

Abstract
Barley stripe mosaic virus (BSMV) contains three positive-sense, single-stranded genomic RNAs, designated α, β, and γ, that encode seven major proteins and one minor translational readthrough protein. Three proteins (αa, βa, and γa) are translated directly from the genomic RNAs and the remaining proteins encoded on RNAβ and RNAγ are expressed via three subgenomic messenger RNAs (sgRNAs). sgRNAβ1 directs synthesis of the triple gene block 1 (TGB1) protein. The TGB2 protein, the TGB2′ minor translational readthrough protein, and the TGB3 protein are expressed from sgRNAβ2, which is present in considerably lower abundance than sgRNAβ1. A third sgRNA, sgRNAγ, is required for expression of the γb protein. We have used deletion analyses and site-specific mutations to define the boundaries of promoter regions that are critical for expression of the BSMV sgRNAs in infected protoplasts. The results reveal that the sgRNAβ1 promoter encompasses positions −29 to −2 relative to its transcription start site and is adjacent to a cis-acting element required for RNAβ replication that maps from −107 to −74 relative to the sgRNAβ1 start site. The core sgRNAβ2 promoter includes residues −32 to −17 relative to the sgRNAβ2 transcriptional start site, although maximal activity requires an upstream hexanucleotide sequence residing from positions −64 to −59. The sgRNAγ promoter maps from −21 to +2 relative to its transcription start site and therefore partially overlaps the γa gene. The sgRNAβ1, β2, and γ promoters also differ substantially in sequence, but have similarities to the putative homologous promoters of other Hordeiviruses. These differences are postulated to affect competition for the viral polymerase, coordination of the temporal expression and abundance of the TGB proteins, and constitutive expression of the γb protein.
Introduction
The synthesis of subgenomic messenger RNAs (sgRNAs) is a common strategy employed by positive-sense RNA viruses to mediate expression and regulation of 3′ proximal open reading frames (ORFs) on multicistronic genomic RNAs. Three general mechanisms have been proposed for sgRNA synthesis Choi and White 2002, White 2002. The most commonly accepted mechanism, internal initiation from negative-sense RNA templates (Miller et al., 1985), appears to be operating during sgRNA synthesis by the Bromoviruses Siegel et al 1997, Siegel et al 1998, Alfamoviruses (Van Der Kuyl et al., 1990), Turnip crinkle virus (TCV) (Wang and Simon, 1997), and several other viruses (Miller and Koev, 2000). In contrast, a discontinuous mechanism of transcription that produces sgRNAs with 5′ and 3′ sequences identical to the genomic RNAs has been reported for the large Coronaviruses (Sawicki and Sawicki, 1998) and Arterioviruses (Pasternak et al., 2001). These sgRNAs arise from minus-strand subgenomic templates produced via polymerase jumping during transcription from genomic RNA templates. A third mechanism, premature termination, has been reported for Flock house virus (FHV) (Zhong and Ruekert, 1993; Lindenbach et al., 2000), Red clover necrotic mosaic virus (RCNMV) (Sit et al., 1998), and Tomato bushy stunt virus (TBSV) (reviewed by White, 2002). In these cases, truncated negative-sense RNAs that had been generated by premature termination at regions of secondary structure of genomic RNA are postulated to serve as templates for the positive-sense sgRNAs. In the case of FHV and TBSV, cis-acting sequences within the genomic RNA are postulated to mediate long distance interactions that contribute to premature polymerase termination during transcription of the negative-sense templates (White, 2002), whereas in the case of RCNMV, termination during transcription from the RNA 1 template requires base pairing of trans-activator sequences residing on RNA 2 with trans-activator binding sequences on RNA 1 (Sit et al., 1998). Other less well-defined examples of premature termination appear to occur during synthesis of two classes of 5′ coterminal sgRNAs appearing in Citrus tristiza virus (CTV) infected plants (Che et al., 2001). Although the possible functions of the CTV sgRNAs have not been resolved, the synthesis of the two species appears to be regulated because they exhibit some differences in the timing of their appearance and they are the earliest and the most abundant sgRNAs synthesized.
The core sgRNA promoters have been mapped and characterized in a variety of plant RNA viruses that synthesize either single or multiple sgRNAs (Miller and Koev, 2000). These promoters range in size from less than 30 nucleotides (nt) Johnston and Rochon 1995, Wang et al 1999 to nearly 150 nt Van Der Vossen et al 1995, Koev and Miller 2000, and they normally reside upstream of, or encompass only a few nucleotides downstream of, the transcription initiation site. However, a few promoters include substantial regions downstream of the transcription initiation site Balmori et al 1993, Koev and Miller 2000. For example, the Beet necrotic yellow vein virus (BNYVV) RNA3 sgRNA promoter occupies more than 100 nt downstream of the transcription start site (Balmori et al., 1993) and the Barley yellow dwarf virus (BYDV) sgRNA2 promoter resides entirely within the sgRNA. In addition to this complexity and variation among RNA viruses, long distance interactions have also been noted in several viruses (Miller and Keov, 2000). In the case of Tobacco mosaic virus (TMV), the 3′ untranslated region (UTR) contains three pseudoknot structures whose ectopic placement appears to redistribute polymerase activity to the closest upstream sgRNA promoter (Shivprasad et al., 1999). In contrast, optimal activity of the TBSV sgRNA2 promoter requires long distance cis interactions with complementary upstream sequences separated by more than 1000 nt Zhang et al 1999, Choi and White 2002.
Promoter elements within viruses expressing more than one sgRNA can also vary considerably in their core sequences, and in their sizes and positioning relative to the transcription start sites of the sgRNAs. In one well-studied case, the BYDV sgRNA1 promoter has been mapped from −75 to +21 relative to its transcription initiation site, while the sgRNA2 and sgRNA3 promoters map between positions +1 and +143, and −6 and +38, respectively (Koev and Miller, 2000). Thus, the BYDV sgRNA1 and sgRNA3 promoters occupy sequences that overlap the transcription initiation site, whereas the sgRNA2 promoter resides entirely within the sgRNA transcript. Aside from a conserved hexanucleotide shared between the sgRNA1 and sgRNA2 promoters, little or no similarity exists between the three promoters. Several lines of evidence suggest that both primary sequence and secondary structure, including two stem loop structures, function during the regulation of BYDV sgRNA1 synthesis.
In contrast to the well-defined viruses described above, only rudimentary information is available to define sequences affecting expression of the sgRNAs of a number of other viruses, including Barley stripe mosaic virus (BSMV). BSMV is a Hordeivirus whose genome is divided into three positive-sense, single-stranded RNAs, designated α, β, and γ (Fig. 1). The α and γ RNAs are required for replication, while RNAβ is essential for cell-to-cell movement. The replicase proteins αa and γa are translated directly from their respective genomic RNAs (Petty et al., 1990). In addition, RNAγ also encodes a small cysteine-rich protein, designated γb, that is translated from sgRNAγ and is dispensable for BSMV replication. The coat protein, βa, is translated directly from RNAβ and, following an intergenic region, the overlapping viral movement genes are arranged in a “triple gene block” (TGB). The three major TGB proteins (TGB1, 2, and 3) and one minor protein, TGB2′, are translated from two sgRNAs, designated sgRNAβ1 and sgRNAβ2. The TGB1 protein is expressed from sgRNAβ1, while the other overlapping proteins TGB2, TGB2′, and TGB3 are translated from sgRNAβ2 (Zhou and Jackson, 1996b). The TGB2′ protein is a translational readthrough product of the TGB2 ORF, and the TGB3 protein is translated by leaky scanning of the TGB2 start codon (Zhou and Jackson, 1996b). The transcription initiation sites of the three BSMV sgRNAs have been mapped and their relative abundance has been determined Gustafson et al 1987, Zhou and Jackson 1996b. Northern blot analyses of nucleic acids extracted from BSMV-infected protoplasts have demonstrated that sgRNAγ and sgRNAβ1 accumulate to high levels, whereas sgRNAβ2 is present in much lower abundance Zhou and Jackson 1996a, Zhou and Jackson 1996b. Limited evidence from protoplasts also indicates that sgRNAβ1 and β2 expression is regulated temporally and that sgRNAγ is expressed constitutively (Zhou and Jackson, 1996b).
Fig. 1.
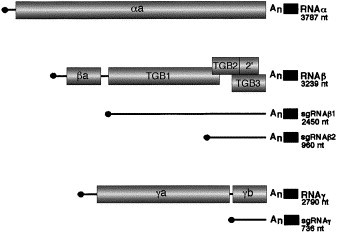
Illustration of the BSMV genomic and subgenomic RNAs. RNAα serves as the mRNA for the αa protein, which contains the capping and helicase subunits of the RNA-dependent RNA polymerase (RdRp). RNAβ encodes the βa protein (coat protein) and the “triple gene block” composed of the TGB1, TGB2, TGB2′, and TGB3 proteins. The βa protein is translated from the genomic RNA; the TGB1 protein is expressed from sgRNAβ1 and the TGB2, TGB2′, and TGB3 proteins are translated from sgRNAβ2. The genomic RNAγ serves as a messenger for translation of the γa polymerase subunit of the RdRp and encodes the γb protein, which is expressed from sgRNAγ. All genomic and subgenomic RNAs are capped (black circle) at the 5′ terminus, contain an internal poly (A) tail (An), and possess a conserved tRNA-like structure (black rectangle) at the 3′ terminus.
To begin to determine how expression of the three BSMV sgRNAs may be mediated, we have delineated the boundaries of the promoters based on the expression of sgRNAs in infected protoplasts. In addition, we have mapped a cis-acting element adjacent to the sgRNAβ1 promoter that is required for RNAβ replication (Zhou and Jackson, 1996b). The three BSMV sgRNA promoters have also been repositioned into β and γ RNAs that either contain or lack the native promoters. The results indicate that the analogous BSMV sgRNA promoters have some sequence similarity to predicted promoter regions of other Hordeiviruses. Nevertheless, the three sgRNA promoters do not share extensive sequence similarity, nor are recognizable common structural elements evident. Although the BSMV core promoter sequences failed to function when inserted ectopically into RNAβ and RNAγ, core promoters introduced with additional flanking sequences were active, but this activity was dependent on the context into which the sequences were inserted.
Results
Mapping sequences required for expression of sgRNAγ
In a previous study, we had determined that the sgRNAγ transcription start site begins at nt 2054 on RNAγ (Gustafson et al., 1987). This site resides within the 42-nt intergenic region between the γa and γb ORFs (Fig. 1). To analyze the sgRNAγ promoter, BY-2 protoplasts were transfected with in vitro transcripts, and the RNAs synthesized during infection were evaluated by Northern blot analyses. Because the γa stop codon is located 18 nt upstream of the sgRNAγ transcriptional start site, we predicted that the sgRNAγ promoter would overlap the γa ORF. The γa protein encodes the viral polymerase and hence is essential for viral replication. Therefore, engineering deletions to map the sgRNAγ promoter could have interfered with the γa ORF and might have affected replication. To circumvent this problem, protoplasts were transfected with two RNAγ derivatives. One RNA, γKpnI/HpaI, provided a source of the γa replicase protein and also contained a 350-nt deletion (from positions 2112 to 2461 on RNAγ) to remove the majority of the γb ORF, and a second derivative designed to assess promoter activity contained deletions engineered within the putative sgRNAγ promoter (Fig. 2A). The deletion present in γKpnI/HpaI RNA allowed us to distinguish between the two RNAγ derivatives based on their sizes. Protoplasts were transfected with RNAα, RNAγ, and RNAγKpnI/HpaI, and nucleic acid was extracted from protoplasts at 20 h posttransfection. Each of the three RNAs replicated to wild-type (wt) levels in protoplasts and synthesized the predicted sgRNAs (data not shown). Therefore, in subsequent mapping experiments, protoplasts were transfected with RNAα, RNAγKpnI/HpaI, and an RNAγ derivative that contained a deletion within the putative sgRNAγ promoter.
Fig. 2.
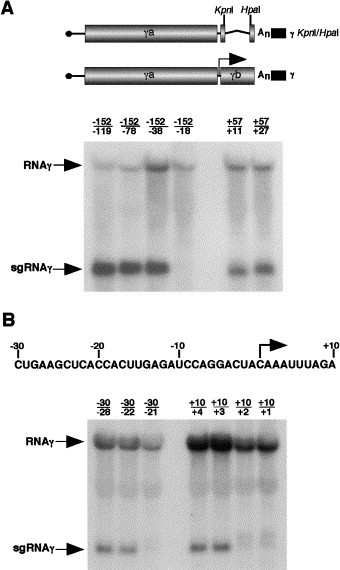
Identification of the boundaries of the sgRNAγ promoter. (A) The schematic illustration shows the two γ RNAs used in conjunction with RNAα to transfect protoplasts. The γ KpnI/HpaI RNA provided a source of the γa protein, while the second RNAγ was used to construct deletions within the region surrounding the sgRNAγ transcription start site (note arrow). Tobacco BY-2 protoplasts were cotransfected with the α and γ KpnI/HpaI RNAs plus RNAγ derivatives with deletions originating at either −152 or +57 relative to the sgRNAγ transcription start site. The deletions are shown above the blot. Total nucleic acid was extracted 20 h posttransfection, separated on 1% agarose gels, and transferred to nylon membranes. Northern blot analyses were conducted with a γ-specific riboprobe designed to detect the presence of the genomic RNAγ and sgRNAγ. The probe was derived from the KpnI/HindIII (2111 to 2444 nt) fragment of RNAγ and hence does not hybridize to the genomic or sgRNAs generated from the γ KpnI/HpaI RNA. (B) Analysis of RNAγ derivatives containing small deletions in the region spanning −30 to +10 relative to the sgRNAγ transcription start site. The sequence shows the intergenic region in the negative sense, with the arrow representing the sgRNAγ transcription start site.
To identify starting points for analysis of the sgRNAγ promoter region, two RNAγ derivatives were constructed. One contained a large-scale deletion in the γa ORF from nt 593 to 1899 and the other had a smaller deletion from positions 2111 to 2262 in the γb ORF. When these RNAs were transfected individually into protoplasts along with RNAα and RNAγKpnI/HpaI, both of the deletion derivatives were able to replicate and synthesize sgRNAγ at levels comparable to wt RNAγ (data not shown). These results indicate that the deleted regions are not required for sgRNAγ synthesis and that they do not contain regulatory elements that affect replication. Therefore, a more refined analysis of the sgRNAγ promoter was initiated by evaluation of deletions beginning at position −152 (position 1902 on RNAγ) and extending to positions −119, −78, −38, and −18 upstream of the transcription start site. Two additional deletions beginning at position +57 and extending to +27 and +11 were also generated. To specifically visualize the replication of these RNAγ derivatives and the presence of sgRNAγ, a 32P-labeled riboprobe was used that is complementary to the KpnI/HpaI region that had been eliminated in the γKpnI/HpaI RNA used to mediate replication of the test RNAs. This probe thus recognizes only the RNAγ and sgRNAγ derivatives designed to assess promoter activity. As shown in Fig. 2A, Northern blot analysis revealed that each of the RNAγ test derivatives were able to replicate in trans and that the γRNAs containing the deletions −152/−119, −152/−78, −152/−38, +57/+27, and +57/+11 were able to synthesize sgRNAγ. However, when the region between −152 and −18 was deleted, sgRNA synthesis was not evident. Therefore, these results indicated that the sgRNAγ promoter resides between positions −38 and +11 relative to the sgRNAγ transcriptional start site.
To analyze the sgRNAγ promoter more precisely, smaller deletions were generated in the region between positions −30 and +10 (Fig. 2B). The γ RNAs containing the deletions −30/−28, −30/−22, −30/−21, +10/+4, +10/+3, +10/+2, and +10/+1 were transfected into protoplasts along with the α and γ KpnI/HpaI RNAs. Northern blot analysis demonstrated that the RNAs containing deletions −30/−28 and −30/−22 replicated and were capable of synthesizing sgRNAγ, but that removal of the residue at position −21 eliminated sgRNAγ synthesis (Fig. 2B). Our interpretation of these results is that the C at position −22 is dispensable for sgRNAγ synthesis, but that the A at position −21 is required for promoter activity. The RNAs used for deletion analysis starting at position +10 were all able to replicate, but only the +10 to +4 or +3 deletions were able to synthesize sgRNAγ (Fig. 2B). When the next residue at +2 was deleted, sgRNAγ was not evident, and hence, the A at position +2 is required for sgRNAγ synthesis, whereas the A at +3 is dispensable. Therefore, these results indicate that the sgRNAγ promoter boundaries map to positions −21 and +2 relative to the sgRNAγ transcription start site. The sgRNAγ promoter thus overlaps the sgRNAγ transcription initiation site and the last two codons of the γa polymerase protein.
Defining the sgRNAβ1 promoter and the RNAβ cis-acting element boundaries
RNAβ is dispensable for replication and therefore, substantial deletions could be engineered into the RNA to facilitate analysis of the sgRNAβ1 promoter. The sgRNAβ1 transcription start site had previously been mapped to nt 789 in the 118-nt intergenic region between the βa and TGB1 ORFs (Zhou and Jackson, 1996b). In addition, we had identified a cis-acting element required for RNAβ replication within this intergenic region (Zhou and Jackson, 1996a). Therefore, careful consideration of the cis-acting element had to be taken into account because of the possibility that the sgRNAβ1 promoter boundaries might reside within this region. Hence, we devised a strategy to identify the cis element and to map the sgRNAβ1 promoter by transfecting BY-2 protoplasts with the α and γ RNAs and an RNAβ mutant to evaluate promoter activity. Northern blot analysis with a β-specific probe was then used to assess replication and sgRNAβ1 synthesis.
Identification of the sgRNAβ1 promoter initially was complicated because the 18S rRNA masked sgRNAβ1 in Northern blots (data not shown). To circumvent this problem, we constructed mutations in a cDNA clone that contained a 1427-nt deletion (βS/B) which removed the majority of the TGB ORFs. This deletion did not affect RNAβ replication, in agreement with our previous results (Zhou and Jackson, 1996a) and also clearly resolved sgRNAβ1 and the rRNAs to permit detection of sgRNAβ1 (data not shown). It is important to note that the βS/B RNA deletion also eliminated the sgRNAβ2 promoter to mitigate possible competition effects that might have affected synthesis of sgRNAβ1. To define the starting points for mapping the sgRNAβ1 promoter, we used two RNAβ derivatives. One of these, βΔ1.6, contained a deletion from position 295 to 633, and the other, βΔ2.0, eliminated nts 802 to 1375 on the genomic RNA sequence. Northern blot analysis of RNA isolated from protoplasts transfected with the α and γ RNAs and the βΔ1.6 or βΔ2.0 RNAs indicated that the deletions had no discernable effect on sgRNAβ1 synthesis (data not shown). Therefore, the BstBI site at −156 (RNAβ nt 633) and the NcoI site at +14 (nt 802) relative to the sgRNAβ1 transcription start site were used as starting points for analysis.
Initially, deletions were generated from −156 to −107, −74 and −34 relative to the sgRNAβ1 transcription initiation site. As shown in Fig. 3A, the −156/−107 deletion mutant was able to replicate and to synthesize sgRNAβ1, but the −156/−74 and −156/−34 deletions reduced replication to barely detectable levels. To confirm the replication of RNAs α and γ, the blots were stripped and reprobed with α- and γ-specific probes (data not shown). These results indicated that the −156/−74 and −156/−34 deletions destroyed a cis-element within the intergenic region that is required for replication (Zhou and Jackson, 1996a). To further define the RNAβ cis-acting element, two additional derivatives were constructed that contained deletions spanning −107/−74 and −74/−34. Northern blot analysis using a β-specific probe revealed that replication of −107/−74, similar to that of the −156/−74 and −156/−34 mutants, was undetectable. However, the −74/−34 deletion mutant replicated to low levels and produced small amounts of sgRNAβ1 (Fig. 3A). These results suggest that the cis-acting element required for RNAβ replication resides within the 118-nt intergenic region between positions −107 and −74. However, sequences within the −74 to −34 region also contribute to efficient replication of RNAβ.
Fig. 3.
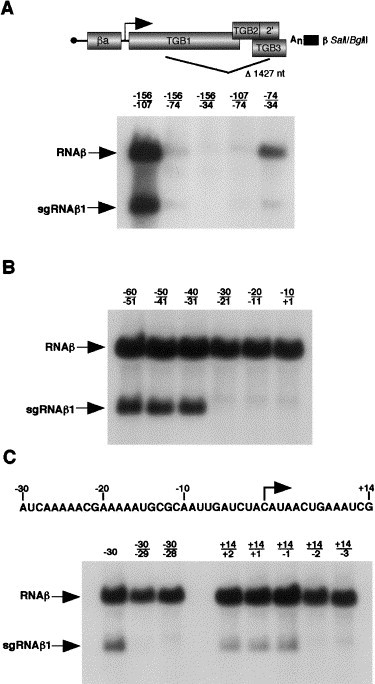
Delineating the boundaries of the sgRNAβ1 promoter and the RNAβ cis- acting element. Protoplasts were transfected with wt α and γ RNAs plus RNAβ derivatives containing the 1427 nt S/B (β SalI/BglII) deletion. Total nucleic acid was extracted and blotted as described in Fig. 2, except that the blots were probed with a β-specific riboprobe (β NcoI/SalI). The numbers above the lanes correspond to deletions at positions relative to the sgRNAβ1 transcription start site. (A) Large-scale deletion mapping of the sgRNAβ1 promoter and the RNAβ cis-acting element required for replication. (B) Deletion mapping of the sgRNAβ1 promoter using 10-nt deletions at positions corresponding to the transcription start site. (C) Fine-scale mapping of the sgRNAβ1 promoter. The sequence represents the negative-sense orientation of the intergenic region between the βa and TGB1 ORFs which contains the sgRNAβ1 promoter. The arrow represents the sgRNAβ1 transcription start site.
The sgRNAβ1 promoter activity was examined in more detail by engineering 10-nt deletions in βS/B from positions −60 to +1 relative to the sgRNAβ1 transcription start site. Northern blot analyses revealed that the −60/−51, −50/−41, and −40/−31 deletions did not affect genomic RNA replication and abundant amounts of sgRNAβ1 were synthesized. However, when the −30/−21, −20/−11, or −10/+1 regions were deleted, genomic RNA replicated to wt levels but sgRNAβ1 failed to accumulate (Fig. 3B). To further resolve the nature of the active sgRNA promoter element, we engineered smaller deletions from positions −30 to +14. As shown in Fig. 3C, when the A residue at −30 was deleted, sgRNAβ1 was evident, but deletion of the adjacent U at −29 effectively eliminated promoter activity without obvious effects on replication of the genomic RNAs. In contrast, deletions that spanned +14/+2, +14/+1, and +14/−1 retained promoter activity (Fig. 3C). However, when the next residue, a U at −2, was deleted, promoter activity was disrupted. These results suggest that the core promoter sequence required for sgRNAβ1 synthesis resides immediately upstream of the sgRNAβ1 transcription start site between positions −29 and −2. However, an alternative possibility that needs further exploration is that the +14/+1 deletion could have generated an alternative transcription initiation site at the upstream C residue. Similar alternative sites have previously been identified in in vitro studies with the Brome mosaic virus sgRNA promoter (Stawicki and Kao, 1999).
Delineating the sequences required for sgRNAβ2 expression
The transcription start site of sgRNAβ2 has previously been identified within the 3′ end of the TGB1 ORF at position 2279 in RNAβ and is 1490 nt downstream of the sgRNAβ1 transcription start site (Zhou and Jackson, 1996b). Since sgRNAβ2 is expressed at very low levels in infected protoplasts Zhou and Jackson 1996a, Zhou and Jackson 1996b, we attempted to increase its abundance to improve the ease and reproducibility of promoter analysis. For this purpose, the clone β1−34/+14 was created by removing nucleotides 755 through 802 (−34/+14 relative to the sgRNAβ1 transcription start site) on RNAβ (Fig. 4A). As expected, this 48-nt deletion abrogated sgRNAβ1 synthesis and also resulted in easily detectable levels of sgRNAβ2 without affecting RNAβ replication (data not shown). This result implies that competition between the two promoters has a major role in regulating the differential rates of synthesis of the two sgRNAs.
Fig. 4.
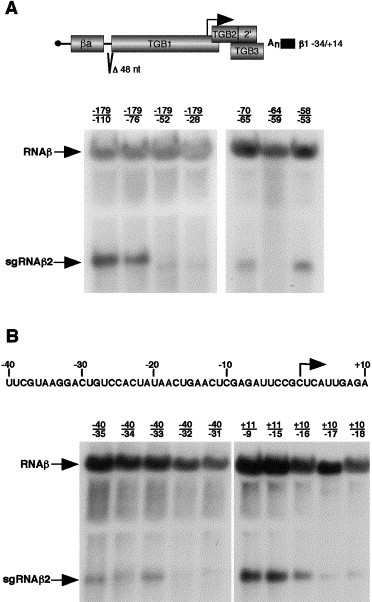
Defining the boundaries of the sgRNAβ2 promoter. Protoplasts were transfected with the α and γ genomic RNAs and RNAβ derivatives containing a 48-nt deletion (β1−34/+14) to eliminate sgRNAβ1 promoter activity. The deletions are numbered according to the sgRNAβ2 transcription start site, which is illustrated by the arrow. Total nucleic acids were extracted at 20 h posttransfection and processed as described in Fig. 2, except that hybridizations were performed with a β-specific riboprobe (β SspI/BglII) derived from sequence upstream of the sgRNA2 promoter region. (A) Large-scale deletion mapping of the sgRNAβ2 promoter is shown on the blot to the left. To define the region between −70 and −53 further, 6-nt deletions were constructed and are shown on the blot to the right. (B) Small-scale mapping of the sgRNAβ2 promoter. The sequence represents the negative-sense region of the TGB1 ORF that contains the sgRNAβ2 promoter.
To initially define sequences flanking the sgRNAβ2 promoter, two large deletions were generated in the β1−34/+14 clone that eliminated RNAβ positions 1705 to 2109 and 2287 to 2434. RNAs containing these deletions were individually transfected into protoplasts along with the α and γ RNAs, and nucleic acids were extracted at 20 h posttransfection. Northern blot analyses using a β-specific probe revealed the presence of sgRNAβ2 in both derivatives, indicating that neither of the deletions affected sgRNAβ2 promoter activity (data not shown). Therefore, we began to focus on the region between −179 (nt 2109) and +9 (nt 2288) relative to the sgRNAβ2 transcription initiation site.
Analysis of four deletions extending from position −179 to positions −110, −76, −52, and −28 relative to the transcription start site revealed that the mutant RNAs were able to replicate in protoplasts, but only mutants containing the deletions −179/−110 or −179/−76 were able to direct synthesis of sgRNAβ2 (Fig. 4A). More detailed analyses performed on the region between −70 and −52 demonstrated that the −70/−65 and −58/−53 mutants transcribed sgRNAβ2, but that promoter activity was destroyed by the −64/−59 deletion (Fig. 4A). These results thus indicate that an upstream element residing between −64 and −59 relative to the sgRNAβ2 transcription start site is required for sgRNAβ2 promoter activity. However, in the presence of this element, deletions between positions −40/−32 and +10/−17 nearly eliminated sgRNAβ2 synthesis (Fig. 4B). In contrast, Northern blot analyses revealed that deletions between −52/−47, −46/−41, −40/−35, −40/−34, and −40/−33 had only minor effects on RNAβ replication and sgRNAβ2 synthesis in transfected protoplasts (Fig. 4B and data not shown). Similarly, the deletions +11/−9, +11/−15, and +10/−16 were able to replicate and synthesize sgRNAβ2. These results demonstrate that the sgRNAβ2 promoter maps from the G at −32 to the C at −17 and hence does not encompass the transcriptional start site.
Ectopic expression of the sgRNA promoters
The sgRNAβ1, sgRNAβ2, and sgRNAγ promoter fragments were initially inserted into RNAβ to determine whether they could function ectopically and to evaluate their competition with a native sgRNA promoter present on the same RNA. For this purpose, fragments encompassing each of the three sgRNA promoters were PCR-amplified and inserted into RNAβ β S/B or β1−34/+14 at position 1134 (344 nt downstream of the sgRNAβ1 promoter). The β S/B clone provided a background that contained the native sgRNAβ1 promoter (Fig. 5A), while the β1−34/+14 clone lacked the sgRNAβ1 promoter (Fig. 5B). Two fragments that encompassed the sgRNAβ1 promoter were inserted ectopically into the two RNAβ derivatives. The larger fragment β1a consisted of 266 nt amplified from positions 599 to 864 on the genomic RNA and a smaller 150-nt fragment β1b was derived from positions 715 to 864. A 295-nt fragment (β2) overlapping the sgRNAβ2 promoter was derived from positions 2080 to 2374, and a 276-nt fragment (γ) was amplified from positions 1864 to 2139 to encompass the sgRNAγ promoter.
Fig. 5.
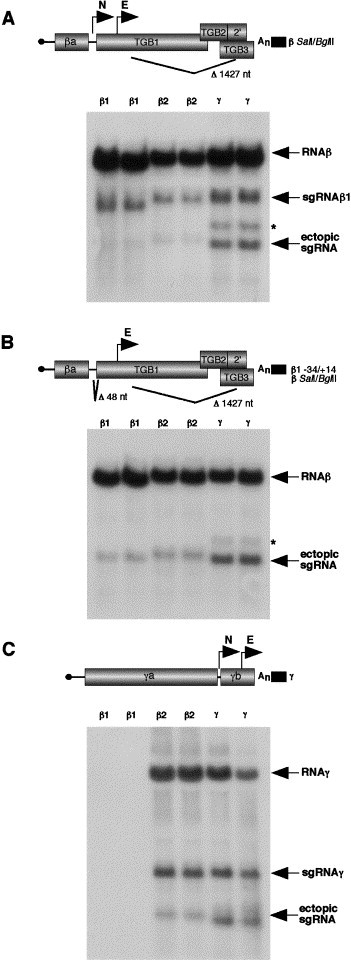
Ectopic expression of the sgRNAγ, sgRNAβ1, and sgRNAβ2 promoters in RNAβ and RNAγ derivatives. The designations on the genomic RNAs are as described in the legend to Fig. 1. N and E represent the native promoters and the ectopic promoter insertion sites, respectively. Protoplast transfections and RNA blots were carried out as described in Fig. 2. (A) Activity of the 150 nt sgRNAβ1 (β1), 294 nt sgRNAβ2 (β2), and 275 nt sgRNAγ (γ) fragments inserted at nt 1134 of the β S/B (β SalI/BglII) RNA containing the native sgRNAβ1 promoter. Duplicate tubes of BY2 protoplasts were transfected with RNAs α and γ, and either the βS/B+150 nt sgβ1, the βS/B+sgβ2, or the βS/B+sgγ RNA derivatives. RNA blots were probed with a β-specific riboprobe (βc-2785). (B) Expression of the sgRNAβ1, sgRNAβ2, and sgRNAγ promoters in the RNAβ S/B derivative (β1−34/+14), which contains a 48-nt deletion inactivating the native sgRNAβ1 promoter. (C) Activity of the sgRNAβ1, sgRNAβ2, and sgRNAγ promoters inserted into the γb ORF at nt 2339 of RNAγ. Protoplasts were coinoculated with RNAα and RNAγ containing the promoter fragments and extracted at 20 hpi. RNA blots were probed with a riboprobe that anneals to the conserved 3′ end of BSMV RNAs and therefore should detect all genomic and sgRNAs. Asterisks along the side of A and B refer to a consistently observed band of unknown origin that originated from the sgRNAγ promoter insertions.
Northern blot analysis of RNA isolated from protoplasts transfected with RNAs α and γ, and RNAβ derivatives containing the native sgRNAβ1 promoter with the smaller sgβ1 (150 nt), sgβ2, or the sgγ fragment insertions at position 1134 revealed that the β RNAs were able to replicate and synthesize sgRNAβ1 from the native promoter (Fig. 5A). However, transcription from the ectopic sgRNAβ1 and sgRNAβ2 promoters could be detected only after protracted exposure of the blots (Fig. 5A, β1 and β2 lanes, and data not shown). The RNAβ derivative containing the 48-nt deletion in the intergenic region was able to replicate and to express small amounts of the ectopic sgRNAs from the smaller 150-nt sgRNAβ1 fragment and sgRNAβ2 (Fig. 5B, β1 and β2 lanes), but both the RNAβ derivatives failed to replicate when the larger 266-nt sgRNAβ1 fragment was inserted (data not shown). In marked contrast, the 276-nt ectopic RNAγ fragment was active following insertion into both of the RNAβ derivatives and ectopic sgRNAs of the appropriate sizes were easily detectable in both backgrounds (Figs. 5A and B, γ lanes). These results demonstrate that the three BSMV sgRNA promoters can function ectopically in RNAβ, but that the extent of their activity varies depending on their flanking sequences and the genomic RNA context into which they are inserted. The results also provide evidence that the ectopic sgRNAγ promoter competes for the BSMV replicase complex more efficiently than an ectopic sgRNAβ1 or sgRNAβ2 promoter when positioned downstream of the wt sgRNAβ1 promoter.
Each of the four promoter fragments (β1a, β1b, β2, and γ) were also inserted into the γ cDNA clone at position 2339, which is located 285 nt downstream of the transcription start site of the native sgRNAγ (Fig. 5C). As seen with RNAβ, the presence of the sgRNAβ2 fragment (β2) and the sgRNAγ promoter derivative (γ) permitted abundant RNAγ replication and synthesis of native sgRNAγ (Fig. 5C, β2 and γ lanes). Both the sgRNAβ2 and the sgRNAγ promoters were also able to direct synthesis of easily detectable amounts of ectopic sgRNA. Again, transcription from the β2 promoter was less than half of that from the native sgRNAγ promoter, and the ectopic sgRNAγ promoter also appeared to be substantially more active than the corresponding sgRNAβ2 promoter. However, RNAγ replication was nearly eliminated when either the smaller 150-nt sgRNAβ1 promoter (β1, Fig. 5C) or the larger 266-nt sgRNAβ1 promoter fragments were inserted (data not shown). These results suggest that the ectopic sgRNAβ2 promoter competes less efficiently for the BSMV replicase complex than the wt sgRNAγ promoter. The deleterious effects of the sgRNAβ1 fragment insertions in RNAγ also suggest that sequence, context effects, and perhaps competition for replicase can have drastic effects on replication.
To determine if the three minimal sgRNA promoters were sufficient for ectopic transcription, we inserted each of them into position 1134 of the RNAβ β S/B derivative and the 48-nt deletion derivative (β1−34/+14). Similar insertions were made into RNAγ at position 2339. Ectopic sgRNA expression was not detected following protoplast transfection with any of the derivatives, although the genomic RNAs were able to replicate (data not shown). This result suggests that although the minimal promoter sequences identified by deletion analysis are required for activity in their native context, each of the promoters requires additional flanking sequences to provide optimal promoter activity from the ectopic positions tested in these experiments.
Sequence comparisons of the three BSMV sgRNA promoter regions
The comparisons shown in Fig. 6A indicate that the three BSMV sgRNA promoters share little obvious sequence relatedness. We also conducted a more refined gapped alignment and were still unable to detect major blocks of similarity (data not shown). Additional comparisons of the 238 nt at the 3′ common termini of the genomic RNAs and at the diverse 3′ ends of the three minus-strand RNAs also indicated that the four replication promoters within the genomic RNAs do not contain substantial regions of similarity to the three BSMV sgRNA promoters (data not shown). However, comparisons of the three sgRNA promoters of BSMV with the putative promoters of Poa semilatent virus (PSLV) and Lychnis ringspot virus (LRSV) revealed that the analogous Hordeivirus sgRNA promoters share a number of blocks of identical sequence (Fig. 6B). Furthermore, the BSMV and PSLV sequences share a higher degree of similarity with each other than either does with LRSV. In the case of the BSMV sgRNAγ promoter, nearly 80% of the PSLV residues are identical (19/24) to the BSMV sequence, whereas a gapped alignment between BSMV and LRSV indicated a considerably lower degree of similarity (14/24 residues) within the aligned region. These results buttress the existing strong evidence for a common origin of the Hordeiviruses and support biological evidence, suggesting that BSMV and PSLV are more closely related to each other than to LRSV (Hunter et al., 1989).
Fig. 6.

Comparison of the BSMV sgRNA promoter sequences. (A) Sequence of the three BSMV sgRNA promoters in the minus-sense orientation. The numbers above the sequence correspond to nucleotide positions relative to the respective transcription initiation sites. The underlined sequences are required for sgRNA synthesis. (B) Alignment of the sgRNAγ, sgRNAβ1, and sgRNAβ2 promoter regions of the Hordeiviruses BSMV, PSLV, and LRSV. Regions with the highest sequence similarity are shown in red. Note that the transcription start sites for the PSLV and LRSV sgRNA promoters have not been defined experimentally, although their initiation sites have been predicted previously (Savenkov et al., 1998). Alignments were performed using the MegAlign program associated with the Lasergene software package. (C) Alignment of the sgRNAβ1, sgRNAβ2, and sgRNAγ promoter regions of BSMV with mapped (TMV and PVX) or putative (BNYVV, PCV, PMTV, and PVM) sgRNA promoter regions of other viruses. Regions identified to have high sequence similarity are shown with residues identical to the BSMV sequence highlighted in red. Alignments were performed using the MegAlign program associated with the Lasergene software package.
To obtain additional information about the relationship of the Hordeivirus sgRNA promoters, sequence comparisons were conducted with other TGB-containing viruses. Initial comparisons using the NCBI BLAST program failed to reveal significant matches with other viral promoter sequences. To obtain more definitive comparisons, we analyzed the 300 nucleotides surrounding the start codons of the TGB proteins of Beet necrotic yellow vein virus, Peanut clump virus (PCV), Potato mop top virus (PMTV), Potato virus M (PVM), and Potato virus X (PVX). No obvious similarity was detected in comparisons of the sgRNAβ2 promoter regions, and only short blocks of common sequence were noted in the regions surrounding the putative sgRNAβ1 promoters. These common blocks primarily consisted of strings of A residues located at various positions upstream of the translational start site and hence they are unlikely to represent conserved promoter elements. Thus, the results indicate that the sequence conservation noted among the TGB proteins of the Hordeiviruses does not extend to the analogous TGB promoter regions of more distantly related viruses.
We also conducted comparisons of the BSMV sgRNAγ promoter with putative promoter regions of other viruses. Interestingly, the PCV p15, which encodes a cysteine-rich protein with some similarity to BSMV γb, and the BNYVV TGB1 sequences have a high degree of similarity to the BSMV sgRNAγ promoter. In these cases, 16 (PCV) or 14 (BNYVV) residues of 24 are identical to the BSMV sgRNAγ sequence. These regions of similarity include two blocks (ACCACUU and ACUA) within the 20 residues upstream of the transcriptional start site of the BSMV sgRNAγ, and they surprisingly share more than 60% identity (15/24 residues) with a region of the TMV 30 K movement protein promoter (Grdzelishvili et al., 2000). Despite these intriguing similarities, the significance, if any, of these common sequences is conjectural and additional analyses are needed to determine their importance in sgRNA synthesis.
Discussion
During the BSMV infection cycle, the three sgRNAs vary in abundance and in their timing of expression (Zhou and Jackson, 1996a). Early in infection, the predominant species are sgRNAγ, which appears to be relatively constant throughout replication, and sgRNAβ1, whose abundance decreases as infection progresses. In contrast, the abundance of sgRNAβ2 is considerably lower than that of sgRNAβ1, but both sgRNAs appear to have similar patterns of temporal expression. These results suggest that the timing and the relative expression levels of the sgRNAs may be important in regulating aspects of the infection cycle (Zhou and Jackson, 1996a). Therefore, understanding the nature of the sgRNA promoters and the requirements for regulated expression of the mRNAs under their control will contribute substantially toward elucidating events required for systemic invasion and disease development in host plants. As a step toward such understanding, we have delineated the boundaries of the three BSMV sgRNA promoters.
The sgRNAγ, sgRNAβ1, and sgRNAβ2 promoters map to positions −21 to +2, −29 to −2, and −32 to −17 relative to their transcription start sites, respectively. RNAβ also requires an enhancer-like element located between −64 to −59 for production of sgRNAβ2. These three promoters are <50 nt in size and are located in close proximity to their respective transcription initiation sites. The sgRNAγ promoter lies primarily upstream of its transcription initiation site but includes the +1 nt that corresponds to the transcription start site and an additional nucleotide at the +2 position. The BSMV sgRNAβ1 and sgRNAβ2 promoters, which reside upstream of their transcription initiation sites, differ from other plant virus sgRNA promoters previously characterized because the sgRNAβ1 and sgRNAβ2 promoters appear not to overlap their transcription start sites (Miller and Koev, 2000). Independent experiments have demonstrated the importance of the +1 nt in the sgRNA promoters of a number of well-characterized viruses. For example, in vitro studies using the BMV proscript system to elucidate the minimal requirements for replicase recognition of the BMV sgRNA promoter have shown that changing the +1 nt of the promoter from a C to a G decreases sgRNA synthesis by more than 90% (Siegel et al., 1997). The importance of the +1 nt has also been shown in another Tricornavirus where changing the C to a U abolished AMV sgRNA promoter activity (Van Der Vossen et al., 1995). Similarly, when the +1 nt of the TMV MP sgRNA promoter was changed from a C to a G, sgRNA accumulation was below the limits of detection (Grdzelishvili et al., 2000). In contrast, transversion of the +1 nucleotides of the BYDV sgRNA2 and sgRNA3 promoters had no effect on sgRNA accumulation, even though both promoters span this region (Koev and Miller, 2000). Although the sgRNAγ promoter overlaps its transcription start site, we have not determined whether nucleotide substitutions at this position affect sgRNA synthesis.
Unlike many RNA viruses, the BSMV sgRNA promoters do not share substantial blocks of sequence identity within their core regions (Fig. 6A). However, the sgRNAγ and sgRNAβ2 promoters do appear to be more closely related to each other than either is to the sgRNAβ1 promoter. This probably is an anomaly due to nucleotide composition within the promoters because the sgRNAβ1 promoter sequence is particularly rich in A and U residues (73%), whereas the sgRNAβ2 and sgRNAγ promoters have a more uniform distribution of bases with an A/U content of just over 50%. In analyses comparing the sgRNAβ1 promoter with either the sgRNAβ2 or the sgRNAγ promoter, we could align no more than two identical residues in a row. However, for the sgRNAβ2 and sgRNAγ promoters, a block of five identical nucleotides (CCACU) can be aligned within the 26 residues upstream of their transcription start sites.
The three BSMV sgRNA promoters align well with the putative sgRNA promoter regions of the less well-characterized Hordeiviruses, PSLV and LRSV, whose sgRNA transcription initiation sites have not been mapped. Comparison of the three BSMV sgRNA promoters with the analogous promoters of the other Hordeiviruses reveals over 75% sequence similarity (Fig. 6B and Savenkov et al., 1998). Two blocks of identical residues were identified in the sgRNAβ1 promoter regions (AAAAAU and CUAC). Only one block of conserved sequence was observed for the sgRNAβ2 promoter (CCACU), and this same block is present in BSMV and LRSV sgRNAγ promoters. Among the three Hordeiviruses, the sgRNAγ promoter displayed the greatest degree of similarity because it contained three blocks of identical sequence of three nucleotides or longer (GAAGCU, ACCA, and ACU) as well as individual conserved residues. Additional analyses of the less conserved regions failed to indicate preferences for particular purine or pyrimidine residues. These results indicate that the abundance and timing of transcription of the three Hordeivirus sgRNAs, β1, β2, and γ, may be regulated by promoter sequences that are only distantly related. Alternatively, it is possible that short and/or long distance structural interactions that are not evident in our analyses may have profound roles in promoter timing and activity.
It appears that the sgRNAβ2 promoters of BSMV and PSLV are sufficiently similar for recognition by the BSMV replicase because in vivo studies have revealed that BSMV can support synthesis of PSLV sgRNAβ2 (Solovyev et al., 1999). In these studies, a hybrid BSMV RNAβ containing PSLV TGB1, TGB2, and most of the TGB3 sequence was able to replicate and move through the vasculature to uninoculated leaves in plants coinoculated with the α and γ RNAs (Solovyev et al., 1999). However, when a hybrid BSMV RNAβ containing the LRSV TGB1, TGB2, and TGB3 regions analogous to the PLSV sequence was coinoculated with the α and γ RNAs, the hybrid virus was unable to infect the same common hosts. These results suggest that the BSMV replicase complex can recognize the related PSLV sgRNAβ2 promoter, but it either cannot recognize the LRSV sgRNAβ2 promoter or the LRSV TGB proteins do not form sufficiently compatible interactions to mediate BSMV movement.
In comparisons of the BSMV sgRNA promoters with the analogous sequences of non-Hordeivirus TGB promoters, we were unable to detect a strong correlation between the putative promoter sequences directing sgRNAs encoding specific TGB proteins. For example, no obvious similarity was evident between the sgRNAβ1 promoter and the regions upstream of the PMTV and PCV TGB1 genes, and only limited similarity was detected with the regions upstream of the TGB1 genes of BNYVV, PVM, and PVX. Moreover, an equivalent degree of limited similarity was also observed in the regions upstream of the unrelated BNYVV p14 and the PCV TGB2 genes. Likewise, the sgRNAγ promoter shares small blocks of common sequence with the region upstream of the PCV p15 gene, which encodes a protein closely related to the γb protein, but the promoter also has similarity to the regions upstream of the unrelated BNYVV TGB1 and TMV p30 genes. The BSMV, BNYVV, PVM, and PCV replicase proteins belong to the supergroup 3 tobamo lineage of RNA-dependent RNA polymerases (Koonin and Dolja, 1993), so in these cases it is tempting to speculate that the promoter sequences coevolved in response to the polymerase specificity rather than as a block consisting of the promoter and its associated protein. However, the RdRp proteins of BMV and PVX are also classified in the supergroup 3 tobamo lineage, yet their sgRNA promoter sequences share no apparent similarity to the BSMV promoters. Therefore, it is difficult to make a case for promoter evolution based on either promoter protein constraints or polymerase similarity.
We performed computer analyses to search for common secondary structural elements within or flanking the minimal BSMV core promoters, but were unable to identify common elements. This is surprising because in other viruses, sgRNA promoter recognition by replicase complexes often requires primary sequence as well as secondary structural elements. These requirements may facilitate diverse interactions between replicase complexes and promoters with limited sequence similarity. In BYDV, both RNA sequence and secondary structure are essential for promoter activity (Koev et al., 1999). The BYDV sgRNA1 promoter folds into two stem loops downstream of its transcription initiation site, but comparisons with the predicted structures of the sgRNA2 and sgRNA3 promoters reveal that they possess very different sized stem loops downstream of their respective transcription initiation sites (Koev and Miller, 2000). The TMV MP sgRNA promoter also requires a stem loop structure for promoter activity that will tolerate substantial variation in primary sequence without major effects on sgRNA synthesis (Grdzelishvili et al., 2000). In addition, chemical and enzymatic probing of the 1.45-kb sgRNA promoter of TCV has confirmed the presence of a 96-nt hairpin structure, although only a 21-nt hairpin and a 9-nt flanking single-stranded region comprise the minimal promoter (Wang et al., 1999). Thus, our comparisons suggest that BSMV promoters differ from those of a number of other viruses in that conservation of primary sequence rather than conserved structure may have a predominant role in promoter activity.
The available evidence indicates that sequences flanking the core promoter and positioning of the promoter contribute substantially to the transcription of the BSMV sgRNAs. In the case of sgRNAβ2, expression increased substantially upon removal of a 48-nt sequence required for sgRNAβ1 promoter activity. This result suggests that competition for replicase has an important role in mediating the levels of expression of sgRNAβ1 and sgRNAβ2. The ectopic expression experiments support this hypothesis because both the ectopic sgRNAγ and the sgRNAβ2 promoters directed transcription more efficiently in constructs lacking the wt promoters than in constructs with wt promoters. Positioning also appears to have variable effects on promoter activity. Accumulation of sgRNAs transcribed from promoters located close to the 3′ terminus of the genomic-sense RNA was less abundant than those of the wt sgRNA from an upstream promoter. When the sgRNAγ, sgRNAβ1, and sgRNAβ2 promoters were placed downstream of the wt sgRNAβ1 promoter in RNAβ, the ectopic sgRNAs were considerably reduced in comparisons with the wt sgRNAβ1 (Fig. 5). However, in RNAγ, the ectopic sgRNAγ promoter strength appeared to be similar to that of the wt sgRNAγ promoter. On the other hand, when the ectopic sgRNAβ2 promoter was placed downstream of the wt sgRNAγ promoter, the ectopic sgRNA accumulated to much lower levels than the wt sgRNAγ (Fig. 5C). In contrast to the larger regions encompassing the BSMV sgRNA promoters, the minimal BSMV promoter sequences were unable to function ectopically. These results suggest that positioning, flanking sequences, and, in the case of sgRNAβ2, an upstream enhancer element may contribute substantially to promoter strength. In particular, the reduction in activity resulting from placement of the BSMV sgRNA promoters near the 3′ end of the genomic RNA contrasts with many examples reported from other RNA viruses where more 3′ located ectopic promoters are preferentially transcribed French and Ahlquist 1988, Boccard and Baulcombe 1993, Wang and Simon 1997, Koev and Miller 2000.
The majority of viruses that contain multiple sgRNA promoters on the same RNA shows a strong correlation between the abundance of their sgRNAs and their proximity to the 3′ end of the genomic RNA Kelly et al 1994, Wang and Simon 1997, Grdzelishvili et al 2000. However, BSMV sgRNAβ1 and sgRNAβ2 promoters appear not to share this correlation because sgRNAβ2, which is proximal to the 3′ terminus of RNAβ, accumulates to much lower levels than the upstream sgRNAβ1. This pattern of synthesis for TGB sgRNAs has also been demonstrated for Potato virus X (Verchot et al., 1998) and is likely the case for the multicomponent Benyviruses, Furoviruses, Pomoviruses, and Pecluviruses and the monopartite Carlaviruses, Allexiviruses, and Foveaviruses (van Regenmortel et al., 2000), which contain related TGBs. During replication of BSMV, PVX, and BNYVV, two sgRNAs are generated for translation of the overlapping TGB proteins. The relatively high abundance of sgRNA1 reflects the accumulation of the TGB1 protein during the early stages of infection, whereas the 3′ proximal sgRNAβ2 is often difficult to detect Zhou and Jackson 1996a, Zhou and Jackson 1996b, Verchot et al 1998. It is possible that several evolutionary constraints may contribute to the low abundance of sgRNAβ2 and the analogous sgRNA in other TGB-containing viruses. One possibility is that the relative abundance of the proteins encoded by the sgRNAs may be important for the movement complex function. Alternatively, because the sgRNAβ2 promoter resides within the TGB1 ORF, optimal promoter strength may have been constrained by necessities for TGB1 protein function.
Due to the compact nature of plant RNA viruses, sequences residing within the same region of the genome often have evolved to perform multiple functions. This is especially evident in BSMV because the sgRNAβ1 core promoter is proximal to a cis-acting element required for RNAβ replication, and also for the sgRNAγ and sgRNAβ promoters, which overlap regions encoding the γa and TGB1 ORFs, respectively. Since the BSMV sgRNA promoter regions are multifunctional, it is likely that the replicase complex has coevolved with viral cis-acting elements to permit recognition of the three different sgRNA promoters and to discriminate between the distinct elements at the 3′ ends of the positive- and negative-sense RNAs. These varied interactions may also help explain the temporal regulation of the sgRNA promoters in the same virus. For example, the diverse promoter–replicase interactions within a virus may be facilitated by varied complexes of replicase and host proteins with distinct promoter elements. Such interactions may well mediate the strength and temporal activities of the BSMV sgRNA promoters.
Materials and methods
Recombinant plasmids
The α, β (β42SpI), and γ (γ42) cDNA clones used in this study were derived from the BSMV ND18 strain (Petty et al., 1988). To define the starting points for mapping the sgRNAγ promoter, the γ cDNA clone was digested with KpnI and BsmI, the 3′ overhangs were removed with T4 DNA polymerase, and the vector was religated to generate the clone γΔKpnI/BsmI. The γΔE/E cDNA clone constructed by Zhou and Jackson (1996a) was also used. To generate large-scale deletions in the sgRNAγ promoter, SpeI sites were engineered by site-directed mutagenesis (Kunkel, 1985) at positions −119, −78, −38, −18, and +11 relative to the sgRNAγ transcription start site at 2054 nt using the oligonucleotides listed in Table 1. The resulting clones, γSpeI-119, γSpeI-78, γSpeI-38, and γSpeI-18 were digested with ClaI (−152) and SpeI, while the γSpeI+11 clone was digested with KpnI (+57) and SpeI. The digested clones were treated with T4 DNA polymerase to generate blunt ends and religated. These clones were designated γ−152/−119, γ−152/−78, γ−152/−38, γ−152/−18, and γ+57/+11. The clone γ+57/+27 was constructed by digesting the γ42BamHI clone (Petty et al., 1990) with BamHI and KpnI, blunting with T4 DNA polymerase, and religating. Smaller deletions within the sgRNAγ promoter region were engineered by site-directed mutagenesis using the γ cDNA clone to create deletions ranging in size from 2 to 10 nts in the −30 to +10 regon surrounding the transcription start site. Throughout the course of this work, deletions or site-directed mutations were sequenced to ensure that the desired changes had been introduced.
Table 1.
Synthetic oligonucleotides used in this study
| Name | Primer sequence | Description |
|---|---|---|
| γSpeI−119 | GCTATTTCTGCACTAGTTTCTTTATG | sgRNAγ promoter large-scale mapping |
| γSpeI−78 | GACTTTAATAAGTACTAGTTGCTGTTTAATTG | sgRNAγ promoter large-scale mapping |
| γSpeI−38 | GAAATTTGTGGATAAACTAGTGAGAAAAGAC | sgRNAγ promoter large-scale mapping |
| γSpeI−18 | GAAAAGACTTCGACTAGTGAACTCTAGG | sgRNAγ promoter large-scale mapping |
| γSpeI+11 | GTTTAAATCTACTAGTTTTACCTTCGC | sgRNAγ promoter large-scale mapping |
| β1KpnI−107 | CCAGATGCCGAGGTACCCGTGACCTGCTG | sgRNAβ1 promoter large-scale mapping |
| β1KpnI−74 | GCGGTAAAAGGGGTACCATATGTATC | sgRNAβ1 promoter large-scale mapping |
| β1KpnI−34 | TATCTATTTTCGGTACCTTTTAGTTTTTG | sgRNAβ1 promoter large-scale mapping |
| β2+9SmaI | GGCGAGTAACTCCCGGGGTTATTCTGCAA | sgRNAβ2 promoter large-scale mapping |
| β2KpnI−110 | GGAAAGCTTTAGCAGGGTACCATTTGCGTTTAG | sgRNAβ2 promoter large-scale mapping |
| β2KpnI−76 | CTTTGAGCAGACAGGTACCGAAGTTAATCATC | sgRNAβ2 promoter large-scale mapping |
| β2KpnI−52 | GTTAATCATCAGGGCGGTACCGGAAATTCGTC | sgRNAβ2 promoter large-scale mapping |
| β2KpnI−28 | ATTCGTCAAGCATTGGTACCAGGTGATATTGAC | sgRNAβ2 promoter large-scale mapping |
| 5′sgγBamHI | GGATCCGGTGCTTGATGCTTTGGATAAG | PCR amplification of sgRNAγ promoter |
| 3′sgγBamHI | GGATCCCCACAGTAAGTACTTGTAGTTAAG | PCR amplification of sgRNAγ promoter |
| 5′sgβ1BamHI | GGATCCGAGTGCGAGACTCCCAGTG | PCR amplification of sgRNAβ1 promoter |
| 3′sgβ1BamHI | GGATCCCACCTTTCACTGAATCAG | PCR amplification of sgRNAβ1 promoter |
| 5′sgβ2BamHI | GGATCCGTGCCCTGGCAATTGATGTGCAAG | PCR amplification of sgRNAβ2 promoter |
| 3′sgβ2BamHI | GGATCCCGACACCGATTCCGGCGACAATTGG | PCR amplification of sgRNAβ2 promoter |
| β1134BamHI | GCTCAACCGAGTAGGATCCATGACCTTAAAGG | Engineer BamHI site from nts 1132–1137 in RNAβ |
| γ2339BamHI | CCAAAAGCATGCGGATCCGTATGATTCAC | Engineer BamHI site from nts 2338–2343 in RNAγ |
| 5′sgβ1−74BamHI | GGATCCCATATGTATCTTATTTATT | PCR amplification of sgRNAβ1 promoter |
| BSMV3′SpeI | GGGAAGACCACTAGTCATGCAAGC | Change MluI site to SpeI at BSMV RNAγ3′ end |
Note. Restriction sites are in bold.
To identify the starting points for mapping the sgRNAβ1 promoter, we used the βΔ1.6 and βΔ2.0 clones (Petty and Jackson, 1990). Refined mapping of the sgRNAβ1 promoter was performed using a β cDNA clone (βS/B) containing a 1427-nt deletion from SalI to BglII that removes part of the TGB1, TGB2, and TGB3 coding regions. This deletion was engineered to increase the separation between the sgRNAβ1 and the 18S ribosomal RNA in agarose gels. KpnI sites were introduced into the βS/B cDNA clone at positions −107, −74, and −34 relative to the sgRNAβ1 transcription start site at 789 nt using site-directed mutagenesis and β1-specific oligonucleotides (Table 1). The resulting clones were digested with BstBI (−156) and KpnI, blunted with T4 DNA polymerase, and religated to generate the β1−156/−107, β1−156/−74, and β1−156/−34 clones. Deletions were engineered in the sgRNAβ1 promoter region of the βS/B clone by site-directed mutagenesis to produce the β1−60/−51, β1−50/−41, β1−40/−31, β1−30/−21, β1−20/−11, and β1−10/+1 clones with a series of sequential deletions from positions −60 to +1. More detailed resolution of the sgRNAβ1 promoter region was obtained by site-directed mutagenesis of the βS/B cDNA clone to generate the clones β1−30, β1−30/−29, β1−30/−28, β1 +14/+2, β1,+14/+1, β1+14/−1, β1+14/−2, and β1+14/−3.
To define the cis-acting element required for RNAβ replication, two KpnI sites were created at −107 and −74, or −74 and −34, relative to the transcription start site using site-directed mutagenesis. These clones were digested with KpnI and religated to produce the clones β1−107/−74 and β1−74/−34.
The sgRNAβ2 promoter region was analyzed in a β cDNA background that contained a deletion between −34 and +14 relative to the sgRNAβ1 transcription start site and hence lacked the sgRNAβ1 promoter. Clone β1−34/+14 was constructed by digesting the β1−34KpnI clone with KpnI and NcoI, blunting with T4 DNA polymerase, and religating. Since the sgRNAβ2 transcription start site is located at nt 2279, the starting points for mapping the sgRNAβ2 promoter were defined with the clone βΔPstI/EcoRI, which lacks nts 1705 to 2109, and β2 + 9/SspI, lacking nts 2290 to 2434. Clone βΔPstI/EcoRI was generated from the β1−34/+14 clone by digestion with PstI and EcoRI, blunting with T4 DNA polymerase, and religation. Clone β2 + 9/SspI was engineered by introducing a SmaI site at position +9 relative to the sgRNAβ2 transcription start site by site-directed mutagenesis with the oligonucleotide β2 + 9SmaI (Table 1). The resulting derivative was partially digested with SspI, digested with SmaI, and religated to generate the β2 + 9/SspI clone. For large-scale mapping of the sgRNAβ2 promoter, the β1−34/+14 cDNA clone was used in conjunction with the β2-specific oligonucleotides listed in Table 1 to engineer unique KpnI sites upstream of the sgRNAβ2 promoter at positions −110, −76, −52, and −28. These clones were subsequently digested with EcoRI (−179) and KpnI, blunted with T4 DNA polymerase, and religated to generate the clones β2−179/−110, β2−179/−76, β2−179/−52, and β2−179/−28.
Six nucleotide deletions from −70 to −35 relative to the sgRNAβ2 transcription start site were generated by site-directed mutagenesis of clone β1−34/+14 to produce the clones β2−70/−65, β2−64/−59, β2−58/−53, β2−52/−47, β2−46/−41, and β2−40/−35. More refined mapping of the sgRNAβ2 promoter was facilitated by generating the clones β2 −40/−35, β2−40/−34, β2−40/−33, β2−40/−32, β2−40/−31, β2+11/−9, β2 + 10/−15, β2 + 10/−16, β2 + 10/−17, and β2 + 10/−18 by site-directed mutagenesis of the β1−34/+14 cDNA clone.
To determine if the three sgRNA promoters can function ectopically, regions surrounding the sgRNAγ (276 nt), sgRNAβ2 (295 nt), or the sgRNAβ1 promoter (256 and 150 nt) were PCR amplified. These reactions were carried out with the oligonucleotides listed in Table 1 to introduce BamHI sites at the 5′ and 3′ termini. A BamHI site was engineered in the βS/B clone at nt 1132–1137, by site-directed mutagenesis with the β1134BamHI oligonucleotide (Table 1). The BamHI site was then introduced into RNAβ derivatives that either contained or lacked the sgRNAβ1 promoter (β1−34/+14). In addition, a BamHI site was engineered into the γ cDNA clone (containing the sgRNAγ promoter) and the γ−30/−21 cDNA clone (lacking the wt sgRNAγ promoter) at nt 2338–2343 with the γ2239BamHI oligonucleotide (Table 1). These intermediate plasmids were then digested with BamHI and ligated to each of the amplified subgenomic promoters (sgRNAγ, sgRNAβ1, and sgRNAβ2) to generate clones β1+sgγ, β1+sgβ1, β1+sgβ2, β+sgγ, β+sgβ1, β+sgβ2, γ1+sgγ, γ1+sgβ1, γ1+sgβ2, γ+sgγ, γ+sgβ1 and γ+sgβ2. Since the sgRNAβ1 promoter contains a MluI site, the γ +sgβ1 and γ + sgβ1 clones were linearized for in vitro transcription reactions at a SpeI site introduced at their 3′ termini with the BSMV3′SpeI oligonucleotide (Table 1).
Protoplast isolation and transfection
Protoplasts from the BY-2 tobacco cell suspension line were prepared and transfected as described previously (Watanabe et al., 1982). To map the sgRNAγ promoter, protoplasts were transfected with in vitro transcripts (Petty et al., 1988) generated from the cDNA clones α and γ KpnI/HpaI (Zhou and Jackson, 1996a), plus one of the modified γ clones described above. For analysis of the sgRNAβ1 and sgRNAβ2 promoters, protoplasts were transfected with in vitro transcripts from the α and γ cDNA clones, plus the appropriate modified β cDNA clone. Prior to use in in vitro transcription reactions, the β cDNA clones were linearized with SpeI, and the α and γ cDNA clones were linearized with MluI, except for the γ1+sgβ1 and γ+sgβ1 clones which were linearized with SpeI.
RNA extraction and analysis
RNA was extracted from BY-2 protoplasts at 20 h posttransfection as described previously (Zhou and Jackson, 1996a). For Northern blot analysis, 5 μg of total nucleic acid was separated on 1% agarose TBE gels and blotted onto Hybond NX (Amersham Pharmacia Biotech) nylon membranes. Prehybridization, hybridization, and washes were performed as recommended in the manufacturer’s instructions.
RNAβ- and RNAγ-specific [32P]-UTP-labeled riboprobes were generated by in vitro transcription reactions using T7 or T3 RNA polymerase (Zhou and Jackson, 1996a). Portions of the β (2434 to 2785 nt, 802 to 1375 nt) or γ (2111 to 2444 nt) cDNA clones were introduced into pBluescript KS+ (Stratagene) or pGem5Zf+ (Promega, Madison, WI) plasmids and linearized prior to being used in in vitro transcription reactions. An additional RNAβ-specific-riboprobe (2785–2985 nt) was generated by linearizing the βc-2785 clone (D.M. Lawrence and A.O. Jackson, unpublished results) with BamHI prior to use in in vitro transcription reactions.
Acknowledgements
We thank Erin Duncan for technical assistance and Teresa Rubio and Sharon Fodor for comments on the manuscript. This research was supported by USDA Competitive Grant 97-35303-4572 and by a grant from Torrey Mesa Research Institute, Syngenta Research and Technology. J.B. was the recipient of a National Science Foundation Graduate Research Fellowship.
References
- Balmori E., Gilmer D., Richards K., Guilley H., Jonard G. Mapping the promoter for subgenomic RNA synthesis on the beet necrotic yellow vein virus RNA 3. Biochimie. 1993;75:517–521. doi: 10.1016/0300-9084(93)90056-x. [DOI] [PubMed] [Google Scholar]
- Boccard F., Baulcombe D. Mutational analysis of cis-acting sequences and gene function in RNA3 of cucumber mosaic virus. Virology. 1993;193:563–578. doi: 10.1006/viro.1993.1165. [DOI] [PubMed] [Google Scholar]
- Che X., Piestun D., Mawassi M., Yang G., Satyanarayana T., Gowda S., Dawson W.O., Bar-Joseph M. 5′-Coterminal subgenomic RNAs in citrus tristeza virus-infected cells. Virology. 2001;283:374–381. doi: 10.1006/viro.2001.0880. [DOI] [PubMed] [Google Scholar]
- Choi I.R., White K.A. An RNA activator of subgenomic mRNA1 transcription in tomato bushy stunt virus. J. Biol. Chem. 2002;277:3760–3766. doi: 10.1074/jbc.M109067200. [DOI] [PubMed] [Google Scholar]
- French R., Ahlquist P. Characterization and engineering of sequences controlling in vivo synthesis of brome mosaic virus subgenomic RNA. J. Virol. 1988;62:2411–2420. doi: 10.1128/jvi.62.7.2411-2420.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grdzelishvili V.Z., Chapman S.N., Dawson W.O., Lewandowski D.J. Mapping of the tobacco mosaic virus movement protein and coat protein subgenomic RNA promoters in vivo. Virology. 2000;275:177–192. doi: 10.1006/viro.2000.0511. [DOI] [PubMed] [Google Scholar]
- Gustafson G., Hunter B., Hanau R., Armour S.L., Jackson A.O. Nucleotide sequence and genetic organization of barley stripe mosaic virus RNA γ. Virology. 1987;158:394–406. doi: 10.1016/0042-6822(87)90211-x. [DOI] [PubMed] [Google Scholar]
- Hunter B.G., Smith J., Fattoh F., Jackson A.O. Relationship of lychnis ringspot virus to barley stripe mosaic virus and poa semilatent virus. Intervirology. 1989;30:18–26. doi: 10.1159/000150072. [DOI] [PubMed] [Google Scholar]
- Johnston J.C., Rochon D.M. Deletion analysis of the promoter for the cucumber necrosis virus 0.9-kb subgenomic RNA. Virology. 1995;214:100–109. doi: 10.1006/viro.1995.9950. [DOI] [PubMed] [Google Scholar]
- Kelly L., Gerlach W.L., Waterhouse P.M. Characterization of the subgenomic RNAs of an Australian isolate of barley yellow dwarf luteovirus. Virology. 1994;202:565–573. doi: 10.1006/viro.1994.1378. [DOI] [PubMed] [Google Scholar]
- Koev G., Miller W.A. A positive-strand RNA virus with three very different subgenomic RNA promoters. J. Virol. 2000;74:5988–5996. doi: 10.1128/jvi.74.13.5988-5996.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koev G., Mohan B.R., Miller W.A. Primary and secondary structural elements required for synthesis of barley yellow dwarf virus subgenomic RNA1. J. Virol. 1999;73:2876–2885. doi: 10.1128/jvi.73.4.2876-2885.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koonin E.V., Dolja V.V. Evolution and taxonomy of positive-strand RNA viruses: implications of comparative analysis of amino acid sequences. Crit. Rev. Biochem. Mol. Biol. 1993;28:375–430. doi: 10.3109/10409239309078440. [DOI] [PubMed] [Google Scholar]
- Kunkel T.A. Rapid and efficient site-specific mutagenesis without phenotypic selection. Proc. Natl. Acad. Sci. USA. 1985;82:488–492. doi: 10.1073/pnas.82.2.488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindenbach B.D., Sgro J.Y., Ahlquist P. Long-distance base pairing in flock house virus RNA1 regulates subgenomic RNA3 synthesis and RNA2 replication. J. Virol. 2002;76:3905–3919. doi: 10.1128/JVI.76.8.3905-3919.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller W.A., Dreher T.W., Hall T.C. Synthesis of brome mosaic virus subgenomic RNA in vitro by internal initiation on minus-sense genomic RNA. Nature. 1985;313:68–70. doi: 10.1038/313068a0. [DOI] [PubMed] [Google Scholar]
- Miller W.A., Koev G. Synthesis of subgenomic RNAs by positive-strand RNA viruses. Virology. 2000;273:1–8. doi: 10.1006/viro.2000.0421. [DOI] [PubMed] [Google Scholar]
- Pasternak A.O., van den Born E., Spaan W.J., Snijder E.J. Sequence requirements for RNA strand transfer during nidovirus discontinuous subgenomic RNA Synthesis. EMBO J. 2001;20:7220–7228. doi: 10.1093/emboj/20.24.7220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petty I.T.D., French R., Jones R.W., Jackson A.O. Identification of barley stripe mosaic virus genes involved in viral RNA replication and movement. EMBO J. 1990;96:3453–3457. doi: 10.1002/j.1460-2075.1990.tb07553.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petty I.T.D., Hunter B.G., Jackson A.O. A novel strategy for one-step cloning of full-length cDNA and its application to the genome of barley stripe mosaic virus. Gene. 1988;74:423–432. doi: 10.1016/0378-1119(88)90175-8. [DOI] [PubMed] [Google Scholar]
- Petty I.T.D., Jackson A.O. Mutational analysis of barley stripe mosaic virus RNA β. Virology. 1990;179:712–718. doi: 10.1016/0042-6822(90)90138-h. [DOI] [PubMed] [Google Scholar]
- Savenkov E.I., Solovyev A.G., Morozov S.Y. Genome sequences of poa semilatent and lychnis ringspot hordeiviruses. Arch. Virol. 1998;143:1379–1393. doi: 10.1007/s007050050382. [DOI] [PubMed] [Google Scholar]
- Sawicki S.G., Sawicki D.L. A new model for coronavirus transcription. Adv. Exp. Med. Biol. 1998;44:215–219. doi: 10.1007/978-1-4615-5331-1_26. [DOI] [PubMed] [Google Scholar]
- Shivprasad S., Pogue G.P., Kewandowski D.J., Hidalgo J., Donson J., Grill L.K., Dawson W.O. Heterologous sequences greatly affect foreign gene expression in tobacco mosaic virus-based vectors. Virology. 1999;255:312–323. doi: 10.1006/viro.1998.9579. [DOI] [PubMed] [Google Scholar]
- Siegel R.W., Adkins S., Kao C.C. Sequence-specific recognition of a subgenomic RNA promoter by a viral RNA polymerase. Proc. Natl. Acad. Sci. USA. 1997;94:11238–11243. doi: 10.1073/pnas.94.21.11238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siegel R.W., Bellon L., Beigelman L., Kao C.C. Moieties in an RNA promoter specifically recognized by a viral RNA-dependent RNA polymerase. Proc. Natl. Acad. Sci. USA. 1998;95:11613–11618. doi: 10.1073/pnas.95.20.11613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sit T.L., Vaewhongs A.A., Lommel S.A. RNA-mediated trans-activation of transcription from a viral RNA. Science. 1998;281:829–832. doi: 10.1126/science.281.5378.829. [DOI] [PubMed] [Google Scholar]
- Solovyev A.G., Savenkov E.I., Grdzelishvili V.Z., Kalinina N.O., Morozov S.Y., Schiemann J., Atabekov J.G. Movement of hordeivirus hybrids with exchanges in the triple gene block. Virology. 1999;253:278–287. doi: 10.1006/viro.1998.9528. [DOI] [PubMed] [Google Scholar]
- Stawicki S.S., Kao C.C. Spatial perturbations within an RNA promoter specifically recognized by a viral RNA-dependent RNA Polymerase (RdRp) reveal that RdRp can adjust its promoter binding sites. J. Virol. 1999;73:198–204. doi: 10.1128/jvi.73.1.198-204.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Der Kuyl A.C., Langereis K., Houwing C.J., Jaspars E.M.J., Bol J.F. Cis-acting elements involved in replication of alfalfa mosaic virus RNA in vitro. Virology. 1990;176:346–354. doi: 10.1016/0042-6822(90)90004-b. [DOI] [PubMed] [Google Scholar]
- Van Der Vossen E.A.G., Notenboom T., Bol J.F. Characterization of sequences controlling the synthesis of alfalfa mosaic virus subgenomic RNA in vitro. Virology. 1995;212:663–672. doi: 10.1006/viro.1995.1524. [DOI] [PubMed] [Google Scholar]
- van Regenmortel M.H.V., Fauquet C.M., Bishop D.H.L., Carstens E.B., Estes M.K., Lemon S.M., Maniloff J., Mayo M.A., McGeoch D.J., Pringle C.R., Wickner R.B. The Seventh Report of the International Committee on Taxonomy of Viruses. Academic Press; San Diego: 2000. Virus Taxonomy: The taxonomy and nomenclature of viruses; pp. 899–922. and 969–989. [Google Scholar]
- Verchot J., Angell S.M., Baulcombe D.C. In vivo translation of the triple gene block of potato virus X requires two subgenomic mRNAs. J. Virol. 1998;72:8316–8320. doi: 10.1128/jvi.72.10.8316-8320.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J., Carpenter C.D., Simon A.E. Minimal sequence and structural requirements of a subgenomic RNA promoter for turnip crinkle virus. Virology. 1999;253:327–336. doi: 10.1006/viro.1998.9538. [DOI] [PubMed] [Google Scholar]
- Wang J., Simon A.E. Analysis of the two subgenomic RNA promoters for turnip crinkle virus in vivo and in vitro. Virology. 1997;232:174–186. doi: 10.1006/viro.1997.8550. [DOI] [PubMed] [Google Scholar]
- Watanabe Y., Ohno T., Okada Y. Virus multiplication in tobacco protoplasts inoculated with tobacco mosaic virus RNA encapsulated in large unilamellar vesicular liposomes. Virology. 1982;120:478–480. doi: 10.1016/0042-6822(82)90048-4. [DOI] [PubMed] [Google Scholar]
- White K.A. The premature termination model: a possible third mechanism for subgenomic mRNA transcription in (+)-strand RNA viruses. Virology. 2002;304:147–154. doi: 10.1006/viro.2002.1732. [DOI] [PubMed] [Google Scholar]
- Zhang G., Slowinski V., White K.A. Subgenomic mRNA regulation by a distal RNA element in a (+)-strand RNA virus. RNA. 1999;5:550–561. doi: 10.1017/s1355838299982080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhong W., Ruekert R.R. Flock house virus: down-regulation of subgenomic RNA3 synthesis does not involve coat protein and is targeted to synthesis of its positive strand. J. Virol. 1993;67:2716–2722. doi: 10.1128/jvi.67.5.2716-2722.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou H., Jackson A.O. Analysis of cis-acting elements required for replication of barley stripe mosaic virus RNAs. Virology. 1996;219:150–160. doi: 10.1006/viro.1996.0232. [DOI] [PubMed] [Google Scholar]
- Zhou H., Jackson A.O. Expression of the barley stripe mosaic virus RNA β “triple gene block”. Virology. 1996;216:367–379. doi: 10.1006/viro.1996.0072. [DOI] [PubMed] [Google Scholar]