

Abstract
Helicases are crucial participants in many types of DNA repair reactions, including homologous recombination. The properties of these enzymes can be assayed by traditional bulk biochemical analysis; however, these types of assays cannot directly access some types of information. In particular, bulk biochemical assays cannot readily access information that may be obscured in population averages. Single-molecule assays offer the potential advantage of being able to visualize the molecules in question in real time, thus providing direct access to questions relating to translocation velocity, processivity, and insights into how helicases may behave on different types of substrates. Here, we describe the use of single-stranded DNA (ssDNA) curtains as an assay for directly viewing the behavior of the Saccharomyces cerevisiae Srs2 helicase on single molecules of ssDNA. When used with total internal reflection fluorescence microscopy, these methods can be used to track the binding and movements of individual helicase complexes, and allow new insights into helicase behaviors at the single-molecule level.
1. INTRODUCTION
1.1. Homologous Recombination
Homologous recombination (HR) is essential for maintaining genome integrity, and HR defects are directly linked to human cancers and cancer-prone syndromes (Kass, Moynahan, & Jasin, 2016; Malkova & Haber, 2012; Moynahan & Jasin, 2010; Prakash, Zhang, Feng, & Jasin, 2015). HR allows for the repair of double-stranded DNA breaks (DSBs) and is also necessary for the recovery of stalled or collapsed replication forks (Heyer, 2015; Jasin & Rothstein, 2013; Mazon, Mimitou, & Symington, 2010; Paques & Haber, 1999; Symington, Rothstein, & Lisby, 2014).
HR is promoted by the RAD52 epistasis group of genes, which were originally identified in Saccharomyces cerevisiae as mutants defective in DNA repair (Paques & Haber, 1999; Symington et al., 2014). During HR, single-stranded DNA (ssDNA), derived from the nucleolytic processing of a DSB or collapsed replication fork, is quickly bound by RPA (replication protein A), which is a conserved heterotrimeric eukaryotic protein complex that removes ssDNA secondary structure, protects ssDNA from nucleolytic degradation, and serves as a platform from DNA damage signaling (Chen & Wold, 2014). With the aid of Rad52, RPA is replaced by the ATP-dependent DNA-binding protein Rad51, which forms an extended right-handed helical filament on ssDNA, and the resulting nucleoprotein filament is referred to as the presynaptic complex (Kowalczykowski, 2015). Pairing of the presynaptic complex with a homologous dsDNA template results in displacement of the noncomplementary strand from the duplex to generate a D-loop (Kowalczykowski, 2015). The resulting intermediate can be processed via one of several alternative pathways, all of which can allow for the repair of the DSB using information derived from the donor template (Paques & Haber, 1999; Symington et al., 2014) (Fig. 1).
Fig. 1.
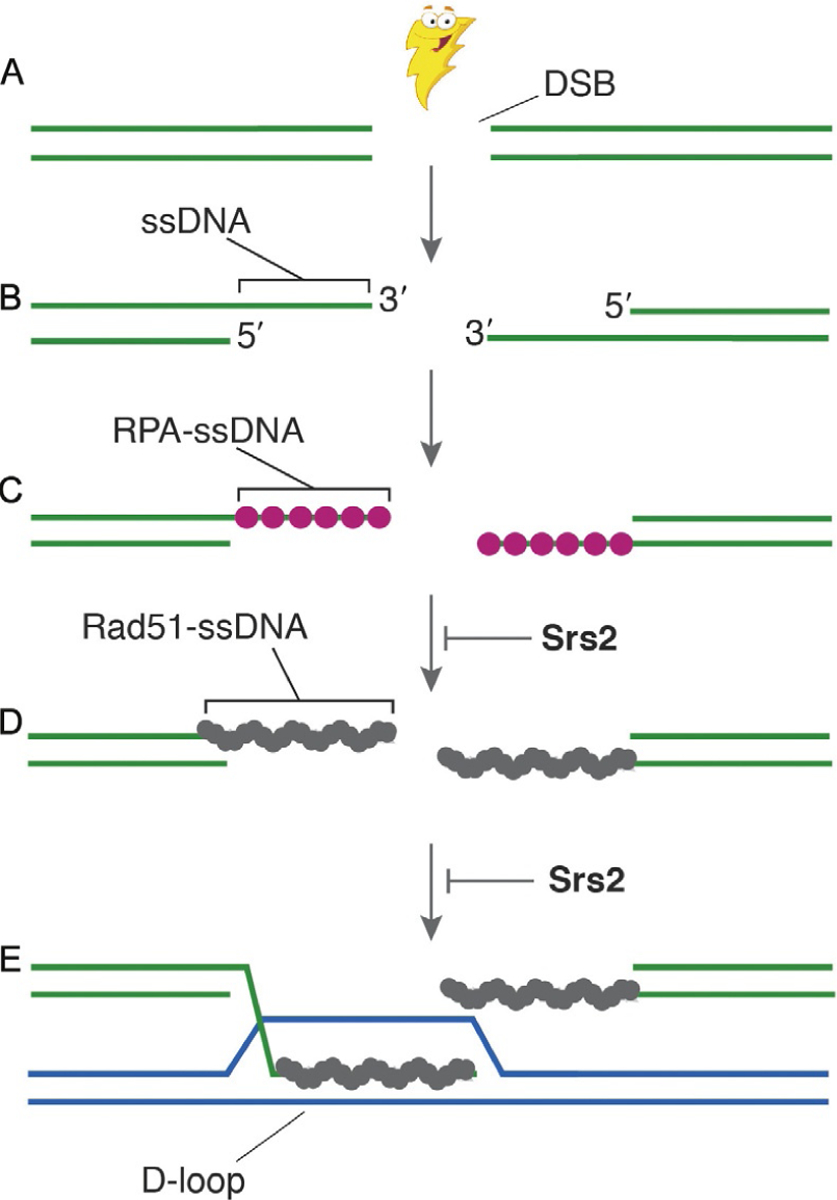
The role of Srs2 in HR. Here, we show a highly simplified overview of the early stages of HR and indicate the steps at which Srs2 has been implicated to act upon recombination intermediates. (A) In this simplified schematic, HR begins with the formation of a double-stranded break (DSB). (B) The break is then processed by 5′→3′ resection, resulting in the formation of long 3′ ssDNA overhangs. (C) These ssDNA over-hangs serve as a binding platform for replication protein A (RPA), which is necessary to protect the ssDNA against degradation and is also required for the removal of secondary structure to enable efficient assembly of the Rad51 filaments. (D) The RPA is then replaced by the Rad51 to yield Rad51-ssDNA filaments, which are also referred to as presynaptic complexes. (E) The presynaptic complex then performs the homology search to locate a double-stranded DNA molecule with sequence complementarity to the ssDNA that is bound by Rad51. Once homology has been identified, Rad51 catalyzes a strand invasion reaction to generate a D-loop intermediate in which the presynaptic ssDNA is paired with its complement, and the noncomplementary is displaced. The 3′ terminus of the invading ssDNA within the D-loop can now serve as a primer for the DNA synthesis reactions that are required for downstream steps in the HR pathway. As highlighted in the figure, Srs2 has been reported to act at two key stages of this pathway, by either dismantling the Rad51 filaments (Krejci et al., 2003; Veaute et al., 2003), or by disrupting early strand invasion intermediates (Liu et al., 2017).
1.2. Srs2 Functions and Activities During HR
There are approximately 95 helicases encoded within the human genome, and these proteins participate in nearly all aspects of nucleic acid metabolism, including the DNA repair pathways that are necessary for maintenance of genome stability (Bernstein, Gangloff, & Rothstein, 2010; Branzei & Szakal, 2017; Brosh, 2013; Croteau, Popuri, Opresko, & Bohr, 2014; Niu & Klein, 2017). For instance, HR is held in check by many regulatory mechanisms, including those mediated through the action of DNA helicases. Indeed, several helicases promote genome stability and accurate recombination by functioning as antirecombinases that prevent aberrant HR by dismantling inappropriate or toxic recombination intermediates (Bernstein et al., 2010; Branzei & Szakal, 2017; Brosh, 2013; Croteau et al., 2014; Niu & Klein, 2017). It is now thought that in many instances the normal behavior of these HR-related helicases is to disrupt recombination intermediates, rather than to simply separate duplex nucleic acids (Bernstein et al., 2010; Branzei & Szakal, 2017; Brosh, 2013; Croteau et al., 2014; Niu & Klein, 2017). The importance of these enzymes is underscored by helicase mutations that give rise to human diseases such as Rothmund–Thomson syndrome and Fanconi anemia, which are both characterized by genome instability and increased incidence of cancer (Bernstein et al., 2010; Brosh, 2013; Croteau et al., 2014).
The S. cerevisiae protein Srs2 is a superfamily 1 (SF1) helicase and antirecombinase that is required for genome integrity, and deletion of the SRS2 gene results in an increase in recombination frequency (Niu & Klein, 2017). Srs2 is considered a prototypical antirecombinase due to its well-characterized ability to remove Rad51 filaments from ssDNA (Fig. 1) (Antony et al., 2009; Krejci et al., 2003; Marini & Krejci, 2010; Niu & Klein, 2017; Qiu et al., 2013; Sasanuma, Furihata, Shinohara, & Shinohara, 2013; Vasianovich et al., 2017; Veaute et al., 2003). The importance of Srs2 for genome integrity was revealed in genetic studies showing that Δsrs2 Δsgs1 and Δsrs2 ΔRad54 double mutants, which are synthetic lethal, presumably due the accumulation of toxic recombination intermediates (Ira, Malkova, Liberi, Foiani, & Haber, 2003; Klein, 2001). Srs2 is homologous to the bacterial UvrD, PcrA, and Rep helicases, which are also thought to promote the removal of RecA from ssDNA (Marini & Krejci, 2010; Niu & Klein, 2017; Park et al., 2010; Petrova et al., 2015). Although Srs2 homologs have not yet been identified in humans, the mammalian protein FBH1 is a potential candidate, and FBH1 can also remove Rad51 from ssDNA (Simandlova et al., 2013). A growing body of evidence suggests similar antirecombinase regulatory roles might be filled by other helicases, including RECQ1, RECQ5, BLM (Sgs1 in yeast), FANCM (Mph1 in yeast), FANCJ, and RTEL1 (Bernstein et al., 2010; Branzei & Szakal, 2017; Brosh, 2013; Heyer, Ehmsen, & Liu, 2010). Thus, these types of helicases play crucial regulatory roles as antirecombinases in many different organisms.
1.3. Single-Molecule Studies of Helicase Activities
Much of our knowledge of the helicases involved in HR comes from a combination of genetic and bulk biochemical studies. Indeed, Srs2 was originally identified in yeast as a suppressor of radiation-sensitive mutations in the error-prone repair pathway, and its deletion leads to a hyperrecombination phenotype (Niu & Klein, 2017; Palladino & Klein, 1992; Rong, Palladino, Aguilera, & Klein, 1991). Comparison of the Srs2 amino acid sequence revealed that the gene was highly homologous to the bacterial DNA helicases UvrD and Rep, and subsequent biochemical studies demonstrated that the protein has robust DNA-dependent ATP hydrolysis activity and helicase activity (Rong & Klein, 1993). Biochemical studies revealed the remarkable finding that Srs2 could strip Rad51 from ssDNA, providing detailed mechanistic insights into its antirecombinase activity (Krejci et al., 2003; Veaute et al., 2003). Further genetic analysis indicated that Srs2 suppressed crossovers, suggesting that it was capable of disrupting strand invasion intermediates (Ira et al., 2003), which was recently substantiated by in vitro biochemical analysis (Liu et al., 2017). Together, these studies illustrate the importance of genetics and biochemistry for defining protein functions in HR.
Single-molecule (SM) studies have the potential to further our understanding of HR by providing even more detailed insights into reaction mechanisms, and these methods are particularly beneficial for reactions that involve heterogeneous populations, transient intermediates, or both, as is often the case with reactions involving helicases. Indeed, SM methods have proven deeply insightful for understanding helicases, and some examples include SM studies of the RecBCD complex (Bianco et al., 2001; Spies et al., 2003), which is involved in DNA end processing in Escherichia coli, the bacterial Rep, UvrD, and PcrA helicases (Comstock et al., 2015; Myong, Rasnik, Joo, Lohman, & Ha, 2005; Park et al., 2010), the archaeal helicase XPD (Honda, Park, Pugh, Ha, & Spies, 2009), and the eukaryotic helicases Srs2 and Pif1 (Qiu et al., 2013; Sokoloski, Kozlov, Galletto, & Lohman, 2016). Studies of RecBCD relied primarily upon wide-field epi-illumination of bead-trapped DNA molecules, whereas most of the other helicases have been studied by smFRET. To help expand this tool box of methods available for SM studies of helicases involved in HR, we have recently applied ssDNA curtain assays to study Srs2 activities at the SM level (De Tullio et al., 2017; Kaniecki et al., 2017).
2. METHODS
2.1. Overview of ssDNA Curtains for Studying Protein–ssDNA Interactions
We have developed DNA curtains as a tool for real-time visualization of protein–nucleic acid interactions at the SM level using total internal reflection fluorescence microscopy (TIRFM) (Fig. 2A) (Fazio, Visnapuu, Wind, & Greene, 2008; Gorman, Fazio, Wang, Wind, & Greene, 2010; Graneli, Yeykal, Prasad, & Greene, 2006). In brief, DNA curtains are prepared by first depositing metal barriers and anchors on the surface of a fused silica microscope slide by electron-beam lithography. The slide is then coated with a fluid lipid bilayer, which prevents nonspecific surface adsorption and provides a mobile platform for anchoring DNA molecules through a biotin–streptavidin linkage. Buffer flow is used to push the DNA molecules into the barriers where they all align with one another (Fazio et al., 2008; Graneli et al., 2006). If desired, the second DNA end can be attached to a downstream anchor point (Fig. 2B–D) (Gorman, Fazio, et al., 2010). This approach can be used with dsDNA or ssDNA and allows for the direct visualization of hundreds of individual DNA molecules by TIRFM, thus offering a flexible experimental platform that can be adapted to the study of many types of protein–DNA interactions (Duzdevich et al., 2015; Gorman et al., 2012; Gorman, Plys, Visnapuu, Alani, & Greene, 2010; Lee, Finkelstein, Arciszewska, Sherratt, & Greene, 2014; Lee et al., 2015; Qi et al., 2015; Redding et al., 2015; Silverstein, Gibb, & Greene, 2014; Sternberg, Redding, Jinek, Greene, & Doudna, 2014; Wang et al., 2013). We have recently provided detailed descriptions of the TIRFM instrumentation used for these assays, the nanofabrication methods for making patterned slide surfaces with electron-beam lithography, and the procedures for preparing microfluidic flow cells (Greene, Wind, Fazio, Gorman, & Visnapuu, 2010; Ma, Steinfeld, & Greene, 2017; Qi & Greene, 2016). Here, we describe methods for how ssDNA curtains can be used to study the S. cerevisiae helicase Srs2 as it acts upon ssDNA substrates bound by either Rad51 or RPA. Included in this information are details on fluorescent protein purification, ssDNA substrate preparation, and further details on bilayer deposition and ssDNA curtain preparation. We then provide a detailed overview on experiments using fluorescently tagged or unlabeled Srs2, and analysis of the resulting data.
Fig. 2.
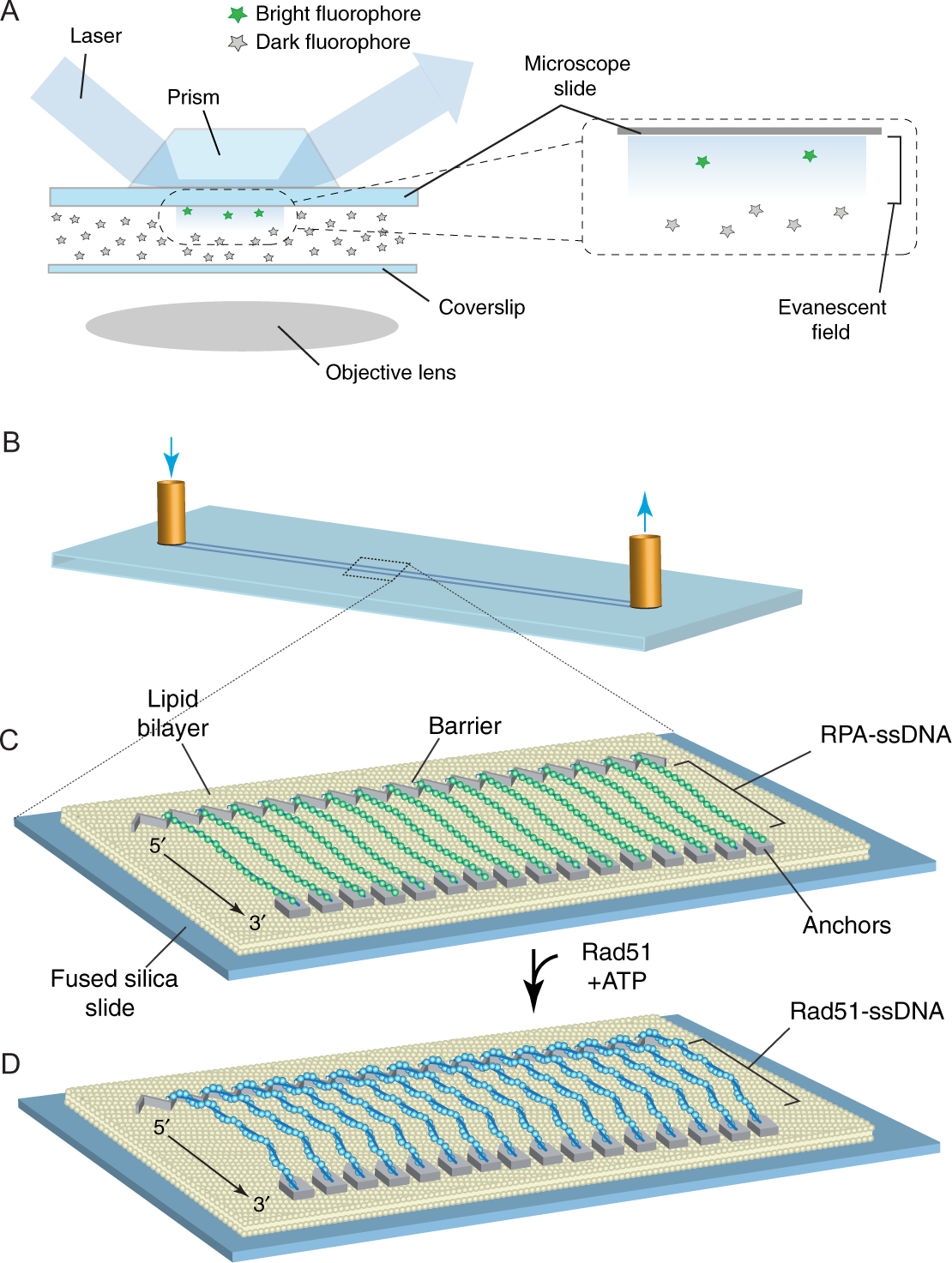
Schematic overviews of TIRFM and ssDNA curtains. (A) This illustration shows a simplified depiction of a typical prism-type TIRFM (Axelrod, 1989). Note: We use prism-type TIRFM setups because they allow for observation of reactions anchored to the microscope slide glass, as opposed to the coverslip, which is visualized in objective-type TIRFM. The benefit of prism-type TIRFM is that the thicker microscope slides are more robust and can be easily modified by electron-beam lithography, and then repeated cleaned and reused (Greene et al., 2010; Ma et al., 2017). (B) Schematic illustration of a flow cell used for sample analysis by TIRFM. (C) Schematic showing a double-tethered ssDNA curtain. In this example, the surface is coated with a lipid bilayer, which is disrupted at defined locations by metallic patterns that have been deposited by electron-beam lithography (Greene et al., 2010; Ma et al., 2017). The zigzag-shaped upstream pattern is used for alignment of the ssDNA, and the pentagonal anchor points allow for nonspecific adsorption of the downstream ends of the RPA-ssDNA molecules. In this configuration, the RPA-ssDNA can be viewed even in the absence of buffer flow. Similar patterns, but omitting the anchor points, can be used to set up single-tethered ssDNA curtains, in which case constant buffer flow is necessary to keep the molecules within the evanescent field for visualization. (D) Rad51-ssDNA filaments can be assembled by injecting Rad51 into the sample chamber in the presence of ATP, and assembly of these filaments coincides with the loss of GFP signal due to dissociation of the GFP-tagged RPA (Qi et al., 2015). Panels (B)–(D) were reproduced with permission from Qi, Z., Redding, S., Lee, J. Y., Gibb, B., Kwon, Y., Niu, H., et al. (2015). DNA sequence alignment by microhomology sampling during homologous recombination. Cell, 160, 856–869. doi: https://doi.org/10.1016/j.cell.2015.01.029.
2.2. Protein and ssDNA Preparation
A key aspect of using ssDNA curtains for studying the activities of Srs2 and other HR-related proteins is the need to prepare and purify fluorescently tagged proteins. For much of our work on HR we rely upon either unlabeled proteins, or proteins that are labeled with GFP or mCherry. Although GFP and mCherry are perhaps not as bright and photostable as many organic fluorophores, they offer the advantage of providing a homogenous protein preparation where all of the proteins are labeled. Moreover, GFP-tagged versions of many HR proteins have already been evaluated in in vivo studies, including GFP-Srs2 and GFP-RPA, thus validating their biological functions (Burgess et al., 2009; Lisby, Barlow, Burgess, & Rothstein, 2004; Lisby & Rothstein, 2015). For GFP-labeled proteins that have not yet been tested, genetic assays are readily available to validate biological activities. Here, we will describe the procedures for producing GFP- or mCherry-labeled Srs2 and RPA; note that similar procedures also work for the unlabeled version of these proteins. For ssDNA curtains using Rad51, we rely upon unlabeled version of the protein, as GFP-tagged Rad51 is not functional in vivo (Lisby et al., 2004); procedures for purifying unlabeled S. cerevisiae Rad51 have been described elsewhere (Sung & Stratton, 1996).
2.2.1. Purification of GFP-Tagged Srs2
GFP- or mCherry-tagged Srs2 is expressed from a pET15b vector in E. coli Rosetta2 (DE3) cells (Novagen). In this instance, the N-terminus of the protein bears the GFP or mCherry fusion and also has a 9xHis tag that is used for purification. The GFP gene contains the A206K mutation to prevent GFP dimerization (Zacharias, Violin, Newton, & Tsien, 2002), and we typically work with Srs2898, which retains antirecombinase activity in vitro, but is less prone to aggregation compared to the full-length construct (Antony et al., 2009). Unlabeled Srs2898 is expressed from the pET11c vector.
Bacteria are freshly transformed, and then single colonies grown on LB-agar plates (supplemented with 100μg/mL carbenicillin and 40μg/mL chloramphenicol) are selected and grown in 25mL culture for ~3–5h. Then 1mL of this culture is used to inoculate 1L of LB medium supplemented with 100μg/mL carbenicillin and 40μg/mL chloramphenicol, and cultures are grown at 37°C with continuous shaking until reaching an OD600 of ~1.0. The temperature is then reduced to 16°C before initiating protein expression by the addition of 0.1–0.5mM isopropyl-β-D-thiogalactopyranoside (IPTG).
Cells are grown for an additional 20h at 16°C with slow shaking (80 RPM). Cells are then harvested by centrifugation, and the cell pellet is frozen at −80°C.
The frozen cell pellet is thawed at 37°C and resuspended in cell breakage buffer containing 40mM NaHPO4 (pH 7.5), 600mM KCl, 5% glycerol, 10mM imidazole (pH 7.8), 0.1mM TCEP (Tris(2-carboxyethyl)phosphine hydrochloride; Sigma, Cat. No. C4706), 0.05% Tween-20, 10μM E-64 (Sigma-Aldrich, Cat. No. E3132), 100μM AEBSF (4-(2-aminoethyl)benzenesulfonyl fluoride hydrochloride; Sigma-Aldrich, Cat. No. A8456), 1mM benzamidine, and 1mM PMSF (phenylmethanesulfonyl fluoride; Sigma-Aldrich, Cat. No. P7626). The cells are then lysed by sonication on ice, and the lysate was clarified by ultracentrifugation at 25,000rpm for 30min.
The clarified lysate (~30–40mL) is then incubated for 30min with a Talon metal affinity resin (5mL bed volume per liter of culture; Clontech, Cat. No. 635503) equilibrated with Buffer Nickel A (40mM NaHPO4 [pH 7.5], 300mM KCl, 5% glycerol, 15mM imidazole, 0.02% Tween-20, 1mM benzamidine, 1mM PMSF, 0.125% myoinositol). Before elution, the column is washed extensively with Buffer Nickel A.
Proteins are eluted from the Talon metal affinity column with a step of Buffer Nickel A containing 400mM imidazole(pH7.8).Immediately after elution, the sample is adjusted to 5mM EDTA (pH 8) and 1mM TCEP.
The eluate is then dialyzed in SnakeSkin Dialysis Tubing (10,000 MWCO; Thermo Scientific, Cat. No. 68100) against 1L of Heparin Buffer (20mM NaHPO4 [pH 7.5], 100mM KCl, 5% glycerol, 0.01% Tween-20, 1mM TCEP, 2mM EDTA, 0.125% myoinositol) for 1.5h, with 1L buffer changes every 30min.
The dialyzed eluate is then loaded onto a 5mL HiTrap Heparin column (GE Lifesciences, Cat. No. 17-0406-01) equilibrated with Heparin Buffer, and proteins are eluted with a single step of Heparin Buffer containing 500mM KCl.
The HiTrap Heparin-purified fraction (~4mL) is then dialyzed in SnakeSkin Tubing (10,000 MWCO) against Storage buffer (40mM NaHPO4 [pH 7.5], 300mM KCl, 10% glycerol, 0.01% Tween-20, 1mM TCEP, 0.5mM EDTA, 0.125% myoinositol) for 2h at 4°C.
The sample is then applied to a Superdex 200 size exclusion column previously equilibrated with Storage buffer.
Peak fractions from the Superdex 200 corresponding to monomeric Srs2 are then pooled, flash frozen in liquid nitrogen, and stored in small aliquots at −80°C. The frozen aliquots should be small enough that each can be used for a single set of experiments to avoid multiple freeze–thaw cycles.
2.2.2. Purification of GFP-Tagged RPA
We use a GFP- or mCherry-tagged RPA to remove secondary structure from the ssDNA so that the molecules can be easily extended by buffer flow, and the fluorescent proteins also allow us to visualize the ssDNA with no need for any additional DNA-labeling dyes (Gibb, Silverstein, Finkelstein, & Greene, 2012). In addition, RPA-ssDNA is the physiological substrate for the early stages of HR in eukaryotes (Bianco, Tracy, & Kowalczykowski, 1998; Wold, 1997). S. cerevisiae RPA is a heterotrimeric protein complex comprised of Rfa1, Rfa2, and Rfa3 subunits, and we used a tagged version of RPA which bears the GFP or mCherry fusion on the C-terminus of the Rfa1 separated by a 7-alanine linker; note that this GFP-tagged version of RPA retains biological function in vivo (Lisby et al., 2004).
6xHis-tagged S. cerevisiae RPA-eGFP and RPA-mCherry encoded in pET11d plasmid are expressed in E. coli Rosetta2 (DE3) cells (Novagen). The cells are grown in LB supplemented with 100μg/mL carbenicillin and 40μg/mL chloramphenicol at 37°C to an OD600 of ~1. The growth temperature is then reduced to 16°C and protein expression is induced by the addition of 0.5mM IPTG, and the culture is grown for an additional 10–15h at 16°C.
Cells are harvested by centrifugation, and the cell pellet is frozen at −80°C.
The cell pellet is then thawed and resuspended in cell lysis buffer (50mM NaHPO4, 250mM KCl, 5mM imidazole [pH 7.9], 5% glycerol, 0.1mM TCEP, 0.02% Tween-20, 10μM E-64, 100μM AEBSF, 1mM benzamidine, 1mM PMSF, and myoinositol 0.25%), and lysed by sonication. The lysate is clarified by ultracentrifugation at 25,000rpm for 30min.
The clarified lysate is then applied in batch to a Talon metal affinity resin (5mL volume bed per liter of culture; Clontech, Cat. No. 635503) previously washed with Buffer A (30mM NaHPO4 [pH 7.5], 250mM KCl, 5% glycerol, 10mM imidazole, 0.02% Tween-20, 0.1mM TCEP, 1mM benzamidine, 1mM PMSF, 0.125% myoinositol), and incubated with rotation for 30min at 4°C.
The column is extensively washed with ~200mL of Buffer A, and the bound RPA is then eluted with Buffer A containing 400mM imidazole (pH 7.8).
The eluted protein is then dialyzed against buffer Superdex (30mM NaHPO4 [pH 7.5], 250mM KCl, 10% glycerol, 0.02% Tween-20, 1mM TCEP, 0.5mM EDTA, 0.25% myoinositol) using a Float-A-Lyzer dialysis device (MWCO 3.5kDa; Spectrum Labs, Cat. No. G235065).
The dialyzed fraction is concentrated to a final volume of ~4mL using centrifugation (Vivaspin 6, 5000 MWCO; GE Healthcare, Cat. No. 28-9322-94), and is injected onto a Superdex 200 column. The peak containing trimeric RPA is pooled and concentrated using centrifugation (Vivaspin 6; GE Healthcare).
The final RPA-eGFP or RPA-mCherry concentrations can be determined from the absorbance of the eGFP or mCherry chromophores at 488nm (ε488 =55,000cm−1 M−1) or 587nm (ε587nm =72,000cm −1 M−1), respectively. The presence of the three subunits of RPA can be verified by analysis using SDS/PAGE 4%–20% gradient gels (Invitrogen, Cat. No. XP00122BOX) with Coomassie staining.
The final protein concentration should be ~10–20μM. The purified protein is then flash frozen in liquid nitrogen and stored at −80°C in small aliquots to avoid freeze–thaw cycles.
2.2.3. Preparation of ssDNA
An important benefit of ssDNA curtain assays is that they allow the use of long ssDNA substrates, on the order of ≥40,000 nucleotides in length. Use of this long ssDNA is important because TIRFM imaging allows one to visualize along the entire contour lengths of the molecules and thus obtain spatial and temporal information from many different regions at once. These ssDNA substrates are prepared by rolling circle replication (RCR) using a circular ssDNA template and a biotinylated primer (Fig. 3A) (Gibb et al., 2012; Ma et al., 2017), as described below.
Fig. 3.
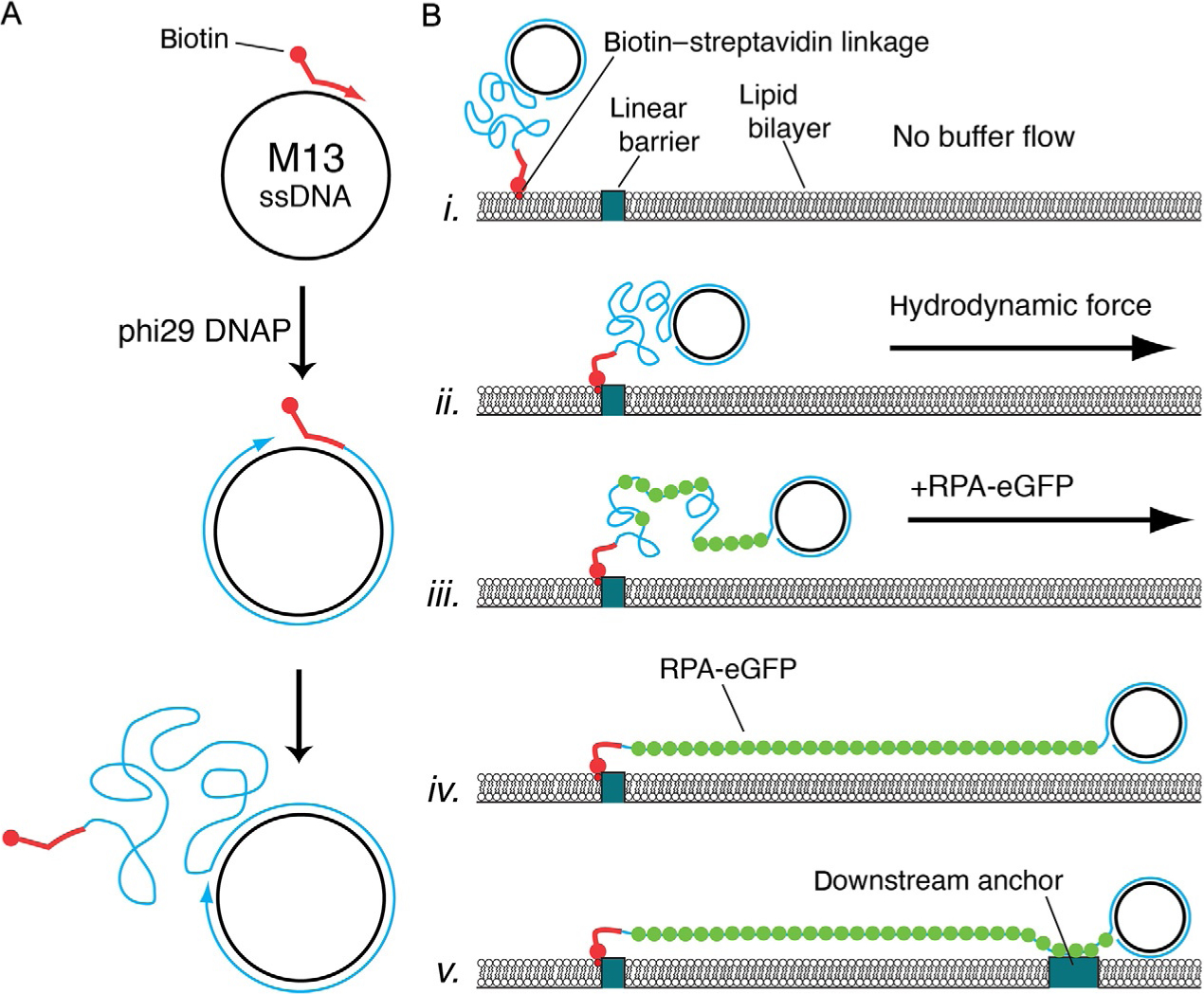
Schematic of ssDNA preparation and anchoring procedures. (A) The biotinylated ssDNA substrate is generated by rolling circle replication (RCR) using a biotinylated input primer, a circular ssDNA template such as M13mp18, and phi29 DNA polymerase (DNAP) (Gibb et al., 2012; Ma et al., 2017). (B) For single-tethered ssDNA curtains, the biotinylated ssDNA is first anchored to the bilayer, and the ssDNA is then aligned at the nanofabricated barriers through the application of buffer flow. RPA-GFP is then introduced into the flow cell to label the ssDNA and remove secondary structure. As RPA-GFP enters the sample chamber, the ssDNA becomes visible and gradually unfolds and extends parallel to the bilayer (Gibb et al., 2012; Ma et al., 2017). For double-tethered curtains, the RPA-ssDNA is nonspecifically adsorbed to exposed anchor points downstream from barriers (Gibb et al., 2012; Ma et al., 2017). Panels (A) and (B) were adapted with permission from Gibb, B., Silverstein, T. D., Finkelstein, I. J., & Greene, E. C. (2012). Single-stranded DNA curtains for real-time single-molecule visualization of protein-nucleic acid interactions. Analytical Chemistry, 84, 7607–7612. Copyright (2012) American Chemical Society.
The biotinylated primer is annealed to a circular M13 DNA template in a 100μL reaction containing: 10mM Tris–HCl (pH 8.0), 50mM NaCl, 10mM MgCl2, 10μg (89.4nM) of M13mp18 (New England Biolabs, Cat. No. N4040S), and 20nM primer (5′—BIOTEG—TTT TTT TTT TTT TTT TTT TTT TTT TTT TTT GTA AAA CGA CGG CCA GT). The annealing mix is placed in a ~95°C water bath for ~5min, and the water bath is then placed on a benchtop and allowed to cool slowly to room temperature. Alternatively, the annealing mix can be placed in a thermocycler and slowly cooled from 95°C to 4°C, over 30min. The annealing reactions are then diluted with buffer containing 10mM Tris–HCl (pH 8.0), 50mM NaCl, and 5mM MgCl2 to a total volume of 300μL and can be stored at −20°C until use.
We typically make a fresh preparation of ssDNA for each ssDNA curtain experiment using RCR. RCR reactions are prepared using 10μL of 5× reaction buffer (250mM Tris–HCl [pH 7.5], 20mM DTT, 50mM ammonium sulfate, and 50mM MgCl2), 1μL annealed M13 template (see above), 2μL of 10mM dNTP mix, 1μL purified ϕ29 polymerase (10μM stock) (Gibb et al., 2012), and 36μL water for a final sample volume of 50μL. Samples are mixed by gentle pipetting to avoid shearing the DNA.
RCR reactions are then incubated at 30°C for 25min, and used immediately after preparation. Alternatively, we have also prepared RCR reactions in a final volume of 500μL. These larger reactions can then be divided into smaller aliquots, flash frozen on liquid nitrogen stored at −80°C.
2.3. Procedures for ssDNA Curtain Assembly
An important aspect of DNA curtain experiments is the requirement for a supported lipid bilayer, which is deposited onto the nanopatterned surface of the sample chamber; detailed descriptions of nanofabrication and flow cell assembly procedures have been described elsewhere (Greene et al., 2010; Ma et al., 2017). The bilayer passivates the surface to minimize nonspecific adsorption of proteins, and the bilayer also serves as a mobile anchor point for tethering the 5- ends of the ssDNA substrates. Tethering is achieved by spiking the bilayer with a small fraction of biotinylated lipids to which the biotinylated ssDNA molecules are anchored through a biotin–streptavidin linkage (Fig. 3B).
2.3.1. Liposome Stock Solutions
A liposome stock solution is required to deposit the lipid bilayer onto the sample surface, and here we describe basic procedures for preparation of the liposome stock. It should be noted that if the lipid bilayer is not fluid, then the tethered DNA molecules cannot be aligned into DNA curtains. Therefore, we encourage persons making lipid bilayers for the first time to prepare a control bilayer that is spiked with a small fraction (≤1%) of fluorescently tagged lipids (e.g., rhodamine-PE; l-α-phosphatidylethanolamine-N-(lissamine rhodamine B sulfonyl) (ammonium salt), Avanti Polar Lipids Inc., Cat. No. 810146C), and a simple FRAP (fluorescence recovery after photo-bleaching) measurement can be used to determine whether the lipids within the bilayer are mobile (Graneli et al., 2006).
Steps described that require the use of chloroform should be performed in an approved fume hood by laboratory personnel trained in the proper use of organic solvents.
Lipid stocks are prepared by dissolving the following components in 10mL of chloroform: 1g DOPC (1,2-dioleoyl-sn-glycero-3-phosphocholine) (Avanti Polar Lipids, Inc., Cat. No. 850375P), 100mg PEG-2000 DOPE (18:1 PEG2000:1,2-dioleoyl-sn-glycero-3-phosphoethanolamine-N-[methoxy(polyethylene glycol)-2000] (ammonium salt)) (Avanti Polar Lipids, Inc., Cat. No.880130P), and 5mg biotinylated DOPE (1,2-dioleoyl-sn-glycero-3-phosphoethanolamine-N-(cap biotinyl) (sodium salt)) (Avanti Polar Lipids, Inc., Cat. No. 870273P). The dissolved lipid mixtures can then be stored at −20°C.
Clean an organic solvent-compatible syringe (e.g., Hamilton, Cat. No. 91022) by flushing the syringe with chloroform. Then use the syringe to transfer 200μL of the lipid stock to a 2-mL glass vial (National Scientific, Cat. No. C4015).
Apply a gentle stream of nitrogen gas to the sample to slowly evaporate the chloroform from the lipid stock; this step should take several minutes. The pressure of the nitrogen stream can be increased once the chloroform has visibly evaporated to help ensure speed evaporation of the chloroform. The uncapped glass vial can then be placed under vacuum overnight to ensure complete removal of all traces of chloroform. When complete, the lipid stock should appear as a solid residue visible on the side of the glass vial.
2mL of lipid buffer (10mM Tris–HCl [pH 8.0], 100mM NaCl) is then added to the dried lipid residue. The dry lipids are resuspended into the buffer with the help of an automatic pipette. The sample should be incubated at room temperature for approximately 1h to help hydrate the lipids. Following this incubation, the sample is then vortexed until all the dried lipids are suspended into solution as judged by visual inspection. At this stage, the solution will appear turbid.
The dissolved lipid mixture is then transferred to a 5-mL polypropylene culture tube (Falcon, Cat. No. 35-2058) and sonicated in an ice bath using a microtip sonicator (Misonix S-4000). Sonication is performed in 5–10s bursts, with 1-min intervals between bursts. Sonicate until the turbid lipid solution becomes clear—this typically takes ~3–5min of total sonication time.
After sonication, the resulting liposomes should be filtered through a 0.22-μm nylon syringe filter (Fisherbrand, Cat. No. 09-720-3). This liposome stock solution is ready for use and can be stored for approximately 30 days at 4°C.
2.3.2. Lipid Bilayers and ssDNA Substrate Attachment
Here, we describe how the bilayer is deposited onto the flow cell surface and how the ssDNA is attached to the bilayer. Note that if air bubbles pass through the sample chambers, they will destroy the lipid bilayer; in our experience, disruption of the bilayer due to passage of an air bubble through the sample chamber is a common failure point. Therefore, extreme care should always be taken to prevent any air from entering the flow cell, and drop-to-drop connections must be used for all tubing and syringe attachments. Degassing buffers prior to use can help minimize bubble accumulation and are highly recommended.
Make two pieces of tubing to connect the syringes to the flow cell ports (Greene et al., 2010; Ma et al., 2017). For this, cut ~8cm of tubing (IDEX, Cat. No. 1907L), and attach a connector (IDEX, Cat. No.F-333NX) to the end of one piece of tubing—this end will be connected to the flow cell. On the other end of the tubing, place first one connector (IDEX, Cat. No. F-300X) and then attach a female-to-female Luer peek (IDEX, Cat. No. P-659)—this end will be attached to the syringe.
Fill two 3-mL plastic syringes (BD, Cat. No. 309657) with degassed MilliQ water. Remove all air bubbles from the syringe, and then connect the tubing to the syringe. Push some water into the tubing removing all the bubbles, and then connect one tubing with its attached syringe to the inlet port of the flow cell. Next, connect the second tubing with its attached syringe to the outlet port of the flow cell.
Once both syringes are connected, gently push 1mL of water through the flow cell by hand and use the two syringes to gently flush the water back and forth through the sample chamber. This procedure helps remove any air bubbles from the sample chamber prior to bilayer deposition.
Next, fill two 10-mL plastic syringes (BD, Cat. No. 309604) with 5 mL of lipid buffer (10mM Tris–HCl [pH 8], 100mM NaCl). Disconnect one of the 3-mL syringes from the flow cell, gently push water from the remaining 3-mL syringe through the sample chamber, and then make a drop-to-drop connection with one of the 10-mL syringes. Next, disconnect the remaining 3-mL syringe, and replace it with the second 10-mL syringe, again being sure to make drop-to-drop connections. Then slowly flush the buffer back and forth several times until no bubbles are present in the flow cell.
Mix 40μL of the liposome stock solution with 960μL of lipid buffer (10mM Tris–HCl [pH 8.0], 100mM NaCl). Fill a 1-mL plastic syringe (BDH, Cat. No. 309659) with the lipid solution. Next, replace one of the 10-mL syringes with this 1-mL syringe using drop-to-drop connections. Gently push ~200μL of the mixture into the flow cell, and then repeat this process every 5–8min until all of the liposome mixture is used. After the final injection, wait 5–8min more, and wash the flow cell with 3mL of lipid buffer. Then incubate the flow cell for ~20min at room temperature to allow formation of the bilayer.
Any areas of the flow cell surface that remain exposed after bilayer deposition are then blocked by gently flushing 3mL of HR buffer (30mM Tris–Ac [pH 7.5], 50mM KCl, 5mM MgAc, 1mM DTT, 0.3mg/mL BSA) through the chamber and the flow cell is incubated for an additional ~5min at room temperature.
The biotinylated lipids must now be saturated with streptavidin to provide anchor points for the biotinylated ssDNA (see Section 2.2.3). For this, mix 980μL of HR buffer with 25μL of streptavidin stock mix (1mg/mL; Sigma-Aldrich, Cat. No. S0677) and slowly flush the solution through the sample chamber in one step.
Carefully flush the sample chamber with 3mL of HR buffer to remove all traces of free streptavidin. This step is crucial because it may not be possible to attach the biotinylated ssDNA to the surface if any free streptavidin remains in solution.
The sample chamber is now ready for addition of the ssDNA sample. Dilute the RCR reaction (30–50μL) by the addition of 1mL of HR buffer and then slowly push ~200μL of the diluted ssDNA sample through the flow cell. Note: This injection should be in the direction that pushes the ssDNA into the barriers (e.g., from left to right in Fig. 3B). Repeat this process every 5min until all of the solution is used. Following the final ssDNA injection, replace the 1-mL syringe with a new 3-mL syringe containing HR buffer.
Once complete, the flow cell can be mounted onto the microscope stage and connected to a sample injection system; our group uses a simple injection system comprised of a syringe pump (KD Scientific KDS-201) and a high-pressure switch valve, which can be used for sample injections (IDEX Health & Science, MXP9900-000) (Greene et al., 2010; Ma et al., 2017). While attaching the flow cell to the injection system, it is again absolutely essential to use drop-to-drop connections to avoid inadvertently injecting air bubbles through the sample chamber. The first step in the connection process is to disconnect the inlet syringe from tubing and connect the tubing to a union body (IDEX, Cat. No. P-760–01). Make a drop-to-drop connection with the sample delivery system (0.2mL/min). After that, disconnect the outlet syringe and close the system. Flush the flow cell with HR buffer at 0.5mL/min for 5min to push the ssDNA molecules into the barriers. Stop the flow, and then incubate for 10min to help ensure that the ssDNA is evenly distributed along the barriers. After this incubation period the flow cell is ready for injection of RPA (see Section 2.3.3).
2.3.3. Using RPA-GFP to Visualize ssDNA
We use GFP- or mCherry-tagged RPA to both fluorescently label the tethered ssDNA molecules and to help remove the ssDNA secondary structure prior to testing the activities of other HR proteins (Gibb et al., 2012). The use of GFP-RPA offers the additional benefit that RPA-ssDNA is the physiological relevant substrate for early steps in HR (Chen & Wold, 2014; Wold, 1997). Here, we outline a typical procedure for labeling and extending double-tethered ssDNA curtains with RPA-GFP or RPA-mCherry. It should be noted that the same procedures can be used with unlabeled RPA, but the ssDNA will not be visible by TIRFM until another fluorescently tagged ssDNA binding protein, such as the mediator protein Rad52, is injected into the sample chamber (Gibb et al., 2014).
The stock solution of S. cerevisiae RPA-GFP taken from the −80°C freezer is directly diluted into 30mL of HR buffer at room temperature. RPA has a nanomolar binding affinity for ssDNA (Wold, 1997), so a final concentration of 0.1nM RPA-GFP is typically sufficient to label and extend the tethered ssDNA molecules.
The RPA-GFP sample is then flushed through the sample chamber containing tethered ssDNA molecules at a rate of 0.8mL/min for approximately 5–10min. The unbound ssDNA is not visible, but will become labeled almost immediately upon injecting RPA-GFP. The ssDNA will initially appear as highly compacted fluorescent foci due to the presence of extensive secondary structure, but the molecules will extend over the course of the RPA injection as this secondary structure is disrupted (Fig. 3B).
Shortly after beginning the RPA buffer flow, a 500μL pulse of 6 M urea is flushed through the sample chamber at a flow rate of 0.8mL/min. This pulse of 6 M urea does not affect the lipid bilayer or disrupt the biotin-streptavidin linkages, and is used to help remove any residual ssDNA secondary structure, ϕ29 DNA polymerase, or M13 circular ssDNA template. The urea is then flushed out of the sample chamber by continuous buffer flow at a flow rate of 0.8mL/min—this high flow rate helps ensure that the RPA-ssDNA molecules will adhere to the downstream anchor points (see Figs. 2C and 3B).
The location and the quality of the RPA-bound ssDNA curtains can now be verified by visual inspection using TIRFM, and once their locations have been determined, the RPA-ssDNA molecules are ready for subsequent experimental analysis.
2.4. Visualizing Srs2 Translocation Activity Using ssDNA Curtains
Here, we provide detailed examples of experimental methods for visualizing the behavior of either unlabeled or GFP-tagged Srs2 as it acts upon Rad51-bound ssDNA, and we also provide methods for visualizing the behavior of GFP- or mCherry-tagged Srs2 as it acts upon ssDNA bound by fluorescent RPA. The experiments described below utilize a prism-type total TIRFM microscope (Nikon) equipped with a 488-nm laser (Coherent Sapphire, 200mW) and a 561-nm laser (Coherent Sapphire, 200mW), which can be used to excite GFP and mCherry fluorescent proteins, respectively.
2.4.1. Visualizing Srs2 as It Acts Upon Rad51-ssDNA
The following procedure describes how to prepare Rad51 filaments and visualize Srs2 translocation beginning with double-tethered ssDNA curtains bound by RPA-GFP (see Section 2.3.3). These assays can be used with unlabeled Srs2, and in this case, the movement of Srs2 along the Rad51-ssDNA can be revealed by the rebinding of fluorescent RPA (Fig. 4A). Alternatively, fluorescently tagged Srs2 can be visualized as fluorescent molecules that bind to and translocate along the Rad51-ssDNA, while the tracts of Rad51 that are removed from the DNA can be concurrently visualized by the reappearance of fluorescent RPA (Fig. 4B).
Fig. 4.
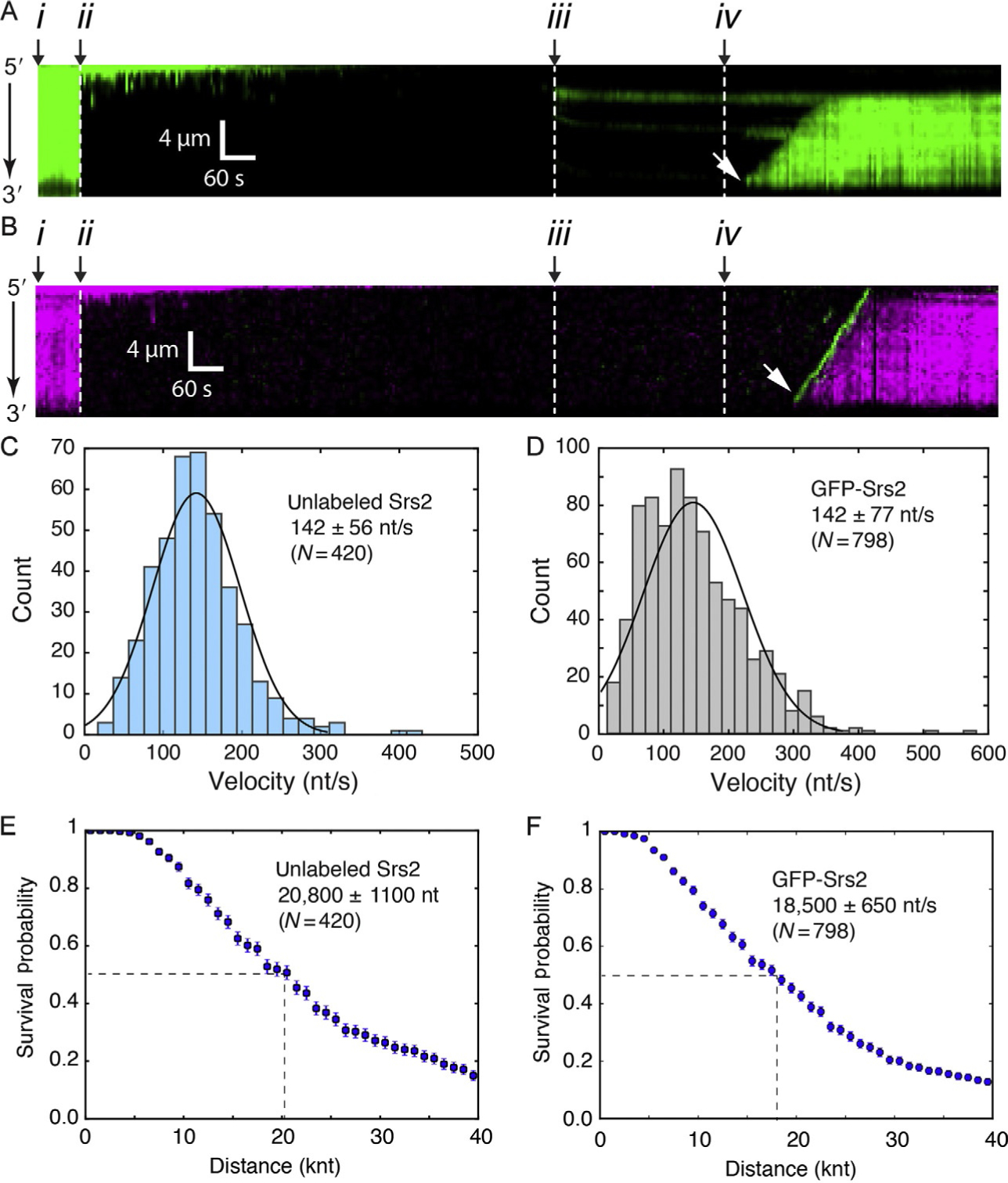
Examples of Srs2 translocation on single Rad51-ssDNA molecules. (A) Examples of kymographs for double-tethered ssDNA curtain assays for Srs2898 and GFP-RPA. (B) Examples of kymographs for double-tethered ssDNA curtain assays for GFP-Srs2898 and mCherry-RPA. In (A) and (B): (i) corresponds to the start of the experiment, in which the ssDNA is coated with fluorescent RPA; (ii) indicates assembly of the Rad51 filaments upon injection of Rad51 and ATP; (iii) free Rad51 is then flushed from the sample chamber; (iv) Srs2 in injected into the sample chamber; and (v) the white arrowheads highlight the beginning of Srs2 translocation events. (C) Velocity distributions for unlabeled Srs2898 acting upon Rad51-ssDNA in the presence of RPA-GFP. (D) Velocity distributions for GFP-Srs2898 acting upon Rad51-ssDNA in the presence of RPA-mCherry. Survival probability of (E) unlabeled Srs2898 and (F) GFP-Srs2898 acting on Rad51-ssDNA. Dashed lines in (E) and (F) highlight the values at which half of the complexes stop translocating. Error bars in the survival probability plots represent s.d. calculated from bootstrap analysis. Panels reproduced with permission from Kaniecki, K., De Tullio, L., Gibb, B., Kwon, Y.- H., Sung, P., & Greene, E. C. (2017). Dissociation of Rad51 presynaptic complexes and heteroduplex DNA joints by tandem assemblies of Srs2. Cell Reports, 21(11), 3166–3177.
Rad51 filament assembly is initiated by injecting 1–2μM S. cerevisiae Rad51 in HR buffer containing 2mM ATP in the absence of RPA. Rad51 filament formation can proceed in the presence of RPA, but is inhibited with increasing concentrations of RPA in solution.
The sample is then incubated in the absence of flow for 10–15min at 32°C to allow for Rad51 filament assembly. If using fluorescent RPA, the Rad51 assembly reaction can be monitored by visual inspection of the RPA-GFP, which will be displaced from the ssDNA upon Rad51 binding.
Buffer flow is then resumed with HR buffer containing 2mM ATP and 100pM RPA-GFP at 0.2mL/min for 3min to flush away any remaining free Rad51. The Rad51 filaments will remain stable so long as ATP is present in the buffer, so the RPA-GFP present in the buffer should not bind to the ssDNA, but rather serves to verify that the Rad51 filaments remain intact.
A 150μL inline loop containing a sample of Srs2 (unlabeled, GFP- or mCherry-tagged, as desired; typically, at a concentration of 100pM) diluted in the same HR buffer flowing through the sample chamber is opened, and the ssDNA molecules are observed by TIRFM while continuously flushing with HR buffer. This procedure results in a short pulse of Srs2 activity, coinciding with the 150μL injection, and free Srs2 will quickly be flushed from the sample chamber by the continuous buffer flow. This procedure restricts the number of Srs2 molecules that bind to the ssDNA during the initial injection, thus facilitating data analysis (see Section 2.5) by helping to ensure that a relatively small number of translocation trajectories will occur on any given ssDNA molecule.
Note that for experiments using the combination of GFP and mCherry, we use a two-color prism-type TIRFM system (Nikon) equipped with a 488-nm laser (Coherent Sapphire, 200mW) for detecting GFP and a 561-nm laser (Coherent Sapphire, 200mW) for detecting mCherry. For visualizing RPA-GFP, we typically use ~20% (~40mW) of the laser power (488 nm) for sample illumination. To detect single translocating mCherry-Srs2 complexes, we typically employ between 50% and 75% (~100–150mW) of the laser power (561nm). To avoid the bleed-through from the green into the red channel during image acquisition, we use a custom-built shuttering system in which the image from the green (GFP) and the red (mCherry) channels are recorded independently, and the green and red images are offset separating the acquisition of each other by 100ms. With this shutter system, when one camera records the red channel image, the green laser is shuttered off, and vice versa.
For some experiments, it may be desirable to utilize dark RPA and dark Rad51. In these situations, the ssDNA will not be visible until bound by fluorescent Srs2, so researchers must rely upon control experiments previously performed using the fluorescent RPA to verify that all of the above procedure work as expected. It is also helpful in these cases to use ssDNA prepared from a larger RCR reaction that has been aliquoted and flash frozen and stored at −80°C (see Section 2.2.3). Use of the frozen stock helps to avoid any batch-to-batch variability in the RCR ssDNA preparation, thereby ensuring that more reliable amounts of ssDNA are present on the flow cell surface.
2.4.2. Visualizing Srs2 Binding and Translocation on RPA-ssDNA
The following procedure describes how to visualize Srs2 translocation on double-tethered ssDNA curtains bound by RPA-GFP (i.e., in the absence of Rad51). Again, this procedure begins with the RPA-ssDNA curtains that are described in Section 2.3.3. With this procedure, fluorescently tagged Srs2 can be visualized as fluorescent molecules that bind to and translocate along the fluorescent RPA-ssDNA (Fig. 5A). Srs2 removes RPA from the ssDNA, and the displaced RPA is continuously replenished by the free RPA in the sample buffer. However, as noted below, the experiment can also be modified to monitor the removal of fluorescent RPA from the ssDNA (Fig. 5B).
Fig. 5.
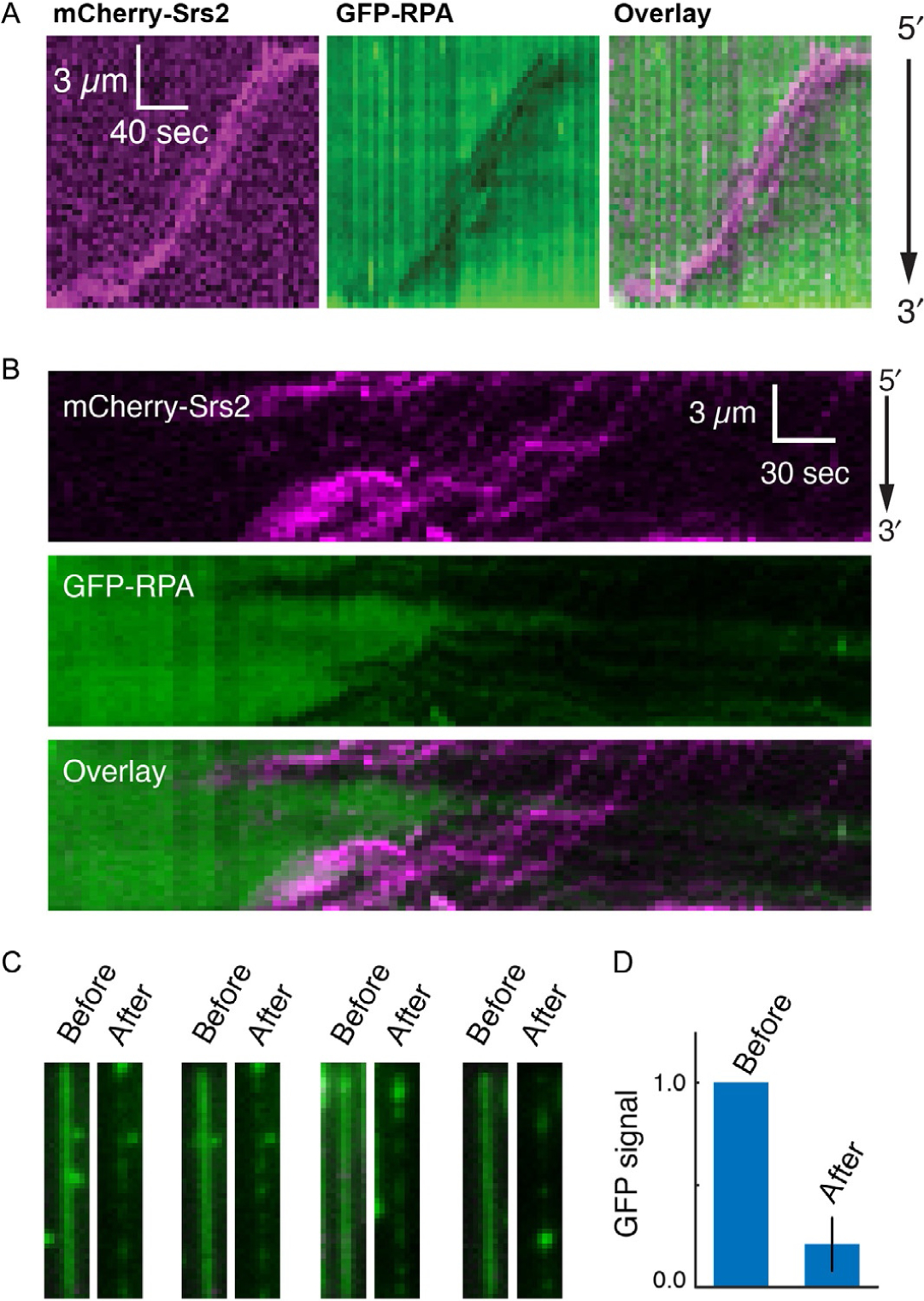
Examples of Srs2 translocation on single molecules of RPA-ssDNA. (A) Kymographs demonstrating the 3′ → 5′ movement of mCherry-Srs2 (magenta) along GFP-RPA-bound ssDNA (green). The orientation of the ssDNA is indicated, and the images show the mCherry-Srs2 fluorescence signal, the GFP-RPA fluorescence signal, and the overlaid fluorescence signals, as indicated. (B) mCherry-Srs2 translocation on GFP-RPA-bound ssDNA when free GFP-RPA is absent from solution. The three kymographs show the mCherry-Srs2 fluorescence signal (top), the GFP-RPA signal (middle), and the overlaid images (bottom), as indicated. (C) Images of GFP-RPA-bound ssDNA before and after injection of mCherry-Srs2. (D) Fractional loss of normalized GFP-RPA signal (integrated over entire ssDNA molecules; N =20) due to the action of Srs2. Panels reproduced with permission from De Tullio, L., Kaniecki, K., Kwon, Y.- H., Crickard, J. B., Sung, P., & Greene, E. C. (2017). Yeast Srs2 helicase promotes redistribution of single-stranded DNA-bound RPA and Rad52 in homologous recombination regulation. Cell Reports, 21(3), 570–577.
Experiments for mCherry-Srs2 translocation on RPA-GFP-bound ssDNA are performed in HR buffer (30mM Tris–Ac [pH 7.5], 50mM KCl, 5mM MgAc, 1mM DTT, 0.3mg/mL BSA, and 2mM ATP), and reactions are performed in flow cells maintained at 32°C, as described (Ma et al., 2017). Note that in this particular protocol, RPA-GFP is maintained in the buffer at a concentration of 100pM, which ensures that the ssDNA is visible throughout the experiment.
For experiments using the combination of GFP and mCherry, we use the two-color prism-type TIRFM system (Nikon) equipped with a 488-nm laser (Coherent Sapphire, 200mW) for detecting GFP, a 561-nm laser (Coherent Sapphire, 200mW) for detecting mCherry, and the custom-built shuttering system to eliminate signal bleed-through (see Section 2.4.1).
To visualize mCherry-Srs2 binding and translocation, inject a 150μL aliquot of mCherry-Srs2 (typically 100pM) in HR buffer while maintaining a constant flow rate of 0.2mL/min. This procedure results in a 45-s injection window during which mCherry-Srs2 can bind to the RPA-ssDNA molecules; after this time period the HR buffer behind the injection loop does not contain any Srs2. Therefore, any unbound mCherry-Srs2 will be flushed from the sample chamber. This produce helps minimize the number of mCherry-Srs2 binding events, which makes interpretation of the resulting data much easier (see Section 2.5).
Begin recording data ~2min before mCherry-Srs2 enters the sample chamber, and continue data collection for a total of 20min (300 frames total). For a typical experiment, data can be collected at a rate of 1 frame per 4s with 100ms integration time, and the laser should be shuttered between each acquired image to minimize photobleaching. These acquisition rates can be adjusted as deemed necessary by the user.
As an alternative to the procedure described above, one can also assay the ability of mCherry-Srs2 to remove RPA-GFP from the ssDNA (Fig. 5B–D). For this, RPA-GFP is simply omitted from the HR buffer that is flushed through the sample chamber following the 150μL injection of mCherry-Srs2. It should however be noted that the ssDNA can readily break when RPA is absent, making these types measurements more challenging.
2.5. Data Analysis
In the following sections, we describe general procedures for analyzing Srs2 data from ssDNA curtain experiments using measurements of GFP-Srs2 translocating on Rad51-ssDNA in the presence of free RPA-mCherry as an example (see Fig. 4B). Similar procedures can be used to analyze the behavior of dark Srs2 on Rad51-ssDNA in the presence of RPA-GFP (see Fig. 4A), or mCherry-Srs2 on RPA-ssDNA (see Fig. 5), and pertinent details of these procedures are also highlighted below. The general work flow for these analysis procedures is shown in Fig. 6.
Fig. 6.
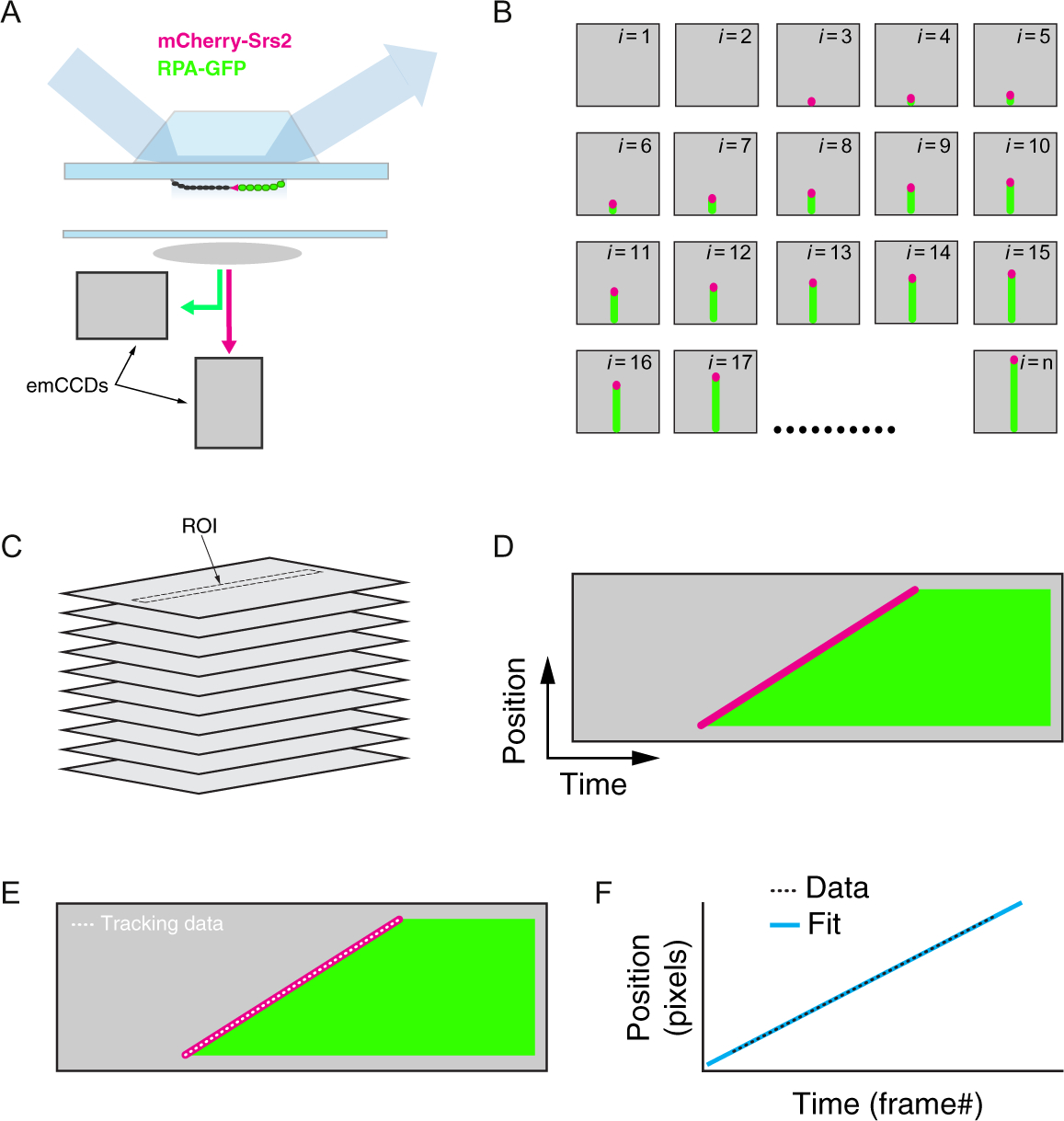
Work flow for data analysis procedures for two-color analysis of Srs2 trajectories. (A) Schematic illustration of the TIRFM system for analysis of Srs2-mCherry (shown in magenta) translocation on dark Rad51-ssDNA filaments (shown in black) in the presence of RPA-GFP (shown in green); color coding for mCherry-Srs2 (magenta) and RPA-GFP (green) is the same throughout the figure. (B) Cartoon illustration of a hypothetic set of raw TIFF files representing the initial binding of Srs2-mCherry to the Rad51-ssDNA (beginning in frame i =1) and following its movement along the ssDNA over the course of an entire experiment (ending in frame i = n). (C) The raw TIFF files are then used to generate a tiff stack (i.e., a movie). (D) Two-color kymographs are then extracted from these movies with NIH ImageJ based upon user-defined ROIs (regions of interest), which represent 1-pixel-wide areas encompassing individual ssDNA molecules from within the ssDNA curtain. (E) The NeuronJ plugin in NIH ImageJ is then used to track the movement of Srs2 along the ssDNA. (F) The resulting x,y values in pixels are converted into nucleotides and time, respectively, and linear fits to the data are used to determine translocation velocities. For each movie, steps (D–F) are repeated until all of the Srs2 trajectories have been analyzed.
2.5.1. Generating Kymographs From Wide-Field Images
All TIRFM data are collected as raw TIFF files using Nikon NIS Elements software, and then these raw TIFF files are converted into kymographs for data analysis (Fig. 6A–D).
First, the raw TIFF files are used to create a tiff stack (i.e., movie) showing the entire field of view using Fiji software (ImageJ 1.48b, Wayne Rasband; National Institutes of Health, USA) (Fig. 6B and C).
The resulting tiff stack is then used to generate kymographs that illustrate the events which take place on individual ssDNA molecules during the experimental observation period (Fig. 6D). Kymographs are generated using the “Reslice” function in Fiji. For this, a 1-pixel-wide region of interest (ROI) is superimposed on a selected ssDNA molecule from within the tiff stack, and the corresponding information for every image within the entire tiff stack is compiled as single kymograph (e.g., Figs. 4A, B, 5A, B, and 6D). Within each kymograph, the y-axis reflects the spatial information along the length of the ssDNA, and x-axis represents time.
The resulting kymographs can then be used to determine the sites at which Srs2 initiates translocation, translocation velocities (Fig. 4C and D), and the processivity of each translocating protein (Fig. 4E and F).
2.5.2. Analysis of Srs2 Translocation Trajectories
All analyses of Srs2 translocation are performed from the kymographs that are generated as described above.
Individual dark Srs2 trajectories were manually analyzed by selecting the starting and ending point of a “wedge” of fluorescent RPA (e.g., see Fig. 4A). These wedges are evaluated as linear traces to estimate velocity, whereas fluorescent Srs2 trajectories are tracked in NIH ImageJ, as described below.
To analyze fluorescently tagged Srs2 trajectories (e.g., Fig. 4B), we use the NIH ImageJ plugin NeuronJ (Meijering et al., 2004). NeuronJ is a semiautomated algorithm that can be used to define and track contiguous changes in signal intensity, also called “ridges” or “ridge pixels,” in a two-dimensional image. In the case of GFP- or mCherry-tagged Srs2, the ridges are the bright fluorescence signals observed for the moving molecules of Srs2 (e.g., see Figs. 4B and 6E).
NeuronJ processes 8-bit-type images. So before starting, the kymographs are transformed from 16-bit to 8-bit images. The user defines a starting and end points within the image, and then the program defines and tracks the ridge pixels, and records the x,y-coordinates of each trajectory (Fig. 6F). For our analysis, we define the end points as either when the Srs2 signal disappears (i.e., dissociation) or when the slope of the Srs2 trajectory reaches a plateau (i.e., Srs2 stalls). In the case of Srs2 kymographs, the output for the y-axis represents distance in pixels. The output for the x-coordinate is in pixels, and each pixel represents a single frame from the original tiff stack from which the kymograph was constructed.
The x-coordinate values are readily converted from pixel values to units of time. For instance, assuming that data are collected using 5s shuttering, then a kymograph that started at x =10 (where x corresponds to the x-axis pixel value) and ending when x = 20 would have activity that occurred over a 50-s time period.
The y-coordinate pixel values are converted into nucleotide values based upon the estimated lengths of the protein-bound ssDNA substrates. These calculated estimates will differ for Rad51-ssDNA and RPA-ssDNA, which are anticipated to be extended to differing lengths (Qi et al., 2015). For Rad51-ssDNA, this corresponds to ~725 nucleotides (nt) of Rad51-ssDNA per pixel. This calculation was made based upon the observation that dsDNA with homology to a single location within a single repetition of M13mp18 ssDNA displayed peak-to-peak distances of 2.7 μm. A single repetition of M13mp18 ssDNA is 7249nt and each pixel within our 60× objective is 0.27 μm. Thus, there are 7249nt of Rad51-ssDNA within 10 pixels. For RPA-ssDNA, we assumed that 1 pixel corresponds to ~1087 nucleotides of ssDNA-RPA filament. This value is based on the observation that the length of ssDNA is 1.5× times shorter when is bound by RPA than when it is bound to Rad51 (Gibb et al., 2014). Therefore, it must be emphasized that the apparent velocities when converted from pixels/s to nt/s values will depend upon these assumed contour lengths of the extended Rad51-ssDNA and RPA-ssDNA complexes within the ssDNA curtains.
Once these conversions are completed, translocation velocity (in nt/s) can be calculated from the kymographs based on the slope of each Srs2 trajectory (Fig. 6F), which is readily obtained by fitting the tracking data to a linear equation. The resulting data can be presented as distribution histograms to obtain the mean and standard deviation for the Srs2 translocation velocities under any given experimental condition (e.g., Fig. 4C and D).
Srs2 processivity can also be defined from the same analysis of the kymographs (e.g., Fig. 4E and F). The distance of each translocation event is defined as the total length in nucleotides from each initial Srs2 binding position to the end of the translocation trajectory and defined by the location where Srs2 either dissociated from the ssDNA, photobleached, or stopped moving. The resulting values are used to generate survival probability plots, where the apparent processivity values reflect the distance at which 50% of the Srs2 complexes dissociate, photobleach, or stop moving. Note that the reported error bars for the survival probability correspond to standard deviation calculated by bootstrap analysis using a custom Python script that has been reported elsewhere (Qi & Greene, 2016).
3. CONCLUSIONS AND FUTURE DIRECTIONS
Helicases play crucial roles in all aspects of nucleic acid metabolism, and mutations in these important motor proteins can give rise to severe genetic disorders and cancer-prone syndromes (Bernstein et al., 2010; Branzei & Szakal, 2017; Brosh, 2013). Here, we have described assays that can be used to visualize the behaviors of the S. cerevisiae helicase and antirecombinase Srs2 as it acts upon long ssDNA substrates bound by either Rad51 or RPA. We anticipate that relatively simple modifications of the procedures described here will allow these protocols to be applied to many other types of helicases and motor proteins that act upon ssDNA. We can also envision some technical improvements that may increase the utility of this approach. In particular, the long ssDNA molecules used in these assays are inherently more challenging to work with than dsDNA, which is a relatively stiff molecule that is well behaved in flow. Therefore, although we can directly measure distances in micron or pixels, we can only estimate the lengths (in nucleotides) of the ssDNA molecules under observation. One way to overcome this problem may be to include fluorescent fiduciary markers at known locations along the ssDNA, which would allow for more accurately measure the lengths of Rad51-ssDNA and RPA-ssDNA rather than relying upon length estimates. In addition, the methodologies described here for tracking Srs2 movement rely upon relatively simple tracking procedure found in the NIH ImageJ plugin NeuronJ. This approach is fast and relatively simple, so it is very suitable for many types of investigations. However, it cannot be used to decipher high-resolution features of the trajectories—for instance, we can clearly see examples of pauses and changes in velocity in the Srs2 kymographs. Analysis of these detailed features would require implementation of a more intensive particle tracking algorithm that could be utilized for tracking the progress of GFP- or mCherry-tagged Srs2.
ACKNOWLEDGMENTS
We thank J. Brooks Crickard and Upasana Roy for comments on the manuscript. This research was funded by an NIH grant R35GM118026 (E.C.G.). L.D.T. was funded by a PEW Latin American postdoctoral fellowship, the Williams Foundation, and by a program for Assistant Researchers, CONICET, Argentina.
REFERENCES
- Antony E, Tomko EJ, Xiao Q, Krejci L, Lohman TM, & Ellenberger T (2009). Srs2 disassembles Rad51 filaments by a protein-protein interaction triggering ATP turnover and dissociation of Rad51 from DNA. Molecular Cell, 35, 105–115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Axelrod D (1989). Total internal reflection fluorescence microscopy. Methods in Cell Biology, 30, 245–270. [DOI] [PubMed] [Google Scholar]
- Bernstein KA, Gangloff S, & Rothstein R (2010). The RecQ DNA helicases in DNA repair. Annual Review of Genetics, 44, 393–417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bianco PR, Brewer LR, Corzett M, Balhorn R, Yeh Y, Kowalczykowski SC, et al. (2001). Processive translocation and DNA unwinding by individual RecBCD enzyme molecules. Nature, 409, 374–378. [DOI] [PubMed] [Google Scholar]
- Bianco PR, Tracy RB, & Kowalczykowski SC (1998). DNA strand exchange proteins: A biochemical and physical comparison. Frontiers in Bioscience, 3, D570–D603. [DOI] [PubMed] [Google Scholar]
- Branzei D, & Szakal B (2017). Building up and breaking down: Mechanisms controlling recombination during replication. Critical Reviews in Biochemistry and Molecular Biology, 52, 381–394. [DOI] [PubMed] [Google Scholar]
- Brosh RM Jr. (2013). DNA helicases involved in DNA repair and their roles in cancer. Nature Reviews. Cancer, 13, 542–558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burgess RC, Lisby M, Altmannova V, Krejci L, Sung P, & Rothstein R (2009). Localization of recombination proteins and Srs2 reveals anti-recombinase function in vivo. The Journal of Cell Biology, 185, 969–981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen R, & Wold MS (2014). Replication protein A: Single-stranded DNA’s first responder: Dynamic DNA-interactions allow replication protein A to direct single-strand DNA intermediates into different pathways for synthesis or repair. Bioessays, 36, 1156–1161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Comstock MJ, Whitley KD, Jia H, Sokoloski J, Lohman TM, Ha T, et al. (2015). Protein structure. Direct observation of structure-function relationship in a nucleic acid-processing enzyme. Science, 348, 352–354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Croteau DL, Popuri V, Opresko PL, & Bohr VA (2014). Human RecQ helicases in DNA repair, recombination, and replication. Annual Review of Biochemistry, 83, 519–552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Tullio L, Kaniecki K, Kwon Y-H, Crickard JB, Sung P, & Greene EC (2017). Yeast Srs2 helicase promotes redistribution of single-stranded DNA-bound RPA and Rad52 in homologous recombination regulation. Cell Reports, 21, 570–577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duzdevich D, Warner MD, Ticau S, Ivica NA, Bell SP, & Greene EC (2015). The dynamics of eukaryotic replication initiation: Origin specificity, licensing, and firing at the single-molecule level. Molecular Cell, 58, 483–494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fazio T, Visnapuu ML, Wind S, & Greene EC (2008). DNA curtains and nanoscale curtain rods: High-throughput tools for single molecule imaging. Langmuir, 24, 10524–10531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gibb B, Silverstein TD, Finkelstein IJ, & Greene EC (2012). Single-stranded DNA curtains for real-time single-molecule visualization of protein-nucleic acid interactions. Analytical Chemistry, 84, 7607–7612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gibb B, Ye LF, Kwon Y, Niu H, Sung P, & Greene EC (2014). Protein dynamics during presynaptic-complex assembly on individual single-stranded DNA molecules. Nature Structural & Molecular Biology, 21, 893–900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gorman J, Fazio T, Wang F, Wind S, & Greene EC (2010). Nanofabricated racks of aligned and anchored DNA substrates for single-molecule imaging. Langmuir, 26, 1372–1379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gorman J, Plys AJ, Visnapuu ML, Alani E, & Greene EC (2010). Visualizing one-dimensional diffusion of eukaryotic DNA repair factors along a chromatin lattice. Nature Structural & Molecular Biology, 17, 932–938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gorman J, Wang F, Redding S, Plys AJ, Fazio T, Wind S, et al. (2012). Single-molecule imaging reveals target-search mechanisms during DNA mismatch repair. Proceedings of the National Academy of Sciences of the United States of America, 109, E3074–E3083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graneli A, Yeykal CC, Prasad TK, & Greene EC (2006). Organized arrays of individual DNA molecules tethered to supported lipid bilayers. Langmuir, 22, 292–299. [DOI] [PubMed] [Google Scholar]
- Greene EC, Wind S, Fazio T, Gorman J, & Visnapuu ML (2010). DNA curtains for high-throughput single-molecule optical imaging. Methods in Enzymology, 472, 293–315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heyer WD (2015). Regulation of recombination and genomic maintenance. Cold Spring Harbor Perspectives in Biology, 7, a016501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heyer WD, Ehmsen KT, & Liu J (2010). Regulation of homologous recombination in eukaryotes. Annual Review of Genetics, 44, 113–139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Honda M, Park J, Pugh RA, Ha T, & Spies M (2009). Single-molecule analysis reveals differential effect of ssDNA-binding proteins on DNA translocation by XPD helicase. Molecular Cell, 35, 694–703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ira G, Malkova A, Liberi G, Foiani M, & Haber JE (2003). Srs2 and Sgs1-Top3 suppress crossovers during double-strand break repair in yeast. Cell, 115, 401–411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jasin M, & Rothstein R (2013). Repair of strand breaks by homologous recombination. Cold Spring Harbor Perspectives in Biology, 5, a012740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaniecki K, De Tullio L, Gibb B, Kwon Y-H, Sung P, & Greene EC (2017). Dissociation of Rad51 presynaptic complexes and heteroduplex DNA joints by tandem assemblies of Srs2. Cell Reports, 21, 3166–3177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kass EM, Moynahan ME, & Jasin M (2016). When genome maintenance goes badly awry. Molecular Cell, 62, 777–787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klein HL (2001). Mutations in recombinational repair and in checkpoint control genes suppress the lethal combination of srs2Delta with other DNA repair genes in Saccharomyces cerevisiae. Genetics, 157, 557–565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kowalczykowski SC (2015). An overview of the molecular mechanisms of recombinational DNA repair. Cold Spring Harbor Perspectives in Biology, 7, a016410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krejci L, Van Komen S, Li Y, Villemain J, Reddy MS, Klein H, et al. (2003). DNA helicase Srs2 disrupts the Rad51 presynaptic filament. Nature, 423, 305–309. [DOI] [PubMed] [Google Scholar]
- Lee JY, Finkelstein IJ, Arciszewska LK, Sherratt DJ, & Greene EC (2014). Single-molecule imaging of FtsK translocation reveals mechanistic features of protein-protein collisions on DNA. Molecular Cell, 54, 832–843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee JY, Terakawa T, Qi Z, Steinfeld JB, Redding S, Kwon Y, et al. (2015). DNA recombination. Base triplet stepping by the Rad51/RecA family of recombinases. Science, 349, 977–981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lisby M, Barlow JH, Burgess RC, & Rothstein R (2004). Choreography of the DNA damage response: Spatiotemporal relationships among checkpoint and repair proteins. Cell, 118, 699–713. [DOI] [PubMed] [Google Scholar]
- Lisby M, & Rothstein R (2015). Cell biology of mitotic recombination. Cold Spring Harbor Perspectives in Biology, 7, a016535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu J, Ede C, Wright WD, Gore SK, Jenkins SS, Freudenthal BD, et al. (2017). Srs2 promotes synthesis-dependent strand annealing by disrupting DNA polymerase delta-extending D-loops. eLife, 6, e22195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma CJ, Steinfeld JB, & Greene EC (2017). Single-stranded DNA curtains for studying homologous recombination. Methods in Enzymology, 582, 193–219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malkova A, & Haber JE (2012). Mutations arising during repair of chromosome breaks. Annual Review of Genetics, 46, 455–473. [DOI] [PubMed] [Google Scholar]
- Marini V, & Krejci L (2010). Srs2: The “Odd-Job Man” in DNA repair. DNA Repair, 9, 268–275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mazon G, Mimitou EP, & Symington LS (2010). SnapShot: Homologous recombination in DNA double-strand break repair. Cell, 142, 646. [DOI] [PubMed] [Google Scholar]
- Meijering E, Jacob M, Sarria JC, Steiner P, Hirling H, & Unser M (2004). Design and validation of a tool for neurite tracing and analysis in fluorescence microscopy images. Cytometry. Part A, 58, 167–176. [DOI] [PubMed] [Google Scholar]
- Moynahan ME, & Jasin M (2010). Mitotic homologous recombination maintains genomic stability and suppresses tumorigenesis. Nature Reviews Molecular Cell Biology, 11, 196–207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Myong S, Rasnik I, Joo C, Lohman TM, & Ha T (2005). Repetitive shuttling of a motor protein on DNA. Nature, 437, 1321–1325. [DOI] [PubMed] [Google Scholar]
- Niu H, & Klein HL (2017). Multifunctional roles of Saccharomyces cerevisiae Srs2 protein in replication, recombination and repair. FEMS Yeast Research, 17, fow111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palladino F, & Klein HL (1992). Analysis of mitotic and meiotic defects in Saccharomyces cerevisiae SRS2 DNA helicase mutants. Genetics, 132, 23–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paques F, & Haber JE (1999). Multiple pathways of recombination induced by double-strand breaks in Saccharomyces cerevisiae. Microbiology and Molecular Biology Reviews, 63, 349–404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park J, Myong S, Niedziela-Majka A, Lee KS, Yu J, Lohman TM, et al. (2010). PcrA helicase dismantles RecA filaments by reeling in DNA in uniform steps. Cell, 142, 544–555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petrova V, Chen SH, Molzberger ET, Tomko E, Chitteni-Pattu S, Jia H, et al. (2015). Active displacement of RecA filaments by UvrD translocase activity. Nucleic Acids Research, 43, 4133–4149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prakash R, Zhang Y, Feng W, & Jasin M (2015). Homologous recombination and human health: The roles of BRCA1, BRCA2, and associated proteins. Cold Spring Harbor Perspectives in Biology, 7, a016600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qi Z, & Greene EC (2016). Visualizing recombination intermediates with single-stranded DNA curtains. Methods, 105, 62–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qi Z, Redding S, Lee JY, Gibb B, Kwon Y, Niu H, et al. (2015). DNA sequence alignment by microhomology sampling during homologous recombination. Cell, 160, 856–869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qiu Y, Antony E, Doganay S, Koh HR, Lohman TM, & Myong S (2013). Srs2 prevents Rad51 filament formation by repetitive motion on DNA. Nature Communications, 4, 2281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Redding S, Sternberg SH, Marshall M, Gibb B, Bhat P, Guegler CK, et al. (2015). Surveillance and processing of foreign DNA by the Escherichia coli CRISPR-Cas system. Cell, 163, 854–865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rong L, & Klein HL (1993). Purification and characterization of the SRS2 DNA helicase of the yeast Saccharomyces cerevisiae. The Journal of Biological Chemistry, 268, 1252–1259. [PubMed] [Google Scholar]
- Rong L, Palladino F, Aguilera A, & Klein HL (1991). The hyper-gene conversion hpr5–1 mutation of Saccharomyces cerevisiae is an allele of the SRS2/RADH gene. Genetics, 127, 75–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sasanuma H, Furihata Y, Shinohara M, & Shinohara A (2013). Remodeling of the Rad51 DNA strand-exchange protein by the Srs2 helicase. Genetics, 194, 859–872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silverstein TD, Gibb B, & Greene EC (2014). Visualizing protein movement on DNA at the single-molecule level using DNA curtains. DNA Repair (Amst), 20, 94–109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simandlova J, Zagelbaum J, Payne MJ, Chu WK, Shevelev I, Hanada K, et al. (2013). FBH1 helicase disrupts RAD51 filaments in vitro and modulates homologous recombination in mammalian cells. The Journal of Biological Chemistry, 288, 34168–34180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sokoloski JE, Kozlov AG, Galletto R, & Lohman TM (2016). Chemo-mechanical pushing of proteins along single-stranded DNA. Proceedings of the National Academy of Sciences of the United States of America, 113, 6194–6199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spies M, Bianco PR, Dillingham MS, Handa N, Baskin RJ, & Kowalczykowski SC (2003). A molecular throttle: The recombination hotspot chi controls DNA translocation by the RecBCD helicase. Cell, 114, 647–654. [DOI] [PubMed] [Google Scholar]
- Sternberg SH, Redding S, Jinek M, Greene EC, & Doudna JA (2014). DNA interrogation by the CRISPR RNA-guided endonuclease Cas9. Nature, 507, 62–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sung P, & Stratton SA (1996). Yeast Rad51 recombinase mediates polar DNA strand exchange in the absence of ATP hydrolysis. The Journal of Biological Chemistry, 271, 27983–27986. [DOI] [PubMed] [Google Scholar]
- Symington LS, Rothstein R, & Lisby M (2014). Mechanisms and regulation of mitotic recombination in Saccharomyces cerevisiae. Genetics, 198, 795–835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vasianovich Y, Altmannova V, Kotenko O, Newton MD, Krejci L, & Makovets S (2017). Unloading of homologous recombination factors is required for restoring double-stranded DNA at damage repair loci. The EMBO Journal, 36, 213–231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Veaute X, Jeusset J, Soustelle C, Kowalczykowski SC, Le Cam E, & Fabre F (2003). The Srs2 helicase prevents recombination by disrupting Rad51 nucleoprotein filaments. Nature, 423, 309–312. [DOI] [PubMed] [Google Scholar]
- Wang F, Redding S, Finkelstein IJ, Gorman J, Reichman DR, & Greene EC (2013). The promoter-search mechanism of Escherichia coli RNA polymerase is dominated by three-dimensional diffusion. Nature Structural & Molecular Biology, 20, 174–181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wold MS (1997). Replication protein A: A heterotrimeric, single-stranded DNA-binding protein required for eukaryotic DNA metabolism. Annual Review of Biochemistry, 66, 61–92. [DOI] [PubMed] [Google Scholar]
- Zacharias DA, Violin JD, Newton AC, & Tsien RY (2002). Partitioning of lipid-modified monomeric GFPs into membrane microdomains of live cells. Science, 296, 913–916. [DOI] [PubMed] [Google Scholar]