

Abstract
Sepsis is a heterogeneous clinical syndrome that is commonly complicated by acute kidney injury (sepsis-AKI). Currently, no approved pharmacological therapies exist to either prevent sepsis-AKI or to treat sepsis-AKI once it occurs. A growing body of evidence supports a connection between red blood cell (RBC) biology and sepsis-AKI. Elevated levels of circulating cell-free hemoglobin (CFH) released from RBCs during hemolysis are common during sepsis and can contribute to sepsis-AKI through several mechanisms including tubular obstruction, nitric oxide depletion, oxidative injury, and pro-inflammatory signaling. A number of potential pharmacological therapies targeting CFH in sepsis have been identified including haptoglobin, hemopexin, and acetaminophen, and early phase clinical trials suggest that acetaminophen may have beneficial effects on lipid peroxidation and kidney function in patients with sepsis. Bedside measurement of CFH levels may facilitate predictive enrichment for future clinical trials of CFH-targeted therapeutics. However, rapid and reliable bedside tests for plasma CFH will be required for such trials to move forward.
Keywords: Sepsis, acute kidney injury, cell-free hemoglobin, biomarkers, hemolysis, acetaminophen
1. Introduction
Sepsis, the clinical syndrome of organ dysfunction and immune dysregulation in response to infection,1 remains a leading cause of both hospitalization and inpatient mortality in the United States,2 and is associated with high hospital costs, morbidity, and mortality.3 Acute kidney injury (AKI) is a common complication of sepsis and septic shock, with a reported incidence of 10 to 65% among patients admitted to an intensive care unit (ICU) with sepsis.4,5 The development of AKI during sepsis (sepsis-AKI) carries both prognostic and therapeutic implications. Sepsis-AKI is associated with increased hospital costs, prolonged hospitalization, and increased mortality both in the ICU and at 90 days.4,5 Furthermore, as many antibiotics and other common pharmaceuticals are either cleared from the body via the kidneys or are directly nephrotoxic, AKI often complicates the treatment of sepsis by forcing clinicians to either modify or discontinue important therapeutic drugs.
The pathophysiology of sepsis-AKI is incompletely understood.6 Hemodynamic effects cannot fully explain sepsis-AKI as it can occur in the absence of overt hypotension,4,7 and only a minority of sepsis-AKI patients have acute tubular necrosis (ATN) on histopathology.8 Currently, no approved pharmacological therapies exist to either prevent sepsis-associated AKI or to treat AKI once it occurs.9 Over the last decade, multiple lines of evidence have converged to support the hypothesis that plasma cell free hemoglobin (CFH) may contribute to the pathogenesis of organ dysfunction including AKI during sepsis.10–14 In this review, we discuss the biology of CFH as it pertains to sepsis and other critical illness states, the renal effects of CFH both in animal models and in humans, and potential therapies to mitigate the nephrotoxic effects of CFH during sepsis.
2. Red blood cell biology during sepsis
2.1. Overview
In mammals, nearly all hemoglobin (Hb) is contained within circulating red blood cells (RBCs). Hb that escapes the RBC cytoplasm because of hemolysis, termed cell-free hemoglobin (CFH), is tightly regulated. Mammals have several mechanisms to rapidly scavenge and remove CFH and its byproducts from circulation, including the plasma proteins haptoglobin15 and hemopexin.16 Circulating CFH is not detectable in any significant quantities under normal physiologic conditions. However, many acute and chronic medical states can increase circulating CFH levels. These include genetic conditions such as sickle cell disease and the thalassemias, parasitic infections from the Plasmodium and Babesia genera, auto-immune disorders, hemodialysis, and mechanical circulatory support devices such as left ventricular assist devices, cardiopulmonary bypass, and extracorporeal membrane oxygenation. Sepsis also exposes the RBC to multiple physiologic stressors that increase risk of hemolysis and release of CFH into circulation.17
2.2. CFH release during disseminated intravascular coagulation
Perhaps the most readily recognized cause of hemolysis during sepsis is disseminated intravascular coagulation (DIC), which commonly complicates sepsis.18 DIC is an acquired microangiopathy characterized by systemic activation of both the coagulation and fibrinolytic cascades leading to formation of microvascular thrombi and consumption of circulating platelets and coagulation factors.19 The formation of fibrin strands results in mechanical shearing of RBCs, resulting in intravascular hemolysis and release of CFH into the circulation.18,19 The degree of hemolysis can be profound in septic patients with DIC, often necessitating multiple RBC transfusions to compensate for ongoing losses both from hemolysis and bleeding.19 Although septic patients with DIC can develop clinically significant elevations in CFH levels, there are other mechanisms that contribute to hemolysis during sepsis, and the absence of DIC does not preclude high levels of circulating CFH.11
2.3. RBC deformability
During sepsis, RBCs undergo changes in cellular geometry and deformability.20,21 Deformability, a cellular property that characterizes a cell’s ability to change shape during flow,20,21 is an important property of RBCs that facilitates laminar flow in high-flow vessels with high shear rate and allows RBCs to navigate through small end-organ capillary networks and the interendothelial slits of the spleen.20 Deformability of RBCs is decreased during sepsis21 resulting in more rigid RBCs that are prone to injury during transit through small vessels and to destruction by the reticuloendothelial system. These reductions in RBC deformability can be induced by changes in 2,3 diphosphoglycerate levels,22 alterations in glucose metabolism,23 depletion of nitric oxide,24 exposure to reactive oxygen species,25 and interactions with white blood cells,24 all of which occur during sepsis.
2.4. RBC suicidal death during sepsis
Cytokine signaling during sepsis can trigger suicidal RBC death, termed eryptosis. In an experimental model, Kempe et al. reported that exposure of RBCs from healthy human donors to plasma from septic adult patients resulted in increased RBC lysis compared to exposure of RBCs to plasma from non-septic healthy adults.26 The increased hemolysis was accompanied by increases in RBC cytosolic Ca2+ activity and ceramide formation, leading to increased exposure of phosphatidylserine on the RBC plasma membrane, which triggered eryptosis and release of CFH.26 Furthermore, exposure of donor RBCs to sphingomyelinase derived from Staphylococcus aureus also triggered RBC lysis along the same pathway.26 Under normal physiologic conditions, phosphatidylserine exposure during eryptosis promotes binding of eryptotic RBCs to receptors expressed by splenic macrophages.27 This event facilitates phagocytosis of RBCs for controlled degradation,27 which prevents release of CFH into circulation.17 However, this pathway may be impaired during sepsis. Phosphatidylserine also triggers RBC binding to endothelial cells,28 activation of the coagulation cascade,29 and increases RBC aggregation,28 mechanisms that contribute to uncontrolled hemolysis during acute sickle cell crises and to the thrombophilia of inherited hemoglobin disorders.28,29 Uncontrolled activation of these eryptosis signaling pathways during sepsis may overwhelm the endogenous capacity to remove RBCs with phosphatidylserine exposure, thus promoting uncontrolled hemolysis and release of CFH into circulation.
2.5. Pathogen-specific contributors to CFH release
Pathogen-specific factors may also contribute to hemolysis in sepsis. Parasitic infections by Plasmodium spp. and Babesia spp., the causative organisms of malaria and babesiosis, directly cause hemolysis as a result of their intraerythrocytic life cycle.30,31 These organisms can induce both acute hemolysis with large elevations in plasma CFH levels32,33 as well as chronic low-grade hemolysis leading to chronic CFH exposure.31 Many gram-negative and gram-positive bacterial species can produce pore-forming toxins (PFTs) during infection.34 PFTs are a structurally diverse class of toxin proteins that are secreted by the invading pathogens, bind to target host cells, then perforate the host cell plasma membrane, which disrupts cell homeostasis and causes subsequent cell death.34 Among these, the hemolysins produced by Staphylococcus aureus, Enterococcus spp., and Escherichia coli are notable for their ability to induce RBC hemolysis in vitro.34 Although it is unclear if these hemolysins always lead to clinically significant hemolysis in vivo, these toxins commonly serve as critical virulence factors in invasive bacterial infections.
3. Cell free hemoglobin is elevated during sepsis
Two recent studies have reported on CFH levels in septic adults.11,12 In a study of 391 adult ICU patients with sepsis, Janz et al. observed that plasma CFH was detectable in 81% of patients even in the absence of overt hemolytic disease or DIC.11 Higher levels of CFH were independently associated with increased risk of in-hospital mortality when controlling for age, comorbid medical conditions, and severity of illness.11 Similar findings were reported by Adamzik et al. who measured CFH levels in 161 critically ill adults with sepsis.12 A higher CFH level (defined as plasma CFH concentration above the cohort median) was associated with increased risk of 30-day mortality, and the CFH level had better predictive performance for mortality than plasma procalcitonin, sequential organ failure assessment (SOFA) score, or simplified acute physiology scale II (SAPS-II) score.12 In both studies, even relatively low levels of plasma CFH were associated with increased risk of death.
Similar findings have been observed in experimental animal models. Larsen et al. examined the role of CFH using a cecal ligation and puncture (CLP) murine polymicrobial sepsis model.10 Elevated plasma CFH levels only occurred following “high grade” CLP (using a cecal ligature resulting in 80–90% lumen occlusion and multiple cecal punctures), whereas elevated CFH levels did not occur following “low grade” CLP (with 50–60% lumen occlusion and a single cecal puncture).10 Notably, the administration of exogenous heme, the iron-containing porphyrin subunit of hemoglobin, to mice treated with “low grade” CLP resulted in a marked increase in markers of organ dysfunction and increased mortality,10 indicating that heme can potentiate the toxic systemic effects of sepsis. Furthermore, mice with genetic knockout of heme-oxygenase 1 (Hmox-1), a rate-limiting enzyme in the metabolic degradation of heme and an important antioxidant enzyme, also had increased organ dysfunction and death.10 Taken together with the data from human studies, it is clear that sepsis on its own can induce release of CFH, and high CFH levels can serve as a biomarker of increased risk of organ dysfunction and death.
4. Mechanisms of renal toxicity by cell-free hemoglobin
The release of CFH into the circulation can have multiple toxic effects on the kidney (Table 1).35
Table 1.
Potential mechanisms of hemoglobin renal toxicity in sepsis-AKI
| Tubular obstruction |
|---|
| Extracellular hemoglobin forms small (32kD) αβ-chain heterodimers that are freely filtered by the glomerulus |
| Breakdown of filtered hemoglobin in urinary space releases heme |
| Heme accumulation forms pigment casts in urinary space |
| Tubular obstruction promotes proximal tubular endocytosis of heme and tubular epithelial injury |
| Extravascular translocation of hemoglobin |
| Hemoglobin αβ-heterodimers can translocate through the vascular walls of peritubular capillaries |
| Breakdown of hemoglobin releases free heme and causes iron deposition in the renal interstitium |
| Reaction with nitric oxide (NO) |
| Consumes NO leading to vasoconstriction and endothelial dysfunction |
| Reaction of oxyhemoglobin with NO generates ferric (Fe3+) hemoglobin |
| Generation of reactive oxygen species |
| Ferric (Fe3+) and ferryl (Fe4+) iron hemoglobin outside of |
| Globin radicals can drive lipid and protein peroxidation |
| Free heme oxidizes LDL, promoting inflammation and cytotoxicity |
| Increased oxidative stress promotes hemolysis and more CFH release |
| Heme-mediated immune signaling |
| Heme potentiates neutrophilic inflammation and inhibits neutrophil apoptosis |
| Heme can bind TLR4 triggering pro-inflammatory signaling |
4.1. Tubular obstruction
Much of the CFH released during hemolysis dissociates into αβ-heterodimers than can pass through the glomerular filtration barrier and are excreted unchanged in the urine.36 Breakdown of CFH in the urinary space releases heme, which in sufficient quantities can accumulate into pigment casts that can cause tubular obstruction.37,38 Similar tubular obstruction with pigment casts is also observed during rhabdomyolysis, when release of large quantities of the hemoprotein myoglobin into the circulation leads to heme accumulation in the urinary space.37,38 Rhabdomyolysis is discussed in more detail later in this review. Although tubular obstruction from pigment casts is often transient and its effects on filtration pressures are unclear,37 even transient periods of stasis may promote increased endocytosis of toxic metabolites by the proximal tubular epithelium, resulting in hemosiderin deposition in the surrounding interstitium, tubular epithelial injury, and tubular necrosis.37,38
4.2. Extravascular translocation and release of free heme
CFH αβ-heterodimers can also directly translocate through the peritubular capillaries and accumulate in the renal interstitium, resulting in detectable CFH levels in renal lymphatics in experimental animal models.39 CFH translocation into the renal interstitium may result in prolonged retention of CFH in the kidneys even after CFH has cleared from the plasma. Breakdown of the globin protein outside of the RBC releases the iron-complexed porphyrin heme into the circulation or extravascular space (free heme). The complexed iron in free heme can react with cell membranes, plasma proteins, and lipids, and the heme ring can serve as a ligand for other molecular signaling pathways as described below.
4.3. Oxidative stress and reactive oxygen species generation
CFH represents a significant source of oxidative stress due to the chemical reactivity of the iron moiety of heme. The RBC cytoplasm normally serves as a reducing environment that maintains the heme-complexed iron in a reduced ferrous (Fe2+) state.40 Once released outside of the RBC cytoplasm, the iron moiety can undergo oxidation to form the more reactive ferric (Fe3+) and ferryl (Fe4+) states.40,41 These highly reactive oxidized iron species can in turn drive formation of other reactive oxygen species including lipid peroxides40,41 and F2-isoprostanes,40,41 leading to increased oxidative stress and cellular injury. Furthermore, since the RBC itself is sensitive to oxidative stress, CFH-mediated formation of reactive oxygen species can induce a positive feedback loop that promotes RBC plasma membrane instability, leading to increased hemolysis and release of more CFH.25
4.4. Nitric oxide consumption
CFH also consumes nitric oxide in regions of both high and low oxygen tension,42,43 with the resultant NO depletion leading to vasoconstriction and endothelial dysfunction.42,43 This mechanism is thought to drive the acute hypertensive response observed in massive hemolysis as well as chronic vascular disease in patients with chronic hemolytic disorders like sickle cell anemia.42 Sepsis has variable effects on macrovascular and microvascular renal blood flow (RBF)44,45,46 and both vasoconstriction and vasodilation have been observed simultaneously in different vascular beds. Inducible nitric oxide synthetases (iNOS) and NO signaling are markedly upregulated in the kidney during sepsis and have critical roles in renal control of microvascular function and local blood flow.47,48 Although pharmacological inhibition of iNOS (decreasing local NO in the kidney) may improve renal capillary perfusion in experimental animal models,47,48 the diffuse nature of CFH-mediated NO depletion may also overwhelm the kidney’s ability to regulate RBF and glomerular filtration at the level of the glomerular afferent and efferent arterioles, leading to impaired control of microvascular perfusion and worsened renal injury.
4.5. CFH-mediated immune dysregulation
Lastly, the byproducts CFH can interact with the immune system through multiple pathways that contribute to immune dysregulation. Heme can potentiate neutrophilic inflammation by stimulating neutrophil chemotaxis,49 elaboration of the reactive oxidative burst,49 and inhibiting neutrophil apoptosis.50 CFH also can interact with the innate immune system through Toll-like receptors (TLR).51,52 In particular, CFH can bind to pathogen-associated molecular patterns (PAMPs) such as lipopolysaccharide (LPS) and lipoteichoic acid (LTA) which can then modulate the binding of these PAMPs to TLR-4 and TLR-2, respectively, altering expression of downstream cytokines.52 Free heme can also directly bind to TLR4.51,53 Although it is not clear if these pathways are relevant to CFH-mediated AKI in humans, TLR4 is constitutively expressed in renal vascular walls and may also be present on the apical membrane of renal tubular epithelial cells,54 and pharmacological blockade or genetic deletion of TLR4 signaling can protect against renal injury in various experimental models of sepsis-AKI.54 Therefore, heme-TLR4 interactions may play an important role in sepsis-AKI when plasma CFH is elevated.
5. Renal toxicity of circulating hemoproteins in experimental models
Several lines of evidence from experimental animal models suggest that CFH and free heme can exert nephrotoxic effects and are reviewed here.
5.1. Myoglobin and rhabdomyolysis
There is clear evidence that elevated levels of circulating hemoproteins other than hemoglobin can induce AKI. Conditions such as major crush injury, extreme physical exertion, malignant hyperthermia, or severe electrolyte disturbances can trigger massive skeletal muscle myocyte death, termed rhabdomyolysis, resulting in release of large quantities of the hemoprotein myoglobin into circulation.37,41 Myoglobin is a monomeric heme-containing globin protein found in skeletal and heart muscle that is structurally related to hemoglobin and serves as an oxygen storage protein for myocytes.55 Because of its structural and chemical similarities to CFH, elevated circulating myoglobin exerts nephrotoxic effects though mechanisms similar to CFH including tubular obstruction by heme casts, deposition of iron in the renal interstitium, and generation of lipid peroxidation products.41,56,57 In rodent models, both direct venous perfusion with exogenous myoglobin56 and intramuscular injection with 50% glycerol to induce muscle necrosis41,57 consistently induce AKI, particularly in conjunction with other predisposing conditions including volume depletion, renal ischemia, or metabolic acidosis.56 Although myoglobin shares may common mechanistic effects with CFH, some features, particularly interactions with TLRs, appear to be more relevant to CFH and have not been described for myoglobin.
5.2. CFH infusion and transfusion-associated hemolysis
Several studies have examined the effects of CFH in isolation on renal function in canine, guinea pig, and rodent animal models. Direct intravenous infusion of exogenous CFH results in hemoglobinuria and an acute hypertensive response in dogs and guinea pigs43,58 as does exposure to CFH from transfusion of stored blood in guinea pigs.59 These hypertensive changes are accompanied by vascular endothelial dysfunction and subendothelial vascular injury.59 Transfusion with older stored RBCs reliably leads to increased release of CFH and heme compared to transfusion with younger fresh RBCs both in guinea pig and canine models using massive exchange transfusion59,60 and in a mouse model of hemorrhagic shock with RBC resuscitation.61 In both settings, the release of CFH results in renal histopathologic changes including increased deposition of free iron,58,59,61, lipid peroxidation,58 and renal tubular dilation and necrosis.58,59,61 It is also accompanied by increased kidney expression of biochemical markers of renal tubular injury such as neutrophil gelatinase-associated lipocalin (NGAL) and kidney injury molecule-1 (KIM-1).61 The impact on glomerular filtration is less clear, as elevated CFH levels decreased glomerular filtration as measured by serum creatinine in some models,59 while in other models CFH did not affect glomerular filtration.60,61 Interestingly, several groups have shown that these renal changes could be abrogated by augmenting plasma haptoglobin (Hp) levels to capture and sequester CFH before it could reach the kidneys, either by pre-treating dogs with glucocorticoids to induce hepatic expression of endogenous Hp58 or by directly infusing additional exogenous Hp.58,59,61
5.3. CFH during sepsis
The effects of free heme and CFH on renal function during sepsis have been specifically tested in two experimental animal models.10,13 As described above, Larsen et al. reported the effects of free heme in a murine CLP model. Transgenic mice deficient in Hmox1 (Hmox1−/−) were less able to metabolize circulating free heme and CFH compared to wild type (Hmox1+/+) mice. Compared to Hmox1+/+ mice, Hmox1−/− mice had increased evidence of renal injury as indicated by increased tubular epithelial necrosis on renal histology and decreased glomerular filtration as indicated by increased blood urea nitrogen (BUN) levels following CLP-induced sepsis.10 Furthermore, intraperitoneal administration of exogenous heme to septic Hmox1+/+ mice resulted in increased markers of renal injury, and blocking free heme with exogenous hemopexin was protective against renal injury.10
Shaver et al. tested the effects of CFH on kidney injury in a murine polymicrobial peritonitis sepsis model.13 Polymicrobial peritonitis was induced using an intraperitoneal injection of cecal slurry (IP CS) from a donor mouse, a method which recapitulates most features of the more common CLP model,62 while facilitating more reproducible control over the inflammatory response and degree of lethality as the dose of cecal slurry is more easily titrated than CLP.62 Treatment with a sub-lethal dose of IP CS resulted in modest elevations in CFH levels at 24 hours13 which were accompanied by similarly modest kidney mRNA expression of renal epithelial injury markers NGAL and KIM-1, but without substantial changes in GFR at 24 or 48 hours.13 In contrast, simultaneous treatment with both IP CS and intravenous CFH to augment CFH levels resulted in markedly increased NGAL and KIM-1 mRNA expression. This was accompanied by histologic evidence of renal tubular injury with tubular distortion and detachment of tubular epithelial cells from the basement membrane, findings not observed in mice treated with IP CS alone. The IP CS + IV CFH mice were also the only treatment group to experience decreases in GFR over the 48-hour period following treatment.13
Taken together, the data from model animal studies using myoglobin, CFH in isolation, and CFH during sepsis strongly support the hypothesis that heme and CFH are nephrotoxic, and that CFH potentiates acute kidney injury during sepsis.
6. Clinical evidence for renal toxicity of cell-free hemoglobin in conditions other than sepsis
The relationship between CFH and AKI has been studied extensively in conditions other than sepsis, which may provide additional insight into the potential impact of CFH in sepsis-AKI.
6.1. Cardiopulmonary Bypass Surgery
The clinical effects of CFH on renal function are perhaps best characterized in the setting of cardiopulmonary bypass (CPB) to facilitate cardiac surgery.63–65 Hemolysis is a well-recognized consequence of CPB,66 and is caused by several factors including direct mechanical RBC injury from contact with non-endothelial surfaces such as tubing and pumps, interaction with the gas-permeable membrane of the oxygenator, activation of the coagulation cascade, and RBC transfusion to replace blood loss during the procedure.66 Plasma CFH levels above 50 mg/dL are routinely encountered during CPB,63,64 and associations between CFH levels during or immediately following surgery and risk of subsequent postoperative AKI have been noted in both adults63,67 and children.64 A study by Billings et al. merits particular note as the investigators examined the relationship between CFH levels, oxidative stress, and AKI.67 CPB patients who experienced post-operative AKI had higher peak CFH levels as well as higher plasma levels of the oxidative stress markers F2-isoprostanes and isofurans during the surgical procedure.67 Moreover, peak CFH levels were correlated with higher peak plasma isofuran levels during the surgery, both in patients with and without AKI. Lastly, patients with AKI had sustained elevations in urine isofuran levels for up to two days postoperatively, suggesting the AKI patients experienced sustained periods of increased oxidative stress.67 Taken together, this data supports the contention that CFH-mediated oxidative stress can play a significant role in driving AKI in these patients.
6.2. Transfusion-associated hemolysis
Although clinical studies of the renal effects of RBC transfusion in critically ill adults have reported variable results,68,69 hemolysis during RBC transfusion has clear effects on systemic and pulmonary vascular endothelial function. In a study of 18 healthy volunteers comparing autologous transfusion of 5-day-old RBCs versus 42-day-old RBCs, Risbano et al. reported that transfusion with older blood increased post-transfusion hemolysis, with resultant depletion of plasma NO and impaired vascular endothelial function as measured by acetylcholine-dependent forearm blood flow.70 Berra et al. reported that transfusion with 40-day-old RBCs similarly led to greater increases in mean pulmonary artery pressure compared with 3-day-old RBCs in 14 obese but otherwise healthy volunteers.71 These observations may have clinical relevance in sepsis-AKI as endothelial dysfunction is a prominent feature of sepsis even in the absence of RBC transfusions,72 and normal renal homeostasis depends on tight regulation of endothelial barrier function and microvascular perfusion.73 Therefore, it is reasonable to hypothesize that CFH-mediated disruption of normal endothelial function during sepsis could affect the renal microvasculature. However, this mechanism has not yet been directly evaluated in sepsis-AKI.
6.3. Sickle Cell Anemia
The inborn genetic hemoglobinopathies including sickle cell disease (SCD) and the thalassemias are among the most common monogenetic disorders in the world.74,75 These disorders are frequently characterized by chronic hemolysis and punctuated by episodes of acute hemolysis that quickly overwhelms endogenous CFH processing mechanisms.75 The kidney is particularly vulnerable to injury in SCD because of the tendency for HbS to polymerize in the renal medulla, where the partial pressure of oxygen is low, leading to vascular occlusion by sickled RBCs and subsequent renal infarction with papillary necrosis.75 Although mechanisms including endothelial dysfunction, volume depletion, medications, and acute infection all contribute to AKI in SCD,75 the presence and degree of an SCD patient’s “hemolysis phenotype” also appears to influence renal outcomes. Degree of hemolysis as measured either by biochemical markers or drop in hemoglobin concentration are associated with increased risk of AKI in both children and adults presenting with acute vaso-occlusive crises.76,77 Furthermore, hemoglobinuria, a marker of chronic ongoing hemolysis, was associated with increased incidence of chronic kidney disease (CKD) as well as progression of CKD in a large longitudinal study of African-American adults with SCD.78
6.4. Malaria
Malaria is another condition where CFH release can exert substantial effects on the kidney.33 Blackwater fever, a syndrome of malaria-induced massive intravascular hemolysis, hemoglobinuria, and acute renal failure, has been recognized for over a century.79 Increased hemolysis in malaria has been associated with abnormal renal function,33 although most studies have not specifically examine the correlation between CFH and AKI. Most recently, there has been interest in understanding the mechanistic relationship between parasite burden, CFH release, and renal function. In an observational study of 107 patients with severe malaria in Bangladesh, Plewes et al. found that CFH levels were higher in patients with AKI compared to those without AKI.32 Unsurprisingly, higher CFH levels were also correlated with higher parasite burdens and other biochemical markers of hemolysis. Perhaps more interesting, the investigators also noted that among those patients admitted to the hospital initially without AKI, increased oxidative stress markers (plasma F2-isoprostanes and isofurans) and decreased RBC deformability predicted subsequent creatinine elevation and need for hemodialysis.32 This observed relationship between the mechanistic factors that contribute to abnormal CFH biology and AKI risk provides further evidence that CFH is driving the increased risk of AKI in severe malaria patients.
7. Targeting cell-free hemoglobin in sepsis-induced acute kidney injury
Since the release of CFH into circulation is a common feature of sepsis and can cause renal dysfunction in experimental models, CFH represents an obvious therapeutic target for sepsis-AKI, with several potential mechanistic approaches. Potential strategies to mitigate the effects of CFH during sepsis-AKI include limiting release of CFH, increasing scavenging of CFH, and decreasing CFH-mediated oxidative injury.
7.1. Limiting CFH release
Prevention of hemolysis, and thus prevention of the release of CFH from the RBC, is an attractive target. However as discussed previously, multiple physiologic processes influence CFH release during sepsis, and many of these factors are not clearly amenable to pharmacological intervention.17 Therefore, few studies specifically targeting the mechanisms of hemolysis during sepsis have been reported.
Limiting RBC transfusions to when hemoglobin concentration falls below 7.0 g/dL (restrictive transfusion) may reduce exposure to transfusion-associated hemolysis, and thus may indirectly mitigate CFH-mediated renal injury during sepsis. However, data on the renal effects of transfusion thresholds in sepsis are limited.69 A recent meta-analysis of transfusion threshold in sepsis or septic shock identified only two randomized trials that reported on renal failure at 28 days following admission.69 Neither study reported on AKI, change in creatinine, or other renal biomarkers.69 No significant differences were noted between patients assigned to restrictive or liberal strategies, although these conclusions are limited by the small samples and low event rates, with a total of only 83 events in 1277 patients studied.69 Nevertheless, since a restrictive transfusion threshold both conserves a scarce resource and does not increase risk of other adverse outcomes,69 limiting RBC transfusion is a reasonable strategy to mitigate risk of CFH exposure.
7.2. Haptoglobin
Release of sufficient CFH into the vascular compartment during sepsis can overwhelm endogenous scavenger mechanisms. Therefore, supplementation with exogenous scavenger proteins could prevent CFH from causing downstream effects. Haptoglobin (Hp) is the primary endogenous scavenger of CFH in mammals.15 A dimeric plasma protein synthesized by the liver, Hp binds irreversibly to CFH.35 The resultant CFH-Hp complex binds to the CD163 receptor present on monocytes and macrophages,80 triggering endocytosis and clearance of CFH from the circulation by the reticuloendothelial system.35 Hp is an acute phase reactant protein in humans, leading to variable Hp levels in sepsis.81,82 In a study of 387 patients with severe sepsis, lower plasma Hp levels were independently associated with increased hospital mortality.82 Notably, the relationship between plasma Hp level and mortality was only observed in patients with elevated CFH levels, whereas Hp level did not affect mortality in patients without elevated CFH,82 further suggesting that circulating Hp is critical for limiting the toxicity of CFH during sepsis.
Several experimental studies support a potential therapeutic role for supplemental Hp to mitigate CFH-mediated renal injury. As noted previously, augmenting plasma Hp levels with either glucocorticoids58 or exogenous Hp59,61 abrogates the renal toxicity of CFH in animal models of transfusion-associated hemolysis. Using a canine model of severe S. aureus pneumonia, Remy et al. showed that purified human Hp improved survival, and reduced vasodilatory shock, lung injury scores and circulating levels of non-transferrin bound iron compared to infusion of human albumin.60 The beneficial effects of supplemental Hp were accentuated in animals also treated with large-volume exchange transfusion to increase circulating CFH levels,60 suggesting that the benefit of haptoglobin supplementation was related primarily to improved scavenging of CFH. Mean creatinine levels were lower among the Hp-treated group at several time-points, although the differences were limited by small sample size and not statistically significant.60
Purified human Hp is commercially available for clinical use in Japan and is approved for treatment of severe hemolysis during extracorporeal cardiopulmonary bypass, severe burn injuries, or following massing transfusion. Several uncontrolled trials in Japan have suggested that intraoperative haptoglobin supplementation during CPB can reduce risk of subsequent postoperative AKI,83,84 but prospective trials have been limited by small sample sizes. To date, no clinical trials have tested haptoglobin supplementation in patients with sepsis.
7.3. Hemopexin
As noted previously, breakdown of CFH can release the iron-complexed porphyrin heme (free heme) into circulation. As free heme can cause toxicity in its own right, it necessitates its own clearance mechanisms. Hemopexin (Hpx) serves as the primary endogenous scavenger of free heme in humans.16,85 Hpx is a glycoprotein synthesized by the liver and is normally present in the circulation in high concentrations.16,85 Although other plasma proteins including albumin and lipoproteins of LDL or HDL particles can also bind porphyrins, Hpx has the highest binding affinity for heme of any protein (K < 10−13M).35,85 Once bound to Hpx, heme is delivered primarily to the liver, preventing the pro-oxidant and pro-inflammatory effects of heme.86 Heme binding by Hpx also prevents heme from intercalating in the plasma cell membrane where it can drive lipid peroxidation.87 Binding of Hpx to the CD91 receptor on hepatocytes and phagocytic cells triggers endocytosis of the Hpx-heme complex, subsequent degradation of heme by HO-1, and degradation or recycling of Hpx.88
In contrast to Hp, the role of Hpx in the acute phase response remains controversial.85,89 In several observational studies of critically ill septic adults, non-survivors of sepsis had lower plasma Hpx levels than survivors,10,82 although many sepsis patients still have Hpx levels within the reference range of 0.5 to 1.5 mg/dL.16,85 Hpx supplementation has been explored in experimental animal models. As noted previously, Larsen et al. found that Hpx supplementation mitigated renal injury in mice with intraperitoneal sepsis, whereas treatment with equivalent amounts of non-heme-binding IgG did not improve renal injury.10 In contrast, Graw et al. found that Hpx supplementation did not prevent renal injury in a mouse model of hemorrhagic shock with massive RBC resuscitation.61 Currently, Hpx has not been tested in clinical trials of sepsis and is not available for clinical use.
7.4. Acetaminophen
The common analgesic and antipyretic acetaminophen (APAP) has also been identified as a potential therapy for CFH-mediated organ injury. Although traditionally prescribed for its effects on cyclooxygenase-prostaglandin pathways, at clinically relevant doses, APAP can reduce ferryl (Fe4+) complexed iron in CFH to the less reactive ferric (Fe3+) species.41,90 The specificity of APAP for Ferryl-CFH reduction is due to structural similarity between the heme moiety of CFH and the peroxidase moiety of cyclooxygenase.41 This was experimentally demonstrated in a rat rhabdomyolysis model of AKI. Boutaud et al. observed that treatment with APAP before or after induction of rhabdomyolysis reduced markers of oxidative stress, reduced heme-to-protein cross-linking from peroxide reactions, and mitigated AKI (Figure 1).41 APAP’s unique hemoprotein reductant activity is not present in non-specific anti-oxidants such as vitamin E and N-acetyl cysteine at doses that have been studied in clinical sepsis.91,92 Thus, APAP has the potential to target CFH-mediated organ dysfunction in sepsis where other antioxidants have failed.
Figure 1.
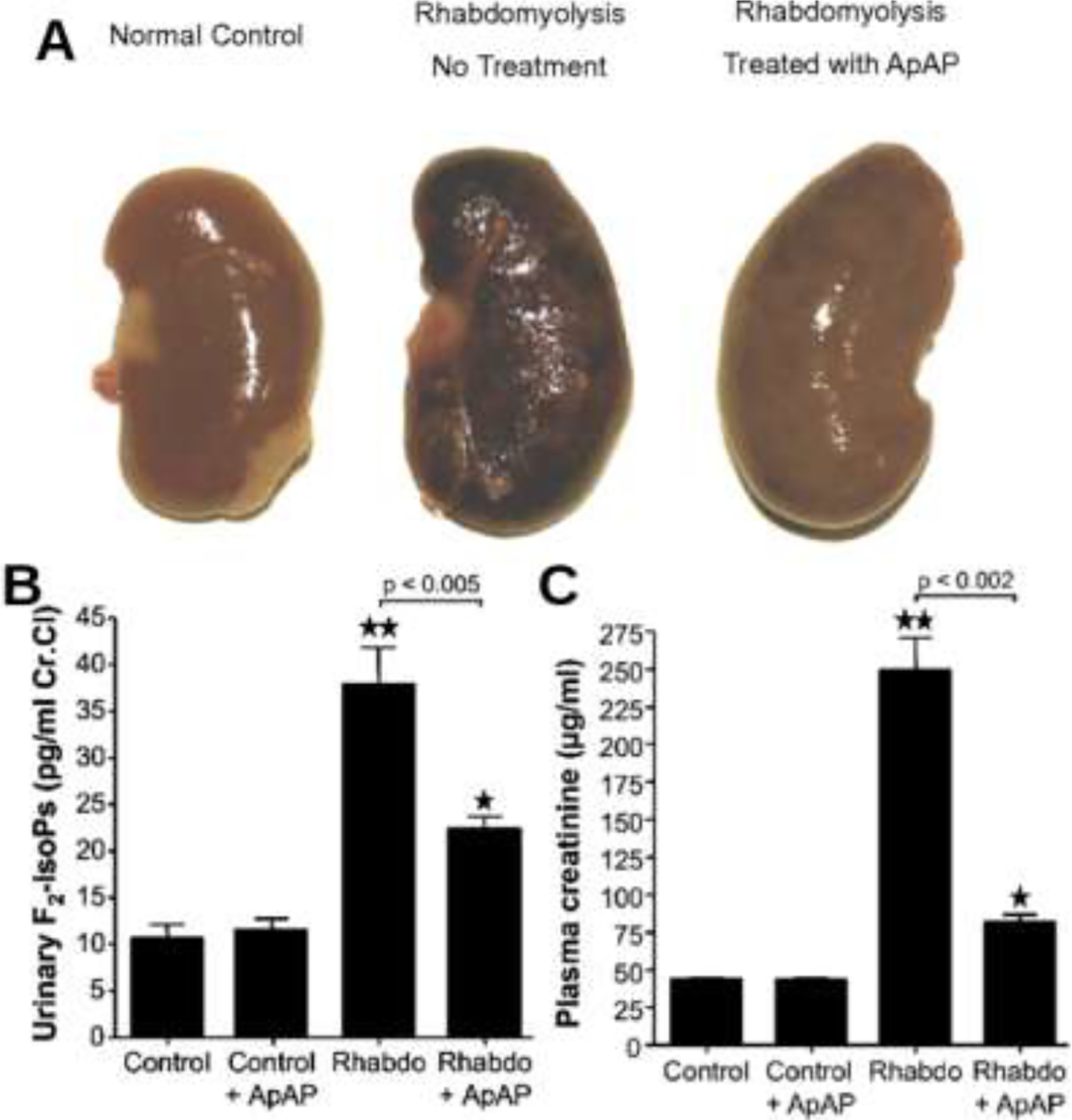
Renoprotective effects of acetaminophen in rats with rhabdomyolysis. (A) Gross examination of a representative kidney from a normal rat (Left), a rat with rhabdomyolysis (Center), and a rat with rhabdomyolysis treated with acetaminophen (APAP) (Right). Effect of treatment with APAP on reactive oxygen species (B) and on kidney function (C) in normal rats (controls), in normal rats treated with APAP (control + ApAP), in rats with rhabdomyolysis (Rhabdo), and in rats with rhabdomyolysis treated with APAP (Rhabdo + ApAP). (B) Urinary excretion of F2-isoprostanes. **, P < 0.0001 for Rhabdo vs. controls; *, P < 0.005 for Rhabdo + APAP vs. controls. (C) Plasma creatinine levels. **, P < 0.0001 for Rhabdo vs. controls; *, P = 0.0002 for Rhabdo + APAP vs. controls. For both figures, the brackets indicate that APAP significantly reduced urinary F2-isoprostanes and plasma creatinine compared with Rhabdo alone. (n ≥ 6 in each group). Abbreviations: ApAP – Acetaminophen. F2-IsoPs − F2-isoprostanes. Reproduced with permission.41
Several small clinical trials have tested APAP specifically for mitigating CFH-mediated organ injury.65,93–95 Collectively, these trials suggest a beneficial effect of APAP on hemoglobin-mediated oxidative injury and AKI. APAP demonstrated favorable effects on plasma levels of lipid peroxidation products in two clinical trials in children or adults undergoing cardiac surgery with CPB.65,95 Although no differences in post-operative AKI were noted in these trials, they were limited by small sample sizes and low event rates.65,95 Janz et al. reported a phase 2a randomized placebo-controlled clinical trial of APAP in 40 patients with severe sepsis and elevated plasma CFH measured at enrollment. Patients who received enteral APAP at 1 gram every 6 hours for 3 days had significantly reduced oxidative stress as measured by plasma F -isoprostanes.93 APAP-treated patients also had lower serum creatinine levels on days 3 and 4 after randomization as well as lower peak serum creatinine through the remainder of the hospitalization (Figure 2).93 Finally, in a randomized clinical trial in 61 adults with severe falciparum malaria in Bangladesh and Thailand, Plewes et al. showed a clear beneficial effect of APAP on both lipid peroxidation and AKI as measured by relative change in serum creatinine over the first 72 hours after enrollment (Figure 3).94 The renal benefits of APAP were confined to patients with higher plasma CFH levels (> 4.5 mg/dL) as well as those with higher levels of lipid peroxidation products on enrollment (Figure 3).94 Also interestingly, a secondary analysis using pharmacokinetic-pharmacodynamic (PK–PD) mixture model showed that higher total APAP exposure over 72 hours correlated with a lower probability of declining renal function.94
Figure 2.

Creatinine measurements over study period in the Acetaminophen for the Reduction of Oxidative injury in Severe Sepsis (ACROSS) Trial. In patients never receiving any type of renal replacement therapy during or after the study (n = 36), creatinine levels were similar at baseline (study day 0) and were significantly reduced in the acetaminophen group on study day 3, the day following study completion (day 4), through the remainder of hospitalization, and at discharge or death. P values represent between-group comparisons at each time point. Circles and squares represent medians of placebo and acetaminophen groups, respectively. Whiskers represent interquartile ranges (between 25th and 75th percentiles). Table under figure represents number of patients (n) at each time point. Reproduced with permission.93
Figure 3.

Effect of acetaminophen on serum creatinine in randomized trial of severe falciparum malaria. Points represent mean percent change in creatinine from baseline at 12, 24, 36, 60, 48, and 72 hours in the entire cohort (A) and in patients stratified by level of intravascular hemolysis (B and C). Plasma cell-free hemoglobin (CFH) ≥45000 ng/mL (B); plasma CFH <45000 ng/mL (C). Frequencies in rows below figures represent number of patients (n) at each time point. P value represents overall treatment effect. Abbreviations: Cr, creatinine; CFH, cell-free hemoglobin. Reproduced under the terms of the Creative Commons CC BY license from reference,94 which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Notably, all of the aforementioned trials specifically targeted patients with either predicted or measured elevations of plasma CFH. This stands in contrast to the HEAT Trial, a large multi-center randomized clinical trial of APAP for ICU patients with fever and suspected infection, which did not observe any differences in renal outcomes or days free from renal failure.96 The HEAT Trial specifically tested the hypothesis that allowing “permissive hyperthermia” (by avoiding APAP for fever) would improve outcomes compared to standard treatment of fever with APAP. The trial did not report CFH or myoglobin levels, and the incidence of conditions associated with elevated circulating hemoproteins such as clinical rhabdomyolysis or DIC were either low or not reported.96 Furthermore, the total APAP exposure of the treatment group was lower (median 8 grams over 2 days) than in the trials by Janz et al. and Plewes et al. (12 grams over 3 days).93,94 Therefore, although APAP may not be beneficial in an unselected ICU population, the HEAT trial does not exclude the potential value of APAP for CFH-mediated organ dysfunction. Collectively, these data highlight both the potential use for plasma CFH levels as a predictive biomarker for sepsis-AKI, and that any future clinical trials of a pharmacological agent targeting CFH during sepsis will need to consider specific enrichment for patients with elevated CFH levels.
7.5. Other strategies
Several other therapeutic agents have the potential to target CFH-mediated injury in sepsis but are less well studied. Vitamin C (ascorbic acid) has hemoprotein reductant activity and can prevent the increase in endothelial permeability induced by exposure of human endothelial cells to CFH.97 Vitamin C has also shown some promise in observational studies of sepsis,98 but whether this is related to effects on circulating CFH is not known. Notably, the recently published CITRIS-ALI study, a randomized trial of intravenous vitamin C in patients with sepsis-induced acute respiratory distress syndrome, demonstrated no differences in renal outcomes between patients treated with vitamin C or placebo despite significant differences in mortality.99 Therapies that target free iron might also be beneficial in the setting of elevated CFH levels in sepsis,100 however no studies have been reported in humans to date.
7.6. Current barriers to therapeutics targeting CFH
In order to use circulating CFH levels as a tool for predictive enrichment in clinical trials of CFH-targeting therapies, a rapid and reliable point-of-care measurement of circulating CFH is needed. Currently, there is no rapid bedside test available for CFH. Although HemoCue® America markets a point-of-care device that can measure low levels of CFH (HemoCue® Plasma/Low Hb System), this device is optimized for blood banking rather than measurement of the relatively low levels of plasma cell-free hemoglobin that have been documented in sepsis patients.11,94 Although other methods are available for measurement of plasma cell-free hemoglobin such as an enzyme-linked immunosorbent assay,94 none are yet rapidly available at the bedside for clinical trial enrollment.
8. Conclusions
Sepsis is a heterogeneous clinical syndrome that has as yet defied all attempts to identify effective pharmacological therapies. Sepsis-AKI is similarly in need of effective therapies as it remains associated with higher costs, longer hospital stays, and worse clinical outcomes. A growing body of evidence suggests that an elevated level of circulating CFH is a feature of some, but not all patients with sepsis, and CFH may play a role in the pathophysiology of sepsis-AKI through a variety of injurious mechanisms. Several potential therapies to target CFH in sepsis have been identified including haptoglobin, hemopexin and acetaminophen. Bedside measurement of CFH levels may facilitate predictive enrichment for future therapeutic trials of these CFH-targeted therapeutics such that only patients with elevated CFH, who would be most likely to benefit, would be enrolled. However, rapid, accurate bedside tests for plasma CFH will need to be developed in order for such trials to move forward.
Financial support:
This work was supported by the National Institutes of Health grants T15LM007450 for Dr Kerchberger, and R01HL135849, K24HL103836 for Dr Ware.
Abbreviations
- CFH
cell-free hemoglobin
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflict of interest statement: Dr. Ware reports consulting fees from CSL Behring, Bayer and Quark Pharmaceuticals as well as a research contract with CSL Behring (current) and past research contracts with Boehringer Ingelheim and Global Blood Therapeutics, all unrelated to the current manuscript.
References
- 1.Singer M, Deutschman CS, Seymour CW, et al. The Third International Consensus Definitions for Sepsis and Septic Shock (Sepsis-3). JAMA. 2016;315(8):801–810. doi: 10.1001/jama.2016.0287 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hall MJ, Levant S, Carol JD Trends in Inpatient Hospital Deaths: National Hospital Discharge Survey, 2000–2010. Hyattsville, MD: National Center for Health Statistics; 2013. https://www.cdc.gov/nchs/products/databriefs/db118.htm. Accessed September 15, 2019. [Google Scholar]
- 3.Angus DC, Linde-Zwirble WT, Lidicker J, Clermont G, Carcillo J, Pinsky MR. Epidemiology of severe sepsis in the United States: Analysis of incidence, outcome, and associated costs of care. Crit Care Med. 2001;29(7):1303. [DOI] [PubMed] [Google Scholar]
- 4.Bagshaw SM, Uchino S, Bellomo R, et al. Septic Acute Kidney Injury in Critically Ill Patients: Clinical Characteristics and Outcomes. Clin J Am Soc Nephrol. 2007;2(3):431–439. doi: 10.2215/CJN.03681106 [DOI] [PubMed] [Google Scholar]
- 5.Bagshaw SM, Lapinsky S, Dial S, et al. Acute kidney injury in septic shock: clinical outcomes and impact of duration of hypotension prior to initiation of antimicrobial therapy. Intensive Care Med. 2009;35(5):871–881. doi: 10.1007/s00134-008-1367-2 [DOI] [PubMed] [Google Scholar]
- 6.Morrell ED, Kellum JA, Pastor-Soler NM, Hallows KR. Septic acute kidney injury: molecular mechanisms and the importance of stratification and targeting therapy. Crit Care. 2014;18. doi: 10.1186/s13054-014-0501-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Murugan R, Karajala-Subramanyam V, Lee M, et al. Acute kidney injury in non-severe pneumonia is associated with an increased immune response and lower survival. Kidney Int. 2010;77(6):527–535. doi: 10.1038/ki.2009.502 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Langenberg C, Bagshaw SM, May CN, Bellomo R. The histopathology of septic acute kidney injury: a systematic review. Crit Care. 2008;12(2):R38. doi: 10.1186/cc6823 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Peerapornratana S, Manrique-Caballero CL, Gómez H, Kellum JA. Acute kidney injury from sepsis: current concepts, epidemiology, pathophysiology, prevention and treatment. Kidney Int. 2019;0(0). doi: 10.1016/j.kint.2019.05.026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Larsen R, Gozzelino R, Jeney V, et al. A Central Role for Free Heme in the Pathogenesis of Severe Sepsis. Sci Transl Med 2010;2(51):51ra71. doi: 10.1126/scitranslmed.3001118 [DOI] [PubMed] [Google Scholar]
- 11.Janz DR, Bastarache JA, Peterson JF, et al. Association Between Cell-Free Hemoglobin, Acetaminophen, and Mortality in Patients With Sepsis: An Observational Study. Crit Care Med 2013;41(3):784–790. doi: 10.1097/CCM.0b013e3182741a54 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Adamzik M, Hamburger T, Petrat F, Peters J, Groot H de, Hartmann M. Free hemoglobin concentration in severe sepsis: methods of measurement and prediction of outcome. Crit Care. 2012;16(4):R125. doi: 10.1186/cc11425 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Shaver CM, Paul MG, Putz ND, et al. Cell-free hemoglobin augments acute kidney injury during experimental sepsis. Am J Physiol-Ren Physiol. 2019;317(4):F922–F929. doi: 10.1152/ajprenal.00375.2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Janz DR, Zhao Z, Koyama T, et al. Longer storage duration of red blood cells is associated with an increased risk of acute lung injury in patients with sepsis. Ann Intensive Care. 2013;3(1):33. doi: 10.1186/2110-5820-3-33 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wicher KB, Fries E. Haptoglobin, a hemoglobin-binding plasma protein, is present in bony fish and mammals but not in frog and chicken. Proc Natl Acad Sci. 2006;103(11):4168–4173. doi: 10.1073/pnas.0508723103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Muller-Eberhard U Hemopexin. N Engl J Med. 1970;283(20):1090–1094. doi: 10.1056/NEJM197011122832007 [DOI] [PubMed] [Google Scholar]
- 17.Effenberger-Neidnicht K, Hartmann M. Mechanisms of Hemolysis During Sepsis. Inflammation. 2018;41(5):1569–1581. doi: 10.1007/s10753-018-0810-y [DOI] [PubMed] [Google Scholar]
- 18.Okamoto K, Tamura T, Sawatsubashi Y. Sepsis and disseminated intravascular coagulation. J Intensive Care. 2016;4(1):23. doi: 10.1186/s40560-016-0149-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Levi M, Toh CH, Thachil J, Watson HG. Guidelines for the diagnosis and management of disseminated intravascular coagulation. Br J Haematol. 2009;145(1):24–33. doi: 10.1111/j.1365-2141.2009.07600.x [DOI] [PubMed] [Google Scholar]
- 20.Piagnerelli M, Boudjeltia KZ, Vanhaeverbeek M, Vincent J-L. Red blood cell rheology in sepsis. Intensive Care Med. 2003;29(7):1052–1061. doi: 10.1007/s00134-003-1783-2 [DOI] [PubMed] [Google Scholar]
- 21.Baskurt OK, Gelmont D, Meiselman HJ. Red Blood Cell Deformability in Sepsis. Am J Respir Crit Care Med. 1998;157(2):421–427. doi: 10.1164/ajrccm.157.2.9611103 [DOI] [PubMed] [Google Scholar]
- 22.Suzuki Y, Nakajima T, Shiga T, Maeda N. Influence of 2,3-diphosphoglycerate on the deformability of human erythrocytes. Biochim Biophys Acta BBA -Biomembr. 1990;1029(1):85–90. doi: 10.1016/0005-2736(90)90439-U [DOI] [PubMed] [Google Scholar]
- 23.Jägers J, Brauckmann S, Kirsch M, Effenberger-Neidnicht K. Moderate glucose supply reduces hemolysis during systemic inflammation. J Inflamm Res. 2018;11:87–94. doi: 10.2147/JIR.S155614 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Korbut R, Gryglewski RJ. Nitric oxide from polymorphonuclear leukocytes modulates red blood cell deformability in vitro. Eur J Pharmacol. 1993;234(1):17–22. doi: 10.1016/0014-2999(93)90700-R [DOI] [PubMed] [Google Scholar]
- 25.Powell RJ, Machiedo GW, Rush BFJ, Dikdan G. Oxygen free radicals: Effect on red cell deformability in sepsis. Crit Care Med. 1991;19(5):732. [PubMed] [Google Scholar]
- 26.Kempe DS, Akel A, Lang PA, et al. Suicidal erythrocyte death in sepsis. J Mol Med. 2007;85(3):273–281. doi: 10.1007/s00109-006-0123-8 [DOI] [PubMed] [Google Scholar]
- 27.Arias CF, Arias CF. How do red blood cells know when to die? R Soc Open Sci 4(4):160850. doi: 10.1098/rsos.160850 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Setty BNY, Kulkarni S, Stuart MJ. Role of erythrocyte phosphatidylserine in sickle red cell–endothelial adhesion. Blood. 2002;99(5):1564–1571. doi: 10.1182/blood.V99.5.1564 [DOI] [PubMed] [Google Scholar]
- 29.Setty BNY, Kulkarni S, Rao AK, Stuart MJ. Fetal hemoglobin in sickle cell disease: relationship to erythrocyte phosphatidylserine exposure and coagulation activation. Blood. 2000;96(3):1119–1124. [PubMed] [Google Scholar]
- 30.Vannier E, Krause PJ. Human Babesiosis. N Engl J Med. 2012;366(25):2397–2407. doi: 10.1056/NEJMra1202018 [DOI] [PubMed] [Google Scholar]
- 31.Roberts DJ, Casals-Pascual C, Weatherall DJ. The Clinical and Pathophysiological Features of Malarial Anaemia In: Compans RW, Cooper MD, Honjo T, et al. , eds. Malaria: Drugs, Disease and Post-Genomic Biology. Current Topics in Microbiology and Immunology. Berlin, Heidelberg: Springer Berlin Heidelberg; 2005:137–168. doi: 10.1007/3-540-29088-5_6 [DOI] [PubMed] [Google Scholar]
- 32.Plewes K, Kingston HWF, Ghose A, et al. Cell-free hemoglobin mediated oxidative stress is associated with acute kidney injury and renal replacement therapy in severe falciparum malaria: an observational study. BMC Infect Dis. 2017;17(1):313. doi: 10.1186/s12879-017-2373-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Yeo TW, Lampah DA, Tjitra E, et al. Relationship of Cell-Free Hemoglobin to Impaired Endothelial Nitric Oxide Bioavailability and Perfusion in Severe Falciparum Malaria. J Infect Dis. 2009;200(10):1522–1529. doi: 10.1086/644641 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Parker MW, Feil SC. Pore-forming protein toxins: from structure to function. Prog Biophys Mol Biol. 2005;88(1):91–142. doi: 10.1016/j.pbiomolbio.2004.01.009 [DOI] [PubMed] [Google Scholar]
- 35.Schaer DJ, Buehler PW, Alayash AI, Belcher JD, Vercellotti GM. Hemolysis and free hemoglobin revisited: exploring hemoglobin and hemin scavengers as a novel class of therapeutic proteins. Blood. 2013;121(8):1276–1284. doi: 10.1182/blood-2012-11-451229 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Urbaitis BK, Razynska A, Corteza Q, Fronticelli C, Bucci E. Intravascular retention and renal handling of purified natural and intramolecularly cross-linked hemoglobins. J Lab Clin Med. 1991;117(2):115–121. [PubMed] [Google Scholar]
- 37.Zager RA. Rhabdomyolysis and myohemoglobinuric acute renal failure. Kidney Int. 1996;49(2):314–326. doi: 10.1038/ki.1996.48 [DOI] [PubMed] [Google Scholar]
- 38.Sakthirajan R, Dhanapriya J, Varghese A, et al. Clinical profile and outcome of pigment-induced nephropathy. Clin Kidney J. 2018;11(3):348–352. doi: 10.1093/ckj/sfx121 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Matheson B, Razynska A, Kwansa H, Bucci E. Appearance of dissociable and cross-linked hemoglobins in the renal hilar lymph. J Lab Clin Med. 2000;135(6):459–464. doi: 10.1067/mlc.2000.106458 [DOI] [PubMed] [Google Scholar]
- 40.Reeder BJ, Svistunenko DA, Cooper CE, Wilson MT. The Radical and Redox Chemistry of Myoglobin and Hemoglobin: From In Vitro Studies to Human Pathology. Antioxid Redox Signal. 2004;6(6):954–966. doi: 10.1089/ars.2004.6.954 [DOI] [PubMed] [Google Scholar]
- 41.Boutaud O, Moore KP, Reeder BJ, et al. Acetaminophen inhibits hemoprotein-catalyzed lipid peroxidation and attenuates rhabdomyolysis-induced renal failure. Proc Natl Acad Sci. 2010;107(6):2699–2704. doi: 10.1073/pnas.0910174107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Reiter CD, Wang X, Tanus-Santos JE, et al. Cell-free hemoglobin limits nitric oxide bioavailability in sickle-cell disease. Nat Med. 2002;8(12):1383–1389. doi: 10.1038/nm1202-799 [DOI] [PubMed] [Google Scholar]
- 43.Donadee C, Raat NJH, Kanias T, et al. Nitric Oxide Scavenging by Red Blood Cell Microparticles and Cell-Free Hemoglobin as a Mechanism for the Red Cell Storage Lesion. Circulation. 2011;124(4):465–476. doi: 10.1161/CIRCULATIONAHA.110.008698 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Langenberg C, Bellomo R, May C, Wan L, Egi M, Morgera S. Renal blood flow in sepsis. Crit Care. 2005;9(4):R363–R374. doi: 10.1186/cc3540 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Bagshaw SM, Langenberg C, Wan L, May CN, Bellomo R. A systematic review of urinary findings in experimental septic acute renal failure. Crit Care Med. 2007;35(6):1592. doi: 10.1097/01.CCM.0000266684.17500.2F [DOI] [PubMed] [Google Scholar]
- 46.Post EH, Kellum JA, Bellomo R, Vincent J-L. Renal perfusion in sepsis: from macro- to microcirculation. Kidney Int. 2017;91(1):45–60. doi: 10.1016/j.kint.2016.07.032 [DOI] [PubMed] [Google Scholar]
- 47.Tiwari MM, Brock RW, Megyesi JK, Kaushal GP, Mayeux PR. Disruption of renal peritubular blood flow in lipopolysaccharide-induced renal failure: role of nitric oxide and caspases. Am J Physiol-Ren Physiol. 2005;289(6):F1324–F1332. doi: 10.1152/ajprenal.00124.2005 [DOI] [PubMed] [Google Scholar]
- 48.Wu L, Mayeux PR. Effects of the Inducible Nitric-Oxide Synthase Inhibitor l-N6-(1-Iminoethyl)-lysine on Microcirculation and Reactive Nitrogen Species Generation in the Kidney following Lipopolysaccharide Administration in Mice. J Pharmacol Exp Ther. 2007;320(3):1061–1067. doi: 10.1124/jpet.106.117184 [DOI] [PubMed] [Google Scholar]
- 49.Graça-Souza AV, Arruda MAB, Freitas MS de, Barja-Fidalgo C, Oliveira PL. Neutrophil activation by heme: implications for inflammatory processes. Blood. 2002;99(11):4160–4165. doi: 10.1182/blood.V99.11.4160 [DOI] [PubMed] [Google Scholar]
- 50.Arruda MA, Rossi AG, Freitas MS de, Barja-Fidalgo C, Graça-Souza AV. Heme Inhibits Human Neutrophil Apoptosis: Involvement of Phosphoinositide 3-Kinase, MAPK, and NF-κB. J Immunol. 2004;173(3):2023–2030. doi: 10.4049/jimmunol.173.3.2023 [DOI] [PubMed] [Google Scholar]
- 51.Figueiredo RT, Fernandez PL, Mourao-Sa DS, et al. Characterization of Heme as Activator of Toll-like Receptor 4. J Biol Chem. 2007;282(28):20221–20229. doi: 10.1074/jbc.M610737200 [DOI] [PubMed] [Google Scholar]
- 52.Lee SK, Ding JL. A Perspective on the Role of Extracellular Hemoglobin on the Innate Immune System. DNA Cell Biol. 2013;32(2):36–40. doi: 10.1089/dna.2012.1897 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Belcher JD, Chen C, Nguyen J, et al. Heme triggers TLR4 signaling leading to endothelial cell activation and vaso-occlusion in murine sickle cell disease. Blood. 2014;123(3):377–390. doi: 10.1182/blood-2013-04-495887 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Anderberg SB, Luther T, Frithiof R. Physiological aspects of Toll-like receptor 4 activation in sepsis-induced acute kidney injury. Acta Physiol Oxf Engl 2017;219(3):573–588. doi: 10.1111/apha.12798 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Hardison RC. Evolution of Hemoglobin and Its Genes. Cold Spring Harb Perspect Med. 2012;2(12). doi: 10.1101/cshperspect.a011627 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Heyman SN, Rosen S, Fuchs S, Epstein FH, Brezis M. Myoglobinuric acute renal failure in the rat: a role for medullary hypoperfusion, hypoxia, and tubular obstruction. J Am Soc Nephrol. 1996;7(7):1066–1074. [DOI] [PubMed] [Google Scholar]
- 57.Moore KP, Holt SG, Patel RP, et al. A Causative Role for Redox Cycling of Myoglobin and Its Inhibition by Alkalinization in the Pathogenesis and Treatment of Rhabdomyolysis-induced Renal Failure. J Biol Chem. 1998;273(48):31731–31737. doi: 10.1074/jbc.273.48.31731 [DOI] [PubMed] [Google Scholar]
- 58.Boretti FS, Buehler PW, D’Agnillo F, et al. Sequestration of extracellular hemoglobin within a haptoglobin complex decreases its hypertensive and oxidative effects in dogs and guinea pigs. J Clin Invest. 2009;119(8):2271–2280. doi: 10.1172/JCI39115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Baek JH, D’Agnillo F, Vallelian F, et al. Hemoglobin-driven pathophysiology is an in vivo consequence of the red blood cell storage lesion that can be attenuated in guinea pigs by haptoglobin therapy. J Clin Invest. 2012;122(4):1444–1458. doi: 10.1172/JCI59770 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Remy KE, Cortés-Puch I, Solomon SB, et al. Haptoglobin improves shock, lung injury, and survival in canine pneumonia. JCI Insight. 2018;3(18):e123013. doi: 10.1172/jci.insight.123013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Graw JA, Mayeur C, Rosales I, et al. Haptoglobin or Hemopexin Therapy Prevents Acute Adverse Effects of Resuscitation After Prolonged Storage of Red Cells. Circulation. 2016;134(13):945–960. doi: 10.1161/CIRCULATIONAHA.115.019955 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Wynn JL, Scumpia PO, Delano MJ, et al. Increased mortality and altered immunity in neonatal sepsis produced by generalized peritonitis. Shock. 2007;28(6):675–683. doi: 10.1097/shk.0b013e3180556d09 [DOI] [PubMed] [Google Scholar]
- 63.Vermeulen Windsant IC, Snoeijs MG, Hanssen SJ, et al. Hemolysis is associated with acute kidney injury during major aortic surgery. Kidney Int. 2010;77(10):913–920. doi: 10.1038/ki.2010.24 [DOI] [PubMed] [Google Scholar]
- 64.Kim-Campbell N, Gretchen C, Callaway C, et al. Cell-free plasma hemoglobin and male gender are risk factors for AKI in low risk children undergoing cardiopulmonary bypass. Crit Care Med. 2017;45(11):e1123–e1130. doi: 10.1097/CCM.0000000000002703 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Billings FT, Petracek MR, Roberts II LJ, Pretorius M. Perioperative Intravenous Acetaminophen Attenuates Lipid Peroxidation in Adults Undergoing Cardiopulmonary Bypass: A Randomized Clinical Trial. PLoS ONE. 2015;10(2). doi: 10.1371/journal.pone.0117625 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Vercaemst L Hemolysis in Cardiac Surgery Patients Undergoing Cardiopulmonary Bypass: A Review in Search of a Treatment Algorithm. J Extra Corpor Technol. 2008;40(4):257–267. [PMC free article] [PubMed] [Google Scholar]
- 67.Billings FT, Ball SK, Roberts LJ, Pretorius M. Postoperative Acute Kidney Injury is Associated with Hemoglobinemia and an Enhanced Oxidative Stress Response. Free Radic Biol Med. 2011;50(11):1480–1487. doi: 10.1016/j.freeradbiomed.2011.02.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Habib RH, Zacharias A, Schwann TA, et al. Role of hemodilutional anemia and transfusion during cardiopulmonary bypass in renal injury after coronary revascularization: Implications on operative outcome. Crit Care Med. 2005;33(8):1749. doi: 10.1097/01.CCM.0000171531.06133.B0 [DOI] [PubMed] [Google Scholar]
- 69.Hirano Y, Miyoshi Y, Kondo Y, Okamoto K, Tanaka H. Liberal versus restrictive red blood cell transfusion strategy in sepsis or septic shock: a systematic review and meta-analysis of randomized trials. Crit Care. 2019;23(1):262. doi: 10.1186/s13054-019-2543-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Risbano MG, Kanias T, Triulzi D, et al. Effects of Aged Stored Autologous Red Blood Cells on Human Endothelial Function. Am J Respir Crit Care Med. 2015;192(10):1223–1233. doi: 10.1164/rccm.201501-0145OC [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Berra L, Pinciroli R, Stowell CP, et al. Autologous Transfusion of Stored Red Blood Cells Increases Pulmonary Artery Pressure. Am J Respir Crit Care Med. 2014;190(7):800–807. doi: 10.1164/rccm.201405-0850OC [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Shapiro NI, Schuetz P, Yano K, et al. The association of endothelial cell signaling, severity of illness, and organ dysfunction in sepsis. Crit Care. 2010;14(5):R182. doi: 10.1186/cc9290 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Kumar S, Molitoris BA. Renal Endothelial Injury and Microvascular Dysfunction in Acute Kidney Injury. Semin Nephrol. 2015;35(1):96–107. doi: 10.1016/j.semnephrol.2015.01.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.WHO Human Genomics in Global Health. Genes and human diseases. World Health Organization. http://www.who.int/genomics/public/geneticdiseases/en/. Accessed October 13, 2019. [Google Scholar]
- 75.Rees DC, Williams TN, Gladwin MT. Sickle-cell disease. The Lancet. 2010;376(9757):2018–2031. doi: 10.1016/S0140-6736(10)61029-X [DOI] [PubMed] [Google Scholar]
- 76.Baddam S, Aban I, Hilliard L, Howard T, Askenazi D, Lebensburger JD. Acute kidney injury during pediatric sickle cell vaso-occlusive pain crisis. Pediatr Nephrol Berl Ger. 2017;32(8):1451–1456. doi: 10.1007/s00467-017-3623-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Sklar AH, Perez JC, Harp RJ, Caruana RJ. Acute renal failure in sickle cell anemia. Int J Artif Organs. 1990;13(6):347–351. [PubMed] [Google Scholar]
- 78.Saraf SL, Zhang X, Kanias T, et al. Haemoglobinuria is associated with chronic kidney disease and its progression in patients with sickle cell anaemia. Br J Haematol. 2014;164(5):729–739. doi: 10.1111/bjh.12690 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Prout WT. Practical Notes On The Treatment Of Blackwater Fever. Br Med J. 1907;2(2445):1324–1327. [Google Scholar]
- 80.Kristiansen M, Graversen JH, Jacobsen C, et al. Identification of the haemoglobin scavenger receptor. Nature. 2001;409(6817):198–201. doi: 10.1038/35051594 [DOI] [PubMed] [Google Scholar]
- 81.Philip AGS, Hewitt JR. Early Diagnosis of Neonatal Sepsis. Pediatrics. 1980;65(5):1036–1041. [PubMed] [Google Scholar]
- 82.Janz DR, Bastarache JA, Sills G, et al. Association between haptoglobin, hemopexin and mortality in adults with sepsis. Crit Care. 2013;17:R272. doi: 10.1186/cc13108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Nomura K, Kurosawa H, Hashimoto K, et al. Hemolytic Renal Damage during Cardiopulmonary Bypass and the Preventive Effect of Haptoglobin. Jpn J Cardiovasc Surg. 1993;22(5):404–408. doi: 10.4326/jjcvs.22.404 [DOI] [Google Scholar]
- 84.Kubota K, Egi M, Mizobuchi S. Haptoglobin Administration in Cardiovascular Surgery Patients: Its Association With the Risk of Postoperative Acute Kidney Injury. Anesth Analg. 2017;124(6):1771. doi: 10.1213/ANE.0000000000002093 [DOI] [PubMed] [Google Scholar]
- 85.Delanghe JR, Langlois MR. Hemopexin: a review of biological aspects and the role in laboratory medicine. Clin Chim Acta. 2001;312(1):13–23. doi: 10.1016/S0009-8981(01)00586-1 [DOI] [PubMed] [Google Scholar]
- 86.Hvidberg V, Maniecki MB, Jacobsen C, Højrup P, Møller HJ, Moestrup SK. Identification of the receptor scavenging hemopexin-heme complexes. Blood. 2005;106(7):2572–2579. doi: 10.1182/blood-2005-03-1185 [DOI] [PubMed] [Google Scholar]
- 87.Miller YI, Smith A, Morgan WT, Shaklai N. Role of Hemopexin in Protection of Low-Density Lipoprotein against Hemoglobin-Induced Oxidation. Biochemistry. 1996;35(40):13112–13117. doi: 10.1021/bi960737u [DOI] [PubMed] [Google Scholar]
- 88.Smith A, Hunt RC. Hemopexin joins transferrin as representative members of a distinct class of receptor-mediated endocytic transport systems. Eur J Cell Biol. 1990;53(2):234–245. [PubMed] [Google Scholar]
- 89.Tolosano E, Altruda F. Hemopexin: Structure, Function, and Regulation. DNA Cell Biol. 2002;21(4):297–306. doi: 10.1089/104454902753759717 [DOI] [PubMed] [Google Scholar]
- 90.González-Sánchez MI, Manjabacas MC, García-Carmona F, Valero E. Mechanism of Acetaminophen Oxidation by the Peroxidase-like Activity of Methemoglobin. Chem Res Toxicol. 2009;22(11):1841–1850. doi: 10.1021/tx9002512 [DOI] [PubMed] [Google Scholar]
- 91.Szakmany T, Hauser B, Radermacher P. N-acetylcysteine for sepsis and systemic inflammatory response in adults. Cochrane Database Syst Rev 2012;12(9):CD006616. doi: 10.1002/14651858.CD006616.pub2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Heyland D, Muscedere J, Wischmeyer PE, et al. A randomized trial of glutamine and antioxidants in critically ill patients. N Engl J Med. 2013;368(16):1489–1497. doi: 10.1056/NEJMoa1212722 [DOI] [PubMed] [Google Scholar]
- 93.Janz DR, Bastarache JA, Rice TW, et al. Randomized, Placebo-controlled Trial of Acetaminophen for the Reduction of Oxidative Injury in Severe Sepsis: The ACROSS Trial. Crit Care Med 2015;43(3):534–541. doi: 10.1097/CCM.0000000000000718 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Plewes K, Kingston HWF, Ghose A, et al. Acetaminophen as a Renoprotective Adjunctive Treatment in Patients With Severe and Moderately Severe Falciparum Malaria: A Randomized, Controlled, Open-Label Trial. Clin Infect Dis. 2018;67(7):991–999. doi: 10.1093/cid/ciy213 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Simpson SA, Zaccagni H, Bichell DP, et al. Acetaminophen Attenuates Lipid Peroxidation in Children Undergoing Cardiopulmonary Bypass. Pediatr Crit Care Med 2014;15(6):503. doi: 10.1097/PCC.0000000000000149 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Young P, Saxena M, Bellomo R, et al. Acetaminophen for Fever in Critically Ill Patients with Suspected Infection. N Engl J Med. 2015;373(23):2215–2224. doi: 10.1056/NEJMoa1508375 [DOI] [PubMed] [Google Scholar]
- 97.Kuck JL, Bastarache JA, Shaver CM, et al. Ascorbic acid attenuates endothelial permeability triggered by cell-free hemoglobin. Biochem Biophys Res Commun. 2018;495(1):433–437. doi: 10.1016/j.bbrc.2017.11.058 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Marik PE, Khangoora V, Rivera R, Hooper MH, Catravas J. Hydrocortisone, Vitamin C, and Thiamine for the Treatment of Severe Sepsis and Septic Shock: A Retrospective Before-After Study. Chest. 2017;151(6):1229–1238. doi: 10.1016/j.chest.2016.11.036 [DOI] [PubMed] [Google Scholar]
- 99.Fowler AA, Truwit JD, Hite RD, et al. Effect of Vitamin C Infusion on Organ Failure and Biomarkers of Inflammation and Vascular Injury in Patients With Sepsis and Severe Acute Respiratory Failure: The CITRIS-ALI Randomized Clinical Trial. JAMA. 2019;322(13):1261–1270. doi: 10.1001/jama.2019.11825 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Ritter C, Andrades ME, Reinke A, Menna-Barreto S, Moreira JCF, Dal-Pizzol F. Treatment with N-acetylcysteine plus deferoxamine protects rats against oxidative stress and improves survival in sepsis. Crit Care Med 2004;32(2):342. doi: 10.1097/01.CCM.0000109454.13145.CA [DOI] [PubMed] [Google Scholar]