

Abstract
Background and Purpose
Roflupram improves cognition and limits neuroinflammation in the brain. However, the beneficial effects of roflupram on Parkinson's disease (PD) remain unknown. Therefore, we aimed to elucidate the pharmacological effects and mechanisms of action of ROF in experimental models of PD.
Experimental Approach
We used an in vitro PD model of SH‐SY5Y cells exposed to 1‐methyl‐4‐phenylpyridinium iodide (MPP+). Cell viability and apoptosis were analysed via the MTT assay and flow cytometry. Mitochondrial morphology, mitochondrial respiratory capacity, and ROS were measured by a mitochondrial tracker, Seahorse Analyzer, and a MitoSOX‐Red dye. For in vivo PD model, behavioural tests, Nissl staining, and immunohistochemistry were used to evaluate protection by roflupram. The levels of TH, cAMP response element‐binding protein (CREB), and PPARγ coactivator‐1α (PGC‐1α) were analysed by western blotting.
Key Results
Roflupram decreased MPP+‐induced apoptosis in SH‐SY5Y cells and human dopaminergic neurons. Roflupram also increased mitochondrial respiratory capacity, decreased ROS production, and restored mitochondrial morphology. Roflupram reversed the MPP+‐induced reductions of phosphorylated CREB, PGC‐1α and TH. These protective effects were blocked by the PKA inhibitor H‐89 or by PGC‐1α siRNA. In mice treated with MPTP, roflupram significantly improved motor functions. Roflupram prevented both dopaminergic neuronal loss and the reduction of phosphorylated CREB and PGC‐1α in the substantia nigra and striatum.
Conclusion and Implications
Roflupram protected dopaminergic neurons from apoptosis via the CREB/PGC‐1α pathway in PD models. Hence, roflupram has potential as a protective drug in the treatment of PD.
Abbreviations
- CREB
cAMP response element‐binding protein
- FCCP
carbonyl cyanide 4‐(trifluoromethoxy)phenylhydrazone
- MMP
mitochondrial membrane potential
- MPP+
1‐methyl‐4‐phenylpyridinium iodide
- MTT
3‐(4,5‐dimethylthiazol‐2‐yl)‐2,5‐diphenyltetrazolium bromide
- MPTP
1‐methyl‐4‐phenyl‐1,2,3,6‐tetrahydropyridine
- OCR
oxygen consumption rate
- PD
Parkinson's disease
- PGC‐1α
PPARγ coactivator‐1α
- SN
substantia nigra
- TMRE
tetramethylrhodamine ethyl ester perchlorate
What is already known
Roflupram is a PDE4 inhibitor with low emetic potential and excellent blood–brain barrier permeability.
Roflupram reverses cognition deficit in APP/PS1 mice and inhibits neuroinflammation by inducing autophagy.
What does this study add
Roflupram protects dopaminergic neurons from apoptosis and ameliorates motor deficits in models of Parkinson's disease.
Roflupram improves mitochondrial function by activating CREB and increasing PGC‐1α expression.
What is the clinical significance
Roflupram may be clinically effective in preventing and treating Parkinson's disease.
1. INTRODUCTION
Parkinson's disease (PD) is the second most prevalent neurodegenerative disease in the world. The features of PD include loss of dopaminergic neurons in the substantia nigra (SN), formation of Lewy bodies, overproduction of ROS, and mitochondrial dysfunction (Migdalska‐Richards & Schapira, 2016). These pathological features lead to clinical manifestations of static tremor, bradykinesia, myotonia, and abnormal posture in patients with PD (Jankovic, 2008). Currently available drugs cannot prevent the PD‐induced degenerative process of dopaminergic neurons in the SN (Dauer & Przedborski, 2003), and they usually produce intolerable side effects. For example, after several years of https://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=3639 therapy, patients become progressively more disabled due to the appearance of motor fluctuations and abnormal involuntary movements (Picconi, Hernandez, Obeso, & Calabresi, 2018). Additionally, levodopa may further promote neuronal cell death due to the overproduction of ROS (Kostrzewa, Kostrzewa, & Brus, 2002). Other drugs, such as dopaminergic agonists, can also produce a range of side effects, including gastrointestinal reactions and psychological effects (Stocchi, Torti, & Fossati, 2016). Therefore, there are still criticisms and unmet needs for antiparkinsonian therapy. As such, it is necessary to design and discover novel compounds with greater efficacy and safety to treat PD.
The https://www.guidetopharmacology.org/GRAC/FamilyDisplayForward?familyId=260 enzymes constitute a group within the family of phosphodiesterases hydrolysing https://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=2352. Inhibition of PDE4 increases levels of cAMP, thereby activating https://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=1694&familyId=284&familyType=ENZYME and the cAMP response element‐binding protein (CREB). CREB is a transcription factor that promotes the transcription of downstream targets, including PPARγ coactivator‐1α (https://www.guidetoimmunopharmacology.org/GRAC/ObjectDisplayForward?objectId=593; Wu et al., 2006). We have previously demonstrated a protective role of PDE4 inhibitors in several neurological disease models (Zhong et al., 2018; Zhong et al., 2019). Regarding the role of PDE4 inhibitors in the treatment of PD, it has been reported that inhibition of PDE4 protects dopaminergic neurons and prevents deficits of spatial working memory in patients with PD (Niccolini et al., 2017; Yang, Calingasan, Lorenzo, & Beal, 2008), but its mechanism of action remains unclear. Moreover, clinical use of existing PDE4 inhibitors are mostly limited by their serious side effects, such as nausea and vomiting (Richter, Menniti, Zhang, & Conti, 2013). For example, in Phase II clinical trials for the treatment of depression, the canonical PDE4 inhibitor https://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=5260 was shown to induce intolerable nausea and vomiting (Dyke & Montana, 2002). Roflupram (also known as Zl‐n‐91 or FFPM) is a selective PDE4 inhibitor (Zheng et al., 2008), and its chemical structure is shown in Figure 1a. This compound is a potent PDE4 inhibitor with higher selectivity for https://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=13004, https://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=13012, and https://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=13034 than that shown by rolipram (Guo et al., 2017). Additionally, roflupram penetrates the blood–brain barrier after oral administration and induces little or no emetic response (Guo et al., 2017). We have previously found that roflupram reverses cognitive deficits in amyloid precursor protein/presenilin 1 (APP/PS1) transgenic mice through activation of the cAMP/PKA/CREB pathway and via suppression of inflammasome activation (Guo et al., 2017; You et al., 2017). These data indicated that roflupram had potential for the treatment of other neurodegenerative diseases. However, the potential protective effects of roflupram in PD models is largely unknown.
Figure 1.
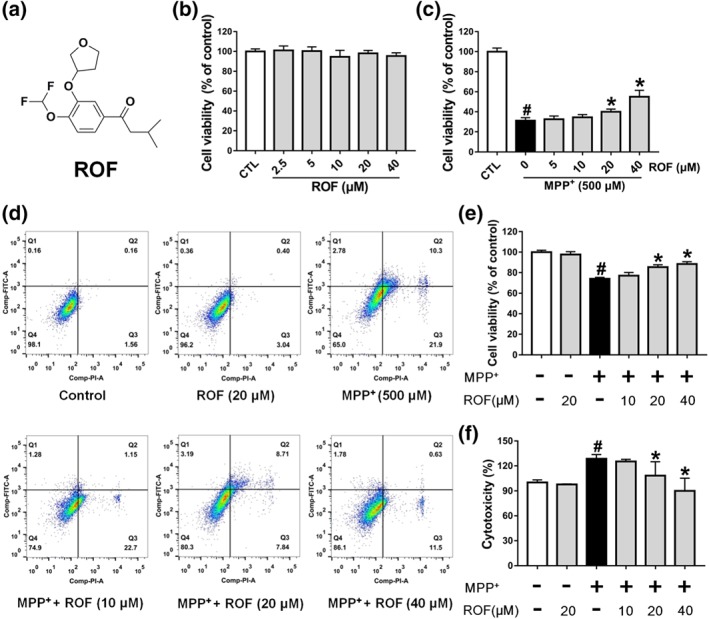
Roflupram increases cell viability of MPP+‐induced cells. (a) The chemical structure of roflupram (ROF). (b) SH‐SY5Y cells were treated with various concentrations of roflupram for 48 hr to evaluate the cytotoxic effects on cells. (c) The cells were pretreated with different concentrations of roflupram for 1 hr, and then treated with 500 μM of MPP+ for 48 hr to measure cell viability with MTT assay. (d and e) The cells were pretreated with various concentrations of roflupram 1 hr earlier and were then exposed to 500 μM of MPP+ for 24 hr; cell viability was measured using flow cytometry. Representative graphs of flow cytometric outputs for each group are shown in (d), and statistical analysis of flow cytometry is presented in (e). (f) The cells were treated with various concentrations of roflupram for 1hr and were then stimulated with 500 μM of MPP+ for 48 hr to measure LDH activity in the medium. Data are presented as mean ± SD (n = 5) and represent five independent experiments. # P < .05, significantly different from control group. *P < .05, significantly different from MPP+‐treated group
Activated CREB enhances the expression of PGC‐1α (Herzig et al., 2001) and PGC‐1α plays a pivotal role in regulating mitochondrial biogenesis and resistance to ROS (Ciron et al., 2015; St‐Pierre et al., 2006). Transgenic mice over‐expressing PGC‐1α in dopaminergic neurons are more resistant to neurotoxicity induced by 1‐methyl‐4‐phenyl‐1,2,3,6‐tetrahydropyridine (MPTP; Mudo et al., 2012). On the basis of these findings, we sought to determine if roflupram would protect dopaminergic neurons against 1‐methyl‐4‐phenylpyridinium iodide (https://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=4568)/MPTP‐induced neuronal degeneration and whether the CREB/PGC‐1α pathway contributed to the protective effects of roflupram. We found that roflupram promoted the survival of SH‐SY5Y cells and human dopaminergic neurons in culture, exposed to MPP+. Moreover, roflupram improved behavioural performance and attenuated the loss of dopaminergic neurons in both the SN and striatum of an MPTP‐induced mouse model of PD in vivo. Mechanistically, the neuroprotective effects of roflupram may partly involve the up‐regulation of the CREB/PGC‐1α pathway. Taking our results together, we suggest that roflupram is a promising compound for preventing the degradation of dopaminergic neurons in patients with PD.
2. METHODS
2.1. Animals
All animal care and experimental procedures were performed in accordance with the NIH Guide for the Care and Use of Laboratory Animals (NIH, revised 1996) and approved by the Laboratory Animal Ethics Committee of Southern Medical University (Guangzhou, China). Animal studies are reported in compliance with the ARRIVE guidelines (Kilkenny et al., 2010) and with the recommendations made by the British Journal of Pharmacology (McGrath & Lilley, 2015).
Adult male C57BL/6J mice (RRID:IMSR_JAX:000664, 8 weeks old, 22–25 g, specific pathogen free) were obtained from the Laboratory Animal Centre of Southern Medical University (Guangzhou, China). Mice were housed five per cage (plastic cages) with conventional bedding in the animal facility of Southern Medical University. The mice were adapted to feed for a week before the formal experiments and housed in a standard feeding environment (50–70% humidity, 23–25°C, 12–12 hr light/dark cycle with lights) where they could eat and consume water at will.
Experiments were designed according to the principles of the 3Rs, (replacement, refinement, and reduction) to minimize animal pain and reduce the number of mice used in our experiments. Mice were killed after behavioural tests by an overdose of inhaled 5% isoflurane. The sample size calculations were conducted in our pilot study to determine the lowest number of mice needed to detect a significant difference in the level of https://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=1713 between MPTP‐treated mice and (MPTP + ROF)‐treated mice. Using the two‐tailed test, a sample size of five mice per group was needed to detect a difference with 95% confidence and 80% power. Based on the power calculation, 10 mice were allocated to each experimental group to evaluate behavioural performance. After the behavioural tests, five mice in each group were used for biochemical analysis and the remainder in each group were used for the immunostaining. No mice were excluded from statistical analysis. A total of 40 mice were used in the experiments described here.
2.2. Experimental design for MPTP mouse model of PD
Mice were randomly divided into four groups (10 mice per group). On the first day of the experiment, the mice in the vehicle group and the MPTP group were given solvent (10 ml·kg‐1 ) intragastrically (i.g.), and the other two groups were given roflupram (1.0 mg·kg−1, i.g.) or rolipram (2.5 mg·kg−1, i.g.) in the same volume. On the second day, 2 hr after roflupram or rolipram were administered, the mice in the vehicle group were given saline (10 ml·kg‐1 , i.p.) and the other three groups were given MPTP (30 mg·kg−1, i.p.) in the same volume. The treatment schedule in the next 4 days was the same as the second day. Starting on the seventh day of the experiment, no further treatments were given to any of the mice for 10 days. Behavioural experiments were carried out after the 10 treatment‐free days. The tissues were collected 24 hr after the end of the behavioural tests, from mice overdosed with 5% isoflurane. This schedule is illustrated in Figure 6a. This model of PD in mice replicates biochemical and histological changes in humans suffering from PD (Heikkila, Hess, & Duvoisin, 1984).
2.3. Cell culture
The SH‐SY5Y cell line used in our experiments was derived from the American Type Culture Collection (ATCC, Cat#CRL‐2266, RRID:CVCL_0019). The cells were cultured in culture flasks containing 5 ml of culture medium, and the culture medium included Nutrient Mixture Ham's F‐12 with 1% penicillin/streptomycin and 10% (v/v) FBS. The culture condition of incubator was 95% air and 5% CO2 at 37°C, and the cells were passaged at 48 hr per interval.
The Lund Human Mesencephalic (LUHMES) cell line used in our study was derived from American Type Culture Collection (ATCC, Cat#CRL‐2927, RRID:CVCL_B056). The cell culture and differentiation protocol have been described previously (Schildknecht et al., 2013; Scholz et al., 2011). Briefly, conditionally immortalized cells were cultured and maintained in DMEM/F12 supplemented with N2 supplement, 2‐mM https://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=723 and 40 ng/ml https://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=4924 at 37°C with 5% CO2. For differentiation, the cells were seeded in a culture flask pre‐coated with 1 μg·ml−1 fibronectin and 50 μg·ml−1 poly‐l‐ornithine (PLO) in proliferation medium. After 24 hr, the medium was replaced with differentiation medium, composed of advanced DMEM/F12 with N2 supplement, 2‐mM l‐glutamine, 2.25‐μM tetracycline, 2 ng·ml−1 recombinant human https://www.guidetopharmacology.org/GRAC/LigandDisplayForward?tab=biology&ligandId=4940, and 1‐mM dibutyryl 3′,5′‐cAMP. During the process of cell differentiation, the medium was changed with fresh differentiation medium every other day. After 6 days of differentiation, LUHMES cells are characterized by high expression of TH and are used as a model of human dopaminergic neurons (Krug et al., 2014; Scholz et al., 2011).
2.4. Cell viability assay
SH‐SY5Y cells were cultured in 96‐well plates at 1 × 104 cells per well overnight at 37°C with 95% air and 5% CO2. Then, the cell culture solution was discarded, and the culture medium containing different concentrations of roflupram was added to each well for 1‐hr incubation. The concentrations of roflupram used are shown in the Figure legends. Subsequently, the cells were exposed to 500 μM of MPP+ for 48 hr. Finally, the culture medium was discarded and MTT solution (0.5 mg·ml−1) was added to each well for 4 hr. After the MTT solution was replaced with 150 μl of DMSO, the mixture was shaken for 10 min and was then analysed photometrically at a wavelength of 570 nm in a microplate reader (Synergy HT; BioTek, Broadview, IL).
2.5. LDH assay
SH‐SY5Y cells were cultured in 96‐well plates at 1 × 104 cells per well overnight. The cells were pretreated with roflupram for 1 hr and then were exposed to 500 μM of MPP+ for 48 hr; the 96‐well plates were centrifuged for 5 min in a plate centrifuge at 400× g. Then, 120 μl of the supernatant from each well was transferred to a new 96‐well plate. LDH activity in this supernatant was assayed with a LDH Cytotoxicity Assay Kit (Beyotime, Shanghai, China)., according to the manufacturer's instructions. The absorbance values in the wells were measured at both 490 and 600 nm.
2.6. Flow cytometric analysis of apoptosis
SH‐SY5Y cells were cultured in 6‐well plates at 2 × 105 cells per well and were incubated overnight. Then, the culture medium was discarded, and the medium containing different concentrations of roflupram was added and incubated for 1 hr. The concentrations of roflupram used are shown in the Figure legends. Subsequently, the cells were exposed to 500 μM of MPP+ for 24 hr. The cell supernatant was then removed, and the cells were digested and centrifuged at 134 x g for 5 min. Then, 500 μl of binding buffer and 5 μl of Annexin V‐FITC were mixed and added to the centrifuge tube; subsequently, 5 μl of PI was also mixed into the centrifuge tube. Finally, these samples were then subjected to flow cytometry (BD FACS‐Verse™ Flow Cytometer, BD Biosciences, San Jose, CA, USA).
2.7. ATP analysis
The cells were cultured in 6‐well plates at 2 × 105 cells per well overnight. The cells were then treated with roflupram (20 μM) for 1h and then with MPP+ (500 μM) for 24 hr. Then, the cells were lysed in a lysis buffer (200 μL per well) for 10 min and centrifuged at 12,000× g for 5 min at 4°C. The supernatant was collected for subsequent measurement of ATP. The ATP in samples (20‐μl) of the supernatant was determined with an ATP Assay kit (Beyotime), using a a micropipettor (P20, Gilson Inc., Villiers le Bel, France) for rapid mixing. The chemiluminescence (as RLU) was then determined in a microplate reader (Synergy HT; BIOTEK, Broadview, IL). The protein concentration of the sample was determined by the BCA protein assay kit, following the manufacturer's instructions. The relative ATP level was calculated as follows: relative ATP level = ATP value/protein value.
2.8. Mitochondrial membrane potential (MMP) measurement
SH‐SY5Y cells were cultured in confocal dishes at a density of 1 × 105 cells·ml−1. The next day, the cells were treated with roflupram (20 μM) 1 hr before incubation with 500 μM of MPP+ for 24 hr. Then the culture medium was discarded and the cells were washed three times with PBS, for 5 min each time. The tetramethylrhodamine ethyl ester perchlorate (TMRE; 50 nM) dye was added to the confocal dishes, incubated for 20 min in the dark, and then washed three times with PBS for 5 min each time. Finally, the confocal dishes were placed in an inverted confocal microscope (ECLIPSE Ti, Nikon, Tokyo, Japan) for imaging.
2.9. Determination of ROS
SH‐SY5Y cells were cultured in 24‐well plates at a density of 1.2 × 105 cells·ml−1 and were allowed to adhere overnight. After treatment with 20 μM roflupram for 1hr and 500 μM of MPP+ for 24 hr, the cells were washed three times with PBS for 5 min each time. MitoSOX‐Red (2 μM) dye was added to each well, was incubated for 1 hr in the dark at 37°C, and was then washed three times with PBS for 5 min each time. Finally, the plates were placed under a fluorescent microscope (ECLOPSE Ti‐U, Nikon, Tokyo, Japan) for imaging.
2.10. Immunostaining
SH‐SY5Y cells were seeded in confocal dishes at a density of 1.2 × 105 cells·ml−1. Then, the cells pretreated with roflupram (20 μM; 1hr) were incubated with 500 μM of MPP+ for 48 hr. After washing with PBS three times, for 5 min each time; 300 μl of 4% paraformaldehyde was added to each dish for 20 min at 37°C, and the dishes were then washed three times with PBS. Then, 300 μl of 0.2% Triton X‐100 was added to each dish for 10 min at room temperature and then was washed three times with PBS for 5 min each time. The cells were then incubated in 300 μl of 5% BSA solution (in PBS) for 50 min at room temperature; then, the cells were incubated with anti‐cleaved caspase‐3 antibody solution (1:200) overnight at 4°C. On the next day, the wells of the confocal dishes were washed three times with PBS for 5 min each time. After the washed PBS was discarded, the secondary antibody solution (1:200) was added and incubated for 4 hr in the dark at 4°C. After incubation, the secondary antibody was removed, and the samples were then washed three times with PBS for 5 min each time. Subsequently, Hoechst 33342 (2 μg·ml−1) was incubated in the dishes for 15 min and washed with PBS, and the samples were then examined under an inverted confocal microscope. The immuno‐related procedures used comply with the recommendations made by the British Journal of Pharmacology.
2.11. Transfection of siRNA
Specific siRNA for PGC‐1α (siPGC‐1α) were obtained from Shanghai GenePharma Co. (Shanghai, China). The sequences were as follows: sense: 5′‐GGCACGCAAUCCUAUUCA UTT‐3′; antisense: 5′‐AUGAAUAGGAUUGCGUGCCTT‐3′. Scrambled siRNA sequences were used as the negative control. The SH‐SY5Y cells were cultured in 6‐well plates at a density of 1 × 105 cells·ml−1 and allowed to adhere overnight. According to the manufacturer's instructions, Lipofectamine 2000 (Invitrogen, Carlsbad, CA, USA) was mixed with Opti‐MEM and mixed with siRNA (50 nM) for 20 min. Then, the mixture was added to each well. Six hours after transfection, culture medium was removed and replaced with a fresh medium. The cells were used for further analysis 24 hr after transfection.
2.12. Analysis of mitochondrial morphology
The cells were cultured in confocal dishes at a density of 1.2 × 105 cells·ml−1 and were allowed to adhere overnight. After treatment with roflupram (20 μM) for 1 hr and 500 μM of MPP+ for 24 hr, the culture medium was discarded, and the cells were washed three times with PBS for 5 min each time. Subsequently, Mitotracker Red (500 nM, 200 μl) dye was added to each dish, incubated for 1 hr in the dark at 37°C, and then washed with PBS three times for 5 min each time. Finally, the confocal dishes were placed under an inverted confocal microscope for observation and image capturing, and the images were analysed using an image analysis system (Image‐Pro Plus, version 6.0) with the Mitochondrial Network Analysis (MiNA) toolset. MiNA is freely available at https://github.com/ScienceToolkit/MiNA. The analysis and statistics of the mitochondrial captured images were performed according to a previously published article (Valente, Maddalena, Robb, Moradi, & Stuart, 2017).
2.13. Western blotting
Brain tissues (striatum and substantia nigra) in RIPA buffer (10 ml per g of tissue) supplemented with a protease and phosphatase inhibitor cocktail (10 μl per ml of buffer; Sigma‐Aldrich) were homogenized using an ultrasonic cell disruptor (VCX130, Sonics, NEWTOWN, CT, USA) at 130 W, 20 KHz for 2 s, interval 10 s, followed by 5 cycles, on ice. The samples were then centrifuged at 20,000x g for 15 min at 4°C, and the supernatant collected. Protein in the supernatant was measured with the BCA protein assay, according to the manufacturer's instructions. The samples of supernatant were then boiled at 95°C for 10 min, and 20 μl of each sample was used to load 40 μg of protein for electrophoresis in 10% SDS‐PAGE. After electrophoresis, the protein was transferred to PVDF membranes (Millipore, #ISEQ00010); transferring was performed under constant pressure at 86 V for 90 min. Subsequently, 5% non‐fat dry milk prepared with Tris‐Buffered Saline Tween‐20 (TBST: 25mM Tris, 140 mM NaCl, 3 mM KCl, 0.1% Tween‐20, pH‐7.4) was used for blocking at room temperature for 2 hr. 50 ml of TBST containing 5% non‐fat milk powder were used for eachmembrane. Subsequently, the membranes were washed four times for 5 min each time with TBST and the primary antibodies were incubated at 4°C overnight. Then, the membranes were washed four times for 5 min each time with TBST, and the secondary antibodies were incubated at 4°C for 4 hr. Finally, the membranes were washed four times for 5 min each time, and chemiluminescent reagent was added dropwise for exposure. The grey value of the protein in the exposed membranes were analysed by image analysis system (Image‐Pro Plus, version 6.0).
2.14. Measurement of mitochondrial respiratory capacity
Mitochondrial respiratory capacity of SH‐SY5Y cells was detected using a Seahorse XF‐24 Analyzer (Seahorse Bioscience, Billerica, MA, USA) by measuring the oxygen consumption rate (OCR). The cells were treated according to the manufacturer's recommended protocol. Briefly, the OCR was measured by the sequential addition of oligomycin (an ATP synthase inhibitor, 1 μM), phenylhydrazone (FCCP, a mitochondrial respiration uncoupler, 0.5 μM), and rotenone (a complete respiratory inhibitor, 2 μM). This protocol is normally used for the determination of basal OCR, ATP‐linked respiration, and maximal respiration (Tomkova, Misuth, Lenkavska, Miskovsky, & Huntosova, 2018). After the measurement of mitochondrial respiratory capacity, the cells were lysed, and the total protein was quantified using the BCA protein assay. The final result was normalized against the protein content of the corresponding well.
2.15. Real‐time PCR for mtDNA copy number analysis
Total DNA was extracted from SH‐SY5Y cells using TIANamp Genomic DNA Kit (TIANGEN BIOTECH, Beijing, China). Real‐time PCR analysis using LC480 SYBR Green IMaster (Roche, Basel, Switzerland) was performed on a real‐time PCR system (Roche, Basel, Switzerland). The ratio of mitochondria DNA (mtDNA, represented by the https://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=1376) to nuclear DNA (represented by the https://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=2608) was shown as relative mtDNA copy number. The primers for the Cox2 gene were as follows: Cox2‐forward, 5′‐CAGTCCCCTCCCTAGGACTT‐3′; Cox2‐reverse, 5′‐TTTCAGAGCATTGGCCATAGAA‐3′. The primers for the Fasn gene were as follows: Fasn‐forward, 5′‐AGGATATGGAGAGGGCTGGT‐3′; Fasn‐reverse, 5′‐ACCCAAGCATCATTTTCGTC‐3′.
2.16. Pole test
The mice were acclimated for 30 min in the behavioural testing laboratory before the experiment. A straight wooden rod with a diameter of 0.8 cm and a height of 60 cm was used. There was a small wooden ball at the top of the rod, which was covered with gauze to prevent the mouse from slipping. Mice were placed on top of the straight rod, and we recorded the time taken for the mouse to climb along the wood to the bottom of the rod. Mice were trained for 2 days before formal behavioural testing. Each training session consisted of three trials.
2.17. Rotarod test
Mice were trained for 3 days prior to formal testing. Mice were placed on a rotarod at a speed of 20 r.p.m. from slow to fast, and they were able to learn to walk freely within 5 min, after which the training was over. At the time of the formal experiment, the mice were placed on a rotarod with a rolling speed of 12 r.p.m., and the time for which the mice stayed on the rotarod were observed and recorded.
2.18. Gait dynamics
Gait dynamic analysis is a well‐established method to monitor motor defects in MPTP‐treated mice (Amende et al., 2005). In the present study, 2 days before the formal behavioural test, mice were trained daily and placed on a running belt at a speed of 20 cm·s−1 for approximately 30 s. When the mice were able to walk smoothly on the treadmill belt, they were removed, and the training was over. When the mice were formally tested for behaviour, the mice were placed in a walking compartment with a width of 7 cm and a length of 30 cm. The gait analysis system continuously imaged the underside of the mice. The mice walked for about 5 s on the transparent treadmill belt at a speed of 30 cm·s−1. A video of stable walking of each mouse was analysed by a gait software system (DigiGait Analysis Software Version 14.5). The parameters of stride length, stride duration, ataxia coefficient, and stride frequency of the limbs of the mice were obtained and analysed as previously described (Goldberg, Hampton, McCue, Kale, & Meshul, 2011).
2.19. Nissl staining
The brains were removed from paraformaldehyde for paraffin embedding, and brain sections with the striatum and SN were sliced to a thickness of 4 μm. The paraffin sections were dewaxed, washed with water, stained in toluidine blue dye solution for 5 min, washed with water, and then baked. The sections were cleaned with xylene and sealed with neutral gum. Slices were placed under a microscope for observation and imaging. Nissl‐positive neurons in the SN and striatum regions were counted.
2.20. Immunohistochemistry
Paraffin sections of the SN and striatum (4 μm ) were deparaffinized, washed with water, and subjected to antigen retrieval using citrate antigen repair buffer (pH 6.0). The sections were placed in a 3% hydrogen peroxide solution and incubated at room temperature for 25 min in the dark. After which, they were washed three times for 5 min each time on a decolorizing shaker. The tissue was evenly covered with 3% BSA dropwise in the culture circle and was incubated at room temperature for 30 min. The blocking solution was gently removed, and an anti‐https://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=1243 primary antibody solution was added dropwise to the sections, and the sections were placed in a wet box and incubated at 4°C overnight. The slides were placed in PBS (pH 7.4) and washed three times on a decolorizing shaker for 5 min each wash. After the sections were dried, the secondary antibody covered with the corresponding species of the primary antibody was added dropwise and incubated for 50 min at room temperature. After the slices were subjected to 3,3′‐diaminobenzidine (DAB) development, the sheets were dehydrated. Positive antibody expression in the sections was dark brown, and images were collected under a microscope. Numbers of TH‐immunoreactive neurons were counted in the SN, and the intensity of dopaminergic neuron fibres in the striatum was quantified using ImageJ respectively.
2.21. Immunofluorescent staining
The brains were fixed in 4% paraformaldehyde for 24 hr, and thendehydrated in 20% sucrose solution until the tissues sank to the bottom of thecontainer and then 30% sucrose solution until subsidence at 4°C. The brains were removed from 30% sucrose for frozen sectioning of the SN and striatum, Brains were cryosectioned at 20 µm by cryostat (Leica, CM1860) and the sections were washed three times with 1× TBS for 10 min each wash. Then, 5% goat serum containing 0.3% Triton 100‐X TBST was used for blocking for 1 hr at room temperature, and sections were incubated overnight at 4°C in a dilution of the primary antibody. The sections were washed three times with TBST, and the secondary antibody solution was incubated in the dark for 1 hr at room temperature. The sections were then washed three times with TBST for 10 min each time. Hoechst 33342 (2 μg·ml−1) was added for staining, and the mixture was allowed to stand at room temperature for 15 min. Finally, the sections were washed three times with TBST for 10 min each time. The tissue sections were attached to glass slides, mounted with mounting medium, and, after waiting for the mounting medium to solidify, were placed under a microscope for observation and imaging.
2.22. Data and statistical analysis
The data and statistical analysis comply with the recommendations of the British Journal of Pharmacology on experimental design and analysis in pharmacology (Curtis et al., 2018). For all studies, experimenters were blind to the specific groups, the treatment conditions, and the treatments of the trained/tested mice. Quantitative analysis of fluorescence OD values and protein expression were normalized to the mean of the control group or negative control group to allow for unwanted sources of variation. The bands used for statistical analysis in western blots were normalized to GAPDH or total protein. The data in the paper are presented as mean ± SD, and the n represents the number of animals or tissue samples or cell preparations. Statistical comparison calculations were performed using SPSS 19.0 software. Each experiment was performed independently five times. Statistical significance analysis was performed using one‐way ANOVA and Bonferroni post hoc test when F achieved P < .05, and there was no significant variance inhomogeneity. Values of P < .05 were considered to show statistically significant differences.
2.23. Materials
Roflupram (#SML2106) was purchased from Sigma‐Aldrich (St. Louis, MO, USA), and the purity was determined to be above 98% by HPLC analysis. Rolipram (#S1403) was obtained from Selleck (Houston, TX, USA), and the purity was determined to be above 98% by HPLC analysis. For in vitro experiments, roflupram was dissolved in DMEM/F12 medium containing 0.1% DMSO. In animal experiments, roflupram and rolipram were suspended in vehicle (6% Solutol HS‐15, 1% DMSO, 40% hydroxypropyl‐β‐cyclodextrin) to provide working solutions.
Other materials were supplied as follows. DMSO (#D2650), 1‐methyl‐4‐phenylpyridine iodide (MPP+; #D048), and 3‐(4,5‐dimethyl‐2‐ thiazolyl)‐2,5‐diphenyl‐2‐H‐tetrazolium bromide (MTT; #M2128) were bought from Sigma‐Aldrich (St. Louis, MO, USA). Cell counting kit‐8 (CCK‐8; #CK04) was obtained from Dojindo Corporation (Tokyo, Japan). Oligomycin (#495455), carbonyl cyanide 4‐(trifluoromethoxy)phenylhydrazone (FCCP; #C2920), poly‐l‐ornithine (#P3655), fibronectin human plasma (#F0895), and rotenone (#R8875) were bought from Sigma‐Aldrich (St. Louis, MO, USA). 1‐Methyl‐4‐phenyl‐1,2,3,6‐tetrahydropyridine (MPTP) hydrochloride (#S4732) and https://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=5983 (#S1582) were purchased from Selleck (Houston, TX, USA). Tetramethylrhodamine, ethyl ester, perchlorate (TMRE; #T669), MitoSOX‐Red (#M36008), and Hoechst 33342 (#H1399) were obtained from Thermo Fisher Scientific, Inc. (Waltham, MA, USA). Annexin V‐FITC/PI double staining cell apoptosis detection kit (#KGA108) was purchased from KeyGen Biotech (Nanjing, China). LDH Cytotoxicity Assay Kit (#C0017) and ATP Assay Kit (#S0026) were obtained from Beyotime Institute of Biotechnology (Shanghai, China). BCA protein assay kit (#23225) and Mitotracker Red (#M7512) were bought from Thermo Fisher Scientific, Inc. (Waltham, MA, USA). Culture medium, FBS (#10099141) and PBS (#10010023) were purchased from Gibco (Carlsbad, CA, USA). TIANamp Genomic DNA Kit (#DP304) was obtained from TIANGEN BIOTECH (Beijing, China). LC480 SYBR GreenIMaster (#4887352001) was purchased from Roche (Basel, Switzerland). Anti‐GAPDH (#2118, RRID:AB_561053) and anti‐cleaved caspase‐3 (#9661S, RRID:AB_2341188) were bought from Cell Signaling Technology (Danvers, USA). Anti‐TH (#ab152, RRID:AB_390204), anti‐p‐CREB (Ser133, #06‐519, RRID:AB_310153), and anti‐T‐CREB (#AB3006, RRID:AB_91283) were obtained from Merck‐Calbiochem (St. Louis, MO, USA). Anti‐PGC‐1α (#ab54481, RRID:AB_881987) was purchased from Abcam (Cambridge, MA, USA). Lipofectamine 2000 (#11668019) and lipofectamine 3000 (#L3000015) were the products from Invitrogen (Invitrogen, Carlsbad, CA, USA).
2.24. Nomenclature of targets and ligands
Key protein targets and ligands in this article are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY (Harding et al., 2018), and are permanently archived in the Concise Guide to PHARMACOLOGY 2019/20 (Alexander et al., 2019).
3. RESULTS
3.1. Roflupram increases cell viability and protects against apoptosis in SH‐SY5Y cells exposed to MPP +
To investigate whether roflupram would be protective towards dopaminergic cells, we first treated SH‐SY5Y cells with roflupram alone for 48 hr and measured cell viability using an MTT assay. Treatment with roflupram (2.5–40 μM) alone had no effect on cell viability (Figure 1b). We then investigated the protective effect of roflupram. As shown in Figure 1c, MPP+ significantly reduced cell viability, while roflupram reversed the impairment mediated by MPP+ and increased cell viability in a concentration‐dependent manner. The results from the flow cytometry assay showed that MPP+ enhanced apoptosis significantly, while roflupram reduced the ratio of apoptotic cells at both early and late stages of apoptosis (Figure 1d,e). To further confirm the protective effect of ROF, we analysed the activity of LDH released into the culture medium. LDH activity was markedly elevated after exposure to MPP+ and this increase was blocked by pretreatment with roflupram for 1hr (Figure 1f). Cytochemical staining for cleaved caspase‐3 was also performed to examine the extent of cellular apoptosis. Consistently, roflupram decreased the expression of cleaved caspase‐3 in a dose‐dependent manner (Figure S1). These results suggest that roflupram was neuroprotective against the effects of MPP+ in SH‐SY5Y cells.
3.2. Roflupram reverses mitochondrial damage induced by MPP +
Our previous studies indicated that inhibition of PDE4 in SH‐SY5Y cells reduces the level of ROS (Zhong et al., 2019). As mitochondria are the major organelle producing ROS inside cells, we next investigated whether roflupram affected the morphology and functions of mitochondria. A specific mitochondrial tracker was used to assess morphology (Figure 2a). The numbers of networks/individuals, mitochondrial fluorescence intensity, mitochondrial footprints, and number of mitochondria were analysed (Figure 2b–f); MPP+ treatment destroyed the mitochondrial network and caused a decrease in the absolute number of networks per cell. Treatment with roflupram (20 μM) increased the number of networks (Figure 2b). Mitochondrial‐related indicators such as the number of individuals, mitochondrial fluorescence intensity, mitochondrial footprint, and number of mitochondria also decreased. After treatment with roflupram (20 μM) for 24 hr, these mitochondrial morphological parameters were increased (Figure 2c–f). To investigate the effects of roflupram on the alteration of MMP, the cells were stained with a TMRE fluorescent dye. As shown in Figure 2g,h, pretreatment with roflupram attenuated the loss of MMP. We then measured the production of ROS originating from mitochondria (mtROS). As shown in Figure 2i,j, roflupram decreased the MitoSOX fluorescence in the presence of MPP+. We also measured the levels of ATP and mtDNA in cells. The results showed that pretreatment with roflupram antagonized the effects of MPP+ and increased the levels of both mtDNA and ATP (Figure 2k–l). Similarly, treatment with roflupram significantly increased basal and maximum mitochondrial respiration in SH‐SY5Y cells challenged with MPP+ (Figure 2m,n), suggesting that roflupram increased the oxidative capacity of mitochondria. These data indicate that roflupram attenuated MPP+‐induced impairment and ameliorated both the morphology and function of mitochondria.
Figure 2.
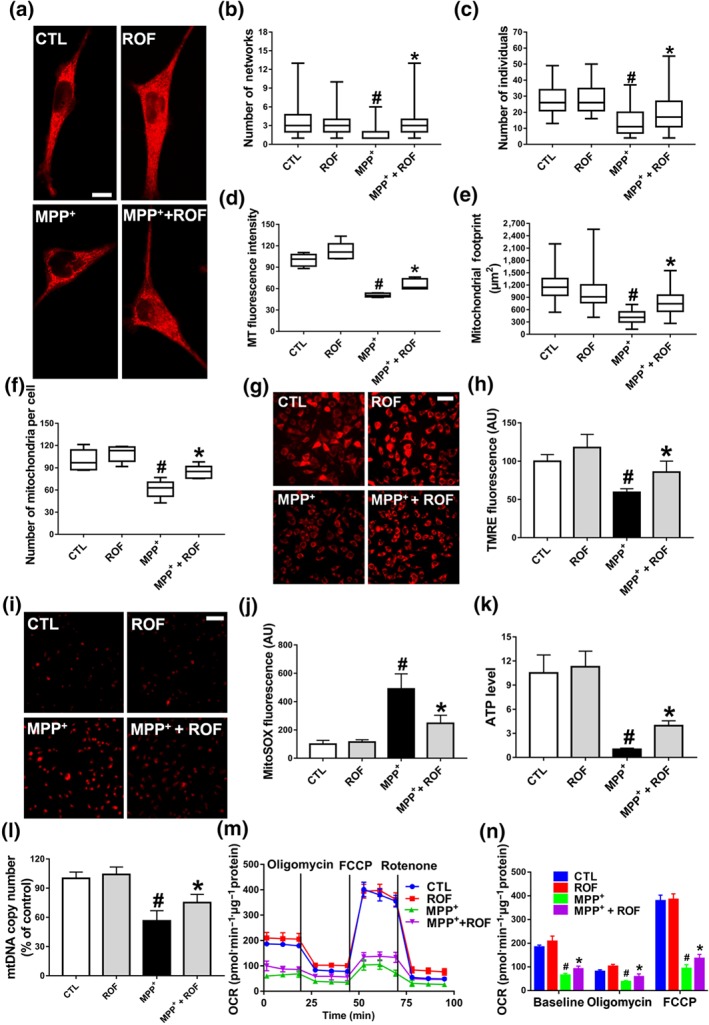
Effect of roflupram on mitochondrial morphology and function of MPP+‐treated cells. (a) SH‐SY5Y cells were pretreated with roflupram (ROF; 20 μM) for 1 hr and were then treated with 500 μM of MPP+ for 24 hr. Mitotracker Red was added to mark mitochondria. Scale bar = 10 μm. Statistical analysis was performed on 100 cells in each group to obtain five data sets; the number of networks (b), number of individuals (c), MT fluorescence intensity (d), mitochondrial footprint (e), and the number of mitochondria per cell (f). After treating the cells with roflupram for 1 hr, 500 μM of MPP+ was added for 24 hr to measure mitochondrial membrane potential (MMP) (g) and oxidative stress (i), and statistical analyses were determined via TMRE fluorescence (h) and MitoSOX fluorescence (j). (k) The cells were pretreated with roflupram (20 μM) for 1 hr and then treated with 500 μM of MPP+ for 24 hr. ATP levels were detected with an ATP Assay Kit. (l) mtDNA copy number was measured in SH‐SY5Y cells by real‐time PCR. (m) Oxygen consumption rate (OCR) was measured in SH‐SY5Y cells using the Seahorse XF‐24 Analyzer in response to the sequential administration of oligomycin (1 μM), FCCP (0.5 μM) and rotenone (2 μM). (n) Quantification of OCR in panel (M). In (a, g, i) the scale bar = 500 μm. Data are from five independent experiments and are presented as box and whisker plots in (b, c, d, e, f), with medians, quartiles and ranges shown. In (h, j, k, l, m, n), data are shown as means ± SD. # P < .05, significantly different from control group. *P < .05, significantly different from MPP+‐treated group
3.3. Roflupram enhances the level of phosphorylated CREB and the expression of PGC‐1α
Our previous studies have shown that inhibition of PDE4 in neuronal cells increases the level of cAMP and thereby activates the PKA/CREB signalling pathway (Guo et al., 2017; Zhong et al., 2018). PGC‐1α is a target of CREB and is a key factor in regulating the biogenesis and function of mitochondria (Ciron et al., 2015; Herzig et al., 2001). Thus, we were interested in investigating whether roflupram exerts its protective effects via CREB/PGC‐1α signalling. As shown in Figure 3a, we found that MPP+ decreased the protein expression of TH in SH‐SY5Y cells and that roflupram counteracted the effects of MPP+. We then found that the level of phosphorylated CREB was significantly decreased in response to MPP+ stimulation, compared with that of the control. Additionally, the phosphorylation of CREB was increased as the result of roflupram treatment (Figure 3c,d). Similar results were observed in terms of the protein level of PGC‐1α (Figure 3e,f). These data suggest that roflupram normalized CREB activation and increased PGC‐1α expression to ameliorate MPTP‐induced cellular deficits.
Figure 3.
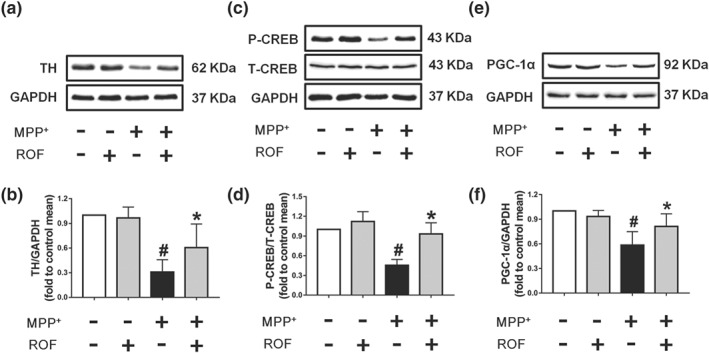
Effect of roflupram on the expression of related proteins of MPP+‐treated cells. SH‐SY5Y cells were pretreated with roflupram (ROF; 20 μM) for 1 hr and then treated with 500 μM of MPP+ for 24 hr. After the cells were processed, the expression of TH (a), p‐CREB (c), and PGC‐1α (e) were measured by western blotting. (b) Densitometric quantification of TH/GAPDH in (a). (d) Densitometric quantification of p‐CREB/T‐CREB in (c). (f) Densitometric quantification PGC‐1α/GAPDH in (e). Data are presented as mean ± SD (n = 5) and represent five independent experiments. # P < .05, significantly different from control group. *P < .05, significantly different from MPP+‐treated group
3.4. Activation of PKA/CREB/PGC‐1α signalling is essential for roflupram‐mediated neuroprotection against MPP +‐mediated cellular damage
To investigate the signalling pathway responsible for the protective effect of roflupram, SH‐SY5Y cells were pretreated with the PKA inhibitor, H‐89, for 1 hr, and were then exposed to MPP+ in the presence of roflupram. As shown in Figure 4a, roflupram reversed the MPP+‐induced reduction of phosphorylated CREB and PGC‐1α, while H‐89 significantly blocked the stimulatory role of roflupram on the expression of PGC‐1α and phosphorylation of CREB (Figure 4a–c). We then asked whether PKA/CREB and PGC‐1α mediate the neuroprotective effects of roflupram. As shown in Figure 4d,e, roflupram reversed the inhibitory effects of MPP+ on MMP, while H‐89 blocked the stimulatory role of roflupram on MMP. Simultaneously, we determined cell viability using an LDH assay. Although H‐89 alone had no toxic effect in SH‐SY5Y cells, it blocked the protective effects of roflupram and decreased LDH release (Figure 4f). We then determined the involvement of PGC‐1α. The cells were transfected with PGC‐1α specific siRNA or a negative control. The knock down efficiency was confirmed by western blot (Figure S2). Our results showed that knocking down PGC‐1α attenuated the effects of roflupram on MMP (Figure 4g,h). In accordance with this finding, knocking down PGC‐1α abolished the protective effect of roflupram (Figure 4i). We then performed a rescue experiment with over‐expression of PGC‐1α in SH‐SY5Y cells. A PGC‐1α plasmid was transfected into the siRNA‐treated SH‐SY5Y cells. We found that over‐expression of PGC‐1α reversed the reduction of the MMP induced by MPP+ and PGC‐1α siRNA (Figure 4j–l). Taken together, these data imply that roflupram protected SH‐SY5Y cells against MPP+‐induced neuronal cell injury through activation of the PKA/CREB/PGC‐1α pathway.
Figure 4.
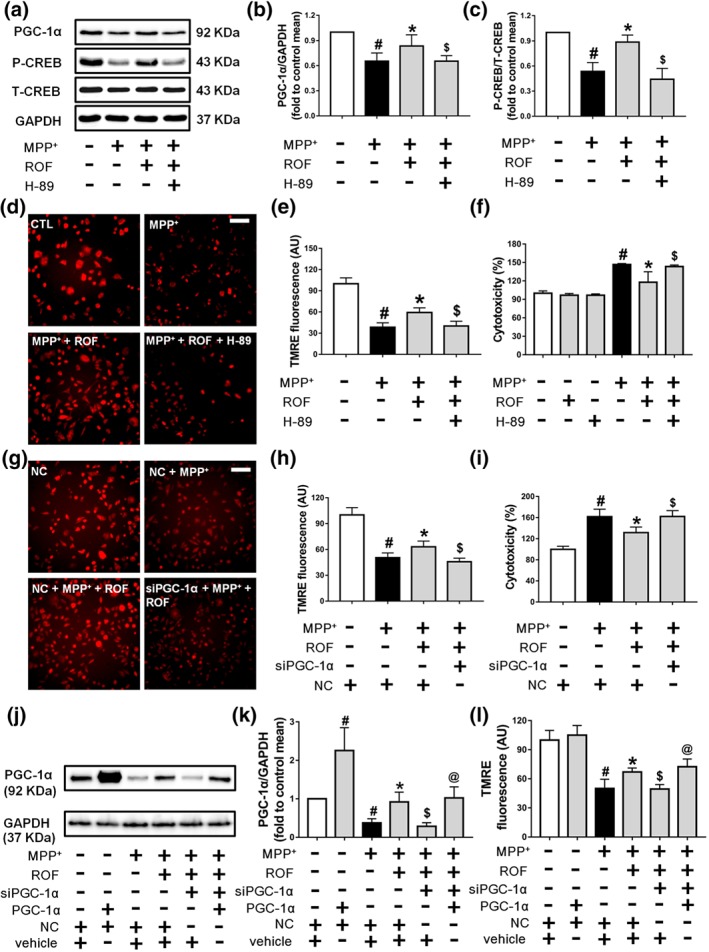
The CREB/PGC‐1α pathway is involved in the protective effects of roflupram in MPP+‐treated cells. (a) SH‐SY5Y cells were pretreated with H‐89 (10 μM) for 1 hr and then treated with roflupram (ROF; 20 μM) for 1 hr; after that, 500 μM of MPP+ was added for 24 hr. After the cells were processed, the expression levels of PGC‐1α, p‐CREB, and total CREB were measured by western blotting. (b and c) Densitometric quantification of PGC‐1α/GAPDH (b) in (a) and p‐CREB/T‐CREB (c) in (a). (d) The cells were pretreated with H‐89 (10 μM) for 1 hr and then treated with roflupram (20 μM) for 1 hr. Subsequently, 500 μM of MPP+ was added to stimulate with cells for 24 hr. After treatment, the cells were washed with PBS and dyed with TMRE (50 nM) for 20 min. Finally, the cells were viewed under a fluorescence microscope. Scale bar = 500 μm. (e) Statistical analysis of fluorescent photographs in (d). (f) The cells were pretreated with H‐89 (10 μM) for 1 hr and then treated with roflupram (20 μM) for 1 hr; after that, 500 μM of MPP+ was added for 48 hr. After treatment, LDH activity in the medium was measured. (g) Transfected cells were pretreated with ROF (20 μM) for 1 hr, and then, 500 μM of MPP+ was added for 24 hr. After the cells were processed, the cells were dyed with TMRE (50 nM) for 20 min. Finally, the cells were placed under a fluorescent microscope for imaging. Scale bar = 500 μm. (h) Statistical analysis of fluorescent pictures in (g). (i) Transfected cells were pretreated with ROF (20 μM) for 1 hr, and 500 μM of MPP+ was added for 48 hr. After treatment, LDH activity in the medium was detected. (j) SH‐SY5Y cells were transfected for 24 hr with PGC‐1α plasmid and with PGC‐1α siRNA or random siRNA. Transfected cells were then treated with MPP+ for an additional 24 hr. The expression of PGC‐1α was measured by western blotting. (k) Densitometric quantification of PGC‐1α/GAPDH in (j). (l) After transfection and treatment with MPP+, the cells were dyed with TMRE (50 nM) for 20 min before imaging under a fluorescent microscope. Fluorescent pictures were analysed statistically. Data are presented as mean ± SD (n = 5) and represent five independent experiments. # P < .05, significantly different from control group or negative control group. *P < .05, significantly different from MPP+‐treated group. $ P < .05, significantly different from versus MPP+ + ROF‐treated group. @ P < .05, significantly different from siPGC‐1α + MPP+ + ROF‐treated group
3.5. Roflupram is protective against MPP +‐mediated cellular damage in a model of human dopaminergic neurons
We then investigated whether roflupram could exert similar effects in human dopaminergic neurons. Our model used the LUHMES cells, which are conditionally immortalized mesencephalic neuronal precursors that can be differentiated to fully post‐mitotic human dopaminergic neurons (Efremova et al., 2015). After 6 days of differentiation, TH was robustly expressed in the model human dopaminergic neurons (Figure 5a,b). We then found that roflupram (5–40 μM) did not affect the viability of these neuronal cells (Figure 5c). As shown in Figure S3, MPP+‐induced a comparable level of toxicity at 5 μM (56% cell viability reduction). Therefore, we used this concentration to test the neuroprotective effects of roflupram and found that ROF protected human dopaminergic neurons from MPP+‐induced toxicity, as shown by decreased LDH release and increased cell viability and ATP production (Figure 5d–f).
Figure 5.
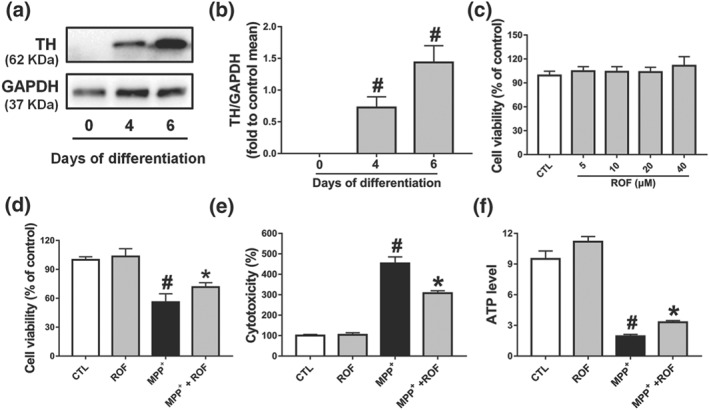
The protective effects of roflupram in the (LUHMES cells + MPP+) model. (a) Measurement of TH expression by western blotting on Days 0, 4, and 6 of differentiation. (b) Densitometric quantification of TH/GAPDH in (a). (c) Differentiated cells (d6) were incubated with various concentrations of roflupram (ROF) for 48 hr; CCK‐8 assay was used to evaluate the cytotoxic effects of roflupram on cells. Differentiated cells (d6) were pretreated with roflupram (20 μM) for 1 hr and then treated with 5 μM of MPP+ for an additional 48 hr. After treatment, cell viability was measured by CCK‐8 assay (d); LDH activity in the medium was measured by a LDH Cytotoxicity Assay Kit (e), and ATP levels were measured with an ATP Assay Kit (f). Data are presented as mean ± SD (n = 5) and represent five independent experiments. # P < .05, significantly different from control group. *P < .05, significantly different from MPP+‐treated group
3.6. Roflupram improves motor deficits in MPTP‐treated mice
We next investigated the protective effects of roflupram in a mouse model of PD. The known PDE4 inhibitor rolipram was used as a positive control and the experimental schedule is shown in Figure 6a. In the pole test, compared with that of mice in the vehicle group, mice in the MPTP‐treated group took much more time to turn downward and climb from the top of the pole to the bottom. Treatment with roflupram or rolipram ameliorated such motor deficits (Figure 6b). Representative pole test videos of MPTP‐treated mice with and without roflupram treatment is shown in Movie S1. Similarly, in the rotarod test, mice treated with roflupram or rolipram spent significantly longer on the rotarod, compared to mice injected with MPTP alone (Figure 6c, Movie S2). Additionally, we used a gait dynamic analysis system to investigate the effects of roflupram treatment on limb coordination and motor coordination in MPTP‐induced mice (Movie S3). As shown in Figure 6d–g, the length of one paw across the given stride was defined as the stride length, which was significantly shorter in MPTP‐treated mice, than that of vehicle‐treated mice. Stride length was increased after administration of roflupram or rolipram, indicating an increase in gait stability (Figure 6d). The stride frequency was determined by measuring the number of complete steps per paw per second, which was increased in the MPTP‐treated mice and was decreased after treatment with either roflupram or rolipram (Figure 6e). The ataxia coefficient was increased in MPTP‐induced mice, indicating an increase in gait instability compared to that of vehicle‐treated mice. Both roflupram and rolipram ameliorated gait inconsistency caused by MPTP (Figure 6f). Similarly, both ROF and rolipram treatments alleviated the reduction in stride duration in MPTP‐treated mice (Figure 6g). These results indicate that treatment with either roflupram or rolipram improved limb coordination and motor coordination in MPTP‐treated mice.
Figure 6.
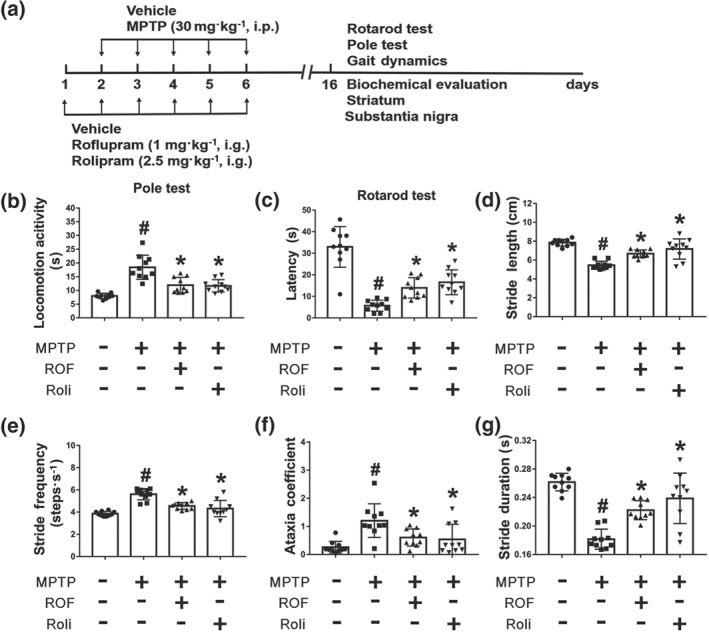
Behavioural deficits of MPTP‐treated mice are ameliorated by treatment with roflupram or rolipram. (a) Schedule of behavioural experiments. (b) Roflupram (ROF) or rolipram (Roli) ameliorated locomotor behaviour in MPTP‐treated mice. (c) Roflupram or rolipram improved the performance of MPTP‐treated mice in the rotarod test. In (d, e, f, g), the mice walked for about 5 s on a running belt at a speed of 30 cm·s−1. The parameters of stride length (d), stride frequency (e), ataxia coefficient (f), and stride duration (g) of the mice were obtained for statistical analysis. Gait results indicated that roflupram or rolipram improved limb mobility in MPTP‐treated mice. Data are presented as individual values with means ± SD (n = 10 mice per group). # P < .05, significantly different from vehicle group. *P < .05, significantly different from MPTP‐treated group
3.7. ROF promotes dopaminergic neuronal survival in both the SN and striatum in MPTP‐treated mice
Next, we explored whether the neuroprotective effect of roflupram was associated with a decreased loss of dopaminergic neurons in the SN and striatum. MPTP treatment markedly decreased Nissl‐positive cells in the SN and either roflupram or rolipram reversed the effect of MPTP and increased the number of neurons in this area (Figure 7a,b). We then explored the effects of MPTP and roflupram on the number of dopaminergic neuron bodies. As shown in Figure 7c–e, MPTP induced a significant loss of TH‐positive neurons in the SN. In contrast, treatment with roflupram clearly reduced the MPTP‐induced loss of TH‐positive cells. Rolipram exhibited a similar effect to that of roflupram. The expression of TH in mice treated with MPTP was lower compared with that in mice treated with vehicle. As expected, both roflupram and rolipram increased the level of TH significantly (Figure 7f,g).
Figure 7.
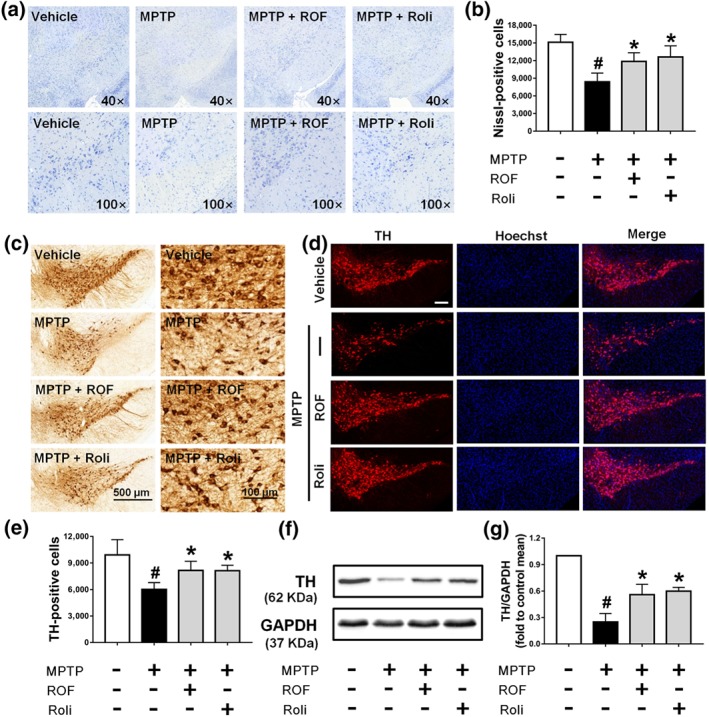
Roflupram (ROF) or rolipram (Roli) reduce the loss of TH in the SN of MPTP‐treated mice. (a) Nissl staining of the SN of mice in different samples under the microscope. (b) Number of Nissl‐positive cells, as exhibited in (a). (c) Immunohistochemistry of the SN of mice. (e) Number of TH‐positive cells, as exhibited in (c). (d) Tissue immunofluorescent staining of the SN of mice. Scale bar = 200 μm. (f) The SN of each mouse was analysed and the expression of TH was measured by western blotting. (g) Densitometric quantification of TH/GAPDH in (f). Data are presented as mean ± SD (n = 5) and represent five independent experiments. # P < .05, significantly different from vehicle group. *P < .05, significantly different from MPTP‐treated group
In addition to the SN, we also examined the loss of dopaminergic neuron fibres in the striatum. We found that roflupram increased the number of Nissl‐positive neurons (Figure 8a,b) and TH‐positive dopaminergic neuron fibres (Figure 8c–e) in the striatum. Additionally, roflupram enhanced the protein expression of TH in the striatum (Figure 8f–g). These results indicate that roflupram improved the survival of dopaminergic neurons in the context of MPTP‐induced PD in mice.
Figure 8.
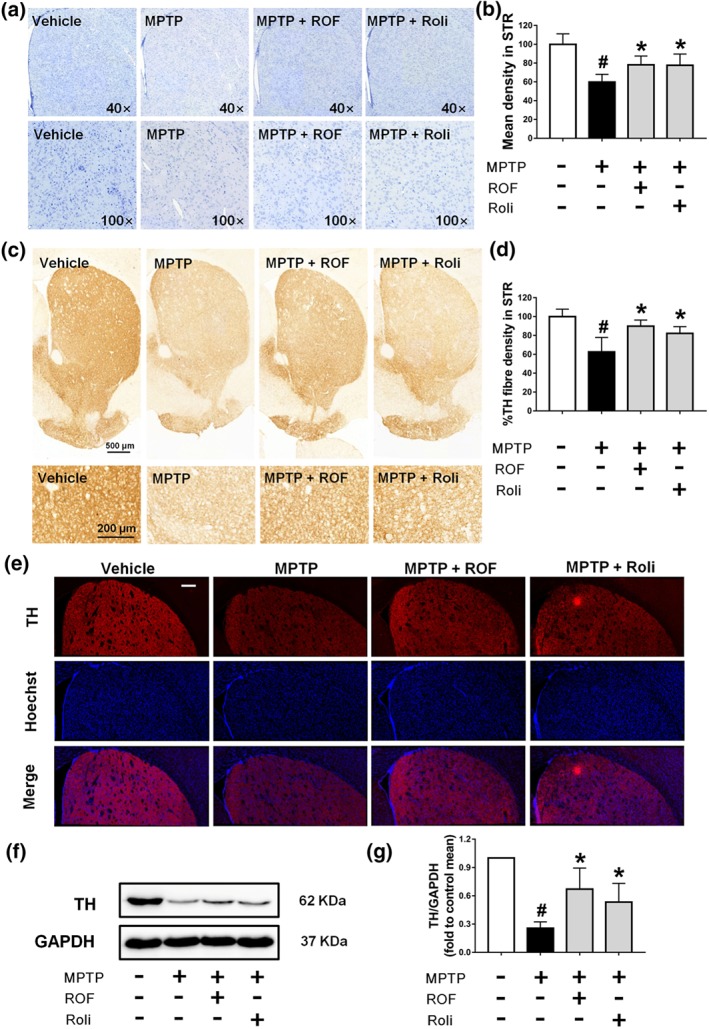
Roflupram (ROF) or rolipram (Roli) reduce the loss of TH in the striatum of MPTP‐treated mice. (a) Observation of Nissl staining results in the striatum in multiple samples. (b) The mean density of Nissl staining in the striatum (STR) was measured from the samples in (a). (c) Immunohistochemistry of the striatum of mice. (d) TH fibre density in the striatum was analysed from (c). (e) Tissue immunofluorescent staining of the striatum of mice. Scale bar = 200 μm. (f) The striatum of each mouse was analysed and the expression of TH measured by western blotting. (g) Densitometric quantification of TH/GAPDH in (f). Data are presented as mean ± SD (n = 5) and represent five independent experiments. # P < .05, significantly different from vehicle group. *P < .05, significantly different from MPTP‐treated group
3.8. Roflupram promotes the phosphorylation of CREB and expression of PGC‐1α in both the SN and striatum in MPTP‐treated mice
We next investigated the levels of phosphorylated CREB and PGC‐1α in the brains of MPTP‐challenged mice. Dopaminergic neuron bodies were identified by TH co‐labelling, and all type of cells in the SN were stained with Hoechst 33342 (Figure 9a). A significant decrease in PGC‐1α immunofluorescence was observed in the SN of mice treated with MPTP compared with that of the control SN. PGC‐1α and TH staining were markedly increased after treatment with roflupram or rolipram (Figure 9a). These results were consistent with the data that the protein level of PGC‐1α was increased significantly in mice treated with roflupram (Figure 9b). Roflupram treatment also clearly increased the fluorescence of p‐CREB, compared with that in mice treated with MPTP only (Figure 9c). Western blotting analysis also showed that the phosphorylation of CREB in roflupram‐treated mice was significantly enhanced, compared with that in mice only treated with MPTP (Figure 9d). The changes in p‐CREB and PGC‐1α in the striatum were similar to those in the SN. Roflupram treatment increased the protein expression of PGC‐1α in the striatum (Figure 10a,b). Additionally, the phosphorylation of CREB was enhanced significantly (Figure 10c,d). In addition, roflupram significantly enhanced the level of ATP in the SN of mice subjected to MPTP (Figure S5). The above data suggest that roflupram exerted a neuroprotective effect accompanied by up‐regulation of PGC‐1α and p‐CREB in an MPTP‐induced mouse model of PD.
Figure 9.
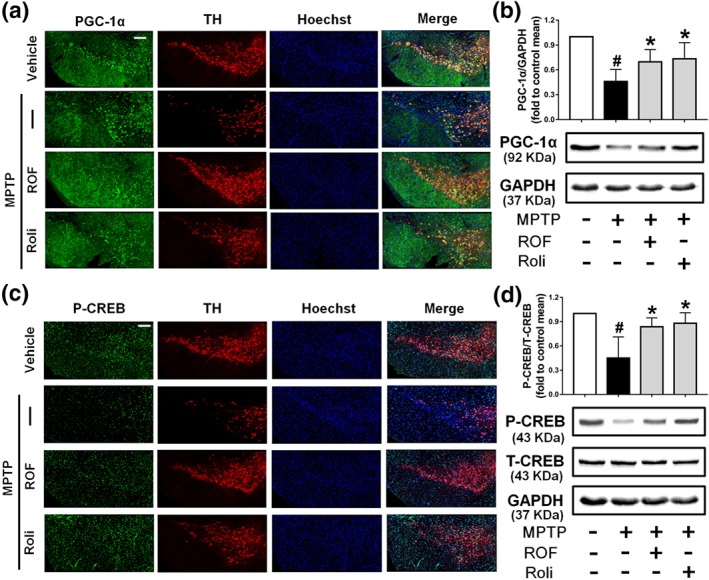
Roflupram (ROF) or rolipram (Roli) regulate the CREB/PGC‐1α pathway in the SN of MPTP‐treated mice. (a) Tissue immunofluorescence staining of the SN of mice was performed to detect the expression of PGC‐1α. Scale bar = 200 μm. (b) The SN of mice was analysed and the expression of PGC‐1α were measured by western blotting. (c) Tissue immunofluorescent staining of the SN of mice was carried out to measure the expression of p‐CREB. Scale bar = 200 μm. (d) The SN of mice was analysed and the expression of p‐CREB and total CREB was measured by western blotting. Data are presented as mean ± SD (n = 5) and represent five independent experiments. # P < .05, significantly different from vehicle group. *P < .05, significantly different from MPTP‐treated group
Figure 10.
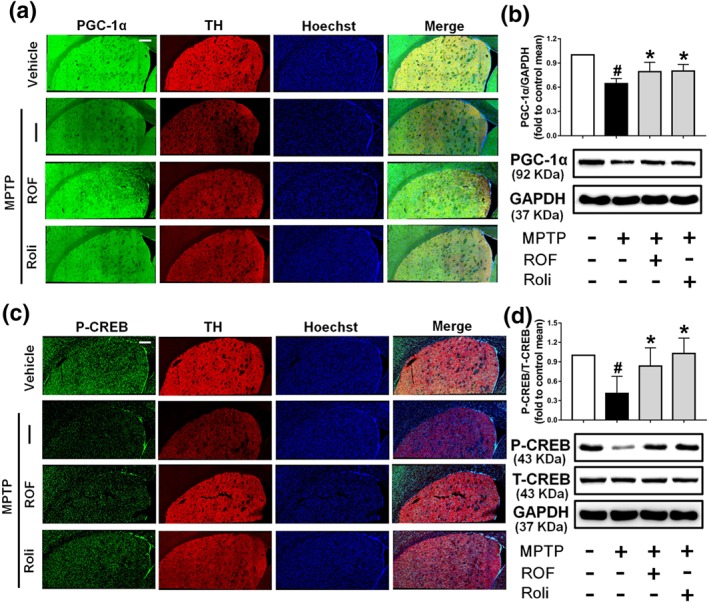
Roflupram (ROF) or rolipram (Roli) regulate the CREB/PGC‐1α pathway in the striatum of MPTP‐treated mice. (a) Tissue immunofluorescent staining of the striatum of mice was performed to detect the expression of PGC‐1α. Scale bar = 200 μm. (b) The striatum of each mouse was obtained, and the expression of PGC‐1α was measured by western blotting. (c) Tissue immunofluorescent staining of the striatum of mice was carried out to measure the expression of p‐CREB. Scale bar = 200 μm. (d) The striatum of each mouse was obtained, and the expression levels of p‐CREB and total CREB were measured by western blotting. Data are presented as mean ± SD (n = 5) and represent five independent experiments. # P < .05, significantly different from vehicle group. *P < .05, significantly different from MPTP‐treated group
4. DISCUSSION
The major finding of the current study is that inhibition of PDE4 by roflupram protects dopaminergic neurons against apoptosis in models of PD. Interestingly, we also noticed that MPP+ treatment resulted in nuclear accumulation of cleaved caspase‐3, while roflupram reversed the effect of MPP+ (Figure S1). It has been proposed that the nuclear translocation of caspase‐3 is mediated by an active transport (Kamada, Kikkawa, Tsujimoto, & Hunter, 2005). Activated caspase‐3 cleaves its nuclear substrates, resulting in DNA fragmentation and chromatin condensation (Luo et al., 2010). Our data suggested that roflupram blocked the pro‐apoptotic effects of MPP+ in dopaminergic neurons. As a selective inhibitor of PDE4, the protective effect of roflupram against PD has not been reported previously. In view of the many advantages of roflupram over the currently available PDE4 inhibitors, we were interested in investigating the protective role of ROF in PD models.
First, compared with that of the first‐generation PDE4 inhibitor rolipram, roflupram is a more potent inhibitorof PDE4. Roflupram exhibits lower IC50 values (56.2 nM) against PDE4CAT (core catalytic domains of human PDE4), than that of rolipram (2480 nM). In addition, roflupram also shows nanomolar IC50 values against PDE4A (31 nM), PDE4B (42.9 nM), and PDE4D (11.8 nM; Guo et al., 2017), showing the potency of roflupram as a PDE4 inhibitor. Second, compared with roflumilast, roflupram crosses the blood–brain barrier easily (Guo et al., 2017), which makes it possible to develop roflupram as a candidate drug for the treatment of neurological disorders. Following permeation of the blood‐brain barrier, roflupram had less or no emetic potential, compared with that of canonical PDE4 inhibitors (Guo et al., 2017). Finally, roflupram is effective in improving cognition in APP/PS1 mice (Guo et al., 2017). Importantly, PD is accompanied by non‐motor dysfunctions, including cognitive deficits. Hence, we hypothesized that roflupram might ameliorate cognitive deficits in PD. Additionally, inhibition of inflammation is beneficial for the amelioration of PD symptoms (Kim et al., 2018). Our previous studies have shown that roflupram decreased the production of IL‐1β and TNF‐α (Guo et al., 2017; You et al., 2017). What is more, our recent studies indicated that roflupram promoted autophagy in neuronal cells (You et al., 2017). Appropriate autophagy helps to clean out damaged cells and toxins, which is beneficial for neuronal survival. Hence, we also predicted that PD patients would benefit from both the anti‐inflammatory and autophagy‐enhancing effects of roflupram.
Mitochondrial dysfunction plays an important role in the progression of PD (Burbulla et al., 2017; Scholpa, Lynn, Corum, Boger, & Schnellmann, 2018). Damaged mitochondria can negatively affect the physiological functions of neurons. For example, impaired mitochondria produce excessive ROS that can destroy the cell membrane and nuclear membrane (Hedde, Hanks, Schmidt, & Hughes, 2017). Mitochondria also influence calcium homeostasis inside and outside of cells, which directly affects neuronal functions (Surmeier, 2018). In addition, impaired mitochondria produce less ATP, which will subsequently create problems with axonal transport and neurotransmission (Smith & Gallo, 2018). All of these aspects can affect the survival and functions of dopamine‐producing neurons and, thus, contribute to the development of PD. In our previous studies, we found that inhibition of PDE4 in SH‐SY5Y cells could counteract the decrease of MMP induced by MPP+ (Zhong et al., 2018). We also found that inhibition of intracellular PDE4 activity in neurons increased the production of ATP under the condition of oxygen–glucose deprivation (Xu et al., 2019). These findings drew our attention to the regulation of mitochondrial function by PDE4. Our present results showed that inhibition of PDE4 ameliorated mitochondrial morphology (Figure 2). Several possible biological functions of mitochondrial networks and mitochondrial footprints have been proposed, such as improved quality control, an increased ATP synthesis rate, and protection against apoptotic stressors (Briston et al., 2018; Hoitzing, Johnston, & Jones, 2015). These morphological changes were paralleled with improved biological functions of mitochondria, including increased OCR, mtDNA, MMP, and ATP production (Figure 2). We would like to point out that IC50 value of ROF against PDE4 is 56.2 nM (Guo et al., 2017), while in the present study, the neuroprotective effects of ROF were observed at a concentration of 20 μM, which is clearly higher that its IC50 value. The detailed mechanisms underlying these neuroprotective effects need further investigation. The effects of PDE4 inhibition on the activities of other PDE family members, especially PDE7 and PDE8, will probably need to be taken into consideration.
We then investigated the potential intracellular mechanisms of roflupram protection. PGC‐1α is highly expressed in neuronal cells (Canto & Auwerx, 2009). Physiologically, PGC‐1α functions as a regulator of mitochondrial biogenesis, ATP production, and mitophagy, thereby maintaining the normal biological functions of mitochondria (Johri, Chandra, & Flint Beal, 2013). Interestingly, inhibition of PDE4 increases cAMP levels in neurons, thereby activating the PKA/CREB signalling pathway (Richter et al., 2013). Of note, the promoter of PGC‐1α contains the binding site for CREB (the sequence is TGACGTCA; Fernandez‐Marcos & Auwerx, 2011). CREB phosphorylation increases the transcription of PGC‐1α (Wu et al., 2006). Activation of the CREB/PGC‐1α signalling pathway in dopaminergic neurons contributes to an increased cell viability and this pathway has been proposed as a promising therapeutic target for the treatment of PD (Kang et al., 2017; Makela et al., 2016). These findings are consistent with data obtained in the present studies. Roflupram induced the activation of CREB, as shown by the increase in the phosphorylation of CREB at Ser133 (Figure 3c,d). In addition to CREB, we also found that the protein expression of PGC‐1α was increased (Figure 3e,f). Interestingly, as shown in Figure 1c,e,f, 20‐μM roflupram exerted around 10% cytoprotection, while roflupram produced much higher restoration of the levels of phosphorylated CREB and PGC‐1α. This observation suggests that inhibition of CREB/PGC‐1α pathway is not the only mechanism used by MPP+ to destroy neuronal cells. For example, MPP+ induced neurotoxicity by promoting an overload of misfolded proteins in PD (Lehtonen et al., 2016). MPP+ may also induce neuronal injury through up‐regulating the expression of cation channels (Sun et al., 2018). In this study, we confirmed that roflupram did activate the CREB/PGC‐1α pathway in neuronal cells treated with MPP+, but that such effects were insufficient to totally block the toxicity induced by MPP+. Combination of roflupram with other neuroprotective agents may be needed for complete block of the effects of MPP+.
To further confirm the involvement of PKA/CREB, we then employed the PKA inhibitor H‐89 to block the activation of CREB mediated by cAMP/PKA and to confirm the regulation by CREB of the expression of PGC‐1α (Figure 4a–c). The results showed that H‐89 decreased both the phosphorylation of CREB and the expression of PGC‐1α, indicating that PGC‐1α was regulated by PKA/CREB. In accordance with these findings, H‐89 and PGC‐1α siRNA blocked the reduction of both MMP and cell viability. Hence, roflupram mediated its protective effect through activating the PKA/CREB/PGC‐1α signalling pathway. Most importantly, roflupram showed similar neuroprotection in human dopaminergic neurons, similar to that in SH‐SY5Y cells (Figure 5). These findings further support the conclusion that roflupram conferred neuroprotection against MPP+‐triggered neuronal cell death.
Consistent with these in vitro data, treatment with roflupram in mice challenged with MPTP improved motor behavioural performance and decreased the loss of dopaminergic neurons induced by MPTP. Importantly, the phosphorylation of CREB and expression of PGC‐1α were enhanced by roflupram in both the SN and striatum. These findings support the hypothesis that PDE4 might be a potential target for PD treatment and that CREB/PGC‐1α is involved in the protective effect of roflupram. It is noteworthy that PGC‐1α regulates mitochondrial production and functions in several ways. For example, PGC‐1α is involved in mitophagy (Yeo, Kang, Gomez‐Cabrera, Vina, & Ji, 2019). Appropriate autophagy is beneficial for the survival of dopaminergic neurons (Hu, Chen, Zhang, & Ma, 2017). Our present study mainly focused on the regulation by PDE4 inhibition of the CREB/PGC‐1α pathway and its role in neuroprotection against MPP+/MPTP. Further studies are needed to determine the involvement of mitochondrial biogenesis and autophagy in the protective effect of roflupram.
In conclusion, our results revealed that roflupram prevented MPP+/MPTP‐induced degeneration of dopaminergic neurons, ameliorated motor dysfunctions, increased TH expression, and restored mitochondrial function. These findings indicate that roflupram is probably a promising drug candidate for treating PD. Further research and preclinical studies are needed to systematically investigate the potential of roflupram for the treatment of PD.
AUTHOR CONTRIBUTIONS
J.Z., W.D., Y.Q., J.X. (Jinfeng Xie), and J.X. (Jiao Xiao) performed the experiments and analysed the data. J.Z., H.W., and J.P. drafted the manuscript. H.W. and J.P. designed research project, supervised the experiments, and critically reviewed the manuscript. All authors have given their final approval for the manuscript.
CONFLICT OF INTEREST
The authors declare that they have no conflict of interest.
DECLARATION OF TRANSPARENCY AND SCIENTIFIC RIGOUR
This Declaration acknowledges that this paper adheres to the principles for transparent reporting and scientific rigour of preclinical research as stated in the BJP guidelines for https://bpspubs.onlinelibrary.wiley.com/doi/full/10.1111/bph.14207, https://bpspubs.onlinelibrary.wiley.com/doi/full/10.1111/bph.14208, and https://bpspubs.onlinelibrary.wiley.com/doi/full/10.1111/bph.14206, and as recommended by funding agencies, publishers and other organizations engaged with supporting research.
Supporting information
Movie S1. Representative pole test videos of MPTP‐treated mice with and without ROF. Mice were placed on top of the straight rod and we recorded the time that the mice climbed along the wood to the bottom of the rod.
Movie S2. Representative rotarod test videos of MPTP‐treated mice with and without ROF. The mice were placed on a rotarod with a rolling speed of 12 rpm, and the duration that the mice stayed on the rotarod were observed and recorded.
Movie S3. Representative pole test videos of MPTP‐treated mice with and without ROF. The mice were placed in a walking compartment with a width of 7 cm and a length of 30 cm. A high‐speed camera at the lower end of the transparent treadmill belt continuously captured the walking images of the mice. The mice walked for about 5 s on the running belt at a speed of 30 cm/s. Digital paw prints and dynamic gait signals for each of the 4 limbs were generated by the gait dynamic system.
Figure S1. ROF decreases the level of cleaved caspase‐3 in SH‐SY5Y cells treated with MPP+. The SH‐SY5Y cells were pretreated with various concentrations of ROF for 1 h, and then stimulated with 500 μM of MPP+ for 48 h. Immunofluorescence was performed with anti‐cleaved caspase‐3 (green). Hoechst was used to stain the nuclei (blue). The stained cells were photographed using an inverted confocal microscope. Scale bar = 50 μm.
Figure S2. Verification of siRNA‐mediated knock down of PGC‐1α in SH‐SY5Y cells. (A) Knocking‐down efficiency of PGC‐1α siRNA (siPGC‐1α) (50 nM) in SH‐SY5Y cells was verified via detecting the protein expression of PGC‐1α by Western blotting. (B) Densitometric quantification of PGC‐1α/GAPDH in (A). (C) SH‐SY5Y cells were transfected with PGC‐1α siRNA or random siRNA for 24 h. Transfected cells were then treated with MPP+ (500 μM) and ROF (20 μM) for an additional 24 h. the expression level of PGC‐1α was detected by Western blotting. (D) Densitometric quantification of PGC‐1α/GAPDH in (C). (D) Densitometric quantification of PGC‐1α/GAPDH in (C). Data are presented as mean ± SD (n = 5). #P < 0.05 versus negative control (NC) group. *P < 0.05 versus MPP+‐treated group. $P < 0.05 versus MPP+ + ROF‐treated group.
Figure S3. MPP+ could decrease cell viability of LUHMES cells in a concentration‐dependent manner. After 6 days of differentiation, LUHMES cells were treated with MPP+ (2.5–40 μM) for 48 h, and the cell viability of LUHMES was reduced in a concentration‐dependent manner. MPP+‐induced a comparable level of toxicity at a concentration of 5 μM (56% cell viability reduction). Data are presented as mean ± SD (n = 5) and represent five independent experiments. #P < 0.05 versus control group.
Figure S4. ROF treatments improved limb coordination and motor coordination in MPTP‐treated mice. The mice were walked for about 5 sec on a transparent treadmill belt at a speed of 30 cm/s. The gait analysis system continuously imaged the underside of the mice. Digital paw prints and dynamic gait signals for each of the 4 limbs were generated by the gait dynamic system (DigiGait Analysis Software Version 14.5). Representative of the hind limbs of mice in each group is shown.
Figure S5. ROF increases the level of ATP in the SN of MPTP‐induced mice. The concentration of ATP in the SN of MPTP‐induced mice was measured by an enzyme‐linked immunoassay kit, and the value was measured by a luminometer. Data are presented as mean ± SD (n = 5). #P < 0.05 versus vehicle group. *P < 0.05 versus MPTP‐treated group.
ACKNOWLEDGEMENTS
This work was supported by National Natural Science Foundation of China (81773698), the Program for Changjiang Scholars and Innovative Research Team in University (IRT_16R37), Science and Technology Program of Guangdong (2018B030334001 and 2015B020211007), and Science and Technology Program of Guangzhou (201604020112).
Zhong J, Dong W, Qin Y, et al. Roflupram exerts neuroprotection via activation of CREB/PGC‐1α signalling in experimental models of Parkinson's disease. Br J Pharmacol. 2020;177:2333–2350. 10.1111/bph.14983
Jiahong Zhong and Wenli Dong have equal contribution to this work.
Contributor Information
Jiangping Xu, Email: jpx@smu.edu.cn.
Haitao Wang, Email: wht821@smu.edu.cn.
REFERENCES
- Alexander, S. P. H. , Fabbro, D. , Kelly, E. , Mathie, A. , Peters, J. A. , Veale, E. L. , … CGTP Collaborators (2019). THE CONCISE GUIDE TO PHARMACOLOGY 2019/20: Enzymes. British Journal of Pharmacology, 176, S297–S396. 10.1111/bph.14752 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amende, I. , Kale, A. , McCue, S. , Glazier, S. , Morgan, J. P. , & Hampton, T. G. (2005). Gait dynamics in mouse models of Parkinson's disease and Huntington's disease. Journal of Neuroengineering and Rehabilitation, 2, 20 10.1186/1743-0003-2-20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Briston, T. , Stephen, J. M. , Thomas, L. W. , Esposito, C. , Chung, Y. L. , Syafruddin, S. E. , … Ashcroft, M. (2018). VHL‐mediated regulation of CHCHD4 and mitochondrial function. Frontiers in Oncology, 8, 388 10.3389/fonc.2018.00388 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burbulla, L. F. , Song, P. , Mazzulli, J. R. , Zampese, E. , Wong, Y. C. , Jeon, S. , … Krainc, D. (2017). Dopamine oxidation mediates mitochondrial and lysosomal dysfunction in Parkinson's disease. Science, 357(6357), 1255–1261. 10.1126/science.aam9080 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Canto, C. , & Auwerx, J. (2009). PGC‐1α, SIRT1 and AMPK, an energy sensing network that controls energy expenditure. Current Opinion in Lipidology, 20(2), 98–105. 10.1097/MOL.0b013e328328d0a4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ciron, C. , Zheng, L. , Bobela, W. , Knott, G. W. , Leone, T. C. , Kelly, D. P. , & Schneider, B. L. (2015). PGC‐1α activity in nigral dopamine neurons determines vulnerability to α‐synuclein. Acta Neuropathologica Communications, 3, 16 10.1186/s40478-015-0200-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Curtis, M. J. , Alexander, S. , Cirino, G. , Docherty, J. R. , George, C. H. , Giembycz, M. A. , … Ahluwalia, A. (2018). Experimental design and analysis and their reporting II: Updated and simplified guidance for authors and peer reviewers. British Journal of Pharmacology, 175(7), 987–993. 10.1111/bph.14153 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dauer, W. , & Przedborski, S. (2003). Parkinson's disease: Mechanisms and models. Neuron, 39(6), 889–909. 10.1016/S0896-6273(03)00568-3 [DOI] [PubMed] [Google Scholar]
- Dyke, H. J. , & Montana, J. G. (2002). Update on the therapeutic potential of PDE4 inhibitors. Expert Opinion on Investigational Drugs, 11(1), 1–13. 10.1517/13543784.11.1.1 [DOI] [PubMed] [Google Scholar]
- Efremova, L. , Schildknecht, S. , Adam, M. , Pape, R. , Gutbier, S. , Hanf, B. , … Leist, M. (2015). Prevention of the degeneration of human dopaminergic neurons in an astrocyte co‐culture system allowing endogenous drug metabolism. British Journal of Pharmacology, 172(16), 4119–4132. 10.1111/bph.13193 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernandez‐Marcos, P. J. , & Auwerx, J. (2011). Regulation of PGC‐1α, a nodal regulator of mitochondrial biogenesis. The American Journal of Clinical Nutrition, 93(4), 884S–890S. 10.3945/ajcn.110.001917 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldberg, N. R. , Hampton, T. , McCue, S. , Kale, A. , & Meshul, C. K. (2011). Profiling changes in gait dynamics resulting from progressive 1‐methyl‐4‐phenyl‐1,2,3,6‐tetrahydropyridine‐induced nigrostriatal lesioning. Journal of Neuroscience Research, 89(10), 1698–1706. 10.1002/jnr.22699 [DOI] [PubMed] [Google Scholar]
- Guo, H. , Cheng, Y. , Wang, C. , Wu, J. , Zou, Z. , Niu, B. , … Xu, J. (2017). FFPM, a PDE4 inhibitor, reverses learning and memory deficits in APP/PS1 transgenic mice via cAMP/PKA/CREB signaling and anti‐inflammatory effects. Neuropharmacology, 116, 260–269. 10.1016/j.neuropharm.2017.01.004 [DOI] [PubMed] [Google Scholar]
- Harding, S. D. , Sharman, J. L. , Faccenda, E. , Southan, C. , Pawson, A. J. , Ireland, S. , … NC‐IUPHAR (2018). The IUPHAR/BPS guide to pharmacology in 2018: Updates and expansion to encompass the new guide to immunopharmacology. Nucleic Acids Research, 46(D1), D1091–D1106. 10.1093/nar/gkx1121 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hedde, J. R. , Hanks, A. N. , Schmidt, C. J. , & Hughes, Z. A. (2017). The isozyme selective phosphodiesterase‐4 inhibitor, ABI‐4, attenuates the effects of lipopolysaccharide in human cells and rodent models of peripheral and CNS inflammation. Brain, Behavior, and Immunity, 64, 285–295. 10.1016/j.bbi.2017.04.015 [DOI] [PubMed] [Google Scholar]
- Heikkila, R. E. , Hess, A. , & Duvoisin, R. C. (1984). Dopaminergic neurotoxicity of 1‐methyl‐4‐phenyl‐1,2,5,6‐tetrahydropyridine in mice. Science, 224(4656), 1451–1453. 10.1126/science.6610213 [DOI] [PubMed] [Google Scholar]
- Herzig, S. , Long, F. , Jhala, U. S. , Hedrick, S. , Quinn, R. , Bauer, A. , … Montminy, M. (2001). CREB regulates hepatic gluconeogenesis through the coactivator PGC‐1. Nature, 413(6852), 179–183. 10.1038/35093131 [DOI] [PubMed] [Google Scholar]
- Hoitzing, H. , Johnston, I. G. , & Jones, N. S. (2015). What is the function of mitochondrial networks? A theoretical assessment of hypotheses and proposal for future research. BioEssays: News and Reviews in Molecular, Cellular and Developmental Biology, 37(6), 687–700. 10.1002/bies.201400188 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu, Z. Y. , Chen, B. , Zhang, J. P. , & Ma, Y. Y. (2017). Up‐regulation of autophagy‐related gene 5 (ATG5) protects dopaminergic neurons in a zebrafish model of Parkinson's disease. The Journal of Biological Chemistry, 292(44), 18062–18074. 10.1074/jbc.M116.764795 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jankovic, J. (2008). Parkinson's disease: Clinical features and diagnosis. Journal of Neurology, Neurosurgery, and Psychiatry, 79(4), 368–376. 10.1136/jnnp.2007.131045 [DOI] [PubMed] [Google Scholar]
- Johri, A. , Chandra, A. , & Flint Beal, M. (2013). PGC‐1α, mitochondrial dysfunction, and Huntington's disease. Free Radical Biology & Medicine, 62, 37–46. 10.1016/j.freeradbiomed.2013.04.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamada, S. , Kikkawa, U. , Tsujimoto, Y. , & Hunter, T. (2005). Nuclear translocation of caspase‐3 is dependent on its proteolytic activation and recognition of a substrate‐like protein(s). The Journal of Biological Chemistry, 280(2), 857–860. 10.1074/jbc.C400538200 [DOI] [PubMed] [Google Scholar]
- Kang, H. , Khang, R. , Ham, S. , Jeong, G. R. , Kim, H. , Jo, M. , … Shin, J. H. (2017). Activation of the ATF2/CREB‐PGC‐1α pathway by metformin leads to dopaminergic neuroprotection. Oncotarget, 8(30), 48603–48618. 10.18632/oncotarget.18122 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kilkenny, C. , Browne, W. , Cuthill, I. C. , Emerson, M. , Altman, D. G. , & Group NCRRGW (2010). Animal research: Reporting in vivo experiments: The ARRIVE guidelines. British Journal of Pharmacology, 160(7), 1577–1579. 10.1111/j.1476-5381.2010.00872.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim, H. W. , Lee, H. S. , Kang, J. M. , Bae, S. H. , Kim, C. , Lee, S. H. , … Moon, J. (2018). Dual effects of human placenta‐derived neural cells on neuroprotection and the inhibition of neuroinflammation in a rodent model of Parkinson's disease. Cell Transplantation, 27(5), 814–830. 10.1177/0963689718766324 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kostrzewa, R. M. , Kostrzewa, J. P. , & Brus, R. (2002). Neuroprotective and neurotoxic roles of levodopa (L‐DOPA) in neurodegenerative disorders relating to Parkinson's disease. Amino Acids, 23(1–3), 57–63. 10.1007/s00726-001-0110-x [DOI] [PubMed] [Google Scholar]
- Krug, A. K. , Gutbier, S. , Zhao, L. , Poltl, D. , Kullmann, C. , Ivanova, V. , … Leist, M. (2014). Transcriptional and metabolic adaptation of human neurons to the mitochondrial toxicant MPP+ . Cell Death & Disease, 5, e1222 10.1038/cddis.2014.166 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lehtonen, S. , Jaronen, M. , Vehvilainen, P. , Lakso, M. , Rudgalvyte, M. , Keksa‐Goldsteine, V. , … Goldsteins, G. (2016). Inhibition of excessive oxidative protein folding is protective in MPP+ toxicity‐induced Parkinson's disease models. Antioxidants & Redox Signaling, 25(8), 485–497. 10.1089/ars.2015.6402 [DOI] [PubMed] [Google Scholar]
- Luo, M. , Lu, Z. , Sun, H. , Yuan, K. , Zhang, Q. , Meng, S. , … Zhai, Z. (2010). Nuclear entry of active caspase‐3 is facilitated by its p3‐recognition‐based specific cleavage activity. Cell Research, 20(2), 211–222. 10.1038/cr.2010.9 [DOI] [PubMed] [Google Scholar]
- Makela, J. , Tselykh, T. V. , Kukkonen, J. P. , Eriksson, O. , Korhonen, L. T. , & Lindholm, D. (2016). Peroxisome proliferator‐activated receptor‐gamma (PPARγ) agonist is neuroprotective and stimulates PGC‐1α expression and CREB phosphorylation in human dopaminergic neurons. Neuropharmacology, 102, 266–275. 10.1016/j.neuropharm.2015.11.020 [DOI] [PubMed] [Google Scholar]
- McGrath, J. C. , & Lilley, E. (2015). Implementing guidelines on reporting research using animals (ARRIVE etc.): New requirements for publication in BJP . British Journal of Pharmacology, 172(13), 3189–3193. 10.1111/bph.12955 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Migdalska‐Richards, A. , & Schapira, A. H. (2016). The relationship between glucocerebrosidase mutations and Parkinson disease. Journal of Neurochemistry, 139(Suppl 1), 77–90. 10.1111/jnc.13385 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mudo, G. , Makela, J. , Di Liberto, V. , Tselykh, T. V. , Olivieri, M. , Piepponen, P. , … Lindholm, D. (2012). Transgenic expression and activation of PGC‐1α protect dopaminergic neurons in the MPTP mouse model of Parkinson's disease. Cellular and Molecular Life Sciences: CMLS, 69(7), 1153–1165. 10.1007/s00018-011-0850-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niccolini, F. , Wilson, H. , Pagano, G. , Coello, C. , Mehta, M. A. , Searle, G. E. , … Politis, M. (2017). Loss of phosphodiesterase 4 in Parkinson disease: Relevance to cognitive deficits. Neurology, 89(6), 586–593. 10.1212/WNL.0000000000004201 [DOI] [PubMed] [Google Scholar]
- Picconi, B. , Hernandez, L. F. , Obeso, J. A. , & Calabresi, P. (2018). Motor complications in Parkinson's disease: Striatal molecular and electrophysiological mechanisms of dyskinesias. Movement Disorders: Official Journal of the Movement Disorder Society, 33(6), 867–876. 10.1002/mds.27261 [DOI] [PubMed] [Google Scholar]
- Richter, W. , Menniti, F. S. , Zhang, H. T. , & Conti, M. (2013). PDE4 as a target for cognition enhancement. Expert Opinion on Therapeutic Targets, 17(9), 1011–1027. 10.1517/14728222.2013.818656 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schildknecht, S. , Karreman, C. , Poltl, D. , Efremova, L. , Kullmann, C. , Gutbier, S. , … Leist, M. (2013). Generation of genetically‐modified human differentiated cells for toxicological tests and the study of neurodegenerative diseases. ALTEX, 30(4), 427–444. 10.14573/altex.2013.4.427 [DOI] [PubMed] [Google Scholar]
- Scholpa, N. E. , Lynn, M. K. , Corum, D. , Boger, H. A. , & Schnellmann, R. G. (2018). 5‐HT1F receptor‐mediated mitochondrial biogenesis for the treatment of Parkinson's disease. British Journal of Pharmacology, 175(2), 348–358. 10.1111/bph.14076 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scholz, D. , Poltl, D. , Genewsky, A. , Weng, M. , Waldmann, T. , Schildknecht, S. , & Leist, M. (2011). Rapid, complete and large‐scale generation of post‐mitotic neurons from the human LUHMES cell line. Journal of Neurochemistry, 119(5), 957–971. 10.1111/j.1471-4159.2011.07255.x [DOI] [PubMed] [Google Scholar]
- Smith, G. M. , & Gallo, G. (2018). The role of mitochondria in axon development and regeneration. Developmental Neurobiology, 78(3), 221–237. 10.1002/dneu.22546 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stocchi, F. , Torti, M. , & Fossati, C. (2016). Advances in dopamine receptor agonists for the treatment of Parkinson's disease. Expert Opinion on Pharmacotherapy, 17(14), 1889–1902. 10.1080/14656566.2016.1219337 [DOI] [PubMed] [Google Scholar]
- St‐Pierre, J. , Drori, S. , Uldry, M. , Silvaggi, J. M. , Rhee, J. , Jager, S. , … Spiegelman, B. M. (2006). Suppression of reactive oxygen species and neurodegeneration by the PGC‐1 transcriptional coactivators. Cell, 127(2), 397–408. 10.1016/j.cell.2006.09.024 [DOI] [PubMed] [Google Scholar]
- Sun, Y. , Sukumaran, P. , Selvaraj, S. , Cilz, N. I. , Schaar, A. , Lei, S. , & Singh, B. B. (2018). TRPM2 promotes neurotoxin MPP+/MPTP‐induced cell death. Molecular Neurobiology, 55(1), 409–420. 10.1007/s12035-016-0338-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Surmeier, D. J. (2018). Determinants of dopaminergic neuron loss in Parkinson's disease. The FEBS Journal, 285(19), 3657–3668. 10.1111/febs.14607 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tomkova, S. , Misuth, M. , Lenkavska, L. , Miskovsky, P. , & Huntosova, V. (2018). In vitro identification of mitochondrial oxidative stress production by time‐resolved fluorescence imaging of glioma cells. Biochimica et Biophysica Acta, Molecular Cell Research, 1865(4), 616–628. 10.1016/j.bbamcr.2018.01.012 [DOI] [PubMed] [Google Scholar]
- Valente, A. J. , Maddalena, L. A. , Robb, E. L. , Moradi, F. , & Stuart, J. A. (2017). A simple ImageJ macro tool for analyzing mitochondrial network morphology in mammalian cell culture. Acta Histochemica, 119(3), 315–326. 10.1016/j.acthis.2017.03.001 [DOI] [PubMed] [Google Scholar]
- Wu, Z. , Huang, X. , Feng, Y. , Handschin, C. , Feng, Y. , Gullicksen, P. S. , … Stevenson, S. C. (2006). Transducer of regulated CREB‐binding proteins (TORCs) induce PGC‐1α transcription and mitochondrial biogenesis in muscle cells. Proceedings of the National Academy of Sciences of the United States of America, 103(39), 14379–14384. 10.1073/pnas.0606714103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu, B. , Wang, T. , Xiao, J. , Dong, W. , Wen, H. Z. , Wang, X. , … Wang, H. (2019). FCPR03, a novel phosphodiesterase 4 inhibitor, alleviates cerebral ischemia/reperfusion injury through activation of the AKT/GSK3β/β‐catenin signaling pathway. Biochemical Pharmacology, 163, 234–249. 10.1016/j.bcp.2019.02.023 [DOI] [PubMed] [Google Scholar]
- Yang, L. , Calingasan, N. Y. , Lorenzo, B. J. , & Beal, M. F. (2008). Attenuation of MPTP neurotoxicity by rolipram, a specific inhibitor of phosphodiesterase IV. Experimental Neurology, 211(1), 311–314. 10.1016/j.expneurol.2007.02.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yeo, D. , Kang, C. , Gomez‐Cabrera, M. C. , Vina, J. , & Ji, L. L. (2019). Intensified mitophagy in skeletal muscle with aging is downregulated by PGC‐1α overexpression in vivo. Free Radical Biology & Medicine, 130, 361–368. 10.1016/j.freeradbiomed.2018.10.456 [DOI] [PubMed] [Google Scholar]
- You, T. , Cheng, Y. , Zhong, J. , Bi, B. , Zeng, B. , Zheng, W. , … Xu, J. (2017). Roflupram, a phosphodiesterase 4 inhibitior, suppresses inflammasome activation through autophagy in microglial cells. ACS Chemical Neuroscience, 8(11), 2381–2392. 10.1021/acschemneuro.7b00065 [DOI] [PubMed] [Google Scholar]
- Zheng, S. , Kaur, G. , Wang, H. , Li, M. , Macnaughtan, M. , Yang, X. , … Ke, H. (2008). Design, synthesis, and structure‐activity relationship, molecular modeling, and NMR studies of a series of phenyl alkyl ketones as highly potent and selective phosphodiesterase‐4 inhibitors. Journal of Medicinal Chemistry, 51(24), 7673–7688. 10.1021/jm701635j [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhong, J. , Xie, J. , Xiao, J. , Li, D. , Xu, B. , Wang, X. , … Wang, H. (2019). Inhibition of PDE4 by FCPR16 induces AMPK‐dependent autophagy and confers neuroprotection in SH‐SY5Y cells and neurons exposed to MPP+‐induced oxidative insult. Free Radical Biology & Medicine, 135, 87–101. 10.1016/j.freeradbiomed.2019.02.027 [DOI] [PubMed] [Google Scholar]
- Zhong, J. , Yu, H. , Huang, C. , Zhong, Q. , Chen, Y. , Xie, J. , … Wang, H. (2018). Inhibition of phosphodiesterase 4 by FCPR16 protects SH‐SY5Y cells against MPP+‐induced decline of mitochondrial membrane potential and oxidative stress. Redox Biology, 16, 47–58. 10.1016/j.redox.2018.02.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Movie S1. Representative pole test videos of MPTP‐treated mice with and without ROF. Mice were placed on top of the straight rod and we recorded the time that the mice climbed along the wood to the bottom of the rod.
Movie S2. Representative rotarod test videos of MPTP‐treated mice with and without ROF. The mice were placed on a rotarod with a rolling speed of 12 rpm, and the duration that the mice stayed on the rotarod were observed and recorded.
Movie S3. Representative pole test videos of MPTP‐treated mice with and without ROF. The mice were placed in a walking compartment with a width of 7 cm and a length of 30 cm. A high‐speed camera at the lower end of the transparent treadmill belt continuously captured the walking images of the mice. The mice walked for about 5 s on the running belt at a speed of 30 cm/s. Digital paw prints and dynamic gait signals for each of the 4 limbs were generated by the gait dynamic system.
Figure S1. ROF decreases the level of cleaved caspase‐3 in SH‐SY5Y cells treated with MPP+. The SH‐SY5Y cells were pretreated with various concentrations of ROF for 1 h, and then stimulated with 500 μM of MPP+ for 48 h. Immunofluorescence was performed with anti‐cleaved caspase‐3 (green). Hoechst was used to stain the nuclei (blue). The stained cells were photographed using an inverted confocal microscope. Scale bar = 50 μm.
Figure S2. Verification of siRNA‐mediated knock down of PGC‐1α in SH‐SY5Y cells. (A) Knocking‐down efficiency of PGC‐1α siRNA (siPGC‐1α) (50 nM) in SH‐SY5Y cells was verified via detecting the protein expression of PGC‐1α by Western blotting. (B) Densitometric quantification of PGC‐1α/GAPDH in (A). (C) SH‐SY5Y cells were transfected with PGC‐1α siRNA or random siRNA for 24 h. Transfected cells were then treated with MPP+ (500 μM) and ROF (20 μM) for an additional 24 h. the expression level of PGC‐1α was detected by Western blotting. (D) Densitometric quantification of PGC‐1α/GAPDH in (C). (D) Densitometric quantification of PGC‐1α/GAPDH in (C). Data are presented as mean ± SD (n = 5). #P < 0.05 versus negative control (NC) group. *P < 0.05 versus MPP+‐treated group. $P < 0.05 versus MPP+ + ROF‐treated group.
Figure S3. MPP+ could decrease cell viability of LUHMES cells in a concentration‐dependent manner. After 6 days of differentiation, LUHMES cells were treated with MPP+ (2.5–40 μM) for 48 h, and the cell viability of LUHMES was reduced in a concentration‐dependent manner. MPP+‐induced a comparable level of toxicity at a concentration of 5 μM (56% cell viability reduction). Data are presented as mean ± SD (n = 5) and represent five independent experiments. #P < 0.05 versus control group.
Figure S4. ROF treatments improved limb coordination and motor coordination in MPTP‐treated mice. The mice were walked for about 5 sec on a transparent treadmill belt at a speed of 30 cm/s. The gait analysis system continuously imaged the underside of the mice. Digital paw prints and dynamic gait signals for each of the 4 limbs were generated by the gait dynamic system (DigiGait Analysis Software Version 14.5). Representative of the hind limbs of mice in each group is shown.
Figure S5. ROF increases the level of ATP in the SN of MPTP‐induced mice. The concentration of ATP in the SN of MPTP‐induced mice was measured by an enzyme‐linked immunoassay kit, and the value was measured by a luminometer. Data are presented as mean ± SD (n = 5). #P < 0.05 versus vehicle group. *P < 0.05 versus MPTP‐treated group.