

Abstract
Herein, we present the cathodic paths of the Group-7 metal complex [Re(3,3′-DHBPY)(CO)3Cl] (3,3′-DHBPY = 3,3′-dihydroxy-2,2′-bipyridine) producing a moderately active catalyst of electrochemical reduction of CO2 to CO. The combined techniques of cyclic voltammetry and IR/UV–vis spectroelectrochemistry have revealed significant differences in the chemistry of the electrochemically reduced parent complex compared to the previously published Re/4,4′-DHBPY congener. The initial irreversible cathodic step in weakly coordinating THF is shifted toward much less negative electrode potentials, reflecting facile reductive deprotonation of one hydroxyl group and strong intramolecular hydrogen bonding, O–H···O–. The latter process occurs spontaneously in basic dimethylformamide where Re/4,4′-DHBPY remains stable. The subsequent reduction of singly deprotonated [Re(3,3′-DHBPY-H+)(CO)3Cl]− under ambient conditions occurs at a cathodic potential close to that of the Re/4,4′-DHBPY-H+ derivative. However, for the stabilized 3,3′-DHBPY-H+ ligand, the latter process at the second cathodic wave is more complex and involves an overall transfer of three electrons. Rapid potential step electrolysis induces 1e–-reductive cleavage of the second O–H bond, triggering dissociation of the Cl– ligand from [Re(3,3′-DHBPY-2H+)(CO)3Cl]2–. The ultimate product of the second cathodic step in THF was identified as 5-coordinate [Re(3,3′-DHBPY-2H+)(CO)3]3–, the equivalent of classical 2e–-reduced [Re(BPY)(CO)3]−. Each reductive deprotonation of the DHBPY ligand results in a redshift of the IR ν(CO) absorption of the tricarbonyl complexes by ca. 10 cm–1, facilitating the product assignment based on comparison with the literature data for corresponding Re/BPY complexes. The Cl– dissociation from [Re(3,3′-DHBPY-2H+)(CO)3Cl]2– was proven in strongly coordinating butyronitrile. The latter dianion is stable at 223 K, converting at 258 K to 6-coordinate [Re(3,3′-DHBPY-2H+)(CO)3(PrCN)]3–. Useful reference data were obtained with substituted parent [Re(3,3′-DHBPY)(CO)3(PrCN)]+ that also smoothly deprotonates by the initial reduction to [Re(3,3′-DHBPY-H+)(CO)3(PrCN)]. The latter complex ultimately converts at the second cathodic wave to [Re(3,3′-DHBPY-2H+)(CO)3(PrCN)]3– via a counterintuitive ETC step generating the 1e– radical of the parent complex, viz., [Re(3,3′-DHBPY)(CO)3(PrCN)]. The same alternative reduction path is also followed by [Re(3,3′-DHBPY-H+)(CO)3Cl]− at the onset of the second cathodic wave, where the ETC step results in the intermediate [Re(3,3′-DHBPY)(CO)3Cl]•– further reducible to [Re(3,3′-DHBPY-2H+)(CO)3]3– as the CO2 catalyst.
Short abstract
Cyclic voltammetry and IR spectroelectrochemistry were applied to unravel impact of internal hydrogen bonding on the cathodic path of [Re(3,3′-DHBPY)(CO)3Cl]. Following the initial reductive deprotonation, a strong hydrogen bond between the oxyanion substituents leads to a cathodic behavior markedly different from both its congener [Re(4,4′-DHBPY)(CO)3Cl] and reference [Re(BPY)(CO)3Cl]. The ultimate reduction product, Re(3,3′-DHBPY-2H+)(CO)3]3−, is catalytically active in the reductive disproportionation of CO2 to CO.
Introduction
The environmental impact of anthropogenic CO2 emissions has become one of the biggest concerns of modern science. Solar fuels, produced efficiently from either electrocatalytic or photocatalytic CO2 reduction, offer a tantalizing route toward sustainable energy production.1−3 Many transition metal complexes have been studied as possible precursors to catalytically active species. The goal is to accomplish multiple electron–proton transfers required to transform CO2 not only to CO and formic acid (2e–) but also to highly desirable high-density fuels such as acetone (4e–) or methanol (6e–).4−14 One of the classical complexes, [Re(BPY)(CO)3Cl] (BPY = 2,2′-bipyridine), and the family of its plentiful derivatives have been well-established in the literature as the subject of numerous studies over the past few decades.15−24 However, the scarcity of noble metals such as rhenium has led to a refocusing of efforts toward complexes of the more Earth-abundant metals. In Group-7, manganese is the most promising candidate, and the analogous Mn-BPY complexes have recently been shown to be also active toward CO2 in the presence of Brønsted or Lewis acids.25−27 Since then the wealth of literature regarding the catalyst family of manganese α-diimine complexes has grown considerably.28−38 Despite the clear move in the literature toward these Earth-abundant metals, rhenium complexes still play a critical role as model or reference systems, especially as other avenues of research are pioneered, including the study of so-called proton-responsive ligands acting as local sources of the protons consumed along the catalytic CO2 reduction path.39−44 The idea of introducing an intramolecular source of protons proximal to the site of CO2 coordination and concomitant reduction to improve turnover rates has received a great deal of attention. In 2012, iron tetraphenylporphyrin (FeTPP) was modified by placing phenolic groups in all ortho/ortho′ positions, which led to a significant increase in the observed catalytic activity, attributed to the local proton sources now present.39 Another efficient iron-based catalyst for selective reduction of CO2 to formate in aqueous solution is the reduced hydride cluster [HFe4N(CO)12]−.45 Recently, a dinuclear rhenium complex, [{Re(CO)3Cl}2(μ-L)](L = 4-tert-butyl-2,6-bis(6-(1H-imidazol-2-yl)-pyridin-2-yl)phenol), has been reported by Siewert and co-workers to show an increased catalytic activity for the CO production in DMF/H2O relative to the activity of [Re(BPY)(CO)3Cl] in MeCN/H2O.40 In the absence of CO2, the dinuclear complex undergoes two 2e– cathodic steps. The first 2e– reduction is L-based, triggering homolytic N–H bond cleavage at the imidazole site and reductive liberation of H2. The reductive deprotonation of imidazole (ImH) to imidazolate (Im–) has also been demonstrated by Hartl and co-workers in the related complexes [Re(BPY)(CO)3(ImH)]+ and [Re(PHEN)(CO)3(ImH)]+ (PHEN = 1,10-phenanthroline).41,42 The second 2e– reduction of the dinuclear Re complex triggers O–H bond cleavage at the phenol group and is accompanied by the loss of the Cl– ligand and concomitant metal-centered reduction. It is in the latter step where a catalytic response is observed in the presence of CO2. In addition to this, the complex [Mn(PDBPY)(CO)3Br] (PDBPY = 4-phenyl-6-(phenyl-2,6-diol)-2,2′-bipyridine) shows extreme sensitivity toward added-acid strength as well an atypical reduction pathway for tricarbonyl Mn-BPY complexes, presenting with suppressed dimer formation in acetonitrile.43 Interestingly, alternative positions of the phenolic groups from ortho in the latter complex to meta/para in related [Mn(PTBPY)(CO)3Br] (PTBPY = 4-phenyl-6-(phenyl-3,4,5-triol)-2,2′-bipyridine) resulted in a different cathodic path under Ar (dimerization, absent reductive deprotonation of the phenolic groups), as well as a reduced catalytic reactivity toward CO2.43 Recently, the study has been extended to the corresponding Re complexes showing a higher selectivity for formate.44 Fujita and co-workers studied the cathodic behavior of two proton-responsive complexes [Re(x,x′-DHBPY)(CO)3Cl] (x = 4 and 6, DHBPY = dihydroxy-2,2′-bipyridine).46 In the case of Re/4,4′-DHBPY, an enhanced catalytic activity relative to reference Re/BPY was reported, while Re/6,6′-DHBPY proved to be inactive toward CO2 reduction. Both Re/x,x′-DHBPY complexes undergo two closely spaced, 1e–-reductive deprotonations, R1 at Ep,c = −1.56 V and R2 at Ep,c = −1.76 V vs Ag/AgCl for x = 4 (Scheme 1). The intimate mechanism has been suggested to involve initial protonation of the singly reduced 4,4′-DHBPY ligand followed by H2 and proton dissociation to give [Re(4,4′-DHBPY-H+)(CO)3Cl]−. The three separate ν(CO) absorption bands corresponding to the parent Cs facial geometry, become red-shifted by about 10 cm–1 per 1e– reduction. This value indicates that the Re-BPY metallacycle and, therefore, also the π-back-donation to the carbonyl ligands are barely affected by lowering the oxidation state. The moderate ν(CO) shift is explained by the formation of the reductively deprotonated 4,4′-DHBPY ligand acting as a stronger σ-donor. Dissociation of the axial Cl– ligand was proposed to accompany the second reductive-deprotonation step R2, yielding a 5-coordinate complex, [Re(4,4′-DHBPY-2H+)(CO)3]− (Scheme 1). However, the split ν(CO) band pattern is not in favor of such an assignment, as fluxional 5-coordinate tricarbonyls generally show two (A1+ E) absorption bands. Electrocatalytic reduction of CO2 with Re/4,4′-DHBPY becomes triggered by the cathodic step R3 at Ep,c= −2.03 V vs Ag/AgCl rather than preceding R2 (see Figure 6 in ref (46)), selectively transforming CO2 to CO with a faradaic efficiency of 95% ± 2% in dry DMF. Notably, Fujita and co-workers claimed [Re(4,4′-DHBPY-2H+)(CO)3]− to be the catalyst and did not monitor the electron transfer process R3 and underlying structural transformation by IR spectroelectrochemistry; the molecular and electronic structure of the catalytically active species has therefore remained uncertain. Scheme 1 presents it as the five-coordinate dianion, [Re(4,4′-DHBPY-2H+)(CO)3]2–, an equivalent of the radical [Re(4,4′-di-tBu-BPY)(CO)3], but it also can be the corresponding trianion, [Re(4,4′-DHBPY-2H+)(CO)3]3–, an equivalent of [Re(4,4′-di-tBu-BPY)(CO)3]−, the well-established catalyst with two added electrons delocalized over the π-system of the Re–N∩N(BPY) metalacycle.47 This study will address the cathodic activation of a related double-deprotonated precatalyst. Notably, Fujita and co-workers46 did not involve in their study the close complex with the 3,3′-dihydroxy-2,2′-bipyridine (3,3′-DHBPY) ligand. The photophysical properties of this complex and interesting photochemistry of its deprotonated anionic form were reported in the literature by Vogler and co-workers.48,49 The parent Re/3,3′-DHBPY complex was shown to undergo single deprotonation with excess KOH or in dimethylformamide (DMF). Interestingly, the deprotonated anionic product, [Re(3,3′-DHBPY-H+)(CO)3Cl]−, with the Re–N∩N metallacycle strongly absorbing at 416 nm, converts on exposure to light of λ > 400 nm to a linkage isomer with Re–N∩O– coordination (Scheme 2). Although rarely encountered for Re tricarbonyl complexes, this photoisomerization behavior bears some similarity with the so-called “roll-over” pathway common to several rhodium-50,51 and platinum-based52 C–H functionalization catalysts. In the original reports,48,49 it remains unclear whether the chelating Re–N∩O– coordination induces dissociation of the axial Cl– ligand (not shown in Scheme 2). This concomitant step cannot be excluded, as documented by another complex with a bidentate N∩O– ligand at the Re(CO)3+ core, which binds pyridine in the axial position instead of Cl–.53 The objective of this comparative study was to ascertain whether the metallacycle isomerization could also be induced electrochemically, on further reduction of [Re(3,3′-DHBPY-H+)(CO)3Cl]−. Otherwise, the goal was to unravel the impact of the initial deprotonation step on the cathodic behavior of the complex and the potential of the identified reduction products to trigger catalytic reduction of carbon dioxide.
Scheme 1. Reduction Path of [Re(4,4′-DHBPY)(CO)3Cl].

Involving two successive 1e–-reductive deprotonation steps, R1 and R2.
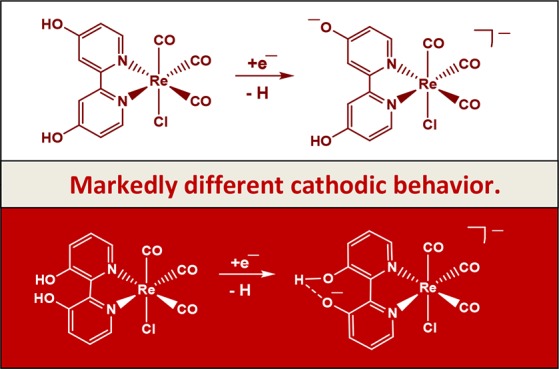
Scheme 2. Reaction Pathway of [Re(3,3′-DHBPY)(CO)3Cl].

Involving facile chemical deprotonation of one hydroxyl group at 3,3′-DHBPY by addition of excess KOH to yield [Re(3,3′-DHBPY-H+)(CO)3Cl]− stabilized by intraligand hydrogen bonding. Exposure to the light of λ > 400 nm triggers the “roll-over” isomerization, resulting in the Re–N∩O– metallacycle. Addition of HCl at any stage will reverse the process.
Experimental Section
Materials and Syntheses
Organic solvents were freshly distilled prior to use under an inert atmosphere of nitrogen from an appropriate drying agent: Na/benzophenone for tetrahydrofuran (THF) and CaH2 for butyronitrile (PrCN). The supporting electrolyte, Bu4NPF6 (Acros-Organics), was recrystallized twice from absolute ethanol. It was dried under vacuum at 373 K for 5 h and for an additional 20 min just prior to electrochemical measurements carried out under an atmosphere of dry argon. Standard Schlenk techniques were applied to all procedures. 1H NMR spectra were recorded on a Bruker Nanobay spectrometer (400 MHz). For the electrocatalytic studies (cyclic voltammetry/infrared spectroelectrochemistry (IR-SEC)), the electrolyte was saturated with CO2 by bubbling it on a frit at atmospheric pressure for ca. 10 min. Chemical deprotonation of [Re(3,3′-DHBPY)(CO)3Cl] was carried out using solid sodium bis(trimethylsilyl)amide (NaHMDS, 95%, Sigma-Aldrich).
Synthesis of [Re(3,3′-DHBPY)(CO)3Cl]
A sample of [Re(3,3′-DHBPY)(CO)3Cl] was prepared according to the literature procedures.48 The product identity and purity were confirmed by molecular absorption spectroscopies. IR (THF): ν(CO) at 2018, 1914, and 1893 cm–1. UV–vis (THF): λmax = 350 nm. 1H NMR (400 MHz, CD3CN) δ 8.75 (2H, d, J = 5.2), 7.72 (2H, d, J = 8.8), 7.53 (2H, dd, J = 13.6, 3.2). For comparison, the 1H NMR spectrum of [Re(4,4′-DHBPY)(CO)3Cl] shows similar resonances at δ 8.66 (d), 7.69 (d), and 7.03 (dd). The broad signal of the phenolic protons at δ > 9 (ref (46)) was not observed due to some moisture in acetonitrile-d3.
Synthesis of [Re(3,3′-DHBPY)(CO)3(OTf)] (OTf = CF3SO3–) and Reactivity toward PrCN
[Re(3,3′-DHBPY)(CO)3Cl] (100 mg, 0.20 mol) and AgOTf (64 mg, 0.25 mol) were dissolved under an inert atmosphere in the dark in freshly distilled anhydrous THF (10 mL). The solution was stirred for 2 h at room temperature. The white precipitate of AgCl was then filtered off, and the solution was collected in a Schlenk vessel purged with argon. Hexane (4 × 10 mL) was added slowly to precipitate a yellow solid. The solvent was evaporated to dryness to collect a dry but extremely hydroscopic yellow powder in a moderate yield (33.6%). IR (THF): ν(CO) at 2032, 1928, 1910 cm–1. Rapid thermal conversion of [Re(3,3′-DHBPY)(CO)3(OTf)] to [Re(3,3′-DHBPY)(CO)3(PrCN)]+ takes place in PrCN. IR (PrCN): ν(CO) at 2039 and 1933 br cm–1. 1H NMR (400 MHz, CD3CN) δ 8.78 (2H, d, J = 5.2), 7.85 (2H, d, J = 8.8), 7.65 (2H, dd, J = 13.6, 3.2). This facile ligand exchange was utilized to prepare in situ electrochemical samples with the latter cationic complex.
Methods
Cyclic Voltammetry
Cyclic voltammograms (CVs) were recorded with a Metrohm Autolab PGSTAT302N potentiostat operated with the Nova 1.9 software. The air-tight single-compartment electrochemical cell housed a 0.42 mm2 platinum microdisc working electrode [polished between scans with a 0.25 μm diamond paste (Kemet International Ltd.)], a coiled platinum wire counter electrode, and a silver wire pseudoreference electrode. The ferrocene/ferrocenium (Fc/Fc+) redox couple served as an internal standard for voltammetric measurements. Solutions contained 10–1 M Bu4NPF6 and 10–3 M analyte. The cell was chilled to 195 K by submersion in an acetone/dry ice slurry.
IR and UV–Vis Spectroelectrochemistry
IR SEC experiments were performed using a Bruker Vertex 70v FT-IR spectrometer equipped with a DLaTGS detector or linked to an external Bio-Rad FTS 60 MCT detector (with the sample compartment adapted for low-temperature spectroelectrochemistry). UV–vis SEC experiments were conducted with a Scinco S-3100 diode-array spectrophotometer (200–1100 nm). The in situ electrolyses were carried out using an air-tight OTTLE cell.54 The cell was equipped with a platinum minigrid (32 wires per cm) working and auxiliary electrodes, a Ag microwire pseudoreference electrode, and optically transparent CaF2 windows. The course of the spectroelectrochemical experiment was monitored by thin-layer cyclic voltammetry controlled with a PalmSens EmStat3 potentiostat operated with the PSTrace5 software. Low-temperature spectroelectrochemical measurements were carried out with a cryostatted OTTLE cell.55 Solutions contained 3 × 10–1 M Bu4NPF6 and 3 × 10–3 M analyte.
Coulometry
To quantify the electrocatalytic reactions reported in this work, controlled potential electrolysis was conducted under Ar and CO2 in two electrochemical cells. The first one, controlled by a PalmSens3 potentiostat, was a pear-shaped cell, equipped with a Pt-microdisc (2 mm, diameter) working electrode, a Pt-wire counter electrode separated from the bulk solution by a Vycor tip, and an Ag-wire pseudoreference electrode. The second one, controlled by a BioLogic SP-200 potentiostat, was an H-shaped cell with three compartments separated at the bottom by dense glass frits and housing the Pt-foil working and counter electrodes with the geometric surface areas of 2 × 0.96 cm2 and 2 × 0.5 cm2, respectively, and a coiled Ag wire pseudoreference electrode. In both cases, the working and reference electrodes were immersed in a CO2-saturated THF solution containing 3 mM [Re(3,3′-DHBPY)(CO)3Cl], 0.5 M Bu4NPF6, and 3 mM ferrocene (as a CV potential reference). To prevent anodic THF polymerization, the solution surrounding the counter electrode was 0.5 M Bu4NPF6 in CO2-saturated acetonitrile. The solutions were stirred continuously during the measurements. The turnover numbers and faradaic efficiencies were quantified by gas chromatography (GC)-headspace analysis. On the time scale of the experiments, only the pear-shaped cell was suitable for recording accurate faradic efficiency values, while the H-shaped cell, due to diffusion of CO between the compartments and not into the headspace, was only used to determine accurate turnover numbers. Thus, the data were combined to give the appreciable overall picture. The gas chromatography was conducted on an Agilent 6890N instrument equipped with a 5 Å molecular sieve column (ValcoPLOT, 30 m length, 0.53 mm inner diameter) and a pulsed discharge detector (D-3-I-HP, Valco Vici), with helium as the carrier gas. H2 and CO were quantified by using a custom-made calibrant gas (500 ppm of H2, 200 ppm of CH4, and 200 ppm of CO in CO2 as the balance gas).
Results and Discussion
Cyclic Voltammetry
Cyclic Voltammetry of [Re(3,3′-DHBPY)(CO)3Cl] in THF at Variable Temperature
The cathodic behavior of [Re(3,3′-DHBPY)(CO)3Cl] differs significantly from that of its congener with the 4,4′-DHBPY ligand. In THF, the title complex exhibits two irreversible cathodic waves (Figure 1). The first wave, R1, at Ep,c = −1.01 V vs Fc/Fc+ is very broad, indicating an irreversible, slow electron transfer process. Notably, the initial reduction of [Re(4,4′-DHBPY)(CO)3Cl] is shifted significantly more negatively, by ca. 1 V (Table 1). This extraordinary difference can be ascribed to a strong stabilizing effect of the single reductive deprotonation resulting in a hydrogen bond between the BPY oxyanion substituents (Scheme 2).56 The following cathodic step, R2′, appears as a quasi-reversible cathodic wave at a much more negative potential of Ep,c = −2.38 V vs Fc/Fc+. The large potential gap, reaching nearly 1.4 V, is in stark contrast to the CVs recorded for the 4,4′-DHBPY and 6,6′-DHBPY derivatives and their singly (selectively) deprotonated forms, where the separation between first and second cathodic waves is merely 200 mV.
Figure 1.
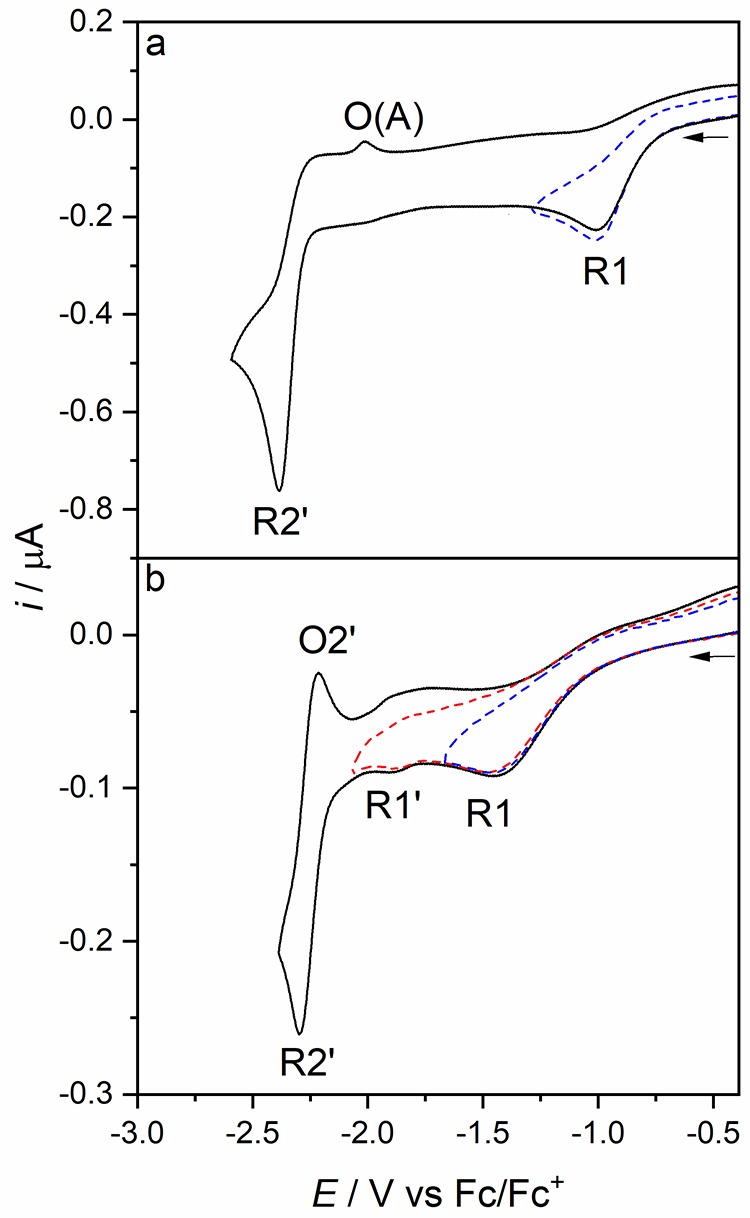
CVs of [Re(3,3′-DHBPY)(CO)3Cl] in THF/Bu4NPF6. Experimental conditions: Pt microdisc, 298 K (a) and 195 K (b), v = 100 mV s–1.
Table 1. Cyclic Voltammetry of [Re(3,3′-DHBPY)(CO)3Cl] and Related DHBPY Complexes.
| complex | solvent | cathodic wave | Ep,c (V vs Fc/Fc+) |
|---|---|---|---|
| [Re(3,3′-DHBPY)(CO)3Cl] | THF | R1 | –1.01 |
| –1.40a | |||
| [Re(3,3′-DHBPY)(CO)3Cl] | R1* | –1.90a,b | |
| [Re(3,3′-DHBPY)(CO)3Cl]•– | R2 | –2.38 | |
| [Re(3,3′-DHBPY-H+)(CO)3Cl]− | R2′ | –2.38 | |
| –2.30a | |||
| [Re(3,3′-DHBPY-H+)(CO)3Cl]−c | R2* | –2.16 | |
| [Re(3,3′-DHBPY)(CO)3Cl] [Re(3,3′-DHBPY-H+)(CO)3Cl]− | PrCN | R1 | –0.84 |
| –1.22a | |||
| R2′ | –2.30 | ||
| –2.25a | |||
| [Re(3,3′-DHBPY)(CO)3(PrCN)]+ | PrCN | R1 | –0.93 |
| [Re(3,3′-DHBPY-H+)(CO)3(PrCN)] | R2′ | –2.07 | |
| [Re(4,4′-DHBPY)(CO)3Cl]d,e | DMF | R1 | –1.98 |
| [Re(4,4′-DHBPY-H+)(CO)3Cl]− | R2 | –2.18 | |
| [Re(4,4′-DHBPY-2H+)(CO)3]− | R3 | –2.45 |
Measured at 195 K.
No concomitant reductive deprotonation of the 3,3′-DHBPY ligand taking place. E1/2= −1.87 V.
Prepared by chemical deprotonation of [Re(3,3′-DHBPY)(CO)3Cl] with 1 equiv of NaHMDS.
Measured by Fujita and co-workers.46
No deprotonation of the parent complex in DMF was reported
At room temperature, R1 is totally irreversible with any counter wave absent even at scan rates in excess of 1 V s–1 (Figure S1). Additionally, turning the scan positively beyond R2′ does not generate a well-defined counterwave O2′ (Figure 1a) but reveals a new anodic wave, O(A), at Ep,a= −2.01 V vs Fc/Fc+, which becomes better perceptible at higher scan rates (Figure S2). The origin of both phenomena will be analyzed in the following IR spectroelectrochemical section.
On cooling to 195 K, the cathodic CV of [Re(3,3′-DHBPY)(CO)3Cl] in THF changes notably (Figure 1b). First, R1 is shifted negatively by ca. 400 mV (Ep,c = −1.45 V vs Fc/Fc+) and significantly broadened. A new, minor cathodic wave R1* appears at Ep,c = −1.90 V vs Fc/Fc+. At higher scan rates, R1* grows relative to R1 and becomes reversible (Figures S3 and S4) at ν < 500 mV s–1. The reversible nature of the process on the subsecond time scale suggests that R1* corresponds to the reduction of parent [Re(3,3′-DHBPY)(CO)3Cl] to the corresponding nondeprotonated radical anion. It will be shown in the spectroelectrochemical section that this process contributes to the overall cathodic path of the title complex even at ambient temperature. Sweeping the electrode potential toward R2′ at 195 K located this wave at Ep,c = −2.30 V vs Fc/Fc+ (Figures 1b and S3). Its shape and overall reversibility (indicated by the anodic counter wave O2′ and the unity peak/current ratio) points to a diffusion-controlled 1e– cathodic process on the (sub)second CV time scale that does not trigger any significant chemical transformation of [Re(3,3′-DHBPY-H+)(CO)3Cl]− generated at R1 (see the “IR and UV–Vis Spectroelectrochemistry” section). This temperature-controlled behavior reveals increased stability of [Re(3,3′-DHBPY-H+)(CO)3Cl]2– (both O–H and Re–Cl bonds, in contrast to the Re/4,4′-DHBPY congener) ascribed to the strong hydrogen bonding in the ligand redox couple [3,3′-DHBPY-H+]−/[3,3′-DHBPY•–-H+]2– that hinders the second reductive proton elimination to give [3,3′-DHBPY-2H+]2–.
Cyclic Voltammetry of [Re(3,3′-DHBPY)(CO)3Cl] and [Re(3,3′-DHBPY)(CO)3(PrCN)]+ in PrCN at Variable Temperature
The cathodic behavior of [Re(3,3′-DHBPY)(CO)3Cl] was investigated by cyclic voltammetry also in butyronitrile, PrCN (Figure 2). At room temperature, the totally irreversible wave R1 is shifted less negatively to Ep,c = −0.84 vs Fc/Fc+ in this electrolyte compared to THF (Table 1). The second cathodic wave R2′ is shifted by merely 80 mV, which reveals the stability of [Re(3,3′-DHBPY-H+)(CO)3Cl]− in the potentially coordinating solvent. Apart from the moderate potential shifts, the CV response does not deviate significantly from that recorded in THF. This also applies for the CV scan at 195 K (Figure 2b), where only R1* is absent.
Figure 2.

CVs of [Re(3,3′-DHBPY)(CO)3Cl] in PrCN/Bu4NPF6. Experimental conditions: Pt microdisc, T = 298 K (a) and 195 K (b), v = 100 mV s–1.
Compared to [Re(4,4′-DHBPY)(CO)3Cl] in DMF (Table 1) the cathodic wave R3 is absent or unresolved in the CV responses of [Re(3,3′-DHBPY)(CO)3Cl] both in THF (see above) and PrCN. This is understandable at 195 K where the Re–Cl bond remains stable upon the 1e– reduction generating intact [Re(3,3′-DHBPY•–-H+)(CO)3Cl]2– (Figures 1b and 2b). The reductively double-deprotonated complex [Re(3,3′-DHBPY-2H+)(CO)3Cl]2–, which is supposed to further reduce at an R3 potential, is not formed on the short time scale of the CV experiment due to the strong intramolecular hydrogen bonding in the single-deprotonated precursor.56 At ambient temperature, the irreversible cathodic wave R2′ in fact encompasses several secondary EC processes resulting from the instability of [Re(3,3′-DHBPY-2H+)(CO)3Cl]2–. These deductions have been corroborated by IR spectroelectrochemical experiments described in the following section. Direct comparison of the two Re/x,x′-DHBPY (x = 3 and 4) complexes in DMF is not possible as [Re(3,3′-DHBPY)(CO)3Cl] instantly deprotonates in this basic solvent.
Separate substitution of the axial chloride ligand in [Re(3,3′-DHBPY)(CO)3Cl] with PrCN to form the corresponding cationic complex impacts strongly the cathodic process at R2′, causing its positive shift by 230 mV. The less negative electrode potential agrees with the reduction of less basic [Re(3,3′-DHBPY-H+)(CO)3(PrCN)] formed at R1 from singly reduced [Re(3,3′-DHBPY)(CO)3(PrCN)]+ (Figure 3, Table 1). This observation supports the stability of [Re(3,3′-DHBPY-H+)(CO)3Cl]− in PrCN when formed from parent [Re(3,3′-DHBPY)(CO)3Cl] at R1 (Figure 2a).
Figure 3.

CV of [Re(3,3′-DHBPY)(CO)3(PrCN)]+ in PrCN/Bu4NPF6. Experimental conditions: Pt microdisc, T = 298 K, v = 100 mV s–1.
Cyclic Voltammetry of [Re(3,3′-DHBPY-H+)(CO)3Cl]– in THF at Ambient Temperature
The CV of [Re(3,3′-DHBPY-H+)(CO)3Cl]− prepared in situ in THF by chemical deprotonation of [Re(3,3′-DHBPY)(CO)3Cl] with NaHMDS is highly revealing (Figure 4). As expected, the cathodic wave R1 of the neutral parent complex is absent. However, instead of the single cathodic wave R2′ assigned deliberately to the deprotonated anionic complex formed electrochemically at R1 (Figure 1a), there are two close-lying waves, the first one at Ep,c = −2.16 V denoted as R2*, followed by R2 at Ep,c = −2.38 V which coincides with R2′ and is also allied to the anodic wave O(A) recorded on the reverse potential scan. This cathodic behavior is echoed in the corresponding IR spectroelectrochemical experiment presented and thoroughly discussed in the following section, where [Re(3,3′-DHBPY-H+)(CO)3Cl]− is fully transformed already at R2* but not to an anticipated double-deprotonated product. Instead, the IR monitoring revealed the formation of [Re(3,3′-DHBPY)(CO)3Cl]•–, similar to the reversible reduction at 195 K of parent [Re(3,3′-DHBPY)(CO)3Cl] at R1* (Table 1). This surprising transformation corresponds to a net-zero-electron, electron-transfer catalytic (ETC) process, which agrees with the rather low peak current at R2*. The cathodic wave R2 in Figure 4 can be then ascribed to the collective subsequent reduction of [Re(3,3′-DHBPY)(CO)3Cl]•– and [Re(3,3′-DHBPY-H+)(CO)3Cl]− diffusing from the bulk solution. The ETC process is reminiscent, for example, to the 1e– reduction of [Re(DBSQ)(CO)3(PPh3)] (DBSQ = 3,5-di-tBu-1,2-benzosemiquinone anion) which produces isoelectronic [Re(DBSQ)(CO)2(PPh3)2] instead of [Re(DBCat)(CO)3(PPh3)]− (DBCat = 3,5-di-tBu-1,2-catecholate dianion).57
Figure 4.
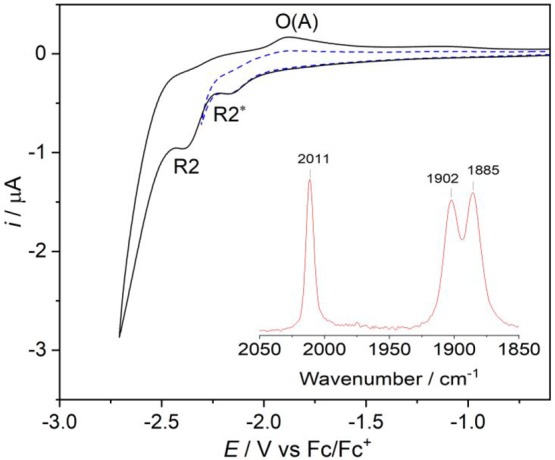
CV of [Re(3,3′-DHBPY-H+)(CO)3Cl]− in THF/Bu4NPF6. Experimental conditions: Pt microdisc, T = 298 K, v = 100 mV s–1. Inset: IR spectrum of the analyte solution, indicating the complete deprotonation of one −OH group by NaHMDS.
Infrared and UV–Vis Spectroelectrochemistry (SEC)
Reduction of [Re(3,3′-DHBPY)(CO)3Cl] in THF at Ambient Temperature
At cathodic potentials coinciding with R1, [Re(3,3′-DHBPY)(CO)3Cl] in the thin-solution layer of the OTTLE cell undergoes O–H bond cleavage (reductive deprotonation) yielding [Re(3,3′-DHBPY-H+)(CO)3Cl]− (Figure 5a,b and Scheme 3). The UV–vis spectral changes (Figure 5b) reveal the characteristic replacement of the Re-to-BPY MLCT band at 351 nm with a new, likely intraligand (DHBPY-H+) absorption band at 425 nm, as reported by Vogler and co-workers49 for the chemical reaction of [Re(3,3′-DHBPY)(CO)3Cl] with solid KOH in wet acetonitrile (Scheme 2). In the infrared CO-stretching region (Figure 5a), a small but perceptible redshift of the three ν(CO) absorption bands (2A′ + A′′; Cs geometry) by ca. 10 cm–1 from the parent complex is observed. The reference wavenumber values for [Re(4,4′-DHBPY-H+)(CO)3Cl]− are indeed very close (Table 2). These data confirm that the added electron does not enter the π*(BPY) LUMO as in the archetypal case of the 1e– reduction of [Re(BPY)(CO)3Cl].24,58,59 The assignment of the reduction product at R1 as [Re(3,3′-DHBPY-H+)(CO)3Cl]− is strongly supported by the independent chemical deprotonation of the parent complex by the added base, NaHMDS (Figures 4 and S5). It is noteworthy that instant single-deprotonation was also encountered49 in DMF (Table 2) where the neutral congener [Re(4,4′-DHBPY)(CO)3Cl] remains stable. The close proximity of the hydroxyl substituents in [Re(3,3′-DHBPY-H+)(CO)3Cl]− enables strong hydrogen bonding,48,49 providing a significant thermodynamic driving force to form readily a stable deprotonated complex, as also reflected in the much lower R1 overpotential compared to the initial reduction of [Re(4,4′-DHBPY)(CO)3Cl].
Figure 5.

Cathodic IR (a, c) and UV–vis (b, d) spectroelectrochemistry: (a, b) 1e–-reductive deprotonation of [Re(3,3′-DHBPY)(CO)3Cl] (↓) to [Re(3,3′-DHBPY-H+)(CO)3Cl]−(↑) at R1; (c, d) rapid potential-step conversion of [Re(3,3′-DHBPY-H+)(CO)3Cl]− (↓) to 5-coordinate [Re(3,3′-DHBPY-2H+)(CO)3]3– (↑) in an unresolved 3e– process at 200 mV beyond the onset of R2′. Conditions: an OTTLE cell, THF/Bu4NPF6, T = 298 K.
Scheme 3. Cathodic Pathways of [Re(3,3′-DHBPY)(CO)3Cl] in THF.

First 1e–-reductive deprotonation at R1 followed by rapid second 1e–-reductive deprotonation at R2′, concomitant chloride dissociation, and 2e– reduction of the 5-coordinate transient complex delocalized over the BPY–Re(CO)3 π-system. The alternative (potential-controlled) ETC pathway (at R2*, or the onset of R2′) produces the corresponding radical anion of the parent complex.
Table 2. Infrared and UV–Vis Absorption Data for [Re(3,3′-DHBPY)(CO)3Cl] and [Re(3,3′-DHBPY)(CO)3(PrCN)]+ and Their Reduction Products.
| complex | solvent | ν(CO) (cm–1) | λmax (nm) |
|---|---|---|---|
| [Re(BPY)(CO)3Cl] | THFa | 2019, 1917, 1895 | |
| [Re(3,3′-DHBPY)(CO)3Cl] | THF | 2018, 1914, 1893 | 351 |
| PrCN | 2020, 1915, 1897 | 353 | |
| [Re(4,4′-di-tBu-BPY)(CO)3Cl] | DMFb | 2018, 1913, 1890 | |
| [Re(3,3′-DHBPY-H+)(CO)3Cl]− | THF | 2011, 1902, 1886 | 425 |
| PrCN | 2012, 1903, 1885 | 426 | |
| THFc | 2011, 1902, 1885 | ||
| DMFd | 2011, 1902, 1882 | 425 | |
| [Re(BPY)(CO)3Cl]•– | THF | 1996, 1883, 1868a | |
| 1996, 1882, 1867f | 361, 486 sh, 511f | ||
| Re(3,3′-DHBPY)(CO)3Cl]•– | THF | 1994, 1878, 1866 | |
| [Re(4,4′-di-tBu-BPY)(CO)3Cl]•– | DMFb | 1992, 1878, 1860 | |
| [Re(3,3′-DHBPY-2H+)(CO)3Cl]2– | THFe | 2002, 1883, 1873 | |
| PrCNg | 2002, 1885, 1867 | ||
| [Re(3,3′-DHBPY-2H+)(CO)3]3– | THF | 1921, 1818 br | 354, 553, 654 |
| [Re(3,3′-DHBPY)(CO)3(OTf)] | THF | 2032, 1928, 1910 | |
| [Re(3,3′-DHBPY)(CO)3(PrCN)]+ | PrCN | 2039, 1933 br | |
| [Re(BPY)(CO)3(PrCN)]+ | PrCNa | 2041, 1937 br | |
| [Re(3,3′-DHBPY)(CO)3(PrCN)] | PrCN | 2004, 1890 br | |
| [Re(BPY)(CO)3(PrCN)] | PrCNa | 2010, 1895 br | |
| [Re(3,3′-DHBPY-H+)(CO)3(PrCN)] | PrCN | 2031, 1928 br | |
| [Re(3,3′-DHBPY-2H+)(CO)3(PrCN)]− | PrCNg | 2023, unresolved | |
| [Re(3,3′-DHBPY-2H+)(CO)3(PrCN)]2– | PrCN | 1986, 1874 br | |
| [Re(3,3′-DHBPY-2H+)(CO)3(PrCN)]3– | PrCN | 1967, 1849, 1835 | |
| [Re(4,4′-DHBPY)(CO)3Cl]b | DMF | 2019, 1911, 1893 | |
| [Re(4,4′-DHBPY-H+)(CO)3Cl]−b | DMF | 2012, 1900, 1881 | |
| [Re(4,4′-DHBPY-2H+)(CO)3Cl]2–b | DMF | 2002, 1886, 1865 |
Reproduced from ref (24).
Reported by Fujita and co-workers.46
Formed by single deprotonation of the parent complex with NaHMDS.
Formed by instantaneous deprotonation of the parent complex in DMF.
Observed only as a transient in THF in a potential-step experiment.
T = 253 K. Reproduced from ref (59).
T = 223 K.
The subsequent reduction of electrochemically generated [Re(3,3′-DHBPY-H+)(CO)3Cl]− occurs at the cathodic wave R2′ (Figure 1a), proceeding via two alternative paths (Scheme 3) controlled by the mode of tuning the cathodic potential on the spectroelectrochemical time scale: either a rapid potential step (a) beyond R2′ and (b) at R2′ (b) or (c) slow potential sweep through R2′. This cathodic course is remarkably more complex than indicated by the reversible CV response in THF/Bu4NPF6 at 195 K (Figure 1b). The intramolecular hydrogen bond in [Re(3,3′-DHBPY-H+)(CO)3Cl]− is strong enough to extend the lifetime of the BPY-reduced species compared to the Re/4,4′-DHBPY congener but the second reductive elimination of hydrogen triggers structural changes monitored by IR spectroscopy, using information from selected reference systems.
In the first instance, the results from the potential step experiment (a) at 200 mV beyond R2′ are presented. The cathodic electrolysis at the overpotential produces in just a few seconds a single detectable species with IR ν(CO) bands markedly shifted to 1921 and 1818 br cm–1, which strongly absorbs in the visible spectral region (Figure 5c,d, respectively, and Table 2). This product is assigned as a formally Re0-BPY– 5-coordinate complex [Re(3,3′-DHBPY-2H+)(CO)3]3–. The ν(CO) intensity pattern and the large redshift correspond with the properties of the well-known58 fluxional complex [Re(BPY)(CO)3]− absorbing in THF at 1947 and 1843 cm–1. The additional >20 cm–1 downward shift seen for [Re(3,3′-DHBPY-2H+)(CO)3]3– complies with the preceding double reductive deprotonation of DHBPY, increasing the π-back-donation to the carbonyl ligands. The identification of this 5-coordinate structure also offers an explanation as to the origin of the O(A) counterwave seen in the CV on the reverse anodic scan (Figure 1a). This may be considered now as the reoxidation of this 5-coordinate complex. While encountered for isoelectronic 5-coordinate tricarbonyl complexes, such as [Mn(BPY)(CO)3]− or [W(BPY)(CO)3]2–,26,60 the O(A) signature has never been reported16,24 for either [Re(tBu-BPY)(CO)3]− or [Re(BPY)(CO)3]−, but it is our strong expectation that if such a wave were detected then it would also occur in this region.
In case (b), where the course of the thin-layer spectroelectrochemical experiment is not stepped but rapidly scanned through R2′ to the maximum faradaic current (Figure 6), an intermediate species is detected in consort with the ultimate 5-coordinate complex. On comparison with the Re/4,4′-DHBPY congener (Table 2), this transient is assigned as the double-deprotonated species [Re(3,3′-DHBPY-2H+)(CO)3Cl]2– (ν(CO) at 2002, 1885, and 1868 cm–1). Differently from Re/4,4′-DHBPY, the double-deprotonated species is unstable at room temperature, decaying via concerted Cl– dissociation and electron transfer to [Re(3,3′-DHBPY-2H+)(CO)3]3–. It will be shown later that [Re(3,3′-DHBPY-2H+)(CO)3Cl]2– can be stabilized at low temperature in coordinating butyronitrile, which supports the intact Re–Cl still present in this intermediate 6-coordinate complex, in line with the well-resolved 2A′ + A′′ ν(CO) band pattern.
Figure 6.
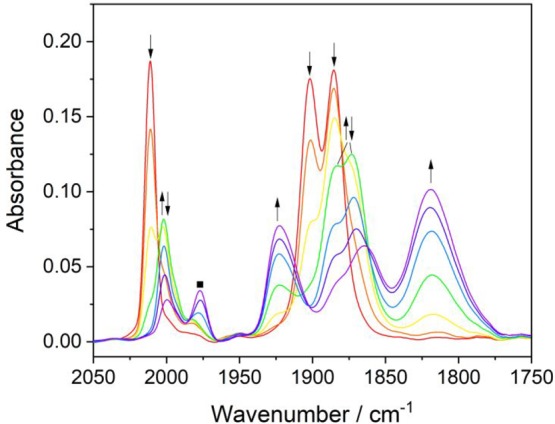
Cathodic IR spectroelectrochemistry at R2′ as [Re(3,3′-DHBPY-H+)(CO)3Cl]−(↓) undergoes 1e– reductive deprotonation to transient [Re(3,3′-DHBPY-2H+)(CO)3Cl]2– (↑↓) and its concomitant decay to [Re(3,3′-DHBPY-2H+)(CO)3]3– (↑). Conditions: an OTTLE cell, THF/Bu4NPF6, T = 298 K. The label ■ corresponds to an unassigned side product.
In the slow potential sweep experiment (c), the R2′ wave due to the reduction of [Re(3,3′-DHBPY-H+)(CO)3Cl]− is gradually scanned through, adding additional complexity to the overall mechanistic description. The strength of the internal hydrogen bond is of key importance to the single-deprotonated anion, and the activation energy for the second deprotonation is therefore quite high. In a very limited potential range, this allows for a situation where the primary BPY-reduced species, [Re(3,3′-DHBPY•–-H+)(CO)3Cl]2– does not undergo the second reductive deprotonation to [Re(3,3′-DHBPY-2H+)(CO)3Cl]2– (experiment (b)) but converts to a more stable product absorbing in the ν(CO) region at 1994, 1878, and 1866 cm–1 (Figure 7). These wavenumbers can be assigned to the nondeprotonated radical anion of the title complex, [Re(3,3′-DHBPY)(CO)3Cl]•–. The revealing evidence for this rather surprising assignment comes from the comparison with the equivalent complexes [Re(BPY)(CO)3Cl]•– (ν(CO) at 1998, 1885, and 1867 cm–1)24 or [Re(4,4′-di-tBu-BPY)(CO)3Cl]•– (ν(CO) at 1992, 1878, and 1860 cm–1),46 taking into account the relative donor power of the BPY substituents (H, OH, and tBu). While [Re(3,3′-DHBPY)(CO)3Cl]•– is highly unstable at R1, where the facile single reductive deprotonation takes place (see above), it persists in the limited potential interval between the onset of R2′ and its own reduction coinciding with the R2′ maximum, which ultimately converts it to [Re(3,3′-DHBPY-2H+)(CO)3]3– (Figure 7). The transformation of [Re(3,3′-DHBPY-H+)(CO)3Cl]− to [Re(3,3′-DHBPY)(CO)3Cl]•– is a zero-electron ETC reaction that kinetically dominates over the double reductive deprotonation of the 3,3′-DHBPY ligand. The latter process is triggered only by the subsequent electrochemical reduction of [Re(3,3′-DHBPY)(CO)3Cl]•–, monitored accurately by the thin-layer cyclovoltammetric response. Thus, different from the Re/4,4′-DHBPY congener, double-deprotonated [Re(3,3′-DHBPY-2H+)(CO)3Cl]2– can only be observed at ambient temperature at potentials more negative than the reduction potential of [Re(3,3′-DHBPY)(CO)3Cl]•– (experiment (b) in this section). The partly resolved electronic absorption of [Re(3,3′-DHBPY)(CO)3Cl]•– in the visible spectral region (around 480 nm) is shown in Figure S6.
Figure 7.
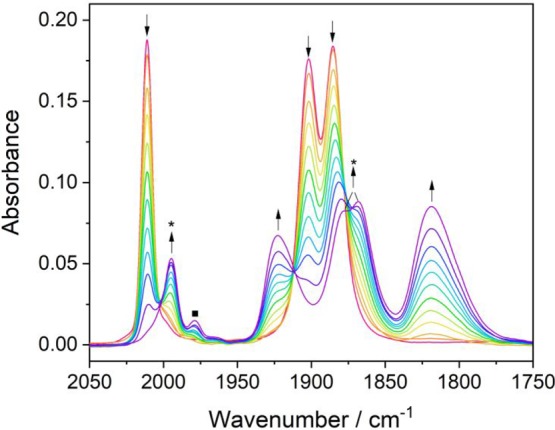
Cathodic potential-sweep IR spectroelectrochemistry at R2′ where starting [Re(3,3′-DHBPY-H+)(CO)3Cl]−(↓) undergoes 1e– reduction and ETC conversion to [Re(3,3′-DHBPY)(CO)3Cl]•– (*) and the subsequent reduction of the latter intermediate to [Re(3,3′-DHBPY-2H+)(CO)3]3– (↑). Conditions: an OTTLE cell, THF/Bu4NPF6, T = 298 K. The label ■ corresponds to an unassigned side product.
Summarizing the striking results in this section, the cathodic behavior of [Re(3,3′-DHBPY-H+)(CO)3Cl]− in THF at R2′ comprises a range of different processes. The initial 1e– reduction gives [Re(3,3′-DHBPY•–-H+)(CO)3Cl]2–, stable at low temperature on the CV time scale, which converts by an ETC step at low overpotentials (the onset of R2′) to [Re(3,3′-DHBPY)(CO)3Cl]•– or undergoes the second DHBPY deprotonation at higher overpotentials to generate reactive [Re(3,3′-DHBPY-2H+)(CO)3Cl]2–. In both cases, the ultimate reduction product at R2′ is the 5-coordinate π-delocalized complex [Re(3,3′-DHBPY-2H+)(CO)3]3–, the double-deprotonated equivalent of 2e–-reduced [Re(BPY)(CO)3]−.
The following sections present cathodic IR SEC experiments with [Re(3,3′-DHBPY)(CO)3Cl] conducted in THF following its chemical deprotonation and at low temperature in PrCN to collect more supporting evidence for the intriguing cathodic path in Scheme 3.
Reduction of [Re(3,3′-DHBPY-H+)(CO)3Cl]− in THF at Ambient Temperature
The electrochemical reduction of chemically single-deprotonated [Re(3,3′-DHBPY-H+)(CO)3Cl]− (Figure 8) strongly resembles the potential sweep IR spectroelectrochemical experiment at R2′ described in the preceding section (Figure 7). Notably, as visualized in the corresponding CV (Figure 4), the separation of the initial cathodic wave (R2*) is more distinct, allowing the ETC-product [Re(3,3′-DHBPY)(CO)3Cl]•– to be generated separately (Figure 8a) prior to its conversion to [Re(3,3′-DHBPY-2H+)(CO)3]3– at R2 (Figure 8b). The parallel UV–vis monitoring of these two steps is presented in Figure S6. Given the facile deprotonation of the title complex at R1 to [Re(3,3′-DHBPY-H+)(CO)3Cl]−, the formation of [Re(3,3′-DHBPY)(CO)3Cl]•– with “restored” 3,3′-DHBPY in the radical anionic state may be considered surprising. The initial formation of [Re(3,3′-DHBPY•–-H+)(CO)3Cl]2– at R2* (or the onset of R2′) was expected to trigger the second reductive deprotonation generating 1/2 H2 and [Re(3,3′-DHBPY-2H+) (CO)3Cl]2–. However, this process is apparently unfavorable for the clashing two phenoxide groups in the 3,3′-positions and there is a limited chance to delocalize the negative charges onto the ring nitrogens, differently from the Re/4,4′-DHBPY congener (Scheme 1). The singly reduced complex [Re(3,3′-DHBPY)(CO)3Cl]•– can be generated at R1* (Figures S3 and S4; T = 195 K) but the formation of the strong intramolecular hydrogen bond in [Re(3,3′-DHBPY-H+)(CO)3Cl]− is favored, and the radical anion of the title complex is then only restored at R2* (or the onset of R2′) where the hydrogen bond gets disrupted by the reduction localized at the π*(BPY) LUMO. Obviously, this thermodynamic advantage is lost by the subsequent reduction of [Re(3,3′-DHBPY)(CO)3Cl]•– at more negative R2 (Figure 4), which leads directly to double-deprotonated [Re(3,3′-DHBPY-2H+)(CO)3]3–, and no [Re(3,3′-DHBPY-2H+)(CO)3Cl]2– is detectable at ambient temperature during the potential sweep.
Figure 8.
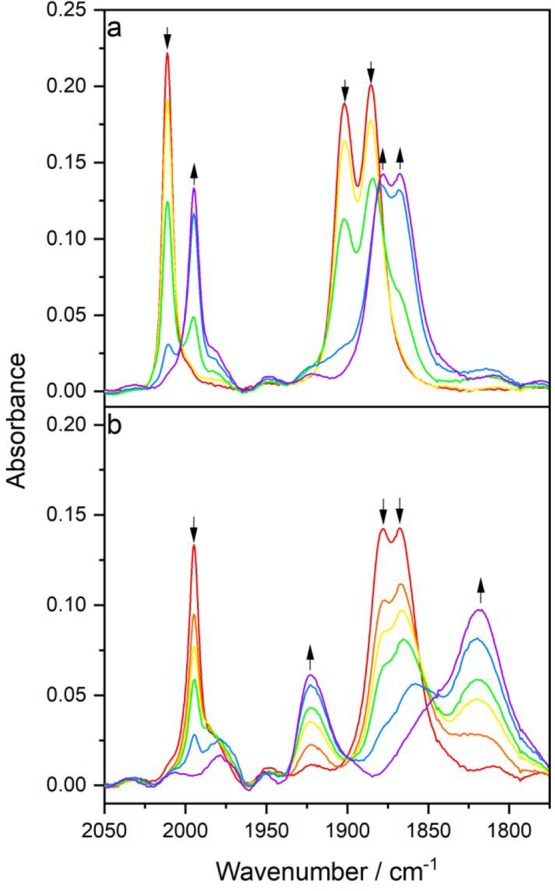
Cathodic IR spectroelectrochemistry showing (a) ETC-conversion of [Re(3,3′-DHBPY-H+)(CO)3Cl]− (↓) at R2* (cf. Figure 4) to [Re(3,3′-DHBPY)(CO)3Cl]•– (↑) as a net-zero-electron process and (b) subsequent reduction of [Re(3,3′-DHBPY)(CO)3Cl]•– (↓) at R2 to [Re(3,3′-DHBPY-2H+)(CO)3]3– (↑). Conditions: an OTTLE cell, THF/Bu4NPF6, T = 298 K.
Reduction of [Re(3,3′-DHBPY)(CO)3Cl] in PrCN at Variable Temperature
IR SEC experiments with [Re(3,3′-DHBPY)(CO)3Cl] in PrCN at 223 K (Figure 9) and 258 K (Figure 10) complement the description of the tangled cathodic path in THF at ambient temperature (Figures 1 and 6–8). The first cathodic step at R1 also results in PrCN in the instant formation of [Re(3,3′-DHBPY-H+)(CO)3Cl]− independent of the temperature. Only the reduction process at R2′ (Figure 2) depends strongly on the electrolyte temperature. At 223 K, the Re–Cl bond remains sufficiently stable in the reduced state to allow the direct observation of the double-deprotonated complex, [Re(3,3′-DHBPY-2H+)(CO)3Cl]2–, with ν(CO) absorption bands at 2002, 1885, and 1866 cm–1 (Figure 9). Notably, for Re/4,4′-DHBPY the corresponding dianionic complex is stable already at room temperature,46 exhibiting comparable ν(CO) wavenumbers (Table 2, considered in this work still as 6-coordinate, with the Re–Cl bond intact). As the IR SEC experiment at 223 K progresses, there is evidence for the formation of a small amount of a secondary product absorbing at 1967, 1849, and 1835 cm–1 (Figure 9). The latter compound becomes the dominant observable product already during the reduction of [Re(3,3′-DHBPY-H+)(CO)3Cl]− at R2′ when the electrolyte temperature is elevated to 258 K; its precursor [Re(3,3′-DHBPY-2H+)(CO)3Cl]2– is almost not perceptible during the IR spectral monitoring at this temperature (Figure 10). This development is consistent with the cleavage of the Re–Cl bond in [Re(3,3′-DHBPY-2H+)(CO)3Cl]2– and coordination of a PrCN solvent molecule in the vacated position. Indeed, the above ν(CO) wavenumbers can be ascribed to the 6-coordinate (solvento) analogue of 5-coordinate [Re(3,3′-DHBPY-2H+)(CO)3]3– seen at room temperature in weakly coordinating THF (Figures 5–7), that is, [Re(3,3′-DHBPY-2H+)(CO)3(PrCN)]3– (Table 2). The reference complex [Re(BPY)(CO)3(PrCN)]−, lacking the two donor phenoxide groups, consistently absorbs in the IR ν(CO) region at higher wavenumbers, viz., 1980, 1861, and 1851 cm–1; the stability of the PrCN-coordinated Re center increases by lowering the temperature.24
Figure 9.
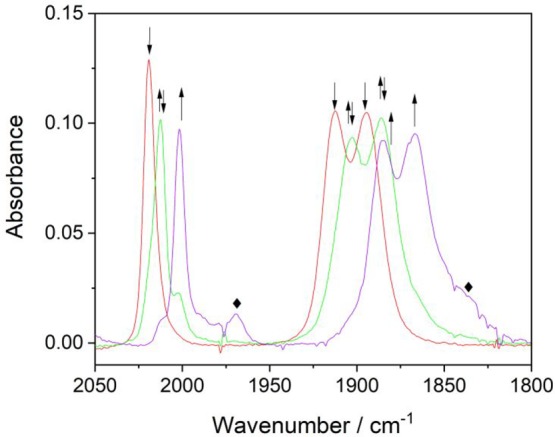
Cathodic IR spectroelectrochemistry of [Re(3,3′-DHBPY)(CO)3Cl] (↓) in PrCN/Bu4NPF6 at 223 K. The parent complex undergoes two successive reductive deprotonations yielding [Re(3,3′-DHBPY-H+)(CO)3Cl]− (↑↓) at R1, and [Re(3,3′-DHBPY-2H+)(CO)3Cl]2– (↑) at R2′. The label ⧫ indicates the presence of a small amount of [Re(3,3′-DHBPY-2H+)(CO)3(PrCN)]3– as a secondary product resulting from the dissociation of the Re–Cl bond in the double-deprotonated precursor complex.
Figure 10.
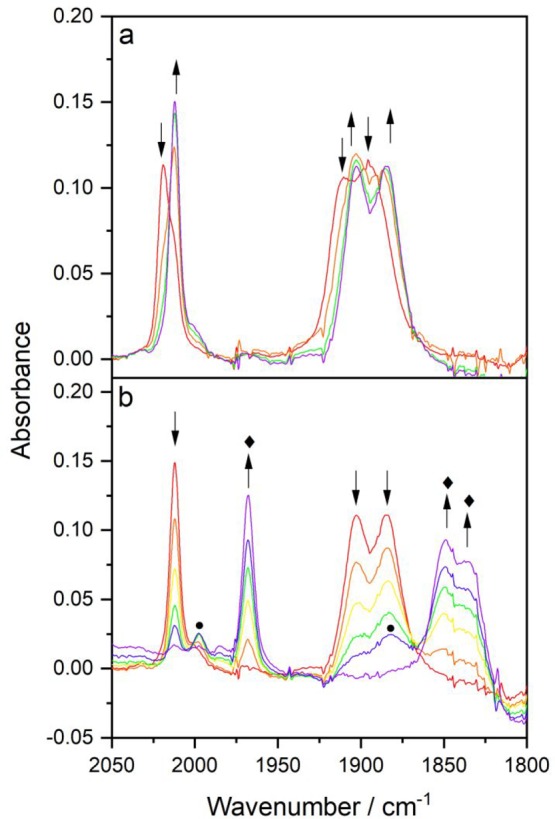
Cathodic IR spectroelectrochemistry of [Re(3,3′-DHBPY)(CO)3Cl] (↓) in PrCN/Bu4NPF6 at 258 K. (a) Conversion of the parent complex to [Re(3,3′-DHBPY-H+)(CO)3Cl]− (↑) at R1. (b) Reductive transformation of [Re(3,3′-DHBPY-H+)(CO)3Cl]− (↓) at R2′ to transient [Re(3,3′-DHBPY-2H+)(CO)3Cl]2–, denoted by ●, and dominant [Re(3,3′-DHBPY-2H+)(CO)3(PrCN)]3–, denoted by ⧫.
At room temperature, [Re(3,3′-DHBPY-H+)(CO)3Cl]− formed at R1 (Figure S7a,b) becomes further reduced at R2′ to give a mixture of products (Figure S7c), starting with the radical anion of the parent complex, [Re(3,3′-DHBPY)(CO)3Cl]•–, also detectable in THF along the ETC path (see above). Importantly, no [Re(3,3′-DHBPY-2H+)(CO)3Cl]2– seems to be formed in this case (cf. Figure 8b). The subsequent 6-coordinate, double-deprotonated reduction product, [Re(3,3′-DHBPY-2H+)(CO)3(PrCN)]3–, which replaces 5-coordinate analogue [Re(3,3′-DHBPY-2H+)(CO)3]3– in the PrCN electrolyte, appears to coexist initially with its 1e–-oxidized form, [Re(3,3′-DHBPY-2H+)(CO)3(PrCN)]2– (ν(CO) at 1986 and 1874 br cm–1). For comparison, the reference radical [Re(BPY)(CO)3(PrCN)] with less donating BPY•– absorbs in the ν(CO) region at 2010 and 1895 br cm–1.24 At the 1e– BPY-reduced level, the additional double deprotonation of the 3,3′-DHBPY ligand in [Re(3,3′-DHBPY-2H+)(CO)3(PrCN)]2– shifts the ν(CO) bands to smaller wavenumbers again by ca. 20 cm–1. The PrCN coordination can be distinguished by the local C3v symmetry at the Re center and the two ν(CO) absorption bands belonging to the A1 + E modes. Toward the end of the cathodic step at R2′, radical species [Re(3,3′-DHBPY-2H+)(CO)3(PrCN)]2– was further reduced to the 6-coordinate trianion. Different from the situation at 258 K, [Re(3,3′-DHBPY-2H+)(CO)3(PrCN)]3– formed in an equilibrium with 5-coordinate [Re(3,3′-DHBPY-2H+)(CO)3]3– (Figure S7d).
Reduction of [Re(3,3′-DHBPY)(CO)3(PrCN)]+ in PrCN at Variable Temperature
The reductive deprotonation of the 3,3′-DHBPY ligand was also investigated at the more electron-deficient Re center in the complex [Re(3,3′-DHBPY)(CO)3(PrCN)]+ (ν(CO) at 2039 and 1933 br cm–1) prepared instantly in the PrCN electrolyte from the precursor [Re(3,3′-DHBPY)(CO)3(OTf)] (absorbing in THF at ν(CO) 2032, 1928, and 1910 cm–1). The cationic complex undergoes smooth reductive deprotonation at R1 (Figure 3), producing [Re(3,3′-DHBPY-H+)(CO)3(PrCN)], as testified by the characteristic <10 cm–1 shift to smaller ν(CO) wavenumbers (Figure 11a, Table 2). On the basis of this observation, the facile reductive deprotonation of 3,3′-DHBPY hardly depends on the axial ligand. It also proves that after the first reduction at R1 singly deprotonated 3,3′-DHBPY-H+ remains chelated in the N,N′-coordination mode (the local C3v symmetry), since the ν(CO) A1 + E pattern of the precursor is preserved. The subsequent reduction of [Re(3,3′-DHBPY-H+)(CO)3(PrCN)] at R2′ is again decidedly more complex, initially generating a new species absorbing in the IR ν(CO) region at 2004 and 1890 br cm–1 (Figure 11b). These wavenumbers are too small to correspond to the reductively double-deprotonated complex [Re(3,3′-DHBPY-2H+)(CO)3(PrCN)]− (Table 2). In analogy with the above ETC path of [Re(3,3′-DHBPY-H+)(CO)3Cl]− at R2′ producing [Re(3,3′-DHBPY)(CO)3Cl]•– (Scheme 3), we assign the intermediate as nondeprotonated radical [Re(3,3′-DHBPY)(CO)3(PrCN)] existing in the limited cathodic potential range between the onset and the maximum of R2′ where it reduces further (Scheme 4). In support of this assignment, [Re(BPY)(CO)3(PrCN)] with the weaker BPY donor absorbs at slightly larger wavenumbers, viz., 2010 and 1895 br cm–1.24 The ultimate cathodic step encompassed in the potential scan at R2′ is the conversion of [Re(3,3′-DHBPY)(CO)3(PrCN)] into the double-deprotonated complex with the 2e–-reduced aromatic BPY system, viz., [Re(3,3′-DHBPY-2H+)(CO)3(PrCN)]3–, and its 1e–-oxidized form (Figure 11b), reflecting the redox equilibrium observed already at the terminal stage of the cathodic path of [Re(3,3′-DHBPY)(CO)3Cl] in PrCN (Figure S7c,d).
Figure 11.
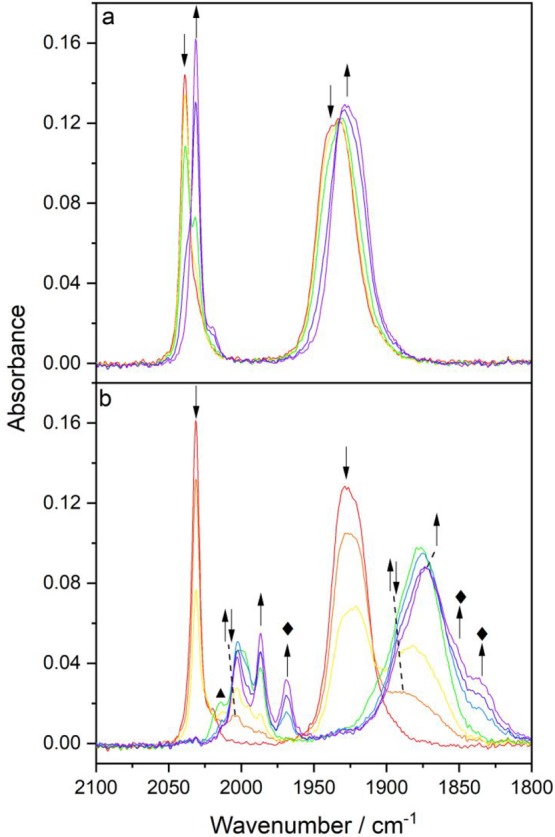
Cathodic IR spectroelectrochemistry of [Re(3,3′-DHBPY)(CO)3PrCN]+ in PrCN/Bu4NPF6 at 298 K. (a) 1e– reduction of the parent complex (↓) at R1 producing deprotonated [Re(3,3′-DHBPY-H+)(CO)3(PrCN)] (↑). (b) Subsequent reduction of [Re(3,3′-DHBPY-H+)(CO)3(PrCN)] (↓) at R2′ to give initially the 2e–-reduced ETC-species [Re(3,3′-DHBPY)(CO)3(PrCN)] (↑↓), which further converts cathodically to the double-deprotonated and 2e–-reduced 6-coordinate species, [Re(3,3′-DHBPY-2H+)(CO)3(PrCN)]3–, denoted by ⧫, in redox equilibrium with its 1e–-oxidized radical form, [Re(3,3′-DHBPY-2H+)(CO)3(PrCN)]2– (↑). The label ▲ denotes an unassigned reduced intermediate.
Scheme 4. Cathodic Pathway of [Re(3,3′-DHBPY)(CO)3(PrCN)]+ in PrCN.

First 1e–-reductive deprotonation at R1 followed by the ETC pathway at the onset of R2′ that produces the corresponding radical of the parent complex, [Re(3,3′-DHBPY•–)(CO)3(PrCN)]. The ultimate 2e–-reductive double deprotonation at R2′ is triggered by the concomitant reduction of the ETC product, resulting in 6-coordinate [Re(3,3′-DHBPY-2H+)(CO)3(PrCN)]2– and its reduced trianionic form isolelectronic with [Re(BPY)(CO)3(PrCN)]−.
The electrochemical reduction of [Re(3,3′-DHBPY)(CO)3(PrCN)]+ in PrCN at 223 K (Figure S8) follows the same path as described above at ambient temperature (Scheme 4). The only notable difference is the appearance of the reductively double-deprotonated complex [Re(3,3′-DHBPY-2H+)(CO)3(PrCN)]−, although only as a minor intermediate when reducing [Re(3,3′-DHBPY-H+)(CO)3(PrCN)] at R2′ (Figure S8b). It is important to reiterate that analogous [Re(3,3′-DHBPY-2H+)(CO)3Cl]2– was stable under the same conditions (Figure 9).
Cyclic Voltammetry and IR Spectroelectrochemistry of [Re(3,3′-DHBPY)(CO)3Cl] in CO2-saturated THF
The cathodic wave R1 of [Re(3,3′-DHBPY)(CO)3Cl], slightly shifted to Ep,c = −1.09 V vs Fc/Fc+ and further broadened in the THF electrolyte saturated with CO2, was passed without any significant increase in the peak current compared to the CV scan under argon (Figure 12). However, the second wave R2′ near −2.40 V vs Fc/Fc+ led to a high current increase, indicative of catalytic CO2 reduction. This behavior coincides with the generation of the 2e–-reduced 5-coordinate complex [Re(3,3′-DHBPY-2H+)(CO)3]3– (Scheme 3, Figures 5c,d), the double-deprotonated equivalent of [Re(BPY)(CO)3]− as one of the iconic catalysts for the CO2 conversion to CO.58 We cannot confirm an active role of transient 5-coordinate species [Re(3,3′-DHBPY-2H+)(CO)3]− in the catalytic mechanism, as proposed by Fujita and co-workers for [Re(4,4′-DHBPY-2H+)(CO)3]− (see Scheme 1).46 According to the ν(CO) pattern (Figures 6 and 9), the detectable double-deprotonated complex is still 6-coordinate, viz., [Re(3,3′-DHBPY-2H+)(CO)3Cl]2–. The Cl– dissociation at elevated temperature results in weakly coordinating THF in ultimate cathodic generation of [Re(3,3′-DHBPY-2H+)(CO)3]3– that is isoelectronic with [Re(BPY)(CO)3]−, each phenoxide group lowering the ν(CO) wavenumbers by ca. 10 cm–1 compared to the unsubstituted BPY ligand.
Figure 12.
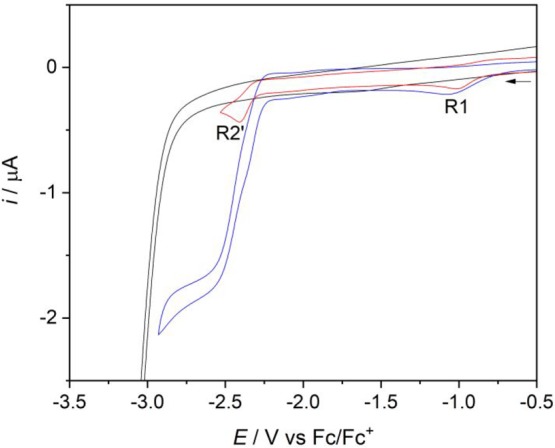
CVs of [Re(3,3′-DHBPY)(CO)3Cl] in THF/Bu4NPF6 saturated with argon (red curve) and CO2 (blue curve). The black curve represents the background CV of CO2 dissolved in the electrolyte. Experimental conditions: Pt microdisc, T = 298 K, v = 100 mV s–1.
Cathodic IR spectroelectrochemistry of [Re(3,3′-DHBPY)(CO)3Cl] in the THF electrolyte saturated with CO2 was conducted to probe the catalytic process triggered at the cathodic wave R2′ (Figure 13). Passing the initial cathodic wave R1 reveals again the smooth transformation of the parent complex to [Re(3,3′-DHBPY-H+)(CO)3Cl]− while the 13CO2 satellite absorption remains intact (Figure 13a). The situation changes at R2′ where the 13CO2 band rapidly drops, and the absorption of free CO at 2135 cm–1 is clearly detectable together with that of free HCO3– at 1675 and 1641 cm–1 (Figure 13b).61 At the same time, the ν(CO) bands of [Re(3,3′-DHBPY-H+)(CO)3Cl]− have only slightly decreased, reflecting the efficient catalytic transformation of CO2 to CO. As expected, the catalyst of the latter process, viz., [Re(3,3′-DHBPY-2H+)(CO)3]3– generated at R2′ (Figure 7), is not observable in the presence of CO2 dissolved in the electrolyte. The strong catalytic performance triggered by the reduction of [Re(3,3′-DHBPY-H+)(CO)3Cl]− resembles [Re(BPY)(CO)3Cl] in acetonitrile, where 2e–-reduced [Re(BPY)(CO)3]− is already formed at the cathodic wave of the parent complex.24
Figure 13.
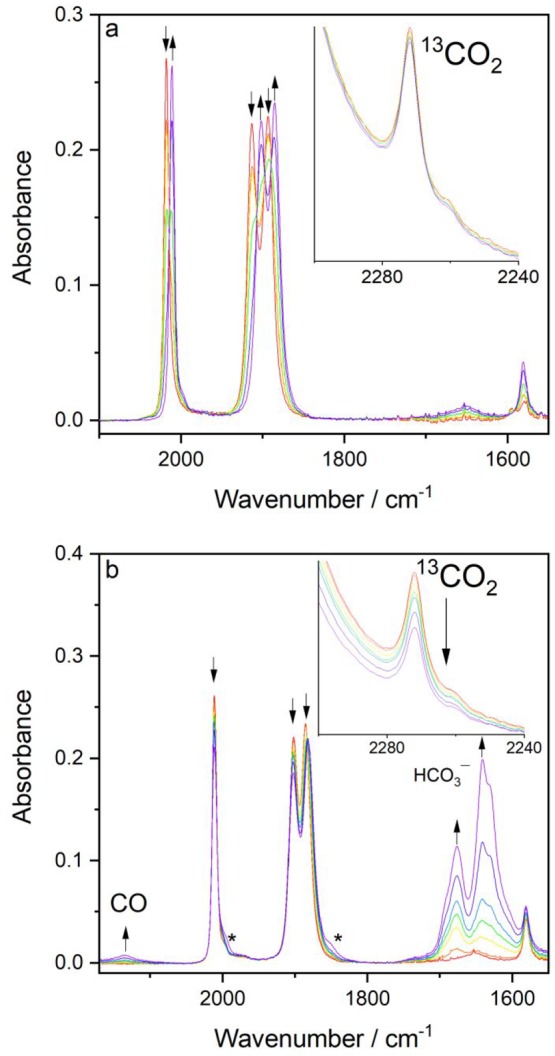
IR spectroelectrochemistry of [Re(3,3′-DHBPY)(CO)3Cl] in CO2-saturated THF/Bu4NPF6. (a) Reduction at R1, reductive deprotonation forming [Re(3,3′-DHBPY-H+)(CO)3Cl]− (↑). (b) Reduction at R2′, efficient electrocatalytic conversion of CO2 to CO, with a minor appearance of free bicarbonate. Inset: the reference 13CO2 satellite band. Asterisk indicates a minor amount of [Re(3,3′-DHBPY)(CO)3Cl]•– (cf. Figure 7).
Coulometry of [Re(3,3′-DHBPY)(CO)3Cl] in CO2-saturated THF
In order to verify the catalytic activity revealed by cyclic voltammetry and IR SEC, bulk electrolysis measurements (Figures S9 and S10) were carried out under CO2 at the catalytic potential of −2.6 V vs Fc/Fc+, using foil and microdisc Pt working electrodes for the 3 mM catalyst precursor in THF/0.5 M TBAPF6. Two different cells were used to accurately determine the faradic efficiency (at the small Pt microdisc) and turnover numbers (at the larger Pt foil). The results have revealed that the current enhancement under CO2 is due to its catalytic conversion to CO, consistent with the reductive disproportionation mechanism: 2CO2 + 2e– → CO + CO32–.15,33 By GC-headspace analysis, an excellent selectivity for CO2 reduction was observed. After 1 h, the total faradaic efficiency was 83% (82% for CO and 1% for H2), with a turnover number of 2.3 (increasing to 3.8 after 3 h). At the smaller microdisc electrode, roughly 0.31 C passed in 1 h (Figure S9); at the same time, the charge passed at the larger foil electrode was roughly 11 C (Figure S10). The corresponding control experiment under argon, using the foil electrode, produced only trace amounts of CO detected by GC and only 0.43 C passed (Figure S11).
Conclusions
Much like the earlier reported46 complex [Re(4,4′-DHBPY)(CO)3Cl], also [Re(3,3′-DHBPY)(CO)3Cl] undergoes facile reductive deprotonation generating [Re(3,3′-DHBPY-H+)(CO)3Cl]−. The stabilizing strong intraligand hydrogen bonding in 3,3′-DHBPY-H+ is the most likely reason for the unusually large positive shift of the reduction potential, Ep,c (R1), and the instantaneous deprotonation in basic DMF compared to the other two isomers. The Cl– ligand in [Re(3,3′-DHBPY-H+)(CO)3Cl]− can be replaced with PrCN. However, this substitution reaction does not occur along the cathodic path, and [Re(3,3′-DHBPY-H+)(CO)3Cl]− remains stable in PrCN under ambient conditions. However, the second reductive deprotonation at Ep,c (R2′) produces [Re(3,3′-DHBPY-2H+)(CO)3Cl]2– that is only stable at sufficiently low temperature (223 K in PrCN). The 6-coordinate geometry and the preserved Re–Cl bond in the latter dianion are strongly supported by the 2A′ + A′′ ν(CO) band pattern (Cs geometry). The ν(CO) wavenumbers in the [Re(x,x′-DHBPY-2H+)(CO)3Cl]2– (x = 3 and 4) series are very similar. For x = 4, the reductively double-deprotonated species was formulated46 as a (stable) 5-coordinate complex [Re(4,4′-DHBPY-2H+)(CO)3]− (Scheme 1); however, this formulation does not seem to correspond with its solution IR spectra that do not reflect the expected fluxionality of the Re(CO)3 moiety and instead show only a modest low-energy shift of the ν(CO) wavenumbers, viz., ca. 10 cm–1 per each deprotonating step. No evidence for [Re(3,3′-DHBPY-2H+)(CO)3]− has also been obtained at room temperature in weakly coordinating THF. Instead, the cleavage of the Re–Cl bond at the cathodic wave R2′ induces a concomitant transfer of two extra electrons placed in the aromatic BPY system of the 3,3′-DHBPY-2H+ ligand. The ultimate reduction product, 5-coordinate [Re(3,3′-DHBPY-2H+)(CO)3]3–, reveals a high degree of π-delocalization of the added electron pair over the Re tricarbonyl unit, similar to the bonding situation in reference species24,58 [Re(BPY)(CO)3]−. The alternative cathodic transformation of [Re(3,3′-DHBPY-H+)(CO)3Cl]− to [Re(3,3′-DHBPY-2H+)(CO)3]3– at R2′ begins with an ETC process at the onset of the wave, which counterintuitively produces the radical anion of the parent complex, Re(3,3′-DHBPY)(CO)3Cl]•–. A similar cathodic path is followed by the cationic derivative [Re(3,3′-DHBPY)(CO)3(PrCN)]+ that smoothly reductively deprotonates at R1 to [Re(3,3′-DHBPY-H+)(CO)3(PrCN)], followed by the ETC step at R2′ generating the radical [Re(3,3′-DHBPY)(CO)3(PrCN)]. In the ultimate reduction step encompassed by the R2′ sequence in PrCN, 6-coordinate [Re(3,3′-DHBPY-2H+)(CO)3(PrCN)]3– replaces the 5-coordinate trianion seen in weakly coordinating THF. This also applies for the cathodic path of [Re(3,3′-DHBPY)(CO)3Cl] in PrCN at low temperature. Finally, [Re(3,3′-DHBPY-2H+)(CO)3]3– was found in THF to catalyze 2e–-reductive disproportionation of CO2 to CO with 82% current efficiency, as revealed by IR spectroelectrochemistry and coulometry combined with gas chromatography. The modest catalytic performance places precursor [Re(3,3′-DHBPY)(CO)3Cl] between more efficient Re/4,4′-DHBPY and poorly performing Re/6,6′-DHBPY congeners.46 This difference can be ascribed to higher nucleophilicity of the nitrogen atoms in chelated [4,4′-DHBPY-2H+]2– (see the resonance structure in Scheme 1) compared to [3,3′-DHBPY-2H+]2–.56
Acknowledgments
This work was jointly funded by EPSRC-DTP (GS16-014). and Spectroelectrochemistry Reading, a spin-out company of the University of Reading. The authors acknowledge the University of Reading for the NMR access in the Chemical Analysis Facility and the Stephenson Institute for Renewable Energy, University of Liverpool, who provided access to their analytical equipment and expertise in conducting the coulometric experiments.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.inorgchem.0c00263.
Electrochemical data with independent numbering (PDF)
The authors declare no competing financial interest.
Supplementary Material
References
- Morris A. J.; Meyer G. J.; Fujita E. Molecular Approaches to the Photocatalytic Reduction of Carbon Dioxide for Solar Fuels. Acc. Chem. Res. 2009, 42, 1983–1994. 10.1021/ar9001679. [DOI] [PubMed] [Google Scholar]
- Gust D.; Moore T. A.; Moore A. L. Solar Fuels via Artificial Photosynthesis. Acc. Chem. Res. 2009, 42, 1890–1898. 10.1021/ar900209b. [DOI] [PubMed] [Google Scholar]
- Armaroli N.; Balzani V. Solar Electricity and Solar Fuels: Status and Perspectives in the Context of the Energy Transition. Chem. Eur. J. 2016, 22, 32–57. 10.1002/chem.201503580. [DOI] [PubMed] [Google Scholar]
- Benson E. E.; Kubiak C. P.; Sathrum A. J.; Smieja J. M. Electrocatalytic and Homogeneous Approaches to Conversion of CO2 to Liquid Fuels. Chem. Soc. Rev. 2009, 38, 89–99. 10.1039/B804323J. [DOI] [PubMed] [Google Scholar]
- Savéant J.-M. Molecular Catalysis of Electrochemical Reactions. Mechanistic Aspects. Chem. Rev. 2008, 108, 2348–2378. 10.1021/cr068079z. [DOI] [PubMed] [Google Scholar]
- Kobayashi K.; Tanaka K. Approach to Multi-Electron Reduction beyond Two-Electron Reduction of CO2. Phys. Chem. Chem. Phys. 2014, 16, 2240–2250. 10.1039/c3cp52635f. [DOI] [PubMed] [Google Scholar]
- Windle C. D.; Perutz R. N. Advances in Molecular Photocatalytic and Electrocatalytic CO2 Reduction. Coord. Chem. Rev. 2012, 256, 2562–2570. 10.1016/j.ccr.2012.03.010. [DOI] [Google Scholar]
- Elgrishi N.; Chambers M. B.; Wang X.; Fontecave M. Molecular Polypyridine-Based Metal Complexes as Catalysts for the Reduction of CO2. Chem. Soc. Rev. 2017, 46, 761–796. 10.1039/C5CS00391A. [DOI] [PubMed] [Google Scholar]
- Grice K. A. Carbon Dioxide Reduction with Homogenous Early Transition Metal Complexes: Opportunities and Challenges for Developing CO2 Catalysis. Coord. Chem. Rev. 2017, 336, 78–95. 10.1016/j.ccr.2017.01.007. [DOI] [Google Scholar]
- Dalle K. E.; Warnan J.; Leung J. J.; Reuillard B.; Karmel I. S.; Reisner E. Electro- and Solar-Driven Fuel Synthesis with First Row Transition Metal Complexes. Chem. Rev. 2019, 119, 2752–2875. 10.1021/acs.chemrev.8b00392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang C.; Nichols A. W.; Machan C. W. A Look at Periodic Trends in D-Block Molecular Electrocatalysts for CO2 Reduction. Dalton Trans. 2019, 48, 9454–9468. 10.1039/C9DT00491B. [DOI] [PubMed] [Google Scholar]
- Francke R.; Schille B.; Roemelt M. Homogeneously Catalyzed Electroreduction of Carbon Dioxide - Methods, Mechanisms, and Catalysts. Chem. Rev. 2018, 118, 4631–4701. 10.1021/acs.chemrev.7b00459. [DOI] [PubMed] [Google Scholar]
- Mizukawa T.; Tsuge K.; Nakajima H.; Tanaka K. Selective Production of Acetone in the Electrochemical Reduction of CO2 Catalyzed by a Ru-Naphthyridine Complex. Angew. Chem., Int. Ed. 1999, 38, 362–363. . [DOI] [PubMed] [Google Scholar]
- Tanaka K. Metal-Catalyzed Reversible Conversion between Chemical and Electrical Energy Designed towards a Sustainable Society. Chem. Rec. 2009, 9, 169–186. 10.1002/tcr.200800039. [DOI] [PubMed] [Google Scholar]
- Machan C. W.; Chabolla S. A.; Kubiak C. P. Reductive Disproportionation of Carbon Dioxide by an Alkyl-Functionalized Pyridine Monoimine Re(I) fac-Tricarbonyl Electrocatalyst. Organometallics 2015, 34, 4678–4683. 10.1021/acs.organomet.5b00406. [DOI] [Google Scholar]
- Smieja J. M.; Kubiak C. P. Re(Bipy-tBu)(CO)3Cl - Improved Catalytic Activity for Reduction of Carbon Dioxide: IR-Spectroelectrochemical and Mechanistic Studies. Inorg. Chem. 2010, 49, 9283–9289. 10.1021/ic1008363. [DOI] [PubMed] [Google Scholar]
- Stanton C. J.; Machan C. W.; Vandezande J. E.; Jin T.; Majetich G. F.; Schaefer H. F.; Kubiak C. P.; Li G.; Agarwal J. Re(I) NHC Complexes for Electrocatalytic Conversion of CO2. Inorg. Chem. 2016, 55, 3136–3144. 10.1021/acs.inorgchem.6b00079. [DOI] [PubMed] [Google Scholar]
- Hawecker J.; Lehn J. M.; Ziessel R. Electrocatalytic Reduction of Carbon Dioxide Mediated by Re(Bipy)(CO)3Cl (Bipy = 2,2′-Bipyridine). J. Chem. Soc., Chem. Commun. 1984, 984, 328–330. 10.1039/C39840000328. [DOI] [Google Scholar]
- Sullivan B. P.; Bolinger C. M.; Conrad D.; Vining W. J.; Meyer T. J. One- and Two-Electron Pathways in the Electrocatalytic Reduction of CO2 by fac-Re(Bpy)(CO)3Cl (Bpy = 2,2′-Bipyridine). J. Chem. Soc., Chem. Commun. 1985, 985, 1414–1416. 10.1039/C39850001414. [DOI] [Google Scholar]
- Keith J. A.; Grice K. A.; Kubiak C. P.; Carter E. A. Elucidation of the Selectivity of Proton-Dependent Electrocatalytic CO2 Reduction by fac-Re(Bpy)(CO)3Cl. J. Am. Chem. Soc. 2013, 135, 15823–15829. 10.1021/ja406456g. [DOI] [PubMed] [Google Scholar]
- Vollmer M. V.; Machan C. W.; Clark M. L.; Antholine W. E.; Agarwal J.; Schaefer H. F.; Kubiak C. P.; Walensky J. R. Synthesis, Spectroscopy, and Electrochemistry of (α-Diimine)M(CO)3Br, M = Mn, Re, Complexes: Ligands Isoelectronic to Bipyridyl Show Differences in CO2 Reduction. Organometallics 2015, 34, 3–12. 10.1021/om500838z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crawley M. R.; Kadassery K. J.; Oldacre A. N.; Friedman A. E.; Lacy D. C.; Cook T. R. Rhenium(I) Phosphazane Complexes for Electrocatalytic CO2 Reduction. Organometallics 2019, 38, 1664–1676. 10.1021/acs.organomet.9b00138. [DOI] [Google Scholar]
- Chabolla S. A.; MacHan C. W.; Yin J.; Dellamary E. A.; Sahu S.; Gianneschi N. C.; Gilson M. K.; Tezcan F. A.; Kubiak C. P. Bio-Inspired CO2 Reduction by a Rhenium Tricarbonyl Bipyridine-Based Catalyst Appended to Amino Acids and Peptidic Platforms: Incorporating Proton Relays and Hydrogen-Bonding Functional Groups. Faraday Discuss. 2017, 198, 279–300. 10.1039/C7FD00003K. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson F. P. A.; George M. W.; Hartl F.; Turner J. J. Electrocatalytic Reduction of CO2 Using the Complexes [Re(Bpy)(CO)3L]n (n = + 1, L = P(OEt)3, CH3CN; n = 0, L = Cl–, OTf–; Bpy = 2,2′-Bipyridine; OTf– = CF3SO3) as Catalyst Precursors: Infrared Spectroelectrochemical Investigation. Organometallics 1996, 15, 3374–3387. 10.1021/om960044+. [DOI] [Google Scholar]
- Smieja J. M.; Sampson M. D.; Grice K. A.; Benson E. E.; Froehlich J. D.; Kubiak C. P. Manganese as a Substitute for Rhenium in CO2 Reduction Catalysts: The Importance of Acids. Inorg. Chem. 2013, 52, 2484–2491. 10.1021/ic302391u. [DOI] [PubMed] [Google Scholar]
- Bourrez M.; Molton F.; Chardon-Noblat S.; Deronzier A. [Mn(Bipyridyl)(CO)3Br]: An Abundant Metal Carbonyl Complex as Efficient Electrocatalyst for CO2 Reduction. Angew. Chem., Int. Ed. 2011, 50, 9903–9906. 10.1002/anie.201103616. [DOI] [PubMed] [Google Scholar]
- Grills D. C.; Ertem M. Z.; McKinnon M.; Ngo K. T.; Rochford J. Mechanistic Aspects of CO2 Reduction Catalysis with Manganese-Based Molecular Catalysts. Coord. Chem. Rev. 2018, 374, 173–217. 10.1016/j.ccr.2018.05.022. [DOI] [Google Scholar]
- Agarwal J.; Shaw T. W.; Stanton C. J.; Majetich G. F.; Bocarsly A. B.; Schaefer H. F. NHC-Containing Manganese(I) Electrocatalysts for the Two-Electron Reduction of CO2. Angew. Chem., Int. Ed. 2014, 53, 5152–5155. 10.1002/anie.201311099. [DOI] [PubMed] [Google Scholar]
- Lam Y. C.; Nielsen R. J.; Gray H. B.; Goddard W. A. A Mn Bipyrimidine Catalyst Predicted To Reduce CO2 at Lower Overpotential. ACS Catal. 2015, 5, 2521–2528. 10.1021/cs501963v. [DOI] [Google Scholar]
- Stanbury M.; Compain J.-D.; Chardon-Noblat S. Electro and Photoreduction of CO2 Driven by Manganese-Carbonyl Molecular Catalysts. Coord. Chem. Rev. 2018, 361, 120–137. 10.1016/j.ccr.2018.01.014. [DOI] [Google Scholar]
- Sampson M. D.; Nguyen A. D.; Grice K. A.; Moore C. E.; Rheingold A. L.; Kubiak C. P. Manganese Catalysts with Bulky Bipyridine Ligands for the Electrocatalytic Reduction of Carbon Dioxide: Eliminating Dimerization and Altering Catalysis. J. Am. Chem. Soc. 2014, 136, 5460–5471. 10.1021/ja501252f. [DOI] [PubMed] [Google Scholar]
- Machan C. W.; Stanton C. J.; Vandezande J. E.; Majetich G. F.; Schaefer H. F.; Kubiak C. P.; Agarwal J. Electrocatalytic Reduction of Carbon Dioxide by Mn(CN)(2,2′-Bipyridine)(CO)3: CN Coordination Alters Mechanism. Inorg. Chem. 2015, 54, 8849–8856. 10.1021/acs.inorgchem.5b01715. [DOI] [PubMed] [Google Scholar]
- Sampson M. D.; Kubiak C. P. Manganese Electrocatalysts with Bulky Bipyridine Ligands: Utilizing Lewis Acids To Promote Carbon Dioxide Reduction at Low Overpotentials. J. Am. Chem. Soc. 2016, 138, 1386–1393. 10.1021/jacs.5b12215. [DOI] [PubMed] [Google Scholar]
- Spall S. J. P.; Keane T.; Tory J.; Cocker D. C.; Adams H.; Fowler H.; Meijer A. J. H. M.; Hartl F.; Weinstein J. A. Manganese Tricarbonyl Complexes with Asymmetric 2-Iminopyridine Ligands: Toward Decoupling Steric and Electronic Factors in Electrocatalytic CO2 Reduction. Inorg. Chem. 2016, 55, 12568–12582. 10.1021/acs.inorgchem.6b01477. [DOI] [PubMed] [Google Scholar]
- Neri G.; Walsh J. J.; Teobaldi G.; Donaldson P. M.; Cowan A. J. Detection of Catalytic Intermediates at an Electrode Surface during Carbon Dioxide Reduction by an Earth-Abundant Catalyst. Nat. Catal. 2018, 1, 952–959. 10.1038/s41929-018-0169-3. [DOI] [Google Scholar]
- Clark M. L.; Ge A.; Videla P. E.; Rudshteyn B.; Miller C. J.; Song J.; Batista V. S.; Lian T.; Kubiak C. P. CO2 Reduction Catalysts on Gold Electrode Surfaces Influenced by Large Electric Fields. J. Am. Chem. Soc. 2018, 140, 17643–17655. 10.1021/jacs.8b09852. [DOI] [PubMed] [Google Scholar]
- Sinopoli A.; La Porte N. T.; Martinez J. F.; Wasielewski M. R.; Sohail M. Manganese Carbonyl Complexes for CO2 Reduction. Coord. Chem. Rev. 2018, 365, 60–74. 10.1016/j.ccr.2018.03.011. [DOI] [Google Scholar]
- Riplinger C.; Carter E. A. Influence of Weak Brønsted Acids on Electrocatalytic CO2 Reduction by Manganese and Rhenium Bipyridine Catalysts. ACS Catal. 2015, 5, 900–908. 10.1021/cs501687n. [DOI] [Google Scholar]
- Costentin C.; Drouet S.; Robert M.; Savéant J. M. A Local Proton Source Enhances CO2 Electroreduction to CO by a Molecular Fe Catalyst. Science 2012, 338, 90–94. 10.1126/science.1224581. [DOI] [PubMed] [Google Scholar]
- Wilting A.; Stolper T.; Mata R. A.; Siewert I. Dinuclear Rhenium Complex with a Proton Responsive Ligand as a Redox Catalyst for the Electrochemical CO2 Reduction. Inorg. Chem. 2017, 56, 4176–4185. 10.1021/acs.inorgchem.7b00178. [DOI] [PubMed] [Google Scholar]
- Zeng Q.; Messaoudani M.; Vlček A.; Hartl F. Temperature-Dependent Reduction Pathways of Complexes fac-[Re(CO)3(N-R-Imidazole)(1,10-Phenanthroline)]+ (R = H, CH3). Electrochim. Acta 2013, 110, 702–708. 10.1016/j.electacta.2013.02.018. [DOI] [Google Scholar]
- Zeng Q.; Messaoudani M.; Vlček A.; Hartl F. Electrochemical Reductive Deprotonation of an Imidazole Ligand in a Bipyridine Tricarbonyl Rhenium(I) Complex. Eur. J. Inorg. Chem. 2012, 2012, 471–474. 10.1002/ejic.201101100. [DOI] [Google Scholar]
- Franco F.; Cometto C.; Nencini L.; Barolo C.; Sordello F.; Minero C.; Fiedler J.; Robert M.; Gobetto R.; Nervi C. Local Proton Source in Electrocatalytic CO2 Reduction with [Mn(Bpy-R)(CO)3Br] Complexes. Chem. Eur. J. 2017, 23, 4782–4793. 10.1002/chem.201605546. [DOI] [PubMed] [Google Scholar]
- Rotundo L.; Garino C.; Priola E.; Sassone D.; Rao H.; Ma B.; Robert M.; Fiedler J.; Gobetto R.; Nervi C. Electrochemical and Photochemical Reduction of CO2 Catalyzed by Re(I) Complexes Carrying Local Proton Sources. Organometallics 2019, 38, 1351–1360. 10.1021/acs.organomet.8b00588. [DOI] [Google Scholar]
- Taheri A.; Thompson E. J.; Fettinger J. C.; Berben L. A. An Iron Electrocatalyst for Selective Reduction of CO2 to Formate in Water: Including Thermochemical Insights. ACS Catal. 2015, 5, 7140–7151. 10.1021/acscatal.5b01708. [DOI] [Google Scholar]
- Manbeck G. F.; Muckerman J. T.; Szalda D. J.; Himeda Y.; Fujita E. Push or Pull? Proton Responsive Ligand Effects in Rhenium Tricarbonyl CO2 Reduction Catalysts. J. Phys. Chem. B 2015, 119, 7457–7466. 10.1021/jp511131x. [DOI] [PubMed] [Google Scholar]
- Clark M. L.; Cheung P. L.; Lessio M.; Carter E. A.; Kubiak C. P. Kinetic and Mechanistic Effects of Bipyridine (Bpy) Substituent, Labile Ligand, and Brønsted Acid on Electrocatalytic CO2 Reduction by Re(Bpy) Complexes. ACS Catal. 2018, 8, 2021–2029. 10.1021/acscatal.7b03971. [DOI] [Google Scholar]
- Kunkely H.; Vogler A. Optical Properties of (3,3′-Dihydroxy-2,2′-Bipyridine) Tricarbonyl-Rhenium(I) Chloride. Absorption and Emission Spectra. Inorg. Chim. Acta 2003, 343, 357–360. 10.1016/S0020-1693(02)01175-1. [DOI] [Google Scholar]
- Vogler A.; Shenderovich I. G. Photochemistry of Deprotonated Rhenium(I) (3,3′-Dihydroxy-2,2′-Bipyridine) Tricarbonyl Chloride. Photoisomerization at the Chelate in Basic Solution. Inorg. Chim. Acta 2014, 421, 496–499. 10.1016/j.ica.2014.07.011. [DOI] [Google Scholar]
- Ghorai D.; Dutta C.; Choudhury J. Switching of “Rollover Pathway” in Rhodium(III)-Catalyzed C-H Activation of Chelating Molecules. ACS Catal. 2016, 6, 709–713. 10.1021/acscatal.5b02540. [DOI] [Google Scholar]
- Hong S. Y.; Kwak J.; Chang S. Rhodium-Catalyzed Selective C-H Functionalization of NNN Tridentate Chelating Compounds via a Rollover Pathway. Chem. Commun. 2016, 52, 3159–3162. 10.1039/C5CC09960A. [DOI] [PubMed] [Google Scholar]
- Maidich L.; Dettori G.; Stoccoro S.; Cinellu M. A.; Rourke J. P.; Zucca A. Electronic and Steric Effects in Rollover C-H Bond Activation. Organometallics 2015, 34, 817–828. 10.1021/om500681u. [DOI] [Google Scholar]
- Czerwieniec R.; Kapturkiewicz A.; Anulewicz-Ostrowska R.; Nowacki J. ReI(CO)3+ Complexes with N∩O– Bidentate Ligands. J. Chem. Soc., Dalton Trans. 2002, 18, 3434–3441. 10.1039/B204020D. [DOI] [Google Scholar]
- Krejčik M.; Daněk M.; Hartl F. Simple Construction of an Infrared Optically Transparent Thin-Layer Electrochemical Cell: Applications to the Redox Reactions of Ferrocene, Mn2(CO)10 and Mn(CO)3(3,5-Di-t-Butyl-Catecholate)−. J. Electroanal. Chem. Interfacial Electrochem. 1991, 317, 179–187. 10.1016/0022-0728(91)85012-E. [DOI] [Google Scholar]
- Hartl F. Spectroscopic Characterization of Some Unstable Ortho-Semiquinone and Ortho-Quinone Complexes of Mn(I) by Variable-Temperature Thin-Layer Spectroelectrochemistry at Optically Transparent Electrodes. Inorg. Chim. Acta 1995, 232, 99–108. 10.1016/0020-1693(94)04395-C. [DOI] [Google Scholar]
- Himeda Y.; Onozawa-Komatsuzaki N.; Sugihara H.; Arakawa H.; Kasuga K. Half-Sandwich Complexes with Dihydroxy Polypyridine: Water-Soluble, Highly Efficient Catalysts for Hydrogenation of Bicarbonate Attributable to Electron-Donating Ability of Oxyanion on Catalyst Ligand. Organometallics 2004, 23, 1480–1483. 10.1021/om030382s. [DOI] [Google Scholar]
- Hartl F.; Vlček A. Rhenium(I) Carbonyl Dioxolene Complexes: Electrochemical and Spectroelectrochemical (Resonance Raman, UV-Vis, IR) Study of [Re(CO)3L(Diox)]z and [Re(CO)2(PPh3)2(Diox)]z (L = CO, PPh3, P-DPPE, THF, Ph3PO, Me2CO, Py; z = −1, 0, +1) Redox Series. Inorg. Chem. 1992, 31, 2869–2876. 10.1021/ic00039a037. [DOI] [Google Scholar]
- Stor G. J.; Hartl F.; Van Outersterp J. W. M.; Stufkens D. J. Spectroelectrochemical (IR, UV/Vis) Determination of the Reduction Pathways for a Series of [Re(CO)3(α-Diimine)L’]0/+ (L’ = Halide, OTf– THF, MeCN, n-PrCN, PPh3, P(OMe)3) Complexes. Organometallics 1995, 14, 1115–1131. 10.1021/om00003a013. [DOI] [Google Scholar]
- Záliš S.; Consani C.; Nahhas A. El; Cannizzo A.; Chergui M.; Hartl F.; Vlček A. Origin of Electronic Absorption Spectra of MLCT-Excited and One-Electron Reduced 2,2′-Bipyridine and 1,10-Phenanthroline Complexes. Inorg. Chim. Acta 2011, 374, 578–585. 10.1016/j.ica.2011.02.084. [DOI] [Google Scholar]
- Tory J.; Setterfield-Price B.; Dryfe R. A. W.; Hartl F. [M(CO)42,2′-Bipyridine)] (M = Cr, Mo, W) Complexes as Efficient Catalysts for Electrochemical Reduction of CO2 at a Gold Electrode. ChemElectroChem 2015, 2, 213–217. 10.1002/celc.201402282. [DOI] [Google Scholar]
- Cheng S. C.; Blaine C. A.; Hill M. G.; Mann K. R. Electrochemical and IR Spectroelectrochemical Studies of the Electrocatalytic Reduction of Carbon Dioxide by [Ir2(Dimen)4]2+ (Dimen = 1,8-Diisocyanomenthane). Inorg. Chem. 1996, 35, 7704–7708. 10.1021/ic9512385. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.