

Abstract
The aetiology of asthma associated with viral infection is complex. The dynamics that contribute to disease pathogenesis are multifactorial and involve overlapping molecular and cellular mechanisms, particularly the immune response to respiratory virus infection or allergen sensitization. This review summarizes the evidence associated with factors that may contribute to the development or exacerbation of asthma including age, host factors, genetic polymorphisms, altered immune responses, and aspects of viral antigen expression. This review also provides an important perspective of key events linked to the development of asthmatic disease and related pulmonary inflammation from human and animal studies, and discusses their relationship as targets for disease intervention strategies.
Keywords: Asthma, Pulmonary inflammation, Viral aetiology, Molecular mechanism, Cellular mechanism
Nomenclature
- ADRβ2
β2 adrenergic receptor
- AHR
airway hyper-responsiveness
- AOAH
acyloxyacyl hydroxylase
- BALF
bronchoalveolar lavage fluid
- CC16
Clara cell secretory protein 16
- CD
cluster designation
- CGRP
calcitonin gene-related peptide
- COPD
chronic obstructive pulmonary disease
- CTL
cytotoxic T lymphocytes
- DC
dendritic cells
- ECP
eosinophil cationic proteins
- FCɛR1α
high-affinity IgE receptor
- F protein
fusion glycoprotein
- G protein
G attachment glycoprotein
- GMCSF
granulocyte-macrophage colony stimulating factor
- HLA-DRB1
human leukocyte antigen DRB1
- hMPV
human metapneumovirus
- ICAM-1
intracellular cell-adhesion molecule 1
- IFN
interferon
- IgE
immunoglobulin E
- IL
interleukin
- IL-4R
IL-4 receptor
- LA-α
lymphotoxin-alpha
- LT
leukotriene
- LPS
lipopolysaccharide
- MBP
eosinophil major basic protein
- NGF
nerve growth factor
- NK
natural killer cells
- NKT
natural killer T cells
- NO
nitric oxide
- NS
non-structural
- OVA
ovalbumin
- Treg
regulatory T cells
- RNAi
RNA interference
- RSV
respiratory syncytial virus
- RV
rhinovirus
- TGF-β
transforming growth factor-beta
- Th1
T helper 1
- Th2
T helper 2
- TLR
Toll like receptor
- TNF-α
tumour necrosis factor-alpha
1. Introduction
Asthma is a chronic, reversible inflammatory disorder of the airways characterized by laboured breathing due to variable and reversible airway flow limitation. Asthmatics have sensitive airways that react to stimuli with inflammation, swelling of the airway lining, muscle tightening and mucus production; all of which make breathing more difficult. The symptoms can be one or more of the following: wheezing, coughing, tightness in the chest and shortness of breath. The prevalence or disease burden of asthma varies from country to country. It is estimated that there are approximately 300 million affected individuals worldwide [1]. Annual worldwide deaths from asthma have been estimated at 250,000. The highest prevalence of asthma is in the UK, Australia, New Zealand and Ireland [1]. In Australia, 12% of children and young adults aged 0–24 years have asthma [2] and expenditure on asthma in 2000 was estimated at AUS$693 million [3]. In the USA, 20 million people are affected by asthma and the total direct and indirect costs of asthma exceeded US$11.3 billion in 1998 [4].
The underlying causes of asthma are not well understood but many factors are recognized as contributing to the disease, including an atopic (allergic) disposition (often a family history of allergies and asthma), certain genetic mutations, exertion, irritants (including smoking, indoor and outdoor air pollutants), both inhaled and food allergens (e.g. house dust mites, mould spores, pollens, peanuts, egg), and viral infections [3]. In people with underlying asthma, respiratory viruses frequently trigger exacerbation of the disease, with 80–90% of exacerbations in children associated with respiratory infections [5] and up to two thirds of cases in adults [5]. Respiratory viruses are also thought to have a role in people becoming sensitive to allergens and developing asthma [9], [10].
Asthma is a multifactorial disease, with the genetics of the host interacting with a variety of environmental triggers. Viral infections are one of the environmental triggers, having been implicated both in the induction of the disease and, more frequently, in its exacerbation. A number of viruses are associated with the exacerbation of asthma in children and adults. The most common viruses detected in connection with the symptoms of asthma are rhinoviruses (RV); however, human respiratory syncytial virus (RSV), human metapneumovirus (hMPV), parainfluenza and influenza viruses, adenoviruses and coronaviruses are also thought to contribute to the exacerbation of asthma [5], [6]. The characteristics of the main viruses are listed in Table 1 . For some of these viruses, there is evidence from both epidemiological and experimental studies to confirm the role of a viral infection in exacerbating asthma, while for other viruses the evidence is less clear.
Table 1.
Viruses commonly implicated in the exacerbation of asthma
| Name | Classification | Virus structure | Genetic material | Clinical disease | Viral proteins that directly modulate host immune response |
|---|---|---|---|---|---|
| RV | Family: Picornaviridae | Protein capsid with ‘canyon’ binding site. The canyon regions contain the binding site of the cellular receptor, ICAM-1 | Non-segmented single molecule of linear positive sense RNA | Most frequent cause of common cold (responsible for 30–50% of cases). Second most common virus to cause bronchiolitis. Incidence highest in children <5 years | Not known |
| Genus: Rhinovirus | Genome (7.2 kb) | ||||
| Species: Human rhinovirus | |||||
| RSV | Family: Paramyxoviridae | Enveloped with F and G surface proteins. F protein: virus penetration and syncytium formation. G protein: virus attachment | Non-segmented single molecule of linear negative sense RNA | Major cause of lower respiratory tract infection in young children <5 years. Major cause of pneumonia and bronchiolitis in infants under 1 year of age | NS1 and NS2 proteins—interference of interferon system. G protein: Chemokine mimicry—inhibits the recruitment of cytotoxic cells. Drives Th2 immune response |
| Genus: Pneumovirus | Genome (15 kb) | ||||
| Species: Human respiratory syncytial virus | |||||
| hMPV | Family: Paramyxoviridae | Pleomorphic, enveloped virus with helical nucleocapsid and F and G surface proteins. Similar to RSV, hMPV F and G proteins may be required for infection | Non-segmented single molecule of linear negative sense RNA | Significant cause of lower respiratory tract infection in young children <5 years. Clinical disease similar to RSV | Not known |
| Genus: Metapneumovirus | Genome (13 kb) smaller than other pneumoviruses such as RSV | ||||
| Species: Human metapneumovirus | |||||
| Influenza virus | Family: Orthomyxoviridae | Enveloped with haemagglutinin (H) and Neuraminidase (N) proteins. Influenza A viruses are divided into subtypes based on these two surface proteins | 8 segments of negative strand RNA | Major cause of respiratory infection. In serious cases, influenza causes pneumonia | NS1—interference of interferon system |
| Genus: Influenza virus | Genome (12–14 kb) | ||||
| Species: Influenza virus A | |||||
| Influenza virus B | |||||
| Influenza virus C | |||||
F, fusion glycoprotein; G, G attachment glycoprotein; H, haemagglutinin; hMPV, human metapneumovirus; N, neuraminidase; ICAM-1, intracellular cell adhesion molecule 1; NS, non-structural; RV, rhinovirus; RSV, respiratory syncytial virus.
The immune responses triggered by viral infections and allergens are well established (Fig. 1 ). This review focuses on the association between viruses and the exacerbation of asthma and outlines current understanding of the molecular and cellular mechanisms involved.
Fig. 1.

Immune responses triggered by viral infection and allergens. (A) During the early stages of a viral infection, antigen presenting cells (APCs: macrophages, DCs) become activated and secrete IL-12. Viral peptide is presented by APCs to naïve T cells in association with MHC class I and co-stimulatory signals (B7 and CD28). The production of IL-12 and the binding of antigen-MHC molecules commit the differentiation of naïve T cells to the Th1 cell subset that secretes Th1 cytokines including IFN-γ. The cytokine contributes to the activation of macrophage (make more IL-12), B cells (make IgG2a) and cytotoxic T cell (kill infected cells). An IFN-γ dominated microenvironment inhibits the development of a Th2 cell subset. Together, these responses result in the resolution of the infection in the airways. (B) In the early phase of allergen exposure, cross linking of antigen-specific IgE on the surface of mast cells results in the activation and release of mediators that cause bronchoconstriction and inflammation. Activated mast cells also produce IL-4 that commits naïve T cells to the Th2 subset as well as B cell isotype class switching to IgE production. In addition, antigenic peptide is presented to naïve T cells by APCs in the context of MHC class II and co-stimulatory signals. In an IL-4 dominated microenvironment, this triggers the differentiation of naïve T cells to Th2 cell subset that generates Th2 cytokines (IL-4, IL-5, IL-10 and IL-13). These cytokines are responsible for orchestrating the late phase of the allergic response. IL-4 and IL-13 contribute to mast cell activation and the synthesis of IgE. IL-5 is implicated in eosinophilia and is known to stimulate these cells, resulting in degranulation and release of toxic basic proteins (e.g. ECP, MBP). IL-10 inhibits APCs, therefore preventing IL-12 from initiating a Th1 immune response.
2. Inflammatory cascade associated with asthma
The inflammatory cascade associated with asthma involves mast cells, DC, T cells, eosinophils, macrophages, fibroblasts and neutrophils (Table 2 ). When the inflammation intensifies, the airways become very sensitive to provoking stimuli and airway hyper-responsiveness (AHR) develops. The mast cells degranulate and they, and other cells, release chemical mediators into the lower respiratory tract that prolong the response and cause contraction of the bronchial smooth muscles (Table 2). These actions, and airway swelling, mucus secretion and inflammation, contribute to the bronchoconstriction and airway obstruction seen in asthma attacks.
Table 2.
Cells and soluble mediators involved in viral exacerbation of asthma
| Cells/mediators | Mechanism/s of action |
|---|---|
| Mast cells | • Activated by allergens |
| • Release mediators (e.g. histamines, leukotrienes, prostaglandins) that result in bronchoconstriction of the airways and AHR | |
| Eosinophils | • Release basic proteins (e.g. ECP, MBP) that can result in injury of airway epithelial cells |
| T cells | • Release of Th2 cytokines (e.g. IL-4, IL-5, IL-10, IL-13) that are responsible for eosinophil maturation and degranulation, IgE production and mast cell activation |
| • Strong Th2 cell activity may also be due to low numbers of regulatory T cells (Tregs) | |
| • IL-13 producing CD8+ T cells (Tc2) are thought to play a major role in the development of AHR | |
| B cells | • In the presence of antigen (allergen) and IL-4, B cells differentiate into plasma cells secreting IgE |
| Macrophages | • May be activated by allergens through low-affinity IgE receptors |
| • Release inflammatory mediators | |
| Dendritic cells | • Take up allergens from the airways and interact with T cells |
| • Stimulate production of Th2 cells from naïve T cells | |
| Antibody (IgE) | • Binds to Fc receptors on mast cells, eosinophils and macrophages |
| • Induces degranulation of mast cells causing release of mediators | |
| Chemokines | • Produced by immune cells (e.g. T, B, macrophages, DCs) and non-immune cells (e.g. fibroblast, epithelial cells) |
| • Important in the recruitment of inflammatory cells into the airways | |
| • Selected chemokines recruit specific cell type. (e.g. eotaxin is selective for eosinophils; MDC is selective for Th2 cells) | |
| Cysteinyl leukotrienes | • Mainly secreted by mast cells and eosinophils |
| • Strong bronchoconstrictors | |
| • Pro-inflammatory mediators | |
| Cytokines | • Produced by immune (e.g. T, B, macrophages, DCs) and non-immune cells (e.g. fibroblast, epithelial cells) |
| • Major player in the inflammatory response in asthma | |
| • TNF-α: required for enhancing the inflammatory response; GMCSF: required for eosinophil survival; IL-5: required for eosinophil differentiation and survival; IL-4: required for Th2 cell differentiation and IgE formation | |
| Histamine | • Produced by mast cells |
| • Contributes to bronchoconstriction and to the inflammatory response | |
| Prostaglandin | • Derived predominantly from mast cells |
| • Bronchoconstrictor | |
| • Involved in Th2 cell recruitment to the airways | |
| Neural factors | • Derived from neural signalling pathways |
| • Neuropeptides: somatostatin, CGRP, substance P, neurokinin A | |
| • NGF upregulates receptor for substance P | |
| • Contribute to bronchoconstriction, AHR and inflammation |
AHR, airway hyper-responsiveness; CGRP, calcitonin gene-related peptide; ECP, eosinophil cationic proteins; MBP, eosinophil major basic protein; GMCSF, granulocyte-macrophage colony stimulating factor; MDC, macrophage-derived chemokine; NGF, nerve growth factor; TNF-α, Tumour necrosis factor alpha.
3. Factors in developing viral exacerbation of asthma
A number of factors are known to affect the likelihood of asthma being exacerbated in response to a viral infection (Fig. 2 ). These factors include the age at which the first viral infection occurs, the gender, genetic makeup of the host, the ability of the virus to persistently infect or to become latent in the host and viral antigens.
Fig. 2.
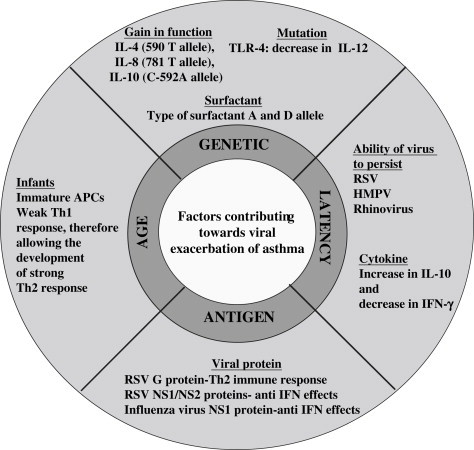
Factors known to affect the likelihood of asthma being exacerbated in response to a viral infection. These factors are responsible for skewing the immune response towards a Th2 biased immune response.
3.1. Age of initial viral infection
Viral infections that occur early in life appear to have an important role in shaping immunological development. If initial infection occurs early in life, the T helper 1 (Th1) response may be weak, allowing the development of stronger Th2 responses to challenges later in life. Mice initially infected with RSV as neonates were more likely to have a Th2 type response upon secondary infection, whereas those first infected when older were more likely to have a Th1 type response [7]. One interpretation of these findings is that RSV infection in neonates suppresses the development of the Th1 immune response and a Th2 response occurs. In contrast, if the initial infection is delayed for several weeks, a Th1 immune response is mounted which lacks the pathogenic Th2 component associated with asthma. There is some evidence that infection at one stage in life may lead to a reduction in atopy while, at another stage, it may potentiate the emergence of allergies [8] and that early severe RSV infections in infancy leads to allergic asthma later in life [9], [10]. However, this issue is controversial.
3.2. Gender
Boys are more likely to have severe asthma than girls; however at puberty (about age 13–14) the ratios reverse and many more adult women are admitted to hospital for asthma than adult men [11]. The reasons for this switch in susceptibility are not known but this observation suggests a potential role for sex hormones in the development of asthma.
3.3. Host genetics
A predisposition towards developing asthma runs in families, indicating a genetic component to the disease that has been confirmed by studies in twins [12]. Specific genes have been associated with asthma in nearly 200 studies, with 64 different genes associated with asthma or atopy identified in one study [13]. Some of these genes have repeatedly been found to be associated with asthma, including the Th2 cytokines IL-4 and IL-13, human leukocyte antigen DRB1 (HLA-DRB1), TNF-α, lymphotoxin-alpha (LA-α), high-affinity IgE receptor (FCɛR1α) and the IL-4 receptor (IL-4R). The gene β2 adrenergic receptor (ADRβ2) has been associated with asthma severity [13].
Other genes associated with atopy and asthma include, for atopy, pattern recognition receptor CD14 (receptor for endotoxins such as lipopolysaccharide (LPS)) and granulocyte-macrophage colony stimulating factor (GMCSF); for asthma, Clara cell secretory protein (CC16), CD14, acyloxyacyl hydroxylase (AOAH), TNF-α, leukotriene C4 (LTC4) synthase, IL-10 and IL-8; and, for asthma severity, ADRβ2 [14], [15]. Host genetics is also thought to affect the severity of viral infections and the type of immune response to the infection. For example, specific alleles of pulmonary surfactant genes have been associated with more severe RSV infections in infants [16]. Surfactant proteins line the alveolar surface of the lungs and are essential for normal respiratory function. Other genetic variations have been shown to affect the expression of cytokines in response to viral infections. A variation causing an increase in IL-8 transcription was found to lead to enhanced susceptibility to RSV induced bronchiolitis [17] and a variant affecting transcription of IL-4 was found to be associated with severe RSV disease and elevated levels of IgE [18]. Similarly, variations in genes for IL-10 have been identified, with those people heterozygous for the gene found to be less likely to develop RSV bronchiolitis [19], and two distinct mutations in the gene for Toll like receptor 4 (TLR-4) have been found to be associated with severe RSV bronchiolitis [20]. An association between differences in IL-13 genes and severe RSV infection has also been found [21]. Environmental factors, such as viral infections, may impact on genetically predisposed individuals to affect the development of asthma and the viral exacerbation of asthma.
3.4. Latency or persistence
A number of respiratory viruses have been found to persist in the host, despite the host mounting an immune response. For example, hMPV can persist in the lungs of mice for up to 60 days, despite the presence of neutralizing antibody [22]. In mice, RSV was found to persist in the lungs for more than 100 days, despite normal cytotoxic T-cell responses and normal RSV specific antibodies [23] and viral proteins have been found in guinea pigs 6 weeks after RSV infection, despite the presence of neutralizing antibodies [24].
The evidence about respiratory viral persistence in humans is less clear, with several recent studies finding directly opposing results. A study by Wilkinson et al. found that RSV persisted in patients with chronic obstructive pulmonary disease (COPD) [25], while Falsey et al. found no RSV persistence associated with COPD [26]. Studies on viral persistence with the other main virus associated with the exacerbation of asthma, RV, have also produced opposing results. In one study, 95% of adults with an exacerbation of asthma were found to be clear of RV 6 weeks after the exacerbation [27], whereas a study in children found that more than 40% still had detectable RV after 6 weeks and this viral persistence was associated with more severe acute exacerbations of asthma [28].
A possible mechanism for immune evasion is persistence at a site that avoids immune system surveillance. With respiratory viruses, such a site may be the pulmonary neurons and RSV infection of nerve cells has recently been demonstrated [29]. In addition, a role for cytokine in viral persistence was demonstrated in mice by Alvarez and Tripp. They showed that the persistent presence of hMPV is associated with increased IL-10 expression and weak CTL activity [30]. It is currently not clear whether respiratory viruses persist in the host, despite a functional immune response. Further characterization is required to understand the possible relationships between respiratory viral latency, the host's immune responses and the exacerbation of asthma.
3.5. Viral antigen
It is well established that RSV G protein can influence the immune response in murine models. The G protein seems to elicit pre-dominantly a Th2 driven response characterized by lung eosinophilia and local production of IL-4 and IL-5 [31]. Prior sensitization with a recombinant vaccinia virus expressing RSV G protein leads to a Th2 driven augmented disease, contrasting with the usual Th1 response seen in primary viral infections [31]. Studies have also shown that the non-structural proteins of RSV and influenza virus are capable of modulating the immune response through their ability to inhibit IFN responses in the infected host [31].
4. Neural pathways in the viral exacerbation of asthma
Released mediators derived from neural signalling pathways contribute to the bronchoconstriction associated with asthma. Studies in humans and animals have shown that an intact nervous system contributes to virus-induced AHR. In a guinea pig model, cutting the vagus nerve reduced airway responsiveness to histamines in animals previously infected with parainfluenza virus [32]. In humans, virus induced AHR was also vagally mediated [33]. In guinea pigs, rats and cats, the efferent, parasympathetic bronchoconstrictor limb of the cough and bronchoconstriction reflexes was enhanced during acute viral illness [32], [34].
Acetylcholine is released from the nerve fibres that innervate smooth muscle in the airways and binds to M3 muscarinic receptors on the smooth muscle and causes bronchoconstriction. This is normally limited because acetylcholine also binds to M2 muscarinic receptors on neurons that inhibit further release of acetylcholine [35]. Dysfunction of these M2 receptors is thought to contribute to AHR during viral infections [34], [36]. A number of factors are thought to cause this receptor dysfunction including viral neuraminidase [37], endogenous tachykinins [35], induced inflammatory cell products, such as eosinophil cationic proteins (ECP) [37], eosinophil major basic protein (MBP) [38], IFNs from macrophages or macrophage-stimulated T cells or nitric oxide (NO) released from virus-activated macrophages [39]. The effects on the M2 receptor function are transient and normal functions are restored several weeks after resolution of the acute infection [34].
Stimulation of the nerve fibres during viral infection augments the release of neuropeptides, including somatostatin, calcitonin gene-related peptide (CGRP) and the tachykinins substance P and neurokinin A. A number of these neuropeptides are associated with symptoms of the exacerbation of asthma. For example, CGRP can cause eosinophilia in rat lungs [40], substance P and neurokinin A are associated with an increase in eosinophil numbers in the airways [41], and substance P activates eosinophils and mast cells and enhances the degranulation of eosinophils and the release of histamines from mast cells [42]. CGRP also inhibits the production of Th1 cytokines, therefore unbalancing the Th1/Th2 equation in favour of a Th2 response [43].
A role for the neurotrophin nerve growth factor (NGF) in viral induced asthma is suggested by results finding that NGF is increased in RSV infected rats, compared to control animals [44]. NGF up-regulates the expression of the high affinity receptor for substance P (the NK1 receptor) and substance P contributes to the neurogenic inflammation seen in RSV-infected airways [45]. NGF expression in the lungs normally declines as animals age but RSV infection interferes with this decline [44]. NGF may lead to short- and long-term changes in the distribution and development of nerves and changes in the reactivity of sensory nerves in the respiratory tract [44] and may also contribute to AHR, inflammation and the activation of recruited eosinophils and mast cells [46].
5. Immune mechanisms of virus induced asthma exacerbations
In understanding the mechanisms underlying viral exacerbation of asthma, it is helpful to observe models of primary virus infection. Respiratory viruses including parainfluenza, influenza and RSV have been shown to induce airway inflammation and lung function changes in rodent models. In RSV infection in mice, airway inflammation and the development of AHR are dependent on the presence of the Th2 cytokines IL-5 [47] and IL-13 [48] and not on IFN-γ, the most abundant cytokine produced during RSV infection. In addition, inflammation and lung function changes in this model depend on CD8+ T cells [49]. Taken together this suggests that type 2 cytokine producing CD8+ T cells may drive virus induced reactive airway disease. Further, respiratory viral infections lead to a marked expansion of mature DC in the lung [50]. These mature lung DC have a strong capacity to activate both naïve and memory T cells and to induce their proliferation. In a model of influenza infection, maturation of lung DC resulted in strong immunogenicity of an otherwise non-immunogenic antigen [50]. Increases in numbers of lung DC, which arise from local precursors in the lung, outlast the resolution of disease in RSV infection [51]. Importantly, pulmonary DC can stimulate T-cell responses in the lung in situ without migration to the regional lymph nodes, favouring Th2 and Tc2 responses [52]. These observations suggest that respiratory viral infections lead to a prolonged period of increased antigen presentation in the airways resulting in de novo and memory T-cell responses not only to the virus but also to unrelated antigens including allergens.
In addition to studies of primary infections, models studying the interactions between respiratory viral infections and allergen sensitization are essential in understanding the mechanisms of virus induced asthma exacerbations. Many small animal models were designed to reveal the pathogenic mechanisms behind the enhancement of allergic sensitization by respiratory virus infections; the increased airway inflammation and responsiveness resulting from allergic airway sensitization following respiratory viral infection and, in those with established allergic airway sensitization, the increased airway inflammation and responsiveness due to respiratory viral infections. These studies show that the immune responses to allergen sensitization and respiratory viral infections interact to cause persistent inflammation and AHR, symptomatic of the asthmatic response (Fig. 2) [53].
Many studies have been done in animals sensitized to different allergens and then infected with respiratory viruses, a model that mimics viral exacerbation of asthma in sensitized individuals. In one study, guinea pigs were sensitized with ovalbumin (OVA) and challenged with RSV and those that underwent both treatments were found to have more severe symptoms of AHR and inflammation (Fig. 3 ) [53]. Similar results were also found in mice sensitized to OVA and then infected with RSV [54], mice sensitized to OVA and challenged with murine cytomegalovirus [55], and mice sensitized to the house dust mite Dermatophagoides farinae then infected with RSV [56].
Fig. 3.

Immunological effects of viral infection in atopic lung. RSV infection of allergic mice (exposed to OVA) mimics viral infection (e.g. RSV, RV) in an atopic individual (young child or elderly). RSV infection in an atopic person results in further amplification of the Th2 immune response. These aberrant immune responses contribute to exacerbated airway pathology with persistent wheezing and asthma symptoms.
Experiments have been performed to determine the contribution of released inflammatory mediators on AHR following viral infection. IL-4 is important in the exacerbation of asthma but the response is not solely dependent on IL-4 and IL-5 has been observed to increase in virally infected sensitized mice and has been directly associated with the viral exacerbation of asthma [57]. IL-10 is thought to be necessary for the development of allergic AHR [58], however, its role in viral exacerbation of AHR is less certain. The role of IL-10 is complex, inhibiting IL-5 production, eosinophilic inflammation and chemotaxis but potentially inducing a decrease in Th1 cytokine production [59]. Mice deficient in IL-10 that were both sensitized (to OVA) and infected with RSV did develop AHR including eosinophilic inflammation, Th2 cytokine production and goblet cell hyperplasia. Inhibition of IL-13 has been shown to significantly reduce AHR in sensitized and infected mice, to the level of that seen in mice only infected or only sensitized [60]. These factors all appear to contribute in varying extents to the viral exacerbation of asthma.
The cysteinyl leukotrienes (CysLTs) such as LTC4, LTD4 and LTE4 are products of activated eosinophils, basophils, mast cells and macrophages. These potent inflammatory mediators have a diverse range of biological activities, including the ability to exacerbate asthma by inducing the contraction of bronchial smooth muscle [61]. LTC4 has been found at elevated levels in the nasopharyngeal secretions of children during the acute phase of RSV infection, with higher levels in patients with lower respiratory tract involvement than in those with upper respiratory illness alone [62]. LTE4 has been found to be elevated in urine samples collected from patients with asthma exacerbations [63]. CysLT levels have also been found to be increased in the lower airways during RSV bronchiolitis, although their concentrations are lower than those in acute asthma [64]. In mice, RSV infection has been found to produce a marked increase in CysLTs in the BALF and lung tissue, resulting in the recruitment of neutrophils and lymphocytes into the airways and AHR [65]. CysLTs have been found to have multiple effects in animals, including the induction of Th2 responses in the lungs through effects on DCs and cytokine generation, the recruitment and activation of cells such as eosinophils and mast cells, and inflammation [63]. Interestingly, an LT synthesis blocker prevents the development of RSV induced lung function changes in mice [66] and montelukast, a CysLT antagonist, seems to reduce post-bronchiolitis wheeze in small children [67].
6. Relevance of animal studies to the human asthma model
Bronchial reactivity to stimulation by histamine has been found to be increased in asthmatic patients following infection with influenza A virus [68]. Human RV infection in adults with mild asthma often has little effect on the lower respiratory tract or lung function [69]; however, when infection in asthmatics is followed by allergen challenge, AHR and eosinophilia in BALF have been observed for up to a month following challenge [70]. Deliberate infection with RV has found that asthmatic symptoms tended to peak later during infection and were more pronounced and persistent during the later stages of infection [71]. Although nasal and upper respiratory symptoms were similar between asthmatics with high IgE levels, asthmatics with low IgE levels and control subjects before infection, asthmatics with higher IgE levels had more symptoms of lower respiratory inflammation before and during infection. Asthmatics with high IgE levels also had lower lung function results before infection, while the results for the asthmatics with low IgE levels were similar to the controls. However, the lung function tests results did not vary significantly during the course of RV infection [71].
A similar study of people with allergic rhinitis (hay fever) found that RV infection increased AHR and the inflammatory reaction to allergen challenge. Before RV infection, most subjects developed only short-term responses to the allergen; however, during RV infection nearly all subjects developed late asthmatic reactions [70]. These late asthmatic reactions were independent of changes in airway reactivity or the size of the immediate response to allergen challenge, and the propensity to develop them persisted for up to 4 weeks after virus infection [70].
A similar study found that the allergic response changed from nearly all subjects having only an immediate response before infection to a majority of subjects having both an immediate and late asthmatic reaction during or following RV infection [72]. Changes associated with RV infection and subsequent allergen challenge include an immediate increase in the release of histamine and the recruitment of eosinophils into the airways within a 48 h period [73].
A study of peripheral blood mononuclear cells from patients with atopic asthma and normal controls found that exposure to RV induced the production of IFN-γ, IL-12, IL-10 and IL-13 in both groups, with significantly lower levels of IFN-γ and IL-12 and higher levels of IL-10 in the cells from asthmatics than the cells from normal subjects. IL-4 was induced only in the asthmatic group and the IFN-γ/IL-4 ratio was more than three times lower in this group. This suggests that the normal type 1 immune response to RV infection is defective in atopic asthmatic individuals, with infection inducing a Th2-immune response similar to their response to allergens [74].
7. Outlook on prevention and therapy
Vaccines and therapeutics have the potential for eliminating or reducing the synergy between viral infections and asthma, as well as decreasing unrecognized but associated disease burden and health care costs. Vaccination is the mainstay of prophylaxis for respiratory viruses; however, for some viruses such as RSV and RV, developing safe and effective vaccines has been difficult. Despite the need for anti-viral drugs as an adjunct to vaccines, few are available that are effective. Therefore, novel and new approaches are highly desirable. Recently, Mahalingam and Tindle have demonstrated the efficacy of an hMPV CTL epitope vaccine that triggers a strong Th1 immune response which was associated with accelerated viral clearance in a murine model [75]. In addition, RNA interference (RNAi), a naturally occurring process for controlling gene expression that occurs through a process mediated by short interfering RNA (siRNA) molecules, has been shown to have the remarkable ability to silence RSV replication both in vitro and in vivo (Tripp et al., unpublished data). These approaches can be used to target host cell genes to silence aspects of the inflammatory pathway that include Th1 or Th2 cytokines, chemokines or proteins that regulate their expression. Thus, disease intervention strategies that target the virus and/or host response are rational approaches for breaking the link between the synergy of virus infection and asthma.
Acknowledgements
S.M. and L.S. are recipients of the Australian National Health and Medical Research Council (NHMRC) R. Douglas Wright and NHMRC Peter Doherty Fellowships respectively. S.M. acknowledges support from the NHMRC for research on respiratory viral infections (Project Grant #399701). K.M. acknowledges support from NHMRC programme grant #224207. R.A.T. acknowledges the Georgia Research Alliance for their continued support. J.S. acknowledges support from the Wellcome Trust (Senior Fellowship Grant #067454). We thank Mr Timothy Lewis for excellent editorial assistance.
References
- 1.The International Study of Asthma and Allergies in Childhood (ISAAC) Steering Committee Worldwide variation in prevalence of symptoms of asthma, allergic rhino conjunctivitis and atopic eczema. Lancet. 1998;351:1225–1232. [PubMed] [Google Scholar]
- 2.Australian Bureau of Statistics (ABS) National Health Survey: Summary of Results. 2004–2005:1–92. [Google Scholar]
- 3.Woolcock Institute of Medical Research/Australian Institute of Health and Welfare; 2005. Asthma in Australia: Australian centre for asthma monitoring. (Asthma series 2). Cat. No. ACM 6. [Google Scholar]
- 4.National Institutes of Health: National Heart, Lung, and Blood Institute; 1999. Data Fact Sheet: Asthma Statistics. 1–4. [Google Scholar]
- 5.Johnston S., Pattemore P., Sanderson G., Smith S., Lampe F. Community study of role of viral infections in exacerbations of asthma in 9–11 year old children. BMJ. 1995;310:1225–1229. doi: 10.1136/bmj.310.6989.1225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Schwarze J., Gelfand E.W. Respiratory viral infections as promoters of allergic sensitization and asthma in animal models. Eur. Respir. J. 2002;19:341–349. doi: 10.1183/09031936.02.00254302. [DOI] [PubMed] [Google Scholar]
- 7.Culley F., Pollot J., Openshaw P. Age at first viral infection determines the pattern of T- cell mediated disease during reinfect ion in adulthood. J. Exp. Med. 2002;196:1381–1386. doi: 10.1084/jem.20020943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Landau L. Paediatric basis of adult lung disease. Paediatr. Respir. Rev. 2006;7:S251–S254. doi: 10.1016/j.prrv.2006.04.186. [DOI] [PubMed] [Google Scholar]
- 9.Sigurs N., Gustafsson P., Bjarnason R., Lundgberg F., Schmidt S. Severe Respiratory Syncytial Virus bronchiolitis in infancy and asthma and allergy at age 13. Am. J. Respir. Crit. Care Med. 2005;171:137–141. doi: 10.1164/rccm.200406-730OC. [DOI] [PubMed] [Google Scholar]
- 10.Sigurs N., Bjarnason R., Sigurbergsson F., Kjellman B., Björkstén B. Asthma and immunoglobulin E antibodies after respiratory syncytial virus: a prospective cohort study with matched controls. Pediatrics. 1995;95:500–505. [PubMed] [Google Scholar]
- 11.Johnston N.W., Sears M.R. Asthma exacerbations 1: Epidemiology. Thorax. 2006;61:722–728. doi: 10.1136/thx.2005.045161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Duffy D.L., Martin N.G., Battistutta D., Hopper J.L., Mathews J.D. Genetics of asthma and hay fever in Australian twins. Am. Rev. Respir. Dis. 1990;142:1351–1358. doi: 10.1164/ajrccm/142.6_Pt_1.1351. [DOI] [PubMed] [Google Scholar]
- 13.Hoffjan S., Nicolae D., Ober C. Association studies for asthma and atopic diseases: a comprehensive review of the literature. Respir. Res. 2003;4:14–26. doi: 10.1186/1465-9921-4-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Puthothu B., Krueger M., Heinze J., Forster J., Heinzmann A. Impact of IL-8 and IL-8-Receptor alpha polymorphisms on the genetics of bronchial asthma and severe RSV infections. Clin. Mol. Allergy. 2006;4:2. doi: 10.1186/1476-7961-4-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Barnes K.C., Grant A., Gao P., Baltadjieva D., Berg T., Chi P., Zhang S., Zambelli-Weiner A., Ehrlich E., Zardkoohi O., Brummet M.E., Stockton M., Watkins T., Gao L., Gittens M., Wills-Karp M., Cheadle C., Beck L.A., Beaty T.H., Becker K.G., Garcia J.G., Mathias R.A. Polymorphisms in the novel gene acyloxyacyl hydroxylase (AOAH) are associated with asthma and associated phenotypes. J. Allergy Clin. Immunol. 2006;118:70–77. doi: 10.1016/j.jaci.2006.03.036. [DOI] [PubMed] [Google Scholar]
- 16.Lofgren J., Ramet M., Renko M., Marttila R., Hallman M. Association between surfactant protein A gene locus and severe respiratory syncytial infection in infants. J. Infect. Dis. 2002;185:283–289. doi: 10.1086/338473. [DOI] [PubMed] [Google Scholar]
- 17.Hacking D., Knight J., Rocket K., Brown H., Frampton J., Kwiatkowski D.P., Hull J., Udalova I.A. Increased in vivo transcription of an IL-8 haplotype associated with respiratory syncytial virus disease- susceptibility. Genes Immun. 2004;5:274–282. doi: 10.1038/sj.gene.6364067. [DOI] [PubMed] [Google Scholar]
- 18.Choi E., Lee H., Yoo T., Chanock S. A common haplotype of interleukin 4 gene IL-4 is associated with severe respiratory syncytial virus disease in Korean children. J. Infect. Dis. 2002;186:1207–1211. doi: 10.1086/344310. [DOI] [PubMed] [Google Scholar]
- 19.Hoebee B., Rietveld E., Bont L., van Oosten M., Hodemaekers H., Nagelkerke N.J., Neijens H.J., Kimpen J.L., Kimman T.G. Influence of promoter variants of IL-10, IL-9 and tumour necrosis factor-alpha genes on respiratory syncytial virus bronchiolitis. J. Infect. Dis. 2004;189:239–247. doi: 10.1086/380908. [DOI] [PubMed] [Google Scholar]
- 20.Tal G., Mandelberg A., Dalal I. Association between common toll-like receptor 4 mutations and severe respiratory syncytial virus disease. J. Infect. Dis. 2004;189:2057–2063. doi: 10.1086/420830. [DOI] [PubMed] [Google Scholar]
- 21.Puthothu B., Krueger M., Forster J., Heinzmann A. Association between severe respiratory syncytial virus infection and IL-13/IL-4 haplotypes. J. Infect. Dis. 2006;193:438–441. doi: 10.1086/499316. [DOI] [PubMed] [Google Scholar]
- 22.Alvarez R., Harrod K., Shieh W., Zaki S., Tripp R. Human metapneumovirus persists in BALB/c mice despite the presence of neutralizing antibodies. J. Virol. 2004;78:14003–14011. doi: 10.1128/JVI.78.24.14003-14011.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Schwarze J., O'Donnell D., Rohwedder A., Openshaw P. Latency and persistence of Respiratory Syncytial Virus despite T cell immunity. Am. J. Respir. Crit. Care Med. 2004;169:801–805. doi: 10.1164/rccm.200308-1203OC. [DOI] [PubMed] [Google Scholar]
- 24.Riedel F., Oberdieck B., Streckert H., Philippou S., Krusat T., Marek W. Persistence of airway hyperresponsiveness and viral antigen following respiratory syncytial virus bronchiolitis in young guinea pigs. Eur. Respir. J. 1997;10:639–645. [PubMed] [Google Scholar]
- 25.Wilkinson T., Donaldson G., Johnston S., Openshaw P., Wedzicha J. Respiratory syncytial virus, airway inflammation, and FEV1 decline in patients with chronic obstructive pulmonary disease. Am. J. Respir. Crit. Care Med. 2006;173:871–876. doi: 10.1164/rccm.200509-1489OC. [DOI] [PubMed] [Google Scholar]
- 26.Falsey A., Formica M., Hennessey P., Criddle M., Sullender W., Walsh E.E. Detection of respiratory syncytial virus in adults with chronic obstructive pulmonary disease. Am. J. Respir. Crit. Care Med. 2006;173:639–643. doi: 10.1164/rccm.200510-1681OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ferreiraa A., Williamsa Z., Donningera H., van Schalkwyka E., Bardin P.G. Rhinovirus is associated with severe asthma exacerbations and raised nasal interleukin-12 respiration. Respiration. 2002;69:136–142. doi: 10.1159/000056316. [DOI] [PubMed] [Google Scholar]
- 28.Kling S., Donninger H., Williams Z., Vermeulen J., Weinberg E., Latiff K., Ghildyal R., Bardin P. Persistence of rhinovirus RNA after asthma exacerbation in children. Clin. Exp. Allergy. 2005;35:672–678. doi: 10.1111/j.1365-2222.2005.02244.x. [DOI] [PubMed] [Google Scholar]
- 29.Li X., Fu Z., Alvarez R., Henderson C., Tripp R. Respiratory syncytial virus infects neuronal cells and processes that innervate the lung by a process involving RSV G protein. J. Virol. 2006;80:537–540. doi: 10.1128/JVI.80.1.537-540.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Alvarez R., Tripp R.A. The immune response to human metapneumovirus is associated with aberrant immunity and impaired virus clearance in BALB/c mice. J. Virol. 2005;79:5971–5978. doi: 10.1128/JVI.79.10.5971-5978.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Mahalingam S., Schwarze J., Zaid A., Nissen M., Sloots T., Tauro S., Storer J., Alvarez R., Tripp R.A. Perspective on the host response to human metapneumovirus infection: what can we learn from respiratory syncytial virus infections? Microbes Infect. 2006;8:285–293. doi: 10.1016/j.micinf.2005.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Buckner C., Songsiridej V., Dick E., Busse W. In vivo and in vitro studies on the use of the guinea pig as a model for virus-provoked airway hyperreactivity. Am. Rev. Respir. Dis. 1985;132:305–310. doi: 10.1164/arrd.1985.132.2.305. [DOI] [PubMed] [Google Scholar]
- 33.Empey D.W., Laitinen L.A., Jacobs L., Gold W.M., Nadel J.A. Mechanisms of bronchial hyperreactivity in normal subjects following upper respiratory tract infection. Am. Rev. Respir. Dis. 1976;113:523–527. doi: 10.1164/arrd.1976.113.2.131. [DOI] [PubMed] [Google Scholar]
- 34.Sorkness R., Clough J.J., Castleman W., Lemanske R. Virus-induced airway obstruction and parasympathetic hyperresponsiveness in adult rats. Am. J. Respir. Crit. Care Med. 1994;150:28–34. doi: 10.1164/ajrccm.150.1.8025764. [DOI] [PubMed] [Google Scholar]
- 35.Jacoby D.B. Virus-induced asthma attacks. J. Aerosol Med. 2004;17:169–173. doi: 10.1089/0894268041457156. [DOI] [PubMed] [Google Scholar]
- 36.Fryer A., Jacoby D. Parainfluenza virus infection damages inhibitory M2 muscarinic receptors on pulmonary parasympathetic nerves in the guinea pig. Br. J. Pharmacol. 1991;102:267–271. doi: 10.1111/j.1476-5381.1991.tb12164.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Fryer A., Jacoby D. Function of pulmonary M2 muscarinic receptors in antigen-challenged guinea pigs is restored by heparin and poly-L-glutamate. J. Clin. Invest. 1992;90:2292–2298. doi: 10.1172/JCI116116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Evans C.M., Fryer A.D., Jacoby D.B., Gleich G., Costello R. Pre-treatment with antibody to eosinophil major basic protein prevents hyperresponsiveness by protecting neuronal Ms muscarinic receptor function. J. Clin. Invest. 1997;100:2254–2262. doi: 10.1172/JCI119763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lee A.M., Fryer A.D., van Rooijen N., Jacoby D.B. Role of macrophages in virus-induced airway hyperresponsiveness and neuronal M2 muscarinic receptor dysfunction. Am. J. Physiol. Lung Cell Mol. Physiol. 2004;286:1255–1259. doi: 10.1152/ajplung.00451.2003. [DOI] [PubMed] [Google Scholar]
- 40.Bellibas S.E. The effect of human calcitonin gene-related peptide on eosinophil chemotaxis in the rat airway. Peptides. 1996;17:563–564. doi: 10.1016/0196-9781(96)00027-7. [DOI] [PubMed] [Google Scholar]
- 41.Braunstein G., Fajac I., Lacronique J., Frossard N. Clinical and inflammatory responses to exogenous tachykinins in allergic rhinitis. Am. Rev. Respir. Dis. 1991;144:630–635. doi: 10.1164/ajrccm/144.3_Pt_1.630. [DOI] [PubMed] [Google Scholar]
- 42.Heaney L.G., Cross L.J., Stanford C.F., Ennis M. Substance, P induces histamine release from human pulmonary mast cells. Clin. Exp. Allergy. 1995;25:179–186. doi: 10.1111/j.1365-2222.1995.tb01024.x. [DOI] [PubMed] [Google Scholar]
- 43.Kawamura N., Tamura H., Obana S., Wenner M., Ishikawa T., Nakata A., Yamamoto H. Differential effects of neuropeptides on cytokine production by mouse helper T cell subsets. Neuroimmunomodulation. 1998;5:9–15. doi: 10.1159/000026321. [DOI] [PubMed] [Google Scholar]
- 44.Hu C., Wedde-Beer K., Auais A., Rodriguez M., Piedimonte G. Nerve growth factor and nerve growth factor receptors in respiratory syncytial virus-infected lungs. Am. J. Physiol. Lung Cell Mol. Physiol. 2002;283:494–502. doi: 10.1152/ajplung.00414.2001. [DOI] [PubMed] [Google Scholar]
- 45.Piedimonte G., Rodriguez M., King K., McLean S., Jiang X. Respiratory syncytial virus upregulates expression of the substance P receptor in rat lungs. Am. J. Physiol. Lung Cell Mol. Physiol. 1999;277:831–840. doi: 10.1152/ajplung.1999.277.4.L831. [DOI] [PubMed] [Google Scholar]
- 46.Nassenstein C., Schulte-Herbruggen O., Renz H., Braun A. Nerve growth factor: The central hub in the development of allergic asthma? Eur. J. Pharmacol. 2006;533:195–206. doi: 10.1016/j.ejphar.2005.12.061. [DOI] [PubMed] [Google Scholar]
- 47.Schwarze J., Cieslewicz G., Hamelmann E., Joetham A., Schultz L.D., Lamers M.C., Gelfand E.W. IL-5 and eosinophils are essential for the development of airway hyperresponsiveness following acute respiratory syncytial virus infection. J. Immunol. 1999;162:2997–3004. [PubMed] [Google Scholar]
- 48.Tekkanat K.K., Maassab H.F., Cho D.S., Lai J.J., John A., Berlin A., Kaplan M.H., Lukacs N.W. IL-13-induced airway hyperreactivity during respiratory syncytial virus infection is STAT6 dependent. J. Immunol. 2001;166:3542–3548. doi: 10.4049/jimmunol.166.5.3542. [DOI] [PubMed] [Google Scholar]
- 49.Schwarze J., Cieslewicz G., Joetham A., Ikemura T., Hamelmann E., Gelfand E.W. CD8 T cells are essential in the development of respiratory syncytial virus-induced lung eosinophilia and airway hyperresponsiveness. J. Immunol. 1999;162:4207–4211. [PubMed] [Google Scholar]
- 50.Brimnes M.K., Bonifaz L., Steinman R.M., Moran T.M. Influenza virus-induced dendritic cell maturation is associated with the induction of strong T cell immunity to a coadministered, normally nonimmunogenic Protein. J. Exp. Med. 2003;198:133–144. doi: 10.1084/jem.20030266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Wang H., Peters N., Laza-Stanca V., Nawroly N., Johnston S.L., Schwarze J. Local CD11c+ MHC II precursors give rise to pulmonary myeloid dendritic cells and are depleted in the process after RSV infection. J. Immunol. 2006;177:2536–2542. doi: 10.4049/jimmunol.177.4.2536. [DOI] [PubMed] [Google Scholar]
- 52.Constant S.L., Brogdon J.L., Piggott D.A., Herrick C.A., Visintin I., Ruddle N.H., Bottomly K. Resident lung antigen-presenting cells have the capacity to promote Th2 T cell differentiation in situ. J. Clin. Invest. 2002;110:1441–1448. doi: 10.1172/JCI16109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Robinson P., Hegele R., Schellenberg R. Allergic sensitization increases airway reactivity in guinea pigs with respiratory syncytial virus bronchiolitis. Clin. Immunol. 1997;100:492–498. doi: 10.1016/s0091-6749(97)70141-6. [DOI] [PubMed] [Google Scholar]
- 54.Makela M., Tripp R., Dakama A. Prior airway exposure to allergen increases virus-induced airway hyperresponsiveness. J. Allergy Clin. Immunol. 2003;112:861–869. doi: 10.1016/s0091-6749(03)02020-7. [DOI] [PubMed] [Google Scholar]
- 55.Wu C., Puddington L., Whiteley H., Yiamouyiannis C., Schramm C., Mohammadu F., Thrall R.S. Murine Cytomegalovirus infection alters Th1/Th2 cytokine expression, decreases airway eosinophilia and enhances mucus production in allergic airway disease. J. Immunol. 2001;167:2798–2807. doi: 10.4049/jimmunol.167.5.2798. [DOI] [PubMed] [Google Scholar]
- 56.Matsuse H., Behera A., Kumar M., Rabb H. Recurrent respiratory syncytial virus infections in allergen sensitized mice lead to persistent airway inflammation and hyperresponsiveness. J. Immunol. 2000;164:6583–6592. doi: 10.4049/jimmunol.164.12.6583. [DOI] [PubMed] [Google Scholar]
- 57.Barends M., Van Oosten M., De Rond C.G., Dormans J.A., Osterhaus A.D. Timing of infection and prior immunization with respiratory syncytial virus in RSV-enhanced allergic inflammation. J. Infect. Dis. 2004;189:1866–1872. doi: 10.1086/386341. [DOI] [PubMed] [Google Scholar]
- 58.Mäkelä M.J., Kanehiro A., Borish L., Dakhama A., Loader J., Joetham A., Xing Z., Jordana M., Larsen G.L., Gelfand E.W. IL-10 is necessary for the expression of airway hyperresponsiveness but not pulmonary inflammation after allergic sensitization. Proc. Natl. Acad. Sci. U.S.A. 2000;97:6007–6012. doi: 10.1073/pnas.100118997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Makela M., Kanehiro A., Borish L. The failure of IL-10 deficient mice to develop airway hyperresponsiveness is overcome by respiratory syncytial virus infection in allergen sensitized/challenged mice. Am. J. Respir. Crit. Care Med. 2002;165:824–831. doi: 10.1164/ajrccm.165.6.2105062. [DOI] [PubMed] [Google Scholar]
- 60.Won Park J., Taube C., Yang E., Joetham A. Respiratory syncytial virus induced airway hyperresponsiveness is independent of IL-13 compared with that induced by allergen. J. Allergy Clin. Immunol. 2003;112:1078–1087. doi: 10.1016/j.jaci.2003.08.046. [DOI] [PubMed] [Google Scholar]
- 61.Kanaoka Y., Boyce J. Cysteinyl leukotrienes and their receptors: cellular distribution and function in immune and inflammatory responses. J. Immunol. 2004;173:1503–1510. doi: 10.4049/jimmunol.173.3.1503. [DOI] [PubMed] [Google Scholar]
- 62.Volovitz B., Welliver R., De Castro G., Krystofik D., Ogra P. The release of leukotrienes in the respiratory tract during infection with respiratory syncytial virus: role in obstructive airway disease. Pediatr. Res. 1988;24:504–507. doi: 10.1203/00006450-198810000-00018. [DOI] [PubMed] [Google Scholar]
- 63.Drazen J.M., O'Brien J., Sparrow D., Weiss S.T., Martins M.A., Israel E., Fanta C.H. Recovery of leukotriene E4 from the urine of patients with airway obstruction. Am. Rev. Respir. Dis. 1992;146:104–108. doi: 10.1164/ajrccm/146.1.104. [DOI] [PubMed] [Google Scholar]
- 64.Kim C.K., Koh J.Y., Han T.H., Kim do K., Kim B.I., Koh Y.Y. Increased levels of BAL cysteinyl leukotrienes in acute RSV bronchiolitis. Acta Pediatr. 2006;95:479–485. doi: 10.1080/08035250600554268. [DOI] [PubMed] [Google Scholar]
- 65.Fullmer J.J., Khan A.M., Elidemir O., Chiappetta C., Stark J.M., Colasurdo G.N. Role of cysteinyl leukotrienes in airway inflammation and responsiveness following RSV infection in BALB/c mice. Pediatr. Allergy Immunol. 2005;16:593–601. doi: 10.1111/j.1399-3038.2005.00248.x. [DOI] [PubMed] [Google Scholar]
- 66.Welliver R.C., 2nd, Hintz K.H., Glori M., Welliver R.C., Sr. Zileuton reduces respiratory illness and lung inflammation, during respiratory syncytial virus infection, in mice. J. Infect. Dis. 2003;187:1773–1779. doi: 10.1086/375277. [DOI] [PubMed] [Google Scholar]
- 67.Bisgaard H. Study Group on Montelukast and Respiratory Syncytial Virus. A randomized trial of montelukast in respiratory syncytial virus postbronchiolitis. Am. J. Respir. Crit. Care Med. 2003;167:379–383. doi: 10.1164/rccm.200207-747OC. [DOI] [PubMed] [Google Scholar]
- 68.Laitinen L., Kava T. Bronchial reactivity following uncomplicated influenza A infection in healthy subjects and in asthmatic patients. Eur. J. Respir. Dis. 1980;106(Suppl):51–58. [PubMed] [Google Scholar]
- 69.Fleming E., Little F., Schnurr D., Avila P., Wong H., Liu J., Yagi S., Boushey H.A. Rhinovirus-16 colds in healthy and in asthmatic subjects. Similar changes in upper and lower airways. Am. J. Crit. Care Med. 1999;160:100–108. doi: 10.1164/ajrccm.160.1.9808074. [DOI] [PubMed] [Google Scholar]
- 70.Lemanske R., Dick E., Swenson C., Vrtis R., Busse W. Rhinovirus upper respiratory infection increases airway hyperreactivity and late asthmatic reactions. J. Clin. Invest. 1989;83:1–10. doi: 10.1172/JCI113843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Zambrano J., Carper H., Rakes G., Patrie J., Murphy D., Platts-Mills T.A., Hayden F., Gwaltney J., Hatley T.K., Owens A.M., Heymann P. Experimental rhinovirus challenges in adults with mild asthma: response to infection in relation to IgE. J. Allergy Clin. Immunol. 2003;111:1008–1016. doi: 10.1067/mai.2003.1396. [DOI] [PubMed] [Google Scholar]
- 72.Calhoun W., Swenson C., Dick E., Schwartz L., Lemanske R., Busse W. Experimental rhinovirus 16 infection potentiates histamine release after antigen bronchoprovocation in allergic subjects. Am. Rev. Respir. Dis. 1991;144:1267–1273. doi: 10.1164/ajrccm/144.6.1267. [DOI] [PubMed] [Google Scholar]
- 73.Calhoun W., Dick E., Schwartz L., Busse W. A common cold virus, rhinovirus 16, potentiates airway inflammation after segmental antigen bronchoprovocation in allergic subjects. J. Clin. Invest. 1994;94:2200–2208. doi: 10.1172/JCI117581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Papadopoulos N., Stanciu L., Papi A., Holgate S., Johnston S. A defective Type 1 response to rhinovirus in atopic asthma. Thorax. 2002;57:328–332. doi: 10.1136/thorax.57.4.328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Mahalingam S., Herd K., Mackay I.M., Nissen M., Sloots T.P., Tindle R.W. Cytotoxic T-lymphocyte epitope vaccination protects against human metapneumovirus infection and disease in mice. J. Virol. 2006;80:2034–2044. doi: 10.1128/JVI.80.4.2034-2044.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]