

Abstract
Background
Jellyfish belong to the phylum Cnidaria, which occupies an important phylogenetic location in the early-branching Metazoa lineages. The jellyfish Rhopilema esculentum is an important fishery resource in China. However, the genome resource of R. esculentum has not been reported to date.
Findings
In this study, we constructed a chromosome-level genome assembly of R. esculentum using Pacific Biosciences, Illumina, and Hi-C sequencing technologies. The final genome assembly was ∼275.42 Mb, with a contig N50 length of 1.13 Mb. Using Hi-C technology to identify the contacts among contigs, 260.17 Mb (94.46%) of the assembled genome were anchored onto 21 pseudochromosomes with a scaffold N50 of 12.97 Mb. We identified 17,219 protein-coding genes, with an average CDS length of 1,575 bp. The genome-wide phylogenetic analysis indicated that R. esculentum might have evolved more slowly than the other scyphozoan species used in this study. In addition, 127 toxin-like genes were identified, and 1 toxin-related “hub” was found by a genomic survey.
Conclusions
We have generated a chromosome-level genome assembly of R. esculentum that could provide a valuable genomic background for studying the biology and pharmacology of jellyfish, as well as the evolutionary history of Cnidaria.
Keywords: jellyfish, Rhopilema esculentum, whole-genome sequencing, chromosome-level assembly, toxin-like genes
Data Description
Background
Jellyfish belong to the phylum Cnidaria, which occupies an important phylogenetic location and is one of the earliest branching Metazoa lineages [1]. The jellyfish Rhopilema esculentum (Kishinouye, 1891), an edible species in the class Scyphozoa (also named "true jellyfish"), is widely distributed in the seas around China, Japan, and Korea [2], and it is one of the most abundant fishery animals in these locations. R. esculentum has been exploited as food for thousands of years and has been gaining more attention recently because of its pharmacological properties [3]. In contrast to many other jellyfish species that have drawn public attention because of their harmful blooms [4], the population of R. esculentum has declined in recent years as a result of overfishing [2]. The stock enhancement and aquaculture of R. esculentum have been initiated to meet the expanding market demand, which accounts for ∼82,280 tons per year, generating US $122,800,000 worth of profit per year for the Chinese economy [5]. The lack of genomic resource has limited the phylogenetic study of jellyfish and the investigation of their many specific characteristics. Recently, several genome assemblies have been reported for the medusozoan species, including the moon jellyfish (Aurelia aurita) [6, 7], the giant Nomura's jellyfish (Nemopilema nomurai) [8], the upside-down jellyfish (Cassiopea xamachana) [9], the hydrozoan jellyfish Clytia hemisphaerica [10], Morbakka virulenta [7], Alatina alata [9], and Calvadosia cruxmelitensis [9]. However, no chromosome-level reference genome has been reported for the class Scyphozoa, and at present, there is very limited information about the genome architecture of R. esculentum. In the present study, we sequenced the chromosome-level genome of R. esculentum and assembled and annotated it to improve our understanding of the evolutionary and pharmacology characteristics of jellyfish.
Sample and sequencing
One cultured R. esculentum (NCBI:txid499914) individual was collected from Yingkou, Liaoning Province, China (Fig. 1). After starving for 2 days, the epidermis tissue was sampled, and genomic DNA was extracted using a TIANamp Marine Animal DNA Kit (Tiangen, Beijing, China) and then directly used for the genomic DNA sequencing. The genomic DNA was sheared using a sonication device, and the resulting fragments were used for the construction of short-insert paired-end (PE) libraries. Short-insert libraries with a size of 500 bp were constructed in accordance with the instructions in the Illumina library preparation kit. All libraries were sequenced on an Illumina HiSeq2500 platform (Illumina, San Diego, CA, USA) with 150-bp PE. In total, ∼22.6 Gb (80×) of raw data were generated, and 20.03 Gb (71×) of clean data were filtered by FastQC (FastQC, RRID:SCR_014583) v0.11.2 (Supplementary Table S1). The genomic DNA used for sequencing was also sheared to yield ∼20 kb fragments for the construction of Pacific Biosciences (PacBio) libraries. DNA fragments of <7 kb were filtered using BluePippin (Sage Science, Beverly, MA, USA). The filtered DNA was then converted into the proprietary SMRTbell library using the PacBio DNA Template Preparation Kit. In total, 39.76 Gb (140×) of quality-filtered data with a mean length of 7196 bp were obtained from the PacBio Sequel platform (Supplementary Table S1).
Figure 1:

Picture of the jellyfish R. esculentum captured in Yingkou, Liaoning Province, China.
Genome size and heterozygosity estimation
The distribution of k-mer frequency, also known as the k-mer spectrum, is widely used for the estimation of genome size. We used a jellyfish software based on a k-mer distribution [11] to estimate the genome size with high-quality reads >Q20 from short-insert libraries (500 bp). We obtained a k-mer (k = 17) depth distribution from the jellyfish analysis and clearly observed the peak depth from the distribution data. We obtained a genome size estimation of 290 Mb and a heterozygosity of 1.68% by GenomeScope v1.0.0 (Supplementary Fig. S1) [12]. A total of 54.4% of the genome was predicted to be non-repetitive sequences.
Genome assembly and annotation
In the present study, the long reads of PacBio sequencing data were used to solve the high level of heterozygosity, which is one of the main challenges in the assembly of marine invertebrate genomes [13, 14]. The genome assembly was performed using the software wtdbg2 with default parameters [15]. The assembly sequences were then polished using Quiver (SMRT Analysis v2.3.0) with default parameters. To achieve higher continuity and accuracy for the assembled genome, 5 rounds of iterative error correction were performed with the Illumina clean genome data using in-house script. Finally, a genome of 275.42 Mb was assembled, with 760 contigs and a contig N50 size of 1.13 Mb (Table 1 and Supplementary Fig. S2).
Table 1:
Statistics of the assembly and annotation of R. esculentum genome
| Genome feature | Value |
|---|---|
| Genome assembly | |
| Total length (Mb) | 275.42 |
| Contig N50 (Mb) | 1.13 |
| Longest contig (Mb) | 6.59 |
| Contig number | 760 |
| GC content (%) | 36.25 |
| Pseudochromosome number | 21 |
| Scaffold N50 (Mb) | 12.97 |
| Genome annotation | |
| Gene number | 17,219 |
| Gene density (per 100 kb) | 62.52 |
| CDS mean length (bp) | 1,575 |
| Exon mean length (bp) | 198.8 |
| Intron mean length (bp) | 987.2 |
| Exon number per gene | 7.92 |
| Exon GC content (%) | 42.29 |
Both RepeatModeler (RepeatModeler, RRID:SCR_015027) and RepeatMasker (RepeatMasker, RRID:SCR_012954) [16] were used to perform the de novo identification and masking of repeat sequences. To ensure the integrity of the genes in subsequent analysis, all repeat sequences, except for the low-complexity or simple repeats, were masked in this analysis because some of the low-complexity or simple repeats could be found in the genes. Finally, 29.23% of the assembled bases (80,495,815 bp) were masked (Supplementary Table S2). Of these, 9.93% could be annotated with known repeat families, and 19.30% were unclassified repeats.
The identification of protein-coding regions and the prediction of genes were performed using a combination of ab initio prediction, homology-based prediction, and transcriptome-based prediction methods. The ab initio gene prediction was conducted with Augustus (Augustus: Gene Prediction, RRID:SCR_008417) version 2.5.5 [17], GlimmerHMM (GlimmerHMM, RRID:SCR_002654) version 3.0.1 [18], and SNAP15 [19] to predict the coding genes. For the homology-based prediction, homologous proteins of several Cnidarian species (myxosporean [Thelohanellus kitauei], coral [Stylophora pistillata and Orbicella faveolata], hydrozoan [Hydra vulgaris], sea anemone [Exaiptasia pallida], and the Cnidaria EST database) were downloaded from NCBI and aligned with our assembled genome. Then, GeneWise (GeneWise, RRID:SCR_015054) version 2.2.0 [20, 21] was used to generate the gene structures based on the homology alignments. For transcriptome-based prediction, 60 individuals of 4 development periods (scyphistoma, strobili, ephyra, and juvenile medusa) were collected. Five individuals were pooled and 3 replicates were set for each development period analysis. The transcriptome of samples were sequenced using the Illumina HiSeq2500 platform (154.6 Gb clean reads, PE-250) (Supplementary Table S3) and the resulting sequences were mapped to the genome assembly using TopHat (TopHat, RRID:SCR_013035) version 2.0.8 [22]. Cufflinks (Cufflinks, RRID:SCR_014597) version 2.1.1 [23, 24] was then used to identify the spliced transcripts in the gene models. All the gene evidence predicted from the above 3 approaches were integrated by EvidenceModeler (EVM) [25] into a weighted and non-redundant consensus of the gene structures. A total of 17,219 genes, with an average CDS length of 1,575 bp, were finally predicted to be present in the genome of R. esculentum (Table 1). All the gene sequences were searched using BLASTP with an E-value of 1e−5 against several public databases, including NR [26], GO (Supplementary Fig. S3) [27], Swiss [28], KOG (Supplementary Fig. S4) [29], and KEGG [30], to obtain the functional annotation. A total of 16,713 genes (97.1%) were successfully mapped to ≥1 database, and 8,880 genes were annotated in all 4 databases (E-value < 1e−5) (Supplementary Fig. S5).
Quality assessment
We first aligned all the Illumina genome reads against the R. esculentum assembled genome using BWA (BWA, RRID:SCR_010910), version 0.7.17, to evaluate the coverage of the genome. The percentage of aligned reads was estimated to be 99.81%. BUSCO (BUSCO, RRID:SCR_015008), version 3.0.2 [31], was then used to evaluate the integrity of the genome (Supplementary Table S4). The values of core gene estimation were calculated as follows: C: 97.0% (S: 92.1%, D: 5.0%), F: 1.7%, M: 1.3%, n: 303, where C, S, D, F, M, and n indicate complete BUSCOs, complete and single-copy BUSCOs, complete and duplicated BUSCOs, fragmented BUSCOs, missing BUSCOs, and total BUSCO groups searched, respectively (Supplementary Table S5). The results indicated that the assembly covered most of the genetic regions, further confirming the assembly quality of the R. esculentum genome.
Pseudochromosome construction
Hi-C experiments were used for the chromosome assembly of R. esculentum. The whole-body homogenate of 1 R. esculentum was fixed in 1% (vol/vol) formaldehyde and was then used to prepare the Hi-C libraries. Nuclei extraction and permeabilization, chromatin digestion, and proximity-ligation treatments were performed as previously described [32]. The DNA was digested overnight (12 h) with 200 U of the restriction enzyme MboIat 37°C with shaking. The libraries were sequenced on the Illumina X-TEN platform (San Diego, CA, USA) with 2 × 150 bp reads. They were independently analysed in the HiC-Pro pipeline (default parameters and LIGATION_SITE = GATC) [33]. A total of 23.96 Gb of trimmed reads were obtained, accounting for ∼82-fold coverage of the R. esculentum genome. The 3D-DNA was used to assign the order and orientation of each group [34]. The contact maps were plotted using HiCPlotter software [35]. Finally, 260.17 Mb (94.46%) of the assembly was anchored onto 21 pseudochromosomes, which was in agreement with the karyotype (2n = 42) of R. esculentum [36] (Fig. 2, Supplementary Fig. S6, and Supplementary Table S6). This chromosome-level assembly resulted in a scaffold N50 of 12.97 Mb.
Figure 2:

Schematic representation of the genomic characteristics of R. esculentum. Track A: 21 pseudochromosomes of R. esculentum genome (Mb). Track B: Protein-coding genes present in the scaffolds. Red represents genes on forward strand, and green, genes on reverse strand. Track C: Distribution of gene density with sliding windows of 1 Mb. Higher density is shown in darker red color. Track D: Distribution of GC content in the genome. Track E: Distribution of repeats in the genome. Track F: Schematic presentation of major interchromosomal relationships.
Phylogenetic analysis
To examine the evolutionary relationships among R. esculentum and other species, the whole protein sequences of R. esculentum and 12 other species (Supplementary Table S7) were analysed, including species from Ctenophora (ctenophore [Mnemiopsis leidyi]), Porifera (demosponge [Amphimedon queenslandica]), Placozoa (Trichoplax adhaerens), Cnidaria (jellyfish [R. esculentum and A. aurita], Hydrozoa (H. vulgaris), coral (S. pistillata), sea anemone (Nematostella vectensis), Protostomia (Lophotrochozoa [Pacific oyster (Crassostrea gigas)], Ecdysozoa [cladoceran (Daphnia pulex)]), and Deuterostomia (Echinodermata [sea urchin (Strongylocentrotus purpuratus)], Hemichordata [acorn worm (Saccoglossus kowalevskii)], Chordata [zebrafish (Danio rerio)]). All protein models of the 12 other species were obtained from Ensembl or NCBI. Orthologous alignment analysis was performed using OrthoMCL (OrthoMCL DB: Ortholog Groups of Protein Sequences, RRID:SCR_007839) [37]. In detail, the protein-coding genes from the above-sequenced genomes were aligned with each other using the BLASTP program [38]. Similarity in the pair-wise sequence alignments generated by BLASTP was used as distance parameters for gene family clustering by MCL with an inflation value of 1.5.
A set of 32,138 gene families were eventually identified among the other 12 species, of which 2,092 families were present in all 13 species (Fig. 3 and Supplementary Table S8). A total of 335 selected single-copy orthologous genes were aligned using MUSCLE (MUSCLE, RRID:SCR_011812) v3.6 [39] and then concatenated into a single multiple sequence alignment through an in-house Perl script. A maximum likelihood phylogeny was reconstructed using RAxML (RAxML, RRID:SCR_006086) [40] (Fig. 4). The phylogenetic results supported the monophyly of R. esculentum, A. aurita, and H. vulgaris. The PROTGAMMAJTT model was used for RaXML analyses [40]. The divergence times of M. leidyivs A. aurita, S. purpuratusvs A. aurita, and D. reriovs N. vectensis were retrieved from the time tree [41] and used as the fossil calibration. R8s was used to calculate the divergence time of each node in the phylogenetic tree [42]. We dated the divergence time of R. esculentum and H. vulgaris to ∼501.71 million years ago, consistent with previous studies [43]. To compare the jellyfish genomic traits with those of the other 12 species, we performed a comparative genomic analysis for all 13 species using CAFE software (Supplementary Table S9) [44]. Twenty-seven gene families were found to be significantly expanded and another 27 gene families were found to be significantly contracted in R. esculentum (P < 0.05) (Supplementary Tables S10 and S11). Interestingly, the gene families enriched in the GO category of transmembrane transport were significantly expanded, and the relative GO sub-categories included drug transmembrane transport, drug transmembrane transporter activity, ion transmembrane transporter activity, and amino acid transmembrane transporter activity. The action of venom, an important characteristic of jellyfish species, may contribute to gene expansion in the transmembrane transport category [45, 46].
Figure 3:
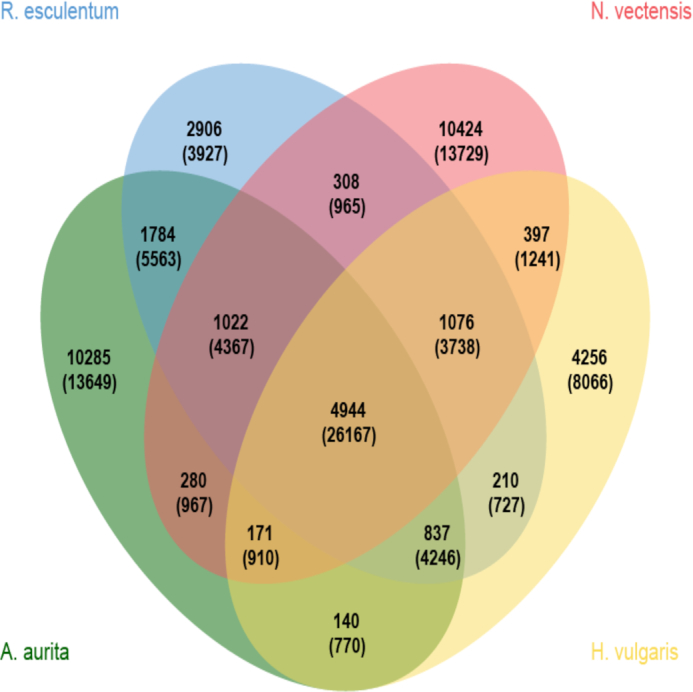
Venn diagram of the protein-coding orthologues shared among R. esculentum, H. vulgaris, N. vectensis, and A. aurita. Each number represents the number of gene families, and the number in parentheses is the number of genes.
Figure 4:

Phylogenetic analysis of R. esculentum and other metazoan species. The numbers of gene gains (plus sign) and gene losses (minus sign) are shown on the branches, which are also displayed as pie plots (green: gene gain; red: gene loss; blue: gene persistence). The divergence times are dated and displayed below the phylogenetic tree.
A comparative genomic analysis was performed for the 4 jellyfish species in the class Scyphozoa (including R. esculentum, A. aurita, N. nomurai, and C. xamachana) and H. vulgaris (used as outgroup, and to calculate the divergence time). A total of 244 unique gene families were identified in R. esculentum using BLASTP with an E-value of 1e−5 in the NR database. It was surprising that more than half of those (136 unique gene families) were best annotated with Anthozoa species in the NR database. It was suggested that the 136 unique gene families were not from the split of R. esculentum but from the ancestor of Anthozoa and Scyphozoa. This result implied that some gene families that were possessed by the last common ancestor of Anthozoans and Scyphozoans were kept by the Anthozoan species and R. esculentum but were lost in A. aurita, N. nomurai, C. xamachana, and H. vulgaris. This was also supported by the phylogenetic analysis of the 13 species, in which R. esculentum was found to exhibit fewer gene gains (331) and fewer gene losses (294) compared with H. vulgaris (513 gains and 666 losses) and A. aurita (696 gains and 962 losses) (Fig. 4). This indicated that R. esculentum might have evolved more slowly than the other scyphozoan species used in this study.
Analysis of toxin-like genes in jellyfish
Jellyfish is one important lineage of extant venomous animals [47, 48]. The venom is injected into the victim or prey when triggered to discharge. Jellyfish stings are dangerous to swimmers and fishermen because they can cause local oedema, vesicular eruption, shock, and even death [49, 50]. The venom of jellyfish consists of polypeptides, enzymes, and some non-protein bioactive components [48], such as neurotoxins, myotoxins, hemolytic toxins, and cardiotoxins [51]. The venom constituents of jellyfish have been investigated by pharmacological studies in recent years. Omics analyses, especially transcriptomic and proteomic analyses, have been used to conduct large-scale identification of toxins and related genes from jellyfish, and many putative toxins have been identified [47, 50–53]. However, owing to the limitation of genome information and sampling [50], the overall understanding of toxin-like genes is limited, which may be responsible for the lack of consistency among the results obtained from previous studies [53]. Here, we conducted a genomic survey of toxin-like genes in the assembled R. esculentum genome.
In step 1, all the genes of R. esculentum were screened using BLASTP with a cutoff E-value of 1e−10 against the database of the animal toxin annotation project (Tox-Prot) in UniProt. In step 2, according to the best hits of gene annotations of NR, Uniprot, and Tox-Prot, the genes that were consistently annotated as toxin-like genes were then chosen. In step 3, to make the pool of venom-related genes more complete, we checked all the gene annotations of the jellyfish and picked out the genes where the annotations were consistent with the annotations in the database of Tox-Prot and were not identified in the first 2 steps. These genes were also considered as toxin-like genes.
There were 127 toxin-like genes identified, including 60 metalloproteinases, 18 phospholipases, 13 nucleases and nucleotidases, 13 peptidases and inhibitors, 12 genes with toxin activity, and 11 other venom-related genes (Table 2). It is not surprising that metalloproteases were the most abundant group of toxins because they are widely considered to be a key toxic component in various venomous animals, such as spiders [54], snakes [55], scorpions [56], and jellyfish [50, 57]. Metalloprotease can interfere with blood coagulation and induce necrosis. Metalloprotease is always associated with the symptoms of stings, such as swelling, myonecrosis, inflammation, and blister formation [48, 53].
Table 2:
Summary of all the identified toxin-like genes from the genome of the jellyfish R. esculentum
| Gene | Copy No. | Description | Family | Reported in jellyfish |
|---|---|---|---|---|
| Phospholipase A2 | 9 | Phospholipase A2 activity | Phospholipase A2 family | Yes |
| Acidic phospholipase A2 PA4 | 4 | Phospholipase A2 activity | Phospholipase A2 family | Yes |
| Phospholipase A2 isozymes PA3A/PA3B/PA5 | 4 | Phospholipase A2 activity | Phospholipase A2 family | Yes |
| Putative phospholipase B-like 2 | 1 | Hydrolase activity | Phospholipase B-like family | Yes |
| Zinc metalloproteinase nas | 39 | Metalloendopeptidase activity | Yes | |
| Disintegrin and metalloproteinase | 21 | Metalloendopeptidase activity | Yes | |
| Ectonucleotide pyrophosphatase/phosphodiesterase | 8 | Nuclease activity | Nucleotide pyrophosphatase/phosphodiesterase family | Yes |
| 5′-Nucleotidase | 5 | 5′-Nucleotidase activity | 5′-Nucleotidase family | Yes |
| Serine carboxypeptidase | 1 | Serine-type carboxypeptidase activity | Peptidase S10 family | Yes |
| Serine protease | 7 | Serine-type endopeptidase activity | Peptidase S1 family | Yes |
| Prothrombin | 2 | Serine-type endopeptidase activity | Peptidase S1 family | Yes |
| Dipeptidyl peptidase 9 | 1 | Serine-type peptidase activity | Peptidase S9B family | Yes |
| Kunitz-type serine protease inhibitor | 1 | Serine-type endopeptidase inhibitor activity | Venom Kunitz-type family | Yes |
| Cystatin | 1 | Cysteine-type endopeptidase inhibitor activity | Cystatin family | Yes |
| Plancitoxin-1 | 3 | Toxin activity | DNase II family | Yes |
| Ryncolin | 6 | Toxin activity | Ficolin lectin family | Yes |
| Toxin TX | 2 | Toxin activity | Jellyfish toxin family | Yes |
| Trpa1 | 1 | Toxin activity | (High similarity with α-latrotoxin-Lt1a) | Yes |
| Peroxiredoxin-4 | 2 | Protein homodimerization activity | Peroxiredoxin family | Yes |
| Glutaminyl-peptide cyclotransferase-like protein | 1 | Glutaminyl-peptide cyclotransferase activity | Glutaminyl-peptide cyclotransferase family | Yes |
| Lysosomal acid lipase/cholesteryl ester hydrolase | 1 | Lipase activity | Lipase family | Yes |
| Trehalase | 1 | α-trehalase activity | Glycosyl hydrolase 37 family | Yes |
| Acetylcholinesterase | 1 | Acetylcholinesterase activity | Type-B carboxylesterase/lipase family | Yes |
| Lysosomal acid phosphatase | 1 | Acid phosphatase activity | Histidine acid phosphatase family | No |
| Reticulocalbin | 1 | Calcium ion binding | CREC family | No |
| Translationally controlled tumour protein homolog | 1 | Calcium ion binding | TCTP family | Yes |
| Hyaluronidase-1 | 2 | Hyaluronan synthase activity | Glycosyl hydrolase 56 family | Yes |
Full gene names are provided in Supplementary Table S12.
Phospholipases comprise the second most abundant group of toxins. Various forms of phospholipases have been identified, such as phospholipase A2, acidic phospholipase A2 PA4, phospholipase A2 isozymes PA3A/PA3B/PA5, and putative phospholipase B-like 2. Phospholipases are ubiquitous in the venom of many poisonous animals and they exhibit various degrees of toxicity, among which hemolytic activity is the most striking [51]. High levels of phospholipase A2 activity have been observed in the tentacles of scyphozoan and cubozoan species [51, 58] and are presumably involved in defence and in the capturing of prey [48]. In the present study, 9 copies of phospholipase were found in a tandem fashion located on 3 loci of the genome.
Two copies of “jellyfish toxin,” also called "cubozoan-related porins," were also found. The jellyfish toxins have been observed in high abundance in cubozoan venoms [52] and they have also been reported in other medusozoans, such as Scyphozoans [51], Hydrozoans [59], and Anthozoans [60]. They are potent and rapid-acting toxins, having both hemolytic and pore-forming activities [48, 51]. Compared with the high abundance in cubozoans, where as many as 15 isoforms of the jellyfish toxin were found in Chironex fleckeri, the relatively fewer copies found in scyphozoan species may be linked to the less severe stings inflicted by these species of jellyfish [52].
Two new toxins were identified: reticulocalbin and lysosomal acid phosphatase. These toxins have not been reported in jellyfish. Reticulocalbin is known to have calcium ion–binding activity. Its role in venom is still unclear, although it was speculated to play a potentially unknown role in prey incapacitation by binding with phospholipase A2 [61, 62]. Lysosomal acid phosphatase is an orthologue of venom acid phosphatase, which is an acidic heat-labile protein with carbohydrate IgE binding epitopes [63]. It is mostly found in the honeybee and has been implicated in allergic reaction [63–65]. The discovery of these toxin-coding genes in R. esculenum would add to a growing understanding of the composition of jellyfish venoms. When compared with the venom composition of the jellyfish N. nomurai (also named Stomolophus meleagris), a species closely related to R. esculentum, it was noted that 2 types of main toxins were lost in R. esculentum, including a serine protease inhibitor (only 1 copy found) and a potassium channel inhibitor ShK [50]. They are known to block the activities of trypsin and plasmin and to function as neurotoxins [50]. The different compositions of the venom may account for the different symptoms after the sting. For instance, R. esculentum sting always causes strong pruritus compared with stings of other jellyfish species [66].
Interestingly, 8 toxin-like genes were located closely on contig 521 as a “hub,” including 4 PLA2s, 2 ENPP5s, 1 TRPA1, and 1 SLC47A1 (Table 3). The functions of toxin-like genes in the hub included phospholipase A2 activity, nuclease activity, toxin activity, and toxin extrusion. In addition, according to the chromosome-level analysis, contig 747 and contig 751 were located on the 2 sides of contig 521 and contained 5 and 3 toxin-like genes, respectively. These 3 contigs were arranged in chromosome 7 (3,691,690–13,486,489 bp) as a head-to-tail tandem, forming a bigger hub. The neighbouring genes have been shown to co-express rather than express independently [67, 68]. Thus, we speculated that contig747-contig521-contig751 tandem on chromosome 7 may play important roles in the formation and function of venom in R. esculentum. Further studies are needed to clarify their specific functions.
Table 3:
Structure of the toxin-related hub on contig 521

|
In summary, we have sequenced and assembled the genome of R. esculentum at the chromosome level. The obtained genome data will provide a valuable resource for conducting further study on R. esculentum and other Cnidarian species.
Availability of Supporting Data and Materials
The raw genome sequencing data obtained by Illumina and PacBio platform are available via NCBI with accession Nos. SRR8617500 and SRR8617499, respectively (BioProject accession No. PRJNA523480). The raw sequencing data of the transcriptome are available via NCBI with accession No. SRR8401786-SRR8401797 (BioProject accession No. PRJNA512552). Supporting data are available via the GigaScience GigaDB repository [69].
Additional Files
Supplementary Fig. S1: k-mer estimation of the genome size of R. esculentum.
Supplementary Fig. S2: Contig length distribution of the assembled genome of R. esculentum.
Supplementary Fig. S3: GO analysis and functional classification of the protein coding genes in R. esculentum.
Supplementary Fig. S4: KOG analysis and functional classification of the protein coding genes in R. esculentum.
Supplementary Fig. S5: Venn diagram of the statistics of the functional annotation.
Supplementary Fig. S6: Interaction frequency distribution of Hi-C links among chromosomes of R. esculentum.
Supplementary Table S1: Statistics of the clean data of Illumina and PacBio sequencing for R. esculentum.
Supplementary Table S2: Statistics of the repeat elements of R. esculentum genome assembly indicated by both RepeatModeler and RepeatMasker software.
Supplementary Table S3: Summary of the transcriptome sequenced data of R. esculentum.
Supplementary Table S4: Core gene estimation for the R. esculentum genome assembly obtained using BUSCO.
Supplementary Table S5: BUSCO scores of gene model and trinity assembly of R. esculentum.
Supplementary Table S6: Quantity of the contigs anchored with Hi-C.
Supplementary Table S7: Information of the 12 representative species that were used in the analysis of evolutionary relationships.
Supplementary Table S8: Summary of the orthologous gene clusters analysed in 13 species that were used in the analysis of evolutionary relationships.
Supplementary Table S9: Gene family analysis performed with CAFE.
Supplementary Table S10: Annotations of the significantly expanded gene families of R. esculentum.
Supplementary Table S11: Annotations of the significantly contracted gene families of R. esculentum.
Supplementary Table S12: Abbreviations and full names of the genes used in this study.
Joseph F. Ryan -- 10/31/2019 Reviewed
Lucas Leclère -- 11/10/2019 Reviewed
Lucas Leclère -- 3/12/2020 Reviewed
David A. Gold -- 11/19/2019 Reviewed
Competing Interests
The authors declare that they have no competing interests.
Abbreviations
BLAST: Basic Local Alignment Search Tool; bp: base pairs; BUSCO: Benchmarking Universal Single-Copy Orthologs; BWA: Burrows-Wheeler Aligner; CAFE: computational analysis of gene family evolution; CDS: coding domain sequence; Gb: gigabase pairs; GC: guanine-cytosine; GO: Gene Ontology; kb: kilobase pairs; KEGG: Kyoto Encyclopedia of Genes and Genomes; KOG: Eukaryotic Orthologous Groups; Mb: megabase pairs; NCBI: National Center for Biotechnology Information; NR: Non-Redundant database; PacBio: Pacific Biosciences; PE: paired-end; RAxML: Randomized Axelerated Maximum Likelihood; tRNA: transfer RNA.
Ethics Statement
This study was approved by the Animal Care and Use Committee of Liaoning Ocean and Fisheries Science Research Institute. This study did not involve endangered or protected species.
Authors' Contributions
Z.Z. and Yunfeng Li designed the project. M.T. and Yulong Li collected the samples. Y.P., C.H., and Y.D. extracted the genomic DNA. L.G., Y.S., and Y.P. participated in data analyses. L.G. and Y.S. wrote the manuscript. All authors have read and approved the final manuscript.
Funding
This work was supported by the National Natural Science Foundation of China (31,302,173; 31,602,156; 31,602,155); the Science and Technology Program of Liaoning Province, China (2,013,203,001); the Natural Science Foundation of Liaoning Province, China (20,180,551,158); the Scientific Research Program of Ocean and Fisheries Administration of Liaoning Province, China (201,827); and Liaoning Science Public Welfare Research Fund Project (20,180,015).
References
- 1. Dunn CW, Hejnol A, Matus DQ, et al.. Broad phylogenomic sampling improves resolution of the animal tree of life. Nature. 2008;452(7188):745. [DOI] [PubMed] [Google Scholar]
- 2. Dong Z, Liu D, Keesing JK. Contrasting trends in populations of Rhopilema esculentum and Aurelia aurita in Chinese waters. In: Pitt KA, Lucas CH, eds. Jellyfish Blooms. Springer; 2014:207–18. [Google Scholar]
- 3. Zhuang Y, Hou H, Zhao X, et al.. Effects of collagen and collagen hydrolysate from jellyfish (Rhopilema esculentum) on mice skin photoaging induced by UV irradiation. J Food Sci. 2009;74(6):H183–H8. [DOI] [PubMed] [Google Scholar]
- 4. Dong Z, Liu D, Keesing JK. Jellyfish blooms in China: dominant species, causes and consequences. Mar Pollut Bull. 2010;60(7):954–63. [DOI] [PubMed] [Google Scholar]
- 5. Ministry of Agriculture Bureau of Fisheries. 2018 China Fisheries Statistical Yearbook. Beijing: China Agriculture Publishing Company; 2018. [Google Scholar]
- 6. Gold DA, Katsuki T, Li Y, et al.. The genome of the jellyfish Aurelia and the evolution of animal complexity. Nat Ecol Evol. 2019;3(1):96. [DOI] [PubMed] [Google Scholar]
- 7. Khalturin K, Shinzato C, Khalturina M, et al.. Medusozoan genomes inform the evolution of the jellyfish body plan. Nat Ecol Evol. 2019;3(5):811. [DOI] [PubMed] [Google Scholar]
- 8. Kim H-M, Weber JA, Lee N, et al.. The genome of the giant Nomura's jellyfish sheds light on the early evolution of active predation. BMC Biol. 2019;17(1):28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Ohdera A, Ames CL, Dikow RB, et al.. Box, stalked, and upside-down? Draft genomes from diverse jellyfish (Cnidaria, Acraspeda) lineages: A latina alata (Cubozoa), Calvadosia cruxmelitensis (Staurozoa), and Cassiopea xamachana (Scyphozoa). GigaScience. 2019;8(7), doi: 10.1093/gigascience/giz069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Leclère L, Horin C, Chevalier S, et al.. The genome of the jellyfish Clytia hemisphaerica and the evolution of the cnidarian life-cycle. Nat Ecol Evol. 2019;3(5):801. [DOI] [PubMed] [Google Scholar]
- 11. Marçais G, Kingsford C. A fast, lock-free approach for efficient parallel counting of occurrences of k-mers. Bioinformatics. 2011;27(6):764–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Vurture GW, Sedlazeck FJ, Nattestad M, et al.. GenomeScope: fast reference-free genome profiling from short reads. Bioinformatics. 2017;33(14):2202–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Zhang X, Sun L, Yuan J, et al.. The sea cucumber genome provides insights into morphological evolution and visceral regeneration. PLoS Biol. 2017;15(10):e2003790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Zhang G, Fang X, Guo X, et al.. The oyster genome reveals stress adaptation and complexity of shell formation. Nature. 2012;490(7418):49–54. [DOI] [PubMed] [Google Scholar]
- 15.wtdbg2. https://github.com/ruanjue/wtdbg2. Accessed date: 2018-10. [Google Scholar]
- 16.RepeatMasker. http://www.repeatmasker.org. Accessed date: 2018-10. [Google Scholar]
- 17. Stanke M, Diekhans M, Baertsch R, et al.. Using native and syntenically mapped cDNA alignments to improve de novo gene finding. Bioinformatics. 2008;24(5):637–44. [DOI] [PubMed] [Google Scholar]
- 18. Majoros WH, Pertea M, Salzberg SL. TigrScan and GlimmerHMM: two open source ab initio eukaryotic gene-finders. Bioinformatics. 2004;20(16):2878–9. [DOI] [PubMed] [Google Scholar]
- 19. Korf I. Gene finding in novel genomes. BMC Bioinformatics. 2004;5(1):59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Birney E, Durbin R. Using GeneWise in the Drosophila annotation experiment. Genome Res. 2000;10(4):547–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Birney E, Clamp M, Durbin R. GeneWise and genomewise. Genome Res. 2004;14(5):988–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Trapnell C, Pachter L, Salzberg SL. TopHat: discovering splice junctions with RNA-Seq. Bioinformatics. 2009;25(9):1105–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Cufflinks. http://cufflinks.cbcb.umd.edu/. Accessed date: 2018-11. [Google Scholar]
- 24. Trapnell C, Roberts A, Goff L, et al.. Differential gene and transcript expression analysis of RNA-seq experiments with TopHat and Cufflinks. Nat Protoc. 2012;7(3):562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Haas BJ, Salzberg SL, Zhu W, et al.. Automated eukaryotic gene structure annotation using EVidenceModeler and the Program to Assemble Spliced Alignments. Genome Biol. 2008;9(1):1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Benson DA, Karsch-Mizrachi I, Lipman DJ, et al.. GenBank. Nucleic Acids Res. 2005;33(suppl 1):D34–D8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Gene Ontology Consortium. Gene Ontology annotations and resources. Nucleic Acids Res. 2012;41(D1):D530–D5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Bairoch A, Apweiler R. The SWISS-PROT protein sequence database and its supplement TrEMBL in 2000. Nucleic Acids Res. 2000;28(1):45–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Tatusov RL, Fedorova ND, Jackson JD, et al.. The COG database: an updated version includes eukaryotes. BMC Bioinformatics. 2003;4(1):41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Kanehisa M, Goto S. KEGG: Kyoto Encyclopedia of Genes and Genomes. Nucleic Acids Res. 2000;28(1):27–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Waterhouse RM, Seppey M, Simão FA, et al.. BUSCO applications from quality assessments to gene prediction and phylogenomics. Mol Biol Evol. 2017;35(3):543–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Zhu W, Hu B, Becker C, et al.. Altered chromatin compaction and histone methylation drive non-additive gene expression in an interspecific Arabidopsis hybrid. Genome Biol. 2017;18(1):157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Servant N, Varoquaux N, Lajoie BR, et al.. HiC-Pro: an optimized and flexible pipeline for Hi-C data processing. Genome Biol. 2015;16:259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Dudchenko O, Batra SS, Omer AD, et al.. De novo assembly of the Aedes aegypti genome using Hi-C yields chromosome-length scaffolds. Science. 2017;356(6333):92–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Akdemir KC, Chin L. HiCPlotter integrates genomic data with interaction matrices. Genome Biol. 2015;16(1):198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Guo P. The karyotype of Rhopilcma esculenta. J Fish China. 1994;18(3):253–5. [Google Scholar]
- 37. Li L, Stoeckert CJ, Roos DS. OrthoMCL: identification of ortholog groups for eukaryotic genomes. Genome Res. 2003;13(9):2178–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Altschul SF, Madden TL, Schäffer AA, et al.. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 1997;25(17):3389–402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Edgar RC. MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004;32(5):1792–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Stamatakis A. RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics. 2014;30(9):1312–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.TimeTree. http://www.timetree.org. Accessed date: 2019-05. [Google Scholar]
- 42. Sanderson MJ. r8s: inferring absolute rates of molecular evolution and divergence times in the absence of a molecular clock. Bioinformatics. 2003;19(2):301–2. [DOI] [PubMed] [Google Scholar]
- 43. Park E, Hwang D-S, Lee J-S, et al.. Estimation of divergence times in cnidarian evolution based on mitochondrial protein-coding genes and the fossil record. Mol Phylogenet Evol. 2012;62(1):329–45. [DOI] [PubMed] [Google Scholar]
- 44. De Bie T, Cristianini N, Demuth JP, et al.. CAFE: a computational tool for the study of gene family evolution. Bioinformatics. 2006;22(10):1269–71. [DOI] [PubMed] [Google Scholar]
- 45. Grishin EV. Neurotoxin from black widow spider venom. Structure and function. Adv Exp Med Biol. 1996;391:231–6. [DOI] [PubMed] [Google Scholar]
- 46. Meldolesi J, Scheer H, Madeddu L, et al.. Mechanism of action of α-latrotoxin: the presynaptic stimulatory toxin of the black widow spider venom. Trends Pharmacol Sci. 1986;7:151–5. [Google Scholar]
- 47. Jaimes-Becerra A, Chung R, Morandini AC, et al.. Comparative proteomics reveals recruitment patterns of some protein families in the venoms of Cnidaria. Toxicon. 2017;137:19–26. [DOI] [PubMed] [Google Scholar]
- 48. Jouiaei M, Yanagihara A, Madio B, et al.. Ancient venom systems: a review on Cnidaria toxins. Toxins. 2015;7(6):2251–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Lee H, Jung E, Kang C, et al.. Scyphozoan jellyfish venom metalloproteinases and their role in the cytotoxicity. Toxicon. 2011;58(3):277–84. [DOI] [PubMed] [Google Scholar]
- 50. Li R, Yu H, Xue W, et al.. Jellyfish venomics and venom gland transcriptomics analysis of Stomolophus meleagris to reveal the toxins associated with sting. J Proteomics. 2014;106:17–29. [DOI] [PubMed] [Google Scholar]
- 51. Liu G, Zhou Y, Liu D, et al.. Global transcriptome analysis of the tentacle of the jellyfish Cyanea capillata using deep sequencing and expressed sequence tags: Insight into the toxin- and degenerative disease-related transcripts. PLoS One. 2015;10(11):e0142680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Brinkman DL, Jia X, Potriquet J, et al.. Transcriptome and venom proteome of the box jellyfish Chironex fleckeri. BMC Genomics. 2015;16(1):407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Li R, Yu H, Yue Y, et al.. Combined proteomics and transcriptomics identifies sting-related toxins of jellyfish Cyanea nozakii. J Proteomics. 2016;148:57–64. [DOI] [PubMed] [Google Scholar]
- 54. Trevisan-Silva D, Gremski LH, Chaim OM, et al.. Astacin-like metalloproteases are a gene family of toxins present in the venom of different species of the brown spider (genus Loxosceles). Biochimie. 2010;92(1):21–32. [DOI] [PubMed] [Google Scholar]
- 55. Markland Jr FS, Swenson S. Snake venom metalloproteinases. Toxicon. 2013;62:3–18. [DOI] [PubMed] [Google Scholar]
- 56. Brazón J, Guerrero B, D'Suze G, et al.. Fibrin(ogen)olytic enzymes in scorpion (Tityus discrepans) venom. Comp Biochem Physiol B Biochem Mol Biol. 2014;168:62–9. [DOI] [PubMed] [Google Scholar]
- 57. Jouiaei M, Casewell NR, Yanagihara AA, et al.. Firing the sting: chemically induced discharge of cnidae reveals novel proteins and peptides from box jellyfish (Chironex fleckeri) venom. Toxins. 2015;7(3):936–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Nevalainen TJ, Peuravuori HJ, Quinn RJ, et al.. Phospholipase A2 in cnidaria. Comp Biochem Physiol B Biochem Mol Biol. 2004;139(4):731–5. [DOI] [PubMed] [Google Scholar]
- 59. Brinkman DL, Konstantakopoulos N, McInerney BV, et al.. Chironex fleckeri (box jellyfish) venom proteins: expansion of a cnidarian toxin family that elicits variable cytolytic and cardiovascular effects. J Biol Chem. 2014;289(8):4798–812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Jouiaei M, Sunagar K, Federman Gross A, et al.. Evolution of an ancient venom: recognition of a novel family of cnidarian toxins and the common evolutionary origin of sodium and potassium neurotoxins in sea anemone. Mol Biol Evol. 2015;32(6):1598–610. [DOI] [PubMed] [Google Scholar]
- 61. Dodds DN, Schlimgen AK, Lu SY, et al.. Novel reticular calcium binding protein is purified on taipoxin columns. J Neurochem. 1995;64(5):2339–44. [DOI] [PubMed] [Google Scholar]
- 62. Margres MJ, McGivern JJ, Wray KP, et al.. Linking the transcriptome and proteome to characterize the venom of the eastern diamondback rattlesnake (Crotalus adamanteus). J Proteomics. 2014;96:145–58. [DOI] [PubMed] [Google Scholar]
- 63. Hoffman D, Weimer E, Sakell R, et al.. Sequence and characterization of honeybee venom acid phosphatase. J Allergy Clin Immun. 2005;115(2):S107. [Google Scholar]
- 64. Grunwald T, Bockisch B, Spillner E, et al.. Molecular cloning and expression in insect cells of honeybee venom allergen acid phosphatase (Api m 3). J Allergy Clin Immun. 2006;117(4):848–54. [DOI] [PubMed] [Google Scholar]
- 65. Kim BY, Jin BR. Molecular characterization of a venom acid phosphatase Acph-1-like protein from the Asiatic honeybee Apis cerana. J Asia-Pac Entomol. 2014;17(4):695–700. [Google Scholar]
- 66. Kawahara M, Uye S, Burnett J, et al.. Stings of edible jellyfish (Rhopilema hispidu m, Rhopilema esculentum and Nemopilema nomurai) in Japanese waters. Toxicon. 2006;48(6):713–6. [DOI] [PubMed] [Google Scholar]
- 67. Michalak P. Coexpression, coregulation, and cofunctionality of neighboring genes in eukaryotic genomes. Genomics. 2008;91(3):243–8. [DOI] [PubMed] [Google Scholar]
- 68. Lercher MJ, Blumenthal T, Hurst LD. Coexpression of neighboring genes in Caenorhabditis elegans is mostly due to operons and duplicate genes. Genome Res. 2003;13(2):238–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Li Y, Gao L, Pan Y, et al.. Supporting data for "Chromosome-level reference genome of the jellyfish Rhopilema esculentum.". GigaScience Database. 2020. 10.5524/100720. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Joseph F. Ryan -- 10/31/2019 Reviewed
Lucas Leclère -- 11/10/2019 Reviewed
Lucas Leclère -- 3/12/2020 Reviewed
David A. Gold -- 11/19/2019 Reviewed