

Abstract
While it is now possible to identify and genetically fingerprint the causative agents of emerging viral diseases, often with extraordinary speed, suitable therapies cannot be developed with equivalent speed, because drug discovery requires information that goes beyond knowledge of the viral genome. Peptides, however, may represent a special opportunity.
For all enveloped viruses, fusion between the viral and the target cell membrane is an obligatory step of the life cycle. Class I fusion proteins harbor regions with a repeating pattern of amino acids, the heptad repeats (HRs), that play a key role in fusion, and HR‐derived peptides such as enfuvirtide, in clinical use for HIV, can block the process. Because of their characteristic sequence pattern, HRs are easily identified in the genome by means of computer programs, providing the sequence of candidate peptide inhibitors directly from genomic information.
Moreover, a simple chemical modification, the attachment of a cholesterol group, can dramatically increase the antiviral potency of HR‐derived inhibitors and simultaneously improve their pharmacokinetics. Further enhancement can be provided by dimerization of the cholesterol‐conjugated peptide. The examples reported so far include inhibitors of retroviruses, paramyxoviruses, orthomyxoviruses, henipaviruses, coronaviruses, and filoviruses. For some of these viruses, in vivo efficacy has been demonstrated in suitable animal models.
The combination of bioinformatic lead identification and potency/pharmacokinetics improvement provided by cholesterol conjugation may form the basis for a rapid response strategy, where development of an emergency cholesterol‐conjugated therapeutic would immediately follow the availability of the genetic information of a new enveloped virus. Copyright © 2014 European Peptide Society and John Wiley & Sons, Ltd.
Keywords: emerging viral disease, emerging virus, bioterrorism, emergency and preparedness response, viral fusion, fusion inhibitor, cholesterol conjugation, dimerization, peptide antiviral
Peptides may represent a special opportunity against emerging viral diseases. Peptides derived from the viral fusion proteins, which can interfere with virus entry into the host cell, can be identified directly from genomic information. When conjugated with cholesterol, their potency is dramatically increased and pharmacokinetics simultaneously improved. Further enhancement is provided by dimerization. Cholesterol conjugation may form the basis for a rapid response strategy, where an emergency therapy would be developed based on genomic information of a new enveloped virus.

The Threat of Emerging Viral Diseases
The spectrum of infectious diseases that threaten human health is rapidly broadening: The number of previously unknown conditions that have emerged since 1970 exceeds 40, with a new disease discovered on average more than once a year 1, 2. According to a 2008 World Health Organization (WHO) report, between the years 2002 and 2007, a record of 1100 worldwide epidemic events occurred 3. A comprehensive examination of episodes of emerging infectious disease outbreaks between 1940 and 2004, after controlling for reporting bias, came to the conclusion that the incidence of the events has increased significantly over time 4.
For viral diseases in particular, in addition to the constant fear of a new wave of pandemic influenza, the list of emerging or reemerging viruses includes HIV, hantavirus, Lassa virus, Marburg virus, hepatitis C virus, dengue virus, Rift Valley fever virus, Ebola virus, Nipah virus, Hendra virus, West Nile virus, severe acute respiratory syndrome (SARS) coronavirus, avian influenza virus, human polyomavirus, adenovirus 14, Chikungunya virus, and most recently the Middle East respiratory syndrome coronavirus 5. At the time when this manuscript was being finalized, the largest and longest outbreak of Ebola virus was still spreading 6. The latter virus, together with Nipah virus, hantavirus, Lassa virus, and Marburg virus, is on the Centers for Disease Control and Prevention list of bioterrorism agents. For most of these viruses, the therapeutic options are limited if not completely absent.
Prevention and Therapy of Emerging Viral Diseases
The best description of the features of an outbreak of a novel virus comes from the SARS pandemic of 2002/2003 – the first major emerging disease threat of the 21st century. In the short period between the WHO global alert (March 2003) and the WHO announcement that the pandemic was over (5 July 2003), SARS affected 8098 people, caused 774 deaths, disrupted international travel, and cost huge business losses. SARS presented many of the features of the most feared pandemic diseases: a respiratory disease that is transmitted from person to person, with a long asymptomatic incubation period, and symptoms very similar to other commonplace illnesses.
At the time of its first appearance, its causative agent (SARS‐associated coronavirus) was unknown, and there was neither a diagnostic test nor a specific treatment. The international response to the new threat was extremely efficacious, leading to the isolation of SARS‐CoV 7 and sequencing of its genome in the record time of just 2 weeks 8, 9.
The measures that proved efficacious in containing the disease were isolation, quarantine, travel surveillance, increased personal hygiene, and personal protection equipment (masks, gloves, and eyeglasses). As to pharmacological treatments, however, a WHO expert panel review concluded that it was not possible to determine whether any of the treatments used for SARS‐infected patients did some benefit and suggested that some treatments might have actually been harmful 10.
As the SARS case vividly illustrates, our ability to quickly isolate and genetically fingerprint the causative agent of new viral diseases is not matched by an ability to develop suitable treatments. This is not surprising, because drug discovery typically requires additional information to that simply deriving from knowledge of the viral genome.
A Role for Peptides: Inhibition of Viral Fusion
Against this background, peptides may represent a special case, because for enveloped viruses, it is generally possible to identify the sequence of candidate peptide inhibitors directly from genomic information.
For all enveloped viruses, fusion between the viral and the target cell membrane represents an obligatory step of the life cycle. Interfering with this process is a well‐established therapeutic strategy 11 that has led to the development of the peptide fusion inhibitor enfuvirtide (Fuzeon®, also known as T20, Roche, Basel, Switzerland), which is in clinical use for HIV 12.
Viral fusion is mediated by specialized proteins, which harbor a structural motif that consists of a repeating pattern of seven amino acids: the heptad repeat (HR). The most prevalent class (class I) of fusion proteins typically has two HR regions: the first one close to the N‐terminus (HRN), adjacent to the fusion peptide, and the second one close to the C‐terminus (HRC), immediately preceding the transmembrane domain. It is generally accepted that in the key intermediate of viral fusion, the so‐called prehairpin intermediate, bridging the viral and cell membranes, the HRN and HRC regions are separated, and the HRN forms a trimeric coiled coil (Figure 1). Folding of the HRC onto the HRN trimer leads to the formation of a six‐helical bundle (6HB), and in this process, the two membranes are brought in close apposition, which leads to their fusion 11 (Figure 1). Peptides corresponding to the sequence of the HR regions can bind to the prehairpin intermediate, prevent its transition to the 6HB, and block fusion (Figure 2(A)). This is the mechanism of inhibition of enfuvirtide 12, which applies to several other peptides derived from the HR regions of many viruses 13, 14, 15, 16, 17, 18, 19, 20, 21, 22, 23, 24, 25, 26, 27, 28, 29.
Figure 1.

Viral fusion, exemplified by HIV. The fusion protein of HIV, gp41, is schematically drawn at the top of the figure. Gp41 features two heptad repeat (HR) regions, which are separated in the prehairpin intermediate. The fusion peptide (FP), N‐terminal to the HRN, anchors the protein into the target cell membrane. The protein is also anchored in the viral membrane by the transmembrane (TM) region, C‐terminal to the HRC. Refolding of the intermediate leads to formation of a six‐helix bundle (6HB), bringing the two membranes in close contact and driving fusion.
Figure 2.
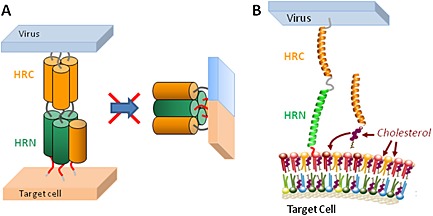
Inhibition of viral fusion. (A) HR‐derived peptides such as C34 block fusion by binding to the prehairpin intermediate and preventing the intramolecular refolding to the 6HB. (B) A cholesterol‐conjugated HR‐derived peptide concentrates in the cholesterol‐rich membrane domains, which are the sites where fusion occurs.
Prediction of Fusion Inhibitors from the Viral Genome
The characteristic sequence pattern of the HR provides an important advantage for the development of peptide‐based antivirals, because HR can be easily identified through computer programs (e.g. LearnCoil 30 or MultiCoil 31, 32). This was the case for SARS‐CoV, where HR regions were immediately identified 33 and led to the development of peptide inhibitors 27, 29, in the absence of any structural information on the fusion protein. This came later and largely confirmed the predictions 34, 35, 36, 37.
From Fusion Inhibitors to Antiviral Drugs
This pathway to drug development offers the opportunity to develop a specific antiviral in a very short timeframe. However, not all HR‐derived peptides display the same antiviral potency as the HIV fusion inhibitor enfuvirtide. A major factor at play is the difference in fusion kinetics: viruses that unlike HIV transition through the hairpin intermediate very rapidly offer a shorter window of opportunity for peptide inhibitors that, accordingly, show reduced potency 38, 39, 40.
If the peptide lead derived from genomic information has insufficient potency, it is necessary to modify it through peptide engineering strategies that, although often successful 18, 41, 42, 43, 44, 45, 46, 47, 48, 49, may be time consuming. An alternative approach, which does not require any change in the native HR sequence, exploits the role of cholesterol in viral fusion.
Role of Cholesterol in Viral Fusion
Cholesterol plays a key role in the fusion and budding of enveloped viruses 50, 51, 52, 53, 54, 55, 56, 57, 58, 59, with HIV as the most studied case. It has been shown that cholesterol depletion from virus‐infected cells suppresses virus production 60 and that depletion of cholesterol from either virions or target cells inhibits virus–cell fusion 61, 62, 63, 64. HIV is able to influence the cholesterol metabolism of the host cell 65, 66, 67, 68, and its lipid membrane is highly enriched in cholesterol 69. Moreover, a link has been established between low levels of cholesterol in antigen‐presenting cells and the status of long‐term nonprogressor, a rare infected individual who controls disease progression in the absence of antiretroviral therapy 70, 71.
The role of cholesterol in viral fusion is related to its role in membrane fluidity and in particular in the formation of lipid rafts 60, 72, 73, 74, 75, 76. Accordingly, the composition of the lipid membrane of HIV is highly enriched in cholesterol and sphingomyelin 69, 74, 75. Moreover, CD4, the primary receptor for HIV, is enriched in lipid rafts 77.
Exploiting cholesterol conjugation for fusion inhibitor design
In 2009, Ingallinella et al. proposed that addition of a cholesterol group to an HR‐derived peptide would augment its affinity for membranes and in particular for the cholesterol‐rich lipid rafts. The resulting increase in local concentration at the site of action would translate into enhanced antiviral potency (Figure 2(B)). Application of this concept to the HR‐derived peptide C34 yielded C34‐Chol, where the cholesterol group was attached, via a thioether bond, to the side chain of a Cys residue added to the C‐terminus, with an intervening Gly–Ser–Gly spacer. Cholesterol conjugation brought a dramatic improvement of antiviral potency: Depending on the viral strain tested, C34‐Chol was 25‐ to 100‐fold more potent than unconjugated C34 and 50‐ to 400‐fold more potent than enfuvirtide 78. No such improvement was apparent when cholesterol was substituted with palmitic acid 78, a lipid that is both a poorer membrane anchor and is not enriched in lipid rafts 79.
Numerous examples have since accumulated that cholesterol conjugation can improve the antiviral potency of HR‐derived peptides from many enveloped viruses – these include in vitro and in vivo studies (Table 1). Cholesterol conjugation has also been successfully applied to a covalent inhibitor derived from C34, where an isothiocyanate group on the side chain of Asp632 targets Lys574 in HRN 80.
Table 1.
Viruses for which cholesterol conjugation increases the antiviral potency of a heptad repeat‐derived peptide inhibitor
Interestingly, in line with the hypothesized mechanism of action of cholesterol conjugation, another lipid that is enriched in the HIV lipidome, dihydrosphingosine 69, also augments potency when conjugated to an HR peptide; in this case, one derived from the HRN instead of the HRC 81.
Cholesterol‐conjugated peptides show improved pharmacokinetics and are active in vivo
Cholesterol conjugation also solves an outstanding problem of peptide therapeutics, the short in vivo half‐life because of enzyme degradation and rapid clearance 82. An effective way to improve peptide pharmacokinetics (PK) is conjugation to lipids 82, which drive binding to serum proteins. The most typical derivatization is with long chain fatty acids, but derivatization with cholesterol has also been explored 83, 84, 85.
In a comparative subcutaneous PK study in mice at the concentration of 3.5 mg/kg, C34 was undetectable in plasma after 6 h, while 130 nm of C34‐Chol was still detectable 24 h after the injection: a concentration ≈300‐fold higher than the IC90 measured against multiple HIV‐1 strains 78.
In another example, a single dose of 2 mg/kg of a cholesterol‐conjugated inhibitor of Nipah virus, administered intraperitoneally to golden hamsters, was sufficient to achieve a plasma concentration >100‐fold higher than the in vitro IC50 for 24 h 86.
A single intramuscular injection of 1.6 mg/kg of a cholesterol‐conjugated inhibitor of Newcastle disease virus (NDV) administered to chickens yielded a plasma concentration of 210 nm at 24 h and ≈120 nm after 48 h, to be compared with the in vitro IC50 = 8.1 nm and IC90 = 13 nm 87.
Overall, the PK of a cholesterol‐conjugated inhibitor is expected to be at least comparable with – and likely better than – the PK of enfuvirtide, which is administered twice daily by subcutaneous injection 88.
Dimerization to complement cholesterol conjugation
One further possibility to potentiate the activity of HR‐derived peptide inhibitors is to exploit multimerization, another modification that can be implemented on the native sequence of the peptide. Multimerization brings an avidity effect 89, which effectively reduces the koff of the inhibitor fusion protein complex and increases the potency of fusion 90, 91 and entry 92 inhibitors. The effect of cholesterol conjugation, which increases the kon by facilitating the encounter of the peptide with the viral target protein, is complementary: The two modifications can therefore work in concert.
A reagent that can simultaneously dimerize the peptide and install a cholesterol group onto it is described in Figure 3 93. Trimers or higher‐order multimers could be prepared by the same strategy, but the maximum benefit of multimerization is achieved at the level of a dimer 90, 91. The reagent is stable upon prolonged storage at −20 °C. Notably, the same cysteine‐containing precursor peptide used to produce the monomeric cholesterol‐conjugated peptide can be reacted with the cholesterol dimer‐forming moiety: This enables parallel synthesis of the cholesterol‐conjugated monomer and dimer (Figure 3), which can then also be tested in parallel to establish the optimal inhibitor.
Figure 3.
Parallel cholesterol conjugation. Reaction of the same peptide precursor with off‐the‐shelf reagents 1 and 2 yields the monomeric and dimeric cholesterol‐conjugated inhibitors. The bromoacetyl functionality was unsuitable for reagent 2 93.
Using the reagent in Figure 3, dimeric cholesterol‐conjugated inhibitors were prepared for HIV 93, Nipah 93, and measles 94 virus (MV), which were more potent and effective than the corresponding cholesterol‐conjugated monomers. Potentiation of a trimeric HIV peptide inhibitor 91 by cholesterol conjugation 95 has independently been reported by another laboratory.
Scope of Cholesterol Conjugation
Since its discovery in 2009, the scope of cholesterol conjugation has considerably increased, and it now includes examples for retroviruses, paramyxoviruses, coronaviruses, orthomyxoviruses, henipaviruses, and filoviruses (Table 1).
For some of these viruses, in vivo efficacy has been demonstrated in a suitable animal model 86, 87, 94. For example, Porotto et al. showed that once‐daily intraperitoneal administration of a cholesterol‐conjugated inhibitor of Nipah virus to golden hamsters, an established model of infection, effectively prevented what would otherwise be fatal Nipah virus encephalitis. Prophylactic treatment was 100% efficacious when initiated 2 days before viral challenge and 80% efficacious when initiated concurrently with the infection. Notably, in light of the highly lethal nature of this virus, treatment 2 days after infection led to survival of 40% of the animals 86.
The same laboratory used intranasal infection of MV in suckling mice expressing the human SLAM transgene as a model to evaluate the efficacy of cholesterol‐conjugated HR‐derived peptides. Infection with MV causes a lethal acute neurological syndrome, which was completely prevented, in 100% of the animals, by prophylactic once‐daily administration of a cholesterol‐conjugated dimer derived from the sequence of the MV HRC domain 94.
Li et al. used cholesterol conjugation to improve the potency of their previously described inhibitor of NDV 96. Intramuscular injection of the cholesterol‐conjugated peptide 1 day before virus infection, and then every 3 days, protected 70% of the chickens from different serotypes of NDV 87. Moreover, treatment of the animals 2 days after infection resulted in 50% protection 87.
Two interesting features of these studies deserve further comment: First, in the aforementioned three studies, the peptide was detectable in the brain 24 h after administration 86, 87, 94, indicating that cholesterol conjugation may enable penetration of the blood–brain barrier, a difficult feat for drugs in general and for biologics in particular 97.
Second, the modest but observable degree of protection observed at 2 days postexposure (40% in the Porotto et al. 86 and 50% in the Li et al. 87 study) is comparable with the results reported for monoclonal antibodies against Ebola virus in macaques (50% efficacy when dosed 2 days after exposure) 98, 99, including the ZMapp antibody cocktail 99, which is being used as an emergency therapeutic in the latest Ebola outbreak 100.
It must be noted though that all the examples reported so far come from viruses featuring class I fusion proteins. While being the most prevalent class, it does not include major pathogens such as hepatitis C virus and dengue virus, both belonging to class II, and herpes simplex virus belonging to class III.
Class I proteins are mainly α‐helical and form 6HBs as the postfusion structure, as exemplified by HIV (Figure 1). Class II fusion proteins are characterized by trimers of hairpins composed of β‐structures 101, with a fusion loop located at the tip of an elongated β‐sheet, in place of a fusion peptide upstream of an α‐helix. Class III proteins form trimers of hairpins by combining a central α‐helical trimeric core, similar to class I proteins, with fusion loops structurally similar to class II proteins 101. Despite these structural differences, all fusion proteins adopt a hairpin structure 102 that is susceptible to inhibition.
For example, although the entry of herpes viruses (featuring a class III fusion protein) is a complex process, not yet fully clarified, in which multiple glycoproteins are involved, several laboratories have reported that peptides derived from the HR regions of gB and gH of herpes simplex virus type 1, bovine herpes virus type 1, and human cytomegalovirus are able to inhibit infection 103.
For dengue virus, helical domains have been identified in the stem region of E protein (a class II protein) that are critical for virus assembly and entry 104. Peptide inhibitors derived from this membrane‐binding region have been described 105, 106, 107, one of which inhibits all four virus serotypes 107. Moreover, de novo computational design of peptide inhibitors has been successful 108, 109. The work by Xu et al. in particular 109 indicates how the same computational approach may yield candidate peptide inhibitors for fusion proteins of all classes.
Given the ubiquitous importance of cholesterol‐rich lipid rafts in viral fusion, it is expected that cholesterol conjugation may enhance the antiviral potency also for peptides derived from class II and III fusion proteins. However, no data in this regard have yet been reported.
Perspective: A Rapid Response Strategy to Emerging Viral Diseases
The combination of bioinformatic lead identification (lead ID) and potency/PK improvement provided by cholesterol conjugation may form the basis for a rapid response strategy to emerging viral diseases (Figure 4). The project would begin immediately after the genome of the new pathogen is made available. The lead ID phase would consist in the bioinformatic analysis (e.g. via LearnCoil 30 or MultiCoil 31, 32) of the newly sequenced genome to identify the HR regions; it could last a few days. As discussed in the previous section, the computational identification of inhibitory peptides for viruses utilizing class II or III fusion proteins would be less straightforward, and the number of candidate peptides to be tested likely larger. However, given the high throughput of parallel solid‐phase peptide synthesis and the modularity of the cholesterol conjugation reaction, the initial number of target sequences should not represent a key limiting factor in the lead optimization (lead opt) phase. In lead opt, candidate peptides would be synthesized in the form of suitable precursors for cholesterol conjugation and reacted with the reagents shown in Figure 3 or similar ones. The cholesterol‐conjugated monomers and dimers would be tested for antiviral activity. In the logic of off‐the‐shelf reagents for immediate use, variations of the reagents of Figure 3 to optimize the length of the linker joining the monomers might provide considerable increase in potency 89, as shown by Kay et al. 91. Haloacetyl or maleimide‐functionalized cores with polyethylene glycol spacers of different length could all be reacted with the same precursor to rapidly select the most potent inhibitor.
Figure 4.
Rapid response strategy based on cholesterol‐conjugated fusion inhibitors. From the sequenced genome of the emerging virus, the HR regions are identified bioinformatically. This corresponds to the lead identification (lead ID) phase. In the lead optimization (lead opt) phase, candidate HR peptides are synthesized in the form of suitable precursors for cholesterol conjugation and reacted with the reagents shown in Figure 3. The cholesterol‐conjugated monomer and dimer are tested for antiviral activity. The lead opt cycle continues until a peptide is found potent enough to be moved to large‐scale production.
The lead opt cycle would continue until a peptide was found, potent enough to be moved to large‐scale production. With a relatively small number of candidate peptides, and parallel synthesis and conjugation, it is expected that the lead opt phase would be rapid, optimally a few weeks. Large‐scale production of the candidate peptide therapeutic could begin immediately, because the protocol for lab‐scale and large‐scale solid‐phase peptide synthesis are essentially the same. Preclinical evaluation (formulation, animal PK to predict the human dose) could proceed in parallel with the good manufacturing practices batch production. Abbreviated preclinical toxicology, in connection with the ‘animal rule’ 110, would be justified in an emergency situation.
Conclusions: A Role for Peptides in the Response to Emerging Viral Diseases
The strategy outlined in the previous section could provide in a very short timeframe a cholesterol‐conjugated therapeutic specific for the emerging virus. Although not necessarily the optimal drug for the new disease, it would represent a key complement to preventive measures and could enable better control of the initial outbreak. Further research efforts may then lead to a substitute therapy with increased efficacy and/or better ease of administration.
One could even envisage setting up a core laboratory that routinely runs the lead ID and lead opt stages on newly identified viruses of potential concern, in advance of an actual outbreak of disease. The limited resources necessary could be part of the Emergency and Preparedness Response set out by the Centers for Disease Control and Prevention (http://emergency.cdc.gov/) or the Global Alert and response by WHO (http://www.who.int/csr/en/) for preparing for and responding to public health emergencies.
Biography
Antonello Pessi obtained his Chemistry Laurea degree at the University of Rome. After a short stay in the laboratory of R. Sheppard at LMB (Cambridge, UK), he held positions of increasing responsibility at ENIRICERCHE, SCLAVO (now part of GSK), and IRBM, the Italian site of Merck Research Laboratories, where he spent 20 years. Under his direction, the Peptide Centre of Excellence, the centralized Merck Unit for peptide medicinal chemistry, provided a number of preclinical candidates to the company pipeline. In 2009, he founded PeptiPharma, which offers consulting and contract research on therapeutic peptides. He is also the CSO of JV Bio, a start‐up focused on clinical development of antiviral peptides and antibodies. He is the recipient of the 2005 Leonidas Zervas Award of the European Peptide Society and author of more than 160 publications and 40 patents. His current academic interest, in collaboration with CEINGE, a nonprofit consortium of the University of Naples, is the design of antibacterial peptides.

Pessi, A. (2015), Cholesterol‐conjugated peptide antivirals: a path to a rapid response to emerging viral diseases. J. Pept. Sci., 21, 379–386. doi: 10.1002/psc.2706.
Special issue of contributions presented at the 14th Naples Workshop on Bioactive Peptides “The renaissance era of peptides in drug discovery”, June 12–14, 2014, Naples.
References
- 1. Snowden FM. Emerging and reemerging diseases: a historical perspective. Immunol. Rev. 2008; 225: 9–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Pekosz A, Glass GE. Emerging viral diseases. Md. Med. 2008; 9(11): 13–16. [PMC free article] [PubMed] [Google Scholar]
- 3. WHO . The World Health Report 2007. Geneva, 2007.
- 4. Jones KE, Patel NG, Levy MA, Storeygard A, Balk D, Gittleman JL, Daszak P. Global trends in emerging infectious diseases. Nature 2008; 451: 990–993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. de Groot RJ, Baker SC, Baric RS, Brown CS, Drosten C, Enjuanes L, Fouchier RAM, Galiano M, Gorbalenya AE, Memish ZA, Perlman S, Poon LLM, Snijder EJ, Stephens GM, Woo PCY, Zaki AM, Zambon M, Ziebuhr J. Middle East respiratory syndrome coronavirus (MERS‐CoV): announcement of the coronavirus study group. J. Virol. 2013; 87: 7790–7792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Hayden EC. World struggles to stop Ebola. Nature 2014; 512: 355–356. [DOI] [PubMed] [Google Scholar]
- 7. Drosten C, Gunther S, Preiser W, van der Werf S, Brodt H‐R, Becker S, Rabenau H, Panning M, Kolesnikova L, Fouchier RAM, Berger A, Burguiere A‐M, Cinatl J, Eickmann M, Escriou N, Grywna K, Kramme S, Manuguerra J‐C, Muller S, Rickerts V, Sturmer M, Vieth S, Klenk H‐D, Osterhaus ADME, Schmitz H, Doerr HW. Identification of a novel coronavirus in patients with severe acute respiratory syndrome. New Engl. J. Med. 2003; 348: 1967–1976. [DOI] [PubMed] [Google Scholar]
- 8. Rota PA, Oberste MS, Monroe SS, Nix WA, Campagnoli R, Icenogle JP, Penaranda S, Bankamp B, Maher K, Chen M‐H, Tong S, Tamin A, Lowe L, Frace M, DeRisi JL, Chen Q, Wang D, Erdman DD, Peret TCT, Burns C, Ksiazek TG, Rollin PE, Sanchez A, Liffick S, Holloway B, Limor J, McCaustland K, Olsen‐Rasmussen M, Fouchier R, Gunther S, Osterhaus ADME, Drosten C, Pallansch MA, Anderson LJ, Bellini WJ. Characterization of a novel coronavirus associated with severe acute respiratory syndrome. Science 2003; 300: 1394–1399. [DOI] [PubMed] [Google Scholar]
- 9. Marra MA, Jones SJM, Astell CR, Holt RA, Brooks‐Wilson A, Butterfield YSN, Khattra J, Asano JK, Barber SA, Chan SY, Cloutier A, Coughlin SM, Freeman D, Girn N, Griffith OL, Leach SR, Mayo M, McDonald H, Montgomery SB, Pandoh PK, Petrescu AS, Robertson AG, Schein JE, Siddiqui A, Smailus DE, Stott JM, Yang GS, Plummer F, Andonov A, Artsob H, Bastien N, Bernard K, Booth TF, Bowness D, Czub M, Drebot M, Fernando L, Flick R, Garbutt M, Gray M, Grolla A, Jones S, Feldmann H, Meyers A, Kabani A, Li Y, Normand S, Stroher U, Tipples GA, Tyler S, Vogrig R, Ward D, Watson B, Brunham RC, Krajden M, Petric M, Skowronski DM, Upton C, Roper RL. The genome sequence of the SARS‐associated coronavirus. Science 2003; 300: 1399–1404. [DOI] [PubMed] [Google Scholar]
- 10. Stockman LJ, Bellamy R, Garner P. SARS: systematic review of treatment effects. PLoS Med. 2006; 3: e343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Eckert DM, Kim PS. Mechanisms of viral membrane fusion and its inhibition. Annu. Rev. Biochem. 2001; 70: 777–810. [DOI] [PubMed] [Google Scholar]
- 12. LaBonte J, Lebbos J, Kirkpatrick P. Enfuvirtide. Nat. Rev. Drug Discov. 2003; 2: 345–346. [DOI] [PubMed] [Google Scholar]
- 13. Ni L, Zhu J, Zhang J, Yan M, Gao GF, Tien P. Design of recombinant protein‐based SARS‐CoV entry inhibitors targeting the heptad‐repeat regions of the spike protein S2 domain. Biochem. Biophys. Res. Commun. 2005; 330: 39–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Young JK, Hicks RP, Wright GE, Morrison TG. Analysis of a peptide inhibitor of paramyxovirus (NDV) fusion using biological assays, NMR, and molecular modeling. Virology 1997; 238: 291–304. [DOI] [PubMed] [Google Scholar]
- 15. Bossart KN, Mungall BA, Crameri G, Wang LF, Eaton BT, Broder CC. Inhibition of Henipavirus fusion and infection by heptad‐derived peptides of the Nipah virus fusion glycoprotein. Virol. J. 2005; 2: 57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. D'Ursi AM, Giannecchini S, Esposito C, Alcaro MC, Sichi O, Armenante MR, Carotenuto A, Papini AM, Bendinelli M, Rovero P. Development of antiviral fusion inhibitors: short modified peptides derived from the transmembrane glycoprotein of feline immunodeficiency virus. ChemBioChem 2006; 7: 774–779. [DOI] [PubMed] [Google Scholar]
- 17. Gao GF. (ed.). Peptide inhibitors targeting virus‐cell fusion in class I enveloped viruses. Combating the Threat of Pandemic Influenza John Wiley & Sons, Inc.: Hoboken, NJ, 2007; 226–246. [Google Scholar]
- 18. Harrison RS, Shepherd NE, Hoang HN, Ruiz‐Gomez G, Hill TA, Driver RW, Desai VS, Young PR, Abbenante G, Fairlie DP. Downsizing human, bacterial, and viral proteins to short water‐stable alpha helices that maintain biological potency. Proc. Natl. Acad. Sci. U. S. A. 2010; 107: 11686–11691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Yao Q, Compans RW. Peptides corresponding to the heptad repeat sequence of human parainfluenza virus fusion protein are potent inhibitors of virus infection. Virology 1996; 223: 103–112. [DOI] [PubMed] [Google Scholar]
- 20. Rapaport D, Ovadia M, Shai Y. A synthetic peptide corresponding to a conserved heptad repeat domain is a potent inhibitor of Sendai virus–cell fusion: an emerging similarity with functional domains of other viruses. EMBO J. 1995; 14: 5524–5531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Mizukoshi F, Baba K, Goto Y, Setoguchi A, Fujino Y, Ohno K, Oishi S, Kodera Y, Fujii N, Tsujimoto H. Antiviral activity of membrane fusion inhibitors that target gp40 of the feline immunodeficiency virus envelope protein. Vet. Microbiol. 2009; 136: 155–159. [DOI] [PubMed] [Google Scholar]
- 22. Steffen I, Pohlmann S. Peptide‐based inhibitors of the HIV envelope protein and other class I viral fusion proteins. Curr. Pharm. Des. 2010; 16: 1143–1158. [DOI] [PubMed] [Google Scholar]
- 23. Miller EH, Harrison JS, Radoshitzky SR, Higgins CD, Chi X, Dong L, Kuhn JH, Bavari S, Lai JR, Chandran K. Inhibition of Ebola virus entry by a C‐peptide targeted to endosomes. J. Biol. Chem. 2011; 286: 15854–15861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Lambert DM, Barney S, Lambert AL, Guthrie K, Medinas R, Davis DE, Bucy T, Erickson J, Merutka G, Petteway SR, Jr . Peptides from conserved regions of paramyxovirus fusion (F) proteins are potent inhibitors of viral fusion. Proc. Natl. Acad. Sci. U. S. A. 1996; 93: 2186–2191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Porotto M, Doctor L, Carta P, Fornabaio M, Greengard O, Kellogg GE, Moscona A. Inhibition of Hendra virus fusion. J. Virol. 2006; 80: 9837–9849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Wild TF, Buckland R. Inhibition of measles virus infection and fusion with peptides corresponding to the leucine zipper region of the fusion protein. J. Gen. Virol. 1997; 78: 107–111. [DOI] [PubMed] [Google Scholar]
- 27. Bosch BJ, Martina BE, Van Der Zee R, Lepault J, Haijema BJ, Versluis C, Heck AJ, De Groot R, Osterhaus AD, Rottier PJ. Severe acute respiratory syndrome coronavirus (SARS‐CoV) infection inhibition using spike protein heptad repeat‐derived peptides. Proc. Natl. Acad. Sci. U. S. A. 2004; 101: 8455–8460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Liu IJ, Kao CL, Hsieh SC, Wey MT, Kan LS, Wang WK. Identification of a minimal peptide derived from heptad repeat (HR) 2 of spike protein of SARS‐CoV and combination of HR1‐derived peptides as fusion inhibitors. Antiviral Res. 2009; 81: 82–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Liu S, Xiao G, Chen Y, He Y, Niu J, Escalante CR, Xiong H, Farmar J, Debnath AK, Tien P, Jiang S. Interaction between heptad repeat 1 and 2 regions in spike protein of SARS‐associated coronavirus: implications for virus fusogenic mechanism and identification of fusion inhibitors. Lancet 2004; 363: 938–947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Singh M, Berger B, Kim PS. LearnCoil‐VMF: computational evidence for coiled‐coil‐like motifs in many viral membrane‐fusion proteins. J. Mol. Biol. 1999; 290: 1031–1041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Trigg J, Gutwin K, Keating AE, Berger B. MultiCoil2: predicting coiled coils and their oligomerization states from sequence in the twilight zone. PLoS One 2011; 6: e23519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Wolf E, Kim PS, Berger B. MultiCoil: a program for predicting two‐ and three‐stranded coiled coils. Proc. Natl. Acad. Sci. U. S. A. 1997; 6: 1179–1189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Zeng FY, Chan CW, Chan MN, Chen JD, Chow KY, Hon CC, Hui KH, Li J, Li VY, Wang CY, Wang PY, Guan Y, Zheng B, Poon LL, Chan KH, Yuen KY, Peiris JS, Leung FC. The complete genome sequence of severe acute respiratory syndrome coronavirus strain HKU‐39849 (HK‐39). Exp. Biol. Med. 2003; 228: 866–873. [DOI] [PubMed] [Google Scholar]
- 34. Tripet B, Howard MW, Jobling M, Holmes RK, Holmes KV, Hodges RS. Structural characterization of the SARS‐coronavirus spike S fusion protein core. J. Biol. Chem. 2004: 279: 20836–20849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Deng Y, Liu J, Zheng Q, Yong W, Lu M. Structures and polymorphic interactions of two heptad‐repeat regions of the SARS virus S2 protein. Structure 2006; 14: 889–899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Ingallinella P, Bianchi E, Finotto M, Cantoni G, Eckert DM, Supekar VM, Bruckmann C, Carfi A, Pessi A. Structural characterization of the fusion‐active complex of severe acute respiratory syndrome (SARS) coronavirus. Proc. Natl. Acad. Sci. U. S. A. 2004; 101: 8709–8714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Xu Y, Lou Z, Liu Y, Pang H, Tien P, Gao GF, Rao Z. Crystal structure of severe acute respiratory syndrome coronavirus spike protein fusion core. J. Biol. Chem. 2004; 279: 49414–49419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Reeves JD, Gallo SA, Ahmad N, Miamidian JL, Harvey PE, Sharron M, Pohlmann S, Sfakianos JN, Derdeyn CA, Blumenthal R, Hunter E, Doms RW. Sensitivity of HIV‐1 to entry inhibitors correlates with envelope/coreceptor affinity, receptor density, and fusion kinetics. Proc. Natl. Acad. Sci. U. S. A. 2002; 99: 16249–16254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Aguilar HC, Aspericueta V, Robinson LR, Aanensen KE, Lee B. A quantitative and kinetic fusion protein‐triggering assay can discern distinct steps in the Nipah virus membrane fusion cascade. J. Virol. 2010; 84: 8033–8041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Porotto M, Yokoyama CC, Orefice G, Kim HS, Aljofan M, Mungall BA, Moscona A. Kinetic dependence of paramyxovirus entry inhibition. J. Virol. 2009; 83: 6947–6951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Yao X, Chong H, Zhang C, Waltersperger S, Wang M, Cui S, He Y. Broad antiviral activity and crystal structure of HIV‐1 fusion inhibitor sifuvirtide. J. Biol. Chem. 2012; 287: 6788–6796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Chong H, Yao X, Qiu Z, Sun J, Zhang M, Waltersperger S, Wang M, Liu SL, Cui S, He Y. Short‐peptide fusion inhibitors with high potency against wild‐type and enfuvirtide‐resistant HIV‐1. FASEB J. 2013; 27: 1203–1213. [DOI] [PubMed] [Google Scholar]
- 43. Dwyer JJ, Wilson KL, Davison DK, Freel SA, Seedorff JE, Wring SA, Tvermoes NA, Matthews TJ, Greenberg ML, Delmedico MK. Design of helical, oligomeric HIV‐1 fusion inhibitor peptides with potent activity against enfuvirtide‐resistant virus. Proc. Natl. Acad. Sci. U. S. A. 2007; 104: 12772–12777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Oishi S, Ito S, Nishikawa H, Watanabe K, Tanaka M, Ohno H, Izumi K, Sakagami Y, Kodama E, Matsuoka M, Fujii N. Design of a novel HIV‐1 fusion inhibitor that displays a minimal interface for binding affinity. J. Med. Chem. 2008; 51: 388–391. [DOI] [PubMed] [Google Scholar]
- 45. Otaka A, Nakamura M, Nameki D, Kodama E, Uchiyama S, Nakamura S, Nakano H, Tamamura H, Kobayashi Y, Matsuoka M, Fujii N. Remodeling of gp41‐C34 peptide leads to highly effective inhibitors of the fusion of HIV‐1 with target cells. Angew. Chem. Int. Ed. Engl. 2002; 41: 2937–2940. [DOI] [PubMed] [Google Scholar]
- 46. Judice JK, Tom JY, Huang W, Wrin T, Vennari J, Petropoulos CJ, McDowell RS. Inhibition of HIV type 1 infectivity by constrained alpha‐helical peptides: implications for the viral fusion mechanism. Proc. Natl. Acad. Sci. U. S. A. 1997; 94: 13426–13430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Sia SK, Carr PA, Cochran AG, Malashkevich VN, Kim PS. Short constrained peptides that inhibit HIV‐1 entry. Proc. Natl. Acad. Sci. U. S. A. 2002; 99: 14664–14669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Bird GH, Madani N, Perry AF, Princiotto AM, Supko JG, He X, Gavathiotis E, Sodroski JG, Walensky LD. Hydrocarbon double‐stapling remedies the proteolytic instability of a lengthy peptide therapeutic. Proc. Natl. Acad. Sci. U. S. A. 2010; 107: 14093–14098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. He Y, Xiao Y, Song H, Liang Q, Ju D, Chen X, Lu H, Jing W, Jiang S, Zhang L. Design and evaluation of sifuvirtide, a novel HIV‐1 fusion inhibitor. J. Biol. Chem. 2008; 283: 11126–11134. [DOI] [PubMed] [Google Scholar]
- 50. Lu Y, Liu DX, Tam JP. Lipid rafts are involved in SARS‐CoV entry into Vero E6 cells. Biochem. Biophys. Res. Commun. 2008; 369: 344–349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Daya M, Cervin M, Anderson R. Cholesterol enhances mouse hepatitis virus‐mediated cell fusion. Virology 1988; 163: 276–283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Vashishtha M, Phalen T, Marquardt MT, Ryu JS, Ng AC, Kielian M. A single point mutation controls the cholesterol dependence of Semliki Forest virus entry and exit. J. Cell Biol. 1998; 140: 91–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Danthi P, Chow M. Cholesterol removal by methyl‐beta‐cyclodextrin inhibits poliovirus entry. J. Virol. 2004; 78: 33–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Petersen J, Drake MJ, Bruce EA, Riblett AM, Didigu CA, Wilen CB, Malani N, Male F, Lee FH, Bushman FD, Cherry S, Doms RW, Bates P, Briley K, Jr. The major cellular sterol regulatory pathway is required for Andes virus infection. PLoS Pathog. 2014; 10: e1003911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Li GM, Li YG, Yamate M, Li SM, Ikuta K. Lipid rafts play an important role in the early stage of severe acute respiratory syndrome‐coronavirus life cycle. Microbes Infect. 2007; 9: 96–102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Chang TH, Segovia J, Sabbah A, Mgbemena V, Bose S. Cholesterol‐rich lipid rafts are required for release of infectious human respiratory syncytial virus particles. Virology 2012; 422: 205–213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Carette JE, Raaben M, Wong AC, Herbert AS, Obernosterer G, Mulherkar N, Kuehne AI, Kranzusch PJ, Griffin AM, Ruthel G, Cin PD, Dye JM, Whelan SP, Chandran K, Brummelkamp TR. Ebola virus entry requires the cholesterol transporter Niemann–Pick C1. Nature 2011; 477: 340–343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Hambleton S, Steinberg SP, Gershon MD, Gershon AA. Cholesterol dependence of varicella‐zoster virion entry into target cells. J. Virol. 2007; 81: 7548–7558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Takeda M, Leser GP, Russell CJ, Lamb RA. Influenza virus hemagglutinin concentrates in lipid raft microdomains for efficient viral fusion. Proc. Natl. Acad. Sci. U. S. A. 2003; 100: 14610–14617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Ono A, Freed EO. Plasma membrane rafts play a critical role in HIV‐1 assembly and release. Proc. Natl. Acad. Sci. U. S. A. 2001; 98: 13925–13930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Mañes S, del Real G, Lacalle RA, Lucas P, Gómez‐Moutón C, Sánchez‐Palomino S, Delgado R, Alcamí J, Mira E, Martínez AC. Membrane raft microdomains mediate lateral assemblies required for HIV‐1 infection. EMBO Rep. 2000; 1: 190–196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Guyader M, Kiyokawa E, Abrami L, Turelli P, Trono D. Role for human immunodeficiency virus type 1 membrane cholesterol in viral internalization. J. Virol. 2002; 76: 10356–10364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Viard M, Parolini I, Sargiacomo M, Fecchi K, Ramoni C, Ablan S, Ruscetti FW, Wang JM, Blumenthal R. Role of cholesterol in human immunodeficiency virus type 1 envelope protein‐mediated fusion with host cells. J. Virol. 2002; 76: 11584–11595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Liao Z, Cimakasky LM, Hampton R, Nguyen DH, Hildreth JE. Lipid rafts and HIV pathogenesis: host membrane cholesterol is required for infection by HIV type 1. AIDS Res. Hum. Retroviruses 2001; 17: 1009–1019. [DOI] [PubMed] [Google Scholar]
- 65. van 't Wout AB, Swain JV, Schindler M, Rao U, Pathmajeyan MS, Mullins JI, Kirchhoff F. Nef induces multiple genes involved in cholesterol synthesis and uptake in human immunodeficiency virus type 1‐infected T cells. J. Virol. 2005; 79: 10053–10058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Mujawar Z, Rose H, Morrow MP, Pushkarsky T, Dubrovsky L, Mukhamedova N, Fu Y, Dart A, Orenstein JM, Bobryshev YV, Bukrinsky M, Sviridov D. Human immunodeficiency virus impairs reverse cholesterol transport from macrophages. PLoS Biol. 2006; 4: e365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Zheng Y‐H, Plemenitas A, Fielding CJ, Peterlin BM. Nef increases the synthesis of and transports cholesterol to lipid rafts and HIV‐1 progeny virions. Proc. Natl. Acad. Sci. U. S. A. 2003; 100: 8460–8465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Cui HL, Grant A, Mukhamedova N, Pushkarsky T, Jennelle L, Dubrovsky L, Gaus K, Fitzgerald ML, Sviridov D, Bukrinsky M. HIV‐1 Nef mobilizes lipid rafts in macrophages through a pathway that competes with ABCA1‐dependent cholesterol efflux. J. Lipid Res. 2012; 53: 696–708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Brugger B, Glass B, Haberkant P, Leibrecht I, Wieland FT, Krausslich HG. The HIV lipidome: a raft with an unusual composition. Proc. Natl. Acad. Sci. U. S. A. 2006; 103: 2641–2646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Rappocciolo G, Jais M, Piazza P, Reinhart TA, Berendam SJ, Garcia‐Exposito L, Gupta P, Rinaldo CR. Alterations in cholesterol metabolism restrict HIV‐1 trans infection in nonprogressors. mBio 2014; 5: e01031–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Prasad VR, Bukrinsky MI. New clues to understanding HIV nonprogressors: low cholesterol blocks HIV trans infection. mBio 2014; 5: e01396–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Nguyen DH, Hildreth JEK. Evidence for budding of human immunodeficiency virus type 1 selectively from glycolipid‐enriched membrane lipid rafts. J. Virol. 2000; 74: 3264–3272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Blanchette CD, Lin W‐C, Ratto TV, Longo ML. Galactosylceramide domain microstructure: impact of cholesterol and nucleation/growth conditions. Biophys. J. 2006; 90: 4466–4478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Aloia RC, Jensen FC, Curtain CC, Mobley PW, Gordon LM. Lipid composition and fluidity of the human immunodeficiency virus. Proc. Natl. Acad. Sci. U. S. A. 1988; 85: 900–904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Aloia RC, Tian H, Jensen FC. Lipid composition and fluidity of the human immunodeficiency virus envelope and host cell plasma membranes. Proc. Natl. Acad. Sci. U. S. A. 1993; 90: 5181–5185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Ono A, Freed EO. Role of lipid rafts in virus replication. Adv. Virus Res. 2005; 64: 311–358. [DOI] [PubMed] [Google Scholar]
- 77. Chernomordik LV, Frolov VA, Leikina E, Bronk P, Zimmerberg J. The pathway of membrane fusion catalyzed by influenza hemagglutinin: restriction of lipids, hemifusion, and lipidic fusion pore formation. J. Cell Biol. 1998; 140: 1369–1382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Ingallinella P, Bianchi E, Ladwa NA, Wang Y‐J, Hrin R, Veneziano M, Bonelli F, Ketas TJ, Moore JP, Miller MD, Pessi A. Addition of a cholesterol group to an HIV‐1 peptide fusion inhibitor dramatically increases its antiviral potency. Proc. Natl. Acad. Sci. U. S. A. 2009; 106: 5801–5806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Resh MD. Membrane targeting of lipid modified signal transduction proteins. Subcell. Biochem. 2004; 37: 217–232. [DOI] [PubMed] [Google Scholar]
- 80. Zhao L, Tong P, Chen YX, Hu ZW, Wang K, Zhang YN, Zhao DS, Cai LF, Liu KL, Zhao YF, Li YM. A multi‐functional peptide as an HIV‐1 entry inhibitor based on self‐concentration, recognition, and covalent attachment. Org. Biomol. Chem. 2012; 10: 6512–6520. [DOI] [PubMed] [Google Scholar]
- 81. Ashkenazi A, Viard M, Unger L, Blumenthal R, Shai Y. Sphingopeptides: dihydrosphingosine‐based fusion inhibitors against wild‐type and enfuvirtide‐resistant HIV‐1. FASEB J. 2012; 26: 4628–4636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Zhang L, Bulaj G. Converting peptides into drug leads by lipidation. Curr. Med. Chem. 2012; 19: 1602–1618. [DOI] [PubMed] [Google Scholar]
- 83. Madsen K, Knudsen LB, Agersoe H, Nielsen PF, Thogersen H, Wilken M, Johansen NL. Structure–activity and protraction relationship of long‐acting glucagon‐like peptide‐1 derivatives: importance of fatty acid length, polarity, and bulkiness. J. Med. Chem. 2007; 50: 6126–6132. [DOI] [PubMed] [Google Scholar]
- 84. Pocai A, Carrington PE, Adams JR, Wright M, Eiermann G, Zhu L, Du X, Petrov A, Lassman ME, Jiang G, Liu F, Miller C, Tota LM, Zhou G, Zhang X, Sountis MM, Santoprete A, Capito E, Chicchi GG, Thornberry N, Bianchi E, Pessi A, Marsh DJ, SinhaRoy R. Glucagon‐like peptide 1/glucagon receptor dual agonism reverses obesity in mice. Diabetes 2009; 58: 2258–2266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Santoprete A, Capito E, Carrington PE, Pocai A, Finotto M, Langella A, Ingallinella P, Zytko K, Bufali S, Cianetti S, Veneziano M, Bonelli F, Zhu L, Monteagudo E, Marsh DJ, Sinharoy R, Bianchi E, Pessi A. DPP‐IV‐resistant, long‐acting oxyntomodulin derivatives. J. Pept. Sci. 2011; 17: 270–280. [DOI] [PubMed] [Google Scholar]
- 86. Porotto M, Rockx B, Yokoyama CC, Talekar A, DeVito I, Palermo LM, Liu J, Cortese R, Lu M, Feldmann H, Pessi A, Moscona A. Inhibition of Nipah virus infection in vivo: targeting an early stage of paramyxovirus fusion activation during viral entry. PLoS Pathog. 2010; 6: e1001168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Li C‐G, Tang W, Chi X‐J, Dong Z‐M, Wang X‐X, Wang X‐J. A cholesterol tag at the N terminus of the relatively broad‐spectrum fusion inhibitory peptide targets an earlier stage of fusion glycoprotein activation and increases the peptide's antiviral potency in vivo . J. Virol. 2013; 87: 9223–9232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Fung HB, Guo Y. Enfuvirtide: a fusion inhibitor for the treatment of HIV infection. Clin. Ther. 2004; 26: 352–378. [DOI] [PubMed] [Google Scholar]
- 89. Mack ET, Snyder PW, Perez‐Castillejos R, Bilgiçer B, Moustakas DT, Butte MJ, Whitesides GM. Dependence of avidity on linker length for a bivalent ligand‐bivalent receptor model system. J. Am. Chem. Soc. 2011; 134: 333–345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Welch BD, VanDemark AP, Heroux A, Hill CP, Kay MS. Potent D‐peptide inhibitors of HIV‐1 entry. Proc. Natl. Acad. Sci. U. S. A. 2007; 104: 16828–16833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Welch BD, Francis JN, Redman JS, Paul S, Weinstock MT, Reeves JD, Lie YS, Whitby FG, Eckert DM, Hill CP, Root MJ Kay MS Design of a potent D‐peptide HIV‐1 entry inhibitor with a strong barrier to resistance. J. Virol. 2010; 84: 11235–11244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Keeffe JR, Gnanapragasam PNP, Gillespie SK, Yong J, Bjorkman PJ, Mayo SL. Designed oligomers of cyanovirin‐N show enhanced HIV neutralization. Proc. Natl. Acad. Sci. U. S. A. 2011; 108: 14079–14084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Pessi A, Langella A, Capitò E, Ghezzi S, Vicenzi E, Poli G, Ketas T, Mathieu C, Cortese R, Horvat B, Moscona A, Porotto M. A general strategy to endow natural fusion‐protein‐derived peptides with potent antiviral activity. PLoS One 2012; 7: e36833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Welsch JC, Talekar A, Mathieu C, Pessi A, Moscona A, Horvat B, Porotto M. Fatal measles virus infection prevented by brain‐penetrant fusion inhibitors. J. Virol. 2013; 87: 13785–13794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Francis JN, Redman JS, Eckert DM, Kay MS. Design of a modular tetrameric scaffold for the synthesis of membrane‐localized D‐peptide inhibitors of HIV‐1 entry. Bioconjug. Chem. 2012; 23: 1252–1258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Wang XJ, Li CG, Chi XJ, Wang M. Characterisation and evaluation of antiviral recombinant peptides based on the heptad repeat regions of NDV and IBV fusion glycoproteins. Virology 2011; 416: 65–74. [DOI] [PubMed] [Google Scholar]
- 97. Pardridge WM. Drug transport across the blood–brain barrier. J. Cereb. Blood Flow Metab. 2012; 32: 1959–1972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Qiu X, Audet J, Wong G, Pillet S, Bello A, Cabral T, Strong JE, Plummer F, Corbett CR, Alimonti JB, Kobinger GP. Successful treatment of Ebola virus‐infected cynomolgus macaques with monoclonal antibodies. Sci. Transl. Med. 2012; 4: 138ra81. [DOI] [PubMed] [Google Scholar]
- 99. Pettitt J, Zeitlin L, Kim do H, Working C, Johnson JC, Bohorov O, Bratcher B, Hiatt E, Hume SD, Johnson AK, Morton J, Pauly MH, Whaley KJ, Ingram MF, Zovanyi A, Heinrich M, Piper A, Zelko J, Olinger GG. Therapeutic intervention of Ebola virus infection in rhesus macaques with the MB‐003 monoclonal antibody cocktail. Sci. Transl. Med. 2013; 5: 199ra113. [DOI] [PubMed] [Google Scholar]
- 100. Mullard A. Experimental Ebola drugs enter the limelight. The Lancet 2014; 384: 649. [DOI] [PubMed] [Google Scholar]
- 101. Weissenhorn W, Hinz A, Gaudin Y. Virus membrane fusion. FEBS Lett. 2007; 581: 2150–2155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Kielian M, Rey FA. Virus membrane‐fusion proteins: more than one way to make a hairpin. Nat. Rev. Microbiol. 2006; 4: 67–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. Galdiero S, Falanga A, Tarallo R, Russo L, Galdiero E, Cantisani M, Morelli G, Galdiero M. Peptide inhibitors against herpes simplex virus infections. J. Pept. Sci. 2013; 19: 148–158. [DOI] [PubMed] [Google Scholar]
- 104. Lin S‐R, Zou G, Hsieh S‐C, Qing M, Tsai W‐Y, Shi P‐Y, Wang W‐K. The helical domains of the stem region of dengue virus envelope protein are involved in both virus assembly and entry. J. Virol. 2011; 85: 5159–5171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. Schmidt AG, Yang PL, Harrison SC. Peptide inhibitors of dengue‐virus entry target a late‐stage fusion intermediate. PLoS Pathog. 2010; 6: e1000851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106. Schmidt AG, Yang PL, Harrison SC. Peptide inhibitors of flavivirus entry derived from the E protein stem. J. Virol. 2010; 84: 12549–12554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107. Lok S‐M, Costin JM, Hrobowski YM, Hoffmann AR, Rowe DK, Kukkaro P, Holdaway H, Chipman P, Fontaine KA, Holbrook MR, Garry RF, Kostyuchenko V, Wimley WC, Isern S, Rossmann MG, Michael SF. Release of dengue virus genome induced by a peptide inhibitor. PLoS One 2012; 7: e50995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108. Costin JM, Jenwitheesuk E, Lok S‐M, Hunsperger E, Conrads KA, Fontaine KA, Rees CR, Rossmann MG, Isern S, Samudrala R, Michael SF. Structural optimization and de novo design of dengue virus entry inhibitory peptides. PLoS Negl. Trop. Dis. 2010; 4: e721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109. Xu Y, Rahman NA, Othman R, Hu P, Huang M. Computational identification of self‐inhibitory peptides from envelope proteins. Proteins 2012; 80: 2154–2168. [DOI] [PubMed] [Google Scholar]
- 110. Mitchell WH, Goldenthal KL. New drug and biological drug products; evidence needed to demonstrate effectiveness of new drugs when human efficacy studies are not ethical or feasible. Final rule. Fed. Regist. 2002; 67: 37988–37998. [PubMed] [Google Scholar]
- 111. Porotto M, Yokoyama CC, Palermo LM, Mungall B, Aljofan M, Cortese R, Pessi A, Moscona A. Viral entry inhibitors targeted to the membrane site of action. J. Virol. 2010; 84: 6760–6768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112. Lee KK, Pessi A, Gui L, Santoprete A, Talekar A, Moscona A, Porotto M. Capturing a fusion intermediate of influenza hemagglutinin with a cholesterol‐conjugated peptide, a new antiviral strategy for influenza virus. J. Biol. Chem. 2011; 286: 42141–42149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113. Higgins CD, Koellhoffer JF, Chandran K, Lai JR. C‐peptide inhibitors of Ebola virus glycoprotein‐mediated cell entry: effects of conjugation to cholesterol and side chain–side chain crosslinking. Bioorg. Med. Chem. Lett. 2013; 23: 5356–5360. [DOI] [PMC free article] [PubMed] [Google Scholar]