed] [Google Scholar]
- Koppel K, Bratt G, Schulman S, Bylund H, Sandstrom E. 2002. Hypofibrinolytic state in HIV‐1‐infected patients treated with protease inhibitor‐containing highly active antiretroviral therapy. J Acquir Immune Defic Syndr 29:441–449. [DOI] [PubMed] [Google Scholar]
- Kortepeter MG, Bausch DG, Bray M. 2011. Basic clinical and laboratory features of filoviral hemorrhagic feverJ. Infect Dis 204 Suppl 3:S810–S816. [DOI] [PubMed] [Google Scholar]
- Korteweg C, Gu J. 2008. Pathology, molecular biology, and pathogenesis of avian influenza A (H5N1) infection in humans. Am J Pathol 172:1155–1170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kowdley KV. 2005. Hematologic side effects of interferon and ribavirin therapy. J Clin Gastroenterol 39:S3–S8. [DOI] [PubMed] [Google Scholar]
- Kuiken T, Rimmelzwaan GF, Van Amerongen G, Osterhaus AD. 2003. Pathology of human influenza A (H5N1) virus infection in cynomolgus macaques (Macaca fascicularis). Vet Pathol 40:304–310. [DOI] [PubMed] [Google Scholar]
- Kunz S. 2009. The role of the vascular endothelium in arenavirus haemorrhagic fevers. Thromb Haemost 102:1024–1029. [DOI] [PubMed] [Google Scholar]
- Kurugol Z, Vardar F, Ozkinay F, Kavakli K, Cetinkaya B, Ozkinay C. 2001. Lupus anticoagulant and protein S deficiency in a child who developed disseminated intravascular coagulation in association with varicella. Turk J Pediatr 43:139–142. [PubMed] [Google Scholar]
- Kurugol Z, Vardar F, Ozkinay F, Kavakli K, Ozkinay C. 2000. Lupus anticoagulant and protein S deficiency in otherwise healthy children with acute varicella infection. Acta Paediatr 89:1186–1189. [DOI] [PubMed] [Google Scholar]
- Laine O, Makela S, Mustonen J, Huhtala H, Szanto T, Vaheri A, Lassila R, Joutsi‐Korhonen L. 2010. Enhanced thrombin formation and fibrinolysis during acute Puumala hantavirus infection. Thromb Res 126:154–158. [DOI] [PubMed] [Google Scholar]
- Laine O, Joutsi‐Korhonen L, Makela S, Mikkelsson J, Pessi T, Tuomisto S, Huhtala H, Libraty D, Vaheri A, Karhunen P, Mustonen J. 2011. Polymorphisms of PAI‐1 and platelet GP Ia may associate with impairment of renal function and thrombocytopenia in Puumala hantavirus infection. Thromb Res. 129:611–615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee M. 1987. Coagulopathy in patients with hemorrhagic fever with renal syndrome. J Korean Med Sci 2:201–211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee N, Hui D, Wu A, Chan P, Cameron P, Joynt GM, Ahuja A, Yung MY, Leung CB, To KF, Lui SF, Szeto CC, Chung S, Sung JJ. 2003. A major outbreak of severe acute respiratory syndrome in Hong Kong. N Engl J Med 348:1986–1994. [DOI] [PubMed] [Google Scholar]
- Levi M. 2007. Disseminated intravascular coagulation. Crit Care Med 35:2191–2195. [DOI] [PubMed] [Google Scholar]
- Levi M, van der Poll T, Buller HR. 2004. Bidirectional relation between inflammation and coagulation. Circulation 109:2698–2704. [DOI] [PubMed] [Google Scholar]
- Levi M, van der PT, Schultz M. 2012. New insights into pathways that determine the link between infection and thrombosis. Neth J Med 70:114–120. [PubMed] [Google Scholar]
- Levy GA, Liu M, Ding J, Yuwaraj S, Leibowitz J, Marsden PA, Ning Q, Kovalinka A, Phillips MJ. 2000. Molecular and functional analysis of the human prothrombinase gene (HFGL2) and its role in viral hepatitis. Am J Pathol 156:1217–1225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li X, Jeffers LJ, Garon C, Fischer ER, Scheffel J, Moore B, Reddy KR, Demedina M, Schiff ER. 1999. Persistence of hepatitis C virus in a human megakaryoblastic leukaemia cell line. J Viral Hepat 6:107–114. [DOI] [PubMed] [Google Scholar]
- Lijfering WM, Sprenger HG, van Son WJ, van der Meer J. 2007. Mesenteric vein thrombosis associated with primary cytomegalovirus infection: A case report. Blood Coagul Fibrinolysis 18:509–511. [DOI] [PubMed] [Google Scholar]
- Lijfering WM, de Vries AP, Veeger NJ, van Son WJ, Bakker SJ, van der Meer J. 2008. Possible contribution of cytomegalovirus infection to the high risk of (recurrent) venous thrombosis after renal transplantation. Thromb Haemost 99:127–132. [DOI] [PubMed] [Google Scholar]
- Lipinski S, Bremer L, Lammers T, Thieme F, Schreiber S, Rosenstiel P. 2011. Coagulation and inflammation. Molecular insights and diagnostic implications. Hamostaseologie 31:94–102, 104. [DOI] [PubMed] [Google Scholar]
- Liu ZH, Wei R, Wu YP, Lisman T, Wang ZX, Han JJ, Ren DL, Chen B, Xia ZL, Chen B, Zhu Z, Zhang Y, Cui X, Hu HT, de Groot PG, Xu WB. 2005. Elevated plasma tissue‐type plasminogen activator (t‐PA) and soluble thrombomodulin in patients suffering from severe acute respiratory syndrome (SARS) as a possible index for prognosis and treatment strategy. Biomed Environ Sci 18:260–264. [PubMed] [Google Scholar]
- Liu Z, Gao M, Han Q, Fang J, Zhao Q, Zhang N. 2008. Intensity of platelet beta(3) integrin in patients with hemorrhagic fever with renal syndrome and its correlation with disease severity. Viral Immunol 21:255–262. [DOI] [PubMed] [Google Scholar]
- Lutteke N, Raftery MJ, Lalwani P, Lee MH, Giese T, Voigt S, Bannert N, Schulze H, Kruger DH, Schonrich G. 2010. Switch to high‐level virus replication and HLA class I upregulation in differentiating megakaryocytic cells after infection with pathogenic hantavirus. Virology 405:70–80. [DOI] [PubMed] [Google Scholar]
- Magro CM, Crowson AN, Dawood M, Nuovo GJ. 2002. Parvoviral infection of endothelial cells and its possible role in vasculitis and autoimmune diseases. J Rheumatol 29:227–1235. [PubMed] [Google Scholar]
- Mahanty S, Bray M. 2004. Pathogenesis of filoviral haemorrhagic fevers. Lancet Infect Dis 4:487–498. [DOI] [PubMed] [Google Scholar]
- Mairuhu AT, Mac Gillavry, Setiati MR, Soemantri TE, ten A, Cate H, Brandjes DP, van Gorp EC. 2003. Is clinical outcome of dengue‐virus infections influenced by coagulation and fibrinolysis? A critical review of the evidence. Lancet Infect Dis 3:33–41. [DOI] [PubMed] [Google Scholar]
- Manco‐Johnson MJ, Nuss R, Key N, Moertel C, Jacobson L, Meech S, Weinberg A, Lefkowitz J. 1996. Lupus anticoagulant and protein S deficiency in children with postvaricella purpura fulminans or thrombosis. J Pediatr 128:319–323. [DOI] [PubMed] [Google Scholar]
- Mangia A, Margaglione M, Cascavilla I, Gentile R, Cappucci G, Facciorusso D, Grandone E, Di Minno G, Rizzetto M, Andriulli A. 1999. Anticardiolipin antibodies in patients with liver disease. Am J Gastroenterol 94:2983–2987. [DOI] [PubMed] [Google Scholar]
- Marsden PA. 2006. Inflammation and coagulation in the cardiovascular system: The contribution of influenza. Circ Res 99:1152–1153. [DOI] [PubMed] [Google Scholar]
- Marsden PA, Ning Q, Fung LS, Luo X, Chen Y, Mendicino M, Ghanekar A, Scott JA, Miller T, Chan CW, Chan MW, He W, Gorczynski RM, Grant DR, Clark DA, Phillips MJ, Levy GA. 2003. The Fgl2/fibroleukin prothrombinase contributes to immunologically mediated thrombosis in experimental and human viral hepatitis. J Clin Invest 112:58–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martina BE, Koraka P, Osterhaus AD. 2009. Dengue virus pathogenesis: An integrated view. Clin Microbiol Rev 22:564–581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marty AM, Jahrling PB, Geisbert TW. 2006. Viral hemorrhagic fevers. Clin Lab Med 26:345–386, viii. [DOI] [PubMed] [Google Scholar]
- Maslo C, Peraldi MN, Desenclos JC, Mougenot B, Cywiner‐Golenzer C, Chatelet FP, Jacomet C, Rondeau E, Rozenbaum W, Sraer JD. 1997. Thrombotic microangiopathy and cytomegalovirus disease in patients infected with human immunodeficiency virus. Clin Infect Dis 24:350–355. [DOI] [PubMed] [Google Scholar]
- Mason A, Wick M, White H, Perrillo R. 1993. Hepatitis B virus replication in diverse cell types during chronic hepatitis B virus infection. Hepatology 18:781–789. [DOI] [PubMed] [Google Scholar]
- Mauad T, Hajjar LA, Callegari GD, da Silva LF, Schout D, Galas FR, Alves VA, Malheiros DM, Auler JO, Jr. , Ferreira AF, Borsato MR, Bezerra SM, Gutierrez PS, Caldini ET, Pasqualucci CA, Dolhnikoff M, Saldiva PH. 2010. Lung pathology in fatal novel human influenza A (H1N1) infection. Am J Respir Crit Care Med 181:72–79. [DOI] [PubMed] [Google Scholar]
- Mayer JL, Beardsley DS. 1996. Varicella‐associated thrombocytopenia: Autoantibodies against platelet surface glycoprotein V. Pediatr Res 40:615–619. [DOI] [PubMed] [Google Scholar]
- McSorley J, Shapiro L, Brownstein MH, Hsu KC. 1974. Herpes simplex and varicella‐zoster: Comparative histopathology of 77 cases. Int J Dermatol 13:69–75. [DOI] [PubMed] [Google Scholar]
- Miller HC, Stephan M. 1993. Hemorrhagic varicella: A case report and review of the complications of varicella in children. Am J Emerg Med 11:633–638. [DOI] [PubMed] [Google Scholar]
- Mukhopadhyay S, Philip AT, Stoppacher R. 2010. Pathologic findings in novel influenza A (H1N1) virus (“swine flu”) infection: Contrasting clinical manifestations and lung pathology in two fatal cases. Am J Clin Pathol 133:380–387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muramoto Y, Ozaki H, Takada A, Park CH, Sunden Y, Umemura T, Kawaoka Y, Matsuda H, Kida H. 2006. Highly pathogenic H5N1 influenza virus causes coagulopathy in chickens. Microbiol Immunol 50:73–81. [DOI] [PubMed] [Google Scholar]
- Neau D, Bonnet F, Viallard JF, Longy‐Boursier M, Le Bras M. 1997. Thrombotic thrombocytopenic purpura and cytomegalovirus infection in an immunocompetent adult. Clin Infect Dis 25:1495–1496. [DOI] [PubMed] [Google Scholar]
- Nicholson AC, Hajjar DP. 1998. Herpesvirus in atherosclerosis and thrombosis: Etiologic agents or ubiquitous bystanders? Arterioscler Thromb Vasc Biol 18:339–348. [DOI] [PubMed] [Google Scholar]
- Nicholson AC, Hajjar DP. 1999. Herpesviruses and thrombosis: Activation of coagulation on the endothelium. Clin Chim Acta 286:23–29. [DOI] [PubMed] [Google Scholar]
- Niewold TB, Bundrick JB. 2006. Disseminated intravascular coagulation due to cytomegalovirus infection in an immunocompetent adult treated with plasma exchange. Am J Hematol 81:454–457. [DOI] [PubMed] [Google Scholar]
- Opal SM. 2003. Interactions between coagulation and inflammation. Scand J Infect Dis 35:545–554. [DOI] [PubMed] [Google Scholar]
- Othman M, Labelle A, Mazzetti I, Elbatarny HS, Lillicrap D. 2007. Adenovirus‐induced thrombocytopenia: The role of von Willebrand factor and P‐selectin in mediating accelerated platelet clearance. Blood 109:2832–2839. [DOI] [PubMed] [Google Scholar]
- Panzer S, Seel E, Brunner M, Kormoczi GF, Schmid M, Ferenci P, Peck‐Radosavljevic M. 2006. Platelet autoantibodies are common in hepatitis C infection, irrespective of the presence of thrombocytopenia. Eur J Haematol 77:513–517. [DOI] [PubMed] [Google Scholar]
- Papatheodoridis GV, Papakonstantinou E, Andrioti E, Cholongitas E, Petraki K, Kontopoulou I, Hadziyannis SJ. 2003. Thrombotic risk factors and extent of liver fibrosis in chronic viral hepatitis. Gut 52:404–409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Passos AM, Treitinger A, Spada C. 2010. An overview of the mechanisms of HIV‐related thrombocytopenia. Acta Haematol 124:13–18. [DOI] [PubMed] [Google Scholar]
- Pekic S, Cvijovic G, Stojanovic M, Kendereski A, Micic D, Popovic V. 2005. Hypopituitarism as a late complication of hemorrhagic fever. Endocrine 26:79–82. [DOI] [PubMed] [Google Scholar]
- Persoons MC, Stals FS, van Dam Mieras MC, Bruggeman CA. 1998. Multiple organ involvement during experimental cytomegalovirus infection is associated with disseminated vascular pathology. J Pathol 184:103–109. [DOI] [PubMed] [Google Scholar]
- Petaja J. 2011. Inflammation and coagulation. An overview. Thromb Res 127:S34–S37. [DOI] [PubMed] [Google Scholar]
- Peters CJ, Liu CT, Anderson GW, Jr. , Morrill JC, Jahrling PB. 1989. Pathogenesis of viral hemorrhagic fevers: Rift Valley fever and Lassa fever contrasted. Rev Infect Dis 11:S743–S749. [DOI] [PubMed] [Google Scholar]
- Phinney PR, Fligiel S, Bryson YJ, Porter DD. 1982. Necrotizing vasculitis in a case of disseminated neonatal herpes simplex infection. Arch Pathol Lab Med 106:64–67. [PubMed] [Google Scholar]
- Poland SD, Rice GP, Dekaban GA. 1995. HIV‐1 infection of human brain‐derived microvascular endothelial cells in vitro. J Acquir Immune Defic Syndr Hum Retrovirol 8:437–445. [DOI] [PubMed] [Google Scholar]
- Prieto J, Yuste JR, Beloqui O, Civeira MP, Riezu JI, Aguirre B, Sangro B. 1996. Anticardiolipin antibodies in chronic hepatitis C: Implication of hepatitis C virus as the cause of the antiphospholipid syndrome. Hepatology 23:199–204. [DOI] [PubMed] [Google Scholar]
- Pryzdial EL, Wright JF. 1994. Prothrombinase assembly on an enveloped virus: Evidence that the cytomegalovirus surface contains procoagulant phospholipid. Blood 84:3749–3757. [PubMed] [Google Scholar]
- Rand ML, Wright JF. 1998. Virus‐associated idiopathic thrombocytopenic purpura. Transfus Sci 19:253–259. [DOI] [PubMed] [Google Scholar]
- Rice J, Resar LM. 1998. Hematologic abnormalities associated with influenza A infection: A report of 3 cases. Am J Med Sci 316:401–403. [DOI] [PubMed] [Google Scholar]
- Richmond JK, Baglole DJ. 2003. Lassa fever: Epidemiology, clinical features, and social consequences. BMJ 327:1271–1275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberts PJ, Cummins D, Bainton AL, Walshe KJ, Fisher‐Hoch SP, McCormick JB, Gribben JG, Machin SJ, Linch DC. 1989. Plasma from patients with severe Lassa fever profoundly modulates f‐met‐leu‐phe induced superoxide generation in neutrophils. Br J Haematol 73:152–157. [DOI] [PubMed] [Google Scholar]
- Rollin PE, Bausch DG, Sanchez A. 2007. Blood chemistry measurements and D‐Dimer levels associated with fatal and nonfatal outcomes in humans infected with Sudan Ebola virus. J Infect Dis 196:S364–S371. [DOI] [PubMed] [Google Scholar]
- Ruf W. 2004. Emerging roles of tissue factor in viral hemorrhagic fever. Trends Immunol 25:461–464. [DOI] [PubMed] [Google Scholar]
- Ruiz‐Irastorza G, Khamashta MA, Hughes GR. 2002. Hughes syndrome crosses boundaries. Autoimmun Rev 1:43–48. [DOI] [PubMed] [Google Scholar]
- Sainokami S, Abe K, Ishikawa K, Suzuki K. 2005. Influence of load of hepatitis A virus on disease severity and its relationship with clinical manifestations in patients with hepatitis A. J Gastroenterol Hepatol 20:1165–1175. [DOI] [PubMed] [Google Scholar]
- Scheurlen W, Ramasubbu K, Wachowski O, Hemauer A, Modrow S. 2001. Chronic autoimmune thrombopenia/neutropenia in a boy with persistent parvovirus B19 infection. J Clin Virol 20:173–178. [DOI] [PubMed] [Google Scholar]
- Schnittler HJ, Feldmann H. 2003. Viral hemorrhagic fever—A vascular disease? Thromb Haemost 89:967–972. [PubMed] [Google Scholar]
- Schouten M, Wiersinga WJ, Levi M, van der Poll T. 2008. Inflammation, endothelium, and coagulation in sepsis. J Leukoc Biol 83:536–545. [DOI] [PubMed] [Google Scholar]
- Schouten M, Sluijs KF, Gerlitz B, Grinnell BW, Roelofs JJ, Levi MM, van't Veer C. 2010. Activated protein C ameliorates coagulopathy but does not influence outcome in lethal H1N1 influenza: A controlled laboratory study. Crit Care 14:R65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seitsonen E, Hynninen M, Kolho E, Kallio‐Kokko H, Pettila V. 2006. Corticosteroids combined with continuous veno‐venous hemodiafiltration for treatment of hantavirus pulmonary syndrome caused by Puumala virus infection. Eur J Clin Microbiol Infect Dis 25:261–266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sene D, Piette JC, Cacoub P. 2008. Antiphospholipid antibodies, antiphospholipid syndrome and infections. Autoimmun Rev 7:272–277. [DOI] [PubMed] [Google Scholar]
- Sengul S, Bozkus Y, Kutlay S, Keven K, Erturk S, Erbay B. 2006. Acute cytomegalovirus infection complicated by venous thrombosis in a renal transplant recipient. Transplant Proc 38003A:3116–3117. [DOI] [PubMed] [Google Scholar]
- Shachor‐Meyouhas Y, Kassis I. 2010. Petechial rash with pandemic influenza (H1N1) infection. Pediatr Infect Dis J 29:480. [DOI] [PubMed] [Google Scholar]
- Smeeth L, Cook C, Thomas S, Hall AJ, Hubbard R, Vallance P. 2006. Risk of deep vein thrombosis and pulmonary embolism after acute infection in a community setting. Lancet 367:1075–1079. [DOI] [PubMed] [Google Scholar]
- Song JS, Min CH, Kang E, Yu SH. 1999. Expression of ICAM‐1 on the Hantaan virus‐infected human umbilical vein endothelial cells. Korean J Intern Med 14:47–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sonmez M, Aydin K, Durmus A, Sucu N, Yilmaz M, Akdogan E, Koksal I, Ovali E, Omay SB. 2007. Plasma activity of thrombin activatable fibrinolysis inhibitor in Crimean‐Congo hemorrhagic fever. J Infect 55:184–187. [DOI] [PubMed] [Google Scholar]
- Sosothikul D, Seksarn P, Pongsewalak S, Thisyakorn U, Lusher J. 2007. Activation of endothelial cells, coagulation and fibrinolysis in children with Dengue virus infection. Thromb Haemost 97:627–634. [PubMed] [Google Scholar]
- Soto‐Abraham MV, Soriano‐Rosas J, Diaz‐Quinonez A, Silva‐Pereyra J, Vazquez‐Hernandez P, Torres‐Lopez O, Roldan A, Cruz‐Gordillo A, Alonso‐Viveros P, Navarro‐Reynoso F. 2009. Pathological changes associated with the 2009 H1N1 virus. N Engl J Med 361:2001–2003. [DOI] [PubMed] [Google Scholar]
- Squizzato A, Gerdes VE, Buller HR. 2005. Effects of human cytomegalovirus infection on the coagulation system. Thromb Haemost 93:403–410. [DOI] [PubMed] [Google Scholar]
- Squizzato A, Ageno W, Cattaneo A, Brumana N. 2007. A case report and literature review of portal vein thrombosis associated with cytomegalovirus infection in immunocompetent patients. Clin. Infect. Dis. 44:e13–e16. [DOI] [PubMed] [Google Scholar]
- Squizzato A, Gerdes VE. 2012. Viral hepatitis and thrombosis: a narrative review Semin . Thromb. Hemost. 38:530–534. [DOI] [PubMed] [Google Scholar]
- Suh DC, Park JS, Park SK, Lee HK, Chang KH. 1995. Pituitary hemorrhage as a complication of hantaviral disease. AJNR Am J Neuroradiol 16:175–178. [PMC free article] [PubMed] [Google Scholar]
- Sundberg E, Hultdin J, Nilsson S, Ahlm C. 2011. Evidence of disseminated intravascular coagulation in a hemorrhagic fever with renal syndrome‐scoring models and severe illness. PLoS ONE 6:e21134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sutherland MR, Raynor CM, Leenknegt H, Wright JF, Pryzdial EL. 1997. Coagulation initiated on herpesviruses. Proc Natl Acad Sci USA 94:13510–13514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sutherland MR, Friedman HM, Pryzdial EL. 2007. Thrombin enhances herpes simplex virus infection of cells involving protease‐activated receptor 1. J Thromb Haemost 5:1055–1061. [DOI] [PubMed] [Google Scholar]
- Sutherland MR, Ruf W, Pryzdial EL. 2012. Tissue factor and glycoprotein C on herpes simplex virus type 1 are protease‐activated receptor 2 cofactors that enhance infection. Blood 119:3638–3645. [DOI] [PubMed] [Google Scholar]
- Tanaka M, Kamijo T, Koike K, Ueno I, Nakazawa Y, Kurokawa Y, Sakashita K, Komiyama A, Fujisawa K. 2003. Specific autoantibodies to platelet glycoproteins in Epstein‐Barr virus‐associated immune thrombocytopenia. Int J Hematol 78:168–170. [DOI] [PubMed] [Google Scholar]
- Tanir G, Aydemir C, Tuygun N, Kaya O, Yarali N. 2005. Immune thrombocytopenic purpura as sole manifestation in a case of acute hepatitis A. Turk J Gastroenterol 16:217–219. [PubMed] [Google Scholar]
- Terada H, Baldini M, Ebbe S, Madoff MA. 1966. Interaction of influenza virus with blood platelets. Blood 28:213–228. [PubMed] [Google Scholar]
- Uthman IW, Gharavi AE. 2002. Viral infections and antiphospholipid antibodies. Semin Arthritis Rheum 31:256–263. [DOI] [PubMed] [Google Scholar]
- van de Weg CA, van Gorp EC, Supriatna M, Soemantri A, Osterhaus AD, Martina BE. 2012. Evaluation of the 2009 WHO dengue case classification in an Indonesian pediatric cohort. Am J Trop Med Hyg 86:166–170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van der Poll T, de Boer JD, Levi M. 2011. The effect of inflammation on coagulation and vice versa. Curr Opin Infect Dis 24:273–278. [DOI] [PubMed] [Google Scholar]
- Van der Poll T, Levi M. 2012. Crosstalk between inflammation and coagulation: The lessons of sepsis. Curr Vasc Pharmacol. Epub ahead of print. jan 2012. [DOI] [PubMed] [Google Scholar]
- van Gorp EC, Suharti C, ten Cate H, Dolmans WM, van der Meer JW, ten Cate JW, Brandjes DP. 1999. Review: Infectious diseases and coagulation disorders. J Infect Dis 180:176–186. [DOI] [PubMed] [Google Scholar]
- van Gorp EC, Minnema MC, Suharti C, Mairuhu AT, Brandjes DP, ten Cate H, Hack CE, Meijers JC. 2001. Activation of coagulation factor XI, without detectable contact activation in dengue haemorrhagic fever. Br J Haematol 113:94–99. [DOI] [PubMed] [Google Scholar]
- van Gorp EC, Setiati TE, Mairuhu AT, Suharti C, Cate HH, Dolmans WM, van der Meer JW, Hack CE, Brandjes DP. 2002. Impaired fibrinolysis in the pathogenesis of dengue hemorrhagic fever. J Med Virol 67:549–554. [DOI] [PubMed] [Google Scholar]
- van Hal S, Senanayake S, Hardiman R. 2005. Splenic infarction due to transient antiphospholipid antibodies induced by acute Epstein‐Barr virus infection. J Clin Virol 32:245–247. [DOI] [PubMed] [Google Scholar]
- van Ommen CH, van Wijnen M, de Groot FG, van der Horst CM, Peters M. 2002. Postvaricella purpura fulminans caused by acquired protein S deficiency resulting from antiprotein S antibodies: Search for the epitopes. J Pediatr Hematol Oncol 24:413–416. [DOI] [PubMed] [Google Scholar]
- van Steijn JH, van Tol KM, van Essen LH, Gans RO. 2000. Disseminated intravascular coagulation as an unusual presentation of an Epstein‐Barr virus infection. Neth J Med 57:169–171. [DOI] [PubMed] [Google Scholar]
- Venkata C, Sampathkumar P, Afessa B. 2010. Hospitalized patients with 2009 H1N1 influenza infection: The Mayo Clinic experience. Mayo Clin Proc 85:798–805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Violi F, Ferro D, Basili S, Artini M, Valesini G, Levrero M, Cordova C. 1995. Increased rate of thrombin generation in hepatitis C virus cirrhotic patients. Relationship to venous thrombosis. J Investig Med 43:550–554. [PubMed] [Google Scholar]
- Violi F, Ferro D, Basili S. 1997. Hepatitis C virus, antiphospholipid antibodies, and thrombosis. Hepatology 25:782. [DOI] [PubMed] [Google Scholar]
- Visser MR, Tracy PB, Vercellotti GM, Goodman JL, White JG, Jacob HS. 1988. Enhanced thrombin generation and platelet binding on herpes simplex virus‐infected endothelium. Proc Natl Acad Sci USA 85:8227–8230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Visseren FL, Bouwman JJ, Bouter KP, Diepersloot RJ, de Groot PH, Erkelens DW. 2000. Procoagulant activity of endothelial cells after infection with respiratory viruses. Thromb Haemost 84:319–324. [PubMed] [Google Scholar]
- Weksler BB. 2007. Review article: The pathophysiology of thrombocytopenia in hepatitis C virus infection and chronic liver disease. Aliment Pharmacol Ther 26:13–19. [DOI] [PubMed] [Google Scholar]
- Whitaker AN, Bunce I, Graeme ER. 1974. Disseminated intravascular coagulation and acute renal failure in influenza A2 infection. Med J Aust 2:196–201. [DOI] [PubMed] [Google Scholar]
- Wills BA, Oragui EE, Stephens AC, Daramola OA, Dung NM, Loan HT, Chau NV, Chambers M, Stepniewska K, Farrar JJ, Levin M. 2002. Coagulation abnormalities in dengue hemorrhagic fever: Serial investigations in 167 Vietnamese children with Dengue shock syndrome. Clin Infect Dis 35:277–285. [DOI] [PubMed] [Google Scholar]
- Wiwanitkit V. 2008. Hemostatic disorders in bird flu infection. Blood Coagul Fibrinolysis 19:5–6. [DOI] [PubMed] [Google Scholar]
- Wolf K, Tsakiris DA, Weber R, Erb P, Battegay M. 2002. Antiretroviral therapy reduces markers of endothelial and coagulation activation in patients infected with human immunodeficiency virus type 1. J Infect Dis 185:456–462. [DOI] [PubMed] [Google Scholar]
- Wong RS, Wu A, To KF, Lee N, Lam CW, Wong CK, Chan PK, Ng MH, Yu LM, Hui DS, Tam JS, Cheng G, Sung JJ. 2003. Haematological manifestations in patients with severe acute respiratory syndrome: Retrospective analysis. BMJ 326:1358–1362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woodhouse PR, Khaw KT, Plummer M, Foley A, Meade TW. 1994. Seasonal variations of plasma fibrinogen and factor VII activity in the elderly: Winter infections and death from cardiovascular disease. Lancet 343:435–439. [DOI] [PubMed] [Google Scholar]
- Woodroffe SB, Kuan S. 1998. Human cytomegalovirus infection induces mRNA expression and secretion of plasminogen inhibitor type‐1 in endothelial cells. J Med Virol 55:268–271. [DOI] [PubMed] [Google Scholar]
- World Health Organization (WHO) . Dengue Hemorrhagic fever: diagnosis, treatment, prevention and control, 2nd edn. Geneva. 1997. [Google Scholar]
- Wu YP, Wei R, de Groot PG. 2003. SARS in Hong Kong. N Engl J Med 349:708–709. [DOI] [PubMed] [Google Scholar]
- Wu YP, Wei R, Liu ZH, Chen B, Lisman T, Ren DL, Han JJ, Xia ZL, Zhang FS, Xu WB, Preissner KT, de Groot PG. 2006. Analysis of thrombotic factors in severe acute respiratory syndrome (SARS) patients. Thromb Haemost 96:100–101. [DOI] [PubMed] [Google Scholar]
- Xu T, Qiao J, Zhao L, Wang G, He G, Li K, Tian Y, Gao M, Wang J, Wang H, Dong C. 2006. Acute respiratory distress syndrome induced by avian influenza A (H5N1) virus in mice. Am J Respir Crit Care Med 174:1011–1017. [DOI] [PubMed] [Google Scholar]
- Yamazaki M, Asakura H, Kawamura Y, Ohka T, Endo M, Matsuda T. 1991. Transient lupus anticoagulant induced by Epstein‐Barr virus infection. Blood Coagul Fibrinolysis 2:771–774. [DOI] [PubMed] [Google Scholar]
- Yang M, Ng MH, Li CK. 2005. Thrombocytopenia in patients with severe acute respiratory syndrome (review). Hematology 10:101–105. [DOI] [PubMed] [Google Scholar]
- Youd P, Main J, Jackson E. 2003. Cytomegalovirus infection and thrombosis: A causative association? J Infect 46:141–142. [DOI] [PubMed] [Google Scholar]
- Yuste JR, Prieto J. 2003. Anticardiolipin antibodies in chronic viral hepatitis. Do they have clinical consequences? Eur J Gastroenterol Hepatol 15:717–719. [DOI] [PubMed] [Google Scholar]
- Zeniya M, Fukata H, Toda G. 1995. Thrombomodulin expression of sinusoidal endothelial cells in chronic viral hepatitis. J Gastroenterol Hepatol 10:S77–S80. [DOI] [PubMed] [Google Scholar]
- Zhang YZ, He YW, Dai YA, Xiong Y, Zheng H, Zhou DJ, Li J, Sun Q, Luo XL, Cheng YL, Qin XC, Tian JH, Chen XP, Yu B, Jin D, Guo WP, Li W, Wang W, Peng JS, Zhang GB, Zhang S, Chen XM, Wang Y, Li MH, Li Z, Lu S, Ye C, de J, Xu J. 2012. Hemorrhagic fever caused by a novel Bunyavirus in China: Pathogenesis and correlates of fatal outcome. Clin Infect Dis 54:527–533. [DOI] [PubMed] [Google Scholar]