

Abstract
Tula virus is a member of the Hantavirus genus of the family Bunyaviridae. Viruses of this family have an unusual pattern of intracellular maturation at the ER–Golgi compartment. We recently found that Tula virus, similar to several other hantaviruses, is able to induce apoptosis in cultured cells [Li, X.D., Kukkonen, S., Vapalahti, O., Plyusnin, A., Lankinen, H., Vaheri, A., 2004. Tula hantavirus infection of Vero E6 cells induces apoptosis involving caspase 8 activation. J. Gen. Virol. 85, 3261–3268.]. However, the cellular mechanisms remain to be clarified. In this study, we demonstrate that the progressive replication of Tula virus in Vero E6 cells initiates several death programs that are intimately associated with ER stress: (1) early activation of ER-resident caspase-12; (2) phosphorylation of Jun NH2-terminal kinase (JNK) and its downstream target transcriptional factor, c-jun; (3) induction of the pro-apoptotic transcriptional factor, growth arrest- and DNA damage-inducible gene 153, or C/EBP homologous protein (Gadd153/chop); and (4) changes in the ER-membrane protein BAP31 implying cross-talk with the mitochondrial apoptosis pathway. Furthermore, we confirmed that a sustained ER stress was induced marked by an increased expression of an ER chaperone Grp78/BiP. Taken together, we have identified involvement of ER stress-mediated death program in Tula virus-infected Vero E6 cells which provides a new approach to understand the mechanisms in hantavirus-induced apoptosis.
Keywords: Endoplasmic reticulum (ER) stress, Grp78/BiP, Caspase-12, Jun NH2-terminal kinase (JNK), Growth arrest- and DNA damage-inducible gene 153 or CAATT enhancer-binding protein, C/EBP homologous protein (Gadd153/chop)
Introduction
Hantaviruses (family Bunyaviridae) are known to cause two severe human diseases: hemorrhagic fever with renal syndrome (HFRS) and hantavirus pulmonary syndrome (HPS). Hantaviruses are enveloped, spherical, negative-stranded RNA viruses with three genomic segments: small (S), medium (M), and large (L), which encode the nucleocapsid protein (N), two envelope proteins (Gn and Gc), and the RNA polymerase/transcriptase (L protein), respectively (Plyusnin et al., 1996, Schmaljohn, 1996, Shi and Elliott, 2004).
The maturation of glycoproteins of most Bunyaviridae including hantaviruses usually takes place at the region from endoplasmic reticulum (ER) to Golgi apparatus (Matsuoka et al., 1994, Spiropoulou, 2001, Spiropoulou et al., 2003). If overexpressed, the Gn envelope protein of some hantavirus members has an unusual tendency to form aggregates that may resemble aggresomes in cells (Ruusala et al., 1992, Spiropoulou et al., 2003). In this regard, Gn of hantavirus genus is similar to glycoproteins of hepatitis C virus (HCV) (Choukhi et al., 1999), some coronaviruses (Rottier, 1995) and Bunyamwera virus (Nakitare and Elliott, 1993). It has been suggested that aggresome-like structures result from the accumulation of misfolded proteins (Kopito, 2000).
The ER is a eukaryotic organelle characterized by its extensive membranous network. It acts not only as a principal site for posttranslational modifications, folding, oligomerization of the newly synthesized membrane and secretory proteins, but also as a major signal transduction compartment in the cell. Perturbations in the ER homeostasis will initiate an ER stress response pathway, which is evolutionarily conserved from yeast to human (Harding et al., 2002, Kaufman et al., 2002). ER stress may be divided into two classes: unfolded protein response (UPR) and ER-overload response (EOR). The former is represented by a marked increase in ER-localized proteins such as glucose-regulated protein 78 (Grp78/BiP) or 94 (Grp94). The latter is characterized by activation of the NF-κB pathway.
In certain circumstances, severe or prolonged ER stress will lead to cell death (Breckenridge et al., 2003, Oyadomari et al., 2002) through initiation of downstream death programs such as activation of a unique ER-located caspase-12 (Lamkanfi et al., 2004), phosphorylation of NH2-terminal Jun kinase (JNK), and induction of growth arrest- and DNA damage-inducible gene 153, also called C/EBP homologous protein (Gadd153/chop) (Oyadomari and Mori, 2004). Recently, a number of enveloped RNA viruses, such as hepatitis C virus (HCV), Japanese encephalitis virus (JEV), and influenza A virus, have been shown to induce programmed cell death through an ER stress-mediated mechanism (Pavio et al., 2003, Su et al., 2002, Tardif et al., 2002, Waris et al., 2002).
The cellular mechanism for hantavirus-induced apoptosis in cultured cells remains to be defined (Akhmatova et al., 2003, Kang et al., 1999, Li et al., 2004, Markotic et al., 2003). In this report, we show that Tula virus infection induces ER stress–response pathway marked by an increased expression of Grp78/BiP and subsequently leads to activation of several ER stress-mediated apoptotic programs.
Results
Tula virus infection activates several ER-stress-associated apoptotic programs
We have recently demonstrated that Tula virus infection causes apoptotic cell death of Vero E6 cells (Li et al., 2004). In order to determine whether ER stress may play a role in it, we analyzed several apoptotic pathways known to be associated with ER.
Activation of caspase-12
Caspase activation is a sequential event carried out by the initiator and effector caspases (Chang and Yang, 2000). We have shown that Tula virus-induced apoptosis is caspase-mediated and can be blocked by a broad caspase peptide inhibitor z-VAD-fmk (Li et al., 2004). In that study, we observed that activated caspase-8 started to appear on the fourth day post-infection (p.i.). However, the questions remained, how does caspase-8 get activated and where do the signals come from. Taking into account the prominent role of ER in the replication of Tula virus, we tested several known players that are associated with ER stress. Caspase-12 is known as an ER-resident caspase that may cooperate with caspase-8 during ER stress (Lamkanfi et al., 2004, Nakagawa et al., 2000). Therefore, we thought that caspase-12 is another likely candidate to activate the Tula hantavirus-induced caspase cascade. Fig. 1 shows that caspase-12 was cleaved evidently from the inactive pro-enzyme to the active one starting on the third day p.i., which was at least 1 day earlier than the activation of caspase-8 and caspase-3. This indicates that caspase-12 might be the initiator caspase responsible for transduction of the death signal from the ER.
Fig. 1.
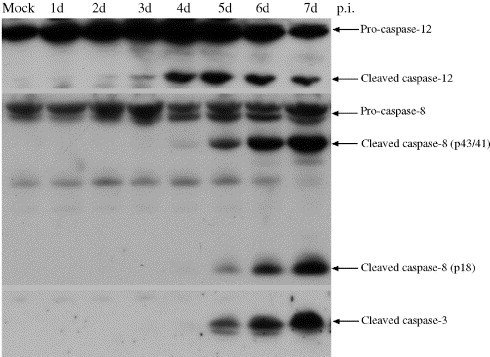
Capsase-12 is activated earlier than caspase-8 and caspase-3. Vero E6 cell lysates following TULV infection on the indicated day p.i. were collected for immunoblotting and analyzed with antibodies against caspase-12, caspase-8, and caspase-3, respectively. The cleavage of caspase-12 took place on the fourth day p.i., while caspase-8 and caspase-3 were activated on the fifth day p.i.
Activation of JNK pathway
It is known that activation of the JNK pathway is required for apoptosis in certain circumstances (Takeda et al., 2003). In response to ER stress, the JNK pathway is activated through the complex of IRE1-TRAF2-ASK1 (Nishitoh et al., 2002). In view of the pivotal role of JNK in apoptosis and ER stress, we were prompted to investigate its role in the model of Tula virus-induced cell death. In Vero E6 cells the endogenous expression of JNK (1 and 2) is relatively high (Fig. 2A, lower). Tula virus infection led to apparent phosphorylation of JNK1, and to a much less extent also JNK2 on the fifth day p.i., as also confirmed by calf intestinal alkaline phosphatase (CIP) treatment (Fig. 2A, upper), but the level of the non-phosphorylated form of JNKs remained almost unchanged. We further analyzed a major downstream substrate of JNK, c-jun. Interestingly, we noticed that the endogenous level of c-jun was undetectable in mock-treated control Vero E6 cells. However, as the infection proceeded, both forms of c-jun, the non-phosphorylated and phosphorylated, were dramatically increased (Fig. 2B). This can be explained by that phosphorylated JNK1 stabilizes c-jun after a stress. In contrast, JNK2 destabilizes c-jun protein for degradation under the non-stimulating conditions (Ronai, 2004, Sabapathy et al., 2004). Furthermore, the importance of activation of JNK pathway on induction of apoptosis was confirmed by the observation of reduced PARP cleavage after adding JNK inhibitor II (Fig. 2C). These results indicate that activation of the JNK pathway may contribute significantly to the Tula virus-induced apoptosis in Vero E6 cells.
Fig. 2.
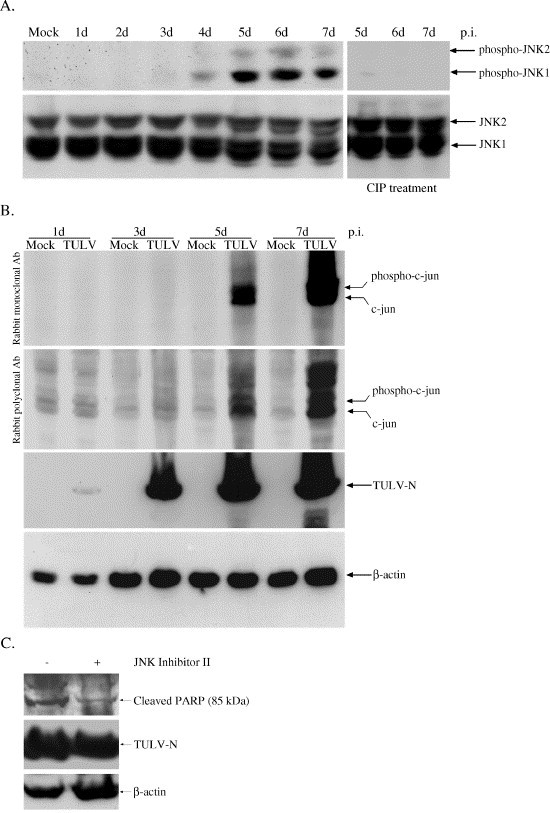
Activation of JNK pathway. (A) Mock-treated or Tula virus-infected cell lysates were resolved in SDS–PAGE and transferred onto nitrocellulose, the membranes were then treated (on the right) or untreated with CIP. (B) TULV-infected or mock Vero E6 cell lysates were collected at the indicated day p.i. and analyzed with antibodies against c-jun (rabbit monoclonal and rabbit polyclonal). Infection was monitored with antibody against nucleocapsid protein. β-actin was used as a loading control. (C) JNK inhibitor II SP600126 (50 μM, added every other day) blocked the cleavage of PARP in Vero E6 cells on the fifth day post-Tula virus infection.
Increased protein and mRNA of Gadd153/chop
We (Li et al., 2004) and others (Kang et al., 1999) have shown that the Bcl-2 protein level is down-regulated during hantavirus infection. We wanted to know the cellular mechanism that may explain the decreased production of Bcl-2 protein. It has been reported that growth arrest- and DNA damage-inducible gene 153 or CAATT enhancer-binding protein, C/EBP homologous protein (Gadd153/chop) sensitizes cells to ER stress-mediated cell death by down-regulating Bcl-2 mRNA (McCullough et al., 2001). So, we turned to evaluate the role of Gadd153/chop in Tula virus-infected Vero E6 cells. Fig. 3 shows that chop was indeed increased transcriptionally (Figs. 3B and C) and translationally (Fig. 3A) following Tula virus infection in Vero E6 cells.
Fig. 3.
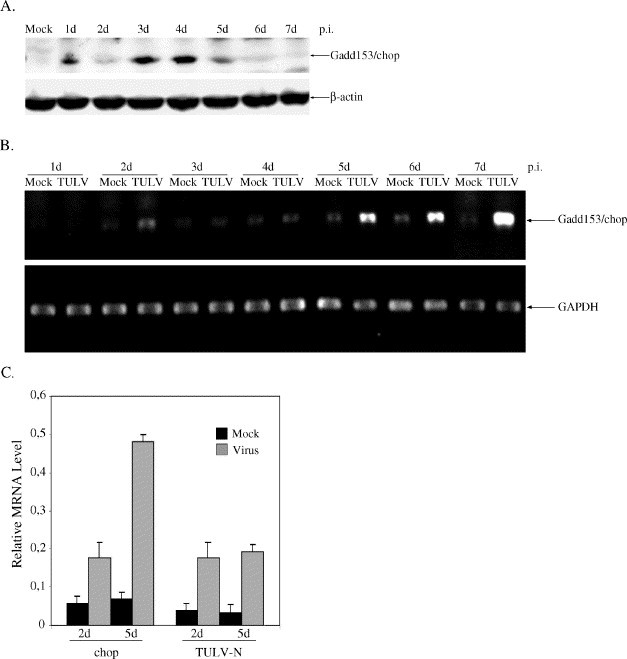
Gadd153/chop is induced during TULV infection. (A) Increased protein level of Gadd153/chop following infection. (B) Induction of Gadd153/chop at the late stage of TULV infection shown by conventional RT-PCR. (C) Real-time PCR confirms the transcriptional regulation of Gadd153/chop on the fifth day of TULV infection. β-actin (in A) and GAPDH (in B and C) were as controls.
Effects of Tula virus infection on ER membrane protein BAP31
BAP31, a ubiquitously expressed integral ER-membrane protein, regulates apoptosis through association with Bcl-2, Bcl-XL, or pro-caspase-8. NH2-terminal membrane embedded fragment p20 is a cleavage product of BAP31 by activated caspase-8 in response to extrinsic apoptotic stimuli via death receptor such as TNF-R1. Prolonged expression of p20 fragment alone is enough to induce caspase activation and apoptosis through mitochondria-mediated pathway (Breckenridge et al., 2003, Ng and Shore, 1998, Ng et al., 1997). It is interesting to point out that in the kidney, BAP31 staining was prominent in the proximal tubules while glomeruli were BAP31-negative (Manley and Lennon, 2001). The clinical findings in HFRS and HPS patients suggest that tubuli are the most affected region in the kidney. Here we sought to evaluate the role of BAP31 in the Tula virus-infected Vero E6 cells. Both the full-length and p20 fragment of BAP31 were down-regulated and readily detectable by mouse monoclonal antibody (8DIIE) and rabbit polyclonal antibody throughout the time course of Tula virus infection (Fig. 4 ). In addition, a novel protein band migrating at about 10 kDa was recognized by the rabbit polyclonal antibody against BAP31. It is worthwhile to note that the time course of appearance of this band correlated well with the activation of caspase-8, as already shown in Fig. 1. So, these results indicate that the dramatic change of BAP31 protein may play a critical role in Tula virus-induced apoptosis.
Fig. 4.
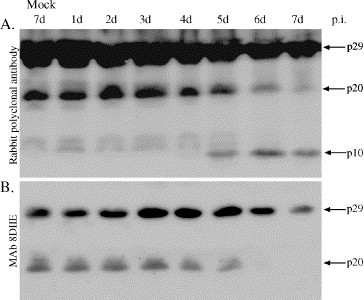
Changes in the integral ER membrane protein BAP31 shown by immunoblot. (A) Rabbit polyclonal antibody against BAP31 shows the decreased amount of the full-length and p20 fragment of Bap31 while a p10 fragment of Bap31 started to accumulate. (B) A specific monoclonal antibody against BAP31 confirms the down-regulation of the full-length of and p20 fragment BAP31.
Tula virus infection activates ER stress marked by the induction of Grp78/BiP
We have suspected that hantavirus replication might initiate a sustained ER-stress response that might eventually trigger downstream apoptotic pathways. To test this hypothesis, we sought for the evidence on ER stress by analyzing the Tula virus-infected Vero E6 cell lysates that were collected at different time points. In Fig. 5A, we demonstrate that Tula hantavirus infection induced the production of Grp78/BiP protein more specifically than other stress factors like Grp94, protein disulfide bond isomerase (PDI), calreticulin, or the cytosolic chaperone HSP70. Grp78/BiP started to increase on the third day and reached its peak on the seventh day p.i. This correlated with the progressive viral replication shown by immunoblot against the nucleocapsid protein of Tula virus (Fig. 5A). Grp78/BiP is generally considered an ER-stress marker protein that is regulated at the transcriptional level. As expected, Grp78/BiP mRNA was elevated following infection shown by conventional RT-PCR (Fig. 5B) and real-time PCR (Fig. 5C). In addition, to rule out additional factors other than virus replication itself for the sustained ER-chaperone induction, we used UV-inactivated virus, as well as ribavirin and interferon-alpha (IFN-α), efficient antiviral agents against hantaviruses in vitro and in vivo (Severson et al., 2003). In the absence of viral replication with UV radiation, we could not see the induction of Grp78/BiP (data not shown). Similarly, decreasing viral replication by treatment of Vero E6 cells with either ribavirin or IFN-α reduced the level of BiP/Grp78 (Fig. 5D). Taken together, these data suggest that progressive replication is required for Tula virus-induced ER stress and that Tula virus-infection induces up-regulation of Grp78/BiP in Vero E6 cells.
Fig. 5.
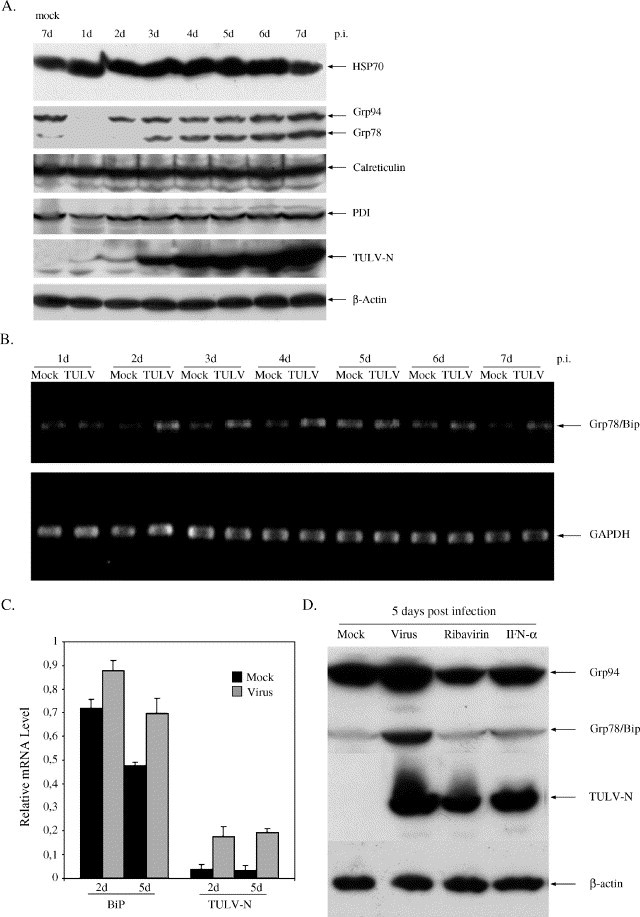
Increased protein and RNA expression of Grp78/BiP during Tula virus infection. (A) Grp78/BiP protein expression. (B) Increased mRNA of Grp78/BiP shown by semi-quantitive RT-PCR. (C) Increased mRNA of Grp78/BiP shown by real-time PCR. (D) Replication is required for ER stress. β-actin (in A and D) and GAPDH (in B and C) were as controls.
Discussion
At present, biochemical and cellular processes in the replication cycle of hantaviruses are still poorly characterized. To some extent, this has limited our efforts to understand molecular mechanisms in the pathogenesis of HFRS and HPS. We sought to approach this issue by utilizing a recently discovered model on Tula virus-induced apoptosis of Vero E6 cells (Li et al., 2004). We now show that the replication of Tula virus is able to induce ER-stress response marked by the increased expression of ER-located chaperone Grp78/BiP. Multiple downstream apoptotic signaling pathways are subsequently activated and ultimately lead to death of all infected Vero E6 cells by apoptosis. Here, a model is suggested in Fig. 6 to summarize the Tula virus-induced ER stress-signaling pathway. We propose that misfolding hantavirus glycoproteins lead to ER stress. Its seems possible that the programs downstream of ER stress may involve pathways operating not only in initiation and execution of apoptosis, but also in a proinflammatory response.
Fig. 6.
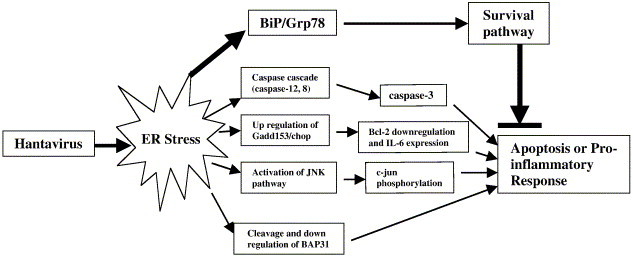
The diagram shows a hypothesized model for Tula hantavirus-induced apoptosis.
ER-stress signaling is initiated from ER lumen by unfolded or misfolded proteins. Generally speaking, protein misfolding is a fact of life (Kopito, 2000). This may be especially true for many enveloped RNA viruses for which the ER serves as the primary site of replication. In order to maximize production of viral proteins, viruses must demand coordinated ER function, but they also have to cope with the ER-stress response of host cells. Envelope glycoproteins encoded by Flaviviridae such as Japanese encephalitis virus, HCV, and bovine viral diarrhea virus (BVDV) can accumulate to huge amounts in the lumen of ER (Jordan et al., 2002, Su et al., 2002, Tardif et al., 2002). It has been shown that overexpression of enveloped viral components alone in cultured cells is enough to induce ER stress-mediated apoptosis (Kalkeri et al., 2001, Liberman et al., 1999). Moreover, HCV envelope proteins exist as high molecular weight aggregates that are not recognized by conformation-specific antibodies, suggesting that they exist in nonnative conformations (Choukhi et al., 1999). Currently, at least three lines of evidence indicate that there may exist a significant amount of misfolded Gn protein of hantavirus within infected cells. Firstly, it has been noticed for a long time that the Gn protein of hantavirus tends to aggregate into a high molecular weight complex which is prominent in SDS-containing buffer (Ruusala et al., 1992, Spiropoulou et al., 2003, Vapalahti et al., 1996). Secondly, mature Gn and Gc proteins surprisingly remain in the high mannose type of glycosylation and associate with the ER chaperones calnexin and calreticulin, an implication of the failure in passing quality control (Shi and Elliott, 2004). Since Gn is hardly found on the plasma membrane, these findings suggest the possibility that a significant portion of misfolded Gn protein is retained on the way from ER, ER–Golgi intermediate, to cis–Golgi (Shi et al., 2004). It has also been suggested that even the misfolded protein can move to ER–Golgi intermediate and to cis–Golgi and recycle back to ER in complex with Grp78/BiP (Hammond and Helenius, 1994). Therefore, it is tempting to speculate that the maturation of hantavirus might be regulated by signals emanated from the ER lumen where unfolded or unassembled viral glycoproteins are present.
We have also shown that sustained ER stress may eventually contribute to apoptosis of Vero E6 cells induced by Tula hantavirus. Normally, ER stress starts first with a survival signal by transcriptional induction of ER-localized chaperones. The mechanism of the switch to apoptotic program during ER stress is currently poorly understood. We found that Tula virus infection activates several apoptosis pathways downstream of ER stress.
We believe that our findings could contribute to understanding of the molecular mechanisms involved in hantavirus induction of proinflammatory cytokines like TNF-α or IL-6. For example, caspase-12 has recently been linked with inflammatory and innate immune response to endotoxins (Saleh et al., 2004); it is well known that JNK activation of transcription factor activator protein 1 (AP-1) through phosphorylation of c-jun is required for expression of TNF-α, IL-2, and IFN-γ (Bennett et al., 2001, Foletta et al., 1998), in which TNF-α is an important mediator in HFRS (Linderholm et al., 1996, Temonen et al., 1996); Gadd153/chop is directly responsible for up-regulation of IL-6 (Hattori et al., 2003), which plays an important role in the pathogenesis of HFRS and HPS (Linderholm et al., 1996, Mäkelä et al., 2004). Our observation of the induction of Gadd153/chop by Tula virus infection may provide a cellular mechanism for IL-6 production. Further studies are needed to elucidate the relationship between hantavirus and maturation of pro-inflammatory cytokines. In summary, we think that our present findings in this report should provide new approaches to understand the molecular mechanisms of pathogenesis of hantavirus infections.
Material and methods
Antibodies and other reagents
Rabbit polyclonal antibodies against phosphorylated or non-phosphorylated form of JNK were from Promega and Santa Cruz Biotechnology Inc., respectively. Mouse monoclonal antibody against Gadd153/Chop was from Santa Cruz Biotechnology Inc., rabbit polyclonal antibody against caspase-12 from BioVision, rabbit polyclonal antibody against caspase-3, mouse monoclonal antibody anti-caspase-8, and rabbit monoclonal anti-c-jun antibody from Cell Signalling Biotechnology. Rabbit polyclonal anti-c-jun antibody was from Oncogene Research Products and mouse monoclonal antibody against β-actin from Sigma. Rabbit polyclonal and monoclonal antibodies against BAP31 have been described previously (Määttä et al., 2000). Rabbit polyclonal antibodies against Puumala hantavirus N and glycoproteins Gn and Gc have been described previously (Vapalahti et al., 1995). JNK inhibitor II SP600126 was from Calbiochem.
Virus and cell cultures
Tula hantavirus Moravia strain 5302 (Vapalahti et al., 1996) was propagated in Vero E6 cells. The Tula virus preparation used to infect cells had a titer of 4.0 × 105 focus forming units per ml. Vero E6 cells (green monkey kidney cell line) were grown in MEM with 10% heat-inactivated fetal calf serum, glutamine, 100 IU/ml of penicillin, and 100 μg/ml of streptomycin at 37 °C in a humidified atmosphere containing 5% CO2. The culture medium of Tula virus-infected cells was found to be mycoplasma-free when tested with a highly sensitive PCR ELISA-based mycoplasma detection kit (Roche).
UV irradiation of virus
A stock of virus in a lidless 3 cm in diameter culture dish was irradiated at 254 nm, using a 30-W UV lamp at room temperature at the distance of 10 cm. The exposure time was about 30 min. Then UV-treated and untreated virus stocks were used to infect cell monolayers.
RNA isolation and RT-PCR
Mock-treated and -infected Vero E6 cells were grown in 3 cm in diameter culture dishes. Cells were collected into 1 ml TriPure RNA isolation reagent (Roche) each day until the 8th day p.i. and stored at −70 °C. Total RNA was isolated following the instructions provided by the manufacturer (Roche). Subsequently, an equal amount of RNA sample (about 5 μg) from each day was applied to generate cDNA, with which as template conventional PCR was performed. The primer pairs for respective genes were as follows: Grp78/BiP forward—5′GCA GCT GCT ATT GCT TAT GG, reverse—5′TTG AGC TCT TCA AAT TTG GC; Gadd153/Chop forward—5′AGA GAT GGC AGC TGA GTC AT, reverse—5′ TCA TGC TTG GTG CAG ATT CAC; glyceraldehydes-3-phosphate dehydrogenase (GAPDH) forward—5′AGT CAA CGG ATT TGG TCG TA, reverse—5′AGG GGT GCT AAG CAG TTG GT. The entire experiment was repeated at least three times using RNA samples made independently. To obtain the relative expression level of each sample, the target's quantity was divided by the respective GAPDH quantity.
Immunoblotting
Briefly, mock-treated and -infected cell monolayers (about 1 × 107 cells) were harvested by scraping with a rubber policeman and washed with 10 ml of ice-cold phosphate-buffered saline (PBS) twice. Next, cells were immediately lysed into 200 μl Laemmli Sample Buffer (non-reduced) and sonicated and centrifuged at maximal speed. The protein concentration in the resulting supernatant was determined using BCA Protein Assay Kit from Pierce. After that, β-mercaptoethanol was added into each sample, which was denatured at 95 °C for 5 min and stored at −20 °C. 50 μg of protein was analyzed by 10% SDS–PAGE and immunoblotted according to standard protocols. For studying JNK activation, after the Western transfer, the membrane was further treated with calf intestinal alkaline phosphatase (CIP) (New England Biolabs) for about 2 h at 37 °C.
Acknowledgments
We thank Dr. Erkki Hölttä and Dr. Esa Kuismanen for discussion and providing antibodies. We are grateful to Ms. Leena Kostamovaara for expert technical assistance. This work was supported by grants from Academy of Finland, EU grants (QLK2-CT-1999-01119 and QLK2-CT-2002-01358), Magnus Ehrnrooth Foundation, and Sigrid Jusélius Foundation, Helsinki, Finland.
Contributor Information
Xiao-Dong Li, Email: Xiaodong.Li@Helsinki.Fi.
Hilkka Lankinen, Email: Hilkka.Lankinen@Helsinki.Fi.
References
- Akhmatova N.K., Yusupova R.S., Khaiboullina S.F., Sibiryak S.V. Lymphocyte apoptosis during hemorrhagic fever with renal syndrome. Russ. J. Immunol. 2003;8:37–46. [PubMed] [Google Scholar]
- Bennett B.L., Sasaki D.T., Murray B.W., O'Leary E.C., Sakata S.T., Xu W., Leisten J.C., Motiwala A., Pierce S., Satoh Y., Bhagwat S.S., Manning A.M., Anderson D.W. SP600125, an anthrapyrazolone inhibitor of Jun N-terminal kinase. Proc. Natl. Acad. Sci. U. S. A. 2001;98:13681–13686. doi: 10.1073/pnas.251194298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Breckenridge D.G., Germain M., Mathai J.P., Nguyen M., Shore G.C. Regulation of apoptosis by endoplasmic reticulum pathways. Oncogene. 2003;22:8608–8618. doi: 10.1038/sj.onc.1207108. [DOI] [PubMed] [Google Scholar]
- Chang H.Y., Yang X. Proteases for cell suicide: functions and regulation of caspases. Microbiol. Mol. Biol. Rev. 2000;64:821–846. doi: 10.1128/mmbr.64.4.821-846.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choukhi A., Pillez A., Drobecq H., Sergheraert C., Wychowski C., Dubuisson J. Characterization of aggregates of hepatitis C virus glycoproteins. J. Gen. Virol. 1999;80:3099–3107. doi: 10.1099/0022-1317-80-12-3099. [DOI] [PubMed] [Google Scholar]
- Foletta V.C., Segal D.H., Cohen D.R. J. Leukocyte Biol. 1998;63:139–152. doi: 10.1002/jlb.63.2.139. [DOI] [PubMed] [Google Scholar]
- Hammond C., Helenius A. Quality control in the secretory pathway: retention of a misfolded viral membrane glycoprotein involves cycling between the ER, intermediate compartment, and Golgi apparatus. J. Cell Biol. 1994;126:41–52. doi: 10.1083/jcb.126.1.41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harding H.P., Calfon M., Urano F., Novoa I., Ron D. Transcriptional and translational control in the Mammalian unfolded protein response. Annu. Rev. Cell Dev. Biol. 2002;18:575–599. doi: 10.1146/annurev.cellbio.18.011402.160624. [DOI] [PubMed] [Google Scholar]
- Hattori T., Ohoka N., Hayashi H., Onozaki K. C/EBP homologous protein (CHOP) up-regulates IL-6 transcription by trapping negative regulating NF-IL6 isoform. FEBS Lett. 2003;541:33–39. doi: 10.1016/s0014-5793(03)00283-7. [DOI] [PubMed] [Google Scholar]
- Jordan R., Nikolaeva O., Wang L., Conyers B., Mehta A., Block T.M., Dwek R.A. Inhibition of host ER glucosidase activity prevents Golgi processing of virion-associated bovine viral diarrhea virus E2 glycoproteins and reduces infectivity of secreted virions. Virology. 2002;295:10–19. doi: 10.1006/viro.2002.1370. [DOI] [PubMed] [Google Scholar]
- Kalkeri G., Khalap N., Garry R.F., Fermin C.D., Srikanta D. Hepatitis C virus protein expression induces apoptosis in HepG2 cells. Virology. 2001;282:26–37. doi: 10.1006/viro.2000.0835. [DOI] [PubMed] [Google Scholar]
- Kang J.I., Park S.H., Lee P.W., Ahn B.Y. Apoptosis is induced by hantaviruses in cultured cells. Virology. 1999;264:99–105. doi: 10.1006/viro.1999.9896. [DOI] [PubMed] [Google Scholar]
- Kaufman R.J., Scheuner D., Schroder M., Shen X., Lee K., Liu C.Y., Arnold S.M. The unfolded protein response in nutrient sensing and differentiation. Nat. Rev., Mol. Cell Biol. 2002;3:411–421. doi: 10.1038/nrm829. [DOI] [PubMed] [Google Scholar]
- Kopito R.R. Aggresomes, inclusion bodies and protein aggregation. Trends Cell Biol. 2000;10:524–530. doi: 10.1016/s0962-8924(00)01852-3. [DOI] [PubMed] [Google Scholar]
- Lamkanfi M., Kalai M., Vandenabeele P. Caspase-12: an overview. Cell Death Differ. 2004;11:365–368. doi: 10.1038/sj.cdd.4401364. [DOI] [PubMed] [Google Scholar]
- Li X.D., Kukkonen S., Vapalahti O., Plyusnin A., Lankinen H., Vaheri A. Tula hantavirus infection of Vero E6 cells induces apoptosis involving caspase 8 activation. J. Gen. Virol. 2004;85:3261–3268. doi: 10.1099/vir.0.80243-0. [DOI] [PubMed] [Google Scholar]
- Liberman E., Fong Y., Selby M.J., Choo Q., Cousens L., Houghton M., Yen T.S.B. Activation of the grp78 and grp94 promoters by hepatitis C virus E2 envelope protein. J. Virol. 1999;73:3718–3722. doi: 10.1128/jvi.73.5.3718-3722.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Linderholm M., Ahlm C., Settergren B., Waage A., Tarnvik A. Elevated plasma levels of tumor necrosis factor (TNF)-alpha, soluble TNF receptors, interleukin (IL)-6, and IL-10 in patients with hemorrhagic fever with renal syndrome. J. Infect. Dis. 1996;173:38–43. doi: 10.1093/infdis/173.1.38. [DOI] [PubMed] [Google Scholar]
- Määttä J., Hallikas O., Welti S., Hilden P., Schroder J., Kuismanen E. Limited caspase cleavage of human BAP31. FEBS Lett. 2000;484:202–206. doi: 10.1016/s0014-5793(00)02159-1. [DOI] [PubMed] [Google Scholar]
- Mäkelä S., Mustonen J., Ala-Houhala I., Hurme M., Koivisto A.M., Vaheri A., Pasternack A. Urinary excretion of interleukin-6 correlates with proteinuria in acute Puumala hantavirus-induced nephritis. Am. J. Kidney Dis. 2004;43:809–816. doi: 10.1053/j.ajkd.2003.12.044. [DOI] [PubMed] [Google Scholar]
- Manley H.A., Lennon V.A. Endoplasmic reticulum membrane-sorting protein of lymphocytes (BAP31) is highly expressed in neurons and discrete endocrine cells. J. Histochem. Cytochem. 2001;49:1235–1243. doi: 10.1177/002215540104901005. [DOI] [PubMed] [Google Scholar]
- Markotic A., Hensley L., Geisbert T., Spik K., Schmaljohn C. Hantaviruses induce cytopathic effects and apoptosis in continuous human embryonic kidney cells. J. Gen. Virol. 2003;84:2197–2202. doi: 10.1099/vir.0.19090-0. [DOI] [PubMed] [Google Scholar]
- Matsuoka Y., Chen S.Y., Compans R.W. A signal for Golgi retention in the bunyavirus G1 glycoprotein. J. Biol. Chem. 1994;269:22565–22573. [PubMed] [Google Scholar]
- McCullough K.D., Martindale J.L., Klotz L.O., Aw T.Y., Holbrook N.J. Gadd153 sensitizes cells to endoplasmic reticulum stress by down-regulating Bcl2 and perturbing the cellular redox state. Mol. Cell. Biol. 2001;21:1249–1259. doi: 10.1128/MCB.21.4.1249-1259.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakagawa T., Zhu H., Morishima N., Li E., Xu J., Yankner B.A., Yuan J. Caspase-12 mediates endoplasmic-reticulum-specific apoptosis and cytotoxicity by amyloid-beta. Nature. 2000;403:98–103. doi: 10.1038/47513. [DOI] [PubMed] [Google Scholar]
- Nakitare G.W., Elliott R.M. Expression of the Bunyamwera virus M genome segment and intracellular localization of NSm. Virology. 1993;195:511–520. doi: 10.1006/viro.1993.1402. [DOI] [PubMed] [Google Scholar]
- Ng F.W., Shore G.C. Bcl-XL cooperatively associates with the Bap31 complex in the endoplasmic reticulum, dependent on procaspase-8 and Ced-4 adaptor. J. Biol. Chem. 1998;273:3140–3143. doi: 10.1074/jbc.273.6.3140. [DOI] [PubMed] [Google Scholar]
- Ng F.W., Nguyen M., Kwan T., Branton P.E., Nicholson D.W., Cromlish J.A., Shore G.C. p28 Bap31, a Bcl-2/Bcl-XL- and procaspase-8-associated protein in the endoplasmic reticulum. J. Cell Biol. 1997;139:327–338. doi: 10.1083/jcb.139.2.327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishitoh H., Matsuzawa A., Tobiume K., Saegusa K., Takeda K., Inoue K., Hori S., Kakizuka A., Ichijo H. ASK1 is essential for endoplasmic reticulum stress-induced neuronal cell death triggered by expanded polyglutamine repeats. Genes Dev. 2002;16:1345–1355. doi: 10.1101/gad.992302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oyadomari S., Mori M. Roles of CHOP/GADD153 in endoplasmic reticulum stress. Cell Death Differ. 2004;11:381–389. doi: 10.1038/sj.cdd.4401373. [DOI] [PubMed] [Google Scholar]
- Oyadomari S., Araki E., Mori M. Endoplasmic reticulum stress-mediated apoptosis in pancreatic beta-cells. Apoptosis. 2002;7:335–345. doi: 10.1023/a:1016175429877. [DOI] [PubMed] [Google Scholar]
- Pavio N., Romano P.R., Graczyk T.M., Feinstone S.M., Taylor D.R. Protein synthesis and endoplasmic reticulum stress can be modulated by the hepatitis C virus envelope protein E2 through the eukaryotic initiation factor 2alpha kinase PERK. J. Virol. 2003;77:3578–3585. doi: 10.1128/JVI.77.6.3578-3585.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plyusnin A., Vapalahti O., Vaheri A. Hantaviruses: genome structure, expression and evolution. J. Gen. Virol. 1996;77:2677–2687. doi: 10.1099/0022-1317-77-11-2677. [DOI] [PubMed] [Google Scholar]
- Ronai Z. JNKing revealed. Mol. Cell. 2004;15:843–844. doi: 10.1016/j.molcel.2004.09.011. [DOI] [PubMed] [Google Scholar]
- Rottier P.J.M. The coronaviruses membrane glycoprotein. In: Siddell S.G., editor. The Coronaviridae. Plenum Press; New York: 1995. pp. 73–113. [Google Scholar]
- Ruusala A., Persson R., Schmaljohn C.S., Pettersson R.F. Coexpression of the membrane glycoproteins G1 and G2 of Hantaan virus is required for targeting to the Golgi complex. Virology. 1992;186:53–64. doi: 10.1016/0042-6822(92)90060-3. [DOI] [PubMed] [Google Scholar]
- Saleh M., Vaillancourt J.P., Graham R.K., Huyck M., Srinivasula S.M., Alnemri E.S., Steinberg M.H., Nolan V., Baldwin C.T., Hotchkiss R.S., Buchman T.G., Zehnbauer B.A., Hayden M.R., Farrer L.A., Roy S., Nicholson D.W. Differential modulation of endotoxin responsiveness by human caspase-12 polymorphisms. Nature. 2004;429:75–79. doi: 10.1038/nature02451. [DOI] [PubMed] [Google Scholar]
- Sabapathy K., Hochedlinger K., Nam S.Y., Bauer A., Karin M., Wagner E.F. Distinct roles for JNK1 and JNK2 in regulating JNK Activity and c-jun-dependent cell proliferation. Mol. Cell. 2004;15:713–725. doi: 10.1016/j.molcel.2004.08.028. [DOI] [PubMed] [Google Scholar]
- Schmaljohn C.S. In: The Bunyaviridae. Elliott RM, editor. Plenum Press; New York: 1996. pp. 63–90. [Google Scholar]
- Severson W.E., Schmaljohn C.S., Javadian A., Jonsson C.B. Ribavirin causes error catastrophe during Hantaan virus replication. J. Virol. 2003;77:481–488. doi: 10.1128/JVI.77.1.481-488.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spiropoulou C.F. Hantavirus maturation. Curr. Top. Microbiol. Immunol. 2001;256:33–46. doi: 10.1007/978-3-642-56753-7_3. [DOI] [PubMed] [Google Scholar]
- Spiropoulou C.F., Goldsmith C.S., Shoemaker T.R., Peters C.J., Compans R.W. Sin Nombre virus glycoprotein trafficking. Virology. 2003;308:48–63. doi: 10.1016/s0042-6822(02)00092-2. [DOI] [PubMed] [Google Scholar]
- Shi X., Elliott R.M. Analysis of N-linked glycosylation of Hantaan virus glycoproteins and the role of oligosaccharide side chains in protein folding and intracellular trafficking. J. Virol. 2004;78:5414–5422. doi: 10.1128/JVI.78.10.5414-5422.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi X., Lappin D.F., Elliott R.M. Mapping the Golgi targeting and retention signal of Bunyamwera virus glycoproteins. J. Virol. 2004;78:10793–10802. doi: 10.1128/JVI.78.19.10793-10802.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Su H.L., Liao C.L., Lin Y.L. Japanese encephalitis virus infection initiates endoplasmic reticulum stress and an unfolded protein response. J. Virol. 2002;76:4162–4171. doi: 10.1128/JVI.76.9.4162-4171.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takeda K., Matsuzawa A., Nishitoh H., Ichijo H. Roles of MAPKKK ASK1 in stress-induced cell death. Cell Struct. Funct. 2003;28:23–29. doi: 10.1247/csf.28.23. [DOI] [PubMed] [Google Scholar]
- Temonen M., Mustonen J., Helin H., Pasternack A., Vaheri A., Holthofer H. Cytokines, adhesion molecules, and cellular infiltration in nephropathia epidemica kidneys: an immunohistochemical study. Clin. Immunol. Immunopathol. 1996;78:47–55. doi: 10.1006/clin.1996.0007. [DOI] [PubMed] [Google Scholar]
- Tardif K.D., Mori K., Siddiqui A. Hepatitis C virus subgenomic replicons induce endoplasmic reticulum stress activating an intracellular signaling pathway. J. Virol. 2002;76:7453–7459. doi: 10.1128/JVI.76.15.7453-7459.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waris G., Tardif K.D., Siddiqui A. Endoplasmic reticulum (ER) stress: hepatitis C virus induces an ER-nucleus signal transduction pathway and activates NF-kappaB and STAT-3. Biochem. Pharmacol. 2002;64:1425–1430. doi: 10.1016/s0006-2952(02)01300-x. [DOI] [PubMed] [Google Scholar]
- Vapalahti O., Kallio-Kokko H., Narvanen A., Julkunen I., Lundkvist A., Plyusnin A., Lehvaslaiho H., Brummer-Korvenkontio M., Vaheri A., Lankinen H. Human B-cell epitopes of Puumala virus nucleocapsid protein, the major antigen in early serological response. J. Med. Virol. 1995;46:293–303. doi: 10.1002/jmv.1890460402. [DOI] [PubMed] [Google Scholar]
- Vapalahti O., Lundkvist A., Kukkonen S.K., Cheng Y., Gilljam M., Kanerva M., Manni T., Pejcoch M., Niemimaa J., Kaikusalo A., Henttonen H., Vaheri A., Plyusnin A. Isolation and characterization of Tula virus, a distinct serotype in the genus Hantavirus, family Bunyaviridae. J. Gen. Virol. 1996;77:3063–3067. doi: 10.1099/0022-1317-77-12-3063. [DOI] [PubMed] [Google Scholar]