

Abstract
Since its first emergence in 1998 in Malaysia, Nipah virus (NiV) has become a great threat to domestic animals and humans. Sporadic outbreaks associated with human‐to‐human transmission caused hundreds of human fatalities. Here, we collected all available NiV sequences and combined phylogenetics, molecular selection, structural biology and receptor analysis to study the emergence and adaptive evolution of NiV. NiV can be divided into two main lineages including the Bangladesh and Malaysia lineages. We formly confirmed a significant association with geography which is probably the result of long‐term evolution of NiV in local bat population. The two NiV lineages differ in many amino acids; one change in the fusion protein might be involved in its activation via binding to the G protein. We also identified adaptive and positively selected sites in many viral proteins. In the receptor‐binding G protein, we found that sites 384, 386 and especially 498 of G protein might modulate receptor‐binding affinity and thus contribute to the host jump from bats to humans via the adaption to bind the human ephrin‐B2 receptor. We also found that site 1645 in the connector domain of L was positive selected and involved in adaptive evolution; this site might add methyl groups to the cap structure present at the 5′‐end of the RNA and thus modulate its activity. This study provides insight to assist the design of early detection methods for NiV to assess its epidemic potential in humans.
Importance.
NiV was identified by the World Health Organization (WHO) as a likely cause of a future pandemic. In South and Southeast Asia, it has already been transmitted several times from bats to humans with the resulting outbreaks being associated with human‐to‐human transmission and a high mortality rate. Using all available sequence data, we performed a combined bioinformatics study to analyse its adaptive evolution. We also identified amino acids in many viral proteins that might be associated with the host jump from bats to humans. The results obtained can assist the implementation of surveillance systems in the affected countries.
1. INTRODUCTION
Emerging infectious diseases (EIDs) are a great threat to public health worldwide. Around 60%–80% of human and mammal EIDs originate from wildlife, a typical example being henipavirus‐related lethal neurologic and respiratory diseases that originated from bat viruses (He, Zhao, et al., 2019; Li et al., 2018; Su et al., 2016). Nipah virus (NiV) is a deadly paramyxovirus which is naturally harboured by several species of bats belonging to the genus Pteropus. NiV disease first emerged in Southeast Asia in September 1998 and by December 1999, the epidemic had caused 283 human encephalitis cases and 109 deaths in Malaysia and 11 cases with encephalitis or respiratory symptoms and 1 death in Singapore (Chua, 2003; Chua et al., 2000). Since 2001, frequent outbreaks of NiV occurred in India, Bangladesh and the Philippines adding up to 666 human infections, 388 deaths and a mortality rate of around 60% according to the World Health Organization (WHO) and other recent reports (Arunkumar et al., 2018; Ching et al., 2015; Chua, 2003; Sourimant & Plemper, 2016). Of note, in February 2018, NiV infection was listed by the WHO as one of the priority diseases posing a public health risk. In May 2018, a NiV outbreak was reported in Kerala, India, with 23 identified human cases (18 laboratory‐confirmed cases) and 21 deaths, being the third NiV outbreak known to occur in India (Arunkumar et al., 2018). Unlike the initial outbreaks in Malaysia and Singapore, human‐to‐human transmission played an important role in the spread of NiV during the outbreaks in India, Bangladesh and the Philippines (Arankalle et al., 2011; Arunkumar et al., 2018; Chadha et al., 2006; Ching et al., 2015; Gurley et al., 2007; Luby et al., 2009). An epidemiological investigation on Bangladesh human cases of Nipah virus infection during 2001–2007 shows that more than half of human infections caused by human‐to‐human transmission (Luby et al., 2009). In addition to bats of the genus Pteropus, NiV also naturally infects animals more closely related to humans, such as pigs, goats, horses, dogs and cats (AbuBakar et al., 2004; Ching et al., 2015; Chua, 2003; Chua et al., 2000). This wide host range may be due to the two cell receptors for NiV, ephrin‐B2 and ephrin‐B3, which are highly conserved across many species (Bonaparte et al., 2005; Negrete et al., 2005, 2006; Xu, Broder, & Nikolov, 2012). Due to its high lethality, the lack of effective vaccines or treatments and the re‐emergence of deadly zoonotic NiV in South and Southeast Asia that suggested human‐to‐human transmission, greater outbreaks of NiV might be possible in the future.
Given the ongoing infections of humans, NiV is considered to have pandemic potential. When stablishing in a new host, NiV has to adapt to novel conditions, a process that provides strong selection pressure. Little is known about the NiV genomic changes required for its transmission to humans, in line with the lack of knowledge on common genetic ‘host jump’ rules from bats to humans or to other mammals. Here, we combined phylogenetic with selection analysis and structural biology to understand the role of different NiV lineages in interspecies transmission and the role of adaptive evolution during transmission from bats to new hosts in relation to structural and functional changes. Moreover, we investigate a possible increase in pathogenicity and the ability for human transmission and the genetic and evolutionary dynamics of NiV from the Bangladesh and Malaysia lineages.
2. MATERIAL AND METHODS
2.1. Sequence data set
All the NiV sequences available in December 2018 in National Center for Biotechnology Information (NCBI) GenBank database (https://www.ncbi.nlm.nih.gov/genbank/) were included in the analysis. After deleting sequences from unknown sources or too short in length, the sequence dataset included 17 full‐genome sequences, 113 nucleocapsid (N) coding sequences, 20 phosphoprotein (P) coding sequences, 23 matrix protein (M) coding sequences, 19 fusion protein (F) coding sequences, 21 glycoprotein (G) coding sequences and 16 polymerase protein (L) coding sequences (Table S1). The sampling dates ranged from 1999 to 2018.
2.2. Sequence alignment and model selection
Sequences were aligned using MUSCLE and manually adjusted within the MEGA software (version 7) (Edgar, 2004; Kumar, Stecher, & Tamura, 2016). The best fit nucleotide substitution models were detected using the IQ‐tree software (version 1.6.5) according to the Bayesian information criterion (BIC) score (Lam‐Tung, Schmidt, Arndt, & Bui Quang, 2015). The TempEst software (version 1.5.1) was used to analyse the root to tips distances against time (Rambaut, Lam, Max Carvalho, & Pybus, 2016).
2.3. Recombination analysis
To detect potential recombination events, 17 aligned genomic sequences and all N, P, M, F, G and L coding sequences were submitted to the Recombination Detection Program 4 (RDP4) (D. P. Martin, Murrell, Golden, Khoosal, & Muhire, 2015). Seven different methods including RDP (Martin & Rybicki, 2000), GENECONV (Padidam, Sawyer, & Fauquet, 1999), Chimaera (Posada & Crandall, 2001), MaxChi (Smith, 1992), BootScan (Martin, Posada, Crandall, & Williamson, 2005), SiScan (Gibbs, Armstrong, & Gibbs, 2000) and 3Seq (Boni, Posada, & Feldman, 2007) with default settings were used for recombination signal detection. The highest acceptable p‐value was set to 0.05. Only recombination results confirmed by four or more methods are displayed. Recombination events were further identified using SimPlot software (version 3.5.1) (Lole et al., 1999).
2.4. Phylogenetic and evolution analysis
Maximum likelihood (ML) trees were constructed in RAxML software (version 8.4.10) (Stamatakis, 2014) using the general time‐reversible plus gamma (GTR + G) distribution model or the Hasegawa‐Kishino‐Yano model plus gamma (HKY + G) distributed rate heterogeneity nucleated substitution models and 1,000 bootstraps. In addition, maximum clade credibility (MCC) trees were reconstructed using BEAST software (version 1.8.4) (Drummond & Andrew, 2007), with the GTR + G, uncorrelated lognormal relaxed clock and coalescent: Bayesian SkyGrid model chosen according to Bayes factor and Marginal Likelihood methods (Li et al., 2018). The tip dates were estimated according to the time of virus isolation or sequencing with the format of year‐mouth. Markov Chain Monte Carlo (MCMC) sampling was run for 1 × 108 generations, with trees and posteriors sampled every 1 × 104 steps. Two independent runs were combined using LogCombiner (He, Auclert, et al., 2019). The final tree was summarized using Tree Annount software and displayed using FigTree (version 1.4.7).
2.5. Geographical correlation
The Bayesian Tip‐Significance testing software (BaTS) was used to analyse the correlation between each NiV sequence and geographical location (Parker, Rambaut, & Pybus, 2008). The NiV geographic structure was defined according to countries, including Malaysia, Bangladesh, India, Cambodia and Thailand. The association index (AI) and parsimony score (PS) statistics were calculated using the MCC trees of NiV N gene. When the p‐values of AI and PS were less than .05, the correlation between NiV and geographical distribution was considered significant (He, Li, et al., 2019).
2.6. Amino acid differences and structural and function changes
To locate positively selected and adaptive sites in the L, G and F protein and all the amino acid differences between two virus lineages in the G and F proteins, we created figures with PyMol (Molecular Graphics System, version 2.0 Schrödinger, LLC, https://pymol.org/2/). For G, we used the pdb file 3D12, which contains the structure of the ectodomain of NiV G protein (residues 71–602) bound to the mouse ephrin‐B3 (residues 30–170) (Xu et al., 2008) and the pdb file 2VSM which is the structure of G (residues 188–606) bound to the human ephrin‐B2 receptor (residues 31–170) (Bowden, Aricescu, et al., 2008). The prefusion structure of the F protein was visualized using the pdb file 5EVM (Xu et al., 2015). Since no post‐fusion structure of NiV F is available, we used the structure of F from the related paramyxovirus Newcastle Disease Virus (pdb file 3MAW) (Swanson et al., 2010), which has ~50% amino acid similarity with NiV F. Likewise, since no structure of L from NiV (or from any other paramyxovirus) is available, we used the structure of L from Vesicular Stomatitis virus (pdb file 5A22) (Liang et al., 2015) and identified the positively selected and adaptive sites by sequence alignment. Determination of the distance of a salt bridge was done with the measurement wizard tool of the PyMol software.
2.7. Selection and adaptive evolution analysis
Selection analysis was performed by uploading the ML trees and the sequences to DATAMONKEY (http://www.datamonkey.org). The fixed effects likelihood (FEL), single‐likelihood ancestor counting (SLAC), fast unconstrained Bayesian approximation (FUBAR), mixed effects model of evolution (MEME) were the algorithms used to identify sites under selection. Significance level was set with p‐value threshold of 0.1 for FEL, SLAC and MEME and with posterior probability of 0.9 for FUBAR. A site detected by more than two algorithms was considered under selection. An adaptive branch‐site REL test for episodic diversification (aBSREL) was used to detect positively selected branches (Kosakovsky Pond & Frost, 2005; Murrell et al., 2013; Murrell et al., 2012; Smith et al., 2015). We also split the N, P, G, F, L, M genes into the Bangladesh and the Malaysian lineages and reconstructed the common ancestor amino acid sequence of each lineage independently. The ML method implemented in CODEML of PAML (version 4.8) was used to reconstruct the ancestral amino acid state. Potential adaptive sites were defined as changes which dominated in another (non‐bat) host, sequences that were different from other dominant amino acids in pigs and ancestral amino acids. The association of potential adaptive sites and phenotype (host jump) using counts of dominant amino acids in pigs and other host sequences was determined using the chi‐square test. The statistical significance was tested using the method described by He et al. (article In press).
2.8. Comparison of amino acid differences between mammalian and bat receptors
Given that two bat species from the Pteropus genus (P. alecto and P. vampyrus) from where NiV were isolated revealed no amino acid differences in the ephrin‐B2 and no differences in the part of ephrin‐B3 that is present in the crystal structure, we can assume that the two receptors are highly conserved between bat species. Ephrin‐B2 and ephrin‐B3 sequences from the Pteropus vampyrus bat (NP_001292125.1 and ABV44497.1) were aligned with the human ephrin‐B2 and the mouse ephrin‐B3 sequences, respectively. Only a few amino acid differences were found, which were labelled in the structure of their mammalian ortholog.
3. RESULTS
3.1. NiV recombination, phylogenetic and evolutionary dynamics analysis
Only one possible recombination event occurred in NiV genomic sequences was listed by RDP4. Further analysis performed by SimPlot 3.5.1 indicated that the event was a false‐positive result (data not shown). Since there was no recombination event interfering the construction of phylogenetic trees, all full‐genome sequences and sequences of each gene were used to reconstruct ML trees. N gene tree has apparently more complex structure than full‐genome tree since more N sequences were used in the analysis. In both ML trees, NiV all could be divided into two main lineages: the Bangladesh and Malaysia lineages, and only NiV from the Bangladesh lineage circulate in India and Bangladesh while in Malaysia only NiV from the Malaysia lineage circulate (Figure 1), as reported previously (Lo et al., 2012; Lo Presti et al., 2016; Rahman et al., 2010). Analysis based on a large number of N coding sequences (Figure 1b) revealed human and bat‐derived NiV in Bangladesh lineage and a more complicated structure in the Malaysia lineage including viruses derived from multiple hosts (bat, swine and human), which is corresponding to the different transmission modes of two lineages (Av et al., 2018). Of note, NiV belonging to different lineages was observed in local bat population of Thailand and Cambodia, and this phenomenon in Thailand was previously reported while NiV from Cambodia bats was formerly thought to only belong to Malaysian lineage, which could be the result of the analysis of additional Cambodia bats sequences obtained in 2013 (Lo et al., 2012; Reynes et al., 2005; Wacharapluesadee et al., 2010, 2005, 2016). In addition, NiV could be divided into these two lineages based on ML trees reconstructed based on other coding sequences (Fig [Link], [Link]), which is similar to the results of previous studies (Lo et al., 2012). BaTS analysis revealed that the NiV p‐value of AI and PS was less than .05 (Table 1) and apart from the MC p‐value of Cambodia which was 1, the p‐values of other countries were less than .05. This is consistent with the structure of phylogenetic trees and indicates a significant geographic association.
Figure 1.
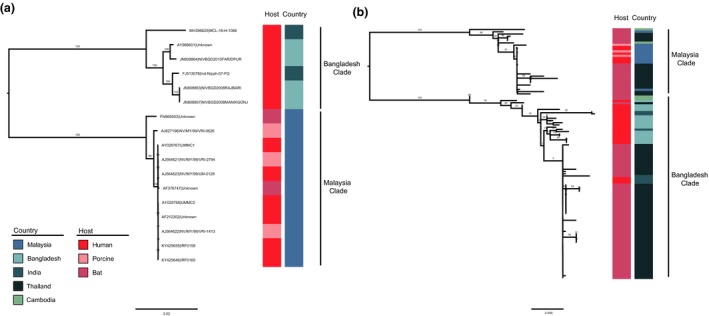
Maximum likelihood (ML) trees of NiV full genomes (a) and the N gene (b). The ML trees were reconstructed in RAxML (version 8.4.10) using the GTR + G distribution model with 1,000 bootstraps. Host and country of NiV isolates are indicated with inner and outer coloured rectangular boxes, respectively [Colour figure can be viewed at http://www.wileyonlinelibrary.com/]
Table 1.
Bayesian Tip‐association Significance testing (BaTS) of NiV
| Statistic | Observed mean | Lower 95% CI | Upper 95% CI | Null mean | Lower 95% CI | Upper 95% CI | Significance |
|---|---|---|---|---|---|---|---|
| AI | 0.57 | 0.32 | 0.82 | 3.11 | 2.57 | 3.55 | 0.00 |
| PS | 7.13 | 7.00 | 8.00 | 19.47 | 17.10 | 21.53 | 0.00 |
| MC (Malaysia) | 10.00 | 10.00 | 10.00 | 1.90 | 1.25 | 2.95 | 0.01 |
| MC (Cambodia) | 1.00 | 1.00 | 1.00 | 1.01 | 1.00 | 1.07 | 1.00 |
| MC (Thailand) | 6.00 | 6.00 | 6.00 | 1.68 | 1.23 | 2.29 | 0.01 |
| MC (Bangladesh) | 3.99 | 4.00 | 4.00 | 1.63 | 1.17 | 2.21 | 0.01 |
| MC (India) | 2.00 | 2.00 | 2.00 | 1.06 | 1.00 | 1.20 | 0.01 |
This article is being made freely available through PubMed Central as part of the COVID-19 public health emergency response. It can be used for unrestricted research re-use and analysis in any form or by any means with acknowledgement of the original source, for the duration of the public health emergency.
Next, we reconstructed the NiV evolution dynamics based on the N gene. Based on the MCC tree, we can conclude that the two NiV lineages were associated with independent epidemics (Figure 2). The time to the most recent common ancestor (tMRCA) of NiV was estimated to be 1992.63 (95% HPD: 1985.55–1997.58) (Table 2). The tMRCA of the Malaysia lineage was the year 1994.44 (95% HPD: 1989.03–1998.01) while for the Bangladesh lineage was 2000.01 (95% HPD: 1995.60‐2002.55). Additionally, the NiV evolutionary rate was 1.10 × 10–3 substitutions/site/year (95% HPD: 7.34 × 10–4–1.50 × 10–3 substitutions/site/year) based on the N gene. In particular, the Bangladesh lineage had a mean 6.50 × 10–3 substitutions/site/year (95% HPD: 6.03 × 10–9–1.60 × 10–2 substitutions/site/year) while the Malaysia lineage had a mean 1.43 × 10–2 substitutions/site/year (95% HPD: 4.98 × 10–8–6.40 × 10–2 substitutions/site/year). To understand the population size of NiV, the Bayesian SkyGrid coalescent was reconstructed. We found that the population size of NiV fluctuated in the past 20 years, but overall it has remained at the same level.
Figure 2.
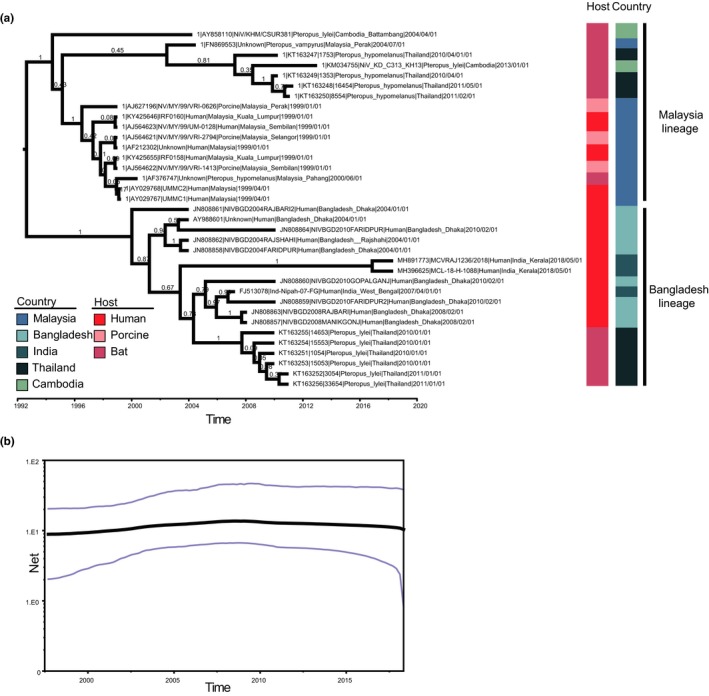
Maximum clade credibility (MCC) tree and skygrid plot based on the N gene. (a) The MCC tree was reconstructed using BEAST (version 1.8.4). The GTR + G distribution model and the coalescent: Bayesian skygrid model with a total chain length of 1 × 109 and sampled every 1 × 104 times. Host and country of NiV isolates are indicated with inner and outer coloured rectangular boxes, respectively. (b) Skygrid plot of NiV. The grey line is the mean value while the blue line is 95% HPD [Colour figure can be viewed at http://www.wileyonlinelibrary.com/]
Table 2.
tMRCA and substitution rate of NiV based on N gene
| Virus/genotype | tMRCA (year) | Substitution rate (substitutions/site/year) | ||
|---|---|---|---|---|
| Mean | 95%HPD | Mean | 95%HPD | |
| NiV | 1992.63 | [1985.55, 1997.58] | 1.10E−03 | [7.37E−04, 1.50E−03] |
| Malaysia | 1994.44 | [1989.03, 1998.01] | 1.43E−02 | [4.98E−08, 6.40E−02] |
| Bangladesh | 2000.01 | [1995.60, 2002.55] | 6.50E−03 | [6.03E−09, 1.60E−02] |
The time of most recent common ancestor (tMRCA).
This article is being made freely available through PubMed Central as part of the COVID-19 public health emergency response. It can be used for unrestricted research re-use and analysis in any form or by any means with acknowledgement of the original source, for the duration of the public health emergency.
3.2. Amino acid differences between the Bangladesh and Malaysia lineages
The G protein is a type II membrane protein consisting of an N‐terminal intraviral domain (~50 residues), one transmembrane region (~20 residues), a helical stalk region (~100 residues) and a head domain (residues 176–603) that folds into a ß‐propeller with six blades surrounding a central cavity (Bowden, Crispin, et al., 2008; Xu et al., 2008). The head region binds to the cellular receptor ephrin‐B2 or ephrin‐B3 (Bonaparte et al., 2005; Bowden, Aricescu, et al., 2008; Bowden, Crispin, et al., 2008; Negrete et al., 2005, 2006; Xu et al., 2008). It is currently believed that receptor‐binding transduces a signal to the stalk region which then activates the viral F protein leading to conformational changes that result in membrane fusion. In virus particles, G forms a tetrameric spike during intracellular transport and at the cell surface might interact with the F protein (Bose, Jardetzky, & Lamb, 2015). The amino acid locations, which vary between the Bangladesh and the Malaysia lineage of bat‐derived viruses and are neither adaptive nor positive selected sites are shown in Table S2 and Figure 3a as magenta sticks within the head domain of G (blue cartoon) bound to ephrin‐B3 (green cartoon). Most of them are located at the proposed interaction surface between G monomers and thus might contribute to oligomerization of G (Bowden, Crispin, et al., 2008). N481 is part of the used glycosylation site N481NT (Bowden, Crispin, et al., 2008), which is exchanged to D in all strains from the Bangladesh lineage and in three out of 13 strains from the Malaysia lineage which therefore lack a carbohydrate at this site.
Figure 3.
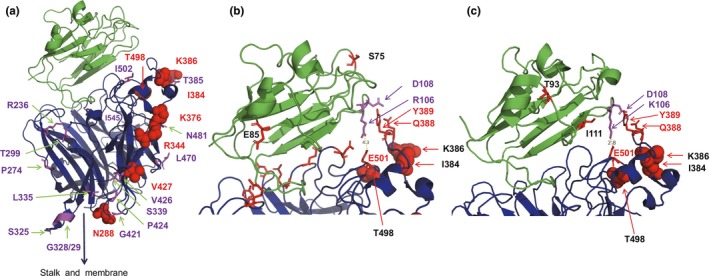
(a) Structure of the ectodomain of the NiV G protein (blue) bound to the ephrin‐B3 receptor (green). Adaptive amino acids are shown as red spheres. Amino acid differences between viruses of the two lineages are depicted as magenta sticks. (b) Receptor‐binding site of G (blue) bound to ephrin‐B3 (green). Amino acids in G forming salt bridges and hydrogen bonds with mouse ephrin‐B3 are shown as red sticks. E501 forms a salt bridge with R106 and Q388 and Y389 form hydrogen bonds with D108. Adaptive amino acids are shown as red spheres. The amino acids which are different in bat ephrin‐B3 (S75, E85) are labelled as red sticks in the ephrin structure. 4.3 indicates the distance between E501 and R106 in Angstrom. (c) Receptor‐binding site of G (blue) bound to ephrin‐B2 (green). E501 forms a salt bridge with K106 and Q388 and Y389 form hydrogen bonds with D108. Adaptive amino acids are shown as red spheres. The amino acids which are different in bat ephrin‐B2 (T93, I111) are labelled as red sticks in the ephrin structure. In addition, K106 is exchanged in bat ephrin‐B2 by an R and thus to the same amino acid present in ephrin‐B3. 2.8 indicates the distance between E501 and K106 in Angstromss [Colour figure can be viewed at http://www.wileyonlinelibrary.com/]
The F protein, which forms a trimer, is a typical type I transmembrane protein, which is proteolytically cleaved by cathepsin L into the N‐terminal F2 subunit and the larger F1 subunit which carries the fusion peptide, the transmembrane region and a short C‐terminal cytoplasmic tail (Xu et al., 2015). The F protein of the Bangladesh and Malaysia lineages differ in seven amino acids. Residues 2, 6, 9, 11 and 19 are located in the cleaved signal peptide and thus are unlikely to play a role for the function of F. The other two residues are highlighted as green spheres in the prefusion structure of the F protein (Figure 4a), which consists of a globular head composed of three domains (DI, DII and DIII), followed by a C‐terminal stalk. DI and the Ig‐like fold DII are implicated in interactions with the G protein. Residue 273 is located in the DIII region in the globular head domain. Of particular interest is residue 42, which is part of domain II of the F2 subunit. It is located very close to the fusion peptide; the distance to I122, the C‐terminal residue of the fusion peptide, is only ~5 Å (Figure 4b). Furthermore, residue 42 is also part of the strap region composed of β‐sheets which is implicated in interactions with the G protein.
Figure 4.
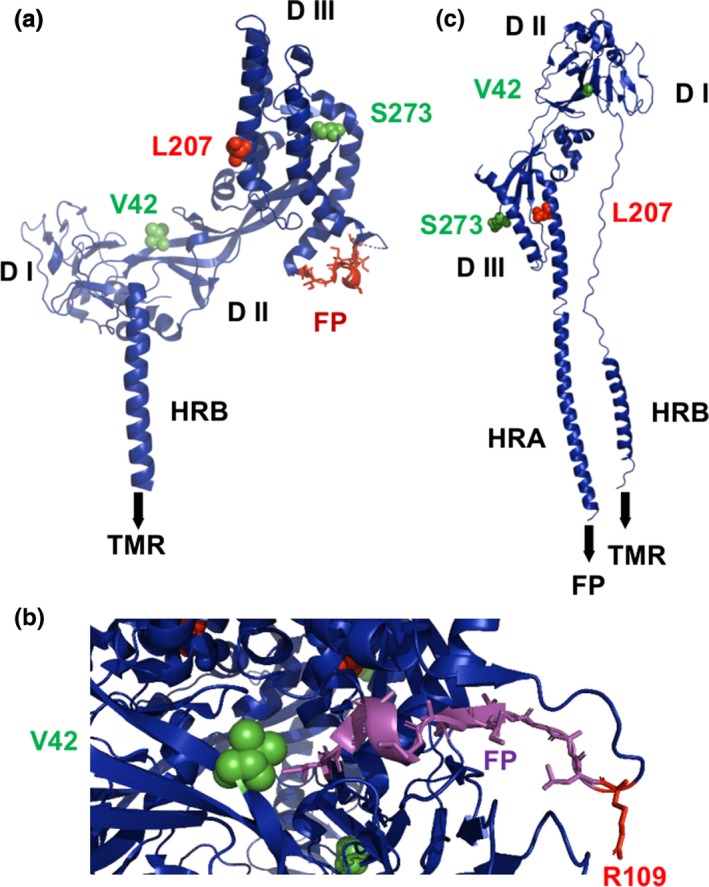
(a) Structure of a monomer of the ectodomain of the prefusion structure of the NiV F protein shown as a cartoon. The adaptive amino acid 207 is shown as red spheres. Amino acid differences between viruses of the two lineages are depicted as green spheres. The fusion peptide (FP) is shown as orange sticks. DI, DII and DIII are the individual domains. HRB: heptad repeat B. TMR denotes the position of the transmembrane region, which is not present in the crystal structure. (b) Location of residue 42 near the fusion peptide in the trimeric structure of F. R109 is the protease cleavage site, the last residue of the F2 subunit. (c) Post‐fusion structure of Newcastle Disease virus F protein with the adaptive amino acid 207 and the amino acid differences between viruses of the two lineages as green spheres. Note that parts of the DII domain underwent large conformational changes that led to the formation of the heptad repeat region A (HRA) that forms with HRB a six‐helix bundle that is characteristic for the post‐fusion conformation of type I viral fusion proteins [Colour figure can be viewed at http://www.wileyonlinelibrary.com/]
To analyse where these amino acids are located in the post‐fusion structure of F, we labelled the analog residues in the F protein of Newcastle Disease virus. Upon activation, the F protein undergoes large‐scale refolding, mostly in DIII. The heptad repeat region A (HRA) extends into a long α‐helix, which forms a six‐helix bundle with the HRB region of the stalk. The regions of F that contain residues 42 and 273 do not refold after activation of F (Figure 4c). However, residue 42 and the fusion peptide in the post‐fusion structure of F are very far apart, at the opposite side of the molecule.
3.3. Selection analysis and adaptive sites
Residues 437 in P, 241 in M, 207 in F, 20 in G and 1645 in L were identified to be under positive selection (Table 3) by MEME and FUBAR. However, FEL only found positive selection at residue 1645 in L. Next, we identified the adaptive sites for transmission of NiV from bats to humans independently in the Bangladesh and Malaysia lineages. Of note, we found only one adaptive site, 436, in the N protein. Although six sites in G (288, 344, 376, 384, 386, 427 and 498) and site 1645 in L were not significantly associated with cross‐species adaption, we also considered them as adaptive sites because the Bangladesh lineage had only one or none sequence from an infected bat (Table 4).
Table 3.
Positively selected sites in P, M, F, G, L coding sequences of NiV
| Gene | Site | FEL | SLAC | FUBAR | MEME | ||||
|---|---|---|---|---|---|---|---|---|---|
| dN‐dS | p‐value | dN‐dS | p‐value | dN‐dS | Post.Pro | w+ | p‐value | ||
| P | 437 | 0 | 1 | 7.58 | 0.52 | 11.35 | 0.90 | >100 | 0.03 |
| M | 241 | 0 | 1 | 0.09 | 0.74 | 31.93 | 0.93 | >100 | 0 |
| L | 1645 | 6.17 | 0.05 | 9.71 | 0.47 | 25.22 | 0.94 | >100 | 0.09 |
| G | 20 | 1.85 | 0.32 | 8.27 | 0.49 | 17.78 | 0.91 | >100 | 0.04 |
| F | 207 | 0 | 1 | 9.88 | 0.45 | 15.09 | 0.91 | >100 | 0 |
| N | None | ||||||||
This article is being made freely available through PubMed Central as part of the COVID-19 public health emergency response. It can be used for unrestricted research re-use and analysis in any form or by any means with acknowledgement of the original source, for the duration of the public health emergency.
Table 4.
Adaptive sites in NiV in each coding genes
| Site | From (bat) | To (human) | |
|---|---|---|---|
| G protein | 376 | T | K |
| G protein | 288 | N | S |
| G protein | 344 | K | M |
| G protein | 384 | I | V |
| G protein | 386 | E | K |
| G protein | 427 | V | I |
| G protein | 498 | K | T |
| L protein | 1645 | F | S |
| N protein | 436 | I | M |
This article is being made freely available through PubMed Central as part of the COVID-19 public health emergency response. It can be used for unrestricted research re-use and analysis in any form or by any means with acknowledgement of the original source, for the duration of the public health emergency.
In the G protein, the positively selected site 20 is located in the cytoplasmic tail. The amino acid present at this site varies among (and is even deleted) some virus strains. In general, amino acids in cytoplasmic tails affect intracellular transport of G, support membrane fusion and are believed to be involved in interactions with the peripheral matrix proteins that are required for virus assembly (Sawatsky, Bente, Czub, & von Messling, 2016).
Figure 3a depicts the location of the adaptive sites (labelled as red spheres) in the structure of G bound to the ephrin‐B3 receptor. All of them are located at one side at the surface of the molecule opposite to the proposed interaction surface between G monomers (Sawatsky et al., 2016). N288 (which is not a carbohydrate attachment site), R344, K376 and V427 are located too far away from the receptor‐binding site on top of the molecule, and thus, these amino acid changes are unlikely to affect binding to ephrin‐B3. K376 might modulate the proposed signalling caused by binding of G to the receptor from the head to the stalk domain which activates the fusion activity of the F protein (Wong et al., 2017). Residue 289 has been shown to change upon selection of G‐mutants with a monoclonal antibody, and N288 might thus be part of this antibody epitope. Likewise, residues 384 and 386 are also part of an antibody epitope (White et al., 2005). To our knowledge, no functions have been associated with the other amino acids.
The adaptive sites I384, K386 and T498 do not directly interact with ephrin‐B3, but are located close to the receptor‐binding site, which is depicted in higher magnification in Figure 3b. A loop in the structure of ephrin forms a shallow but extensive protein–protein interaction surface that is buried deeply in a hydrophobic pocket on the G surface. These hydrophobic interactions are assisted by amino acids (shown as red sticks) which form salt bridges or hydrogen bonds with ephrin‐B3. Especially interesting in this regard is E501, which forms a salt bridge with R106 in ephrin‐B3 and Q388 and Y389 that form hydrogen bonds with D108 (Xu et al., 2008). The same network of interactions is also involved in binding of ephrin‐B2, except that R106 is replaced by K (Bowden, Aricescu, et al., 2008). Thus, mutations at residues 384, 386 and 498 located in close proximity to these sites might affect the strength of the G‐receptor interaction. Two sites that differ between the two virus lineages (T385 and I502, which are A and V, respectively, in the Bangladesh lineage) are also located in this region, but they have not been identified as adaptive sites by our analysis. The positively selected site 207 is located in the interior of the head domain of the F protein within a α‐helix of the DIII domain. This region of the molecule does not refold, and no function has been associated with this residue (Figure 4).
We used the cryo‐EM structure of the homologous L protein from Vesicular Stomatitis virus (Liang et al., 2015) to locate the positively selected and adaptive site 1645. Sequence alignment identified that residue T1471 of L of VSV occupies the homologues position. This residue is located inside the connector domain, which connects the N‐terminal catalytic sites (RNA‐dependent RNA polymerase and capping domain) with the C‐terminal methyl transferase activity of L (Figure 5). More precisely, T1471 is located near the methyl transferase domain in a loop between two α‐helices of the eight‐helix bundle connector domain.
Figure 5.
Cartoon structure of the L protein of Vesicular Stomatitis Virus showing the location of T1471, which is at the same position as amino acid 1645 of NiV L protein. The domains of L are coloured in blue (RNA‐dependent RNA polymerase, RdRp, residues 35–865), green (capping domain cap, 866–1334), yellow (connector domain, CD, 1358–1557), orange (methyl transferase, MT, 1598–1892); and red (C‐terminal domain, CTD, 1893–2109). T1471 (purple sphere) is part of the connector domain, but close to the methyl transferase domain [Colour figure can be viewed at http://www.wileyonlinelibrary.com/]
3.4. Comparison of amino acid differences between bat and other mammalian receptors
Based on previous structural analyses of G protein and its interaction with ephrin‐B2 and ephrin‐B3 (Bowden, Aricescu, et al., 2008; Bowden, Crispin, et al., 2008; Xu et al., 2008), we then asked whether there are amino acid differences between the bat and the other mammalian ephrin‐B2 and ephrin‐B3 receptors that might require changes in G in order to adapt from bat receptors to human receptors. Alignment of the other mammalian and bat ephrin‐B3 sequences revealed two amino acid changes: S75 changes to N in bat and E85 is replaced by G in bat (labelled as red sticks in Figure 3b). Although these are non‐conservative changes, they are too far away from the part of ephrin‐B3 contacting G and thus are unlikely to affect virus binding. In ephrin‐B2, three conservative amino acid differences became apparent: T93 is replaced by S, I111 by V and K106 by R in the bat receptor (labelled as red sticks in Figure 3c, except K106 which is a purple stick). Especially, interesting is residue 106, which forms a salt bridge with E510 in G. This salt bridge is also present in the structure of G bound to ephrin‐B3, but here a K instead of an R is present. Both of these basic and positively charged amino acids are apparently capable to form a salt bridge with E501, but since the side chain of K is larger than the side chain of R, the distance of this salt bridge is shorter (2.8 Å) in G bound to mammalian ephrin‐B2 compared with G bound to mammalian ephrin‐B3 (4.3 Å). In the bat ephrin‐B2, the shorter R is present and thus the adaptive sites I384, K386 and especially T498 might modulate the interaction between G and ephrin‐B2 which might be required to facilitate the host jump from bats to humans.
4. DISCUSSION
Since its emergence in 1998–1999 in Malaysia, NiV has reappeared in several South and Southeast Asian countries (Figure S2), including India, Bangladesh and the Philippines, leading to severe human infections associated with a high mortality rate. Fruit bats (Genus Pteropus) are known to be the natural host and reservoir of NiV, but cross‐species transmission is common. However, different NiV genotypes underwent different evolutionary paths. Moreover, different genotypes are associated with different fatality rates and transmission characteristics (Sharma, Kaushik, Kumar, Yadav, & Kaushik, 2019), but no study reported on genome differences between genotypes and whether adaptive evolution occurred during NiV spillover from bats to other mammals, especially to humans. Hence, we integrated a variety of bioinformatics methods, from genomic alterations to systematic analysis, phylogeography, selection and adaptive analysis to understand the characteristic of NiV.
Firstly, based on ML trees of full‐genome and structural protein‐coding sequences, we confirmed that NiV can be divided into two main lineages, the Bangladesh and Malaysia lineages. Since there is a significant geographical relationship between NiV and the epidemic countries, this characteristic is probably a result of a long‐term evolution of NiV in local bat population. Interestingly, Thailand and Cambodia, as two countries locate between Malaysia and Bangladesh, different lineages of NiV were identified in local bat population. And epidemiological investigations on NiV in Philippines, Indonesia, East Timor, three countries which are close to Malaysia, also suggest the sequences of NiV that circulates in their counties are more similar to Malaysia lineage NiV, which is consistent with our results (Breed et al., 2013; Ching et al., 2015; Sendow et al., 2013). Bats, the second largest order of mammals after rodents, harbour more than 200 types of viruses, including many that are highly pathogenic for humans (e.g. rabies virus, Ebola virus, severe acute respiratory syndrome coronavirus (SARS‐CoV), Hendra virus (HeV). NiV circulates within bat populations via close mutual contact when bats crowd together. Moreover, bats can fly over long distances and thus can spread the virus from country to country. The bats of the genus Pteropod have long‐range flight capabilities, suggesting that neighbouring countries, such as China should also strengthen NiV surveillance (Figure S2).
The tMRCA was later than previously reported probably due to the larger number of analysed sequences (Sun, Jia, Liang, Chen, & Liu, 2018). On the other hand, the NiV evolutionary rate was similar to some other important zoonotic RNA viruses such as Ebola virus (Yi‐Gang et al., 2015). Thus, NiV is highly variable, which makes disease prevention, control and vaccine development difficult. Of note, we found a significant relationship between NiV and geography, except for Cambodia, which may be due to the limited number of available sequences from this region. This indicates that once NiV is epidemic in one area, it differentiates into a new lineage that adapts to the local background. This adaption also causes a fast rate of evolution.
Given that different NiV lineages differ in their ability for human‐to‐human transmission, we conducted mutation, selection and adaptive evolution analysis and related changes to structural and functional modifications in particular for the G protein and its receptors, ephrin‐B2 and/or ephrin‐B3. None of the G protein adaptive sites are in direct contact with amino acids of ephrin‐B2 or ephrin‐B3. However, adaptive amino acids were identified near the second interaction site which comprises a hydrogen‐bonding network between Q388 and Y389 in G and the negatively charged D108 in both ephrin‐B2 and ephrin‐B3 and a salt bridge between negatively charged E501 in G and a basic residue at position 106 in ephrin. Interestingly, the identity of the basic amino acid varies: in bats and other mammalians ephrin‐B3 and in the bat ephrin‐B2 it is an arginine, whereas in the mammalian ephrin‐B2 a lysine is present. Since the side chain of lysine is longer, the distance between E501 in G and the basic residue in ephrin becomes shorter and as a consequence probably the strength of the interaction increases. We speculate that for bat‐derived NiV to adapt to the human ephrin‐B2 receptor the adaptive sites at position 384, 386 and especially 498 might modulate the receptor‐binding affinity and might thus contribute to a host jump. Interestingly, the G protein of a henipavirus recently isolated from a bat in Africa does not contain this second interaction site since the amino acids at an equivalent position to Q388 and Y389 do not form hydrogen bonds with D108 in ephrin. As a consequence, the G protein of African bat henipavirus shows decreased ephrin‐B2 binding relative to the G protein of NiV (Lee et al., 2015). Thus, hydrogen bonds at the secondary binding site impart affinity and stabilize the receptor‐bound complex and even small differences in receptor‐binding can translate into significant differences in the efficiency of infection.
On the other hand, the function of the positively selected site 207 in the F protein and the variant amino acid 273 between two lineages is hard to predict. However, the F protein contains another interesting amino acid difference between the two NiV lineages. Residue 42 is located within the so‐called strap region that is supposed to be the binding site for the receptor‐bound G protein and also very close to the fusion peptide in the trimeric structure of F (Figure 3). Residue 42 is a valine in the Malaysia lineage of the bat‐derived virus, but an isoleucine in the Bangladesh lineage which is also hydrophobic with a larger side chain. It is thus tempting to speculate that residue 42 affects the exposure of the fusion peptide after activation of F by binding to the G protein and thus virus entry into cells.
The positively selected and adaptive site in the L protein is located in the connector domain, which consists of a bundle of eight helices. No specific function has been assigned to this domain; however, it seems to play an organizational role in positioning or spacing the catalytic domains. Both sides of the connector domain contain unstructured linkers, which are supposed to bind to the P protein and might modulate binding. Alternatively, since position 1,471 is located close to the methyl transferase domain, which adds methyl groups to the cap structure present at the 5´‐end of the RNA, residue 1,471 might modulate this activity.
The genetic polymorphisms of NiV may be associated with virus circulation, infectivity and antigenic variability. When NiV jump from bats to humans, they face new selection pressures from their new host environment. In particular, antigenic variability is critical to escape the host immune response. Three of the adaptive sites we found in the G protein, residues 288, 384 and 386, are part of a known antibody epitope (White et al., 2005). NiV adaptation to humans probably depends on the stepwise accumulation of potentiating mutations that favour the emergence of a particular adaptive mutation, similar to the mutations described for the adaption of avian influenza virus to humans (Imai et al., 2018; Su et al., 2017). Therefore, the mutational panel provided here might be very useful as an early detection system for transitional stages in the NiV evolution before it acquires full pandemic potential. We also identified several amino acid changes between two virus lineages which may affect receptor binding and hence transmission. The application of these findings is invaluable not just for veterinarians/virologists but also for public health officers, as the threat of a more serious NiV pandemic is real.
CONFLICT OF INTEREST
The authors declare no conflict of interest.
ETHICAL STATEMENT
Our paper is an evolution and bioinformatics analysis paper, all the date come from genBank, we no need to add the ethical statement about the sample.
Supporting information
ACKNOWLEDGEMENTS
This work was financially supported by the National Key Research and Development Program of China (2017YFD0500101), the Natural Science Foundation of Jiangsu Province (BK20170721), the China Association for Science and Technology Youth Talent Lift Project (2017‐2019), the Fundamental Research Funds for the Central Universities (Y0201600147) and the Priority Academic Program Development of Jiangsu Higher Education Institutions.
Li K, Yan S, Wang N, et al. Emergence and adaptive evolution of Nipah virus. Transbound Emerg Dis. 2020;67:121–132. 10.1111/tbed.13330
Li and Yan are co‐first author.
Contributor Information
Michael Veit, Email: Michael.Veit@fu-berlin.de.
Shuo Su, Email: shuosu@njau.edu.cn.
REFERENCES
- AbuBakar, S. , Chang, L. Y. , Ali, A. R. , Sharifah, S. H. , Yusoff, K. , & Zamrod, Z. (2004). Isolation and molecular identification of Nipah virus from pigs. Emerging Infectious Diseases, 10(12), 2228–2230. 10.3201/eid1012.040452 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arankalle, V. A. , Bandyopadhyay, B. T. , Ramdasi, A. Y. , Jadi, R. , Patil, D. R. , Rahman, M. , … Mishra, A. C. (2011). Genomic characterization of Nipah virus, West Bengal, India. Emerging Infectious Diseases, 17(5), 907–909. 10.3201/eid1705.100968 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arunkumar, G. , Chandni, R. , Mourya, D. T. , Singh, S. K. , Sadanandan, R. , Sudan, P. , … Ghafur, A. (2018). Outbreak investigation of Nipah Virus Disease in Kerala, India, 2018. The Journal of Infectious Diseases, 219(12), 1867–1878. 10.1093/infdis/jiy612 [DOI] [PubMed] [Google Scholar]
- Av, R. , Sadanandan, S. , Thulaseedharan, N. K. , Kg, S. K. , Pallivalappil, B. , & As, A. K. (2018). Nipah Virus Infection. Journal of the Association of Physicians of India, 66(12), 58–60. [PubMed] [Google Scholar]
- Bonaparte, M. I. , Dimitrov, A. S. , Bossart, K. N. , Crameri, G. , Mungall, B. A. , Bishop, K. A. , … Broder, C. C. (2005). Ephrin‐B2 ligand is a functional receptor for Hendra virus and Nipah virus. Proceedings of the National Academy of Sciences of the United States of America, 102(30), 10652–10657. 10.1073/pnas.0504887102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boni, M. F. , Posada, D. , & Feldman, M. W. (2007). An exact nonparametric method for inferring mosaic structure in sequence triplets. Genetics, 176(2), 1035–1047. 10.1534/genetics.106.068874 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bose, S. , Jardetzky, T. S. , & Lamb, R. A. (2015). Timing is everything: Fine‐tuned molecular machines orchestrate paramyxovirus entry. Virology, 479, 518–531. 10.1016/j.virol.2015.02.037 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bowden, T. A. , Aricescu, A. R. , Gilbert, R. J. , Grimes, J. M. , Jones, E. Y. , & Stuart, D. I. (2008). Structural basis of Nipah and Hendra virus attachment to their cell‐surface receptor ephrin‐B2. Nature Structural & Molecular Biologyss, 15(6), 567–572. 10.1038/nsmb.1435 [DOI] [PubMed] [Google Scholar]
- Bowden, T. A. , Crispin, M. , Harvey, D. J. , Aricescu, A. R. , Grimes, J. M. , Jones, E. Y. , & Stuart, D. I. (2008). Crystal structure and carbohydrate analysis of Nipah virus attachment glycoprotein: A template for antiviral and vaccine design. Journal of Virology, 82(23), 11628–11636. 10.1128/JVI.01344-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Breed, A. C. , Meers, J. , Sendow, I. , Bossart, K. N. , Barr, J. A. , Smith, I. , … Field, H. E. (2013). The distribution of henipaviruses in Southeast Asia and Australasia: Is Wallace's line a barrier to Nipah virus? PLoS ONE, 8(4), e61316 10.1371/journal.pone.0061316 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chadha, M. S. , Comer, J. A. , Lowe, L. , Rota, P. A. , Rollin, P. E. , Bellini, W. J. , … Mishra, A. C. (2006). Nipah virus‐associated encephalitis outbreak, Siliguri, India. Emerging Infectious Diseases, 12(2), 235–240. 10.3201/eid1202.051247 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ching, P. K. G. , de los Reyes, V. C. , Sucaldito, M. N. , Tayag, E. , Columna‐Vingno, A. B. , Malbas, F. F. , … Foxwell, A. R. (2015). Outbreak of henipavirus infection, Philippines, 2014. Emerging Infectious Diseases, 21(2), 328–331. 10.3201/eid2102.141433 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chua, K. B. (2003). Nipah virus outbreak in Malaysia. Journal of Clinical Virology, 26(3), 265–275. 10.1016/S1386-6532(02)00268-8 [DOI] [PubMed] [Google Scholar]
- Chua, K. B. , Bellini, W. J. , Rota, P. A. , Harcourt, B. H. , Tamin, A. , Lam, S. K. , … Mahy, B. W. (2000). Nipah virus: A recently emergent deadly paramyxovirus. Science, 288(5470), 1432–1435. [DOI] [PubMed] [Google Scholar]
- Drummond, A. J. , & Andrew, R. (2007). BEAST: Bayesian evolutionary analysis by sampling trees. Bmc Evolutionary Biology, 7(1), 214 10.1186/1471-2148-7-214 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edgar, R. C. (2004). MUSCLE: A multiple sequence alignment method with reduced time and space complexity. BMC Bioinformatics, 5, 113 10.1186/1471-2105-5-113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gibbs, M. J. , Armstrong, J. S. , & Gibbs, A. J. (2000). Sister‐scanning: A Monte Carlo procedure for assessing signals in recombinant sequences. Bioinformatics, 16(7), 573–582. 10.1093/bioinformatics/16.7.573 [DOI] [PubMed] [Google Scholar]
- Gurley, E. S. , Montgomery, J. M. , Hossain, M. J. , Bell, M. , Azad, A. K. , Islam, M. R. , … Breiman, R. F. (2007). Person‐to‐person transmission of Nipah virus in a Bangladeshi community. Emerging Infectious Diseases, 13(7), 1031–1037. 10.3201/eid1307.061128 [DOI] [PMC free article] [PubMed] [Google Scholar]
- He, W. , Auclert, L. Z. , Zhai, X. , Wong, G. , Zhang, C. , Zhu, H. , … Su, S. (2019). Interspecies transmission, genetic diversity, and evolutionary dynamics of pseudorabies virus. The Journal of Infectious Diseases, 219(11), 1705–1715. 10.1093/infdis/jiy731 [DOI] [PubMed] [Google Scholar]
- He, W. , Li, G. , Zhu, H. , Shi, W. , Wang, R. , Zhang, C. , … Su, S. (2019). Emergence and adaptation of H3N2 canine influenza virus from avian influenza virus: An overlooked role of dogs in interspecies transmission. Transboundary and Emerging Diseases, 66(2), 842–851. 10.1111/tbed.13093 [DOI] [PubMed] [Google Scholar]
- He, W. , Zhao, J. , Xing, G. , Li, G. , Wang, R. , Wang, Z. , … Zhou, J. (2019). Genetic analysis and evolutionary changes of Porcine circovirus 2. Molecular Phylogenetics and Evolution, 139, 106520 10.1016/j.ympev.2019.106520 [DOI] [PubMed] [Google Scholar]
- Imai, H. , Dinis, J. M. , Zhong, G. , Moncla, L. H. , Lopes, T. J. S. , McBride, R. , … Kawaoka, Y. (2018). Diversity of influenza A(H5N1) viruses in Infected humans, Northern Vietnam, 2004–2010. Emerging Infectious Diseases, 24(7), 1128–1238. 10.3201/eid2407.171441 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kosakovsky Pond, S. L. , & Frost, S. D. (2005). Not so different after all: A comparison of methods for detecting amino acid sites under selection. Molecular Biology and Evolution, 22(5), 1208 10.1093/molbev/msi105 [DOI] [PubMed] [Google Scholar]
- Kumar, S. , Stecher, G. , & Tamura, K. (2016). MEGA7: Molecular evolutionary genetics analysis version 7.0 for bigger datasets. Molecular Biology and Evolution, 33(7), 1870 10.1093/molbev/msw054 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lam‐Tung, N. , Schmidt, H. A. , Arndt, V. H. , & Bui Quang, M. (2015). IQ‐TREE: A fast and effective stochastic algorithm for estimating maximum‐likelihood phylogenies. Molecular Biology and Evolution, 32(1), 268–274. 10.1093/molbev/msu300 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee, B. , Pernet, O. , Ahmed, A. A. , Zeltina, A. , Beaty, S. M. , & Bowden, T. A. (2015). Molecular recognition of human ephrinB2 cell surface receptor by an emergent African henipavirus. Proceedings of the National Academy of Sciences of the United States of America, 112(17), E2156–2165. 10.1073/pnas.1501690112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, G. , He, W. , Zhu, H. , Bi, Y. , Wang, R. , Xing, G. , … Su, S. (2018). Origin, Genetic Diversity, and Evolutionary Dynamics of Novel Porcine Circovirus 3. Advanced Science, 5(9), 1800275 10.1002/advs.201800275 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang, B. O. , Li, Z. , Jenni, S. , Rahmeh, A. A. , Morin, B. M. , Grant, T. , … Whelan, S. P. J. (2015). Structure of the L protein of vesicular stomatitis virus from electron cryomicroscopy. Cell, 162(2), 314–327. 10.1016/j.cell.2015.06.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lo, M. K. , Lowe, L. , Hummel, K. B. , Sazzad, H. M. S. , Gurley, E. S. , Hossain, M. J. , … Rota, P. A. (2012). Characterization of Nipah virus from outbreaks in Bangladesh, 2008–2010. Emerging Infectious Diseases, 18(2), 248–255. 10.3201/eid1802.111492 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lo Presti, A. , Cella, E. , Giovanetti, M. , Lai, A. , Angeletti, S. , Zehender, G. , & Ciccozzi, M. (2016). Origin and evolution of Nipah virus. Journal of Medical Virology, 88(3), 380–388. 10.1002/jmv.24345 [DOI] [PubMed] [Google Scholar]
- Lole, K. S. , Bollinger, R. C. , Paranjape, R. S. , Gadkari, D. , Kulkarni, S. S. , Novak, N. G. , … Ray, S. C. (1999). Full‐length human immunodeficiency virus type 1 genomes from subtype C‐infected seroconverters in India, with evidence of intersubtype recombination. Journal of Virology, 73(1), 152–160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luby, S. P. , Hossain, M. J. , Gurley, E. S. , Ahmed, B.‐N. , Banu, S. , Khan, S. U. , … Rahman, M. (2009). Recurrent Zoonotic Transmission of Nipah Virus into Humans, Bangladesh, 2001–2007. Emerging Infectious Diseases, 15(8), 1229–1235. 10.3201/eid1508.081237 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin, D. P. , Murrell, B. , Golden, M. , Khoosal, A. , & Muhire, B. (2015). RDP4: Detection and analysis of recombination patterns in virus genomes. Virus Evolution, 1(1), vev003 10.1093/ve/vev003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin, D. P. , Posada, D. , Crandall, K. A. , & Williamson, C. (2005). A modified bootscan algorithm for automated identification of recombinant sequences and recombination breakpoints. AIDS Research and Human Retroviruses, 21(1), 98–102. 10.1089/aid.2005.21.98 [DOI] [PubMed] [Google Scholar]
- Martin, D. , & Rybicki, E. (2000). RDP: Detection of recombination amongst aligned sequences. Bioinformatics, 16(6), 562–563. 10.1093/bioinformatics/16.6.562 [DOI] [PubMed] [Google Scholar]
- Murrell, B. , Moola, S. , Mabona, A. , Weighill, T. , Sheward, D. , Pond, S. L. K. , & Scheffler, K. (2013). FUBAR: A fast, unconstrained Bayesian AppRoximation for inferring selection. Molecular Biology and Evolution, 30(5), 1196–1205. 10.1093/molbev/mst030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murrell, B. , Wertheim, J. O. , Moola, S. , Weighill, T. , Scheffler, K. , & Pond, S. L. K. (2012). Detecting individual sites subject to episodic diversifying selection. Plos Genetics, 8(7), e1002764 10.1371/journal.pgen.1002764 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Negrete, O. A. , Levroney, E. L. , Aguilar, H. C. , Bertolotti‐Ciarlet, A. , Nazarian, R. , Tajyar, S. , & Lee, B. (2005). EphrinB2 is the entry receptor for Nipah virus, an emergent deadly paramyxovirus. Nature, 436(7049), 401–405. 10.1038/nature03838 [DOI] [PubMed] [Google Scholar]
- Negrete, O. A. , Wolf, M. C. , Aguilar, H. C. , Enterlein, S. , Wang, W. , Mühlberger, E. , … Lee, B. (2006). Two key residues in ephrinB3 are critical for its use as an alternative receptor for Nipah virus. PLoS Path, 2(2), 78–86. 10.1371/journal.ppat.0020007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Padidam, M. , Sawyer, S. , & Fauquet, C. M. (1999). Possible emergence of new geminiviruses by frequent recombination. Virology, 265(2), 218–225. 10.1006/viro.1999.0056 [DOI] [PubMed] [Google Scholar]
- Parker, J. , Rambaut, A. , & Pybus, O. G. (2008). Correlating viral phenotypes with phylogeny: Accounting for phylogenetic uncertainty. Infection Genetics & Evolution, 8(3), 239–246. 10.1016/j.meegid.2007.08.001 [DOI] [PubMed] [Google Scholar]
- Posada, D. , & Crandall, K. A. (2001). Evaluation of methods for detecting recombination from DNA sequences: Computer simulations. Proceedings of the National Academy of Sciences of the United States of America, 98(24), 13757–13762. 10.1073/pnas.241370698 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rahman, S. A. , Hassan, S. S. , Olival, K. J. , Mohamed, M. , Chang, L. Y. , & Hassan, L. … Henipavirus Ecology Research, G (2010). Characterization of Nipah virus from naturally infected Pteropus vampyrus bats, Malaysia. Emerging Infectious Diseases, 16(12), 1990–1993. 10.3201/eid1612.091790 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rambaut, A. , Lam, T. T. , Max Carvalho, L. , & Pybus, O. G. (2016). Exploring the temporal structure of heterochronous sequences using TempEst (formerly Path‐O‐Gen). Virus Evolution, 2(1), vew007 10.1093/ve/vew007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reynes, J.‐M. , Counor, D. , Ong, S. , Faure, C. , Seng, V. , Molia, S. , … Sarthou, J.‐L. (2005). Nipah Virus in Lyle's Flying Foxes, Cambodia. Emerging Infectious Diseases, 11(7), 1042–1047. 10.3201/eid1107.041350 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sawatsky, B. , Bente, D. A. , Czub, M. , & von Messling, V. (2016). Morbillivirus and henipavirus attachment protein cytoplasmic domains differently affect protein expression, fusion support and particle assembly. Journal of General Virology, 97(5), 1066–1076. 10.1099/jgv.0.000415 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sendow, I. , Ratnawati, A. , Taylor, T. , Adjid, R. M. A. , Saepulloh, M. , Barr, J. , … Field, H. (2013). Nipah Virus in the Fruit Bat Pteropus vampyrus in Sumatera, Indonesia. PLoS ONE, 8(7), e69544 10.1371/journal.pone.0069544 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma, V. , Kaushik, S. , Kumar, R. , Yadav, J. P. , & Kaushik, S. (2019). Emerging trends of Nipah virus: A review. Reviews in Medical Virology, 29(1), e2010 10.1002/rmv.2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith, J. M. (1992). Analyzing the mosaic structure of genes. Journal of Molecular Evolution, 34(2), 126–129. 10.1007/BF00182389 [DOI] [PubMed] [Google Scholar]
- Smith, M. D. , Wertheim, J. O. , Weaver, S. , Murrell, B. , Scheffler, K. , & Pond, S. L. K. (2015). Less is more: An adaptive branch‐site random effects model for efficient detection of episodic diversifying selection. Molecular Biology and Evolution, 32(5), 1342–1353. 10.1093/molbev/msv022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sourimant, J. , & Plemper, R. K. (2016). Organization, function, and therapeutic targeting of the morbillivirus RNA‐dependent RNA polymerase complex. Viruses, 8(9), 251 10.3390/v8090251 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stamatakis, A. (2014). RAxML version 8: A tool for phylogenetic analysis and post‐analysis of large phylogenies. Bioinformatics, 30(9), 1312–1313. 10.1093/bioinformatics/btu033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Su, S. , Gu, M. , Liu, D. , Cui, J. , Gao, G. F. , Zhou, J. Y. , & Liu, X. F. (2017). Epidemiology, evolution, and pathogenesis of H7N9 influenza viruses in five epidemic waves since 2013 in China. Trends in Microbiology, 25(9), 713–728. 10.1016/j.tim.2017.06.008 [DOI] [PubMed] [Google Scholar]
- Su, S. , Wong, G. , Shi, W. , Liu, J. , Lai, A. C. K. , Zhou, J. , … Gao, G. F. (2016). Epidemiology, genetic recombination, and pathogenesis of coronaviruses. Trends in Microbiology, 24(6), 490–502. 10.1016/j.tim.2016.03.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun, B. , Jia, L. , Liang, B. , Chen, Q. , & Liu, D. (2018). Phylogeography, transmission, and viral proteins of Nipah virus. Virologica Sinica, 33(5), 385–393. 10.1007/s12250-018-0050-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swanson, K. , Wen, X. , Leser, G. P. , Paterson, R. G. , Lamb, R. A. , & Jardetzky, T. S. (2010). Structure of the Newcastle disease virus F protein in the post‐fusion conformation. Virology, 402(2), 372–379. 10.1016/j.virol.2010.03.050 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wacharapluesadee, S. , Boongird, K. , Wanghongsa, S. , Ratanasetyuth, N. , Supavonwong, P. , Saengsen, D. , … Hemachudha, T. (2010). A longitudinal study of the prevalence of Nipah virus in Pteropus lylei bats in Thailand: Evidence for seasonal preference in disease transmission. Vector‐Borne and Zoonotic Diseases, 10(2), 183–190. 10.1089/vbz.2008.0105 [DOI] [PubMed] [Google Scholar]
- Wacharapluesadee, S. , Lumlertdacha, B. , Boongird, K. , Wanghongsa, S. , Chanhome, L. , Rollin, P. , … Hemachudha, T. (2005). Bat Nipah virus, Thailand. Emerging Infectious Diseases, 11(12), 1949–1951. 10.3201/eid1112.050613 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wacharapluesadee, S. , Samseeneam, P. , Phermpool, M. , Kaewpom, T. , Rodpan, A. , Maneeorn, P. , … Hemachudha, T. (2016). Molecular characterization of Nipah virus from Pteropus hypomelanus in Southern Thailand. Virology Journal, 13, 53 10.1186/s12985-016-0510-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- White, J. R. , Boyd, V. , Crameri, G. S. , Duch, C. J. , van Laar, R. K. , Wang, L. F. , & Eaton, B. T. (2005). Location of, immunogenicity of and relationships between neutralization epitopes on the attachment protein (G) of Hendra virus. Journal of General Virology, 86, 2839–2848. 10.1099/vir.0.81218-0 [DOI] [PubMed] [Google Scholar]
- Wong, J. J. W. , Young, T. A. , Zhang, J. , Liu, S. , Leser, G. P. , Komives, E. A. , … Jardetzky, T. S. (2017). Monomeric ephrinB2 binding induces allosteric changes in Nipah virus G that precede its full activation. Nature Communications, 8(1), 781 10.1038/s41467-017-00863-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu, K. , Broder, C. C. , & Nikolov, D. B. (2012). Ephrin‐B2 and ephrin‐B3 as functional henipavirus receptors. Seminars in Cell & Developmental Biology, 23(1), 116–123. 10.1016/j.semcdb.2011.12.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu, K. , Chan, Y.‐P. , Bradel‐Tretheway, B. , Akyol‐Ataman, Z. , Zhu, Y. , Dutta, S. , … Nikolov, D. B. (2015). Crystal structure of the pre‐fusion Nipah virus fusion glycoprotein reveals a novel hexamer‐of‐trimers assembly. PLoS Path, 11(12), e1005322 10.1371/journal.ppat.1005322 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu, K. , Rajashankar, K. R. , Chan, Y. P. , Himanen, J. P. , Broder, C. C. , & Nikolov, D. B. (2008). Host cell recognition by the henipaviruses: Crystal structures of the Nipah G attachment glycoprotein and its complex with ephrin‐B3. Proceedings of the National Academy of Sciences of the United States of America, 105(29), 9953–9958. 10.1073/pnas.0804797105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yi‐Gang, T. , Wei‐Feng, S. , Di, L. , Jun, Q. , Long, L. , Xiao‐Chen, B. , … Ming, N. (2015). Genetic diversity and evolutionary dynamics of Ebola virus in Sierra Leone. Nature, 524(7563), 93–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials