

Abstract
Despite intensive efforts, a considerable proportion of colorectal cancer (CRC) patients develop local recurrence and distant metastasis. Stomatin-like protein 2 (SLP-2), a member of the highly conserved stomatin superfamily, is upregulated across cancer types. However, the biological and functional roles of SLP-2 remain elusive in CRC. Here, we report that high SLP-2 expression was found in CRC tissues and was linked to tumor progression and tumor cell differentiation. Additionally, high SLP-2 expression correlated with poor overall survival (OS) in CRC patients (p < 0.001). SLP-2 knockout (SLP-2KO), generated by CRISPR/Cas9, reduced cell growth, migration, and invasion; induced apoptosis in CRC cells; and reduced tumor xenograft growth in vivo. A 181-compound library screening showed that SLP-2KO produced resistance to JAK2 inhibitors (NVP-BSK805 and TG-101348) and a PIM1 inhibitor (SGI-1776), revealing that the JAK2-STAT3-PIM1 oncogenic pathway was potentially controlled by SLP-2 in CRC. In vitro and in vivo, TG-101348 combined with SGI-1776 was synergistic in CRC (combination index [CI] < 1). Overall, our findings suggest that SLP-2 controls the JAK2-STAT3-PIM1 oncogenic pathway, offering a rationale for a novel therapeutic strategy with combined SGI-1776 and TG-101348 in CRC. Additionally, SLP-2 may be a prognostic marker and biomarker for sensitivity to JAK2 and PIM1 inhibitors.
Keywords: stomatin-like protein 2, JAK2, STAT3, PIM1, colorectal cancer
Graphical Abstract
Liu and colleagues show that enhanced SLP-2 expression correlates with tumor progression in CRC. SLP-2 knockout by CRISPR/Cas9 inhibits CRC cell growth. PIM1i SGI-1776 and JAK2i TG-101348 are successfully identified as SLP-2-linked drugs after compound library screening. Co-treatment with the two drugs produces synergistic effect, which provides a promising therapy in CRC.
Introduction
Colorectal cancer (CRC) is one of the leading causes of cancer-related morbidity worldwide. As the clinical outcomes of CRC patients widely vary, the use of stratified treatment options remains limited, and resistance to conventional therapies frequently occurs at advanced stages, the mortality of CRC patients remains high.1 Therefore, the development and identification of novel genes or proteins that are prognostic markers and molecular therapeutic targets in CRC remain crucial.
Stomatin-like protein 2 (SLP-2), encoded by STOML2, is a member of the highly conserved stomatin superfamily. SLP-2 was first identified as a membrane protein in erythrocytes and was later found to be widely expressed in mammalian tissues.2,3 SLP-2 is considered a major mitochondrial inner membrane protein and is involved in the regulation of ion channel permeability, mechanoreception, and lipid domain organization. Recent studies revealed that SLP-2 is differentially expressed in diverse human malignancies and plays an oncogenic role in tumor occurrence and progression.4, 5, 6, 7, 8, 9, 10 However, the underlying mechanism of SLP-2 has yet to be clearly demonstrated. A previous study demonstrated that SLP-2 promotes non-small-cell lung cancer (NSCLC) cell proliferation through the β-catenin pathway.7 Moreover, SLP-2 may regulate the tumor invasion ability of glioma via the nuclear factor κB/matrix metalloproteinase-9 (NF-κB/MMP9) pathway, while in head and neck squamous cell carcinoma, the interleukin-6 (IL-6)-STAT3 pathway is reported to be regulated by SLP-2, modulating cell proliferation and motility.6,10
To gain new insights into the role of SLP-2 in CRC, we first sought to examine the mRNA and protein levels of SLP-2 in Chinese patients with colorectal tumors. We found that SLP-2 expression was progressively upregulated with tumor progression and was correlated with inferior outcomes in CRC patients. Additionally, to further understand the effects of SLP-2 on the functional and regulatory mechanisms of CRC, a series of studies was performed, and we showed that SLP-2 affects CRC cell proliferation and xenograft growth in vitro and in vivo, respectively. Moreover, a 181-compound library screen showed that SLP-2 knockout (SLP-2KO) produced the resistance of CRC cells to JAK2 and PIM inhibitors, which suggests the potential involvement of the JAK2-STAT3-PIM1 oncogenic pathway in the functional mechanism of SLP-2 in CRC. Taken together, our data demonstrate the prognostic value of SLP-2 and a potential working mechanism for SLP-2 and suggest a novel combinatorial therapy targeting JAK2 and PIM1 in CRC.
Results
Expression of SLP-2 in CRC Tissues and Its Associations with the Clinicopathological Features of CRC Patients
To investigate the role of SLP-2 in the carcinogenesis of CRC, we first examined SLP-2 on both the mRNA and protein levels in tumors and paired adjacent nontumor tissues. As shown in Figure 1A, SLP-2 mRNA expression was nine times higher in the 74 tumors than in the matched adjacent nontumor tissues from CRC patients, as analyzed by quantitative real-time reverse transcription PCR (RT-PCR). Western blot analyses confirmed that SLP-2 protein expression was higher in tumors than in paired adjacent nontumor tissues (Figure 1B). Furthermore, the immunohistochemical (IHC) staining of SLP-2 was performed in colorectal adenomas (n = 50), high-grade intraepithelial neoplasias (HGINs; n = 50), invasive carcinoma, and paired adjacent nontumor tissues (n = 491), and we found that the proportion of tumors with high SLP-2 expression progressively increased when nontumor tissues progressed to invasive carcinoma (Figure 1C), which suggests that SLP-2 may be associated with tumor progression. The localization of SLP-2 expression was cytoplasmic, and representative staining showed the negative, weak, moderate, and strong expressions of SLP-2 in nontumor tissue, adenoma, HGIN, and invasive carcinoma, respectively (Figure 1D). Tumors with high SLP-2 expression (n = 223) were associated with clinicopathological features that were indicative of a more aggressive phenotype, which included the depth of tumor invasion, lymphatic and/or venous invasion, nodal involvement, distant metastasis, and tumor, node, metastasis (TNM) staging (Table S3). As shown in Figures 1E and 1F, a significantly higher proportion of poorly differentiated CRC than of highly and moderately differentiated invasive carcinomas displayed increased SLP-2 staining intensity. No significant difference was found regarding patient age, gender, carcinoembryonic antigen (CEA) levels, and histological type between tumors with low and high SLP-2 expression (Table S3). Additionally, log-rank analyses revealed that patients with high SLP-2 expression had significantly shorter overall survival (OS) and progression-free survival (PFS) (p < 0.0001; Figures 1G and 1H) than patients with low SLP-2 expression. Multivariate Cox regression analyses further confirmed that high SLP-2 expression, similar to other prognostic factors, such as age, distant metastasis, and TNM stage, was an independent prognostic factor for CRC (hazard ratio, 0.469; p = 0.006) (Table S4).
Figure 1.

High SLP-2 Expression Correlates with Tumor Progression and Poor Prognosis in CRC
(A) Quantitative assessment of the SLP-2 transcript in 74 CRC and matched adjacent normal tissue samples. (B) Representative western blot analysis of SLP-2 protein levels in five paired CRC and matched adjacent normal tissue samples. (C) Stacked bar plots showing the percentage of patients with high or low SLP-2 protein expression in colorectal adenoma, HGIN, invasive carcinoma, and matched adjacent nontumor tissue samples. (D) Representative micrographs of SLP-2 protein expression in nontumor, adenoma, HGIN, and invasive carcinoma (scale bars, 100 μm). (E) Representative micrographs of SLP-2 protein expression in highly, moderately and poorly differentiated CRC (scale bars, 50 μm). (F) Stacked bar plots indicating the number of patients with high or low SLP-2 protein expression that had highly, moderately, and poorly differentiated CRC. (G and H) OS (G) and PFS (H) of CRC patients with low (red line) or high (blue line) SLP-2 expression. Error bars represent the mean ± SEM. ∗p < 0.05, ∗∗∗p < 0.001, two-tailed, unpaired t tests. N & Non-T, nontumor tissue; T, tumor; Ade, adenomas; HGIN, high-grade intraepithelial neoplasia; Ica, invasive carcinoma; Dif, differentiation; PFS, progression-free survival; OS, overall survival.
SLP-2KO Arrests CRC Cell Growth In Vitro and In Vivo
Given the correlation of high SLP-2 expression with poor prognosis in CRC, we sought to investigate the functional impacts of SLP-2 in vitro. Western blot analysis showed that SLP-2 was differentially expressed in CRC cells (Figure 2A). HCT116 and SW480 cells were selected to generate isogenic models by using CRISPR/Cas9-mediated SLP-2KO. Isogenic models were confirmed by western blotting for SLP-2 expression, and SLP-2KO in HCT116 and SW480 resulted in the significant inhibition of cell growth (Figure 2B) and colony formation (Figures 2C and 2D) compared to cells that were transfected with empty vector (EV) in the two cell lines. To investigate whether the growth suppression in CRC cells that was caused by SLP-2KO was partially due to the induction of apoptosis, we examined the enzymatic activity of caspase-3/7 in HCT116SLP-2KO and SW480SLP-2KO cells. EV-transfected cells served as a control for normalization. We observed a significant increase in the activities of caspase-3/7 (approximately 2 times higher than those in control cells) in HCT116SLP-2KO and SW480SLP-2KO cells (Figure 2E). In accordance with the in vitro findings, SLP-2KO significantly inhibited CRC xenograft growth compared to that of the control cells (Figure 2F). Additionally, all tumors from the two groups were dissected, fixed, and stained with hematoxylin and eosin (H&E) and the proliferation index Ki-67. Consistent with the findings in CRC clinical samples, morphological analysis suggested that SLP-2 was correlated with tumor differentiation, as SLP-2KO induced adenoid differentiation in samples from HCT116SLP-2KO xenografts (Figure 2G). IHC analyses showed that the proportion of Ki-67 expression in the SLP-2KO group decreased by 34% compared to that in the control (EV) group (Figures 2G and 2H).
Figure 2.

SLP-2KO Inhibits Tumor Cell Growth In Vitro and In Vivo
(A) Representative western blot analysis of SLP-2 protein levels in seven colorectal cell lines. GAPDH was used as a protein loading control. (B) Cell growth curves of HCT116SLP-2KO and SW480SLP-2KO cells compared with EV-transfected cells. The knockout efficiency of each cell line was assessed by western blot analysis. All western blots were representative of at least two repeats. (C) Representative images of the colony formation assay of HCT116SLP-2KO, HCT116EV, SW480SLP-2KO, and SW480EV cells. (D) The bar plots indicate the number of colonies of each cell line. (E) Caspase-3/7 enzymatic activity was measured using an Apo-ONE homogeneous caspase-3/7 assay in HCT116SLP-2KO, HCT116EV, and SW480SLP-2KO, and SW480EV cells. (F) Tumor volumes over 28 days in an orthotopic model of HCT116SLP-2KO and HCT116EV cells. (G) Representative micrographs of H&E staining (left) and Ki67 expression (right) in HCT116SLP-2KO and HCT116EV tumors. Scale bars, 100 μm. (H) Scatter dot plots indicate the percentage of Ki67 positivity in HCT116SLP-2KO and HCT116EV tumors (n = 5). Error bars represent the mean ± SEM. ∗∗∗p < 0.001, two-tailed, unpaired t tests. EV, empty vector px458; SLP-2KO, SLP-2 knockout; sg1, single-guide RNA-1; sg1, single-guide RNA-2; H&E, hematoxylin & eosin.
SLP-2KO Inhibits Migration and Invasion in CRC Cells
Next, we investigated whether SLP-2KO inhibited the migration and invasion of HCT116 and SW480 cells. A wound-healing assay was performed to evaluate the effect of SLP-2KO on cancer cell migration. As shown in Figures 3A and 3B, compared with control cells transfected with EV, SLP-2KO significantly inhibited HCT116 and SW480 cell migration by up to 66% and 58%, respectively. In addition, an invasion assay was performed in Transwell chambers containing inserts with 8-μm pores, the upper surfaces of which were coated with Matrigel. HCT116 and SW480 cells were suspended in medium containing 1% FBS and plated in the upper chambers. After 12 h of culture, SLP-2KO sharply impeded HCT116 and SW480 cell invasion by up to 55% and 60%, respectively (Figures 3C and 3D). Furthermore, we determined whether SLP-2KO inhibited MMP family members, which are key regulators of tumor invasion, and we found that SLP-2KO downregulated MMP2 protein expression to a large extent (Figure 3E).
Figure 3.

SLP-2KO Inhibits Cell Migration and Invasion in CRC Cells
(A) Representative images of the migration of HCT116SLP-2KO, HCT116EV, SW480SLP-2KO, and SW480EV cells, as analyzed using a wound-healing assay at 0 and 24 h. Magnification: 100×. (B) The bar plots indicate the wounded area of each cell line. (C) Representative images of the invasion of HCT116SLP-2KO, HCT116EV, SW480SLP-2KO, and SW480EV cells, as analyzed using a Transwell assay at 12 h after seeding. Magnification: 100×. (D) The bar plots indicate the number of invaded cells of each cell line. Error bars in (B) and (D) indicate mean ± SEM (n = 3). ∗∗∗p < 0.001, two-tailed, unpaired t tests. (E) Representative western blot analysis of MMP2 in HCT116SLP-2KO, HCT116EV, SW480SLP-2KO, and SW480EV cells. GAPDH was used as a protein loading control.
Cell-Based Screening Assay Identifies SLP-2-Related Compounds in CRC
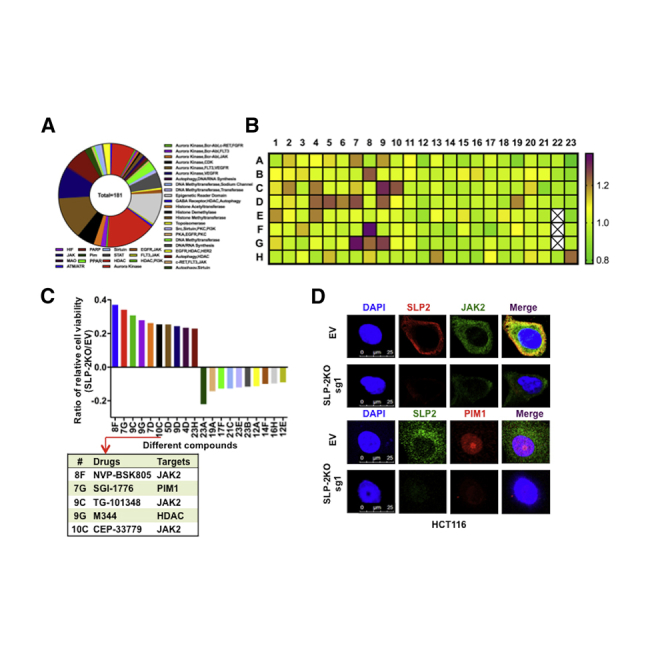
The importance of SLP-2, which is correlated with poor prognosis and promoted cell growth across cancer types, prompted us to investigate whether SLP-2 could serve as a therapeutic target or a biomarker to predict drug efficacy in CRC.8,9,11, 12, 13 Therefore, we performed high-throughput drug screening, which included 181 drugs that were either promising novel anticancer agents or traditional chemotherapeutic agents (Figure 4A), in isogenic HCT116 cells. After 72 h, cell viability was determined with alamarBlue assays and normalized to vehicle-treated cells to examine the differences in drug response. As shown in Figure 4B, the heatmap ranks the ratios of the relative cell viability (HCT116SLP-2KO/HCT116EV) for different drugs. The top 10 drugs that had antagonistic or synergistic effects with SLP-2KO in HCT116 cells were selected (Figure 4C). Of the top five drugs that were antagonistic to SLP-2KO, three drugs targeted JAK2 and one targeted PIM1, a downstream signaling regulator of the JAK2-STAT3 pathway.12, 13, 14 The efficacy of selective JAK2 inhibitors (NVP-BSK805 and TG-101348) and a pan-PIM kinase inhibitor (SGI-1776) has been shown in many cancers; in addition, TG-101348 and SGI-1776 were examined in different clinical trials.15, 16, 17, 18, 19 Our findings from this study suggested that SLP-2 levels might correlate with the sensitivity of JAK2 and PIM1 inhibitors in CRC. To confirm the association between SLP-2 and the efficacy of SGI-1776 and TG-101348, SW480 and HCT116 isogenic cells generated by EV (px458) and SLP-2KO small guide RNA (sgRNA) transfection were treated with drugs, and the half maximal inhibitory concentration (IC50) values were calculated. Consistently, HCT116SLP-2KO and SW480SLP-2KO cells exhibited 1.8- to 2.5-fold more resistance to TG-101348 and 2.5- to 3-fold more resistance to SGI-1776, respectively (Figure 4D). A colony formation assay further validated that SLP-2KO conferred CRC cell resistance to both drugs. As shown in Figure 4E, SLP-2KO increased the number of colonies grown from both HCT116 and SW480 cells treated with TG-101348 (1 μM) or SGI-1776 (2.5 μM) compared to cells transfected with EV.
Figure 4.

Screen for Antineoplastic Drugs and Therapeutic Targets in SLP-2KO CRC Cells
(A) A pile plot showing 181 agents with 34 targets. (B) Heatmap demonstrating the response of HCT116SLP-2KO cells to each of the 181 agents tested in the chemical screen. Each box represents a drug. All drugs had a maximum dose of 2.5 μM. Drug responses are shown as gradations from purple (sensitivity) to green (resistance). White boxes indicate the internal control, DMSO. (C) The bar plots show the ratio of the relative cell viability with the treatment of 10 drugs that had antagonistic or synergistic effects with SLP-2KO in HCT116 cells (left). The table shows the targets of the top five drugs that were antagonistic to SLP-2KO in HCT116 cells. (D) Dose-response curves of HCT116SLP-2KO, HCT116EV, SW480SLP-2KO, and SW480EV cells treated with the indicated drugs (x axis) at 0.001, 0.01, 0.1, 1, or 10 μM or with 0.1% DMSO (control). Cell viability was assessed at 72 h. Data are expressed relative to the control. (E) Colony formation assay of HCT116SLP-2KO and HCT116EV cells treated with 2.5 μM SGI-1776 and 1 μM TG-101348 and of SW480SLP-2KO and SW480EV cells treated with 2.5 μM SGI-1776 and 1 μM TG-101348 in six-well plates (left). Bar plots indicate the number of colonies (right). Error bars represent the mean ± SEM. ∗∗p < 0.01; ∗∗∗p < 0.001, two-tailed, unpaired t tests.
SLP-2 Regulates the JAK2-STAT3-PIM1 Oncogenic Pathway
The correlation of the SLP-2 levels with the sensitivity of inhibitors targeting JAK2 and PIMs also implies that the oncogenic JAK2-STAT-PIMs were potentially controlled by SLP-2 in CRC. To examine this hypothesis, we used quantitative real-time RT-PCR to examine the mRNA expression of JAK2 and PIM1-3 in isogenic cells and found that SLP-2KO significantly downregulated the mRNA levels of JAK2 and PIM1, but not of PIM2 and PIM3, in both HCT116 and SW480 cells (Figure 5A). Consistently, the results from western blots revealed that SLP-2KO resulted in a marked decrease in the levels of JAK2 and a subsequent decrease in phosphorylated STAT3 (p-STAT3) as well as PIM1 in CRC cells with SLP-2KO compared to EV-transfected cells. However, the total STAT3 level and the level of another key component of JAK2 signaling, STAT5 (total or phosphorylated levels), showed subtle changes (Figure 5B). In addition, immunofluorescence analysis demonstrated that SLP-2 and JAK2 were coexpressed in the cytoplasm, PIM1 was localized to the nucleus, and SLP-2KO resulted in an apparent decrease in JAK2 and PIM1 in HCT116SLP-2KO and SW480SLP-2KO cells compared to EV-transfected cells (Figure 5C). Therefore, the aforementioned findings established that the JAK2-STAT3-PIM1 oncogenic pathway was controlled by SLP-2 in CRC.
Figure 5.

SLP-2 Regulates the JAK2-STAT3-PIM1 Oncogenic Pathway in CRC
(A) Quantitative real-time RT-PCR analysis of JAK2, PIM1, PIM2, and PIM3 mRNA expression in HCT116 and SW480 cells that were transduced with SLP-2KO-sg1 and -2. Data are presented as the mean ± SEM and were normalized to GAPDH expression. y-axis, fold change versus GAPDH and EV (px48)-transfected cells. (B) Western blot analysis of HCT116SLP-2KO, SW480SLP-2KO, and EV (px458) control cells for SLP-2, JAK2, total STAT3, p-STAT3, total STAT5, p-STAT5, and PIM1. GAPDH was used as a loading control. (C) Immunofluorescence analysis of JAK2 and PIM1 in HCT116SLP-2KO, SW480SLP-2KO, and EV (px48) control cells. The signals were detected with a Leica confocal imaging system. Scale bars, 25 μm.
TG-101348 Combined with SGI-1776 Is Highly Synergistic in CRC Cells and Leads to a Marked Tumor Growth Delay in a Xenograft Model of CRC
Based on the association of SLP-2 and the sensitivity of CRC cells to TG-101348 and SGI-1776, we continued to examine whether the dual targeting of JAK2 and PIM1 could be a novel therapeutic strategy in CRC. We first exposed HCT116 and SW480 cells to different concentrations of SGI-1776 and TG-101348 and analyzed the synergistic effects by determining the cell viability. As shown in Figure 6A, the combination of SGI-1776 and TG-101348 potently inhibited HCT116 and SW480 cell growth, as shown by the strong synergistic effects (combination index [CI] < 1.0). Consistently, compared with the effects of either inhibitor alone, the combination of SGI-1776 and TG-101348 suppressed cell viability by approximately 48% to 80% in HCT116 cells and by 31% to 55% in SW480 cells (Figures S1A and S1B), respectively; and the combination treatment also suppressed colony formation (Figures 6B and 6C). In addition, the combination treatment induced the apoptosis of HCT116 and SW480 cells, as shown by increased caspase-3/7 activity (Figure 6D) and enhanced PARP and caspase-3 cleavage. However, no changes in BCL-2 and Bax were observed in HCT116 and SW480 cells that were treated with either the individual drugs or the drug combination (Figure 6E).
Figure 6.

The Combination of TG-101348 with SGI-1776 Is Synergistic in CRC
(A) Percentage of growth inhibition of single-agent TG-101348 or SGI-1776 and combination treatment in HCT116 and SW480, as indicated by heatmaps (left). Synergism of growth inhibition by combined treatment with TG-101348 and SGI-1776 in HCT116 and SW480 cells, as analyzed by an isobologram analysis (right). (B) A colony formation assay was performed with HCT116 and SW480 cells treated with DMSO, 1 μM TG-101348, 2.5 μM SGI-1776, and the combination treatment. (C) Bar plots indicate the number of colonies. (D) Caspase-3/7 enzymatic activity was measured using an Apo-ONE homogeneous caspase-3/7 assay in HCT116 and SW480 cells treated with DMSO, 1 μM TG-101348, 2.5 μM SGI-1776, and the combination treatment. (E) Representative western blot analysis of cleaved caspase-3, cleaved PARP, BCL-2, and Bax protein expression in HCT116 and SW480 cells treated with DMSO, 1 μM TG-101348, 2.5 μM SGI-1776, and the combination treatment. GAPDH was used as a protein loading control. (F) Tumor volumes over 29 days in an orthotopic model of HCT116 cells treated with DMSO, TG-101348, SGI-1776, or the combination treatment. Representative images are of tumor tissues that were recovered from each group. (G) Representative micrographs of Ki67 expression in tumors treated with DMSO, TG-101348, SGI-1776, or the combination treatment. Scale bars, 100 μm. (H) Scatter dot plots indicate the percentage of Ki67 positivity in tumors from each group. Error bars represent the mean ± SEM. ∗∗p < 0.01; ∗∗∗p < 0.001, two-tailed, unpaired t tests.
Moreover, we subsequently examined the in vivo efficacy of combinatorial treatment with TG-101348 and SGI-1776 using a human CRC xenograft mouse model that was generated by using HCT116 cells. The growth delay observed in the groups treated with the inhibitor combination was significantly longer than that observed in the groups treated with each inhibitor alone. TG-101348 (120 mg/kg) and SGI-1776 (75 mg/kg) inhibited tumor growth by ∼58% and ∼48%, respectively, compared with DMSO. Significantly, the combination treatment dramatically inhibited tumor growth by ∼76% (TG-101348/SGI-1776, day 29; p < 0.001) (Figure 6F). In addition to the tumor volume, we also assessed the expression of the proliferation index Ki-67 by IHC analysis and found that the inhibitor combination decreased the ratio of Ki-67 expression compared to the single-agent treatments (Figures 6G and 6H), which confirmed our in vitro findings. Taken together, these findings suggest that the combination of TG-101348 and SGI-1776 is a promising therapeutic strategy for CRC.
Discussion
Despite intensive efforts, a considerable number of CRC patients develop local recurrence and distant metastasis; hence, the exploration of novel therapeutic targets and the screening of related compounds are needed. In this study, we found that enhanced SLP-2 expression, which was also noted elsewhere, is linked to tumor invasion, distant metastasis, and an inferior outcome in patients with CRC.15, 16, 17, 18, 19, 20 By integrating data from functional studies, we showed that SLP-2 promotes CRC cell growth in vitro and in vivo. Several reports have confirmed the oncogenic roles of SLP-2 via different mechanisms in multiple tumors; however, the precise mechanisms of SLP-2 that initiate tumorigenesis and the exploitation of SLP-2 as an actionable target in CRC have yet to be clearly investigated.21,22
In a compound library screen, we observed that HCT116SLP-2KO cells were more resistant to JAK2 and PIM inhibitors than HCT116 cells transfected with EV (px458). Importantly, after further in vitro and in vivo validation, we examined whether SLP-2 was a potential biomarker for CRC sensitivity to the JAK2 inhibitor TG-101348 and the pan-PIM inhibitor SGI-1776. TG-101348 (also known as SAR302503) is a selective JAK2 inhibitor that has been used in an advanced clinical trial in patients with refractory solid tumors as well as in many basic scientific studies of solid tumors with the activation of JAK2/STAT3.23 Additionally, programmed death ligand (PD-L1, CD274), an interferon (IFN)-gamma/JAK2-inducible gene, could be abrogated by TG-101348 in NSCLC.20,24 Here, we reported that the efficacy of TG-101348 was linked to SLP-2 levels and that single-agent TG-101348 treatment also inhibited CRC cells. In findings similar to those in Hodgkin’s lymphoma, we confirmed that tumor growth was delayed by treatment with TG-101348 in CRC mouse xenografts.25 In addition to TG-101348, HCT116SLP-2KO cells were also resistant to another JAK2 inhibitor, NVP-BSK805, but the cell viability was barely affected in the subsequent validation in CRC cells (data not shown). SGI-1776, a small-molecule inhibitor of the PIM1, PIM2, and PIM3 kinases, induced apoptosis in human leukemia/lymphoma, such as mantle cell lymphoma, multiple myeloma, chronic lymphocytic leukemia, and acute myeloid leukemia cell lines; additionally, studies with mouse xenografts and acute myeloid leukemia cell lines showed that this compound had little toxicity to normal human lymphocytes.26, 27, 28, 29 Recently, major progress was reported with SGI-1776 in solid tumors, as SGI-1776 significantly inhibited the xenograft growth of triple-negative breast cancer with high PIM1 and MYC expression but did not inhibit the xenograft growth of receptor-positive breast cancer with low MYC expression and moderate PIM1 expression.30 In the present study, HCT116 cells with SLP-2KO were resistant to SGI-1776, and its efficacy was further evaluated in vitro and in vivo. As treatment with TG-101348 or SGI-1776 alone showed limited efficacy, we investigated a combinatorial therapeutic strategy with these two drugs in CRC. Consequently, pharmacological JAK2 inhibition by TG-101348 synergized with the pan-PIM inhibitor SGI-1776 to promote the killing of CRC cells.
Although SLP-2 promotes tumor cell survival and is highly expressed in CRC, the exact function and mechanisms are unclear. Different signaling pathways, such as mitogen-activated protein kinase (MAPK) and Wnt/β-catenin, were examined to elucidate how SLP-2 controls tumor cell survival.7,8 Phylogenetic analysis showed that SLP-2 was acquired through mitochondrial endosymbiosis and is involved in the regulation of ion channel permeability, mechanoreception, and lipid domain organization.31 In addition, SLP-2 binds to the mitochondrial lipid cardiolipin, suggesting a relationship between SLP-2 and the mitochondrial apoptosis pathway. A recent study reported that SLP-2 affects the mitochondrial apoptosis pathway in cervical cancer cells.23 In this study, compound library screening showed that SLP-2 correlated with the sensitivity of inhibitors targeting JAK2 and PIMs, which also implied that the oncogenic JAK2-STAT-PIM pathway was potentially controlled by SLP-2 in CRC. The data from in vivo assays suggest that SLP-2 regulates JAK2 and subsequently regulates the phosphorylation levels of STAT3, but not STAT5, in CRC cells. Notably, the JAK2-STAT3 signaling pathway mediates tumor growth and invasion, while the inhibition of this pathway induced cell apoptosis via the mitochondrial pathway in CRC cells.32,33 Moreover, the analysis of the targets of the pan-PIM inhibitor SGI-1776 showed that the oncogenic kinase PIM1, but not PIM2 or -3, was dramatically regulated by the JAK2-STAT3 pathway in CRC, which was consistent with the results noted elsewhere.13
In conclusion, screening with compound libraries in cells with or without candidate gene editing is an underutilized strategy to identify biomarkers for tumor sensitivity to targeted agents and to identify rational and synergistic drug-drug combinations. In this article, we identified the synergistic interaction of TG-101348, a selective JAK2 inhibitor, and SGI-1776, a pan-PIM inhibitor that was affected by the SLP-2 level, with this technique. Additionally, our data showed that SLP-2 could serve as a prognostic marker in patients with CRC and that SLP-2 promotes tumor malignant phenotypes via the JAK2-STAT3-PIM1 oncogenic pathway.
Materials and Methods
Patient Samples
After obtaining written informed consent from patients, pathological reports of patients diagnosed between 2004 and 2014 were retrieved from the archives of the Department of Pathology at Jinshan Hospital of Fudan University and Renji Hospital, School of Medicine, Shanghai Jiao Tong University. For IHC assays, 491 formalin-fixed and paraffin-embedded CRC tissues and matched adjacent nontumor tissues, as well as 50 adenomas and 50 HGINs, from patients who underwent surgical resection or biopsy were collected. For quantitative real-time RT-PCR assays, 74 fresh CRC tissues and matched adjacent nontumor tissues were immediately immersed in liquid nitrogen after surgical resection and were then frozen at −80°C until RNA and protein extraction. Patient follow-up and clinicopathological features were retrieved, such as patient age, sex, CEA levels, histological type, tumor differentiation, lymph node and distant metastases, and TNM stage. All samples were used in accordance with the guidelines of the Internal Review and Ethics Boards of the Fudan University Jinshan Hospital and Renji Hospital, School of Medicine, Shanghai Jiao Tong University and were conducted in accordance with the generally accepted guidelines for the use of human materials.
Quantitative Real-Time RT-PCR
Total RNA was extracted from tissues and cultured cells using an RNeasy Mini Kit (QIAGEN, Hilden, Germany) according the standard protocol provided by the manufacturer. cDNA was synthesized via reverse transcription using SuperScrip IV VILO Master Mix (Invitrogen, Carlsbad, CA, USA) according to the manufacturer’s instructions. Then, quantitative real-time RT-PCR was carried out using TaqMan probes (Applied Biosystems, San Diego, CA, USA) that were specific for SLP-2, JAK2, PIM1, PIM2, and PIM3; the values were normalized to the expression of GAPDH (an endogenous control) and were analyzed with the 2−ΔΔCt method.
IHC Analysis
IHC analysis was performed as previously described.5 Briefly, sections were deparaffinized in xylene and hydrated with a series of graded alcohols, and antigen recovery was performed with citrate buffer (pH 6.0) in a microwave oven (95°C–100°C). After blocking with 10% normal goat serum, sections were incubated with an anti-SLP-2 rabbit polyclonal antibody (ProteinTech, Chicago, IL, USA; 1:300) or an anti-Ki-67 mouse monoclonal antibody (Santa Cruz Biotechnology, Dallas, TX, USA; 1:300) at 4°C overnight. Then, sections were incubated with an anti-rabbit secondary antibody (Dako, Glostrup, Denmark) at room temperature for 30 min. The signal was detected using a 3′3-diaminobenzidine (DAB) kit (Dako). Finally, after counterstaining with a hematoxylin solution (Sigma-Aldrich, St. Louis, MO, USA), SLP-2 and Ki-67 immunostaining were reviewed under a light microscope. A negative control was generated by replacing the primary antibody with 0.1% PBS.
Using a scoring system that we previously established, SLP-2 protein expression was independently evaluated and scored by two pathologists who were blinded to the clinicopathological features and patient outcomes.9 The proportion of positive cells and the intensity of SLP-2 immunostaining were assessed. The proportion of positive cells was classified as 0 (0%, no positive staining), 1 (≤25% positive cells), 2 (26%–50% positive cells), 3 (51%–75% positive cells), or 4 (>75% positive cells). The intensity of the immunostaining was scored as 0 (no staining), 1 (weak), 2 (moderate), or 3 (strong). The final staining index (SI) was defined as the sum of the scores of the positivity and immunostaining intensity. The SI of SLP-2 was stratified into 4 levels: −, scores 0–2; +, scores 3–4; ++, scores 5–6; and +++, score of 7. Finally, we defined “−” and “+” as SLP-2 negative and low, respectively; and “++” and “+++” as SLP-2 high.
Cell Lines and Treatments
The CRC cell lines Caco-2, HT-29, LOVO (SIBS, Shanghai, China), SW480, SW620, HCT116, and HCT8 (American Type Culture Collection [ATCC], Manassas, VA, USA) were cultured according to the standard protocols provided by ATCC. All cell lines were tested and authenticated by short tandem repeat (STR) assays (CBTCCCAS, China). NVP-BSK805 (S2686), TG-101348 (S2736), and SGI-1776 (S2198) were purchased from Selleckchem (Houston, TX, USA), and dimethyl sulfoxide (DMSO) (Sigma-Aldrich, St. Louis, MO, USA) was used for pharmacological inhibition in vitro and in vivo.
CRISPR/Cas9 Genome Editing
Three sgRNA sequences were designed to knockout SLP-2, and the sequencing primers used in this study are provided in Table S1. Guide RNA (gRNA) sequences were cloned into the dual Cas9/gRNA expression vector pSpCas9(BB)-2A-GFP (px458, Addgene 48138, Cambridge, MA, USA) according to published protocols.34 After validation by Sanger sequencing, the cloned vectors and EV, which served as a control, were transduced into CRC cells. Single GFP+ cells were sorted into RPMI 1640 with 20% fetal bovine serum (FBS) and were cultured to grow single-cell colonies. Western blotting was used to confirm the deletion of SLP-2 in transfected CRC cells.
Compound Library Screens
A panel of 181 drugs (Selleckchem) was selected for screening the drug sensitivity of CRC cells. The majority of these compounds are currently in clinical trials, and detailed information is provided in Table S2. Compounds (10 mM in DMSO) were diluted to a final concentration of 2.5 μM and were plated in duplicate in a 96-well format. Each cell line was plated at a density of 5,000 cells per well and was incubated at 37°C in a humidified chamber with 5% CO2 for 3 days. Cell viability was assessed using alamarBlue (Invitrogen) according to the manufacturer’s instructions, and measurements were performed on a SpectraMax M5 reader (Molecular Devices, San Jose, CA, USA).
Western Blot Analysis
Total protein lysates were prepared from frozen tissue samples or harvested CRC cells using NP40 lysis buffer (Invitrogen) containing the protease inhibitor phenylmethylsulphonyl fluoride (PMSF; Invitrogen) and a protease inhibitor cocktail (Invitrogen). Western blot analysis was conducted as previously described.35 Following standard protocols, membranes were blotted with anti-SLP-2 (ProteinTech, 1:2,000), JAK2, STAT3, p-STAT3, STAT5, phosphorylated STAT5 (p-STAT5), MMP2, and GAPDH (Cell Signaling Technology, Beverley, MA, USA) at a dilution of 1:1,000 and anti-PIM1 (Novus, Littleton, CO, USA) at a dilution of 1:1,000, followed by horseradish peroxidase (HRP)-conjugated secondary antibodies. Bands were detected by enhanced chemiluminescence (GE Healthcare, Uppsala, Sweden).
Cell Viability and Cell Growth Curves
Cell viability assays were used to determine the IC50 values of different drugs. Briefly, each cell line (5,000 cells per well) was seeded in complete growth media in 96-well plates in triplicate and incubated with drugs for 3 days, alamarBlue (Invitrogen) was added, and analysis was performed as described for the compound screening assays. To generate cell growth curves, CRC cells that underwent genome editing were plated in duplicate in six-well plates at a density of 25,000 cells per well and were counted by using trypan blue (Invitrogen) every other day.
Colony Formation
A colony formation assay was performed as previously described.36 Briefly, each cell line (1 × 104 cells per well) was seeded in six-well plates. The medium was replaced every 3 days. At day 21, the growth medium was removed, and viable colonies were stained with 0.005% crystal violet solution (Sigma-Aldrich). Colonies were counted using Fiji (ImageJ). Experiments were performed in triplicate.
Caspase-3/7 Assay
Each cell line was plated in triplicate at a density of 5,000 cells per well (white, clear-bottomed 96-well plates) and was incubated at 37°C in a humidified chamber with 5% CO2 overnight. The caspase-3/7 enzymatic activities of each cell line were measured using an Apo-ONE homogeneous caspase-3/7 assay (Promega, Madison, WI, USA) according to the manufacturer’s recommendations.
Wound-Healing Assay
A wound-healing assay was performed as previously described.35 Cells were serum starved for 16 h in 2% horse serum with the indicated media without epidermal growth factor (EGF) and were then trypsinized, seeded (15 × 105) in triplicate in six-well plates and cultured overnight. A scratch was generated in the cell layer using a pipette tip. Images were obtained at 0 and 24 h after wounding. The percentage of wound closure was assessed by the reduction in the scratch width and was plotted.
Transwell Invasion Assay
Transwell membranes were pretreated with Matrigel (BD Biosciences, San Jose, CA, USA). Cells were serum starved for 16 h in serum-free medium and were then harvested and resuspended in the indicated media supplemented with 1% FBS at a density of 3 × 105 cells per milliliter. A total of 0.5 mL cells from each group was added to the upper chamber, and medium containing 10% FBS was added to the lower chamber as a chemoattractant. After incubation for 12 h, the surface of the upper chamber was cleared with cotton swabs. Staining solution containing 4% formaldehyde and hematoxylin was applied to fix and stain the migrated cells adhered on the surface of the lower membrane. Cells were counted and photographed at 100× magnification in five randomly chosen fields.
Immunofluorescence
The immunofluorescence analysis of each cell line was performed as previously described.37 Cells were sparsely seeded on slides and cultured overnight and then were fixed and permeabilized in 4% paraformaldehyde (PFA) and 0.5% Triton X-100, respectively. After 1 h of blocking in 10% goat serum, primary antibodies (the same antibodies that were used in western blot assays) were added and incubated overnight at 4°C, and Alexa-Fluor-conjugated secondary antibodies (1:500) were added and incubated for 1 h at room temperature. The slides were mounted with Fluoroshield Mounting Medium with DAPI (Abcam, Cambridge, MA, USA). Confocal analysis was performed with the Leica SP5 DM confocal microscopy system. Experiments were performed in triplicate.
Mouse Studies
All animal experiments with cell line xenografts were performed in accordance with the guidelines approved by the Animal Ethics Boards of Renji Hospital, School of Medicine, Shanghai Jiao Tong University. CRC xenografts were generated as previously described.35 Male 6- to 8-week-old NSG mice were randomly divided into two groups (n = 5 per group). A total of 5 × 106 HCT116SLP-2KO-sg1 or HCT116EV cells in 100 μL PBS mixed with 100 μL Matrigel (BD Biosciences, CA, USA) were subcutaneously injected into both flanks of nude mice. Tumor volume was measured in two dimensions every 3 days, starting at after 9 days of implantation, and was used to calculate the volume as V = ab2/2. At 29 days after tumor implantation, tumors were excised and fixed in 4% neutral PFA solution for morphological and IHC analyses.
To investigate the potential pharmacological inhibition of tumor growth in vivo with a JAK2 inhibitor combined with a PIM1 inhibitor, xenografts were generated as described earlier. When the tumors reached 68–185 mm3, the mice were randomly divided into four groups (n = 5 per group). The mice in each group were treated as follows: with vehicle control, SGI-1776 (75 mg/kg daily via oral gavage), TG-101348 (120 mg/kg twice daily via oral gavage), or both inhibitors for 16 days.24,25,30 In addition to tumor volume measurements, morphological and IHC analyses were also performed. The procedures at the end of the experiment were performed as previously described.11,35 For implantation, the mice were anesthetized with an intraperitoneal (i.p.) injection of ketamine/xylazine (87/13 mg/mL) at 1 mL/kg. Finally, all mice were euthanized by cervical dislocation while under anesthesia.
Statistical Analysis
Statistical analyses were performed using GraphPad Prism v.7.0 (GraphPad Software, La Jolla, CA, USA). The data are presented as the mean ± the standard error of the mean (SEM) and were analyzed using unpaired two-tailed Student’s t tests or one-way analysis of variance (ANOVA). The correlations of SLP-2 expression with clinicopathological characteristics were analyzed by using Spearman’s correlation analysis. Kaplan-Meier survival curves were plotted, and a log-rank test was performed. For drug-synergy assessments, the CI was calculated using CompuSyn v.1.0 (ComboSyn, Paramus, NJ, USA). Two-sided p values < 0.05 were considered significant, and a CI < 1.0 was considered to indicate synergism.
Author Contributions
All authors participated in the design and interpretation of the experiments and the data analysis. Q.L., A.L., and L.W. conducted the experiments. A.L. and X.X. contributed to the generation of figures and wrote the manuscript, and Z.L. contributed to data analysis and the revision of the manuscript. W.H., L.Z., X.S., and X.Y. collected the clinical information from the CRC patients and performed IHC assays. C.W. and Z.L. were responsible for cell culture and gene editing assays with CRISPR/Cas9. H.Z. and S.L. designed and conducted the animal assays. All authors read and approved the final manuscript.
Conflicts of Interest
The authors declare no competing interests.
Acknowledgments
We acknowledge Dr. Tai (Department of Pathology, Memorial Sloan Kettering Cancer Center, New York, NY, USA) for providing technical advice about gene editing with CRISPR/Cas9. We acknowledge Dr. Somwar (Human Oncology and Pathogenesis Program, Memorial Sloan Kettering Cancer Center, New York, NY, USA) for his valuable assistance with compound library screening. This work was supported by grants from the Natural Science Foundations of Shanghai, China (nos. 17ZR1416900 and 19ZR1476600) and the Shanghai Municipal Health Commission (no. 2016JQ003).
Footnotes
Supplemental Information can be found online at https://doi.org/10.1016/j.omto.2020.03.010.
Contributor Information
Xiuying Xiao, Email: xiaoxiuying2002@163.com.
Zebing Liu, Email: zebing1311@yahoo.com.
Supplemental Information
References
- 1.Sveen A., Kopetz S., Lothe R.A. Biomarker-guided therapy for colorectal cancer: strength in complexity. Nat. Rev. Clin. Oncol. 2020;17:11–32. doi: 10.1038/s41571-019-0241-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Wang Y., Morrow J.S. Identification and characterization of human SLP-2, a novel homologue of stomatin (band 7.2b) present in erythrocytes and other tissues. J. Biol. Chem. 2000;275:8062–8071. doi: 10.1074/jbc.275.11.8062. [DOI] [PubMed] [Google Scholar]
- 3.Christie D.A., Lemke C.D., Elias I.M., Chau L.A., Kirchhof M.G., Li B., Ball E.H., Dunn S.D., Hatch G.M., Madrenas J. Stomatin-like protein 2 binds cardiolipin and regulates mitochondrial biogenesis and function. Mol. Cell. Biol. 2011;31:3845–3856. doi: 10.1128/MCB.05393-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hájek P., Chomyn A., Attardi G. Identification of a novel mitochondrial complex containing mitofusin 2 and stomatin-like protein 2. J. Biol. Chem. 2007;282:5670–5681. doi: 10.1074/jbc.M608168200. [DOI] [PubMed] [Google Scholar]
- 5.Zhou C., Li Y., Wang G., Niu W., Zhang J., Wang G., Zhao Q., Fan L. Enhanced SLP-2 promotes invasion and metastasis by regulating Wnt/β-catenin signal pathway in colorectal cancer and predicts poor prognosis. Pathol. Res. Pract. 2019;215:57–67. doi: 10.1016/j.prp.2018.10.018. [DOI] [PubMed] [Google Scholar]
- 6.Qu H., Jiang W., Wang Y., Chen P. STOML2 as a novel prognostic biomarker modulates cell proliferation, motility and chemo-sensitivity via IL6-Stat3 pathway in head and neck squamous cell carcinoma. Am. J. Transl. Res. 2019;11:683–695. [PMC free article] [PubMed] [Google Scholar]
- 7.Yang C.T., Li J.M., Li L.F., Ko Y.S., Chen J.T. Stomatin-like protein 2 regulates survivin expression in non-small cell lung cancer cells through β-catenin signaling pathway. Cell Death Dis. 2018;9:425. doi: 10.1038/s41419-018-0461-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ma W., Xu Z., Wang Y., Li W., Wei Z., Chen T., Mou T., Cheng M., Luo J., Luo T. A positive feedback loop of slp2 activates mapk signaling pathway to promote gastric cancer progression. Theranostics. 2018;8:5744–5757. doi: 10.7150/thno.28898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Liu Z., Yang Y., Zhang Y., Ye X., Wang L., Xu G. Stomatin-like protein 2 is associated with the clinicopathological features of human papillary thyroid cancer and is regulated by TGF-β in thyroid cancer cells. Oncol. Rep. 2014;31:153–160. doi: 10.3892/or.2013.2833. [DOI] [PubMed] [Google Scholar]
- 10.Song L., Liu L., Wu Z., Lin C., Dai T., Yu C., Wang X., Wu J., Li M., Li J. Knockdown of stomatin-like protein 2 (STOML2) reduces the invasive ability of glioma cells through inhibition of the NF-κB/MMP-9 pathway. J. Pathol. 2012;226:534–543. doi: 10.1002/path.3008. [DOI] [PubMed] [Google Scholar]
- 11.Liu Z., Cai Y., Yang Y., Li A., Bi R., Wang L., Shen X., Wang W., Jia Y., Yu B. Activation of MET signaling by HDAC6 offers a rationale for a novel ricolinostat and crizotinib combinatorial therapeutic strategy in diffuse large B-cell lymphoma. J. Pathol. 2018;246:141–153. doi: 10.1002/path.5108. [DOI] [PubMed] [Google Scholar]
- 12.Zemskova M., Sahakian E., Bashkirova S., Lilly M. The PIM1 kinase is a critical component of a survival pathway activated by docetaxel and promotes survival of docetaxel-treated prostate cancer cells. J. Biol. Chem. 2008;283:20635–20644. doi: 10.1074/jbc.M709479200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Liu K., Gao H., Wang Q., Wang L., Zhang B., Han Z., Chen X., Han M., Gao M. Hispidulin suppresses cell growth and metastasis by targeting PIM1 through JAK2/STAT3 signaling in colorectal cancer. Cancer Sci. 2018;109:1369–1381. doi: 10.1111/cas.13575. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 14.Bellon M., Lu L., Nicot C. Constitutive activation of Pim1 kinase is a therapeutic target for adult T-cell leukemia. Blood. 2016;127:2439–2450. doi: 10.1182/blood-2015-11-685032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zhou T., Georgeon S., Moser R., Moore D.J., Caflisch A., Hantschel O. Specificity and mechanism-of-action of the JAK2 tyrosine kinase inhibitors ruxolitinib and SAR302503 (TG101348) Leukemia. 2014;28:404–407. doi: 10.1038/leu.2013.205. [DOI] [PubMed] [Google Scholar]
- 16.Xia Z., Knaak C., Ma J., Beharry Z.M., McInnes C., Wang W., Kraft A.S., Smith C.D. Synthesis and evaluation of novel inhibitors of Pim-1 and Pim-2 protein kinases. J. Med. Chem. 2009;52:74–86. doi: 10.1021/jm800937p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Anizon F., Shtil A.A., Danilenko V.N., Moreau P. Fighting tumor cell survival: advances in the design and evaluation of Pim inhibitors. Curr. Med. Chem. 2010;17:4114–4133. doi: 10.2174/092986710793348554. [DOI] [PubMed] [Google Scholar]
- 18.Brault L., Gasser C., Bracher F., Huber K., Knapp S., Schwaller J. PIM serine/threonine kinases in the pathogenesis and therapy of hematologic malignancies and solid cancers. Haematologica. 2010;95:1004–1015. doi: 10.3324/haematol.2009.017079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Chang M., Kanwar N., Feng E., Siu A., Liu X., Ma D., Jongstra J. PIM kinase inhibitors downregulate STAT3(Tyr705) phosphorylation. Mol. Cancer Ther. 2010;9:2478–2487. doi: 10.1158/1535-7163.MCT-10-0321. [DOI] [PubMed] [Google Scholar]
- 20.Ikeda S., Okamoto T., Okano S., Umemoto Y., Tagawa T., Morodomi Y., Kohno M., Shimamatsu S., Kitahara H., Suzuki Y. PD-L1 is upregulated by simultaneous amplification of the PD-L1 and JAK2 genes in non-small cell lung cancer. J. Thorac. Oncol. 2016;11:62–71. doi: 10.1016/j.jtho.2015.09.010. [DOI] [PubMed] [Google Scholar]
- 21.Liu D., Zhang L., Shen Z., Tan F., Hu Y., Yu J., Li G. Increased levels of SLP-2 correlate with poor prognosis in gastric cancer. Gastric Cancer. 2013;16:498–504. doi: 10.1007/s10120-013-0232-3. [DOI] [PubMed] [Google Scholar]
- 22.Cao W., Zhang B., Liu Y., Li H., Zhang S., Fu L., Niu Y., Ning L., Cao X., Liu Z., Sun B. High-level SLP-2 expression and HER-2/neu protein expression are associated with decreased breast cancer patient survival. Am. J. Clin. Pathol. 2007;128:430–436. doi: 10.1309/C6X54HRB580EP2NQ. [DOI] [PubMed] [Google Scholar]
- 23.Kim B., Kim H.S., Kim S., Haegeman G., Tsang B.K., Dhanasekaran D.N., Song Y.S. Adipose stromal cells from visceral and subcutaneous fat facilitate migration of ovarian cancer cells via IL-6/JAK2/STAT3 pathway. Cancer Res. Treat. 2017;49:338–349. doi: 10.4143/crt.2016.175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Pitroda S.P., Stack M.E., Liu G.F., Song S.S., Chen L., Liang H., Parekh A.D., Huang X., Roach P., Posner M.C. JAK2 inhibitor SAR302503 abrogates PD-L1 expression and targets therapy-resistant non-small cell lung cancers. Mol. Cancer Ther. 2018;17:732–739. doi: 10.1158/1535-7163.MCT-17-0667. [DOI] [PubMed] [Google Scholar]
- 25.Hao Y., Chapuy B., Monti S., Sun H.H., Rodig S.J., Shipp M.A. Selective JAK2 inhibition specifically decreases Hodgkin lymphoma and mediastinal large B-cell lymphoma growth in vitro and in vivo. Clin. Cancer Res. 2014;20:2674–2683. doi: 10.1158/1078-0432.CCR-13-3007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Yang Q., Chen L.S., Neelapu S.S., Miranda R.N., Medeiros L.J., Gandhi V. Transcription and translation are primary targets of Pim kinase inhibitor SGI-1776 in mantle cell lymphoma. Blood. 2012;120:3491–3500. doi: 10.1182/blood-2012-02-412643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Cervantes-Gomez F., Chen L.S., Orlowski R.Z., Gandhi V. Biological effects of the Pim kinase inhibitor, SGI-1776, in multiple myeloma. Clin. Lymphoma Myeloma Leuk. 2013;13(Suppl 2):S317–S329. doi: 10.1016/j.clml.2013.05.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Chen L.S., Redkar S., Bearss D., Wierda W.G., Gandhi V. Pim kinase inhibitor, SGI-1776, induces apoptosis in chronic lymphocytic leukemia cells. Blood. 2009;114:4150–4157. doi: 10.1182/blood-2009-03-212852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Chen L.S., Redkar S., Taverna P., Cortes J.E., Gandhi V. Mechanisms of cytotoxicity to Pim kinase inhibitor, SGI-1776, in acute myeloid leukemia. Blood. 2011;118:693–702. doi: 10.1182/blood-2010-12-323022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Horiuchi D., Camarda R., Zhou A.Y., Yau C., Momcilovic O., Balakrishnan S., Corella A.N., Eyob H., Kessenbrock K., Lawson D.A. PIM1 kinase inhibition as a targeted therapy against triple-negative breast tumors with elevated MYC expression. Nat. Med. 2016;22:1321–1329. doi: 10.1038/nm.4213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zanon A., Kalvakuri S., Rakovic A., Foco L., Guida M., Schwienbacher C., Serafin A., Rudolph F., Trilck M., Grünewald A. SLP-2 interacts with Parkin in mitochondria and prevents mitochondrial dysfunction in Parkin-deficient human iPSC-derived neurons and Drosophila. Hum. Mol. Genet. 2017;26:2412–2425. doi: 10.1093/hmg/ddx132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Schulz-Heddergott R., Stark N., Edmunds S.J., Li J., Conradi L.C., Bohnenberger H., Ceteci F., Greten F.R., Dobbelstein M., Moll U.M. Therapeutic ablation of gain-of-function mutant p53 in colorectal cancer inhibits stat3-mediated tumor growth and invasion. Cancer Cell. 2018;34:298–314.e7. doi: 10.1016/j.ccell.2018.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Du W., Hong J., Wang Y.C., Zhang Y.J., Wang P., Su W.Y., Lin Y.W., Lu R., Zou W.P., Xiong H., Fang J.Y. Inhibition of JAK2/STAT3 signalling induces colorectal cancer cell apoptosis via mitochondrial pathway. J. Cell. Mol. Med. 2012;16:1878–1888. doi: 10.1111/j.1582-4934.2011.01483.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ran F.A., Hsu P.D., Wright J., Agarwala V., Scott D.A., Zhang F. Genome engineering using the CRISPR-Cas9 system. Nat. Protoc. 2013;8:2281–2308. doi: 10.1038/nprot.2013.143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Liu Z., Wei P., Yang Y., Cui W., Cao B., Tan C., Yu B., Bi R., Xia K., Chen W. BATF2 deficiency promotes progression in human colorectal cancer via activation of HGF/MET signaling: a potential rationale for combining MET inhibitors with IFNs. Clin. Cancer Res. 2015;21:1752–1763. doi: 10.1158/1078-0432.CCR-14-1564. [DOI] [PubMed] [Google Scholar]
- 36.Li A., Liu Z., Li M., Zhou S., Xu Y., Xiao Y., Yang W. HDAC5, a potential therapeutic target and prognostic biomarker, promotes proliferation, invasion and migration in human breast cancer. Oncotarget. 2016;7:37966–37978. doi: 10.18632/oncotarget.9274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Geyer F.C., Li A., Papanastasiou A.D., Smith A., Selenica P., Burke K.A., Edelweiss M., Wen H.C., Piscuoglio S., Schultheis A.M. Recurrent hotspot mutations in HRAS Q61 and PI3K-AKT pathway genes as drivers of breast adenomyoepitheliomas. Nat. Commun. 2018;9:1816. doi: 10.1038/s41467-018-04128-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.