

Abstract
Infectious Bronchitis virus (IBV) genotype Q1 was detected for the first time in China in 1996, and then spread worldwide. The first report of Q1 genotype in Italy occurred in 2011 and a deep molecular investigation of a Q1 isolated in Italy in 2013 has led to speculation regarding the origin of this genotype. Phylogenetic analysis of the S1 sequence of a Q1 Italian strain revealed a close relationship with sequences of the 624I strains circulating in Italy in the early 1990s and this led to the idea that 624I was an ancestor of the Q1 genotype. Despite the fact that most heterogeneity of IBVs occurs in the S1 gene, the sequence analysis of this gene alone was not sufficient to confirm or deny this hypothesis. In the present study, an Italian 624I (gammaCoV/AvCov/Ck/Italy/IP14425/96) was fully sequenced for the first time and compared to all available complete Q1 genome sequences. This analysis confirmed the genetic correlation between GammaCoV/AvCov/Ck/Italy/IP14425/96 and Q1 strains, suggesting a common origin between 624I and Q1 genotypes.
1. INTRODUCTION
Infectious bronchitis (IB) is an avian disease distributed worldwide that represents one of the most persistent health problems of the commercial poultry industry (Cook, Jackwood, & Jones, 2012; de Wit, Cook, & van der Heijden, 2011). It is caused by a Gammacoronavirus called Infectious Bronchitis Virus (IBV) and has a positive sense single‐stranded 27.6 kb RNA genome (Jackwood & De Wit, 2013). The IBV genome can evolve rapidly by mutation and recombination events, resulting in the emergence of new IBV variants which sometimes confer minimal or negligible cross‐protection (de Wit et al., 2011). A majority of such variants cause a transitory problem, which then disappear or remain confined into a specific geographical region. However, a few variants can persist and spread to new areas where they continue to cause disease (Jackwood, 2012; de Wit et al., 2011).
Recently, genotypes of Asian origin have spread to cause worldwide disease and major economic losses (de Wit et al., 2011). IBV Q1 is one such genotype. It was first detected in China in 1996 (Yu et al., 2001) and then reported in Asia, Middle East, Europe, and South America (Ababneh, Dalab, Alsaad, & Al‐Zghoul, 2012; Huang, Lee, Cheng, & Wang, 2004; Jackwood, 2012; Marandino et al., 2015; Rimondi et al., 2009). In Italy, the Q1 genotype was reported for the first time in 2011 after causing an outbreak of disease associated with respiratory signs, increased mortality, kidney lesions, and proventriculitis (Toffan et al., 2013). Since then, the genotype has been continuously detected in Italy (Massi et al., 2015). Phylogenetic analysis performed using full or partial S1 sequences showed a high identity (>99%) with Chinese Q1 isolates (Franzo et al., 2015; Massi et al., 2015; Toffan et al., 2013). At a similar period, a high identity (94.1%) between those strains and strains belonging to the 624I genotype was observed (Franzo et al., 2015; Massi et al., 2015), such that the recently proposed new IBV nomenclature based on the S1 sequence placed them in the same lineage (GI‐16) (Valastro et al., 2016).
IBV 624I had been reported for the first time in Italy in 1993 (Capua, Gough, Mancini, Casaccia, & Weiss, 1994; Capua et al., 1999), producing disease associated with kidney lesions and drop in egg production in breeders and layers (Capua, Grasso, Ferdinandi, Weiss, & Casaccia, 1996). This genotype continued to be detected in Italy until 2004 and then reappeared from 2010 when it was again detected in a few broiler farms affected by respiratory disease located in different areas of Italy. In the following years, the number of detections increased (Massi, 2013) but since 2013 the 624I genotype has not been further detected (Massi et al., 2015).
A recent retrospective study carried out on 123 IBV strains isolated in Italy between 1963 and 1989 revealed that 624I genotype had not only circulated long before its first reporting in 1993, but that in fact it has been one of the major IBV genotypes circulating in the Country at that time (Taddei et al., 2012). Evidence of the presence of this genotype has also been found in Slovenia, where several 624I strains were isolated between 1991 and 1999 (Krapez, Slavec, Barlic‐Maganja, & Rojs, 2010), in Poland and South Africa (Capua et al., 1999) and eventually in Russia where 624I genotype was reported in 2002 (Bochkov, Batchenko, Shcherbakova, Borisov, & Drygin, 2006).
The high identity observed between Q1 and 624I genotypes raises questions regarding their possible related origins. IBV 624I has been hypothesized to be an ancestor of the Q1 genotype (Franzo et al., 2015; Massi, 2013), but unfortunately the unavailability of any 624I full genome sequence did not allow such final conclusions to be drawn.
In the present study, an IBV 624I was fully sequenced and phylogenetic analysis was performed both using a dataset based on available IBV full‐length genome sequences and, due to the larger number of published sequences, a dataset based on full S1 gene. In addition, recombination analysis was carried out using the complete IBV 624I and Q1 strains.
2. MATERIALS AND METHODS
2.1. Virus
IBV 624I strain was isolated in 1996 during a disease outbreak in chicken farms located in Northern Italy. The virus was isolated in specific pathogen‐free (SPF) chicken eggs and the 3th passage was propagated in SPF chicken embryo tracheal organ cultures (TOC). After isolation, the virus underwent serological analysis, resulting as belonging to the 624I serotype. In this study, this virus is named gammaCoV/AvCov/Ck/Italy/IP14425/96.
2.2. RNA extraction, RT‐PCR, and sequencing
The RNA was extracted from the supernatant of infected TOC using Qiamp viral RNA mini kit (Qiagen, Hilden, Germany) following the manufacture's protocol. Viral RNA was firstly retro‐transcribed using Super Script III enzyme (Invitrogen, Carlsbad, CA, USA), then amplified using Ranger enzyme (Bioline, London, UK) according to the manufacturer's instructions. Retrotranscription, amplification, and sequencing were carried out using primers previously designed for IBV full genome sequencing (Franzo et al., 2015; Listorti et al., 2017). Where primers did not work due to sequence differences, new primers were designed based on the newly determined sequences flanking those genome regions (Table 1). Sequencing was performed by Source BioScience (Nottingham, UK). Each genome fragment was sequenced twice. Where gammaCoV/AvCov/Ck/Italy/IP14425/96a sequence differed from those of Q1 strains, the locations were sequenced again, starting with a new retrotranscription of the region.
Table 1.
Primers used for reverse transcription, PCR and sequencing
| RT | Sequence 5′‐3′ | Sequencing | Sequences 5′‐3′ |
|---|---|---|---|
| 2.06neg | tttagtaaaaagaccacc | 0.67+ | cctaaggattacgctgatgcttttgc |
| 4.26neg | catacttttgcgcatc | 1.26+ | ttcgcaggaacttgtcttgcaagc |
| 6.18neg | agaaaacctacaccag | 2.52+ | tagaggaatgtcacagcttggtgc |
| 8.18neg | gtaaagaatgtactaaac | 3.04+ | tacaccaatgtcacagcttggtgc |
| 10.10neg | cacagttgtgtgcactaactcaaag | 3.10+ | ctctcgatgttgtgaatttaccatctgg |
| 12.10neg | ctccataagaatcctg | IB4.6+a | gtacggatgaagtaatagaagcttc |
| 14.13neg | taaaacttggttgttcc | 4.80+ | tgattgtgatgttgtgaatttaccatctgg |
| 16.10neg | ttcacataaagcatcaac | IB5.15nega | catcagtatcaggtgttaacttataag |
| QX18.10neg | catagaagaagaatggcatagctttc | 5.32+ | ctattagtcttagggcaatatggg |
| 19.73neg | caaaatgcattactcgc | IB5.70+a | gtgtggtttatttacacaagtaatccag |
| 22.51neg | catatcttctttttgacc | 6.53+ | gttaaacctacagcatatgcttacc |
| 24.08neg | tttgaatcattaaacagac | IB6.7nega | ctaatcgttctgaaagtgcctgatcaag |
| 26.24neg | ccaagatacatttccag | IB7.15+a | ctttataacaagatctggtgctaaac |
| 27.89neg | Ttgctctaactctatac | 7.17+ | aatgctcctccggtagtatggaag |
| Dta‐Adaptnegb | gcatctcgaggcttgtggcttttttttttttttttttttta | IB8.2nega | caacccaaactagcattattgtaaacac |
| Dtc‐Adaptnegb | gcatctcgaggcttgtggctttttttttttttttttttttc | 8.60+ | gtatgatggcaacgagtttgttgg |
| Dtg‐Adaptnegb | gcatctcgaggcttgtggctttttttttttttttttttttg | IB8.61nega | taccaacaaactcgttgccatc |
| PCR | Sequence 5′‐3′ | 9.26+ | cctgtcactatgcgttctaatggtac |
| 0.06+ | gcgctagatttccaacttaacaaaacg | 10.60+ | ggtaaatccacctaaaactgtgtggg |
| 2.03neg | gacttgcgaaacaagatgccaaatgcc | 11.13+ | gaagttagatagcatggagagacg |
| 1.92+ | tggaggcttgcatatggaaaagtgcg | 12.64+ | gacttaaagtcagaagtaacagctg |
| 4.20neg | ggtataaagaggatttctttatcctcaagatcatg | 13.18+ | gatctcctcaagtatgattatactgagg |
| 4,10+ | cggaggatggtgttaaataccgc | QX13.90+ | gaggtgacgtctaaatattttgaatg |
| 6.08neg | caaataatattagaaagaccaaataaagccaattcc | 14.69+ | caaggtcttgtagcagatatttctgg |
| 5.90+a | gactatggtaaagactcatttgacg | 15.25+ | aagtgttgctatgaccatgtcatgc |
| 8.05neg | cctggtttagtatactcacatacactacc | IB15.60+a | ggtgcagcttggtgattttacctttg |
| 7.97+ | cctaatggtgttaggcttatagttcc | IB16.40nega | ctataaccttgaaacactgacgtg |
| 10.10neg | gtactaaagactacaggatcataccattg | QX16.80neg | gaatcagctgtaacacagaatataac |
| 10.02+ | cagttattattggagtttgtgctgaag | 16.62+ | catgaaagtggctcagcctacaac |
| 12.08neg | gaatcctgatccggagttggacttggc | 17.18+ | caacatgttttataacacgtgatgaggc |
| 12.01+ | gtggcagcaggtaatcaacctttagg | 18.02neg | ctgcttgacattgggtactactggattc |
| 14.12neg | ataaaacttggttgttccaataactacagg | 18.18+ | taacctacctggttgtaatggtgg |
| 14.05+ | gtgtctatcctttctactatgactaataggc | 18.71+ | gaagagaaatattcgcacactgcc |
| 16.09neg | cacataaagcatcaacagctgcatgag | 19.67nega | gtattgacagagttgtgtatactttggc |
| QX15.06+ | gatgattgcactcgcatagtacctc | 19.46+ | gtaacagtgtcaattgattaccatagc |
| QX18.10neg | catagaagaagaatggcatagctttc | 19.97+ | gatagccaataatggcaatgatgacg |
| 17.62+a | tactcaggcttatgcttgttggaagc | IB20.40+a | cattgtttatagtgtctcttttgtttgcac |
| 19.67neg | gtattgacagagttgtgtatactttgcc | IB21.70nega | gtcacaaattgcccttatgtaagttatgg |
| 19.46+ | gtaacagtgtcaattgattaccatagc | IB21.95+a | ccagcagtttgtagtttctggtgg |
| 22.26neg | tccatacgcgtttgtatgtactcatctg | IB23.60nega | cacgtgcagtgatgtagtaactacc |
| IB21.95+ | ccagcagtttgtagtttctggtgg | QX24.20neg | ctacttacactgtttcaattgttttctc |
| IB24.07neg | gaatcattaaacagactttttaggtctg | IB24.70+a | gtactcttggtactgaacaagcag |
| 23.99+ | cattatgcctctaatgagtaagtgtgg | IB25.60nega | ctcttgaaaagagagcatgaaacaaagagg |
| 26.24nega | ccaagatacatttccag | QX27.20a,b | cctacatgtctatcgccaggg |
| 26.02+ | gaaaagcgcgaatttatctgagagaagg | ||
| 27.83neg | catagccaattaaacttaacttaaactaaaatttagctc | ||
| 26.49+ | gatagccaagatggtatagtgtggg | ||
| Adapt negb | gcatctcgagggttgtggc |
Primer names generally indicate approximate binding positions in the 624I genome.
Coding and anticoding sense primers are labelled + and neg, respectively.
aIndicates primers designed for the 624I IBV strain sequencing based on the newly determined flanking sequences. bIndicates primers used for the retrotranscription, amplification, and sequencing of the 3′END of the genome.
This article is being made freely available through PubMed Central as part of the COVID-19 public health emergency response. It can be used for unrestricted research re-use and analysis in any form or by any means with acknowledgement of the original source, for the duration of the public health emergency.
2.3. RT‐PCR of the 3′ END of genome
3′ end of the genome was determined using a 3′RACE protocol previously described (Laconi et al., 2016). Briefly, RT was performed with a primer containing 20 Ts followed by an adaptor sequence at its 5′ terminus. This was amplified by PCR using two primers, one within the end of the genome and one matching the adaptor (Table 1). These PCR products were sequenced towards the polyA tail.
2.4. Sequences analysis and comparison
Chromatograms were analysed using the program Chromas (http://technelysium.com.au/wp/chromas/) and sequences aligned using BioEdit (http://www.mbio.ncsu.edu/bioedit/bioedit.html) against the genome of IBV strains gammaCoV/Ck/Italy/I2022/13 (KP780179) and CK/CH/LDL/97I (JX195177). Open Reading Frame (ORF) prediction was carried out using ORFfinder program (https://www.ncbi.nlm.nih.gov/orffinder/).
2.5. Complete genome sequences analysis
A dataset containing 313 complete genome sequences (nt) of IBV was downloaded from ViPr, an open source bioinformatics database and analysis resource for virology research. To minimize the computational load, cd‐hit‐est test of the CD‐HIT Suite (Li & Godzik, 2006) was used to cluster sequences that shared over 98% identity and a prototype sequence within each cluster was selected. After clustering, 187 representative sequences remained. Sequences were aligned using ClustaW and phylogenetic analysis was carried out with MEGA7 software (Kumar, Stecher, & Tamura, 2016) using Maximum Likelihood method with Tamura‐Nei substitution model and 1,000 bootstrap replicates to assess the robustness of the branches.
2.6. S1 gene sequences analysis
A dataset containing all available complete or nearly complete S1 gene sequences (at least 1,000 bp) was downloaded from ViPr. After clustering, 320 sequences remained. The sequences were aligned using ClustalW method, and a phylogenetic tree was constructed using the Maximum Likelihood method with Kimura 2 parameter substitution model and 1,000 bootstrap replicates to assign confidence level to the branches in MEGA 7 software. Based on these results, a subset of sequences clustering with gammaCoV/AvCov/Ck/Italy/IP14425/96 was selected and expanded to its original number of taxa. Sequences belonging to this dataset were aligned with ClustalW method, and a phylogenetic tree was reconstructed using the parameters previously described.
2.7. Recombination event analysis
Presence of past recombination events for strain gammaCoV/AvCov/Ck/Italy/IP14425/96 was evaluated using RDP4 software (http://web.cbio.uct.ac.za/~darren/rdp.html)(Martin; Martin, Murrell, Khoosal, & Muhire, 2017). Occurrence of possible recombination events was also evaluated for the available Q1 full genome sequences: gammaCoV/Ck/Italy/I2022/13 (KP780179), an Italian isolate from 2013 and CK/CH/LDL/97I (JX195177), a Chinese isolate from 1997. The Kimura 2 parameter substitution model with a window size of 200 nucleotides and a step size of 20 nucleotides was used to calculate the pairwise percentage of identity between gammaCoV/AvCov/Ck/Italy/IP14425/96, the Q1 strains and 13 complete genome sequences of relevant strains, selected on the previous phylogenetic analysis. Phylogenetic analysis was performed for those genome portions where a sharp change in percentage of identity strongly suggested recombination events using a dataset including all the sequences available for the given regions.
3. RESULTS
3.1. Genome organization of strain gammaCoV/AvCov/Ck/Italy/IP14425/96
A consensus sequence of 27.573 bp was obtained (minimum coverage 2X), with the 5′ UTR incomplete by approximately 100 nt. The ORF analysis predicted 13 ORFs and revealed the following genome organization: 5′UTR‐1a‐1b‐S‐3a‐3b‐E‐M‐4b‐4c‐5a‐5b‐N‐6b‐3′UTR (Table 2). The same genome organization was observed for viruses gammaCoV/Ck/Italy/I2022/13, CK/CH/LDL/97I and UY/09/CA/01, all belonging to the Q1 genotype (Table 2).
Table 2.
Genomic organization of 624I and Q1 strains
| Gene | Genome position | |||
|---|---|---|---|---|
| gammaCoV/AvCoV/Ck/Italy/P14425/96 | gammaCoV/Ck/Italy/I2022/13 | CK/CH/LDL/97I | UY/09/CA/01 | |
| 1a | 433‐12288 | 432‐12290 | 433‐12291 | 432‐12254 |
| 1ab | 12363‐20321 | 12365‐20323 | 12366‐20324 | 12329‐20287 |
| S | 20272‐23772 | 20274‐23774 | 20275‐23775 | 20238‐23711 |
| 3a | 23772‐23945 | 23774‐23947 | 23775‐23948 | 23738‐23911 |
| 3b | 23945‐24136 | 23947‐24141 | 23938‐24142 | 23911‐24105 |
| E | 24117‐24404 | 24122‐24430 | 24123‐24431 | 24086‐24394 |
| M | 24394‐25074 | 24423‐25103 | 24424‐25104 | 24387‐25067 |
| 4b | 25075‐25359 | 25104‐25388 | 25105‐25377 | 25068‐25352 |
| 4c | 25280‐25450 | 25309‐25470 | 25310‐25387 | 25273‐25434 |
| 5a | 25434‐25631 | 25454‐25651 | 25466‐25663 | 25418‐25614 |
| 5b | 25628‐25876 | 25648‐25896 | 25660‐25908 | 25612‐25860 |
| N | 25819‐27045 | 25839‐27068 | 25851‐27080 | 25803‐27032 |
| 6b | 27054‐27278 | 27058‐27222 | 27089‐27403 | 27041‐27265 |
Strains belonging to the two genotypes show the same genome organization.
Accessory genes 4b, 4c, and 6b have been identified in the genome of all the four viruses.
This article is being made freely available through PubMed Central as part of the COVID-19 public health emergency response. It can be used for unrestricted research re-use and analysis in any form or by any means with acknowledgement of the original source, for the duration of the public health emergency.
3.2. Accession number
Sequence of the IBV strain gammaCoV/AvCov/Ck/Italy/IP14425/96 was submitted to the GenBank database and the following accession number was assigned: MG021194.
3.3. Phylogenetic analysis of full genomes of IBV strains
The phylogenetic analysis of the 187 representative full IBV genome sequences demonstrated that gammaCoV/AvCov/Ck/Italy/IP14425/96 clustered together with Q1 strains gammaCoV/Ck/Italy/I2022/13, CK/CH/LDL/97I and UY/09/CA/01, occupying a basal position in the specific cluster (Figure 1). In the same clade, the Uruguayan strain UY/11/CA/18 (MF421320), was also present, previously ascribed to the SAI genotype (Lineage G‐11) (Figure 1) (Marandino et al., 2015).
Figure 1.
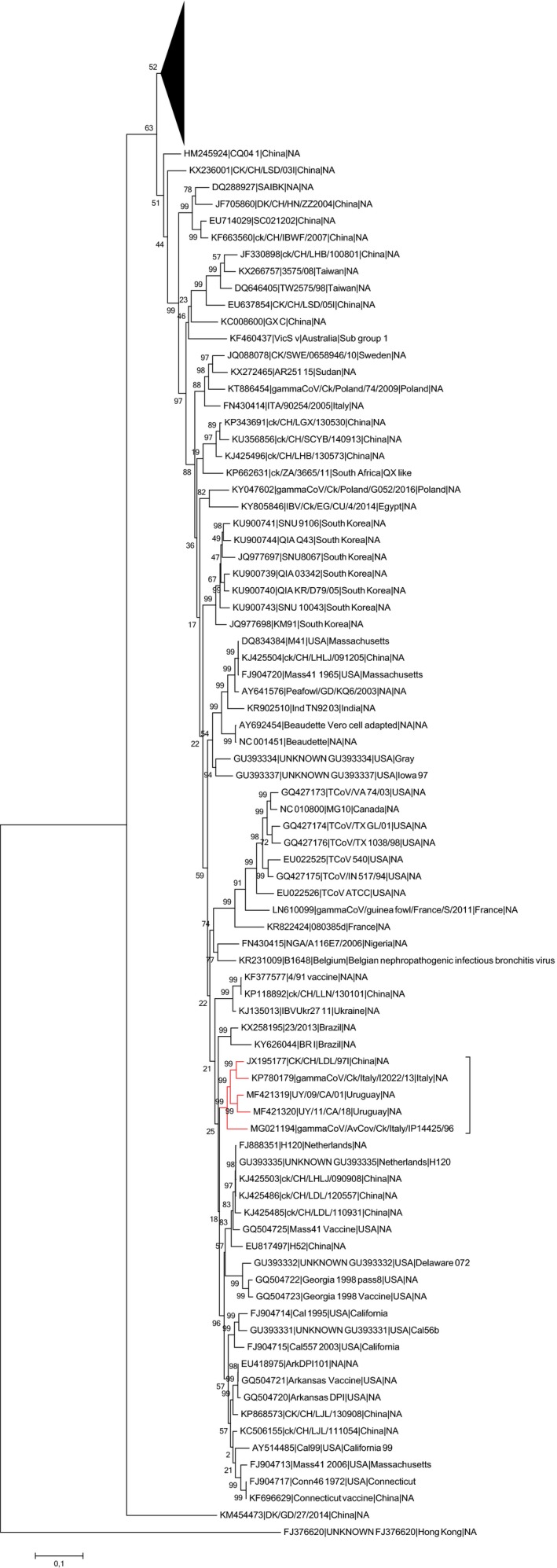
Phylogenetic tree based on Maximum Likelihood method with Tamura‐Nei substitution model constructed using 187 representative complete IBV and TCoV genomes. For easiness of representation, the upper part of the phylogenetic tree was compressed and the strains considered in the present study marked in red [Colour figure can be viewed at http://wileyonlinelibrary.com]
GammaCoV/AvCov/Ck/Italy/IP14425/96 624I strain showed the highest sequence identity with the Italian Q1 strain gammaCoV/Ck/Italy/I2022/13 (p‐distance 0.054), while the percentage of identity slightly decreased when the virus was compared to the Chinese (p‐distance 0.058) and the Uruguayan (p‐distance 0.062) Q1 strains.
3.4. Phylogenetic analysis of full S1 sequences
Phylogenetic analysis using a dataset characterized by 320 representative full S1 sequences showed that gammaCoV/AvCov/Ck/Italy/IP14425/96 clustered together with strains previously identified as 624/I genotype isolated in Italy in the late ‘80s and early ‘90s and strains belonging to Q1 genotype, isolated in Italy, in China, in Taiwan, and South America (Figure 2a). The highest sequence identity was observed with strain 624I/94/JQ901492.1 (p‐distance = 0.036) while the identity was lower when compared to Q1 strains (data not shown).
Figure 2.

(a) Phylogenetic tree based on Maximum Likelihood method with Kimura‐2 model constructed using 320 S1 gene complete sequences. For easiness of representation, strain reported in the present study and closely related strains, are marked in red. (b) Expanded phylogenetic tree based on Maximum Likelihood method with Kimura‐2 model constructed using S1 complete sequences closely related to gammaCoV/AvCov/Ck/Italy/IP14425/96 [Colour figure can be viewed at http://wileyonlinelibrary.com]
The subtree obtained with the expanded dataset shows three clades, of which one contained all 624/I strains and occupied a basal position with respect to the others. In the remaining two clades, Q1 Italian, Chinese and some of the Taiwanese strains, cluster together, while the remaining Q1 Taiwan strains and all South American Q1 strains form a distinctive phylogenetic group (Figure 2b).
3.5. Recombination analysis
Recombination analysis was performed to assess the possible recombinant nature of the 624I strain gammaCoV/AvCov/Ck/Italy/IP14425/96 and of the Q1 strains gammaCoV/Ck/Italy/I2022/13 and CK/CH/LDL/97I.
Possible recombination events were identified in the 1a gene sequence of the strain CK/CH/LDL/97I with a H120 vaccine strain (FJ888351) (Figure 3a). Neither gammaCoV/Ck/Italy/I2022/13 nor gammaCoV/AvCov/Ck/Italy/IP14425/96 Q1 strains showed a similar recombination event in the 1a gene (Figures 3b,c).
Figure 3.
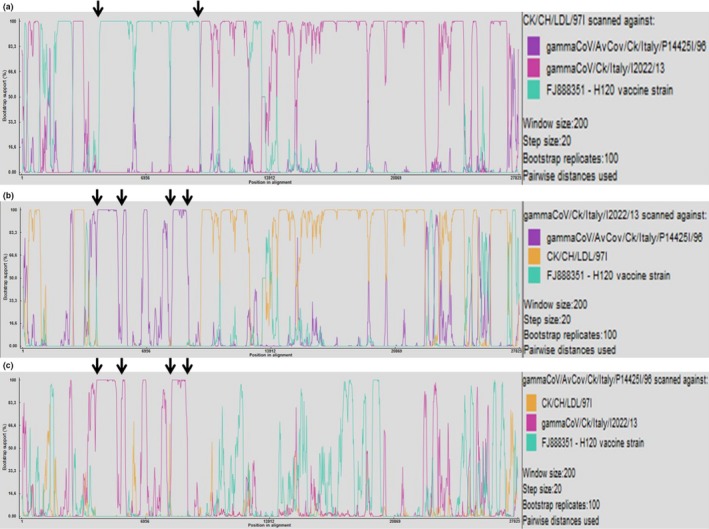
RDP screenshots displaying the possible recombination events associated with CK/CH/LDL/97I (a), gammaCoV/Ck/Italy/I2022/13 (b), and gammaCoV/AvCov/Ck/Italy/P14425/96 (c) Each panel displays the pairwise identities among the possible recombinant and its putative parents. Crossover sites indicating sharp changes in pairwise identity are pointed out by arrows. (a) Comparisons among the putative recombinant CK/CH/LDL/97I show sharp changes in the pairwise identity within the 1a gene, H120 vaccine strain FJ888351 has been identified as putative parental strains for that genomic region. (b) Comparison among the putative recombinant gammaCoV/Ck/Italy/I2022/13 shows sharp changes in the pairwise identity within the 1a gene; strain gammaCoV/AvCov/Ck/Italy/P14425I/96 has been identified as major parental strain for that region. (c) Comparison among the putative recombinant gammaCoV/AvCov/Ck/Italy/P14425/96 shows sharp changes in the pairwise identity within the 1a gene; strain gammaCoV/Ck/Italy/I2022/13 has been identified as major parental strain for that genomic region [Colour figure can be viewed at http://wileyonlinelibrary.com]
A phylogenetic analysis was performed considering only the 1a gene, revealing that CK/CH/LDL/97I clustered with H120 and Mass strains, while gammaCoV/AvCov/Ck/Italy/IP14425/96 and gammaCoV/Ck/Italy/I2022/13 form a distinctive clade together with a QX‐like Italian strain (ITA/90254/2005 ‐ FN430414) (Figure 4).
Figure 4.

Phylogenetic tree based on Maximum Likelihood method with Tamura‐Nei substitution constructed model using 187 representative IBV and TCoV 1a genes. For easiness of representation, the upper part of the phylogenetic tree was compressed and the strains considered in the present study marked in red [Colour figure can be viewed at http://wileyonlinelibrary.com]
4. DISCUSSION
The genome of the IBV strain gammaCoV/AvCov/Ck/Italy/IP14425/96 isolated in Italy in 1996 was fully sequenced and this represents the first report of a full genome sequencing of a virus belonging to the 624I genotype. The isolate shows a genome organization slightly different when compared to the genome organization of most IBVs previously reported (5′UTR‐1a‐1b‐S‐3a‐3b‐E‐M‐5a‐5b‐N‐3′UTR) (Cavanagh, 2005), since ORF analysis showed the presence of accessory genes 4b, 4c, and 6b already reported for TCoV and other IBVs (Abolnik, 2015; Hewson, Ignjatovic, Browning, Devlin, & Noormohammadi, 2011). It is not clear whether the scarcity of reports of presence of the accessory genes 4b, 4c, and 6b in IBVs is due to their absence in some genomes; or whether it depends on algorithms and software used by other authors for those ORF's detections. A recent ORF analysis of the genome of the Q1 strain gammaCoV/Ck/Italy/I2022/13, (Marandino et al., 2017) did not support the presence of ORF 6b in contrast to the results presented here. On the contrary, a recent study confirmed the expression of the 4b protein after M41 IBV infection in vitro (Bentley, Keep, Armesto, & Britton, 2013). IBV accessory genes 3a, 3b, 5a, and 5b are known to be not necessary for viral replication, but several studies demonstrated their involvement in the pathogenicity of the virus (van Beurden et al., 2017; Kint, Dickhout et al., 2015; Kint, Fernandez‐Gutierrez et al., 2015, 2016). A similar function might be hypothesized also for genes 4b, 4c, and 6b, especially in the light that the 4b homologous gene in the MERS‐CoV has been reported as an antagonist of type I interferon response (Yang et al., 2013) and that the 6b homologue in SARS‐CoV was shown to be able to induce apoptosis (Ye, Wong, Li, & Xie, 2008). More studies need to be done to improve the knowledge on these three accessory genes, in particular whether they are peculiar of certain genotypes and whether their expression influences the pathogenicity or the tropism of the virus.
Phylogenetic analyses performed using two different datasets, one built with IBV complete genome sequences and one built with IBV complete or nearly complete S1 gene sequences, showed that 624I and Q1 genotypes clustered together. Our findings, strongly suggest a common origin between the two genotypes. The basal location of the 624I strain in both the phylogenetic trees, coupled with the epidemiological data available, suggests that this genotype might have played a role in the emergence of Chinese Q1. This model requires long‐distance intercontinental dispersion of the 624I genotype and this possibility is supported by its ability to circulate for extended periods within the same country (Taddei et al., 2012) and beyond a geographical area (Capua et al., 1999; Krapez et al., 2010). Unfortunately, there is no comprehensive model explaining the intercontinental dispersal of the 624I genotype. Some hypothesis can be proposed, such as migratory birds, illegal trading and poultry movement. Albeit speculative, these hypotheses seem to be supported by the detection of 624I genotype in Russia, which represents an intermediate position between Europe and the Far East (Bochkov et al., 2006). Further investigations are needed since, despite the IBV worldwide dissemination has been observed and accepted for other genotypes (Franzo et al., 2017), the mechanism behind this evidence is not fully understood.
The recombination analysis showed that the Chinese strain CK/CH/LDL/97I underwent recombination with a H120 vaccine strain, which has been previously demonstrated to be involved in recombination events leading to reversion to virulence and the emerge of new genotypes in China (Zhang et al., 2010). The identification of such a recombination event within the 1a gene might explain the relatively high genetic diversity between 624I strain gammaCoV/AvCov/Ck/Italy/IP14425/96 and Q1 Chinese strain CK/CH/LDL/97I, two viruses isolated only one year apart.
The absence of such recombination in the genome of Q1 Italian strain gammaCoV/Ck/Italy/I2022/13 suggests that not all Q1 strains emerged as a result of a recombination event with a H120 strain. The absence of a recombination event in the 1a gene of Q1 Italian strain gammaCoV/Ck/Italy/I2022/13 might indicate that Chinese and Italian Q1 strains are the result of independent evolutions from the 624I genotype. However, the huge differences in field conditions and therefore genetic pressures between the two countries, together with the phylogenetic results based on S1 gene sequences in which Q1 Italian strains cluster together with Chinese and Taiwanese strains, make this hypothesis highly unlikely.
Taken as a whole, the data presented in this study suggest the 624I genotype to be the ancestor of Q1.
Laconi A, Listorti V, Franzo G, et al. Molecular characterization of whole genome sequence of infectious bronchitis virus 624I genotype confirms the close relationship with Q1 genotype. Transbound Emerg Dis. 2019;66:207–216. 10.1111/tbed.13000
REFERENCES>
- Ababneh, M. , Dalab, A. E. , Alsaad, S. , & Al‐Zghoul, M. (2012). Presence of infectious bronchitis virus strain CK/CH/LDL/97I in the Middle East. ISRN Veterinary Science, 2012, 201721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abolnik, C. (2015). Genomic and single nucleotide polymorphism analysis of infectious bronchitis coronavirus. Infection, Genetics and Evolution, 32, 416–424. 10.1016/j.meegid.2015.03.033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bentley, K. , Keep, S. M. , Armesto, M. , & Britton, P. (2013). Identification of a noncanonically transcribed subgenomic mRNA of infectious bronchitis virus and other gammacoronaviruses. Journal of Virology, 87, 2128–2136. 10.1128/JVI.02967-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Beurden, S. J. , Berends, A. J. , Kramer‐Kuhl, A. , Spekreijse, D. , Chenard, G. , Philipp, H. C. , … Verheije, M. H. (2017). A reverse genetics system for avian coronavirus infectious bronchitis virus based on targeted RNA recombination. Virology Journal, 14, 109 10.1186/s12985-017-0775-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bochkov, Y. A. , Batchenko, G. V. , Shcherbakova, L. O. , Borisov, A. V. , & Drygin, V. V. (2006). Molecular epizootiology of avian infectious bronchitis in Russia. Avian Pathology, 35, 379–393. 10.1080/03079450600921008 [DOI] [PubMed] [Google Scholar]
- Capua, I. , Gough, R. E. , Mancini, M. , Casaccia, C. , & Weiss, C. (1994). A ‘Novel’ infectious bronchitis strain infecting broiler chickens in Italy. Journal of Veterinary Medicine Series B, 41(2), 83–89. 10.1111/j.1439-0450.1994.tb00211.x [DOI] [PubMed] [Google Scholar]
- Capua, I. , Grasso, G. , Ferdinandi, S. , Weiss, C. , & Casaccia, C. (1996). Studies on outbreak of nephritis and of drop in egg production associated with infectious bronchitis strain 624/I. Zootecnia International, 1, 49–51. [Google Scholar]
- Capua, I. , Minta, Z. , Karpinska, E. , Mawditt, K. , Britton, P. , Cavanagh, D. , & Gough, R. E. (1999). Co‐circulation of four types of infectious bronchitis virus (793/B, 624/I, B1648 and Massachusetts). Avian Pathology, 28, 587–592. 10.1080/03079459994380 [DOI] [PubMed] [Google Scholar]
- Cavanagh, D. (2005). Coronaviruses in poultry and other birds. Avian Pathology, 34, 439–448. 10.1080/03079450500367682 [DOI] [PubMed] [Google Scholar]
- Cook, J. K. , Jackwood, M. , & Jones, R. C. (2012). The long view: 40 years of infectious bronchitis research. Avian Pathology, 41, 239–250. 10.1080/03079457.2012.680432 [DOI] [PubMed] [Google Scholar]
- Franzo, G. , Listorti, V. , Naylor, C. J. , Lupini, C. , Laconi, A. , Felice, V. , … Cecchinato, M. (2015). Molecular investigation of a full‐length genome of a Q1‐like IBV strain isolated in Italy in 2013. Virus Research, 210, 77–80. 10.1016/j.virusres.2015.07.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franzo, G. , Massi, P. , Tucciarone, C. M. , Barbieri, I. , Tosi, G. , Fiorentini, L. , … Moreno, A. (2017). Think globally, act locally: Phylodynamic reconstruction of infectious bronchitis virus (IBV) QX genotype (GI‐19 lineage) reveals different population dynamics and spreading patterns when evaluated on different epidemiological scales. PLoS ONE, 12, e0184401 10.1371/journal.pone.0184401 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hewson, K. A. , Ignjatovic, J. , Browning, G. F. , Devlin, J. M. , & Noormohammadi, A. H. (2011). Infectious bronchitis viruses with naturally occurring genomic rearrangement and gene deletion. Archives of Virology, 156, 245–252. 10.1007/s00705-010-0850-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang, Y. P. , Lee, H. C. , Cheng, M. C. , & Wang, C. H. (2004). S1 and N gene analysis of avian infectious bronchitis viruses in Taiwan. Avian Diseases, 48, 581–589. 10.1637/7186-033004R [DOI] [PubMed] [Google Scholar]
- Jackwood, M. W. (2012). Review of infectious bronchitis virus around the world. Avian Diseases, 56, 634–641. 10.1637/10227-043012-Review.1 [DOI] [PubMed] [Google Scholar]
- Jackwood, M. , & de Wit, J. J. (2013). Infectious Bronchitis In Swayne G., McDougald L. R., Nolan L. K., Suarez D. L. & Nair V. L. (Eds.), Diseases of Poultry (pp. 139–159), 13th edn, Ames, IA: Blackwell Publishing Professional. [Google Scholar]
- Kint, J. , Dickhout, A. , Kutter, J. , Maier, H. J. , Britton, P. , Koumans, J. , … Forlenza, M. (2015). Infectious bronchitis coronavirus inhibits STAT1 signaling and requires accessory proteins for resistance to type I interferon activity. Journal of Virology, 89, 12047–12057. 10.1128/JVI.01057-15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kint, J. , Fernandez‐Gutierrez, M. , Maier, H. J. , Britton, P. , Langereis, M. A. , Koumans, J. , … Forlenza, M. (2015). Activation of the chicken type I interferon response by infectious bronchitis coronavirus. Journal of Virology, 89, 1156–1167. 10.1128/JVI.02671-14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kint, J. , Langereis, M. A. , Maier, H. J. , Britton, P. , van Kuppeveld, F. J. , Koumans, J. , … Forlenza, M. (2016). Infectious Bronchitis Coronavirus Limits Interferon Production by Inducing a Host Shutoff That Requires Accessory Protein 5b. Journal of Virology, 90, 7519–7528. 10.1128/JVI.00627-16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krapez, U. , Slavec, B. , Barlic‐Maganja, D. , & Rojs, O. Z. (2010). Molecular analysis of infectious bronchitis viruses isolated in Slovenia between 1990 and 2005: A retrospective study. Virus Genes, 41, 414–416. 10.1007/s11262-010-0528-x [DOI] [PubMed] [Google Scholar]
- Kumar, S. , Stecher, G. , & Tamura, K. (2016). MEGA7: Molecular evolutionary genetics analysis Version 7.0 for bigger datasets. Molecular Biology and Evolution, 33, 1870–1874. 10.1093/molbev/msw054 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laconi, A. , Clubbe, J. , Falchieri, M. , Lupini, C. , Cecchinato, M. , Catelli, E. , … Naylor, C. J. (2016). A comparison of AMPV subtypes A and B full genomes, gene transcripts and proteins led to reverse‐genetics systems rescuing both subtypes. Journal of General Virology, 97, 1324–1332. 10.1099/jgv.0.000450 [DOI] [PubMed] [Google Scholar]
- Li, W. , & Godzik, A. (2006). Cd‐hit: A fast program for clustering and comparing large sets of protein or nucleotide sequences. Bioinformatics, 22, 1658–1659. 10.1093/bioinformatics/btl158 [DOI] [PubMed] [Google Scholar]
- Listorti, V. , Laconi, A. , Catelli, E. , Cecchinato, M. , Lupini, C. , & Naylor, C. J. (2017). Identification of IBV QX vaccine markers : Should vaccine acceptance by authorities require similar identifications for all live IBV vaccines? Vaccine, 35, 5531–5534. 10.1016/j.vaccine.2017.06.021 [DOI] [PubMed] [Google Scholar]
- Marandino, A. , Pereda, A. , Tomas, G. , Hernandez, M. , Iraola, G. , Craig, M. I. , … Perez, R. (2015). Phylodynamic analysis of avian infectious bronchitis virus in South America. Journal of General Virology, 96, 1340–1346. 10.1099/vir.0.000077 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marandino, A. , Tomas, G. , Panzera, Y. , Greif, G. , Parodi‐Talice, A. , Hernandez, M. , … Perez, R. (2017). Whole‐genome characterization of Uruguayan strains of avian infectious bronchitis virus reveals extensive recombination between the two major South American lineages. Infection, Genetics and Evolution, 54, 245–250. 10.1016/j.meegid.2017.07.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin, D. P. , Murrell, B. , Khoosal, A. , & Muhire, B. (2017). Detecting and Analyzing Genetic Recombination Using RDP4. Methods in Molecular Biology, 1525, 433–460. 10.1007/978-1-4939-6622-6 [DOI] [PubMed] [Google Scholar]
- Massi, P. (2013). Situazione epidemiologica della Bronchite Infettiva in Italia. Rivista di Medicina Veterinaria, Speciale 2013, 13–20. [Google Scholar]
- Massi, P. , Barbieri, I. , Fiorentini, L. , Casadio, M. , Parigi, M. , & Tosi, G. (2015). Analisi molecolare di ceppi del Virus della Bronchite Infettiva aviare negli anni 2013 e 2014. considerazioni sui genotipi circolanti in Italia e in altri paesi europei ed extra‐europei. Atti del LIV convegno annuale della Societa’ Italiana di Patologia Aviare, Forlì, 1, 220–229. [Google Scholar]
- Rimondi, A. , Craig, M. I. , Vagnozzi, A. , Konig, G. , Delamer, M. , & Pereda, A. (2009). Molecular characterization of avian infectious bronchitis virus strains from outbreaks in Argentina (2001‐2008). Avian Pathology, 38, 149–153. 10.1080/03079450902737821 [DOI] [PubMed] [Google Scholar]
- Taddei, R. , Tosi, G. , Boniotti, M. B. , Casadio, M. , Fiorentini, L. , Fabbi, M. , & Massi, P. (2012) Caratterizzazione molecolare di ceppi del virus della bronchite infettiva aviare isolati in Italia tra il 1963 ed il 1989. Proceedings of 51 Convegno annuale Societa’ Italiana Patologia Aviare (SIPA) : 11‐12 Ottobre 2012, Salsomaggiore Terme (PR) 332‐341.
- Toffan, A. , Bonci, M. , Bano, L. , Bano, L. , Valastro, V. , Vascellari, M. , … Terregino, C. (2013). Diagnostic and clinical observation on the infectious bronchitis virus strain Q1 in Italy. Veterinaria Italiana, 49, 347–355. [DOI] [PubMed] [Google Scholar]
- Valastro, V. , Holmes, E. C. , Britton, P. , Fusaro, A. , Jackwood, M. W. , Cattoli, G. , & Monne, I. (2016). S1 gene‐based phylogeny of infectious bronchitis virus: An attempt to harmonize virus classification. Infection, Genetics and Evolution, 39, 349–364. 10.1016/j.meegid.2016.02.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Wit, J. J. S. , Cook, J. K. , & van der Heijden, H. M. (2011). Infectious bronchitis virus variants: A review of the history, current situation and control measures. Avian Pathology, 40, 223–235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang, Y. , Zhang, L. , Geng, H. , Deng, Y. , Huang, B. , Guo, Y. , … Tan, W. (2013). The structural and accessory proteins M, ORF 4a, ORF 4b, and ORF 5 of Middle East respiratory syndrome coronavirus (MERS‐CoV) are potent interferon antagonists. Protein & Cell, 4, 951–961. 10.1007/s13238-013-3096-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ye, Z. , Wong, C. K. , Li, P. , & Xie, Y. (2008). A SARS‐CoV protein, ORF‐6, induces caspase‐3 mediated, ER stress and JNK‐dependent apoptosis. Biochimica et Biophysica Acta, 1780, 1383–1387. 10.1016/j.bbagen.2008.07.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu, L. , Jiang, Y. , Low, S. , Wang, Z. , Nam, S. H. , Liu, W. , & Kwang, J. (2001). Characterization of three infectious bronchitis virus isolates from china associated withproventriculus in vaccinated chickens. Avian Diseases, 45, 416–424. 10.2307/1592981 [DOI] [PubMed] [Google Scholar]
- Zhang, Y. , Wang, H. N. , Wang, T. , Fan, W. Q. , Zhang, A. Y. , Wei, K. , … Yang, X. (2010). Complete genome sequence and recombination analysis of infectious bronchitis virus attenuated vaccine strain H120. Virus Genes, 41, 377–388. 10.1007/s11262-010-0517-0 [DOI] [PMC free article] [PubMed] [Google Scholar]