

Abstract
Mycobacterium tuberculosis (Mtb) has plagued humanity for tens of thousands of years, yet still remains a threat to human health. Its pathology is largely associated with pulmonary tuberculosis with symptoms including fever, hemoptysis, and chest pain. Mtb, however, also manifests in other extrapulmonary organs, such as the pleura, bones, gastrointestinal tract, central nervous system, and lymph nodes. Compared to the knowledge of pulmonary tuberculosis, extrapulmonary pathologies of Mtb are quite understudied. Lymph node tuberculosis is one of the most common extrapulmonary manifestations of tuberculosis, and presents significant challenges in its diagnosis, management, and treatment due to its elusive etiologies and pathologies. The objective of this review is to overview the current understanding of the tropism and pathogenesis of Mtb in endothelial cells of the extrapulmonary tissues, particularly, in lymph nodes. Lymphatic endothelial cells (LECs) are derived from blood vascular endothelial cells (BECs) during development, and these two types of endothelial cells demonstrate substantial molecular, cellular and genetic similarities. Therefore, systemic comparison of the differential and common responses of BECs vs. LECs to Mtb invasion could provide new insights into its pathogenesis, and may promote new investigations into this deadly disease.
Keywords: Extrapulmonary tuberculosis, Vascular endothelial cells, Lymphatic endothelial cells, host-directed chemotherapy
Introduction
Mtb has developed sophisticated strategies to subvert host immune defenses and thereby live in the body over a prolonged period of time. Mtb has no classical toxin or virulent factor and instead its key virulence factor is the complex cell wall with a massive peptidoglycan-arabinogalactan core and thick mycolic acid lipid layer. These uncommon lipids within the cell wall are thought to play a major role in a number of signaling pathways for host immune evasion (1, 2). Although some Mtb carriers remain asymptomatic for a long time, Mtb infection eventually leads to active tuberculosis in the lungs (pulmonary), and/or in an extrapulmonary fashion, including the lymph nodes, gastrointestinal tract, bones, joints, and much more (3).
Lymphadenitis is the most common extrapulmonary manifestation of tuberculosis (4). Afflicted patients can be asymptomatic or present with fever, weight loss, fatigue, and/or night sweats. Although the precise pathogenesis into the lymph nodes is not completely understood, it is believed that Mtb engulfed by macrophages is drained through the lymphatic vessels, eventually reaching the lymph nodes. Cytokines and lymphokines from infected macrophages cause monocyte aggregation and recruitment of naïve macrophages. As the recruited immune cells improve their ability to kill off Mtb, the granulomas develop a caseous center containing necrotic tissue, with immune cells on the periphery surrounded by fibroblasts (5, 6). Granulomas are the compact, organized aggregates of immune cells and serve as a hallmark of tuberculosis. Details are described in the following sections.
LECs and BECs line lymphatic vessels and blood vessels, respectively, and their ultrastructure and functions are highly similar. Blood vessels deliver oxygen and nutrients throughout the body, while lymphatic vessels drain interstitial fluid, transport immune cells, and absorb lipids from the intestines. The homeobox transcriptional factor PROX1 is expressed early during embryonic development, and functions as a master gene for regulating the induction of lymphatic differentiation from the blood vascular system (7, 8). Inhibition of PROX1 expression blocks lymphatic differentiation from venous endothelial cells, lymphatic expression, and lymph valve development. Because of this close histogenetic relationship between these two types of endothelial cells, the gene expression profiles between BECs and LECs are very similar, expressing several common cellular markers, such as CD31, VEGFR-2, and VE-Cadherin. Some genes, however, are selectively expressed in either of two cell types. For example, VEGFR-1 and CD34 are predominantly expressed in BECs, while LYVE1, PROX1, VEGFR-3 and PDPN are more abundantly expressed in LECs (9–11). However, the presence or absence of these markers on BECs and LECs are often obscure and varied; their expressions depend on hierarchical vascular beds, anatomical locations and structures, pathophysiological conditions, and developmental stages, thus demonstrating the heterogeneity and functional specialization of these two endothelial cell types (Table 1). In this article, we will discuss the development of these two endothelial cell types, Mtb pathogenesis, and Mtb interactions with BECs and LECs. We will raise new questions about Mtb tropism for these cells and call for future investigations.
Table. 1.
Differential and common characteristics between LECs and BECs
| LECs | BECs | Common | Ref. | |
|---|---|---|---|---|
| Surface markers | FLT4, PROX1, LYVE1, PDPN, FOXC2, ACKR2, NRP2, CCL21 | CD34, ENG, NRP1, MCAM, ROBO4, ITGAV, FLT1 | CDH5 PECAM1 KDR DLL4 | (11, 82–105) |
| Structure |
|
|
|
|
| Function |
|
|
|
Mycobacterium tuberculosis
Mtb is the etiological agent of tuberculosis, a scourge of an illness mainly affecting the respiratory system. Tuberculosis remains the leading cause of human mortality with an estimated 10.4 million new cases and 1.6 million annual deaths (12). A major barrier to the efficient control of the tuberculosis pandemic is the lack of rapid and simple treatments, especially because effective chemotherapy was developed around 50 years ago (13, 14). Current tuberculosis chemotherapies are lengthier and more complicated than for virtually any other bacterial infection, and are thus associated with high rates of non-compliance and treatment failure. Reduced efficacy of tuberculosis chemotherapies has given rise to the emergence of multidrug-resistant (MDR) and extensively drug-resistant (XDR) tuberculosis. This has led to a new paradigm in tuberculosis drug development, termed host-directed therapeutic strategy, which involves modulation of host responses to destabilize Mtb adaptation and thereby improve pathogen eradication (15, 16). Thus, interactions between the host immune system and invading Mtb can be a rich source of host-directed therapy targets, but knowledge is still scant.
Pulmonary tuberculosis is the primary form of tuberculosis and is initiated by inhalation of Mtb-containing droplets into the alveoli. Alveolar macrophages are the first immune cells that infiltrate the site of infection of the Mtb bacilli triggering the invasion through the subtending epithelium; they then attract more powerful inflammatory lymphocytes by cytokine secretion (17–19). Infection with Mtb also leads to the recruitment of mononuclear cells from neighboring blood vessels, and to the formation of granulomas that are highly vacuolized at the initial stages (20–22). Upon Mtb infection, human macrophages rapidly turn on an angiogenic program, allowing mycobacterial dissemination through blood vessels, the formation of which is mainly triggered by vascular endothelial growth factor (VEGF) in an Mtb RD1 (region of difference)-dependent manner. The RD1 locus of Mtb includes genes that encode: early secreted antigenic target (ESAT-6), ESAT-6 secretion system-1 (ESX-1), and culture filtrate protein-10 (CFP-10) (23). ESX-1 secretion system and its secreted substrates ESAT-6, and CFP-10 are well known to play significant roles in Mtb virulence and interactions with the host immune system. As such, Bacillus Calmette-Guérin (BCG), a tuberculosis vaccine strain that lacks the RD1 genetic locus, and the RD1-deficient Mtb strains are poor inducers of VEGF (24). On the contrary, hyper-virulent Mtb strains, such as those in the Beijing/W lineage, induce more VEGF secretion than the less virulent H37Rv, the most prevalent strain of tuberculosis in research laboratory, indicating the potential association between the expression of optimal virulence and development of extrapulmonary TB disease (25).
The host response to pulmonary tuberculosis results in granuloma formation, a structure consisting of concentric layers of infected macrophages, epithelioid cells, and multinucleated giant cells surrounded by activated lymphatic vessels (26). As it matures, the granuloma becomes less abundant in blood vessels, and the central core of the granuloma is necrotized while an extracellular matrix becomes fibrous.
Since granuloma formation is observed in both active and latent forms of pulmonary tuberculosis, the extent of granuloma formation is known to have no direct association with effectiveness of immune defense. Although the infected Mtb bacilli are not cleared, granulomas are generally considered to be host-protective structures (27). However, this notion has been challenged in recent years with studies in a zebrafish model infected with Mycobacterium marinum where the mycobacterial growth rate was enhanced as granuloma continued to form, a process that normally happens only at the early stages (28). Furthermore, in the same zebrafish model, macrophages infected with Mtb was shown to promote recruitment of uninfected naïve macrophages, which facilitate apoptosis of infected cells and trigger bacterial replication (28). Consequently, macrophages actively participate in mycobacterial dissemination. Mtb, phagocytosed by macrophages, frequently escapes from established granulomas, migrating through blood vessels to lymphatic organs (a source of extrapulmonary tuberculosis) and various tissues where they serve as sources for new granuloma formation (26, 29).
The ability of Mtb to disseminate via the bloodstream, and consequently affecting other organs is well described (24). Extrapulmonary tuberculosis represents about 20% of all tuberculosis cases in immunocompetent patients, and over 50% of cases in immunocompromised individuals (30, 31). Some observations using mouse model have supported the idea that extrapulmonary tuberculosis may result from a dissemination of Mtb from primary tuberculosis (32). Consistent with animal models, while primary tuberculosis affects any part of the organ, secondary tuberculosis aroused from the reactivation of primary tuberculosis usually damages mainly the upper lobes of the lungs in human patients (33). This differential localization suggests the important role of early hematogenous dissemination of the primary tuberculosis Mtb bacilli during the establishment of primary infection. As such, understanding the host-pathogen relationship can help guide new drug discovery and drug delivery platforms, including host immunopathology pathways that affect Mtb recognition in the form of both pulmonary and extrapulmonary tuberculosis.
Pathogen-associated molecular pattern and ECs interactions
Innate immune recognition and surveillance are directed by specific interactions with microbial components that are conserved within a given class of bacterial pathogens. These components are termed pathogen-associated molecular patterns (PAMPs). Mtb has evolved a range of immune evasion strategies to circumvent phagosome maturation, thereby allowing to replicate within the host. This capability is also achieved through PAMPs. Therefore, Mtb PAMPs constitute a critical component for the pathogenicity of microorganisms and serve molecular signatures of pathogen classes (34).
Mtb presents various classes of PAMPs on the cell envelope including phthiocerol dimycocerosate (PDIM), trehalose-6,6’-dimycolates (TDM), and lipoarabinomannan (LAM) (35–37). PDIM is the glycolipid PAMP that was first identified as a virulent factor of Mtb. PDIM-deficient Mtb strain showed significantly attenuated replication in guinea pig and mouse models (38–41). A recent study supported this notion by showing that PDIM-deficient Mtb is less phagocytosed by human LECs (hLEC) due to defective membrane rupture of phagosome (42). TDM is another major glycolipid that is shown to interact with host macrophage inducible c-type lectin (Mincle). Thereby, TDM induces the secretion of pro-inflammatory cytokines such as IL-6, IL-12, and TNF-α and triggers the granuloma formation identified in mouse and rabbit models (43–45). Among these PAMPs, LAM is the major component that modulates the host immune system and allows Mtb to infect macrophages, dendritic cells (DCs), and endothelial cells (ECs) (25, 46–49). LAM is a mycobacterial cell-wall lipoglycan, and is structurally composed of a mannosyl-phosphatidyl-myo-inositol anchor (MPI), a polysaccharide backbone (composed of D-mannan and D-arabinan), and caps (Fig. 1). LAM is classified into three subgroups depending upon cap structures: Mannose-capped (ManLAM), Phospho-myo-inositol capped (PILAM), and non-capped LAM (AraLAM) (Fig. 1). Mtb interactions with the host immune system are determined by the cap structure and thereby associated with the virulence (35, 50).
Fig. 1.
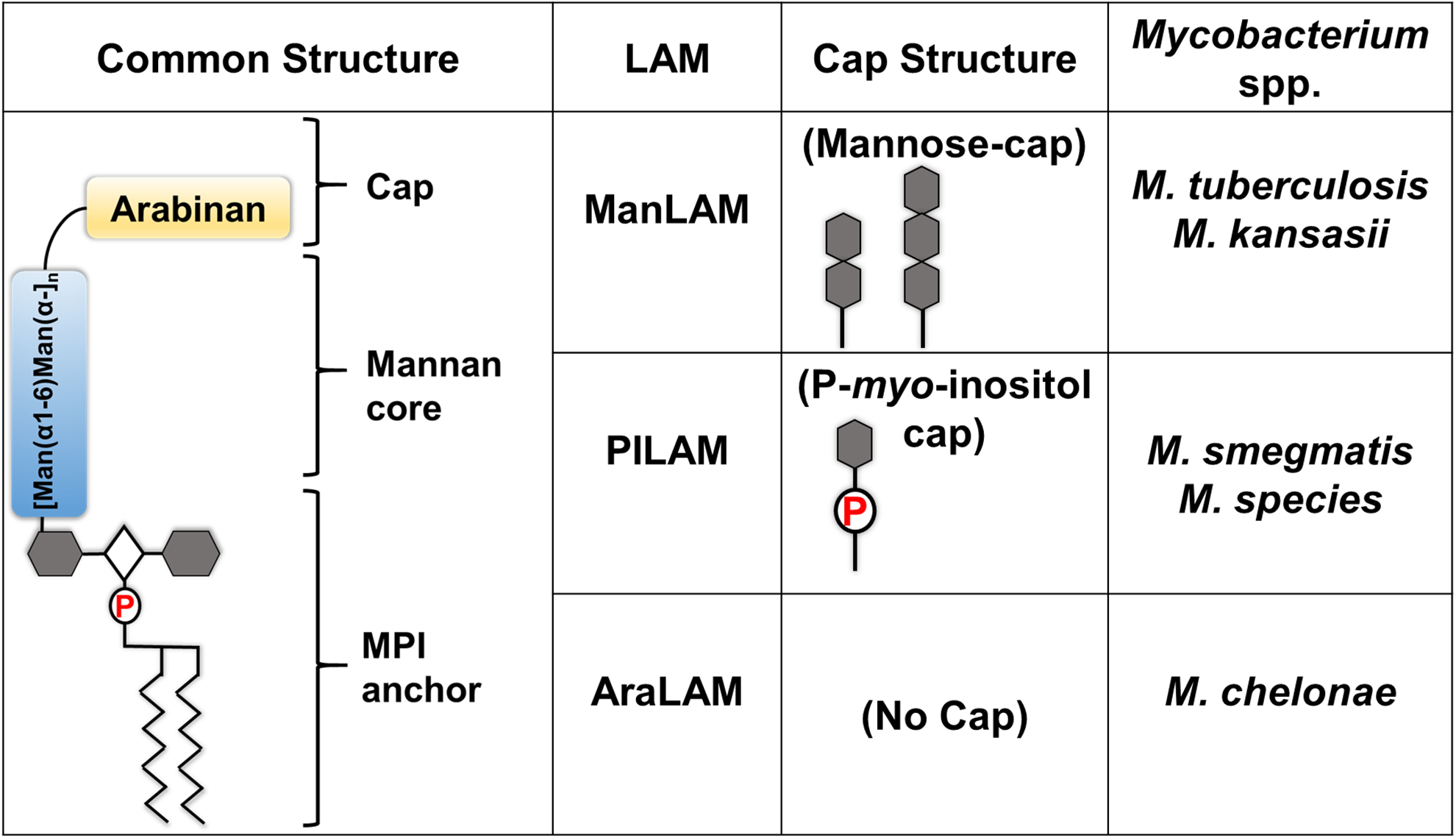
Various lipoarabinomannan (LAM) structures characterized in Mycobacterial spp.
ManLAM was identified as the major PAMP and virulence factor of Mtb among LAMs. Controversially, BCG vaccine strain was also shown to contain structurally similar ManLAM (51) and consequently, severe complications of BCG vaccination include suppurative lymphadenitis (52). The incident rate of lymphadenitis due to BCG vaccination is largely influenced by the immunization technique, vaccine dose, and age and/or physique of the vaccinees. ManLAM of Mtb modulates host immune systems by affecting phagosome-lysosome fusion, dendritic cell (DC) maturation, CD4+ T cell activation, and recruitment in uninfected macrophages. Accordingly, ManLAM can trigger the production of a panel of cytokines, such as IL-10 and IL-37 by DCs and type II alveolar epithelial cells, respectively, with reciprocal suppression of IL-12 secretion from DCs (53–56). Therefore, ManLAM of Mtb may be a crucial factor to initiate dissemination from the lung to other organs.
ManLAM is known to interact with CD44, Mannose receptors (MRs), and DC-specific intracellular adhesion molecule-3 grabbing nonintegrin (DC-SIGN) (54, 57). MRs serve as the specialized pattern recognition receptors (PRRs) that are prevalent on macrophages, DCs, and ECs. Some studies revealed that, apart from some specialized cell types in the kidney, trachea, and retina, MR expression of ECs appears to be restricted to certain organs that function in antigen uptake or presentation, such as the sinus-lining cells of the liver, spleen, and lymph nodes (58). ECs are the internal barrier of the stroma and blood vessels, and contribute to the host immune responses (59). Unlike other cell types, the interactions between Mtb and ECs have rarely been studied in detail. Some studies using mouse model have shown that immune cells infected with Mtb can be disseminated from the site of infection to the lymph nodes through hematogenous circulation, where free Mtb bacilli sensitize CD4+ T lymphocytes in an Mtb-specific antigen dependent manner in order to proliferate and induce inflammatory cytokine secretion (60, 61). Indeed, in active and latent tuberculosis individuals, Mtb DNA was found in both lung tissue and spleen ECs; these results indicate that ECs can act as reservoirs for Mtb in extrapulmonary tuberculosis (62, 63). Studies of the interactions between Mtb and ECs may yield a greater understanding of Mtb because it will allow us to link primary pulmonary tuberculosis and both extrapulmonary and secondary tuberculosis, ultimately uncovering a new source of host-directed therapeutic targets.
Mtb and BEC Interactions
In pulmonary tuberculosis, while macrophages serve as the primary immune cells that phagocytose foreign agents, BECs are also susceptible to Mtb invasion. When comparing the Human Lung Microvascular Endothelial Cell line (HULEC) to the Human foreskin Microvascular Endothelial Cell line (HMEC-1) in vitro, HULEC has twice the association with the attachment, internalization, and replication of Mtb than that of HMEC-1 (64). Not only are Mtb bacilli internalized by HULEC, but the presence of bacterial septa within the Mtb bacillus indicated active replication and coalescence in the vacuoles, terminating in host cell lysis. Indeed, unlike Mtb within HULEC, Mtb was shown to be defect in replication within the HMEC-1 culture, and large numbers of bacteria, bacterial septa, and cell lysis were not observed (64). The difference in the internalization processes between HULEC and HMEC-1 may also be due to differences in cell surface markers, such as various adhesion molecules (64). Although Mtb is known to be internalized by BECs, this investigation also demonstrates that Mtb infection is not specific to BECs and that internalization does not guarantee Mtb proliferation.
Mtb pathogenesis in vivo and in vitro has also shown a variety of interactions with other immune cells that indirectly affect BECs. Infection of human macrophages with Mtb was shown to upregulate genes involved in angiogenesis in an RD1-dependent manner. Angiogenesis is a process of forming new blood vessels from the existing ones. Numerous factors have been identified to promote and regulate the angiogenic process (65, 66). Among them, VEGF family members (VEGF-A to D) are known to regulate angiogenesis and/or lymphangiogenesis by directly affecting BECs and LECs, respectively. VEGF-A and VEGF-B activate VEGFR-1 and VEGFR-2, whereas VEGF-C and VEGF-D stimulate VEGFR-2 and VEGFR-3 (65, 66). VEGF-A is highly expressed by infected macrophages when compared to uninfected cells (24). Additionally, the expression of the ribonuclease/angiogenin inhibitor (RNH) gene, an inhibitor of the angiogenic factor angiogenin, was downregulated in Mtb infected macrophages and thereby synergistically promoted angiogenesis. Collectively, macrophage-induced angiogenesis in SCID mice aided Mtb dissemination due to development of highly vascularized structures similar to the structure formed in early granulomas. As such, treating the mice with an anti-VEGF antibody markedly suppressed dissemination of Mtb to the spleen and lungs, with weaker effects seen in the draining lymph nodes (24). Blocking VEGFR-2 with a monoclonal antibody also significantly suppressed Mtb dissemination to the extrapulmonary organs such as spleen, while having a weaker effect on dissemination to the lungs. Interestingly, blocking VEGFR-1 did not show any effects on both intracellular viability and Mtb dissemination to other organs (24). Mtb infected macrophages were also known to mobilize endothelial progenitor cells (EPCs), possibly by cytokine secretion, to differentiate into new blood vessels at the site of infection (24).
An analysis of serum levels of VEGF in patients with pulmonary tuberculosis demonstrated that those with active pulmonary tuberculosis had significantly higher VEGF serum levels compared to those with latent form of tuberculosis or acute bronchitis (67). Furthermore, decreases in the serum levels of VEGF positively correlate with poor prognosis (67). Comparative examination of granuloma formation using Mtb and the BCG vaccine strain also demonstrated that vascularity was an important factor in Mtb virulence and survival (68). Interestingly, assessing microvessel distribution in granulomatous regions of the lung in human samples and a rabbit model indicated that VEGF was observed to be highly expressed in the inner regions of granulomas compared to granuloma peripheries or normal lung tissue. However, microvessel density is relatively high in the periphery but becomes gradually lower, resulting in an almost avascular central region (69). Moreover, vessels were also collapsed or compressed with low pericyte coverage. Indeed, administration of a fluorescent small molecule dye demonstrated intense signaling in normal lung tissue but lower intensity in the granulomas, with a substantial intensity decrease toward the necrotic centers. Treating human and rabbit granulomas with bevacizumab, anti-VEGF antibody, led to normal vasculature formation and/or distribution, decreased hypoxic fractions of the granulomas, and increased intensity of the dye delivery within the granulomas, demonstrating a potential avenue for future drug therapies against Mtb (69).
In another study using a zebrafish model infected with M. marinum, angiogenesis was enhanced via VEGF-A induction as granuloma formation matured (70). Examination of these newly synthesized vessels demonstrated that nuclei in the existing intersegmental vessels left toward sites of infection with sprouting from arterial and venous intersegmental vessels, confirming the mode of vascular elongation. Persistent stimulus of M. marinum infection was required for angiogenesis to occur, and ESX-1 deficient M. marinum strains resulted in a reduction in granuloma formation. ESX-1 is a secretion system that exports effector proteins that allow Mtb to evade the host immune response and to escape from the phagolysosome into the cytosol. Its absence thus severely attenuates its virulence (71). Furthermore, VEGFR inhibition in zebrafish demonstrated reduced vascular permeability, which would limit bacterial dissemination via the vasculature (70).
Along with VEGF-A, angiopoietin-2 (ANG-2) has also been shown to be an important component of angiogenesis in Mtb pathogenesis. Using both human and zebrafish granulomas, ANG-2 was proven to antagonize the stability of ANG-1 binding to endothelial specific receptor tyrosine kinase 2 (TIE2), a receptor for both ANG-2 and ANG-1 (72). This ANG-2 mediated antagonization of TIE2 causes decreased stability and increased permeability of the blood vessel, which would promote bacterial dissemination. Furthermore, vascular endothelial-protein tyrosine phosphatase (VE-PTP), a phosphatase specific to TIE2, regulates TIE2 downstream signaling by inhibiting its dephosphorylation (73). Inhibition of VE-PTP activates the TIE2 receptor even in the presence of high ANG-2 antagonism. Analysis of human granulomas demonstrated significantly higher ANG-2 secretion from macrophages and stromal cells, compared to macrophages in unrelated areas of lung tissue. Additionally, analyses of M. marinum infected zebrafish larvae and adults demonstrated attenuated growth and dissemination of M. marinum when the VE-PTP pathway was inhibited (72). Taken together, these results indicate that manipulating the effects of ANG-2 and VE-PTP on TIE2 may be another source of host-directed therapies.
Mtb and LEC Interactions
LECs have recently been shown to be another important niche within the host for Mtb replication and dissemination. An in vitro infection model using hLECs has shown that Mtb exploits the host endocytosis and autophagy pathways for infiltration (25, 32). Mtb resides in the LECs within lymph node granulomas, and its internalization is heavily dependent on the MRs expressed on the cell surface (74). When LECs were pretreated with mannan, a ligand for MRs, there was a significant reduction in internalization of Mtb (25, 32). Once inside LECs, a small fraction of Mtb is mobilized into autophagosomes, vesicles that are marked for degradation of cytoplasmic contents, or the cytosol, by escaping from initial phagosomes in an RD1 dependent manner. Therefore, the autophagy pathway allows a small fraction of Mtb to replicate inside LECs. Thus, infection with the RD1-deficient Mtb strain or BCG vaccine strain exhibits a severe attenuation in replication capability within LECs, as most of the RD1-deficient Mtb remains in the phagosome or phagolysosome. Activation of hLECs by interferon gamma (IFN-γ) suppresses Mtb replication within the cytosol, mainly due to the induced local concentration of nitric oxide via activation of endothelial NOS (eNOS) and the specific eNOS is colocalized with the Mtb infected hLECs (32, 75, 76). Furthermore, the autophagy pathway in IFN-γ activated hLECs is responsible for negatively regulating bacterial growth while inactivated hLECs increases Mtb growth. In response to this dual-purposed autophagy pathway, Mtb partitions its population into two groups: one resides within the autophagosome, and the other is within the cytosol. This bet-hedging strategy helps Mtb increase its chances to survive in response to the host immune system because if one population of Mtb fails, the second population can flourish. Further investigation will be needed for a better understanding of this survival strategy (25).
Another study investigated the micronodules in the perilymphatic area of the lung in human adults with pulmonary tuberculosis. CT scans were used to detect invading Mtb bacilli and showed significantly lower amount of positive acid-fast bacilli smears, consolidation and cavitation in the perilymphatic regions compared to the centrilobular area of the lungs of pulmonary tuberculosis patients (77). This finding suggested that Mtb may use drainage to lymph nodes as a means of dissemination, with further investigations needed to identify the mechanism. Mtb-specific T cells were recruited not only at the pulmonary lymph nodes after Mtb drainage, but also at peripheral sites such as the spleen, suggesting a crucial role of lymphatic vessels and lymph nodes in the manifestation of extrapulmonary tuberculosis (32, 78) Separate studies using guinea pigs showed that lymphatic vessels in the lungs serve as the earliest site of Mtb infection and can develop into pulmonary lymphangitis several days post-infection, indicating that the lymphatic system not only plays a key role in extrapulmonary dissemination of Mtb, but also serves as one of the primary sites of Mtb infection (32, 79).
Mycobacterial granulomas have been shown to induce lymphangiogenesis (32, 80). LECs of mice infected with M. bovis exhibited phenotypic changes similar to those that occur during inflammation, such as colocalization with Intercellular adhesion molecule-1 (ICAM-1) and vascular cell adhesion molecule-1 (VCAM-1) (32, 80). Furthermore, granulomatous lymphangiogenesis was shown to be reliant on the VEGFC-VEGFR3 pathway, and that chemical inhibition of this pathway led to decreased lymphangiogenesis. Lymphangiogenesis was also demonstrated to have a key role in the immune system response to BCG infection. Not only does lymphangiogenesis increase access to peripheral immune sites for granuloma antigens, but also mediates T-cell proliferation and its adequate immune responses. Thus, functional inactivation of VEGFR3 with MAZ51 inhibited lymphangiogenesis and T-cell proliferation in the lung lymph nodes of mice infected with Mtb (80).
Closing Remarks and Relevant Questions
In summary, both the lymphatic and blood vasculature, in addition to macrophages could serve as important cell niche for the growth and survival of Mtb. Mtb not only infects BECs and LECs, but also promotes angiogenesis and lymphangiogenesis, which eventually contribute to Mtb dissemination and establishment of extrapulmonary tuberculosis. Macrophages and ECs share abundant common features in the context of Mtb pathogenesis. Both cells are developmentally derived from hemangioblasts and share common molecular markers (81). They play critical roles in activation of innate and acquired immune responses, which are essential to control and eliminate the invading Mtb. Macrophages and ECs encounter Mtb at the initial stage of infection. However, while macrophages are mobile, dynamic, and highly phagocytic, ECs are rather inert, passive and non-phagocytic. Whereas macrophages have been extensively studied as a major host cells that respond Mtb infection, the pathogenic interaction between Mtb and ECs are understudied. In this study, we reviewed the current knowledge of the interaction of Mtb with ECs and aimed to compare and contrast the similar and differential features of BECs vs. LECs. We also propose that the VEGF signaling pathways should be a vital component in interaction between ECs and Mtb, potentially serving as a source for host-directed therapeutic targets. We sincerely hope that our discussion could trigger new investigations on my important questions as below regarding the endothelial lineage-specific interaction of Mtb with the blood and lymphatic systems.
What are the mechanistic bases underlying the differential responses between BECs and LECs against Mtb infection?
What other signaling pathways are utilized by Mtb for the infection and growth in BECs and LECs?
Can any significant findings on Mtb-induced angiogenic and lymphangiogenic responses be translated into novel treatments for tuberculosis, such as by co-opting them as avenues for drug delivery instead of Mtb dissemination?
Mtb executes immune-evasion within BECs and LECs in an RD1 dependent manner. How do we use this immune-evasion strategy to develop more effective vaccine strains against both pulmonary and extrapulmonary tuberculosis?
Does inhibition of the VEGF/VEGFR pathways destabilize granuloma formation, reactivate latent Mtb within the granuloma, and induce antibiotic diffusion and/or efficacy to better target invading Mtb?
Acknowledgements
The authors gratefully acknowledge the support of Donald E. & Delia Baxter Foundation (to H.E) and National Institutes of Health (to Y.K.H).
Footnotes
The authors disclose no conflict of interest with this work.
References
- 1.Brennan PJ. Structure, function, and biogenesis of the cell wall of Mycobacterium tuberculosis. Tuberculosis (Edinb). 2003;83(1–3):91–7. [DOI] [PubMed] [Google Scholar]
- 2.Barry CE 3rd. Interpreting cell wall ‘virulence factors’ of Mycobacterium tuberculosis. Trends Microbiol. 2001;9(5):237–41. [DOI] [PubMed] [Google Scholar]
- 3.Lee JY. Diagnosis and treatment of extrapulmonary tuberculosis. Tuberc Respir Dis (Seoul). 2015;78(2):47–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Behr MA, Waters WR. Is tuberculosis a lymphatic disease with a pulmonary portal? Lancet Infect Dis. 2014;14(3):250–5. [DOI] [PubMed] [Google Scholar]
- 5.Quast TM, Browning RF. Pathogenesis and clinical manifestations of pulmonary tuberculosis. Dis Mon. 2006;52(11–12):413–9. [DOI] [PubMed] [Google Scholar]
- 6.Handa U, Mundi I, Mohan S. Nodal tuberculosis revisited: a review. J Infect Dev Ctries. 2012;6(1):6–12. [DOI] [PubMed] [Google Scholar]
- 7.Oliver G, Alitalo K. The lymphatic vasculature: recent progress and paradigms. Annu Rev Cell Dev Biol. 2005;21:457–83. [DOI] [PubMed] [Google Scholar]
- 8.Oliver G, Srinivasan RS. Endothelial cell plasticity: how to become and remain a lymphatic endothelial cell. Development. 2010;137(3):363–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hong YK, Harvey N, Noh YH, Schacht V, Hirakawa S, Detmar M, et al. Prox1 is a master control gene in the program specifying lymphatic endothelial cell fate. Dev Dyn. 2002;225(3):351–7. [DOI] [PubMed] [Google Scholar]
- 10.Hirakawa S, Hong Y-k, Harvey N, Schacht V, Detmar M. Identification of Vascular Lineage-Specific Genes by Transcriptional Profiling of Isolated Blood Vascular and Lymphatic Endothelial Cells. The American Journal of Pathology. 2003;162(2):575–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kriehuber E, Breiteneder-Geleff S, Groeger M, Soleiman A, Schoppmann SF, Stingl G, et al. Isolation and characterization of dermal lymphatic and blood endothelial cells reveal stable and functionally specialized cell lineages. J Exp Med. 2001;194(6):797–808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.WHO. Global tuberculosis report. 2017.
- 13.Sandhu GK. Tuberculosis: current situation, challenges and overview of its control programs in India. J Glob Infect Dis. 2011;3(2):143–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Nathan C, Barry CE 3rd. TB drug development: immunology at the table. Immunol Rev. 2015;264(1):308–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wallis RS, Hafner R. Advancing host-directed therapy for tuberculosis. Nat Rev Immunol. 2015;15(4):255–63. [DOI] [PubMed] [Google Scholar]
- 16.Nathan C Fresh approaches to anti-infective therapies. Sci Transl Med. 2012;4(140):140sr2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Russell DG, Cardona PJ, Kim MJ, Allain S, Altare F. Foamy macrophages and the progression of the human tuberculosis granuloma. Nat Immunol. 2009;10(9):943–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ernst JD. The immunological life cycle of tuberculosis. Nat Rev Immunol. 2012;12(8):581–91. [DOI] [PubMed] [Google Scholar]
- 19.Cadena AM, Fortune SM, Flynn JL. Heterogeneity in tuberculosis. Nat Rev Immunol. 2017;17(11):691–702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Tsai MC, Chakravarty S, Zhu G, Xu J, Tanaka K, Koch C, et al. Characterization of the tuberculous granuloma in murine and human lungs: cellular composition and relative tissue oxygen tension. Cell Microbiol. 2006;8(2):218–32. [DOI] [PubMed] [Google Scholar]
- 21.Ulrichs T, Kaufmann SH. New insights into the function of granulomas in human tuberculosis. J Pathol. 2006;208(2):261–9. [DOI] [PubMed] [Google Scholar]
- 22.Kaufmann SH. How can immunology contribute to the control of tuberculosis? Nat Rev Immunol. 2001;1(1):20–30. [DOI] [PubMed] [Google Scholar]
- 23.Guinn KM, Hickey MJ, Mathur SK, Zakel KL, Grotzke JE, Lewinsohn DM, et al. Individual RD1-region genes are required for export of ESAT-6/CFP-10 and for virulence of Mycobacterium tuberculosis. Mol Microbiol. 2004;51(2):359–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Polena H, Boudou F, Tilleul S, Dubois-Colas N, Lecointe C, Rakotosamimanana N, et al. Mycobacterium tuberculosis exploits the formation of new blood vessels for its dissemination. Sci Rep. 2016;6:33162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lerner TR, de Souza Carvalho-Wodarz C, Repnik U, Russell MR, Borel S, Diedrich CR, et al. Lymphatic endothelial cells are a replicative niche for Mycobacterium tuberculosis. J Clin Invest. 2016;126(3):1093–108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Guirado E, Schlesinger LS. Modeling the Mycobacterium tuberculosis Granuloma - the Critical Battlefield in Host Immunity and Disease. Front Immunol. 2013;4:98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Seiscento M, Vargas FS, Acencio MM, Teixeira LR, Capelozzi VL, Sales RK, et al. Pleural fluid cytokines correlate with tissue inflammatory expression in tuberculosis. Int J Tuberc Lung Dis. 2010;14(9):1153–8. [PubMed] [Google Scholar]
- 28.Volkman HE, Clay H, Beery D, Chang JC, Sherman DR, Ramakrishnan L. Tuberculous granuloma formation is enhanced by a mycobacterium virulence determinant. PLoS Biol. 2004;2(11):e367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Pagan AJ, Ramakrishnan L. The Formation and Function of Granulomas. Annu Rev Immunol. 2018. [DOI] [PubMed] [Google Scholar]
- 30.Kwan CK, Ernst JD. HIV and tuberculosis: a deadly human syndemic. Clin Microbiol Rev. 2011;24(2):351–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kim CH, Lim JK, Lee DH, Yoo SS, Lee SY, Cha SI, et al. Outcomes of standard and tailored anti-tuberculosis regimens in patients with tuberculous pleural effusion. Int J Tuberc Lung Dis. 2016;20(11):1516–21. [DOI] [PubMed] [Google Scholar]
- 32.Saeed S, Quintin J, Kerstens HH, Rao NA, Aghajanirefah A, Matarese F, et al. Epigenetic programming of monocyte-to-macrophage differentiation and trained innate immunity. Science. 2014;345(6204):1251086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Fujita J, Higa F, Tateyama M. Radiological findings of mycobacterial diseases. J Infect Chemother. 2007;13(1):8–17. [DOI] [PubMed] [Google Scholar]
- 34.Medzhitov R, Janeway C, Jr. The Toll receptor family and microbial recognition. Trends Microbiol. 2000;8(10):452–6. [DOI] [PubMed] [Google Scholar]
- 35.Nigou J, Gilleron M, Puzo G. Lipoarabinomannans: from structure to biosynthesis. Biochimie. 2003;85(1–2):153–66. [DOI] [PubMed] [Google Scholar]
- 36.Daffe M, Laneelle MA. Distribution of phthiocerol diester, phenolic mycosides and related compounds in mycobacteria. J Gen Microbiol. 1988;134(7):2049–55. [DOI] [PubMed] [Google Scholar]
- 37.Welsh KJ, Hunter RL, Actor JK. Trehalose 6,6’-dimycolate--a coat to regulate tuberculosis immunopathogenesis. Tuberculosis (Edinb). 2013;93 Suppl:S3–9. [DOI] [PubMed] [Google Scholar]
- 38.Goren MB, Brokl O, Schaefer WB. Lipids of putative relevance to virulence in Mycobacterium tuberculosis: phthiocerol dimycocerosate and the attenuation indicator lipid. Infect Immun. 1974;9(1):150–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Cox JS, Chen B, McNeil M, Jacobs WR, Jr. Complex lipid determines tissue-specific replication of Mycobacterium tuberculosis in mice. Nature. 1999;402(6757):79–83. [DOI] [PubMed] [Google Scholar]
- 40.Camacho LR, Ensergueix D, Perez E, Gicquel B, Guilhot C. Identification of a virulence gene cluster of Mycobacterium tuberculosis by signature-tagged transposon mutagenesis. Mol Microbiol. 1999;34(2):257–67. [DOI] [PubMed] [Google Scholar]
- 41.Day TA, Mittler JE, Nixon MR, Thompson C, Miner MD, Hickey MJ, et al. Mycobacterium tuberculosis strains lacking surface lipid phthiocerol dimycocerosate are susceptible to killing by an early innate host response. Infect Immun. 2014;82(12):5214–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lerner TR, Queval CJ, Fearns A, Repnik U, Griffiths G, Gutierrez MG. Phthiocerol dimycocerosates promote access to the cytosol and intracellular burden of Mycobacterium tuberculosis in lymphatic endothelial cells. BMC Biol. 2018;16(1):1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Donnachie E, Fedotova EP, Hwang SA. Trehalose 6,6-Dimycolate from Mycobacterium tuberculosis Induces Hypercoagulation. Am J Pathol. 2016;186(5):1221–33. [DOI] [PubMed] [Google Scholar]
- 44.Ishikawa E, Ishikawa T, Morita YS, Toyonaga K, Yamada H, Takeuchi O, et al. Direct recognition of the mycobacterial glycolipid, trehalose dimycolate, by C-type lectin Mincle. J Exp Med. 2009;206(13):2879–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Schoenen H, Bodendorfer B, Hitchens K, Manzanero S, Werninghaus K, Nimmerjahn F, et al. Cutting edge: Mincle is essential for recognition and adjuvanticity of the mycobacterial cord factor and its synthetic analog trehalose-dibehenate. J Immunol. 2010;184(6):2756–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Randall PJ, Hsu NJ, Quesniaux V, Ryffel B, Jacobs M. Mycobacterium tuberculosis infection of the ‘non-classical immune cell’. Immunol Cell Biol. 2015;93(9):789–95. [DOI] [PubMed] [Google Scholar]
- 47.Schlesinger LS, Kaufman TM, Iyer S, Hull SR, Marchiando LK. Differences in mannose receptor-mediated uptake of lipoarabinomannan from virulent and attenuated strains of Mycobacterium tuberculosis by human macrophages. J Immunol. 1996;157(10):4568–75. [PubMed] [Google Scholar]
- 48.Tailleux L, Pham-Thi N, Bergeron-Lafaurie A, Herrmann JL, Charles P, Schwartz O, et al. DC-SIGN induction in alveolar macrophages defines privileged target host cells for mycobacteria in patients with tuberculosis. PLoS Med. 2005;2(12):e381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Tailleux L, Schwartz O, Herrmann JL, Pivert E, Jackson M, Amara A, et al. DC-SIGN is the major Mycobacterium tuberculosis receptor on human dendritic cells. J Exp Med. 2003;197(1):121–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Dao DN, Kremer L, Guerardel Y, Molano A, Jacobs WR Jr., Porcelli SA, et al. Mycobacterium tuberculosis lipomannan induces apoptosis and interleukin-12 production in macrophages. Infect Immun. 2004;72(4):2067–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Prinzis S, Chatterjee D, Brennan PJ. Structure and antigenicity of lipoarabinomannan from Mycobacterium bovis BCG. J Gen Microbiol. 1993;139(11):2649–58. [DOI] [PubMed] [Google Scholar]
- 52.Govindarajan KK, Chai FY. BCG Adenitis-Need for Increased Awareness. Malays J Med Sci. 2011;18(2):66–9. [PMC free article] [PubMed] [Google Scholar]
- 53.Fratti RA, Chua J, Vergne I, Deretic V. Mycobacterium tuberculosis glycosylated phosphatidylinositol causes phagosome maturation arrest. Proc Natl Acad Sci U S A. 2003;100(9):5437–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Huang Z, Zhao GW, Gao CH, Chi XW, Zeng T, Hu YW, et al. Mannose-capped Lipoarabinomannan from Mycobacterium tuberculosis induces IL-37 production via upregulating ERK1/2 and p38 in human type II alveolar epithelial cells. Int J Clin Exp Med. 2015;8(5):7279–87. [PMC free article] [PubMed] [Google Scholar]
- 55.Mahon RN, Sande OJ, Rojas RE, Levine AD, Harding CV, Boom WH. Mycobacterium tuberculosis ManLAM inhibits T-cell-receptor signaling by interference with ZAP-70, Lck and LAT phosphorylation. Cell Immunol. 2012;275(1–2):98–105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Sweet L, Singh PP, Azad AK, Rajaram MV, Schlesinger LS, Schorey JS. Mannose receptor-dependent delay in phagosome maturation by Mycobacterium avium glycopeptidolipids. Infect Immun. 2010;78(1):518–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Sun X, Pan Q, Yuan C, Wang Q, Tang XL, Ding K, et al. A Single ssDNA Aptamer Binding to Mannose-Capped Lipoarabinomannan of Bacillus Calmette-Guerin Enhances Immunoprotective Effect against Tuberculosis. J Am Chem Soc. 2016;138(36):11680–9. [DOI] [PubMed] [Google Scholar]
- 58.Groger M, Holnthoner W, Maurer D, Lechleitner S, Wolff K, Mayr BB, et al. Dermal microvascular endothelial cells express the 180-kDa macrophage mannose receptor in situ and in vitro. J Immunol. 2000;165(10):5428–34. [DOI] [PubMed] [Google Scholar]
- 59.Sumpio BE, Riley JT, Dardik A. Cells in focus: endothelial cell. Int J Biochem Cell Biol. 2002;34(12):1508–12. [DOI] [PubMed] [Google Scholar]
- 60.Roberts LL, Robinson CM. Mycobacterium tuberculosis infection of human dendritic cells decreases integrin expression, adhesion and migration to chemokines. Immunology. 2014;141(1):39–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Blomgran R, Ernst JD. Lung neutrophils facilitate activation of naive antigen-specific CD4+ T cells during Mycobacterium tuberculosis infection. J Immunol. 2011;186(12):7110–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Hernandez-Pando R, Jeyanathan M, Mengistu G, Aguilar D, Orozco H, Harboe M, et al. Persistence of DNA from Mycobacterium tuberculosis in superficially normal lung tissue during latent infection. Lancet. 2000;356(9248):2133–8. [DOI] [PubMed] [Google Scholar]
- 63.Barrios-Payan J, Saqui-Salces M, Jeyanathan M, Alcantara-Vazquez A, Castanon-Arreola M, Rook G, et al. Extrapulmonary locations of mycobacterium tuberculosis DNA during latent infection. J Infect Dis. 2012;206(8):1194–205. [DOI] [PubMed] [Google Scholar]
- 64.Mehta PK, Karls RK, White EH, Ades EW, Quinn FD. Entry and intracellular replication of Mycobacterium tuberculosis in cultured human microvascular endothelial cells. Microb Pathog. 2006;41(2–3):119–24. [DOI] [PubMed] [Google Scholar]
- 65.Saharinen P, Eklund L, Pulkki K, Bono P, Alitalo K. VEGF and angiopoietin signaling in tumor angiogenesis and metastasis. Trends Mol Med. 2011;17(7):347–62. [DOI] [PubMed] [Google Scholar]
- 66.Lohela M, Bry M, Tammela T, Alitalo K. VEGFs and receptors involved in angiogenesis versus lymphangiogenesis. Curr Opin Cell Biol. 2009;21(2):154–65. [DOI] [PubMed] [Google Scholar]
- 67.Matsuyama W, Hashiguchi T, Matsumuro K, Iwami F, Hirotsu Y, Kawabata M, et al. Increased serum level of vascular endothelial growth factor in pulmonary tuberculosis. Am J Respir Crit Care Med. 2000;162(3 Pt 1):1120–2. [DOI] [PubMed] [Google Scholar]
- 68.Ridley MJ, Heather CJ, Brown I, Willoughby DA. Experimental epithelioid cell granulomas, tubercle formation and immunological competence: an ultrastructural analysis. J Pathol. 1983;141(2):97–112. [DOI] [PubMed] [Google Scholar]
- 69.Datta M, Via LE, Kamoun WS, Liu C, Chen W, Seano G, et al. Anti-vascular endothelial growth factor treatment normalizes tuberculosis granuloma vasculature and improves small molecule delivery. Proc Natl Acad Sci U S A. 2015;112(6):1827–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Oehlers SH, Cronan MR, Scott NR, Thomas MI, Okuda KS, Walton EM, et al. Interception of host angiogenic signalling limits mycobacterial growth. Nature. 2015;517(7536):612–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Groschel MI, Sayes F, Simeone R, Majlessi L, Brosch R. ESX secretion systems: mycobacterial evolution to counter host immunity. Nat Rev Microbiol. 2016;14(11):677–91. [DOI] [PubMed] [Google Scholar]
- 72.Oehlers SH, Cronan MR, Beerman RW, Johnson MG, Huang J, Kontos CD, et al. Infection-Induced Vascular Permeability Aids Mycobacterial Growth. J Infect Dis. 2017;215(5):813–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Fachinger G, Deutsch U, Risau W. Functional interaction of vascular endothelial-protein-tyrosine phosphatase with the angiopoietin receptor Tie-2. Oncogene. 1999;18(43):5948–53. [DOI] [PubMed] [Google Scholar]
- 74.Schlesinger LS. Macrophage phagocytosis of virulent but not attenuated strains of Mycobacterium tuberculosis is mediated by mannose receptors in addition to complement receptors. J Immunol. 1993;150(7):2920–30. [PubMed] [Google Scholar]
- 75.Desvignes L, Ernst JD. Interferon-gamma-responsive nonhematopoietic cells regulate the immune response to Mycobacterium tuberculosis. Immunity. 2009;31(6):974–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Flynn JL, Chan J, Triebold KJ, Dalton DK, Stewart TA, Bloom BR. An essential role for interferon gamma in resistance to Mycobacterium tuberculosis infection. J Exp Med. 1993;178(6):2249–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Ko JM, Park HJ, Kim CH. Clinicoradiologic evidence of pulmonary lymphatic spread in adult patients with tuberculosis. AJR Am J Roentgenol. 2015;204(1):38–43. [DOI] [PubMed] [Google Scholar]
- 78.Chackerian AA, Alt JM, Perera TV, Dascher CC, Behar SM. Dissemination of Mycobacterium tuberculosis is influenced by host factors and precedes the initiation of T-cell immunity. Infect Immun. 2002;70(8):4501–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Basaraba RJ, Smith EE, Shanley CA, Orme IM. Pulmonary lymphatics are primary sites of Mycobacterium tuberculosis infection in guinea pigs infected by aerosol. Infect Immun. 2006;74(9):5397–401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Harding J, Ritter A, Rayasam A, Fabry Z, Sandor M. Lymphangiogenesis is induced by mycobacterial granulomas via vascular endothelial growth factor receptor-3 and supports systemic T-cell responses against mycobacterial antigen. Am J Pathol. 2015;185(2):432–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.de Bruijn M The hemangioblast revisited. Blood. 2014;124(16):2472–3. [DOI] [PubMed] [Google Scholar]
- 82.Kaipainen A, Korhonen J, Mustonen T, van Hinsbergh VW, Fang GH, Dumont D, et al. Expression of the fms-like tyrosine kinase 4 gene becomes restricted to lymphatic endothelium during development. Proc Natl Acad Sci U S A. 1995;92(8):3566–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Adams RH, Alitalo K. Molecular regulation of angiogenesis and lymphangiogenesis. Nat Rev Mol Cell Biol. 2007;8(6):464–78. [DOI] [PubMed] [Google Scholar]
- 84.Albelda SM, Muller WA, Buck CA, Newman PJ. Molecular and cellular properties of PECAM-1 (endoCAM/CD31): a novel vascular cell-cell adhesion molecule. J Cell Biol. 1991;114(5):1059–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Baluk P, Fuxe J, Hashizume H, Romano T, Lashnits E, Butz S, et al. Functionally specialized junctions between endothelial cells of lymphatic vessels. J Exp Med. 2007;204(10):2349–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Banerji S, Ni J, Wang SX, Clasper S, Su J, Tammi R, et al. LYVE-1, a new homologue of the CD44 glycoprotein, is a lymph-specific receptor for hyaluronan. J Cell Biol. 1999;144(4):789–801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Breiteneder-Geleff S, Soleiman A, Kowalski H, Horvat R, Amann G, Kriehuber E, et al. Angiosarcomas express mixed endothelial phenotypes of blood and lymphatic capillaries: podoplanin as a specific marker for lymphatic endothelium. Am J Pathol. 1999;154(2):385–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Brooks PC, Clark RA, Cheresh DA. Requirement of vascular integrin alpha v beta 3 for angiogenesis. Science. 1994;264(5158):569–71. [DOI] [PubMed] [Google Scholar]
- 89.Dagenais SL, Hartsough RL, Erickson RP, Witte MH, Butler MG, Glover TW. Foxc2 is expressed in developing lymphatic vessels and other tissues associated with lymphedema-distichiasis syndrome. Gene Expr Patterns. 2004;4(6):611–9. [DOI] [PubMed] [Google Scholar]
- 90.Duff SE, Li C, Garland JM, Kumar S. CD105 is important for angiogenesis: evidence and potential applications. FASEB J. 2003;17(9):984–92. [DOI] [PubMed] [Google Scholar]
- 91.Fong GH, Rossant J, Gertsenstein M, Breitman ML. Role of the Flt-1 receptor tyrosine kinase in regulating the assembly of vascular endothelium. Nature. 1995;376(6535):66–70. [DOI] [PubMed] [Google Scholar]
- 92.Fonsatti E, Sigalotti L, Arslan P, Altomonte M, Maio M. Emerging role of endoglin (CD105) as a marker of angiogenesis with clinical potential in human malignancies. Curr Cancer Drug Targets. 2003;3(6):427–32. [DOI] [PubMed] [Google Scholar]
- 93.Friedlander M, Theesfeld CL, Sugita M, Fruttiger M, Thomas MA, Chang S, et al. Involvement of integrins alpha v beta 3 and alpha v beta 5 in ocular neovascular diseases. Proc Natl Acad Sci U S A. 1996;93(18):9764–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Hirakawa S, Hong YK, Harvey N, Schacht V, Matsuda K, Libermann T, et al. Identification of vascular lineage-specific genes by transcriptional profiling of isolated blood vascular and lymphatic endothelial cells. Am J Pathol. 2003;162(2):575–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Hong YK, Lange-Asschenfeldt B, Velasco P, Hirakawa S, Kunstfeld R, Brown LF, et al. VEGF-A promotes tissue repair-associated lymphatic vessel formation via VEGFR-2 and the alpha1beta1 and alpha2beta1 integrins. FASEB J. 2004;18(10):1111–3. [DOI] [PubMed] [Google Scholar]
- 96.Kato S, Itonaga I, Ji RC, Miura M. Enzyme triple staining for differentiation of lymphatics from venous and arterial capillaries. Lymphology. 1996;29(1):15–9. [PubMed] [Google Scholar]
- 97.Magnussen A, Kasman IM, Norberg S, Baluk P, Murray R, McDonald DM. Rapid access of antibodies to alpha5beta1 integrin overexpressed on the luminal surface of tumor blood vessels. Cancer Res. 2005;65(7):2712–21. [DOI] [PubMed] [Google Scholar]
- 98.Nibbs RJ, Kriehuber E, Ponath PD, Parent D, Qin S, Campbell JD, et al. The beta-chemokine receptor D6 is expressed by lymphatic endothelium and a subset of vascular tumors. Am J Pathol. 2001;158(3):867–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Noguera-Troise I, Daly C, Papadopoulos NJ, Coetzee S, Boland P, Gale NW, et al. Blockade of Dll4 inhibits tumour growth by promoting non-productive angiogenesis. Nature. 2006;444(7122):1032–7. [DOI] [PubMed] [Google Scholar]
- 100.Okada Y, Yano K, Jin E, Funahashi N, Kitayama M, Doi T, et al. A three-kilobase fragment of the human Robo4 promoter directs cell type-specific expression in endothelium. Circ Res. 2007;100(12):1712–22. [DOI] [PubMed] [Google Scholar]
- 101.Pusztaszeri MP, Seelentag W, Bosman FT. Immunohistochemical expression of endothelial markers CD31, CD34, von Willebrand factor, and Fli-1 in normal human tissues. J Histochem Cytochem. 2006;54(4):385–95. [DOI] [PubMed] [Google Scholar]
- 102.Ridgway J, Zhang G, Wu Y, Stawicki S, Liang WC, Chanthery Y, et al. Inhibition of Dll4 signalling inhibits tumour growth by deregulating angiogenesis. Nature. 2006;444(7122):1083–7. [DOI] [PubMed] [Google Scholar]
- 103.St Croix B, Rago C, Velculescu V, Traverso G, Romans KE, Montgomery E, et al. Genes expressed in human tumor endothelium. Science. 2000;289(5482):1197–202. [DOI] [PubMed] [Google Scholar]
- 104.Wigle JT, Oliver G. Prox1 function is required for the development of the murine lymphatic system. Cell. 1999;98(6):769–78. [DOI] [PubMed] [Google Scholar]
- 105.Yuan L, Moyon D, Pardanaud L, Breant C, Karkkainen MJ, Alitalo K, et al. Abnormal lymphatic vessel development in neuropilin 2 mutant mice. Development. 2002;129(20):4797–806. [DOI] [PubMed] [Google Scholar]