

Abstract
Japanese encephalitis virus (JEV) nonstructural protein 5 (NS5) exhibits a Type I interferon (IFN) antagonistic function. This study characterizes Type I IFN antagonism mechanism of NS5 protein, using proteomic approach. In human neuroblastoma cells, NS5 expression would suppress IFNβ‐induced responses, for example, expression of IFN‐stimulated genes PKR and OAS as well as STAT1 nuclear translocation and phosphorylation. Proteomic analysis showed JEV NS5 downregulating calreticulin, while upregulating cyclophilin A, HSP 60 and stress‐induced‐phosphoprotein 1. Gene silence of calreticulin raised intracellular Ca2+ levels while inhibiting nuclear translocalization of STAT1 and NFAT‐1 in response to IFNβ, thus, indicating calreticulin downregulation linked with Type I IFN antagonism of JEV NS5 via activation of Ca2+/calicineurin. Calcineurin inhibitor cyclosporin A attenuated NS5‐mediated inhibition of IFNβ‐induced responses, for example, IFN‐sensitive response element driven luciferase, STAT1‐dependent PKR mRNA expression, as well as phosphorylation and nuclear translocation of STAT1. Transfection with calcineurin (vs. control) siRNA enhanced nuclear translocalization of STAT1 and upregulated PKR expression in NS5‐expressing cells in response to IFNβ. Results prove Ca2+, calreticulin, and calcineurin involvement in STAT1‐mediated signaling as well as a key role of JEV NS5 in Type I IFN antagonism. This study offers insights into the molecular mechanism of Type I interferon antagonism by JEV NS5.
Keywords: Calreticulin, Interferon, Japanese encephalitis virus, Microbiology, Non‐structural protein 5, STAT1
Abbreviations
- CsA
cyclosporine A
- DEN
dengue
- IFN
interferon
- JEV
Japanese encephalitis virus
- MEM
minimum essential medium
- NS5
nonstructural protein 5
- Q‐TOF
quadruple‐TOF
- TBEV
tick‐borne encephalitis
- WNV
West Nile
- YF
yellow fever
1. Introduction
Japanese encephalitis virus (JEV) is a member of the Flaviviridae family comprising important human pathogens: dengue (DEN), yellow fever (YF), St. Louis encephalitis, West Nile (WNV), and tick‐borne encephalitis (TBEV) virus 1, 2. JEV infection involves the nervous system, for example, basal ganglia, brainstem, cerebellum, cerebral cortex, spinal cord, and thalamus 3, 4, 5. JEV causes severe CNS disease—for example, poliomyelitis‐like acute flaccid paralysis, aseptic meningitis, encephalitis 6—with 30% fatality rate. Around half the survivors show severe neurological sequelae 6. About 50 000 Japanese encephalitis cases with 10 000 deaths occur annually in East and Southeast Asia, along with northern Australia 1, 7.
The single, long ORF of JEV genome encodes structural proteins capsid (C), membrane (prM/M), and envelope (E) plus seven nonstructural proteins (NS1, NS2A, NS2B, NS3, NS4A, NS4B, NS5) 2. Mature viral proteins arise from proteolytic processing of this single polyprotein by viral NS2B‐NS3 and variant cellular proteases 2. NS5, multifunctional protein containing RNA‐dependent RNA polymerase and methyltransferase activity, is involved in viral RNA replication 8. Short hairpin RNA targeting JEV NS5 gene reduces levels of viral RNA genome and proteins, specifically and efficiently inhibiting JEV replication 9. JEV NS5 protein suppresses STAT1 nuclear translocation and tyrosine phosphorylation of Tyk2 and SATA1 that arise in response to IFN by blocking IFN‐stimulated (where IFN is interferon) JAK‐STAT signaling 10. WNV NS5 protein prevents phosphorylated STAT1 accumulation and blocks IFN‐dependent gene expression, then rescues growth of a highly IFN‐sensitive virus in the presence of IFN treatment 11. DEN NS5 inhibits IFN‐alpha, not IFN‐gamma, signaling via binding with and blocking phosphorylation of STAT2 12, 13. TBEV NS5 inhibits STAT1 phosphorylation in response to IFN via binding interaction with post synaptic density protein‐95/drosophila disc large tumor suppressor‐1/zonula occludens‐1 protein scribble (hScrib) affecting IFN Type I and II mediated JAK‐STAT signaling 8, 14, 15. Studies demonstrate flavivirus NS5 proteins as potent IFN antagonists of variant mechanisms.
Clinical, neurophysiological, and radiological examination of Japanese encephalitis patients has indicated the presence of JEV antigens and genomes in the basal ganglia, brainstem, cerebellum, cerebral cortex, spinal cord, and thalamus 3, 4, 5. TE671 neural cells derived from medulloblastoma serve to rate pathogenesis of the measles virus 16, 17. JEV NS2B‐NS3 protease demonstrably induces TE671 human medulloblastoma apoptosis 18, 19. IFN‐stimulated gene 15 (ISG15) overexpression in TE671 cells caused phosphorylation of IRF‐3 (Ser396), JAK2 (Tyr1007/1008), and STAT1 (Tyr701 and Ser727), along with activation of STAT1‐dependent genes, such as IRF‐3, IFN‐β, IL‐8, PKR and OAS, correlating with inhibited JEV replication 20. This study uses proteomic analysis of TE671 cell expressing JEV NS5 to identify unique protein profiling involved in IFN antagonism of JEV NS5 protein. Specific inhibitors of calreticulin/calcineurin pathway show reversal of STAT1‐mediated Type I IFN‐induced response in NS5‐expressing cells, correlating with inhibitory effects on JEV replication in cells and virus‐induced apoptosis.
2. Materials and methods
2.1. Viruses and cells
This study used JEV strain T1P1, as described earlier 21. BHK‐21 cells used for plaque assays were maintained in the minimum essential medium (MEM) with 10% FBS. Human TE671 medulloblastoma cells for transfection with empty vector and NS5‐expressing vector were grown in MEM with 2 mM l‐glutamine, 1 mM sodium pyruvate, and 10% FBS.
2.2. Expression of recombinant JEV NS5 protein and immunofluorescent staining
JEV NS5 Flag‐tagged pCR3.1 and its empty control vectors were supplied by Dr. Yi‐Ling Lin (Genomics Research Center, Academia Sinica, Taiwan), as described in a prior report 21. TE671 cells at 60–90% confluency in 6‐well plates were transfected with a mixture of GenePorter reagent and recombinant vector containing NS5 gene or empty vector for 5 h, then maintained in 2 mL of MEM containing 20% bovine serum for 2 days, as described previously 22. Transfected cell line was selected with a long‐term incubation of MEM containing 10% FBS and 800 μg/mL of G418. To detect NS5 expression in TE671 cells by immunofluorescent assay, transfected cells were fixed with cold methanol, incubated with diluted mouse anti‐Flag tag mAb for 2 h, followed by FITC‐conjugated anti‐mouse IgG antibodies for 2 h. Cells were subsequently stained with DAPI (Sigma) for 10 min. After three washings in PBS, photographs of cells were taken by immunofluorescent microscopy. To analyze subcellular localization of STAT1 and NFAT‐1, transfected cells were stained with primary antibodies anti‐STAT1 or NFAT‐1 (Cell Signaling), followed by incubation with secondary antibodies FITC‐conjugated anti‐mouse IgG plus DAPI, as described above. To quantify nuclear translocalization of STAT1 and NFAT‐1, green fluorescence intensity of 100 random cells and their nuclei in images were measured by Image Pro 6 software (Media Cybernetics), results expressed as fluorescence intensity ratio of nuclear area to cell area in counted cells.
2.3. Western blot
To assess NS5 expression, lysates of NS5‐expressing and vector control cell lines were resolved by SDS‐PAGE and transferred to NC, as we previously reported 23. Resulting blots were blocked with 5% skim milk and reacted with anti‐Flag tag mAb as probe; immune complexes were detected with HRP‐conjugated goat anti‐mouse IgG antibodies, followed by ECL detection (Amersham Pharmacia Biotech). To rate protein profiling in response to IFNβ, both NS5‐expressing and vector control cells were harvested postincubation with or without 1000 U/mL IFNβ for 2 days prior to Western blot with properly diluted antibodies: mouse anti‐Flag tag, anti‐cyclophilin A (Cell Signaling), anti‐calreticulin, and anti‐β actin antibodies (Invitrogen). For analyzing signal transduction in response to IFNβ and cyclosporin A (CsA), both NS5‐expressing and vector control cells were harvested postincubation with or without 1000 U/mL IFNβ and 10 μg/mL CsA for 30, 60, and 120 min prior to Western blot with rabbit anti‐STAT1 and antiphospho STAT1 (Tyr701) antibodies (Cell Signaling).
2.4. Transient transfection, dual‐luciferase reporter assay, and RNA interference
For IFN signaling pathway assay, dual‐luciferase reporter system with cis‐reporter plasmid pISRE‐Luc containing IFN‐sensitive response element (ISRE) and internal control reporter pRluc‐C1 was carried out, as we reported earlier 23. JEV NS5‐expressing and empty vector control cells were transfected with dual cis‐reporter plasmids using GenePorter reagent, then treated with or without 1000 U/mL IFNβ (Merck‐Serono). After 4‐h incubation, activity of firefly and Renilla luciferase was measured by dual Luciferase Reporter Assay System (Promega) and TROPIX TR‐717 Luminometer (Applied Biosystems). For silencing calreticulin and calcineurin, NS5‐expressing and vector control cells were transfected with control siRNA, ON‐TARGETplus Human CALR (811) siRNA (SMARTpool L‐008197‐00‐0005), or ON‐TARGETplus Human PPP3CA (5530) siRNA (SMARTpool L‐008300‐00‐0005). After 2‐day incubation, cells underwent further experiments, for example, mRNA expression of ISGs by real‐time PCR and subcellular localization of STAT1 and NFAT‐1.
2.5. Quantification of mRNA levels of IFNβ‐induced genes, using real time RT‐PCR
NS5‐expressing and vector control cells were transfected with control, calreticulin, and calcineurin siRNA, then treated with 1000 U/mL IFNβ or 10 μg/mL CsA for 4‐h incubation. Total RNAs from lysate of each experiment were isolated by a PureLink Micro‐to‐Midi Total RNA Purification System Kit (Invitrogen), 1000 ng of total RNA for cDNA synthesis using oligo dT primer and SuperScript III reverse transcriptase kit (Invitrogen). Two‐step RT‐PCR with SYBR Green I gauged mRNA levels of IFNβ‐induced genes. Primer pairs included 5′‐CAACCAGCGGTTGACT TTTT‐3′ and 5′‐ATCCAGGAAGGCAAACTGAA‐3′ for PKR, 5′‐GATGTGGTTAG GTTTATAGCTG‐3′ and 5′‐TTGGG GGTTAGGTTTCTGCCTTT‐3′ for OAS, 5′‐CT GCTGCGGCCCTTGTTATT‐3′ and 5′‐CATGGGCTGGGACCTGACGGTGA AG‐3′ for ISG15, 5′‐CCTGCCACCCAAGAAGATAAAG‐3′ and 5′‐GTTCCCACTCTCCGTCCATC‐3′ for calreticulin, 5′‐GATGCTGGTAAATGTC CTCAAC‐3′ and 5′‐CACACTCTCACTCTCTTCTCTG‐3′ for calcineurin, 5′‐TGCT GCCTCCAAGAACACAA‐3′ and 5′‐TGTAGAACTGCCGG AGCACA‐3′ for IL‐4, and 5′‐AGCCACATCGCTCAGACAC‐3′ and 5′‐GCCCAATACGACCAA ATCC‐3′ for housekeeping gene glyceraldehyde 3‐phosphate dehydrogenase (GAPDH). Real‐time PCR reaction contained 2.5 μL of cDNA (reverse transcription mixture), 200 nM of each primer in SYBR Green I master mix (LightCycler TaqMAn Master, Roche Diagnostics); it was performed by amplification protocol consisting of 1 cycle at 50°C for 2 min, 1 cycle at 95°C for 15 s and 60°C for 1 min. PCR product was quantified by ABI PRISM 7700 sequence detection system (PE Applied Biosystems), relative fold changes of PKR and OAS mRNA normalized by GAPDH mRNA.
2.6. 2DE, in‐gel digestion, and nanoelectrospray MS
2DE, protein spot analysis, in‐gel digestion, nanoelectrospray MS, data interpretation, and database searchgel electrophoresis were performed as we previously reported 24. Briefly, vector control and NS5‐expressing cells with or without 2‐day incubation of 1000 U/mL IFNβ were harvested and dissolved in lysis buffer containing 8 M urea, 4% CHAPS, 2% pH 3–10 nonlinear IPG buffer (GE Healthcare), and Complete, Mini, EDTA‐free protease inhibitor mixture (Roche). A 100 μg of protein sample of each lysate in rehydration buffer (8 M urea, 2% CHAPS, 0.5% IPG buffer pH 3–10 nonlinear, 18 mM dithiothreilol, 0.002% bromophenol blue) was applied to nonlinear Immobiline DryStrips (17 cm, pH 3–10; GE Healthcare). After IEF, gel strips were incubated for 30 min in equilibration solution I (6 M urea, 2% SDS, 30% glycerol, 1% DTT, 0.002% bromophenol blue, 50 mM Tris‐HCl, pH 8.8), then for 30 min in equilibration solution II (6 M urea, 2% SDS, 30% glycerol, 2.5% iodoacetamide, 0.002% bromophenol blue, 50 mM Tris‐HCl, pH 8.8). Gel strips were subsequently transferred to run 12% PAGE. Gels were fixed in 40% ethanol and 10% glacial acetic acid for 30 min, stained with AgNO3 solution for 20 min, and finally scanned by a GS‐800 imaging densitometer with PDQuest software version 7.1.1 (Bio‐Rad). Data from three independent lysates under each experimental condition were used to correct spot intensity graphs and statistical analysis (Microsoft Excel). In silver‐stained gels, each spot of interest showing statistically significant 1.5‐fold rise or fall in spot intensity was sliced, put into the microtube, and washed twice with 50% ACN in 100 mM ammonium bicarbonate buffer (pH 8.0) for 10 min at room temperature. In‐gel digestion method described previously 24 served to recover peptides from gel spots for nanoelectrospray MS. Proteins in spots of interest were identified by Ultimate Capillary LC system (LC Packings, Amsterdam) along with QSTARXL quadruple‐TOF (Q‐TOF) mass spectrometer (Applied Biosystem/MDS Sciex, Foster City, CA). Nanoelectrospray MS and database search were described earlier 25. Protein function and subcellular location were annotated using Swiss‐Prot (http://us.expasy.org/sprot/), proteins categorized according to biological process and pathway via PANTHER classification (http://www.pantherdb.org) described previously 24.
2.7. Detecting intracellular Ca2+ and subcellular localization of STAT1 and NFAT‐1 in siRNA transfected cells
Vector control and NS5‐expressing cells were transfected with control or calreticulin siRNA (SMARTpool L‐008197‐00‐0005, Thermo Scientific) in FBS‐free media overnight, harvested, washed twice with PBS, then incubated with 10 mg/mL FLUO3/AM (Sigma) at 37°C for 30 min in darkroom. After last wash with PBS, cells were analyzed by flow cytometry (excitation at 506 nm and emission at 526 nm; Becton Dickinson FACS Calibur). For subcellular localization assay, siRNA transfected cells were treated with or without 1000 U/mL IFNβ for 4 h, then tested by immunofluorescent staining with rabbit anti‐STAT1 or anti‐NFAT‐1 antibodies (Cell Signaling) described above.
2.8. Effects of calcineurin inhibitor and siRNA on IFNβ‐induced, STAT1‐mediated responses
Since IFNβ induced apoptosis of TE671 cells, vector control and NS5‐expressing cells were used for test their susceptibility to IFNβ, then treated singly or with both 10 μg/mL CsA (calcineurin inhibitor) and 1000 U/mL IFNβ for 48 h. IFNβ‐induced cytopathic effect of both cell types was photographed by light microscopy. Both types were transfected with control and calcineurin siRNA (SMARTpool L‐008300‐00‐ 0005, Thermo Scientific). Effect of CsA and calcineurin siRNA on cells in response to IFNβ was analyzed by assay for ISRE‐driven promoter activity, ISGs gene expression, phosphorylation, and subcellular location of STAT1, performed as described above.
2.9. Extracellular virus yield reduction assays
TE671 cells were infected with JEV at an multiplicity of infection (MOI) of 0.5 in the presence or absence of 500 U/mL IFNβ and/or 1000 ng/mL CsA. At 48 h post infection, cultured supernatants were harvested to calculate virus yield. BHK‐21 cell monolayers were incubated with serial dilution of cultured supernatant at 37°C for 1 h and overlaid with MEM containing 1.1% methylcellulose. After 72 h incubation, monolayers were stained with naphthol blue‐black dye and viral plaques counted.
3. Results
3.1. IFNβ antagonistic ability of JEV NS5 in TE 671 human medulloblastoma cells
Since JEV NS5 protein showed Type I IFN antagonistic ability in BHK‐21 cells 21, we further characterized JEV NS5 inhibitory effect on IFNβ‐induced response in TE671 human medulloblastoma cells. Recombinant plasmid pCR3.1‐JEV NS5 (expressing JEV NS5 with Flag tags) or empty control vector (expressing Flag tags), generated as the prior report 21, were first transfected into TE671 cells, JEV NS5 expression detected by immunofluorescent staining (Fig. 1A) and Western blot (Fig. 1B). Immunofluorescent staining with anti‐Flag antibodies and DAPI found vector‐derived Flag‐tag in both empty vector‐ and pCR3.1‐JEV NS5‐transfected cells (Fig 1A). In particular, most vector‐derived Flag‐tag fusion proteins in pCR3.1‐JEV NS5‐transfected cells were located in nucleus, consistent with prior reports in which NS5 protein of DEN‐2, YF and JEV was detected in nuclei of mammalian cells 26, 27, 28. Western blot of lysates with anti‐Flag‐tag antibodies revealed 103‐kDa band in pCR3.1‐JEV NS5‐transfected (Fig. 1B) but not in empty vector‐transfected cells, proving JEV NS5 stably expressed in human medulloblastoma cells.
Figure 1.
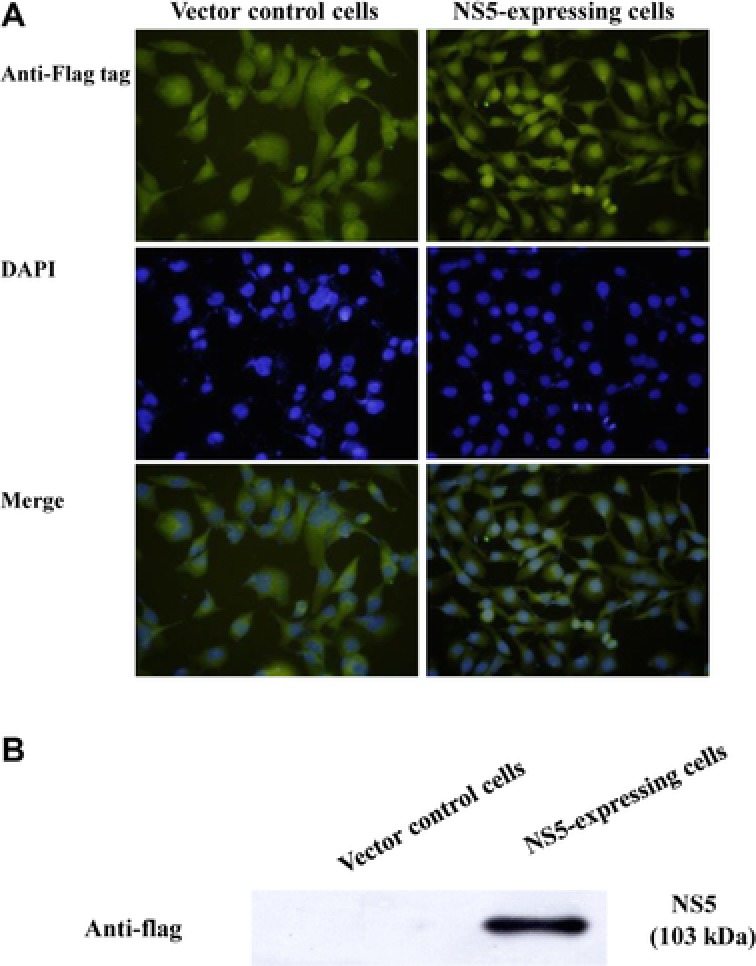
Expression of JEV NS5 protein in TE671 human medulloblastoma cells. Cells transfected with pCR3.1‐Flag (control vector; left) or pCR3.1‐JEV NS5‐Flag (right) were selected by 2‐wk incubation with G418. Expression of Flag‐epitope tagged protein was examined by fluorescence microscopy (A). Also, Western blot to detect expressed NS5 was conducted via anti‐Flag antibody (B). Lysates from cells transfected with pCR3.1‐Flag (lane 1) or pCR3.1‐JEV NS5‐Flag (lane 2) were analyzed by 10% SDS‐PAGE prior to blotting, resulting blots probed with anti‐Flag tag antibody.
Antagonistic ability of JEV NS5 on IFNβ‐induced ISRE‐driven promoter activity was analyzed by dual luciferase reporter assay. IFNβ induced significant increase of ISRE‐driven firefly luciferase activity in vector control cells, 20 times higher than in NS5‐expressing cells (Fig. 2A). Meanwhile, quantitative real‐time RT‐PCR assays indicated induction of ISRE‐driven genes PKR and OAS by IFNβ as approximately 2.5‐ and 3.5‐fold lower in NS5‐expressing cells than in vector controls, respectively (Fig. 2B). Western blot with antiphospho‐Tyr701 STAT1 antibodies also indicated JEV NS5, reducing IFNβ‐induced phosphorylation of STAT1 at Tyr701 (Fig. 2C, lanes 6–8). Results demonstrated JEV NS5 inhibiting IFNβ‐induced responses as well as showing its Type 1 IFN antagonistic activity in human medulloblastoma cells. Results concurred with a previous finding: NS5 blocked STAT1 nuclear translocation and tyrosine phosphorylation in BHK21 cells treated with Type I IFN 21.
Figure 2.
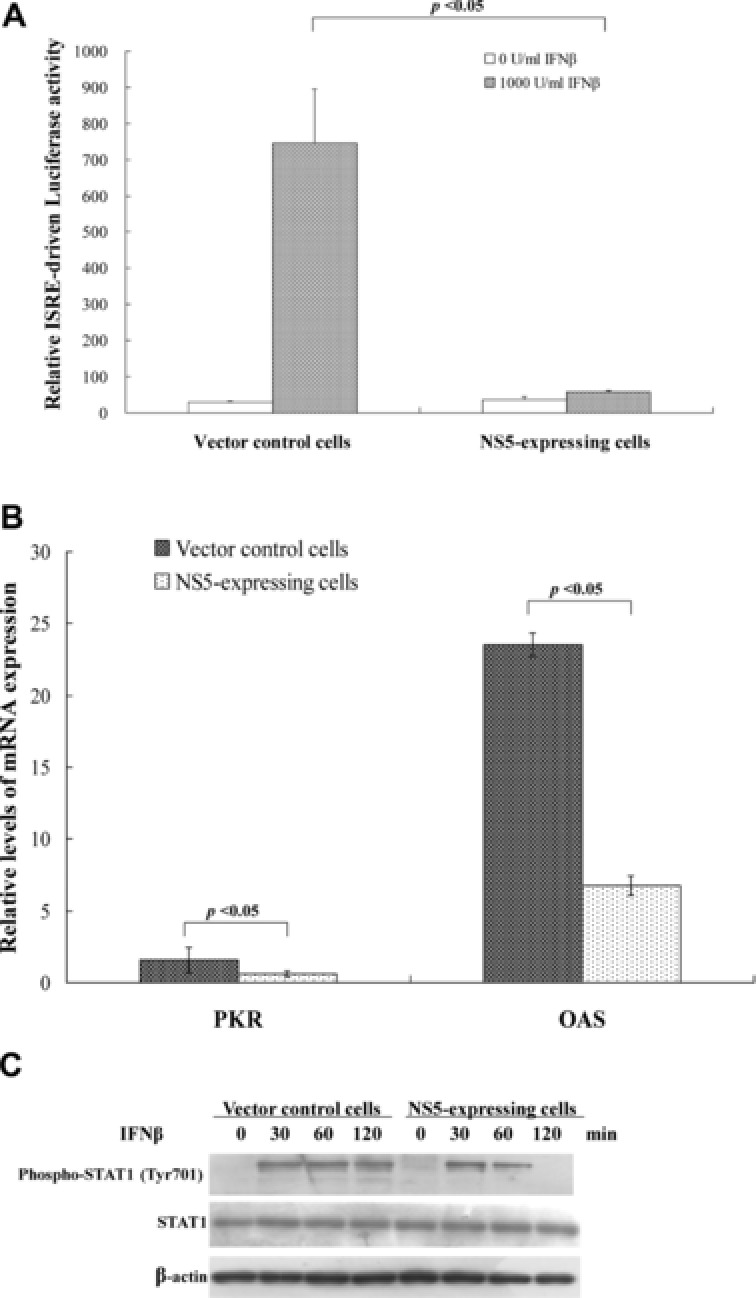
Inhibitory effects of JEV NS5 protein on IFNβ‐induced responses. (A) To analyze ISRE promoter activity, vector control and NS5‐expressing cells were transiently cotransfected with reporter plasmid containing firefly luciferase under control of ISRE and internal control reporter pRluc‐C1. After 4‐h IFNβ treatment, firefly luciferase and renilla luciferase were measured; firefly luciferase activity normalized to renilla luciferase activity is reported. (B) To analyze ISRE‐driven gene expression, vector control and NS5‐expressing cells were treated with or without 1000 U/mL IFNβ for 8 h; relative levels of PKR and OAS mRNAs were gauged by quantitative real‐time PCR, relative fold levels of PKR or OAS mRNA presented as ratio of PKR or OAS mRNA/GAPDH mRNA. (C) To analyze STAT1 phosphorylation, Western blot of lysates from cells treated with IFNβ for 0, 30, 60, or 120 min was performed by anti‐phospho‐STAT1 (Tyr701) and anti‐β actin antibody as internal control.
3.2. Proteomic analysis of IFNβ antagonism by JEV NS5
To identify key proteins inhibiting IFNβ‐induced STAT1‐mediated signaling, protein expression in vector control and NS5‐expressing cells with or without IFNβ was analyzed by 2D electrophoresis and nanoscale capillary LC/ESI Q‐TOF MS to differentiate regulated protein spots (Supporting Information Fig. 1). Table 1 shows unique protein profile of NS5‐expressing cells compared to vector controls. JEV NS5 expression upregulated inosine‐5′‐monophosphate dehydrogenase 2, 60 kDa HSP (Hsp60), heterogeneous nuclear ribonucleoprotein D0, HSP 27 (Hsp27), stress‐induced phosphoprotein 1 (STIP1), peroxiredoxin‐1, and cyclophilin A (Table 1); it downregulated d‐3‐phosphoglycerate dehydrogenase, heterogeneous nuclear ribonucleoprotein H3, T‐complex protein 1 subunit beta, thioredoxin, prohibitin, and calreticulin in human medulloblastoma cells (Table 1).
Table 1.
Up‐ and down‐regulated proteins in NS5‐expressing cells compared to vector control cells
| Spot ID | PANTHER gene ID | Protein identification | MASCOT score | MW/pI | Numbers of peptides identified | Sequence coverage (%) | Molecular function | Biological process | Fold change (±SD) |
|---|---|---|---|---|---|---|---|---|---|
| Upregulated proteins in NS5‐expressing cells compared to vector control cells | |||||||||
| 1 | 3615 | Inosine‐5′‐monophosphate dehydrogenase 2 (IMPDH 2) | 451 | 55.8/6.44 | 11 | 35 | Oxidoreductase | Purine metabolism | 4.16 ± 0.27↑ |
| 4 | 3329 | 60 kDa HSP (Hsp60) | 1145 | 61/5.7 | 15 | 52 | Protein folding, protein complex assembly | 1.71 ± 0.01↑ | |
| 5 | 3184 | Heterogeneous nuclear ribonucleoprotein D0 (hnRNP D0) | 96 | 38.4/7.62 | 2 | 6 | RNA splicing factor, transesterification, structural constituent of ribosome poly(A) RNA binding | mRNA splicing | 3.42 ± 0.05↑ |
| 6 | 3315 | HSP beta‐1 (HSPB1, Hsp27–1) | 458 | 22.8/5.98 | 5 | 66 | Structural molecule | Protein folding, stress response | 1.60 ± 0.32↑ |
| 8 | 5052 | Peroxiredoxin‐1 (PRDX1) | 192 | 22.1/8.27 | 4 | 13 | Oxidoreductase peroxidase | Antioxidation and free radical removal | 3.28 ± 0.18↑ |
| 9 | 10963 | Stress‐induced‐phosphoprotein 1 (STIP1) | 70 | 62.6/6.40 | 6 | 11 | Stress response | 2.08 ± 0.11↑ | |
| 12 | 5478 | Cyclophilin A (CYPA) | 119 | 18.09/7.82 | 11 | 68 | Isomerase | Protein folding, nuclear transport, immunity and defense | 1.72 ± 0.08↑ |
| Downregulated proteins in NS5‐expressing cells compared to vector control cells | |||||||||
| 2 | 26227 | d‐3‐phosphoglycerate dehydrogenase (3‐PGDH) | 424 | 56.6/6.29 | 13 | 33 | Oxidoreductase | Amino acid biosynthesis | 0.01 ± 0.04↓ |
| 3 | 3189 | Heterogeneous nuclear ribonucleoprotein H3 (HNRPH3) | 94 | 36.9/6.37 | 3 | 13 | Structural constituent of ribosome, nucleic acid binding | mRNA splicing | 0.12 ± 0.09↓ |
| 7 | 10576 | T‐complex protein 1 subunit beta (TCP‐1‐beta) | 129 | 57.5/6.01 | 7 | 20 | Chaperonin | 0.60 ± 0.06↓ | |
| 10 | 7295 | Thioredoxin (MTRX) | 86 | 11.7/4.82 | 2 | 19 | Oxidoreductase | Electron transport, sulfur redox metabolism, other intracellular signaling cascade, stress response, apoptosis, cell proliferation and differentiation | 0.27 ± 0.11↓ |
| 11 | 5245 | Prohibitin (PHB) | 72 | 29.8/5.57 | 7 | 37 | DNA replication, cell cycle control, cell proliferation and differentiation | 0.12 ± 0.07↓ | |
| 13 | 811 | Calreticulin precursor (CALR) | 101 | 63.89/4.3 | 8 | 24 | Calcium ion binding | Protein folding | 0.51 ± 0.12↓ |
This article is being made freely available through PubMed Central as part of the COVID-19 public health emergency response. It can be used for unrestricted research re-use and analysis in any form or by any means with acknowledgement of the original source, for the duration of the public health emergency.
To confirm expression profile of vector control and NS5‐expressing cells treated with/without IFNβ, spots in 2D gels, such as thioredoxin, cyclophilin A, and calreticulin, were enlarged, identified using MS/MS analysis and quantified relative expression by Western blotting and real‐time PCR (Fig. 3A to E). Two peptide peaks of thioredoxin (Spot ID 10) with a MASCOT score of 86 and sequence coverage of 19% in MS analysis were sequenced by Q‐TOF MS analysis and identified to match within thioredoxin residues 86–94 and 98–105 (Fig. 3B and C). Lysates of both cells treated with varied IFNβ concentrations were analyzed by Western blot with monoclonal antibodies against cyclophilin A and calreticulin (Fig. 3D), indicating IFNβ treatment increased 18‐kDa immuno‐reactive bands as cyclophilin A in NS5‐expressing, but not vector control cells. On the other hand, IFNβ treatment slightly reduced protein levels of calreticulin in NS5‐expressing compared to vector control cells. Relative calreticulin mRNA levels in both cells were measured by real‐time PCR (Fig. 3E). NS5‐expressing cells had nearly 40% decrease of calreticulin mRNA compared to vector controls. Results confirmed data of 2D/MS.
Figure 3.
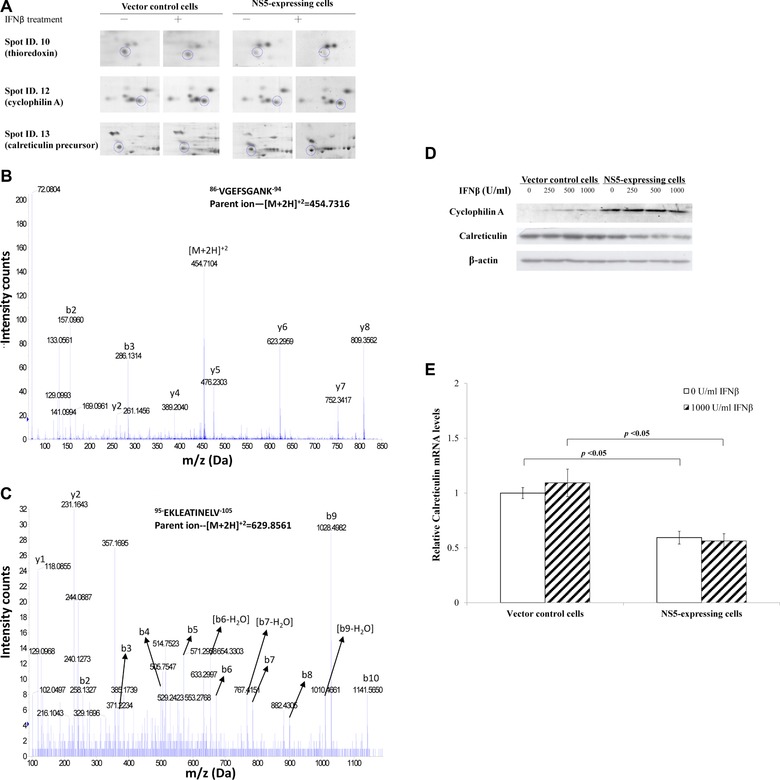
Western blot and MS/MS analysis of protein profiling in NS5‐expressing and vector control cells in response to IFNβ. (A) Enlarged images of cyclophilin A and calreticulin protein spots in 2D gels from vector control and NS5‐expressing cells in response to IFNβ treatment. (B, C) Nanoelectrospray mass spectrum of two charged ion peaks for thioredoxin (spot ID 10) was shown. (D) To analyze cyclophilin A and calreticulin protein levels, vector control and NS5‐expressing cells in the presence or absence of IFNβ were harvested for Western blot with anti‐cyclophilin A, anti‐calreticulin, and anti‐β‐actin antibodies. (E) To quantify relative calreticulin mRNA levels, total RNA extracts from both cells were analyzed by quantitative PCR and normalized by GAPDH mRNA, presented as relative ratio.
Functional classification of these regulatory proteins by PANTHER system indicated upregulated proteins as involved in catalytic (IMPDH2, CYPA, PRDX1), isomerase (CYPA), peroxidase (PRDX1), RNA‐binding (HNRNPD), and antioxidant (PRDX1) activities; downregulated proteins exhibited calcium ion binding activities (CALR), catalytic (MTRX, 3‐PGDH), and structural molecule (HNRNPH3; Table 1). Since cyclophilin A forms the complex with calcineurin and calreticulin is an ER Ca2+ storage protein 29, cyclophilin A upregulation and calreticulin downregulation could link with increase of intracellular Ca2+ and activation of calcineurin‐dependent pathway. Moreover, calcineurin is a Ca2+/calmodulin‐dependent serine/threonine phosphatase that desphosphorylates NFAT1, STAT3, and STAT6 in innate immune response 30, 31, 32. Therefore, calreticulin/Ca2+/calcieurin pathway might be involved in Type I IFN antagonism by JEV NS5.
To test functional role of calreticulin in Type I IFN antagonism of JEV NS5 protein, calreticulin in vector control and NS5‐expressing cells was silenced using siRNAs (Fig. 4A to E). Intracellular Ca2+ levels in cells transfected with both control and calreticulin siRNA were stained with FLUO3/AM, then analyzed by flow cytometry (Fig. 4A). Intracellular Ca2+ levels were 4.9‐fold higher in NS5‐expressing than vector control cells posttransit transfection with control siRNA. Importantly, calreticulin silence by siRNA caused sharp (1.8‐ and 5.5‐fold) increase of Ca2+ levels in NS5‐expressing and vector control cells, respectively. Immunofluorescent analysis indicated IFNβ treatment increasing expression and nuclear localization of STAT1 in vector controls transfected with control siRNA; calreticulin silence decreased STAT1 in nuclei of vector controls (Fig. 4B). By contrast, NS5 expressing cells with or without calreticulin silence showed no significant change of expression and subcellular localization of STAT1 in response to IFNβ (Fig. 4C). To elucidate effect of calreticulin silence on calcineurin‐NFAT signaling, NFAT‐1 nuclear localization in both types of cells was analyzed by immunofluorescent staining with anti‐NFAT‐1 antibodies (Fig. 4D and E). IFNβ treatment induced nuclear translocalization of NFAT‐1 in vector controls; calreticulin silence definitely reduced NFAT‐1 nuclear localization in response to IFNβ, indicating downregulation and gene silence of calreticulin linked with increase of intracellular Ca2+ and reduction of STAT1 and NFAT‐1 nuclear localization, as well as activating Ca2+‐calcineurin‐NFAT signaling.
Figure 4.
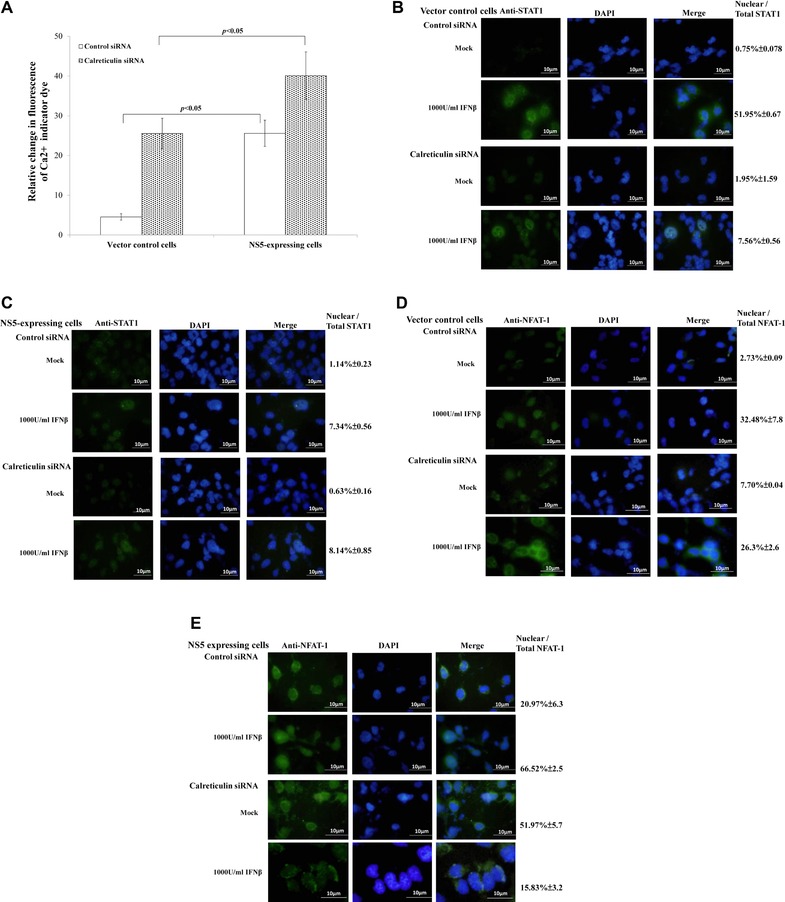
Functional characterization of calreticulin with siRNA‐mediated gene silencing. (A) To detect intracellular Ca2+, cells transfected with control or calreticulin siRNA were harvested 4 h posttreatment with or without IFNβ, stained with FLUO3/AM and analyzed by flow cytometry. To analyze subcellular localization of STAT1 (B, C) and NFAT‐1 (D, E), vector control (B, D) and NS5‐expressing cells (C, E) transfected with control or calreticulin siRNA were tested by immunfluorescent staining with anti‐STAT1 or anti‐NFAT‐1 antibodies.
3.3. Attenuating IFNβ antagonistic ability of JEV NS5 by calcineurin inhibitor CsA
To test the role of Ca2+‐dependent calcineurin in Type I IFN antagonism of JEV NS5, CsA (calcineurin inhibitor) and IFNβ‐induced response in each type of cell were correlated (Fig. 5); IFNβ treatment caused death of vector control, but not NS5‐expressing cells (Fig. 5A). Combined treatment of IFNβ and CsA induced death of both cell types, implying that calcineurin inhibition by CsA attenuates IFNβ antagonistic activity of JEV NS5 protein. In dual‐luciferase reporter system, single CsA treatment activated ISRE‐driven promoter in NS5‐expressing cells, not vector controls (Fig. 5B). Combined treatment of CsA and IFNβ induced higher ISRE‐driven promoter activity than single CsA or IFNβ treatment in NS5‐expressing cells (Fig. 5B). Single CsA treatment had no effect on ISRE‐driven promoter activity in vector controls; combined treatment of CsA and IFNβ showed less ISRE‐driven promoter activity than single IFNβ treatment in vector controls.
Figure 5.
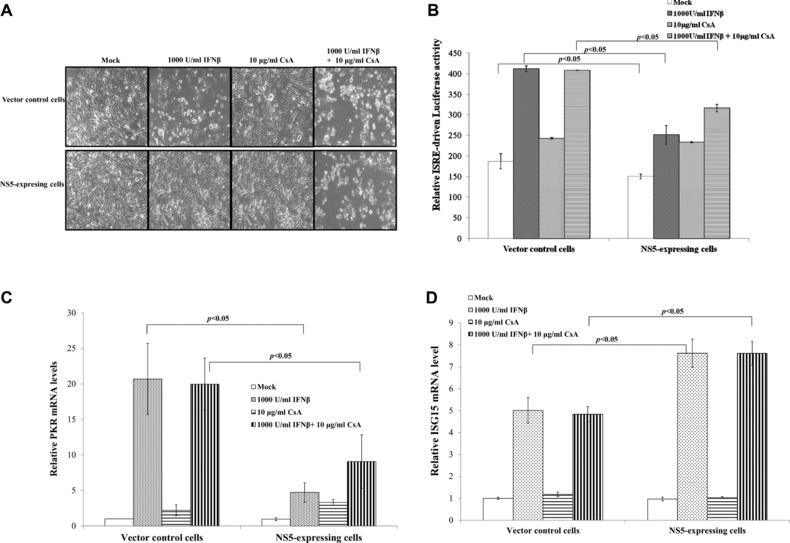
Effects of cyclosporin A (CsA) on NS5‐modulated suppression of IFNβ‐induced response. (A) For cytopathic effect assays, cells were treated singly or with both CsA and IFNβ for 48 h, imaging analyzed by light microscopy. (B) To analyze ISRE‐driven promoter activity, vector control and NS5‐expressing cells were transiently cotransfected with dual‐luciferase reporters, treated singly or with both CsA and IFNβ for 4 h. Firefly and renilla luciferase were measured, with relative firefly luciferase activity normalized to renilla luciferase activity. Relative mRNA levels of PKR (C) and ISG15 (D) were measured by quantitative PCR and normalized by GAPDH mRNA, presented as relative ratio.
Transcriptional factor IRF‐3 bonded with ISRE of ISG15 promoter, then directly induced ISG15 mRNA expression, while STAT1 was involved in PKR gene regulation 33, 34. To analyze association of IRF‐3 and STAT1 with CsA effect on activation of ISRE‐driven promoter, relative mRNA fold change of IRF‐3 dependent gene ISG15 and STAT1 dependent gene PKR was quantified by real‐time PCR. Single IFNβ treatment induced 20.7‐fold upregulation of PKR mRNA in vector controls, only 4.7‐fold increase in NS5‐expressing cells. Combined treatment of CsA and IFNβ had a similar level of PKR mRNA in vector control cells; PKR mRNA expression rose by 9.1‐fold in NS5‐expressing cells (Fig. 5C). Relative PKR mRNA fold change indicated similar pattern as ISRE‐driven promoter activity in response to single or combined treatment (Fig. 5B and C). Single IFNβ treatment raised mRNA levels of ISG15 in NS5‐expressing cells more than in vector controls; combined treatment with CsA and IFNβ upregulated ISG15 mRNA expression, similar to single IFNβ treatment (Fig. 5D). Results suggest calcineurin inhibitor CsA reducing Type I IFN antagonism of JEV NS5 via STAT1‐dependent signal pathways.
3.4. Influence of calcineurin inhibitor CsA on NS5‐suppressed phosphorylation and nuclear translocation of STAT1 in response to Type I IFN
Combined treatment with CsA and IFNβ reduced Type I IFN antagonistic activity of JEV NS5; we hypothesized calcineurin involvement in NS5‐induced IFNβ antagonism on STAT1‐mediated signaling pathway. Tyrosine phosphorylation and nuclear translocation of STAT1 were subsequently examined in vector control and NS5‐expressing cells with or without single or double CsA and IFNβ treatment (Fig. 6). Single treatment of IFNβ yielded marked increase of STAT1 phosphorylation at Tyr701 in vector controls (Fig. 6A, lanes 2–4); tyrosine phosphorylated STAT1 in NS5‐expressing cells was lower than those in vector controls 30‐, 60‐, and 120‐min postsingle treatment of IFNβ (Fig. 6A, lanes 10–12). Combined treatment with CsA and IFNβ elevated phosphorylation level of STAT1 at Tyr701 in NS5‐expressing cells compared to single treatment (Fig. 6A, lanes 14–16 vs. 10–12). Importantly, confocal imaging analysis with anti‐STAT1 antibodies revealed combined treatment of CsA and IFNβ enhancing IFNβ‐induced STAT1 nuclear translocation in NS5‐expressing cells (Fig. 6B and C), supporting elevation of STAT1 phosphorylation at Tyr701 in NS5‐expressing cells by combined IFNβ and CsA treatment. Meanwhile, CsA alone induced a slight amount of STAT1 translocation to nuclei in both cell types, implying calreticulin downregulation activating Ca2+/calcineurin signals, associated with inhibition of STAT1‐mediated signaling in NS5‐induced IFNβ antagonism.
Figure 6.
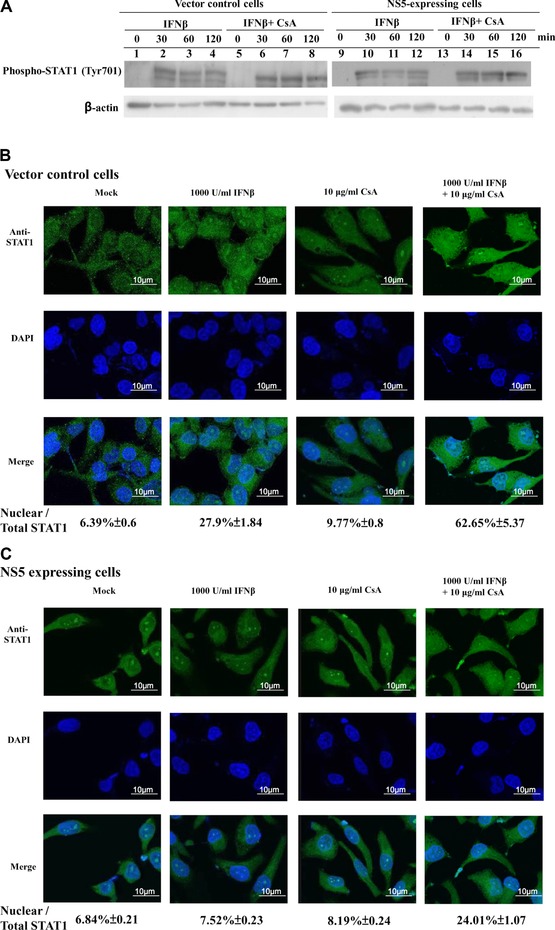
Effect of CsA on IFNβ‐induced phosphorylation and nuclear translocation of STAT1 in NS5‐expressing cells. (A) To analyze tyrosine phosphoryl‐ated STAT1, vector control and NS5‐expressing cells were treated singly or with both IFNβ and cyclosporine A for 0, 30, 60, or 120 min. Lysates were subjected to Western blot, probed with anti‐phospho‐STAT1 (Tyr701). For subcellular location of STAT1, vector control (B) and NS5‐expressing (C) cells were treated singly or with both CsA and IFNβ for 24 h, then washed, fixed, and reacted with anti‐STAT1 and FITC‐conjugated anti‐mouse IgG antibodies. Finally, cells were stained with DAPI for 10 min, imaging analyzed by immunofluorescent microscopy.
3.5. Enhancing IFNβ‐induced response by siRNA‐mediated silence of calcineurin
Besides calcineurin inhibitor CsA, siRNA‐mediated gene silence was performed to affirm the hypothesis, with calcineurin activation linked to suppression of STAT1‐mediated signaling, as responsible for a crucial mechanism of Type I IFN antagonism by JEV NS5 (Supporting Information Fig. 2; Fig. 7A to D). Silencing of calcineurin significantly increased STAT1 nuclear translocalization in NS5‐expressing cells stimulated by IFNβ. Control siRNA did not reduce Type I IFN antagonistic activity of JEV NS5 (Fig. 7B); calcineurin silence induced twofold increase of PKR mRNA over control siRNA in NS5‐expressing cells stimulated by IFNβ (Fig. 7C). Calcineurin‐dependent expression of IL‐4 was tested in NS5‐expressing and vector control cells (Fig. 7D). Due to calcineurin activation by JEV NS5 (Figs. 5 and 6), relative IL‐4 mRNA level in NS5‐expressing cells was nearly 50% higher than vector controls in response to IFNβ (Fig. 7D). siRNA silencing of calcineurin significantly decreased IL‐4 mRNA in both types of cells, even stimulated with IFNβ. Results confirmed activation of Ca2+/calcineurin signaling as a critical mechanism of Type I IFN antagonism by JEV NS5.
Figure 7.
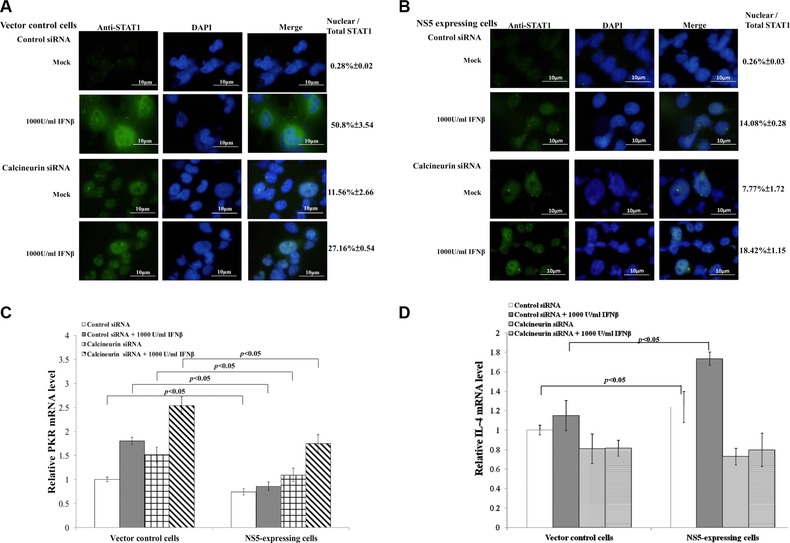
Functional analysis of calcineurin with siRNA‐mediated gene silencing. To analyze subcellular localization of STAT1, vector control (A) and NS5‐expresing cells (B) transfected with control or calreticulin siRNA were tested using immunfluorescent staining with anti‐STAT1 antibodies. Relative mRNA levels of PKR (C) and IL‐4 (D) were measured by quantitative PCR and normalized by GAPDH mRNA, presented as relative ratio.
3.6. Inhibition of in vitro JEV replication by single and combined treatment of cyclosporine A (CsA) and IFNβ
To analyze protein levels of calreticulin, TE671 cells were infected with JEV at MOIs of 0.1, 0.5, 1 and 2, then harvested 48 h post infection; lysate used Western blot with anticalreticulin monoclonal antibodies (Fig. 8A). Calreticulin was downregulated in infected cells by viral dose‐dependent manner, correlating with downregulation of calreticulin by NS5 protein. Inhibitory effect of single and combined treatment of CsA and IFNβ was subsequently determined in vitro by viral replication (Fig. 8B). Single treatment of CsA (1000 ng/mL) significantly reduced JEV plaque titers by 4.3‐fold 48 h post infection; single treatment of IFNβ (500 U/mL) significantly reduced JEV plaque titers by 24.0‐fold 48 h post infection. Combined treatment of CspA and IFNβ potently inhibited JEV plaque titers by 63.4‐fold at 48 h post infection. Annexin V‐FITC/propidium iodide apoptosis staining indicated ratio of late apoptotic/necrotic cells (Annexin V‐FITC positive/propidium iodide positive) at 48 h post infection as 3.2% in mock controls, 28.7% in JEV‐infected cells, 13.2% in infected cells with single IFNβ treatment, 11.1% in infected cells treated with single CsA, and 6.5% in those treated with both CsA and IFNβ (Fig. 8C). Analysis ruled out indirect side effect of combined treatment on apoptosis reducing viral replication. Higher anti‐JEV efficacy by combined CsA and IFNβ versus single treatment concurred with recovery of IFNβ‐induced STAT1 phosphorylation in NS5‐expressing cell by combined treatment of CsA and IFNβ.
Figure 8.
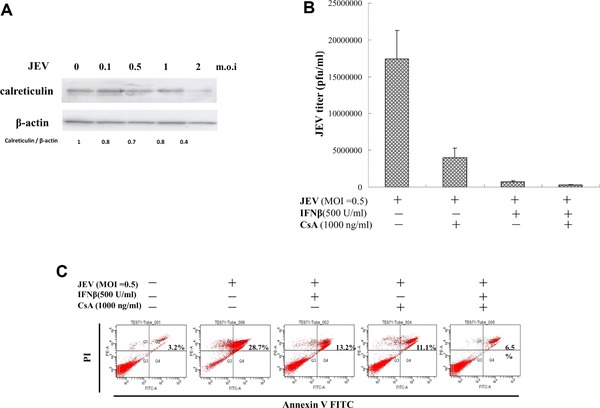
Effect of cyclosporine A on JEV replication in human medulloblastoma cells. (A) To analyze calreticulin expression, TE671 cells were infected with JEV at MOIs of 0.1, 0.5, 1, and 2, then harvested 2 days post infection. Lysates subjected to Western blot were probed with anticalreticulin. (B) TE671 cells were infected with JEV at the MOI of 0.5 and treated with IFNβ alone or both IFNβ and cyclosporine A for 48‐h and cultured supernatants harvested to determine virus yields. BHK‐21 cell monolayers were incubated with serial dilution of cultured supernatant at 37°C for 1 h, then overlaid with MEM medium containing 1.1% methylcellulose. After 3 days of incubation, BHK‐21 cell monolayers were stained with naphthol blue‐black dye and viral plaques counted. (C) For analysis effect of CsA on apoptosis, JEV‐infected, TE671 cells were infected with JEV at MOI of 0.5 and treated with IFNβ alone or with IFNβ plus CsA for 2 days, then harvested, stained with both Annexin V‐FITC and PI, and analyzed for percent of cells in apoptosis, using flow cytometry.
4. Discussion
JEV induces no Type I IFN response (vital to pathogenesis) in cell culture. This study dealt with JEV NS5, previously reported as antagonistic toward members of Type I IFN pathway. We first demonstrated that JEV NS5 expression in human medulloblastoma cells inhibits (a) IFNβ‐induced ISRE promoter activities, (b) IFNβ‐stimulated gene expression, and (c) suppression of IFNβ‐induced nuclear translocation and phosphorylation of STAT1 (Fig. 2). Significant suppression of STAT1 phosphorylation at Tyr701 was responsible for Type I IFN antagonism of JEV NS5 proteins, in agreement with prior studies: WNV and TBEV NS5 proteins suppressing tyrosine phosphorylation of STAT1 and DEN NS5 protein suppressing tyrosine phosphorylation of STAT2, blocking formation of STAT1/STAT1 homo‐ or heterodimers in Type I IFN‐induced JAK/STAT signaling pathway 8, 10, 11, 12, 13, 35. Recently, interaction of TBEV NS5 with PDZ protein scribble (hScrib) was associated with JAK‐STAT interference, vital for an antagonist of IFN response 15.
Proteomics approach to identify NS5‐induced protein profiling indicated downregulation of calreticulin plus upregulation of cyclophilin A, Hsp60, and STIP1, as linked with activation of calcineurin (Fig. 3, Table 1), suggesting the key of host cellular factors in Type I IFN antagonistic function of JEV NS5 protein. Calreticulin is a Ca2+‐binding chaperone in ER, as a key upstream regulator of calcineurin in Ca2+‐signaling cascade 36. NS5 downregulated Ca2+‐binding chaperone calreticulin, confirmed by Western blot and quantitative real‐time PCR (Fig. 3D and E), as correlating with increased release of Ca2+ from ER into cytosol (Fig. 4A), as well as gene silencing of calreticulin raising intracellular Ca2+ levels in controls. Promoter of human calreticulin gene comprised AP‐1, Sp1, and H4TF‐1 sites, plus four CCAAT sequences, binding with c‐Jun/c‐Fos, specific protein 1 (Sp1), H4TF‐1, CAAT/enhancer‐binding protein transcriptional factors 37, 38 regulated by p38 MAPK and ERK1/2. Western blot indicated JEV NS5 protein suppressing phosphorylated levels of ERK1/2, not p38 MAPK, in response to IFNβ (Supporting Information Fig. 3); JEV NS5 protein affects transcriptional activation of calreticulin promoter by reducing ERK1/2 phosphorylation.
Downregulation and siRNA‐mediated silence of calreticulin reduced the nuclear translocalization of NFAT‐1 in response to IFNβ (Fig. 4D and E), indicating activation of calcineurin by calreticulin downregulation and silence. Among upregulated proteins, cyclophilin A is involved in virus replication and virion maturation 39, significantly interacting with calcineurin and inhibited its phosphatase activity in the presence of CsA 40, 41. Hsp60 is one calcineurin‐interacting protein 42. STIP1, an Hsp70/ Hsp90‐organizing protein, as a cochaperone binds to both Hsp70/Hsp90, correlated with calcineurin regulation 43. SARS‐CoV NSP1 overexpression strongly induced production of interleukin 2 by activating calcineurin/NFAT pathway; calcineurin inhibitor CspA strongly inhibited pan‐coronavirus replication in vitro 41. Proteomics results and the above literature survey led us to hypothesize calcineurin as involved in Type I IFN antagonism of JEV NS5. Because induction of ISG15 was IRF‐3‐dependent, PKR expression is STAT1‐dependent in response to Type I IFN 33, 34, IFNβ upregulated expression of ISG15, but not PKR in NS5‐expressing cells (Fig. 5C and D), implying NS5 specifically inhibited STAT1‐mediated signaling. Combined CsA and IFNβ treatment as well as siRNA‐mediated calcineurin silence restored IFNβ‐induced STAT1‐mediated responses in NS5‐expressing cells, for example., PKR mRNA expression, STAT1 phosphrylation at Tyr701, nuclear translocation of STAT1 (Figs. 5, 6, 7). These affirm Type I IFN antagonism of JEV NS5 correlating with downregulation of calreticulin in which activated Ca2+‐dependent calcineurin phosphatase inhibits STAT1‐mediated signaling. HBV‐induced upregulation of calreticulin was identified as antagonizing Type I IFN responses: inhibiting STAT1 phosphorylation and reducing PKR mRNA expression 44. Calreticulin, an ER Ca2+‐binding chaperone, might be involved in viral pathogenesis, offering a potential target for development of antiviral agents.
Combined treatment of CsA and IFNβ proved more effective in suppressing JEV replication than single treatment (Fig. 8). Interestingly, single treatment with CsA slightly induced ISRE‐driven promoter activation, STAT1 nuclear translocation in both cell types, and inhibition of JEV replication in vitro by 4.3‐fold (Figs. 5B, 6B and C, and 8B). This indicated inhibition of calreticulin/calcineurin pathway by CsA eliciting antiviral innate immune responses. CsA with distinct immunosuppressive and anti‐inflammatory mechanisms from IFN antiviral mechanisms sharply reduced IFN antagonistic function of JEV NS5 via inactivation of calcineurin‐mediated pathways. Single and combined treatment of CspA analogs and IFNs should be further tested in vitro and in vivo for evaluating antiviral efficacy.
Cyclophilin A directly bonded with WNV RNA and NS5 while facilitating viral replication, serving as a component of the replication complex 35. Replication of WNV, DEN, and YF viruses in cyclophilin A knockdown cells was less efficient; transsupplying of wild peptidyl‐prolyl isomerases domain of cyclophilin A rescued viral replication in cyclophilin A knockdown cells 35. In HCV replication complex, cyclophilin A was recruited to maintain optimal conformation between RNA‐dependent RNA polymerase and template binding 45. A chaperone system was likewise required for viral replication assembly and RNA binding, for example, interaction of HSP 90 with herpes simplex virus Type 1 polymerase 46 and hepatitis B virus reverse transcriptase 47. Cyclophilin A was suggested as a molecular chaperone to keep flavivirus replication complexes in active conformation. In our laboratory, coimmunoprecipitation assays also proved direct interaction of JEV NS5 with cyclophilin A (data not shown). Likewise, overexpression of cyclophilin A induced IFNβ production, linked with suppressed rotavirus replication via activation of peptidyl‐prolyl isomerases independent but JNK‐dependent signaling 48. Cyclophilin A could thus be crucial for maintaining JEV NS5 functional structure.
Type I IFN antagonism of JEV NS5 protein correlated with suppressing phosphorylation and nuclear translocation of STAT1, linking with calreticulin downregulation and activation of Ca2+/calcineurin‐signaling pathway. Treatment with calcineurin‐specific inhibitor or siRNA increased tyrosine phosphorylation and STAT1 nuclear translocation, upregulating STAT1‐dependent ISG gene expression in NS5‐expressing cells responded to IFNβ. This study indicated correlation between Type I IFN antagonistic activity and proteome profiling induced by JEV NS5 protein, highlighting a pivotal role of JEV NS5 protein in Type I IFN antagonism.
The authors have declared no conflict of interest.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Figure S1. Silver stain of two‐dimensional gel of NS5‐expressing and vector control cells. 100 μg of total protein of lysates from vector control (A), NS5‐expressing (B), IFNβ‐treated vector control (C), or IFNβ‐treated NS5‐expressing (D) cells was applied to 2D gel electrophoresis. After first‐dimensional isoelectric focusing, nonlinear Immobiline DryStrip was transferred to the top of 12% polyacrylamide gels. Finally, gels were fixed, stained with silver nitrate solution, then scanned by GS‐800 imaging densitometer. Protein size markers are shown at the left of each gel (in kDa).
Figure S2. Quantity of relative calcinurin mRNA levels in siRNA‐transfectd cells. Total RNAs extracted from vector control and NS5‐expressing cells transfected with siRNAs were analyzed by quantitative PCR and normalized by GAPDH mRNA, presented as relative ratio.
Figure S3. Western blot analysis of phosphorylated levels of ERK1/2 and p38 MAPK in NS5‐expressing and vector control cells in response to IFNβ. Vector control and NS5‐expressing cells in the presence or absence of IFNβ were harvested for Western blot with anti‐ERK1/2, anti‐phospho‐ERK1/2, anti‐p38 MAPK, anti‐phospho‐p38 MAPK, and anti‐β‐actin antibodies.
Acknowledgments
We thank China Medical University and National Science Council, Taiwan, for financial support (CMU101‐S‐24, CMU98‐P‐03, NSC99‐2628‐B‐039‐006‐MY3, and NSC98‐2324‐B‐039‐006).
Colour Online: See the article online to view Figs. 1, 3, 4, 6–8 in colour
5 References
- 1. Burke, D. S. , Monath, T. P. , Flaviviruses, in: Fields B. N., Knipe D. M., Howley P. M., Griffin D. E. (Eds.), Fields Virology, Vol. 4, Lippincott Williams and Wilkins, Philadelphia, PA, 2001, pp. 1043–1125. [Google Scholar]
- 2. Lindenbach, B. D. , Rice, C. M. , Flaviviridae: the viruses and their replication, in: Knipe D. M., Howley P. M. (Eds.), Fields Virology, Vol. 4. Lippincott Williams & Wilkins, Philadelphia, PA, 2001, pp. 991–1041. [Google Scholar]
- 3. Kalita, J. , Misra, U. K. , Comparison of CT and MRI findings in the diagnosis of Japanese encephalitis. J. Neurol. Sci. 2000, 174, 3–8. [DOI] [PubMed] [Google Scholar]
- 4. Misra, U. K. , Kalita, J. , Goel, D. , Mathur, A. , Clinical, radiological and neurophysiological spectrum of JEV encephalitis and other non‐specific encephalitis during post‐monsoon period in India. Neurol. India. 2003, 51, 55–59. [PubMed] [Google Scholar]
- 5. Liu, T. H. , Liang, L. C. , Wang, C. C. , Liu, H. C. , Chen, W. J. , The blood‐brain barrier in the cerebrum is the initial site for the Japanese encephalitis virus entering the central nervous system. J. Neurovirol. 2008, 14, 514–521. [DOI] [PubMed] [Google Scholar]
- 6. Solomon, T. N. , Dung, M. R. , Kneen, R. , Gainsborough, M. D. , Vaughn, W. , Khanh, V. T. , Japanese encephalitis. J. Neurol. Neurosurg. Psychiatry 2000, 68, 405–415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Tsai, T. F. , Factors in the changing epidemiology of Japanese encephalitis and West Nile fever, in: Saluzzo J. F., Dodet B., eds. Factors in the Emergence of Arbovirus Diseases, Elselvier, Paris, 1997, pp. 179–189. [Google Scholar]
- 8. Park, G. S. , Morris, K. L. , Hallett, R. G. , Bloom, M. E. , Best, S. M. , Identification of residues critical for the interferon antagonist function of Langat virus NS5 reveals a role for the RNA‐dependent RNA polymerase domain. J. Virol. 2007, 81, 6936–6946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Qi, W. B. , Hua, R. H. , Yan, L. P. , Tong, G. Z. et al., Effective inhibition of Japanese encephalitis virus replication by small interfering RNAs targeting the NS5 gene. Virus Res. 2008, 132, 145–151. [DOI] [PubMed] [Google Scholar]
- 10. Lin, R. J. , Chang, B. L. , Yu, H. P. , Liao, C. L. , Lin, Y. L. , Blocking of interferon‐induced Jak‐Stat signaling by Japanese encephalitis virus NS5 through a protein tyrosine phosphatase‐mediated mechanism. J. Virol. 2006, 80, 5908–5918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Laurent‐Rolle, M. , Boer, E. F. , Lubick, K. J. , Wolfinbarger, J. B. et al., The NS5 protein of the virulent West Nile virus NY99 strain is a potent antagonist of type I interferon‐mediated JAK‐STAT signaling. J. Virol. 2010, 84, 3503–3515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Ashour, J. , Laurent‐Rolle, M. , Shi, P. Y. , García‐Sastre, A ., NS5 of dengue virus mediates STAT2 binding and degradation. J. Virol. 2009, 83, 5408–5418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Mazzon, M. , Jones, M. , Davidson, A. , Chain, B. , Jacobs, M. , Dengue virus NS5 inhibits interferon‐alpha signaling by blocking signal transducer and activator of transcription 2 phosphorylation. J. Infect. Dis. 2009, 200, 1261–1270. [DOI] [PubMed] [Google Scholar]
- 14. Best, S. M. , Morris, K. L. , Shannon, J. G. , Robertson, S. J. et al., Inhibition of interferon‐stimulated JAK‐STAT signaling by a tick‐borne flavivirus and identification of NS5 as an interferon antagonist. J. Virol. 2005, 79, 12828–12839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Werme, K. , Wigerius, M. , Johansson, M. , Tick‐borne encephalitis virus NS5 associates with membrane protein scribble and impairs interferon‐stimulated JAK‐STAT signalling. Cell Microbiol. 2008, 10, 696–712. [DOI] [PubMed] [Google Scholar]
- 16. Miller, C. A. , Carrigan, D. R. , Reversible repression and activation of measles virus infection in neural cells. Proc. Natl. Acad. Sci. USA 1982, 79, 1629–1633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Ziegler, R. J. , Stauffer, E. K. , Mumps virus‐induced alterations in cellular excitability during persistent infections. J. Gen. Virol. 1987, 68, 2501–2507. [DOI] [PubMed] [Google Scholar]
- 18. Yang, T. C. , Shiu, S. L. , Chuang, P. H. , Lin, Y. J. et al., Japanese encephalitis virus NS2B‐NS3 protease induces caspase 3 activation and mitochondria‐mediated apoptosis in human medulloblastoma cells. Virus Res. 2009, 143, 77–85. [DOI] [PubMed] [Google Scholar]
- 19. Yang, T. C. , Lai, C. C. , Shiu, S. L. , Chuang, P. H. et al., Japanese encephalitis virus down‐regulates thioredoxin and induces ROS‐mediated ASK1‐ERK/p38 MAPK activation in human promonocyte cells. Microb. Infect. 2010, 12, 643–651. [DOI] [PubMed] [Google Scholar]
- 20. Hsiao, N. W. , Chen, J. W. , Yang, T. C. , Orloff, G. M. et al., ISG15 over‐expression inhibits replication of the Japanese encephalitis virus in human medulloblastoma cells. Antivir. Res. 2010, 85, 504–511. [DOI] [PubMed] [Google Scholar]
- 21. Lin, C. W. , Lin, K. H. , Lyu, P. C. , Chen, W. J. , Japanese encephalitis virus NS2B‐NS3 protease binding to phage‐displayed human brain proteins with the domain of trypsin inhibitor and basic region leucine zipper. Virus Res. 2006, 116, 106–113. [DOI] [PubMed] [Google Scholar]
- 22. Li, S. W. , Lai, C. C. , Ping, J. F. , Tsai, F. J. et al., Severe acute respiratory syndrome coronavirus papain‐like protease suppressed alpha interferon‐induced responses through downregulation of extracellular signal‐regulated kinase 1‐mediated signalling pathways. J. Gen. Virol. 2011, 92, 1127–1140. [DOI] [PubMed] [Google Scholar]
- 23. Lin, C.W. , Cheng, C.W. , Yang, T.C. , Li, S.W. et al., Interferon antagonist function of Japanese encephalitis virus NS4A and its interaction with DEAD‐box RNA helicase DDX42. Virus Res. 2008, 137, 49–55. [DOI] [PubMed] [Google Scholar]
- 24. Lai, C. C. , Jou, M. J. , Huang, S. Y. , Li, S. W. et al., Proteomic analysis of up‐regulated proteins in human promonocyte cells expressing severe acute respiratory syndrome coronavirus 3c‐like protease. Proteomics 2007, 7, 1446–1460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Tsang, K. W. , Ho, P. L. , Ooi, G. C. , Yee, W. K. et al., A Cluster of Cases of Severe Acute Respiratory Syndrome in Hong Kong. N. Engl. J. Med. 2003, 348, 1977–1985. [DOI] [PubMed] [Google Scholar]
- 26. Pryor, M. J. , Rawlinson, S. M. , Butcher, R. E. , Barton, C. L. et al., Nuclear localization of dengue virus nonstructural protein 5 through its importin alpha/beta‐recognized nuclear localization sequences is integral to viral infection. Traffic 2007, 8, 795–807. [DOI] [PubMed] [Google Scholar]
- 27. Buckley, A. , Gaidamovich, S. , Turchinskaya, A. , Gould, E. A. , Monoclonal antibodies identify the NS5 yellow fever virus non‐structural protein in the nuclei of infected cells. J. Gen. Virol. 1992, 73, 1125–1130. [DOI] [PubMed] [Google Scholar]
- 28. Uchil, P. D. , Kumar, A. V. , Satchidanandam, V. , Nuclear localization of flavivirus RNA synthesis in infected cells. J. Virol. 2006, 80, 5451–5464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Groenendyk, J. , Lynch, J. , Michalak, M. , Calreticulin, Ca2+, and calcineurin—signaling from the endoplasmic reticulum. Mol. Cells 2004, 17, 383–389. [PubMed] [Google Scholar]
- 30. Hermann‐Kleiter, N. , Baier, G. , NFAT pulls the strings during CD4+ T helper cell effector functions. Blood 2010, 115, 2989–2997. [DOI] [PubMed] [Google Scholar]
- 31. Moffatt, S. D. , Cockman, M. , Metcalfe, S. M. , STAT 6 up‐regulation by FK506 in the presence of interleukin‐4. Transplantation 2000, 69, 1521–1523. [DOI] [PubMed] [Google Scholar]
- 32. Woetmann, A. , Nielsen, M. , Christensen, S. T. , Brockdorff, J. et al., Inhibition of protein phosphatase 2A induces serine/threonine phosphorylation, subcellular redistribution, and functional inhibition of STAT3. Proc. Natl. Acad. Sci. USA 1999, 96, 10620–10625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Nakaya, T. , Sato, M. , Hata, N. , Asagiri, M. et al., Gene induction pathways mediated by distinct IRFs during viral infection. Biochem. Biophys. Res. Commun. 2001, 283, 1150–1156. [DOI] [PubMed] [Google Scholar]
- 34. Grandvaux, N. , Servant, M. J. , tenOever, B. , Sen, G. C. et al., Transcriptional profiling of interferon regulatory factor 3 target genes: direct involvement in the regulation of interferon‐stimulated genes. J. Virol. 2002, 76, 5532–5539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Qing, M. , Yang, F. , Zhang, B. , Zou, G. et al., Cyclosporine inhibits flavivirus replication through blocking the interaction between host cyclophilins and viral NS5 protein. Antimicrob. Agents Chemother. 2009, 53, 3226–3235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Lynch, J. , Michalak, M. , Calreticulin is an upstream regulator of calcineurin. Biochem. Biophys. Res. Commun. 2003, 311, 1173–1179. [DOI] [PubMed] [Google Scholar]
- 37. McCauliffe, D. P. , Yang, Y. S. , Wilson, J. , Sontheimer, R. D. , Capra, J. D. , The 5’‐flanking region of the human calreticulin gene shares homology with the human GRP78, GRP94, and protein disulfide isomerase promoters. J. Biol. Chem. 1992, 267, 2557–2562. [PubMed] [Google Scholar]
- 38. Waser, M. , Mesaeli, N. , Spencer, C. , Michalak, M. , Regulation of calreticulin gene expression by calcium. J. Cell Biol. 1997, 138, 547–557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Keyes, L. R. , Bego, M. G. , Soland, M. , St‐Jeor, S. , Cyclophilin A is required for efficient human cytomegalovirus DNA replication and reactivation. J. Gen. Virol. 2012, 93, 722–732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Liu, J. , Farmer, J. D., Jr. , Lane, W. S. , Friedman, J. et al., Calcineurin is a common target of cyclophilin‐cyclosporin A and FKBP‐FK506 complexes. Cell 1991, 66, 807–815. [DOI] [PubMed] [Google Scholar]
- 41. Pfefferle, S. , Schöpf, J. , Kögl, M. , Friedel, C. C. et al., The SARS‐coronavirus‐host interactome: identification of cyclophilins as target for pan‐coronavirus inhibitors. PLoS Pathog. 2011, 7, e1002331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Li, W. , Handschumacher, R. E. , Identification of two calcineurin B‐binding proteins: tubulin and heat shock protein 60. Biochim. Biophys. Acta 2002, 1599, 72–81. [DOI] [PubMed] [Google Scholar]
- 43. Someren, J. S. , Faber, L. E. , Klein, J. D. , Tumlin, J. A. , Heat shock proteins 70 and 90 increase calcineurin activity in vitro through calmodulin‐dependent and independent mechanisms. Biochem. Biophys. Res. Commun. 1999, 1260, 619–625. [DOI] [PubMed] [Google Scholar]
- 44. Yue, X. , Wang, H. , Zhao, F. , Liu, S. et al., Hepatitis B virus‐induced calreticulin protein is involved in IFN resistance. J. Immunol. 2012, 189, 279–286. [DOI] [PubMed] [Google Scholar]
- 45. Liu, Z. , Yang, F. , Robotham, J. M. , Tang, H. , Critical role of cyclophilin A and its prolyl‐peptidyl isomerase activity in the structure and function of the hepatitis C virus replication complex. J. Virol. 2009, 83, 6554–6565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Burch, A. D. , Weller, S. K. , Herpes simplex virus type 1 DNA polymerase requires the mammalian chaperone hsp90 for proper localization to the nucleus. J. Virol. 2005, 79, 10740–10749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Stahl, M. , Beck, J. , Nassal, M. , Chaperones activate hepadnavirus reverse transcriptase by transiently exposing a C‐proximal region in the terminal protein domain that contributes to RNA binding. J. Virol. 2007, 81, 13354–13364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. He, H. , Zhou, D. , Fan, W. , Fu, X. et al., Cyclophilin A inhibits rotavirus replication by facilitating host IFN‐I production. Biochem. Biophys. Res. Commun. 2012, 422, 664–669. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Figure S1. Silver stain of two‐dimensional gel of NS5‐expressing and vector control cells. 100 μg of total protein of lysates from vector control (A), NS5‐expressing (B), IFNβ‐treated vector control (C), or IFNβ‐treated NS5‐expressing (D) cells was applied to 2D gel electrophoresis. After first‐dimensional isoelectric focusing, nonlinear Immobiline DryStrip was transferred to the top of 12% polyacrylamide gels. Finally, gels were fixed, stained with silver nitrate solution, then scanned by GS‐800 imaging densitometer. Protein size markers are shown at the left of each gel (in kDa).
Figure S2. Quantity of relative calcinurin mRNA levels in siRNA‐transfectd cells. Total RNAs extracted from vector control and NS5‐expressing cells transfected with siRNAs were analyzed by quantitative PCR and normalized by GAPDH mRNA, presented as relative ratio.
Figure S3. Western blot analysis of phosphorylated levels of ERK1/2 and p38 MAPK in NS5‐expressing and vector control cells in response to IFNβ. Vector control and NS5‐expressing cells in the presence or absence of IFNβ were harvested for Western blot with anti‐ERK1/2, anti‐phospho‐ERK1/2, anti‐p38 MAPK, anti‐phospho‐p38 MAPK, and anti‐β‐actin antibodies.