

Abstract
A number of different human pathogens code for their own enzymes involved in the synthesis of the RNA cap structure. Although the RNA cap structures originating from human and microbial enzymes are often identical, the subunit composition, structure and catalytic mechanisms of the microbial‐encoded enzymes involved in the synthesis of the RNA cap structure are often significantly different from those of host cells. As a consequence, these pathogenic cap‐forming enzymes are potential targets for antimicrobial drugs. During the past few years, experimental studies have started to demonstrate that inhibition of the RNA capping activity is a reasonable approach for the development of antimicrobial agents. The combination of structural, biochemical, and molecular modeling studies are starting to reveal novel molecules that can serve as starting blocks for the design of more potent and specific antimicrobial agents. Here, we examine various strategies that have been developed to inhibit microbial enzymes involved in the synthesis of the RNA cap structure, emphasizing the challenges remaining to design potent and selective drugs. WIREs RNA 2011 2 184–192 DOI: 10.1002/wrna.43
This article is categorized under:
-
1
RNA Processing > Capping and 5' End Modifications
INTRODUCTION
The synthesis and maturation of eukaryotic mRNAs are crucial events for gene expression. During mRNA synthesis, eukaryotic mRNAs undergo a series of critical modifications before being exported to the cytoplasm where they are translated into proteins. These processing events include the addition of a cap structure at the 5′‐terminus, the splicing out of introns, the editing of specific nucleotides, and the acquisition of a poly(A) tail at the 3′‐terminus . The eukaryotic cap structure found at the 5′‐end of mRNAs is critical for the splicing of the cap‐proximal intron, the transport of mRNAs from the nucleus to the cytoplasm, and for both the stability and translation of mRNAs.1 Synthesis of the cap structure occurs co‐transcriptionnally on nascent mRNAs and involves three enzymatic reactions. First, an RNA 5′‐triphosphatase hydrolyzes the gamma‐phosphate at the 5′‐end of the nascent pre‐mRNA to generate a 5′‐diphosphate end. Second, a RNA guanylyltransferase then transfers a GMP moiety to the diphosphate end of the RNA. Finally, using S‐adenosyl‐methionine as a co‐substrate, an RNA (guanine‐N7) methyltransferase catalyzes the transfer of a methyl group to the N‐7 position of the guanine to produce the characteristic m7GpppRNA cap structure.2
A number of different microbial pathogens code for their own enzymes involved in the synthesis of a cap structure. Although the RNA cap structures originating from human and microbial enzymes are often identical, the physical organization of the genes, subunit composition, structure and catalytic mechanisms of the microbial‐encoded enzymes involved in the synthesis of the RNA cap structure are often significantly different from those of host cells. As a consequence, these pathogenic cap‐forming enzymes are potential targets for antimicrobial drugs. Here, we review various strategies that have been developed to inhibit viral, fungal, and protozoan enzymes involved in RNA capping, focusing on the challenges remaining to design potent and selective antimicrobial drugs.
THE RNA TRIPHOSPHATASE AS A DRUG TARGET
Although bacteria and archaea maintained the original cap‐less translation strategy, early eukaryotes have developed, in tandem with the emergence of the 5′‐exoribonucleases, a cap structure at the 5′‐end of their mRNAs, presumably to discriminate between viral and cellular mRNAs. Human cells harbor a metal‐independent RNA triphosphatase (RTase) that belongs to the cysteine phosphatase family and catalyze a two‐step reaction involving a covalent cysteinyl‐phosphoenzyme intermediate.3 On the other hand, the RTases observed in protozoa, eukaryotic viruses, and fungi are completely different both structurally and mechanistically (Figure 1). Catalysis by these metal‐dependent enzymes involves the attack of a water molecule in close proximity to the gamma‐phosphate with no formation of a covalent intermediate. Therefore, the RNA triphosphatase represents an attractive target for the development of novel, potent and low off‐target effect antimicrobial drugs. Although no effective drugs are currently available against pathogenic RTase, their differences with the human RTase can be exploited to develop specific inhibitors. In addition to numerous viral families that encode RTases, other important human pathogens such as Candida albicans, Trypanosoma brucei, and the pathogenic malaria parasite Plasmodium falciparum also encode RTases that could be targeted by antimicrobial drugs.
Figure 1.
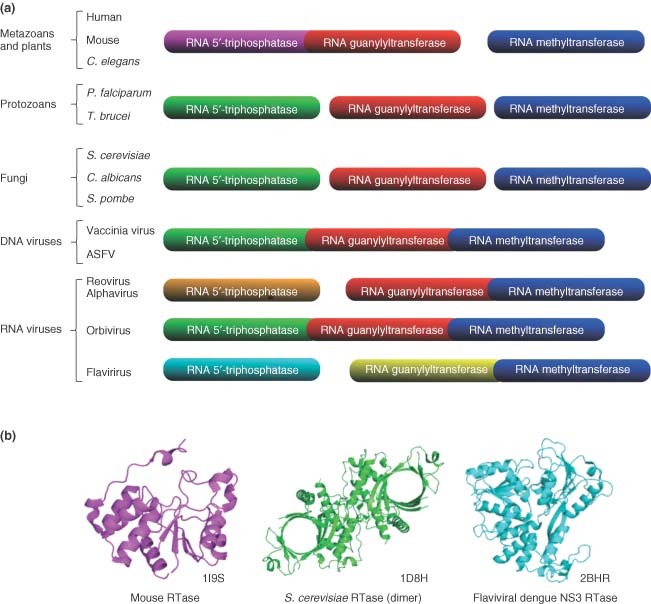
Structural and functional organization of the capping apparatus. (a) Schematic representation of the RNA triphosphatase, RNA guanylyltransferase, and RNA methyltransferase gene organization among different organisms. Both the mechanistical and structural conservation levels are correlated with the color index. (b) Crystal structures of the RNA triphosphatases from mouse (metal independent), S. cerevisiae (metal dependent), and Dengue virus (metal dependent). The corresponding Protein Data Bank (PDB) accession codes are indicated.
PHOSPHATE‐BASED INHIBITORS
Competitive inhibition by the product of a reaction is a common way to impair enzymatic activity. Although experiments have shown that inorganic phosphate (Pi) does not significantly reduce metal‐dependent RTase activity, related compounds have demonstrated interesting inhibitory effects (Table 1). Two independent in vitro studies4,5 on Schizosaccharomyces pombe pcT1P and Chlorella virus cvRtp1 have revealed a reduction of the phosphohydrolase activity in the presence of pyrophosphate (PPi, K i = 400 and 2.4 µM, respectively). Further studies4, 5, 6 using tripolyphosphate (PPPi) have pointed out a greater inhibition of both the pcT1P and cvRtp1 enzymes (K i = 30 and 0.6 µM, respectively) as well as the RTase from West Nile Virus (NS3) which is also strongly inhibited by PPPi (K i = 40 nM). Biochemical studies have shown that cvRtp1 has a greater affinity for PPPi than its natural triphosphorylated RNA substrate.4 As cvRtp1 has a weak tripolyphosphatase activity, it is tempting to speculate that a non‐hydrolyzable PPPi analog would be even more potent. Although the inhibition of the human capping enzyme (HCE) by PPi and PPPi has not been reported, it has to be pointed out that phosphate analogs, such as the FDA‐approved anticytomegalovirus drug foscarnet, have already shown their efficiency in clinic.7 Although PPi and PPPi could not realistically be used as a drug, they could serve as lead compounds for the rational design of novel anti‐RTase drugs. Foscarnet is a succesful example of a PPi structural mimic which has demonstrated both good therapeutical safety and efficicacy. Therefore, a second generation of foscarnet‐based PPi or PPPi analogs can likely be envisioned to efficiently target the RTase of several pathogens.
Table 1.
Inhibitors of Enzymes Involved in the Synthesis of the RNA Cap Structure

NUCLEOTIDE ANALOGS
In contrast to the mammalian RTase, the RTases of several pathogens also possess a nucleotide triphosphatase activity that is catalyzed by the same active site as the one used for the RTase reaction. This raises the interesting possibility of using nucleotide analogs as potential inhibitors. Recent studies on the RTases of Saccharomyces cerevisiae (Cet1) and West Nile virus (NS3) have highlighted a number of synthetic purine analogs (6‐chloropurine‐riboside‐5′‐triphosphate, 6‐methylthioguanosine‐5′‐triphosphate, and 8‐iodo‐guanosine‐5′‐triphosphate) that harbor a high affinity for the active site of the enzymes while being poorly hydrolyzed in comparison with natural nucleotides.8,9 Those compounds harbor very interesting inhibition constants in the sub to low micromolar range (0.4–11 µM) for the Cet1 NTPase activity. As the human capping enzyme is structurally and mechanistically different, namely by its inability to hydrolyze free nucleotides, it is tempting to speculate that those nucleotide analogs will not inhibit the RTase activity in the human capping enzyme. The use of such nucleotide analogs appears as an interesting strategy to inhibit microbial RTases as nucleotide analogs, often marketed as non‐phosphorylated prodrugs for better absorption and distribution, have already proven their efficiency against numerous diseases including cancer, autoimmunity, fungal, and viral infections.
VANADATE‐BASED INHIBITORS
Vanadium is a transition element that is present in a number of oxometalate forms known as vanadates. Orthovanadate (VO is a phosphate analog that has been effectively used as a mechanistic probe of enzymes that catalyze phosphoryl‐transfer reactions. In recent years, the transition state mimic orthovanadate was shown to inhibit the activity of the Chlorella virus RTase (cvRtp1, K
i = 150 µM), whereas decavanadate demonstrated a slightly better inhibitory effect (K
i = 100 µM).10 Interestingly, two other closely related metal‐dependent RTases, namely S. cerevisiae Cet1 and Vaccinia virus D1, remain unaffected by both orthovanadate and decavanadate.10 Further characterization showed that decavanadate is a non‐competitive inhibitor of cvRtp1 and mutagenesis studies revealed that binding of decavanadate does not involve amino acids located in the active site of the enzyme.10 Even if the K
i for the human counterpart is unknown, it is temptating to speculate that this inhibitor will have nearly no effect on the human capping enzyme based on both the structural differences and the allosteric mechanism of action of decavanadate. Although the K
i values of those oxoanion compounds is not in the submicromolar range, the specificity that they harbor for cvRtp1 versus the two closely related Cet1 and D1 proteins is a demonstration that specific inhibition of RTase can also be achieved by using allosteric inhibitors targeting regions located outside of the RTase active site.
is a phosphate analog that has been effectively used as a mechanistic probe of enzymes that catalyze phosphoryl‐transfer reactions. In recent years, the transition state mimic orthovanadate was shown to inhibit the activity of the Chlorella virus RTase (cvRtp1, K
i = 150 µM), whereas decavanadate demonstrated a slightly better inhibitory effect (K
i = 100 µM).10 Interestingly, two other closely related metal‐dependent RTases, namely S. cerevisiae Cet1 and Vaccinia virus D1, remain unaffected by both orthovanadate and decavanadate.10 Further characterization showed that decavanadate is a non‐competitive inhibitor of cvRtp1 and mutagenesis studies revealed that binding of decavanadate does not involve amino acids located in the active site of the enzyme.10 Even if the K
i for the human counterpart is unknown, it is temptating to speculate that this inhibitor will have nearly no effect on the human capping enzyme based on both the structural differences and the allosteric mechanism of action of decavanadate. Although the K
i values of those oxoanion compounds is not in the submicromolar range, the specificity that they harbor for cvRtp1 versus the two closely related Cet1 and D1 proteins is a demonstration that specific inhibition of RTase can also be achieved by using allosteric inhibitors targeting regions located outside of the RTase active site.
RNA GUANYLYLTRANSFERASES AS TARGETS FOR ANTIMICROBIAL DRUGS
Of all the enzymes involved in RNA capping, the RNA guanylyltransferase (GTase) reaction has traditionally been considered a poor candidate as an antimicrobial target because of the high mechanistic and structural conservation of this enzyme across species.11 A common trait of GTases from most eukaryotic viruses and from all known orders of the eukaryotic domain of life is their inclusion in the nucleotidyltransferase superfamily along with DNA and RNA ligases, with which they share mechanistic as well as structural similarities.11 In fact, with the exception of a few viral GTases, most of the characterized enzymes of this family share an acute similarity in their active sites, differing mainly in their protein–protein interaction network. Nonetheless, minor differences between the members of this family can likely be exploited in order to develop antimicrobial agents.
Several inhibitors of the GTase activity have been identified. However, most of these inhibitors display a relatively low specificity toward GTases of pathogens. For instance, recent in vitro studies have shown that foscarnet, an antiviral drug that targets the DNA polymerase of human cytomegalovirus, is a potent inhibitor of the GTase reaction.12 Its mechanism of action is purported to occur through substrate binding inhibition on account of its analogous nature to pyrophosphate (PPi), a product of both the polymerase as well as the RNA guanylyltransferase reaction. Ribavirin, a broad‐spectrum nucleoside analog approved by the FDA as an antiviral for severe respiratory syncytial virus, Hepatitis C, and other viral infections, is another example of a RNA polymerase inhibitor with pleiotropic activities.13 Its incorporation during strand synthesis, leading to chain termination or error catastrophe, is one of the modes of action of this antiviral.13 In its cellular triphosphorylated form, ribavirin can also be used as a substrate by viral GTases.14 However, RNAs capped with ribavirin are relatively inert to methylation by viral RNA (guanine‐N7) methyltransferases, thus resulting in mRNAs which are stable but not efficiently translated into viral proteins.14 The incorporation of ribavirin instead of guanine as a ‘pseudo‐RNA cap’ at the 5′‐end of viral RNAs is a landmark demonstration that the viral RNA cap structure itself could act as an antimicrobial target. The use of guanine‐N7 methylation‐inert cap donor molecules could potentially prove to be an interesting line of research for the development of antimicrobial drugs. However, the ability to synthesize viral RNAs capped by nucleoside analogs in cells and, more importantly, the fate of these RNAs in the cells remain to be assessed. Inhibitors like foscarnet and ribavirin provide a strong basis for the rational design of antiviral drugs. However, on account of the possibility of off‐targets, the risk of major side effects remains. Several issues related to the specificity problem faced by ribavirin and other nucleoside analogs can likely be partially resolved by the development of non‐nucleoside inhibitors.
UNCONVENTIONAL GTases AS DRUG TARGETS
Viruses possessing unconventional capping machineries have been interesting targets for the development of specific GTase inhibitors. For instance, the respiratory syncytial virus (RSV) harbors an unconventional RNA capping apparatus similar to that of the vesicular stomatitis virus (VSV). This capping apparatus differs from mammalian capping enzymes in terms of mechanistic and enzymatic properties.15 In VSV, the viral L protein first hydrolyzed GTP to GDP. Then, the 5′‐RNA:GDP polyribonucleotidyltransferase (PRNTase) activity of the enzyme transfers the 5′‐phosphorylated viral mRNA‐start sequence (L‐pRNA) to GDP to generate the capped Gpp‐pRNA .16 Liuzzi et al., have effectively designed non‐nucleoside competitive inhibitors exhibiting high potency against the capping apparatus of RSV.17 In fact, these inhibitors are large polycyclic aromatic molecules which were initially designed against the RNA‐dependent RNA polymerase activity of the RSV L protein which also harbors the viral RNA capping enzymatic activity. The use of these non‐nucleoside inhibitors leads to the formation of triphosphorylated prematurely terminated RNAs in antiviral assays, by preventing the guanylylation of nascent transcripts, thus effectively targeting the viral RNA capping mechanism rather than the polymerase reaction.17 This class of inhibitors could be a vital starting point for the development of new drugs directed against the RNA capping enzyme of RSV. Therefore, this aforementioned study by Liuzzi and colleagues underlines a major avenue for the design and implementation of drugs directed against RSV. In addition, this work is a proof of concept that unconventional capping enzymes could be targeted for the development of antimicrobial drugs.
In the past few years, divergent GTases that do not possess the consensus catalytic motif found in conventional GTases (KxDG) have been identified in several viral families. Interestingly, several of these enzymes are found in highly pathogenic viruses like Dengue virus, Yellow Fever virus, West Nile virus, Japanese Encephalitis virus, and the negative non‐segmented viruses exemplified by VSV and RSV. Although their mechanism of action is still quite poorly understood, these proteins are potent antiviral targets mainly on account of their divergence from mammalian RNA capping systems. In addition, protein–protein interactions have been shown to be important for the GTase activity of some of those viruses.18 Disruption of these interactions, using peptidic inhibitors, appears as a promising avenue of research for the development of antiflaviviral drugs.
TARGETING THE GTases OF FUNGI AND PROTOZOANS
All characterized GTases of unicellular eukaryotes, excluding their functional organization, bear a strong similarity to their mammalian homologs in terms of structure and mechanism. The most promising avenue of research with regards to the RNA capping machinery for the development of antifungal and antiprotozoan agents is potentially to target essential subunit interactions within the RNA capping complex. In most eukaryotes, the recruitment of the capping machinery to the RNA Pol II pre‐initiation complex is dependent on the phosphorylation status of the carboxy‐terminal domain (CTD) of the polymerase.19 Moreover, in several micro‐organisms, such as S. cerevisiae, the proper recruitment of the various enzymes involved in RNA capping is also fundamentally dependent on protein–protein interactions.20 These interactions not only ensure the presence of the RNA capping machinery in close proximity to the nascent mRNAs, but are also important for the optimal activity and stability of each protein involved in the RNA capping machinery. For instance, in the budding yeast capping machinery, the GTase binds to the phosphorylated CTD, whereas the RTase enzyme lacks this property. However, the interaction of the RTase with the GTase guides the RTase to the site of RNA transcription.20 Inhibitors targeting this interaction could prove to be vital for the development of highly selective antifungal agents. Alternatively, as distinct phosphorylation events of the RNA Pol II CTD are associated with RNA capping subunits recruitment, inhibitors targeting the protein–CTD interface could be an interesting line of research for antifungal development. On account of the different functional organization of the capping subunits in mammalian cells and unicellular organisms, side effects would likely be minimal. However, it should be pointed out that such a strategy might not be useful to target the capping machinery of some organisms. For instance, in C. albicans and related species, the interaction between the various subunits of the capping machinery is not a pre‐requisite for their recruitment to the RNA Pol II initiation complex as they independently interact with the phosphorylated CTD.20 Therefore, only inhibitors targeting the binding of RNA capping subunits to the phosphorylated CTD of the RNA Pol II complex would be of any antimicrobial significance for certain microbial species.
TRANS‐SPLICING IN TRYPANOSOMES AS A DRUG TARGET
Protozoans usually encode yeast‐like capping apparatuses.21 This is the case for the highly pathogenic malaria parasite P. falciparum.21 Therefore, development of chemotherapeutic agents targeted against the RNA capping apparatus would follow the same route as for antifungal agents. However, with regards to trypanosomes, another aspect of the RNA capping chemistry may be targeted. RNA capping of mRNAs in trypanosomes operate through trans‐splicing chemistry, a distinct kind of splicing which involves the capping of a 39‐nucleotide long spliced‐leader RNA (SL RNA) followed by its transfer onto protein encoding RNAs.22 Although this unique mechanism is itself a potential antiprotozoan target, the GTase of these micro‐organisms could also be of interest for drug development as characterization of this enzyme demonstrated an apparent specificity for the SL RNA.23 Competitive inhibitors based on the SL RNA or inhibitors targeting the binding of the SL RNA to the trypanosome RNA guanylyltransferase could potentially be of antimicrobial significance.
MECHANISM‐BASED INHIBITION OF MICROBIAL RNA METHYLTRANSFERASES
During the past few years, the RNA (guanine‐N7) methyltransferase (N7‐MTase) has been a major target for the rationale development of drugs directed against RNA cap synthesis. The reduction in the infectivity of poxviruses and RNA viruses that encode for their own N7‐MTases upon treatment with S‐adenosyl homocysteine (SAH) hydrolase inhibitors demonstrated that cap methylation is a potent drug target.24,25 Inhibitors of cellular SAH hydrolase lead to an increase in intracellular SAH by preventing its cleavage to adenosine and homocysteine. As viral and fungal N7‐MTases are all sensitive to product‐based inhibition, elevated SAH levels is detrimental to the parasite's growth. However, seeking antimicrobial effects through the targeting of intracellular SAH hydrolase inhibitors is indirect and hence, poses specificity problems. This issue can be resolved by directly targeting viral and fungal N7‐MTases.
One of the first identified MTase inhibitor is sinefungin, a natural nucleoside isolated from the cultures of Streptomyces incarnatus and Streptomyces griseolus. On account of its structural analogy to S‐adenosyl‐ methionine (SAM) and SAH, its mode of action has been proven to be directed against RNA (guanine‐N7) and 2′‐O methyltransferases.26 Sinefungin is an inhibitor of numerous MTases including the vaccinia virus RNA N7‐MTase (K i = 12 nM).27 In cellulo, sinefungin inhibits the growth of S. cerevisiae with an IC50 of 22 ng/ml, whereas an isogenic strain, in which the yeast capping apparatus was replaced by the mammalian counterpart, harbored a 5‐fold increased resistance to sinefungin (IC50 112 ng/ml).28,29 Such isogenic yeast strains with fungal versus mammalian capping systems constitute a tremendous tool for rational screens of antifungal drugs that target RNA cap formation in vivo.29 Due to its potent antiparasitic and antileishmaniose effect, sinefungin has been a major starting point for the elaboration of several MTase inhibitors, including the RNA guanine (N7) methyltransferase reaction.30 These inhibitors range from cyclic analogs of sinefungin to base‐modified or amino acid‐modified analogs bearing side chain substitutions. The design of drugs through product‐based inhibition, however, poses serious specificity issues as the human N7‐MTase is also sensitive to inhibition by SAH.
Through mechanism‐based considerations, SAM–nucleotide adducts, which mimic a transition state structure of (guanine‐N7) methylation, have been at the basis for the design of novel molecules of potentially chemotherapeutic interest. Thus, more than 20 years ago, Benghiat et al. designed several multi‐substrate adducts and evaluated their antiviral effects on the vaccinia virus N7‐MTase.31 This study led to the identification of 5′‐deoxy‐5′ (6‐(2‐aminopyrrolo(2,3‐d)‐pyrimidine‐4‐one)methylthio)adenosine (dAPPMA) as a potent antiviral reagent (IC50 = 92 µM). Unfortunately, its inhibition constant for the human RNA N7‐MTase is still unknown.31 The major innovation of this study was the design of a N7‐MTase specific inhibitor through the mimicking of a transition state intermediate, and thus is a major proof of concept that the development of mechanism‐based, low molecular weight specific inhibitors of N7‐Mtase is feasible. However, up until now, no cellular approach has been implemented to evaluate the relative potency of generic molecules based on this identified multi‐substrate adduct inhibitor.
THE RNA METHYLTRANSFERASE OF FLAVIVIRUS AS A DRUG TARGET
In the previous decade, the RNA methyltransferase activity of the NS5 protein of Dengue virus and of other flaviviruses has been at the center of major global initiatives in an effort to develop novel inhibitors. Several innovative approaches spanning from in silico molecular docking to high throughput in vitro screening of library of molecules have been used to define and design molecules of potential antiflaviviral significance. Virtual screening by 2D similarity searching, pharmacophore filtering, and molecular docking for competitive inhibitors of the SAM‐binding site in the NS5 protein of Dengue virus, followed by subsequent in vitro bioassay studies, have led to the identification of a potent inhibitor (Compound 7, IC50 = 60 µM), bearing a novel molecular scaffold, which essentially consists of a linear connection of two simple aromatic cores with a branched cyclic hydrocarbon through amide and carboxylic linkages. The specificity of Compound 7 has not been addressed yet.32 The identification of this novel molecular scaffold bearing a strong affinity to the ligand binding site of the NS5 MTase domain could represent in itself a strong lead for the development of novel antiflaviviral therapeutics. The combination of in silico and biochemical experimentation was also used for the identification of aurintricarboxylic acid (ATA) as another novel‐scaffold high potency inhibitor of the RNA methyltransferase reaction catalyzed by the West Nile Virus (IC50 = 4.2 µM) and the Dengue virus (IC50 = 2.3 µM) NS5 proteins.33 Of noteworthy interest, ATA has also been recently shown to be a potent inhibitor or the SARS‐coronavirus methyltransferases nsp14 (IC50 = 6.4 µM) and nsp10/16 (IC50 = 2.1 µM).34 However, the specificity of ATA still needs to be addressed. Although the aforementioned studies targeted only the SAM‐binding site in their initial pharmacological virtual query, Podvinec et al. used a multistage virtual docking approach against both the SAM‐ and RNA‐binding sites of the methyltransferase domain of the NS5 protein of Dengue virus.35 Subsequent in vitro assays on the putative inhibitors led to the identification of several potent compounds (IC50 < 10 µM). These inhibitors targeted both the RNA‐binding and SAM‐binding sites and thus, laid the basis for a potential multi‐drug approach for the treatment of flaviviral infections. However, no study pertaining to the relative toxicity of these compounds has yet been made available in order to provide an indication of their relative potency as a lead for drug design.
CONCLUSION
Experimental studies are starting to demonstrate that inhibition of the RNA capping activity is a reasonable approach for the development of antimicrobial agents. The combination of structural, biochemical, and molecular modeling studies are revealing novel molecules that can serve as starting blocks for the design of more potent and specific antimicrobial agents. The next few years will undoubtedly bring more informed structure‐based drug discovery programs targeting enzymes involved in RNA cap synthesis.
NOTES
We apologize to those colleagues whose original work could not be cited due to space constraints.
FURTHER READING
- Lescar J, Luo D, Xu T, Sampath A, Lim SP, Canard B, Vasudevan SG. Towards the design of antiviral inhibitors against flaviviruses: the case for the multifunctional NS3 protein from Dengue virus as a target. Antiviral Res 2008, 80:94–101. [DOI] [PubMed] [Google Scholar]
- Shuman S. The mRNA capping apparatus as drug target and guide to eukaryotic phylogeny. Cold Spring Harb Symp Quant Biol 2001, 66:301–312. [DOI] [PubMed] [Google Scholar]
RELATED WIREs ARTICLES
https://doi.org/10.1002/wrna.52
https://doi.org/10.1002/wrna.19
REFERENCES
- 1. Furuichi Y, Shatkin AJ. Viral and cellular mRNA capping: past and prospects. Adv Virus Res 2000, 55:135–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Shuman S. Structure, mechanism, and evolution of the mRNA capping apparatus. Prog Nucleic Acid Res Mol Biol 2001, 66:1–40. [DOI] [PubMed] [Google Scholar]
- 3. Shuman S. What messenger RNA capping tells us about eukaryotic evolution. Nat Rev Mol Cell Biol 2002, 3:619–625. [DOI] [PubMed] [Google Scholar]
- 4. Gong C, Shuman S. Chlorella virus RNA triphosphatase. Mutational analysis and mechanism of inhibition by tripolyphosphate. J Biol Chem 2002, 277:15317–15324. [DOI] [PubMed] [Google Scholar]
- 5. Pei Y, Schwer B, Hausmann S, Shuman S. Characterization of Schizosaccharomyces pombe RNA triphosphatase. Nucleic Acids Res 2001, 29:387–396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Benzaghou I, Bougie I, Picard‐Jean F, Bisaillon M. Energetics of RNA binding by the West Nile virus RNA triphosphatase. FEBS Lett 2006, 580:867–877. [DOI] [PubMed] [Google Scholar]
- 7. Schreiber A, Härter G, Schubert A, Bunjes D, Mertens T, Michel D. Antiviral treatment of cytomegalovirus infection and resistant strains. Expert Opin Pharmacother 2009, 10:191–209. [DOI] [PubMed] [Google Scholar]
- 8. Issur M, Despins S, Bougie I, Bisaillon M. Nucleotide analogs and molecular modeling studies reveal key interactions involved in substrate recognition by the yeast RNA triphosphatase. Nucleic Acids Res 2009, 37:3714–3722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Despins S, Issur M, Bougie I, Bisaillon M. Deciphering the molecular basis for nucleotide selection by the flavivirus RNA helicase. Nucleic Acids Res. In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Bougie I, Bisaillon M. Inhibition of a metal‐dependent viral RNA triphosphatase by decavanadate. Biochem J 2006, 398:557–567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Shuman S, Liu Y, Schwer B. Covalent catalysis in nucleotidyl transfer reactions: essential motifs in Saccharomyces cerevisiae RNA capping enzyme are conserved in Schizosaccharomyces pombe and viral capping enzymes and among polynucleotide ligases. Proc Natl Acad Sci USA 1994, 91:12046–12050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Soulière MF, Perreault JP, Bisaillon M. Kinetic and thermodynamic characterization of the RNA guanylyltransferase reaction. Biochemistry 2008, 47:3863–3874. [DOI] [PubMed] [Google Scholar]
- 13. Graci JD, Cameron CE. Mechanisms of action of ribavirin against distinct viruses. Rev Med Virol 2006, 16:37–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Bougie I, Bisaillon M. The broad spectrum antiviral nucleoside ribavirin as a substrate for a viral RNA capping enzyme. J Biol Chem 2004, 279: 22124–22130. [DOI] [PubMed] [Google Scholar]
- 15. Ogino T, Banerjee AK. Unconventional mechanism of mRNA capping by the RNA‐dependent RNA polymerase of vesicular stomatitis virus. Mol Cell 2007, 25:85–97. [DOI] [PubMed] [Google Scholar]
- 16. Ogino T, Yadav SP, Banerjee AK. Histidine‐mediated RNA transfer to GDP for unique mRNA capping by vesicular stomatitis virus RNA polymerase. Proc Natl Acad Sci USA 2010, 107:3463–3468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Liuzzi M, Mason SW, Cartier M, Lawetz C, McCollum RS, Dansereau N, Bolger G, Lapeyre N, Gaudette Y, Lagacé L, et al. Inhibitors of respiratory syncytial virus replication target cotranscriptional mRNA guanylylation by viral RNA‐dependent RNA polymerase. J Virol 2005, 79:13105–13115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Issur M, Geiss BJ, Bougie I, Picard‐Jean F, Despins S, Mayette J, Hobdey SE, Bisaillon M. The flavivirus NS5 protein is a true RNA guanylyltransferase that catalyzes a two‐step reaction to form the RNA cap structure. RNA 2009, 15:2340–2350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Cho EJ, Takagi T, Moore CR, Buratowski S. mRNA capping enzyme is recruited to the transcription complex by phosphorylation of the RNA polymerase II carboxy‐terminal domain. Genes Dev 1997, 11:3319–3326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Takagi T, Cho EJ, Janoo RT, Polodny V, Takase Y, Keogh MC, Woo SA, Fresco‐Cohen LD, Hoffman CS, Buratowski S. Divergent subunit interactions among fungal mRNA 5′‐capping machineries. Eukaryot Cell 2002, 1:448–457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Ho CK, Shuman S. A yeast‐like mRNA capping apparatus in Plasmodium falciparum. J Biol Chem 2001, 276:46182–46186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Perry KL, Watkins AP, Agabian N. Trypanosome mRNAs have unusual “cap 4” structures acquired by addition of a spliced leader. Proc Natl Acad Sci USA 1987, 84:8190–8194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Zwierzynski TA, Buck GA. In vitro capping in Trypanosoma cruzi identifies and shows specificity for the spliced leader RNA and U‐RNAs. Nucleic Acids Res 1990, 18:4197–4206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. De Clercq E, Cools M. Antiviral potency of adenosine analogues: correlation with inhibition of S‐adenosylhomocysteine hydrolase. Biochem Biophys Res Commun 1985, 129:306–311. [DOI] [PubMed] [Google Scholar]
- 25. Hasobe M, McKee JG, Ishii H, Cools M, Borchardt RT, De Clercq E. Elucidation of the mechanism by which homocysteine potentiates the anti‐vaccinia virus effects of the S‐adenosylhomocysteine hydrolase inhibitor 9‐(trans‐2′,trans‐3′‐dihydroxycyclopent‐4′‐enyl)‐adenine. Mol Pharmacol 1989, 36:490–496. [PubMed] [Google Scholar]
- 26. Pugh CS, Borchardt RT. Effects of S‐adenosylhomocysteine analogues on vaccinia viral messenger ribonucleic acid synthesis and methylation. Biochemistry 1982, 21:1535–1541. [DOI] [PubMed] [Google Scholar]
- 27. Pugh CS, Borchardt RT, Stone HO. Sinefungin, a potent inhibitor of virion mRNA(guanine‐7‐)‐methyltransferase, mRNA(nucleoside‐2′‐)‐methyltransferase, and viral multiplication. J Biol Chem 1978, 253:4075–4077. [PubMed] [Google Scholar]
- 28. Zheng S, Hausmann S, Liu Q, Ghosh A, Schwer B, Lima CD, Shuman S. Mutational analysis of Encephalitozoon cuniculi mRNA cap (guanine‐N7) methyltransferase, structure of the enzyme bound to sinefungin, and evidence that cap methyltransferase is the target of sinefungin's antifungal activity. J Biol Chem 2006, 281:35904–35913. [DOI] [PubMed] [Google Scholar]
- 29. Saha N, Schwer B, Shuman S. Characterization of human, Schizosaccharomyces pombe, and Candida albicans mRNA cap methyltransferases and complete replacement of the yeast capping apparatus by mammalian enzymes. J Biol Chem 1999, 274:16553–16562. [DOI] [PubMed] [Google Scholar]
- 30. Paolantonacci P, Lawrence F, Robert‐Géro M. Differential effect of sinefungin and its analogs on the multiplication of three Leishmania species. Antimicrob Agents Chemother 1985, 28:528–531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Benghiat E, Crooks PA, Goodwin R, Rottman F. Inhibition of vaccinia RNA guanine 7‐methyltransferase by compounds designed as multisubstrate adducts. J Pharm Sci 1986, 75:142–145. [DOI] [PubMed] [Google Scholar]
- 32. Luzhkov VB, Selisko B, Nordqvist A, Peyrane F, Decroly E, Alvarez K, Karlen A, Canard B, Qvist J. Virtual screening and bioassay study of novel inhibitors for dengue virus mRNA cap (nucleoside‐2′O)‐methyltransferase. Bioorg Med Chem 2007, 15:7795–7802 [DOI] [PubMed] [Google Scholar]
- 33. Milani M, Mastrangelo E, Bollati M, Selisko B, Decroly E, Bouvet M, Canard B, Bolognesi M. Flaviviral methyltransferase/RNA interaction: structural basis for enzyme inhibition. Antiviral Res 2009, 83:28–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Bouvet M, Debarnot C, Imbert I, Selisko B, Snijder EJ, Canard B, Decroly E. In vitro reconstitution of SARS‐coronavirus mRNA cap methylation. PLoS Pathog 2010, 6:e1000863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Podvinec M, Lim SP, Schmidt T, Scarsi M, Wen D, Sonntag LS, Sanschagrin P, Shenkin PS, Schwede T. Novel Inhibitors of dengue virus methyltransferase: discovery by in vitro‐driven virtual screening on a desktop computer grid. J Med Chem 2010, 53: 1483–1495. [DOI] [PubMed] [Google Scholar]