

Highlights
-
•
Two pharmacophore models have been developed.
-
•
Virtual screening was performed by the pharmacophore models and docking.
-
•
Six selected hits were discovered to have inhibitory activities.
Keywords: Aminopeptidase N, Pharmacophore modeling, Virtual screening, Molecular docking
Abstract
Inhibitors of aminopeptidase N (APN) have been thought as potential drugs for the treatment of tumor angiogenesis, invasion and metastasis and a considerable number of APN inhibitors have been reported recently. To clarify the essential structure–activity relationship for the APN inhibitors as well as identify new potent leads against APN, pharmacophore models were established using structure- and common feature-based approaches and validated with a database of active and inactive compounds. These validated pharmacophores were then used in database screening for novel virtual leads. The hit compounds were further subjected to molecular docking studies to refine the retrieved hits. Finally, six structurally diverse compounds that showed strong interactions with the key amino acids and the zinc ion were selected for biological evaluation, where two hits showed more than 70% inhibition against APN at 60 μM concentration. The evaluation results show the potential of our screening approach in identifying APN inhibitors.
1. Introduction
Aminopeptidase N (APN), a zinc-dependent type II membrane-bond ectopeptidase, is widely expressed in hematopoietic cells of myeloid origin and non-hematopoietic cells and tissues, such as fibroblasts, brain cells and epithelial cells of the liver, kidney, and intestine [1], [2], [3], [4], [5]. In vivo, APN is involved in the metabolism of angiotensin III in the degradation of nociceptin, in the brain and peripheral organs and in the inactivation of enkephalins, in association with neutral endopeptidase NEP [6], [7], [8]. Moreover, this enzyme has been proved to behave as a receptor for coronaviruses TGEV and 229E in humans and pigs, and to be identical to a human lymphocyte surface cluster differentiation antigen CD13 [9], [10], [11]. APN has been also reported to play a vital role in the invasion of metastatic tumors in vitro [12], [13]. All these findings make APN an important target for therapeutic applications, which require the development of selective and potent inhibitors [14].
Since the first marketed anti-APN drug bestatin has been launched in 1976, more and more APN inhibitors such as probestin, lapstatin and AHPA-Val that have displayed efficient inhibitory ability, have been reported [1]. Bestatin and amastatin have been the most extensively studied. However, recently the clinical use of these APN inhibitors, has been limited due to their low selectivity and important drug resistance [14], [15].
Though many different APN inhibitors have been synthesized and experimentally assessed, there is no information available regarding the discovery of structurally novel leads up to date. Thus, it is important to develop new small molecule inhibitors with novel modes of action. The development of such kind of drug is focused on how to design novel APN inhibitors. Chemical feature-based pharmacophores and virtual library screening may serve as a guide in the design of novel lead candidates. This study aims to construct the chemical feature-based pharmacophore model and identify novel scaffolds with the potential to turn as the new category of APN inhibitors.
In our study, we have successfully used pharmacophore modeling, database screening and molecular docking approach to identify lead candidates with potent inhibitory activities. Pharmacophore models were generated by structure- and common feature-based approach. The pharmacophore models were validated with a database of active and inactive compounds and used as a 3D structural search query to find new potent leads from the database. The binding free energy and molecular interactions with the active site residues were considered as important components to identify the potential leads.
2. Materials and methods
2.1. Structure based approach
The high-resolution crystal structure of human aminopeptidase N (CD13) (PDB ID: 4FYR, 1.91 Å) in complex with bestatin was chosen to generate a structure-based pharmacophore model that can determine the important pharmacophoric features for ligand binding. Binding Site Analysis tool available in Discovery Studio 2.5 (DS 2.5; Accelrys, Inc., San Diego, CA, USA), version 2.5, from Accelrys (San Diego, USA) has been used to create a sphere with a radius that comprises all the catalytically important amino acid residues in the ligand-binding pocket [16]. The Interaction Generation protocol implemented in DS was used to identify hydrogen bond acceptor (HBA), hydrogen bond donor (HBD), hydrophobic (HY) and ring aromatic (RA) features based on the active site residues that are inside the sphere. These features were then clustered and the most representative features were selected and included in the pharmacophore model (Pharmacophore-A).
2.2. Establishment of the common feature-pharmacophore
The selection of a suitable training set is essential for the quality of automatically generated pharmacophore models. A set of 7 structurally diverse aminopeptidase-N inhibitors were selected from literature [17], [18], [19], [20], [21], [22], [23] as the training set to establish the common feature-based pharmacophores. The 2D chemical structures of selected molecules were draw manually using ChemDraw (CambridgeSoft, Cambridge, MA) and subsequently exported to DS. All compounds were minimized to the closest local minimum based on the Charmm-like force field (DS). The HipHop module available in DS was employed to establish the pharmacophore. For the calculation setup, two ligand properties have to be specified. The first property is the principal value that defines the activity level of the molecule. A principal value of 2 indicates all chemical features of a reference compound are considered in building the pharmacophore space during model generation. The second ligand property to define is the MaxOmitFeat value that specifies how many features are allowed to miss for each compound. A MaxOmitFeat value of 1 allows that all but one of the features in the generated pharmacophore must map the molecule. Compounds 1–2 were assigned a principal value of 2 and a MaxOmitFeat value of 0, respectively, while compounds 3–7 were given principal and MaxOmitFeat values of 1. On the basis of an analysis of the chemical features present in the training set structures, hydrogen bond donor, hydrogen bond acceptor, and hydrophobic features were selected for this study. A maximum number of 255 and an energy threshold of 10 kcal/mol above the energy minimum for conformation searching were set for each compound by using Best conformation generation option. Among the 10 possible hypotheses returned, the top ranked hypothesis (Pharmacophore-B) was selected and validated by the Güner–Henry (GH) scoring method.
2.3. Validation and database searching
An external database contains 1100 compounds [[1], [14], [24], [25], [26], [27], [28]] structurally different from the training set molecules. These compounds in the database were used as a test set to validate the discriminative ability of the best pharmacophore model in distinguishing active compounds from the inactive ones. All queries were performed using Ligand Pharmacophore Mapping protocol. The Güner–Henry (GH) scoring method was applied to quantify model selectivity precision of hits and the recall of actives from a dataset containing known actives and inactives. This method includes the percent ratio of actives in the hit list (%A, precision), the percent yield of actives in a database (%Y, recall), the GH score, and the enrichment factor (E). The high is the E value the great is the ability of a pharmacophore in identifying the active compounds. The GH test score ranges from 0 to 1, where a score close to 1 indicates an ideal model. The following is the proposed metrics for analyzing hit lists by a pharmacophore model-based database search:
A, the number of active compounds in the database; D, the number of compounds in the database; Ha, the number of actives in the hit list (true positives); Ht, the number of hits retrieved; %Y, the percentage of known actives in the hit list (recall); %A is the percentage of known active compounds retrieved from the database (precision); GH is the Güner–Henry score and E, the enrichment of the concentration of actives by the model relative to random screening without any pharmacophoric approach [29], [30], [31], [32], [33], [34]. To identify novel hit compounds, the validated pharmacophore model was used as 3D query to screen our in-house database with 35,000 compounds. Ligand Pharmacophore Mapping protocol was applied in database screening. Finally, the retrieved compounds were selected based on with a fit value higher than 3, the binding free energy and molecular interactions with the key amino acids at the active site.
2.4. Molecular docking
The Molecular Operating Environment (MOE) program, version 2008 (Chemical Computing Group Inc, Montreal, Quebec, Canada) was used to perform all molecular docking calculations. The docking compounds were built using the MOE builder interface and subjected to energy minimization and the partial charges were computed using MMFF94x force field [35]. The crystal structure of APN (PDB ID: 4FYR) was prepared for the docking studies where: (i) hydrogen atoms were added to the structure with their standard geometry; (ii) the partial charges were computed using Amber99 force field [35]. The active site was defined with a 6.5 Å radius around the bound inhibitor (bestatin) in the crystal structure of APN. Molecular docking calculations were done using Triangle Matcher placement method with London dG scoring. Based on the binding mode and molecular interactions observed in the active site, the final hits were selected.
2.5. In vitro APN inhibition assay
The IC50 values against APN were determined by using l-Leu-p-nitroanilide as substrate and Microsomal aminopeptidase from Porcine Kidney Microsomes as enzyme in 50 mM PBS (pH 7.2). After adding the detected compounds, the solution with various concentrations was incubated with APN at 37 °C for 5 min. Then the solution of substrate was added into the above mixture, which was incubated for another 30 min at 37 °C. The hydrolysis of the substrate was measured by following the change in the absorbance monitored at 405 nm with a plate reader (Varioskan, Thermo, USA).
3. Results and discussion
A workflow overview of this study including pharmacophore modeling, selection of compounds, and biological testing is provided in Fig. 1 .
Fig. 1.
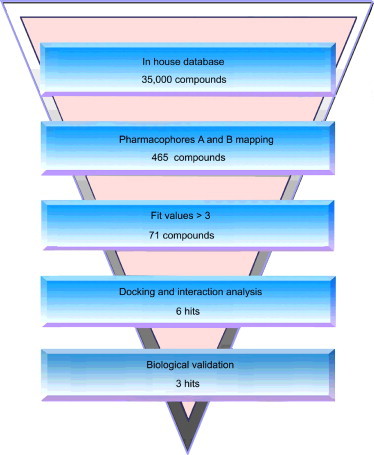
A workflow overview of pharmacophore modeling, selection of compounds, and biological testing.
3.1. Structure based pharmacophore generation
In the present work, the final clustered pharmacophore (Pharmacophore-A) was generated using a structure-based modeling approach. This pharmacophore model was basically composed of one hydrogen-bond acceptor (HBA) and two hydrogen-bond donor (HBD) features. The 3D space and distance constraints of these features are shown in Fig. 2 A. To further include additional information on size and shape of the bestatin-binding pocket of APN, a shape constraint was added to the chemical features of the pharmacophore Hypo1. As displayed in Fig. 3, the carbonyl group of bestatin that formed very important interactions with a zinc ion and Tyr477 in the binding site of APN overlaid the HBA feature of pharmacophore-A. In addition, the hydroxyl group of this molecule showing an ionic interaction with the zinc ion mapped one HBD feature, while the amino group that enabled considerable interactions with water molecules mapped the other HBD feature. The result indicated a fairly good mapping of bestatin on pharmacophore-A with the shape constraint. However, it is also implied that the pharmacophore-A may be utilized to only screen the compounds that have a chemical structure with functional groups closely related to those of bestatin. Thus a qualitative pharmacophore model (a common feature-based model or HipHop algorithm) was very necessary to identify structurally novel leads different from the bestatin.
Fig. 2.
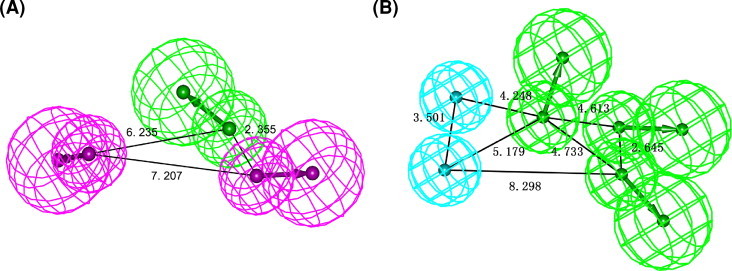
Generated pharmacophore models with distance constraints. (A) Structure-based pharmacophore (Pharmacophore-A); (B) common feature pharmacophore (Pharmacophore-B). Pharmacophore features are color-coded: cyan, hydrophobic (HY); magenta, hydrogen-bond donor (HBD); green, hydrogen-bond acceptor (HBA). (For interpretation of the references to color in this figure legend, the reader is referred to the web version of this article.)
Fig. 3.
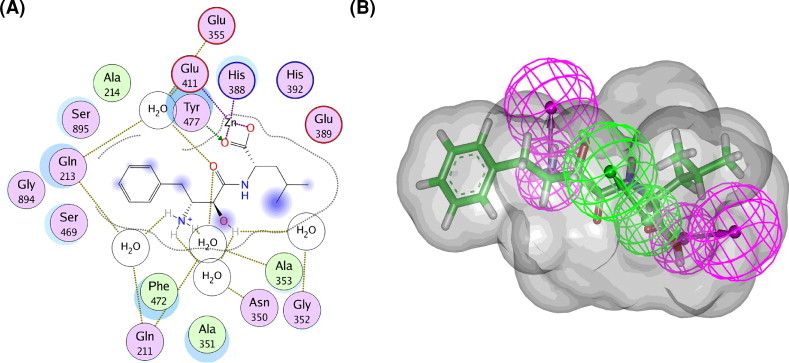
Interaction analysis. (A) 2D interaction diagram for the binding site of APN with the bound inhibitor (bestatin). (B) Overlay of the bestatin upon pharmacophore-A with a shape constraint. Pharmacophore features are color-coded: magenta, hydrogen-bond donor (HBD); green, hydrogen-bond acceptor (HBA). (For interpretation of the references to color in this figure legend, the reader is referred to the web version of this article.)
3.2. Common feature pharmacophore generation
A qualitative pharmacophore model for APN inhibitors was developed using the HipHop algorithm of DS. Based on the principle of structural diversity, the seven most representative APN inhibitors (Fig. 4 ) were selected to be used as a training set in common feature pharmacophore modeling study. All of them have the hydroxyl or carbonyl group as metal (zinc ion) binding region, and a long hydrophobic spacer to bind with the hydrophobic residues in the bestatin-binding site of APN. Ten common feature pharmacophore models were generated using this training set where all of them had similar number of pharmacophoric features “five features” (Table 1 ) yet various orientations and vector directions. Top ranked pharmacophore (Pharmacophore-B) with three hydrogen-bond acceptor (HBA) and two hydrophobic (HY) features was chosen as a best pharmacophore model (Fig. 2B).
Fig. 4.
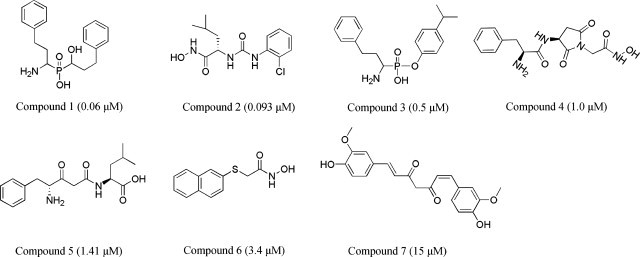
Training set compounds with IC50 values used in common feature pharmacophore generation.
Table 1.
Results of common feature pharmacophore generation.
| Hypos | Features | Rank | Direct hit | Partial hit | Max. fit |
|---|---|---|---|---|---|
| 1 | HY HY HBA HBA HBA | 68.866 | 1110111 | 0001000 | 5 |
| 2 | HY HY HBA HBA HBA | 67.927 | 1110111 | 0001000 | 5 |
| 3 | HY HY HBA HBA HBA | 67.484 | 1110111 | 0001000 | 5 |
| 4 | HY HY HBA HBA HBA | 67.312 | 1110111 | 0001000 | 5 |
| 5 | HY HY HBA HBA HBA | 67.312 | 1110111 | 0001000 | 5 |
| 6 | HY HY HBA HBA HBA | 67.012 | 1110111 | 0001000 | 5 |
| 7 | HY HY HBA HBA HBA | 66.839 | 1110111 | 0001000 | 5 |
| 8 | HY HY HBA HBA HBA | 66.735 | 1110111 | 0001000 | 5 |
| 9 | HY HY HBA HBA HBA | 66.735 | 1110111 | 0001000 | 5 |
| 10 | HY HY HBA HBA HBA | 66.213 | 1110111 | 0001000 | 5 |
3.3. Validation and database searching
An internal database including 1100 compounds was used for the validation process. This database has been created with 1030 inactive compounds and 70 inhibitors with known experimental activity. To validate the ability to distinguish the actives from inactive compounds, the two pharmacophores A and B were used as a 3D query to screen the internal database using Ligand Pharmacophore Mapping protocol in DS. A set of statistical parameters such as total hits (Ht), % ratio of actives, % yield of actives, enrichment factor (E), false negative, false positives, and goodness of hit score (GH) were calculated (Table 2 ). Pharmacophore-A showed an E value of 12.29 by mapping 61 active molecules from the 78 screened compounds from the database. The E value for pharmacophore-B was 13.57 as it has retrieved 57 active compounds among 66 screened molecules from the total of 1100 compounds. The higher is the E value, the greater is the ability of a pharmacophore in identifying the active compounds. This validation results showed that both pharmacophore models A and B were very efficient for database screening. When GH score is higher than 0.7, the model is very good. It was observed to be 0.79 and 0.84 for the pharmacophores A and B, which indicates a good ability to distinguish the active compounds from the inactive ones.
Table 2.
Validation results for the two generated pharmacophore models.
| Parameter | Pharmacophore-A | Pharmacophore-B |
|---|---|---|
| Total molecules in database (D) | 1100 | 1100 |
| Total number of actives in database (A) | 70 | 70 |
| Total hits (Ht) | 78 | 66 |
| Active hits (Ha) | 61 | 57 |
| % Yield of actives[(Ha/Ht) × 100] | 78.21% | 86.36% |
| % Ratio of actives [(Ha/A) × 100] | 87.14% | 81.42% |
| Enrichment factor (E) [(Ha × D)/(Ht × A)] | 12.29 | 13.57 |
| False negatives [A-Ha] | 9 | 13 |
| False positives [Ht-Ha] | 17 | 9 |
| Goodness of hit score (GH)a | 0.79 | 0.84 |
The two validated pharmacophore models were used as a query to screen our in-house database consisting of 35,000 compounds. As a result, 465 molecules were selected to be screened by the models A and B. To reduce the number of false positives and false negatives, the 71 retrieved compounds with a fit value higher than 3 were further subjected to docking studies.
3.4. Molecular docking
A molecular docking study could provide more insight into understanding the protein–ligand interactions and the structural features in the active site of protein. Beginning of the docking, it is very necessary to validate the docking reliability. The ligand (bestatin) of the crystal structured APN–bestatin complex (PDB ID: 4FYR) was adopted as a template to perform the validation. The ligand bestatin was flexibly redocked to the binding site of APN and the docking conformation corresponding to the lowest energy score was selected as the most probable binding conformation. As a result, the redocked bestatin was almost at the same position in the active site of the crystal of APN complex, suggesting a high docking reliability of Triangle Matcher placement method with London dG scoring (Fig. 5). Therefore, the docking protocol could be extended to search the binding conformations of the retrieved hits from the database.
Fig. 5.
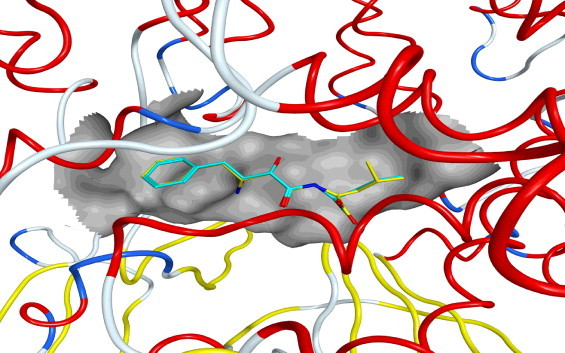
Molecular overlay of the docking and original positions of the bound inhibitor (bestatin) in the active sits of APN. The compounds are color-coded: yellow, the docking position of bestatin; cyan, the original position of bestatin. (For interpretation of the references to color in this figure legend, the reader is referred to the web version of this article.)
To further refine the retrieved hits and also to remove the false positives, the 71 retrieved compounds were docked into the bestatin-binding site of APN using the validated docking parameters. Upon visual inspection of 10 top ranked compounds, six hits showing key interactions with the zinc ion were selected for biological validation.
3.5. Biological activities of retrieved molecules from databases
To validate the reliability of two generated pharmacophore hypotheses, all the hits (hits 1–6) were tested for in vitro inhibition activity against APN. The biological activity testing results are reported as % inhibition and are shown in Table 3 . Hits 2 and 3 selected by pharmacophore-B (Fig. 6A) exhibited more than 70% inhibition of APN, while hit 1 selected by pharmacophore-A (Fig. 6B) inhibited 56% of APN; none of hits 4–6 selected by pharmacophore-A displayed APN inhibition more than 50%. The two hits retrieved by pharmacophore-B showed higher activities than hits retrieved by pharmacophore-A. Based on all of these data, pharmacophore-B with two hydrophobic and three hydrogen bond acceptor features is chosen as a good representative computer-generated model for identifying new APN inhibitors.
Table 3.
Hits selected for biological validation: Docking scores, fit values and APN inhibition at 60 μmol/L concentration.
| Hits | Structure | Docking score (kal/mol) | Pharmacophore (fit value) |
% Inhibition | |
|---|---|---|---|---|---|
| A | B | ||||
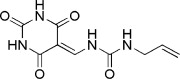 |
|||||
| 1 | −15.9568 | 3.4792 | 56% | ||
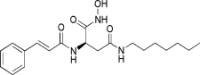 |
|||||
| 2 | −17.5095 | 4.9429 | 71% | ||
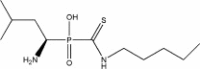 |
|||||
| 3 | −17.7841 | 4.9586 | 89% | ||
 |
|||||
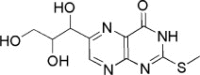
| 4 | −15.8006 | 3.1543 | <50 | ||
 |
|||||
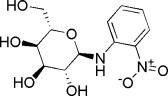
| 5 | −15.2629 | 3.1351 | <50 | ||
 |
|||||
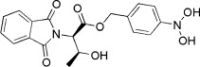
| 6 | −15.2066 | 3.0792 | <50 | ||
 |
|||||
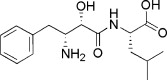
| Bestatin | 75% | ||||
Fig. 6.
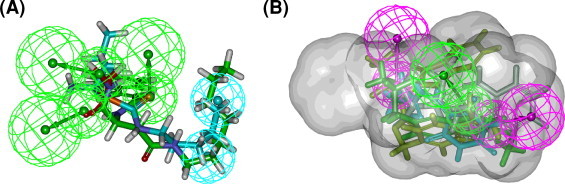
Pharmacophore mapping of six hits. (A) Hits 1 and 4–6 mapped on pharmacophore-A; (B) hits 2 and 3 mapped on pharmacophore-B. Pharmacophore features are color-coded: cyan, hydrophobic (HY); magenta, hydrogen-bond donor (HBD); green, hydrogen-bond acceptor (HBA). (For interpretation of the references to color in this figure legend, the reader is referred to the web version of this article.)
As shown in Table 3, the activity profile of six hits correlate with the fit values computed from the hypothesis and the docking sores. The most active hits are found to have a high docking score and fit value. Among four hits identified by pharmacophore-A, hit 1 showed the best inhibition rate (56%), fit value (3.4792), and docking score (−15.9568). Similarly, hit 3 screened by pharmacophore-B, displayed a stronger inhibition (89%), higher fit value (4.9586) and better docking score (−17.7841 kcal/mol) than hit 2. Overall, the virtual and experimental data demonstrates that our screening approach has good potential in identifying APN inhibitors.
3.6. Computational studies of the binding modes
To further study the binding modes of the two most potent hits (hits 2 and 3) to APN, it was docked into the bestatin-binding site of APN. The 2D interaction map of hit 2 with the active site of APN is represented in Fig. 7 A. The phenyl ring of hit 2 exhibited a cation-π stacking interaction with Arg381, while the nitrogen and oxygen atoms formed very important interactions with zinc ion and water molecules in the active site of APN. In addition, it established hydrophobic interactions with Phe472, Phe896 and Ala351. As shown in Fig. 7B, the phosphinic acid group of hit 3 displayed a vital ionic interaction with zinc ion and hydrogen-bond interactions with Tyr477 and water molecules and two nitrogen atoms were engaged in hydrogen-bond interactions with Glu389 or water molecules. Moreover, this molecule also showed hydrophobic interactions with Ala353, Phe472, Ala351, Met354 and Val385. The understanding of the interactions between APN and the hits would be helpful to rationally design novel and potent APN inhibitors.
Fig. 7.
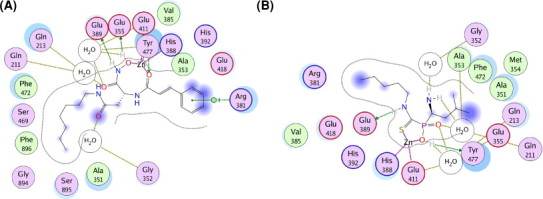
2D (two-dimensional) ligand-protein interaction diagrams for the binding site of APN with hits 2 and 3. (A) Hit 2. (B) Hit 3. The active site residues are represented as follows: polar residues in light purple, hydrophobic residues in green, acidic residues with a red contour ring, basic residues with a blue contour ring. Green and blue arrows indicate hydrogen bonding to side chain and backbone atoms respectively. The cation-π stacking interactions are represented in green dotted lines. Light-blue “halos” around residues indicate the degree of interaction with ligand atoms (larger and darker halos means more interaction). (For interpretation of the references to color in this figure legend, the reader is referred to the web version of this article.)
4. Conclusion
In the present work, two pharmacophore models A and B were developed using structure- and common feature-based approaches. Both the pharmacophore models have provided the first insight into hypothetical ligand binding requirements for APN inhibitors. The validated results of in vitro enzymatic inhibitions on APN imply that pharmacophore-B composed of five features is better than pharmacophore-A in identifying potent APN inhibitors. The inhibition rates of the hits correlate with the fit values computed from the hypothesis and the docking scores. It is possible that searching of other commercial databases may result in the identification of other potentially more active APN inhibitors. We have shifted hits obtained from virtual screening to in vivo studies, the outcomes of which will be reported in the near future.
Acknowledgement
The authors are grateful to Natural Science Foundation Committee of China (NSFC 81220108012, 61335007, 81371684, 81000666, 81171395 and 81328012) for financial support.
Contributor Information
Miaomiao Niu, Email: niumm2012@126.com.
Fengzhen Wang, Email: 392839342@qq.com.
Fang Li, Email: 735313568@qq.com.
Yaru Dong, Email: zhuxituan00@126.com.
Yueqing Gu, Email: guyueqingsubmission@hotmail.com.
References
- 1.Jin K, Zhang X, Ma C, Xu Y, Yuan Y, Xu W. Novel indoline-2,3-dione derivatives as inhibitors of aminopeptidase N (APN) Bioorg Med Chem. 2013;21:2663–2670. doi: 10.1016/j.bmc.2012.06.024. [DOI] [PubMed] [Google Scholar]
- 2.Piedfer M, Dauzonne D, Tang R, N'Guyen J, Billard C, Bauvois B. Aminopeptidase-N/CD13 is a potential proapoptotic target in human myeloid tumor cells. FASEB J. 2011;25:2831–2842. doi: 10.1096/fj.11-181396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Dixon J, Kaklamanis L, Turley H, Hickson ID, Leek RD, Harris AL. Expression of aminopeptidase-n (CD 13) in normal tissues and malignant neoplasms of epithelial and lymphoid origin. J Clin Pathol. 1994;47:43–47. doi: 10.1136/jcp.47.1.43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Raynaud F, Bauvois B, Gerbaud P, Evain-Brion D. Characterization of specific proteases associated with the surface of human skin fibroblasts, and their modulation in pathology. J Cell Physiol. 1992;151:378–385. doi: 10.1002/jcp.1041510219. [DOI] [PubMed] [Google Scholar]
- 5.Piela-Smith TH, Korn JH. Aminopeptidase N: a constitutive cell-surface protein on human dermal fibroblasts. Cell Immunol. 1995;162:42–48. doi: 10.1006/cimm.1995.1049. [DOI] [PubMed] [Google Scholar]
- 6.Zini S, Fournie-Zaluski MC, Chauvel E, Roques BP, Corvol P, Llorens-Cortes C. Identification of metabolic pathways of brain angiotensin II and III using specific aminopeptidase inhibitors: predominant role of angiotensin III in the control of vasopressin release. Proc Natl Acad Sci U S A. 1995;93:11968–11973. doi: 10.1073/pnas.93.21.11968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Montiel JL, Cornille F, Roques BP, Noble F. Nociceptin/orphanin FQ metabolism: role of aminopeptidase and endopeptidase 24.15. J Neurochem. 1997;68:354–361. doi: 10.1046/j.1471-4159.1997.68010354.x. [DOI] [PubMed] [Google Scholar]
- 8.Roques BP, Noble F, Daugé V, Fournié-Zaluski MC, Beaumont A. Neutral endopeptidase 24.11: structure, inhibition, and experimental and clinical pharmacology. Pharmacol Rev. 1993;45:87–146. [PubMed] [Google Scholar]
- 9.Look AT, Ashmun RA, Shapiro LH, Peiper SC. Human myeloid plasma membrane glycoprotein CD13 (gp150) is identical to aminopeptidase N. J Clin Invest. 1989;83:1299–1307. doi: 10.1172/JCI114015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Delmas B, Gelfi J, L'Haridon R, Vogel LK, Sjöström H, Norén O. Aminopeptidase N is a major receptor for the entero-pathogenic coronavirus TGEV. Nature. 1992;357:417–420. doi: 10.1038/357417a0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Yeager CL, Ashmun RA, Williams RK, Cardellichio CB, Shapiro LH, Look AT. Human aminopeptidase N is a receptor for human coronavirus 229E. Nature. 1992;357:420–422. doi: 10.1038/357420a0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Saiki I, Fujii H, Yoneda J, Abe F, Nakajima M, Tsuruo T. Role of aminopeptidase N (CD13) in tumor-cell invasion and extracellular matrix degradation. Int J Cancer. 1993;54:137–143. doi: 10.1002/ijc.2910540122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Menrad A, Speicher D, Wacker J, Herlyn M. Biochemical and functional characterization of aminopeptidase N expressed by human melanoma cells. Cancer Res. 1993;53:1450–1455. [PubMed] [Google Scholar]
- 14.Chen H, Roques BP, Fournié-Zaluski MC. Design of the first highly potent and selective aminopeptidase N (EC 3.4.11.2) inhibitor. Bioorg Med Chem Lett. 1999;9:1511–1516. doi: 10.1016/S0960-894X(99)00219-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Jin K, Zhang X, Ma C, Xu Y, Yuan Y, Xu W. Novel indoline-2,3-dione derivatives as inhibitors of aminopeptidase N (APN) Bioorg Med Chem. 2013;21:2663–2670. doi: 10.1016/j.bmc.2012.06.024. [DOI] [PubMed] [Google Scholar]
- 16.Thangapandian S, John S, Sakkiah S, Lee KW. Ligand and structure based pharmacophore modeling to facilitate novel histone deacetylase 8 inhibitor design. Eur J Med Chem. 2010;45:4409–4417. doi: 10.1016/j.ejmech.2010.06.024. [DOI] [PubMed] [Google Scholar]
- 17.Lee J, Shim JS, Jung SA, Lee ST, Kwon HJ. N-hydroxy-2-(naphthalene-2-ylsulfanyl)-acetamide, a novel hydroxamic acid-based inhibitor of aminopeptidase N and its anti-angiogenic activity. Bioorg Med Chem Lett. 2005;15:181–183. doi: 10.1016/j.bmcl.2004.10.010. [DOI] [PubMed] [Google Scholar]
- 18.Ma C, Jin K, Cao J, Zhang L, Li X, Xu W. Novel leucine ureido derivatives as inhibitors of aminopeptidase N (APN) Bioorg Med Chem. 2013;21:1621–1627. doi: 10.1016/j.bmc.2013.01.068. [DOI] [PubMed] [Google Scholar]
- 19.Grzywa R, Sokol AM, Sieńczyk M, Radziszewicz M, Kościołek B, Carty MP. New aromatic monoesters of alpha-aminoaralkylphosphonic acids as inhibitors of aminopeptidase N/CD13. Bioorg Med Chem. 2010;18:2930–2936. doi: 10.1016/j.bmc.2010.02.056. [DOI] [PubMed] [Google Scholar]
- 20.Ma C, Li X, Liang X, Jin K, Cao J, Xu W. Novel β-dicarbonyl derivatives as inhibitors of aminopeptidase N (APN) Bioorg Med Chem Lett. 2013;23:4948–4952. doi: 10.1016/j.bmcl.2013.06.058. [DOI] [PubMed] [Google Scholar]
- 21.Li Q, Fang H, Wang X, Hu G, Wang Q, Xu W. Novel potent 2,5-pyrrolidinedione peptidomimetics as aminopeptidase N inhibitors. Design, synthesis and activity evaluation. Bioorg Med Chem Lett. 2012;22:850–853. doi: 10.1016/j.bmcl.2011.12.048. [DOI] [PubMed] [Google Scholar]
- 22.Shim JS, Kim JH, Cho HY, Yum YN, Kim SH, Park HJ. Irreversible inhibition of CD13/aminopeptidase N by the antiangiogenic agent curcumin. Chem Biol. 2003;10:695–704. doi: 10.1016/s1074-5521(03)00169-8. [DOI] [PubMed] [Google Scholar]
- 23.Grzywa R, Oleksyszyn J, Salvesen GS, Drag M. Identification of very potent inhibitor of human aminopeptidase N (CD13) Bioorg Med Chem Lett. 2010;20:2497–2499. doi: 10.1016/j.bmcl.2010.02.111. [DOI] [PubMed] [Google Scholar]
- 24.Ma C, Jin K, Cao J, Zhang L, Li X, Xu W. Novel leucine ureido derivatives as inhibitors of aminopeptidase N (APN) Bioorg Med Chem. 2013;21:1621–1627. doi: 10.1016/j.bmc.2013.01.068. [DOI] [PubMed] [Google Scholar]
- 25.Zhang X, Fang H, Zhu H, Wang X, Zhang L, Li M. Novel aminopeptidase N (APN/CD13) inhibitors derived from 3-phenylalanyl-N'-substituted-2,6-piperidinedione. Bioorg Med Chem. 2010;18:5981–5987. doi: 10.1016/j.bmc.2010.06.078. [DOI] [PubMed] [Google Scholar]
- 26.Su L, Jia Y, Zhang L, Xu Y, Fang H, Xu W. Design, synthesis and biological evaluation of novel amino acid ureido derivatives as aminopeptidase N/CD13 inhibitors. Bioorg Med Chem. 2012;20:3807–3815. doi: 10.1016/j.bmc.2012.04.035. [DOI] [PubMed] [Google Scholar]
- 27.Grzywa R, Oleksyszyn J. First synthesis of alpha-aminoalkyl-(N-substituted)thiocarbamoyl-phosphinates: inhibitors of aminopeptidase N (APN/CD13) with the new zinc-binding group. Bioorg Med Chem Lett. 2008;18:3734–3736. doi: 10.1016/j.bmcl.2008.05.050. [DOI] [PubMed] [Google Scholar]
- 28.Su L, Fang H, Yang K, Xu Y, Xu W. Design, synthesis and biological evaluation of novel L-lysine ureido derivatives as aminopeptidase N inhibitors. Bioorg Med Chem. 2011;19:900–906. doi: 10.1016/j.bmc.2010.11.066. [DOI] [PubMed] [Google Scholar]
- 29.Lu SH, Wu JW, Liu HL, Zhao JH, Liu KT, Chuang CK. The discovery of potential acetylcholinesterase inhibitors: a combination of pharmacophore modeling, virtual screening, and molecular docking studies. J Biomed Sci. 2011;18:8. doi: 10.1186/1423-0127-18-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.John S, Thangapandian S, Arooj M, Hong JC, Kim KD, Lee KW. Development, evaluation and application of 3D QSAR Pharmacophore model in the discovery of potential human renin inhibitors. BMC Bioinformatics. 2011;12(Suppl. 14):S4. doi: 10.1186/1471-2105-12-S14-S4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Arooj M, Thangapandian S, John S, Hwang S, Park JK, Lee KW. 3D QSAR pharmacophore modeling, in silico screening, and density functional theory (DFT) approaches for identification of human chymase inhibitors. Int J Mol Sci. 2011;12:9236–9264. doi: 10.3390/ijms12129236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lipinski CA, Lombardo F, Dominy BW, Feeney PJ. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv Drug Deliv Rev. 2001;46:3–26. doi: 10.1016/s0169-409x(00)00129-0. [DOI] [PubMed] [Google Scholar]
- 33.Dearden JC. In silico prediction of drug toxicity. J Comput Aided Mol Des. 2003;17:119–127. doi: 10.1023/a:1025361621494. [DOI] [PubMed] [Google Scholar]
- 34.Sutherland JJ, Raymond JW, Stevens JL, Baker TK, Watson DE. Relating molecular properties and in vitro assay results to in vivo drug disposition and toxicity outcomes. J Med Chem. 2012;55:6455–6466. doi: 10.1021/jm300684u. [DOI] [PubMed] [Google Scholar]
- 35.Purushottamachar P, Patel JB, Gediya LK, Clement OO, Njar VC. First chemical feature-based pharmacophore modeling of potent retinoidal retinoic acid metabolism blocking agents (RAMBAs): identification of novel RAMBA scaffolds. Eur J Med Chem. 2012;47:412–423. doi: 10.1016/j.ejmech.2011.11.010. [DOI] [PMC free article] [PubMed] [Google Scholar]