

SUMMARY
Acute lung injury (ALI) is a lethal inflammatory lung disorder whose incidence is on the rise. Alveolar macrophages normally act to resolve inflammation, but when dysregulated they can provoke ALI. We demonstrate that monocyte-derived macrophages (CD11b+ macrophages) recruited into the airspace upregulate the anti-inflammatory function of alveolar macrophages by suppressing their stimulator of type 1 interferon gene (STING) signaling. Depletion of CD11b+ macrophages in mice (macrophagedep mice) after endotoxin or after Pseudomonas aeruginosa causes expansion of the inflammatory alveolar macrophage population, leading to neutrophil accumulation, irreversible loss of lung vascular barrier function, and lethality. We show that CD11b+ macrophages suppress alveolar macrophage-STING signaling via sphingosine kinase-2 (SPHK2) generation of sphingosine-1-phosphate (S1P). Thus, adoptive transfer of wild-type (WT) or STING−/−, but not SPHK2−/−, CD11b monocytes from murine bone marrow into injured macrophagedep mice rescue anti-inflammatory alveolar macrophages and reverse lung vascular injury. SPHK2-induced S1P generation in CD11b+ macrophages has the potential to educate alveolar macrophages to resolve ALI.
In Brief
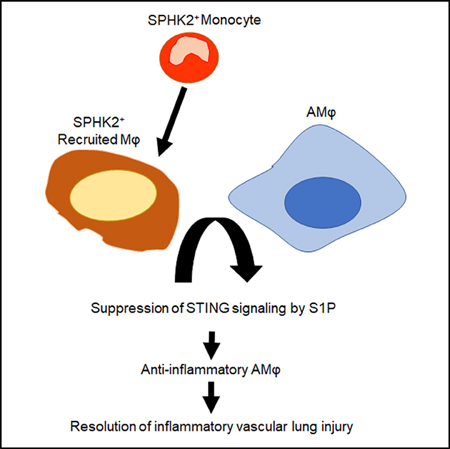
Joshi et al. demonstrate an essential role of SPHK2+ monocyte-derived CD11b+ macrophages, which are recruited to the airspace, in promoting anti-inflammatory function of alveolar macrophages during lung injury. They show that S1P generated by recruited SPHK2+-CD11b+ macrophages suppresses STING signaling in alveolar macrophages to resolve inflammatory injury.
Graphical Abstract
INTRODUCTION
Macrophages, the most abundant immune cells in many tissues, including the lung, have the vital task of restoring tissue homeostasis after triggering inflammatory signaling (Gautier et al., 2012; Wynn et al., 2013). Macrophages initiate host defense, upon sensing pathogens, through pro-inflammatory cytokine generation and neutrophil recruitment via a pathway involving activation of the transcription factor nuclear factor κB (NF-κB) by cell-surface Toll-like receptor 4 (TLR4) (Mogensen, 2009; Newton and Dixit, 2012). Suppression of this inflammatory signaling pathway by macrophages in a timely manner is critical for reinstatement of tissue homeostasis. Impairment of this homeostasis leads to acute lung injury (ALI) due to the accumulation of protein-rich fluid and leukocytes in the alveolar space (Matthay et al., 2012; Randolph, 2009).
STING (stimulator of interferon [IFN] genes), a transmembrane homodimer located in the ER (endoplasmic reticulum) membrane, has recently emerged as a potent inducer of macrophage inflammatory signaling following tissue injury (Barber, 2015). STING is activated upon binding of the second messenger, cyclic GMP-AMP (cGAMP), produced through catalysis of double-stranded cellular DNA by cGAS (cGAMP synthase) (Cai et al., 2014; Li et al., 2013). Activated STING then translocates to the Golgi apparatus where it binds to and activates TANK-binding kinase 1 (TBK1) and IFN regulatory factor 3 (IRF3) via a phosphorylation-dependent mechanism, leading to generation of type 1 IFN (Barber, 2015; Chen et al., 2016). The cause of the protracted lung vascular inflammatory signaling that is the hallmark of ALI remains a central but unanswered question in lung biology. Consequently, we considered the possible role of STING activation and suppression in macrophages in triggering inflammatory lung injury and repair, respectively.
The lung has two major subsets of macrophages, namely, CD11c+/Siglec-F+ alveolar macrophages and CD11b+/Siglec-F− monocyte-derived macrophages (also referred as recruited macrophages) (Byrne et al., 2016; Johnston et al., 2012; Misharin et al., 2013; Murray and Wynn, 2011; Schyns et al., 2018). Alveolar macrophages have been shown to trigger pro-inflammatory cytokine generation, leading to neutrophil accumulation, but subsequently to orchestrate tissue repair (Ward, 2003; Westphalen et al., 2014). Uncontrolled pro-inflammatory signaling by alveolar macrophages can compromise vascular barrier repair, thereby provoking ALI (Duan et al., 2012; Westphalen et al., 2014). Evidence indicates that the population of CD11b+ macrophages expands during resolution of lung injury (McCubbrey et al., 2016; Zaynagetdinov et al., 2013). Whether recruited CD11b+ macrophages play a role in regulating alveolar macrophage anti-inflammatory function following tissue injury remains elusive. In this study, we depleted CD11b+ monocytes/macrophages following injury, using a well-established line of transgenic mice carrying the CD11b-diphtheria toxin (DT) receptor (DTR), to elucidate their role in regulating alveolar macrophage function and resolution of ALI.
We show that depletion of CD11b+ macrophages in mice after endotoxin or after Pseudomonas aeruginosa (PA) induced alveolar macrophage expansion and that these alveolar macrophages produced pro-inflammatory cytokines, including IFN-β, in a long-lasting manner, leading to neutrophil accumulation, irreversible loss of lung vascular barrier function, and lethality. Adoptive transfer of CD11b+ monocytes from wild-type (WT) murine bone marrow into injured macrophagedep mice (mice depleted of CD11b+ macrophages) rescued the anti-inflammatory function of alveolar macrophages and reversed lung vascular injury by suppressing STING signaling. We show that CD11b+ macrophages suppressed alveolar macrophage-STING signaling via sphingosine kinase-2 (SPHK2)-mediated generation of sphingosine-1-phosphate (S1P) and thereby resolved inflammatory vascular injury.
RESULTS
Depletion of CD11b+ Macrophages following Injury Impairs Resolution of Lung Vascular Inflammatory Injury
To determine the role of CD11b+ macrophages in regulating alveolar macrophage homeostatic function following injury and thereby in promoting resolution of lung injury, we activated alveolar macrophages by exposing CD11b-DTR mice to nebulized endotoxin (lipopolysaccharide [LPS]) followed by an intraperitoneal (i.p.) injection of DT (hereafter referred to as macrophagedep mice) (Figure S1A). In CD11b-DTR transgenic mice, human DTR is controlled by the CD11b promoter. DT administration specifically induces apoptosis in the monocytic lineage, causing its depletion without affecting other murine cells (Arnold et al., 2007; Dhaliwal et al., 2012; Stoneman et al., 2007). CD11b-DTR mice receiving PBS served as a paired control. In control mice, LPS increased inflammatory vascular injury within 4 h as indicated by the enhanced generation of pro-inflammatory cytokines, such as interleukin (IL)-6, IL-1β, and IFN-β (Figures 1A–1C), neutrophil accumulation (Figure 1D), transendothelial albumin extravasation, and lung wet/dry weight ratio (Figures 1E–1G). These responses then returned to near basal levels within 24 h after LPS in control mice (Figures 1A–1G). However, cytokines (Figures 1A–1C), neutrophil accumulation (Figure 1D), and lung edema failed to resolve in macrophagedep mice even at 48 h (Figures 1E–1G). qPCR analysis of IL-6, IL-1β, and IFN-β similarly showed a transient increase in these cytokines following LPS challenge in WT lungs but not in macrophagedep lungs (Figures S1B–S1D).
Figure 1. CD11b+ Macrophages Are Required for Preventing Pro-inflammatory Cytokine Generation from Alveolar Macrophages and Thereby Resolution of Inflammatory Vascular Injury after Endotoxin Challenge.
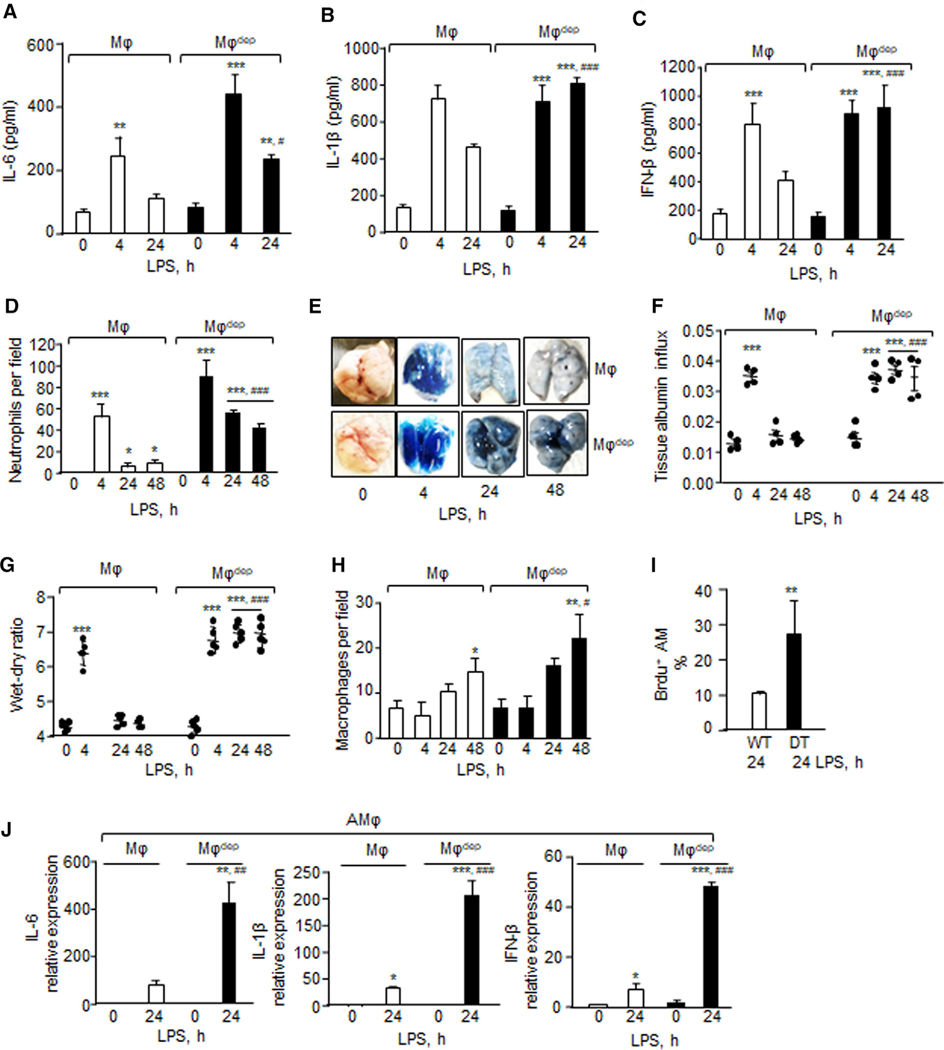
(A–C) At indicated times lungs were harvested, homogenized and supernatant were used to quantify IL-6 (A), IL1β (B), and IFN-β levels (C) using an ELISA kit (n = 4 mice/group).
(D) Neutrophil count was performed (per field) on hematoxylin and eosin-stained bronchoalveolar lavage at indicated times (n = 3 mice/group).
(E–G) 30 min before sacrificing the mice at indicated times, (E) Evans blue-labeled albumin was injected retro-orbitally into macrophages (Mφ) or macrophagedep mice. (F) albumin influx (n = 4 mice/group) and (G) the lung wet/dry ratio (n = 5 mice/group) were determined. A representative image of Evans blue accumulation in the lung is shown in E from experiments that were repetated four times.
(H) Macrophages were counted in hematoxylin and eosin-stained bronchoalveolar lavage at indicated times (n = 3 mice/group).
(I) BrdU (5-bromo-2’-deoxyuridine) was injected i.t. 4 h before sacrificing the mice. Lungs were digested with collagenase and stained with CD11c and BrdU antibodies (n = 3 mice/group).
(J) Lung cells were stained with CD11c, CD45, and Siglec-F antibodies and cytokine expression in alveolar macrophages (CD11c+/CD45+/Siglec-F+) were determined using qPCR, taking GAPDH as control in (n = 4 mice/group). Alveolar macrophages were sorted twice independently.
(A–D) and (F–J) show mean ± SD. *p < 0.05, **p < 0.01, ***p < 0.001 relative to macrophages or macrophagedep mice receiving vehicle alone; #p < 0.05, ##p < 0.01,###p < 0.001 relative to macrophage group after receiving LPS at the indicated time. Analysis was performed using one-way ANOVA followed by Tukey’s multiple comparisons test.
Fluorescence-activated cell sorting (FACS) analysis of control mouse lungs revealed two major macrophage populations consisting of 54% CD11c+ (alveolar macrophages) and 44% CD11b+ (non-alveolar) at 0 h (Figure S1E). We also noted a third macrophage population expressing both CD11b+ and CD11c+ cell-surface markers and representing 3.4% of the macrophage population (Figure S1E). During resolution of lung injury, at 24 h the CD11c+ macrophage population was reduced to 3% while CD11c+/CD11b+ macrophages increased to 31% (Figure S1E). Also, CD11b+ macrophages expanded to 66% during resolution of lung injury at 24 h. While lungs receiving DT showed a 73% reduction in the CD11b+ macrophage population at 24 h after LPS challenge (Figure S1E), DT had no effect on the CD11b+/ CD11c+ population. Intriguingly, we also found that alveolar macrophages proliferated in the lungs as well as bronchoalveolar lavage (BAL) at 24 h after injury in macrophagedep mice (Figures 1H, 1I, S1F, and S1G). Therefore, we next performed FACS analysis in BAL obtained from unexposed WT or LPS-exposed WT and macrophagedep lungs to determine whether DT depleted CD11b+ cells that are recruited in the airspace. We found that DT did not alter CD11b+ macrophages remaining in the lungs after lavage (77 ± 3.5 versus 85.8 ± 1.9) (Figure S1E). DT depleted CD11b+ macrophages in BAL from 28.8% ± 0.2% to 15.3% ± 0.5%). DT also decreased the CD11b+/CD11c+ population that remained in the lungs after lavage (19.7 ± 1.7 to 5.3 ± 0.7) and BAL (61.8% ± 4.2% to 37.2% ± 5.5%) (Figure S1E). Thus, we interpret these findings to mean that DT predominantly targets CD11b+ macrophages that are recruited in the airspace, some of which acquire CD11b+/CD11c+ lineage. We also found that alveolar macrophages (CD45+/CD11c+/Siglec-F+) sorted from macrophagedep lungs showed higher expression of pro-inflammatory cytokines than did control lungs (Figure 1J). Granulocyte-macrophage colony-stimulating factor (GM-CSF), which is known to induce alveolar macrophage expansion and neutrophil recruitment (Becher et al., 2016), was also increased both at protein and mRNA levels in macrophagedep lungs (Figures S1H and S1I). At 48 h, CD11b+ macrophages remained 25% lower in CD11b-DTR lungs receiving DT than in control lungs (data not shown). These results demonstrate that depletion of CD11b+ macrophages following injury lead to alveolar macrophage proliferation and hyperactivation, thereby impairing the tissue repair process. Consistent with this observation, in control mice, BAL protein and myeloperoxidase (MPO) activity increased at 4 h and then declined to near basal levels at 24 and 48 h (Figures S1J and S1K). However, BAL protein and MPO activity remained consistently elevated in CD11b+ macrophagedep mice after injury (Figures S1J and S1K). We confirmed that DT injection in WT mice did not augment lung injury after LPS challenge (data not shown).
Pneumonia is a key factor compromising patient survival in ALI (Broquet et al., 2017). Thus, we administered 1 × 104 colonyforming units (CFU) of PA intratracheally (i.t.) to DTR mice followed by injection of DT to assess the effect of CD11b+ macrophage depletion on the development of PA-induced ALI (Figure S2A). Following 24 h of PA infection, DT similarly induced 62% depletion of CD11b+ macrophages in CD11b-DTR mice, which was associated with a 60% expansion of alveolar macrophages (Figure S2B). At each time point after PA infection, macrophagedep lungs showed markedly increased pro-inflammatory cytokine protein levels as well as mRNA (Figures 2A–2C and S2C–S2E) and lung edema formation compared to control mice (Figure 2D). In control mice, inflammatory cytokines and lung edema returned to baseline at 96 h while these responses persisted in macrophagedep lungs (Figures 2A–2D and S2C–S2E). These findings demonstrate that CD11b+ macrophages are recruited in airspace and play a key role in promoting resolution of ALI by educating alveolar macrophages to acquire anti-inflammatory function in a timely manner.
Figure 2. Role of CD11b+ Macrophages in Regulating Pseudomonas aeruginosa-Induced Lung Inflammatory Injury and Lethality.

(A–C) Lungs receiving 1 × 104 CFU of P. aeruginosa (PA) i.t. were harvested at the indicated times and homogenized, and supernatants were used to quantify IL-6 (A), IL-1β (B), and IFN-β (C) levels using an ELISA kit (n = 4 mice/group).
(D) Lung edema determined as described in Figure 1G. Plot shows individual values (n = 5 mice/group).
(E) Mouse survival was assessed every 6–12 h after 1 × 106 CFU of PA instillation (n = 10 mice/group).
(A–E) are shown as mean ± SD from experiments that were repeated three times independently. *p < 0.05, ***p < 0.001 relative to unexposed macrophages (Mφ) or macrophagedep mice; ###p < 0.001 relative to macrophage group after PA challenge at indicated times. Analysis was performed using one-way ANOVA followed by Tukey’s multiple comparisons test.
Persistent inflammatory vascular injury occurring after pneumonia is linked with lethality in patients in the hospital setting (Delano and Ward, 2016). Thus, we next investigated the role of CD11b+ macrophage suppression of lung vascular inflammatory injury in augmenting mouse survival after pneumonia. We administered 1 × 106 CFU of PA i.t. in macrophagedep mice and assessed survival of these mice for 5 days. PA produced 100% mortality within 24 h in macrophagedep mice while WT mice remained normal for up to 5 days (Figure 2E).
Since STING activated upon tissue injury can prolong inflammatory responses by inducing IFN-β generation (Burdette et al., 2011; Ivashkiv and Donlin, 2014), we assessed whether STING activation after tissue injury caused long-lasting vascular inflammatory injury in macrophagedep lungs in addition to canonical activation of NF-κB activity (Mogensen, 2009). Thus, we determined the activities of TBK1, IRF3, and NF-κB-p65 in macrophagedep lungs. We found that LPS augmented the phosphorylation of TBK1 and IRF3 in a sustained manner, i.e., phosphorylation was increased at 4 h and remained near this level for the next 24 h in macrophagedep lungs (Figures 3A, S2F, and S2G). However, LPS transiently increased TBK1 and IRF3 phosphorylation in control lungs (Figures 3A, S2F, and S2G). LPS also enhanced NF-κB-p65 phosphorylation, a readout of NF-κB activation (Kwon et al., 2016), in a sustained manner in macrophagedep lungs compared to control lungs (Figures 3A and S2H). Compared to control lungs, PA similarly enhanced TBK1 and IRF3 phosphorylation at each time point in macrophagedep lungs (Figures S2I–S2K). Also, PA more markedly augmented NF-κBp65 phosphorylation at 12 and 72 h in macrophagedep lungs than in control lungs (Figure S2L).
Figure 3. CD11b+ Macrophage Depletion Persistently Induces STING Signaling in Alveolar Macrophages, Causing Irreversible Lung Vascular Inflammatory Injury.
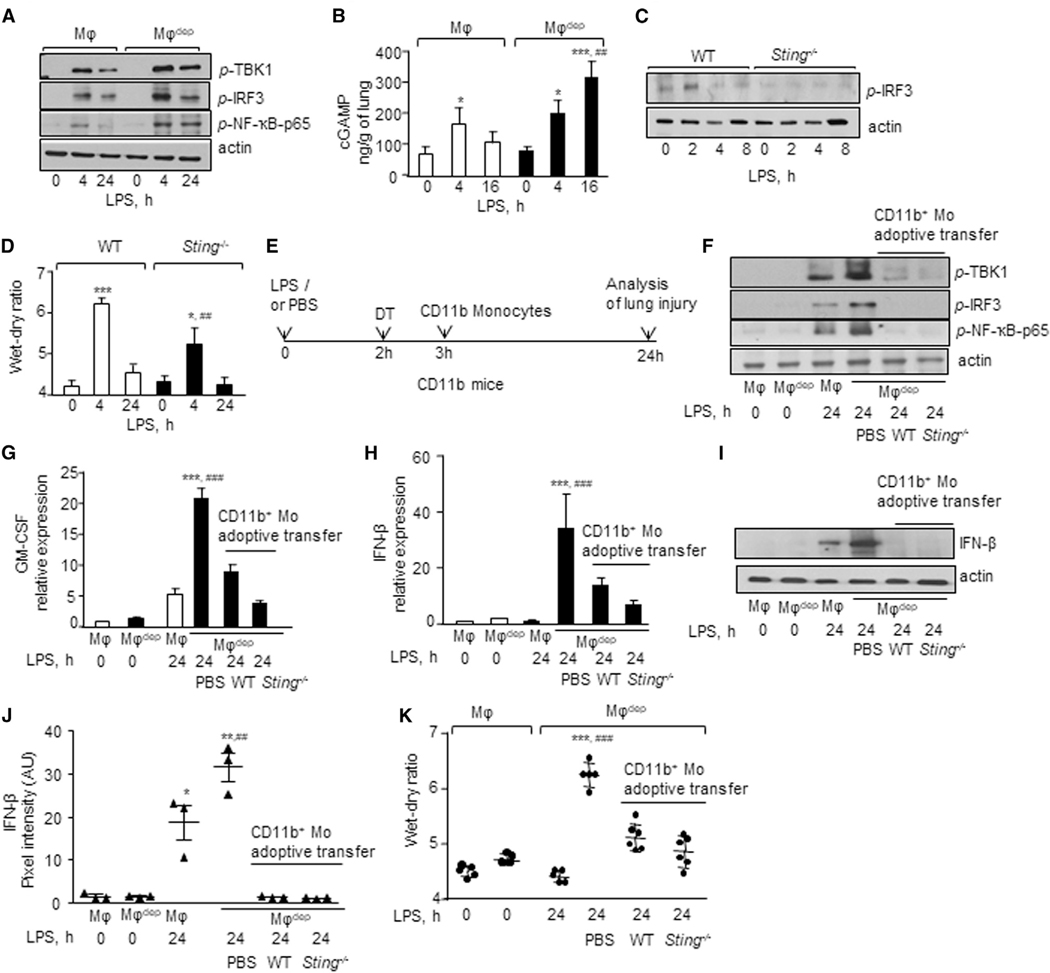
(A) Phosphorylation of TBK1, IRF3, and p65 subunit of NF-κB in lung lysates of mice after LPS challenge was determined using indicated antibodies. Actin was used as a loading control. Experiments were repeated three times independently.
(B) cGAMP was extracted from lungs and quantified by liquid chromatography-mass spectrometry (LC-MS). Data are represented as mean ± SD from experiments that were repeated twice independently (n = 4 mice/group). *p < 0.05, ***p < 0.001 relative to untreated macrophage (MF) and macrophagedep mice; ##p < 0.01 relative to macrophage mice after a 16-h LPS challenge.
(C) WT and Sting−/− BMDMs were exposed to LPS, and IRF3 phosphorylation was determined as in (A). A representative immunoblot is shown from experiments that were independently repeated three times.
(D) Wet/dry ratio in naive or LPS-exposed WT and Sting−/− lungs was determined as in Figure 1G. n = 5 mice/group. Data are represented as mean ± SD from experiments that were repeated two times independently (n = 4 mice/group). *p < 0.05, ***p < 0.001 relative to WT and Sting−/− untreated mice; ##p < 0.01 relative to WT mice after a 4-h LPS challenge.
(E) Protocol shows timing of adoptive transfer of bone marrow CD11b monocytes (CD11b Mo) i.v. in macrophagedep mice after LPS challenge and DT injection. Lung injury was determined at 24 h as in Figure 1G. Macrophagedep mice receiving PBS in lieu of cells and macrophagedep mice challenged with or without LPS served as controls.
(F) Phosphorylation of indicated proteins was determined without or with adoptive transfer of cells and LPS challenge as in (A). A representative immunoblot is shown from experiments that were independently repeated three times.
(G and H) GM-CSF (G) and IFN-β (H) RNA expression without or with adoptive transfer of cells and LPS challenge using qPCR (n = 4 mice/group).
(I and J) A representative immunoblot shows alteration in IFN-β expression after LPS challenge at indicated times. Actin was used as a loading control. Plot in J shows individual pixel intensities of IFN-β expressed as arbitrary units (a.u.) along with mean and SD (n = 3 mice/group).
(K) Lung wet/dry weight ratio without or with adoptive transfer of cells and LPS challenge (n = 5 mice/group).
Data in (G), (H), (J), and (K) are represented as mean ± SD from experiments that were repeated two to three times independently. *p < 0.05, **p < 0.01, ***p < 0.001 relative to untreated macrophages and macrophagedep mice; ##p < 0.01, ###p < 0.001 relative to macrophagedep mice receiving LPS for 24 h or PBS after a 24-h LPS challenge. Data were analyzed by one-way ANOVA followed by Tukey’s multiple comparisons test.
CD11b+ Macrophages Promote Resolution of Inflammatory Vascular Injury by Suppressing STING Signaling in Alveolar Macrophages
STING is activated upon binding cGAMP, a second messenger generated by cGAS (Chen et al., 2016). Thus, we determined cGAMP concentration in control and macrophagedep lungs following LPS and PA injury. Interestingly, we found that LPS induced cGAMP transiently in control mice, while it augmented cGAMP generation in macrophagedep lungs (Figure 3B). Similarly, PA-challenged macrophagedep lungs showed markedly increased cGAMP levels following injury (Figure S2M). Depletion of CD11b+ macrophages alone in CD11b-DTR mice did not increase lung cGAMP concentration (control versus DT-treated CD11b-DTR lungs: 0 h after DT, 114.01 ± 24.29 ng/g; 8 h after DT, 114.01 ± 41.14 ng/g; and 24 h after DT, 143.42 ± 48.25 ng/g).
To confirm that STING activation augmented lung vascular injury, we exposed STING-null mice to nebulized LPS and assessed the lung wet/dry ratio. In addition, we isolated bone marrow-derived macrophages (BMDMs) from WT or STING-null mice to assess IRF3 phosphorylation. As expected, LPS failed to induce IRF3 phosphorylation in Sting−/− BMDMs (Figures 3C and S2N). Compared to WT mice, loss of STING markedly decreased lung edema formation at the peak phase of lung injury (Figure 3D), indicating that STING plays a critical role in extending inflammatory lung injury. Next, we addressed whether suppression of STING signaling in CD11b+ macrophages would promote the pro-resolution role of alveolar macrophages in reinstating tissue-fluid homeostasis. Thus, we adoptively transferred CD11b+ monocytes isolated from Tomato (Td)-WT or Sting−/−mouse bone marrow (BM monocytes) i.v. into macrophagedep mice after lung injury (Figure 3E) and compared their effects in suppressing inflammatory cytokine generation and lung edema after LPS challenge. Macrophagedep mice injected with PBS served as controls. Interestingly, we observed that adoptive transfer of WT as well as Sting−/− BM monocytes inhibited STING signaling in macrophagedep lungs as indicated by inhibition of phosphorylation of TBK1, IRF3, and NF-κB-p65 (Figures 3F and S2O–S2Q), as well as reductions in GM-CSF and IFN-β expression (Figures 3G–3J) and BAL neutrophils (Figure S3A). Adoptive transfer of WT or STING-null BM monocytes in macrophagedep mice also resolved lung edema (Figure 3K). However, macrophagedep mice receiving PBS continued to show exaggerated GM-CSF and IFN-β generation, lung edema and neutrophil accumulation in the BAL (Figures 3G–3K and S3A). Adoptively transferred Td+/CD11b+ monocytes were apparent in the BAL of macrophagedep lungs and expressed the macrophage marker CD68 (Figure S3B), indicating that adoptively transferred CD11b+ cells transitioned into airspace. These data suggest that CD11b+ macrophages in airspace specified alveolar macrophage anti-inflammatory function by suppressing STING signaling, thereby reinstating lung homeostasis.
SPHK2 Generation of S1P in CD11b+ Macrophages Suppresses STING Activity in Alveolar Macrophages to Promote Resolution of Lung Vascular Inflammatory Injury
S1P suppresses lung inflammatory vascular injury in various murine models of lung injury (Natarajan et al., 2013; Szczepaniak et al., 2008; Tauseef et al., 2008). Sphingosine kinases (SPHK1 and SPHK2) convert sphingosine into S1P and modulate inflammatory responses in macrophages (Pyne et al., 2017; Strub et al., 2011; Xiong et al., 2013). Therefore, we tested the hypothesis that S1P generated by SPHK regulates STING activity. We stimulated WT, Sphk1−/−, and Sphk2−/− BMDMs with LPS and assessed whether loss of these kinases altered IFN-β generation by LPS and thus STING activity. We found that LPS induced IFN-β protein and mRNA levels in WT BMDMs at 4 h, which returned to near basal levels by 8 h (Figures 4A and S4A). However, LPS-induced IFN-β remained significantly elevated in SPHK2-null BMDMs even at 8 h (Figures 4A and S4A). In contrast, loss of SPHK1 suppressed LPS-induction of IFN-β in BMDMs (Figure S4B). Consistent with this, we found that LPS augmented the phosphorylation of TBK1, IRF3, and NF-κB-p65 in WT BMDMs at 2–4 h, but the phosphorylation gradually declined toward basal levels in the next 8 h (Figures 4B and S4C–S4E). LPS also induced the phosphorylation of TBK1 and NF-κB-p65 at 2 h in Sphk2−/− BMDMs, but the phosphorylation persisted up to 8 h (Figures 4B and S4C–S4E).
Figure 4. SPHK2 Suppresses Macrophage STING Signaling.

(A) BMDMs isolated from indicated mice were exposed to LPS and IFN-β level was determined as in Figure 1C. Data are represented as mean ± SD from three independent experiments. **p < 0.01, ***p < 0.001 relative to unstimulated WT or SPHK2-null BMDMs; ###p < 0.001 relative to WT BMDMs after an 8-h LPS challenge.
(B) Phosphorylation of TBK1, IRF3, and p65 subunit of NF-κB in indicated BMDMs as in Figure 3A. A representative immunoblot is shown from experiments that were independently repeated three times.
(C) BMDM lysates containing equal amount of protein were incubated with ATP and sphingosine at 37°C. After 1 h, the reaction was stopped and S1P was extracted. S1P levels were determined using an ELISA kit. SPHK activity was calculated as pmol/min/μg of protein. Experiments were performed twice independently using duplicate samples. Data are represented as mean ± SD from two independent experiments. *p < 0.05, ***p < 0.001 relative to unstimulated WT or SPHK2-null BMDMs; #p < 0.05 relative to LPS-exposed WT BMDMs.
(D) BMDMs were pretreated with vehicle or SPHK2 inhibitor ABC294640 (5 μM) for 1 h followed by stimulation with LPS for the indicated times. Phosphorylation of indicated proteins was determined. A representative immunoblot is shown from experiments that were independently repeated three times.
(E) U937 human monocytic cells were differentiated into macrophages by exposing them to phorbol myristate acetate (PMA) (100 ng/mL) for 48 h, deprived of serum for 1 h, and followed by the addition of 1 μg/mL LPS for determining phosphorylation of indicated proteins. A representative immunoblot is shown from experiments that were independently repeated three times.
(F) BMDMs stimulated with or without LPS were stained with anti-STING antibody followed by FITC-labeled secondary antibody and DAPI. A representative confocal image is shown from experiments that were repeated two times. Scale bars, 10 μm.
(G and H) BMDMs permeabilized with digitonin for 30 min were stimulated with 10 μM cGAMP for the indicated times and lysed. Phosphorylation was determined as in Figure 3A (G) while IFN-β levels were quantified as in Figure 1C (H). A representative immunoblot is shown from experiments that were independently repeated three times. Data in (H) are represented as mean ± SD from two independent experiments. **p < 0.01, ***p < 0.001 relative to unstimulated WT or SPHK2-null BMDMs; ##p < 0.01 relative to WT BMDMs after cGAMP challenge.
(I) BMDMs were transfected with WT SPHK2 or control vector. After 48 h, cells were stimulated with LPS to determine the phosphorylation of NF-κB-p65, TBK1, and IRF3. The western blot was then stripped and immunoblotted with GFP to assess SPHK2 expression. A representative immunoblot is shown from experiments that were independently repeated at least three times.
(J) BMDM-DNA fragments were purified and immunoprecipitated (IP) with immunoglobulin G (IgG) or antibody against NF-κB, and the resulting chromatin fragments were subjected to PCR amplification using primers spanning SPHK2 promoter. PCR gel in right shows NF-κB enrichment on the SPHK2 promoter using ChIP. qPCR was performed using primers specific for SPHK2. Data are represented as mean ± SD from three independent experiments. *p < 0.05 relative to unstimulated WT BMDMs. Data were analyzed using one-way ANOVA followed by Tukey’s multiple comparisons test.
Based on the above studies, we next determined the effect of LPS on SPHK activity in WT or SPHK2-null BMDMs. We found that under basal conditions SPHK activity in macrophages was 159 pmol/min/μg protein (Figure 4C). LPS challenge decreased the SPHK activity by ~40% in WT macrophages at 4 h. SPHK activity returned toward basal levels at 8 h (Figure 4C). The basal SPHK activity was not significantly different between WT and SPHK2-null BMDMs (Figure 4C); however, compared to WT BMDMs, LPS significantly reduced SPHK activity further in SPHK2-null BMDMs both at 4 and 8 h (Figure 4C). We confirmed that loss of SPHK2 in BMDMs had no effect on SPHK1 expression (Figures S4F and S4G). Next, we also inhibited SPHK2 activity in WT BMDMs or human macrophages (U937 cell line) using a SPHK2-specific inhibitor, ABC294640 (Xu et al., 2018; Xun et al., 2015) and observed that inhibition of SPHK2 in WT BMDMs or human macrophages similarly augmented TBK1, IRF3, and NF-κBp65 phosphorylation (Figures 4D, 4E, and S4H–S4M). To further rule out the contribution of SPHK1 activity in altering these responses, we also inhibited SPHK1 activity in SPHK2-null BMDMs using a specific inhibitor, PF-543 (Zhang et al., 2014). As expected, inhibition of SPHK1 activity in SPHK2-null BMDMs failed to alter LPS induction of STING signaling (Figures S4N–S4P).
STING localizes on the ER and redistributes to Golgi upon activation (Ishikawa and Barber, 2008). Thus, we assessed whether SPHK2 interfered with LPS-induced STING localization using confocal imaging. We found that in control cells, STING was perinuclear but redistributed to the cell periphery upon LPS stimulation at 4 h and returned to the basal state at 8 h (Figure 4F). Intriguingly, in SPHK2-null BMDMs, STING redistribution to the cell periphery persisted for up to 8 h after LPS stimulation (Figure 4F). We also immunoprecipitated STING in HEK cells treated without or with the SPHK2 inhibitor to assess whether inhibition of SPHK2 promoted STING interaction with TBK1, leading to its activation. As expected, inhibition of SPHK2 induced STING interaction with TBK1 (Figures S5A–S5C), corroborating the data in SPHK2-null BMDMs shown above.
In other studies, we stimulated WT and SPHK2-null BMDMs with a STING agonist, cGAMP. cGAMP also augmented the phosphorylation of TBK1, NF-κB-p65, and IRF3 (Figures 4G and S5D–S5F) as well as IFN-β protein and mRNA in SPHK2-null BMDMs as compared to WT BMDMs (Figures 4H and S5G). Taken together, these findings demonstrate the indispensable role of SPHK2 in suppressing STING signaling.
To assess the causal role of SPHK2 in suppressing STING activity, we rescued SPHK2 expression in SPHK2-null BMDMs. We found that restoring SPHK2 expression in SPHK2-null BMDMs restored TBK1 and NF-κB-p65 phosphorylation to the level seen in WT BMDMs (Figures 4I, S5H, and S5I). We also depleted STING in WT and Sphk2−/− BMDMs and determined the effect of STING depletion on LPS-induced IFN-β expression. Depletion of STING in SPHK2-null BMDMs rescued IFN-β production to the level seen in WT BMDMs (Figures S5J and S5K).
Given that LPS transiently decreased SPHK2 activity while augmenting STING signaling in the WT setting and the inhibition of SPHK2 in WT BMDMs induced long-lasting STING activity, we considered the possibility that SPHK2 is activated after injury downstream of TLR4 (Natarajan et al., 2013). The SPHK2 promoter contains three NF-κB binding sites (Dreos et al., 2015). Thus, we performed chromatin immunoprecipitation (ChIP) assays using an NF-κB (p65) antibody. Interestingly, we found that LPS induced a 2- to 3-fold increase in NF-κB binding to the SPHK2 promoter in WT BMDMs (Figure 4J). These findings indicate that post-injury NF-κB induces SPHK2 expression, which then dampens STING activity to prevent a long-lasting increase in inflammatory cytokine generation and thereby resolving inflammation.
We next sorted alveolar macrophages and CD11b+ macrophages from naive WT and SPHK2-null lungs to assess whether differences in SPHK2 activity in CD11b+ macrophages versus alveolar macrophages accounted for affecting the alveolar macrophage pro-resolving role. First, we assessed SPHK1 and SPHK2 protein expression. Interestingly, we found a 2-fold higher expression of SPHK2 in CD11b+ macrophages than alveolar macrophages (Figures 5A–5C). In contrast, SPHK1 was expressed to the same extent in both macrophage subsets (Figures 5A–5C). Next, we determined SPHK activity and S1P generation in macrophages isolated from BAL of WT or Sphk2−/− mice without or with LPS challenge. SPHK activity and S1P levels were significantly reduced after 4 h with LPS challenge in WT BAL macrophages but returned toward basal levels after 24 h of LPS challenge (Figures 5D and 5E). Compared to WT BAL macrophages, SPHK activity and S1P levels were significantly decreased in SPHK2-null BAL macrophages basally (Figures 5D and 5E). At all times after LPS challenge, SPHK activity and S1P levels remained persistently lower in SPHK2-null BAL macrophages than in WT BAL macrophages (Figures 5D and 5E).
Figure 5. Adoptive Transfer of SPHK2-Null CD11b Monocytes Fail to Resolve Inflammatory Vascular Injury in CD11b+ Macrophage-Depleted Mice.
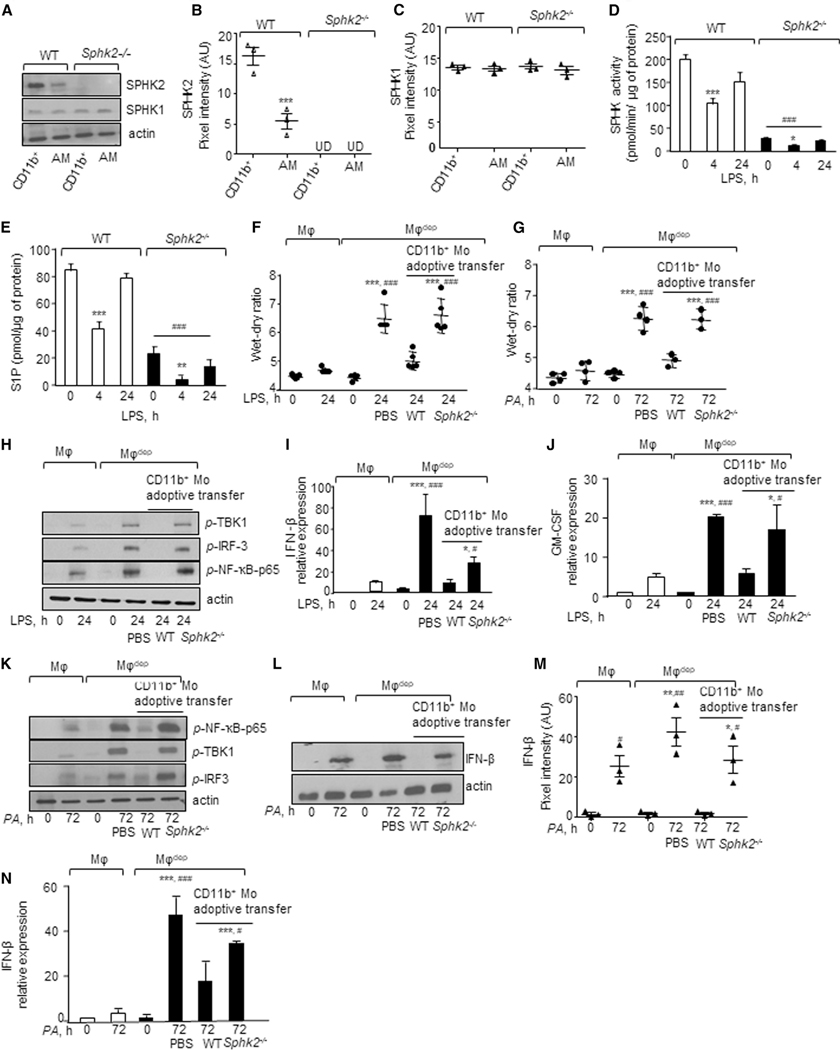
(A–C) At indicated times, lungs were digested with collagenase and stained with CD11c, CD11b, CD45, CD64, and Ly6G antibodies. Alveolar macrophages (CD11c+/CD45+/CD64+/CD11b/LY6G) and recruited macrophages (CD11b+/CD45+/CD64+/CD11c/LY6G) were sorted and lysed, and the expression of SPHK1 and SPHK2 was determined (A) using antibodies. Plots in B and C show individual pixel intensities of indicated proteins (expressed as a.u.) with mean ± SD (n = 4 mice/group). ***p < 0.05 relative to WT CD11b+ macrophages.
(D) BAL macrophages (MF) were isolated from indicated mice and times after LPS challenge and incubated with ATP and sphingosine at 37°C. SPHK activity was determined as in Figure 4C. Experiments were performed twice independently using duplicate samples.
(E) BAL macrophages were lysed and S1P levels were determined using an ELISA kit. Each experiment was performed twice independently using duplicate samples (n = 2).
Data in (D) and (E) are represented as mean ± SD. **p < 0.01, ***p < 0.001 relative to unstimulated WT or SPHK2-null BMDMs; ###p < 0.001 relative to LPS-exposedWT BMDMs.
(F and G) CD11b-DTR mice were first exposed to (F) LPS by inhalation or (G) i.t. PA by instillation followed by DT as described in Figure 3E. Thereafter, 2 × 106 CD11b+ monocytes isolated from bone marrow of WT or Sphk2−/− mice were injected i.v. into CD11b-DTR mice and the lung wet/dry weight ratio was determined. Mice receiving PBS served as controls. Shown are individual data with mean ± SD (n = 5 mice/group). ***p < 0.001 relative to unexposed macrophages or macrophagedep mice or 24-h LPS-/72-h PA exposed macrophagedep mice; ###p < 0.001 relative to macrophagedep mice receiving 24-h LPS/72-h PA and WT CD11b monocytes.
(H) Lung lysates without or with LPS challenge and CD11b monocyte adoptive transfer were processed for determining phosphorylation of indicated proteins as in Figure 3A. A representative immunoblot is shown from experiments that were independently repeated three times.
(I and J) Lungs from indicated mice were homogenized at the indicated times without or with LPS challenge or adoptive transfer, and expression of (I) IFN-β and (J) GM-CSF was determined using qPCR (n = 4). GAPDH was used as internal control.
(K) Lung lysates from indicated mice without or with PA challenge and monocyte adoptive transfer were processed for determining phosphorylation as described in Figure 3A. A representative immunoblot is shown from experiments that were independently repeated three times.
(L–N) Lungs lysates without or with PA challenge or adoptive transfer from indicated mice were processed for determining IFN-β expression at the level of protein (L) (n = 3 mice/group) or mRNA (N) (n = 4 mice/group). Plot in M shows individual pixel intensity for IFN-β protein expressed as arbitrary units (a.u.) along with mean and SD.
Data in (I), (J), (M), and (N) are expressed as mean ± SD from two or three independent experiments. *p < 0.05, **p < 0.01, ***p < 0.001 relative to unexposed macrophages or macrophagedep mice or 24-h LPS-/72-h PA exposed macrophagedep mice; #p < 0.05, ##p < 0.01, ###p < 0.001 relative to 24-h LPS-/72-h PA exposed macrophagedep mice receiving WT CD11b monocytes. All data were analyzed using one-way ANOVA followed by Tukey’s multiple comparisons test and an unpaired t test with Welch’s correction.
We then adoptively transferred WT or Sphk2-null BM monocytes into macrophagedep mice following injury to demonstrate that SPHK2 suppression of STING activity in CD11b+ macrophages is required to rescue the alveolar macrophage pro-resolving role. We found that compared to WT CD11b+ BM monocytes, adoptive transfer of Sphk2−/− BM monocytes failed to resolve LPS- or PA-induced lung inflammatory injury in macrophagedep mouse lungs (Figures 5F and 5G). Also, phosphorylation of NF-κB-p65, TBK1, and IRF3 and expression of IFN-b and GM-CSF remained persistently elevated in the lungs (Figures 5H–5N and S6A–S6F).
S1P Blocks cGAMP Activation of STING
S1P can pull down S1P-binding proteins (Strub et al., 2011; Urtz et al., 2015). Based on our findings that SPHK2 activity was required for suppressing LPS-induced STING signaling and that cGAMP increased STING signaling in SPHK2-null BMDMs, we assessed the possibility that SPHK2 generates S1P, which then binds STING to block its activity. Thus, we used lysates from WT BMDMs without or with LPS stimulation and incubated them with S1P biotin immobilized on streptavidin beads. Pulldown studies showed that STING co-immunoprecipitated with S1P on S1P-conjugated beads but not in control beads in LPS-treated BMDMs (Figures 6A and S6G). Therefore, the results confirm that S1P interacts with STING.
Figure 6. SPHK2-Generated S1P Inhibits STING Activity.

(A) WT BMDM lysates were immunoprecipitated with streptavidin or S1P-conjugated streptavidin beads. Immunocomplexes and total lysates were then immunoblotted with anti-STING antibody.
(B) Expression of IFN-β following stimulation with 10 μM cGAMP as described in Figure 4F. GAPDH expression was used as an internal control. n = 3 mice/group.
(C) Lysates from HEK cells transducing vector, HA-tagged full-length STING, or HA-tagged STING-CTD were immunoprecipitated using S1P streptavidin beads. Lysates and immunocomplexes were immunoblotted with HA antibody to determine the S1P interaction with STING.
(D) Binding poses of S1P to STING-CTD. (i), (ii), and (iii) indicate overplayed binding poses of S1P (PyMOL native atom colors) with cGAMP (blue) with STING-CTD (light pick) (PDB: 4KSY) in resting, closed (PDB: 4LOJ), and open conformation (PDB:4EF4), respectively.
In (A) and (C), a representative immunoblot is shown from experiments that were independently repeated three times. The data in (B) are represented as mean ± SD from two to three independent experiments. *p < 0.05, ***p < 0.001 relative to unstimulated WT or SPHK2-null BMDMs; ###p < 0.001 relative to S1P plus cGAMP-stimulated BMDMs. Data were analyzed using one-way ANOVA followed by Tukey’s multiple comparisons test.
Next, we assessed whether the interaction of S1P with STING inhibited cGAMP activation of inflammatory signaling. Thus, we pretreated WT BMDMs or SPHK2-null BMDMs with S1P followed by addition of cGAMP. Interestingly, we found that treatment of SPHK2-null BMDMs with S1P reduced IFN-β levels to the levels seen in WT BMDMs (Figure 6B), indicating that the interaction of S1P with STING inhibited cGAMP activation of inflammatory signaling.
cGAMP binds to the C terminus of STING to induce STING signaling (Lau et al., 2015). We used lysates from HEK cells transducing hemagglutinin (HA) full-length STING (1–379 aa) or the C terminus of STING (HA-STING-CTD [carboxy-terminal domain]: aa 149–379-STING) and assessed whether S1P interacted with CTD-STING. While CTD-STING was expressed to a lower extent than full-length (FL)-STING in HEK cells, it did interact with S1P (Figures 6C, S6H, and S6I).
We next sought to investigate the possible modes of S1P binding to STING-CTD by performing molecular docking (Trott and Olson, 2010). First, we docked S1P to monomeric STING-CTD (Figure 6D) without its natural ligand (2’,3’-cGAMP) to predict the binding site in an unbiased manner. The lowest energy binding pose showed that S1P can occupy a similar binding pocket to the one used by 2’,3’-cGAMP (Figure 6Di). Next, we investigated possible binding sites of S1P in the context of dimeric STING-CTD. This domain dimerizes under physiological conditions and adopts different conformations depending on the type of bound ligand (Gao et al., 2013; Ouyang et al., 2012). For example, STING-CTD acquires an open flexible conformation in the presence of c-di-GMP whereas it acquires a closed order conformation in the presence of 2’,3’-cGAMP (Gao et al., 2013). Docking of S1P to the closed conformation of the STING-CTD dimer showed that this ligand can occupy the binding pocket of cGAMP (Figure 6Dii). In contrast, S1P exhibited two possible modes of binding within the open conformation of dimeric STING-CTD. A competitive mode occupies the binding site of c-di-GMP (Figure 6Diii) and an allosteric mode occupies a binding site on the interface between the two monomeric units of STING-CTD (Figure S6J).
DISCUSSION
In this study, we identified a previously unknown, cell-intrinsic SPHK2→S1P pathway in CD11b+ macrophages that blocks STING signaling in alveolar macrophages, thereby resolving lung vascular inflammatory injury. Our findings indicate that, following injury, SPHK2 expressing CD11b+ macrophages recruited to the airspace interfere with cGAMP amplification of TBK1, IRF3, and NF-κB activities in alveolar macrophages, thus dampening generation of type 1 IFN and other cytokines and thereby alveolar macrophage expansion and hyperactivation to promote resolution of lung vascular inflammatory injury.
Tissue-resident alveolar macrophages (CD11c+/Siglec-F+) have been extensively studied both in relationship to induction and resolution of ALI (Westphalen et al., 2014). We showed that CD11b+ macrophages expanded during resolution of lung injury, consistent with previous studies (McCubbrey et al., 2016; Zaynagetdinov et al., 2013). Importantly, our data show that conditional depletion of CD11b+ macrophages after LPS and PA challenge impaired resolution of lung injury and rendered the mice highly susceptible to pneumonia-induced lethality. An important question arises whether the effects of CD11b+ (Mac-1 promoter) depletion can only be ascribed to depletion of macrophages or whether neutrophils are involved. However, our data show that DT depleted CD11b+ macrophages, but enhanced neutrophilic recruitment, indicating the specific role of CD11b+ macrophages in suppressing inflammatory lung injury.
A unique property of alveolar macrophages is their ability to rapidly trigger the inflammatory response, which allows for efficient pathogen killing (Bosmann et al., 2013; Byrne et al., 2016; Ward, 2003; Westphalen et al., 2014). Uncontrolled pro-inflammatory signaling in alveolar macrophages can compromise vascular barrier function, thereby provoking ALI (Duan et al., 2012; Westphalen et al., 2014). Evidence also suggests that monocytes can play both pro-inflammatory and anti-inflammatory roles (Duan et al., 2012). We showed that depletion of CD11b+ macrophages led to alveolar macrophage proliferation with enhanced GM-CSF, IFN-β, and IL-6 generation, leading to neutrophil accumulation and long-lasting vascular injury. Thus, our studies demonstrate that, during injury, CD11b+ macrophages educate alveolar macrophages to become anti-inflammatory, thereby preventing sustained inflammatory lung injury. In line with this idea, we showed that adoptively transferred CD11b+ monocytes became recruited macrophages in the airspace of macrophagedep mice that rescued alveolar macrophage anti-inflammatory function, thereby reversing lung injury. Studies suggest that monocytes can differentiate into alveolar macrophages (Byrne et al., 2016). Evidence also indicates that CD11b+/CD11c+ populations represent a prealveolar macrophage lineage (Gibbings et al., 2017; Schneider et al., 2014). However, we cannot rule out the possibility that recruited CD11b+ macrophages in the airspace acquire the alveolar macrophage signature, as we showed that DT also targeted CD11b+/CD11c+ populations in the alveolar space.
A key finding of this study was that depletion of CD11b+ macrophages markedly upregulated cGAMP levels during lung injury. We also showed that STING-null macrophages failed to show IRF3 phosphorylation and these lungs showed attenuated lung injury following LPS challenge. Upon sensing pathogens, cytokines released from alveolar macrophages can modulate the death of lung parenchymal cells depending on insult and timing (Hiruma et al., 2018; Makris et al., 2017). DNA liberated upon cell death can activate cGAS (Burdette et al., 2011; Ishikawa and Barber, 2008), which in turn converts GMP and AMP into the second messenger cGAMP to activate STING (Cai et al., 2014). We showed that LPS and PA transiently increased cGAMP generation in control lungs while increasing cGAMP levels in macrophagedep lungs, resulting in enhanced TBK1, IRF3, and NF-κB-p65 phosphorylation. These findings indicate that recruited CD11b+ macrophages in the airspace exert timely control over cGAMP-STING signaling in alveolar macrophages. Macrophages also exhibit extreme plasticity and can acquire either an inflammatory or anti-inflammatory phenotype depending on the inflammatory insult (Wynn et al., 2013). We showed that under normal conditions, alveolar macrophages transiently acquire an inflammatory phenotype due to reversible STING activity, enabling alveolar macrophages to trigger host defense and promote tissue repair after injury. However, in macrophagedep lungs alveolar macrophages remained inflammatory. Adoptive transfer of CD11b+ monocytes from WT or STING−/− mice into macrophagedep mice reversed alveolar macrophage inflammatory signaling. Our findings therefore demonstrate that airspace CD11b+ macrophages play an important role in suppressing alveolar macrophage-STING signaling, thereby leading to resolution of inflammatory lung injury.
While STING can be inactivated through a post-translational mechanism (Konno et al., 2013), our findings uncovered the fundamental role of CD11b+ macrophages in suppressing STING signaling in alveolar macrophages through SPHK2 activity. We showed that SPHK2 expression was higher in CD11b+ macrophages than alveolar macrophages. Furthermore, we showed that SPHK2 was required for maintaining SPHK activity and S1P levels in macrophages following LPS injury. Adoptive transfer of WT CD11b+ BM monocytes blocked alveolar macrophage-STING activity in macrophagedep mice whereas transfer of SPHK2-deficient CD11b+ BM monocytes failed to do so. Hence, alveolar macrophages remain inflammatory, leading to non-resolvable lung injury. Consistent with this, we showed that SPHK2, but not SPHK1, was required for LPS-stimulated S1P generation and that inhibition of SPHK2 kinase activity enhanced STING signaling in WT BMDMs as well as human macrophages. We further showed that S1P bound to STING and reduced cGAMP activation of IFN-β generation. Our modeling experiments revealed the ability of S1P to bind STING-CTD either allosterically or competitively with natural ligands. These findings imply that S1P inhibits STING-cGAMP signaling through mechanisms involving either stabilization of the STING conformation or competitive inhibition of cGAMP binding via occupation of its binding pocket in the monomeric or dimeric form of the protein. S1P can suppress inflammatory signaling by ligating the S1P receptors and altering downstream signaling (Natarajan et al., 2013; Szczepaniak et al., 2008; Tauseef et al., 2008). While we cannot rule out this possibility, our observations made in this study clearly indicate that S1P can compete with cGAMP or allosterically inhibit cGAMP binding to STING, thereby blocking downstream signaling.
A question arises why SPHK2 but not SPHK1 inhibited STING signaling even though both generate S1P. Evidence indicates that SPHK1-synthesized S1P functions by activating plasmalemmal S1P receptors (Natarajan et al., 2013; Spiegel and Milstien, 2011; Tauseef et al., 2008), while SPHK2-mediated S1P generation regulates cellular functions in a localized manner (Park et al., 2013; Zhang et al., 2013). We showed that SPHK2 suppresses STING redistribution, indicating compartmentalized action of SPHK2 in targeting ER-STING signaling. Consistent with this concept, inhibition of SPHK1 expression in SPHK2-null BMDMs had no effect on STING activity.
Both post-translational and transcriptional mechanisms can induce SPHK2 activity. Studies have shown that extracellular signal-regulated kinase (ERK)/mitogen-activated protein kinase (MAPK) phosphorylate SPHK2, increasing SPHK activity (Bryan et al., 2008; Hait et al., 2007) However, we showed that LPS transiently decreased SPHK activity in WT BMDMs or macrophages in association with increase in STING signaling. SPHK2 activity then returned to its basal level during the resolution phase of inflammatory injury. We also showed that NF-κB binds to the SPHK2 promoter and that the extent of binding increases upon LPS stimulation, thus corroborating previous findings that LPS can activate SPHK2 transcriptionally (Wadgaonkar et al., 2009). Therefore, we infer from these findings that upon activation by TLR4, NF-κB induces SPHK2 activity, which acts as a negative feedback mechanism to block STING activity by generating S1P and thereby permits rapid resolution of inflammatory injury by alveolar macrophages.
To date, there is limited understanding of the role of CD11b+ macrophages, which co-exist with alveolar macrophages in the lung, in regulating alveolar macrophage function and thereby ALI. Findings from this study have identified airspace-recruited CD11b+ macrophages as a key suppressor of STING activity in alveolar macrophages via SPHK2, which in turn leads to resolution of lung injury. Whether SPHK2+/CD11b+-recruited macrophages take on the function of alveolar macrophages or themselves differentiate into alveolar macrophages has yet to be elucidated. ALI caused by Gram-negative bacteria, such as PA, produces significant mortality, especially in elderly patients (Chakraborty et al., 2017; Rubenfeld et al., 2005). We propose that delivery of SPHK2+/CD11b+ macrophages into the airspace represents an attractive option as a form of cell therapy to accelerate alveolar macrophage anti-inflammatory function and thereby recovery from ALI.
STAR★METHODS
Detailed methods are provided in the online version of this paper and include the following:
LEAD CONTACT AND MATERIALS AVAILABILITY
Further information and requests for resources and reagents should be directed to and will be fulfilled by the corresponding author, Dr. Dolly Mehta (dmehta@uic.edu).
This study did not generate new unique reagents.
EXPERIMENTAL MODELS AND SUBJECT DETAILS
Animals
All animal experiments were approved by the Institutional Animal Care and Use Committee of the University of Illinois. CD11b-DTR, Sphk2−/−, Sting−/− and C57Blk/6J mice breeding pairs were initially obtained from Jackson Laboratory (Farmington, CT, USA) and bred at the University of Illinois at Chicago. Mouse colonies were maintained in a pathogen-free housing facility at the University. All experiments were performed in both male and female that were between 6–8 weeks old.
Cells
U-937, a human monocyte cell lines were used, before experiments, the cells were exposed to PMA (100ng/ml) for 48 h to differentiate them to macrophages.
Bacteria
GFP tagged Pseudomonas aeruginosa (GFP-PA01 strain) was used to induce lung injury in mice.
METHODS DETAILS
Macrophage Depletion
DT (25 μg/kg body weight) was injected i.p.in CD11b-DTR mice following LPS or PA exposure. Mice were sacrificed at the indicated time to assess their role in inflammatory vascular injury.
Induction and Assessment of Lung Vascular Permeability
Mice were exposed to a nebulized solution of lyophilized E. coli LPS (1 mg/ml) of Sigma Aldrich # 2880, dissolved in sterile saline for 1 h in a plexiglass box. Pseudomonas aeruginosa was injected intratracheally (i.t.). Briefly, mice were anesthetized with i.p. ketamine (100 mg/kg) and xylazine (12 mg/kg), following which 40 μL of PBS containing 1.0 × 104 CFU of PA was injected into trachea using a 27G needle. After a 15-minute recovery period, mice were returned to their respective cages. Extravasation of Evans blue and lung wet-dry weight ratio were determined to quantify lung injury as described previously (Rayees et al., 2019).
Assessment of Bronchoalveolar Lavage Fluid (BALF) Protein
BAL was isolated by rinsing the lungs three time with 1.0 mL of ice-cold phosphate-buffered saline (PBS). Cells were centrifuged at 1000 rpm for 5 min at 4°C. The supernatant of the first lavage was used for protein analysis.
FACS Analysis
Lung tissues were minced and enzymatically digested with 1 mg/mL collagenase A (Roche, New York, NY) for 50 min at 37°C. Digested tissue was forced through metal canula and passed through a 75-μm nylon filter to obtain single-cell suspensions. The red blood cells were lysed in lung, lavaged lung and BAL using lysis buffer and cell suspensions were washed with FACS buffer. Cells were re-suspended in FACS buffer and incubated with Fc blocking antibody for 30 minutes, to prevent binding of nonspecific FcγRIII/II. Cells were then labeled with indicated antibodies (as a cocktail: Anti-CD11b, anti-CD11c, anti-CD45 anti-Gr1 anti-Siglec F and anti-MHCII), for 30 minutes on ice. Samples were washed and analyzed using LSR-Fortessa (Beckman Coulter) and data were processed using Flow Jo software (TreeStar, Inc). All antibodies used for flow cytometry were anti-mouse antigens. BrdU 75mg/kg i.p. was administered four hours before the sacrifice of mice. Lung isolation and processed as mentioned above. After incubation with above antibodies cells were fixed with IC fixation buffer (Invitrogen). Fixed cells were permeabilized with permeabilization buffer and incubated for half an hour with anti-BrdU antibody in permeabilization buffer. Finally, cells were washed and processed using Cytoflex flow cytometer (Beckman Coulter).
Adoptive Transfer of Macrophages
Bone marrow cells were harvested from the femur and tibia of indicated mice. The CD11b+ bone marrow monocyte population was isolated using MACS column and magnetically labeled CD11b MicroBeads (Miltenyi Biotec), according to the manufacturer’s protocols. Cells were counted, and 2.0 × 106 cells were injected intra retro-orbitally into the CD11b-DTR mice. PBS was used as a vehicle control.
Isolation and Culture of Bone Marrow-Derived Macrophages
Mouse bone marrow derived macrophages (BMDMs) were isolated by flushing the femur and tibia with RPMI media, containing 1% antibiotic/antimycotic, 10% FBS and 25 ng/ml M-CSF. The cells were incubated at 37 °C in 5% CO2 incubator after isolation. On third day, the media of the culture plates were replaced with M-CSF free media. The cells were further incubated for 2 days at 37 °C in a 5% CO2 incubator. Cells were serum starved with 0.1% FBS containing RPMI media for one hour before experiment, followed by addition of LPS (1 μg/ml) with 1% FBS containing media (Rayees et al., 2019).
Myeloperoxidase (MPO) Assay
Lungs perfused with PBS were weighed and frozen at –80°C for a period of a week within which MPO activity was determined. MPO activity is expressed as units of MPO activity where one unit was defined as change in OD at 460 nm per milligram of protein per minute.
Quantitative Real-Time Reverse Transcriptase-PCR
Total RNA was isolated from the macrophages or lungs using TRIzol® reagent (Invitrogen Inc, Carlsbad, CA) according to the manufacturer’s instructions. RNA was quantified using Biodrop and reverse transcription reaction was carried out using specific primers described in supplement section as per published protocols (Rayees et al., 2019). The primer used in the study was as follows: mouse IFN-β forward 5’-ATAAGCAGCTCCAGCTCCAA-3’ reverse: 5’ CTGTCTGCTGGTGGAGTTCA-3’; mouse IL-1β forward: 5’-GGGCTGCTTCCAAACCTTTG-3’ reverse: 5’-TGATACTGCCTGCCTGAAGCTC-3’; mouse IL-6 Forward: 5’ AGTCCGGA GAGGAGACTTCA-3’ reverse: 5’-TTGCCATTGCACAACTCTTT-3’; mouse GAPDH forward: 5’-TACCCCCAATGTGTCCGTCGTG-3’ reverse: 5’-CCTTCAGTGGGCCCTCAGATGC-3’; Human IFN-β Forward: 5’-AAACTCATGAGCAGTCTGCA-3’ Reverse: 5’ AGGA GATCTTCAGTTTCGGAGG-3’; human IL1b Forward: 5’-AAATACCTGTGGCCTTGGGC-3’ reverse: 5’-TTGGGATCTACACTCTC CAGCT-3’; Human Il-6 forward: 5’-GTAGCCGCCCCACACAGA-3’ reverse: 5’-CATGTCTCCTTTCTCAGGGCTG-3’; Human GAPDH forward: 5’-CGAGATCCCTCCAAAATCAA-3’ reverse: 5’-TTCACACCCATGACGAACAT-3’
Immunofluorescence
BMDM seeded on coverslips were washed with PBS, fixed in 4% paraformaldehyde and stained with desired antibody. Imaging was performed using a LSM880 confocal microscope under an oil objective of 63x.
Immunoblotting
Cells or lungs were lysed in RIPA buffer [10mM-Tris Cl (pH 7.4), 150mM NaCl, 1mM EDTA, 1mM EGTA, 0.5% NP 40, 1.0% Triton X-100, 1mM phenyl methyl sulfonyl fluoride (PMSF), 1mM Na3VO4, 25μl/μg protease inhibitor]. In some experiments cell lysates were directly prepared using Laemmli buffer. The lysates were immunoblotted using indicated antibodies: anti-STING, anti-phosphoTBK1, anti-phospho-NFκB p65, anti-TBK1 (all 1:1000 dilutions). As a secondary antibody, anti-rabbit-IgG-HRP (1:2000 dilution) (Santa Cruz Biotechnology) was used. ECL signal was recorded on the ChemiDoc XRS Biorad Imager and data were analyzed with ImageJ. β-actin was used as an experimental control.
We used ImageJ software (ImageJ version 1.44, 32-bit) to quantify pixel intensity for protein band of interest in each experiment. Briefly, after converting each blot into digital image viz., TIF file (most preferred format), a rectangular area containing a band of maximum intensity of a Region of Interest (ROI) was selected. This ROI was then used to measure mean integrated density and area for background and protein band of interest within each experimental condition. Mean integrated density was divided by the area of the ROI to determine pixel intensity. The pixel intensity of background was then subtracted from the pixel intensity of protein band of interest. These data were imported to Graph Pad Prism version 7.0 (Graph Pad Software, La Jolla, CA) to generate individual plots showing distribution of pixel intensity from multiple replicates and to carry out statistical analysis. A one-way ANOVA analysis was carried out for each set of experiment (p < 0.05 is significant) followed by Tukey’s multiple comparison test.
cGAMP Extraction and Measurement Using LC-MS
cGAMP level was measured in the lung sample according to Gao et al. (Gao et al., 2015). Briefly, after harvesting the lung from experimental mice, the lungs were snap-frozen in liquid nitrogen and minced with scissor in cold 80% (v/v) methanol with 2% (v/v) acetic acid. Minced samples were stored at −80°C. For the analysis, samples were thawed on ice and 0.45 pmol cyclic-di-AMP was added as an internal control. Samples were homogenized with a tissue homogenizer (DREMEL Multi Pro) for one minute with intermittent pulse of 30 s. Homogenates were centrifuged at 10000 × g for 10 min after which samples were further extracted twice using 20% (v/v) methanol and 2% (vol/vol) acetic acid. cGAMP was eluted by solid phase extraction (SPE) using HyperSep Aminopropyl SPE 3.0 mL (500mg) Columns (Thermo Scientific). Before drawing the extract, columns were first activated by methanol (100%) and washed twice with 2% (v/v) acetic acid. Finally, the c-GAMP was eluted with 4% (v/v) ammonium hydroxide in 80% methanol. The eluents were spin-vacuumed to dryness by using nitrogen evaporator (N-EVAP™ 111) and reconstituted in HPLC-grade water then transferred to autosampler vials for MS analyses to Metabolomics & Proteomics Facility in Research Resources Center of University of Illinois at Chicago.
Isolation of Bronchoalveolar Macrophages
BAL obtained from 3 mice was pooled and centrifuged. Cell pellet was suspended in RPMI media containing 10% serum, poured into 60 mm plastic culture dishes. After 2h, non-adherent cells were removed by washing with PBS. Adherent cells were detached and cytospin was performed to confirm the presence of macrophages.
S1P Analysis in BAL Macrophages
BAL macrophages from unexposed or LPS exposed were lysed. S1P was determined using S1P ELISA kit (MyBiosource.com, MBS2700637) according to the manufacturer instruction. Briefly, macrophages were rinsed gently with cold PBS ×2 and detached using 1% trypsin. Cells were then centrifuged at 1,000×g for 5 minutes, washed three time with PBS and re-suspended in lysis buffer (150 mM NaCl, 10% v/v glycerol, 50 mM HEPES pH 7.4, 0.05% triton-x 100, 2 mM sodium vanadate (Na3VO4), 10 mM NaF and 1 mM EDTA along with 1 μM S1P lyase inhibitor and 10 ml/ml protease inhibitor cocktail). S1P concentration was determined using known standards provided by the manufacturer.
SPHK Activity
Sphingosine kinase activity was assessed by measuring the conversion of sphingosine (Amersham Biosciences, Piscataway, NJ, USA) to S1P using ATP and cell lysates. Macrophages (BMDM or BAL) were washed ×3 using cold PBS and lysed using fifty microliter of lysis buffer (150 mM NaCl, 10% v/v glycerol, 50 mM HEPES pH 7.4, 0.05% triton-x 100, 2 mM sodium vanadate (Na3VO4), 10 mM NaF and 1 mM EDTA along with 1 μM S1P lyase inhibitor and 10 μl/ml protease inhibitor cocktail). After two freeze thaw cycles, cells were sonicated. Cell lysates containing 10 μg of protein was incubated with 5 μM of sphingosine and 10 μM of cold ATP at 37°C. After 1h, S1P was extracted using acidic chloroform and methanol as described by Bligh and Dyer (1959). S1P levels were determined using ELISA kit as a readout of SPHK activity.
Chromatin Immunoprecipitation (ChIP) Assay
Protein-DNA complex (100–120 mg) was immunoprecipitated with the antibody against NF-κB. DNA fragments were collected by phenol–chloroform–isoamyl alcohol extraction, followed by ethanol precipitation, and then resuspended in 14 μL nuclease water for PCR. Promoter region of SPHK2 targeted a 152 bp fragment was quantified by Syber green-based real time quantitative PCR (q-PCR) using ViiA7 (Applied Biosystem, Foster City, CA). Normal rabbit IgG was used as negative antibody control and DNA from the input (20–40 μg protein-DNA complexes) was used as an internal control.
Molecular Docking
The 2D chemical structure of sphingosine 1- phosphate (S1P) was drawn using ChemBioDraw ultra 13.0. The 3D structure was minimized using the MM2 force field and converted to pdb format using Chem3D Pro 13.0 (PerkinElmer Informatics). Autodock 4.4.6/AutoDock Tools 1.5.6 was utilized to prepare the input files of STING-CTD (PDB ID#:4KSY), 4LOJ (Gao et al., 2013), and 4EF4 (Ouyang et al., 2012) and S1P for docking by converting the pdb format into pdbqt format. This process included adding polar hydrogen bonds to the protein, choosing torsions and bond type for S1P, and determining the grid map for the protein using 1.0 Å spacing. Then AutoDock Vina (Trott and Olson, 2010) was used to dock S1P to STING-CTD and generating a standard nine poses of the ligand that correspond to a different binding sites in the protein. The output files were visualized using Pymol (Chaudhari and Li, 2015). The log files containing RMSD and binding energy values were used to guide in choosing a representative binding pose. The preferred binding pose was overlaid with the original STING-CTD X-ray structures containing the natural ligand to prepare the figures using Pymol (Chaudhari and Li, 2015).
QUANTIFICATION AND STATISTICAL ANALYSIS
Results are expressed as means ± SD from two to three independent experiments. Statistical significance was assessed by one-way ANOVA followed by Tukey’s multiple comparisons test and unpaired t test with Welch’s correction using Graph Pad Prism version 7.0 (Graph Pad Software, La Jolla, CA).
DATA AND CODE AVAILABILITY
This study did not generate any datasets or code.
Supplementary Material
KEY RESOURCE TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Anti-Mouse CD45.1 (clone 30F11) | Thermo Fisher Scientific | Cat # 11–0451; RRID: AB_465050 |
| Anti-Mouse CD45.1 (clone 30F11) | Thermo Fisher Scientific | Cat# 25–0451-82; RRID: AB_469625 |
| Anti-Mouse CD64 (Clone x54–5/7.1) | BioLegend | Cat # 139306; RRID: AB_11219391 |
| Anti-Mouse CD11b (Clone M1/70) | Thermo Fisher Scientific | Cat# 17–0112-82; RRID: AB_469343 |
| Anti-Mouse CD11c (Clone N418) | Thermo Fisher Scientific | Cat#11–0114-82; RRID: AB_464940 |
| Anti-Mouse Ly6c (Clone HK1.4) | BioLegend | Cat# 128012; RRID: AB_1659241 |
| Anti-Mouse Ly6g (Clone 1AB-LY6G) | Thermo Fisher Scientific | Cat# 48–5931-82; RRID: AB_1548788 |
| Anti-Mouse SiglecF (Clone E50–2440) | BD Bioscience | Cat# 552126; RRID: AB_394341 |
| Anti-Mouse BrdU (Clone BU20-a) | BioLegend | Cat# 339808; RRID: AB_10895898 |
| Rabbit Polyclonal anti-TMEM 173/STING | Proteintech | Cat# 19851–1-AP; RRID: AB_10665370 |
| C-terminal Polyclonal anti-SPHK-2 | Millipore | Cat# ABS 527 |
| Rabbit Monoclonal anti-TBK (D1B4) | Cell Signaling | Cat# 3504S; RRID: AB_2255663 |
| Rabbit Monoclonal anti-p-NFkB p65 | Cell Signaling | Cat# 3033S; RRID: AB_331284 |
| Rabbit Monoclonal anti-p-IRF3 (ser 396) | Cell Signaling | Cat# 29047S; RRID: AB_2773013 |
| Mouse Monoclonal anti-NFkB-p65 (F-6) | Santa Cruz Biotechnology | Cat# SC-8008; RRID: AB_628017 |
| Rabbit Monoclonal anti-GFP (D5.1) | Cell Signaling | Cat# 2956S; RRID: AB_1196615 |
| Rabbit Monoclonal anti-SPHK1 (M-209) | Santa Cruz Biotechnology | Cat# SC-48825; RRID: AB_2195835 |
| Mouse Monoclonal anti-actin (BA3R) | Thermo Fisher Scientific | Cat# MA5–15739; RRID: AB_10979409 |
| Mouse Monoclonal anti-IFN-β | BioLegend | Cat# 508102; RRID: AB_315505 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Trizol | Life Techlologies | Cat# 15596–026 |
| Digitonin | Sigma Aldrich | Cat# D141–100MG |
| 2′ −3′cGAMP | Invivogen | Cat# t1r1-nacga23 |
| ATP, disodium salt hydrate | Sigma Aldrich | Cat# FLAAS-IVL |
| GTP, disodium salt hydrate | Sigma Aldrich | Cat# G7127–10MG |
| M-CSF | Sigma Aldrich | Cat# M6518 |
| Lipopolysaccharide | Sigma Aldrich | Cat#2880 |
| BrdU | Sigma Aldrich | Cat# 19160 |
| Critical Commercial Assays | ||
| CD11b Cell Isolation Kit, Mouse | Miltenyi Biotec | Cat# 130–049-601 |
| S1P ELISA Kit, Mouse | MyBiosource.com | Cat# MBS2700637 |
| IL-6 ELISA Kit, Mouse | Signosis | Cat# EA-2206 |
| IL-1β ELISA Kit, Mouse | Signosis | Cat# EA-2508 |
| IFN-β ELISA Kit, Mouse | Signosis | Cat# CUS-EA-2001 |
| GM-CSF ELISA Kit Mouse | Bio-Rad | Cat# Custom made |
| Experimental Models: Cell Lines | ||
| U937 | Gifted by Dr Kostandin Pajcini UIC, IL | N/A |
| Pseudomonas aeruginosa (GFP-PA01) | Gifted by Dr Shekhar Reddy UIC, IL | N/A |
| Experimental Models: Organisms/Strains | ||
| Mouse: CD11b-DTR | Jackson Laboratory | # 008547 |
| Mouse: SPHK1 | Jackson Laboratory | # 019095 |
| Mouse: SPHK2 | Jackson Laboratory | # 019140 |
| Mouse: STING | Jackson Laboratory | # 017537 |
| Mouse: C57BL/6J | Jackson Laboratory | # 000664 |
| Oligonucleotides | ||
| Primers, see STAR methods Quantitative Real-time Reverse Transcriptase-PCR section | This paper | N/A |
| Recombinant DNA | ||
| STING, SPHK2-GFP and GFP cDNA | Prof. Stuart Pitson, Center of Cancer Biology, University of South Australia |
N/A |
| STING-flag | Prof. Susan Ross, Dept of microbiology and immunology, UIC, IL-USA | N/A |
| Software and Algorithms | ||
| GraphPad Prism Version 7.0 | GraphPad Software Inc | https://www.graphpad.com/scientificsoftware/prism/ |
| Zen Lite | Zeiss Inc | https://www.zeiss.com/microscopy/us/products/microscope-software/zen-lite.html |
| ImageJ | ImageJ | https://imagej.nih.gov/ij/download.html |
| FlowJo V10 | Flowjo | https://www.flowjo.com/ |
Highlights.
CD11b+ macrophage depletion following infection blocks resolution of inflammatory injury
Resulting hyperactivation of STING signaling in alveolar macrophages causes inflammatory injury
S1P generated by SPHK2 in CD11b+ macrophages suppresses alveolar macrophage-STING signaling
Such suppression of STING signaling by CD11b+ macrophages resolves vascular injury
ACKNOWLEDGMENTS
We thank Dr. Stuart Pitson (University of South Australia) for providing SPHK2 cDNA and Drs. Daniel B. Stetson (University of Washington) and Susan Ross (University of IL) for providing STING cDNA, and Dr. Kostandin Pajcini (University of Illinois) and Dr. S Banerjee (University of Illinois) for his advice and help on FACS analysis. This work was supported by National Institutes of Health grants HL77806, HL060678, HL84153, and HL137179.
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental Information can be found online at https://doi.org/10.1016/j.celrep.2020.02.112.
DECLARATION OF INTERESTS
The authors declare no competing interests.
REFERENCES
- Arnold L, Henry A, Poron F, Baba-Amer Y, van Rooijen N, Plonquet A, Gherardi RK, and Chazaud B (2007). Inflammatory monocytes recruited after skeletal muscle injury switch into antiinflammatory macrophages to support myogenesis. J. Exp. Med. 204, 1057–1069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barber GN (2015). STING: infection, inflammation and cancer. Nat. Rev. Immunol. 15, 760–770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Becher B, Tugues S, and Greter M (2016). GM-CSF: from growth factor to central mediator of tissue inflammation. Immunity 45, 963–973. [DOI] [PubMed] [Google Scholar]
- Bligh EG, and Dyer WJ (1959). A rapid method of total lipid extraction and purification. Can. J. Biochem. Physiol. 37, 911–917. [DOI] [PubMed] [Google Scholar]
- Bosmann M, Grailer JJ, Russkamp NF, Ruemmler R, Zetoune FS, Sarma JV, and Ward PA (2013). CD11c+ alveolar macrophages are a source of IL-23 during lipopolysaccharide-induced acute lung injury. Shock 39, 447–452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Broquet A, Jacqueline C, Davieau M, Besbes A, Roquilly A, Martin J, Caillon J, Dumoutier L, Renauld J-C, Heslan M, et al. (2017). Interleukin-22 level is negatively correlated with neutrophil recruitment in the lungs in a Pseudomonas aeruginosa pneumonia model. Sci. Rep. 7, 11010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bryan L, Kordula T, Spiegel S, and Milstien S (2008). Regulation and functions of sphingosine kinases in the brain. Biochim. Biophys. Acta 1781, 459–466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burdette DL, Monroe KM, Sotelo-Troha K, Iwig JS, Eckert B, Hyodo M, Hayakawa Y, and Vance RE (2011). STING is a direct innate immune sensor of cyclic di-GMP. Nature 478, 515–518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Byrne AJ, Maher TM, and Lloyd CM (2016). Pulmonary macrophages: a new therapeutic pathway in fibrosing lung disease? Trends Mol. Med. 22, 303–316. [DOI] [PubMed] [Google Scholar]
- Cai X, Chiu Y-H, and Chen ZJ (2014). The cGAS-cGAMP-STING pathway of cytosolic DNA sensing and signaling. Mol. Cell 54, 289–296. [DOI] [PubMed] [Google Scholar]
- Chakraborty K, Raundhal M, Chen BB, Morse C, Tyurina YY, Khare A, Oriss TB, Huff R, Lee JS, St Croix CM, et al. (2017). The mito-DAMP cardiolipin blocks IL-10 production causing persistent inflammation during bacterial pneumonia. Nat. Commun. 8, 13944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chaudhari R, and Li Z (2015). PyMine: a PyMOL plugin to integrate and visualize data for drug discovery. BMC Res. Notes 8, 517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Q, Sun L, and Chen ZJ (2016). Regulation and function of the cGAS-STING pathway of cytosolic DNA sensing. Nat. Immunol. 17, 1142–1149. [DOI] [PubMed] [Google Scholar]
- Delano MJ, and Ward PA (2016). Sepsis-induced immune dysfunction: can immune therapies reduce mortality? J. Clin. Invest. 126, 23–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dhaliwal K, Scholefield E, Ferenbach D, Gibbons M, Duffin R, Dorward DA, Morris AC, Humphries D, MacKinnon A, Wilkinson TS, et al. (2012). Monocytes control second-phase neutrophil emigration in established lipopolysaccharide-induced murine lung injury. Am. J. Respir. Crit. Care Med. 186, 514–524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dreos R, Ambrosini G, Périer RC, and Bucher P (2015). The Eukaryotic Promoter Database: expansion of EPDnew and new promoter analysis tools. Nucleic Acids Res. 43, D92–D96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duan M, Li WC, Vlahos R, Maxwell MJ, Anderson GP, and Hibbs ML (2012). Distinct macrophage subpopulations characterize acute infection and chronic inflammatory lung disease. J. Immunol. 189, 946–955. [DOI] [PubMed] [Google Scholar]
- Gao P, Ascano M, Zillinger T, Wang W, Dai P, Serganov AA, Gaffney BL, Shuman S, Jones RA, Deng L, et al. (2013). Structure-function analysis of STING activation by c[G(2’,5’)pA(3’,5’)p] and targeting by antiviral DMXAA. Cell 154, 748–762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao D, Li T, Li X-D, Chen X, Li Q-Z, Wight-Carter M, and Chen ZJ (2015). Activation of cyclic GMP-AMP synthase by self-DNA causes autoimmune diseases. Proc. Natl. Acad. Sci. USA 112, E5699–E5705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gautier EL, Shay T, Miller J, Greter M, Jakubzick C, Ivanov S, Helft J, Chow A, Elpek KG, Gordonov S, et al. ; Immunological Genome Consortium (2012). Gene-expression profiles and transcriptional regulatory pathways that underlie the identity and diversity of mouse tissue macrophages. Nat. Immunol. 13, 1118–1128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gibbings SL, Thomas SM, Atif SM, McCubbrey AL, Desch AN, Danhorn T, Leach SM, Bratton DL, Henson PM, Janssen WJ, and Jakubzick CV (2017). Three unique interstitial macrophages in the murine lung at steady state. Am. J. Respir. Cell Mol. Biol. 57, 66–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hait NC, Bellamy A, Milstien S, Kordula T, and Spiegel S (2007). Sphingosine kinase type 2 activation by ERK-mediated phosphorylation. J. Biol.Chem. 282, 12058–12065. [DOI] [PubMed] [Google Scholar]
- Hiruma T, Tsuyuzaki H, Uchida K, Trapnell BC, Yamamura Y, Kusakabe Y, Totsu T, Suzuki T, Morita S, Doi K, et al. (2018). IFN-β improves sepsis-related alveolar macrophage dysfunction and postseptic acute respiratory distress syndrome-related mortality. Am. J. Respir. Cell Mol. Biol. 59, 45–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishikawa H, and Barber GN (2008). STING is an endoplasmic reticulum adaptor that facilitates innate immune signalling. Nature 455, 674–678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ivashkiv LB, and Donlin LT (2014). Regulation of type I interferon responses. Nat. Rev. Immunol. 14, 36–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnston LK, Rims CR, Gill SE, McGuire JK, and Manicone AM (2012). Pulmonary macrophage subpopulations in the induction and resolution of acute lung injury. Am. J. Respir. Cell Mol. Biol. 47, 417–426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Konno H, Konno K, and Barber GN (2013). Cyclic dinucleotides trigger ULK1 (ATG1) phosphorylation of STING to prevent sustained innate immune signaling. Cell 155, 688–698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwon H-J, Choi G-E, Ryu S, Kwon SJ, Kim SC, Booth C, Nichols KE, and Kim HS (2016). Stepwise phosphorylation of p65 promotes NF-κB activation and NK cell responses during target cell recognition. Nat. Commun. 7, 11686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lau L, Gray EE, Brunette RL, and Stetson DB (2015). DNA tumor virus oncogenes antagonize the cGAS-STING DNA-sensing pathway. Science 350, 568–571. [DOI] [PubMed] [Google Scholar]
- Li X, Shu C, Yi G, Chaton CT, Shelton CL, Diao J, Zuo X, Kao CC, Herr AB, and Li P (2013). Cyclic GMP-AMP synthase is activated by double-stranded DNA-induced oligomerization. Immunity 39, 1019–1031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Makris S, Paulsen M, and Johansson C (2017). Type I interferons as regulators of lung inflammation. Front. Immunol. 8, 259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matthay MA, Ware LB, and Zimmerman GA (2012). The acute respiratory distress syndrome. J. Clin. Invest. 122, 2731–2740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCubbrey AL, Barthel L, Mould KJ, Mohning MP, Redente EF, and Janssen WJ (2016). Selective and inducible targeting of CD11b+ mononuclear phagocytes in the murine lung with hCD68-rtTA transgenic systems. Am. J. Physiol. Lung Cell. Mol. Physiol. 311, L87–L100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Misharin AV, Morales-Nebreda L, Mutlu GM, Budinger GR, and Perlman H (2013). Flow cytometric analysis of macrophages and dendritic cell subsets in the mouse lung. Am. J. Respir. Cell Mol. Biol. 49, 503–510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mogensen TH (2009). Pathogen recognition and inflammatory signaling in innate immune defenses. Clin. Microbiol. Rev. 22, 240–273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murray PJ, and Wynn TA (2011). Protective and pathogenic functions of macrophage subsets. Nat. Rev. Immunol. 11, 723–737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Natarajan V, Dudek SM, Jacobson JR, Moreno-Vinasco L, Huang LS, Abassi T, Mathew B, Zhao Y, Wang L, Bittman R, et al. (2013). Sphingosine-1-phosphate, FTY720, and sphingosine-1-phosphate receptors in the pathobiology of acute lung injury. Am. J. Respir. Cell Mol. Biol. 49, 6–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newton K, and Dixit VM (2012). Signaling in innate immunity and inflammation. Cold Spring Harb. Perspect. Biol. 4, a006049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ouyang S, Song X, Wang Y, Ru H, Shaw N, Jiang Y, Niu F, Zhu Y, Qiu W, Parvatiyar K, et al. (2012). Structural analysis of the STING adaptor protein reveals a hydrophobic dimer interface and mode of cyclic di-GMP binding. Immunity 36, 1073–1086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park K, Elias PM, Shin K-O, Lee Y-M, Hupe M, Borkowski AW, Gallo RL, Saba J, Holleran WM, and Uchida Y (2013). A novel role of a lipid species, sphingosine-1-phosphate, in epithelial innate immunity. Mol. Cell. Biol. 33, 752–762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pyne NJ, Adams DR, and Pyne S (2017). Sphingosine kinase 2 in autoimmune/inflammatory disease and the development of sphingosine kinase 2 inhibitors. Trends Pharmacol. Sci. 38, 581–591. [DOI] [PubMed] [Google Scholar]
- Randolph AG (2009). Management of acute lung injury and acute respiratory distress syndrome in children. Crit. Care Med. 37, 2448–2454. [DOI] [PubMed] [Google Scholar]
- Rayees S, Joshi JC, Tauseef M, Anwar M, Baweja S, Rochford I, Joshi B, Hollenberg MD, Reddy SP, and Mehta D (2019). PAR2-mediated cAMP generation suppresses TRPV4-dependent Ca2+ signaling in alveolar macrophages to resolve TLR4-induced inflammation. Cell Rep. 27, 793–805.e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rubenfeld GD, Caldwell E, Peabody E, Weaver J, Martin DP, Neff M, Stern EJ, and Hudson LD (2005). Incidence and outcomes of acute lung injury. N. Engl. J. Med. 353, 1685–1693. [DOI] [PubMed] [Google Scholar]
- Schneider C, Nobs SP, Kurrer M, Rehrauer H, Thiele C, and Kopf M (2014). Induction of the nuclear receptor PPAR-γ by the cytokine GM-CSF is critical for the differentiation of fetal monocytes into alveolar macrophages. Nat. Immunol. 15, 1026–1037. [DOI] [PubMed] [Google Scholar]
- Schyns J, Bureau F, and Marichal T (2018). Lung interstitial macrophages: past, present, and future. J. Immunol. Res. 2018, 5160794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spiegel S, and Milstien S (2011). The outs and the ins of sphingosine-1-phosphate in immunity. Nat. Rev. Immunol. 11, 403–415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stoneman V, Braganza D, Figg N, Mercer J, Lang R, Goddard M, and Bennett M (2007). Monocyte/macrophage suppression in CD11b diphtheria toxin receptor transgenic mice differentially affects atherogenesis and established plaques. Circ. Res. 100, 884–893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strub GM, Paillard M, Liang J, Gomez L, Allegood JC, Hait NC, Maceyka M, Price MM, Chen Q, Simpson DC, et al. (2011). Sphingosine-1-phosphate produced by sphingosine kinase 2 in mitochondria interacts with prohibitin 2 to regulate complex IV assembly and respiration. FASEB J. 25, 600–612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szczepaniak WS, Zhang Y, Hagerty S, Crow MT, Kesari P, Garcia JG, Choi AM, Simon BA, and McVerry BJ (2008). Sphingosine 1-phosphate rescues canine LPS-induced acute lung injury and alters systemic inflammatory cytokine production in vivo. Transl. Res. 152, 213–224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tauseef M, Kini V, Knezevic N, Brannan M, Ramchandaran R, Fyrst H, Saba J, Vogel SM, Malik AB, and Mehta D (2008). Activation of sphingosine kinase-1 reverses the increase in lung vascular permeability through sphingosine-1-phosphate receptor signaling in endothelial cells. Circ. Res. 103, 1164–1172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trott O, and Olson AJ (2010). AutoDock Vina: improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 31, 455–461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Urtz N, Gaertner F, von Bruehl M-L, Chandraratne S, Rahimi F, Zhang L, Orban M, Barocke V, Beil J, Schubert I, et al. (2015). Sphingosine 1-phosphate produced by sphingosine kinase 2 intrinsically controls platelet aggregation in vitro and in vivo. Circ. Res. 117, 376–387. [DOI] [PubMed] [Google Scholar]
- Wadgaonkar R, Patel V, Grinkina N, Romano C, Liu J, Zhao Y, Sammani S, Garcia JG, and Natarajan V (2009). Differential regulation of sphingosine kinases 1 and 2 in lung injury. Am. J. Physiol. Lung Cell. Mol. Physiol. 296, L603–L613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ward P (2003). Acute lung injury: how the lung inflammatory response works. Eur. Respir. J. Suppl. 44, 22s–23s. [DOI] [PubMed] [Google Scholar]
- Westphalen K, Gusarova GA, Islam MN, Subramanian M, Cohen TS, Prince AS, and Bhattacharya J (2014). Sessile alveolar macrophages communicate with alveolar epithelium to modulate immunity. Nature 506, 503–506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wynn TA, Chawla A, and Pollard JW (2013). Macrophage biology in development, homeostasis and disease. Nature 496, 445–455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiong Y, Lee HJ, Mariko B, Lu Y-C, Dannenberg AJ, Haka AS, Maxfield FR, Camerer E, Proia RL, and Hla T (2013). Sphingosine kinases are not required for inflammatory responses in macrophages. J. Biol. Chem. 288, 32563–32573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu L, Jin L, Yang B, Wang L, Xia Z, Zhang Q, and Xu J (2018). The sphingosine kinase 2 inhibitor ABC294640 inhibits cervical carcinoma cell growth. Oncotarget 9, 2384–2394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xun C, Chen M-B, Qi L, Tie-Ning Z, Peng X, Ning L, Zhi-Xiao C, and Li-Wei W (2015). Targeting sphingosine kinase 2 (SphK2) by ABC294640 in-hibits colorectal cancer cell growth in vitro and in vivo. J. Exp. Clin. Cancer Res. 34, 94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zaynagetdinov R, Sherrill TP, Kendall PL, Segal BH, Weller KP, Tighe RM, and Blackwell TS (2013). Identification of myeloid cell subsets in murine lungs using flow cytometry. Am. J. Respir. Cell Mol. Biol. 49, 180–189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang L, Urtz N, Gaertner F, Legate KR, Petzold T, Lorenz M, Mazharian A, Watson SP, and Massberg S (2013). Sphingosine kinase 2 (Sphk2) regulates platelet biogenesis by providing intracellular sphingosine 1-phosphate (S1P). Blood 122, 791–802. [DOI] [PubMed] [Google Scholar]
- Zhang Y, Berka V, Song A, Sun K, Wang W, Zhang W, Ning C, Li C, Zhang Q, Bogdanov M, et al. (2014). Elevated sphingosine-1-phosphate promotes sickling and sickle cell disease progression. J. Clin. Invest. 124, 2750–2761. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
This study did not generate any datasets or code.