

ABSTRACT
Antimicrobial peptides (AMPs) are an integral part of the innate immune defense mechanism of many organisms. Due to the alarming increase of resistance to antimicrobial therapeutics, a growing interest in alternative antimicrobial agents has led to the exploitation of AMPs, both synthetic and isolated from natural sources. Thus, many peptide‐based drugs have been the focus of increasing attention by many researchers not only in identifying novel AMPs, but in defining mechanisms of antimicrobial peptide activity as well. Herein, we review the available strategies for the identification of AMPs in human body fluids and their mechanism(s) of action. In addition, an overview of the distribution of AMPs across different human body fluids is provided, as well as its relation with microorganisms and infectious conditions.
Keywords: human antimicrobial peptides, antibacterial, antifungal, antiviral, human biological fluids, peptidomics
1. INTRODUCTION
Human antimicrobial peptides (AMPs) represent approximately 10% of all curated AMPs catalogued to date.1 Human host defense peptides are an intrinsic part of the innate immune system and exhibit a broad activity spectrum against bacteria, fungi, viruses, and parasites (Figs. 1, 2, 3), but also toward mammalian malignant cells, and represent a relatively large group of biomolecules.
Figure 1.
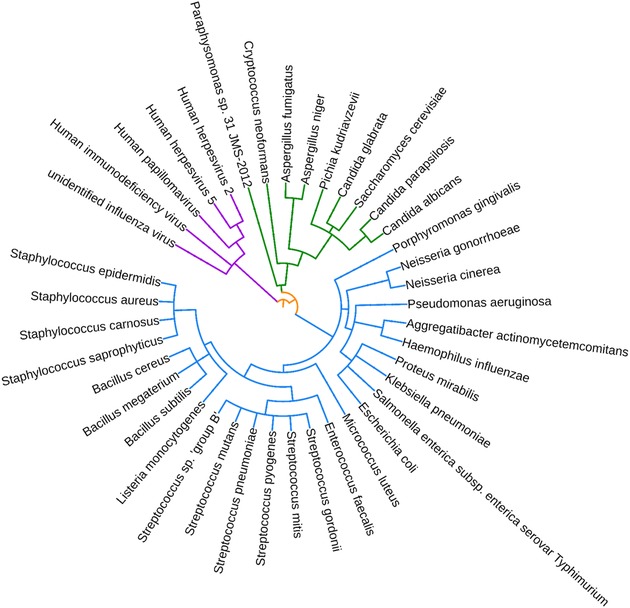
Phylogenetic tree representing all bacterial, viral, and fungal species targeted by human antimicrobial peptides found in body fluids. Created using phyloT.
Figure 2.
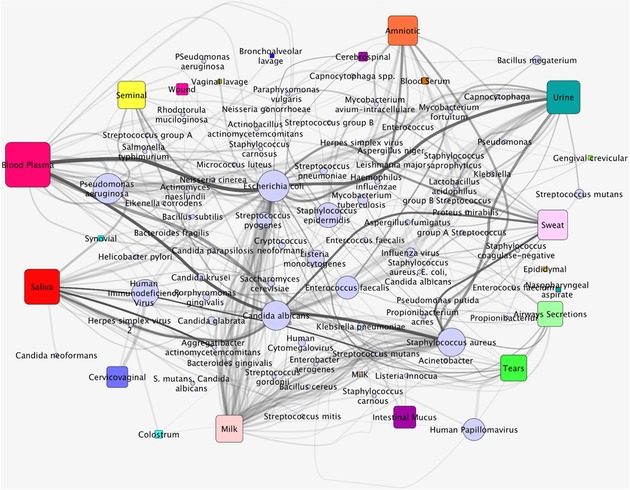
Global network depicting the distribution of human antimicrobial peptides across biological fluids and their microbial targets. Rectangular nodes correspond to biofluids, circular nodes to target species, and each edge correspond to a given gene encoding one or more antimicrobial peptides. The thicker the edge, the stronger is the association of a biofluid to a pathogen, representing increased number of antimicrobial peptides defending against such pathogen in such biofluid. Also, pathogens represented by bigger nodes (e.g., Escherichia coli, Candida albicans, Staphylococcus aureus) represent those that are targeted by more antimicrobial peptides across different biofluids.
Figure 3.
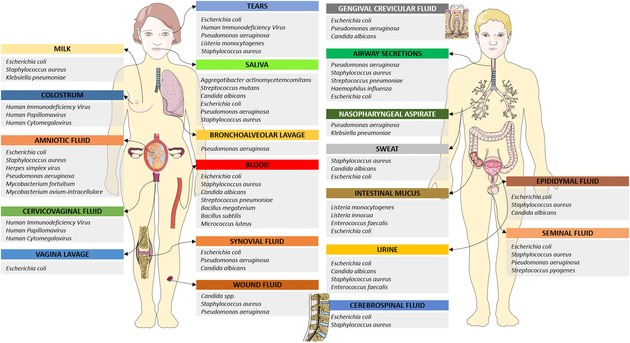
Distribution of species targeted by human antimicrobial peptides throughout human body fluids. Only most represented species are included.
While AMPs can be antibacterial (ABPs), antifungal, antiprotist, antiviral, anticancer, antiparasitic, insecticidal, spermicidal, chemotactic, antioxidant, protease inhibitors, or even exhibit wound healing properties (Supporting Information Table S1), their scope of action overlaps considerably and some peptides show activity at several levels (Fig. 2).1, 2, 3, 4, 5 For instance, on the one hand, human plasma adrenomedullin‐derived peptides are active against multiple bacteria species, human urinary and gingival fluid calcitonin gene‐related peptide displays antimicrobial activity against several bacteria and fungi species, and human neutrophil peptides (HNPs)/defensins isolated from most biological fluids display activity against several bacterial, fungal, and viral species (Fig. 2). On the other hand, dozens of human AMPs known to date display antimicrobial activity against the same human colonizers or pathogens, including Escherichia coli, Pseudomonas aeruginosa, and Staphylococcus aureus (Figs. 2 and 3). Also, it is intriguing to observe that even though some AMPs display broad activity spectra, others seem to be anywhere from species‐ to kingdom‐specific (Fig. 2). While likely to be confounded by observational bias (whereby some species, e.g., E. coli, tend to be tested more frequently than others), this observation also reflects that the activity spectra of AMPs depend not only on their physicochemical properties (which group them according to unique families) but also on target properties and on the environment/biological fluid in which they are found. As the large majority of AMPs identified to date are ABPs (∼83%, Figs. 1, 2, 3), most concepts and examples provided in this review will be based on results from studies focusing on ABPs, closely followed by those derived from studies on antiviral and antifungal peptides. However, the same principles tend to apply across all AMPs.
AMPs are very potent cationic molecules displaying minimum inhibitory concentrations (the minimum concentration that prevents bacterial growth) as low as 1–4 μg/mL,6 which highlights their promise as broad‐spectrum antimicrobial agents. Furthermore, AMPs act more rapidly than conventional antibiotics and are not affected by the typical resistance mechanisms involving conventional drugs, making them very attractive for therapeutic purposes. In addition, AMPs are not mere microbicide agents, as their scope of action may encompass other functions, such as cellular development and immune system modulation. For instance, fetal keratinocytes express significantly more AMPs compared to neonatal and adult keratinocytes, despite the lower degree of exposition to the environment or to pathogenic agents. These elevated levels of AMPs during fetal stages of development are correlated with increased expression of the histone demethylase JMJD3,7 which in turn also plays a key role in several other fundamental processes, including chromatin organization, cell fate commitment, mesodermal, cardiac muscle, and endothelial cell differentiation, and hippocampus development.8, 9 Also, tear fluid human AMPs such as cationic antimicrobial protein 37 and thymosin‐β4 are known to stimulate corneal epithelial cell migration and proliferation in response to injury,10, 11 while lacritin is a glycoprotein that stimulates tear secretion and acts as a mitogen but which, once cleaved, also displays antibacterial activity.12 Thus, AMPs appear to be key mediators involved in several biological processes and to be intrinsically regulated in tandem with other fundamental processes, working together to maintain organic homeostasis. Accordingly, AMPs detain innumerous immune‐modulatory properties, namely cytokine modulation, chemotaxis, angiogenesis stimulation, as well as mast cells’ degranulation and consequent vasodilatation, fibrinolysis inhibition, and tissue injury prevention by inhibiting proteases and by reinforcing wound repair. Further information regarding the interplay of AMPs in the innate immune system as well as their alternative roles is given elsewhere,13, 14, 15 and is beyond the scope of this review. Herein, we will revisit the antimicrobial effects of human AMPs.
2. PRODUCTION AND ELIMINATION OF AMPs
Considering the wide variety of amino acid sequences, structures, mechanisms of action, and performed functions, it is perhaps surprising to discover a high degree of conservation and analogy between mechanisms controlling AMPs formation, regulation, and elimination. In fact, these processes are very dynamic, involving many substrates, precursors, proteases/peptidases, activators, and inhibitors. The majority of AMPs identified in humans appears to result from the cleavage of precursor proteins with other unrelated functions, rather than from the direct transcription and translation into biologically active peptides. Nevertheless, AMPs are an integral element of the innate immune system and not just a product of protein cleavage, representing a defense strategy against invading microorganisms.16 Moreover, as later exemplified, several AMPs resulting from fragmentation of precursor proteins act synergistically with these, in order to fight foreign microorganisms at several defense barriers (Fig. 3).
Bioactive peptides can result from the cleavage of proteins targeted for exportation to the extracellular milieu, where both peptides and precursor protein may play their primary functions. This process requires prior commitment of the precursor proteins to the secretory pathway, which is ensued by the presence of a signal peptide in its sequence.17 Once committed to the secretory pathway, peptides are modified at several locations and time points, beginning in the endoplasmic reticulum, then moving to the Golgi apparatus, traveling inside secretory vesicles, and ending up secreted upon fusion of these vesicles with the cytoplasmic membranes.17, 18
While prokaryotic microorganisms are known to produce AMPs by both gene‐encoded (e.g., microcins and bacteriocins) and ribosome‐independent (e.g., vancomycin and daptomycin) pathways,19, 20, 21 there is no knowledge of ribosome‐independent AMPs formation in humans. Even when an AMP is constitutively produced and secreted, its activity level is not constant, as it may be further modulated by the action of proteolytic enzymes. In contrast with some circular prokaryotic peptides, which have a peptide bond connecting the N‐ and C‐termini, which confers them an enhanced degree of stability, human AMPs are linear in nature and thus considerably more susceptible to proteolytic cleavage.22 Such susceptibility is exploited by a whole set of proteolytic endopeptidases and exopeptidases capable of mediating the formation and elimination of AMPs, including kallikreins, kininases, matrix metalloproteinases, cathepsins, and others.23, 24, 25, 26, 27
Interestingly, human AMPs are subject to the action of both human and bacterial enzymes. Examples of bacterial enzymes capable of modulating human AMPs formation and degradation include metalloproteinase aureolysin and glutamylendopeptidase V8 from S. aureus, elastase from P. aeruginosa, gelatinase from Enterococcus faecalis, and metalloprotease from Proteus mirabilis.28 Moreover, the regulation of human AMPs formation and elimination is made even more complex when one considers the presence of naturally occurring protease inhibitors, which may also present a tissue‐specific expression pattern and modulate the antimicrobial activity of naturally occurring AMPs.29 Lastly, not only peptides are inactivated and eliminated by hydrolysis, they are also intracellularly recycled and cleared through renal filtration or excretion.30, 31
3. SEQUENCES, STRUCTURES, AND PHYSICAL PROPERTIES
AMPs tend to be found as small molecules, generally composed of fewer than 50 amino acid residues and most frequently in the L configuration (left‐handed enantiomer, resulting from the distribution of functional groups around a central carbon atom). Despite mostly being cationic and short in sequence, human AMPs can vary anywhere between 10 and 150 amino acids (human neurokinin A consists of ten, while human RegIIIα consists of 149 amino acids). Most present a hydrophobic content below 60% and their net charge varies between −3 and +20 (β‐amyloid peptide and CXCL9, respectively).32
Furthermore, predicting and rationalizing how any particular AMP accomplishes its biological activity can be done only if its detailed structure–function information is available. Given this information, AMPs tend to be organized into groups, facilitating such rationale, and several criteria can be used to classify and sort AMPs, including origin, size, sequence, biological class, mechanism of action, and secondary structure. Of these, amino acid sequence and secondary structure are the most important factors to take into consideration,33 perhaps because these allow us to predict how a peptide will interact with other molecules.
A rough estimate of all AMPs catalogued to date (Supporting Information Table S2) indicates 14.1% to be α‐helices, followed by β‐sheets (3.7%), and peptides presenting with both helices and β‐sheets (3.7%). However, approximately 60% of all AMPs currently identified do not have a known 3D structure, while only 0.3% have a known structure that is neither a helix nor a β‐sheet. Excluding those that have unknown structures, 99.2% of all AMPs are either α‐helices and/or β‐sheets, suggesting a highly conserved structure for a highly primordial biological function.
Secondary structures of human AMPs may include α‐helices (e.g., LL‐37),34 β‐sheets (e.g., defensin 1),35 and extended peptides (e.g., indolicidin).36 Considering that β‐sheets and α‐helices in polypeptide chains typically contain three to ten amino acids per strand and 3.6 residues per turn, respectively, and that amino acid sequences can be virtually unlimited, one can easily envision that a given peptide can display a multitude of secondary structures and, thus, many diverse conformations (see 37 for protein secondary structures predictive approaches).
Among secondary structures, α‐helices (in addition to being the most frequently encountered in nature) are the most regular and predictable, and, consequently, the most extensively studied. AMPs with linear α‐helices are notably positively charged and amphipathic, but only adopt such secondary conformation upon binding to bacterial membranes.38 Therefore, their antimicrobial activity is directly dependent on the secondary structure.39 In contrast, β‐sheet AMPs possess well‐defined conformations due to the presence of disulfide bridges formed between thiol groups in cysteines. In these peptides, such bridges are often required for antimicrobial activity.40 Contrariwise, bovine lactoferricin was shown to be effective against E. coli 0111 even when such bridges were blocked by pyridylethylation.41
Other human AMPs, such as human β‐defensin 3 (hBD‐3), present both α‐helices and β‐sheets. In the specific case of hBD‐3, disrupting the three stabilizing disulfide bonds does not compromise its antimicrobial activity, but it does diminish its chemotactic properties. 42 Furthermore, the correlation of the conformation with peptides’ antimicrobial activity is not always as straightforward as one could imagine. For example, disruption of hBD‐1's disulfide bonds results in peptides with free cysteines at the carboxy terminal that show increased antimicrobial activity against the pathogenic Candida albicans and the Gram‐positive commensals Bifidobacterium and Lactobacillus.43
Moreover, linear extended peptides do not keep a fixed conformation in solution, and are enriched in one or more amino acids, particularly arginine, proline, and tryptophan. For instance, the AMP histatin found in human saliva is enriched in histidine residues,44 while prophenin is very rich in proline and phenylalanine residues.45
When it comes to loop rich peptides, the presence of many prolines makes an AMP much less likely to form amphipathic structures, adopting what is frequently referred to as polyproline helical type‐II (PPII) structures. These structures are specifically bound by SH3 domains, which are usually found in proteins that interact with other proteins and mediate assembly of specific protein complexes (upon proline‐rich peptides binding).46
Human antibacterial peptides (ABPs), which compose the largest group of human AMPs, exhibit a weighted average net charge of +5.8 per peptide, which is considerably above the average net charge of all AMPs identified to date. Also, despite being intuitive that the more positively charged an AMP is, the more potency it will present,47 human AMPs do not show greater potency or efficacy when compared to those of other species, suggesting that the environment and the presence of different amino acids (D and rare amino acids) also play a significant role.
Notwithstanding the large diversity of AMPs across human biologic fluids (Figs. 2 and 3, Table I), it should be noted that AMPs are much more diverse across all species, than those identified in humans, which present rather conserved properties. In fact, some AMPs from other species are characterized by more intriguing sequences, structures, and physicochemical properties. Case in point, Bacillus subtilis is known to produce several extremely potent lipopeptides (active at concentrations as low as 0.01–0.06 μM) with antimicrobial activity against several species. One of these, gageotetrin A is a very unique anionic AMP that consists of only leucine and glutamic acid residues conjugated with a unique fatty acid, 3‐hydroxy‐11‐methyltridecanoic acid.48 Another example is that of sonorensin from Bacillus sonorensis, a broad‐spectrum AMP from the heterocycloanthracin subfamily, active against both Gram‐negative and Gram‐positive bacteria, including Listeria monocytogenes and S. aureus. Sonorensin displays an amino acid sequence with multiple copies of the same motif, making this peptide rather unique.49 Baceridin is a circular peptide from a plant‐associated Bacillus that is synthesized by ribosome‐independent machinery and bears only six amino acids, 50% in the D configuration.50 Moreover, baceridin is a proteasome inhibitor, compromising cell cycle progression and inducing apoptosis in tumor cells by a p53‐independent pathway.50 Copsin from Coprinopsis cinerea is bactericidal against Enterococcus faecium and L. monocytogenes by interacting with the peptidoglycan precursor lipid II and interfering with the cell wall biosynthesis.51 Curiously, it is modified with a pyroglutamate, which confers a higher degree of thermal stability and resistance toward protease digestion.51
Table I.
Potential Sources of Human Antimicrobial Peptides found in Human Bodily fluidsa
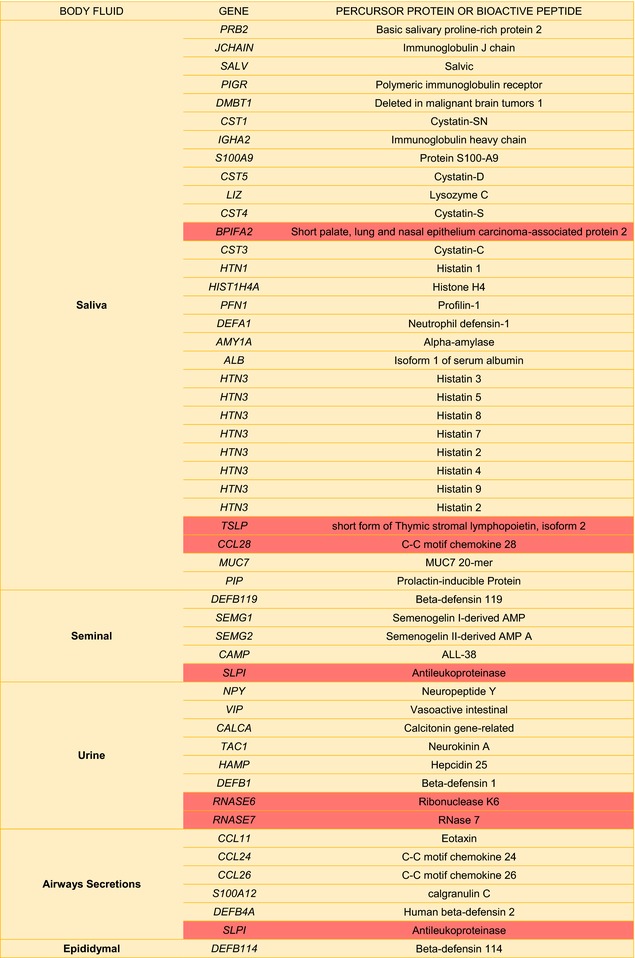
|
|
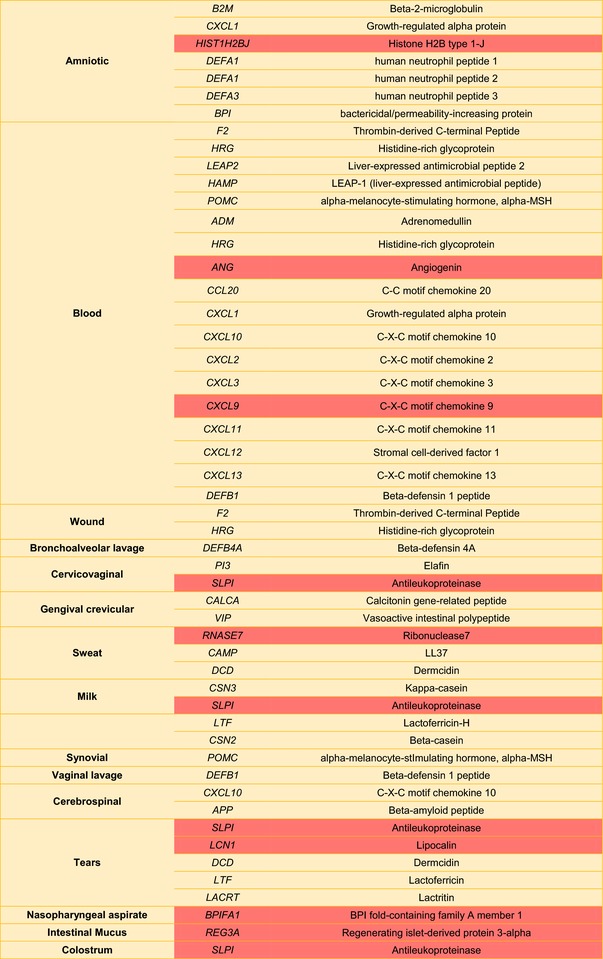
|
Highlighted cells correspond to precursor genes that may also originate peptides with >100 amino acids displaying antimicrobial activity.
This article is being made freely available through PubMed Central as part of the COVID-19 public health emergency response. It can be used for unrestricted research re-use and analysis in any form or by any means with acknowledgement of the original source, for the duration of the public health emergency.
4. MECHANISMS OF ACTION
Even though some display fungicidal and virucidal properties, the chief activity of human AMPs is against Gram‐positive and Gram‐negative bacteria (Figs. 1, 2, 3). Furthermore, while AMPs represent an extremely diverse group of biological active molecules, most share common properties that contribute for their membranolytic or otherwise antimicrobial activity: positive charge, hydrophobicity, and amphiphilicity. Therefore, the mechanism of action of most AMPs involves membrane disruption (Fig. 4), a process that is also required when their translocation is warranted and the respective targets are localized intracellularly.39
Figure 4.
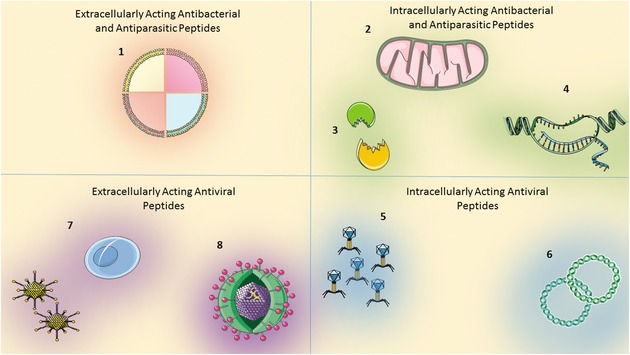
Mechanisms of action of antimicrobial peptides. Antimicrobial peptides may either (1) cluster at the cell surface and cause membrane disruption by several different mechanisms (e.g., barrel staves, carpets, toroidal pores), (2) translocate into cells and impair intracellular organelle machineries, (3) impair protein–protein interactions, enzymatic cascades, and cytosolic signaling pathways, (4) interact with nucleic (not in the case of bacteria) acids, trap replication forks, and compromise nucleic acids as well as protein synthesis, (5) preclude several steps of viral replication, (6) inhibit genetic material trafficking, reverse transcriptase, and viral proteases, (7) block the interaction between virus and host cells (e.g., viral envelope glycoproteins gp120 and gp41 or co‐receptor CXCR4), compromising virus binding and entry, and (8) cause membrane lysis on enveloped viruses. See text for detailed description. Designed using Servier medical Art.
The interaction of AMPs with bacterial membranes depends on the establishment of attractive electrostatic forces between these. Bacterial species are known to carry negatively charged outer membranes due to phosphate groups of the lipopolysaccharides in Gram‐negative bacteria and to the lipoteichoic acids in Gram‐positive bacteria.52 The fact that all Gram‐negative and Gram‐positive bacteria display these type of negatively charged lipids accounts for the lack of specificity of most ABPs, and promotes the attraction between AMPs and bacterial membranes while preventing their binding to most host cells membranes. Moreover, the inherent hydrophobicity and amphiphilicity allow ABPs to form clusters at the membrane's surface once attached.53, 54 At low concentrations (low peptide‐to‐lipid ratio), ABPs tend to adsorb onto the surface, adopting an orientation parallel to the membrane bilayer. However, clustering increases the peptide‐to‐lipid ratio, which subsequently promotes additional clustering. Once a certain peptide‐to‐lipid ratio threshold is reached, ABPs adopt a perpendicular orientation relative to the bacterial membrane, which allows them to insert themselves into the membrane,55 as will be further discussed (Fig. 4).
Peptides can adopt many different secondary structures and conformations, which depend on their amino acids sequence, as well as on the environment. Consequently, different peptides and different environments favor the formation of unique structures, such as barrel staves, carpets, and toroidal pores.40, 56, 57 When the peptides attach and are inserted into the membrane bilayer so that the hydrophobic regions align with the phospholipids’ acyl groups and the hydrophilic regions create the central region of a pore, a barrel stave is formed. In addition, these structures act as a primer for the aggregation of new peptides, thus expanding the pore's diameter.58 Due to ABPs’ cationic sites, membrane crossover would be very energetically unfavorable in their original parallel orientation. However, this orientation changes to a perpendicular one upon ABPs’ clustering, and the peptides are reoriented with their hydrophobic portions facing the membrane acyl groups and their cationic sites facing the interior of the pores. Thus, clustering and pore formation provides a means for membrane disruption and the leakage of intracellular molecules from bacterial cells.59
In contrast, if peptides remain parallel to the membrane surface with their hydrophobic residues facing the membrane while their hydrophilic residues face the surrounding milieu, a “carpet”‐like structure is achieved. With such organization, membrane's rigidity weakens progressively as new peptides are added, culminating in its dissolution and breakup. Mechanistically, this mode of action is less stringent because peptides do not need to adopt a specific structure. However, the necessary concentration of ABPs appears be much higher than that required for the formation of barrel staves.60, 61, 62
For toroidal pores to form, peptides need to aggregate onto the membrane's surface and insert themselves in a perpendicular fashion.63, 64 Subsequently, ABPs are reoriented parallel to the membrane so that polar residues interact with the polar heads of the membrane lipids, thus causing membranes to bend. This induced bending of the bilayer causes the upper and lower leaflets to meet, and allows a mixture of peptides and lipid head groups to line with the interior of the pore, forming toroidal or wormhole structures.60, 63, 65
ABPs may also inhibit bacterial growth and have a bactericidal effect by promoting the clustering of anionic lipids, such as phosphatidylglycerols (PGs) and cardiolipin. For instance, for LL‐37, initially believed to act according to the “carpet” model, there is evidence that its fragments, namely KR‐12, act as a magnet that competes for the negatively charged PGs, promoting their redistribution into “PG‐rich domains.” Such phospholipidic reorganization may disturb cell signaling and compromise the activity of membrane‐bound proteins, namely, the voltage‐dependent potassium channels, which can seriously jeopardize bacteria survival.34 Likewise, N‐acylated peptides derived from lactoferricin were shown to induce defects in E. coli cell division by rearranging the distribution of inner membrane phospholipids, essentially due to the clustering of cardiolipin and other anionic lipids.66
The bias toward positive charges confers AMPs the ability to easily interact not only with biological membranes, but also with other negatively charged molecules (Fig. 4) such as nucleic acids and those bearing phosphate groups.39, 67 In addition, because this type of interaction is largely unspecific, AMPs can exhibit not only a broad‐spectrum activity, but also multiple mechanisms of action (Fig. 3).68, 69 For instance, lactoferricin is known to inhibit ATP‐dependent multidrug efflux pumps as well as DNA, RNA, and protein synthesis.70 Ten human AMPs derived from lactoferricin and lysozyme are thought (by sequence homology) to act by trapping the replication fork and preventing base recombination and DNA repair.71 Furthermore, the presence of many prolines in some peptides (also known as PPII structures as aforementioned) hampers the organization of amphipathic structures.72 These structures favor interactions between peptidic structures and between these and nucleic acids, thus playing a major role in signal transduction and complexes assembly.73 However, PPII structures do not necessarily contain prolines,74 and ABPs rich in arginine residues are also able to translocate across membranes and to interact with nucleic acids and proteins. Hence, both proline and/or arginine‐rich ABPs may be capable of inhibiting prosynthetic signaling pathways and enzymatic activity (Fig. 4), shifting the conformation of cellular structures and activating autolysins. However, this is a speculation that warrants further studying.
In addition to targeting bacterial membranes, human defensins also bind to the peptidoglycan precursor lipid II and inhibit cell wall biosynthetic enzymes in Staphylococci species.75 Moreover, when stabilizing disulfide bridges between conserved cysteine residues in human AMPs with β‐hairpin or β‐sheet conformations are disrupted, the resulting linear peptides still maintain their antimicrobial properties despite losing membranolytic activity.76, 77
The antifungal histatins act at multiple levels by mechanisms of action conserved across the histatin family of AMPs, with histatin 5 the most potent AMP in this family. This AMP binds the yeast transmembrane receptor heat‐shock protein Ssa1/2p, is internalized, and then targets primarily yeast mitochondria.78, 79 By doing so, it leads to the formation of reactive oxygen species (ROS), causes ATP efflux, and inhibits oxidative phosphorylation.80, 81 Simultaneously, histatin 5 also interacts with the potassium transporter TRK1p, causing release of K+ ions and further ATP efflux.82 When the functional domain of histatin 5 was synthetically multiplied, its antifungal activities were significantly potentiated and it became significantly faster‐acting compared to the normal histatin 5.82 Such an observation might prove itself very useful, because it indicates that modulating the number of functional domains per AMP may enhance its antimicrobial activity without requiring the administration of increased quantities of AMP. Histatins may also exert their antimicrobial activities by inhibiting host and microorganismal proteolytic enzymes. Histatin 5 is a strong inhibitor of human matrix metalloproteases 2/9 and metalloproteases from Porphyromonas gingivalis.83 Thus, its inhibitory activity over proteolytic enzymes may attenuate tissue damage and microbial propagation during the onset of periodontal inflammatory disease. Also, by inhibiting the cysteine protease clostripain from Clostridium histolyticum, histatin 5 mitigates the virulence factors produced by this Clostridium species and may therefore alleviate the complications associated with gas gangrene syndrome.84
So far, the above‐mentioned mechanisms of action have been discussed in the context of ABPs or antifungal peptides, and sometimes the same peptide is active against both bacteria and fungi. In turn, the action of antiviral peptides can diverge from these mechanisms and should thus be independently commented (Fig. 4), although some overlap does exist.
Extracellularly, most native antiviral peptides often present as α‐helical structures, have primarily lytic activity on enveloped viruses, and can directly inactivate cell‐free virions (Fig. 4). Human lysozyme fragments are this group's prototype, capable of aggregating on the viral surface and causing disruption of membrane envelopes.85, 86 Similarly, CAP37 peptide appears to exert its antiviral activity by destructing the virus envelope of type I herpes simplex virus (HSV) or even by disrupting the capsids of adenoviruses.87 However, because membrane lipids of enveloped viruses derive from host cell membranes, special attention is required when considering the exploitation of this feature, as antiviral peptides may disrupt host cell membranes indiscriminately.
Alternatively, antiviral peptides may block the interaction between the virus and the host cell (Fig. 4). That is believed to be, at least in part, the mechanism of action of human defensins, θ‐defensin, human α1‐antitrypsin VIRIP, cyclic lactoferricin, and serine protease inhibitor elafin. For instance, it has been demonstrated that all α‐defensins [HNP‐1, HNP‐2, HNP‐3, and HNP‐4 and human α‐defensin (HD)‐5 and HD‐6] and hBD‐3 prevented HSV infection by blocking virus binding and entry. Such antiviral activity was attributed to HNP‐1–3, HD‐5, and hBD‐3's high affinity to HSV's glycoprotein B and HNP‐4, HD‐6, and hBD‐3's avidity toward endogenous heparan sulfate.88 Similarly, protection against HIV is conferred by peptides with lectin properties that can bind viral envelope glycoproteins gp120 and gp41, crucial for viral attachment, fusion of envelopes with host membranes, and subsequent entry into host cells.89 HNP‐4 that binds and blocks the action of gp120, and θ‐defensins (e.g., the pseudogene retrocyclin) together with the human α1‐antitrypsin‐derived 20‐residue viral‐inhibitory peptide both targeting gp41 represent examples of such peptides.90, 91, 92 Likewise, HSV‐2 infection can also be blocked by retrocyclin, which binds tightly to glycoprotein B2.93 The cyclic peptide lactoferricin seems to compete for the ligation to extracellular heparan sulfate or chondroitin sulfate, thus preventing the very first event in cytomegalovirus, HSV, and papillomavirus infection.94, 95, 96 Finally, elafin is thought to act indirectly in host cells by hampering viral attachment and probably by modulating host's antiviral and inflammatory responses toward HSV‐2 infection.97 Synergistically with this mechanism, antiviral peptides can also bind and inhibit the activation of viral co‐receptors, leading to further inhibition of glycoproteins‐mediated viral attachment. Not only that, HBD‐2 and ‐3 inhibit viral infection by promoting the internalization of its co‐receptor CXCR4.98, 99 Virucidal peptides may also preclude several steps of viral replication by interacting with intracellular targets (Fig. 4). This is the case of HNP‐1 and HD‐5, which may prevent HSV replication by binding directly to its DNA.88 Also, HNP‐1 arrests influenza virus replication by inhibiting protein kinase C phosphorylation, an essential step for nuclear trafficking events.100 Human cathelicidin (LL‐37) derived peptides, in turn, inhibit reverse transcriptase and viral proteases, thus preventing integration of the viral genetic load into human DNA.101, 102
5. RESISTANCE MECHANISMS AGAINST ABPs
The few resistance mechanisms against AMPs demonstrated so far among microorganisms have been observed almost exclusively in bacteria. With very few exceptions, bacteria do not seem to develop complete resistance against cationic ABPs, though some strategies do exist among bacterial species for diminishing their potency and efficacy. The difficulty in acquiring resistance may stem mainly from the fact that ABPs target an essential cellular component for bacteria survival, which cannot be easily modulated by the cell without causing significant adverse effects. Therefore, resistance mechanisms are not likely to be a limitation in the therapeutic utilization of ABPs. In light of this scarce resistance mechanisms de novo acquisition, what mechanisms do bacteria naturally develop/possess against ABPs?
First, one should consider that the primary target of ABPs consists of bacterial cell membranes and bear in mind how ABPs depend on the electrostatic interactions between membrane negative charges and positive peptidic residues. Accordingly, bacteria have learned how to modulate cell membrane charges and composition. Gram‐positive bacteria can partially modify the peptidoglycan's teichoic acid and Gram‐negative bacteria can partly change its lipopolysaccharide lipid A moiety, thus compromising this interaction. For instance, D‐alanine‐activating enzyme and D‐alanine transfer protein are known to transport positively charged D‐alanines to the surface anionic teichoic acids in Gram‐positive S. aureus, reducing the negative net charge of its outer membrane.103 Also in S. aureus, the five‐component system GraXSR‐VraFG increases the expression of mprF and dltABCD genes upon exposure to cationic AMPs. When S. aureus strains are mutated so that the regulation of these genes is disturbed, these strains become more susceptible to daptomycin, polymyxin B, HNP, and platelet factor 4‐derived peptide, and less infectious in vivo in a nonclinical endocarditis model.104, 105, 106 The gene product MprF attaches lysine residues, which bear a positively charged ε‐amino group, to the anionic phophatidylglycerols, making these less negatively charged and thus less susceptible to interactions with cationic AMPs.105, 106
The Gram‐negative Vibrio cholerae can increase its resistance to polymyxins more than 100‐fold by modifying its lipopolysaccharides in a process mediated by AlmF.107 The lptA gene product found in gonococcal species, such as the Gram‐negative bacteria Neisseria gonorrhoeae, protects lipopolysaccharide lipid A moieties by attaching phosphoethanolamine, making these bacteria less susceptible to AMPs and increasing their capacity to modulate host's immune response by evading complement‐mediated cellular death.108, 109 Moreover, in N. gonorrhoeae and Neisseria meningitides, the presence of phosphoryl and phosphoethanolamine in lipid A moieties enhances the activation of the NF‐κB pathway and the production of proinflammatory cytokines in human cells, which can amplify their virulence.110 Despite this modification‐dependent virulence, the absence of such modifications in commensal Neisseriae species allow these to colonize and coexist with the human host without inducing bactericidal host immune responses.111 In addition, the adaptation of the composition and, hence, the electrostatic properties of the membrane in order to circumvent the host immune system is not an exclusive phenomenon of pathogenic bacteria. Commensal bacteria of the intestinal tract can remove phosphate groups from their membrane's lipopolysaccharides, decreasing their negative charge and allowing greater survival and proliferation in the gut.112 Similarly to the above‐mentioned paradoxical modification‐dependent virulence, mutated commensal gut microbiota Bacteroides that fail to remove phosphate groups from their lipopolysaccharides are killed during inflammation while normal strains keep their resilience.112 Therefore, there appears to be an inverse relationship between acquired resistance and bacterial virulence, so that induced/acquired modifications render cells less virulent while simultaneously allowing these to more easily colonize and coexist within the human host environment.
Second, bacteria are also able to adapt the fluidity of the membrane by increasing the hydrophobic interactions within their outer membrane, thus hindering pore formation.113
Third, bacteria can express proteases against ABPs or transporters and efflux pumps like ATP‐binding cassette (ABC) transporters to circumvent such innate defense barriers.114, 115 Glu‐C protease produced by S. aureus induces the loss of antibacterial activity of neuroendocrine peptides, and chromofungin, procatestatin, and human catestatin are enzymatically degraded when treated with bacterial supernatants.116 In E. faecalis, bacitracin binds the ABC transporter EF2050‐2049, which interacts with the regulatory domain of the two‐component system EF0926‐27, increasing its expression and conferring resistance against bacitracin.117 Two‐component systems regulate the expression of ABC transporters and are constituted by a transport permease and a sensor kinase. However, at least in B. subtilis, the ABC transporter itself is required for both sensing of and resistance to bacitracin, suggesting these transporters to also function as environmental sensors.115 Lastly, bacteria may also synthesize molecules capable of directly neutralizing ABPs,118 which can provide a way for bacteria to bypass AMPs. However, such resistance mechanisms are very limited and resistance to AMPs does not take place to a large extent.
6. FAMILIES OF AMPs IN HUMAN BODILY FLUIDS
In humans, endogenous AMPs have been found in several fluids throughout the body, as depicted in Figures 2 and 3 as well as in Table I. As noted, multiple AMPs have been found in tears, saliva, milk, blood, urine, sweat (and others not‐so‐well‐explored biofluids), reflecting not only a remarkable versatility but also a huge conservation that reinforces its essential role. The robust knowledge of AMPs on some fluids (e.g., tears, urine, saliva) over others (e.g., amniotic fluid, cerebrospinal fluid) most likely reflects their availability, but different exposure to pathogens from the environment (Figs. 2 and 3) should also account for these discrepancies to a large extent. Additionally, saliva and urine screening is of special interest, since both the oral cavity and the urinary tract represent the main doorways for microbial invasion and colonization, and are thus enriched with host defense peptides. Likewise, milk screening is of high relevance due to the presence of particular proteins and AMPs that will boost neonate's immunity. Despite underrepresented, AMPs from other fluids are equally relevant (Figs. 2 and 3). For instance, AMPs collected from airway secretions, bronchoalveloar lavages, and nasopharyngeal aspirates can be valuable sources of information regarding the physiological response to pathogens of the respiratory tract and may, themselves, constitute raw models for new therapeutic drugs. Also, other than circulating immune cells derived, blood has not been regarded as a rich source of AMPs, but this view can be questioned (Figs. 2 and 3), most likely due to tissue‐derived AMPs’ passage to the blood circulation. However, it should be noted that human fluid reservoirs are not independent/isolated, and, consequently, peptides may be present throughout and cross between, possibly undergoing modifications between such reservoirs. AMPs may be present in fluids as a result of many different processes, including exocytosis, in situ proteolysis, tissue necrosis, and cellular lysis. Therefore, in this section, we will characterize naturally occurring AMPs based on structural and functional similarities rather than their source of collection.
A. Defensins
Defensins are cationic peptides rich in cysteine residues, presenting as β‐sheet structures stabilized by disulfide bridges.4 According to the alignment of these stabilizing disulfide bounds, mammalian defensins are classified into three subclasses, α‐defensins, β‐defensins, and θ‐defensins.
A.1. α‐Defensins
Disulfide bounds of α‐defensins occur between cysteines 2–6, 1–4, and 3–5. These peptides consist of 29–35 amino acids and adopt a triple‐stranded β‐sheet conformation. Human neutrophils express four distinct α‐defensins, known as HNP‐1 to HNP‐4. Despite having been initially isolated from peripheral blood leukocytes, HNP‐1 to HNP‐3 can also be found in bone marrow, spleen, and thymus.119 The remaining two already identified human α‐defensins, HD‐5 and HD‐6, are found only in Paneth cells of the small intestine and in epithelial cells, thus being tissue specific.120 Expressed in the aforementioned cell types, AMPs can also be found in bodily fluids in direct contact with these cells. Among the most recent discoveries, HD‐6 has been found to be active against Bifidobacterium adolescentis and other anaerobic gut commensals, but not (directly) bacteriostatic or bactericidal against most pathogenic bacteria, and may therefore play an important role in maintaining a balance among the microbiota of the gut.121 Similarly, human HD‐5 is particularly potent against the Gram‐positive bacillus Clostridium difficile, one of the most common pathogens responsible for nosocomial infections, whose highly virulent strains often attack small intestine mucosa.122 In addition, α‐defensins may show both cellular and regional specificity throughout the gastrointestinal tract, either secreted or active inside intracellular granules and constitutively expressed or amenable to induction.15, 123, 124 It should also be emphasized that the activity spectrum of α‐defensins is not limited to bacteria and fungi, exhibiting activity against several virus, including HIV, HSV, Adenovirus, and human papillomavirus (HPV), and displaying several unique mechanisms of action (please refer to Section 4.).4 It should be noted that the antimicrobial activity of α‐defensins is influenced even by the difference in one amino acid, such as for HNP‐1 to HNP‐3. In fact, an additional acidic residue in HNP‐3 lowers its effectiveness (most accurately, its potency) against S. aureus, P. aeruginosa, and E. coli. This is explained by the easily donated proton, which makes this defensin less positively charged (at least when considering interaction domains only), thus reinforcing the importance of a positive net charge for the antimicrobial activity of AMPs,125, 126 as evidenced in Section 4..
In terms of disease, α‐defensins are known to have a paramount role, particularly when the demands for defense mechanisms are increased. Accordingly, a remarkable 15‐ to 25‐fold increase in the concentration of HNP‐1 and HNP‐2 (and HNP‐3 to a smaller extent) in human tears as been reported after ocular surgery, an increase that allowed these peptides to reach the required concentrations for antimicrobial activity but that is followed by a decrease once healing has been completed and the demands for immune defenses have returned to baseline.127 Nonetheless, the goal of achieving this increased α‐defensin concentration in tears is not restricted to the enhancement of the immune defenses, as it is also known to promote local cellular proliferation and tissue repair.128 In parallel, salivary α‐defensins are active against Streptococcus mutans, but their secretion has been found deregulated in caries‐positive human subjects.129 In contrast, prolonged physical exercise has been shown to significantly increase the levels of salivary α‐defensins,130 showing that lifestyles may have a considerable impact on our AMP‐based defenses against microorganisms and it is possible to modulated the activity thereof.
A.2. β‐Defensins
Disulfide bounds of β‐defensins occur between cysteines 1–5, 2–4, and 3–6. These defensins are longer than α‐defensins, spanning from 36 to 50 amino acids, but also share a triple‐stranded β‐sheet conformation. The first hBD was isolated from plasma samples and kidneys constitute the major source of hBD‐1.131 Since then, peptides derived from human hBD‐1 have been identified in tears, urine, vaginal lavage, sweat, and blood plasma, and these are effective primarily against E. coli.131, 132, 133, 134 In turn, hBD‐2 was isolated from psoriatic skin samples,135 displaying bactericidal activity toward Gram‐negative E. coli, P. aeruginosa, and C. albicans, and bacteriostatic activity toward Gram‐positive S. aureus.
Despite very effective in vitro, both hBD‐1 and hBD‐2 present attenuated activities in vivo as a result of their sensitivity to physiological salinity. In contrast, hBD‐3 is active against S. aureus and vancomycin‐resistant E. faecium even at physiological salt concentrations.136
The hBD‐119 defensin has been found in seminal plasma, and it has been reported as potential efficacious against E. coli, S. aureus, and C. albicans.137, 138 Furthermore, hBD‐114 has been identified in epididymal fluid and has been shown to regulate lipopolysaccharide‐mediated inflammation toward E. coli, S. aureus, and C. albicans, protecting sperm from motility loss.139 In disease conditions, increased concentrations of hBDs in plasma and bronchoalveolar lavage fluid were reported in patients with diffuse panbronchiolitis. In particular, β‐defensin 4A was suggested to play a role against P. aeruginosa.140 Also, while β‐defensins are expressed throughout the gastrointestinal tract, these seem to have evolved together with changes in human gut bacteria, displaying characteristic expression patterns in response to certain microorganisms. Their expression may be significantly altered in human diseases, most notably in inflammatory bowel diseases (for insightful reviews on this topic, please refer to16, 141 and enhanced in inflammatory conditions of the ocular surface134).
With α‐defensins, the role of the pH for AMPs’ activity is considered. Here, β‐defensins serve to illustrate the role of salt concentration. In fact, tears contain a remarkable concentration of sodium chloride, which inhibits the activity of human AMPs (by ∼40% in the case of hBD‐2, an inhibition that increases up to 90% in the presence whole tear fluid, suggesting the presence of other inhibiting factors).142 Therefore, whenever there is an increase in the demand of AMPs activity, (e.g., upon infection or tissue damage), their concentrations must surge to reach the required protective levels. However, an excessive increase in the concentration of any AMP may lead to tissue damage and local functions may be compromised. For this reason, a complex interplay of AMPs is required at surfaces like the ocular epithelia, which is achieved by a qualitative enrichment, in addition to the expected quantitative increase (for a depiction of the multitude of AMPs found in human tears, please refer to 143).
Similarly to α‐defensins, the activity spectrum of β‐defensins is not limited to bacteria and fungi, and they are active against several virus, namely, HIV, influenza A virus (IAV), respiratory syncytial virus, and vaccinia virus, and display several unique mechanisms of action, depending on their target (see above section on mechanisms of action).4
Lastly, genomic studies have predicted additional β‐defensins to be expressed in human cells, but their presence in human bodily fluids and their antimicrobial activity in vivo is yet to be demonstrated.144
A.3. θ‐Defensins
While α‐ and β‐defensins adopt triple‐stranded β‐sheet conformations, θ‐defensins have shorter sequences and adopt a complete circular conformation, with stabilizing disulfide bounds between cysteines 1–6, 2–5, and 3–4. The peptide rhesus θ‐defensin 1 is the prototype of this more recently discovered family of AMPs. Like α‐ and β‐defensins, θ‐defensins possess broad‐spectrum microbial activity against bacteria and fungi, while also being capable of protecting mononuclear cells from infection by HIV‐1.145, 146 Moreover, these peptides display antiviral activity against HSV, HIV, IAV, and severe acute respiratory syndrome (SARS) coronavirus, acting by several mechanisms, depending on their target.4 Structurally, θ‐defensin peptides are very interesting and very odd, representing the first cyclic peptide family discovered in animals, albeit not in humans. Moreover, their circular structure appears to be required for antimicrobial activity.145 These peptides were first isolated as antimicrobial octadecapeptides expressed in leukocytes of rhesus monkeys, and their formation results from the head‐to‐tail joining of two independent nine‐amino acid peptides derived from truncated pro‐α‐defensins.146
In contrast to the previously described classes of defensins, human θ‐defensins are thought to be completely inactivated, despite the existence of mRNAs encoding at least two θ‐defensins expressed in the bone marrow.145 While genes encoding θ‐defensins in humans also encode premature stop codons (thus hampering θ‐defensin expression), synthetic human θ‐defensins (called retrocyclins) have been shown as promising antiviral agents.147 In fact, retrocyclin‐1 inhibits the entry of HIV, HSV, and IAV into host cells. In other mammals, retrocyclin‐1 also protects from infection by Bacillus anthracis spores and the rhesus θ‐defensin 1 protects mice from SARS coronavirus infection.147
B. Cathelicidins
Cathelicidins are a very diverse group of cationic α‐helical and amphipathic AMPs that exhibit a broad‐spectrum activity against bacteria, fungi, and virus.54 The term “cathelicidin” originally referred only to the entire precursor protein, though currently is commonly used for the whole family, including the resulting peptides with antimicrobial activity.
In contrast to the mammalian trend in which cathelicidins and defensins are the main AMPs, there is only one human gene encoding multiple cathelicidin‐related peptides,144, 148 located on chromosome 3 and expressed in the airways, mouth, tongue, esophagus, epididymis, and small intestine.149, 150 Despite this tissue expression pattern, LL‐37 is constitutively formed in spleen, liver, stomach, intestine, and bone marrow. LL‐37 is highly positively charged (+6 charge at physiological pH 7.4), due to the high content of arginine and lysine amino acids, and adopts an α‐helical structure in solutions with ionic composition similar to that of human plasma.151
While defensins share common structural features, cathelicidin‐related peptides are highly heterogeneous despite deriving from the same precursor. Cathelicidins are characterized by a highly conserved N‐terminal signal peptide (the so‐called “cathelin domain”) and a highly variable C‐terminal antimicrobial domain that can be released after cleavage by proteinases.148, 152 Cathelicidin is cleaved into the antimicrobial peptide LL‐37 by both kallikrein 5 and kallikrein 7 serine proteases, it is upregulated by vitamin D, and has been shown to significantly reduce the risk of death from infection dialysis patients.153 LL‐37 was shown to disrupt bacterial membranes through the formation of toroidal pores and carpet structures,63 and to exhibit chemotactic properties, attracting leukocytes and activating secretion of chemokines. In addition, LL‐37 is internalized by cells, acidified in endosomes, and activates the signaling pathway downstream to Toll‐like receptor 3 by interacting with double‐stranded RNA.154
Profiling of airway fluids collected from premature infants’ tracheal aspirates during infection evidenced the production of LL‐37 even at an early stage of development.155 In these samples, LL‐37 was found in significantly increased concentrations during pulmonary or systemic infections, suggesting this peptide to be as important to avoid infectious processes as to fight already established infections.155
The concentration of LL‐37 has been measured in seminal plasma samples from healthy donors and found to be up to 70‐fold higher than in blood plasma.149 Furthermore, it was found to be attached to spermatozoa, eliciting a possible role in human fertilization or its precursor to be extensively cleaved by the high concentration of serine proteases present in seminal plasma.149
LL‐38, an alternative form of human cathelicidin peptides, contains one more alanine at the N‐terminus than LL‐37 and was initially found to be produced in female vaginal secretions due to the action of gastricsin on sperm precursor cathelicidin.156 Both LL‐37 and LL‐38 are active against bacteria such as E. coli, S. aureus, P. aeruginosa, and Bacillus megaterium, and synergistically with peptides from seminal plasma play an essential defense role protecting the human reproductive system.156, 157 Interestingly, both citrullination and ADP (Adenosine diphosphate)‐ribosylation of arginines can compromise the ability of LL‐37 to prevent endotoxin‐induced sepsis, perhaps because the attachment of these negatively charged molecules reduces the peptide's cationicity or by raising steric hindrance.158, 159
C. Histatins
Histatins are a normal component of human saliva and part of the innate immune system, protecting against a broad array of infectious agents, especially against Candida species, Saccharomyces cerevisiae, Cryptococcus neoformans, and Neurospora crassa.160, 161, 162 There are only two genes enconding histatins (both located on chromosome 4), and they are exclusively expressed in human salivary glands.163 However, other active AMPs resulting from the cleavage of histatins 1 and 3 also exist.164 Histatins 1 and 3 are encoded by genes htn1 and htn3, respectively.165 Histatins 2, 4, and 5–12 are the products of posttranslational proteolytic cleavage of histatins 1 and 3, though histatin 5 can also result from posttranscriptional modification of histatin 3 mRNA.165, 166
Peptides derived from histatin 1 bear distinct domains associated with specific functions, namely an N‐terminal domain with antimicrobial activity and a C‐terminal domain with wound‐healing properties. 167, 168 Histatin 5 has two important metal‐binding motifs, consisting of an amino‐terminal copper (II)/nickel (II)‐binding motif and a zinc (II)‐binding motif, although they can also bind to cobalt (II) ions.169, 170 The ability to coordinate metal ions appears to be important for the stabilization of the secondary structure, particularly in the presence of negatively charged membranes.170 Nevertheless, histatins lack any defined secondary structure and are unordered when in aqueous solutions, assuming α‐helical structures only in organic solutions.171, 172 In addition, histatin 5 binding to copper (II) and nickel (II) ions induces the formation of ROS, which contributes for its fungicidal activity.80, 169
Histatins can bind to a receptor on fungal cell membranes and induce cell death by disrupting the cell cycle and causing nonlytic loss of ATP, as discussed above.173 While histatins are effective, potent, and fast‐acting AMPs against several antimicrobial species, these peptides are nontoxic to human cells and human microflora, which may be due to the fact that their translocation across the membrane is site and species specific.78, 174
Both parotid, submandibular, and sublingual glands secret histatins, and their concentration and secretion have been shown to decrease with age, even when accounted for total protein concentration.175 Moreover, oral candidal infections increase with age, suggesting a link between age‐associated deficiency of human histatins and an increased risk of oral infections.175
D. Dermcidins
Dermcidins are broad‐spectrum anionic AMPs with no apparent homology to other known AMPs. Peptides in this group comprise 110 amino acid residues, which are subsequently processed into smaller but still active ones. Dermcidins are constitutively expressed in human eccrine sweat glands and secreted in sweat, exhibiting antimicrobial activity against common human pathogenic microorganisms such as S. aureus, Staphylococcus epidermidis, E. coli, E. faecalis, and C. albicans.176, 177, 178 This constitutive production contrasts with human defensins and cathelicidins, which are induced during inflammatory and stressful conditions.
A small percentage of human breast cancer cells displays RNA encoding dermcidin, and after oxidative stress induction different types of tumor cells leads to the production of diverse and biologically active proteolytically processed dermcidin peptides.179, 180 Therefore, the upregulation of dermicidins may confer human breast cancer cells a selective advantage compared to nonmalignant cells. In contrast, reduced expression of dermcidins in sweat from atopic dermatitis patients contributes to skin infections and altered skin colonization.181 Hence, the downregulation of dermicidins in atopic dermatitis patients may represents a suppression of an innate and fundamental defense mechanism, which, in turn, could confer an advantage to infectious microorganisms.
Unlike histatins, which are nontoxic to human cells and human microflora (see above), dermicidins can have serious deleterious consequences to the human host. Dermcidin isoform 2 is known as a stress‐induced oxidative protein capable of inhibiting the synthesis of nitric oxide in endothelial cells, insulin in pancreatic islet β‐cells, and hepatic hormone synthesis.182 Also, hypertensive patients show significantly higher levels of plasma dermcidin, but salicylic acid has been shown to reduce dermcidin levels while simultaneously attenuating dermcidin‐mediated insulin inhibition in patients with acute myocardial infarction.183 Therefore, the exploitation of dermcidin isoform 2 for antimicrobial therapeutic purposes is most likely not possible due to severe side effects. Despite this, it may be a promising therapeutic target for hypertension and diabetes management.
In animal models, dermcidin has been shown to induce platelet aggregation and coronary artery disease by activating platelet cyclooxygenase and inhibiting the constitutive form of nitric oxide synthase, respectively, with an efficiency 40 times higher than that of ADP at activating cyclooxygenase.182 Notably, dermcidin was able to stimulate the development of coronary artery disease in one animal model, even at suboptimal concentrations of ADP within 30 min.183
Together, these observations suggest that the ability of AMPs to fight infections depends on their induction by infectious microorganisms, the particular peptides generated, and the local environment/tissue in which they act. Also, biological roles performed by human AMPs are more diverse than immediately expected, playing essential functions other than fighting infectious microorganism(s). In fact, dermcidin‐mediated inhibition of insulin is unique in the sense that no other protein is known to arrest pancreatic insulin synthesis in such way, and similar observations might result in novel and unexpected applications of human AMPs.
E. Hepcidins
AMPs expressed by human liver (LEAP 1 and 2) were initially discovered in human blood, and subsequently found in urine. These peptides are known as hepcidins and are characterized by a high content in cysteine residues (approximately 32% of all residues, with eight disulfide bounds).184
LEAP‐1 or hepcidin‐25 is essential for maintaining iron homeostasis, and, when mutated, leads to severe iron overload and juvenile hemochromatosis.185 Hepcidins are the main regulators of plasma iron concentration and its secretion is promoted during inflammatory responses, particularly by the action of interleukin 6, which is one of the main mediators of the acute phase response.186 As iron bioavailability is a limiting factor for bacterial growth, hepcidins represent an organic mechanism for sequestering iron from invasive pathogens. However, when several iron sources are available, as it happens within the human host, eliminating a single reservoir may not be sufficient to attenuate microorganisms’ virulence. Moreover, bacteria are known to exploit almost every host iron‐binding protein and reservoir and, consequently, human LAEPs/hepcidins are a rather limited source of antimicrobial activity.187 For these reasons, the antimicrobial role of iron sequestration depends not only on hepcidins, but also on several other mediators, such as transferrin, lactoferrin, ceruloplasmin, haptoglobin, and hemopexin.
Mutations responsible for decreasing the levels of hepcidins or compromising the function of other regulators of iron homeostasis, such as the major histocompatibility complex class I like hereditary hemochromatosis protein (HFE), transferrin receptor 2, and ferroportin, lead to increased iron blood levels. When levels are high enough to produce hemochromatosis, individuals are at an increased risk of infectious complications, including bacteremia and meningitis caused by E. coli,188 bacteremic cellulitis, and hemorrhagic bullous skin lesions caused by V. cholera,189 multiple pyogenic abscesses in the liver parenchyma as a result of Yersinia enterocolitica infection,190 meningitis, endocarditis, and pericarditis resulting from infection with L. monocytogenes,191 and fatal septicemia during infection with Vibrio vulnificus.192
In contrast, hemochromatosis can also be associated with decreased macrophage iron storages in the case of HFE‐associated hemochromatosis. This depletion makes these cells more efficient in fighting Salmonella enterica subsp. enterica, serovar Typhimurium, and Mycobacterium tuberculosis infections.193, 194 However, these protective effects of macrophages iron storage depletion have only been demonstrated in animal models and appear to be mediated not by hepcidins, but rather by lipocalin‐2, which reduces the availability of iron for Salmonella, and transferrin and lactoferrin, both of which compromise iron acquisition by M. tuberculosis.193, 194
F. Neuroendocrine AMPs
Neuropeptides and neuroendocrine peptides may exert mitogenic actions through which innate barriers are reinforced and may display neurotransmitter, immunoregulatory, and direct antimicrobial activity.
Vasostatin‐1 was the first natural antifungal N‐terminal chromogranin A derived fragment peptide, having been first isolated from chromaffin secretory granules from bovine adrenal medulla.195 However, its sequence is highly conserved in humans (97% identity homology) and its microbial activity also overlaps, to some extent, with that of bovine origin.196 Irrespectively, human vasostatin‐1 displays antimicrobial activities against Gram‐positive bacteria (Micrococcus luteus, B. megaterium) and a large variety of filamentous fungi (N. crassa, Aspergillus fumigatus, Alternaria brassicola, Nectria haematococca, Fusarium culmorum, Fusarium oxysporum, Trichophyton mentagrophytes) and yeast cells (S. cerevisiae, C. albicans).195 Moreover, many autocrine and paracrine biological activities of chromogranin A, its precursor protein, have been attributed to peptides located along its sequence, which are co‐released with catecholamines by chromaffin cells upon stimulation and can be found in human bodily fluids.197, 198 Therefore, the antimicrobial activity spectrum of vasostatin‐1 may be extended by that of co‐released peptides also formed by chromogranin A proteolytic cleavage, and some of the coproduced peptides are even more potent than vasostatin‐1 itself.195
In addition to the structural requirements for the antimicrobial activity of chromogranin A derived peptides, posttranslational modifications may also modulate their activity. For instance, S‐pyridylethylation (an alkylating modification consisting of the addition of pyridylethyl moieties to the –SH functional groups of cysteine residues that disrupts disulfide bridges) renders it selectively active against B. megaterium but not M. luteus, while oxidation seems to render it inactive against bacteria but still active against fungi. In addition, chromogranin A derived peptides also undergo phosphorylation and glycosylation, which can modulate their physicochemical properties. 195
Currently, vasostatin‐1 is known to be stored in endocrine, neuroendocrine, and neuronal cells, and to be released from stimulated chromaffin and immune cells upon stress. Moreover, the stimulation of polymorphonuclear immune cells induces the processing of chromogranin A and the secretion of vasostatin‐1 and other chromogranin A derived peptides,195 which may account for local inflammation. Lastly, recombinant and synthetic human vasostatin‐derived fragments have been shown to inhibit vascular contraction induced by endothelin‐1, parathyroid hormonal secretion, neuronal survival, expression of neurofilaments, and neuronal GABA uptake.199, 200
Adrenomedullin was first isolated from a human adrenal pheochromocytoma and its plasma concentration is raised under specific physiological conditions, namely, renal failure,201 hypertension,202 and sepsis.203 This peptide has antibacterial activity against several Gram‐positive and Gram‐negative bacteria, including Propionibacterium acnes, S. aureus, M. luteus, P. gingivalis, Actinomyces naeslundii, S. mutans, C. albicans, Eikenella corrodens, Actinobacillus actinomycetemcomitans, Streptococcus pneumoniae, Haemophilus influenzae, Streptococcus pyogenes, Bacteroides fragilis, E. coli, and Helicobacter pylori.204
Human neuropeptide Y displays antimicrobial activity against bacteria and fungi, but truncated fragments are tenfold more potent than the intact precursor neuropeptide Y, perhaps because the net charge of the fragments is more positive than that of the intact neuropeptide.205
Encephalins and their derived peptides may either enhance or inhibit the immune response and the corresponding circulating immune cells/mediators in both humans and animal models.206, 207 as well as their subsequent antimicrobial effects, may thus take place indirectly in immune system modulation. Simultaneously, antibacterial assays have revealed that peptide B/enkelytin (a C‐terminal fragment of proenkephalin A) specifically targets Gram‐positive bacteria, including M. luteus, B. megaterium, M. luteus, and S. aureus, while having no effect over Gram‐negative bacteria.208, 209 Classically, proenkephalin A has been ascribed to the secretory granules from adrenal medullary chromaffin cells and various brain regions, such as the striatum and the pituitary, according to some animal studies.210, 211 Despite this, it has also been demonstrated to be expressed in and secreted by polymorphonuclear neutrophils (accounting for its particular enrichment in wound fluids),208 T and B lymphocytes, macrophages, and mast cells.212, 213 However, it should be noted that the antimicrobial effects of encephalins and their derived peptides result mostly from animal studies and have not been adequately studied in human secretions, despite the high conservation of their sequences across species, which most likely contribute for the similar activity spectrum. Even so, phosphorylation and glycosylation are characteristic of these peptides,208, 214 and it is possible to envision that differences at the level of posttranslational modifications or as a result of gene divergence may result in distinct activity spectra. Interestingly, peptide B/enkelytin is metabolized in vivo to opioid peptides, thus revealing an intricate relationship between innate immunity and pain modulation, and its plasma concentration is increased after coronary artery bypass grafting in humans.215 Because it is expressed in both immune and neuronal cells, it is possible that the same stimuli may act on both cells populations, thereby accounting for immune system modulation at the periphery and alterations in central nervous system signaling. However, the exact roles and mechanisms of this interplay are not clear.
In addition, other AMPs found in epithelial secretions from the gut and the skin, for example, include peptides derived from alpha‐melanocyte‐stimulating hormone, which is active against S. aureus and C. albicans.216 Accordingly, evidences suggest that AMPs like these act by inhibiting mRNA and protein synthesis rather than inducing direct membrane disruption, as is the case for most AMPs.217
G. Nonantimicrobial Proteins Give Rise to Bioactive AMPs
Several cationic proteins display nonenzymatic antimicrobial activity, which is sustained by the resulting cleavage peptides after protein fragmentation. For example, lactoferrin is a glycoprotein enriched in milk and neutrophilic granules, which has the ability to sequester iron and thus prevent bacterial overgrowth.218 When digested by pepsin, lactoferrin yields lactoferricin H (human), an AMP able to neutralize bacterial toxins, regulate gene transcription, inhibit complement activation, viral infection, and tumor overgrowth, thus further extending the antimicrobial properties of its precursor.76
Human chemokines are secreted by macrophages and polymorphonuclear immune cells and, therefore, found in the blood circulation.219 Moreover, during allergic inflammatory responses to bacterial infections of the airways, the eosinophil‐recruiting chemokines eotaxin‐1/CCL11, eotaxin‐2/CCL24, and eotaxin‐3/CCL26 are generated by mast cell proteases, active against several airway pathogens, including S. pneumoniae, S. aureus, H. aeruginosa.220
Peptides resulting from the cleavage of pepsinogen A and C prosequences have also been shown to exert antimicrobial activity. Although these were initially identified from the stomach of bullfrogs, synthesized human pepsinogen C prosequences with similar structural characteristics (amphipathic and helical structures) to these cleavage peptides have also exhibited antimicrobial activity.221
Calcitermin is an AMP that results from the cleavage of a 15 amino acids long sequence from the C‐terminal region of protein calgranulin C, isolated from human airway secretions.222 By adopting an alpha‐helical conformation in bacterial membranes, calcitermin is able to target Gram‐negative bacteria (e.g., E. coli and P. aeruginosa) and fungi (e.g., C. albicans).222 Furthermore, the precursor protein and fragment peptide may act synergistically to protect the human host against foreign microorganisms present in the airways. On the one hand, the cleavage peptide calcitermin inserts into membranes of microorganisms. On the other hand, the precursor protein calgranulin C has a proinflammatory activity. By acting as a danger‐associated molecular pattern molecule and binding to the receptor for advanced glycation end products in innate immune cells, it activates the MAP‐kinase and NF‐κB signaling pathways, leading to the production of proinflammatory cytokines and histamine, upregulation of cell adhesion molecules, recruitment of leukocytes, and degranulation of granulocytes.223, 224
Dermcidin‐derived peptides such as DCDs, SSLs, and LEKs (the letters correspond to the first three amino acids of the corresponding precursor dermcidin protein) are also formed in eccrine sweat glands (but not in apocrine sweat glands) at these body sites with high probability for contact with pathogens (e.g., palms, face, arms) and exhibit antimicrobial activity against a large number of microorganisms, including S. aureus, E. faecalis, E. coli, S. epidermidis, Pseudomonas putida, methicillin‐resistant S. aureus, and rifampicin/isoniazid‐resistant M. tuberculosis.176, 177, 178 Such dermcidin‐derived AMPs are regulated by proteolytic processing at several levels (see Section 2.) and are differentially expressed during inflammatory conditions. 225 These peptides are resistant to proteolytic degradation in sweat up to at least 40 hr and, interestingly, most dermcidin‐derived peptides are anionic in nature. If such distinct properties may lead to different antimicrobial activity spectrum is unclear, though not surprising, as skin represents a harsh and particularly unique environment. As of specific interest, it should be noted that the consensual average skin surface pH has been decreasing in the last few years and is now generally accepted to be below 5. While showering and cosmetic products decrease skin's surface pH, plain tap water can increase the skin pH up to 6 hr.226 Noticeably, negative charges seem to be important for dermcidin‐derived peptides’ antimicrobial activity, in contrast with what would be expected for AMPs. In fact, most dermcidin‐derived peptides are naturally anionic, but low pH (high H+ concentration) leads to an increase in their net charge (toward positivity). Therefore, while an acidic skin actually keeps the resident bacterial flora attached to it, a more alkaline skin compromises such attachment, suggesting that the more negatively charged such skin peptides are (alkaline skin), the more efficient these become.226
H. Large Antimicrobial Human Peptides
Despite the average length of 32.76 residues and net charge of +3.21 for the close to 3000 AMPs already identified, human AMPs tend to be longer and more positively charged. AMPs with more than 100 amino acids have been identified across the six kingdoms of life. Curiously, most (approximately half) appear to be of human origin, presenting as amino acid sequences of 127 residues on average and suggesting large AMPs to be a human feature. Moreover, compared to all the remaining AMPs, large human AMPs tend to be biased toward leucine (8.2%) and lysine (10.5%) residues, displaying nonpolar aliphatic and positively charged side groups, respectively (Table I). Therefore, large human AMPs are enriched with amino acids that promote distinguishing features of AMPs: high overall positive charge, hydrophobicity domains, and amphiphilicity. Nevertheless, this bias toward lysine residues is neither unique to human AMPs nor more pronounced among these. For instance, the amphibian cathelicidin RC‐1 is composed by 32% lysine residues, and its homologous snake crotalicidin contains 38% lysine residues, showing high potency against S. pyogenes, Acinetobacter baumannii, E. faecalis, S. aureus, E. coli, Klebsiella pneumoniae, and P. aeruginosa.227
As evidenced in Table I, large human AMPs have been found in a variety of bodily fluids, including tears, saliva, urine, airways secretions, breast milk, blood, sweat, and epididymal fluids. In addition, other large AMPs are also expected to exist despite not constitutively found in biologic fluids, due to their inducible nature. For instance, Paneth cells in the small intestine release AMPs‐rich granules upon stimulation by cholinergic or bacterial stimuli,228 and granulocytes are known to contain AMPs in granules destined for extracellular secretion.229, 230 Therefore, as the synthesis and secretion of large AMPs may not be constitutive but amenable to induction by microbial macromolecules and inflammatory cytokine stimuli, several other large AMPs may yet to be catalogued. Hence, exploration of novel large antimicrobial human peptides is an ongoing work that may yield novel therapeutic agents.
7. PEPTIDOMICS OF NATIVE PEPTIDES IN HUMAN BODILY FLUIDS
Peptidomics is the complete study or global analysis of the peptides present in a biological sample, at a given time, under a particular condition. 231, 232 The identification of naturally occurring AMPs in human biological fluids is a laborious, daunting, and sometimes unsuccessful task. Such naturally occurring peptides can directly result from gene encoding, from passage to bodily fluids as a result of cellular damage and renewal, or from extracellular proteolytic processing of precursor proteins. The latter, considered to be the most prevalent, hampers the identification and interpretation of biologically active AMPs because precursor proteins may themselves display antimicrobial activity, but may also be cleaved into multiple functional and nonfunctional peptides.233, 234 Moreover, owing to the large dependency of AMPs on their sequence and 3D structure, these peptides should not be subject to mass spectrometry (MS) based approaches employing enzymatic or chemical digestion, neither to any in vitro step that may cause their fragmentation. Instead, top‐down proteomic strategies are preferred. Classically, different peptides fractions have been separated and tested for their biological activity, often without ever addressing the peptides’ biological/antimicrobial activity, their sequence, target, or hypothetical mechanism(s) of action. However, current strategies are more focused, precise, and informative, capable of addressing all these parameters, sometimes in a single study (see below).129, 235, 236
Human biological fluids consist of complex samples, containing lipids, salts, proteins, carbohydrates, and other molecules that make the study of endogenous peptides extremely difficult. Consequently, enrichment and separation steps are almost always required. Of these biomolecules, proteins and larger peptides as well as salts are the most complicated to separate from the target peptides. Luckily, salts can sometimes be removed in the same procedures applied for protein removal. First and foremost, proteases/peptidases have to be inactivated, which can be done by denaturing procedures (e.g., microwaving, boiling) or by adding protease/peptidase inhibitors.237, 238 However, both of these can modify the peptide structure and, thus, these steps can be avoided when samples are resistant enough to further protease/peptidase activity. This is the case for urine samples, for example, as, when collected, have long been subject to proteolytic processing in the bladder. Proteins are frequently removed by selective precipitation, most commonly by organic solvent (e.g., acetone) or acidic (e.g., trichloroacetic acid) precipitation, which also removes salts (left in the supernatant).236, 239 However, precipitation‐based proteins removal is not complete and may lead to the formation of peptide aggregates, which are consequently lost in the precipitate.240
Since peptides can present a great variety of sizes, sequences, structures, hydrophobicity properties, net charges, and other characteristics, their isolation is complex. Still, such complexity allows for isolation steps to take place at multiple dimensions, increasing peptides isolation efficiency and accuracy. For instance, peptides can first be isolated based on their size and charge,241, 242 bias toward specific amino acids (e.g., cysteines, tryptophans, methionines),243, 244, 245 or on the presence of post‐translational modifications (PTMs) (e.g., phosphopeptides, glycopeptides).246, 247 Still, because of the aforementioned limitations, the search for endogenous AMPs in human bodily fluids has mostly relied on the identification of candidate AMPs by chromatographic techniques (e.g., high‐performance liquid chromatography, HPLC) and sequencing (e.g., Edman sequencing), followed by their de novo chemical synthesis and in vitro antimicrobial activity testing. For these reasons, the identification and in vivo testing of novel AMPs is often time consuming and requires multiple studies/phases and resources. In order to mitigate this problem, candidate human AMPs have come to be first predicted in multiple ways, including mining of the entire human genome for particular motifs known to confer antimicrobial activity, and screening of human peptidomes (e.g., salivary, urinary) in order to find homologous peptides to other known AMPs. Accordingly, several libraries and databases are currently available for data comparison, and some search programs can even combine both previously obtained data and de novo information.248
Even so, the identification of AMPs benefits the most from combinations of chromatographic and immunological techniques, genomics and proteomics. While genomics and proteomics both allow the prediction of novel AMPs and have the potential for contributing with a larger number of putative AMPs, peptides with proven in vivo antimicrobial activity have to be more accurately identified by combinations of chromatographic and immunological techniques, chemical sequencing and antimicrobial assays. Though immunoassays can detect the intended target in a variety of complex biological samples, these techniques do not provide an integrative view of the peptidome, can suffer from cross‐reactivity or lack of specificity issues and require previous knowledge of the target structure.249, 250 Moreover, this issue becomes a particular problem when considering that shorter or longer peptides resulting from the same precursor protein may all contain the same epitopes and thus be recognized as the same peptide fragment despite exhibiting distinct biological activities.
Electrospray ionization (ESI)236 and matrix‐assisted laser desorption ionization (MALDI)251 have been the most frequently and successfully applied ionization techniques in the field of bodily fluids peptidomics. In line with these, peptides are most frequently previously separated by LC‐based251 and capillary electrophoresis (CE) based approaches, allowing different peptide fractions to be independently separated.252 From this point onwards, almost any mass analyzer can be used, although in practice quadrupole time‐of‐flight (Q‐TOF) and ion traps (IT) instruments are the most frequently utilized ones in peptidomics, especially in exploratory studies.253, 254, 255 In turn, Fourier transform‐ion cyclotron resonance‐MS is more suitable for targeted/confirmatory analysis when dealing with fewer peptides.256 Because these apparatuses allow tandem MS to be performed, peptides can be readily sequenced in addition to their identification, circumventing the above‐mentioned limitations concerning immunological assays and largely simplifying peptidomics workflows.
In summary, when aiming at the isolation of a particular peptide, with predicted or known charge and molecular weight, an MS‐based procedure can theoretically be very straightforward. First, fluid samples require (sometimes multiple) centrifugation steps to remove cells, cellular debris, wastes, and possible contaminants. Then, proteins and peptides in the supernatant are separated and fractionated by multidimensional chromatographic techniques. Within these procedures, proteins and peptides can be initially fractionated by gel filtration (size dimension), followed by cation‐exchange (charge dimension) fractionation. Cation‐exchange chromatography uses a negatively charged ion exchange resin with affinity for molecules with net positive charge. A salt gradient is used to separate the peptides of interest from other bound peptides, based on their predicted isoelectric point. Finally, peptides can be subject to MS analysis. They are typically ionized by ESI or MALDI and subsequently analyzed in a Q‐TOF, IT, or hybrid LTQ. As AMPs tend to be highly basic peptides due to a bias toward arginine and lysine residues, these have a propensity to be particularly enriched when applying cation exchange chromatography based techniques.257 However, because biological fluids are complex matrices, these sometimes require multiple fractionation, isolation, and extraction steps. These are laborious optimization techniques for the detection and quantification of AMPs and, despite their inherent complexity, they are often based in not always accurate functional and structural predictions.
A recent approach has proved itself capable for the isolation of peptides present in bodily fluids that can bind directly to certain bacterial membrane components, such as lipopolysaccharide, and induce an inflammatory response against these.129, 235 After obtaining a bacterial homogenate and extracting membrane components with an organic solvent such as n‐butanol, functionalized beads with N‐hydroxysuccinimidyl‐sepharose (an agarose used in affinity and protein chromatography) are conjugated with these membrane bacterial components and incubated with a human biological fluid (e.g., saliva, urine). Then, peptides bound to the conjugated beads are eluted and analyzed in a LTQ‐Orbitrap hybrid Fourier transform apparatus. Moreover, functionalized beads conjugated with bacterial membrane components can be tested in vivo for their capacity to induce the production of inflammatory mediators. Thus, this exploratory approach could result in the identification of potential human AMPs with selective properties capable of binding bacterial membranes and inducing an inflammatory response.
Recently, Dallas and colleagues have attained the most complete and prolific analysis of naturally occurring peptides with potential antimicrobial activity in human milk by nano‐LC quadrupole‐TOF tandem mass spectrometry (MS/MS), while searching against libraries of human milk peptides and known AMPs.236 In order to remove milk peptides produced by in vitro proteolysis, fresh milk derived peptides were compared to control samples of the same origin but immediately subject to boiling. As boiling denatures proteases, in vitro proteolysis does not take place and peptides present in these controls could then be confidently assigned in the original analysis. Moreover, masses fragmented in the first round of MS/MS were excluded from subsequent rounds of MS.236 The authors realized that more than 80% of the +300 unique naturally occurring peptides identified with 99% confidence have yet unknown functions, which serves to illustrate how much there is still to discover regarding the biological roles of human endogenous peptides in biological fluids. Nevertheless, the antimicrobial activity of the milk peptides (41 predicted AMPs) was confirmed in vivo by radial diffusion assays against the Gram‐negative E. coli and the Gram‐positive S. aureus.236 Furthermore, this study reinforced the observation that the background against which mass spectra are searched seems to be a more critical factor than the mass analyzer or the ionization technique used, when it comes to both the number of peptides identified and the confidence by which these can be assigned.
In the same study, authors have emphasized and demonstrated the importance of avoiding in vitro hydrolysis of AMPs in human bodily fluids, which can be attained by adding protease inhibitors after sample collection or by adequately accounting for this phenomenon, as the authors did by using an appropriate control. In contrast, not all bodily fluids seem to be susceptible to this degree of hydrolysis, at least not to the same extent. Urine, for instance, is thought to be more stable and not to require treatment with protease inhibitors.258 However, by the time urine is collected it has already remained stagnant for considerably longer periods of time, and, hence, probably already extensively exposed to the action of proteases/peptidases inherently present in urine. Therefore, how intact urinary AMPs actually are should always be questionable and these should ideally be interpreted considering the background of hydrolytic enzymes present in the same urine sample.
Also questionable is the inhibition of proteolysis in seminal plasma samples. In fact, the liquefaction process of human semen performed by seminin and other proteolytic enzymes is physiological,259 and, therefore, naturally occurring AMPs should be analyzed in samples that were allowed to liquefy without the addition of protease inhibitors. Curiously, seminin, which is the primary agent responsible for liquefaction of the seminal coagulum, is partially destroyed by freezing at −20°C,259 reinforcing the vitality of analyzing fresh samples. Similarly, the degradation of peptide fragments derived from semenogelins in seminal plasma samples is time dependent and responsible for the decline in HIV‐enhancing activity of sperm, once again underscoring the importance of timely performing analyses of peptides in bodily fluids.260
As previously stated, AMPs activity depends largely on their 3D structures, and their correct characterization becomes of paramount importance. The best experimental techniques have been and currently are X‐ray crystallography 261 and nuclear magnetic resonance,262 both of which allow for the study of structure–function relationships, though very expensive. Therefore, currently, these are commonly replaced or complemented by bioinformatics prediction tools whenever possible. Using such tools, the sequence of amino acids of a peptide allows for the accurate and immediate prediction of the presence of secondary structures (e.g., α‐helices, β‐sheets). Then, the overall 3D structure is devised by algorithms employing energy minimization principles and molecular dynamics, sometimes resulting in multiple and not mutually exclusive 3D conformations, which is in accordance with peptides dynamic structure in biological fluids.263, 264 The most common identification software referenced in the literature for peptide/precursor protein identification are SEQUEST and MASCOT.265, 266 Currently, however, there is a considerable need for algorithms and software capable of correctly identifying AMPs and predicting their structure–function relationship.
8. APPLICATION OF AMPs BEYOND CONVENTIONAL PHARMACOLOGICAL THERAPY
AMPs are a promising replacement for conventional antibiotics owing to their effectiveness against multiresistant bacteria and over multiple bacterial species simultaneously, invoking a diverse set of mechanisms that cannot be easily shortcut by bacteria, fungi, and/or virus. An ideal AMP would be (i) highly selective against its target while leaving human host cells unaffected, (ii) not prone to resistance mechanisms, (iii) relatively easy to produce at low costs, and (iv) stable during storage or upon administration. Drug design of such AMPs focuses on peptidic and nonpeptidic mimetic drugs, modulating the hydrophobicity, positivity, and amphiphilicity of endogenous AMPs to increase their antimicrobial potency. However, exploiting AMPs for therapeutic purposes is actually more complex than initially envisioned. While linear AMPs present a simpler design and a more predictable structure–function relationship, circular peptides may allow the design of longer acting and more resistant AMPs. Such peptides have been designed by inserting the intended AMP into other circular peptide (cyclotides) and have been proven efficacious against HIV and inflammatory diseases by circumventing their instability and poor bioavailability.22, 267 Similarly, while humans produce AMPs with L‐amino acids and their antimicrobial activity may depend on the action of endogenous enzymes, which can only act upon L‐type amino acids, producing AMPs with D‐amino acids may render these more resistant to hydrolysis.
Rather than administering exogenous AMPs, it is possible and perhaps more secure and controllable to pharmacologically stimulate the production of endogenous AMPs. For instance, 1,25‐dihydroxycalcypherol (active vitamin D3) has been successfully used to induce the expression of DEFB4 (defensin, beta 4) and CAMP (cationic antimicrobial peptide) genes, the production of both LL‐37 and human β‐defensin 2 in cell cultures of keratinocytes from diabetic foot ulcers and to promote wound healing.268 Accordingly, the persistence of functional milk human κ‐caseinoglycopeptides in plasma of human infants after human milk feeding serves the purpose of exogenously administering AMPs without the requirement for their de novo synthesis.269
AMPs can help preventing biofilm formation and microorganisms’ growth. That is the case of LL‐37 and its fragments, which were already shown to have antibiofilm formation properties against several methicillin‐resistant S. aureus strains and P. aeruginosa, common pathogens of hospital‐acquired infections, as well as against the melioidoisis’ etiological agent Burkholderia pseudomallei and the uropathogenic E. coli.270, 271, 272, 273 Another AMP with antibiofilm formation activity is hBD‐3, found to prevent the development of methicillin‐resistant S. epidermidis and S. aureus biofilms, largely responsible for orthopedic implants associated infections.274 Likewise, AMPs may also inhibit the formation of fungal biofilms. For instance, histatin 5 has been demonstrated to successfully inhibit the growth of C. albicans biofilm on denture acrylic.275 These examples may broaden the applications of AMPs in the clinical and biomedical fields by allowing functionalization of catheters. Alternatively, implants and other medical devices can also reduce bacterial colonization, biofilm formation, and infection rates, presenting long lasting functionality, broad‐spectrum activity, and minimal cytotoxicity against human cells.276, 277
Human AMPs could, theoretically, also be used as biosensors immobilized onto microchips for identification purposes, allowing the identification of microorganisms. However, such applications have only been exploited with animal or synthetic AMPs. These biosensors on electrical impedance alterations resulting from the presence of bacterial or fungal surface elements, or on the specificity of peptide nucleic acid probes against their targets, presenting higher versatility, lower costs, and lower sensitivity limits compared to other conventional methodologies.278, 279, 280 Alternatively, AMPs could be used as drug delivery systems in the form of conjugates in order to accurately target drugs and other agents to tumor sites or intended organs, as typified by monodisperse “endosomolytic” nanoparticles for in vivo gene delivery using intravenous injection, or by functionalized nanoparticles with higher membrane penetration targeted against gliomas.281, 282 Such functionalization may allow for deeper tissue penetration, increased antimicrobial potency, sustained release, and novel routes of administration to be achieved.
9. DISCLOSERS AND CHALLENGES TO OVERCOME
Ever since the isolation of the first peptide from the frog skin in 1983,283 several breakthroughs were made in the isolation, synthesis, and application of AMPs. Still, many challenges are yet to overcome in the field of AMPs peptidomics,284 reflecting mainly their huge diversity and how little is known about their behavior in vivo and their structure–function relationships.
In terms of costs, producing AMPs can be several hundred times more expensive than the production of conventional antibiotics.285 Moreover, AMPs’ design can be very complex and the quest for the perfect AMP a never‐ending search. In fact, considering the possibility of working with only 20 amino acids, a small peptide with ten amino acids could have up to 2010 = 1.02 × 1013 different sequences, a larger peptide with 20 amino acids could present with up to 2020 = 1.05 × 1026 different sequences, and one with 100 amino acids could adopt any one of 20100 = 1.27 × 10130 different sequences. Such possibilities do not even account for the presence of posttranslational modifications. Then, each one of these peptides could shape into several different secondary structures and fall into a wide spectrum of activity, depending on the milieu conditions. Furthermore, in a way to overcome the challenge of peptide stability, one has to consider the possibility to synthesize and to administer AMPs containing D‐amino acids, thus decreasing their susceptibility to hydrolysis by human enzymes.267 To deal with such large amount of amino acid combinations one should mind using peptide libraries and computational models (such as by quantitative structure–activity relationship analysis) in order to narrow large collections to few surrogate AMPs (for a review please see Blondelle and Lohner's paper.286
Another challenge to overcome is the lower AMP's activity in vivo when compared to that observed in vitro. This has largely hampered the development of AMPs as therapeutic agents, as this decrease in activity stems from differences in pH and salt concentrations, the presence of proteases and corresponding inhibitors, and other interacting molecules hindering antimicrobial activity. Accordingly, several AMPs (e.g., Pexiganan,287 Iseganan,288 Neuprex 289 have reached phase II clinical trials only to fail after approval for marketing because they did not evidence superior activity over already marketed conventional antibiotics.
In early 2016, clinicaltrials.gov (a registry and result database of publicly and privately supported clinical studies of human participants conducted around the world) listed few clinical trials involving AMPs. For instance, LL37 is currently being studied for its efficacy in melanoma patients due to its ability to stimulate the immune system, while C16G2 and chromogranin A derived peptides are under scrutiny for their therapeutic potential against dental diseases. Simultaneously, therapeutic strategies are also being studied to augment AMPs’ expression in AMP‐deficient patients. This deficiency is believed to be due to an increase in Th2 lymphocytes derived cytokines, IL‐4, IL‐13, and IL‐10, and a decrease in TNF‐α, IL‐6, IL‐1, and interferon‐γ. One example of such strategy is Pimecrolimus, a calcineurin inhibitor that binds with high affinity to macrophilin‐12, preventing the translocation of nuclear factor of activated T cells to the nucleus and the transcription and release of such inhibitory cytokines (for more up‐to‐date trials, readers can access clinicaltrials.gov).
Another possible caveat concerns the cytotoxicity to mammalian cells when in concentrations above naturally occurring values. When bacteria produce AMPs, they also develop means to avoid their targeting by such peptides, such as efflux pumps and the sequestering of enzymes. However, eukaryotic cells are equally prone to damage if the administration of exogenous AMPs is carried out in high enough concentrations. Therefore, administering AMPs not constitutively produced or at concentrations above those normally found in the human host environment can be toxic and very detrimental to human cells. Furthermore, rapid metabolism and low bioavailability are characteristic of AMPs because these peptides can suffer extensive proteolysis in vivo, thereby accounting for their inactivation and short half‐life.22, 28, 290 Similarly, for peptides to be available at sites distant from their origin, these would have to cross many cellular membranes. However, due to AMPs’ polarity, membrane hydrophobicity may prevent them from doing so.291 Moreover, in contrast to other highly polar mediators (e.g., ionic salts and a few metabolites), endogenous peptides tend not to have a transporter, further compromising their bioavailability in different compartments and the subsequent impossibility to be absorbed through the gastrointestinal tract.
Therefore, the number of possibilities rapidly escalades, and each AMP‐based therapy can be further optimized based on the modulation of chemical properties and engineering of delivery systems. Even acknowledging all these factors, the human host is most certainly an unpredictable environment and the same peptide may behave differently, displaying unique individual pharmacokinetics and pharmacodynamics. On a final note, due to AMPs’ interaction with and dependence on other immune mediators, variability will most likely result from immune status alterations as those frequently found in human diseases, including infectious ones.
Taken together, the above discussed results support AMPs as constituting a paramount group of defense molecules in human biological fluids, being present in most fluids, contributing for the sterility of some of these, but being most notably enriched in biological fluids bathing tissues with the largest load of microorganisms (Fig. 2), such as saliva and urine. Also, while some AMPs seem the display activity spectra against only a handful of microorganisms, others display broad‐spectrum effects. Thus, the expression and activity patterns of human AMPs enriched in biofluids may have suffered selective pressures resulting from exposers to microorganisms specifically present in each fluid (Fig. 2).
Supporting information
Supplementary Table S1: Distribution of Known Antimicrobial Peptides Listed in The Antimicrobial Peptides Database by Activity Spectrum (2016)
Supplementary Table S2: Distribution of Known Antimicrobial Peptides Listed in The Antimicrobial Peptides Database by Structural Information (2016)
ACKNOWLEDGMENTS
This work was financed by national funding from FCT (Fundação para a Ciência e a Tecnologia) through the project UID/BIM/04501/2013, UID/IC/00051/2013. RV thanks FCT for IF/00286/2015 fellowship. The authors report no declarations of interest.
Biographies
Paulo Bastos is a biomedicine graduate student currently enrolled in a Master's program at the Biomedical Research Foundation, Academy of Athens, working on mass spectrometry assays development. He is also still performing research at iBiMED, University of Aveiro, on human diseases and biomarker research. His research background includes the study of host‐microorganism interactions at the Institut Pasteur in Paris. PB is soon joining the Institute of Science and Technology, Klosterneuburg, Austria.
Fábio Trindade has graduated in Biochemistry (2012) and finished his master degree in Clinical Biochemistry in 2014 at the Department of Chemistry of the University of Aveiro. He has done research at the Mass Spectrometry Center in Aveiro mainly focusing in salivary peptidomics and salivary antimicrobial peptides both in health and in chronic periodontitis. Now he is a PhD student in Biomedicine and he is developing research in urine and pericardial fluid proteomics and myocardial reverse remodeling in both iBiMED (Department of Medical Sciences, University of Aveiro) and UnIC (Faculty of Medicine, University of Porto). His main interests are to understand the molecular basis of human diseases, biomarker research and identification of therapeutic targets.
João Pinto da Costa graduated in Biotechnological Engineering at the University of the Algarve (Portugal) in 2005. Soon thereafter, he successfully concluded his Master+s Degree in Medical Diagnostics, awarded by the Cranfield University (UK). During 2006, he was a visiting scientist at Exelixis, Inc. (USA), a development‐stage biotechnology company focusing on the development of novel small molecule therapeutics for the treatment of cancer. From 2007 to 2009, he worked as a research fellow at Cranfield and Aveiro Universities in different fields of research, including mass spectrometry, and applied mycology. In April 2013, he concluded his Ph.D. thesis in Environmental Chemistry. Since then, João has worked in multiple research projects as either a Post‐Doctoral or research fellow at the University of Porto and the University of Aveiro. As of May 2015, João is a member of the Analytical Chemistry Group at the Chemistry Department of the University of Aveiro, where he is currently developing efforts on the monitoring environmental contaminants in real time by autonomous biosensor platform (NFETs). Furthermore, he frequently cooperates with iBiMED research members on proteomics studies focusing on a wide array of topics, ranging from post‐translational protein modifications to fundamental aspects of human ageing.
Rita Ferreira has a degree and PhD in Biochemistry. She is an assistant Professor in the Chemistry Department of the University of Aveiro, where she teaches curricular units from the Bachelor's and Master's degrees in Biochemistry. Rita Ferreira has developed extensive research work in the field of muscle plasticity in pathophysiological conditions like cancer and works in the application of proteomics for the identification of the biological processes underlying muscle plasticity in pathophysiological conditions.
Rui Vitorino has a degree and PhD in Biochemistry. He is Assistant Professor in the Medical Sciences Department of the University of Aveiro. Rui Vitorino started his research work in 2000 with saliva proteomics research. Since then, he extended the application of proteomics to the characterization of other biofluids' proteome and peptidome in order to identify the biological pathways modulated by diseases such as diabetes mellitus and bladder cancer. Currently, he works to a large extent with bioinformatics tools for the identification of enzymes involved in the regulation of proteome remodeling.
Contract grant sponsor: FCT (Fundação para a Ciência e a Tecnologia); Contract grant numbers: UID/BIM/04501/2013 and UID/IC/00051/2013.
REFERENCES
- 1. Wang G, Li X, Wang Z. APD3: The antimicrobial peptide database as a tool for research and education. Nucleic Acids Res 2015;44(D1):D1087–93. gkv1278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Baker MA, Maloy WL, Zasloff M, Jacob LS. Anticancer efficacy of Magainin2 and analogue peptides. Cancer Res 1993;53:3052–3057. [PubMed] [Google Scholar]
- 3. Fehlbaum P, Bulet P, Chernysh S, Briand JP, Roussel JP, Letellier L, Hetru C, Hoffmann JA. Structure‐activity analysis of thanatin, a 21‐residue inducible insect defense peptide with sequence homology to frog skin antimicrobial peptides. Proc Natl Acad Sci USA 1996;93:1221–1225. doi: 10.1073/pnas.93.3.1221 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Gwyer Findlay E, Currie SM, Davidson DJ. Cationic host defence peptides: Potential as antiviral therapeutics. BioDrugs 2013;27(5):479–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Kieffer AE, Goumon Y, Ruh O, Chasserot‐Golaz S, Nullans G, Gasnier C, Aunis D, Metz‐Boutigue MH. The N‐ and C‐terminal fragments of ubiquitin are important for the antimicrobial activities. FASEB J 2003;17:776–778. doi: 10.1096/fj.02-0699fje [DOI] [PubMed] [Google Scholar]
- 6. Hancock RE. Cationic peptides: Effectors in innate immunity and novel antimicrobials. Lancet Infect Dis 2001;1(3):156–64. [DOI] [PubMed] [Google Scholar]
- 7. Gschwandtner M, Zhong S, Tschachler A, Mlitz V, Karner S, Elbe‐Bürger A, Mildner M. Fetal human keratinocytes produce large amounts of antimicrobial peptides: Involvement of histone‐methylation processes. J Invest Dermatol 2014;134:2192–2201. doi: 10.1038/jid.2014.165 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Lan F, Bayliss PE, Rinn JL, Whetstine JR, Wang JK, Chen S, Iwase S, Alpatov R, Issaeva I, Canaani E, Roberts TM, Chang HY, Shi Y. A histone H3 lysine 27 demethylase regulates animal posterior development. Nature 2007;449:689–694. doi: 10.1038/nature06192 [DOI] [PubMed] [Google Scholar]
- 9. Ohtani K, Zhao C, Dobreva G, Manavski Y, Kluge B, Braun T, Rieger MA, Zeiher AM, Dimmeler S. Jmjd3 controls mesodermal and cardiovascular differentiation of embryonic stem cells. Circ Res 2013;113:856–862. doi: 10.1161/CIRCRESAHA.113.302035 [DOI] [PubMed] [Google Scholar]
- 10. Pereira HA, Ruan X, Gonzalez ML, Tsyshevskaya‐Hoover I, Chodosh J. Modulation of corneal epithelial cell functions by the neutrophil‐derived inflammatory mediator CAP37. Investig Opthalmol Vis Sci 2004;45:4284. doi: 10.1167/iovs.03-1052 [DOI] [PubMed] [Google Scholar]
- 11. Sosne G, Chan CC, Thai K, Kennedy M, Szliter EA, Hazlett LD, Kleinman HK. Thymosin beta 4 promotes corneal wound healing and modulates inflammatory mediators in vivo. Exp Eye Res 2001;72:605–608. doi: 10.1006/exer.2000.0985 [DOI] [PubMed] [Google Scholar]
- 12. McKown RL, Frazier C, V. E, Zadrozny KK, Deleault AM, Raab RW, Ryan DS, Sia RK, Lee JK, Laurie GW. A cleavage‐potentiated fragment of tear lacritin is bactericidal. J Biol Chem 2014:289:22172–22182. doi: 10.1074/jbc.M114.570143 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Hancock REW, Diamond G. The role of cationic antimicrobial peptides in innate host defences. Trends Microbiol 2000;8(9):402–10. [DOI] [PubMed] [Google Scholar]
- 14. Hilchie AL, Wuerth K, Hancock REW. Immune modulation by multifaceted cationic host defense (antimicrobial) peptides. Nat Chem Biol 2013;9:761–768. doi: 10.1038/nchembio.1393 [DOI] [PubMed] [Google Scholar]
- 15. Lai Y, Gallo RL. AMPed up immunity: How antimicrobial peptides have multiple roles in immune defense. Trends Immunol 2009;30(3):131–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Ostaff MJ, Stange EF, Wehkamp J. Antimicrobial peptides and gut microbiota in homeostasis and pathology. EMBO Mol Med 2013;5:1465–1483. doi: 10.1002/emmm.201201773 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Iversen L. Neuropeptides: Regulators of physiological processes. Trends Neurosci 1999;22:482. doi: 10.1016/S0166-2236(99)01429-0 [DOI] [Google Scholar]
- 18. Jung LJ, Scheller RH. Peptide processing and targeting in the neuronal secretory pathway. Science 1991;251(80):1330–1335. [DOI] [PubMed] [Google Scholar]
- 19. Miao V, Coeffet‐LeGal MF, Brian P, Brost R, Penn J, Whiting A, Martin S, Ford R, Parr I, Bouchard M, Silva CJ, Wrigley SK, Baltz RH. Daptomycin biosynthesis in Streptomyces roseosporus: Cloning and analysis of the gene cluster and revision of peptide stereochemistry. Microbiology 2005;151:1507–1523. doi: 10.1099/mic.0.27757-0 [DOI] [PubMed] [Google Scholar]
- 20. Salomón Ra, Farías RN. Microcin 25, a novel antimicrobial peptide produced by Escherichia coli . J Bacteriol 1992;174:7428–7435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Yang SC, Lin CH, Sung CT, Fang JY. Antibacterial activities of bacteriocins: Application in foods and pharmaceuticals. Front Microbiol 2014;26(5):241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Aboye TL, Ha H, Majumder S, Christ F, Debyser Z, Shekhtman A, Neamati N, Camarero JA. Design of a novel cyclotide‐based CXCR4 antagonist with anti‐human immunodeficiency virus (HIV)‐1 activity. J Med Chem 2012;55:10729–10734. doi: 10.1021/jm301468k [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Fräki JE. Human skin proteases. Separation and characterization of two acid proteases resembling cathepsin B1 and cathepsin D and of an inhibitor of cathepsin B1. Arch Dermatol Res 1976;255:317–330. [DOI] [PubMed] [Google Scholar]
- 24. Fräki JE, Jansén CT, Hopsu‐Havu VK. Human sweat kallikrein. Biochemical demonstration and chromatographic separation from several other esteropeptidases in the sweat. Acta Dermatol Venereol 1970;50:321–326. [PubMed] [Google Scholar]
- 25. Fröhlich E, Schaumburg‐Lever G, Klessen C. Immunocytochemical and immunoelectron microscopic demonstration of cathepsin B in human malignant melanoma. Br J Dermatol 1995;132:867–875. [DOI] [PubMed] [Google Scholar]
- 26. Hibino T, Takemura T, Sato K. Human eccrine sweat contains tissue kallikrein and kininase II. J Invest Dermatol 1994;102:214–220. [DOI] [PubMed] [Google Scholar]
- 27. Komatsu N, Takata M, Otsuki N, Toyama T, Ohka R, Takehara K, Saijoh K. Expression and localization of tissue kallikrein mRNAs in human epidermis and appendages. J Invest Dermatol 2003;121:542–549. doi: 10.1046/j.1523-1747.2003.12363.x [DOI] [PubMed] [Google Scholar]
- 28. Sieprawska‐Lupa M, Mydel P, Krawczyk K, Wójcik K, Puklo M, Lupa B, Suder P, Silberring J, Reed M, Pohl J, Shafer W, McAleese F, Foster T, Travis J, Potempa J. Degradation of human antimicrobial peptide LL‐37 by Staphylococcus aureus‐derived proteinases. Antimicrob Agents Chemother 2004;48:4673–4679. doi: 10.1128/AAC.48.12.4673-4679.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Zeeuwen PL, Van Vlijmen‐Willems IM, Jansen BJ, Sotiropoulou G, Curfs JH, Meis JF, Janssen JJ, Van Ruissen F, Schalkwijk J. Cystatin M/E expression is restricted to differentiated epidermal keratinocytes and sweat glands: A new skin‐specific proteinase inhibitor that is a target for cross‐linking by transglutaminase. J Invest Dermatol 2001;116:693–701. doi: 10.1046/j.1523-1747.2001.01309.x [DOI] [PubMed] [Google Scholar]
- 30. Bennett HP, McMartin C. Peptide hormones and their analogues: Distribution, clearance from the circulation, and inactivation in vivo. Pharmacol Rev 1978;30:247–292. [PubMed] [Google Scholar]
- 31. Thornberry NA, Weber AE. Discovery of JANUVIA (Sitagliptin), a selective dipeptidyl peptidase IV inhibitor for the treatment of type 2 diabetes. Curr Top Med Chem 2007;7:557–568. doi: 10.2174/156802607780091028 [DOI] [PubMed] [Google Scholar]
- 32. Wang G. Human antimicrobial peptides and proteins. Pharmaceuticals (Basel) 2014;7:545–594. doi: 10.3390/ph7050545 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Van'T Hof W, Veerman ECI, Heimerhorst EJ, Nieuw Amerongen AV. Antimicrobial peptides: Properties and applicability. Biol Chem 2001;382(4):597–619. [DOI] [PubMed] [Google Scholar]
- 34. Wang G. Structures of human host defense cathelicidin LL‐37 and its smallest antimicrobial peptide KR‐12 in lipid micelles. J Biol Chem 2008;283:32637–32643. doi: 10.1074/jbc.M805533200 [DOI] [PubMed] [Google Scholar]
- 35. Hoover DM, Chertov O, Lubkowski J. The structure of human beta‐defensin‐1: New insights into structural properties of beta‐defensins. J Biol Chem 2001;276:39021–39026. doi: 10.1074/jbc.M103830200 [DOI] [PubMed] [Google Scholar]
- 36. Lee DG, Kim HK, Kim SA, Park Y, Park SC, Jang SH, Hahm KS. Fungicidal effect of indolicidin and its interaction with phospholipid membranes. Biochem Biophys Res Commun 2003;305:305–310. doi: 10.1016/S0006-291X(03)00755-1 [DOI] [PubMed] [Google Scholar]
- 37. Pirovano W, Heringa J. Protein secondary structure prediction. Methods Mol Biol 2010;609:327–348. [DOI] [PubMed] [Google Scholar]
- 38. Tossi A, Sandri L, Giangaspero A. Amphipathic, alpha‐helical antimicrobial peptides. Biopolymers 2000;55:4–30. doi: [DOI] [PubMed] [Google Scholar]
- 39. Park CB, Yi KS, Matsuzaki K, Kim MS, Kim SC. Structure‐activity analysis of buforin II, a histone H2A‐derived antimicrobial peptide: The proline hinge is responsible for the cell‐penetrating ability of buforin II. Proc Natl Acad Sci USA 2000;97:8245–8250. doi: 10.1073/pnas.150518097 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Brogden KA. Antimicrobial peptides: Pore formers or metabolic inhibitors in bacteria? Nat Rev Microbiol 2005;3:238–250. doi: 10.1038/nrmicro1098 [DOI] [PubMed] [Google Scholar]
- 41. Bellamy W, Takase M, Yamauchi K, Wakabayashi H, Kawase K, Tomita M. Identification of the bactericidal domain of lactoferrin. Biochim Biophys Acta 1992;1121:130–136. doi: 10.1016/0167-4838(92)90346-F [DOI] [PubMed] [Google Scholar]
- 42. Klüver E, Schulz‐Maronde S, Scheid S, Meyer B, Forssmann W‐G, Adermann K. Structure‐activity relation of human beta‐defensin 3: Influence of disulfide bonds and cysteine substitution on antimicrobial activity and cytotoxicity. Biochemistry 2005;44:9804–9816. doi: 10.1021/bi050272k [DOI] [PubMed] [Google Scholar]
- 43. Schroeder BO, Wu Z, Nuding S, Groscurth S, Marcinowski M, Beisner J, Buchner J, Schaller M, Stange EF, Wehkamp J. Reduction of disulphide bonds unmasks potent antimicrobial activity of human β‐defensin 1. Nature 2011;469:419–423. doi: 10.1038/nature09674 [DOI] [PubMed] [Google Scholar]
- 44. Tsai H, Bobek LA. Human salivary histatins: Promising anti‐fungal therapeutic agents. Crit Rev Oral Biol Med 1998;9:480–497. doi: 10.1177/10454411980090040601 [DOI] [PubMed] [Google Scholar]
- 45. Cézard C, Silva‐pires V, Mullié C, Sonnet P. Antibacterial peptides: A review. Formatex 2011; 02:926–937. [Google Scholar]
- 46. Adzhubei AA, Sternberg MJE, Makarov AA. Polyproline‐II helix in proteins: Structure and function. J Mol Biol 2013;425(12):2100–32. [DOI] [PubMed] [Google Scholar]
- 47. Dathe M, Nikolenko H, Meyer J, Beyermann M, Bienert M. Optimization of the antimicrobial activity of magainin peptides by modification of charge. FEBS Lett 2001;501:146–150. doi: 10.1016/S0014-5793(01)02648-5 [DOI] [PubMed] [Google Scholar]
- 48. Tareq FS, Lee MA, Lee HS, Lee YJ, Lee JS, Hasan CM, Islam MT, Shin HJ. Gageotetrins A‐C, noncytotoxic antimicrobial linear lipopeptides from a marine bacterium Bacillus subtilis . Org Lett 2014;16:928–931. doi: 10.1021/ol403657r [DOI] [PubMed] [Google Scholar]
- 49. Chopra L, Singh G, Choudhary V, Sahoo DK. Sonorensin: An antimicrobial peptide, belonging to the heterocycloanthracin subfamily of bacteriocins, from a new marine isolate, Bacillus sonorensis MT93. Appl Environ Microbiol 2014;80:2981–2990. doi: 10.1128/AEM.04259-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Niggemann J, Bozko P, Bruns N, Wodtke A, Gieseler MT, Thomas K, Jahns C, Nimtz M, Reupke I, Bruser T, Auling G, Malek N, Kalesse M. Baceridin, a cyclic hexapeptide from an epiphytic bacillus strain, inhibits the proteasome. Chembiochem 2014;15:1021–1029. doi: 10.1002/cbic.201300778 [DOI] [PubMed] [Google Scholar]
- 51. Essig A, Hofmann D, Münch D, Gayathri S, Künzler M, Kallio PT, Sahl HG, Wider G, Schneider T, Aebi M. Copsin, a novel peptide‐based fungal antibiotic interfering with the peptidoglycan synthesis. J Biol Chem 2014;289:34953–34964. doi: 10.1074/jbc.M114.599878 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Jenssen H, Hamill P, Hancock REW. Peptide antimicrobial agents. Clin Microbiol Rev 2006;19(3):491–511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Hancock REW, Sahl HG. Antimicrobial and host‐defense peptides as new anti‐infective therapeutic strategies. Nat Biotechnol 2006;24:1551–1557. doi: 10.1038/nbt1267 [DOI] [PubMed] [Google Scholar]
- 54. Zasloff M. Antimicrobial peptides of multicellular organisms. Nature 2002;415:389–395. doi: 10.1038/415389a [DOI] [PubMed] [Google Scholar]
- 55. Melo MN, Castanho MARB. The mechanism of action of antimicrobial peptides: Lipid vesicles vs. bacteria. Front Immunol 2012;3:236. doi: 10.3389/fimmu.2012.00236 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Oren Z, Shai Y. Mode of action of linear amphipathic α‐helical antimicrobial peptides. Pept Sci 1998;47:451–463. doi: [DOI] [PubMed] [Google Scholar]
- 57. Papo N, Shai Y. Exploring peptide membrane interaction using surface plasmon resonance: Differentiation between pore formation versus membrane disruption by lytic peptides. Biochemistry 2003;42:458–466. doi: 10.1021/bi0267846 [DOI] [PubMed] [Google Scholar]
- 58. Yang L, Harroun TA, Weiss TM, Ding L, Huang HW. Barrel‐stave model or toroidal model? A case study on melittin pores. Biophys J 2001;81:1475–1485. doi: 10.1016/S0006-3495(01)75802-X [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Epand RF, Maloy WL, Ramamoorthy A, Epand RM. Probing the “charge cluster mechanism” in amphipathic helical cationic antimicrobial peptides. Biochemistry 2010;49:4076–4084. doi: 10.1021/bi100378m [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Bechinger B, Lohner K. Detergent‐like actions of linear amphipathic cationic antimicrobial peptides. Biochim Biophys Acta 2006;1758:1529–1539. doi: 10.1016/j.bbamem.2006.07.001 [DOI] [PubMed] [Google Scholar]
- 61. Gazit E, Boman A, Boman HGG, Shai Y. Interaction of the mammalian antibacterial peptide cecropin P1 with phospholipid vesicles. Biochemistry 1995;34:11479–11488. [DOI] [PubMed] [Google Scholar]
- 62. Shai Y. Mode of action of membrane active antimicrobial peptides. Pept Sci 2002;66:236–248. doi: 10.1002/bip.10260 [DOI] [PubMed] [Google Scholar]
- 63. Henzler Wildman KA, Lee DK, Ramamoorthy A. Mechanism of lipid bilayer disruption by the human antimicrobial peptide, LL‐37. Biochemistry 2003;42:6545–6558. doi: 10.1021/bi0273563 [DOI] [PubMed] [Google Scholar]
- 64. Ludtke SJ, He K, Heller WT, Harroun TA, Yang L, Huang HW. Membrane pores induced by magainin. Biochemistry 1996;35:13723–13728. doi: 10.1021/bi9620621 [DOI] [PubMed] [Google Scholar]
- 65. Matsuzaki K, Murase O, Fujii N, Miyajima K. Translocation of a channel‐forming antimicrobial peptide, magainin 2, across lipid bilayers by forming a pore. Biochemistry 1995;34:6521–6526. [DOI] [PubMed] [Google Scholar]
- 66. Zweytick D, Japelj B, Mileykovskaya E, Zorko M, Dowhan W, Blondelle SE, Riedl S, Jerala R, Lohner K. N‐acylated peptides derived from human lactoferricin perturb organization of cardiolipin and phosphatidylethanolamine in cell membranes and induce defects in Escherichia coli cell division. PLoS One 2014;9(3):e90228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Deol SS, Domene C, Bond PJ, Sansom MSP. Anionic phospholipid interactions with the potassium channel KcsA: Simulation studies. Biophys J 2006;90;822–830. doi: 10.1529/biophysj.105.071407 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Peschel A, Sahl HG. The co‐evolution of host cationic antimicrobial peptides and microbial resistance. Nat Rev Microbiol 2006;4:529–536. doi: 10.1038/nrmicro1441 [DOI] [PubMed] [Google Scholar]
- 69. Yeaman MR, Yount NY. Mechanisms of antimicrobial peptide action and resistance. Pharmacol Rev 2003;55:27–55. doi: 10.1124/pr.55.1.2 [DOI] [PubMed] [Google Scholar]
- 70. Ulvatne H, Samuelsen Ø, Haukland HH, Krämer M, Vorland LH. Lactoferricin B inhibits bacterial macromolecular synthesis in Escherichia coli and Bacillus subtilis . FEMS Microbiol Lett 2004;237:377–384. doi: 10.1016/j.femsle.2004.07.001 [DOI] [PubMed] [Google Scholar]
- 71. Su LY, Willner DL, Segall AM. An antimicrobial peptide that targets DNA repair intermediates in vitro inhibits Salmonella growth within murine macrophages. Antimicrob Agents Chemother 2010;54:1888–1899. doi: 10.1128/AAC.01610-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Pappu RV, Rose GD. A simple model for polyproline II structure in unfolded states of alanine‐based peptides. Protein Sci 2002;11:2437–2455. doi: 10.1110/Ps.0217402 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Scocchi M, Tossi A, Gennaro R. Proline‐rich antimicrobial peptides: Converging to a non‐lytic mechanism of action. Cell Mol Life Sci 2011;68(13):2317–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Shi Z, Chen K, Lim Z, Sosnick TR, Kallenbach NR. PII structure in the model peptides for unfolded proteins: Studies on ubiquitin fragments and several alanine‐rich peptides containing QQQ, SSS, FFF, and VVV. Proteins 2006;63:312–321. doi: 10.1002/prot.20788 [DOI] [PubMed] [Google Scholar]
- 75. Sass V, Schneider T, Wilmes M, Korner C, Tossi A, Novikova N, Shamova O, Sahl HG. Human beta‐defensin 3 inhibits cell wall biosynthesis in Staphylococci. Infect Immun 2010;78:2793–2800. doi: 10.1128/IAI.00688-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Gifford JL, Hunter HN, Vogel HJ. Lactoferricin: A lactoferrin‐derived peptide with antimicrobial, antiviral, antitumor and immunological properties. Cell Mol Life Sci 2005;62(22):2588–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Ramamoorthy A, Thennarasu S, Tan A, Gottipati K, Sreekumar S, Heyl DL, An FYP, Shelburne CE. Deletion of all cysteines in tachyplesin I abolishes hemolytic activity and retains antimicrobial activity and lipopolysaccharide selective binding. Biochemistry 2006;45:6529–6540. doi: 10.1021/bi052629q [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Edgerton M, Koshlukova SE, Lo TE, Chrzan BG, Straubinger RM, Raj PA. Candidacidal activity of salivary histatins: Identification of a histatin 5‐binding protein on Candida albicans . J Biol Chem 1998;273:20438–20447. doi: 10.1074/jbc.273.32.20438 [DOI] [PubMed] [Google Scholar]
- 79. Sun JN, Li W, Jang WS, Nayyar N, Sutton MD, Edgerton M. Uptake of the antifungal cationic peptide histatin 5 by Candida albicans Ssa2p requires binding to non‐conventional sites within the ATPase domain. Mol Microbiol 2008;70:1246–1260. doi: 10.1111/j.1365-2958.2008.06480.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Helmerhorst EJ, Troxler RF, Oppenheim FG. The human salivary peptide histatin 5 exerts its antifungal activity through the formation of reactive oxygen species. Proc Natl Acad Sci USA 2001;98:14637–14642. doi: 10.1073/pnas.141366998 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Koshlukova SE, Lloyd TL, Araujo MWB, Edgerton M. Salivary histatin 5 induces non‐lytic release of ATP from Candida albicans leading to cell death. J Biol Chem 1999;274:18872–18879. doi: 10.1074/jbc.274.27.18872 [DOI] [PubMed] [Google Scholar]
- 82. Baev D, Rivetta A, Vylkova S, Sun JN, Zeng GF, Slayman CL, Edgerton M. The TRK1 potassium transporter is the critical effector for killing of Candida albicans by the cationic protein, histatin 5. J Biol Chem 2004;279:55060–55072. doi: 10.1074/jbc.M411031200 [DOI] [PubMed] [Google Scholar]
- 83. Gusman H, Travis J, Helmerhorst EJ, Potempa J, Troxler RF, Oppenheim FG. Salivary histatin 5 is an inhibitor of both host and bacterial enzymes implicated in periodontal disease. Infect Immun 2001;69:1402–1408. doi: 10.1128/IAI.69.3.1402-1408.2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Gusman H, Grogan J, Kagan HM, Troxler RF, Oppenheim FG. Salivary histatin 5 is a potent competitive inhibitor of the cysteine proteinase clostripain. FEBS Lett 2001a;489:97–100. doi: 10.1016/S0014-5793(01)02077-4 [DOI] [PubMed] [Google Scholar]
- 85. Epand RM, Epand RF. Bacterial membrane lipids in the action of antimicrobial agents. J Pept Sci 2011;17(5):298–305. [DOI] [PubMed] [Google Scholar]
- 86. Lee‐Huang S, Maiorov V, Huang PL, Ng A, Hee CL, Chang YT, Kallenbach N, Huang PL, Chen HC. Structural and functional modeling of human lysozyme reveals a unique nonapeptide, HL9, with anti‐HIV activity. Biochemistry 2005;44:4648–4655. doi: 10.1021/bi0477081 [DOI] [PubMed] [Google Scholar]
- 87. Gordon YJ, Romanowski EG, Shanks RMQ, Yates KA, Hinsley H, Pereira HA. CAP37‐derived antimicrobial peptides have in vitro antiviral activity against adenovirus and herpes simplex virus type 1. Curr Eye Res 2009;34:241–249. doi: 10.1080/02713680802714066 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Hazrati E, Galen B, Lu W, Wang W, Ouyang Y, Keller MJ, Lehrer RI, Herold BC. Human alpha‐ and beta‐defensins block multiple steps in herpes simplex virus infection. J Immunol 2006;177:8658–8666. doi:https://doi.org/177/12/8658 [pii] [DOI] [PubMed] [Google Scholar]
- 89. Wu Z, Cocchi F, Gentles D, Ericksen, B , Lubkowski J, Devico A, Lehrer RI, Lu W. Human neutrophil alpha‐defensin 4 inhibits HIV‐1 infection in vitro. FEBS Lett 2005;579:162–166. doi: 10.1016/j.febslet.2004.11.062 [DOI] [PubMed] [Google Scholar]
- 90. Cole AL, Yang OO, Warren AD, Waring AJ, Lehrer RI, Cole AM. HIV‐1 adapts to a retrocyclin with cationic amino acid substitutions that reduce fusion efficiency of gp41. J Immunol 2006;176:6900–6905. doi:https://doi.org/176/11/6900 [pii] [DOI] [PubMed] [Google Scholar]
- 91. Gallo SA, Wang W, Rawat SS, Jung G, Waring AJ, Cole AM, Lu H, Yan X, Daly NL, Craik DJ, Jiang S, Lehrer RI, Blumenthal R. Theta‐defensins prevent HIV‐1 Env‐mediated fusion by binding gp41 and blocking 6‐helix bundle formation. J Biol Chem 2006;281:18787–18792. doi: 10.1074/jbc.M602422200 [DOI] [PubMed] [Google Scholar]
- 92. Münch J, Ständker L, Adermann K, Schulz A, Schindler M, Chinnadurai R, Pöhlmann S, Chaipan C, Biet T, Peters T, Meyer B, Wilhelm D, Lu H, Jing W, Jiang S, Forssmann W‐G, Kirchhoff F. Discovery and optimization of a natural HIV‐1 entry inhibitor targeting the gp41 fusion peptide. Cell 2007;129:263–275. doi: 10.1016/j.cell.2007.02.042 [DOI] [PubMed] [Google Scholar]
- 93. Yasin B, Wang W, Pang M, Cheshenko N, Hong T, Waring AJ, Herold BC, Wagar EA, Lehrer RI. Theta defensins protect cells from infection by herpes simplex virus by inhibiting viral adhesion and entry. J Virol 2004;78:5147–5156. doi: 10.1128/JVI.78.10.5147 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Andersen JH, Osbakk SA, Vorland LH, Traavik T, Gutteberg TJ. Lactoferrin and cyclic lactoferricin inhibit the entry of human cytomegalovirus into human fibroblasts. Antiviral Res 2001;51:141–149. doi: 10.1016/S0166-3542(01)00146-2 [DOI] [PubMed] [Google Scholar]
- 95. Jenssen H, Andersen JH, Uhlin‐Hansen L, Gutteberg TJ, Rekdal Ø. Anti‐HSV activity of lactoferricin analogues is only partly related to their affinity for heparan sulfate. Antiviral Res 2004;61:101–109. doi: 10.1016/j.antiviral.2003.09.001 [DOI] [PubMed] [Google Scholar]
- 96. Mistry N, Drobni P, Naslund J, Sunkari VG, Jenssen H, Evander M. The anti‐papillomavirus activity of human and bovine lactoferricin. Antiviral Res 2007;75:258–265. doi: 10.1016/j.antiviral.2007.03.012 [DOI] [PubMed] [Google Scholar]
- 97. Drannik AG, Nag K, Sallenave J‐M, Rosenthal KL. Antiviral activity of trappin‐2 and elafin in vitro and in vivo against genital herpes. J Virol 2013;87:7526–7538. doi: 10.1128/JVI.02243-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Feng Z, Dubyak GR, Lederman MM, Weinberg A. Cutting edge: Human beta defensin 3—A novel antagonist of the HIV‐1 coreceptor CXCR4. J Immunol 2006;177:782–786. doi: 10.4049/jimmunol.177.2.782 [DOI] [PubMed] [Google Scholar]
- 99. Quiñones‐Mateu M, Lederman M, Feng Z, Chakraborty B, Weber J, Rangel H, Marotta M, Mirza M, Jiang B, Kiser P, Medvik K, Sieg S, Weinberg A. Human epithelial beta‐defensins 2 and 3 inhibit HIV‐1 replication. AIDS 2003;17:F39–F48. doi: 10.1097/01.aids.0000096878.73209.4f [DOI] [PubMed] [Google Scholar]
- 100. Salvatore M, Garcia‐Sastre A, Ruchala P, Lehrer RI, Chang T, Klotman ME. Alpha‐defensin inhibits influenza virus replication by cell‐mediated mechanism(s). J Infect Dis 2007;196:835–843. doi:https://doi.org/JID37926 [pii]\r10.1086/521027 [DOI] [PubMed] [Google Scholar]
- 101. Currie SM, Findlay EG, McHugh BJ, Mackellar A, Man T, Macmillan D, Wang H, Fitch PM, Schwarze J, Davidson DJ. The human cathelicidin LL‐37 has antiviral activity against respiratory syncytial virus. PLoS One 2013;8(8):e73659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Wong JH, Legowska A, Rolka K, Ng TB, Hui M, Cho CH, Lam WWL, Au SWN, Gu OW, Wan DCC. Effects of cathelicidin and its fragments on three key enzymes of HIV‐1. Peptides 2011;32:1117–1122. doi: 10.1016/j.peptides.2011.04.017 [DOI] [PubMed] [Google Scholar]
- 103. Peschel A, Otto M, Jack RW, Kalbacher H, Jung G, Götz F. Inactivation of the dlt operon in Staphylococcus aureus confers sensitivity to defensins, protegrins, and other antimicrobial peptides. J Biol Chem 1999;274:8405–8410. doi: 10.1074/jbc.274.13.8405 [DOI] [PubMed] [Google Scholar]
- 104. Cheung AL, Bayer AS, Yeaman MR, Xiong YQ, Waring AJ, Memmi G, Donegan N, Chaili S, Yang SJ. Site‐specific mutation of the sensor kinase GraS in Staphylococcus aureus alters the adaptive response to distinct cationic antimicrobial peptides. Infect Immun 2014;82:5336–5345. doi: 10.1128/IAI.02480-14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. Falord M, Karimova G, Hiron A, Msadeka T. GraXSR proteins interact with the VraFG ABC transporter to form a five‐component system required for cationic antimicrobial peptide sensing and resistance in Staphylococcus aureus . Antimicrob Agents Chemother 2012;56:1047–1058. doi: 10.1128/AAC.05054-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106. Weidenmaier C, Peschel A, Kempf VAJ, Lucindo N, Yeaman MR, Bayer AS. DltABCD‐ and MprF‐mediated cell envelope modifications of Staphylococcus aureus confer resistance to platelet microbicidal proteins and contribute to virulence in a rabbit endocarditis model. Infect Immun 2005;73:8033–8038. doi: 10.1128/IAI.73.12.8033-8038.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107. Henderson JC, Fage CD, Cannon JR, Brodbelt JS, Keatinge‐Clay AT, Trent MS. Antimicrobial peptide resistance of Vibrio cholerae results from an LPS modification pathway related to nonribosomal peptide synthetases. ACS Chem Biol 2014;9:2382–2392. doi: 10.1021/cb500438x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108. Balthazar JT, Gusa A, Martin LE, Choudhury B, Carlson R, Shafer WM. Lipooligosaccharide structure is an important determinant in the resistance of Neisseria gonorrhoeae to antimicrobial agents of innate host defense. Front Microbiol 2011;2. doi: 10.3389/fmicb.2011.00030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109. Packiam M, Yedery RD, Begum AA, Carlson RW, Ganguly J, Sempowski GD, Ventevogel MS, Shafer WM, Jerse AE. Phosphoethanolamine decoration of Neisseria gonorrhoeae lipid A plays a dual immunostimulatory and protective role during experimental genital tract infection. Infect Immun 2014;82:2170–2179. doi: 10.1128/IAI.01504-14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110. Liu M, John CM, Jarvis GA. Phosphoryl moieties of lipid A from Neisseria meningitidis and N. gonorrhoeae lipooligosaccharides play an important role in activation of both MyD88‐ and TRIF‐dependent TLR4‐MD‐2 signaling pathways. J Immunol 2010;185:6974–6984. doi: 10.4049/jimmunol.1000953 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111. John CM, Liu M, Phillips NJ, Yang Z, Funk CR, Zimmerman LI, Griffiss JM, Stein DC, Jarvis GA. Lack of lipid A pyrophosphorylation and functional lptA reduces inflammation by Neisseria commensals. Infect Immun 2012;80:4014–4026. doi: 10.1128/IAI.00506-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112. Cullen TW, Schofield WB, Barry NA, Putnam EE, Rundell EA, Trent MS, Degnan PH, Booth CJ, Yu H, Goodman AL. Gut microbiota. Antimicrobial peptide resistance mediates resilience of prominent gut commensals during inflammation. Science 2015;347:170–175. doi: 10.1126/science.1260580 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113. Groisman EA, Parra‐Lopez C, Salcedo M, Lipps CJ, Heffron F. Resistance to host antimicrobial peptides is necessary for Salmonella virulence. Proc Natl Acad Sci USA 1992;89:11939–11943. doi: 10.1073/pnas.89.24.11939 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114. Cotter PD, Hill C, Ross RP. Bacteriocins: Developing innate immunity for food. Nat Rev Microbiol 2005;3:777–788. doi:https://doi.org/nrmicro1273 [pii]\r10.1038/nrmicro1273 [DOI] [PubMed] [Google Scholar]
- 115. Dintner S, Staroń A, Berchtold E, Petri T, Mascher T, Gebhard S. Coevolution of ABC transporters and two‐component regulatory systems as resistance modules against antimicrobial peptides in Firmicutes bacteria. J Bacteriol 2011;193:3851–3862. doi: 10.1128/JB.05175-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116. Aslam R, Atindehou M, Lavaux T, Haïkel Y, Schneider F, Metz‐Boutigue MH. Chromogranin A‐derived peptides are involved in innate immunity. Curr Med Chem 2012;19:4115–4123. doi: 10.2174/092986712802430063 [DOI] [PubMed] [Google Scholar]
- 117. Gebhard S, Fang C, Shaaly A, Leslie DJ, Weimar MR, Kalamorz F, Carne A, Cook GM. Identification and characterization of a bacitracin resistance network in Enterococcus faecalis . Antimicrob Agents Chemother 2014;58:1425–1433. doi: 10.1128/AAC.02111-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118. Llobet E, Tomás JM, Bengoechea JA. Capsule polysaccharide is a bacterial decoy for antimicrobial peptides. Microbiology 2008;154:3877–3886. doi: 10.1099/mic.0.2008/022301-0 [DOI] [PubMed] [Google Scholar]
- 119. Zhao C, Wang I, Lehrer RI. Widespread expression of beta‐defensin hBD‐1 in human secretory glands and epithelial cells. FEBS Lett 1996;396:319–322. doi: 10.1016/0014-5793(96)01123-4 [DOI] [PubMed] [Google Scholar]
- 120. White SH, Wimley WC, Selsted ME. Structure, function, and membrane integration of defensins. Curr Opin Struct Biol 1995;5:521–527. doi: 10.1016/0959-440X(95)80038-7 [DOI] [PubMed] [Google Scholar]
- 121. Schroeder BO, Ehmann D, Precht JC, Castillo PA, Küchler R, Berger J, Schaller M, Stange EF, Wehkamp J. Paneth cell α‐defensin 6 (HD‐6) is an antimicrobial peptide. Mucosal Immunol 2014;6:1–11. doi: 10.1038/mi.2014.100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122. Furci L, Baldan R, Bianchini V, Trovato A, Ossi C, Cichero P, Cirillo DM. New role for human alpha‐defensin 5 in the fight against hypervirulent Clostridium difficile strains. Infect Immun 2015;83:986–995. doi: 10.1128/IAI.02955-14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123. Salzman NH, Hung K, Haribhai D, Chu H, Karlsson‐Sjöberg J, Amir E, Teggatz P, Barman M, Hayward M, Eastwood D, Stoel M, Zhou Y, Sodergren E, Weinstock GM, Bevins CL, Williams CB, Bos NA. Enteric defensins are essential regulators of intestinal microbial ecology. Nat Immunol 2010;11:76–83. doi: 10.1038/ni.1825 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124. Selsted ME, Ouellette AJ. Mammalian defensins in the antimicrobial immune response. Nat Immunol 2005;6:551–557. doi: 10.1146/annurev-pathol-011811-132427 [DOI] [PubMed] [Google Scholar]
- 125. Schibli DJ, Hunter HN, Aseyev V, Starner TD, Wiencek JM, McCray PB, Tack BF, Vogel HJ. The solution structures of the human beta‐defensins lead to a better understanding of the potent bactericidal activity of HBD3 against Staphylococcus aureus . J Biol Chem 2002;277:8279–8289. doi: 10.1074/jbc.M108830200 [DOI] [PubMed] [Google Scholar]
- 126. Selsted ME, Harwig SS, Ganz T, Schilling JW, Lehrer RI. Primary structures of three human neutrophil defensins. J Clin Invest 1985;76:1436–1439. doi: 10.1172/JCI112121 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127. Zhou L, Huang LQ, Beuerman RW, Grigg ME, Li SFY, Chew FT, Ang L, Stern ME, Tan D. Proteomic analysis of human tears: Defensin expression after ocular surface surgery. J Proteome Res 2004;3:410–416. doi: 10.1021/pr034065n [DOI] [PubMed] [Google Scholar]
- 128. McDermott AM. Defensins and other antimicrobial peptides at the ocular surface. Ocul Surf 2004;2:229–247. doi: 10.1016/S1542-0124(12)70111-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129. Hong SW, Seo DG, Baik JE, Cho K, Yun CH, Han SH. Differential profiles of salivary proteins with affinity to Streptococcus mutans lipoteichoic acid in caries‐free and caries‐positive human subjects. Mol Oral Microbiol 2014;29:208–218. doi: 10.1111/omi.12057 [DOI] [PubMed] [Google Scholar]
- 130. Davison G, Allgrove J, Gleeson M. Salivary antimicrobial peptides (LL‐37 and alpha‐defensins HNP1‐3), antimicrobial and IgA responses to prolonged exercise. Eur J Appl Physiol 2009;106:277–284. doi: 10.1007/s00421-009-1020-y [DOI] [PubMed] [Google Scholar]
- 131. Valore E V, Park CH, Quayle AJ, Wiles KR, McCray PB, Ganz T. Human beta‐defensin‐1: An antimicrobial peptide of urogenital tissues. J Clin Invest 1998;101:1633–1642. doi: 10.1172/JCI1861 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132. Bensch KW, Raida M, Mägert HJ, Schulz‐Knappe P, Forssmann WG. hBD‐1: A novel beta‐defensin from human plasma. FEBS Lett 1995;368:331–335. doi:https://doi.org/0014-5793(95)00687-5 [pii] [DOI] [PubMed] [Google Scholar]
- 133. Maiti S, Patro S, Purohit S, Jain S, Senapati S, Dey N. Effective control of salmonella infections by employing combinations of recombinant antimicrobial human B‐defensins hBD‐1 and hBD‐2. Antimicrob Agents Chemother 2014;58:6896–6903. doi: 10.1128/AAC.03628-14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134. Paulsen FP, Pufe T, Schaudig U, Held‐Feindt J, Lehmann J, Schröder JM, Tillmann BN. Detection of natural peptide antibiotics in human nasolacrimal ducts. Investig Ophthalmol Vis Sci 2001;42:2157–2163. [PubMed] [Google Scholar]
- 135. Stenger S, Hanson DA, Teitelbaum R, Dewan P, Niazi KR, Froelich CJ, Ganz T, Thoma‐Uszynski S, Melián A, Bogdan C, Porcelli SA, Bloom BR, Krensky AM, Modlin RL. An antimicrobial activity of cytolytic T cells mediated by granulysin. Science 1998;282(80):121–125. doi: 10.1126/science.282.5386.121 [DOI] [PubMed] [Google Scholar]
- 136. Harder J, Bartels J, Christophers E, Schroder JM. Isolation and characterization of human beta ‐defensin‐3, a novel human inducible peptide antibiotic. J Biol Chem 2001;276:5707–5713. doi: 10.1074/jbc.M008557200 [DOI] [PubMed] [Google Scholar]
- 137. Rodríguez‐Jiménez FJ, Krause A, Schulz S, Forssmann WG, Conejo‐Garcia JR, Schreeb R, Motzkus D. Distribution of new human beta‐defensin genes clustered on chromosome 20 in functionally different segments of epididymis. Genomics 2003;81:175–183. [DOI] [PubMed] [Google Scholar]
- 138. Schutte B, Mitros J, Bartlett J, Walters J, Jia H, Welsh M, Casavant T, McCray P. Discovery of five conserved β‐defensin gene clusters using a computational search strategy. Proc Natl Acad Sci USA 2002;99:2129–2133. doi:citeulike-article-id:10316344\rdoi:10.1073/pnas.042692699 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139. Yu H, Dong J, Gu Y, Liu H, Xin A, Shi H, Sun F, Zhang Y, Lin D, Diao H. The novel human β‐defensin 114 regulates lipopolysaccharide (LPS)‐mediated inflammation and protects sperm from motility loss. J Biol Chem 2013;288:12270–12282. doi: 10.1074/jbc.M112.411884 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140. Hiratsuka T, Mukae H, Iiboshi H, Ashitani J, Nabeshima K, Minematsu T, Chino N, Ihi T, Kohno S, Nakazato M. Increased concentrations of human beta‐defensins in plasma and bronchoalveolar lavage fluid of patients with diffuse panbronchiolitis. Thorax 2003;58:425–430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141. Wehkamp J, Schmid M, Fellermann K, Stange EF. Defensin deficiency, intestinal microbes, and the clinical phenotypes of Crohn's disease. J Leukoc Biol 2005;77:460–465. doi: 10.1189/jlb.0904543 [DOI] [PubMed] [Google Scholar]
- 142. Huang LC, Redfern RL, Narayanan S, Reins RY, McDermott AM. In vitro activity of human beta‐defensin 2 against Pseudomonas aeruginosa in the presence of tear fluid. Antimicrob Agents Chemother 2007;51:3853–3860. doi: 10.1128/AAC.01317-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143. Azkargorta M, Soria J, Ojeda C, Guzmán F, Acera A, Iloro I, Suárez T, Elortza F. Human basal tear peptidome characterization by CID, HCD, and ETD followed by in silico and in vitro analyses for antimicrobial peptide identification. J Proteome Res 2015;14:2649–2658. doi: 10.1021/acs.jproteome.5b00179 [DOI] [PubMed] [Google Scholar]
- 144. Scheetz T, Bartlett JA, Walters JD, Schutte BC, Casavant TL, McCray PB. Genomics‐based approaches to gene discovery in innate immunity. Immunol Rev 2002;190:137–145. doi: 10.1034/j.1600-065X.2002.19010.x [DOI] [PubMed] [Google Scholar]
- 145. Selsted ME. Theta‐defensins: Cyclic antimicrobial peptides produced by binary ligation of truncated alpha‐defensins. Curr Protein Pept Sci 2004;5:365–371. doi: 10.2174/1389203043379459 [DOI] [PubMed] [Google Scholar]
- 146. Tang YQ, Yuan J, Osapay G, Osapay K, Tran D, Miller CJ, Ouellette AJ, Selsted ME. A cyclic antimicrobial peptide produced in primate leukocytes by the ligation of two truncated alpha‐defensins. Science 1999;286:498–502. doi: 10.1126/science.286.5439.498 [DOI] [PubMed] [Google Scholar]
- 147. Lehrer RI, Cole AM, Selsted ME. Defensins: Cyclic peptides with endless potential. J Biol Chem 2012;287(32):27014–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148. Zanetti M. Cathelicidins, multifunctional peptides of the innate immunity. J Leukoc Biol 2004;75:39–48. doi: 10.1189/jlb.0403147 [DOI] [PubMed] [Google Scholar]
- 149. Malm J, Sorensen O, Persson T, Frohm‐Nilsson M, Johansson B, Bjartell A, Lilja H, Stahle‐Backdahl M, Borregaard N, Egesten A. The human cationic antimicrobial protein (hCAP‐18) is expressed in the epithelium of human epididymis, is present in seminal plasma at high concentrations, and is attached to spermatozoa. Infect Immun 2000;68:4297–4302. doi: 10.1128/IAI.68.7.4297-4302.2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150. Méndez‐Samperio P. The human cathelicidin hCAP18/LL‐37: A multifunctional peptide involved in mycobacterial infections. Peptides 2010;31:1791–1798. doi: 10.1016/j.peptides.2010.06.016 [DOI] [PubMed] [Google Scholar]
- 151. Durr UHN, Sudheendra US, Ramamoorthy A. LL‐37, the only human member of the cathelicidin family of antimicrobial peptides. Biochim Biophys Acta 2006;1758(9):1408–25. [DOI] [PubMed] [Google Scholar]
- 152. Eckmann L. Defence molecules in intestinal innate immunity against bacterial infections. Curr Opin Gastroenterol 2005;21:147–151. [DOI] [PubMed] [Google Scholar]
- 153. Gombart AF, Bhan I, Borregaard N, Tamez H, Camargo CA, Koeffler HP, Thadhani R. Low plasma level of cathelicidin antimicrobial peptide (hCAP18) predicts increased infectious disease mortality in patients undergoing hemodialysis. Clin Infect Dis 2009;48:418–424. doi: 10.1086/596314 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154. Singh D, Vaughan R, Kao CC. LL‐37 peptide enhancement of signal transduction by toll‐like receptor 3 is regulated by pH identification of a peptide antagonist of LL‐37. J Biol Chem 2014;289:27614–27624. doi: 10.1074/jbc.M114.582973 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155. Schaller‐Bals S, Schulze A, Bals R. Increased levels of antimicrobial peptides in tracheal aspirates of newborn infants during infection. Am J Respir Crit Care Med 2002;165:992–995. doi: 10.1164/ajrccm.165.7.200110-020 [DOI] [PubMed] [Google Scholar]
- 156. Zhao H, Lee WH, Shen JH, Li H, Zhang Y. Identification of novel semenogelin I‐derived antimicrobial peptide from liquefied human seminal plasma. Peptides 2008;29:505–511. doi: 10.1016/j.peptides.2008.01.009 [DOI] [PubMed] [Google Scholar]
- 157. Sorensen OE, Gram L, Johnsen AH, Andersson E, Bangsboll S, Tjabringa GS, Hiemstra PS, Malm J, Egesten A, Borregaard N. Processing of seminal plasma hCAP‐18 to ALL‐38 by gastricsin. A novel mechanism of generating antimicrobial peptides in vagina. J Biol Chem 2003;278:28540–28546. doi: 10.1074/jbc.M301608200 [DOI] [PubMed] [Google Scholar]
- 158. Koziel J, Bryzek D, Sroka A, Maresz K, Glowczyk I, Bielecka E, Kantyka T, Pyrć K, Svoboda P, Pohl J, Potempa J. Citrullination alters immunomodulatory function of LL‐37 essential for prevention of endotoxin‐induced sepsis. J Immunol 2014;192:5363–5372. doi: 10.4049/jimmunol.1303062 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159. Picchianti M, Russo C, Castagnini M, Biagini M, Soldaini E, Balducci E. NAD‐dependent ADP‐ribosylation of the human antimicrobial and immune‐modulatory peptide LL‐37 by ADP‐ribosyltransferase‐1. Innate Immun 2015;21:314–321. doi: 10.1177/1753425914536242 [DOI] [PubMed] [Google Scholar]
- 160. Tsai H, Bobek LA. Human salivary histatin‐5 exerts potent fungicidal activity against Cryptococcus neoformans . Biochim Biophys Acta 1997;1336(3):367–9. [DOI] [PubMed] [Google Scholar]
- 161. Vukosavljevic D, Custodio W, Del Bel Cury AA, Siqueira WL. The effect of histatin 5, adsorbed on PMMA and hydroxyapatite, on Candida albicans colonization. Yeast 2012;29:459–466. doi: 10.1002/yea.2925 [DOI] [PubMed] [Google Scholar]
- 162. Xu T, Levitz SM, Diamond RD, Oppenheim FG. Anticandidal activity of major human salivary histatins. Infect Immun 1991;59:2549–2554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163. vanderSpek JC, Wyandt HE, Skare JC, Milunsky A, Oppenheim FG, Troxler RF. Localization of the genes for histatins to human chromosome 4q13 and tissue distribution of the mRNAs. Am J Hum Genet 1989;45:381–387. [PMC free article] [PubMed] [Google Scholar]
- 164. Oppenheim FG, Xu T, McMillian FM, Levitz SM, Diamond RD, Offner GD, Troxler RF. Histatins, a novel family of histidine‐rich proteins in human parotid secretion. Isolation, characterization, primary structure, and fungistatic effects on Candida albicans . J Biol Chem 1988;263:7472–7477. [PubMed] [Google Scholar]
- 165. Sabatini LM, Azen EA. Histatins, a family of salivary histidine‐rich proteins, are encoded by at least two loci (HIS1 and HIS2). Biochem Biophys Res Commun 1989;160:495–502. doi: 10.1016/0006-291X(89)92460-1 [DOI] [PubMed] [Google Scholar]
- 166. Troxler RF, Offner GD, Xu T, Vanderspek JC, Oppenheim FG. Structural relationship between human salivary histatins. J Dent Res 1990;69:2–6. [DOI] [PubMed] [Google Scholar]
- 167. Grogan J, McKnight CJ, Troxler RF, Oppenheim FG. Zinc and copper bind to unique sites of histatin 5. FEBS Lett 2001;491:76–80. doi: 10.1016/S0014-5793(01)02157-3 [DOI] [PubMed] [Google Scholar]
- 168. Gusman H, Lendenmann U, Grogan J, Troxler RF, Oppenheim FG. Is salivary histatin 5 a metallopeptide? Biochim Biophys Acta 2001b;1545:86–95. doi: 10.1016/S0167-4838(00)00265-X [DOI] [PubMed] [Google Scholar]
- 169. Harford C, Sarkar B. Amino terminal Cu(II)‐ and Ni(II)‐binding (ATCUN) motif of proteins and peptides: Metal binding, DNA cleavage, and other properties †. Acc Chem Res 1997;30:123–130. doi: 10.1021/ar9501535 [DOI] [Google Scholar]
- 170. Melino S, Rufini S, Sette M, Morero R, Grottesi A, Paci M, Petruzzelli R. Zn(2+) ions selectively induce antimicrobial salivary peptide histatin‐5 to fuse negatively charged vesicles. Identification and characterization of a zinc‐binding motif present in the functional domain. Biochemistry 1999;38:9626–9633. doi: 10.1021/bi990212c [DOI] [PubMed] [Google Scholar]
- 171. Brewer D, Hunter H, Lajoie G. NMR studies of the antimicrobial salivary peptides histatin 3 and histatin 5 in aqueous and nonaqueous solutions. Biochem Cell Biol 1998;76:247–256. doi: 10.1139/o98-066 [DOI] [PubMed] [Google Scholar]
- 172. Raj PA, Edgerton M, Levine MJ. Salivary histatin 5: Dependence of sequence, chain length, and helical conformation for candidacidal activity. J Biol Chem 1990;265:3898–3905. [PubMed] [Google Scholar]
- 173. Kavanagh K, Dowd S. Histatins: Antimicrobial peptides with therapeutic potential. J Pharm Pharmacol 2004;56:285–289. doi: 10.1211/0022357022971 [DOI] [PubMed] [Google Scholar]
- 174. Mochon AB, Liu H. The antimicrobial peptide histatin‐5 causes a spatially restricted disruption on the Candida albicans surface, allowing rapid entry of the peptide into the cytoplasm. PLoS Pathog 2008;4:e1000190. doi: 10.1371/journal.ppat.1000190 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175. Johnson DA, Yeh CK, Dodds MW. Effect of donor age on the concentrations of histatins in human parotid and submandibular/sublingual saliva. Arch Oral Biol 2000;45:731–740. [DOI] [PubMed] [Google Scholar]
- 176. Lai Y‐P, Peng Y‐F, Zuo Y, Li J, Huang J, Wang L‐F, Wu ZR. Functional and structural characterization of recombinant dermcidin‐1L, a human antimicrobial peptide. Biochem Biophys Res Commun 2005;328:243–250. doi: 10.1016/j.bbrc.2004.12.143 [DOI] [PubMed] [Google Scholar]
- 177. Schittek B, Hipfel R, Sauer B, Bauer J, Kalbacher H, Stevanovic S, Schirle M, Schroeder K, Blin N, Meier F, Rassner G, Garbe C. Dermcidin: A novel human antibiotic peptide secreted by sweat glands. Nat Immunol 2001;2:1133–1137. doi: 10.1038/ni732 [DOI] [PubMed] [Google Scholar]
- 178. Vuong C, Voyich JM, Fischer ER, Braughton KR, Whitney AR, DeLeo FR, Otto M. Polysaccharide intercellular adhesin (PIA) protects Staphylococcus epidermidis against major components of the human innate immune system. Cell Microbiol 2004;6:269–275. doi: 10.1046/j.1462-5822.2004.00367.x [DOI] [PubMed] [Google Scholar]
- 179. Cabal‐Manzano R, Bhargava P, Torres‐Duarte A, Marshall J, Wainer IW. Proteolysis‐inducing factor is expressed in tumours of patients with gastrointestinal cancers and correlates with weight loss. Br J Cancer 2001;84:1599–1601. doi: 10.1054/bjoc.2001.1830 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180. Cunningham TJ, Hodge L, Speicher D, Reim D, Tyler‐Polsz C, Levitt P, Eagleson K, Kennedy S, Wang Y. Identification of a survival‐promoting peptide in medium conditioned by oxidatively stressed cell lines of nervous system origin. J Neurosci 1998;18:7047–7060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181. Rieg S, Steffen H, Seeber S, Humeny A, Kalbacher H, Dietz K, Garbe C, Schittek B. Deficiency of dermcidin‐derived antimicrobial peptides in sweat of patients with atopic dermatitis correlates with an impaired innate defense of human skin in vivo. J Immunol 2005;174:8003–8010. [DOI] [PubMed] [Google Scholar]
- 182. Ghosh R, Maji UK, Bhattacharya R, Sinha AK. The role of dermcidin isoform 2: A two‐faceted atherosclerotic risk factor for coronary artery disease and the effect of acetyl salicylic acid on it. Thrombosis 2012;2012:987932. doi: 10.1155/2012/987932 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183. Ghosh R, Karmohapatra SK, Bhattacharyya M, Bhattacharya R, Bhattacharya G, Sinha AK. The appearance of dermcidin isoform 2, a novel platelet aggregating agent in the circulation in acute myocardial infarction that inhibits insulin synthesis and the restoration by acetyl salicylic acid of its effects. J Thromb Thrombolysis 2011;31:13–21. doi: 10.1007/s11239-010-0515-z [DOI] [PubMed] [Google Scholar]
- 184. Jordan JB, Poppe L, Haniu M, Arvedson T, Syed R, Li V, Kohno H, Kim H, Schnier PD, Harvey TS, Miranda LP, Cheetham J, Sasu BJ. Hepcidin revisited, disulfide connectivity, dynamics, and structure. J Biol Chem 2009;284:24155–24167. doi: 10.1074/jbc.M109.017764 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185. Roetto A, Papanikolaou G, Politou M, Alberti F, Girelli D, Christakis J, Loukopoulos D, Camaschella C. Mutant antimicrobial peptide hepcidin is associated with severe juvenile hemochromatosis. Nat Genet 2003;33:21–22. doi: 10.1038/ng1053 [DOI] [PubMed] [Google Scholar]
- 186. Nicolas G, Chauvet C, Viatte L, Danan JL, Bigard X, Devaux I, Beaumont C, Kahn A, Vaulont S. The gene encoding the iron regulatory peptide hepcidin is regulated by anemia, hypoxia, and inflammation. J Clin Invest 2002;110:1037–1044. doi: 10.1172/JCI200215686 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187. Parrow NL, Fleming RE, Minnick MF. Sequestration and scavenging of iron in infection. Infect Immun 2013;81(10):3503–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188. Christopher GW. Escherichia coli bacteremia, meningitis, and hemochromatosis. Arch Intern Med 1985;145(10):1908. [PubMed] [Google Scholar]
- 189. Fernández JM, Serrano M, De Arriba JJ, Sánchez MV, Escribano E, Ferreras P. Bacteremic cellulitis caused by Non‐01, Non‐0139 Vibrio cholerae: Report of a case in a patient with hemochromatosis. Diagn Microbiol Infect Dis 2000;37:77–80. doi: 10.1016/S0732-8893(99)00153-4 [DOI] [PubMed] [Google Scholar]
- 190. Hopfner M, Nitsche R, Rohr A, Harms D, Schubert S, Folsch UR. Yersinia enterocolitica infection with multiple liver abscesses uncovering a primary hemochromatosis. Scand J Gastroenterol 2001;36:220–224. doi: 10.1080/003655201750066004 [DOI] [PubMed] [Google Scholar]
- 191. Manso C, Rivas I, Peraire J, Vidal F, Richart C. Fatal Listeria meningitis, endocarditis and pericarditis in a patient with haemochromatosis. Scand J Infect Dis 1997;29:308–309. doi: 10.3109/00365549709019049 [DOI] [PubMed] [Google Scholar]
- 192. Bullen JJ, Spalding PB, Ward CG, Gutteridge JM. Hemochromatosis, iron and septicemia caused by Vibrio vulnificus . Arch Intern Med 1991;151:1606–1609. doi: 10.1001/archinte.1991.00400080096018 [DOI] [PubMed] [Google Scholar]
- 193. Nairz M, Theurl I, Schroll A, Theurl M, Fritsche G, Lindner E, Seifert M, Crouch MLV, Hantke K, Akira S, Fang FC, Weiss G. Absence of functional HFE protects mice from invasive Salmonella enterica Serovar Typhimurium infection via induction of lipocalin‐2. Blood 2009;114:3642–3651. doi: 10.1182/blood-2009-05-223354 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194. Olakanmi O, Schlesinger LS, Britigan BE. Hereditary hemochromatosis results in decreased iron acquisition and growth by Mycobacterium tuberculosis within human macrophages. J Leukoc Biol 2007;81:195–204. doi: 10.1189/jlb.0606405 [DOI] [PubMed] [Google Scholar]
- 195. Lugardon K, Raffner R, Goumon Y, Corti A, Delmas A, Bulet P, Aunis D, Metz‐Boutigue MH. Antibacterial and antifungal activities of vasostatin‐1, the N‐terminal fragment of chromogranin A. J Biol Chem 2000;275:10745–10753. doi: 10.1074/jbc.275.15.10745 [DOI] [PubMed] [Google Scholar]
- 196. Konecki DS, Benedum UM, Gerdes HH, Huttner WB. The primary structure of human chromogranin A and pancreastatin. J Biol Chem 1987;262:17026–17030. [PubMed] [Google Scholar]
- 197. Gadroy P, Stridsberg M, Capon C, Michalski JC, Strub JM, Van Dorsselaer A, Aunist D, Metz‐Boutigue MH. Phosphorylation and O‐glycosylation sites of human chromogranin A (CGA79‐439) from urine of patients with carcinoid tumors. J Biol Chem 1998;273:34087–34097. doi: 10.1074/jbc.273.51.34087 [DOI] [PubMed] [Google Scholar]
- 198. Metz‐Boutigue MH, Garcia‐Sablone P, Hogue‐Angeletti R, Aunis D. Intracellular and extracellular processing of chromogranin A. Determination of cleavage sites. Eur J Biochem 1993;217:247–257. doi: 10.1111/j.1432-1033.1993.tb18240.x [DOI] [PubMed] [Google Scholar]
- 199. Aardal S, Helle KB. The vasoinhibitory activity of bovine chromogranin A fragment (vasostatin) and its independence of extracellular calcium in isolated segments of human blood vessels. Regul Pept 1992;41:9–18. doi: 10.1016/0167-0115(92)90509-S [DOI] [PubMed] [Google Scholar]
- 200. Ciesielski‐Treska J, Ulrich G, Taupenot L, Chasserot‐Golaz S, Corti A, Aunis D, Bader MF. Chromogranin A induces a neurotoxic phenotype in brain microglial cells. J Biol Chem 1998;273:14339–14346. doi: 10.1074/jbc.273.23.14339 [DOI] [PubMed] [Google Scholar]
- 201. Ishimitsu T, Nishikimi T, Saito Y, Kitamura K, Eto T, Kangawa K, Matsuo H, Omae T, Matsuoka H. Plasma levels of adrenomedullin, a newly identified hypotensive peptide, in patients with hypertension and renal failure. J Clin Invest 1994;94:2158–2161. doi: 10.1172/JCI117573 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202. Jougasaki M, Wei CM, McKinley LJ, Burnett JC. Elevation of circulating and ventricular adrenomedullin in human congestive heart failure. Circulation 1995;92:286–289. doi: 10.1161/01.CIR.92.3.286 [DOI] [PubMed] [Google Scholar]
- 203. Hirata Y, Mitaka C, Sato K, Nagura T, Tsunoda Y, Amaha K, Marumo F. Increased circulating adrenomedullin, a novel vasodilatory peptide, in sepsis. J Clin Endocrinol Metab 1996;81:1449–1453. doi: 10.1210/jc.81.4.1449 [DOI] [PubMed] [Google Scholar]
- 204. Allaker RP, Zihni C, Kapas S. An investigation into the antimicrobial effects of adrenomedullin on members of the skin, oral, respiratory tract and gut microflora. FEMS Immunol Med Microbiol 1999;23;289–293. doi: 10.1016/S0928-8244(98)00148-5 [DOI] [PubMed] [Google Scholar]
- 205. Shimizu M, Shigeri Y, Tatsu Y, Yoshikawa S, Yumoto N. Enhancement of antimicrobial activity of neuropeptide Y by N‐terminal truncation. Antimicrob Agents Chemother 1998;42:2745–2746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206. Faith RE, Murgo AJ. Inhibition of pulmonary metastases and enhancement of natural killer cell activity by methionine‐enkephalin. Brain Behav Immun 1988;2:114–122. doi: 10.1016/0889-1591(88)90012-8 [DOI] [PubMed] [Google Scholar]
- 207. Ohmori H, Fujii K, Sasahira T, Luo Y, Isobe M, Tatsumoto N, Kuniyasu H. Methionine‐enkephalin secreted by human colorectal cancer cells suppresses T lymphocytes. Cancer Sci 2009;100:497–502. doi: 10.1111/j.1349-7006.2008.01073.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208. Goumon Y, Lugardon K, Kieffer B, Lefevre J‐F, Van Dorsselaer A, Aunis D, Metz‐Boutigue MH. Characterization of antibacterial COOH‐terminal proenkephalin‐A‐derived peptides (PEAP) in infectious fluids: Importance of enkelytin, the antibacterial PEAP209‐237 secreted by stimulated chromaffin cells. J Biol Chem 1998;273:29847–29856. doi: 10.1074/jbc.273.45.29847 [DOI] [PubMed] [Google Scholar]
- 209. Stern AS, Jones BN, Shively JE, Stein S, Undenfriend S. Two adrenal opioid polypeptides: Proposed intermediates in the processing of proenkephalin. Proc Natl Acad Sci USA 1981;78:1962–1966. doi: 10.1073/pnas.78.3.1962 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210. Pittius CW, Kley N, Loeffler JP, Hollt V. Quantitation of proenkephalin A messenger RNA in bovine brain, pituitary and adrenal medulla: Correlation between mRNA and peptide levels. EMBO J 1985;4:1257–1260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 211. Uhl GR, Navia B, Douglas J. Differential expression of preproenkephalin and preprodynorphin mRNAs in striatal neurons: High levels of preproenkephalin expression depend on cerebral cortical afferents. J Neurosci 1988;8:4755–4764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212. Martin J, Prystowsky MB, Angeletti RH. Preproenkephalin mRNA in T‐cells, macrophages, and mast cells. J Neurosci Res 1987;18:82–87. [DOI] [PubMed] [Google Scholar]
- 213. Rosen H, Behar O, Abramsky O, Ovadia H. Regulated expression of proenkephalin A in normal lymphocytes. J Immunol 1989;143:3703–3707. [PubMed] [Google Scholar]
- 214. Martens GJ, Herbert E. Polymorphism and absence of Leu‐enkephalin sequences in proenkephalin genes in Xenopus laevis . Nature 1984;310:251–254. [DOI] [PubMed] [Google Scholar]
- 215. Tasiemski A, Salzet M., Benson H, Fricchione GL, Bilfinger TV, Goumon Y, Metz‐Boutigue MH, Aunis D, Stefano GB. The presence of antibacterial and opioid peptides in human plasma during coronary artery bypass surgery. J Neuroimmunol 2000;109:228–235. [DOI] [PubMed] [Google Scholar]
- 216. Cutuli M, Cristiani S, Lipton JM, Catania A. Antimicrobial effects of alpha‐MSH peptides. J Leukoc Biol 2000;67:233–239. [DOI] [PubMed] [Google Scholar]
- 217. Bhattacharya A, Datta A. Effect of cyclic AMP on RNA and protein synthesis in Candida albicans . Biochem Biophys Res Commun 1977;77:1483–1444. [DOI] [PubMed] [Google Scholar]
- 218. Nibbering PH, Ravensbergen E, Welling MM, Van Berkel LA, Van Berkel PHC, Pauwels EKJ, Nuijens JH. Human lactoferrin and peptides derived from its N terminus are highly effective against infections with antibiotic‐resistant bacteria. Infect Immun 2001;69:1469–1476. doi: 10.1128/IAI.69.3.1469-1476.2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 219. Yang D, Chen Q, Hoover DM, Staley P, Tucker KD, Lubkowski J, Oppenheim JJ. Many chemokines including CCL20/MIP‐3alpha display antimicrobial activity. J Leukoc Biol 2003;74:448–455. doi: 10.1189/jlb.0103024.http [DOI] [PubMed] [Google Scholar]
- 220. Gela A, Kasetty G, Jovic S, Ekoff M, Nilsson G, Morgelin M, Kjellstrom S, Pease JE, Schmidtchen A, Egesten A. Eotaxin‐3 (CCL26) exerts innate host defense activities that are modulated by mast cell proteases. Allergy 2015;70:161–170. doi: 10.1111/all.12542 [DOI] [PubMed] [Google Scholar]
- 221. Minn I, Kim HS, Kim SC. Antimicrobial peptides derived from pepsinogens in the stomach of the bullfrog, Rana catesbeiana . Biochim Biophys Acta 1998;1407:31–39. [DOI] [PubMed] [Google Scholar]
- 222. Cole AM, Kim YH, Tahk S, Hong T, Weis P, Waring AJ, Ganz T. Calcitermin, a novel antimicrobial peptide isolated from human airway secretions. FEBS Lett 2001;504:5–10. doi: 10.1016/s0014-5793(01)02731-4 [DOI] [PubMed] [Google Scholar]
- 223. Wei XY, Armishaw C, Goyette J, Yang Z, Cai H, Alewood P, Geczy CL. Mast cell and monocyte recruitment by S100A12 and its hinge domain. J Biol Chem 2008;283:13035–13043. doi: 10.1074/jbc.M710388200 [DOI] [PubMed] [Google Scholar]
- 224. Yang Z, Yan WX, Cai H, Tedla N, Armishaw C, Di Girolamo N, Wang HW, Hampartzoumian T, Simpson JL, Gibson PG, Hunt J, Hart P, Hughes JM, Perry MA, Alewood PF, Geczy CL. S100A12 provokes mast cell activation: A potential amplification pathway in asthma and innate immunity. J Allergy Clin Immunol 2007;119:106–114. doi: 10.1016/j.jaci.2006.08.021 [DOI] [PubMed] [Google Scholar]
- 225. Rieg S, Seeber S, Steffen H, Humeny A, Kalbacher H, Stevanovic S, Kimura A, Garbe C, Schittek B. Generation of multiple stable dermcidin‐derived antimicrobial peptides in sweat of different body sites. J Invest Dermatol 2006;126:354–365. doi: 10.1038/sj.jid.5700041 [DOI] [PubMed] [Google Scholar]
- 226. Lambers H, Piessens S, Bloem A, Pronk H, Finkel P. Natural skin surface pH is on average below 5, which is beneficial for its resident flora. Int J Cosmet Sci 2006;28:359–370. doi: 10.1111/j.1467-2494.2006.00344.x [DOI] [PubMed] [Google Scholar]
- 227. Falcao CB, De La Torre BG, Pérez‐Peinado C, Barron AE, Andreu D, Rádis‐Baptista G. Vipericidins: A novel family of cathelicidin‐related peptides from the venom gland of South American pit vipers. Amino Acids 2014;46:2561–2571. doi: 10.1007/s00726-014-1801-4 [DOI] [PubMed] [Google Scholar]
- 228. Ouellette AJ, Selsted ME. Paneth cell defensins: Endogenous peptide components of intestinal host defense. FASEB J 1996;10:1280–1289. [DOI] [PubMed] [Google Scholar]
- 229. Rice WG, Ganz T, Kinkade JM, Selsted ME, Lehrer RI, Parmley RT. Defensin‐rich dense granules of human neutrophils. Blood 1987;70:757–765. doi: 10.1182/blood-2014-12-567834 [DOI] [PubMed] [Google Scholar]
- 230. Sørensen O, Arnljots K, Cowland JB, Bainton DF, Borregaard N. The human antibacterial cathelicidin, hCAP‐18, is synthesized in myelocytes and metamyelocytes and localized to specific granules in neutrophils. Blood 1997;90:2796–2803. [PubMed] [Google Scholar]
- 231. Schulte I, Tammen H, Selle H, Schulz‐Knappe P. Peptides in body fluids and tissues as markers of disease. Expert Rev Mol Diagn 2005;5:145–157. doi:https://doi.org/ERM050206 [pii] [DOI] [PubMed] [Google Scholar]
- 232. Svensson M, Sköld K, Svenningsson P, Andren PE. Peptidomics‐based discovery of novel neuropeptides. J Proteome Res 2003;2:213–219. doi: 10.1021/pr020010u [DOI] [PubMed] [Google Scholar]
- 233. Kieffer TJ, Habener JF. The glucagon‐like peptides. Endocr Rev 1999;20(6):876–913. [DOI] [PubMed] [Google Scholar]
- 234. Sørensen OE, Follin P, Johnsen AH, Calafat J, Sandra Tjabringa G, Hiemstra PS, Borregaard N. Human cathelicidin, hCAP‐18, is processed to the antimicrobial peptide LL‐37 by extracellular cleavage with proteinase 3. Blood 2001;97:3951–3959. doi: 10.1182/blood.V97.12.3951 [DOI] [PubMed] [Google Scholar]
- 235. Baik JE, Hong SW, Choi S, Jeon JH, Park OJ, Cho K, Seo DG, Kum KY, Yun CH, Han SH. Alpha‐amylase is a human salivary protein with affinity to lipopolysaccharide of Aggregatibacter actinomycetemcomitans . Mol Oral Microbiol 2013;28:142–153. doi: 10.1111/omi.12011 [DOI] [PubMed] [Google Scholar]
- 236. Dallas DC, Guerrero A, Khaldi N, Castillo PA, Martin WF, Smilowitz JT, Bevins CL, Barile D, German JB, Lebrilla CB. Extensive in vivo human milk peptidomics reveals specific proteolysis yielding protective antimicrobial peptides. J Proteome Res 2013;12:2295–2304. doi: 10.1021/pr400212z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 237. Thongboonkerd V, McLeish KR, Arthur JM, Klein JB. Proteomic analysis of normal human urinary proteins isolated by acetone precipitation or ultracentrifugation. Kidney Int 2002;62:1461–1469. doi: 10.1046/j.1523-1755.2002.00565.x [DOI] [PubMed] [Google Scholar]
- 238. Yang H, Wang X, Liu X, Wu J, Liu C, Gong W, Zhao Z, Hong J, Lin D, Wang Y, Lai R. Antioxidant peptidomics reveals novel skin antioxidant system. Mol Cell Proteomics 2009;8:571–583. doi: 10.1074/mcp.M800297-MCP200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 239. Manes NP, Gustin JK, Rue J, Mottaz HM, Purvine SO, Norbeck AD, Monroe ME, Zimmer JSD, Metz TO, Adkins JN, Smith RD, Heffron F. Targeted protein degradation by Salmonella under phagosome‐mimicking culture conditions investigated using comparative peptidomics. Mol Cell Proteomics 2007;6:717–727. doi: 10.1074/mcp.M600282-MCP200 [DOI] [PubMed] [Google Scholar]
- 240. Polson C, Sarkar P, Incledon B, Raguvaran V, Grant R. Optimisation of protein precipitation based upon effectiveness of protein removal and ionisation effect in liquid chromatography‐tandem mass spectrometry. J Chromatogr B 2003;785:263–275. [DOI] [PubMed] [Google Scholar]
- 241. Schrader M, Schulz‐Knappe P. Peptidomics technologies for human body fluids. Trends Biotechnol 2001;19:S55–S60. doi: 10.1016/S0167-7799(01)01800-5 [DOI] [PubMed] [Google Scholar]
- 242. Yuan X, Desiderio DM. Human cerebrospinal fluid peptidomics. J Mass Spectrom 2005;40:176–181. doi: 10.1002/jms.737 [DOI] [PubMed] [Google Scholar]
- 243. Foettinger A, Leitner A, Lindner W. Selective enrichment of tryptophan‐containing peptides from protein digests employing a reversible derivatization with malondialdehyde and solid‐phase capture on hydrazide beads. J Proteome Res 2007;6:3827–3834. doi: 10.1021/pr0702767 [DOI] [PubMed] [Google Scholar]
- 244. Grunert T, Pock K, Buchacher A, Allmaier G. Selective solid‐phase isolation of methionine‐containing peptides and subsequent matrix‐assisted laser desorption/ionisation mass spectrometric detection of methionine‐ and of methionine‐sulfoxide‐containing peptides. Rapid Commun Mass Spectrom 2003;17:1815–1824. doi: 10.1002/rcm.1110 [DOI] [PubMed] [Google Scholar]
- 245. Xu Y, Cao Q, Svec F, Fréchet JMJ. Porous polymer monolithic column with surface‐bound gold nanoparticles for the capture and separation of cysteine‐containing peptides. Anal Chem 2010;82:3352–3358. doi: 10.1021/ac1002646 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 246. Kaji H, Yamauchi Y, Takahashi N, Isobe T. Mass spectrometric identification of N‐linked glycopeptides using lectin‐mediated affinity capture and glycosylation site‐specific stable isotope tagging. Nat Protoc 2006;1:3019–3027. doi: 10.1038/nprot.2006.444 [DOI] [PubMed] [Google Scholar]
- 247. Thingholm TE, Jørgensen TJD, Jensen ON, Larsen MR. Highly selective enrichment of phosphorylated peptides using titanium dioxide. Nat Protoc 2006;1:1929–1935. doi: 10.1038/nprot.2006.185 [DOI] [PubMed] [Google Scholar]
- 248. Searle BC, Dasari S, Wilmarth PA, Turner M, Reddy AP, David LL, Nagalla SR. Identification of protein modifications using MS/MS de novo sequencing and the OpenSea alignment algorithm. J Proteome Res 2005;4:546–554. doi: 10.1021/pr049781j [DOI] [PubMed] [Google Scholar]
- 249. Deacon CF, Holst JJ. Immunoassays for the incretin hormones GIP and GLP‐1. Best Pract Res Clin Endocrinol Metab 2009;23(4):425–32. [DOI] [PubMed] [Google Scholar]
- 250. Jankowski V, Vanholder R, Van Der Giet M, Tolle M, Karadogan S, Gobom J, Furkert J, Oksche A, Krause E, Tran TNA, Tepel M, Schuchardt M, Schluter H, Wiedon A, Beyermann M, Bader M, Todiras M, Zidek W, Jankowski J. Mass‐spectrometric identification of a novel angiotensin peptide in human plasma. Arterioscler Thromb Vasc Biol 2007;27:297–302. doi: 10.1161/01.ATV.0000253889.09765.5f [DOI] [PubMed] [Google Scholar]
- 251. Trindade F, Amado F, Pinto da Costa J, Ferreira R, Maia C, Henriques I, Colaço B, Vitorino R. Salivary peptidomic as a tool to disclose new potential antimicrobial peptides. J Proteomics 2015;115:49–57. doi: 10.1016/j.jprot.2014.12.004 [DOI] [PubMed] [Google Scholar]
- 252. Herrero M, Ibanez E, Cifuentes A. Capillary electrophoresis‐electrospray‐mass spectrometry in peptide analysis and peptidomics. Electrophoresis 2008;29(10):2418–60. [DOI] [PubMed] [Google Scholar]
- 253. Tucholska M, Florentinus A, Williams D, Marshall JG. The endogenous peptides of normal human serum extracted from the acetonitrile‐insoluble precipitate using modified aqueous buffer with analysis by LC‐ESI‐Paul ion trap and Qq‐TOF. J Proteomics 2010;73:1254–1269. doi: 10.1016/j.jprot.2010.02.022 [DOI] [PubMed] [Google Scholar]
- 254. Tucholska M, Scozzaro S, Williams D, Ackloo S, Lock C, Siu KWM, Evans KR, Marshall JG. Endogenous peptides from biophysical and biochemical fractionation of serum analyzed by matrix‐assisted laser desorption/ionization and electrospray ionization hybrid quadrupole time‐of‐flight. Anal Biochem 2007;370:228–245. doi: 10.1016/j.ab.2007.07.029 [DOI] [PubMed] [Google Scholar]
- 255. Verhaert P, Uttenweiler‐Joseph S, de Vries M, Loboda A, Ens W, Standing KG. Matrix‐assisted laser desorption/ionization quadrupole time‐of‐flight mass spectrometry: An elegant tool for peptidomics. Proteomics 2001;1:118–131. doi: [DOI] [PubMed] [Google Scholar]
- 256. Tian L, Wang Y, Xu D, Gao Y, Wen X, Tian Y. The differential diagnostic model for serous peptidomics in HBV carriers established by MALDI‐TOF‐MS analysis. Clin Biochem 2014;47:56–62. doi: 10.1016/j.clinbiochem.2013.10.016 [DOI] [PubMed] [Google Scholar]
- 257. Osaki T, Sasaki K, Minamino N. Peptidomics‐based discovery of an antimicrobial peptide derived from insulin‐like growth factor‐binding protein 5. J Proteome Res 2011;10:1870–1880. doi: 10.1021/pr101114a [DOI] [PubMed] [Google Scholar]
- 258. Good DM, Zürbig P, Argilés A, Bauer HW, Behrens G, Coon JJ, Dakna M, Decramer S, Delles C, Dominiczak AF, Ehrich JHH, Eitner F, Fliser D, Frommberger M, Ganser A, Girolami MA, Golovko I, Gwinner W, Haubitz M, Herget‐Rosenthal S, Jankowski J, Jahn H, Jerums G, Julian BA, Kellmann M, Kliem V, Kolch W, Krolewski AS, Luppi M, Massy Z, Melter M, Neusüss C, Novak J, Peter K, Rossing K, Rupprecht H, Schanstra JP, Schiffer E, Stolzenburg JU, Tarnow L, Theodorescu D, Thongboonkerd V, Vanholder R, Weissinger EM, Mischak H, Schmitt‐Kopplin P. Naturally occurring human urinary peptides for use in diagnosis of chronic kidney disease. Mol Cell Proteomics 2010;9:2424–2437. doi: 10.1074/mcp.M110.001917 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 259. Tauber PF, Propping D, Schumacher GFB, Zanelevd LJD. Biochemical aspects of the coagulation and liquefaction of human semen. J Androl 1980;1:281–288. doi: 10.1002/j.1939-4640.1980.tb00043.x [DOI] [Google Scholar]
- 260. Roan NR, Liu H, Usmani SM, Neidleman J, Muller JA, Avila‐Herrera A, Gawanbacht A, Zirafi O, Chu S, Dong M, Kumar ST, Smith JF, Pollard KS, Fandrich M, Kirchhoff F, Munch J, Witkowska HE, Greene WC. Liquefaction of semen generates and later degrades a conserved semenogelin peptide that enhances HIV infection. J Virol 2014;88:7221–7234. doi: 10.1128/JVI.00269-14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 261. Benedetti E. X‐ray crystallography of peptides: The contributions of the Italian laboratories. Biopolymers 1996;40:3–44. doi: [DOI] [PubMed] [Google Scholar]
- 262. Haney EF, Vogel HJ. Chapter 1 NMR of antimicrobial peptides. Annu Rep NMR Spectrosc 2009;65:1–51. doi: 10.1016/S0066-4103(08)00201-9 [DOI] [Google Scholar]
- 263. Thévenet P, Shen Y, Maupetit J, Guyon F, Derreumaux P, Tufféry P. PEP‐FOLD: An updated de novo structure prediction server for both linear and disulfide bonded cyclic peptides. Nucleic Acids Res 2012;40:W288–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 264. Thomas A, Deshayes S, Decaffmeyer M, Van Eyck MH, Charloteaux B, Brasseur R. Prediction of peptide structure: How far are we? Proteins Struct Funct Genet 2006;65:889–897. doi: 10.1002/prot.21151 [DOI] [PubMed] [Google Scholar]
- 265. Elias JE, Haas W, Faherty BK, Gygi SP. Comparative evaluation of mass spectrometry platforms used in large‐scale proteomics investigations. Nat Methods 2005;2:667–675. doi: 10.1038/nmeth785 [DOI] [PubMed] [Google Scholar]
- 266. Sadygov RG, Cociorva D, Yates JR. Large‐scale database searching using tandem mass spectra: Looking up the answer in the back of the book. Nat Methods 2004;1:195–202. doi: 10.1038/nmeth725 [DOI] [PubMed] [Google Scholar]
- 267. Wang CK, Gruber CW, Cemazar M, Siatskas C, Tagore P, Payne N, Sun G, Wang S, Bernard CC, Craik DJ. Molecular grafting onto a stable framework yields novel cyclic peptides for the treatment of multiple sclerosis. ACS Chem Biol 2014;9:156–163. doi: 10.1021/cb400548s [DOI] [PMC free article] [PubMed] [Google Scholar]
- 268. Gonzalez‐Curiel I, Trujillo V, Montoya‐Rosales A, Rincon K, Rivas‐Calderon B, De Haro‐Acosta J, Marin‐Luevano P, Lozano‐Lopez D, Enciso‐Moreno JA, Rivas‐Santiago B. 1,25‐Dihydroxyvitamin D3 induces LL‐37 and HBD‐2 production in keratinocytes from diabetic foot ulcers promoting wound healing: An in vitro model. PLoS One 2014;9. doi: 10.1371/journal.pone.0111355 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 269. Chabance B, Jollès P, Izquierdo C, Mazoyer E, Francoual C, Drouet L, Fiat AM. Characterization of an antithrombotic peptide from kappa‐casein in newborn plasma after milk ingestion. Br J Nutr 1995;73:583–590. doi: 10.1079/BJN19950060 [DOI] [PubMed] [Google Scholar]
- 270. Kai‐Larsen Y, Luthje P, Chromek M, Peters V, Wang X, Holm A, Kádas L, Hedlund KO, Johansson J, Chapman MR, Jacobson SH, Romling U, Agerberth B, Brauner A. Uropathogenic Escherichia coli modulates immune responses and its curli fimbriae interact with the antimicrobial peptide LL‐37. PLoS Pathog 2010;6:1–16. doi: 10.1371/journal.ppat.1001010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 271. Kanthawong S, Bolscher JGM, Veerman ECI, Van Marle J, De Soet HJJ, Nazmi K, Wongratanacheewin S, Taweechaisupapong S. Antimicrobial and antibiofilm activity of LL‐37 and its truncated variants against Burkholderia pseudomallei . Int J Antimicrob Agents 2012;39:39–44. doi: 10.1016/j.ijantimicag.2011.09.010 [DOI] [PubMed] [Google Scholar]
- 272. Mishra B, Golla RM, Lau K, Lushnikova T, Wang G. Anti‐Staphylococcal biofilm effects of human cathelicidin peptides. ACS Med Chem Lett 2016;7:117–121. doi: 10.1021/acsmedchemlett.5b00433 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 273. Overhage J, Campisano A, Bains M, Torfs ECW, Rehm BHA, Hancock REW. Human host defense peptide LL‐37 prevents bacterial biofilm formation. Infect Immun 2008;76:4176–4182. doi: 10.1128/IAI.00318-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 274. Zhu C, Tan H, Cheng T, Shen H, Shao J, Guo Y, Shi S, Zhang X. Human β‐defensin 3 inhibits antibiotic‐resistant Staphylococcus biofilm formation. J Surg Res 2013;183:204–213. doi: 10.1016/j.jss.2012.11.048 [DOI] [PubMed] [Google Scholar]
- 275. Pusateri CR, Monaco EA, Edgerton M. Sensitivity of Candida albicans biofilm cells grown on denture acrylic to antifungal proteins and chlorhexidine. Arch Oral Biol 2009;54:588–594. doi: 10.1016/j.archoralbio.2009.01.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 276. Dutta D, Ozkan J, Willcox MDP. Biocompatibility of antimicrobial melimine lenses: Rabbit and human studies. Optom Vis Sci 2014;91:570–581. doi: 10.1097/OPX.0000000000000232 [DOI] [PubMed] [Google Scholar]
- 277. Lim K, Chua RRY, Ho B, Tambyah PA, Hadinoto K, Leong SSJ. Development of a catheter functionalized by a polydopamine peptide coating with antimicrobial and antibiofilm properties. Acta Biomater 2015;15:127–138. doi: 10.1016/j.actbio.2014.12.015 [DOI] [PubMed] [Google Scholar]
- 278. Etayash H, Jiang K, Thundat T, Kaur K. Impedimetric detection of pathogenic gram‐positive bacteria using an antimicrobial peptide from class IIa bacteriocins. Anal Chem 2014;86:1693–1700. doi: 10.1021/ac4034938 [DOI] [PubMed] [Google Scholar]
- 279. Kim H‐J, Brehm‐Stecher BF. Design and evaluation of peptide nucleic acid probes for specific identification of Candida albicans . J Clin Microbiol 2014;53(2):511–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 280. Lillehoj PB, Kaplan CW, He J, Shi W, Ho C‐M. Rapid, electrical impedance detection of bacterial pathogens using immobilized antimicrobial peptides. J Lab Autom 2014;19:42–49. doi: 10.1177/2211068213495207 [DOI] [PubMed] [Google Scholar]
- 281. Ahmad A, Ranjan S, Zhang W, Zou J, Pyykko I, Kinnunen PKJ. Novel endosomolytic peptides for enhancing gene delivery in nanoparticles. Biochim Biophys Acta 2015;1848:544–553. doi: 10.1016/j.bbamem.2014.11.008 [DOI] [PubMed] [Google Scholar]
- 282. Keohane K, Brennan D, Galvin P, Griffin BT. Silicon microfluidic flow focusing devices for the production of size‐controlled PLGA based drug loaded microparticles. Int J Pharm 2014;467:60–69. doi: 10.1016/j.ijpharm.2014.03.051 [DOI] [PubMed] [Google Scholar]
- 283. Hoffmann W, Richter K, Kreil G. A novel peptide designated PYLa and its precursor as predicted from cloned mRNA of Xenopus laevis skin. EMBO J 1983;2:711–714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 284. Pichereau C, Allary C. Therapeutic peptides under the spotlight. Eur Biopharm Rev 2005,88–93. [Google Scholar]
- 285. Marr AK, Gooderham WJ, Hancock RE. Antibacterial peptides for therapeutic use: obstacles and realistic outlook. Curr Opin Pharmacol 2006;6(5):468–72. [DOI] [PubMed] [Google Scholar]
- 286. Lohner SEB, Lohner K. Optimization and high‐throughput screening of antimicrobial peptides. Curr Pharm Des 2010;16(28):3204–11. [DOI] [PubMed] [Google Scholar]
- 287. Lamb HM, Wiseman LR. Pexiganan acetate. Drugs 1998;56(6):1047–52. [DOI] [PubMed] [Google Scholar]
- 288. Trotti A, Garden A, Warde P, Symonds P, Langer C, Redman R, Pajak TF, Fleming TR, Henke M, Bourhis J, Rosenthal DI, Junor E, Cmelak A, Sheehan F, Pulliam J, Devitt‐Risse P, Fuchs H, Chambers M, O'Sullivan B, Ang KK. A multinational, randomized phase III trial of iseganan HCl oral solution for reducing the severity of oral mucositis in patients receiving radiotherapy for head‐and‐neck malignancy. Int J Radiat Oncol Biol Phys 2004;58:674–681. doi: 10.1016/S0360-3016(03)01627-4 [DOI] [PubMed] [Google Scholar]
- 289. Mackin WM. Neuprex XOMA Corp. IDrugs 1998;1:715–723. [PubMed] [Google Scholar]
- 290. Rosenblum JS, Kozarich JW. Prolyl peptidases: A serine protease subfamily with high potential for drug discovery. Curr Opin Chem Biol 2003;7(4):496–504. [DOI] [PubMed] [Google Scholar]
- 291. Egleton RD, Davis TP. Bioavailability and transport of peptides and peptide drugs into the brain. Peptides 1997;18(9):1431–9. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Table S1: Distribution of Known Antimicrobial Peptides Listed in The Antimicrobial Peptides Database by Activity Spectrum (2016)
Supplementary Table S2: Distribution of Known Antimicrobial Peptides Listed in The Antimicrobial Peptides Database by Structural Information (2016)