

Abstract
Viral infections cause many serious human diseases with high mortality rates. New drug‐resistant strains are continually emerging due to the high viral mutation rate, which makes it necessary to develop new antiviral agents. Compounds of plant origin are particularly interesting. The pentacyclic triterpenoids (PTs) are a diverse class of natural products from plants composed of three terpene units. They exhibit antitumor, anti‐inflammatory, and antiviral activities. Oleanolic, betulinic, and ursolic acids are representative PTs widely present in nature with a broad antiviral spectrum. This review focuses on the recent literatures in the antiviral efficacy of this class of phytochemicals and their derivatives. In addition, their modes of action are also summarized.
Keywords: antiviral, betulinic acid, glycyrrhizin, mechanism, pentacyclic triterpenoid
Abbreviations
- BA
betulinic acid
- BVM
bevirimat
- CPE
cytopathic effect
- EA
echinocystic acid
- EB
Epstein–Barr virus
- EV71
human enterovirus 71
- GA
glycyrrhetinic acid
- HBV
hepatitis B virus
- HCV
hepatitis C virus
- HIV
human immunodeficiency virus
- HSV
herpes simplex virus
- OA
oleanolic acid
- OSV
oseltamivir
- PTs
pentacyclic triterpenoids
- RBV
ribavirin
- ROS
reactive oxygen species
- SAR
structure–activity relationship
- SARS
severe acute respiratory syndrome
- UA
ursolic acid
- VSV
vesicular stomatitis virus
1. INTRODUCTION
Viruses exist wherever life is found.1 They are the most common pathogens leading to infectious and fatal human diseases. According to the 2014 World Health Organization report, over 36.9 million people are living with the human immunodeficiency virus (HIV) infection.2 The most recent estimate indicates that hepatitis C virus (HCV) causes chronical infection of approximately 3% of the world's population.3 In addition to those two viral pathogens, humans also constantly encounter threats from many other emerging and reemerging infectious, zoonotic and devastating viruses, such as the highly pathogenic avian influenza A virus (H5N14 and H7N95), severe acute respiratory syndrome coronavirus (SARS‐CoV),6 Middle East respiratory syndrome coronavirus (MERS‐CoV),7 Ebola virus (EBOV),8 and so on. Given the high mutation rate of viruses and their serious threat to public health, there is a high urgency to develop new antiviral drugs to combat these pathogens. Natural products in the roots, stems, barks, leaves, fruits, and seeds of plants have been used as herbal medicines to treat diseases for more than 3000 years in China. They also have played a significant role in modern drug discovery by serving as prototypes of novel drugs. As mentioned by Newman et al.,9 it has been estimated that over 49% of clinically used antitumor drugs from around the 1940s to the end of 2014 are natural products or their derivatives. Natural product‐derived drugs have also been crucial for antiviral drug discovery. From 1981 to 2014, over 40% of antiviral drugs in clinical use are natural products (∼6%) or designed using natural products as prototypes (∼34%).9
Among the natural products, triterpenoids and their relatives, also called the steroids, constitute a very large family of compounds with over 20,000 identified molecules. And many new compounds are being discovered in plants, animals, and fungi every year.10 Most of them exist in nature as free acids, as esters with fatty acids, ferulic acid, etc., or as triterpenoid saponins linked with one or more sugar chains. They are often the main active constituents of many important medicinal plants, such as the ginsenosides in Panax ginseng, glycyrrhizin in Glycyrrhiza uralensis and saikosaponins in Radix Bupleuri.11, 12, 13 Triterpenoids display a variety of structures with nearly 200 different skeletons known from natural sources or enzymatic reactions, although many are variants of a smaller number of common structural types.14 Many pentacyclic triterpenoids (PTs) show a wide range of pharmacological activities, and some are marketed as therapeutic agents or dietary supplements.15 Structurally, PTs have four six‐membered rings called A, B, C, D with ring E being five‐membered or six‐membered. Based on the carbon skeleton, they are divided into six common subgroups: oleanane, lupane, ursane, friedelane, hopane, and gammacerane (Fig. 1).
Figure 1.
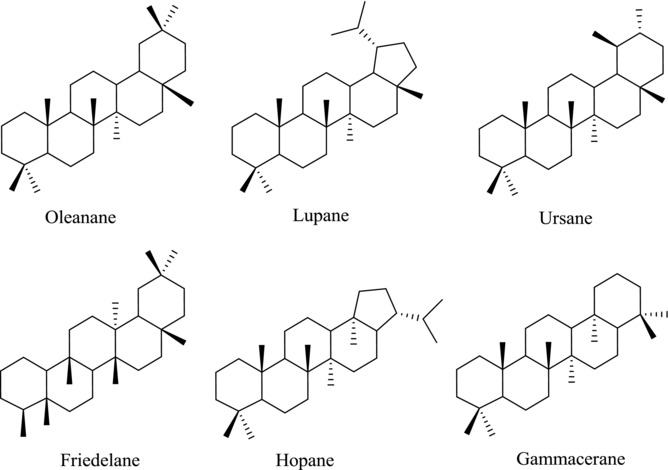
Different types of PT skeletons
PTs are a large class of secondary plant metabolites that are constructed by isoprene (2‐methylbutadiene) (C5H8) units, an abundant natural product in plants.16 The linear triterpene squalene, the major constituent of shark liver oil, is derived from two farnesyl pyrophosphate units. Under the catalysis of squalene epoxidase, squalene is biosynthetically oxidized to 2,3‐oxidosqualene and various cyclic products, yielding the dammarenyl, lupenyl, and baccharenyl cation intermediates that eventually give rise to three major products namely: lupeol, α‐amyrin, and β‐amyrin, respectively (Fig. 2).17 Many PTs have a hydroxyl group at C3, a 12‐ene and a carboxylic acid at C17 or C20. The structural diversity of PTs provides a unique group of agents with a variety of pharmacological activities,18 including antiviral, antitumor, immunomodulatory, anti‐inflammatory, and hepatoprotective activities. Presently, the newly discovered PTs are a subject of annual review, reflecting the increasing importance of this class of compounds.18 The available reviews cover the chemistry,19, 20 antitumor activity,21, 22, 23 anti‐inflammatory activity,24 bioavailability,25 and other pharmacological activity.18
Figure 2.
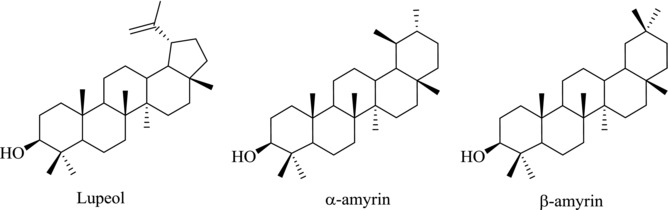
Chemical structures of lupeol, α‐amyrin, and β‐amyrin
Recently, the antiviral activities of PTs are attracting increasing attention. In Japan, Stronger Neo‐Minophagen C™ (SNMC), a preparation containing glycyrrhizin (glycyrrhizic acid, 1) has been used in the treatment of chronic hepatic diseases for over 40 years. Glycyrrhetinic acid (GA, 2) (Fig. 3), a PT derivative of the β‐amyrin type, is obtained from the hydrolysis of glycyrrhizin 1, which is isolated from the herb liquorice and shows inhibition on a variety of viruses including hepatitis B virus (HBV) and HIV.26 In 1989, saponin B1 and B2, two oleanane‐type triterpenoids isolated from soybean seeds, have been reported to have activity against HIV at concentrations greater than 2 and 0.5 mg/mL, respectively.27 Three years later, Chen et al. reported that salaspermic acid (3), another oleanane‐type triterpene derivative isolated from Tripterygium wilfordii, can block the replication of HIV in H9 lymphocytes (IC50: 10 μM).28 Afterwards, Fujioka et al. found that betulinic acid (BA, 4), isolated from the leaves and barks of Syzigium claviflorum, also showed significant activity against HIV (EC50: 1.4 μM).29 Since then, more and more natural PTs have been reported to have antiviral activity. It is well known that lipids, a class of hydrophobic biomolecules, are important for cellular life, especially during virus entry into the host cell. It was reported that the promiscuous antiviral activities of natural PTs might be related to the effect of lipid metabolism.30, 31
Figure 3.
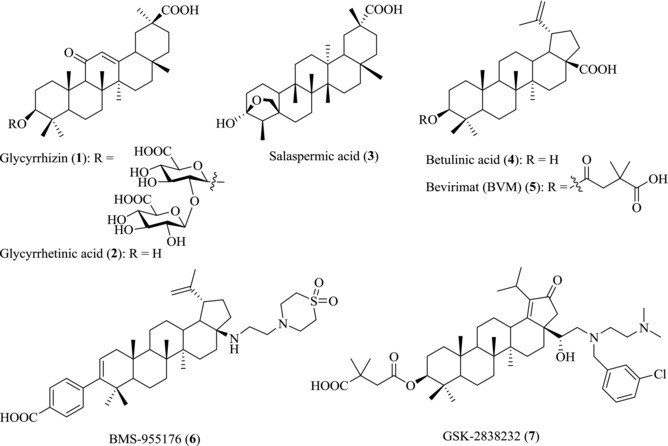
Chemical structures of PTs 1–7
Synthetic chemists become more interested in these scaffolds due to their potential antiviral properties, leading to the production of a variety of PT derivatives through modifications of the C3‐OH, C17‐COOH, and other functional groups. Some of these derivatives are used as clinical drugs for the treatment of liver‐related diseases, while others are at various phases of clinical trials.20 For example, the BA derivative Bevirimat (BVM, 5), also known as 3‐O‐(3′,3′‐dimethylsuccinyl) BA (DSB) or PA‐457, was found to exhibit remarkable anti‐HIV activity against primary and drug‐resistant HIV isolates.32 It represents a unique first in class anti‐HIV compound termed maturation inhibitors (MIs).33 Due to the baseline gag polymorphisms at positions Q369, V370, or T371, which reduced the sensitivity of HIV‐1 to Bevirimat (5), it was halted for further development in 2010.34, 35 Recently, the second class of potent PTs BMS‐955176 (6) and GSK‐2838232 (7) are under development as oral treatment of HIV infection.36, 37, 38, 39 However, up to now, there is no review focusing on the activity of PTs against viral infection.40 The focus of the present review is to summarize the recent advances in the antiviral activities and action models of PTs to aid the antiviral drug development of PTs.
2. PTs AS ANTI‐HIV AGENTS
HIV is a lentivirus that could lead to HIV infection and over time acquired immunodeficiency syndrome (AIDS).41 Because of the lack of CD4‐positive T cells, HIV patients are exposed to life‐threatening opportunistic infections and cancers without normal immune protection.42, 43 Fortunately, HIV‐1 infection can be effectively managed with a long‐term administration of multiple antiviral inhibitors designated as combination antiretroviral therapy (cART).44 Since Zidovudine (ZDV, also known as azidothymidine, AZT) was launched in 1987 as the first anti‐HIV drug that targets HIV reverse transcriptase, so far over 40 formulations have been approved by FDA for HIV therapy. However, none of them is PTs. According to Thomson Reuters, among all chemicals in clinical trials, only four belong to PTs. They are MPC‐9055 (the structure has not been reported in literature), Bevirimat (5), BMS‐955176 (6), and GSK‐2838232 (7). These four compounds are all MIs bearing the scaffold of BA (4). GSK‐2838232 (7), an orally active BA derivative, targets against HIV‐1 maturation process and it is under phase IIa clinical trial. However, the clinical testing of MPC‐9055, Bevirimat (5), and BMS‐955176 (6) have been discontinued.36, 37, 38, 39
Many PTs and their derivatives have been reported to have fair anti‐HIV activity. Most of them usually show weaker anti‐HIV activity than the other clinically available drugs.45 However, they enrich the database of anti‐HIV compounds and provide good lead compounds for further drug development due to their diverse chemical structures. According to the life cycle of HIV, the inhibitors of HIV are divided into entry inhibitor, reverse transcriptase (RTase) inhibitor, protease inhibitor, viral MI, and so on.46 Table 1 summarizes the representative anti‐HIV PTs based on their targets.
Table 1.
Summary of anti‐HIV PTs based on their targets
| Classification | PTs |
|---|---|
| Entry inhibitors | Glycyrrhizin (1) and its analog 8; RPR103611 (9); IC9564 (10) |
| Reverse transcriptase inhibitors | Mimusopic acid (11); 12 |
| Protease inhibitors | UA (13) and its derivatives 14–15; 16 |
| OA (17) and its derivatives 18–20 | |
| Escins Ia (21) and Ib (22) | |
| Maturation inhibitors | MPC‐9055; Bevirimat (5); BMS‐955176 (6); GSK‐2838232 (7); 23 |
| Bifunctional inhibitors | BA derivatives 24–25 |
This article is being made freely available through PubMed Central as part of the COVID-19 public health emergency response. It can be used for unrestricted research re-use and analysis in any form or by any means with acknowledgement of the original source, for the duration of the public health emergency.
2.1. Entry inhibitors
Entry of HIV‐1 into host cells, the first phase of the viral replication cycle, is an ordered multistep process that involves the attachment of virus to the host cell, the binding of virus to the co‐receptor, and the fusion of the cell and viral membranes. HIV‐1 uses multiple pathways for entry,47 such as usurpation of cellular transport pathway,48, 49, 50 surfing along the cell surface.48, 50 These processes provide targets for developing novel entry inhibitors to prevent HIV infection in the first step.
Glycyrrhizin (1), a well‐known triterpenoid saponin isolated from licorice root, has been reported to possess antiHIV‐1 infection activity by blocking the adsorption of HIV‐1 particles onto CD4‐positive T cells.51, 52 Compound 8 (Fig. 4), an analogue of glycyrrhizin with a heteroannular diene structure at the C and D rings, was demonstrated to be as active as glycyrrhizin against HIV‐1 infection. HIV‐1‐induced cytopathogenicity was completely inhibited by this compound at the concentration of 0.16 mM in MT‐4 and MOLT‐4 cells.51 However, the underlying mechanism of this kind of compounds remains unclear.
Figure 4.
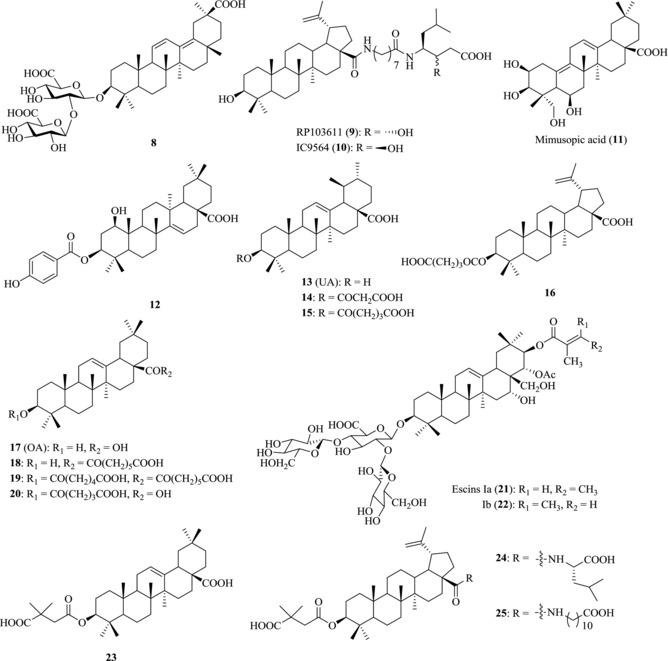
Chemical structures of PTs 8–25 with anti‐HIV activity
RPR103611 (9), a BA derivative with aminooctanoyl amino‐3(S)‐hydroxy‐6‐methylheptanoic acid side chain at C17‐COOH, has been shown to be a potent inhibitor against envelope‐mediated membrane fusion of HIV‐1.53 IC50 values of compound 9 varied in the range of 40–100 nM in most cell systems, with selective indexes in excess of 100. The transmembrane glycoprotein gp41 was initially reported to be the target of RPR103611 (9), because the I84S mutation of gp41 of CXCR4‐dependent HIV‐1 LAI was sufficient for drug resistance against RPR103611 (9).54 Further study illustrated that both the sequence of gp41 loop and the stability of the gp120‐gp41 complex can affect the antiviral efficacy of RPR103611 (9).55
IC9564 (10), the stereoisomer of RPR103611 (9), was found to be equally active with an EC50 value of 0.4 μM.56 Through interacting with gp120 and gp41 and by changing the structure of acceptor gp41, the two BA derivatives 9 and 10 can block the adsorption of HIV to host cells. In addition, compound 9 and its isomer 10 showed the highest activity in the MAGI and fusion assays of H9 lymphocyte.
2.2. RTase inhibitors
HIV RTase catalyzes the replication of virus RNA into DNA. Inhibition of RTase enzyme disrupts this essential process for virus replication and infection. Therefore, RTase has long been an obvious target in the battle against HIV.
Mimusopic acid (11), a triterpene analogue isolated from the seeds of Mimusops elengi, has been reported to possess weak HIV‐RTase inhibitory activity with IC50 at micromolar level.57 Pengsuparp et al. reported that 1‐beta‐hydroxyaleuritolic acid 3‐p‐hydroxybenzoate (12), isolated from the roots of Maprounea africana, also had anti‐HIV‐1 RTase activity with an IC50 value of 3.7 μM.58
2.3. Protease inhibitors
The HIV polyprotein that contains several viral proteins joined together must be cleaved into the individual functional proteins. This cleavage is catalyzed by HIV protease, a symmetric homodimer. Inhibition of HIV protease can prevent the enzyme from cutting the viral protein molecules down to their proper sizes, which makes HIV protease a promising drug target.
Ursolic acid (UA, 13) and its hydrogen malonate (14) isolated from the stems of Cynomorium songaricum have activity in inhibiting HIV‐1 protease with EC50 value at micromolar level. The glutaryl hemiesters 15–16 of BA and UA were shown to have anti‐HIV‐1 protease activity at the concentration of 4 μM.59
Oleanolic acid (OA, 17) from Xanthoceras sorbifolia was found to completely inhibit HIV‐1 protease activity at the concentration of 100 μg/μL.60 Chemical modifications of oleanane‐type triterpene at C3‐OH and/or C17‐COOH increase the anti‐HIV protease activity. The most potent compounds 18–20 showed anti‐HIV protease activity with IC50s in the range of 1.7–4.0 μM.61 In addition, escin Ia (21) and Ib (22), two triterpenes saponins isolated from Aesculus chinensis, also exhibit HIV‐1 protease inhibitory activity.62
2.4. Maturation inhibitors
MIs are an emerging class of anti‐HIV‐1 compounds that can block HIV‐1 maturation by specifically interfering with Gag processing cascade. In the final step of the viral replication cycle, p17 matrix protein (MA), p24 capsid protein (CA), nucleocapsid protein (NC), and p6 protein are released from the Gag polyprotein precursor (Pr55Gag), which represents a novel target for potential intervention of HIV‐1. Inhibition of Gag leads to defective core condensation and the release of noninfectious virus particles from host cells, thus leading to the release of noninfections particles
Bevirimat (5)63 and MPC‐905564 are the first‐generation MIs to be studied in humans. Recently several second‐generation HIV‐1 MIs that show improved potency against viruses containing Bervirimat‐resistant Gag polymorphisms are developed in clinical trials for HIV patients.
The introduction of a 3,3‐dimethylsuccinyl group at the hydroxyl of C3 of OA (17) generated the most potent compound 23 3‐O‐3′,3′‐dimethylsuccinate that has greatly increased the potency without significant cytotoxicity. Compound 23 was shown to have anti‐HIV‐1 activity at subnanomolar concentrations with a therapeutic index of 22,400.65 Similarly, the introduction of a 3,3‐dimethylsuccinyl group at the same position of BA (4) leads to the discovery of Bevirimat (5). Compound 5 greatly increased the anti‐HIV activity of BA with an IC50 value of less than 0.35 nM and a selective index of more than 20,000.66
MPC‐9055 (the structure has not been reported in literature), an orally bioavailable small molecule for HIV therapy, was developed by Myrexis. It blocks HIV‐1 replication by interrupting the processing of the viral capsid protein 25 to p24, which condenses to form the conical core structure surrounding the viral genome. A phase I clinical trial of MPC‐9055 in healthy volunteers was completed in 2008. The primary objective of the study was to evaluate the safety, tolerability, and pharmacodynamics of MPC‐9055. Results show that the overall safety profile was favorable and the observed Pharmacokinetics (PK)/Pharmacodynamics (PD) profile supported continued research and development.67
First‐generation MIs are ineffective against some naturally occurring variations (polymorphisms) in the Gag protease polyprotein; however, the second‐generation inhibitors have been developed to better tolerate gag polymorphisms.68 BMS‐955176 (6) is an orally active second‐generation HIV MI under development by Bristol‐Myers Squibb for the treatment of HIV infection. This compound had been in phase IIb clinical trial, but was discontinued. By blocking the maturation of the virus, it prevents viral reproduction in host CD4 T cells with the EC50 of 1.9 nM against HIV‐1 wild‐type (WT) and the EC50 of 2.7 nM against HIV‐1 WT with the V370A variant of Gag protein. GSK‐2838232 (7), another anti‐HIV inhibitor, is also under clinical investigation.
2.5. Bifunctional inhibitors
BA derivatives with modifications at both C3‐OH and C17‐COOH of BA (4) can block the virus fusion and virus maturation simultaneously as a novel type of bifunctional inhibitors. By combining the anti‐HIV‐1 entry pharmacophore from IC9564 (10) and the anti‐HIV‐1 maturation pharmacophore from Bevirimat (5), bifunctional anti‐HIV compounds N‐[3‐O‐(3′,3′‐dimethylsucciny)‐lup‐20(29)‐en‐28‐oyl]‐l‐leucine (LH15, 25), and N‐[3‐O‐(3′,3′‐dimethylsucciny)‐lup‐20(29)‐en‐28‐oyl]‐11‐aminoundecanoic acid (LH55, 26) were obtained to double strike the life cycle of HIV. They presented the best activity against NL4‐3 with IC50 value of 0.016 and 0.0065 μM, respectively.69
3. PTs AS ANTI‐HCV AGENTS
HCV is an enveloped, positive‐stranded RNA virus in the genus hepacivirus, and belongs to the flaviviridae family. There are at least six distinct genotypes with multiple subtypes in each genotype class, which is important for the prediction of therapy response.70, 71 HCV has an error‐prone replication process for the RNA genome of about 9.6 kb in full length. It encodes a single polyprotein of more than 3000 amino acids, which is cleaved by viral and host enzymes into ten mature individual proteins, including three structural proteins (core, E1, E2) and seven regulatory proteins (p7, NS2, NS3, NS4A, NS4B, NS5A, NS5B).72 According to Thomson Reuters, more than 100 anti‐HCV drugs have been launched. However, none of them is PTs. Only ME 3738 (35), a PT, had been in Phase I clinical trials.73 Table 2 summarizes the representative anti‐HCV PTs based on their targets.
Table 2.
Summary of anti‐HCV PTs based on their targets
| Classification | Pentacyclic triterpenes |
|---|---|
| Entry inhibitors | OA (17) |
| EA (26) and its derivatives (27–32) | |
| Protease inhibitors | OA (17) and its derivatives (33–34) |
| Immunomodulator | ME 3738 (35) |
| Other inhibitor | Glycyrrhizin (1) |
This article is being made freely available through PubMed Central as part of the COVID-19 public health emergency response. It can be used for unrestricted research re-use and analysis in any form or by any means with acknowledgement of the original source, for the duration of the public health emergency.
3.1. Entry inhibitors
It was proposed that HCV glycoproteins could be essential drug targets for the development of HCV entry inhibitors.74, 75, 76, 77 The core structure of E2 protein was solved in 2013. As a result, many agents are investigated as anti‐E2 inhibitors to interrupt the interaction between E2 and CD81, thus inhibiting HCV entry.78, 79 Small molecules are designed and synthesized to mimic the interacting space and hydrophobic feature of the helix D region of CD81, showing promising activity. The inhibitory compounds have no short‐term effect on CD81 expression, do not disrupt CD81 associations with other cell surface proteins, and bind to HCV‐E2 reversibly.80
PTs showed fair activity in anti‐HCV entry assays.81, 82, 83, 84 OA (17) and echinocystic acid (EA, 26) (Fig. 5), two naturally occurring oleanane‐type triterpenes, and their derivatives displayed potent activity against HCV entry. Yu et al. reported that OA (17) only showed weak anti‐HCV entry activity (10 μΜ: ∼65%). However, a small modification of the structure by introduction of a hydroxyl group at C‐16 to provide its analogues 26 leads to significantly enhanced activity with an IC50 value of 1.4 μM in HCVpp model.85 With this lead compound, a comprehensive structure–activity relationship (SAR) study was conducted. Introducing a disaccharide to this lead compound at C17‐COOH leads to the discovery of compound 27, which showed stronger anti‐HCV entry activity with an IC50 of 0.3 μM without the undesired hemolytic effect. Formation of a triterpene dimer of 26 via a linker results in the discovery of compound 28 with an IC50 value of about10 nM.85 A series of bivalent oleanane‐type triterpenes were further designed and synthesized by optimizing the hydrophobicity, rigidity, and length of the linker. It was found that compound 29 exhibited dramatically enhanced activity with an IC50 value in the subnanomolar ranges.86
Figure 5.
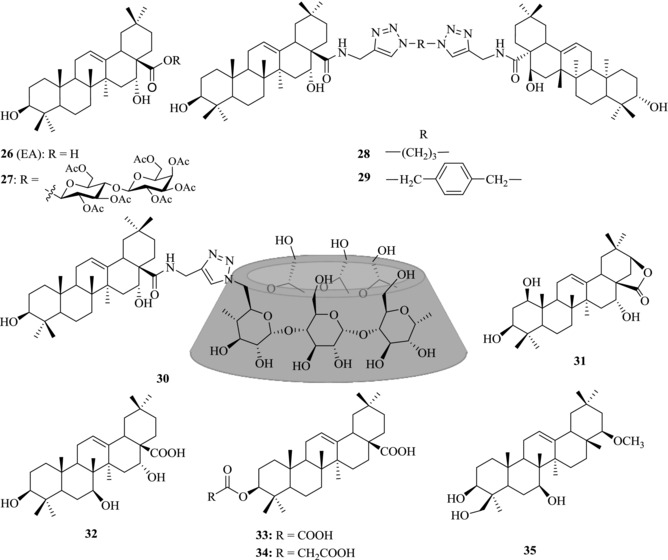
Chemical structures of PTs 26–35 with anti‐HCV activity
It is well known that the low solubility in biological matrixes and the high hydrophobicity of PTs hinder their applications, particularly in the development of therapeutic agents. Given the limited solubility of PTs, a series of triterpene–cyclodextrin (CD) conjugates were synthesized to improve the solubility and maintain the activity of triterpenes.87, 88 Based on HCVpp/VSVGpp entry assays, the IC50 value of compound 30 is 0.25 μM, showing the elevation of water solubility and maintain activity of the anti‐HCV entry.
To elucidate the anti‐HCV pharmacophore of EA (26), a microbial transformation strategy and a ring expansion/opening strategy have been used to explore the SAR.89, 90 Results showed that most of the metabolites generated by microbial transformation did not exhibit improved anti‐HCV activity except compounds 31 and 32, which displayed similar or even a little higher potency than 26 in HCV entry assay. Meanwhile, the ring expansion and ring opening derivatives of EA (26), such as lactones, 3,28‐dioic acids, or pentols, showed no improved anti‐HCV activity. These studies showed that rings A and C of EA are highly conserved and chemical modification of the two rings abolishes the potency of 26, suggesting that the steric hinder effects of the rigid skeleton play an important role in determination of the anti‐HCV entry activity.
In summary, chemical exploration of these triterpene compounds revealed that ring D, right side of ring E, and C17‐COOH are tolerant to modifications, while ring A, B, C and the left side of ring E are highly conserved for the anti‐HCV entry activity. Introducing a hydroxyl group in ring D at C‐16 can enhance the potency of triterpene and remove the hemolytic effect of EA (26) (Fig. 6).
Figure 6.
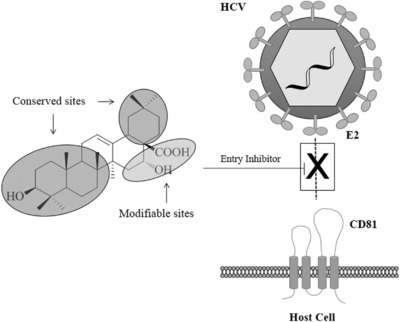
A proposed mechanism for EA (26)‐mediated blocking of HCV entry85
3.2. HCV protease inhibitors
OA (17) was also found to be an HCV protease inhibitor.91 Based on this lead compound, 29 OA derivatives were synthesized and tested for their inhibitory activity on hepatitis C viral protease. The derivatives of dicarboxylic acid hemiesters showed potent activity against HCV protease. Of the dicarboxylic acid hemiesters, two compounds 34 and 35 with relatively short carbon chains showed much lower cytotoxicity than OA but maintained the inhibitory activity on HCV protease.
3.3. Immunomodulator
22β‐methoxyolean‐12‐ene‐3β,24(4β)‐diol (ME 3738, 35), a derivative of soyasapogenol B, inhibits HCV replication by inducing oligoadenylate synthetase 1 gene expression and enhancing the effect of interferon (IFN)‐α to increase IFN‐stimulated gene expression. In vitro assay, 35 reduces HCV core antigen (HCVcAg) and HCV‐RNA levels in mouse livers.92 Moreover, ME 3738 was also found to be an inducer of interleukin 6 (IL‐6). It can stimulate the production of IL‐6 and protect mouse from acute liver injury/failure induced by concanavalin A (Con A) and prevent the development of alcoholic fatty liver.93, 94
Phase I trial of 35 was carried out in 2001 and it showed that ME 3738 strongly reduced positive and negative RNA strand levels, as measured by real‐time Polymerase Chain Reaction (PCR).
3.4. Other HCV inhibitors
Glycyrrhizin (1) has been shown to have the inhibitory activity against various viruses, including HIV, herpes simplex virus (HSV), vesicular stomatitis virus (VSV), HBV, and so on. Compound 1 also showed anti‐HCV protease activity with IC50 value of 16.7 ± 2.4 μM. A synergistic effect between compound 1 and IFN alpha‐2a against HCV has been observed, providing a new treatment along with IFN for HCV infection. It was suggested that the active constituent 1 triggered NFκB pathway and led to induction of IL‐8 secretion and antiviral activity against a series of DNA and RNA viruses.95
4. PTs AS ANTI‐INFLUENZA AGENTS
Influenza viruses belong to the Orthomyxoviridae family, an RNA‐type virus with diverse antigenic characteristics. As common pathogens, influenza viruses can cause serious diseases in humans and animal species. Currently, four types of influenza viruses, influenza viruses A, B, C, and D, have been identified. Among them, types A and B generally caused most of the influenza epidemics and outbreaks, leading to serious impact on public health and economy,96, 97, 98, 99, 100, 101, 102, 103 and type C is generally responsible for sporadic mild upper respiratory symptoms.104, 105, 106 Neuraminidase inhibitors, including oseltamivir and zanamivir, and M2 ion channel inhibitors, including amantadine and rimantadine, are two classes of clinically used drugs against influenza viruses. However, the high mutation rate of influenza virus results in the emergence of influenza virus strains that are resistant to those drugs,107, 108 illustrating the urgent need to develop novel anti‐influenza drugs.
4.1. PT‐glycoconjugates
Glycyrrhizin (1) has been isolated from the roots of licorice, and showed a broad antiviral spectrum.109, 110, 111, 112 Wolkerstorfer et al. investigated the mechanism by which 1 inhibits influenza virus infection and found that compound 1 can interact with the membrane of cells, leading to a decrease of cell endocytotic activity and virus uptake.113 From the lead compound 1, some derivatives were developed. Among them, compounds 36–38 exhibited the most efficient anti‐influenza activity (IC50: 4.3–7.2 μM) (Fig. 7).114 Recently, uralsaponin M (40), a novel triterpenoid saponin isolated from the roots of G. uralensis, was shown to exhibit inhibitory activity against the influenza virus A/WSN/33 (H1N1) in Madin‐Darby canine kidney (MDCK) cells with an IC50 value of 48.0 μM.115
Figure 7.
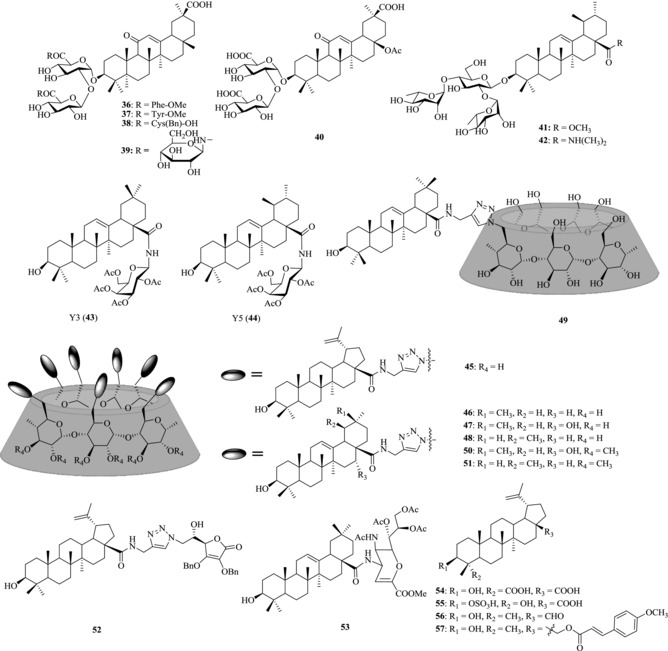
Chemical structures of PTs 36–57 with anti‐influenza activity
Song et al. reported that the conjugates of PTs with chacotriose could inhibit the entry of highly pathogenic H5N1 influenza A virus into MDCK cells in varying degrees.116, 117, 118, 119 Compound 41, a 3‐O‐β‐chacotriosyl substituted methyl ursolate derivative, showed potent anti‐influenza A (H5N1) virus entry activity with IC50 at 6.00–8.54 μM.119 In another work, both the chacotriosyl and the aglycone moiety have been reported to have an important effect on the activity against influenza virus and the anti‐influenza activity could be improved when the disubstituted amide was introduced to the C17‐COOH position.120 The introduction of dimethylamine at the C17‐COOH provides the compound 42, which showed the most potent anti‐influenza activity (IC50: 0.98 μM).116
In view of the broad antiviral spectrum of PTs,85, 86, 87, 89 our group established an anti‐influenza virus drug screening model and screened a mini‐library of PT‐glycoconjugates. Based on a plaque formation inhibition assay, the acetylated galactose–OA conjugate Y3 (43) and the acetylated galactose–UA conjugate Y5 (44), were found to have the best anti‐WSN activity (IC50: 5 μM). In addition, 43 exhibits broad anti‐influenza spectrum with the IC50 ranging from 2.72 to 7.41 μM (Table 3). More importantly, the lead compound 43 shows no tendency to induce resistance of influenza viruses, suggesting that it may have the potential to overcome antiviral drug resistance.
Table 3.
The broad antiviral spectrum of lead compound 43
| IC50 (μM) | ||||||
|---|---|---|---|---|---|---|
| Compound | CC50 (μM) | PR/8 (H1N1)a | LN/1109 (H1N1) | JX/312 (H3N2) | HN/1222 (H3N2) | B/SZ/155 |
| 43 | >200 | 7.41 ± 0.05 | 6.58 ± 1.17 | 2.72 ± 0.35 | 3.18 ± 0.08 | 2.80 ± 0.74 |
| Amantadine | >200 | 7.41 ± 0.21 | 0.44 ± 0.14 | 8.58 ± 1.65 | >200 | >83.24 |
| OSV | >200 | >200 | >200 | 2.06 ± 0.58 | 8.75 ± 4.41 | 91.07 ± 34.51 |
| RBV | >200 | 4.02 ± 1.27 | 5.75 ± 0.35 | 4.19 ± 0.35 | 3.56 ± 0.24 | 1.32 ± 0.60 |
aPR/8 (H1N1), A/Puerto Rico/8/34 (H1N1); LN/1109 (H1N1), A/LiaoNing‐ZhenXing/1109/2010 (H1N1); JX/312 (H3N2), A/JiangXi‐DongHu/312/2006 (H3N2); HN/1222 (H3N2), A/HuNan‐ZhuHui/ 1222/2010 (H3N2); B/SZ/155, B/ShenZhen/155/2005. The PR/8 and LiaoNing/1109 strains belong to H1N1 subtype; the JiangXi/312 and HuNan/1222 strains belong to H3N2 subtypes; the SZ/155 strain belongs to B type.
This article is being made freely available through PubMed Central as part of the COVID-19 public health emergency response. It can be used for unrestricted research re-use and analysis in any form or by any means with acknowledgement of the original source, for the duration of the public health emergency.
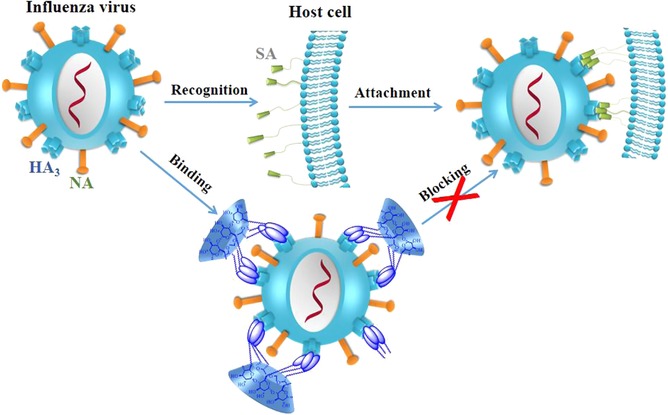
The exploration of the mechanism of action indicated that 43 could block the attachment of influenza virus particles to the membrane of host cells by binding to sialic acid receptor‐binding pocket on the influenza viral HA protein (Fig. 8).
Figure 8.
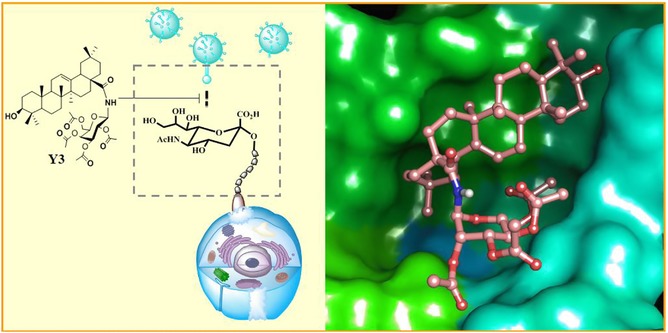
The proposed anti‐influenza mechanism of Y3 (43)121
4.2. PT–CD conjugates
Tri‐ or multivalent ligands show much higher binding affinity to the homotrimeric influenza HA receptor. Recently, our group designed and synthesized a series of multivalent PT‐α‐(β‐, γ‐) CD conjugates (45–48) to enhance the affinity between PTs and HA and thus improve their potency against influenza virus.122, 123 The multivalent OA‐β‐CD conjugate 46 was found to show the most potent antiviral activity against influenza A virus (IC50: 1.6 μM), more potent than the monovalent OA‐β‐CD conjugate 49 by 60‐fold (IC50 > 100 μM). Further study on its anti‐influenza action mechanism showed that the multivalent PT–CD conjugates could also bind to influenza viral HA protein, and thus inhibit the attachment of influenza virus particle to host cells (Fig. 9).
Figure 9.
A proposed mechanism for multivalent PT‐CD conjugates mediated blocking of influenza virus entry122
Recently, per‐O‐methylated CDs and their derivatives have been found to have improved solubility in organic and water solvents.124, 125, 126 On the basis of previous work,122 our group further prepared multivalent PT‐functionalized per‐(2,3‐di‐O‐methyl)‐α‐, β‐, or γ‐CD conjugates and evaluated their in vitro anti‐influenza activities. Two conjugates 50 and 51 were identified to exhibit the highest anti‐influenza virus activity with IC50 at micromolar level (4.7 and 6.5 μM, respectively) and SI > 15.
4.3. Other PTs derivatives
Conjugation of PTs with other bioactive compounds, such as l‐ascorbic acid and sialic acid, have also been used as a strategy to prepare new biological PTs. Conjugates 52 and 53 were found to have the most potent antiviral activity against influenza virus. The IC50 values for them are 8.7 and 8.3 μM, respectively.127, 128, 129, 130
Hong et al. reported that BA (4), isolated from Jujube tree (Zizyphus jujube Mill), exhibited antiviral activity against influenza A/PR/8 virus in vitro and in vivo.131 Two BA derivatives (54 and 55) isolated from Chinese herbal medicine Schefflera heptaphylla were found to have inhibitory activity on H1N1 influenza A virus (IC50 values: 32.2 and 58.4 μM, respectively).132 Tung et al. reported that betulinic aldehyde 56 isolated from Alnus japonica exhibited anti‐influenza effect against influenza A/Korea/KBNP‐0028/2000 virus (IC50: 28.4 μM).133 Furthermore, it was reported that 28‐O‐methoxycynnamoyl betulin 57 showed high activity against H1N1 influenza A virus and low cytotoxicity with a selectivity index of SI >100.134
5. PTs AS ANTI‐SARS AGENTS
SARS is an infectious respiratory disease caused by the SARS‐CoV. Between 2002 and 2003, an outbreak of SARS started in southern China caused a great number of deaths in many countries, which has drawn massive attention worldwide.6 Although SARS is now under control and nearly disappeared, it still could come back. To search for effective inhibitors, more than 10,000 agents including existing drugs, natural products, and synthetic compounds were evaluated for their anti‐SARS activity. A variety of PTs and their derivatives have been shown to have anti‐SARS‐CoV activity.
5.1. Glycyrrhizin and its derivatives
Among PTs, glycyrrhizin (1) and its derivatives were first reported to have the anti‐SARS‐CoV activity by Cinatl et al.110 Inhibitor 1 presented an EC50 of 0.37 mM against the replication of SARS‐CoV with a SI of 67 in Vero‐E6 cells. In addition to inhibiting viral replication, 1 was also able to inhibit the adsorption as well as penetration of the virus at the beginning stage of the life cycle. Compound 1 was more effective when treated after the adsorption process than during the adsorption process (EC50: 2.91 mM vs. 0.73 mM, respectively). Moreover, when given both during and after the adsorption period, it showed the most potent activity. It has been shown that the change of the glycoside chain at C3‐OH and introduction of heterocyclic fragments or amino acids at C20‐COOH into 1 obviously affect the antiviral activity of derivatives. In this case, Hoever et al. evaluated the anti‐SARS‐CoV activity of 15 PT derivatives.111 Compound 58, the most potent derivative showing tenfold increase of the bioactivity, was produced through the introduction of 2‐acetamido‐β‐d‐glucopyranosylamine into the carbohydrate chain of 1 (Fig. 10). It inhibited SARS‐CoV replication with EC50 and CC50 at 40 μM and 3000 μM, respectively. Compound 57 showed similar effect as 58. Compounds 60–64 were effective against SARS‐CoV with EC50 at micromolar level. But these derivatives also demonstrated severe cytotoxicity, resulting in decreased selectivity index ranging from 2 to 5. These results showed that the chemical embellishment by the introduction of CONH bonds into 1 was able to enhance its antivirus effectiveness. Hover also found that 2 (GA), the partial hydrolyzate of 1, exhibited high activity as well as toxicity. Considering the mechanism of their anti‐SARS‐CoV activity, Hover speculated that the entry process of SARS‐CoV into cells was blocked by the attachment of N‐acetylglycosamine with the carbohydrates of the S‐proteins,111 which was demonstrated to be necessary for viral entry into host cells.
Figure 10.
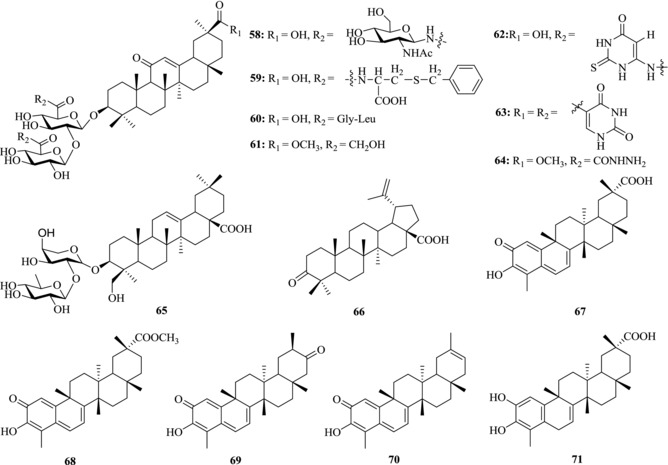
Chemical structures of PTs 58–71 with anti‐SARS activity
Wu et al. found α‐hederin (65), a derivative of 1, showed anti‐SARS‐CoV activity at concentrations of <100 μM. The minimal concentration of inhibition toward SARS‐CoV was 10 μM.135
5.2. BA and betulonic acid
Wen et al. evaluated 221 compounds, including two PTs, 4 and betulonic acid (66), for their anti‐SARS‐CoV activity in Vero‐E6 cells based on a cell‐based cytopathic effect (CPE) assay (Table 4).136 Comparing with 4 whose EC50 value was over 10 μM, the EC50 value of 66 was 0.63 μM, 2.6‐fold lower than that of the positive control (valinomycin, EC50 = 1.63 μM). The CC50 values of compounds 4 and 66 were >100 μM. The SI of compound 66 was 180, indicating that it effectively inhibited viral replication without obvious cytotoxicity. The two compounds were further tested through inhibition assay toward 3CL protease. Compound 4 demonstrated valid inhibitory effect on 3CL protease function with an IC50 value of 10 μM, and its K i value was 8.2 ± 0.7 μM, indicating a competitive inhibition mode of action. However, the IC50 of 66 was >100 μM. Computer docking analysis revealed that 4 was able to occupy the binding pocket of SARS‐CoV 3CL protease. Moreover, the binding was enhanced by the formation of a hydrogen bond between the C3‐OH of 4 and the C = O of Thr24 of the 3CL protease. On the contrary, 66 did not form other intermolecular bonds with the pocket besides the hydrophobic interaction. Their anti‐SARS activity could be the result of a combination of two different antiviral mechanisms. One is via protease inhibition and the other is via blocking SARS‐CoV entry at the post binding step during the fusion of virus particle to host cell membrane, which is also an important and general antiviral mechanism of PTs.
Table 4.
PTs with anti‐SARS‐associated coronavirus activity
| Compound | EC50 (μM) | CC50 (μM) | SI | |
|---|---|---|---|---|
| Ribavirin | >4095 | >4095 | – | |
| 1 | After virus adsorption | 729 | >24,303 | >33 |
| During and after virus adsorption | 365 | >24,303 | >67 | |
| During virus adsorption | 2916 | >24,303 | >8.3 | |
| 2 | >20 | 20 ±5 | – | |
| 4 | >10 | 150 | <15 | |
| 56 | 40 ± 13 | >3000 | >75 | |
| 57 | 35 ± 7 | 1462 ± 50 | 41 | |
| 58 | 139 ± 20 | 215 ± 18 | 2 | |
| 59 | 8 ± 2 | 44 ± 6 | 6 | |
| 60 | 50 ± 10 | 250 ± 19 | 5 | |
| 61 | 5 ± 3 | 15 ± 3 | 3 | |
| 62 | 16 ± 1 | 66 ± 8 | 4 | |
| 63 | 0.63 | 112 | 180 | |
This article is being made freely available through PubMed Central as part of the COVID-19 public health emergency response. It can be used for unrestricted research re-use and analysis in any form or by any means with acknowledgement of the original source, for the duration of the public health emergency.
5.3. Quinone‐methide triterpenes
Ryu and co‐workers isolated four quinone‐methide triterpenes (67–70) from Triterygium regelii. Compound 67 was smoothly converted to its derivatives 71 by hydrogenation under palladium‐carbon catalyst.137 The five compounds were tested for SARS‐CoV 3CL protease inhibitory activity (Table 5). The IC50 values of four separated quinone‐methide triterpenes were below or around 10 μM, but the synthesized 71 with a phenol moiety showed lower activity (IC50 = 21.7 μM). These results demonstrated that the presence of a quinone‐methide moiety exerted relatively significant effect in inhibition. Moreover, all isolated inhibitors had the same mode of competitive inhibition. The K i value of the most potent compound 70 was determined to be 0.8 μM. The hydroxyl group of C3 of 70 formed a firm hydrogen bond with the oxygen atom of the carbonyl group of Cys44 and OH of Thr25 in the pocket, whereas 71 only formed the hydrophobic interaction.
Table 5.
Inhibitory effects of 4, 66–71 on SARS‐CoV 3CL protease
| Compound | IC50 (μM) | Inhibition type |
|---|---|---|
| 4 | 10 | 8.2 ± 0.7 |
| 66 | >100 | – |
| 67 | 10.3 ± 0.2 | Competitive (4.2 ± 0.6) |
| 68 | 5.5 ± 0.7 | Competitive (3.1 ± 0.0) |
| 69 | 9.9 ± 0.1 | Competitive (4.0 ± 0.1) |
| 70 | 2.6 ± 0.3 | Competitive (0.8 ± 0.2) |
| 71 | 21.7 ± 1.9 | – |
This article is being made freely available through PubMed Central as part of the COVID-19 public health emergency response. It can be used for unrestricted research re-use and analysis in any form or by any means with acknowledgement of the original source, for the duration of the public health emergency.
PTs and their derivatives show anti‐SARS‐CoV activity at different steps of the life cycle, which suggested that PTs can block SARS‐CoV replication by a complex mechanism. Though most of them are unclear, SARS‐CoV 3CL protease can be an attractive target for the development of anti‐SARS‐CoV drugs.
6. PTs AGAINST OTHER VIRUSES
6.1. Anti‐HBV activity
HBV, belonging to Hepadnaviridae family, is characterized by its high hepatotropism.138, 139 HBV infection is a global health threat. There are about 240 million HBV carriers worldwide who are at risk of developing liver cancer.140 Several agents have undergone clinical trials to treat HBV infection; however, problems arose in clinical trials have challenged the use of those drugs.140, 141 For example, the nucleotide analogs, such as Lamivudine and Adefovir, and IFN are currently used to treat chronic HBV patients, but the frequent relapse after recovery limited their use.142 Therefore, novel HBV inhibitors are urgently needed.
Glycyrrhizin (1) has been used for treating HBV for many years. In vitro and in vivo experiments have been conducted to test its mechanism of action.143, 144 The study showed that treatment with 1 can inhibit the secretion of hepatitis B surface antigen (HbsAg) in vitro. In addition, Romero et al. reported that 1 has a moderate ability to inhibit the HBV DNA release from cells (IC50: 85 μM).145 Sato et al. determined the SAR of glycyrrhizin (1), GA (2), and GA 3‐O‐monoglucuronide (72) against HBV. Of the three compounds, compound 2 showed the most potent inhibition activity on HbsAg secretion. However, in the guinea pig study, GA (2) was not detected in the liver fraction even after administration of an excessive dose of glycyrrhizin (1), and glycyrrhizin (1) was the main compound in the liver fraction after intravenous administration, suggesting that glycyrrhizin (1) would play an important role in the treatment of patients with chronic hepatitis B.144
The Tibetan herb Potentilla anserine L. has been widely used in China to treat hepatitis B. Compound 73 (Fig. 11) was isolated from the rhizomes of P. anserine L. and has been found to have inhibitory activity on HbsAg and HbeAg secretion and HBV DNA release in vitro (IC50: 90.96, 47.40, and 30.68 μM, respectively). In vivo anti‐HBV study indicated that compound 73 could inhibit HBV DNA replication.146
Figure 11.
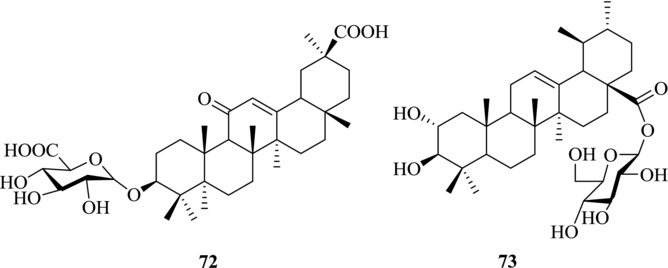
Chemical structures of PTs 72–73 with anti‐HBV activity
Pulsatilla chinensis, another traditional Chinese herb, showed the efficacy of HBV clearance in HBV patient. So far, about 30 components have been isolated from P. chinensis and tested for their HBV clearance activity. Among them, BA (4) and its derivatives were identified as the active components.147, 148 Yao et al. found that BA (4) could inhibit the expression of manganese superoxide dismutase (SOD2) and the subsequent generation of reactive oxygen species (ROS), by which compound 4 could inhibit HBV replication in vitro and in vivo.142
6.2. Anti‐HSV activity
HSV belongs to the Herpesviridae family, which causes serious public health problems149, 150 World Health Organization reports that 67% of the world population under the age of 50 have HSV‐1. Currently, Acyclovir and its derivatives have been approved by FDA to treat HSV infection.151 However, the toxic side effects and the emergence of drug‐resistant HSV virus strains have limited their use.152, 153 Therefore, novel HSV inhibitors are needed.
The medicinal herb Rhus javanica has been reported to have anti‐HSV activity.154, 155 Kurokawa et al. isolated two major anti‐HSV compounds, 66 and moronic acid (74) (Fig. 12), from this herb and evaluated their anti‐HSV activity in vitro and in vivo.156 The effective concentrations for 50% plaque reduction of wild‐type HSV type 1 (HSV‐1) by 66 and 74 were 5.7 μM and 8.6 μM, respectively. The therapeutic index of 74 (10.3–16.3) was larger than that of 66 (6.2). Susceptibility of Acyclovir phosphonoacetic acid‐resistant HSV‐1, thymidine kinase‐deficient HSV‐1, and wild‐type HSV type 2 to 74 was similar to that of the wild‐type HSV‐1, indicating that 74 has a broad antiviral spectrum against HSV.
Figure 12.
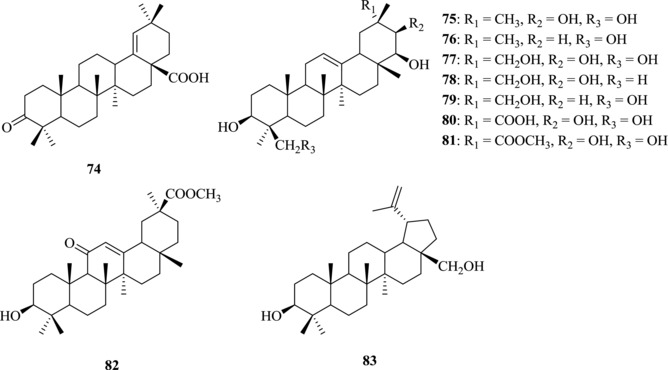
Chemical structures of PTs 74–83 with anti‐HSV activity
Ikeda et al. studied the SAR of 15 oleanane‐type triterpenoids including glycyrrhizin (1) and its sapogenol against HSV‐1.157 The results showed that glycyrrhetic acid (2) and the sapogenol of glycyrrhizin (1) had much more potent anti‐HSV‐1 activity than glycyrrhizin (1). In addition, it was found that soyasapogenol A (75) showed less anti‐HSV activity than soyasapogenol B (76) by 20‐fold, suggesting that hydroxylation at C‐21 might weaken antiviral activity against HSV‐1. It was also found that kudzusapogenol A (77), abrisapogenol B (78), and abrisapogenol C (79) had no anti‐HSV‐1 activity, suggesting that the C‐29 hydroxyl group could abolish the activity against HSV‐1. However, kudzusapogenol B (80) and the methyl esters of 2 (81 and 82) exhibited stronger anti‐HSV activity, suggesting that a methyl ester group at C‐30 could improve the anti‐HSV‐1 activity.
Furthermore, Gong et al. determined the synergistic anti‐HSV‐1 effect of betulin (83) and Acyclovir in a drug combination study.158 The potent and moderate synergistic anti‐HSV‐1 effects were found for Acyclovir and betulin (83) when their concentrations were higher than 4.4 μM and 0.9 μM, respectively. The synergistic anti‐HSV‐2 effects were also found when the concentrations of Acyclovir and betulin were 2.0 μM and 19.0 μM, respectively. The synergistic antiviral effect suggested that the action mechanism of betulin is different from that of Acyclovir. Navid et al. explored the action mechanism and found that the PTs could inhibit the early stage of HSV life cycle.159
6.3. Anti‐EV71 activity
Human enterovirus 71 (EV71) is a single strand RNA virus belonging to Picornaviridae family. It possesses a nonenveloped positive‐sense ssRNA genome. EV71, the major pathogen of hand, foot, and mouth disease (HFMD), can cause epidemic encephalitis and acute flaccid paralysis (AFP), resulting in cardiopulmonary failure and death in babies under 10 years old. Up to now there is still no effective drug to treat EV71 infection.
Glycyrrhizin (1), as one of the main component isolated from Glycyrrhiza spp., has many kinds of antiviral activity. Wang's study first recognized the medicinal effectiveness of 1 against EV71.160 Compound 1 can inhibit EV71 replication in a dose‐dependent manner and the concentrations required for 1 to inhibit EV71 infection were in the millimolar ranges. 1 reduced infectious EV71 production by 34% and 60% at 3 mM and 5 mM, respectively. In addition to the identification of 1 as the antiviral component of G. uralensis against EV71, Wang et al. also revealed the distinct mechanisms of 1 blocking EV71 infection by targeting an event(s) after cell entry.
UA (12) is a PT isolated from the aqueous extract of Ocimum basilicum (also known as sweet basil), an herb commonly used in traditional Chinese medicine. In Chiang's report, 12 demonstrated the strongest activity against EV71 (EC50 = 1.09 μM; SI = 201) in a dose‐dependent manner.161 The SI value of greater than 200, strong activity and low cytotoxicity indicated its potential use for treating infection of EV71 (Table 6). Furthermore, the action of 12 against EV71 was from −2 to 24 hr, indicating that its inhibitory effect occurred in the infection and replication process. Therefore, it may have the potential preventive effect as well as treatment effect.
Table 6.
Anti‐enteroviral activity of UA (12)
| Compound | EC50 (μM) | CC50 (μM) | SI |
|---|---|---|---|
| Ribavirin | >105 ± 16 | >105 ± 16 | – |
| UA | 1.09 ± 0.44 | 220 | 201 |
This article is being made freely available through PubMed Central as part of the COVID-19 public health emergency response. It can be used for unrestricted research re-use and analysis in any form or by any means with acknowledgement of the original source, for the duration of the public health emergency.
Song et al. reported that hederasaponin B (84) (Fig. 13) from Hedera helix has the anti‐EV71 activity.162 Compound 84 could significantly decrease the formation of visible CPE in Vero cells, demonstrating its effective anti‐EV71 C3 and C4a activity without obvious cytotoxicity (Table 7). In addition, western blot assay showed that 84 was able to reduce the expression of viral VP2 protein, thus demonstrating the inhibitory effect of the synthesis of viral capsid protein. By comparison, Ribavirin showed no anti‐EV71 C3 and C4a activity. These results indicated that 84 is a promising drug candidate against various subgenotypes of EV71 because of its broad spectrum of antiviral activity.
Figure 13.
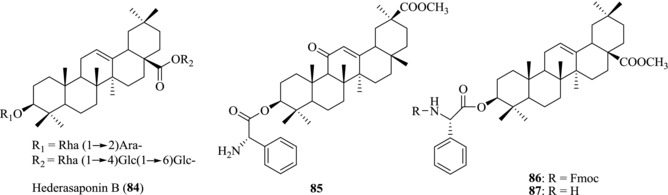
Chemical structures of PTs 84–87 with anti‐EV71 activity
Table 7.
Anti‐EV71 activity of 84 against EV71 C3 and C4a
| EV71 C3 | EV71 C4a | |||
|---|---|---|---|---|
| Compound | EC50 (μM) | CC50 (μM) | EC50 (μM) | CC50 (μM) |
| Ribavirin | – | >39.96 | – | >39.96 |
| 84 | 19.79 ± 10 | >39.96 | 33.38 ± 0.61 | >39.96 |
This article is being made freely available through PubMed Central as part of the COVID-19 public health emergency response. It can be used for unrestricted research re-use and analysis in any form or by any means with acknowledgement of the original source, for the duration of the public health emergency.
Zhao et al. evaluated six newly synthesized PTs for their anti‐EV71 activity. They analyzed their inhibitory effects on the expression of VP1 protein through western blot analysis and reverse transcription (RT) PCR.163 Compounds 85, 86, and 87 demonstrated more potent anti‐EV71 activity than their parent compounds while no significant cytotoxicity was found.
6.4. Anti‐EBV activity
Epstein–Barr virus (EBV) is one of the major pathogens of infectious mononucleosis in humans. It is generally linked with two malignancies, endemic Burkitt's lymphoma and nasopharyngeal carcinoma (NPC). The number of EBV‐associated diseases is increasing, requiring the development of effective vaccines for protection as well as novel antiviral agents for treatment.
Lin et al. reported that glycyrrhizin (1) exhibited activity against EBV replication in superinfected Raji cells in a dose‐dependent manner (IC50 = 0.04 mM, CC50 = 4.8 mM, SI = 120).164 Time of addition studies showed that the anti‐EB effect of 1 was closely associated with an event post virus cell entry rather than the direct inactivation of the virus and the blocking of virus adsorption. The antiviral mechanism appeared to be at the early step of EBV replicative cycle. Different from the mechanism of nucleoside analogs targeting viral DNA polymerase, 1 and its derivatives represent a new type of anti‐EBV agents.
Chang and co‐workers first reported moronic acid (76), which was isolated from the galls of Brazilian propolis and Rhus chinensis, as an agent against EB virus.165 Compared with other triterpenes, 76 has a novel anti‐EB mechanism. During the immediate‐early stage of the lytic cycle of EBV, the transcription of viral lytic genes can be activated by Rta and Zta, two transcription factors expressed by EB virus. It was found that compound 76 can inhibit the functions of Rta by activating a promoter that contains a Rta‐response element, thus reducing the expression of Zta and EA‐D (an EBV early protein) (EC50 = 3.15 μM, CC50 = 46.67 μM, SI = 14.8). Because the target of compound 76 is Rta, one of the important transcription factors of EBV, this compound can be used as a new lead for the development of anti‐EBV drugs.
7. CONCLUSIONS
PTs are a class of plant metabolites with high structural diversity. They provide important sources of lead compounds in drug research and development. Some PTs and their derivatives have been used to prevent and treat chronic hepatic diseases in many countries, such as China and Japan. In addition, they generally show no or weak toxicity. However, the value of PTs in preventing and treating viral diseases has only partially been exploited. Up to now, only Bevirimat (5), BMS‐955176 (6), and GSK‐2838232 (7) have undergone phase IIb clinical trial as HIV MIs. Recently, many research groups have investigated PTs as potential therapeutic agents in the treatment of virus infections. Many natural PTs and their semisynthetic derivatives have been reported to have activity against HIV, HCV, influenza, and other viruses. The investigation of the molecular basis of their antiviral activity is also progressing rapidly. However, their precise targets and action mechanism are usually unclear, which limits their further development. In addition, the poor aqueous solubility is another major drawback for various applications, in particular, the development of drugs. Those intensive explorations will lead to the design of new PTs as potent antiviral inhibitors with better antiviral activity profiles and lower IC50 values.
ACKNOWLEDGMENTS
This work was supported by grants from the National Natural Science Foundation of China (81373271, 81361168002, 81573269, and 21572015) and by National Key Research and Development Program of China (2016YFA0501500). We also like to express our regrets not all relevant researches can be cited in this review due to limited space.
Xiao S, Tian Z, Wang Y, Si L, Zhang L, Zhou D. Recent progress in the antiviral activity and mechanism study of pentacyclic triterpenoids and their derivatives. Med Res Rev. 2018;38:951–976. 10.1002/med.21484
Additional Correspondence
Demin Zhou, State Key Laboratory of Natural and Biomimetic Drugs, School of Pharmaceutical Sciences, Peking University, 38 Xueyuan Road, Haidian District, Beijing 100191, P. R. China.
Email: Deminzhou@bjmu.edu.cn
Dedicated to Professor Lihe Zhang on the occasion of his 80th birthday.
Contributor Information
Sulong Xiao, Email: slxiao@bjmu.edu.cn.
Demin Zhou, Email: Deminzhou@bjmu.edu.cn.
REFERENCES
- 1. Suttle CA. Viruses in the sea. Nature. 2005; 437:356–361. [DOI] [PubMed] [Google Scholar]
- 2. WHO . Http://www.Who.Int/hiv/data/epi_core_july2015.Png.
- 3. Messina JP, Humphreys I, Flaxman A, et al. Global distribution and prevalence of hepatitis C virus genotypes. Hepatology. 2015; 61:77–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Guan Y, Smith GJD. The emergence and diversification of panzootic H5N1 influenza viruses. Virus Res. 2013; 178:35–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Zhu HC, Lam TTY, Smith DK, Guan Y. Emergence and development of H7N9 influenza viruses in China. Curr Opin Virol. 2016;16:106–113. [DOI] [PubMed] [Google Scholar]
- 6. Drosten C, Gunther S, Preiser W, et al. Identification of a novel coronavirus in patients with severe acute respiratory syndrome. New Engl J Med. 2003;348:1967–1976. [DOI] [PubMed] [Google Scholar]
- 7. Zaki AM, van Boheemen S, Bestebroer TM, Osterhaus ADME, Fouchier RAM. Isolation of a novel coronavirus from a man with pneumonia in Saudi Arabia. New Engl J Med. 2012;367:1814–1820. [DOI] [PubMed] [Google Scholar]
- 8. Holmes EC, Dudas G, Rambaut A, Andersen KG. The evolution of Ebola virus: insights from the 2013–2016 epidemic. Nature. 2016;538:193–200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Newman DJ, Cragg GM. Natural products as sources of new drugs from 1981 to 2014. J Nat Prod. 2016;79:629–661. [DOI] [PubMed] [Google Scholar]
- 10. Liby KT, Yore MM, Sporn MB. Triterpenoids and rexinoids as multifunctional agents for the prevention and treatment of cancer. Nat Rev Cancer. 2007;7:357–369. [DOI] [PubMed] [Google Scholar]
- 11. Yang FD, Dong XX, Yin XB, Wang WP, You LT, Ni J. Radix bupleuri: a review of traditional uses, botany, phytochemistry, pharmacology, and toxicology. Biomed Res Int. 2017;2017:7597596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Nuri THM, Yee JCW, Gupta M, Khan MAN, Ming LC. A review of panax ginseng as an herbal medicine. Arch Pharm Pract. 2016;7:61–65. [Google Scholar]
- 13. Hosseinzadeh H, Nassiri‐Asl M. Pharmacological effects of glycyrrhiza spp. And its bioactive constituents: update and review. Phytother Res. 2015;29:1868–1886. [DOI] [PubMed] [Google Scholar]
- 14. Xu R, Fazio GC, Matsuda SPT. On the origins of triterpenoid skeletal diversity. Phytochemistry. 2004;65:261–291. [DOI] [PubMed] [Google Scholar]
- 15. Siddique HR, Saleem M. Beneficial health effects of lupeol triterpene: a review of preclinical studies. Life Sci. 2011;88:285–293. [DOI] [PubMed] [Google Scholar]
- 16. Ruzicka L. The isoprene rule and the biogenesis of terpenic compounds. Experientia. 1953;9:357–367. [DOI] [PubMed] [Google Scholar]
- 17. James JT, Dubery IA. Pentacyclic triterpenoids from the medicinal herb, Centella asiatica (L.) urban. Molecules. 2009;14:3922–3941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Dzubak P, Hajduch M, Vydra D, et al. Pharmacological activities of natural triterpenoids and their therapeutic implications. Nat Prod Rep. 2006;23:394–411. [DOI] [PubMed] [Google Scholar]
- 19. Kvasnica M, Urban M, Dickinson NJ, Sarek J. Pentacyclic triterpenoids with nitrogen‐ and sulfur‐containing heterocycles: synthesis and medicinal significance. Nat Prod Rep. 2015;32:1303–1330. [DOI] [PubMed] [Google Scholar]
- 20. Sheng H, Sun H. Synthesis, biology and clinical significance of pentacyclic triterpenes: a multi‐target approach to prevention and treatment of metabolic and vascular diseases. Nat Prod Rep. 2011;28:543–593. [DOI] [PubMed] [Google Scholar]
- 21. Cichewicz RH, Kouzi SA. Chemistry, biological activity, and chemotherapeutic potential of betulinic acid for the prevention and treatment of cancer and HIV infection. Med Res Rev. 2004;24:90–114. [DOI] [PubMed] [Google Scholar]
- 22. Zhang DM, Xu HG, Wang L, et al. Betulinic acid and its derivatives as potential antitumor agents. Med Res Rev. 2015;35:1127–1155. [DOI] [PubMed] [Google Scholar]
- 23. Salvador JA, Moreira VM, Goncalves BM, Leal AS, Jing Y. Ursane‐type pentacyclic triterpenoids as useful platforms to discover anticancer drugs. Nat Prod Rep. 2012;29:1463–1479. [DOI] [PubMed] [Google Scholar]
- 24. Kashyap D, Sharma A, Tuli HS, Punia S, Sharma AK. Ursolic acid and oleanolic acid: pentacyclic terpenoids with promising anti‐inflammatory activities. Recent Pat Inflamm Allergy Drug Discov. 2016;10:21–33. [DOI] [PubMed] [Google Scholar]
- 25. Furtado NAJC, Pirson L, Edelberg H, et al. Pentacyclic triterpene bioavailability: an overview of in vitro and in vivo studies. Molecules. 2017;22:pii: E400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Pompei R, Flore O, Marccialis MA, Pani A, Loddo B. Glycyrrhizic acid inhibits virus growth and inactivates virus‐particles. Nature. 1979;281:689–690. [DOI] [PubMed] [Google Scholar]
- 27. Nakashima H, Okubo K, Honda Y, Tamura T, Matsuda S, Yamamoto N. Inhibitory effect of glycosides like saponin from soybean on the infectivity of HIV in vitro. AIDS. 1989;3:655–658. [DOI] [PubMed] [Google Scholar]
- 28. Chen K, Kashiwada Y, Zhang DC, et al. Anti‐AIDS agents .6. Salaspermic acid, an anti‐HIV principle from tripterygium‐wilfordii, and the structure‐activity correlation with its related‐compounds. J Nat Prod. 1992;55:340–346. [DOI] [PubMed] [Google Scholar]
- 29. Fujioka T, Kashiwada Y, Kilkuskie RE, et al. Anti‐AIDS agents .11. Betulinic acid and platanic acid as anti‐HIV principles from syzigium‐claviflorum, and the anti‐HIV activity of structurally related triterpenoids. J Nat Prod. 1994;57:243–247. [DOI] [PubMed] [Google Scholar]
- 30. Mazzon M, Mercer J. Lipid interactions during virus entry and infection. Cell Microbiol. 2014;16:1493–1502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Lavie M, Dubuisson J. Interplay between hepatitis C virus and lipid metabolism during virus entry and assembly. Biochimie. 2017;141:62–69. [DOI] [PubMed] [Google Scholar]
- 32. Kashiwada Y, Hashimoto F, Cosentino LM, Chen CH, Garrett PE, Lee KH. Betulinic acid and dihydrobetulinic acid derivatives as potent anti‐HIV agents. J Med Chem. 1996;39:1016–1017. [DOI] [PubMed] [Google Scholar]
- 33. Li F, Goila‐Gaur R, Salzwedel K, et al. Pa‐457: a potent HIV inhibitor that disrupts core condensation by targeting a late step in gag processing. Proc Natl Acad Sci USA. 2003;100:13555–13560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Smith PF, Ogundele A, Forrest A, et al. Phase I and II study of the safety, virologic effect, and pharmacokinetics/pharmacodynamics of single‐dose 3‐O‐(3′,3′‐dimethylsuccinyl)betulinic acid (bevirimat) against human immunodeficiency virus infection. Antimicrob Agents Chemother. 2007;51:3574–3581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Dang Z, Ho P, Zhu L, et al. New betulinic acid derivatives for bevirimat‐resistant human immunodeficiency virus type‐1. J Med Chem. 2013;56:2029–2037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Gulick RM. Choosing initial antiretroviral therapy: current recommendations for initial therapy and newer or investigational agents. Top Antivir Med. 2015;23:128–131. [PMC free article] [PubMed] [Google Scholar]
- 37. Nowicka‐Sans B, Protack T, Lin Z, et al. Identification and characterization of BMS‐955176, a second‐generation HIV‐1 maturation inhibitor with improved potency, antiviral spectrum, and gag polymorphic coverage. Antimicrob Agents Chemother. 2016;60:3956–3969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Olender SA, Taylor BS, Wong M, Wilkin TJ. CROI 2015: advances in antiretroviral therapy. Top Antivir Med. 2015;23:28–45. [PMC free article] [PubMed] [Google Scholar]
- 39. Regueiroren A, Liu Z, Chen Y, et al. Discovery of BMS‐955176, a second generation HIV‐1 maturation inhibitor with broad spectrum antiviral activity. ACS Med Chem Lett. 2016;7:568–572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Paduch R, Kandefer‐Szerszen M. Antitumor and antiviral activity of pentacyclic triterpenes. Mini‐Rev Org Chem. 2014;11:262–268. [Google Scholar]
- 41. Weiss R. How does HIV cause AIDS? Science. 1993;260:1273–1279. [DOI] [PubMed] [Google Scholar]
- 42. Douek DC, Roederer M, Koup RA. Emerging concepts in the immunopathogenesis of AIDS. Ann Rev Med. 2009;60:471–484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. CDC . Opportunistic infections and Kaposi's sarcoma among Haitians in the United States. Conn Med. 1982;46:727–728. [PubMed] [Google Scholar]
- 44. Taiwo B, Hicks C, Eron J. Unmet therapeutic needs in the new era of combination antiretroviral therapy for HIV‐1. J Antimicrob Chemother. 2010;65:1100–1107. [DOI] [PubMed] [Google Scholar]
- 45. Huang L, Chen CH. Molecular targets of anti‐HIV‐1 triterpenes. Curr Drug Targets Infect Disord. 2002;2:33–36. [DOI] [PubMed] [Google Scholar]
- 46. Reeves JD, Piefer AJ. Emerging drug targets for antiretroviral therapy. Drugs. 2005;65:1747–1766. [DOI] [PubMed] [Google Scholar]
- 47. Wilen CB, Tilton JC, Doms RW. HIV: cell binding and entry. Cold Spring Harb Perspect Med. 2012;2:pii: a006866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Lehmann MJ, Sherer NM, Marks CB, Pypaert M, Mothes W. Actin‐ and myosin‐driven movement of viruses along filopodia precedes their entry into cells. J Cell Biol. 2005;170:317–325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Coyne CB, Bergelson JM. Virus‐induced Abl and Fyn kinase signals permit coxsackievirus entry through epithelial tight junctions. Cell. 2006;124:119–131. [DOI] [PubMed] [Google Scholar]
- 50. Sherer NM, Jin J, Mothes W. Directional spread of surface‐associated retroviruses regulated by differential virus‐cell interactions. J Virol. 2010;84:3248–3258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Hirabayashi K, Iwata S, Matsumoto H, et al. Antiviral activities of glycyrrhizin and its modified compounds against human immunodeficiency virus type 1 (HIV‐1) and herpes simplex virus type 1 (HSV‐1) in vitro. Chem Pharm Bull. 1991;39:112–115. [DOI] [PubMed] [Google Scholar]
- 52. Fields AP, Bednarik DP, Hess A, May WS. Human immunodeficiency virus induces phosphorylation of its cell surface receptor. Nature. 1988;333:278–280. [DOI] [PubMed] [Google Scholar]
- 53. Mayaux JF, Bousseau A, Pauwels R, et al. Triterpene derivatives that block entry of human immunodeficiency virus type 1 into cells. Proc Natl Acad Sci USA. 1994;91:3564–3568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Labrosse B, Pleskoff O, Sol N, Jones C, Henin Y, Alizon M. Resistance to a drug blocking human immunodeficiency virus type 1 entry (RPR103611) is conferred by mutations in gp41. J Virol. 1997;71:8230–8236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Labrosse B, Treboute C, Alizon M. Sensitivity to a nonpeptidic compound (RPR103611) blocking human immunodeficiency virus type 1 Env‐mediated fusion depends on sequence and accessibility of the gp41 loop region. J Virol. 2000;74:2142–2150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Sun IC, Chen CH, Kashiwada Y, Wu JH, Wang HK, Lee KH. Anti‐AIDS agents 49. Synthesis, anti‐HIV, and anti‐fusion activities of IC9564 analogues based on betulinic acid. J Med Chem. 2002;45:4271–4275. [DOI] [PubMed] [Google Scholar]
- 57. Sahu NP. Triterpenoid saponins of Mimusops elengi . Phytochemistry. 1996;41:883–886. [DOI] [PubMed] [Google Scholar]
- 58. Pengsuparp T, Cai L, Constant H, et al. Mechanistic evaluation of new plant‐derived compounds that inhibit HIV‐1 reverse transcriptase. J Nat Prod. 1995;58:1024–1031. [DOI] [PubMed] [Google Scholar]
- 59. Ma C, Nakamura N, Miyashiro H, Hattori M, Shimotohno K. Inhibitory effects of constituents from Cynomorium songaricum and related triterpene derivatives on HIV‐1 protease. Chem Pharm Bull. 1999;47:141–145. [DOI] [PubMed] [Google Scholar]
- 60. Ma C, Nakamura N, Hattori M, Kakuda H, Qiao J, Yu H. Inhibitory effects on HIV‐1 protease of constituents from the wood of Xanthoceras sorbifolia . J Nat Prod. 2000;63:238–242. [DOI] [PubMed] [Google Scholar]
- 61. Chao M, Nakamura N, Hattori M. Chemical modification of oleanene type triterpenes and their inhibitory activity against HIV‐1 protease dimerization. Chem Pharm Bull. 2000;48:1681–1688. [DOI] [PubMed] [Google Scholar]
- 62. Yang XW, Zhao J, Cui YX, et al. Anti‐HIV‐1 protease triterpenoid saponins from the seeds of Aesculus chinensis . J Nat Prod. 1999;62:1510–1513. [DOI] [PubMed] [Google Scholar]
- 63. Stoddart CA, Joshi P, Sloan B, et al. Potent activity of the HIV‐1 maturation inhibitor bevirimat in SCID‐hu Thy/Liv mice. PLoS One. 2007;2:e1251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Baichwal V, Austin H, Brown B, et al. Anti‐viral characterization in vitro of a novel maturation inhibitor, MPC‐9055, poster 561. 16th Conference on Retroviruses and Opportunistic Infections, Montreal, Canada; 2009. [Google Scholar]
- 65. Kashiwada Y, Wang HK, Nagao T, et al. Anti‐AIDS agents. 30. Anti‐HIV activity of oleanolic acid, pomolic acid, and structurally related triterpenoids. J Nat Prod. 1998;61:1090–1095. [DOI] [PubMed] [Google Scholar]
- 66. Hashimoto F, Kashiwada Y, Cosentino LM, Chen CH, Garrett PE, Lee KH. Anti‐AIDS agents–XXVII. Synthesis and anti‐HIV activity of betulinic acid and dihydrobetulinic acid derivatives. Bioorg Med Chem. 1997;5:2133–2143. [DOI] [PubMed] [Google Scholar]
- 67. Beelen A, Otto J, Fidler M, et al. Phase 1 single ascending oral dose study of the safety, tolerability, and pharmacokinetics of a novel HIV‐1 maturation inhibitor in HIV negative, healthy volunteers, poster 570. 16th Conference on Retroviruses and Opportunistic Infections, Montreal, Canada; 2009. [Google Scholar]
- 68. Wang D, Lu W, Li F. Pharmacological intervention of HIV‐1 maturation. Acta Pharm Sin B. 2015;5:493–499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Huang L, Yuan X, Aiken C, Chen CH. Bifunctional anti‐human immunodeficiency virus type 1 small molecules with two novel mechanisms of action. Antimicrob Agents Chemother. 2004;48:663–665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. McHutchison JG, Gordon SC, Schiff ER, et al. Interferon alfa‐2b alone or in combination with ribavirin as initial treatment for chronic hepatitis C. N Engl J Med. 1998;339:1485–1492. [DOI] [PubMed] [Google Scholar]
- 71. Poynard T, Marcellin P, Lee SS, et al. Randomised trial of interferon alpha2b plus ribavirin for 48 weeks or for 24 weeks versus interferon alpha2b plus placebo for 48 weeks for treatment of chronic infection with hepatitis C virus. International Hepatitis Interventional Therapy Group (IHIT). Lancet. 1998;352:1426–1432. [DOI] [PubMed] [Google Scholar]
- 72. Lauer GM, Walker BD. Hepatitis C virus infection. N Engl J Med. 2001;345:41–52. [DOI] [PubMed] [Google Scholar]
- 73. Saibara T, Enomoto N, Kaneko S, et al. Clinical efficacy of combination therapy with ME3738 and pegylated interferon‐alpha‐2a in patients with hepatitis C virus genotype 1. Hepatol Res. 2014;44:410–419. [DOI] [PubMed] [Google Scholar]
- 74. Vieyres G, Thomas X, Descamps V, Duverlie G, Patel AH, Dubuisson J. Characterization of the envelope glycoproteins associated with infectious hepatitis C virus. J Virol. 2010;84:10159–10168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Wang W, Guan M, Liu Y, et al. Alanine scanning mutagenesis of hepatitis C virus E2 cysteine residues: insights into E2 biogenesis and antigenicity. Virology. 2014;448:229–237. [DOI] [PubMed] [Google Scholar]
- 76. Gastaminza P, Dryden KA, Boyd B, et al. Ultrastructural and biophysical characterization of hepatitis C virus particles produced in cell culture. J Virol. 2010;84:10999–11009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Lavillette D, Pecheur EI, Donot P, et al. Characterization of fusion determinants points to the involvement of three discrete regions of both E1 and E2 glycoproteins in the membrane fusion process of hepatitis C virus. J Virol. 2007;81:8752–8765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Boucheix C, Rubinstein E. Tetraspanins. Cell Mol Life Sci. 2001;58:1189–1205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Levy S, Shoham T. The tetraspanin web modulates immune‐signalling complexes. Nat Rev Immunol. 2005;5:136–148. [DOI] [PubMed] [Google Scholar]
- 80. VanCompernolle SE, Wiznycia AV, Rush JR, Dhanasekaran M, Baures PW, Todd SC. Small molecule inhibition of hepatitis C virus E2 binding to CD81. Virology. 2003;314:371–380. [DOI] [PubMed] [Google Scholar]
- 81. Baldick CJ, Wichroski MJ, Pendri A, et al. A novel small molecule inhibitor of hepatitis C virus entry. PLoS Pathog. 2010;6:e1001086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Mittapalli GK, Jackson A, Zhao F, et al. Discovery of highly potent small molecule hepatitis C virus entry inhibitors. Bioorg Med Chem Lett. 2011;21:6852–6855. [DOI] [PubMed] [Google Scholar]
- 83. Yang JP, Zhou D, Wong‐Staal F. Screening of small‐molecule compounds as inhibitors of HCV entry. Methods Mol Biol. 2009;510:295–304. [DOI] [PubMed] [Google Scholar]
- 84. Ł Woźnia, S Skąpsk, Marszałek K. Ursolic acid–A pentacyclic triterpenoid with a wide spectrum of pharmacological activities. Molecules. 2015;20:20614–20641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Yu F, Wang Q, Zhang Z, et al. Development of oleanane‐type triterpene as a new class of HCV entry inhibitors. J Med Chem. 2013;56:4300–4319. [DOI] [PubMed] [Google Scholar]
- 86. Yu F, Peng Y, Wang Q, et al. Development of bivalent oleanane‐type triterpenes as potent HCV entry inhibitors. Eur J Med Chem. 2014;77:258–268. [DOI] [PubMed] [Google Scholar]
- 87. Xiao S, Wang Q, Si L, et al. Synthesis and anti‐HCV entry activity studies of beta‐cyclodextrin‐pentacyclic triterpene conjugates. ChemMedChem. 2014;9:1060–1070. [DOI] [PubMed] [Google Scholar]
- 88. Xiao S, Wang Q, Si L, et al. Synthesis and biological evaluation of novel pentacyclic triterpene alpha‐cyclodextrin conjugates as HCV entry inhibitors. Eur J Med Chem. 2016;124:1–9. [DOI] [PubMed] [Google Scholar]
- 89. Wang H, Wang Q, Xiao SL, et al. Elucidation of the pharmacophore of echinocystic acid, a new lead for blocking HCV entry. Eur J Med Chem. 2013;64:160–168. [DOI] [PubMed] [Google Scholar]
- 90. Wang H, Yu F, Peng Y, et al. Synthesis and biological evaluation of ring A and/or C expansion and opening echinocystic acid derivatives for anti‐HCV entry inhibitors. Eur J Med Chem. 2015;102:594–599. [DOI] [PubMed] [Google Scholar]
- 91. Ma CM, Wu XH, Masao H, Wang XJ, Kano Y. HCV protease inhibitory, cytotoxic and apoptosis‐inducing effects of oleanolic acid derivatives. J Pharm Pharm Sci. 2009;12:243–248. [DOI] [PubMed] [Google Scholar]
- 92. Abe H, Imamura M, Hiraga N, et al. ME3738 enhances the effect of interferon and inhibits hepatitis C virus replication both in vitro and in vivo. J Hepatol. 2011;55:11–18. [DOI] [PubMed] [Google Scholar]
- 93. Fukumura A, Tsutsumi M, Tsuchishima M, et al. Effect of the inducer of interleukin‐6 (ME3738) on rat liver treated with ethanol. Alcohol Clin Exp Res. 2007;31:S49–53. [DOI] [PubMed] [Google Scholar]
- 94. Klein C, Wustefeld T, Heinrich PC, Streetz KL, Manns MP, Trautwein C. ME3738 protects from concanavalin A‐induced liver failure via an IL‐6‐dependent mechanism. Eur J Immunol. 2003;33:2251–2261. [DOI] [PubMed] [Google Scholar]
- 95. Ashfaq UA, Masoud MS, Nawaz Z, Riazuddin S. Glycyrrhizin as antiviral agent against hepatitis C virus. J Transl Med. 2011;9:112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. De Clercq E. Antiviral agents active against influenza A viruses. Nat Rev Drug Discov. 2006;5:1015–1025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Webby RJ, Webster RG. Are we ready for pandemic influenza? Science. 2003;302:1519–1522. [DOI] [PubMed] [Google Scholar]
- 98. Kilbourne ED. Influenza pandemics of the 20th century. Emerg Infect Dis. 2006;12:9–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Yuen KY, Wong SS. Human infection by avian influenza a H5N1. Hong Kong Med J. 2005;11:189–199. [PubMed] [Google Scholar]
- 100. Buchy P, Mardy S, Vong S, et al. Influenza A/H5N1 virus infection in humans in Cambodia. J Clin Virol. 2007;39:164–168. [DOI] [PubMed] [Google Scholar]
- 101. Michaelis M, Doerr HW. Of chickens and men: avian influenza in humans. Curr Mol Med. 2009;9:131–151. [DOI] [PubMed] [Google Scholar]
- 102. Yeh JY, Coumar MS, Horng JT, et al. Anti‐influenza drug discovery: structure‐activity relationship and mechanistic insight into novel angelicin derivatives. J Med Chem. 2010;53:1519–1533. [DOI] [PubMed] [Google Scholar]
- 103. Khazeni N, Hutton DW, Collins CI, Garber AM, Owens DK. Health and economic benefits of early vaccination and nonpharmaceutical interventions for a human influenza A (H7N9) pandemic: a modeling study. Ann Intern Med. 2014;160:684–694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. Hause BM, Ducatez M, Collin EA, et al. Isolation of a novel swine influenza virus from Oklahoma in 2011 which is distantly related to human influenza C viruses. PLoS Pathog. 2013;9:e1003176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. Mosnier A, Caini S, Daviaud I, et al. Clinical characteristics are similar across type A and B influenza virus infections. PLoS One. 2015;10:e0136186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106. Poon LL, Song T, Rosenfeld R, et al. Quantifying influenza virus diversity and transmission in humans. Nat Genet. 2016;48:195–200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107. Bright RA, Medina MJ, Xu X, et al. Incidence of adamantane resistance among influenza A (H3N2) viruses isolated worldwide from 1994 to 2005: a cause for concern. Lancet. 2005;366:1175–1181. [DOI] [PubMed] [Google Scholar]
- 108. Moscona A. Global transmission of oseltamivir‐resistant influenza. N Engl J Med. 2009;360:953–956. [DOI] [PubMed] [Google Scholar]
- 109. Baltina LA. Chemical modification of glycyrrhizic acid as a route to new bioactive compounds for medicine. Curr Med Chem. 2003;10:155–171. [DOI] [PubMed] [Google Scholar]
- 110. Cinatl J, Morgenstern B, Bauer G, Chandra P, Rabenau H, Doerr HW. Glycyrrhizin, an active component of liquorice roots, and replication of SARS‐associated coronavirus. Lancet. 2003;361:2045–2046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111. Hoever G, Baltina L, Michaelis M, et al. Antiviral activity of glycyrrhizic acid derivatives against SARS‐coronavirus. J Med Chem. 2005;48:1256–1259. [DOI] [PubMed] [Google Scholar]
- 112. Pompei R, Paghi L, Ingianni A, Uccheddu P. Glycyrrhizic acid inhibits influenza virus growth in embryonated eggs. Microbiologica. 1983;6:247–250. [PubMed] [Google Scholar]
- 113. Wolkerstorfer A, Kurz H, Bachhofner N, Szolar OHJ. Glycyrrhizin inhibits influenza A virus uptake into the cell. Antiviral Res. 2009;83:171–178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114. Baltina LA, Zarubaev VV, Baltina LA, et al. Glycyrrhizic acid derivatives as influenza A/H1N1 virus inhibitors. Bioorg Med Chem Lett. 2015;25:1742–1746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115. Song W, Si L, Ji S, et al. Uralsaponins M‐Y, antiviral triterpenoid saponins from the roots of Glycyrrhiza uralensis . J Nat Prod. 2014;77:1632–1643. [DOI] [PubMed] [Google Scholar]
- 116. Song G, Shen X, Li S, et al. Structure‐activity relationships of 3‐O‐beta‐chacotriosyl ursolic acid derivatives as novel H5N1 entry inhibitors. Eur J Med Chem. 2015;93:431–442. [DOI] [PubMed] [Google Scholar]
- 117. Song G, Shen X, Li S, et al. Structure‐activity relationships of 3‐O‐beta‐chacotriosyl oleanane‐type triterpenoids as potential H5N1 entry inhibitors. Eur J Med Chem. 2016;119:109–121. [DOI] [PubMed] [Google Scholar]
- 118. Li S, Jia X, Shen X, et al. Structure‐activity relationships of 3‐O‐beta‐chacotriosyl oleanic acid derivatives as entry inhibitors for highly pathogenic H5N1 influenza virus. Bioorg Med Chem. 2017;25:4384–4396. [DOI] [PubMed] [Google Scholar]
- 119. Song G, Shen X, Li Y, Zheng Y, Xiong P, Liu S. 3‐O‐β‐chacotriosyl benzyl ursolate inhibits entry of H5N1 influenza virus into target cells. Nan Fang Yi Ke Da Xue Xue Bao. 2015;35:789–794. [PubMed] [Google Scholar]
- 120. Song G, Yang S, Zhang W, et al. Discovery of the first series of small molecule H5N1 entry inhibitors. J Med Chem. 2009;52:7368–7371. [DOI] [PubMed] [Google Scholar]
- 121. Yu M, Si L, Wang Y, et al. Discovery of pentacyclic triterpenoids as potential entry inhibitors of influenza viruses. J Med Chem. 2014;57:10058–10071. [DOI] [PubMed] [Google Scholar]
- 122. Xiao S, Si L, Tian Z, et al. Pentacyclic triterpenes grafted on cd cores to interfere with influenza virus entry: a dramatic multivalent effect. Biomaterials. 2016;78:74–85. [DOI] [PubMed] [Google Scholar]
- 123. Tian Z, Si L, Meng K, et al. Inhibition of influenza virus infection by multivalent pentacyclic triterpene‐functionalized per‐O‐methylated cyclodextrin conjugates. Eur J Med Chem. 2017;134:133–139. [DOI] [PubMed] [Google Scholar]
- 124. Casu B, Reggiani M, Sanderson GR. Methylated cycloamyloses (cyclodextrins) and their inclusion properties. Carbohyd Res. 1979;76:59–66. [Google Scholar]
- 125. Szejtli J. Cyclodextrin and Their Inclusion Complex. Budapest: Akadémiai Kiado; 1982. [Google Scholar]
- 126. Xiao SL, Wang Q, Yu F, et al. Conjugation of cyclodextrin with fullerene as a new class of HCV entry inhibitors. Bioorg Med Chem. 2012;20:5616–5622. [DOI] [PubMed] [Google Scholar]
- 127. Wang H, Xu R, Shi Y, et al. Design, synthesis and biological evaluation of novel L‐ascorbic acid‐conjugated pentacyclic triterpene derivatives as potential influenza virus entry inhibitors. Eur J Med Chem. 2016;110:376–388. [DOI] [PubMed] [Google Scholar]
- 128. Han X, Si LL, Shi YY, et al. Synthesis and in vitro anti‐influenza virus evaluation of novel sialic acid (C‐5 and C‐9)‐pentacyclic triterpene derivatives. Molecules. 2017;22:pii: E1018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129. Shi Y, Si L, Han X, et al. Synthesis of novel pentacyclic triterpene‐neu5ac2en derivatives and investigation of their in vitro anti‐influenza entry activity. MedChemComm. 2017;8:1531–1541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130. Han X, Shi Y, Si L, et al. Design, synthesis and biological activity evaluation of novel conjugated sialic acid and pentacyclic triterpene derivatives as anti‐influenza entry inhibitors. MedChemComm. 2016;7:1932–1945. [Google Scholar]
- 131. Hong EH, Song JH, Kang KB, Sung SH, Ko HJ, Yang H. Anti‐influenza activity of betulinic acid from Zizyphus jujuba on influenza A/PR/8 virus. Biomol Ther. 2015;23:345–349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132. Li Y, Jiang R, Ooi LS, But PP, Ooi VE. Antiviral triterpenoids from the medicinal plant Schefflera heptaphylla . Phytother Res. 2007;21:466–470. [DOI] [PubMed] [Google Scholar]
- 133. Tung NH, Kwon HJ, Kim JH, Ra JC, Kim JA, Kim YH. An anti‐influenza component of the bark of Alnus japonica . Arch Pharm Res. 2010;33:363–367. [DOI] [PubMed] [Google Scholar]
- 134. Kazakova OB, Medvedeva NI, Baikova IP, et al. Synthesis of triterpenoid acylates—an effective reproduction inhibitors of influenza A (H1N1) and papilloma viruses. Bioorg Khim. 2010;36:841–848. [PubMed] [Google Scholar]
- 135. Wu CY, Jan JT, Ma SH, et al. Small molecules targeting severe acute respiratory syndrome human coronavirus. Proc Natl Acad Sci USA. 2004;101:10012–10017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136. Wen CC, Kuo YH, Jan JT, et al. Specific plant terpenoids and lignoids possess potent antiviral activities against severe acute respiratory syndrome coronavirus. J Med Chem. 2007;50:4087–4095. [DOI] [PubMed] [Google Scholar]
- 137. Ryu YB, Park SJ, Kim YM, et al. SARS‐Cov 3CLpro inhibitory effects of quinone‐methide triterpenes from Tripterygium regelii . Bioorg Med Chem Lett. 2010;20:1873–1876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138. Bartholomeusz A, Locarnini S. Hepatitis B virus mutants and fulminant hepatitis B: fitness plus phenotype. Hepatology. 2001;34:432–435. [DOI] [PubMed] [Google Scholar]
- 139. Kalinina T, Riu A, Fischer L, Will H, Sterneck M. A dominant hepatitis B virus population defective in virus secretion because of several S‐gene mutations from a patient with fulminant hepatitis. Hepatology. 2001;34:385–394. [DOI] [PubMed] [Google Scholar]
- 140. Ko C, Michler T, Protzer U. Novel viral and host targets to cure hepatitis B. Curr Opin Virol. 2017;24:38–45. [DOI] [PubMed] [Google Scholar]
- 141. Raj V. Treatment of hepatitis B. Clin Cornerstone. 2001;3:24–36. [DOI] [PubMed] [Google Scholar]
- 142. Yao D, Li H, Gou Y, et al. Betulinic acid‐mediated inhibitory effect on hepatitis B virus by suppression of manganese superoxide dismutase expression. FEBS J. 2009;276:2599–2614. [DOI] [PubMed] [Google Scholar]
- 143. Takahara T, Watanabe A, Shiraki K. Effects of glycyrrhizin on hepatitis B surface antigen: a biochemical and morphological study. J Hepatol. 1994;21:601–609. [DOI] [PubMed] [Google Scholar]
- 144. Sato H, Goto W, Yamamura J, et al. Therapeutic basis of glycyrrhizin on chronic hepatitis B. Antiviral Res. 1996;30:171–177. [DOI] [PubMed] [Google Scholar]
- 145. Romero MR, Efferth T, Serrano MA, et al. Effect of artemisinin/artesunate as inhibitors of hepatitis B virus production in an “in vitro” replicative system. Antiviral Res. 2005;68:75–83. [DOI] [PubMed] [Google Scholar]
- 146. Zhao YL, Cai GM, Hong X, Shan LM, Xiao XH. Anti‐hepatitis B virus activities of triterpenoid saponin compound from Potentilla anserine L. Phytomedicine. 2008;15:253–258. [DOI] [PubMed] [Google Scholar]
- 147. Bi Y, Xu J, Wu X, Ye W, Yuan S, Zhang L. Synthesis and cytotoxic activity of 17‐carboxylic acid modified 23‐hydroxy betulinic acid ester derivatives. Bioorg Med Chem Lett. 2007;17:1475–1478. [DOI] [PubMed] [Google Scholar]
- 148. Ye WC, Ji NN, Zhao SX, et al. Triterpenoids from Pulsatilla chinensis . Phytochemistry. 1996;42:799–802. [DOI] [PubMed] [Google Scholar]
- 149. Roizman B, Whitley RJ. The nine ages of herpes simplex virus. Herpes. 2001;8:23–27. [PubMed] [Google Scholar]
- 150. Whitley RJ, Roizman B. Herpes simplex virus infections. Lancet. 2001;357:1513–1518. [DOI] [PubMed] [Google Scholar]
- 151. De Clercq E. Antiviral drugs in current clinical use. J Clin Virol. 2004;30:115–133. [DOI] [PubMed] [Google Scholar]
- 152. Chen Y, Scieux C, Garrait V, et al. Resistant herpes simplex virus type 1 infection: an emerging concern after allogeneic stem cell transplantation. Clin Infect Dis. 2000;31:927–935. [DOI] [PubMed] [Google Scholar]
- 153. Chakrabarti S, Pillay D, Ratcliffe D, et al. Resistance to antiviral drugs in herpes simplex virus infections among allogeneic stem cell transplant recipients: risk factors and prognostic significance. J Infect Dis. 2000;181:2055–2058. [DOI] [PubMed] [Google Scholar]
- 154. Kurokawa M, Ochiai H, Nagasaka K, et al. Antiviral traditional medicines against herpes simplex virus (HSV‐1), poliovirus, and measles virus in vitro and their therapeutic efficacies for HSV‐1 infection in mice. Antiviral Res. 1993;22:175–188. [DOI] [PubMed] [Google Scholar]
- 155. Nakano M, Kurokawa M, Hozumi T, et al. Suppression of recurrent genital herpes simplex virus type 2 infection by Rhus javanica in guinea pigs. Antiviral Res. 1998;39:25–33. [DOI] [PubMed] [Google Scholar]
- 156. Kurokawa M, Basnet P, Ohsugi M, et al. Anti‐herpes simplex virus activity of moronic acid purified from Rhus javanica in vitro and in vivo. J Pharmacol Exp Ther. 1999;289:72–78. [PubMed] [Google Scholar]
- 157. Ikeda T, Yokomizo K, Okawa M, et al. Anti‐herpes virus type 1 activity of oleanane‐type triterpenoids. Biol Pharm Bull. 2005;28:1779–1781. [DOI] [PubMed] [Google Scholar]
- 158. Gong Y, Raj KM, Luscombe CA, et al. The synergistic effects of betulin with acyclovir against herpes simplex viruses. Antiviral Res. 2004;64:127–130. [DOI] [PubMed] [Google Scholar]
- 159. Heidary Navid M, Laszczyk‐Lauer MN, Reichling J, Schnitzler P. Pentacyclic triterpenes in birch bark extract inhibit early step of herpes simplex virus type 1 replication. Phytomedicine. 2014;21:1273–1280. [DOI] [PubMed] [Google Scholar]
- 160. Wang J, Chen X, Wang W, et al. Glycyrrhizic acid as the antiviral component of Glycyrrhiza uralensis fisch. Against coxsackievirus A16 and enterovirus 71 of hand foot and mouth disease. J Ethnopharmacol. 2013;147:114–121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161. Chiang LC, Ng LT, Cheng PW, Chiang W, Lin CC. Antiviral activities of extracts and selected pure constituents of Ocimum basilicum . Clin Exp Pharmacol Physiol. 2005;32:811–816. [DOI] [PubMed] [Google Scholar]
- 162. Song J, Yeo SG, Hong EH, et al. Antiviral activity of hederasaponin B from Hedera helix against enterovirus 71 subgenotypes C3 and C4a. Biomol Ther. 2014;22:41–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163. Zhao CH, Xu J, Zhang YQ, Zhao LX, Feng B. Inhibition of human enterovirus 71 replication by pentacyclic triterpenes and their novel synthetic derivatives. Chem Pharm Bull. 2014;62:764–771. [DOI] [PubMed] [Google Scholar]
- 164. Lin J‐C. Mechanism of action of glycyrrhizic acid in inhibition of Epstein‐Barr virus replication in vitro. Antiviral Res. 2003;59:41–47. [DOI] [PubMed] [Google Scholar]
- 165. Chang FR, Hsieh YC, Chang YF, Lee KH, Wu YC, Chang LK. Inhibition of the Epstein‐Barr virus lytic cycle by moronic acid. Antiviral Res. 2010;85:490–495. [DOI] [PubMed] [Google Scholar]