

Abstract
Background
Asthma is a complicated chronic inflammatory disorder characterized by airway inflammation and bronchial hyperresponsiveness. Group 2 innate lymphoid cells (ILC2) are tissue-resident innate effector cells that can mediate airway inflammation and hyperresponsiveness through production of IL-5, IL-13 and VEGFA. ILC2 in asthma patients exhibit an activated phenotype. However, molecular pathways that control ILC2 activation are not well understood.
Methods
MYC expression was examined in ILC2 sorted from peripheral blood of healthy controls and asthma patients or cultured with or without activating cytokines. CRISPR knockout technique was used to delete c-Myc in primary murine lung ILC2 or an ILC2 cell line. Cell proliferation was examined, gene expression pattern was profiled by genome-wide microarray analysis, and direct gene targets were identified by Chromatin Immunoprecipitation (ChIP). ILC2 responses, airway inflammation and airway hyperresponsiveness were examined in Balb/c mice challenged with Alternaria extracts, with or without treatment with JQ1.
Results
ILC2 from asthma patients expressed increased amounts of MYC. Deletion of c-Myc in ILC2 results in reduced proliferation, decreased cytokine production, and reduced expression of many lymphocyte activation genes. ChIP identified Stat6 as a direct gene target of c-Myc in ILC2. In vivo inhibition of c-Myc by JQ1 treatment repressed ILC2 activity and suppressed Alternaria-induced airway inflammation and AHR.
Conclusion
c-Myc expression is upregulated during ILC2 activation. c-Myc is essential for ILC2 activation and their in vivo pathogenic effects. These findings suggest that targeting c-Myc may unlock novel strategies to combat asthma or asthma exacerbation.
Keywords: group-2 innate lymphoid cells, airway hyperresponsiveness, c-Myc
Graphical Abstract
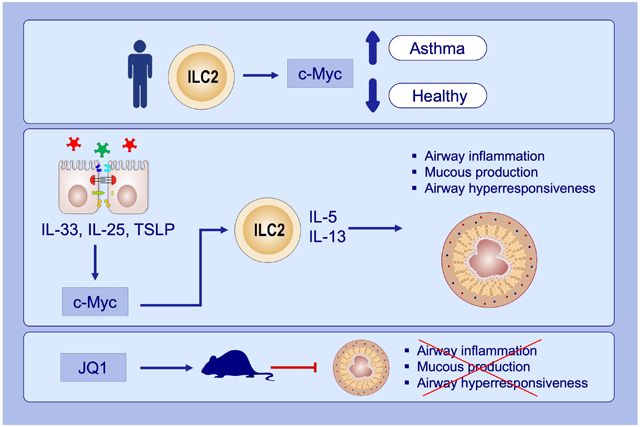
c-Myc expression is upregulated in peripheral blood group 2 innate lymphoid cells (ILC2) of asthma patients.
c-Myc promotes ILC2 activation in response to epithelial-derived cytokines TSLP, IL-33 and IL-25.
Inhibition of c-Myc expression and function by JQ1 (a c-Myc antagonist) treatment represses allergic airway inflammation.
JQ1, (6S)-4-(4-Chlorophenyl)-2,3,9-trimethyl-6H-thieno[3,2-f][1,2,4]triazolo[4,3-a][1,4]diazepine-6-acetic acid 1,1-dimethylethyl ester; TSLP, Thymic stromal lymphopoietin
1. INTRODUCTION
Asthma remains a leading public health concern with unmet therapeutic needs. Asthma is a complicated airway disorder characterized by chronic inflammation, bronchial hyperresponsiveness, and recurrent episodes of symptoms.1, 2 Mounting evidence suggests that both innate and adaptive immune cells play pivotal roles in asthma development and exacerbation.3-6 Recent studies from us and others have indicated the importance of group-2 innate lymphoid cells (ILC2), a unique subset of innate effector cells, in asthma pathogenesis.3, 5, 7-9 ILC2 are a distinct subset of non-circulating tissue-resident cells that are enriched at mucosal barrier sites including the gut and the lung.10 Upon activation, ILC2 are potent producers of type-2 cytokines IL-5 and IL-13 as well as other critical effector molecules such as VEGFA.2, 8 Studies with mouse models suggested that activated ILC2 can induce eosinophil infiltration, promote neutrophilic inflammation, and mediate airway hyperresponsiveness (AHR).8, 11 ILC2 may also promote pathogenic helper 2 T cell responses (Th2) by providing co-stimulatory molecules and presenting antigens.12, 13 ILC2 in asthma patients exhibited an activated phenotype, demonstrated by increased cellularity and enhanced expression of IL-5, IL-13 and VEGFA.8, 14 However, the molecular mechanisms that drive ILC2 activation in asthma are not well understood.
c-Myc is a basic helix-loop-helix transcription factor and pro-oncogene implicated in several cellular processes such as proliferation and differentiation.15, 16 The function of c-Myc in cancer cell lines have been extensively studied, with c-Myc representing one of the most sought-after cancer targets.17-19 However, function and regulation of c-Myc in inflammatory disorders remain poorly understood. c-Myc has been implicated in adaptive T/B lymphocyte development and function. It may control T cell metabolism re-programming during T lymphocyte activation, and its dysregulation may also drive B cell lymphomas.20-23 Whether c-Myc may regulate innate lymphoid cell biology remains unknown.
Here we examined the role of c-Myc in regulating ILC2 responses using samples from human asthma patients as well a mouse model of fungal allergen-induced airway hyperresponsiveness. ILC2 from asthma patients expressed increased amounts of MYC. CRISPR-mediated gene knockout revealed that c-Myc was required for the optimal expression of many lymphocyte activation genes in ILC2, with Stat6 being a direct gene target. More importantly, inhibition of c-Myc repressed ILC2 activity and suppressed ILC2-induced airway inflammation and AHR. Together, our data establish a novel role for c-Myc in controlling ILC2 activation and indicate that targeting c-Myc may unlock novel strategies to combat asthma or asthma exacerbation.
2. MATERIALS AND METHODS
2.1. Study subjects and samples
Informed consent was obtained from all human subjects at the Division of Allergy & Immunology of Albany Medical Center as approved by the Albany Medical Center Institutional Review Board (IRB). Severe asthma as defined according to 2014 ATS guideline of severe asthma. 24 Asthma patients were prescribed asthma medications as described in Table 1. Peripheral blood mononuclear cells (PBMC) from human subjects were isolated by Ficoll gradient centrifugation and ILC2 were sorted by fluorescence activated cell sorting (FACS). mRNA was extracted and qPCR analysis was used to detect gene expression as we previously described.7-9
Table 1.
| Subject | Disease Severity | Sex | Age (year) |
FEV1% | Medication | ||||
|---|---|---|---|---|---|---|---|---|---|
| ICS | LABA | SABD | LTM | Others | |||||
| Healthy Control 1 | M | 29 | |||||||
| Healthy Control 2 | M | 33 | |||||||
| Healthy Control 3 | F | 54 | |||||||
| Healthy Control 4 | F | 37 | |||||||
| Healthy Control 5 | F | 24 | |||||||
| Healthy Control 6 | F | 41 | |||||||
| Healthy Control 7 | F | 39 | |||||||
| Healthy Control 8 | F | 37 | |||||||
| Asthma Patient 1 | severe asthma | M | 55 | 51% | + | + | + | + | anti-IL-5 |
| Asthma Patient 2 | mild asthma | F | 29 | 104% | + | − | + | − | allergen immunotherapy |
| Asthma Patient 3 | mild asthma | F | 55 | 92% | + | − | + | − | anti-IgE, antihistamine |
| Asthma Patient 4 | moderate asthma | M | 13 | 71% | + | + | + | − | antihistamine |
| Asthma Patient 5 | severe asthma | M | 62 | 48% | + | + | + | + | Anti-IgE, antihistamine |
| Asthma Patient 6 | severe asthma | M | 65 | 45% | + | + | + | − | Anti-IgE |
| Asthma Patient 7 | mild asthma | F | 50 | 95% | + | +/− | + | − | antihistamine |
| Asthma Patient 8 | severe asthma | M | 10 | 58% | + | + | + | − | antihistamine |
| Asthma Patient 9 | severe asthma | M | 60 | 73% | + | + | + | − | Anti-IgE |
2.2. Mice, Allergen and cytokine challenge, adoptive transfer and FlexiVent Analysis
Balb/c mice were purchased from Taconic Biosciences. Rag2−/−Il2rg−/− mice and Rag1−/− mice on Balb/c background were purchased from The Jackson Laboratory. Alternaria extracts were purchased from the Greer laboratory. The endotoxin level in Alternaria extracts was around 20 EU/mg by limulus amebocyte lysate assay (WAKO). For Alternaria challenge of wildtype Balb/c mice, 100μg Alternaria alternata extracts were administered to mice intranasally once, 12 hours before FlexiVent analysis. JQ1 (50mg/kg, MedKoo Biosciences) were dissolved in 10% 2-hydroxypropyl-β-cyclodextrin solution. JQ1 or vehicle were injected intraperitoneally twice, at 24 hours before, and at the time of Alternaria challenge. 25 FlexiVent system (SCIREQ) was used to measure airway hyperresponsiveness as we previously described.8 Mouse protocols were approved by the Institutional Animal Care and Use Committee of Albany Medical College.
For adoptive transfer, 105 ILC2 were sorted by FACS from Balb/c mice treated with IL-33 daily for 7 days. The purpose of IL-33 treatment is to obtain sufficient numbers of primary ILC2 for adoptive transfer. 100μg Alternaria alternata extracts (Greer) were administered to mice intranasally 24 hours after ILC2 transfer. JQ1 or vehicle were injected intraperitoneally twice at 24 hours before, and again at the time of Alternaria challenge. 25 Mice were euthanized 12 hours after Alternaria challenge.
For Alternaria challenge of Rag1−/− mice, 100μg Alternaria alternata extracts (Greer) were administered to mice intranasally daily for 3 days. Mice were euthanized one day after the last challenge.
Histology of lung tissue
Lungs were inflated and fixed in 10% Neutral Buffered Formalin (Sigma) overnight, and embedded in paraffin blocks. 5 μm tissue sections were stained with periodic acid-Schiff (PAS). Slides were examined by a pathologist who was blinded to the experimental groups.
2.3. Flow Cytometry and Cell Sorting
Human peripheral blood ILC2 were identified as CD45+Lin−IL-7Rα+CRHT2+ cells. Mouse lung ILC2 were identified as CD45+Lin−Thy1.2+ST2+ cells according to our previous studies.7-9 Lineage antibodies for human cells included anti-CD16 (3G8), anti-CD11c (3.9), anti-CD123 (6H6), anti-CD19 (HIB19), anti-CD3 (UCHT1), anti-CD5 (L17F12), anti-CD94 (DX22), anti-CD34 (581). Additional antibodies used to recognize human ILC2 included anti-CRTH2 (BM16), anti-CD127 (A019D5) and anti-CD117 (104D2). Lineage antibodies for mouse ILC2 included anti-B220 (RA3-6B2), anti-CD3 (2C11), anti-TCRβ (H57), anti-TCRγδ (GL-3), anti-CD11b (M1/70), anti-CD5 (53-7.3). Additional anti-mouse antibodies used included anti-IL-5 (TRFK5), anti-IL-13 (eBio13A), anti-Thy1.2 (53-2.1), anti-Ki67 (16A8), anti-Gr-1 (RB6-8C5), anti-CD11c (N418), anti-CD25 (PC61.5), anti-ST2 (DJ8; MD Bioproducts) and anti-Siglec F (E50-2440; BD Biosciences). Antibodies were purchased from Biolegend or eBioscience, unless specified otherwise. FACS was performed on a FACSAria II (BD Biosciences) and regular flow cytometric analysis was performed on a FACSCanto (BD Biosciences). The Intracellular Cytokine Staining Kit from BD Pharmingen was used to stain intracellular IL-13 and IL-5 as we previously described. 7-9
2.4. Cell culture
Cytokines were purchased from Biolegend or R&D. Freshly isolated human blood ILC2 or mouse lung ILC2 were cultured with α-MEM containing 20% FCS in the presence of the indicated cytokines. A murine ILC2 cell line, ILC2/b6, was described in our previous studies, and maintained with 10ng/ml of IL-2 and IL-33.26 ILC2/b6 cell line was derived from small intestinal laminal propria ILC2. ILC2/b6 cell line cells exhibit the features of activated ILC2. They vigorously proliferate with a doubling time of around 24 hours.26 They constantly produce large amounts of IL-5 and IL-13.26 They have preserved the essential molecular features of primary ILC2, including high expression of GATA3, IL-7Rα, T1/ST2, CD25, and IL-25R.26
2.5. Chromatin immunoprecipitation and CRISPR-mediated gene knockout
Chromatin immunoprecipitation (CHIP) was performed with 2×107 cultured ILC2/b6 cells as we previously described.7 For CRISPR-mediated gene knockout, the following single guide RNA (sgRNA) sequences were used: 5’-GGACGTAGCGACCGCAACAT-3’ (targeting Myc); 5’-TGCGAATACGCCCACGCGATGGG-3’ (non-target control). The sgRNA sequences were previously published by Dr. Feng Zhang’s lab.27 For the CRISPR-mediated gene knockout, error-prone non-homologous end jointing (NHEJ) was used to introduce mutations in the bulk cell population.28,29 sgRNAs were cloned into lenti-CRISPRv2-GFP vectors as we previously described.7 For growth and qPCR assay, freshly isolated primary lung ILC2 was cultured with IL-7, IL-2 and IL-33 for 3 days, and lentiviral transduction was performed by spin-infection as we previously described.7 GFP+ cells were sorted at 48 hours post transduction and growth rate was examined. qPCR was performed with FACS-sorted GFP+ cells, 10 days after lentiviral transduction. For genome-wide microarray and western blot analyses, lentiviral transduction was performed with cultured ILC2/b6 cells as we previously described.7 GFP+ cells were sorted by FACS, 10 days post transduction, and Genome-wide microarray analysis or western blotting was performed.
2.6. Microarray Experiments
Total ILC2 mRNA was extracted and purified by Qiagen RNeasy kits. Microarray experiments were performed at Boston University Microarray and Sequencing Resource. Data were deposited at Gene Expression Omnibus (http://www.ncbi.nlm.nih.gov/geo, GSE129309). Gene Set Enrichment Analysis (GSEA) from microarray data was provided by Boston University Clinical & Translational Science Institute translational bioinformatics consultation service (CTSA grant GRANT U54-TR001012).
2.7. Statistical analysis
Mann Whitney U test or Kruskal-Wallis test was used to compare the difference in mRNA expression levels in sorted ILC2 from human subjects. Student’s t test or ANOVA was used in all other experiments. Data were expressed as mean and SD. P <0.05 was considered statistically significant.
3. RESULTS
3.1. MYC expression in ILC2 is increased in asthma patients
To explore potential molecular regulators of ILC2 in asthma, we sorted ILC2 from the peripheral blood of asthma patients and healthy controls by fluorescence activated cell sorting (FACS) and examined their gene expression by qPCR. Female and male asthma patients with diverse ages and disease severity profiles were recruited (Table 1). Human ILC2 were identified as CD45+Lin−IL-7Rα+CRTH2+ cells (Figure 1A). Post-sorting analysis revealed greater than 95% purity (Figure 1A). We detected MYC expression in ILC2 from both asthma and healthy controls (Figure 1B). However, the expression of MYC in ILC2 of asthma patients was significantly higher than those in healthy controls (P = 0.043, Mann-Whitney U test) (Figure 1B). Great heterogeneity in MYC expression has been observed in the ILC2 of asthma patients (Figure 1B). We next classified asthma patients based on disease severity according to the 2014 ATS guideline of severe asthma (Table 1).24 Strikingly, ILC2 in severe asthma patients, but not those in mild or moderate asthma patients, had significantly increased expression of MYC, compared to those in healthy controls (Figure 1C). Together, ILC2 in asthma patients have increased expression of MYC, which is related to disease severity.
FIGURE 1. ILC2 from asthma patients expressed high level of MYC.

(A) Representative flow cytometry profile of human peripheral blood ILC2s. Human ILC2s were identified as CD45+Lin−IL-7Rα+CRTH2+. (B) mRNA levels of MYC in sorted ILC2 from healthy donors and asthma patients. Data are normalized by GAPDH. (C) mRNA levels of MYC in sorted ILC2 from healthy donors, patients with mild/moderate asthma, and patients with severe asthma. Data were normalized by GAPDH. (D) mRNA levels of MYC in human peripheral blood ILC2s cultured for 5 days with IL-33, IL-25, TSLP or medium alone. Data were normalized by GAPDH. (E) mRNA levels of Myc in mouse lung ILC2 cultured for 5 days with 10ng/ml IL-2 and IL-7, in the presence of the indicated concentration of IL-33. (F) Mouse lung ILC2 cultured for 5 days with 10ng/ml IL-2 and IL-7, in the presence of the indicated concentration of IL-33. IL-13 expression was examined by flow cytometry analysis, and mean fluorescence intensity (MFI) was shown. Data are from 9 healthy donor and 9 asthma patients (A-C), or from 4 healthy donors, pooled from four independent experiments (D), or from 4 mice per group, representative of two independent experiments (E, F). *P < 0.05.
Because ILC2 in asthma patients exhibited an activated phenotype,30 we hypothesized that increased MYC expression was an indicator of ILC2 activation. We therefore cultured sort-purified human blood ILC2 with or without the activating cytokines IL-25, IL-33, or TSLP (Figure 1D). All of these activating cytokines significantly upregulated the expression of MYC in human ILC2, indicating that enhanced MYC expression might be a signature of ILC2 activation (Figure 1D). To verify whether lung-resident ILC2 may also upregulate Myc expression upon activation, we sort-purified ILC2 from the lungs of Balb/c mice and cultured these murine lung-resident ILC2 with or without the stimulating cytokine IL-33 (Figure 1E and F). Mouse lung ILC2 were identified as CD45+Lin−Thy1+T1/ST2+ cells, as previously described (Figure S1). IL-33 upregulated the expression of Myc in mouse lung ILC2 and stimulated their production of IL-13 in a dose-dependent manner (Figure 1E and F). Together, these results suggest that increased MYC expression is an indicator of ILC2 activation.
3.2. c-Myc is required for ILC2 activation
To examine the role of c-Myc in ILC2 activation, we deleted c-Myc in vitro using lentiviral lenti-CRISPRv2-GFP knockout constructs targeting c-Myc or non-target control (NTC). The sgRNA for c-Myc targeted the second exon of c-Myc, around 200bp downstream of the translation start codon (Figure S2A). For these experiments, we used primary ILC2 freshly isolated from the lungs of naïve mice, or ILC2/b6 line cells. ILC2/b6 is an established ILC2 cell line that exhibits the molecular and functional properties of activated ILC2. ILC2/b6 cells express all known ILC2 signature genes, and they vigorously produce IL-5 and IL-13 in vitro.26 Western blot results with ILC2/b6 cells revealed high deletion efficiency of c-Myc in GFP+ cells (Figure 2A). Truncated forms of c-Myc were not detected (Figure S2B). Primary ILC2 can also be efficiency transduced with the CRISPRv2-GFP lentivirus, demonstrated by high percentage of GFP+ cells at 48 hours post transduction (Figure S2C). Notably, knockout of c-Myc drastically suppressed ILC2 proliferation in vitro (Figure 2B). The production of both IL-5 and IL-13 was also significantly reduced in c-Myc deficient ILC2, demonstrating an essential role for c-Myc in promoting ILC2 activation (Figure 2C).
FIGURE 2. c-Myc is required for ILC2 activation in vitro.

(A) ILC2/b6 cells were transduced with lenti-CRSIPRv2-GFP knockout constructs targeting c-Myc or non-target control (NTC). GFP+ cells were sorted on 10 days post transduction, and expression of c-Myc was examined by western blot. (B) Primary mouse ILC2 were isolated from the lungs of naïve mice, cultured with IL-33, IL-7 and IL-2, and transduced with lenti-CRSIPRv2-GFP knockout constructs targeting NTC or c-Myc. Growth of GFP+ cells was calculated as fold change in cell number over 5 days of culture. (C) Expression of IL-5 and IL-13 by GFP+ ILC2 were examined at 10 days after lentiviral transduction. (D) ILC2/b6 cells were transduced with lenti-CRSIPRv2-GFP knockout constructs targeting NTC or c-Myc. Genome-wide microarray was performed with sorted GFP+ cells at 10 days post transduction, and gene set enrichment analyses were performed. (E) Heatmap of representative genes by microarray analysis. (F) Primary mouse ILC2 were isolated from the lungs of naïve mice, cultured with IL-33, IL-7 and IL-2, and transduced with lenti-CRSIPRv2-GFP knockout constructs targeting NTC or c-Myc. mRNA level of Stat6 in GFP+ ILC2 was measured by qPCR analysis at 10 days post transduction. (G) CHIP analysis was performed in ILC2/b6 cells. DNA region lacking c-Myc binding site was used as a negative control. Data are representative of three independent experiments (A), or are from three or four mice per group, representative of two independent experiments (B and C), or are from three biological replicate samples (D and E), or are from 3-4 samples, representative of two to four independent experiments (F and G). *P < 0.05.
Of note, because c-Myc knockout ILC2 did not grow well, we were not able to expand single cell clones of c-Myc deleted ILC2. Hence, the precise mutations could not be examined by regular Sanger sequencing. But our western blotting data demonstrated high deletion efficiency of c-Myc in the bulk ILC2 population (Figure 2A).
To explore the underlying mechanisms by which c-Myc promotes ILC2 activation, we used genome-wide microarray analysis to profile c-Myc dependent genes in ILC2. Due to the rarity of primary ILC2 and the inability of c-Myc deficient cells to proliferate, we used ILC2/b6 cell line to obtain sufficient numbers of cells for unbiased microarray analysis without RNA or cDNA amplification. We transduced the ILC2/b6 cells with lentiviral lenti-CRISPRv2-GFP knockout constructs targeting c-Myc or NTC. We then sort purified GFP+ cells and examined the transcriptomes at 10 days post lentiviral transduction. Interestingly, gene pathway analysis revealed that the expression of lymphocyte activation genes was extensively reduced in c-Myc deficient ILC2 (Figure 2D). The c-Myc dependent genes in ILC2 include the ILC2 master transcription factor Gata3, key ILC2 cytokine receptors Il1rl1 and Il7r, and signaling transduction molecules Pik3ca, Stat6, and Jak2 (Figure 2E). To further identify direct c-Myc targets in ILC2, we searched for promoter regions of the downregulated genes in c-Myc deficient ILC2, and identified a conserved c-Myc binding site at the −120 bp of the promoter region of Stat6. Using primary lung ILC2 isolated from Balb/c mice, we verified that Stat6 expression was indeed dependent on c-Myc in primary lung ILC2 by qPCR (Figure 2F). Chromatin immunoprecipitation demonstrated that c-Myc directly bound to the −120 bp promoter of Stat6 in ILC2 (Figure 2G). Together, c-Myc is required for the expression of many lymphocyte activation genes in ILC2, with Stat6 being a direct gene target.
3.3. c-Myc inhibition represses ILC2 responses in a mouse model of Alternaria-inhalation
To determine the in vivo relevance of c-Myc regulation of ILC2 activation, we challenged mice intranasally with extracts of Alternaria alternata. Alternaria is a fungi allergen that can trigger acute and severe asthma attacks.31-33 Recent study indicates that c-Myc expression and function can be repressed by the inhibition of bromodomain and extra-terminal motif proteins. We used JQ1, a selective and potent bromodomain inhibitor, to investigate the role of c-Myc in promoting ILC2 responses in vivo. JQ1 has a short half-life and excellent pharmacokinetics in vivo.34 Previous study indicates that intraperitoneal administration of JQ1 at the dose of 50mg/kg/day can repress MYC expression and tumor progression in vivo, without affecting the viability of hematopoietic cells.35 Alternaria inhalation rapidly upregulated Myc expression in lung ILC2 within 12 hours, verifying that increased Myc expression is a sensible indicator of ILC2 activation in vivo (Figure 3A). JQ1 treatment reduced Myc expression in ILC2 in Alternaria-challenged mice (Figure 3A). ILC2 in Alternaria-challenged mice exhibited an activated phenotype, demonstrated by increased expression of the proliferation maker Ki67 and enhanced production of IL-5 and IL-13 (Figure 3B-E). JQ1 treatment prevented ILC2 proliferation in Alternaria-challenged mice (Figure 3B and C). JQ1 treatment also drastically repressed production of IL-5 and IL-13 by lung ILC2, suggesting that c-Myc is required for ILC2 activation in vivo (Figure 3D and E). JQ1 treatment also reduced IL-5 release in the lavage (BAL) of Alternaria-challenged mice (Figure 3F). Of note, the concentrations of ILC2-activating cytokines, including IL-33, IL-25 and TSLP, were not altered by JQ1 treatment (Figure 3G). Together, these data indicate a critical role for c-Myc in promoting ILC2 responses in vivo.
FIGURE 3. JQ1 treatment repressed ILC2 activation in vivo.

(A) Balb/c mice were challenged intranasally with Alternaria extracts or PBS and treated intraperitoneally with JQ1 or vehicle control. mRNA levels of Myc in mouse lung ILC2 were examined 12 hours after PBS or Alternaria challenge. (B) Representative flow cytometry profiles depict Ki67 expression by mouse lung ILC2. (C) Percentages of ILC2 in G0 phase. (D) Representative flow cytometry profiles depict expression of IL-5 and IL-13 by lung ILC2 at 12 hours after PBS or Alternaria challenge. (E) Quantification of IL-5 and IL-13 expression by lung ILC2. (F) Concentration of IL-5 in the bronchoalveolar lavage (BAL). (G) Concentrations of IL-33, IL-25 and TSLP in the lung homogenate. (H) FlexiVent analysis of total lung airway resistance (RAW). (I) Representative flow cytometry profiles of eosinophils in the BAL. (J) Number of immune cells in the BAL. Data are from four mice per group, representative of two independent experiments (A), or pooled from three independent experiments (B-E), or from 5 mice per group, pooled from two independent experiments (F-G), or from 4 mice per group, pooled from two independent experiments (H), or from 6 mice per group, pooled from two independent experiments (I and J). *P < 0.05, **P < 0.01.
Of note, as a bromodomain inhibitor, JQ1 may have broader effects other than inhibiting c-Myc expression and function. As an independent approach, we have also deleted c-Myc in purified ILC2 using CRISPR-mediated gene knockout technique (Figure 2). Our results with CRISPR experiments demonstrate an essential cell intrinsic role for c-Myc in ILC2 activation (Figure 2). Together, our work has established a critical role for c-Myc in ILC2 function with multiple lines of evidence, although future work with conditional knockout mice that may specifically delete c-Myc in ILC2 would still be worthwhile.
3.4. c-Myc inhibition represses ILC2-induced airway inflammation and AHR
Our previous work indicated that a single dose of Alternaria can induce airway eosinophilic infiltration and AHR in Balb/c mice, through activation of lung-resident ILC2.7, 8 We thus examined whether blockade of ILC2 activation by c-Myc inhibitors can repress Alternaria-induced airway inflammation and AHR. Alternaria inhalation induced AHR in Balb/c mice within 12 hours and was potently repressed by JQ1 (Figure 3H). Alternaria inhalation also rapidly elicited airway eosinophilic and neutrophilic infiltration in Balb/c mice (Figure 3I and J). JQ1 treatment abolished eosinophilic inflammation, but did not significantly influence neutrophil infiltration in Alternaria-challenged mice (Figure 3I and J). Thus, c-Myc inhibition may repress airway inflammation and AHR induced by acute Alternaria challenge.
To verify whether ILC2-induced airway inflammation and AHR can be repressed by c-Myc inhibition, we next transferred sort-purified lung ILC2 into Rag2−/−Il2rg−/− mice with or without JQ1 treatment (Figure 4A). Transfer of ILC2 enhanced eosinophil infiltration and increased AHR in Alternaria-challenged Rag2−/−Il2rg−/− mice (Figure 4B and C). JQ1 treatment abolished ILC2-induced eosinophilic infiltration and AHR (Figure 4B and C). Thus, JQ1 treatment can specifically repress ILC2-induced airway responses.
FIGURE 4. JQ1 treatment repressed innate-type 2 inflammation.

(A)105 ILC2 were transferred into Rag2−/− Il2rg−/− mice. Cell recovery efficiency was examined by flow cytometry analysis 2 days after adoptive transfer. (B) 105 ILC2 were transferred into Rag2−/− Il2rg−/− mice that were challenged with Alternaria or PBS control, and treated with JQ1 or vehicle control. Number of eosinophils in bronchoalveolar lavage (BAL) was examined by flow cytometry analysis. (C) Airway resistance measured by FlexiVent analysis. (D) Rag1−/− mice were challenged with Alternaria extracts and treated with JQ1 daily for 3 days. Cytokine production of lung ILC2 was examined one day after the last challenge (i.e. 3 days after the first challenge). (E) Mean fluorescence intensity (MFI) of IL-5 and IL-13 in lung ILC2 of Rag1−/− mice. (F) Mucous production was examined by PAS staining one day after the last challenge. (G) Percentage of PAS+ cells per bronchi. (H) Representative flow cytometry profile of BAL cells in Rag1−/− mice challenged with Alternaria or PBS control, and treated with JQ1 or vehicle control. (J) Numbers of BAL eosinophils in Rag1−/− mice challenged with Alternaria or PBS control, and treated with JQ1 or vehicle control. Data are from 4 mice per group, pooled from two independent experiments (A-I) *P < 0.05, **P < 0.01.
We next sought to verify whether c-Myc can repress ILC2 responses independently of adaptive immunity. We challenged Rag1−/− mice with Alternaria extracts daily for 3 days. We then examined ILC2 responses, mucous production and airway inflammation at the day after the last challenge (i.e. 3 days after the first challenge). Notably, Alternaria challenge elicited robust ILC2 responses that were repressed by JQ1 treatment in Rag1−/− mice that lack adaptive immunity (Figure 4D and E). Alternaria challenge also induced mucosal hyperplasia that was potently repressed by JQ1 treatment (Figure 4F and G). JQ1 treatment also abolished eosinophilic infiltration in Alternaria-challenged Rag1−/− mice (Figure 4H and I). Together, these data suggest a critical role for c-Myc in promoting innate type-2 inflammation.
3.4. c-Myc inhibition represses human ILC2 activation
To verify whether human ILC2 activation can be repressed by c-Myc inhibition, we cultured sort-purified human blood ILC2 in the presence or absence of JQ1. We initially used a cytokine cocktail (IL-2, IL-7, IL-25, IL-33) that can stimulate rigorous ILC2 proliferation and activation. JQ1 treatment repressed MYC expression in cultured human ILC2 in a dose-dependent manner (Figure 5A). Proliferation of ILC2 was effectively suppressed by JQ1 treatment (Figure 5B). Cytokine production of ILC2 was also potently inhibited by JQ1 (Figure 5C). We next cultured human ILC2 in the presence of IL-33, IL-25, or TSLP alone. JQ1 treatment effectively repressed cytokine production of ILC2 cultured with any of these stimulating cytokines, suggesting that c-Myc may promote human ILC2 activation regardless of the type of stimulator (Figure 5D). Thus, targeting c-Myc may provide a new avenue to repress human ILC2 responses.
FIGURE 5. JQ1 treatment repressed human ILC2 activation.

(A) 1000 sorted human ILC2 were cultured with 20ng/ml IL-2, IL-7, IL-25, and IL-33, in the presence of the indicated concentrations of JQ1 for 5 days. mRNA levels of MYC were examined by qPCR. (B) Sorted human ILC2 were cultured with 20ng/ml IL-2, IL-7, IL-25, and IL-33 in the presence of the indicated concentration of JQ1. Growth was calculated as fold change of cells number over 5 days of culture. (C) IL-5 and IL-13 production from human ILC2 cultured with IL-2, Il-7, IL-33 and IL-25 for 5 days. (D) 1000 sorted human ILC2 were cultured with 20ng/ml IL-25, IL-33 or TSLP, together with 500nM JQ1 or vehicle control. Cytokine production in the culture supernatant was measured. Data are from 4 donors per group, representative of two to four independent experiments (A-D) *P < 0.05, **P < 0.01.
4. DISCUSSION
In summary, our data establishes c-Myc as an essential molecular driver of ILC2 activation. We show that ILC2 from asthma patients express increased amounts of MYC, and demonstrate that c-Myc is essential for ILC2 activation in vitro and in vivo. We have profiled c-Myc dependent genes in ILC2, and have identified STAT6 as a direct gene target of c-Myc in ILC2. We demonstrate that inhibition of c-Myc by JQ1 treatment can potently block ILC2 activation and suppress innate type-2 inflammation induced by Alternaria fungal allergen. These data collectively demonstrate a critical role for c-Myc in ILC2 activation and indicate that targeting c-Myc may provide new avenues to combat asthma or asthma exacerbation.
Transcriptional regulation of innate lymphoid cell activation remains intriguing but poorly understood. During ILC2 activation, STAT6 might be a key environmental sensor that initiates the activation of lineage-specific enhancers; whereas GATA3 is considered as a master transcriptional regulator.36-40 The capability of c-Myc to directly target STAT6 indicates that c-Myc might be involved in initiating the gene regulatory program and defining the transcriptional landscape during ILC2 activation. c-Myc is also required for the optimal expression of other lymphocyte activation genes such as GATA3 in ILC2. However, conserved binding sites for c-Myc have not been identified in up to −10kb of GATA3 promoter and enhancer, suggesting that c-Myc might indirectly control GATA3 expression. c-Myc is also known to directly target metabolism genes and ribosome biogenesis genes in cancer cell lines, which might influence cell proliferation.41, 42 However, metabolism genes and ribosome genes were not significantly altered in c-Myc deficient ILC2 from our microarray analysis. Thus, the reduced cell proliferation in c-Myc deficient ILC2 probably reflects reduced cellular activation. Indeed, both STAT6 and GATA3 are required for ILC2 activation, and their reduced expression in c-Myc deficient ILC2 may explain diminished proliferation capability of these cells.
Our results also indicate that increased expression of c-Myc is a signature of ILC2 activation in both asthma patients and mouse models of allergic inflammation. The upregulation of c-Myc expression during ILC2 activation is rapid, dramatic and conserved among species. Since c-Myc is required for the expression of a range of other key ILC2 activation genes, c-Myc upregulation is likely one of the earliest molecular events that occur during ILC2 activation. Given the critical role of ILC2 in initiating and amplifying allergic airway inflammation, the appearance of cMychigh ILC2 might underlie the pathogenesis of a subgroup of severe asthma.
Despite well-established function in cancer cells, the role of c-Myc in inflammatory disorders, especially allergic inflammation, has been poorly understood. Our data have revealed a novel role for c-Myc in controlling Alternaria fungal-allergen induced airway inflammation and AHR, by repressing pathogenetic ILC2 responses. This study suggests that targeting c-Myc may unlock novel therapies to repress ILC2 activity in asthma or asthma exacerbation. Of note, asthma is a heterogeneous disease with diverse endotypes and multiple mechanistic pathways. It might be worthwhile to also explore c-Myc function in other asthma-related cell types in future efforts. Such studies might help predict whether c-Myc inhibition might hold promise for most asthma endotypes or whether its efficacy might be restricted to ILC2high endotypes.
Supplementary Material
ACKNOWLEDGEMENTS
This work was supported by National Institutes of Health (NIH) Grants R01HL137813, R01AG057782, and K22AI116728 (to Q.Y.), the Alexandrine and Alexander L. Sinsheimer Scholar Award (to Q.Y.), and R01HL110951, R01HL130304 (to D.D.T.).
Footnotes
CONFLICT OF INTERESTS
The authors declare that they have no relevant conflicts of interest.
REFERENCES
- 1.Castillo JR, Peters SP, Busse WW. Asthma Exacerbations: Pathogenesis, Prevention, and Treatment. J Allergy Clin Immunol Pract 2017;5:918–927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Winkler C, Hochdorfer T, Israelsson E, Hasselberg A, Cavallin A, Thorn K, et al. Activation of group 2 innate lymphoid cells after allergen challenge in asthmatic patients. J Allergy Clin Immunol 2019. [DOI] [PubMed] [Google Scholar]
- 3.Bartemes KR, Kephart GM, Fox SJ, Kita H. Enhanced innate type 2 immune response in peripheral blood from patients with asthma. J Allergy Clin Immunol 2014;134:671–678 e674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lloyd CM, Hessel EM. Functions of T cells in asthma: more than just T(H)2 cells. Nat Rev Immunol 2010;10:838–848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Drake LY, Iijima K, Kita H. Group 2 innate lymphoid cells and CD4+ T cells cooperate to mediate type 2 immune response in mice. Allergy 2014;69:1300–1307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Uwadiae FI, Pyle CJ, Walker SA, Lloyd CM, Harker JA. Targeting the ICOS/ICOS-L pathway in a mouse model of established allergic asthma disrupts T follicular helper cell responses and ameliorates disease. Allergy 2019;74:650–662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Shen X, Liang M, Chen X, Pasha MA, D’Souza SS, Hidde K, et al. Cutting Edge: Core Binding Factor beta Is Required for Group 2 Innate Lymphoid Cell Activation. J Immunol 2019;202:1669–1673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Shen X, Pasha MA, Hidde K, Khan A, Liang M, Guan W, et al. Group 2 innate lymphoid cells promote airway hyperresponsiveness through production of VEGFA. J Allergy Clin Immunol 2018;141:1929–1931 e1924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Yang Q, Ge MQ, Kokalari B, Redai IG, Wang X, Kemeny DM, et al. Group 2 innate lymphoid cells mediate ozone-induced airway inflammation and hyperresponsiveness in mice. J Allergy Clin Immunol 2016;137:571–578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Fan X, Rudensky AY. Hallmarks of Tissue-Resident Lymphocytes. Cell 2016;164:1198–1211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Doherty TA, Broide DH. Airway innate lymphoid cells in the induction and regulation of allergy. Allergol Int 2019;68:9–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Oliphant CJ, Hwang YY, Walker JA, Salimi M, Wong SH, Brewer JM, et al. MHCII-mediated dialog between group 2 innate lymphoid cells and CD4(+) T cells potentiates type 2 immunity and promotes parasitic helminth expulsion. Immunity 2014;41:283–295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Schuijs MJ, Hammad H, Lambrecht BN. Professional and ‘Amateur’ Antigen-Presenting Cells In Type 2 Immunity. Trends Immunol 2019;40:22–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Mindt BC, Fritz JH, Duerr CU. Group 2 Innate Lymphoid Cells in Pulmonary Immunity and Tissue Homeostasis. Front Immunol 2018;9:840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Melnik S, Werth N, Boeuf S, Hahn EM, Gotterbarm T, Anton M, et al. Impact of c-MYC expression on proliferation, differentiation, and risk of neoplastic transformation of human mesenchymal stromal cells. Stem Cell Res Ther 2019;10:73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Luo W, Chen J, Li L, Ren X, Cheng T, Lu S, et al. c-Myc inhibits myoblast differentiation and promotes myoblast proliferation and muscle fibre hypertrophy by regulating the expression of its target genes, miRNAs and lincRNAs. Cell Death Differ 2019;26:426–442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Chen H, Liu H, Qing G. Targeting oncogenic Myc as a strategy for cancer treatment. Signal Transduct Target Ther 2018;3:5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Stine ZE, Walton ZE, Altman BJ, Hsieh AL, Dang CV. MYC, Metabolism, and Cancer. Cancer Discov 2015;5:1024–1039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bouvard C, Lim SM, Ludka J, Yazdani N, Woods AK, Chatterjee AK, et al. Small molecule selectively suppresses MYC transcription in cancer cells. Proc Natl Acad Sci U S A 2017;114:3497–3502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ott G, Rosenwald A, Campo E. Understanding MYC-driven aggressive B-cell lymphomas: pathogenesis and classification. Blood 2013;122:3884–3891. [DOI] [PubMed] [Google Scholar]
- 21.Wang R, Dillon CP, Shi LZ, Milasta S, Carter R, Finkelstein D, et al. The transcription factor Myc controls metabolic reprogramming upon T lymphocyte activation. Immunity 2011;35:871–882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Slack M, Wang T, Wang R. T cell metabolic reprogramming and plasticity. Mol Immunol 2015;68:507–512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Nguyen L, Papenhausen P, Shao H. The Role of c-MYC in B-Cell Lymphomas: Diagnostic and Molecular Aspects. Genes (Basel) 2017;8.24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Chung KF, Wenzel SE, Brozek JL, Bush A, Castro M, Sterk PJ, et al. International ERS/ATS guidelines on definition, evaluation and treatment of severe asthma. Eur Respir J 2014;43:343–373. [DOI] [PubMed] [Google Scholar]
- 25.Shahbazi J, Liu PY, Atmadibrata B, Bradner JE, Marshall GM, Lock RB, et al. The Bromodomain Inhibitor JQ1 and the Histone Deacetylase Inhibitor Panobinostat Synergistically Reduce N-Myc Expression and Induce Anticancer Effects. Clin Cancer Res 2016;22:2534–2544. [DOI] [PubMed] [Google Scholar]
- 26.Zhang K, Xu X, Pasha MA, Siebel CW, Costello A, Haczku A, et al. Cutting Edge: Notch Signaling Promotes the Plasticity of Group-2 Innate Lymphoid Cells. J Immunol 2017;198:1798–1803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sanjana NE, Shalem O, and Zhang F. Improved vectors and genome-wide libraries for CRISPR screening. Nat Methods. 2014;11:783–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ghezraoui H, Piganeau M, Renouf B, Renaud JB, Sallmyr A, Ruis B, et al. Chromosomal translocations in human cells are generated by canonical nonhomologous end-joining. Mol Cell. 2014;55:829–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ran FA, Hsu PD, Lin CY, Gootenberg JS, Konermann S, Trevino AE, et al. Double nicking by RNA-guided CRISPR Cas9 for enhanced genome editing specificity. Cell. 2013;154:1380–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Smith SG, Chen R, Kjarsgaard M, Huang C, Oliveria JP, O’Byrne PM, et al. Increased numbers of activated group 2 innate lymphoid cells in the airways of patients with severe asthma and persistent airway eosinophilia. J Allergy Clin Immunol 2016;137:75–86 e78. [DOI] [PubMed] [Google Scholar]
- 31.Doherty TA, Khorram N, Chang JE, Kim HK, Rosenthal P, Croft M, et al. STAT6 regulates natural helper cell proliferation during lung inflammation initiated by Alternaria. Am J Physiol Lung Cell Mol Physiol 2012;303:L577–588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Snelgrove RJ, Gregory LG, Peiro T, Akthar S, Campbell GA, Walker SA, et al. Alternaria-derived serine protease activity drives IL-33-mediated asthma exacerbations. J Allergy Clin Immunol 2014;134:583–592 e586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kouzaki H, Iijima K, Kobayashi T, O’Grady SM, Kita H. The danger signal, extracellular ATP, is a sensor for an airborne allergen and triggers IL-33 release and innate Th2-type responses. J Immunol 2011;186:4375–4387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Matzuk MM, McKeown MR, Filippakopoulos P, Li Q, Ma L, Agno JE, et al. Small-molecule inhibition of BRDT for male contraception. Cell 2012;150:673–684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Delmore JE, Issa GC, Lemieux ME, Rahl PB, Shi J, Jacobs HM, et al. BET bromodomain inhibition as a therapeutic strategy to target c-Myc. Cell 2011;146:904–917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hoyler T, Klose CS, Souabni A, Turqueti-Neves A, Pfeifer D, Rawlins EL, et al. The transcription factor GATA-3 controls cell fate and maintenance of type 2 innate lymphoid cells. Immunity 2012;37:634–648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zhu J GATA3 Regulates the Development and Functions of Innate Lymphoid Cell Subsets at Multiple Stages. Front Immunol 2017;8:1571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Rothenberg EV. The chromatin landscape and transcription factors in T cell programming. Trends Immunol 2014;35:195–204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Vahedi G, Takahashi H, Nakayamada S, Sun HW, Sartorelli V, Kanno Y, et al. STATs shape the active enhancer landscape of T cell populations. Cell 2012;151:981–993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Yu QN, Guo YB, Li X, Li CL, Tan WP, Fan XL, et al. ILC2 frequency and activity are inhibited by glucocorticoid treatment via STAT pathway in patients with asthma. Allergy 2018;73:1860–1870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Miller DM, Thomas SD, Islam A, Muench D, Sedoris K. c-Myc and cancer metabolism. Clin Cancer Res 2012;18:5546–5553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Dang CV. MYC on the path to cancer. Cell 2012;149:22–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.